

Abstract
An increased interest to expand three-dimensional chemical space for the design of new materials and medicines has created a demand for isosteric replacement groups of commonly used molecular functionality. The structural and chemical properties of chiral S(VI) functional groups provide unique spatial and electronic features compared to their achiral sulfur- and carbon-based counterparts. Manipulation of the S(VI) center to introduce structural variation with stereochemical control has remained a synthetic challenge. The stability of sulfonimidoyl fluorides and the efficiency of sulfur fluorine exchange (SuFEx) chemistry has enabled the development of an enantiopure bifunctional S(VI) transfer reagent (t-BuSF) to overcome current synthetic limitations. Here, this reagent platform serves as a chiral SuFEx template for the rapid asymmetric synthesis of over seventy sulfoximines, sulfonimidoyl fluorides and sulfonimidamides with excellent enantiomeric excess and good overall yields. Furthermore, the practical utility of t-BuSF was demonstrated in the syntheses of enantiopure pharmaceutical intermediates and analogs.
Graphical abstract
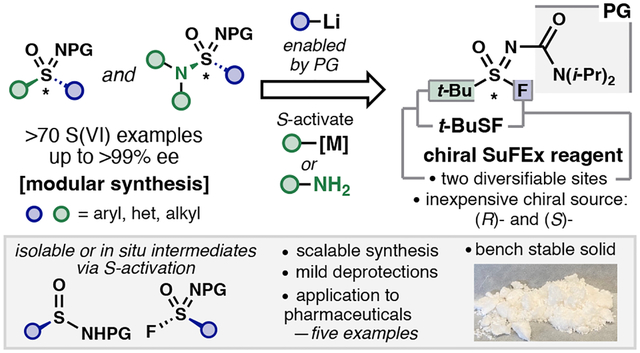
Introduction
Sulfur-containing compounds such as sulfoxides, sulfones, sulfinamides, sulfonamides, sulfinates and sulfonates have been thoroughly explored over the last two centuries and, as a result, can be found in pharmaceuticals, agrochemicals, semiconductors, polymers and a variety of other materials.1–5 Meanwhile, sulfoximines and sulfonimidamides (highlighted in grey, Figure 1A),6–10 have been underrepresented since their discoveries in the early 1900’s.11,12 Within the last ten years sulfonimidoyl compounds and their derivatives have gained attention from the agrochemical and pharmaceutical industries culminating in the development of new pesticides,13 anti-virals,6 anti-cancer agents,9,14,15 osteoporotic treatments,7 and neuroprotective agents.8 More recently, sulfonimidoyl ureas have emerged as promising clinical candidates for targeting NLRP3 which is associated with indications such as autoinflammatory diseases and SARS-CoV-2 (Figure 1A).10,16,17
Figure 1: Chiral S(VI) functional groups and their preparations.
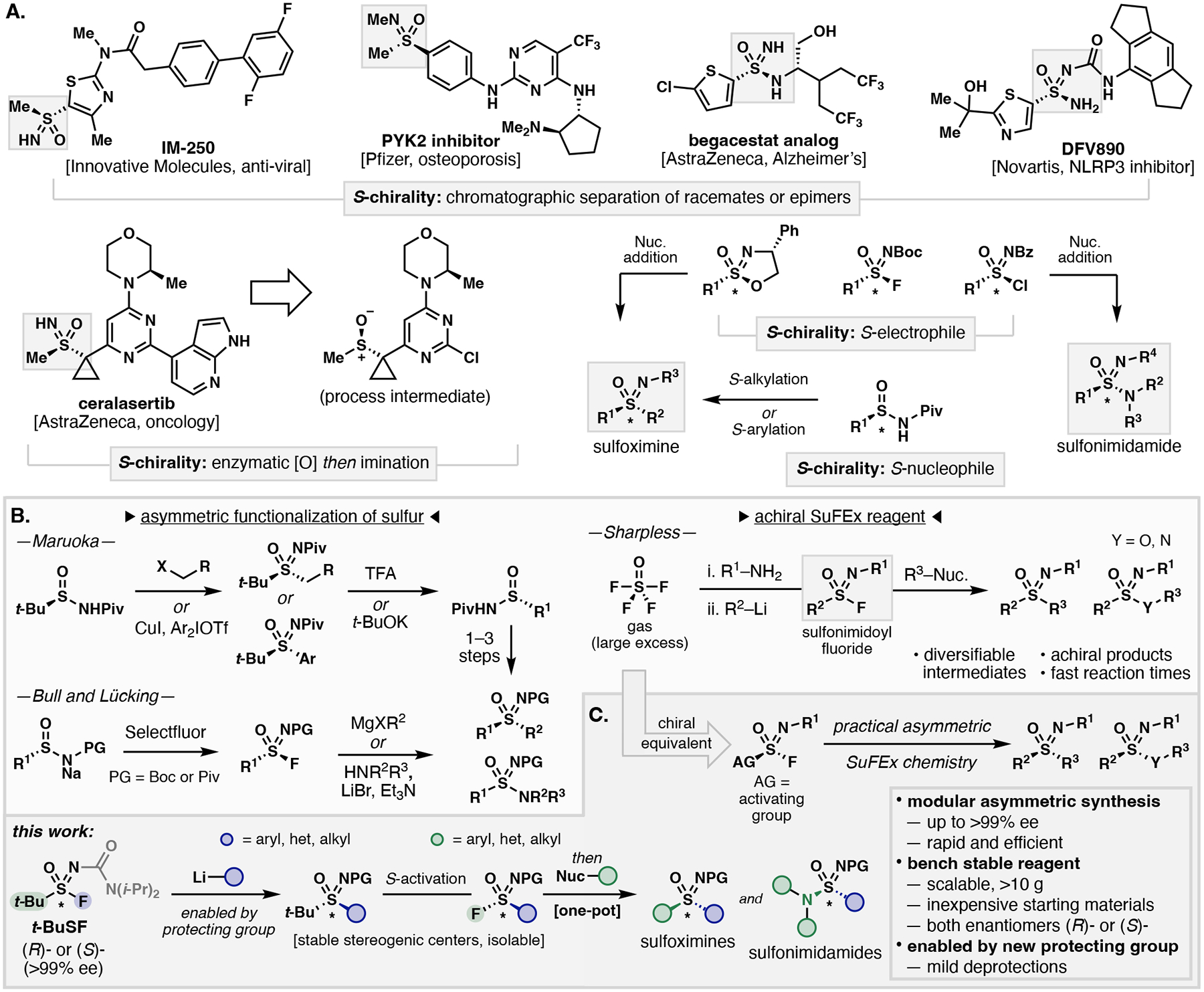
A. Biologically relevant sulfoximines and sulfonimidamides and general strategies used for their asymmetric preparation. B. Methods using S-nucleophile and S-electrophile strategies for the synthesis of sulfonimidoyls. C. Design of an enantiopure sulfonimidoyl transfer reagent for the modular asymmetric synthesis of sulfoximines, sulfonimidoyl fluorides, and sulfonimidamides (this work).
Sulfoximines and sulfonimidamides exhibit unique structural features including multiple hydrogen-bond acceptor and donor modes, stable stereogenic sulfur centers, and high polar surface areas that can provide additional advantages for drug design over their S(VI) counterparts. Pharmaceutical developers such as AstraZeneca, Bayer, and Pfizer have explored sulfoximines and sulfonimidamides as alternatives to sulfones and sulfonamides where, in some instances, their physiochemical and pharmacokinetic properties allowed for advancement into clinical trials.9,14,15 Additionally, sulfoximines and sulfonimidamides have been employed as bioisosteres for carboxylic acids, alcohols and amines.12,18–20 While these aza-S(VI) derivatives serve as pharmacological modulators, sulfonimidoyl fluorides have emerged as promising chemical reactive groups for configurationally stable chiral polymers and future chemical probes owing to their structural variability and increased hydrolytic stability relative to sulfonyl fluorides.21–23
The biological significance of sulfoximines and sulfonimidamides has driven the development of myriad methods for their racemic syntheses via oxidative iminations of S(II) and S(IV) centers,24–26 S(VI) functional group interconversions,27–29 and reagent-based approaches.30–35 In comparison, the preparation of sulfonimidoyl fluorides is much more limited.29,34,37–40 Nearly all enantiomerically enriched sulfoximine and sulfonimidamide pharmaceuticals are prepared as racemates then separated by chromatographic methods to deliver the desired pure stereoisomers (Figure 1A).6–8,10 In general, four main strategies are used to access enantioenriched sulfoximines and sulfonimidamides: 1) chromatographic separation of racemic or epimeric mixtures and chiral resolutions,41–43 2) oxidative imination of sulfoxides and sulfinamides,43–45 3) electrophilic addition to S-nucleophiles (provides sulfoximines only),43,46–48 and 4) nucleophilic addition to S-electrophiles43,49–52 (Figure 1A). However, other elegant strategies such as desymmetrizations and catalytic C-H functionalization-based kinetic resolutions have made considerable progress.43 The development of tert-butyl sulfinyls as linchpin nucleophiles for S-alkylations and S-arylations by Maruoka has provided bifunctional asymmetric routes to sulfoximines (Figure 1B).46–48 During the course of this work, Bull and Lücking prepared enantioenriched sulfonimidoyl fluorides from sulfinyls to access sulfoximines and sulfonimidamides via sulfur fluorine exchange (SuFEx) chemistry with a high degree of stereocontrol (Figure 1B).49–52 Although these methods have advanced the asymmetric synthesis of sulfonimidoyls, a more modular and efficient unified approach is warranted.
