

Abstract
Background
Prevention of diabetic heart myocardial ischemia–reperfusion (IR) injury (MIRI) is challenging. Propofol attenuates MIRI through its reactive oxygen species scavenging property at high doses, while its use at high doses causes hemodynamic instability. Salvianolic acid A (SAA) is a potent antioxidant that confers protection against MIRI. Both propofol and SAA affect metabolic profiles through regulating Adenosine 5‘-monophosphate-activated protein kinase (AMPK). The aim of this study was to investigate the protective effects and underlying mechanisms of low doses of propofol combined with SAA against diabetic MIRI.
Methods
Diabetes was induced in mice by a high-fat diet followed by streptozotocin injection, and MIRI was induced by coronary artery occlusion and reperfusion. Mice were treated with propofol at 46 mg/kg/h without or with SAA at 10 mg/kg/h during IR. Cardiac origin H9c2 cells were exposed to high glucose (HG) and palmitic acid (PAL) for 24 h in the absence or presence of cluster of differentiation 36 (CD36) overexpression or AMPK gene knockdown, followed by hypoxia/reoxygenation (HR) for 6 and 12 h.
Results
Diabetes-exacerbated MIRI is evidenced as significant increases in post-ischemic infarction with reductions in phosphorylated (p)-AMPK and increases in CD36 and ferroptosis. Propofol moderately yet significantly attenuated all the abovementioned changes, while propofol plus SAA conferred superior protection against MIRI to that of propofol. In vitro, exposure of H9c2 cells under HG and PAL decreased cell viability and increased oxidative stress that was concomitant with increased levels of ferroptosis and a significant increase in CD36, while p-AMPK was significantly reduced. Co-administration of low concentrations of propofol and SAA at 12.5 μM in H9c2 cells significantly reduced oxidative stress, ferroptosis and CD36 expression, while increasing p-AMPK compared to the effects of propofol at 25 μM. Moreover, either CD36 overexpression or AMPK silence significantly exacerbated HR-induced cellular injuries and ferroptosis, and canceled propofol- and SAA-mediated protection. Notably, p-AMPK expression was downregulated after CD36 overexpression, while AMPK knockdown did not affect CD36 expression.
Conclusions
Combinational usage of propofol and SAA confers superior cellular protective effects to the use of high-dose propofol alone, and it does so through inhibiting HR-induced CD36 overexpression to upregulate p-AMPK.
Keywords: Diabetic cardiomyopathy, Myocardial ischemia–reperfusion, Salvianolic acid A, AMPK, CD36, Propofol, Diabetes, Palmitic acid
Highlights.
A combination of propofol and salvianolic acid A conferred synergistic protective effects against myocardial ischemia–reperfusion injury in diabetes.
Inhibition of myocardial ischemia–reperfusion injury-induced increase in ferroptosis may represent a major mechanism whereby propofol and salvianolic acid A confer cardioprotection in diabetes.
Propofol combined with salvianolic acid A activated AMPK through inhibiting CD36 under diabetic condition, leading to reduced ferroptosis and cardiomyocyte hypoxia/reoxygenation injury.
The combined application of low-dose propofol and salvianolic acid A to achieve superior cardioprotection to the use of high-dose propofol may effectively avoid hemodynamic instability while increasing its antioxidant properties, which may have significant clinical implications.
Background
People with diabetes, especially those out of glycemic control, are at higher risk of morbidity and mortality after myocardial ischemia during the perioperative period than people without diabetes [1]. People with severe myocardial ischemia or infarction may have to undergo procedures to re-establish coronary blood flow to rescue the ischemic myocardium, but post-ischemic reperfusion also leads to secondary attack, called myocardial ischemic–reperfusion injury (MIRI) [2]. Furthermore, it is widely recognized that diabetic cardiomyopathy (DCM) will further exacerbate MIRI and post-ischemic complications. Meanwhile, the alterations in molecular mechanisms that occur during MIRI are complex and are dependent not only on the severity and types of heart disease present but also on the co-existence of diabetes. Alteration in cardiac energy metabolism in diabetic subjects may adversely affect cardiac susceptibility to MIRI, while myocardial fatty acid (FA) oxidation increases in diabetes but decreases during MIRI due to a limitation of oxygen supply [3].
As one of the main energy sources in the heart, proper FA function/oxidation depends on a number of cofactors involved in oxidative phosphorylation in the mitochondria [4]. Cluster of differentiation 36 (CD36), a major FA transporter protein, belongs to the class B scavenger receptor family and plays a crucial role in the uptake of long-chain FAs (LCFA) [5]. Myocardial metabolic remodeling always occurs prior to structural remodeling in response to external stimuli such as ischemia–reperfusion (IR) and hyperglycemia, CD36 deficiency alleviates DCM and atherosclerosis, while over-expression of CD36 in non-diabetic rodents paradoxically has been shown to ameliorate myocardial IR damage [6]. Although debates still exist, there is no doubt that CD36 may be an important therapeutic target in cardiovascular disease. Also, the therapeutic potential of Adenosine 5′-monophosphate-activated protein kinase (AMPK), a key energy sensor, is widely emphasized for the regulation of various cell death (apoptosis, autophagy, mitophagy, ferroptosis, etc.) and energy metabolism substrates including lipids, glucose and protein [7]. Recent studies have demonstrated that activation of AMPK attenuated post-ischemic cardiac damage in subjects with DCM with concomitant reductions of programmed cell death or mitochondrial fission [8,9]. Interestingly, a study [10] found that reduction in the phosphorylation of AMPK ameliorated oxygen–glucose deprivation and re-oxygenation-triggered neuronal injury via inhibition of autophagy, which is contrary to findings of previous studies.
Ferroptosis, a newly discussed form of programmed cell death, but distinctly different from apoptosis, autophagy and other types of cell death, has been extensively noticed and studied in various diseases [11]. Ferroptosis was evidenced in the heart of type 2 diabetic mice with DCM. For instance, recent studies suggested that enhancing the nuclear factor erythroid 2-Related Factor 2 (Nrf2) / heme oxygenase 1 (HO-1) pathway or activating AMPK conferred cardio-protective effects by reducing ferroptosis in vitro and in vivo [12,13], and Li et al. [14] showed that down-regulation of CD36 ameliorated myocardial injury and decreased the expression level of ferroptosis-related factors in DCM treatment. Meanwhile, multiple studies have provided a variety of potential mechanisms acting on IR injury and support a deleterious role of ferroptosis in cardiomyocytes due to the level of oxidative stress and lipid peroxidation [15,16]. Taken together, although available evidence suggests that both AMPK and CD36, respectively, interact with ferroptosis closely, it is yet hard to know whether AMPK or CD36 may play a leading role in the development of DCM and exacerbate myocardial IR injury.
Studies have shown that propofol counteracts myocardial IR injury by reducing oxidative stress, inflammatory response and cell death, and restoring mitochondrial function [17,18], resulting from its phenolic structure, but these beneficial effects of propofol can only be achieved at a relatively high concentration that is not applicable to clinical settings. High-dose propofol may lead to unstable hemodynamics and can hardly be used to achieve satisfactory cardiac protection in diabetic subjects suffering from MIRI. Salvianolic acid A (SAA), extracted from the roots of a traditional Chinese medicinal herb, Salvia miltiorrhiza, exerts cardio-protective effects against reactive oxygen species (ROS) due to its polyphenolic structure [19]. SAA infused into the heart via a cardiac perfusion device can effectively reduce post-ischemic myocardial injury in rats [20]. Given that increasing the dose of propofol to enhance its antioxidant capacity may cause hemodynamic instability and even end with lethal cardiac shock, we thus proposed a new strategy of combining propofol with SAA for cardio-protection against cardiac injury in diabetic patients suffering from MIRI.
In this study, we hypothesized that the combined administration of propofol and SAA ameliorates ferroptosis and attenuates diabetic MIRI via modulating the CD36/AMPK signaling pathway.
Methods
Type 2 diabetes mellitus model
The animal study was approved by the Committee on the Use of Live Animals, and all experimental protocols and animal handling procedures were conducted according to the recommendations in the principles of animal care. Healthy male C57BL/6 J mice were adapted to a suitable environment for at least 1 week. Then, the mice were fed a high-fat diet (HFD) for 8 weeks followed by intraperitoneal injection of 1% streptozotocin (STZ, Solarbio, China) at a dose of 50 mg/kg once a day for three consecutive days to induce type 2 diabetes mellitus (T2DM). Blood glucose was measured by tail vein blood 7 days later. Mice with fasting glucose levels ≥16.7 mM were considered diabetic.
Establishment of in vivo MIRI and assessment of myocardial infarct size
At 6 weeks after diabetes induction, mice were anesthetized by intraperitoneal injection of pentobarbital (80 mg/kg), tracheal intubation was implemented and they were ventilated with an animal ventilator. For the first part of the in vivo study (part 1), mice were randomly assigned into four groups (n = 6 per group), namely non-diabetic control (NC), T2DM group, non-diabetic control with MIRI group (NC + IR) and T2DM with MIRI group (T2DM + IR). All mice in the MIRI groups underwent 30 min of left anterior descending coronary artery occlusion followed by 120 min of reperfusion. Evans blue and 2,3,5-triphenyltetrazolium chloride (TTC) double staining was used to determine area at risk and infarct area. The Evans blue-stained area (blue), TTC-stained area (red; area at risk) and TTC-negative staining area (pale; infarct myocardium) were photographed and measured with ImageJ. For the second part of the in vivo study (part 2), mice were divided into eight groups (n = 6 per group): NC; T2DM; NC + IR; T2DM + IR; T2DM + IR + SAA2; T2DM + IR + propofol1; T2DM + IR + propofol3; T2DM + IR + propofol1 + SAA2. Here, SAA2 denotes SAA at a chosen dosage based on preliminary study (i.e. SAA1: 5, SAA2: 10 and SAA3: 15 mg/kg/h; see online supplementary data). propofol1 and propofol3 denote propofol dosages based on preliminary study (i.e. propofol1: 46, propofol2: 68, propofol3: 90 and propofol4: 112 mg/kg/h; see online supplementary data). SAA2 (10 mg/kg/h) was continuously infused via jugular vein catheter using a micro-infusion pump starting at 10 min before inducing myocardial ischemia and throughout the period of MIRI. For mice receiving propofol treatment, a loading dose of propofol (12 mg/kg) bolus injection was given 10 min before inducing myocardial ischemia followed by continuous infusion ofpropofol at 46 (propofol1) or 90 (propofol3) mg/kg/h throughout MIRI.
