

ABSTRACT
Chlamydiae are obligate intracellular bacterial pathogens that may cause genital pathology via induction of destructive host immune responses. Human-adapted Chlamydia trachomatis causes inflammatory disease in human hosts but is easily cleared in mice, and mouse-adapted Chlamydia muridarum establishes a productive and pathogenic infection in murine hosts. While numerous anti-chlamydial host resistance factors have been discovered in mice and humans alike, little is known about host factors promoting host fitness independent of host resistance. Here, we show that interferon-inducible immunity-related GTPase M (Irgm) proteins function as such host factors ameliorating infection-associated sequalae in the murine female genital tract, thus characterizing Irgm proteins as mediators of disease tolerance. Specifically, we demonstrate that mice deficient for all three murine Irgm paralogs (pan-Irgm−/−) are defective for cell-autonomous immunity to C. trachomatis, which correlates with an early and transient increase in bacterial burden and sustained hyperinflammation in vivo. In contrast, upon infection of pan-Irgm−/− mice with C. muridarum, bacterial burden is unaffected, yet genital inflammation and scarring pathology are nonetheless increased, demonstrating that Irgm proteins can promote host fitness without altering bacterial burden. Additionally, pan-Irgm−/− mice display increased granulomatous inflammation in genital Chlamydia infection, implicating Irgm proteins in the regulation of granuloma formation and maintenance. These findings demonstrate that Irgm proteins regulate pathogenic immune responses to Chlamydia infection in vivo, establishing an effective infection model to examine the immunoregulatory functions and mechanisms of Irgm proteins.
IMPORTANCE
In response to genital Chlamydia infection, the immune system mounts a proinflammatory response to resist the pathogen, yet inflammation must be tightly controlled to avoid collateral damage and scarring to host genital tissue. Variation in the human IRGM gene is associated with susceptibility to autoinflammatory diseases but its role in ameliorating inflammatory diseases caused by infections is poorly defined. Here, we use mice deficient for all three murine Irgm paralogs to demonstrate that Irgm proteins not only provide host resistance to Chlamydia infections but also limit associated inflammation in the female genital tract. In particular, we find that murine Irgm expression prevents granulomatous inflammation, which parallels inflammatory diseases associated with variants in human IRGM. Our findings therefore establish genital Chlamydia infection as a useful model to study the roles for Irgm proteins in both promoting protective immunity and limiting pathogenic inflammation.
KEYWORDS: Chlamydia, immunity-related GTPases, disease tolerance, interferons, immunopathology, IRGM, sexually transmitted diseases
INTRODUCTION
Chlamydia trachomatis is an obligate intracellular bacterial pathogen that can cause a spectrum of human diseases. Chronic or repeated infection with C. trachomatis serovars D-K can cause genital scarring and pelvic inflammatory disease, infertility, and an increased risk of ectopic pregnancy (1). Ocular serovars A–C cause corneal inflammation and blinding trachoma (2), and serovars L1–L3 can cause lymphogranuloma venereum (3). All of these chlamydial disease syndromes are mediated largely by destructive host immune responses. Genital and ocular chlamydial diseases comprise a massive worldwide public health burden, and no protective vaccine exists (4).
Infectious disease manifestation is determined by protective resistance mechanisms that reduce infectious burden as well as tolerance mechanisms that ameliorate the pathogenic impact of an infection independent of burden relief (5). Animal models of genital Chlamydia infection have proven instrumental in elucidating both protective and pathogenic host immune responses (6). Chlamydia trachomatis is able to persist and cause disease in its preferred human host, but it is readily cleared in murine hosts and generally does not cause disease (7). However, C. muridarum has adapted to subvert murine mechanisms of cell-autonomous immunity and therefore establishes a productive infection in the mouse host (8–10). Genital infection with C. muridarum results in a pathogenic inflammatory response and genital scarring similar to that observed in humans with chlamydial disease (11–13). Pathology in genital C. muridarum infection is dependent on certain murine host immune factors such as CD8+ T cells, neutrophils, IL-1, and TNF-α (6). Therefore, while host immunity is necessary to clear Chlamydia infection and provide disease resistance, regulation of pathogenic immune responses is necessary to prevent genital scarring and thus confer tolerance to Chlamydia disease. Several correlative studies have associated a number of the same immune mediators with human disease in C. trachomatis infection, suggesting that murine C. muridarum infection is a useful model for human chlamydial disease (14–18).
As the pathology in genital Chlamydia infection is mediated largely by host immunity, Chlamydia infection provides a viable model to study mechanisms of immunoregulation. In humans, immunity-related GTPase clade M (IRGM) is a protein whose functions and mechanisms remain to be fully elucidated, which is linked with a variety of inflammatory diseases such as inflammatory bowel disease (19), sepsis (20), and autoimmune thyroid disease (21). Analysis of disease-related IRGM variants mostly correlate with decreased gene expression, implying that IRGM functions to limit inflammation (22). Mechanistically, human IRGM has been implicated in regulating autophagy and may regulate inflammasome activity via selective autophagy of inflammasome machinery (23–25). However, the mechanisms by which variants in IRGM lead to human inflammatory disease remain to be understood.
Mice possess three paralogs of IRGM (Irgm1, Irgm2, and Irgm3), which were initially thought to primarily control cell-autonomous immunity to intracellular pathogens such as C. trachomatis (26). Murine Irgm proteins regulate the localization and activity of a subset of “effector” GKS IRG proteins, which are expressed upon stimulation with the cytokine gamma-interferon (IFNγ) and traffic to intracellular pathogens to mediate their destruction via partially understood mechanisms (27–31). In the absence of Irgm1 and Irgm3, IFNγ stimulation leads to mislocalization and cytoplasmic aggregation of GKS IRGs and a failure to target and restrict the growth of most intracellular pathogens in an IFNγ-dependent manner (30, 32, 33). Irgm1−/− mice are defective for immunity to virtually all intracellular bacterial and protozoal pathogens, while Irgm3−/− mice are susceptible to a more limited set of pathogens (30, 33, 34). Interestingly, for a small number of examined intracellular pathogens such as Listeria monocytogenes, Irgm1−/− mice are defective for IFNγ-mediated resistance while concurrent deletion of Irgm3 in Irgm1/m3−/− mice reverses this susceptibility, implying complex regulatory relationships among Irgm proteins in some contexts (33, 35, 36). With respect to C. trachomatis, both Irgm1−/− and Irgm3−/− mice are defective for IFNγ-mediated resistance to bacterial infection. Irgm1/m3−/− cells are also defective for host resistance in vitro and display increased bacterial burden in vivo early in genital infection, although an enhanced T cell response correlates with clearance of the infection over time (32, 37). The other remaining paralog, Irgm2, has not been studied in Chlamydia infection but only in the context of infection with the protozoan pathogen Toxoplasma gondii, where it acts as a critical host resistance factor (38, 39).