With the introduction of SuFEx chemistry by Sharpless using gaseous thionyl tetrafluoride,34 highly effective syntheses of racemic S(VI) groups in a trifunctional manner have been realized (Figure 1B). Drawing upon the acclamatory use of thionyl tetrafluoride as a SuFEx reagent, it became apparent that the practical limitations of its preparation (>1250 psi) has hindered wide-spread use. We envisioned that a solid, bench-stable chiral sulfonimidoyl fluoride reagent could serve as a modular bifunctional platform to improve synthetic aptitude towards the sulfonimidoyl chemical space with stereocontrol (Figure 1C). Here we disclose the development of a reagent that serves as a chiral SuFEx-hub (t-BuSF) enabled by a novel sulfonimidoyl N–H protecting group and a stereospecific net redox-neutral S-activation strategy of tert-butyl sulfoximines for the asymmetric synthesis of sulfoximines, sulfonimidoyl fluorides, and sulfonimidamides.
Results and discussion
Reagent development.
To address the present drawbacks and limitations associated with the asymmetric synthesis of S(VI) groups, we set out to explore the viability of sequential enantiospecific additions to S(VI) centers. A sulfonimidoyl fluoride was selected to serve as the initial electrophile due to their stability and reactivity profiles. Development of an enantiopure reagent capable of S-bifunctionalization in rapid succesion commenced with tert-butyl sulfinamide 1, serving as a cost-effective source of chirality (R- and S-) and providing an activating group (t-Bu) for later manipulation. A practical one-step synthesis of t-BuSF from sulfinamide 1 and N,N-diisopropyl carbamoyl chloride (ClCON(i-Pr)2) reliably produces an enantiopure crystalline bench-stable solid in high yields and decagram scales (87% yield, >99% ee) (Figure 2A). Over the course of this study, the stability of t-BuSF has been thoroughly examined and shown to be highly stable under ambient conditions for greater than one year with minimal decrease in purity and effectiveness (see Supplementary Information Table S1).
Figure 2:
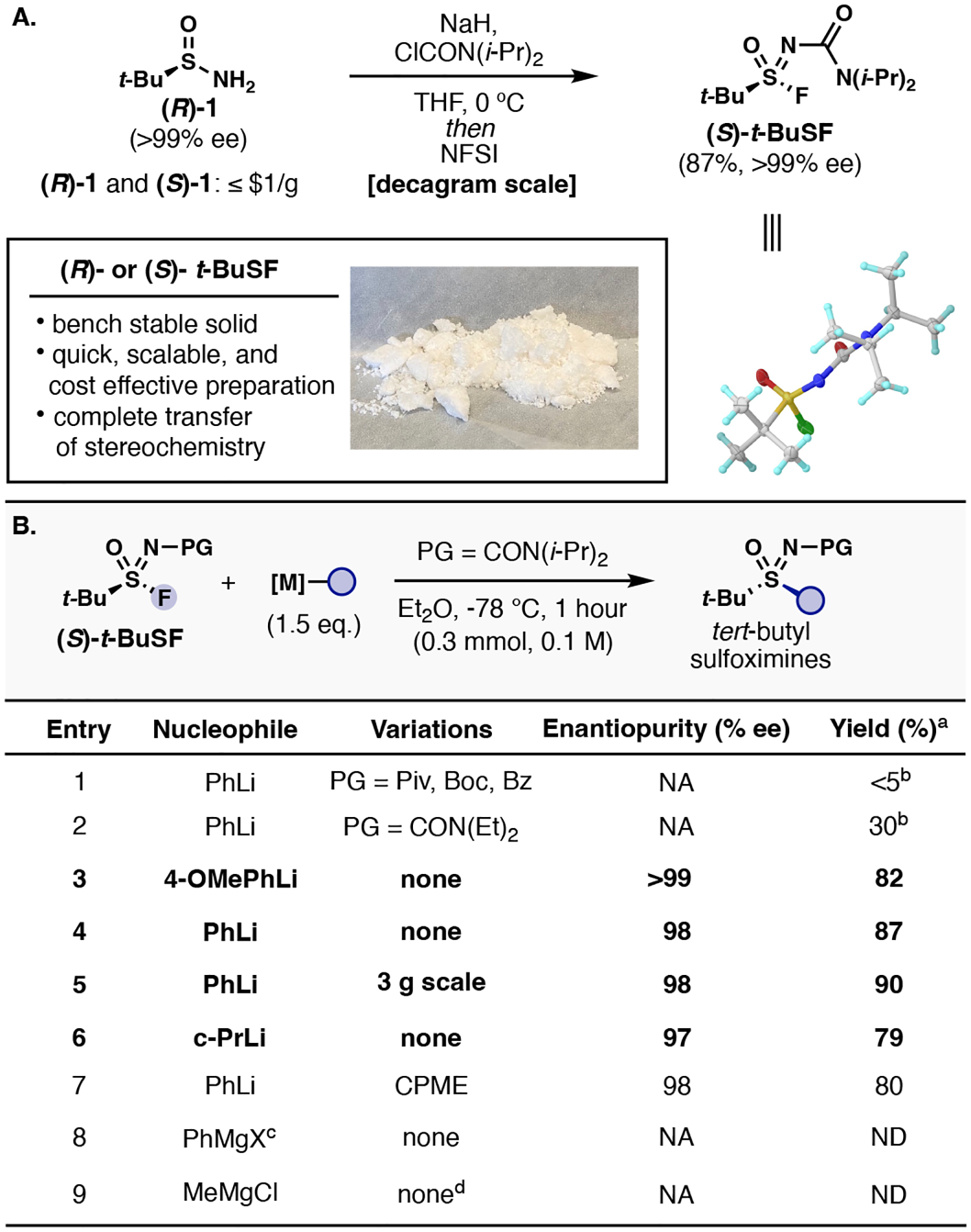
A. Synthesis of enantiopure t-BuSF. B. Reaction investigation and optimization. aIsolated yields. bDetermined by LC–MS. cX = Br, Cl, Cl•LiCl. NA = not available. ND = not detected. See Supporting Information for more details and further discussion.
The choice of protecting group proved to be critical for the first S-functionalization to afford tert-butyl sulfoximines. Commonly employed sulfonimidoyl N–protecting groups such as pivaloyl (Piv), Boc, and benzoyl (Bz) (Fig. 2B, entry 1) were initially examined with organolithium and Grignard reagents under general reaction conditions (Et2O, −78 °C, 1 hour). Nucleophilic displacements of the protecting groups were observed with PhLi to give N–H tert-butyl sulfoximines as the main products. By switching to an N,N-diethyl urea protecting group, a significant increase (<5% to 30%) of the desired sulfoximine was observed (entry 2). To our delight, increasing the steric bulk of the urea with N,N-diisopropyl substituents gave rise to the desired protected tert-butyl sulfoximines in high yield and excellent stereospecificity (entries 3–6). To date, N,N-diisopropyl urea has yet to be described as a protecting group for sulfonimidoyl compounds.