Physiological assessment of cardiac function
Mice were anesthetized with isoflurane and placed on the rail system to maintain body temperature. Echocardiography was performed with a small-animal high-resolution ultrasound system equipped with a 40-MHz single-element transducer (Visual Sonics Vevo 2100, Canada). An M-mode image was acquired at the level of the papillary muscle of the left ventricle from the long-axis view for evaluation of fractional shortening (FS) and the ejection fraction (EF).
Biochemical analysis
Blood collected from mice using an eye enucleation method was further processed by centrifugation at 3000 rpm for 15 min to obtain serum. A biochemical analyser is an instrument that uses the principle of photoelectric colorimetry and biochemical analysis to measure a specific chemical component in body fluids. The levels of lactate dehydrogenase (LDH), triglyceride (TG) and cardiac-type creatine kinase isoenzyme (CK-MB; Donglin Biotechnology, China) in cardiac serum were measured by a chemistry analyzer (HITACHI, Japan).
Histological analysis
The heart was cut in half by performing a transverse slice between the atrioventricular sulcus and the apex. A sample of the apical part was fixed in 4% paraformaldehyde, embedded in paraffin and cut into 5 μm-thick cross-sections. The morphology of the cross-sections was observed with hematoxylin and eosin (HE) staining.
Cell culture and drug administration
H9c2 cells, derived from the left ventricle of Sprague–Dawley rats, were purchased from the Center for Excellence in Molecular Cell Science (Shanghai, China). The growth medium for the cells consisted of Dulbecco’s modified Eagle’s medium, 10% fetal bovine serum and 100 U/ml penicillin and 100 mg/ml streptomycin (Gibco, USA). Cells were cultured in normal glucose (5.6 mM) medium to ~50% density and then transferred into high glucose (HG, 25 mM) and palmitate acid (PAL, 100 μM) medium. After HG and PAL treatment for 24 h, cells were exposed to a hypoxic incubator (37°C, 94% N2, 5% CO2 and 1% O2) with culture medium deprived of glucose and serum. After 6 h of hypoxia, cells were reoxygenated for 12 h in HG and high PAL medium. Propofol at concentrations of 12.5 (P12.5), 25 (P25), 50 (P50) and 100 (P100) μM (Sigma-Aldrich, Germany) or/and SAA at concentrations of 6.25 (S6.25), 12.5 (S12.5), 25 (S25), 50 (S50) and 100 (S100) μM (Solarbio, China) treatments were applied immediately before hypoxia until the end of the experiments.
Cell transfection of small interfering RNA and overexpression
To block the expression of AMPK, AMPK-specific small interfering RNAs (siAMPK) were added to H9c2 cells, which were designed and provided by GenePharma as were their individual controls. In accordance with the manufacturer’s instructions, cell transfection was carried out with Lipofectamine 3000 (Invitrogen, USA). For CD36 overexpression, cells were plated in a 6-well plate at a density of 1.5 × 104 and transfected with pcDNA3.1-Flag-CD36 or empty vector (GenePPL, China) using Lipofectamine 3000. After 6 h, the medium was changed and further procedures were performed. Silencing of AMPK and overexpression of CD36 were confirmed by western blot or real-time quantitative polymerase chain reaction (RT-PCR).
Cell viability assay
Cells were inoculated in a 96-well plate (8 × 103 cells/well) and cultured following the above treatment. Cell viability was assessed by the Cell Counting Kit-8 (CCK-8) kit (Dojindo, Japan). Then, 10 μl of CCK-8 solution was added to each well and incubated for 1 h at 37°C. The absorbance was measured at 450 nm with a micro-plane reader. The cell culture supernatant was added to a 96-well plate, and then fully mixed with LDH reaction buffer at room temperature according to the procedures of the relevant manufacturer (Roche, Switzerland). When the reaction was stopped the absorbance was measured at 450 nm.
Enzyme-linked Immunosorbent Assay (ELISA) assay
CK-MB (Jianglai Biology, China) and 8-hydroxy-2′-deoxyguanosine (8-OHdG, Stressmarq Biosciences Inc, Canada) were quantified in cell-culture supernatant by a double antibody sandwich enzyme-linked immunosorbent assay. They were measured using an ELISA kit by following the manufacturer’s instructions.
Determination of L-glutathione (GSH), oxidized L-glutathione (GSSG), total-superoxide dismutase (T-SOD), malonaldehyde (MDA) and labile iron
H9c2 cells or heart tissue were stained with GSH, GSSG, T-SOD and MDA (Beyotime Biotechnology, China) to detect antioxidant or lipid peroxidation levels, and stained with labile iron (BioVision, USA) to detect Fe2+ levels. All the assays were conducted strictly in accordance with the guidance of the kit manuals.
Flow cytometry assessment for ROS and apoptosis
Intracellular ROS levels were measured using a 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) ROS assay kit (Beyotime, China). An Annexin V-Fluorescein Isothiocyanate (FITC)/propidium iodide (PI) apoptosis detection kit (Beyotime Technology, China) was utilized to test for H9c2 cell apoptosis. Cells were gathered in line with the experimental group and analyzed according to the manufacturer’s protocol. The ROS level and apoptotic index were detected by flow cytometer.
Fluorescent enzyme label analysis
H9c2 cardiomyocytes were plated in 96 well, black-sided plates at 10,000 cells per well in 0.1 ml of maintenance medium at the initiation of the experiment. FerroOrange (Dojindo, Japan), 1 μmol/l, was added to the cells for 30 min while FA uptake was measured by treating differentiated cells with 100 μl of FA dye-loading solution (Sigma-Aldrich, Germany) for 1 h. After that, a multifunctional enzyme labeler was used to measure the fluorescence intensity of each sample.
Measurement of mitochondrial membrane potential and lipid peroxidation
JC-1 (Beyotime Technology, China) is a suitable fluorescent probe for detecting mitochondrial membrane potential (MMP), while C11-BODIPY581/591 (Cayman Chemical, USA) is ideal for detecting lipid peroxides. Briefly, after the completion of the designed therapeutic treatment of H9c2 cells, 10 μM JC-1 staining and C11-BODIPY581/591 were added to the cells and incubated in the dark for 30 min at 37°C and then imaged by an Olympus confocal laser scanning microscope (Olympus, Japan).
RT-PCR analysis
RNA was isolated from the cells using the Trizol reagent and then the RNA samples were treated with DNase for cDNA synthesis. CD36 and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) genes were amplified by RT-PCR.
Adenosine triphosphate assay
Adenosine triphosphate (ATP) content was determined by using an enhanced ATP assay kit (Beyotime Biotechnology) according to the manufacturer’s instructions. H9c2 cells and myocardial tissue were lysed with ATP lysis buffer and centrifuged at 10,000 g for 10 min at 4°C. The supernatants were collected and stored on ice. Before the ATP test, 100 μl of ATP working solution was added to 1.5 ml eppendor tubes and incubated for 5 min at room temperature. Next, the supernatant was transferred to 100 μl of ATP working solution and the amount of luminescence emitted was measured immediately with a luminometer.
Western blotting
Western blot analysis was performed as described previously [17,21] using antibodies against phosphorylated (p)-AMPK, AMPK, GAPDH (Cell Signaling Technology, USA), CD36, arachidonate 15-Lipoxygenase (ALOX15, Abcam, UK) and glutathione peroxidase 4 (GPX4) (Proteintech, China) at 4°C for 12–16 h and the band intensity was analyzed by Image J.
Statistical analysis
Data are expressed as the mean ± standard deviation (SD). All statistical analyses were performed using prism software (version 9.3.1, GraphPad, America). The Shapiro–Wilk test was employed for normality testing. Data were compared between the two groups (i.e. part 1 in vivo study data) using a two-tailed Student’s t-test. Differences among multiple groups were analyzed using one-way analysis of variance (ANOVA), followed by Tukey’s post hoc multiple comparison test. p < 0.05 was considered statistically significant.
Results
Basic characteristics of diabetic mice at the end of experiment
Basic characteristics including body weight, water and food intake, and blood glucose, which were recorded accurately, did not differ between groups before starting HFD treatment (data not shown). However, at the end of the experiment (i.e. following 8 weeks HFD and STZ, and then 6 weeks after diabetes establishment), the basic characteristics of mice in the T2DM group showed obvious diabetic symptoms such as elevated blood glucose, polydipsia and weight loss compared to the NC group, as shown in Table 1 as obtained from part 1 in vivo study. The characteristics of diabetic mice in part 2 of the in vivo study were similar to those of part 1 (data not shown).
Table 1.
Basic characteristics of diabetic mice at the end of the experiment. At the end of the experiment (i.e. following 8 weeks HFD and STZ, and then 6 weeks after diabetes establishment), basic characteristics mice in T2DM group showed obvious diabetic symptoms such as elevated blood glucose, polydipsia and weight loss compared to NC group.
| Group | Body weight (g) | Food intake (g/d) | Water intake (ml/kg/d) | Blood glucose (mmol/l) |
|---|---|---|---|---|
| NC | 25.38 ± 2.22 | 7.62 ± 1.67 | 448.85 ± 35.06 | 7.29 ±1.24 |
| T2DM | 20.75 ± 1.83 | 10.15 ± 1.94* | 750.28 ± 151.65* | 20.58 ± 5.78* |
Two-tailed Student’s t-test. Data are expressed as the mean ± SD, n = 12 per group. NC normal control, T2DM type 2 diabetes mellitus
* p < 0.05 vs NC group
T2DM increased IR-induced injury in mice
To explore the underlying mechanism of MIRI in T2DM, animal models were established by feeding C57BL/6 J male mice HFD for 8 weeks, then injecting them with STZ at 50 mg/kg/d intraperitoneally for three consecutive days and continuing HFD for 6 weeks before subjecting them to IR (consisting of 30 min of coronary ligation followed by 2 h of reperfusion) (Figure 1a, created with BioRender.com). T2DM mice showed an increased susceptibility to IR-induced myocardial damage, represented by increased post-ischemic infarct size (Figure 1b, c) and cardiac dysfunction manifested as reduced left ventricular EF and FS (Figure 1d–f). We used biochemical instrumentation to measure serum levels of LDH (Figure 1g), CK-MB (Figure 1h) and TG (Figure 1i) that IR-treated diabetic mice exhibited higher release levels than controls with diabetes or IR-treated mice. Meanwhile, oxidative damage of diabetic mice was also significantly increased after IR, which was manifested as an increase in 8-OHdG (Figure 1j). HE staining of cardiac sections exhibited extensive myocardial injury including myocardial structure disorder, severe inflammatory exudate, interstitial edema and lesions in the T2DM + IR group (Figure 1k).
