

Abstract
Animals harbor diverse communities of microbes within their gastrointestinal tracts. Phylogenetic relationship, diet, gut morphology, host physiology, and ecology all influence microbiome composition within and between animal clades. Emerging evidence points to host genetics as also playing a role in determining gut microbial composition within species. Here, we discuss recent advances in the study of microbiome heritability across a variety of animal species. Candidate gene and discovery-based studies in humans, mice, Drosophila, Caenorhabditis elegans, cattle, swine, poultry, and baboons reveal trends in the types of microbes that are heritable and the host genes and pathways involved in shaping the microbiome. Heritable gut microbes within a host species tend to be phylogenetically restricted. Host genetic variation in immune- and growth-related genes drives the abundances of these heritable bacteria within the gut. With only a small slice of the metazoan branch of the tree of life explored to date, this is an area rife with opportunities to shed light into the mechanisms governing host–microbe relationships.
Keywords: microbiome, heritability, genome-wide association study, GWAS, mouse, Drosophila, Caenorhabditis elegans, cattle, poultry, agriculture, model organism
INTRODUCTION
Animals harbor ecosystems of microbes on and within their bodies, from bacteria and archaea to protists and fungi, collectively referred to as the “microbiome.” Microbiome research has bloomed in the past 15 years owing to technological advances and growing interest in its association with health and disease. Collaborative efforts like the Earth Microbiome Project and the Human Microbiome Project facilitate exploration of the microbiome (1, 2). Although humans have emerged as the most well-studied animal host, the microbiomes of myriad animals have been characterized to broadly understand the impact of the microbiome on animal health, behavior, and evolutionary history (3–5).
Animal microbiomes are as individualized as their hosts. The total number of resident microbes varies across body sites and species. In animal guts specifically, estimates of microbial counts range from 1013 bacteria in humans, to 1011 microbes per gram in birds, to 107–109 microbes in insects (6–8). Composition varies significantly even within animal classes. Some insect microbiomes consist of only a few crucial gut symbionts, whereas others have diverse microbiomes. Similarly, bird microbiomes differ depending on flight traits (9, 10).
A variety of factors are responsible for the variation in microbiome composition both between and within host species. Host phylogeny significantly shapes the microbiome at broad scales. Trends in core microbiota tend to be restricted within host phylogenetic clades, a finding observed both within mammals and broadly across the animal kingdom (11–13). For some animals, carrying endosymbionts is imperative for their survival. For example, bobtail squid depend on Vibrio fischeri to assist in camouflaging from predators, and corals rely on their endosymbionts to facilitate adaptation to rapidly changing waters (14, 15). Other animals, like Drosophila melanogaster, birds, and bats, show little evidence of a core microbiome, suggesting less reliance on commensal microbes (16–18). Many animals reside in the middle—a core microbiome exists that may provide a fitness benefit but is influenced by dietary variation (12). For example, in mammals, Firmicutes and Bacteroidetes comprise the majority of bacterial abundance in the gut, and variations in composition mirror major dietary patterns (11, 19). Within an individual animal species, similar factors shape interindividual microbiome variation. Diet, social groups, and environmental exposures all determine what microbes colonize within a host, as well as their abundance (20–22).
One emerging factor of interest is the role of host genetics in determining microbiome composition. Host genetics determines a variety of intrinsic host factors that can influence microbial composition in the gut, including immunity, metabolism, and morphology. Identification of host genes and pathways associated with particular components of the microbiome offers a window into the physiological mechanisms important for host–microbiome interactions. Additionally, host genetic control of commensal microbes opens up opportunities for evolutionary pressures to work on the host and microbiome in concert.
Here, we discuss the role of genetic variation in determining animal microbiome composition. We focus on gut microbiomes specifically, as the gut is the most well-studied body site across animals. We discuss methodologies for studying genetics of microbiomes in different systems; present recent advances in human, laboratory, agricultural, and wild animal microbiome genetic research; and highlight host processes found to play similar roles across host species. Finally, we outline promising areas of future study for furthering our understanding of the complex relationships between host genetics, the microbiome, and animal health.
STUDYING GENETICS OF THE MICROBIOME
How to Study the Microbiome
Despite the many ways to characterize microbiomes, the two most common approaches in animal studies include either sequencing a phylogenetic marker gene to examine microbial composition or sequencing all genomic content to analyze potential functional capacity. The first step for both approaches is to extract total DNA from samples—often fecal, cecal, or luminal digesta specimens or aspirates, swabs, and biopsies of tissue.
The most common and cost-effective approach is to sequence a phylogenetic marker gene, most often the 16S ribosomal RNA gene (16S rRNA) from bacteria and archaea. All extant life forms possess genes encoding ribosomal RNA, which consist of both conserved and hypervariable regions intrinsic to their structural features that help assemble the ribosome (23). As such, these conserved regions provide locations for primer binding, and by PCR (polymerase chain reaction) amplification, primers flanking one or more hypervariable regions are used to amplify this intervening region. These amplicons can then be sequenced to high depth using next-generation sequencing platforms for subsequent microbiome analysis.
Although this cost-effective targeted approach paints a rich picture of the taxonomic composition of the microbiome, it has limitations and biases. First, short-read sequencing platforms, such as the commonly used Illumina MiSeq, can sequence only one or two hypervariable regions rather than the full 16S rRNA gene. This limits phylogenetic resolution such that classification of sequences below the genus level cannot be made with high confidence. Second, primers targeting each conserved region have different taxonomic biases (24). As a result, comparing findings from different regions is challenging. Finally, phylogenetic approaches are limited to assessing taxonomic composition only, rather than functional potential. Even with these concerns, 16S amplicon sequencing is a powerful and cost-effective method for assessing taxonomic composition of complex microbiomes in a generally unbiased manner.
Metagenomic sequencing overcomes many of these limitations. With this approach, all DNA in a sample is fragmented, and libraries of these fragments per se, rather than PCR amplicons, are sequenced. The reads are either profiled or assembled to reveal the taxonomic composition and functional potential of the microbiome (25). Although a powerful approach, metagenomic sequencing is far more costly than amplicon sequencing. It can be especially challenging in samples containing high amounts of host DNA. Although many animal microbiome studies rely on 16S data, metagenomic studies shedding light on the functional potential of animal microbiomes are emerging (13, 26).
Once sequence data are generated, various methods are applied to analyze the microbiome. For the 16S-based or other gene amplicon approaches, sequences are either clustered into operational taxonomic units or collapsed into amplicon sequence variants as a proxy for species to assess microbial abundance. For metagenomic sequencing, reads are either profiled directly or assembled into genomes (so-called MAGs—metagenome-assembled genomes) to assign taxonomy or function. In genetic studies of the microbiome, typically the diversity of the microbiome or relative abundances of certain taxa or functional units are treated as the trait of interest. Although these are the current general workflows for microbiome analysis (25, 27), methods are advancing constantly as researchers discover new ways of characterizing microbiomes.
Genetic Approaches
The effect of host genetics on the microbiome is typically examined using three methods: estimating microbial heritability, identifying a relationship between a candidate gene and the microbiome, or applying genome-wide approaches to identify associations between host genetic variation and the microbiome.
Heritability is the degree to which phenotypic variation in a trait within a population is due to host genetic variation, as opposed to environmental influences. In microbiome studies, these traits are usually either the abundances of individual microbes or pathways or microbiome diversity (Figure 1). In animals, heritability is often estimated by comparing the mid-parental value of a trait to the observed trait in the offspring. The more tightly correlated these values are, the more heritable a trait is. One challenge with this approach is discerning between host genetic and maternal effects, as distinguishing between microbes that have been transmitted vertically or selected for genetically can be difficult (28, 29).
Figure 1.