The reaction conditions (solvent and temperature) and nucleophile can impact the stereochemical outcome of the sulfonimidoyl transfer (see Supplementary Information section IIIa. for complete discussion). While 4-methoxylphenyl lithium proceeded with complete inversion of stereochemistry and high yield (>99% ee, 82% yield; entry 3), PhLi led to a slight diminishment in enantiomeric excess (87% yield, 98% ee; entry 4)—yield and stereochemical transfer were maintained upon scale-up (3-gram scale, entry 5). Additionally, cyclopropyl lithium provided the desired secondary aliphatic sulfoximine in good yield and excellent enantiopurity (79% yield, 97% ee; entry 6). Other ethereal solvents such as cyclopropyl methyl ether (CPME), gave comparable yields and identical enantiopurities (entry 7). Surprisingly, Grignard and turbo-Grignard reagents did not deliver the desired tert-butyl sulfoximines (entries 8 and 9). With optimal conditions in hand for the asymmetric transfer of t-BuSF to carbon nucleophiles, we turned our focus towards examining the reaction scope.
Sulfonimidoyl transfer: first S-functionalization.
A comprehensive set of substituted aryllithium nucleophiles were first investigated for reactivity and enantiospecificity trends (Table 1). Thirteen mono-substituted aryllithiums (2a-2n) displayed good overall reactivity ranging from 60% to 90% yields (except for 4-OBn– 2h giving a moderate yield of 40%) and excellent stereochemical transfer (95% to >99% ee). The highest enantiopurities (>99% ee) were among the electron rich substrates, 4-OMe– 2f and 4-BocNH– 2j, however this correlation was not generalized across the substrate scope. Notably, selective C–S bond formation was observed for dianionic aryllithiums containing protic phenol (2i) and carbamate groups (2j)—offering an alternative to protecting group strategies and functional group compatibility. 3-Substituted aryllithiums (2k-n) delivered the target sulfoximines uneventfully in high yields (72–83%) and enantiopurities (95–99% ee). Di-substituted nucleophiles (2o-2t) were well tolerated regardless of the substituent nature or location, providing 67% to 83% yields with 95% to >99% ee. The more sterically encumbered 2-methyl-5-chloro aryllithium (2s) produced similar results (72% yield, 98% ee), while 2-methyl-4-NHBoc (2t) led to a slight decrease in enantiospecificity relative to its mono-substituted 4-NHBoc analog (2j) (>99% to 95% ee).
Table 1: Sulfonimidoyl transfer of t-BuSF to organolithiums.
All reactions were performed at 0.25 mmol scale unless otherwise noted. Isolated yields are reported. Enantiomeric excess (% ee) determined by chiral HPLC. Halogen-Li exchange was performed using aryl bromides and n-BuLi unless otherwise noted. aAryl iodide was used. bOrganolithium was prepared by deprotonation using n-BuLi. ct-BuLi was used.
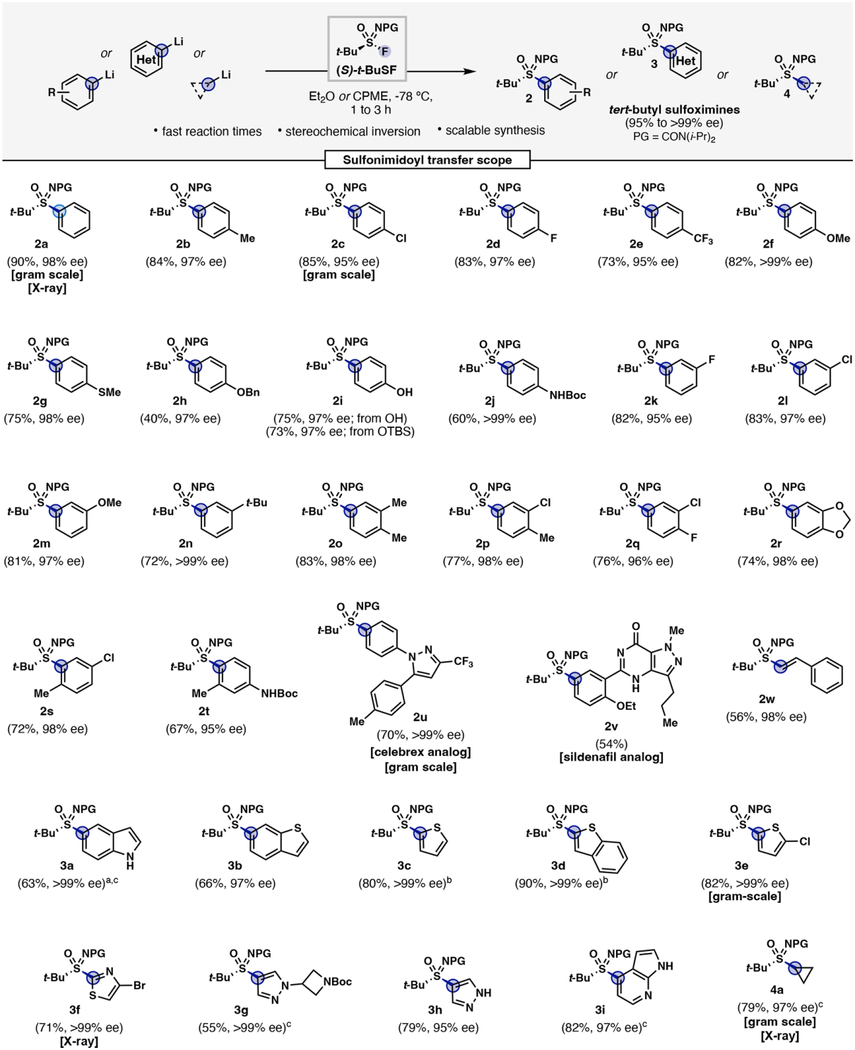
|
Late-stage asymmetric sulfonimidoyl transfer was demonstrated with pharmaceutical relevant scaffolds such as celecoxib and sildenafil to provide 2u and 2v respectively. A vinyl organolithium (2w) was found to be a suitable nucleophile that provides entry into α,β-unsaturated tert-butyl sulfoximines. Heterocyclic S(VI) functionality with defined stereocenters are of high value to the discovery sciences and have proven a formidable synthetic challenge.46 A diverse range of biologically relevant heterocycles undergo sulfonimidoyl transfer smoothly. Bis-lithiation of unprotected 5-iodoindole provided sulfoximine 3a enantiospecifically while a selective lithiation of 5-bromobenzothiophene gave rise to 3b in 97% ee. Deprotonation of thiophene and benzothiophene afforded enantiopure 3c and 3d in high yields. Substituted thiophene (3e) and thiazole (3f) delivered sulfoximines with >99% ee and were amenable to scale-up (>1 g scale). N-Substituted and N–H pyrazoles undergo sulfonimidoyl transfer at the 4-position granting access to 3g and 3h in >99% ee and 95% ee respectively. Synthesis of an S-substituted 7-azaindole sulfoximine (3i) was achieved in high yield and excellent enantiopurity with no observable substitution at the indole nitrogen. Lastly, cyclopropyl lithium was used to prepare secondary aliphatic sulfoximine 4a on gram-scale in 79% yield and 97% ee—improving the synthesis of an N-protected tert-butyl cyclopropyl sulfoximine used in the synthesis of ceralasertib.53 All tert-butyl sulfoximines prepared were found to be stereogenically stable with no observable decomposition under ambient conditions, serving as valuable intermediates for further functionalization.
Sulfonimidoyl transfer: bifunctionalization.
The bifunctional modularity of t-BuSF was enabled by a net redox-neutral de-tert-butylation/fluorination S-activation strategy of tert-butyl sulfoximines to sulfonimidoyl fluorides with complete stereochemical retention (Table 2). S(VI) to S(IV) reduction of tert-butyl sulfoximines with t-BuOK (3 eq.) in THF at 80 °C provides S(IV) sulfinyl urea intermediates (5) that are subsequently S–fluorinated with NFSI at −20 °C to enantiopure S(VI) sulfonimidoyl fluorides (6) in a single step. Although the one-pot reduction/fluorination of tert-butyl sulfoximines is highly efficient and convenient, the sulfinyl urea intermediates can be isolated in excellent yield and enantiopurity if desired (see Supplementary Information section IVb. for details). Due to the known stereogenic liability of sulfinamides (other than tert-butyl substituted),54,55 and to maintain synthetic efficiency, the stereospecific net redox-neutral S-fluorination of tert-butyl sulfoximines is preferred.