Figure 1.

Myocardial IR injury in diabetic mice. (a) Construction of a mouse model of MIRI in T2DM. (b) Representative images for infarct size were detected using TTC and Evans blue staining. (c) Quantitative data for infarct size. (d) Representative M-mode images by echocardiography. (e) Changes of left ventricular EF. (f) Changes of left ventricular FS. (g) Leakage of LDH. (h) Concentrations of CK-MB. (i) Level of TG. (j) Extent of 8-OHdG. (k) Histopathological pictures of heart tissue sections were stained with hematoxylin and eosin (HE) (Scale bar:100 μm). Data are expressed as the mean ± SD, n = 6 per group. *p < 0.05 vs NC group, #p < 0.05 vs T2DM group, $p < 0.05 vs IR group, **p < 0.01 vs NC group, ##p < 0.01 vs T2DM group. HFD high-fat diet, STZ streptozotocin, ip intraperitoneal injection, NC normal control, T2DM type 2 diabetes mellitus, IR ischemia–reperfusion, EF ejection fraction, FS fractional shortening, LDH lactate dehydrogenase, CK-MB cardiac-type creatine kinase isoenzyme, TG triglyceride, 8-OHdG 8-hydroxy-2′-deoxyguanosine
MIRI induced activation of ferroptosis and changes in CD36 and AMPK expression
To identify whether ferroptosis was involved in the pathogenesis of diabetic MIRI, we examined indicators associated with ferroptosis in heart tissues from NC and T2DM mice with or without IR injury. Ferroptosis is characterized by lipid peroxidation, and therefore, we first evaluated the end product of lipid peroxidation MDA. The results showed that DMA level of T2DM + IR group was excessive when compared with those in IR or T2DM groups (Figure 2a). As shown in Figure 2, levels of antioxidant enzyme SOD (Figure 2b) together with GSH (Figure 2c) and GSH/GSSG (Figure 2d), all decreased in diabetic mice and were further exacerbated in the T2DM + IR group, while the myocardial Fe2+ value of mice heart (Figure 2e). Was profoundly up-regulated. Consistent with previous studies, myocardial IR caused a significant reduction in ATP production and this reduction was most severe in the untreated diabetic MIRI group (Figure 2f). Moreover, there was an increase in the expression levels of ALOX15 and a decrease in GPX4 expression (Figure 2g, h) in the T2DM + IR group compared to the T2DM without IR and NC with IR groups. Additionally, western blotting analysis showed that expression of CD36 was significantly increased while p-AMPK/AMPK was significantly decreased in the T2DM group (all p < 0.05, T2DM and NC + IR vs. NC), and these changes were further exacerbated in the T2DM + IR group (Figure 2g, h).
Figure 2.

Ferroptosis is involved in the pathogenesis of diabetic MIRI, along with the protein expression changes of CD36 and AMPK. (a) Degree of MDA. (b) Level of SOD. (c, d) Alterations of GSH and GSH/GSSG levels. (e) Labile iron levels. (f) ATP content. (g) Representative images of protein expression. (h) ALOX15, GPX4, CD36 and p-AMPK/AMPK protein expression. Data are expressed as the mean ± SD, n = 6 per group. *p < 0.05 vs NC group, #p < 0.05 vs T2DM group, $p < 0.05 vs IR group, **p < 0.01 vs NC group. NC normal control, T2DM type 2 diabetes mellitus, IR ischemia–reperfusion, MDA malonaldehyde, SOD superoxide dismutase, GSH L-glutathione, GSSG oxidized L-glutathione, ATP adenosine-triphosphate, ALOX15 arachidonate-15-Lipoxygenase, GPX4 glutathione peroxidase 4, CD36 cluster of differentiation 36, AMPK Adenosine 5‘-monophosphate-activated protein kinase, p-AMPK phosphorylated AMPK
Propofol and SAA reduced post-ischemic myocardial infarction, improved cardiac function, and altered CD36 and AMPK expression in diabetic mice with MIRI
As shown in supplementary Figure S1a-h (see online supplementary material), propofol or SAA effectively reduced the post-ischemic myocardial infarction area, MDA and CK-MB levels in diabetic mice. The effective concentration of SAA ranged from 5 to 15 mg/kg/h, while propofol exerted protective effects at concentrations ranging from 46 to 112 mg/kg/h, and the most significant effective propofol concentration was observed to be 90 mg/kg/h (propofol3). Since diabetic subjects are more sensitive to propofol in terms of hemodynamic inhibition, we used a low dose of propofol (propofol1) in combination with different doses of SAA in order to avoid the potential adverse effects of high doses of propofol (e.g. propofol3), and compared the effects to propofol3. As depicted in supplementary Fig. 1i–l, propofol in combination with different doses of SAA showed different degrees of reversal of diabetic MIRI-induced increases in infarct size, MDA and CK-MB, with the combination of propofol1 and SAA2 acting most similarly to that of high-dose propofol3. We therefore used the combination of propofol1 and SAA2 for the ensuing in vivo (part 2) study.
As shown in Figure 3, pre-treatment with SAA (SAA2) or propofol (propofol1, propofol3) all significantly alleviated MIRI in diabetes, while the combined treatment with propofol1 plus SAA2 showed better effects than individual treatments, reflected as a further significant reduction in myocardial infarct size and further improvement in cardiac function EF compared to the T2DM + IR + propofol3 group, although the left ventricular FS values did not exhibit a statistically significant difference between the two treatment groups. (Figure 3a–e). On average, the post-ischemic myocardial infraction area was 59% (± 5.44) in the diabetic untreated group, and SAA2 treatment alone reduced it to 47% (± 5.0), propofol1 treatment alone reduced it to 46% (± 3.49), while combinational treatment with propofol1 and SAA2 reduced the post-ischemic infarction area to 33% (± 2.88) (Figure 3b), demonstrating a synergistic cardiac protection of propofol1 and SAA2 in diabetes. Consequently, a similar synergistic improvement in post-ischemic cardiac function (EF) was also seen in the PPF1 and SAA2 combinational treatment group (Figure 3d). As depicted in Figure 3, the parameter of ferroptosis was significantly reduced after SAA and/or propofol treatments, manifested as a significant decrease in unstable iron (Figure 3f) accompanied by elevations in GSH and GSH/GSSG levels, with the combined effect of low dose propofol1 and SAA2 similar to that of high dose propofol3 alone (Figure 3g, h). Meanwhile, HE staining of myocardial sections showed that treatment with SAA and/or propofol attenuated the extensive myocardial damage induced by diabetic MIRI, including myocardial structure disorder, severe inflammatory exudate, interstitial edema and lesions (Figure 3i).
Figure 3.

PPF and SAA ameliorated MIRI and reduced labile iron levels in diabetic mice. (a) Representative images for infarct size were detected using TTC and Evans blue staining. (b) Quantitative data for infarct size. (c) Representative M-mode images by echocardiography. (d) Changes of left ventricular EF. (e) Changes of left ventricular FS. (f) Labile iron levels. (g,h) Alterations of GSH and GSH/GSSG levels. (i) Histopathological pictures of heart tissue sections stained with hematoxylin and eosin (HE) (100 μm). Data are expressed as the mean ± SD, n = 6 per group. *p < 0.05 vs NC group, #p < 0.05 vs T2DM group, $p < 0.05 vs IR group, &p < 0.05 vs T2DM + IR group, <p < 0.05 vs T2DM + IR + SAA2 group, >p < 0.05 vs T2DM + IR + PPF1 group, {p < 0.05 vs T2DM + IR + PPF3 group **p <0.01 vs NC group,ns not significant. NC normal control, T2DM type 2 diabetes mellitus, IR ischemia–reperfusion, PPF propofol, SAA salvianolic acid A, EF ejection fraction, FS fractional shortening, GSH L-glutathione, GSSG oxidized L-glutathione
Given the importance of ATP in diabetic MIRI, the protective effect of propofol1 and SAA2 were examined, and results showed that they similarly to propofol3 increased ATP content in T2DM mice induced by IR (Figure 4a). Additionally, we further examined the impact of propofol and/or SAA on protein levels of cardiac ALOX15, GPX4, CD36 and AMPK in the context of diabetic MIRI. Treatment with SAA or propofol significantly attenuated the increases in CD36 and ALOX15 induced by diabetes and MIRI, and increased protein levels of GPX4 and p-AMPK/AMPK, while the combination of low-doses of propofol and SAA (T2DM + IR + propofol1 + SAA2 group) demonstrated a synergistic effect in further reducing diabetes- and MIRI-induced increases in ALOX15, which was comparable to the effect of high-dose propofol (T2DM + IR + propofol3 group). Moreover, propofol1 in combination with SAA2 also further significantly increased post-ischemic cardiac GPX4 and p-AMPK/AMPK expression, as well as significantly decreased ALOX15 and CD36 expression compared to individual treatments (Figure 4b, c).
Figure 4.