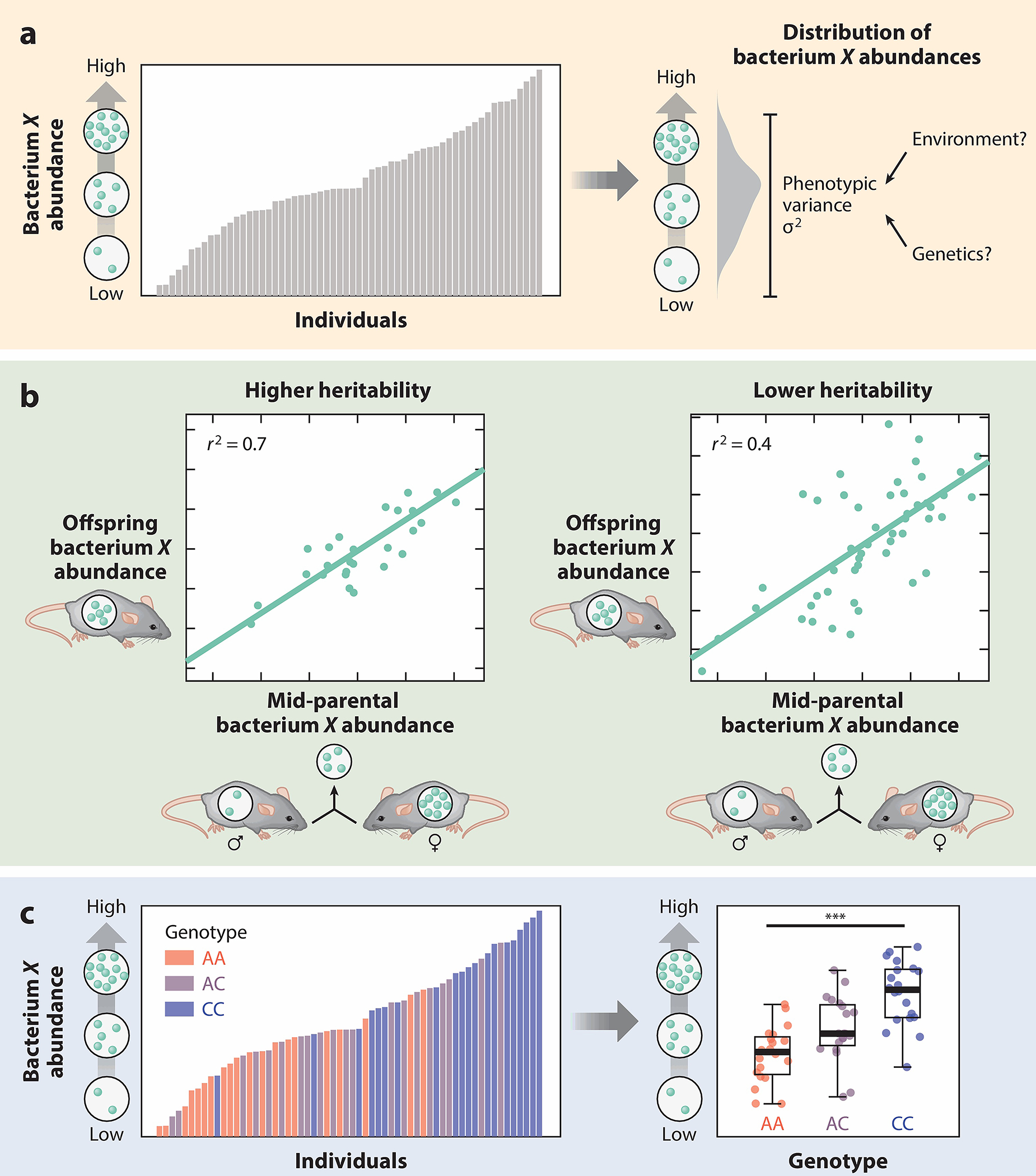
Methodology for studying genetic effects on the microbiome. (a) The abundances of individual taxa in the gut vary across individuals in a population (left). A major open question in microbiome research is to what extent environmental versus genetic factors determine the variability in microbial abundance in a population (right). (b) Heritability of a trait in animal studies is often calculated by comparing offspring trait values (microbial abundance) to the mid-parental traits (the average microbial abundance of the offspring’s parents). Highly heritable microbes show a tighter correlation between offspring and mid-parental abundance measurements (left) compared to less-heritable microbes (right). (c) Quantitative trait locus (QTL) mapping identifies host genetic variation associated with microbial abundance. A significantly associated QTL is identified if bacterial abundance stratifies by genotype class.
Human studies circumvent this issue by calculating heritability from monozygotic (MZ) and dizygotic (DZ) twin pairs (30–32). Because MZ twin pairs have identical genomes, any differences in a trait between twins can be attributed to environmental factors. In contrast, DZ twin pairs share only 50% of their genetic material. By comparing similarities in traits across pairs of MZ twins compared to DZ twins, an estimate of the genetic effects on that trait can be calculated. Because the rates of microbial vertical transmission are not expected to differ between MZ and DZ twin pairs, differences in similarity can be attributed to genetic factors alone. Importantly, heritability is a population-level statistic that can change depending on the amounts of environmental and genetic variation between populations.
A candidate gene approach involves identifying an association between a particular gene of interest and microbes. For example, a candidate gene approach identified the role of MEFV, the gene responsible for familial Mediterranean fever in humans, in determining microbiome composition (33). Although this approach is useful for providing insight into pathways that may contribute to shaping the microbiome, it has limitations. Prior knowledge of potentially important genes or pathways in host–microbe interactions is imperative for a candidate gene approach, limiting discovery opportunities. Additionally, candidate gene studies may not be able to properly characterize the genetic architecture that underlies microbial traits. In many ways, the microbiome can be considered a complex trait. The underlying genetic architecture is likely polygenic, with many genes collectively influencing microbial abundance. Limiting the scope of study to only one gene may explain very little overall trait variation (34).
Genome-wide approaches, like linkage analysis and genome-wide association studies (GWAS), allow for the identification of genetic variation across the entire genome that associates with components of the microbiome. Linkage analysis involves analyzing chromosomal inheritance based on the likelihood that genetic loci are linked. This method is commonly used in animal genetic studies, because crossing two inbred strains allows for the controlled investigation of genetic impact with minimal environmental influence (35). Linkage intervals are often large and contain many genes, and further investigation with fine-mapping approaches is necessary to identify specific causal loci. GWAS, in contrast, explore associations between specific genetic variants and phenotypic traits in large populations of unrelated individuals. Historically, GWAS have been valuable for uncovering the genetic basis for myriad human diseases and traits valuable for agricultural animals (36–38). Recently, they have been used to identify host genetic variants associated with specific microbes (Table 1). Although easier to implement than linkage studies, a major concern of this method is statistical power. Large sample sizes are required to detect significant associations after a multiple-testing penalty is applied to correct for the high numbers of SNPs (single-nucleotide polymorphisms) and taxa combinations typically examined.
Table 1.
Genetic studies of the microbiome in non-human animals
| Type | Animal | Microbiome sample type | Sample size | Study Method | Main Findings | Reference |
|---|---|---|---|---|---|---|
| Mouse | Cecum | 447 | Association analysis between specific microbes and SNPs | Microbiome plasticity to diet is individualized based on host genetics | (60) | |
| Cecum | 599 | GWAS, heritability estimates | Highly heritable (>0.5) taxa (Rikenellaceae, Lachnospiraceae, Clostridiaceae, Oscillospira, Erysipelotrichaceae, Akkermansia) are associated with genes for fat mass, adipose tissue, triglyceride levels, and immunity | (142) | ||
| Feces | 815 | QTL mapping | Taxa (ie. Lactobacillus) are significantly associated with immune-related QTL (IL-22 Irak3), some of which have pleiotropic effects | (56) | ||
| Feces | 34 strains | Linkage analysis | Taxa are significantly associated with immune-related QTL (ie. Irak4, Tgfb3) | (143) | ||
| Feces | 445 | Mediation analysis | 17 SNPs on 7 loci mediate anxiety via numerous taxa (Ruminococcaceae, Clostridiales, Bacteriodaceae) | (133) | ||
| Feces | 64 | 8 strains of CC mice; PERMANOVA; heritability estimates | Diet and genotype together shape the microbiome | (59) | ||
| Feces | 472 | Linkage analysis | Pleiotropic effects of QTL on taxa | (54) | ||
| Feces | 120 | GWAS | Specific taxa (ie. Lactobacillus) are associated with immune-related genes (ie. MHC, Ctnna3, IGF2BP2, Prox1). | (144) | ||
| C. elegans | worm digest | 12–36 worms/strain | 8 strains; PERMANOVA | Changes in immune signaling, especially in the DAF-2 pathway, causes shifts in microbiome composition | (75) | |
| whole body | 3800 | GWAS | Specific taxa (ie. Ochrobactrum) are associated with immune signaling genes (ie. daf-2). | (77) | ||
| Drosophila | Whole body | 515 | GWAS | Taxa are significantly associated with neural genes (ie. dnc, para, Cnx14D) | (69) | |
| Whole body | 36 | Linkage analysis; heritability estimates | Genes involved with growth and development are associated with taxa | (68) | ||
| Agricultural | Cattle | Rumen | 40 | GWAS | Taxa are significantly associated with numerous metabolic-related QTL, some of which have pleiotropic effects | (95) |
| Feces | 228 calves | Linkage analysis (clustering to minimize maternal effect) | Specific taxa (ie. Clostridium, Rikenellaceae) are associated with mucin genes | (99) | ||
| Feces | 278 calves | GWAS; heritability estimates | Oscillospira and Sutterella are highly heritable. Taxa are associated with metabolic (PFKM) and immune genes (CTNNAL1, EPB41L4B). | (98) | ||
| Rumen | 586 | GWAS, heritability estimates | Taxa are significantly associated with immune-related QTL (ie. AKIRIN2) | (125) | ||
| Rumen | 709 | GWAS; heritability estimations | Succinivibrionaceae, Ruminococcus, Clostridiales, and Christensenellaceae are heritable. Specific taxa are significantly associated with growth-related QTL (ie. CDC8, MYH3, RAPH1). | (91) | ||
| Rumen | 691 | GWAS; heritability estimates | Methanogens and Paludibacter are heritable. | (97) | ||
| Rumen | 750 | Heritability estimates | Methanogens are heritable | (94) | ||
| Rumen | 1016 | Association analysis between SNPs and microbes; heritability estimates | 39 core heritable microbes (ie. Ruminococcus, Fibrobacter, Succinovibrionaceae) – 10x more heritable species than humans | (96) | ||
| Rumen | 47 | Heritability estimates | 22 heritable taxa | (100) | ||
| Rumen | 50 live, 68 slaughtered | Regression analysis of sire progeny (maternal effect assumed to be included in residual effects) | Archaea:bacteria ratio correlates with methane emissions, suggesting additive host influence. | (93) | ||
| Swine | Feces | 288 | GWAS | Specific taxa (ie. Akkermansia) are significantly associated with immune and metabolic-related QTL (ie. CHGA) | (123) | |
| Feces | 1205–1283 per timepoint | GWAS; heritability estimates | Specific taxa (ie. Succinivibrionaceae) are associated with growth-related QTL. Succinivibrionaceae is heritable. | (103) | ||
| Colon digesta | 207 | Heritability estimates; G-BLUP and M-BLUP | Several taxa (ie. Lactobacillus) are heritable. M-BLUP is more effective at predicting traits than G-BLUP. | (102) | ||
| Feces | 514 | GWAS; heritability estimates | Specific gut eukaryotes (ie. Blastyocystis, Trichomitus, Neobalantidium, Kazachstania) are significantly associated with numerous immune genes (ie. IL23R, PIK3C3, HNF4A) | (145) | ||
| Feces | 405 | GWAS | Immune genes (ie. PRDM15, STAT1) are key regulators for multiple microbes, suggesting pleiotropic effects | (104) | ||
| Feces | 1183 | Mediation analysis to identify genes that influence fat deposition via the microbiome | Disease-related genetic variants (ie. AK4, SLC43A2, PCK2) are associated with fat traits via microbiome mediation | (134) | ||
| Poultry | Cecum | 144 | Genetic crosses; Linkage analysis; heritability estimates | Clostridium leptum is significantly associated with immune-related genes (C10ORF88) | (124) | |
| Feces | 511 | Genetic crosses of two lines; GWAS | Specific taxa (ie. Methanobacterium) are significantly associated with growth-related QTL. | (111) | ||
| Feces | 258 | Heritability estimates | Selection for body weight traits alters the genetic background, which shapes the microbiome | (110) | ||
| Feces, mucosa | 206 | Association analysis between genetic relatedness and microbes | Correlation between genes and microbes is close to zero; only a few rare microbes are heritable | (113) | ||
| Wild | Wild mouse | Feces, cecum | 50 | GWAS | Several taxa (ie. Odoribacter, Bacteroides) are associated with immune genes (IL12a) | (146) |