Table 2: S-activation and S-functionalization of tert-butyl sulfoximines.
All reactions were performed on 0.1–0.25 mmol scales. Isolated yields are reported. Enantiomeric excess (% ee) determined by chiral HPLC. Diastereomeric excess (de) determined by 1HNMR. Halogen-Mg exchange was performed using aryl iodide unless otherwise noted. aCommercial (Turbo)-Grignard reagent was used. bTurbo-Grignard prepared using i-PrMgCl•LiCl. cAryl bromide was used. dAllyl bromide and Mg0 was used. eNaHMDS was used as base. fTurbo-amide prepared using i-PrMgCl•LiCl. gReaction condition: Et3N, LiBr or NaI, MeCN, heat. hRacemic starting material was used. iUnable to separate by chiral HPLC.
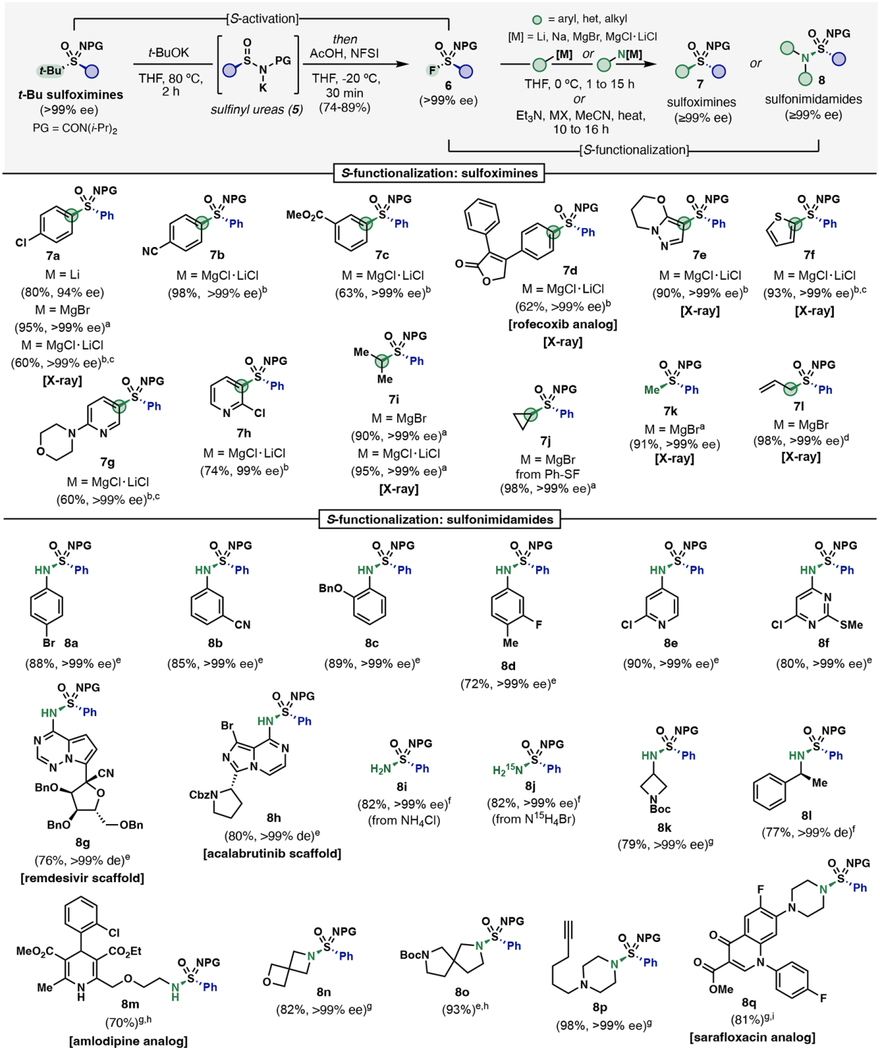
|
The second SuFEx functionalization provides entry to chiral sulfoximines (7) and sulfonimidamides (8) (Table 2). Phenyl sulfonimidoyl fluoride (S6a >99% ee) was used as an electronically neutral model system to demonstrate the reaction scope with carbon and nitrogen nucleophiles. Enantiopure aryl–(hetero)aryl and aryl–alkyl sulfoximines (7a-7l) were prepared via stereospecific addition of Grignards and turbo-Grignards to sulfonimidoyl fluorides 6 at 0 °C in THF. A decrease in enantiopurity was observed with organolithiums regardless of temperature or solvent (7a, 94% ee). Turbo-Grignards were the preferred carbon nucleophiles due to their increased functional group tolerance56 and ease of preparation. For instance, aryl-cyano and -methyl ester were well tolerated to give 7b-7d in good to excellent yield with >99% ee—7d representing a late-stage asymmetric installation of sulfoximines to the rofecoxib scaffold. An enantiopure S(VI) analog of Genentech’s S-pyrazolo sulfonimidoyl urea NLRP3 inhibitor (7e) was prepared in 90% yield. Other heterocycles including thiophene (7f) and pyridines (7g and 7h) also served as good nucleophiles, expanding the heterocyclic sulfoximine chemical space obtainable.
In addition to aromatic nucleophiles, aliphatic Grignard reagents also react enantiospecifically to provide enantiopure aryl–alkyl sulfoximines in high yields. Secondary alkyl sulfoximines 7i and 7j were prepared in excellent yield via an isopropyl Grignard (and turbo-Grignard) and cyclopropyl Grignard respectively. Similarly, primary alkyl substituents were installed enantiospecifically (7k, 7l) and in high yield—with no observable isomerization for allyl sulfoximine 7l.
Enantiospecific fluoride displacement by nitrogen nucleophiles was observed to provide enantiopure secondary, tertiary, and aromatic sulfonimidamides (8) (Table 2). Three different conditions were compatible based on the amine nucleophile. For aromatic amines, use of NaHMDS (2 eq.) at 0 °C in THF affords sulfonimidamides in high yields and >99% ee regardless of the M-HMDS counterion (M = Li, Na, K). A lack of chiral aromatic sulfonimidamide examples within the literature prompted us to emphasize this overlooked sub-class. Anilines bearing 4-Br (8a), 3-CN (8b), 2-OBn (8c), and 4-Me-3-F (8d) substituents undergo fluoride displacement smoothly to the desired sulfonimidamides. Heterocyclic amines such as amino-pyridine (8e) and amino-pyrimidine (8f) introduce common medicinal chemistry motifs to the S(VI) chemical space. An intermediate used in the synthesis of remdesivir was intercepted and sulfonimidoylated, producing analog 8g as a single diastereomer in good yield. Additionally, a sulfonimidamide analog of acalabrutinib (8h) was obtained in 80% yield under the optimized conditions.
Inspired by the results from turbo-Grignard nucleophiles, turbo-amides were explored as nitrogen nucleophiles. Upon treatment of NH4Cl with excess i-PrMgCl·LiCl, turbo-amide H2N-MgCl·LiCl rapidly undergoes fluoride displacement to give 8i in 82% yield and >99% ee. The same method was applied to 15NH4Br to provide the first reported isotopically labeled chiral sulfonimidamide 8j in an identical yield and stereochemical purity. In addition to turbo-amides, the thermal method developed by Bull and Lücking (Et3N, LiBr, MeCN, heat)52 was also compatible with N,N-diisopropyl urea protected sulfonimidoyl fluorides and aliphatic amines.
An amino-substituted N-Boc-azetidine reacted well under thermal conditions when replacing LiBr for NaI as an additive to deliver 8k in 79% yield, whereas the alternative conditions using NaHMDS or turbo-amide resulted in 62% and 71% yields respectively. The chiral turbo-amide derived from (S)-phenylethylamine produced sulfonimidamide 8l as a single diastereomer in good yield. A racemic mixture of amlodipine was found to be a suitable nucleophile under thermal conditions giving rise to sulfonimidamide 8m in 70% yield, as a mixture of epimers. Secondary amine nucleophiles perform exceptionally well with all three general conditions. Spirocyclic sulfonimidamides 8n and 8o were made accessible in high yields using Et3N/LiBr and NaHMDS conditions respectively. When alkynyl linked piperazine of 8p was used a nucleophile, NaHMDS was found to be unsuitable, presumably due to the relative acidity of alkynyl C—H versus N—H of the piperazine. However, Et3N/LiBr provided enantiopure 8p in nearly quantitative yield. Lastly, the thermal SuFEx condition was applied to a methyl ester analog of sarafloxacin granting access to stereogenically pure analog 8q in 81% yield.