While murine Irgm proteins are critical for defense against most intracellular pathogens, humans lack Irgm-regulated effector IRGs and human IRGM has only been shown to contribute to host defense against a small subset of pathogens such as Mycobacterium tuberculosis and not against Toxoplasma or Chlamydia (40, 41). Mouse and human IRGM proteins therefore appear to only share limited functional homology in cell-autonomous immunity; however, murine Irgm proteins—especially Irgm1—have been shown to regulate inflammation in contexts similar to human IRGM. Irgm1−/− mice display susceptibility to intestinal inflammation in multiple contexts (42–44), increased cytokine production upon LPS treatment (45, 46), and type I interferonopathy resembling Sjogren’s syndrome (47), paralleling several human disease syndromes associated with IRGM variants. Irgm1 also has been shown to regulate T cell homeostasis, with Irgm1−/− mice displaying lymphoid collapse upon infection with certain bacterial pathogens including C. trachomatis, although additional deletion of Irgm3 appears to reverse this phenotype (37, 48, 49). On a cellular level, Irgm1 appears to regulate inflammasome activation via selective autophagy, similar to human IRGM (23), and defects in mitophagy can result in spontaneous interferonopathies and linked susceptibility to bacterial infections (47, 50). Irgm2 on the other hand has been shown to limit noncanonical inflammasome activation (51, 52). These findings collectively suggest that, although murine Irgm paralogs are involved in many processes that human IRGM is not, there exists significant functional homology pertaining to immunoregulation.
The dual roles of murine Irgm proteins in orchestrating protective cell-autonomous immunity to intracellular pathogens and regulating inflammation make it difficult to clearly appreciate their roles in shaping the pathophysiology of infections. Furthermore, the interregulatory relationships between the three Irgm proteins—exemplified by the reversal of lymphoid collapse and interferonopathy in Irgm1−/− mice upon concurrent Irgm3 deletion among other phenotypes (33, 36, 37, 47)—obscure the individual roles of Irgm proteins and complicates the homology between mouse and human systems. For this reason, we leverage mice deficient for all three Irgm proteins (pan-Irgm−/−), revealing the collective function of all three Irgm proteins to best model a human system lacking its sole IRGM. Mirroring the finding that IRGM variants are associated with mortality in human Mycobacterium tuberculosis infection (40), pan-Irgm−/− mice were recently shown to display worsened disease and mortality in Mtb infection (53). We employ genital Chlamydia infection in pan-Irgm−/− mice as a model to evaluate protective immunity to this intracellular bacterial pathogen as well as regulation of pathogenic inflammation. We begin by demonstrating that, like Irgm1/m3−/− mice, pan-Irgm−/− mice are defective for cell-autonomous immunity to C. trachomatis (serovar L2) which corresponds with an early increase in bacterial burden in vivo. Compared to wild-type mice, pan-Irgm−/− mice displayed extensively increased inflammation at 6 days post-infection (dpi). To uncouple the increase in bacterial burden from the increase in inflammation, we turned to the mouse-adapted C. muridarum, which evades IRG-mediated cell-autonomous immunity (8, 9). As expected, we observe no difference in C. muridarum burden between wild-type and pan-Irgm−/− mice in vitro or in vivo, yet pan-Irgm−/− mice display increased histologic inflammation at 6 dpi and 45 dpi as well as increased scarring pathology at 25 dpi and 45 dpi. We find that Irgm3 plays a more important role than Irgm1 or Irgm2 in controlling genital inflammation, and we show that excessive genital inflammation in pan-Irgm−/− mice occurs independent of adaptive immunity. These findings demonstrate that Irgm proteins—especially Irgm3—regulate inflammation and immunopathology in genital Chlamydia infection independent of bacterial burden and establish genital Chlamydia infection as a useful model to study Irgm-regulated inflammation in the context of an infection.
RESULTS
Pan-Irgm−/− mice are defective for cell-autonomous immunity to C. trachomatis
We evaluated protective immunity to C. trachomatis in pan-Irgm−/− mice and mice deficient for Irgm2 or doubly deficient for Irgm1 and Irgm3 (Irgm1/m3−/−); all mice were generated on a C57BL/6 background. While IFNγ-primed wild-type mouse embryonic fibroblasts (MEFs) robustly restrict the growth of C. trachomatis, it has been shown that Irgm1/m3−/− MEFs display no IFNγ-mediated restriction of C. trachomatis growth, demonstrating a complete defect in cell-autonomous immunity to this bacterial pathogen (9, 37). The role for Irgm2 in cell-autonomous immunity to C. trachomatis has not been studied, but we and others have shown that Irgm2 promotes cell-autonomous immunity to Toxoplasma (38, 39). We have also shown that pan-Irgm−/− mice are similarly defective for cell-autonomous immunity to Toxoplasma (39). We sought to determine if Irgm2 plays a similar role in cell-autonomous immunity to C. trachomatis. We found that while pan-Irgm−/− MEFs displayed no IFNγ-mediated restriction of C. trachomatis growth, Irgm2−/− MEFs restricted C. trachomatis growth similar to wild-type (Fig. 1A). This finding demonstrates that Irgm2 is largely dispensable for cell-autonomous immunity to C. trachomatis and pan-Irgm−/− MEFs are completely defective for IFNγ-mediated anti-C. trachomatis host resistance, similar to Irgm1/m3−/− MEFs.
Fig 1.