| Baboon | Feces | 585 | Heritability estimates | Most taxa are at least slightly heritable. Longitudinal design and large sample sizes are imperative for reliable heritability estimates. | (L. Grieneisen, M. Dasari, T.J Gould, J.R. Björk, J-C. Grenier, et al., personal communcation) | |
| Coral | Tissue | 100 | 9 colonies; PERMANOVA; Redundancy analysis | Microbes are host-specific and not influenced by environmental stressors like algae, increased carbon dioxide, warmer temperatures, and reduced salinity. | (138) | |
| Sponge | Tissue | 40 | Genotyping of two populations; PERMANOVA | Host genotype shapes overall microbiome composition, but does not have significant impacts on core taxa. | (147) | |
| Stickleback | Gut | 182 | 10 populations; PERMANOVA | More genetically divergent populations will have more divergent microbiomes. | (148) | |
| Gut | 126 | 6 populations; PERMANOVA | Genetically different populations have microbiomes that differ in composition and diversity | (149) |
GENETIC STUDIES OF THE MICROBIOME IN HUMANS
One of the first studies to evaluate whether host genetics played a role in determining microbiome composition was performed in humans (30). Microbiome similarities were higher between MZ twin pairs compared to unrelated individuals via denaturing gradient gel electrophoresis of 16S rRNA gene amplicons. Although only a general measure, this pointed to a role for the host genome in determining microbiome abundance in humans. Further studies in a large twin cohort demonstrated which specific microbes are heritable by examining the gut microbiomes of MZ and DZ twins from the TwinsUK data set (31) and determining which host genetic variants may play a role in that process (32). Specifically, the most heritable taxon identified was the family Christensenellaceae. This taxon co-occurs with other heritable taxa, such as methanogenic archaea, and is protective against obesity phenotypes in humans and mice. Similar analyses of data sets from Korean and Canadian cohorts replicate heritability estimates for Christensenellaceae and methanogenic archaea (39, 40).
With microbiome heritability established, the search commenced for the host genetic variants responsible. GWAS have now been performed in several populations, including those in North America, Europe, and the Middle East (32, 40–47). Several themes, reviewed extensively elsewhere (48–51), have emerged about what types of host genes and processes are associated with the human microbiome.
First, the only well-replicated association to date is between the genetic variant responsible for lactase persistence and the abundance of Bifidobacteria in the gut (32, 41, 47, 52). This association occurs only in individuals who report consuming dairy products, an example of a gene-by-environment interaction involving the microbiome.
Second, other diet- and metabolism-related variants have been implicated. For example, genetic variation in the vitamin D receptor is associated with microbiome composition (44). Several genes responsible for taste are associated with components of the microbiome, including ORA6A2, which is responsible for the soapy taste of cilantro some people experience, and CD36, which is responsible for tasting long-chain fatty acids on the tongue (32).
Finally, immune genes have been consistently found to play a role. Studies of the Human Microbiome Project and TwinsUK data sets have found that immune pathways are enriched in genes significantly associated with gut microbes, with the TwinsUK study specifically noting that the relative abundance of Akkermansia is associated with genetically predicted gene expression of the immune suppressor gene SIGLEC15 in the transverse colon (32, 41). Whereas some of these associations may be specific to humans, such as the association of Bifidobacterium abundance with lactase persistence, similar host physiological processes have been identified in genetic studies of the microbiome across other animals as well.
GENETIC STUDIES OF THE MICROBIOME IN LAB ANIMALS
Studies of organisms reared in a research lab control for environmental exposures, allowing for the genetic effects on the microbiome to be more apparent. Mice, zebrafish, Drosophila, and bobtail squid are feasible model systems for studying host–microbiome interactions at both simple and complex levels (53). A major advantage of microbiome research in these study systems is the already-extensive laboratory infrastructure developed to study host genomes. In addition, it is feasible to maintain a high sample size, maximizing power to identify genome–microbiome associations.
Mice
The mouse is the most well-studied model organism with regard to the effect of the host genome on the microbiome. Compared to other model organisms, such as Drosophila or Caenorhabditis elegans, mice have complex, diverse gut microbiomes (53). Additionally, they have one of the best-studied genomes and a variety of available resources for conducting genetic studies. As a result, mouse studies have pioneered the exploration of animal genome–microbiome interactions through linkage analysis, candidate gene approaches, and association studies. Host genetic effects have a greater influence on microbiome composition than maternal effects, sex, time, litter, housing cage, and even vendor (54–59). A closer investigation into these associations via GWAS and quantitative trait locus (QTL) mapping reveals that some genes have pleiotropic effects on the microbiome (58, 60). More specifically, Leamy et al. (58) found that QTL associated with body weight and fat have pleiotropic effects on microbial taxa. Furthermore, Benson et al. (60) observed that QTL associate with both closely and distantly related taxa, showing the multifaceted means by which genes control the microbiome.
Despite clear evidence that host genetics influences the murine microbiome, environmental effects dominate. In particular, diet plays a larger role than host genetics in shaping the microbiome (61, 62). When analyzed in conjunction, however, host genotype appears to mediate the diet–microbiome relationship. Interactions between host genotype and dietary fat levels significantly impact microbial abundance (58). A closer investigation reveals that host genotype and dietary fat jointly modulate abundances of particular taxa, including Clostridiaceae and Ruminococcaceae (63). Finally, evidence suggests that host genes impact gut microbiome plasticity in response to diet, but more research is needed to identify the specific genes interacting with diet to shape the microbiome (64).
Drosophila
Whereas mice have diverse commensal gut microbiomes, Drosophila melanogaster have a very different relationship to their microbiota. The microbiota recovered from wild Drosophila tend to have relatively low species richness compared to mammals, with anywhere from a few to 30 species within the gut (65). Laboratory stocks of Drosophila typically have even less diverse microbiomes, often composed of only a few taxa (66). Unlike most mammalian systems, Drosophila also possess a transient microbiome. They do not sustain gut microbial communities without constant exposure to new microbes (67). As a result, Drosophila do not possess a core gut microbiome (16). It is not entirely clear how or why Drosophila maintain low levels of gut microbes, although genetic mechanisms and/or transit time potentially plays a role.
One hypothesis is that Drosophila immune systems are overactive, resulting in the inability for gut microbes to colonize. The microbiomes of immune-compromised flies are shaped primarily by genotype, whereas the microbiomes of wild-type flies are independent of host genetics (68). When examining specific genes for associations with the microbiome, several immune-related genes are implicated. Nubbin is the Drosophila homolog of the mammalian transcription factor Oct1/Pou2f1. Loss of this gene in mice increases colon tumor incidence, suggesting a valuable role in immunity (69). In Drosophila, however, it operates as an antimicrobial repressor and promotes microbial colonization (70). Similarly, the intestinal homeobox gene Caudal serves as a global antimicrobial peptide gene regulator through nuclear factor kappa B (71). Perturbing normal expression levels of this gene results in disruption to the normal microbiota, leading to increased abundance of pathogenic taxa.
In addition to the immune system, other genetically determined physiological traits shape the gut microbiome. There are heritable differences in total gut microbial abundance levels between fly lines (72). GWAS implicates genes for neuronal and cellular growth and development, which ultimately shape gut morphology. Considering that gross gut morphology significantly shapes the microbiome (11), it is reasonable that genes relating to interindividual differences in morphology would be associated with specific microbes. Finally, the microbiome is associated with neural genes, although the precise mechanisms have yet to be explored fully (73).
Regardless of the mechanisms responsible for maintaining low microbial levels in the fly gut, genetic variation interacts with the microbiome to influence traits that affect host health. Many aspects of nutrition are the result of interactions of host genetics and the microbiome, including circulating triglyceride, glucose, and protein levels (73). Abundances of Acetobacter tropicalis, for example, are associated with genetic variation in six genes in the Drosophila genome in laboratory conditions where a controlled microbiome is introduced. In turn, flies display triglyceride level differences, but only when A. tropicalis is present, demonstrating the necessary interaction between host genetics and this particular taxon.