The stability of sulfonimidoyl fluorides makes them attractive electrophilic intermediates and has enabled the bifunctionalization of t-BuSF. These synthetic intermediates can be intercepted in situ allowing direct access to enantiopure sulfoximines and sulfonimidamides in a single step from tert-butyl sulfoximines (Table 3A). This streamlined approach expedites the asymmetric target synthesis in just two steps from t-BuSF. Enantiopure S-substituted aryl–aryl sulfoximines (7b, 70%) and aryl–alkyl sulfoximines (7i, 68% and 7k, 74%) can be readily prepared in good yields, while amino (hetero)aryl nucleophiles provided 8a–8d in good to excellent yields. A sulfonimidamide analog of celecoxib (8r) was made available in two steps from t-BuSF, further showcasing the synthetic effectiveness of this method. Tertiary sulfonimidamide (8s) was prepared uneventfully using the corresponding turbo-amide. Overall, the reduced step count, fast reaction times, and stability of tert-butyl sulfoximine precursors, render this a practical and highly efficient method for both target- and diversity-oriented synthesis.
Successful development of t-BuSF hinged on a suitable sulfonimidoyl N–protecting group to provide stability and chemical compatibility for the sulfonimidoyl transfer and S-activation steps. Fortunately, N,N-diisopropyl sulfonimidoyl ureas exhibit the desired structural and chemical properties, and can be readily removed under mild conditions (Figure 3B). Acid-mediated cleavage of the urea with camphor sulfonic acid (CSA) in hexafluoroisopropanol (HFIP) at 60–70 °C furnishes N–H sulfoximines (9a, 9b) and sulfonimidamides (10a, 10b) without erosion of enantiopurity—lower temperatures (40–50 °C) require increased reaction times (>36 hours). Alternatively, a milder hydrolytic deprotection (DMSO/H2O, 80 °C) is applicable for secondary sulfonimidamides (10b). The stereochemical assignments from t-BuSF to N–H sulfonimidoyls were confirmed by single crystal X-ray crystallography using the stepwise synthesis of 9a (see Supplementary Information section VII.).
Figure 3:
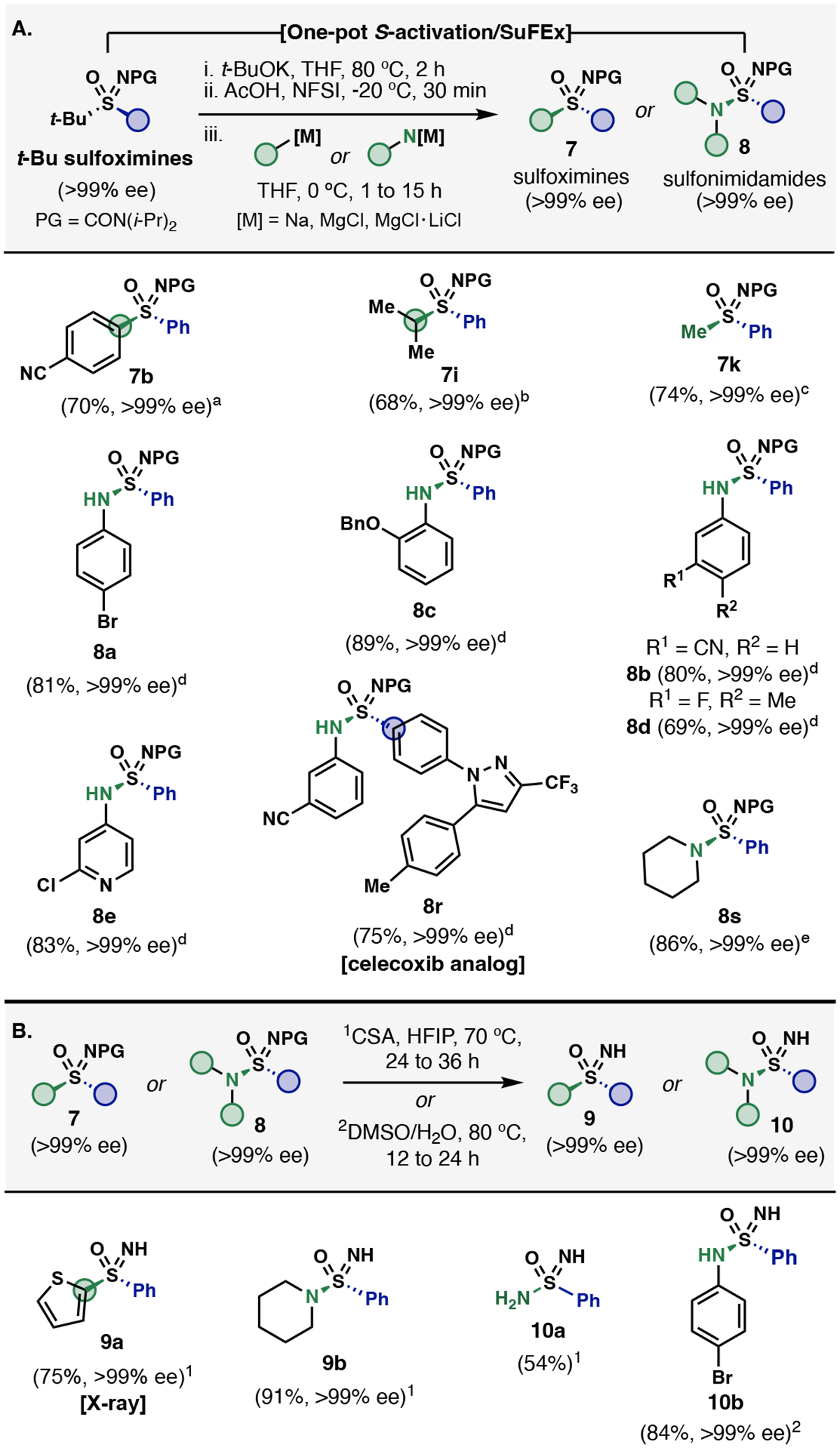
A. One-step synthesis of enantiopure sulfoximines and sulfonimidamides from tert-butyl sulfoximines via S-activation/SuFEx. B. Deprotection of N,N-diisopropyl urea. All reactions were performed on 0.1–0.25 mmol scales. Isolated yields are reported. Enantiomeric excess (% ee) determined by chiral HPLC. aTurbo-Grignard prepared from aryl iodide using i-PrMgCl•LiCl. bi-PrMgCl•LiCl was used. cMeMgCl was used. dNaHMDS was used as base. eTurbo-amide prepared with i-PrMgCl•LiCl.
Synthetic applications.
Five pharmaceutical targets and intermediates were prepared to demonstrate the potential utility of t-BuSF in medicinal chemistry (Figure 4). 4-Chlorophenyl tert-butyl sulfoximine 2c was identified as a common chiral starting material for the synthesis of a recently discovered modulator of the dopamine D1 receptor and a PYK2 inhibitor developed by Pfizer (Figure 4A). Sulfoximine 2c was prepared on a gram-scale using aryllithium 11 to provide enantiopure material in 80% yield after recrystallization. The target sulfoximine 13 was made accessible from two approaches: utilizing sulfonimidoyl fluoride 12 as either an isolable or in situ intermediate. A three-step route (74% yield, >99% ee) provides a sulfonimidoyl fluoride intermediate (12) that serves as a diversifiable point for analog development (highlighted in blue) while a two-step route (71% yield, >99% ee) improves efficiency by reducing step count and purifications. Diverging from sulfoximine 13, Suzuki cross-coupling with boronic acid 14 followed by deprotection gave rise to 15 and establishes a formal route to a dopamine D1 modulator.44 Conversely, an Ullmann coupling between sulfoximine 13 and NH4OH delivered enantiopure 4-aminophenyl protected sulfoximine 16, for the synthesis of Pfizer’s sulfoximinecontaining PYK2 inhibitors.7
Figure 4: Asymmetric synthesis of pharmaceutically relevant compounds and intermediates from t-BuSF.