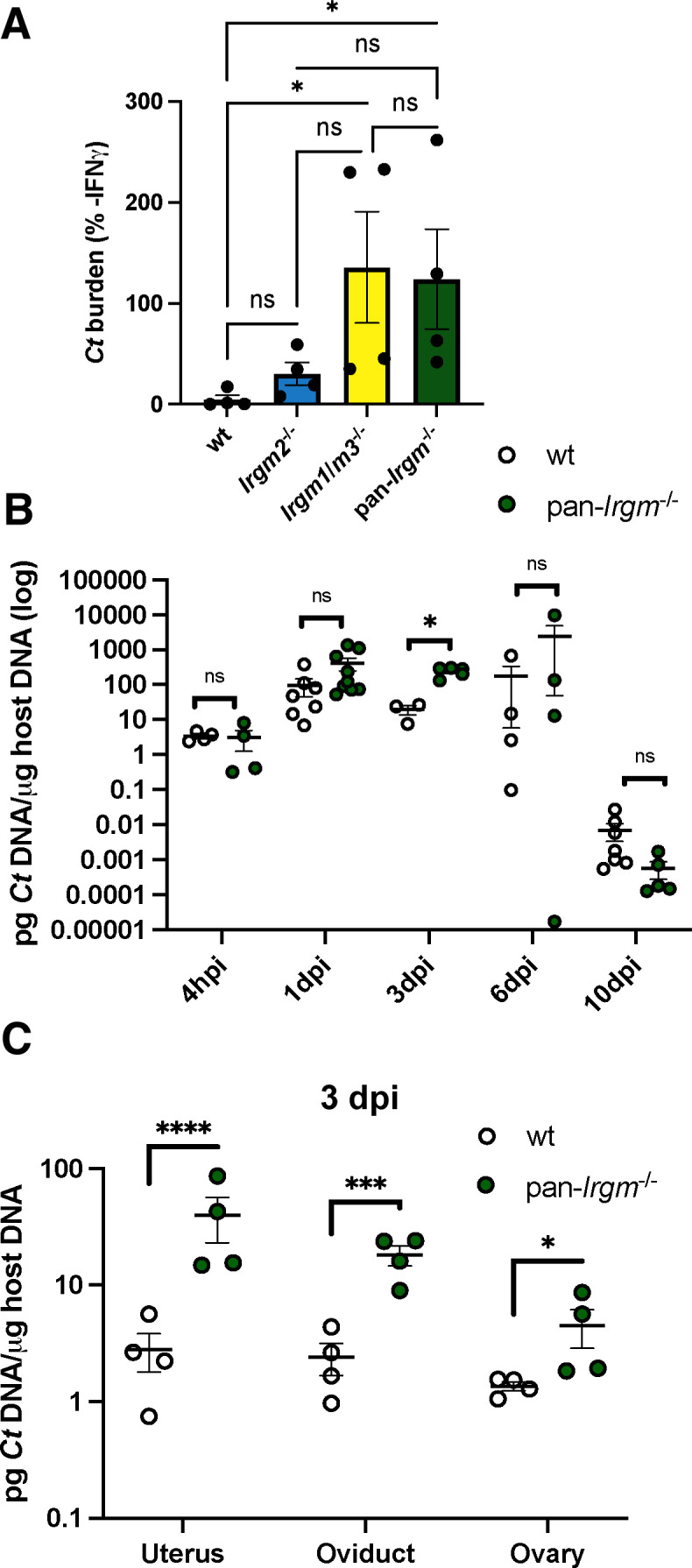
A defect in cell-autonomous immunity correlates with a transient increase in C. trachomatis burden in the female genital tract of pan-Irgm−/− mice. (A) MEFs were primed overnight with 100 U/mL IFNγ prior to infection with C. trachomatis elementary bodies (EBs) at a multiplicity of infection (MOI) of 1:1. DNA was harvested from the infected cells at 24 hpi and bacterial burden was quantified using quantitative real-time polymerase chain reaction (qPCR). Graphs represent the proportion of bacterial burden in IFNγ-primed vs. unprimed cells. Data points represent MEFs derived from separate embryos (n = 4 separate MEF lines). Statistical significance was determined using one-way analysis of variance (ANOVA). (B) Mice were infected transcervically with 5 × 106 C. trachomatis EBs, and female genital tracts were harvested at the indicated timepoints or (C) harvested at 3 dpi and then segmented. DNA was extracted from the harvested organ, and bacterial burden was quantified using qPCR. Each data point represents one mouse. Data in B represent pooled data from one experiment including all timepoints and one additional experiment for 3 dpi and 10 dpi timepoints: 4 hpi n = 4 mice; 1 dpi wild-type n = 7, pan-Irgm−/− n = 9; 3 dpi wild-type n = 3, pan-Irgm−/− n = 5; 6 dpi n = 4; and 10 dpi wild-type n = 7, pan-Irgm−/− n = 5. Data in C represent one experiment (n = 4 mice). Statistical significance was determined using t-test (B, C); *P < 0.05, ***P < 0.0005, and ****P < 0.00005; ns, not significant.
While Irgm1/m3−/− MEFs are completely defective for cell-autonomous immunity to C. trachomatis, Irgm1/m3−/− mice demonstrate an early transient increase in bacterial burden in vivo but ultimately clear the infection with kinetics similar to wild-type mice (37). We next sought to determine the susceptibility of pan-Irgm−/− mice to in vivo genital C. trachomatis infection. We infected mice transcervically with C. trachomatis and tracked bacterial burden over time by qPCR. Similar to previous reports in Irgm1/m3−/− mice (37), pan-Irgm−/− mice displayed increased bacterial burden relative to wild-type mice early in infection at 3 dpi, but bacterial burden is comparable to that of wild-type mice at subsequent timepoints (Fig. 1B). At 3 dpi, we observed an increase in C. trachomatis burden in the uteri, oviducts, and ovaries of infected pan-Irgm−/− mice compared with the wild type (Fig. 1C). Therefore pan-Irgm−/− mice display increased early bacterial burden in vivo that correlates with the expected in vivo timing of the observed in vitro defect in cell-autonomous restriction of bacterial growth, but additional immune mechanisms appear to ultimately compensate for this defect to restore protective immunity as in wild-type mice.
C. trachomatis-infected pan-Irgm−/− mice display increased genital inflammation and frequent granuloma formation
Recent studies have implicated Irgm1 and Irgm2 in the control of inflammation in a variety of disease models (24, 43, 47, 51), but the collective roles of Irgm proteins in regulation of inflammation have only been described in macrophage responses to bacterial LPS and in in vivo Mycobacterium tuberculosis infection (52, 53). Because Chlamydia infection mediates disease predominantly via host inflammatory responses (6), we sought to use genital Chlamydia infection as a model to examine inflammation in pan-Irgm−/− mice. We began by examining genital inflammation at 6 dpi in mice infected with C. trachomatis serovar L2, which causes minimal inflammation and pathology in wild-type mice (54). Consistent with prior reports, at this timepoint, wild-type mice displayed mild inflammation in the uterus and no inflammation in the oviducts and ovaries. Pan-Irgm−/− mice, in contrast, displayed increased uterine inflammation and tissue destruction, as well as significant inflammation in the oviducts and ovaries (Fig. 2A through E). Additionally, there was increased neutrophil influx in pan-Irgm−/− genital tracts compared with the wild type (Fig. 2F and G).
Fig 2.