Caenorhabditis elegans
C. elegans is emerging as a powerful model for host–microbe interactions (74). For decades, C. elegans have been a major player in the fields of development and aging, owing to their fast life cycles and transparency for imaging (75, 76). However, laboratory C. elegans were historically fed simple diets of Escherichia coli, limiting their utility for host–microbe investigations. Recent work has focused on maintaining more natural microbiomes in this organism, opening up opportunities to examine the role of host genetics in shaping these communities. C. elegans microbiomes display variation distinct from their environments, but reproducible within line (77). This points to a possible role of host genetics in determining microbiome composition. Genes in immune and digestive processes likely play a role, as evidenced from gene expression analysis between colonized and axenic C. elegans populations (78) and mutants in immune genes (79). The development of publicly available mock-microbiome panels, such as the CeMbio resource, will provide further insights into which host genes and functions may play a role in this process (80, 81).
Although lab-adapted organisms offer us an opportunity to study genetics in a very controlled and powerful way, there are limitations to these systems. Microbiomes of lab organisms do not necessarily match what is observed in the wild. Drosophila and C. elegans microbiomes in the lab tend to be less diverse than in the wild (65, 82–84). They can be heavily influenced by site-based effects, for example, facility effects across mouse lines (85). Additionally, laboratory lines often have narrower genetic variation than outbred populations, but not necessarily less phenotypic variation (86). This limited view of genetic variation could hamper our abilities to detect associations. Finally, organisms like mice exhibit coprophagic behavior, which can confound genetic estimations if not controlled for properly (87). Nonetheless, model organism studies have provided valuable insight into precisely identifying the genes and processes involved in shaping the microbiome.
GENETIC STUDIES OF THE MICROBIOME IN AGRICULTURAL ANIMALS
Agricultural animals, such as cows, swine, and poultry, provide another powerful system for studying host genetic impacts on the microbiome. Similar to model organisms, there is already an extensive understanding of agricultural animal genetics. Farm animals are the product of decades to millennia of breeding to select for desirable traits, such as greater muscle mass or increased milk production (88). Traditional breeding involved selection by phenotype; however, with improved sequencing technology, researchers began using genomic methods to investigate the genetic and molecular bases for phenotypic variation (89). As a result, and similar to animal models, inbred lines of agricultural animals can provide a powerful, reproducible tool in studies of host genetics.
Major areas of interest in the microbiomes of agricultural animals are for maximizing yields of meat, milk, or fiber; feed efficiency including reduced methane emissions, and improving animal health and infectious disease resistance. The microbiome is associated with weight gain, feed efficiency, and milk and egg production (90–94). Additionally, the microbiome is associated with the general health of an animal and pathogen resistance (95). Naturally, there is interest in whether these outcomes can be selected for via interactions of the microbiome through host genetics.
Ruminants (Cattle and Sheep)
Ruminant animals depend heavily on their microbiome within the reticulo-rumen, proximal to their true stomachs, to properly digest their cellulose-rich diets. As a result, the relationship between ruminants and their microbiomes may be more than simply beneficial—it may be coevolutionary (96). For example, Ruminococcus thrives off complex plant-derived undigestible molecules like cellulose. Cellulose consumption therefore benefits both the host and commensal Ruminococcus (97). Studies in both dairy and beef cattle demonstrate the role genetics plays in determining rumen and hindgut microbiome composition (91, 96, 98–103). Heritable taxa include methanogens, Ruminococcus, Oscillospira, and Succinivibrionaceae. Similar to other vertebrates, genetic variation in immune, mucin, and metabolic genes appears to be responsible for this heritability (103, 104). For some traits, such as methane production, host genetic and microbiome contributions are largely independent (99). In these cases, the host genome and microbiome can be targeted separately to influence the trait. Conversely, for other traits, such as feed efficiency, host genetic variation interacts with the microbiome to determine final levels (96, 98, 105). These examples point to the utility of examining gene-by-environment interactions that include the microbiome.
Swine
Similar to in ruminants, there is a complex interaction between host genetics and the microbiome in swine, which can influence agriculturally relevant traits (106–109). One study in particular developed a novel method for incorporating microbial information into predicting traits (107). In animal breeding, genomic best linear unbiased prediction (G-BLUP) is used to predict host traits from genetic relatedness (110). Camarinha-Silva et al. (107) take this one step further and use microbial relationships to predict host traits (coined M-BLUP). They find that M-BLUP is a better predictor than G-BLUP for all host traits, demonstrating the influence of the microbiome on host phenotype variability. One impactful area of future work could be to build a predictive model that considers the effects of host genetics and the microbiome jointly.
One potential confounding factor for genetic effects in mammalian agricultural animals is vertical transmission via nursing. Microbes can be vertically transmitted via vaginal delivery and suckling and colonize infant guts (111, 112). In piglets, nursing mother identity shapes the microbiome, although whether this influence is due to variation in milk composition or transmission of specific microbes is unclear (113). Although many studies involving infant animals aim to minimize maternal effects by randomizing mother–infant pairs (98, 104), it remains challenging to fully separate maternal and genetic effects in large animal microbiome studies.
Poultry
Although poultry are well studied, we still have a relatively limited understanding of the influence of host genes on the microbiome in poultry owing to the narrow range of genetic backgrounds studied to date. Oftentimes, only a few chicken genotypes are examined for microbiome differences, limiting the discovery space of which genes may play a role. Perhaps unsurprisingly, conflicting reports exist about the role of host genetics on the chicken microbiome. Several studies report that body weight– and growth-related genotypes significantly influence chicken microbiome composition, although the mechanisms by which this occurs remain unclear (114–117). Conversely, others claim there is minimal correlation between host genes and microbes when comparing microbial similarity and genetic relatedness across subjects (118). When examining individual taxa, however, between 5% and 15% genera in the duodenum, jejunum, ileum, cecum, and feces had significant SNP-based heritability estimates, most from Firmicutes and Proteobacteria. Similar to studies in humans and cattle (31, 105), these results further support that the effect of host genetics on the microbiome tends to be targeted and phylogenetically restricted. This suggests that across organisms, host genetics selects for taxa that occupy a particular niche (119). The niche, and the microbes that occupy it, differs depending on the host.
GENETIC STUDIES OF THE MICROBIOME IN WILD ANIMALS
Captivity and domestication shape the microbiomes of nonhuman primates, dogs, and mice (120, 121). It is highly likely that host gene–microbiome interactions differ in natural settings compared to managed systems such as the laboratory or the intensive farming systems used with cattle, swine, and poultry. There are, however, several challenges with studying the genetics of wild animals. First, it is difficult and expensive to collect a sufficient number of genetic and microbiome samples without disturbing the natural environment and/or the animals. Second, some microbes are transmitted within social networks (21, 122, 123). It can be difficult to discern the effects of genetics and social factors when the animals are in high densities. Third, accurately calculating microbiome heritability is challenging, because both microbiome and environmental factors fluctuate naturally over time. Large sample sizes and extensive metadata are especially crucial in this context.
That said, Grieneisen et al. (124) demonstrated recently that it is possible to effectively investigate the genetics of wild baboons while circumventing these issues. The gut microbiomes of 585 wild baboons were examined longitudinally, with approximately 28 samples collected per baboon over a period of 4.5 years. Whereas previous microbiome genetics studies typically involved one sampling time point per subject, this longitudinal design minimizes noise associated with daily variation in microbiome composition. Subsequently, nearly all taxa are heritable, although most of them with only modest heritability of less than 0.15 (124). Interestingly, randomly subsetting the data set to 1,000 samples, similar to sample sizes in human studies (47), weakened heritability estimates. This finding emphasizes the importance of adequate sample size and data density to avoid being underpowered to detect genetic effects.
EMERGING THEMES FROM STUDIES OF MICROBIOME HERITABILITY ACROSS ANIMALS
As more and more studies of microbiome genetics across animals are completed (Figure 2), key themes are emerging. First, regardless of whether an animal’s microbiome consists of only a handful or hundreds of species, host genetics plays a role in determining composition (Figure 3). Although environmental factors tend to have a greater influence than host genetics (47, 62), modestly heritable taxa are identified consistently across organisms (33, 103, 124). Second, heritable taxa tend to be phylogenetically similar within a host species. In humans, heritable taxa tend to fall within the Ruminococcaceae and Lachnospiraceae families (31); in cows, the Bacteroidetes and Firmicutes phyla (105); and in poultry, the Firmicutes and Proteobacteria phyla (118). Finally, certain host genes and processes seem to be implicated across host organisms, including those involved in immunity and growth and development. In the future, greater consideration also needs to be directed to subcommunities residing in the gut mucosa. These communities are known to differ from those in the fecal stream (125) but are also more likely to be strongly influenced by the outputs of host genetics and gene expression.
Figure 2.

The landscape of genetic studies of the microbiome across animals. Genetic studies of the microbiome exist for only a few host taxa across the metazoan tree of life, including humans, mice, and fruit flies. This tree includes 1,000 randomly sampled metazoan taxa from TimeTree.org, displayed by timescale (149). Organisms for which genetic studies have been conducted are highlighted, with gray bars proportional to the number of independent studies completed to date. Major animal clades of interest are indicated by background color on the phylogenetic tree.
Figure 3.

Bacterial genera with evidence of heritability across animals. Bacterial genera with evidence of heritability across humans (orange), mouse (dark orange), cow (pink), pig (purple), chicken (blue), or Drosophila (brown) are listed. To generate the taxonomy, 500 bacterial genera from the Genome Taxonomy Database (GTDB) release 202 were subsampled randomly (150). Major phyla lineages are traced in color and labeled. Genera with evidence of heritability in more than three species are emphasized: Ruminococcus, Anaerostipes, Clostridium, and Streptococcus. Note that not all genera reside in all organisms listed, nor have they necessarily tested for heritability. Absence of detected heritability for any given genus in a host organism should not be interpreted as the evidence of absence of heritability. Plot generated using GraPhlAn (151).