Effects of PPF and SAA on expression of CD36, AMPK and ferroptosis-related proteins and ATP content. (a) ATP content. (b) Representative images of protein expression. (c) ALOX15, GPX4, CD36 and p-AMPK/AMPK protein expression Data are expressed as the mean ± SD, n = 6 per group. *p < 0.05 vs NC group, #p < 0.05 vs T2DM group, $p < 0.05 vs IR group, &p < 0.05 vs T2DM + IR group, <p < 0.05 vs T2DM + IR + SAA2 group, >p < 0.05 vs T2DM + IR + PPF1 group, {p < 0.05 vs T2DM + IR + PPF3 group, ns not significant. NC normal control, T2DM type 2 diabetes mellitus, IR ischemia–reperfusion, PPF propofol, SAA salvianolic acid A, ALOX15 arachidonate-15-lipoxygenase, GPX4 glutathione peroxidase 4, CD36 cluster of differentiation 36, AMPK adenosine 5‘-monophosphate-activated protein kinase, p-AMPK phosphorylated AMPK, ATP adenosine-triphosphate
HG and PAL triggered H9c2 cardiomyocyte injury and oxidative stress which were exacerbated by hypoxic/reoxygenation conditions
Since T2DM patients often suffer from hyperglycemia and hyperlipidemia and are sensitive to MIRI, an in vitro model consisting of exposure of H9c2 cardiomyocytes to HG and PAL (HG + PAL group) before being subjected to hypoxia/reoxygenation (HR) injury was established. As shown in Figure 5, exposure of H9c2 cells to HG and PAL resulted in enhanced vulnerability to HR-triggered cellular injury, characterized by elevated levels of apoptosis and diminished cell viability (Figure 5a, b, c) compared to the control group (NC group). In addition, we found that an increase in the release of LDH (Figure 5d) was further exaggerated by HR conditions (HG + PAL + HR group). To further examine the role of mitochondria, common ROS detected by DCFH-DA was produced in large quantities in the HG + PAL + HR group compared to the HG + PAL group or NC + HR group (Figure 5e, f). Moreover, either HR or HG + PAL significantly impaired MMP while HR under HG + PAL conditions (HG + PAL + HR group) caused the most profound reduction in MMP when compared with the HG + PAL group or the HR group (Figure 5g, h). In line with diabetic MIRI mice, we further provided in vitro proof demonstrating CK-MB and 8-OHdG levels were significantly elevated when cells were exposed to HG + PAL + HR compared to the control group (Figure 5i, j).
Figure 5.

Injuries and oxidative stress were triggered by high levels of glucose (25 mM) and PAL (100 μM) and exacerbated by HR damage in H9c2 cardiomyocytes. (a) Counts of apoptotic cells by flow cytometry. (b) Quantification of apoptotic ratio. (c) Cell viability. (d) Leakage of LDH. (e) ROS levels detected by DCFH-DA fluorescence flow cytometry. (f) Statistical image of mean DCFH-DA fluorescence intensity. (g) MMP assessed by JC-1 staining (Scale bar: 100 μm). (h) Quantification graph of JC-1 staining. (i) Concentrations of CK-MB. (j) Extent of 8-OHdG. Data are expressed as the mean ± SD from three independent experiments each performed in triplicate. *p < 0.05 vs NC group, #p < 0.05 vs HG + PAL group, $p < 0.05 vs NC + HR group, **p < 0.01 vs NC group, ##p < 0.01 vs HG + PAL group. NC normal control, HG high glucose, PAL palmitic acid, HR hypoxia/reoxygenation, LDH lactate dehydrogenase, CK-MB cardiac-type creatine kinase isoenzyme, 8-OHdG 8-hydroxy-2′-deoxyguanosine
HR-treated H9c2 cells resulted in activated and aggravated ferroptosis, reduced mitochondrial function concomitant with reduced AMPK activation, but increased CD36 expression under HG and PAL
To identify the role of ferroptosis in HR-induced cells treated with HG and PAL, we first examined the oxidation–antioxidation system of ferroptosis which was out of balance, evidenced as reductions in T-SOD (Figure 6a), GSH (Figure 6b), GSH/GSSG (Figure 6c) and GPX4 (Figure 6f, g). In the meantime, we found an increase in intracellular Fe2+ levels (Figure 6d) and a decrease in ATP (Figure 6e) content in the HG + PAL group or HR group, which was further exaggerated when both events occurred together (HG + PAL + HR group). ALOX15, a protein involved in the oxidation of polyunsaturated fatty acids (PUFA), was abnormally elevated in the HG + PAL + HR group compared to the NC group (Figure 6f, g). Previous studies suggested that AMPK and CD36 are essential for the maintenance of cardiovascular homeostasis and lipid metabolism [22]. Therefore, we explored the role of AMPK and CD36 in H9c2 cells subjected to HR injury with or without HG + PAL. The results showed that p-AMPK/AMPK was significantly reduced in H9c2 cells exposed to HG + PAL or the HR group (all p < 0.05, HG + PAL or NC + HR vs. NC) and were further significantly reduced when the cells were subjected to HG + PAL and HR together (all p < 0.05, HG + PAL + HR vs. HG + PAL or NC + HR), whereas the opposite trend was observed for CD36 (Figure 6f, g). However, the relationship between AMPK and CD36 at this stage remains unclear. Since the increase of lipid peroxidation is a driver factor of ferroptosis, the cell samples of propofol or/and SAA treatment were analyzed by assay kits. Results showed that mean oxC11-BODIPY fluorescence intensity (Figure 6h, i) detected by fluorescent microscope was gradually increased in the HG + PAL group as compared to the NC group and further exacerbated in the NC + HR and HG + PAL + HR groups, along with augmentation of the levels of MDA (Figure 6j) and mean FA uptake fluorescence intensity (Figure 6k).
Figure 6.

Oxidative stress and ferroptosis in HR-treated H9c2 cardiomyocytes with changes in the expression of AMPK and CD36. (a) Level of T-SOD. (b,c) Alterations of GSH and GSH/GSSG levels. (d) FerroOrange fluorescence intensity. (e) ATP content. (f) Representative images of protein expression. (g) ALOX15, GPX4, CD36 and p-AMPK/AMPK protein expression. (h) Lipid peroxidation measured by C11 BODIPY (Scale bar: 50 μm). (i) Statistical graph of C11 BODIPY. (j) Degree of MDA. (k) FA uptake assay. Data are expressed as the mean ± SD from three independent experiments each performed in triplicate. *p < 0.05 vs NC group, #p < 0.05 vs HG + PAL group, $p < 0.05 vs NC + HR group, **p < 0.01 vs NC group, ##p < 0.01 vs HG + PAL group. NC normal control, HG high glucose, PAL palmitic acid, HR hypoxia/reoxygenation, T-SOD total-superoxide dismutase, GSH L-glutathione, GSSG oxidized L-glutathione, ALOX15 arachidonate-15-lipoxygenase, GPX4 glutathione peroxidase 4, CD36 cluster of differentiation 36, AMPK adenosine 5‘-monophosphate-activated protein kinase, p-AMPK phosphorylated AMPK, MDA malonaldehyde, ATP adenosine-triphosphate
Propofol or SAA respectively and dose-dependently attenuated HR injury in H9c2 cells exposed to HG and PAL conditions
A model combining cells with HR injury and drug therapy was constructed to examine the cardio-protective effects of propofol and SAA in vitro. Different concentrations of propofol or/and SAA were applied respectively to the cells during the whole HR process (Figure 7a, created with BioRender.com). No toxicity was found with various concentrations of propofol and SAA for up to 48 h of the experimental procedures (Figure 7b, e). As shown in Figure 7, we found that propofol and SAA improved cell viability (Figure 7c, d) and T-SOD (Figure 7h, i) along with decreasing LDH (Figure 7f, g) and MDA (Figure 7j, k) in a dose-dependent manner respectively. The effective concentration of propofol ranged from 12.5 to 50 μmol/l, and the most significant concentration was observed at 25 μmol/l, while SAA exerted protective effects in the range from 12.5 to 100 μmol/l, and the lowest effective therapeutic concentration was 12.5 μmol/l.
Figure 7.

Effect of propofol or SAA on HR-induced damage in H9c2 cells under HG and PAL. (a) Construction of an HG and PAL and HR-mediated cell model with or without propofol or SAA. (b,c,d) Cell viability. (e,f,g) Leakage of LDH. (h,i) Level of T-SOD. (j,k)Degree of MDA. Data are expressed as the mean ± SD from three independent experiments each performed in triplicate. *p < 0.05 vs NC group, #p < 0.05 vs HG + PAL group, $p < 0.05 vs NC + HR group, &p < 0.05 vs HG + PAL+HR group, @p < 0.05 vs HG + PAL+HR + P12.5 group, %p < 0.05 vs HG + PAL+HR + P25 group, +p < 0.05 vs HG + PAL+HR + P50 group, ^p < 0.05 vs HG + PAL+HR + S12.5 group, −p < 0.05 vs HG + PAL + HR + S25 group, ※p < 0.05 vs HG + PAL + HR + S50 group, ns not significant. NC normal control, HG high glucose, PAL palmitic acid, HR hypoxia/reoxygenation, P propofol, S salvianolic acid A, D dimethyl sulfoxide, LDH lactate dehydrogenase, MDA malonaldehyde, TSOD total-superoxide dismutase
Propofol and/or SAA attenuated cell damage in H9c2 cells exposed to HR injury under HG and PAL conditions
To avoid the side effects of high doses of propofol, we combined SAA (12.5 μmol/l) with propofol (12.5 μmol/l) in H9c2 cardiomyocytes subjected to HR injury under HG and PAL. As depicted in Figure 8, we found that combination drug group (HG + PAL + HR + P12.5 + S12.5) had increased cell viability (Figure 8a) and decreased levels of LDH (Figure 8b), CK-MB (Figure 8c) and apoptotic ratio (Figure 8d, e), better than the propofol (HG + PAL + HR + P12.5) or SAA (HG + PAL+HR + S12.5) groups, and similar to the effects achieved with a high dose of propofol at 25 μmol/l (HG + PAL + HR + P25). In addition, we tested the protective effect of drugs against oxidative stress. As expected, propofol and SAA protected against HG + PAL + HR-provoked reduction in ROS products (Figure 8f, g) and reversed the increase in 8-OHdG level (Figure 8h). Notably, both P12.5 and S12.5 were effective in down-regulating 8-OHdG, and their combinational use conferred superior effects to the use of P25.
Figure 8.