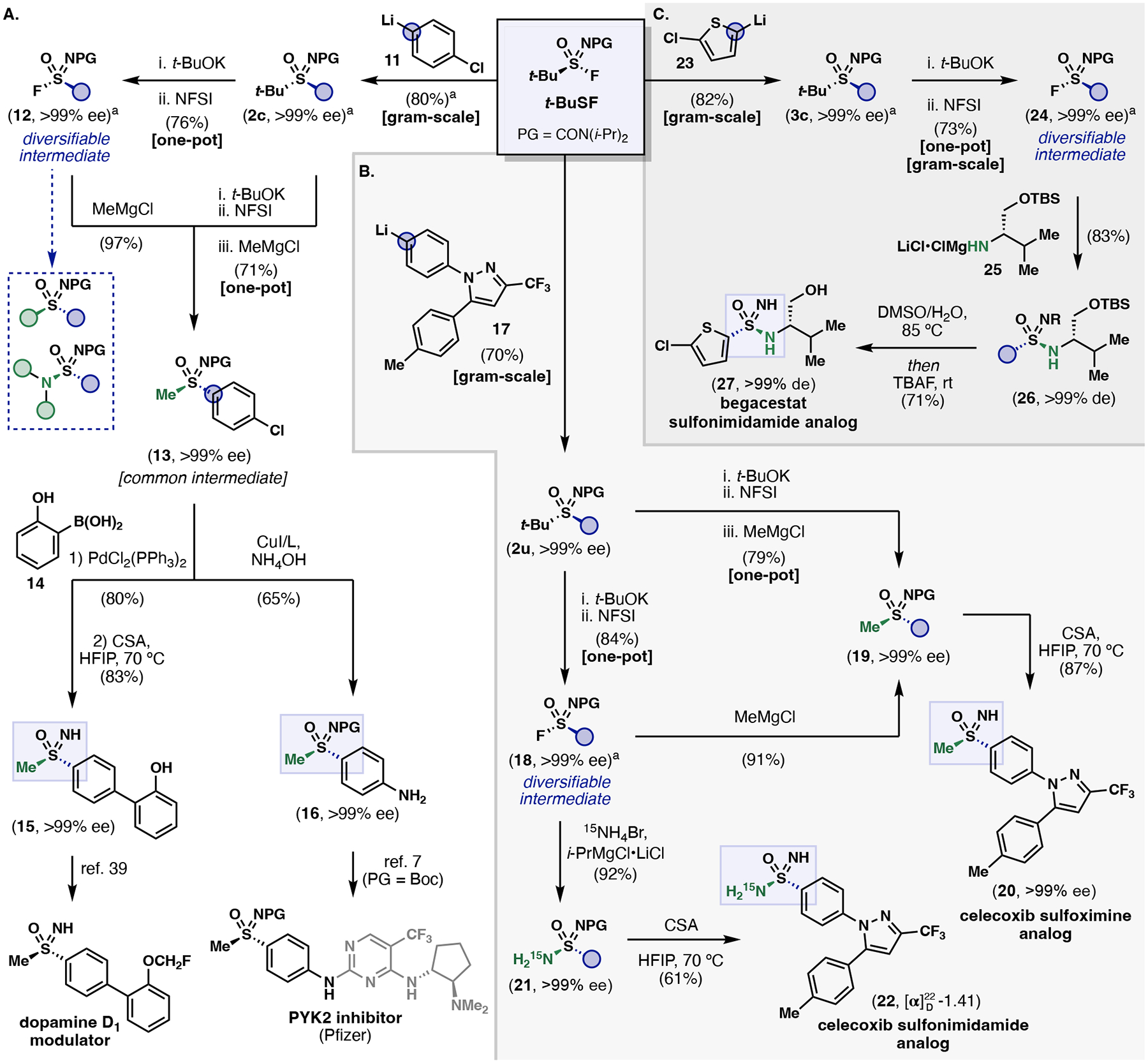
A. Formal syntheses of a dopamine D1 modulator and a synthetic intermediate for the preparation of PYK2 inhibitor from Pfizer. B. Synthesis of enantiopure sulfoximine and sulfonimidamide analogs of celecoxib. C. Synthesis of a begacestat sulfonimidamide analog as a single diastereomer. aYield after recrystallization.
By applying the asymmetric sulfonimidoyl transfer of t-BuSF to 17, a route to enantiopure S(VI) analogs of celecoxib can be achieved (Figure 4B). The first S-functionalization produced 2u on gram-scale and in good yield (70% yield). S-Activation of 2u provides an enantiopure sulfonimidoyl fluoride intermediate 18 (84% yield) which was subsequently treated with MeMgCl to give sulfoximine 19 (91% yield, >99% ee). Alternatively, sulfoximine 19 was obtained in a single step from tert-butyl sulfoximine 2u (79%). Deprotection with CSA led to an enantiopure N–H sulfoximine celecoxib analog (20) in three-steps from t-BuSF. In addition, the first synthesis of an isotopically labeled sulfonimidamide analog of celecoxib was realized via fluoride displacement of 18 with H215N–MgCl·LiCl (92% yield, >99% ee). Upon deprotection, 15N-labeled sulfonimidamide 22 was obtained and to our knowledge, is the first example of a chiral NH-NH2 sulfonimidamide. The specific rotation observed for 22, along with the stereospecific reactivity of urea protected sulfonimidoyl fluorides and deprotection conditions, suggests that the S(VI) center is stereochemically defined.
Potency and selectivity profiles for sulfonimidamide analogs of begacestat are thought to be dependent upon the chirality at the S(VI) stereocenter.8 The reported five-step analog synthesis relied on HPLC purifications with an average yield of less than 12%.8 Overall synthetic efficiency of begacestat sulfonimidamide analogs were improved by employing t-BuSF as a chiral bifunctional linchpin (Figure 4C). A gram-scale sulfonimidoyl transfer to 23 gave enantiopure 3c in high yield. Subsequent S-activation of 3c provided 24 (73% yield, >99% ee) that underwent turbo-amide mediated SuFEx to 26 (83% yield, 99% de). Removal of the protecting groups (DMSO/water, 85 °C then TBAF, rt) produced begacestat analog 27 in good yield. This four-step synthesis delivered the target sulfonimidamide analog as a pure stereoisomer in 35% overall yield from t-BuSF without the need for HPLC separations. The rapid syntheses of celecoxib and begacestat S(VI) analogs further highlights the utility of t-BuSF and the advantages it will offer to discovery programs.
Conclusion
A bench-stable chiral bifunctional S(VI) transfer reagent (t-BuSF) for the asymmetric synthesis of sulfoximines, sulfonimidoyl fluorides and sulfonimidamides has been developed and applied to prepare enantiopure biologically relevant compounds. Sulfonimidoyl transfer to organolithiums with t-BuSF via SuFEx was enabled by the discovery of a new sulfonimidoyl N–H protecting group that provides reagent and product stability, good synthetic compatibility, and is removable under mild conditions. Access to chiral S(VI) building blocks and late-stage intermediates of varying complexity can be achieved in as little as two steps, a stark improvement compared to current asymmetric methods. Additionally, the stereogenic stability of tert-butyl sulfoximines and sulfonimidoyl fluoride intermediates removes the liability associated with sulfinamides. The versatility of t-BuSF has been demonstrated in over seventy examples and applied to five pharmaceutical targets. Given the cost-effectiveness and the chemical space accessible from this reagent platform, it is expected to have positive impacts on the discovery sciences from the development of new medicines and agrochemicals to the discovery of new ligands, organocatalysts and materials. Investigations using t-BuSF to access the remaining S(VI) and S(IV) terrain and alternative S-activation strategies are currently under investigation and will be reported in due course.
Methods
General methods for the synthesis of t-BuSF and bis-functionalization of sulfur with carbon and nitrogen nucleophiles are as follows. More in-depth methods including troubleshooting, graphical procedures, and reagent stability studies can be found in the Supplementary Information.
General synthesis of t-BuSF.
In a suitable septum capped round-bottom flask equipped with a stir bar and argon balloon was added (R)-tert-butyl sulfinamide (1 eq.) followed by THF (0.1–0.2 M) then cooled to 0 °C. NaH (2.5 eq., 60% wt) was added portion-wise then stirred for 20 minutes. ClCON(i-Pr)2 (1 eq.) was added portion-wise then stirred at 0 °C for 1.5 hours. NFSI (1.05 eq.) was added in one portion then stirred at 0 °C for an additional 1 hour. The reaction mixture was diluted with 10% EtOAc in hexanes then filtered through a medium porous sintered glass funnel while rinsing with 10% EtOAc in hexanes. The organic solution was washed with 10% KI aqueous solution (× 3) and brine (× 3). The solvent was dried over anhydrous Na2SO4, filtered and solvents removed under reduced pressure to give a yellow oil. Further purification by silica gel column chromatography using hexanes/EtOAc (0% to 20% EtOAc) provided t-BuSF as a clear colorless oil that solidified to a white crystalline solid under reduced pressure.