The innate immune system can play a key role in shaping the microbiome, by affecting microbial colonization and persistence. It serves as the host’s first line of defense against any foreign object (126). As a result, an important component of the innate immune system is the recognition of an object as self or nonself. Several types of receptors play this role, including Toll-like receptors (TLRs). Some TLRs signal interleukin proteins (IL), which subsequently trigger pro- or anti-inflammatory responses (127, 128).
In the context of host–microbiome interactions, many QTL significantly associated with heritable microbes are found in genes involved in TLR and IL signaling pathways. IL-22, Irak3, and CARD9 genes in mice; CHGA in swine; C10ORF88 in poultry; and AKIRIN2 in cattle were significantly associated with various taxa (60, 129–132). IL-22 is known for its role in the pathogenesis of gastrointestinal diseases (133), so it would be intriguing to further explore its role in shaping the gut microbiome in various animals.
Another component of innate immunity is the family of transforming growth factors (TGFs), and specifically TGF-β. The role of this protein is complex, impacting both innate and adaptive immunity; however, one important function is in regulating macrophages, B cells, and T cells (134). The genes encoding for this protein are significantly associated with microbes in poultry and C. elegans (78, 131), showing another possible avenue by which host genes control the microbiome. The recurring observation of TGFs and TLRs across animal microbiome genetic studies suggests that these genes play an important role in modulating microbe colonization. One potential mechanism could be via recognizing specific microbes and triggering pathways so that only specific taxa remain.
In those animal hosts where it exists, the adaptive immune system plays an important role in managing complex microbial communities (135). Similar to innate immunity, distinguishing between self and nonself is important for microbial identification so that lymphocytes can appropriately produce antibodies and attack pathogens but not commensals. Antigens presented by major histocompatibility complex (MHC) proteins help T cells recognize pathogens (136). Although the role of the MHC as a defense against pathogen invasion is well known, its role in shaping the microbiome is also now being investigated. Variation in MHC haplotypes can shift microbial abundance (44, 137), likely owing to differences in antibody production (138). Although overall genetic background seems to have a greater influence than MHC variation in shaping the microbiome (137), the role the MHC plays in preventing pathogen colonization demonstrates that host genetics may exert some control on the microbiome via the MHC. Immune genes generally seem to control the microbiome by recognizing specific microbes and influencing which can colonize, although the exact mechanisms responsible remain to be elucidated.
Growth and development genes are important factors in determining microbiome heritability. Drosophila potentially control their microbiomes via growth-related genes that influence gut morphology, impacting which microbes can colonize (72). Similarly, methanogens are associated with growth-related genes in cows and poultry (102, 116, 132). This archaeon is known for its role in methanogenesis and in facilitating interspecies hydrogen transfer in the rumen. This ultimately results in increased volatile fatty acid production that can be used as fuel for the host, thus boosting host fitness, albeit at the cost of methane production, a potent greenhouse gas (139). This may also explain the high heritability detected for this taxon, which was found to be above 0.5 (132). Although the biology underpinning these host gene–microbiome associations is not yet clear, such understanding may support the intriguing possibility that the host controls the microbiome for fitness benefits.
FUTURE OPPORTUNITIES
Although the microbiota of animals has been the topic of many studies for more than a century, the scope of microbiome research has focused primarily on humans and preclinical animal models of disease. As such, much still needs to be learned about the biodiversity resident in domesticated and non-domesticated animal species, and new methods are currently in development. One area of interest in the animal microbiome field, for both agriculture and medicine, is the interrelationships between host genetics, the microbiome, and phenotypic traits. Farmers have a vested interest in maximizing the retention of costly feed ingredients in high-quality food and fiber, thereby reducing the environmental footprint of their enterprises and sustaining their profitability and consumer acceptance of their products. Prior studies have generally explored how genes or microbes may directly impact these traits, but whether genes influence traits via direct or indirect influences on the microbiome is still not fully understood. We propose that mediation analysis could address these knowledge gaps. In mediation analysis, a mediator variable is used to identify causal relationships between two associated variables. Within biology, it has been used to investigate pathogenesis (140), but recent studies have explored its application to other traits. Tiezzi et al. (141) identified several genetic variants that affect swine fatness via the microbiome, and Jin et al. (142) found that the microbiome partially mediated the influence of mice genetics on anxiety (141, 142). In humans, Mendelian randomization has been proposed as a way of identifying causal relationships between the microbiome and phenotypes (143), with a successful application demonstrating the causal role of microbially derived short-chain fatty acids on insulin response (144). Further extension of these methods could incorporate information from several mediator variables to account for interactions between microbes.
Our understanding of how host genetics influences the microbiome is also currently limited to only thin slices of animal phylogeny (Figure 2). Perhaps unsurprisingly, most studies to date have been conducted in large, terrestrial mammals, often in human-influenced settings (Table 1). What does microbiome heritability look like across animals, in particular to those populations in the wild? Are there conserved genetic signals across animals of a similar size or with similar digestive physiologies (11)? Considering that heritability measurements depend on environmental variation, how does microbiome heritability change even within a single animal species living in different environments? Two gaps that would be particularly interesting to fill are with flighted animals and marine animals.
Drosophila have largely transient microbiomes, resulting in unusually high compositional variation between individuals and less dependence on having a core microbiome. Interestingly, flighted birds also demonstrate a weak correlation between the microbiome and host phylogeny or diet (17). Bats also exhibit a similar trend, suggesting that the convergent evolution of flight may be associated with the loss of dependence on the microbiome. Considering that Drosophila microbiomes are controlled primarily by genes for growth and development, it would be interesting to see if birds and bats control their microbiome with similar pathways.
Additionally, the ocean is a largely uncharted area of microbiome research, as it is an under-characterized ecosystem with microbes that reside within and alongside hosts. Modes of microbial transmission in a marine context differ substantially from those in the largely terrestrial animals studied to date, which could greatly influence microbiome heritability. Our greatest understanding of marine host gene-microbiomes comes from corals. Corals depend heavily on their endosymbionts to provide nutrients and help them adapt to environmental stressors (145). This symbiotic relationship is tenuous and can result in coral bleaching, a process by which corals lose their endosymbionts and die. Recently, rising ocean temperatures, increased acidification, and other drastic environmental shifts have been disrupting coral microbiomes and wiping out entire reefs (146, 147). As a result, there is considerable interest in investigating what host factors allow for certain microbes to persist and how we could manipulate these factors to improve community stability. So far, research shows that coral microbiomes are highly host specific (148), but more work is necessary to tease out specific pathways involved. Considering the close relationship with their endosymbionts, corals could push our understanding of host gene–microbiome interactions and broad trends across myriad animals.