Propofol and/or SAA reduced H9c2 cells HR injury under HG and PAL. (a) Cell viability. (b) Leakage of LDH. (c) Concentration of CK-MB. (d) Counts of apoptotic cells by flow cytometry. (e) Quantification of apoptotic ratio. (f) ROS levels detected by DCFH-DA fluorescence flow cytometry. (g) Statistical image of mean DCFH-DA fluorescence intensity. (h) Extent of 8-OHdG. Data are expressed as the mean ± SD from three independent experiments each performed in triplicate. *p < 0.05 vs NC group, #p < 0.05 vs HG + PAL group, $p < 0.05 vs NC + HR group, &p < 0.05 vs HG + PAL + HR group, ^p < 0.05 vs HG + PAL + HR + S12.5 group, @p < 0.05 vs HG + PAL + HR + P12.5 group, %p < 0.05 vs HG + PAL + HR + P25 group, ns not significant. NC normal control, HG high glucose, PAL palmitic acid, HR hypoxia/reoxygenation, P propofol, S salvianolic acid A, LDH lactate dehydrogenase, CK-MB cardiac-type creatine kinase isoenzyme, 8-OHdG 8-hydroxy-2′-deoxyguanosine
Propofol and/or SAA alleviated the effects of HR on H9c2 cardiomyocytes by inhibiting ferroptosis, improving mitochondrial function and altering CD36 and AMPK expression under HG and PAL conditions
As shown in Figures 9 and 10, a high dose ofpropofol (HG + PAL +HR + P25 group) and a low concentration of propofol combined with SAA (HG + PAL+HR + P12.5 + S12.5 group), respectively, significantly reversed the adverse changes in MDA (Figure 9a) and FA uptake (Figure 9b) caused by HR injury under HG and PAL, and restored oxidation resistance with an increase in GSH (Figure 9c), GSH/GSSG (Figure 9d) and T-SOD (Figure 9e); both of these groups also yield further protective effects better than administration of propofol or SAA alone. Meanwhile, the high level of intracellular iron (Figure 9f) was suppressed by the above drug treatments. Consistent with previous experiments, we also analyzed CD36, AMPK and ferroptosis protein in vitro after propofol/SAA treatment. As shown in Figure 9g, h, the expression of ferroptosis-related proteins, such as GPX4, was activated by propofol and SAA, while ALOX15 was down-regulated. Besides, activation of CD36 and inhibition of p-AMPK/AMPK (Figure. 9g, l) induced by HG + PAL + HR were also reversed by joint treatment with propofol and SAA. Propofol combined with SAA also had significant impact on lipid peroxidation and mitochondria-related parameters, including diminished mean oxC11-BODIPY fluorescence intensity (Figure 10a, b), and elevated MMP (Figure 10c, d) and ATP content (Figure 10e) in H9c2 cells exposed to HR under HG and PAL. Taken together, our results demonstrated that a low dose of propofol combined with SAA conferred similar cardio-protective effects that were associated with a reduction in ferroptosis as compared to a high dose of propofol alone. However, the potential interplay between propofol and SAA joint treatment and CD36 and AMPK in HG + PAL + HR-induced ferroptosis remains unclear. Thus, we viewed propofol and SAA joint treatment as a new therapeutic strategy and further explored the underlying mechanism in subsequent experiments.
Figure 9.

Cardio-protective effects of propofol and/or SAA treatment in H9c2 cells exposed to HG and PAL. (a) Degree of MDA. (b) FA uptake assay. (c,d) Alterations of GSH and GSH/GSSG levels. (e) Level of T-SOD. (f) FerroOrange fluorescence intensity. (g) Representative images of protein expression. (h) ALOX15, GPX4, CD36, and p-AMPK/AMPK protein expression, respectively. Data are expressed as the mean ± SD from three independent experiments each performed in triplicate. *p < 0.05 vs NC group, #p < 0.05 vs HG + PAL group, $p < 0.05 vs NC + HR group, &p < 0.05 vs HG + PAL + HR group, ^p < 0.05 vs HG + PAL + HR + S12.5 group, @p < 0.05 vs HG + PAL + HR + P12.5 group, %p < 0.05 vs HG + PAL + HR + P25 group, **p < 0.01 vs NC group, ns not significant. NC normal control, HG high glucose, PAL palmitic acid, HR hypoxia/reoxygenation, P propofol, S salvianolic acid A, MDA malonaldehyde, GSH L-glutathione, GSSG oxidized L-glutathione, T-SOD total-superoxide dismutase, ALOX15 arachidonate-15-lipoxygenase, GPX4 glutathione peroxidase 4, CD36 cluster of differentiation 36, AMPK adenosine 5‘-monophosphate-activated protein kinase, p-AMPK phosphorylated AMPK
Figure 10.

Effects of propofol and/or SAA treatment on mitochondrial function and lipid peroxidation in H9c2 cells exposed to HG and PAL. (a) Lipid peroxidation measured by C11 BODIPY (Scale bar: 50 μm). (b) Statistical graph of C11 BODIPY. (c) MMP assessed by JC-1staining (100 μm). (d) Statistical graph of JC-1. (e) ATP content. Data are expressed as the mean ± SD from three independent experiments each performed in triplicate. *p < 0.05 vs NC group, #p < 0.05 vs HG + PAL group, $p < 0.05 vs NC + HR group, &p < 0.05 vs HG + PAL + HR group, ^p < 0.05 vs HG + PAL + HR + S12.5 group, @p < 0.05 vs HG + PAL + HR + P12.5 group, %p < 0.05 vs HG + PAL + HR + P25 group, ns not significant. NC normal control, HG high glucose, PAL palmitic acid, HR hypoxia/reoxygenation, P propofol, S salvianolic acid A, ATP adenosine-triphosphate
Propofol and SAA combination attenuated HR-induced ferroptosis and cell injury in H9c2 cells under HG and PAL conditions through modulation of the CD36/AMPK signaling pathway
To explore whether or not HR-induced injury under HG and PAL conditions was triggered by abnormal signaling of CD36/AMPK and to understand their role in joint propofol and SAA treatment-mediated cellular protection, we knocked out AMPK with a specific siRNA (Figure 11a, b) and overexpressed CD36 (Figure. 11c–e) in H9c2 cells, respectively, before conducting the ensuing experiments. As shown in Figures 11 and 12, cell viability (Figure 11f) was decreased and cell damage indicators including apoptosis rate (Figure 11g, h) and LDH (Figure 11i) were increased, while AMPK was suppressed but CD36 was overexpressed in HR-treated cells exposed to HG and PAL regardless of treatment with propofol and SAA, despite the abovementioned changes all being significantly reversed by propofol and SAA joint treatment. Meanwhile, silencing of AMPK or overexpression of CD36 canceled the anti-oxidative stress effect of propofol combined with SAA in H9c2 cells. The main in vitro manifestations of AMPK gene silencing or CD36 overexpression were increases of 8-OHdG (Figure 11j) and ROS production (Figure 11k, l). Propofol and SAA treatment also increased ATP (Figure 11m) in HR-induced H9c2 cells exposed to HG and PAL, which was abrogated by CD36 overexpression or AMPK knockout. The efficiently of propofol and SAA was related to lipid peroxidation as inferred by observation that silencing of AMPK or overexpression of CD36 reversed the augment in MDA (Figure 11n) and FA uptake (Figure 11o). Moreover, AMPK silence or CD36 overexpression raised the content of Fe2+ (Figure 12a) and ALOX15 expression (Figure 12e, f), as well as decreased the levels of T-SOD (Figure 12b), GSH (Figure 12c), GSH/GSSG (Figure 12d) and GPX4 activity (Figure 12e, f), regardless of propofol and SAA joint treatment. In parallel, the confocal laser microscope showed decreased MMP (Figure 12g, h) and elevated mean oxC11-BODIPY fluorescence intensity (Figure 12i, j) due to AMPK deficiency and CD36 excess. We also observed that the aforementioned inhibition of the ferroptosis index was reversed by AMPK silencing or CD36 overexpression (all p < 0.05, HG + PAL + P12.5 + S12.5 + siAMPK or HG + PAL + P12.5 + S12.5 + CD36-OE vs. HG + PAL + P12.5 + S12.5; Figure 12a–j). More importantly, p-AMPK/AMPK levels were down-regulated after overexpression of CD36, while CD36 expression (Figure 12e, f) did not change with the knockdown of AMPK, indicating that propofol combined with SAA alleviated HR-induced ferroptosis via phosphorylation of AMPK mediated by CD36 in H9c2 cells under HG and PAL, and that CD36 acts upstream of AMPK in the context of cardiomyocyte HR injury under diabetic conditions.
Figure 11.

Overexpression of CD36 or silencing of AMPK canceled the protective effects of propofol and SAA in reducing oxidative stress levels and in increasing cell viability in H9c2 cells damaged by HR under HG and PAL. (a) Representative images of protein expression. (b) p-AMPK/AMPK protein expression. (c) Representative images of protein expression. (d) CD36 protein expression. (e) mRNA expression of CD36. (f) Cell viability. (g) Counts of apoptotic cells by flow cytometry. (h) Quantification of apoptotic ratio. (i) Leakage of LDH. (j) Extent of 8-OHdG. (k) ROS levels detected by DCFH-DA fluorescence flow cytometry. (l) Statistical image of mean DCFH-DA fluorescence intensity. (m)ATP content. (n) Degree of MDA. (o) FA uptake assay. Data are expressed as the mean ± SD from three independent experiments each performed in triplicate. *p < 0.05 vs NC group, &p < 0.05 vs HG + PAL + HR group, ¥p < 0.05 vs HG + PAL + HR + P12.5 + S12.5 group, **p < 0.01 vs NC group, &&p < 0.01 vs HG + PAL + HR group, ¥¥p < 0.01 vs HG + PAL + HR + P12.5 + S12.5 group, ***p < 0.001 vs NC group, ns not significant. NC normal control, HG high glucose, PAL palmitic acid, HR hypoxia/reoxygenation, P propofol, S salvianolic acid A, si small interfering, OE overexpression, AMPK adenosine 5‘-monophosphate-activated protein kinase, p-AMPK phosphorylated AMPK, CD36 cluster of differentiation 36, LDH lactate dehydrogenase, 8-OHdG 8-hydroxy-2′-deoxyguanosine, MDA malonaldehyde, ATP adenosine-triphosphate
Figure 12.