General sulfonimidoyl transfer using t-BuSF.
To a 10 mL flame dried round-bottom flask equipped with a stir bar and argon balloon was added aryl halide (0.375 mmol, 1.5 eq.) followed by anhydrous Et2O (2 mL) then cooled to −78 °C. n-BuLi (0.375 mmol, 1.5 eq., 2.5 M in hexane) was added dropwise and stirred for 1 hour (lithium-halogen exchange) then t-BuSF (66.5 mg, 0.25 mmol, 1 eq.) in Et2O (0.5 mL) was added dropwise. The reaction mixture was stirred at −78 °C for 1 hour. The reaction was quenched with MeOH (0.2 mL) and saturated aqueous NH4Cl (5 mL) then extracted with EtOAc (5 mL × 3), washed with water (10 mL × 3) and brine (10 mL × 3). Dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. Further purification was performed by silica gel column chromatography to give the desired tert-butyl sulfoximines.
General S-activation and S-functionalization of tert-butyl sulfoximines.
To a flame dried round-bottomed flask equipped with a stir bar and argon balloon was added tert-butyl sulfoximine (1 eq.) followed by anhydrous THF (0.3 M). Once dissolved, solid anhydrous t-BuOK (3.0 eq) was added, and the reaction stirred at room temperature for 2–5 minutes, the argon balloon removed, and the reaction heated to 80 °C for 2 hours. The reaction mixture was placed under an atmosphere of argon and cooled to −20 °C with dry ice and acetone bath. AcOH (2 eq) dissolved in anhydrous THF was added slowly to dilute the reaction to 0.1 M then solid NSFI (1 eq.) was added in one portion. The reaction stirred at −20 °C for 30 minutes then either the Grignard or amine nucleophile was added.
Grignard nucleophiles:
The preformed Grignard reagent (2 eq.) was added dropwise and stirred at −20 °C. Upon completion, the reaction was quenched with saturated aqueous NH4Cl solution then extracted with EtOAc (x 3). The combined organic layers were washed with water (x 3) then brine (x 3), dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. Further purification by silica gel column chromatography provided the desired sulfoximines.
Amine nucleophiles:
The amine (1 eq.) and NaHMDS (2 eq.) or preformed turbo-amide (2 eq.) were added dropwise then slowly warmed to room temperature. Upon completion, the reaction was quenched with silica gel, DCM was added, and solvent removed to adsorb the crude material to silica gel. Purification by column chromatography provided the desired sulfonimidamides.
Supplementary Material
Acknowledgements
We gratefully acknowledge the National Institutes of Health (NIGMS R35-GM142577, J.M.L.) for support of this research. This work has also been supported in part by the Chemical Biology Core Facility at the H. Lee Moffitt Cancer Center & Research Institute, an NCI designated Comprehensive Cancer Center (P30-CA076292) and the University of South Florida’s Chemical Purification Analysis and Screening Core Facility (CPAS). We thank H. Lawrence (Moffitt) and Laurent Calcul (USF) for NMR and HRMS support, and Q. Tang (USF) for assistance with X-ray crystallographic analysis.
Footnotes
Competing interests
A patent application naming J.M.L, Z.P.S., and S.T. as inventors has been filed by H. Lee Moffitt Cancer Center & Research Institute, which covers the synthetic methods and development regarding a chiral bifunctional S(VI) reagent for the asymmetric synthesis of sulfur-containing functional groups.
Data availability
All data including experimental procedures, compound characterization data, and stability analysis data are available within the article and its Supplementary Information files. X-Ray crystallographic data for the structures within this article and the Supplementary Information have been deposited with the Cambridge Crystallographic Data Centre. The data can be obtained free of charge from https://www.ccdc.cam.ac.uk/structures/. Compounds with X-ray structures: (S)-t-BuSF (CCDC 2243804), 2a (CCDC 2243801), 3f (CCDC 2243803), 4a (CCDC 2243800), 7a (CCDC 2243808), 7d (CCDC 2243809) 7e (CCDC 2243805), 7f (CCDC 2243806), 7i (CCDC 2243802), 7k (CCDC 2243799), 7l (CCDC 2243807), 9a (CCDC 2243810), S6a (CCDC 2243798).
References
- 1.Scott KA & Njardarson JT Analysis of US FDA-approved drugs containing sulfur atoms. Top. Curr. Chem 376, 5 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Devendar P & Yang GF Sulfur-containing agrochemicals. Top. Curr. Chem 375, 82 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Takimiya K, Osaka I, Mori T & Nakano M Organic semiconductors based on [1]benzothieno[3,2-b][1]benzothiophene substructure. Acc. Chem. Res 47, 1493–1502 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Boyd DA Sulfur and its role in modern materials science. Angew. Chem. Int. Ed 55, 15486–15502 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Yue T-J, Wang L-Y & Ren W-M The synthesis of degradable sulfur-containing polymers: Precise control of structure and stereochemistry. Polym. Chem 12, 6650–6666 (2021). [Google Scholar]
- 6.Gege C, et al. A helicase-primase drug candidate with sufficient target tissue exposure affects latent neural herpes simplex virus infections. Sci. Transl. Med 13, eabf8668 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Walker DP, et al. Sulfoximine-substituted trifluoromethylpyrimidine analogs as inhibitors of proline-rich tyrosine kinase 2 (PYK2) show reduced hERG activity. Bioorg. Med. Chem. Lett 19, 3253–3258 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Sehgelmeble F, et al. Sulfonimidamides as sulfonamides bioisosteres: Rational evaluation through synthetic, in vitro, and in vivo studies with γ-secretase inhibitors. ChemMedChem 7, 396–399 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Foote KM, et al. Discovery and characterization of AZD6738, a potent inhibitor of ataxia telangiectasia mutated and Rad3 related (ATR) kinase with application as an anticancer agent. J. Med. Chem 61, 9889–9907 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Coleman L, Farady C, Gatlik E & Schieker M, Dosing regimen for an NLRP3 inhibitor in the treatment of osteoarthritis. Patent WO 2023/002399 A1 (2023).