CONCLUSION
Across animal species, host genetics plays a role in determining gut microbiome composition. Although the particular microbes affected and host pathways responsible may differ, growing evidence supports the notion that genetically encoded variability in the host determines the abundance of specific taxa residing in the body. As further studies are completed using larger sample sizes and more highly powered methods, and across a wider variety of animals, the mechanisms underlying these relationships will become clear.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Gilbert JA, Jansson JK, Knight R. 2014. The Earth Microbiome project: successes and aspirations. BMC Biol. 12:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, et al. 2012. Structure, function and diversity of the healthy human microbiome. Nature 486(7402):207–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peixoto RS, Harkins DM, Nelson KE. 2021. Advances in microbiome research for animal health. Annu. Rev. Anim. Biosci. 9:289–311 [DOI] [PubMed] [Google Scholar]
- 4.Ezenwa VO, Gerardo NM, Inouye DW, Medina M, Xavier JB. 2012. Animal behavior and the microbiome. Science 338(6104):198–99 [DOI] [PubMed] [Google Scholar]
- 5.Moeller AH, Sanders JG. 2020. Roles of the gut microbiota in the adaptive evolution of mammalian species. Philos. Trans. R. Soc. B 375(1808):20190597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sender R, Fuchs S, Milo R. 2016. Revised estimates for the number of human and bacteria cells in the body. PLOS Biol. 14(8):e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waite DW, Taylor M. 2015. Exploring the avian gut microbiota: current trends and future directions. Front. Microbiol. 6:673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colman DR, Toolson EC, Takacs-Vesbach CD. 2012. Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 21(20):5124–37 [DOI] [PubMed] [Google Scholar]
- 9.Engel P, Moran NA. 2013. The gut microbiota of insects—diversity in structure and function. FEMS Microbiol. Rev. 37(5):699–735 [DOI] [PubMed] [Google Scholar]
- 10.Grond K, Sandercock BK, Jumpponen A, Zeglin LH. 2018. The avian gut microbiota: community, physiology and function in wild birds. J. Avian Biol. 49(11):e01788 [Google Scholar]
- 11.Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, et al. 2008. Evolution of mammals and their gut microbes. Science 320(5883):1647–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Youngblut ND, Reischer GH, Walters W, Schuster N, Walzer C, et al. 2019. Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat. Commun. 10:2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levin D, Raab N, Pinto Y, Rothschild D, Zanir G, et al. 2021. Diversity and functional landscapes in the microbiota of animals in the wild. Science 372(6539):eabb5352. [DOI] [PubMed] [Google Scholar]
- 14.McFall-Ngai MJ, Ruby EG. 1991. Symbiont recognition and subsequent morphogenesis as early events in an animal-bacterial mutualism. Science 254(5037):1491–94 [DOI] [PubMed] [Google Scholar]
- 15.Ainsworth TD, Krause L, Bridge T, Torda G, Raina J-B, et al. 2015. The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J. 9(10):2261–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong AC-N, Chaston JM, Douglas AE. 2013. The inconstant gut microbiota of Drosophila species revealed by 16S rRNA gene analysis. ISME J. 7(10):1922–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song SJ, Sanders JG, Delsuc F, Metcalf J, Amato K, et al. 2020. Comparative analyses of vertebrate gut microbiomes reveal convergence between birds and bats. mBio 11(1):e02901–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hammer TJ, Sanders JG, Fierer N. 2019. Not all animals need a microbiome. FEMS Microbiol. Lett. 366(10):fnz117. [DOI] [PubMed] [Google Scholar]
- 19.Muegge BD, Kuczynski J, Knights D, Clemente JC, González A, et al. 2011. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 332(6032):970–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh RK, Chang H-W, Yan D, Lee KM, Ucmak D, et al. 2017. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 15:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarkar A, Harty S, Johnson KV- A, Moeller AH, Archie EA, et al. 2020. Microbial transmission in animal social networks and the social microbiome. Nat. Ecol. Evol. 4(8):1020–35 [DOI] [PubMed] [Google Scholar]
- 22.Trinh P, Zaneveld JR, Safranek S, Rabinowitz PM. 2018. One Health relationships between human, animal, and environmental microbiomes: a mini-review. Front. Public Health 6:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noller HF, Woese CR. 1981. Secondary structure of 16S ribosomal RNA. Science 212(4493):403–11 [DOI] [PubMed] [Google Scholar]
- 24.Cai L, Ye L, Tong AHY, Lok S, Zhang T. 2013. Biased diversity metrics revealed by bacterial 16S pyrotags derived from different primer sets. PLOS ONE 8(1):e53649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knight R, Vrbanac A, Taylor BC, Aksenov A, Callewaert C, et al. 2018. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 16(7):410–22 [DOI] [PubMed] [Google Scholar]
- 26.Youngblut ND, de la Cuesta-Zuluaga J, Reischer GH, Dauser S, Schuster N, et al. 2020. Large-scale metagenome assembly reveals novel animal-associated microbial genomes, biosynthetic gene clusters, and other genetic diversity. mSystems 5(6):e01045–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goodrich JK, Di Rienzi SC, Poole AC, Koren O, Walters WA, et al. 2014. Conducting a microbiome study. Cell 158(2):250–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baumann P 2005. Biology of bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu. Rev. Microbiol. 59:155–89 [DOI] [PubMed] [Google Scholar]
- 29.Milani C, Mancabelli L, Lugli GA, Duranti S, Turroni F, et al. 2015. Exploring vertical transmission of Bifidobacteria from mother to child. Appl. Environ. Microbiol. 81(20):7078–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zoetendal EG, Akkermans ADL, Akkermans-van Vliet WM, de Visser JAGM, de Vos WM. 2001. The host genotype affects the bacterial community in the human gastrointestinal tract. Microb. Ecol. Health Dis. 13:129–34 [Google Scholar]
- 31.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, et al. 2014. Human genetics shape the gut microbiome. Cell 159(4):789–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goodrich JK, Davenport ER, Beaumont M, Jackson MA, Knight R, et al. 2016. Genetic determinants of the gut microbiome in UK twins. Cell Host Microbe 19(5):731–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khachatryan ZA, Ktsoyan ZA, Manukyan GP, Kelly D, Ghazaryan KA, Aminov RI. 2008. Predominant role of host genetics in controlling the composition of gut microbiota. PLOS ONE 3(8):e3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Altshuler D, Daly MJ, Lander ES. 2008. Genetic mapping in human disease. Science 322(5903):881–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahlqvist E, Hultqvist M, Holmdahl R. 2009. The value of animal models in predicting genetic susceptibility to complex diseases such as rheumatoid arthritis. Arthritis Res. Ther. 11:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Visscher PM, Brown MA, McCarthy MI, Yang J. 2012. Five years of GWAS discovery. Am. J. Hum. Genet. 90(1):7–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, et al. 2017. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101(1):5–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, et al. 2019. The NHGRI-EBI GWAS catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47(D1):D1005–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lim MY, You HJ, Yoon HS, Kwon B, Lee JY, et al. 2017. The effect of heritability and host genetics on the gut microbiota and metabolic syndrome. Gut 66(6):1031–38 [DOI] [PubMed] [Google Scholar]
- 40.Turpin W, Espin-Garcia O, Xu W, Silverberg MS, Kevans D, et al. 2016. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat. Genet. 48:1413–17 [DOI] [PubMed] [Google Scholar]
- 41.Blekhman R, Goodrich JK, Huang K, Sun Q, Bukowski R, et al. 2015. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 16:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kolde R, Franzosa EA, Rahnavard G, Hall AB, Vlamakis H, et al. 2018. Host genetic variation and its microbiome interactions within the Human Microbiome Project. Genome Med. 10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davenport ER, Cusanovich DA, Michelini K, Barreiro LB, Ober C, Gilad Y. 2015. Genome-wide association studies of the human gut microbiota. PLOS ONE 10(11):e0140301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J, Thingholm LB, Skiecevičienė J, Rausch P, Kummen M, et al. 2016. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat. Genet. 48(11):1396–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hughes DA, Bacigalupe R, Wang J, Rühlemann MC, Tito RY, et al. 2020. Genome-wide associations of human gut microbiome variation and implications for causal inference analyses. Nat. Microbiol. 5(9):1079–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurilshikov A, Medina-Gomez C, Bacigalupe R, Radjabzadeh D, Wang J, et al. 2021. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 53(2):156–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, et al. 2018. Environment dominates over host genetics in shaping human gut microbiota. Nature 555(7695):210–15 [DOI] [PubMed] [Google Scholar]
- 48.Davenport ER. 2016. Elucidating the role of the host genome in shaping microbiome composition. Gut Microbes 7(2):178–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goodrich JK, Davenport ER, Waters JL, Clark AG, Ley RE. 2016. Cross-species comparisons of host genetic associations with the microbiome. Science 352(6285):532–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goodrich JK, Davenport ER, Clark AG, Ley RE. 2017. The relationship between the human genome and microbiome comes into view. Annu. Rev. Genet. 51:413–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weissbrod O, Rothschild D, Barkan E, Segal E. 2018. Host genetics and microbiome associations through the lens of genome wide association studies. Curr. Opin. Microbiol. 44:9–19 [DOI] [PubMed] [Google Scholar]
- 52.Bonder MJ, Kurilshikov A, Tigchelaar EF, Mujagic Z, Imhann F, et al. 2016. The effect of host genetics on the gut microbiome. Nat. Genet. 48(11):1407–12 [DOI] [PubMed] [Google Scholar]
- 53.Kostic AD, Howitt MR, Garrett WS. 2013. Exploring host-microbiota interactions in animal models and humans. Genes Dev. 27(7):701–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Korach-Rechtman H, Freilich S, Gerassy-Vainberg S, Buhnik-Rosenblau K, Danin-Poleg Y, et al. 2019. Murine genetic background has a stronger impact on the composition of the gut microbiota than maternal inoculation or exposure to unlike exogenous microbiota. Appl. Environ. Microbiol. 85(18):e00826–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kovacs A, Ben-Jacob N, Tayem H, Halperin E, Iraqi FA, Gophna U. 2011. Genotype is a stronger determinant than sex of the mouse gut microbiota. Microb. Ecol. 61(2):423–28 [DOI] [PubMed] [Google Scholar]