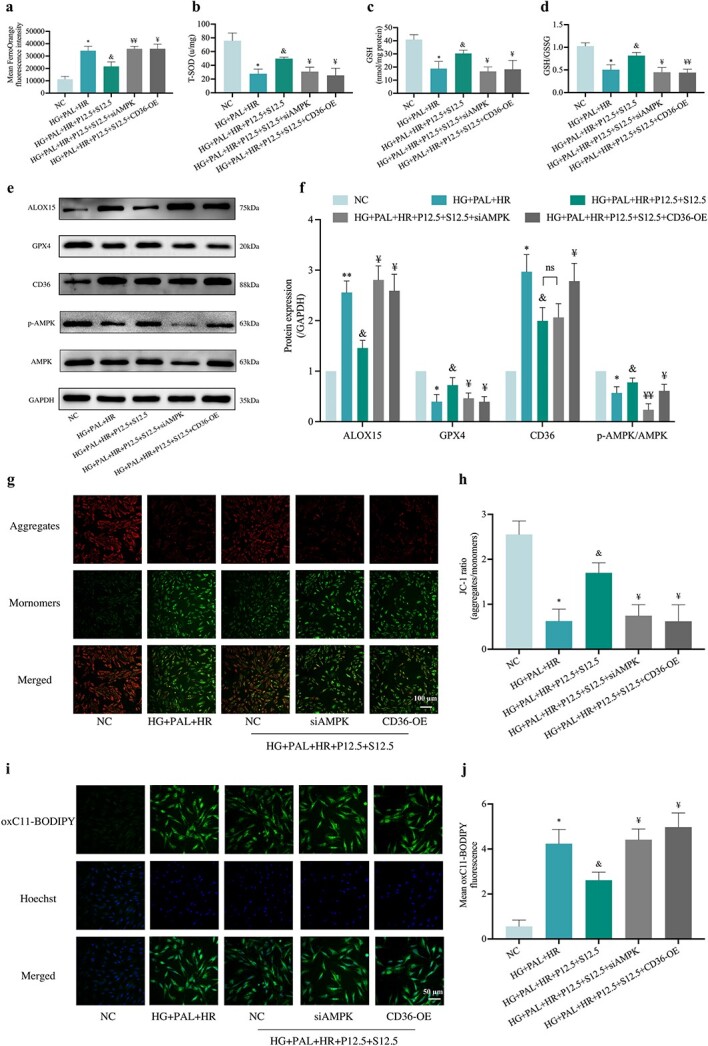
Effect of overexpression of CD36 or silencing of AMPK on the protection of propofol and SAA in H9c2 cells against HR-induced ferroptosis with HG and PAL. (a) FerroOrange fluorescence intensity. (b) Level of T-SOD. (c,d) Alterations of GSH and GSH/GSSG levels. (e) Representative images of protein expression. (f) ALOX15, GPX4, CD36 and p-AMPK/AMPK protein expression. (g) MMP assessed by JC-1 staining (Scale bar:100 μm). (h) Statistical graph of JC-1. (i) Lipid peroxidation measured by C11 BODIPY (50 μm). (j) Statistical graph of C11 BODIPY. Data are expressed as the mean ± SD from three independent experiments each performed in triplicate. *p < 0.05 vs NC group, &p < 0.05 vs HG + PAL + HR group, ¥p < 0.05 vs HG + PAL + HR + P12.5 + S12.5 group, **p < 0.01 vs NC group, ¥¥p < 0.01 vs HG + PAL + HR + P12.5 + S12.5 group, ns not significant. NC normal control, HG high glucose, PAL palmitic acid, HR hypoxia/reoxygenation, P propofol, S salvianolic acid A, si small interfering, OE overexpression, T-SOD total-superoxide dismutase, GSH L-glutathione, GSSG oxidized L-glutathione, ALOX15 arachidonate-15-lipoxygenase, GPX4 glutathione peroxidase 4, CD36 cluster of differentiation 36, AMPK adenosine 5‘-monophosphate-activated protein kinase, p-AMPK phosphorylated AMPK
Discussion
In the present study, the increase in ferroptosis that resulted from excessive oxidative activity and lipid peroxidation was observed both in diabetic mice subjected to MIRI and in HR-treated H9c2 cells under HG and PAL conditions. Further, IR- or HR-triggered cardiomyocyte injury was alleviated by pharmacological intervention using propofol combined with SAA, which was potentially mediated by the inhibition of ferroptosis via the CD36/AMPK pathway.
Diabetic cardiomyopathy is varies distinctly from classic clinical heart failure based on autopsy findings by Rubler et al. [23] in 1972. Myocardial impairments in DCM can be exacerbated by several pathological changes, such as increased inflammatory activity, metabolic disturbance, apoptosis, pyroptosis, autophagic cell death, necroptosis, ferroptosis and mitochondrial dysfunction, etc. [24,25]. Diabetic patients are more susceptible to MIRI with impaired myocardial self-repair [25]. However, the molecular mechanism of MIRI with regards to the severity and types of ischemia is not fully elucidated, and the strategies of therapeutic treatments require further exploration [26]. Clinical and laboratory evidence suggests that down-regulated expression of AMPK leads to cardiac dysfunctional metabolic syndrome in T2DM, and activation of AMPK can attenuate diabetes-associated MIRI [27,28]. Recently, CD36 was found to increase the excessive lipid metabolism of cardiomyocytes, resulting in cellular ferroptosis [14]. The relationship of AMPK and CD36 signaling in ferroptosis appears to be a potential target in DCM-MIRI treatment.
As a multifunctional membrane protein, CD36 is involved in angiogenesis, inflammatory responses, atherosclerotic thrombosis and metabolic diseases including diabetes and obesity [6], and has been shown to respond to external stimuli such as IR and high blood-sugar levels [6,29]. In this study, significantly increased levels of CD36 occurred both in diabetic mice with MIRI and in H9c2 cells subjected to HR under HG and PAL, suggesting an important role of CD36 in mediating the enhanced uptake of LCFA. We found that propofol combined with SAA can significantly reduce the expression of CD36 and thereby inhibit fat uptake. When undergoing mitochondrial β-oxidation, FAs produce abundant ATP and subsequently demonstrate feedback-inhibition of AMPK [30]. Acting as a cellular energy sensor, AMPK has a negative effect on FA metabolism [31]. CD36 was reported to be a negative regulator of AMPK [22] and FA binding to CD36 resulted in inhibition of AMPK. Our study also showed that propofol and SAA increased AMPK phosphorylation and prevented ferroptosis by inhibiting the binding of CD36 to FAs in HR-injured H9c2 cardiomyocytes exposed to HG and PAL. In the meantime, overexpressing CD36 abolished the cardioprotective effects of propofol and SAA, while silencing AMPK showed no significant influence on CD36 expression. According to the present data, ferroptosis might play a critical role in diabetic MIRI, which could be prevented by propofol and SAA treatment via inhibiting CD36 along with activating AMPK.
Myocardial cell death including apoptosis, necrosis and autophagy has been considered key for the pathogenesis of diabetic MIRI [32]. A very recent study shows that MIRI-induced ferroptosis is one of the major outcomes attributable to the severity of post-ischemic myocardial injury both in vivo in a mouse model of MIRI and in vitro in cultured cardiomyocytes subjected to HR [33]. The findings of our current in vivo and in vitro studies are indicative that ferroptosis may be a crucial factor exacerbating MIRI in subjects at a relatively late stage of T2DM, and that excessive oxidative stress induced by MIRI under diabetic conditions serves as an important initiating mechanism. Over-accumulation of iron ions within cells leads to excessive oxidative stress reactions and lipid peroxides, ultimately resulting in cytomembrane rupture and cell death [11,34]. We found that an increase in labile iron levels was accompanied by the pathological changes of MIRI in diabetic mice. Iron overload could lead to lipid peroxidation, which is a critical mechanism in the development of diabetic MIRI. A previous study [35] showed increased expression of ALOX15 and FA intake during ischemia, along with significantly down-regulated expression of GPX4 protein and GSH content in the antioxidant system. These pathophysiological changes together cause cell dysfunction and enhance lipid peroxidation, resulting in ferroptosis during MIRI in diabetes.
Propofol is frequently administered for sedation and anesthesia in cardiac surgery and it has been demonstrated to possess antioxidant properties [36,37]. According to our previous findings, 25 μmol/l of propofol post-treatment effectively reduced apoptosis and autophagy induced by HR injury in H9c2 cells subjected to HG [17]. In the present in vitro study, the concentration of propofol was 12.5 μM, which may correspond to a relatively low level of clinically effective anesthetic blood concentration of 2.25 μg/ml) [38]. Seeing that the blood concentration of propofol for general anesthesia in actual clinical practice is usually within the range of 2–6 μg/ml [38], propofol when used at 25 μM in vitro should correspond to a relatively high dose of propofol used clinically, which may lead to hemodynamic instability and even refractory bradycardia leading to asystole, cardiac shock in patients undergoing surgery, probably due to the high dose of propofol [39]. To facilitate the investigation based on the highly complex mechanisms of diabetic MIRI, we therefore applied a relatively low dose of propofol in combination with SAA which had cardioprotective effects. The present study is the first to report that a combination of 12.5 μmol/l propofol and SAA can inhibit ferroptosis induced by HR in H9c2 myocardial cells subjected to HG and PAL, and the cellular protective effect is even comparable to 25 μmol/l propofol alone. These outcomes provided a potential therapeutic strategy for the prevention and treatment of diabetic MIRI.