Pan-Irgm−/− mice display increased inflammation in genital C. trachomatis infection. (A–G) Mice were infected transcervically with 5 × 106 C. trachomatis EBs, and the female genital tract was harvested at 6 dpi. (A) Gross pathology—arrowhead indicates area of gross inflammation. (B) Genital tracts were fixed, sectioned, and H&E stained. Mildly inflamed wild-type uterine tissue and severely inflamed and edematous pan-Irgm−/− uterus; wild-type ovary and oviduct with no evidence of inflammation and pan-Irgm−/− ovary, oviduct, and mesosalpinx each displaying severe inflammation—arrowheads indicate regions of inflammatory infiltrate or edema. The magnitude of acute and chronic inflammation (C), edema (D), and dilation (E) was graded by a veterinary pathologist (n = 7 mice; each data point represents one uterine horn of an infected mouse). (F) Immunohistochemistry was performed on fixed genital tracts infected with C. trachomatis to label the neutrophil marker myeloperoxidase (MPO—brown). Arrowheads indicate regions or punctae of neutrophilic infiltrate. (G) Flow cytometry on genital tracts infected with C. trachomatis to quantify neutrophils (CD45+Gr1+ live cells; n = 5 mice; each data point represents one entire reproductive tract of an infected mouse). Data represent one experiment that is representative of multiple replicates. Statistical significance was determined using Mann-Whitney test (C–E) or t-test (G); *P < 0.05 and ****P < 0.00005.
Noticeably, uteri of pan-Irgm−/− mice displayed not only signs of acute but also histiocytic (Fig. 3A) as well as granulomatous inflammation (Fig. 3B). While uterine granulomas rarely formed in wild-type mice (1 out of 14), granulomas were found in the uteri of more than half of infected pan-Irgm−/− mice (8 out of 14; Fig. 3C). Collectively, the stark increase in inflammation and tissue destruction in pan-Irgm−/− mice suggests that Irgm proteins regulate genital inflammation in C. trachomatis infection, potentially in addition to their roles in host resistance.
Fig 3.

Pan-Irgm−/− mice display granulomatous inflammation in genital C. trachomatis infection. Mice were infected transcervically with 5 × 106 C. trachomatis EBs and harvested at 6 dpi. (A) High magnification of mild acute inflammation in wild-type uterus and acute and histiocytic inflammation in pan-Irgm−/− uterus. A, acute inflammation; H, histiocytic inflammation. (B) Low magnification of diffuse granulomatous inflammation in pan-Irgm−/− uterus. Arrowheads indicate margins of granulomatous lesion. (C) Quantification of number of uterine horns displaying granulomatous lesions (n = 7 mice). Data represent one experiment that is representative of multiple replicates. Statistical significance was calculated using χ2 test; *P < 0.05.
Irgm proteins do not promote defense against C. muridarum infection
Pathogen load is a major factor that drives inflammation. While C. trachomatis burden was similar between wild-type and pan-Irgm−/− mice at 6 dpi and 10 dpi, there was increased bacterial load at 3 dpi throughout the genital tracts of pan-Irgm−/− mice (Fig. 1). Therefore, Irgm proteins could limit inflammation of the infected female genital tract in part or entirely by promoting cell-autonomous immunity and restriction of bacterial growth. Alternatively, the exacerbation of the inflammatory response seen in pan-Irgm−/− mice could be explained if Irgm proteins additionally attenuated inflammation independent of bacterial burden in chlamydial infection. To be able to test the latter hypothesis, we leveraged the mouse-adapted strain C. muridarum, which evolved to evade IFNγ-mediated cell-autonomous immunity executed by the family of murine IRG proteins (8–10, 32). Confirming that C. muridarum is resistant to Irgm-mediated cell-autonomous immunity, we found that both wild-type and pan-Irgm−/− MEFs displayed similarly limited IFNγ-mediated restriction of C. muridarum growth (Fig. 4A). We next infected wild-type and pan-Irgm−/− mice transcervically with C. muridarum and tracked bacterial burden in vivo over time using qPCR. In contrast to C. trachomatis (Fig. 1), we found no difference in C. muridarum burden in wild-type and pan-Irgm−/− mice at any timepoint (Fig. 4B). These findings demonstrate that Irgm proteins do not play meaningful roles in the clearance of C. muridarum in the female genital tract and provided us with an experimental platform to probe for anti-inflammatory function of Irgm proteins independent of their role as host resistance factors.
Fig 4.

Host resistance to C. muridarum remains unchanged in the absence of all three murine Irgm paralogs. (A) MEFs were primed overnight with IFNγ prior to infection with C. muridarum EBs at an MOI of 1:1. DNA was harvested at 24 hpi, and bacterial burden was quantified using qPCR. Graphs represent the proportion of bacterial burden in IFNγ-primed vs. unprimed cells. Data points represent distinct MEF lines derived from separate embryos (n = 5 separate MEF lines). (B) Mice were infected transcervically with 2.5 × 105 C. muridarum EBs, and genital tracts were harvested at the indicated timepoints. DNA was extracted and bacterial burden was quantified using qPCR. Data represent pooled data from one experiment including all timepoints plus one additional experiment for the 1-dpi timepoint: 4 hpi n = 4 mice; 1 dpi n = 9; 3 dpi n = 4; 6 dpi n = 4; 25 dpi wild-type n = 3, pan-Irgm−/− n = 4; and 45 dpi n = 5. Statistical significance was determined using t-test; ns, not significant.
Pan-Irgm−/− mice display increased inflammation and immunopathology in C. muridarum infection
Because wild-type and pan-Irgm−/− mice displayed indistinguishable levels of bacterial burden in genital C. muridarum infection, any change in inflammation in pan-Irgm−/− mice would necessarily be attributable to the roles for Irgm proteins in the regulation of inflammation. We therefore began by examining inflammation at 6 dpi in transcervical C. muridarum infection. In contrast to C. trachomatis L2 infection, C. muridarum induced significant inflammation and tissue destruction in both wild-type and pan-Irgm−/− mice throughout the genital tract at 6 dpi. In every measure, pan-Irgm−/− mice displayed slightly increased inflammation and markers of tissue destruction, and this difference reached statistical significance for uterine dilation and mesosalpingeal inflammation (Fig. 5A through C). Pan-Irgm−/− mice infected with C. muridarum also displayed large granulomatous lesions (Fig. 5D) and histiocytic inflammation compared with acute, neutrophilic inflammation in wild-type mice (Fig. 5E). While the severity of inflammation already observed in wild-type genital tracts infected with C. muridarum makes it difficult to appreciate increases in inflammation in pan-Irgm−/− mice, the nonetheless statistically significant increases in pathology scores indicate that Irgm proteins dampen inflammation in C. muridarum infection at 6 dpi independent of bacterial burden.