- 11.Lücking U Sulfoximines: A neglected opportunity in medicinal chemistry. Angew. Chem. Int. Ed 52, 9399–9408 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Lücking U Neglected sulfur(VI) pharmacophores in drug discovery: Exploration of novel chemical space by the interplay of drug design and method development. Org. Chem. Front 6, 1319–1324 (2019). [Google Scholar]
- 13.Zhu Y, et al. Discovery and characterization of sulfoxaflor, a novel insecticide targeting sap-feeding pests. J. Agric. Food. Chem 59, 2950–2957 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Siemeister G, et al. Bay 1000394, a novel cyclin-dependent kinase inhibitor, with potent antitumor activity in mono- and in combination treatment upon oral application. Mol. Cancer Ther 11, 2265–2273 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Lücking U, et al. Changing for the better: Discovery of the highly potent and selective CDK9 inhibitor VIP152 suitable for once weekly intravenous dosing for the treatment of cancer. J. Med. Chem 64, 11651–11674 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Agarwal S, et al. Discovery of N-cyano-sulfoximineurea derivatives as potent and orally bioavailable NLRP3 inflammasome inhibitors. ACS Med. Chem. Lett 11, 414–418 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madurka I, et al. DFV890: A new oral NLR3 inhibitor—tested in an early phase 2a randomised clinical trial in patients with covid-19 pneumonia and impaired respiratory function. Infection 51, 641–654 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mäder P & Kattner L Sulfoximines as rising stars in modern drug discovery? Current status and perspective on an emerging functional group in medicinal chemistry. J. Med. Chem 63, 14243–14275 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Chinthakindi PK, et al. Sulfonimidamides in medicinal and agricultural chemistry. Angew. Chem. Int. Ed 56, 4100–4109 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Frings M, Bolm C, Blum A & Gnamm C Sulfoximines from a medicinal chemist’s perspective: Physicochemical and in vitro parameters relevant for drug discovery. Eur. J. Med. Chem 126, 225–245 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Liang D-D, et al. Silicon-free SuFEx reactions of sulfonimidoyl fluorides: Scope, enantioselectivity, and mechanism. Angew. Chem. Int. Ed 59, 7494–7500 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang D-D, Pujari SP, Subramaniam M, Besten M & Zuilhof H Configurationally chiral SuFEx-based polymers. Angew. Chem. Int. Ed 61, e202116158 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mukherjee H, et al. A study of the reactivity of S(VI)–F containing warheads with nucleophilic amino-acid side chains under physiological conditions. Org. Biomol. Chem 15, 9685–9695 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Luisi R & Bull JA Synthesis of sulfoximines and sulfonimidamides using hypervalent iodine mediated NH transfer. Molecules 28, 1120 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu H, Li Z & Bolm C Copper-catalyzed transsulfinamidation of sulfinamides as a key step in the preparation of sulfonamides and sulfonimidamides. Angew. Chem. Int. Ed 57, 15602–15605 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Bizet V, Hendriks CMM & Bolm C Sulfur imidations: Access to sulfimides and sulfoximines. Chem. Soc. Rev 44, 3378–3390 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Chen Y & Gibson J A convenient synthetic route to sulfonimidamides from sulfonamides. RSC Adv. 5, 4171–4174 (2015). [Google Scholar]
- 28.Matos PM & Stockman RA Synthetic approaches and applications of sulfonimidates. Org. Biomol. Chem 18, 6429–6442 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Johnson CR, et al. Preparation and reactions of sulfonimidoyl fluorides. J. Org. Chem 48, 1–3 (1983). [Google Scholar]
- 30.Lo PKT & Willis MC Nickel(II)-catalyzed addition of aryl and heteroaryl boroxines to the sulfinylamine reagent TrNSO: The catalytic synthesis of sulfinamides, sulfonimidamides, and primary sulfonamides. J. Am. Chem. Soc 143, 15576–15581 (2021). [DOI] [PubMed] [Google Scholar]
- 31.Ding M, Zhang Z-X, Davies TQ & Willis MC A silyl sulfinylamine reagent enables the modular synthesis of sulfonimidamides via primary sulfinamides. Org. Lett 24, 1711–1715 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bremerich M, Conrads CM, Langletz T & Bolm C Additions to N-sulfinylamines as an approach for the metal-free synthesis of sulfonimidamides: O-Benzotriazolyl sulfonimidates as activated intermediates. Angew. Chem. Int. Ed 58, 19014–19020 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zasukha SV, et al. Sulfonimidamides and imidosulfuric diamides: Compounds from an underexplored part of biologically relevant chemical space. Chem. Eur. J 25, 6928–6940 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Gao B, Li S, Wu P, Moses JE & Sharpless KB SuFEx chemistry of thionyl tetrafluoride (SOF4) with organolithium nucleophiles: Synthesis of sulfonimidoyl fluorides, sulfoximines, sulfonimidamides, and sulfonimidates. Angew. Chem. Int. Ed 57, 1939–1943 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davies TQ & Willis MC Rediscovering sulfinylamines as reagents for organic synthesis. Chem. Eur. J 27, 8918–8927 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L & Cornella J A unified strategy for arylsulfur(VI) fluorides from aryl halides: Access to Ar-SOF3 compounds. Angew. Chem. Int. Ed 59, 23510–23515 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y, et al. Rapid access to N-protected sulfonimidoyl fluorides: Divergent synthesis of sulfonamides and sulfonimidamides. Org. Lett 23, 3975–3980 (2021). [DOI] [PubMed] [Google Scholar]
- 39.Passia MT, et al. Acid-mediated imidazole-to-fluorine exchange for the synthesis of sulfonyl and sulfonimidoyl fluorides. Org. Lett 24, 8802–8805 (2022). [DOI] [PubMed] [Google Scholar]
- 40.Passia MT, Amer MM, Demaerel J & Bolm C Synthesis of sulfonyl, sulfonimidoyl, and sulfoxyl fluorides under solvent-free mechanochemical conditions in a mixer mill by imidazole-to-fluorine exchange. ACS Sustainable Chemistry & Engineering 11, 6838–6843 (2023). [Google Scholar]
- 41.Kleymann G & Gege C, Preparation of enantiomers of substituted thiazoles as antiviral compounds. Patent WO 2019/068817 A1 (2019).
- 42.Brandt J & Gais H-J An efficient resolution of (±)-S-methyl-S-phenylsulfoximine with (+)-10-camphorsulfonic acid by the method of half-quantities. Tetrahedron Asymmetry 8, 909–912 (1997). [Google Scholar]
- 43.Zhang X, Wang F & Tan C-H Asymmetric synthesis of S(IV) and S(VI) stereogenic centers. JACS Au 3, 700–714 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.García-Cárceles J, et al. 2-(fluoromethoxy)-4′-(s-methanesulfonimidoyl)-1,1′-biphenyl (UCM-1306), an orally bioavailable positive allosteric modulator of the human dopamine D1 receptor for Parkinson’s disease. J. Med. Chem 65, 12256–12272 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang X, Ang ECX, Yang Z, Kee CW & Tan C-H Synthesis of chiral sulfinate esters by asymmetric condensation. Nature 604, 298–303 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aota Y, Kano T & Maruoka K Asymmetric synthesis of chiral sulfoximines via the S-arylation of sulfinamides. J. Am. Chem. Soc 141, 19263–19268 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Aota Y, Kano T & Maruoka K Asymmetric synthesis of chiral sulfoximines through the S-alkylation of sulfinamides. Angew. Chem. Int. Ed 58, 17661–17665 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Maeda Y, et al. Practical asymmetric synthesis of chiral sulfoximines via sulfur-selective alkylation. J. Org. Chem 87, 3652–3660 (2022). [DOI] [PubMed] [Google Scholar]
- 49.Mendonça Matos P, Lewis W, Argent SP, Moore JC & Stockman RA General method for the asymmetric synthesis of N–H sulfoximines via C–S bond formation. Org. Lett 22, 2776–2780 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Yang G-F, et al. Synthesis of chiral sulfonimidoyl chloride via desymmetrizing enantioselective hydrolysis. J. Am. Chem. Soc 145, 5439–5446 (2023). [DOI] [PubMed] [Google Scholar]
- 51.Greed S, Symes O & Bull JA Stereospecific reaction of sulfonimidoyl fluorides with Grignard reagents for the synthesis of enantioenriched sulfoximines. Chem. Commun 58, 5387–5390 (2022). [DOI] [PubMed] [Google Scholar]
- 52.Greed S, et al. Synthesis of highly enantioenriched sulfonimidoyl fluorides and sulfonimidamides by stereospecific sulfur–fluorine exchange (SuFEx) reaction. Chem. Eur. J 26, 12533–12538 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shultz ZP, Scattolin T, Wojtas L & Lopchuk JM Stereospecific α-(hetero)arylation of sulfoximines and sulfonimidamides. Nat. Synth, 1, 170–179 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Booms RE & Cram DJ Stereochemistry of sulfur compounds. III. Radical-chain mechanism for racemization of sulfinamides. J. Am. Chem. Soc 94, 5438–5446 (1972). [Google Scholar]
- 55.Clarke V & Cole ER Sulfenamides and sulfinamides IX exchange reactions of aryl sulfinamides. Phosphorus Sulf Silicon Relat. Elem 92, 45–50 (1994). [Google Scholar]
- 56.Li-Yuan Bao R, Zhao R & Shi L Progress and developments in the turbo Grignard reagent i-PrMgCl·LiCl: A ten-year journey. Chem. Commun 51, 6884–6900 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data including experimental procedures, compound characterization data, and stability analysis data are available within the article and its Supplementary Information files. X-Ray crystallographic data for the structures within this article and the Supplementary Information have been deposited with the Cambridge Crystallographic Data Centre. The data can be obtained free of charge from https://www.ccdc.cam.ac.uk/structures/. Compounds with X-ray structures: (S)-t-BuSF (CCDC 2243804), 2a (CCDC 2243801), 3f (CCDC 2243803), 4a (CCDC 2243800), 7a (CCDC 2243808), 7d (CCDC 2243809) 7e (CCDC 2243805), 7f (CCDC 2243806), 7i (CCDC 2243802), 7k (CCDC 2243799), 7l (CCDC 2243807), 9a (CCDC 2243810), S6a (CCDC 2243798).