In our previous study, treatment with the classical antioxidant N-acetylcysteine, before inducing MIRI by occluding the coronary artery for 30 min followed by 2 h reperfusion in diabetic rats, significantly attenuated MIRI-induced increases in oxidative stress and myocardial infarction [40]. In vitro, HR of H9c2 cells under HG caused a significant increase in oxidative stress and post-hypoxic cell injury, and administration of propofol at concentrations between 12.5 and 50 μmol/l, as used in the current study, significantly reduced HG- and HR-induced increases in ROS production and attenuated post-hypoxic cellular injuries [17]. Yuan et al. found that injection of SAA (10 mg/kg) through the jugular vein 10 min before the start of IR significantly reduced the MIRI-induced increase of infarct size and attenuated cardiomyocyte injury in rats [41]. SAA also significantly attenuated MIRI in rats with T2DM [42]. Likewise, similarly to that seen for the classical antioxidant N-acetylcysteine mentioned above, propofol has been shown to confer protection against MIRI both in normal and in type 2 diabetic rats [43–45]. In our study, propofol (46 mg/kg/h) or SAA (10 mg/kg/h) when used individually through continuous intravenous infusion both significantly reduced post-ischemic oxidative stress and attenuated MIRI in type 2 diabetic mice, which is in line with the abovementioned previous findings. A novel aspect of our current study is that combinational use of low-dose propofol with SAA conferred synergistic cardioprotection against MIRI in diabetes that was associated with the modulation of CD36/AMPK signaling.
Our research demonstrates the critical role of CD36/AMPK signaling in ferroptosis during cardiomyocyte HR injury and in treatment with propofol and SAA in vitro. Our in vivo study demonstrated beneficial and synergistical effects of joint propofol and SAA treatment on cardio-protection against MIRI in type 2 diabetic mice that involves the inhibition of CD36 overexpression and the enhancement of AMPK activation with concomitant reductions in ferroptosis. However, further study is needed to explore the undefined mechanisms of diabetic MIRI, in particular to confirm the relative role of CD36/AMPK signaling among other mechanisms, such as the interplay between CD36 and the glucose transporter GLUT4 during MIRI [46], in joint propofol and SAA treatment-mediated cardioprotection against MIRI in diabetes. The strategy of combined application of low-dose propofol and SAA to alleviate HR-induced cardiomyocyte damage in the case of HG and PAL as well as in in vivo models of MIRI in diabetes could effectively avoid potential hemodynamic instability, the side effect that may be caused by high-dose propofol treatment, while increasing its antioxidant properties. These findings may have clinical implications and serve as valuable evidence for a potential anesthesia strategy in cardiac surgery.
In diabetes, the distribution of CD36 on the cell membrane increases, leading to an increase in LCFA uptake, which enter the mitochondria for aerobic oxidation and inhibit AMPK phosphorylation, and MIRI under diabetic conditions further exacerbates the situation. Deactivation of AMPK can activate de novo synthesis of FAs and subsequent oxidation of PUFAs. Moreover, PUFAs can produce lipid peroxides to trigger ferroptosis under the phospholipid peroxidation of ALOX15. In addition, it is difficult to convert GSH into GSSG to inhibit the production of lipid ROS due to the impairment of GPX4. Thus, it is of importance that the combination of propofol and SAA attenuated ferroptosis caused by diabetic MIRI via the CD36/AMPK pathway (Figure 13, created with BioRender.com).
Figure 13.

Putative mechanistic diagrams of the inhibitory effect of propofol and SAA on ferroptosis in diabetic MIRI. MIRI myocardial ischemia–reperfusion injury, PUFA polyunsaturated fatty acids, GSH L-glutathione, GSSG oxidized L-glutathione, ALOX15 arachidonate-15-lipoxygenase, GPX4 glutathione peroxidase 4, CD36 cluster of differentiation 36, AMPK adenosine 5‘-monophosphate-activated protein kinase
It should be noted that our current study may have the following limitations. Firstly, it is not clear whether the protective effects of propofol and SAA in diabetic MIRI are directly via CD36/AMPK pathway, which may involve more testing such as by use of medicinal chemical probes and mass spectrometry. In addition, although the present study confirms that the propofol and SAA combination exhibits synergistic effects in the protection of diabetic MIRI, their modes of action and whether their configuration has been altered are still unclear. Finally, the internal environment in a human is complex compared with that of a mouse, so whether or not the combined drug is effective for clinical patients needs to be further studied.
Conclusions
The present work demonstrated that propofol and SAA could synergistically inhibit ferroptosis via modulating the CD36/AMPK pathway under diabetic MIRI conditions and that combinational usage of propofol and SAA confers superior cellular protective effects to the use of a high dose of propofol alone through inhibiting HR-induced CD36 overexpression to upregulate p-AMPK.
Supplementary Material
Contributor Information
Jiaqi Zhou, Department of Anesthesiology, Affiliated Hospital of Guangdong Medical University, No. 57, South Renmin Avenue, Zhanjiang, 524000, China; State Key Laboratory of Pharmaceutical Biotechnology, Department of Medicine, The University of Hong Kong, Pok Fu Lam Road, Hong Kong, 999077, China.
Weiyi Xia, Department of Anesthesiology, Affiliated Hospital of Guangdong Medical University, No. 57, South Renmin Avenue, Zhanjiang, 524000, China; Department of Orthopaedics and Traumatology, The University of Hong Kong, Pok Fu Lam Road, Hong Kong, 999077, China.
Jiajia Chen, Department of Anesthesiology, Affiliated Hospital of Guangdong Medical University, No. 57, South Renmin Avenue, Zhanjiang, 524000, China.
Kaijia Han, Department of Anesthesiology, Affiliated Hospital of Guangdong Medical University, No. 57, South Renmin Avenue, Zhanjiang, 524000, China.
Yuxin Jiang, Department of Anesthesiology, Affiliated Hospital of Guangdong Medical University, No. 57, South Renmin Avenue, Zhanjiang, 524000, China.
Anyuan Zhang, Department of Anesthesiology, The Second Affiliated Hospital & Yuying Children's Hospital of Wenzhou Medical University, No. 109 Xueyuan West Road, Wenzhou, Zhejiang, 325027, China.
Dongcheng Zhou, Department of Anesthesiology, Affiliated Hospital of Guangdong Medical University, No. 57, South Renmin Avenue, Zhanjiang, 524000, China.
Danyong Liu, Department of Anesthesiology, Affiliated Hospital of Guangdong Medical University, No. 57, South Renmin Avenue, Zhanjiang, 524000, China.
Jiefu Lin, Department of Anesthesiology, Affiliated Hospital of Guangdong Medical University, No. 57, South Renmin Avenue, Zhanjiang, 524000, China.
Yin Cai, Department of Anesthesiology, Affiliated Hospital of Guangdong Medical University, No. 57, South Renmin Avenue, Zhanjiang, 524000, China; Department of Health Technology and Informatics, The Hong Kong Polytechnic University, No. 11 Yucai Road, hung hom, Kowloon, Hong Kong, 999077, China.
Guanghua Chen, Spinal Division of Orthopedic and Traumatology Center, The Affiliated Hospital of Guangdong Medical University, No. 57 South Renmin Avenue, Zhanjiang 524000, China.
Liangqing Zhang, Department of Anesthesiology, Affiliated Hospital of Guangdong Medical University, No. 57, South Renmin Avenue, Zhanjiang, 524000, China.
Aimin Xu, State Key Laboratory of Pharmaceutical Biotechnology, Department of Medicine, The University of Hong Kong, Pok Fu Lam Road, Hong Kong, 999077, China.
Youhua Xu, Faculty of Chinese Medicine, State Key Laboratory of Quality Research in Chinese Medicine, Macau University of Science and Technology, Avenida WaiLong, Taipa, Macao, 999078, China.
Ronghui Han, Department of Anesthesiology, Affiliated Hospital of Guangdong Medical University, No. 57, South Renmin Avenue, Zhanjiang, 524000, China; Faculty of Chinese Medicine, State Key Laboratory of Quality Research in Chinese Medicine, Macau University of Science and Technology, Avenida WaiLong, Taipa, Macao, 999078, China.
Zhengyuan Xia, Department of Anesthesiology, Affiliated Hospital of Guangdong Medical University, No. 57, South Renmin Avenue, Zhanjiang, 524000, China; State Key Laboratory of Pharmaceutical Biotechnology, Department of Medicine, The University of Hong Kong, Pok Fu Lam Road, Hong Kong, 999077, China.
Abbreviations
ALOX15: Arachidonate 15-lipoxygenase; AMPK: Adenosine 5′-monophosphate-activated protein kinase; ATP: Adenosine triphosphate; CCK-8: Cell Counting Kit-8; CD36: Cluster of differentiation 36; CK-MB: Cardiac-type creatine kinase isoenzyme; DCFH-DA: 2,7-dichlorodihydrofluorescein diacetate; DCM: Diabetic cardiomyopathy; EF: Ejection fraction; ELISA: Enzyme-linked Immunosorbent Assay; FAs: Fatty acids; FITC: Fluorescein Isothiocyanate; FS: Fractional shortening; GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; GPX4: Glutathione peroxidase 4; HE: Hematoxylin and eosin; HFD: High-fat diet; HG: High glucose; HR: Hypoxia/reoxygenation; HO-1: heme oxygenase 1; IR: Ischemia–reperfusion; LCFA: Long-chain fatty acid; LDH: Lactate dehydrogenase; GSH: L-Glutathione; GSSG: Oxidized L-glutathione; MDA: Malonaldehyde; MIRI: Myocardial ischemia–reperfusion injury; MMP: Mitochondrial membrane potential; NC: Non-diabetic control; Nrf2: Nuclear factor erythroid 2-Related Factor 2; 8-OHdG: 8-Hydroxy-2′-deoxyguanosine; PAL: Palmitic acid; p-AMPK: phosphorylated AMPK; PI: propidium iodide; PPF: Propofol; PUFA: Polyunsaturated fatty acids; ROS: Reactive oxygen system; RT-PCR: Real-time quantitative polymerase chain reaction; SAA: Salvianolic acid A; STZ: Streptozotocin; T2DM: Type 2 diabetes mellitus; TG: Triglyceride; T-SOD: Total-superoxide dismutase; TTC: 2,3,5-Triphenyltetrazolium chloride.
Funding
This work was supported by the National Natural Science Fundation of China (81970247, 82270306), Research Grant Council GRF Grant (17118619), and in part by Guangdong Provincial Basic and Applied Basic Science Project (2021A1515011434).