Fig 5.

Increased inflammation in pan-Irgm−/− at 6 dpi in genital C. muridarum infection. Mice were infected transcervically with 2.5 × 105 C. muridarum EBs and harvested at 6 dpi (A–E). (A) Gross pathology of infected genital tracts. Arrowhead denotes region of gross inflammation. (B–E) Genital tracts were fixed, sectioned, and H&E stained. The magnitude of acute and chronic inflammation (B) and dilation (C) was graded by a veterinary pathologist. (D) Low magnification of granulomatous lesion (*) in pan-Irgm−/− uterus. (E) High magnification of acute inflammation in wild-type uterus and acute and histiocytic inflammation in pan-Irgm−/− uterus. A, acute inflammation; H, histiocytic inflammation. Data represent one experiment that is representative of multiple replicates. Wild-type n = 6 mice, pan-Irgm−/− n = 10; each data point represents one uterine horn of an infected mouse. Statistical significance was determined using Mann-Whitney test; *P < 0.05, ns = not significant.
We next examined inflammation at 45 dpi (Fig. 6A), when bacterial burden was reduced substantially compared with 6 dpi (Fig. 4B). At 45 dpi, there was increased inflammation throughout the genital tracts of pan-Irgm−/− mice infected with C. muridarum compared with the wild type. By 45 dpi, there was very little neutrophilic infiltration in the oviducts and mesosalpinges of wild-type mice; however, there was still moderate neutrophil infiltration in the oviducts and mesosalpinges of pan-Irgm−/− mice. Chronic inflammation characterized by lymphocytic infiltration continued in wild-type genital tracts, although to a more significant degree in pan-Irgm−/− uteri and mesosalpinges (Fig. 6B). Histologically, wild-type mice displayed cystic changes with minimal foci of inflammation, while pan-Irgm−/− mice displayed more severe and widespread inflammation that included the oviducts and ovaries (Fig. 6C). These findings demonstrate that pan-Irgm−/− mice display continued acute inflammation and increased chronic inflammation in C. muridarum infection at late timepoints.
Fig 6.

Increased pathology in pan-Irgm−/− mice at later timepoints in genital C. muridarum infection. Mice were infected transcervically with 2.5 × 105 C. muridarum EBs and harvested at 25 dpi or 45 dpi. (A) Gross pathology of infected genital tracts. Scale bars = 0.5 in; arrowheads indicate hydrosalpinx. (B–D) Genital tracts were fixed, sectioned, and H&E stained. (B) The magnitude of acute, chronic, and total acute + chronic inflammation at 45 dpi was graded by a veterinary pathologist (wild-type n = 7 mice, pan-Irgm−/− n = 8). Data represent one experiment that is representative of multiple replicates. (C) Low-magnification images of cystic changes in wild-type uterus and ovary/oviduct and continued inflammation in pan-Irgm−/− uterus and ovary/oviduct. (D) Quantification of hydrosalpinx at 25 dpi and 45 dpi in genital tracts infected with C. muridarum. Data represent pooled data from three experiments (25 dpi n = 12 mice; 45 dpi wild-type n = 21, pan-Irgm−/− n = 22). Each data point represent one uterine horn of an infected mouse. Statistical significance was determined using Mann-Whitney tests (B) or χ2 test (D); *P < 0.05 and **P < 0.005; ns, not significant.
Chlamydia causes genital inflammation that results in tissue scarring and pathology in mouse models (6). We therefore sought to examine the incidence of hydrosalpinx, a reliable marker of Chlamydia-related oviduct scarring, in wild-type and pan-Irgm−/− mice infected with C. muridarum at additional timepoints. We found that there was a substantial increase in the incidence of hydrosalpinx at 25 dpi and 45 dpi in pan-Irgm−/− mice compared with wild-type mice (Fig. 6A and D). This finding demonstrates that Irgm proteins limit scarring pathology and continued inflammation in genital C. muridarum infection and thus define Irgm proteins as mediators of disease tolerance to chlamydial infections.
Irgm3 is the most critical Irgm paralog in controlling genital inflammation in C. muridarum infection
Next, we asked whether these effects were driven by one or a combination of Irgm paralogs. Because Irgm1−/− mice develop spontaneous and often severe interferonopathy as a confounding factor, we instead opted to test Irgm1 function in mice in which type I interferon response had been normalized through co-deletion of Irgm3 (47). In addition to these Irgm1/m3−/− mice, we also infected Irgm2−/− and Irgm3−/− mice transcervically with C. muridarum alongside wild-type and pan-Irgm−/− mice and histologically examined inflammation and inflammatory pathology at 45 dpi. All Irgm-deficient phenotypes demonstrated increased combined acute and chronic inflammation in the uterus of infected mice (Fig. 7A). In many standard metrics, such as acute inflammation alone, chronic inflammation alone, and tissue dilation, it was difficult to clearly separate different Irgm-deficient phenotypes from one another when examining pathology in the uterus, oviduct, and ovaries (Fig. S1A through C). We therefore designed additional pathologic scoring criteria to better characterize phenotypes that were observed but not captured in the standard pathology grading criteria. Grading infected uteri for endometrial gland infiltration and dilation (Fig. 7B), as well as uterine luminal exudate (Fig. 7C), clearly revealed these as pathologic features in Irgm-deficient mice, with Irgm3−/− and pan-Irgm−/− uteri largely presenting as more severe than Irgm2−/− uteri. Irgm1/m3−/− mice generally phenocopied Irgm3−/− mice, indicating that Irgm1 is unlikely to impact the inflammatory response to C. muridarum in the female genital tract. Combining all pathological grading criteria into a combined pathology index revealed the same trend (Fig. 7D). These findings demonstrate that Irgm1 and Irgm2 play more minor roles and Irgm3 plays a more major role in limiting inflammation and tissue distortion and promoting disease tolerance in genital C. muridarum infection.
Fig 7.