- 56.Hoy YE, Bik EM, Lawley TD, Holmes SP, Monack DM, et al. 2015. Variation in taxonomic composition of the fecal microbiota in an inbred mouse strain across individuals and time. PLOS ONE 10(11):e0142825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ericsson AC, Davis JW, Spollen W, Bivens N, Givan S, et al. 2015. Effects of vendor and genetic background on the composition of the fecal microbiota of inbred mice. PLOS ONE 10(2):e0116704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Leamy LJ, Kelly SA, Nietfeldt J, Legge RM, Ma F, et al. 2014. Host genetics and diet, but not immunoglobulin A expression, converge to shape compositional features of the gut microbiome in an advanced intercross population of mice. Genome Biol. 15(12):552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Campbell JH, Foster CM, Vishnivetskaya T, Campbell AG, Yang ZK, et al. 2012. Host genetic and environmental effects on mouse intestinal microbiota. ISME J. 6(11):2033–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Benson AK, Kelly SA, Legge R, Ma F, Low SJ, et al. 2010. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. PNAS 107(44):18933–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang C, Zhang M, Wang S, Han R, Cao Y, et al. 2010. Interactions between gut microbiota, host genetics and diet relevant to development of metabolic syndromes in mice. ISME J. 4(2):232–41 [DOI] [PubMed] [Google Scholar]
- 62.Carmody RN, Gerber GK, Luevano JM, Gatti DM, Somes L, et al. 2015. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 17(1):72–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O’Connor A, Quizon PM, Albright JE, Lin FT, Bennett BJ. 2014. Responsiveness of cardiometabolic-related microbiota to diet is influenced by host genetics. Mamm. Genome 25(11):583–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Parks BW, Nam E, Org E, Kostem E, Norheim F, et al. 2013. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab. 17(1):141–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Adair KL, Wilson M, Bost A, Douglas AE. 2018. Microbial community assembly in wild populations of the fruit fly Drosophila melanogaster. ISME J. 12(4):959–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Douglas AE. 2018. The Drosophila model for microbiome research. Lab. Anim. 47(6):157–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blum JE, Fischer CN, Miles J, Handelsman J. 2013. Frequent replenishment sustains the beneficial microbiome of Drosophila melanogaster. mBio 4(6):e00860–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mistry R, Kounatidis I, Ligoxygakis P. 2017. Interaction between familial transmission and a constitutively active immune system shapes gut microbiota in Drosophila melanogaster. Genetics 206(2):889–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vázquez-Arreguín K, Bensard C, Schell JC, Swanson E, Chen X, et al. 2019. Oct1/Pou2f1 is selectively required for colon regeneration and regulates colon malignancy. PLOS Genet. 15(5):e1007687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dantoft W, Davis MM, Lindvall JM, Tang X, Uvell H, et al. 2013. The Oct1 homolog Nubbin is a repressor of NF-κB-dependent immune gene expression that increases the tolerance to gut microbiota. BMC Biol. 11:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ryu J-H, Kim S-H, Lee H-Y, Bai JY, Nam Y-D, et al. 2008. Innate immune homeostasis by the homeobox gene Caudal and commensal-gut mutualism in Drosophila. Science 319(5864):777–82 [DOI] [PubMed] [Google Scholar]
- 72.Early AM, Shanmugarajah N, Buchon N, Clark AG. 2017. Drosophila genotype influences commensal bacterial levels. PLOS ONE 12(1):e0170332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chaston JM, Dobson AJ, Newell PD, Douglas AE. 2016. Host genetic control of the microbiota mediates the Drosophila nutritional phenotype. Appl. Environ. Microbiol. 82(2):671–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang F, Berg M, Dierking K, Félix M-A, Shapira M, et al. 2017. Caenorhabditis elegans as a model for microbiome research. Front. Microbiol. 8:485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brenner S 1974. The genetics of Caenorhabditis elegans. Genetics 77(1):71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kaletta T, Hengartner MO. 2006. Finding function in novel targets: C. elegans as a model organism. Nat. Rev. Drug Discov. 5(5):387–99 [DOI] [PubMed] [Google Scholar]
- 77.Berg M, Zhou XY, Shapira M. 2016. Host-specific functional significance of Caenorhabditis gut commensals. Front. Microbiol. 7:1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Berg M, Monnin D, Cho J, Nelson L, Crits-Christoph A, Shapira M. 2019. TGFβ/BMP immune signaling affects abundance and function of C. elegans gut commensals. Nat. Commun. 10:604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Taylor M, Vega NM. 2021. Host immunity alters community ecology and stability of the microbiome in a Caenorhabditis elegans model. mSystems 6(2):e00608–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dirksen P, Assié A, Zimmermann J, Zhang F, Tietje A-M, et al. 2020. CeMbio—the Caenorhabditis elegans microbiome resource. G3 10(9):3025–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang F, Weckhorst JL, Assié A, Hosea C, Ayoub CA, et al. 2021. Natural genetic variation drives microbiome selection in the Caenorhabditis elegans gut. Curr. Biol. 31(12):2603–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chandler JA, Lang JM, Bhatnagar S, Eisen JA, Kopp A. 2011. Bacterial communities of diverse Drosophila species: ecological context of a host-microbe model system. PLOS Genet. 7(9):e1002272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Berg M, Stenuit B, Ho J, Wang A, Parke C, et al. 2016. Assembly of the Caenorhabditis elegans gut microbiota from diverse soil microbial environments. ISME J. 10(8):1998–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huggins MA, Sjaastad FV, Pierson M, Kucaba TA, Swanson W, et al. 2019. Microbial exposure enhances immunity to pathogens recognized by TLR2 but increases susceptibility to cytokine storm through TLR4 sensitization. Cell Rep. 28(7):1729–43.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Parker KD, Albeke SE, Gigley JP, Goldstein AM, Ward NL. 2018. Microbiome composition in both wild-type and disease model mice is heavily influenced by mouse facility. Front. Microbiol. 9:1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tuttle AH, Philip VM, Chesler EJ, Mogil JS. 2018. Comparing phenotypic variation between inbred and outbred mice. Nat. Methods 15(12):994–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hildebrand F, Nguyen TLA, Brinkman B, Yunta RG, Cauwe B, et al. 2013. Inflammation-associated enterotypes, host genotype, cage and inter-individual effects drive gut microbiota variation in common laboratory mice. Genome Biol. 14:R4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Andersson L 2001. Genetic dissection of phenotypic diversity in farm animals. Nat. Rev. Genet. 2(2):130–38 [DOI] [PubMed] [Google Scholar]
- 89.Meuwissen T, Hayes B, Goddard M. 2016. Genomic selection: a paradigm shift in animal breeding. Anim. Front. 6(1):6–14 [Google Scholar]
- 90.de Freitas AS, de David DB, Takagaki BM, Roesch LFW. 2020. Microbial patterns in rumen are associated with gain of weight in beef cattle. Antonie van Leeuwenhoek 113(9):1299–312 [DOI] [PubMed] [Google Scholar]
- 91.Guan LL, Nkrumah JD, Basarab JA, Moore SS. 2008. Linkage of microbial ecology to phenotype: correlation of rumen microbial ecology to cattle’s feed efficiency. FEMS Microbiol. Lett. 288(1):85–91 [DOI] [PubMed] [Google Scholar]
- 92.Maltecca C, Bergamaschi M, Tiezzi F. 2020. The interaction between microbiome and pig efficiency: a review. J. Anim. Breed. Genet. 137(1):4–13 [DOI] [PubMed] [Google Scholar]
- 93.Xue M-Y, Sun H-Z, Wu X-H, Liu J-X, Guan LL. 2020. Multi-omics reveals that the rumen microbiome and its metabolome together with the host metabolome contribute to individualized dairy cow performance. Microbiome 8:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Elokil AA, Magdy M, Melak S, Ishfaq H, Bhuiyan A, et al. 2020. Faecal microbiome sequences in relation to the egg-laying performance of hens using amplicon-based metagenomic association analysis. Animal 14(4):706–15 [DOI] [PubMed] [Google Scholar]
- 95.O’Hara E, Neves ALA, Song Y, Guan LL. 2020. The role of the gut microbiome in cattle production and health: Driver or passenger? Annu. Rev. Anim. Biosci. 8:199–220 [DOI] [PubMed] [Google Scholar]
- 96.Li F, Li C, Chen Y, Liu J, Zhang C, et al. 2019. Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle. Microbiome 7:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.La Reau AJ, Meier-Kolthoff JP, Suen G. 2016. Sequence-based analysis of the genus Ruminococcus resolves its phylogeny and reveals strong host association. Microb. Genom. 2(12):e000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Roehe R, Dewhurst RJ, Duthie C-A, Rooke JA, McKain N, et al. 2016. Bovine host genetic variation influences rumen microbial methane production with best selection criterion for low methane emitting and efficiently feed converting hosts based on metagenomic gene abundance. PLOS Genet. 12(2):e1005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Difford GF, Plichta DR, Løvendahl P, Lassen J, Noel SJ, et al. 2018. Host genetics and the rumen microbiome jointly associate with methane emissions in dairy cows. PLOS Genet. 14(10):e1007580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Golder HM, Thomson JM, Denman SE, McSweeney CS, Lean IJ. 2018. Genetic markers are associated with the ruminal microbiome and metabolome in grain and sugar challenged dairy heifers. Front. Genet. 9:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wallace RJ, Sasson G, Garnsworthy PC, Tapio I, Gregson E, et al. 2019. A heritable subset of the core rumen microbiome dictates dairy cow productivity and emissions. Sci. Adv. 5(7):eaav8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang Q, Difford G, Sahana G, Løvendahl P, Lassen J, et al. 2020. Bayesian modeling reveals host genetics associated with rumen microbiota jointly influence methane emission in dairy cows. ISME J. 14(8):2019–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fan P, Nelson CD, Driver JD, Elzo MA, Peñagaricano F, Jeong KC. 2021. Host genetics exerts lifelong effects upon hindgut microbiota and its association with bovine growth and immunity. ISME J. 15:2306–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fan P, Bian B, Teng L, Nelson CD, Driver J, et al. 2020. Host genetic effects upon the early gut microbiota in a bovine model with graduated spectrum of genetic variation. ISME J. 14:302–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sasson G, Ben-Shabat SK, Seroussi E, Doron-Faigenboim A, Shterzer N, et al. 2017. Heritable bovine rumen bacteria are phylogenetically related and correlated with the cow’s capacity to harvest energy from its feed. mBio 8(4):e00703–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Estellé J, Mach N, Ramayo-Caldas Y, Levenez F, Lemonnier G, et al. 2014. The influence of host’s genetics on the gut microbiota composition in pigs and its links with immunity traits. In Proceedings of the 10th World Congress of Genetics Applied to Livestock Production, Vancouver, BC, Aug. 17–22. Champaign, IL: Am. Soc. Anim. Sci. [Google Scholar]