Authors’ contributions
Conception or design of the study: J.Z., R.H. and Z.X.; data collection: J.Z., J.C., K.H. and Y.J.; data analysis and interpretation: J.Z., R.H., J.C., A.Z., D.Z. and D.L.; drafting the article: J.Z., W.X., R.H. and J.L.; critical revision of the article: Y.C., G.C., L.Z., W.X., A.X., Y.X. and Z.X. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All animal experiments in this study were approved by the Committee for Teaching and Research on the Use of Live Animals in Teaching and Research of Guangdong Medical University (Zhanjiang, China)
Conflict of interest
The authors state no conflict of interest.
Data Availability
Data collected and analyzed for the study are available from the corresponding author upon reasonable request.
References
- 1. Htay T, Soe K, Lopez-Perez A, Doan AH, Romagosa MA, Aung K. Mortality and cardiovascular disease in type 1 and type 2 diabetes. Curr Cardiol Rep. 2019;21:45–56. [DOI] [PubMed] [Google Scholar]
- 2. Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007;59:418–58. [DOI] [PubMed] [Google Scholar]
- 3. Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED. Cardiac energy metabolism in heart failure. Circ Res. 2021;128:1487–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab. 2012;15:805–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Glatz JFC, Luiken J. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J Lipid Res. 2018;59:1084–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shu H, Peng Y, Hang W, Nie J, Zhou N, Wang DW. The role of CD36 in cardiovascular disease. Cardiovasc Res. 2022;118:115–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garcia D, Shaw RJ. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell. 2017;66:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu LM, Dong X, Xue XD, Xu S, Zhang X, Xu YL, et al. . Melatonin attenuates diabetic cardiomyopathy and reduces myocardial vulnerability to ischemia-reperfusion injury by improving mitochondrial quality control: role of SIRT6. J Pineal Res. 2021;70:e12698. [DOI] [PubMed] [Google Scholar]
- 9. Xin C, Zhang Z, Gao G, Ding L, Yang C, Wang C, et al. . Irisin attenuates myocardial ischemia/reperfusion injury and improves mitochondrial function through AMPK pathway in diabetic mice. Front Pharmacol. 2020;11:565160:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sun B, Ou H, Ren F, Huan Y, Zhong T, Gao M, et al. . Propofol inhibited autophagy through ca(2+)/CaMKKβ/AMPK/mTOR pathway in OGD/R-induced neuron injury. Mol Med. 2018;24:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. . Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu S, Zhu J, Wu G, Hu Z, Ying P, Bao Z, et al. . 6-Gingerol alleviates Ferroptosis and inflammation of diabetic cardiomyopathy via the Nrf2/HO-1 pathway. Oxidative Med Cell Longev. 2022;2022:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang W, Lu J, Wang Y, Sun P, Gao T, Xu N, et al. . Canagliflozin attenuates lipotoxicity in Cardiomyocytes by inhibiting inflammation and Ferroptosis through activating AMPK pathway. Int J Mol Sci. 2023;24:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li X, Li Z, Dong X, Wu Y, Li B, Kuang B, et al. . Astragaloside IV attenuates myocardial dysfunction in diabetic cardiomyopathy rats through downregulation of CD36-mediated ferroptosis. Phytother Res. 2023;37:3042–56. [DOI] [PubMed] [Google Scholar]
- 15. Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. . Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116:2672–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cai W, Liu L, Shi X, Liu Y, Wang J, Fang X, et al. . Alox15/15-HpETE aggravates myocardial ischemia-reperfusion injury by promoting Cardiomyocyte Ferroptosis. Circulation. 2023;147:1444–60. [DOI] [PubMed] [Google Scholar]
- 17. Han RH, Huang HM, Han H, Chen H, Zeng F, Xie X, et al. . Propofol postconditioning ameliorates hypoxia/reoxygenation induced H9c2 cell apoptosis and autophagy via upregulating forkhead transcription factors under hyperglycemia. Mil Med Res. 2021;8:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shao H, Li J, Zhou Y, Ge Z, Fan J, Shao Z, et al. . Dose-dependent protective effect of propofol against mitochondrial dysfunction in ischaemic/reperfused rat heart: role of cardiolipin. Br J Pharmacol. 2008;153:1641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ho JH, Hong CY. Salvianolic acids: small compounds with multiple mechanisms for cardiovascular protection. J Biomed Sci. 2011;18:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu T, Wu X, Chen Q, Zhu S, Liu Y, Pan D, et al. . The anti-apoptotic and cardioprotective effects of salvianolic acid a on rat cardiomyocytes following ischemia/reperfusion by DUSP-mediated regulation of the ERK1/2/JNK pathway. PLoS One. 2014;9:e102292:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deng F, Wang S, Zhang L, Xie X, Cai S, Li H, et al. . Propofol through upregulating Caveolin-3 attenuates post-hypoxic mitochondrial damage and cell death in H9C2 Cardiomyocytes during Hyperglycemia. Cell Physiol Biochem. 2017;44:279–92. [DOI] [PubMed] [Google Scholar]
- 22. Samovski D, Sun J, Pietka T, Gross RW, Eckel RH, Su X, et al. . Regulation of AMPK activation by CD36 links fatty acid uptake to β-oxidation. Diabetes. 2015;64:353–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30:595–602. [DOI] [PubMed] [Google Scholar]
- 24. Ritchie RH, Abel ED. Basic mechanisms of diabetic heart disease. Circ Res. 2020;126:1501–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wei J, Zhao Y, Liang H, Du W, Wang L. Preliminary evidence for the presence of multiple forms of cell death in diabetes cardiomyopathy. Acta Pharm Sin B. 2022;12:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res. 2015;116:674–99. [DOI] [PubMed] [Google Scholar]
- 27. Coughlan KA, Valentine RJ, Ruderman NB, Saha AK. AMPK activation: a therapeutic target for type 2 diabetes? Diabetes Metab Syndr Obes. 2014;7:241–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang C, Zhu L, Yuan W, Sun L, Xia Z, Zhang Z, et al. . Diabetes aggravates myocardial ischaemia reperfusion injury via activating Nox2-related programmed cell death in an AMPK-dependent manner. J Cell Mol Med. 2020;24:6670–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Glatz JFC, Wang F, Nabben M, Luiken J. CD36 as a target for metabolic modulation therapy in cardiac disease. Expert Opin Ther Targets. 2021;25:393–400. [DOI] [PubMed] [Google Scholar]
- 30. Pepino MY, Kuda O, Samovski D, Abumrad NA. Structure-function of CD36 and importance of fatty acid signal transduction in fat metabolism. Annu Rev Nutr. 2014;34:281–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009;9:407–16. [DOI] [PubMed] [Google Scholar]
- 32. He J, Liu D, Zhao L, Zhou D, Rong J, Zhang L, et al. . Myocardial ischemia/reperfusion injury: mechanisms of injury and implications for management (review). Exp Ther Med. 2022;23:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang R, Dong S, Xia R, Sun M, Sun Y, Ren H, et al. . Kinsenoside mitigates myocardial ischemia/reperfusion-induced ferroptosis via activation of the Akt/Nrf2/HO-1 pathway. Eur J Pharmacol. 2023;956:175985:1–16. [DOI] [PubMed] [Google Scholar]
- 34. Fang X, Ardehali H, Min J, Wang F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat Rev Cardiol. 2023;20:7–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ma XH, Liu JH, Liu CY, Sun WY, Duan WJ, Wang G, et al. . ALOX15-launched PUFA-phospholipids peroxidation increases the susceptibility of ferroptosis in ischemia-induced myocardial damage. Signal Transduct Target Ther. 2022;7:288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Walsh CT. Propofol: milk of amnesia. Cell. 2022;185:4861. [DOI] [PubMed] [Google Scholar]
- 37. Skues MA, Prys-Roberts C. The pharmacology of propofol. J Clin Anesth. 1989;1:387–400. [DOI] [PubMed] [Google Scholar]
- 38. Tao W, Zhang X, Ding J, Yu S, Ge P, Han J, et al. . The effect of propofol on hypoxia- and TNF-α-mediated BDNF/TrkB pathway dysregulation in primary rat hippocampal neurons. CNS Neurosci Ther. 2022;28:761–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kam PC, Cardone D. Propofol infusion syndrome. Anaesthesia. 2007;62:690–701. [DOI] [PubMed] [Google Scholar]
- 40. Wang S, Wang C, Yan F, Wang T, He Y, Li H, et al. . N-Acetylcysteine attenuates diabetic myocardial ischemia reperfusion injury through inhibiting excessive autophagy. Mediat Inflamm. 2017;2017:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yuan X, Xiang Y, Zhu N, Zhao X, Ye S, Zhong P, et al. . Salvianolic acid a protects against myocardial ischemia/reperfusion injury by reducing platelet activation and inflammation. Exp Ther Med. 2017;14:961–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen Q, Xu T, Li D, Pan D, Wu P, Luo Y, et al. . JNK/PI3K/Akt signaling pathway is involved in myocardial ischemia/reperfusion injury in diabetic rats: effects of salvianolic acid a intervention. Am J Transl Res. 2016;8:2534–48. [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Y, Zhang K, Qi X, Yang G, Wang H, Zhang Z, et al. . Effects of propofol on LC3II and mTOR/p-mTOR expression during ischemia-reperfusion myocardium injury in rats with type 2 diabetes mellitus. Exp Ther Med. 2020;19:2441–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang Y, Qi X, Wang C, Zhao D, Wang H, Zhang J. Effects of propofol on myocardial ischemia-reperfusion injury in rats with type-2 diabetes mellitus. Biomed Rep. 2017;6:69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lin C, Sui H, Gu J, Yang X, Deng L, Li W, et al. . Effect and mechanism of propofol on myocardial ischemia reperfusion injury in type 2 diabetic rats. Microvasc Res. 2013;90:162–8. [DOI] [PubMed] [Google Scholar]
- 46. Heather LC, Pates KM, Atherton HJ, Cole MA, Ball DR, Evans RD, et al. . Differential translocation of the fatty acid transporter, FAT/CD36, and the glucose transporter, GLUT4, coordinates changes in cardiac substrate metabolism during ischemia and reperfusion. Circ Heart Fail. 2013;6:1058–66. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data collected and analyzed for the study are available from the corresponding author upon reasonable request.