Increased inflammatory pathology in Irgm- and Rag1-deficient uteri in genital C. muridarum infection at 45 dpi. Mice were infected transcervically with 2.5 × 105 C. muridarum EBs. Genital tracts were harvested at 45 dpi, fixed, sectioned, H&E stained, and graded by a veterinary pathologist. (A) The magnitude of acute and chronic inflammation in the uteri as defined by standard scoring criteria was combined. Additional features such as endometrial glandular pathology (B) and uterine luminal exudates (C) were also included. (A –C) Each data point represents one uterine horn of an infected mouse. (D) A combined uterine pathology index consisting of the sum of acute inflammation, chronic inflammation, dilation, glandular pathology, luminal exudates, and granulomatous inflammation was calculated. Each data point represents combined pathology scores from one entire reproductive tract from an infected mouse. Figure represents pooled data from two independent experiments (wild-type n = 7 mice, Irgm2−/− n = 5, Irgm3−/− n = 12, Irgm1/m3−/− n = 12, pan-Irgm−/− n = 10, and Rag1/pan-Irgm−/− n = 9). Statistical significance was determined using Mann-Whitney tests; *P < 0.05, **P < 0.005, ***P < 0.0005, and ****P < 0.00005.
Increased genital inflammation in pan-Irgm−/− mice infected with C. muridarum is not dependent on adaptive immunity
Irgm proteins limit immune responses by a variety of different mechanisms in different contexts. Specifically, Irgm3 has been shown to regulate inflammation via regulating antigen presentation (55, 56), and Irgm1 and Irgm3 regulate T cell responses in genital C. trachomatis infection (37). Additionally, certain varieties of T cells mediate inflammatory pathology in genital Chlamydia infection (6). We sought to determine whether the increased inflammation observed in pan-Irgm−/− mice was dependent on adaptive immunity. To this end, we generated mice deficient for all three Irgm proteins as well as Rag1, a protein critical for functional T and B cell development (Rag1/pan-Irgm−/−). We included these mice alongside wild-type, Irgm2−/−, Irgm3−/−, Irgm1/m3−/−, and pan-Irgm−/− mice for transcervical C. muridarum infection and histological analysis of genital inflammation at 45 dpi. We found that Rag1/pan-Irgm−/− mice displayed increased inflammation and inflammatory pathology in the uteri, oviducts, and ovaries across all applied metrics, compared with wild-type mice as well as all Irgm knockout models (Fig. 7A; Fig. S1). Because the magnitude of inflammation was radically greater in Rag1/pan-Irgm−/− mice compared with all other groups, they were not included in the expanded grading criteria in Fig. 7B through D. These findings demonstrate that adaptive immune cells such as cytotoxic CD8 T cells are dispensable for the formation of the severe pathology observed in C. muridarum-infected pan-Irgm−/− mice. Rather, our data indicate that adaptive immunity promotes disease tolerance in genital C. muridarum infection in Irgm-deficient mice.
DISCUSSION
Irgm1 and Irgm3 have been shown to be critical regulatory factors for IFNγ-mediated defense against C. trachomatis, but the collective roles of Irgm proteins in host defense and the regulation of inflammation in Chlamydia infection have not been studied. We report that pan-Irgm−/− MEFs are completely deficient for IFNγ-mediated resistance to C. trachomatis, correlating with a moderate transient defect in resistance in vivo and a disproportional increase in genital inflammation at 6 dpi. We show that pan-Irgm−/− and wild-type MEFs and mice are equally resistant to C. muridarum infection, yet pan-Irgm−/− mice display an increase in histologic inflammation at 6 dpi and 45 dpi and an increase in hydrosalpinx at 25 dpi and 45 dpi. We found that these phenotypes were driven more by a deficiency in Irgm3 compared with Irgm2 and Irgm1. These findings demonstrate that Irgm proteins confer resistance to disease caused by C. trachomatis—a pathogen that is susceptible to murine mechanisms of cell-autonomous immunity—and also confer relative disease tolerance to infections with mouse-adapted C. muridarum by controlling destructive inflammatory responses. Genital Chlamydia infection in pan-Irgm−/− mice is therefore an effective infection model to study the roles of Irgm proteins in both disease resistance and disease tolerance.
The phenotypic differences between infection with C. trachomatis and C. muridarum are consistent with existing models of host tropism and susceptibilities to murine immunity. Innate immunity in mice with a functional IRG system has been shown to be sufficient to clear C. trachomatis genital infection (57). The early increase in C. trachomatis burden in vivo correlates well with our finding that pan-Irgm−/− MEFs lack IFNγ-induced cell-autonomous immunity in vitro, which is consistent with a defect in epithelial cells and other non-immune cells to restrict bacterial burden in vivo prior to activation of professional immune cells. Macrophages and neutrophils, which have been shown to kill Chlamydia in vitro, possess additional antimicrobial mechanisms beyond the IRG system including the generation of radical oxygen species (58–61). Robust macrophage and neutrophil responses after 3 dpi, followed by engagement of adaptive immunity after the first week of infection, correlate with the decrease in C. trachomatis burden after 3 dpi and ultimate clearance of infection. In contrast, C. muridarum has adapted to evade cell-autonomous immunity in its preferred rodent host. Because murine cell-autonomous immunity to Chlamydia hinges upon the IRG system, pan-Irgm−/− mice display wild-type-level resistance to C. muridarum infection both in vitro and in vivo. The susceptibilities in vitro and in vivo of pan-Irgm−/− to infection with C. trachomatis and C. muridarum fit elegantly within known models for the host tropism of those two bacterial pathogens.
The increase in inflammation in pan-Irgm−/− mice infected with C. trachomatis compared with the wild type was striking, and while it correlated with the early defect in control of bacterial growth, the magnitude of the increase in pathology appeared out of proportion with the increase in bacterial growth. While we hypothesize that the absence of immunoregulatory Irgm proteins contributes to the observed increase in inflammation, the increased bacterial burden makes it difficult to appreciate the roles for Irgm proteins in promoting disease tolerance in C. trachomatis infection. Genital C. muridarum infection thus provides a useful model in that bacterial burden is unchanged in pan-Irgm−/− compared with wild-type mice. In this system, differences in inflammation and inflammatory pathology are indicative only of the effects of deletion of Irgm proteins on the regulation of inflammation.
Irgm proteins have been shown to regulate inflammation via multiple mechanisms, paralleling roles for human IRGM as demonstrated in cellular studies as well as disease phenotypes associated with genetic IRGM variants. Variants in human IRGM are associated with increased mortality in severe sepsis and increased incidence of inflammatory bowel disease and other autoimmune diseases, and the disease-associated variants are generally correlated with decreased IRGM expression (22, 62). Certain variants in IRGM are also associated with increased incidence of active pulmonary Mycobacterium tuberculosis disease (40). Because IRGM has been shown to facilitate resistance to Mycobacterium tuberculosis in vitro via phagosome-lysosome fusion (41), it is assumed that the increased incidence in mycobacterial disease associated with IRGM variants is caused by a defect in disease resistance. However, our findings in genital Chlamydia infection along with recent studies demonstrating that pan-Irgm−/− mice display altered cytokine profiles and worsened mortality in Mycobacterium tuberculosis infection (53) highlight the possibility that a failure in human IRGM-mediated disease tolerance also contributes to the observed increase in mycobacterial disease in susceptible individuals.