- 107.Camarinha-Silva A, Maushammer M, Wellmann R, Vital M, Preuss S, Bennewitz J. 2017. Host genome influence on gut microbial composition and microbial prediction of complex traits in pigs. Genetics 206(3):1637–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bergamaschi M, Maltecca C, Schillebeeckx C, McNulty NP, Schwab C, et al. 2020. Heritability and genome-wide association of swine gut microbiome features with growth and fatness parameters. Sci. Rep. 10:10134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Reverter A, Ballester M, Alexandre PA, Mármol-Sánchez E, Dalmau A, et al. 2021. A gene co-association network regulating gut microbial communities in a Duroc pig population. Microbiome 9:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Clark SA, van der Werf J. 2013. Genomic best linear unbiased prediction (gBLUP) for the estimation of genomic breeding values. Methods Mol. Biol. 1019:321–30 [DOI] [PubMed] [Google Scholar]
- 111.Jost T, Lacroix C, Braegger CP, Rochat F, Chassard C. 2014. Vertical mother-neonate transfer of maternal gut bacteria via breastfeeding. Environ. Microbiol. 16(9):2891–904 [DOI] [PubMed] [Google Scholar]
- 112.Pannaraj PS, Li F, Cerini C, Bender JM, Yang S, et al. 2017. Association between breast milk bacterial communities and establishment and development of the infant gut microbiome. JAMA Pediatr. 171(7):647–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bian G, Ma S, Zhu Z, Su Y, Zoetendal EG, et al. 2016. Age, introduction of solid feed and weaning are more important determinants of gut bacterial succession in piglets than breed and nursing mother as revealed by a reciprocal cross-fostering model. Environ. Microbiol. 18(5):1566–77 [DOI] [PubMed] [Google Scholar]
- 114.Zhao L, Wang G, Siegel P, He C, Wang H, et al. 2013. Quantitative genetic background of the host influences gut microbiomes in chickens. Sci. Rep. 3:1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Meng H, Zhang Y, Zhao L, Zhao W, He C, et al. 2014. Body weight selection affects quantitative genetic correlated responses in gut microbiota. PLOS ONE 9(3):e89862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ji J, Luo CL, Zou X, Lv XH, Xu YB, et al. 2019. Association of host genetics with intestinal microbial relevant to body weight in a chicken F2 resource population. Poultry Sci. 98(9):4084–93 [DOI] [PubMed] [Google Scholar]
- 117.Ji J, Xu Y, Luo C, He Y, Xu X, et al. 2020. Effects of the DMRT1 genotype on the body weight and gut microbiota in the broiler chicken. Poultry Sci. 99(8):4044–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wen C, Yan W, Sun C, Ji C, Zhou Q, et al. 2019. The gut microbiota is largely independent of host genetics in regulating fat deposition in chickens. ISME J. 13(6):1422–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Felsenstein J 1985. Phylogenies and the comparative method. Am. Nat. 125(1):1–15 [Google Scholar]
- 120.Clayton JB, Vangay P, Huang H, Ward T, Hillmann BM, et al. 2016. Captivity humanizes the primate microbiome. PNAS 113(37):10376–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Reese AT, Chadaideh KS, Diggins CE, Schell LD, Beckel M, et al. 2021. Effects of domestication on the gut microbiota parallel those of human industrialization. eLife 10:e60197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tung J, Barreiro LB, Burns MB, Grenier J-C, Lynch J, et al. 2015. Social networks predict gut microbiome composition in wild baboons. eLife 4:e05224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Moeller AH, Foerster S, Wilson ML, Pusey AE, Hahn BH, Ochman H. 2016. Social behavior shapes the chimpanzee pan-microbiome. Sci. Adv. 2(1):e1500997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Grieneisen L, Dasari M, Gould TJ, Björk JR, Grenier J-C, et al. 2021. Gut microbiome heritability is nearly universal but environmentally contingent. Science 373(6551):181–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, et al. 2014. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15(3):382–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Iwasaki A, Medzhitov R. 2015. Control of adaptive immunity by the innate immune system. Nat. Immunol. 16(4):343–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dunne A, O’Neill LAJ. 2003. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci. STKE 2003(171):re3. [DOI] [PubMed] [Google Scholar]
- 128.Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11(5):373–84 [DOI] [PubMed] [Google Scholar]
- 129.Lamas B, Richard ML, Leducq V, Pham H-P, Michel M-L, et al. 2016. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 22(6):598–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Crespo-Piazuelo D, Migura-Garcia L, Estellé J, Criado-Mesas L, Revilla M, et al. 2019. Association between the pig genome and its gut microbiota composition. Sci. Rep. 9:8791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mignon-Grasteau S, Narcy A, Rideau N, Chantry-Darmon C, Boscher M-Y, et al. 2015. Impact of selection for digestive efficiency on microbiota composition in the chicken. PLOS ONE 10(8):e0135488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Abbas W, Howard JT, Paz HA, Hales KE, Wells JE, et al. 2020. Influence of host genetics in shaping the rumen bacterial community in beef cattle. Sci. Rep. 10:15101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Parks OB, Pociask DA, Hodzic Z, Kolls JK, Good M. 2016. Interleukin-22 signaling in the regulation of intestinal health and disease. Front. Cell Dev. Biol. 3:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kubiczkova L, Sedlarikova L, Hajek R, Sevcikova S. 2012. TGF-β—an excellent servant but a bad master. J. Transl. Med. 10:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.McFall-Ngai M 2007. Care for the community. Nature 445(7124):153. [DOI] [PubMed] [Google Scholar]
- 136.Wieczorek M, Abualrous ET, Sticht J, Álvaro-Benito M, Stolzenberg S, et al. 2017. Major histocompatibility complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front. Immunol. 8:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Khan AA, Yurkovetskiy L, O’Grady K, Pickard JM, de Pooter R, et al. 2019. Polymorphic immune mechanisms regulate commensal repertoire. Cell Rep. 29(3):541–50.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kubinak JL, Stephens WZ, Soto R, Petersen C, Chiaro T, et al. 2015. MHC variation sculpts individualized microbial communities that control susceptibility to enteric infection. Nat. Commun. 6:8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bryant MP. 1979. Microbial methane production—theoretical aspects. J. Anim. Sci. 48(1):193–201 [Google Scholar]
- 140.VanderWeele TJ. 2016. Mediation analysis: a practitioner’s guide. Annu. Rev. Public Health 37:17–32 [DOI] [PubMed] [Google Scholar]
- 141.Tiezzi F, Fix J, Schwab C, Shull C, Maltecca C. 2021. Gut microbiome mediates host genomic effects on phenotypes: a case study with fat deposition in pigs. Comput. Struct. Biotechnol. J. 19:530–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jin X, Zhang Y, Celniker SE, Xia Y, Mao J-H, et al. 2021. Gut microbiome partially mediates and coordinates the effects of genetics on anxiety-like behavior in Collaborative Cross mice. Sci. Rep. 11:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wade KH, Hall LJ. 2020. Improving causality in microbiome research: can human genetic epidemiology help? Wellcome Open Res. 4:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sanna S, van Zuydam NR, Mahajan A, Kurilshikov A, Vich Vila A, et al. 2019. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat. Genet. 51(4):600–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bourne DG, Morrow KM, Webster NS. 2016. Insights into the coral microbiome: underpinning the health and resilience of reef ecosystems. Annu. Rev. Microbiol. 70:317–40 [DOI] [PubMed] [Google Scholar]
- 146.Avila-Magaña V, Kamel B, DeSalvo M, Gómez-Campo K, Enríquez S, et al. 2021. Elucidating gene expression adaptation of phylogenetically divergent coral holobionts under heat stress. Nat. Commun. 12(1):5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Webster NS, Negri AP, Botté ES, Laffy PW, Flores F, et al. 2016. Host-associated coral reef microbes respond to the cumulative pressures of ocean warming and ocean acidification. Sci. Rep. 6(1):19324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Glasl B, Smith CE, Bourne DG, Webster NS. 2019. Disentangling the effect of host-genotype and environment on the microbiome of the coral Acropora tenuis. PeerJ 7:e6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kumar S, Stecher G, Suleski M, Hedges SB. 2017. TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34(7):1812–19 [DOI] [PubMed] [Google Scholar]
- 150.Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, et al. 2018. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36(10):996–1004 [DOI] [PubMed] [Google Scholar]
- 151.Asnicar F, Weingart G, Tickle TL, Huttenhower C, Segata N. 2015. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 3:e1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Org E, Parks BW, Joo JWJ, Emert B, Schwartzman W, et al. 2015. Genetic and environmental control of host-gut microbiota interactions. Genome Res. 25(10):1558–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.McKnite AM, Perez-Munoz ME, Lu L, Williams EG, Brewer S, et al. 2012. Murine gut microbiota is defined by host genetics and modulates variation of metabolic traits. PLOS ONE 7(6):e39191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Snijders AM, Langley SA, Kim Y-M, Brislawn CJ, Noecker C, et al. 2016. Influence of early life exposure, host genetics and diet on the mouse gut microbiome and metabolome. Nat. Microbiol. 2:16221. [DOI] [PubMed] [Google Scholar]
- 155.Ramayo-Caldas Y, Prenafeta-Boldú F, Zingaretti LM, Gonzalez-Rodriguez O, Dalmau A, et al. 2020. Gut eukaryotic communities in pigs: diversity, composition and host genetics contribution. Anim. Microbiome 2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Suzuki TA, Phifer-Rixey M, Mack KL, Sheehan MJ, Lin D, et al. 2019. Host genetic determinants of the gut microbiota of wild mice. Mol. Ecol. 28(13):3197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Griffiths SM, Antwis RE, Lenzi L, Lucaci A, Behringer DC, et al. 2019. Host genetics and geography influence microbiome composition in the sponge Ircinia campana. J. Anim. Ecol. 88(11):1684–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Smith CC, Snowberg LK, Caporaso JG, Knight R, Bolnick DI. 2015. Dietary input of microbes and host genetic variation shape among-population differences in stickleback gut microbiota. ISME J. 9:2515–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Steury RA, Currey MC, Cresko WA, Bohannan BJM. 2019. Population genetic divergence and environment influence the gut microbiome in Oregon threespine stickleback. Genes 10(7):484. [DOI] [PMC free article] [PubMed] [Google Scholar]