Irgm1−/− mice display numerous phenotypes that parallel these inflammatory associations of human IRGM, including an increased susceptibility to intestinal inflammation in multiple models, a Sjogren’s syndrome-like autoimmune phenotype underlined by a type I interferonopathy, and a defect in resistance to mycobacterial infection (41–44, 47, 63, 64). Mechanistically, both human IRGM and murine Irgm1 control canonical inflammasome activity via selective autophagy and have been shown to promote mitochondrial homeostasis (23, 25, 46, 65). In Irgm1−/− cells, mitochondrial instability results in increased inflammatory signaling and type I interferon production (24, 47). Irgm1 also regulates lymphocyte and leukocyte homeostasis, although homologous functions have not been investigated in humans (37, 48, 49). Additionally, Irgm2 has been implicated in controlling noncanonical inflammasome activation upon stimulation with LPS or infection with E. coli (51, 52). Irgm3 contributes to the regulation of immune responses via antigen presentation (55, 56). These mechanisms have also not been investigated in human systems.
There is no known role for human IRGM in Chlamydia infection, and because of the vastly divergent mechanisms of cell-autonomous immunity between mice and humans, it is exceedingly unlikely that human IRGM regulates protective immunity to C. trachomatis, as supported by some experimental evidence (66). However, the parallels in immune phenotypes between mouse and human models of IRGM deficiency strongly support functional homology in immunoregulation across species. It is distinctly possible that human IRGM promotes disease tolerance in Chlamydia infection similar to murine Irgm proteins and that similar parallels may exist in infection with other pathogens such as M. tuberculosis. The mechanisms by which inflammation is dysregulated in pan-Irgm−/− mice could therefore elucidate the roles for human IRGM in human disease.
Several specific immune mechanisms have been implicated in mediating pathology in genital C. muridarum infection, including neutrophils and CD8+ T cells as well as the cytokines IL-1 and TNFα (6). Our finding that pan-Irgm−/− mice display increased hydrosalpinx in C. muridarum infection compared with the wild type indicates that Irgm proteins regulate pathogenic immune responses. For example, it is possible that pan-Irgm−/− mice display increased inflammasome activation or increased signaling via another mechanism of bacterial sensing. It is also possible that Irgm proteins regulate lymphocyte or leukocyte homeostasis, resulting in increased neutrophil or CD8+ T cell activation in pan-Irgm−/− mice. However, our finding that Rag1/pan-Irgm−/− mice have worsened rather than improved pathology in C. muridarum infection compared with pan-Irgm−/− mice demonstrates that adaptive immunity as a whole limits inflammation in these mice. However, deletion of Rag1 concurrently interferes with adaptive responses that are pathogenic (e.g., CD8+ T cells) and protective (e.g., IFNγ-secreting Th1 cells or suppressive regulatory T cells) in C. muridarum infection (6). Further experiments targeting more specific branches of adaptive immunity in Irgm-deficient cells will help shed light on these mechanisms.
The persistence of active acute and chronic inflammation in pan-Irgm−/− mice infected with C. muridarum at 45 dpi despite no increase in bacterial burden throughout infection suggests that Irgm proteins are required for the control and resolution of inflammation. The observed increase in granulomatous inflammation in Chlamydia-infected pan-Irgm−/− mice was particularly striking and intriguing, as Crohn’s disease, which is linked with polymorphisms in IRGM, is characterized by intestinal granulomatous inflammation (67). Increased and persistent granulomatous inflammation could imply defects in the clearance of bacterial antigens, dysregulation of macrophage activity or signaling, or a failure to control lymphocyte activity. While all three Irgm paralogs appear to play distinct roles in controlling immunity to genital C. muridarum infection, our observation that Irgm3-deficient mice displayed essentially the same phenotypic features as pan-Irgm−/− mice (including granulomatous inflammation) suggests that future studies could be directed specifically at Irgm3. Our findings address the collective roles of Irgm proteins in regulating inflammation to promote disease tolerance, establishing genital Chlamydia infection in pan-Irgm−/− mice as an effective model to study these phenotypes, and implicating Irgm proteins in specific inflammatory processes that may suggest parallel roles for human IRGM.
MATERIALS AND METHODS
Cell lines and bacterial strains
Primary MEFs were derived from wild-type C57BL/6, Irgm2−/−, Irgm1/m3−/− and pan-Irgm−/− mice as described previously (52). Briefly, the head and vital organs were removed from E12.5–E14.5 embryos, embryos were minced and digested in 0.25% trypsin (Gibco) at 37C. MEFs and African green monkey kidney Vero cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% heat-inactivated fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 µg/mL). Cells to be used for Chlamydia infection were cultured without antibiotics.
Chlamydia trachomatis serovar L2 expressing GFP and the C. muridarum AR Nigg strain (68) were propagated in Vero cells, and EBs were purified as described (32).
Quantitative real-time polymerase chain reaction
DNA was extracted from cells and tissue using the DNeasy Blood and Tissue DNA extraction kit (Qiagen). Tissue was harvested from infected mice and homogenized using a Power Gen 500 tissue homogenizer (Fisher Scientific) prior to DNA extraction. For quantification of DNA, samples were run in duplicate using TaqMan Fast Advanced Master Mix (Thermo Fisher) according to the manufacturer’s protocol. For quantification of host DNA, primers and fluorescent probes included in the TaqMan Rodent GAPDH Control Reagents kit (Thermo Fisher) were used. For quantification of Chlamydia DNA, custom primers and probes designed to amplify and label the Chlamydia 16S gene were used (forward: 5′-GGAGGCTGCAGTCGAGAATCT-3′; reverse: 5′-TTACAACCCTAGAGCCTTCATCACA-3′; probe 5′-6FAM-TCGTCAGACTTCCGTCCATTGCGA-TAM-3′; Fisher Scientific/Eurofins).
Measurement of in vitro Chlamydia burden
For quantification of Chlamydia burden in vitro, primary MEFs were incubated with or without 100 U/mL IFNγ overnight prior to synchronous infection with Chlamydia EBs by centrifugation at 4°C at an MOI of 1:1. Cells were harvested 24 hpi for DNA extraction and quantitative real-time polymerase chain reaction (qPCR). Chlamydia and host DNA mass were quantified using a standard curve generated using purified host and Chlamydia DNA and the same qPCR reagents used throughout each experiment. Bacterial burden in IFNγ-primed cells was divided by the bacterial burden in the corresponding unprimed cells of the same genotype. Graphs represent pooled data from at least three independent experiments conducted using MEFs derived from different embryos.
Mouse husbandry and in vivo genital Chlamydia infection
All mouse lines were maintained at animal facilities at Duke University Medical Center. Animal protocols were approved by the Institutional Animal Care and Use Committees at Duke University. Wild-type C57BL/6 (Jackson Laboratories), Irgm2−/−, pan-Irgm−/− (52), Irgm3−/−, Irgm1/m3−/− (33), and Rag1/pan-Irgm−/− mice were used. All knockout mouse lines are on a C57BL/6 background as described in the relevant citations. For all experiments, 6–12-week-old mice were injected subcutaneously with 2.5 mg medroxyprogesterone acetate (Depo-Provera, Pfizer) diluted in sterile PBS 1 week prior to infection. Chlamydia EBs were diluted in sucrose-phosphate-glutamate (SPG) buffer (10 mM sodium phosphate, 220 mM sucrose, and 0.50 mM L-glutamic acid). For transcervical infection, 5 µL of diluted Chlamydia was instilled directly into the uterus using a Non-Surgical Embryo Transfer device (Braintree Scientific). For in vivo C. trachomatis infection, 5 × 106 IFU was delivered per mouse, and for in vivo C. muridarum infection, 2.5 × 105 IFU was delivered per mouse. For all mock infections, sterile SPG was used. Infected mice were monitored over the course of each infection.
Evaluation of genital pathology
Genital tracts were removed surgically, and pictures of gross pathology were taken using an iPhone XR. Hydrosalpinx was evaluated visually upon removal of the genital tract. Genital tracts were fixed in 10% formalin before routine processing to paraffin, microtomy at 5 µm, and hematoxylin and eosin staining. Semiquantitative grading of the severity of pathological features was performed by a board-certified veterinary anatomic pathologist (JE) as described (69). Acute inflammation, chronic inflammation, edema, dilation, and fibrosis were graded on the following scale from 0 to 4: 0 = normal; 1 = rare foci of inflammation; 2 = scattered (1–4) aggregates of inflammation or mild diffuse inflammation; 3 = numerous aggregates (>4) of inflammation or moderate diffuse inflammation; and 4 = severe diffuse infiltration or confluence of inflammation. In Fig. 7 and Fig. S1, additional grading criteria were used to assess endometrial glandular inflammation, endometrial exudates, and granulomatous lesions. Left and right genital tracts were graded separately. Scoring was performed in a masked fashion without knowledge of allocation group assignment. Graphs represent the sums of acute and chronic inflammation scores in the left and right sides of each genital tract.
Statistical analysis
Prism 9 software (GraphPad) was used to perform statistical analysis. One-way ANOVA with Tukey’s multiple-comparisons test was used to determine the significance of differences among group means. Fisher’s exact test was used to determine the significance of differences in incidence of hydrosalpinx or granulomatous inflammation between genotypes. The Mann-Whitney U test was used for determining statistical significance for nonparametric data. All graphs represent mean plus or minus standard error of the mean of at least three independent experiments.
ACKNOWLEDGMENTS
We would like to thank members of the Coers lab as well as the labs of Drs. Clare Smith, David Tobin, and Edward Miao for providing valuable feedback.
This work was supported by the National Institutes of Health grant AI103197 (to J.C.) and AI148243 (to GAT and JC). J.C. holds an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Jörn Coers, Email: jorn.coers@duke.edu.
Alan Sher, National Institute of Allergy and Infectious Diseases, Bethesda, Maryland, USA.
DATA AVAILABILITY
The numerical values of all quantified data depicted in data panels in this manuscript are openly available in the Digital Repositories at Duke at https://doi.org/10.7924/r4r214s79
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/mbio.00303-24.
Increased inflammation, tissue distortion, and pathology in Irgm- and Rag1-deficient C. muridarum-infected mice.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Brunham RC, Paavonen J. 2020. Reproductive system infections in women: upper genital tract, fetal, neonatal and infant syndromes. Pathog Dis 78:ftaa023. doi: 10.1093/femspd/ftaa023 [DOI] [PubMed] [Google Scholar]
- 2. Taylor HR, Burton MJ, Haddad D, West S, Wright H. 2014. Trachoma. Lancet 384:2142–2152. doi: 10.1016/S0140-6736(13)62182-0 [DOI] [PubMed] [Google Scholar]
- 3. Mabey D, Peeling RW. 2002. Lymphogranuloma venereum. Sex Transm Infect 78:90–92. doi: 10.1136/sti.78.2.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Braxton JDD, Emerson B. 2018. Sexually transmitted surveillance 2017. Centers for disease control and prevention [Google Scholar]
- 5. McCarville JL, Ayres JS. 2018. Disease tolerance: concept and mechanisms. Curr Opin Immunol 50:88–93. doi: 10.1016/j.coi.2017.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dockterman J, Coers J. 2021. Immunopathogenesis of genital Chlamydia infection: insights from mouse models. Pathog Dis 79:ftab012. doi: 10.1093/femspd/ftab012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perry LL, Feilzer K, Caldwell HD. 1997. Immunity to Chlamydia trachomatis is mediated by T helper 1 cells through IFN-gamma-dependent and -independent pathways. J Immunol 158:3344–3352. doi: 10.4049/jimmunol.158.7.3344 [DOI] [PubMed] [Google Scholar]
- 8. Nelson DE, Virok DP, Wood H, Roshick C, Johnson RM, Whitmire WM, Crane DD, Steele-Mortimer O, Kari L, McClarty G, Caldwell HD. 2005. Chlamydial IFN-γ immune evasion is linked to host infection tropism. Proc Natl Acad Sci U S A 102:10658–10663. doi: 10.1073/pnas.0504198102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Coers J, Bernstein-Hanley I, Grotsky D, Parvanova I, Howard JC, Taylor GA, Dietrich WF, Starnbach MN. 2008. Chlamydia muridarum evades growth restriction by the IFN-γ-inducible host resistance factor Irgb10. J Immunol 180:6237–6245. doi: 10.4049/jimmunol.180.9.6237 [DOI] [PubMed] [Google Scholar]
- 10. Finethy R, Coers J. 2016. Sensing the enemy, containing the threat: cell-autonomous immunity to Chlamydia trachomatis. FEMS Microbiol Rev 40:875–893. doi: 10.1093/femsre/fuw027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shah AA, Schripsema JH, Imtiaz MT, Sigar IM, Kasimos J, Matos PG, Inouye S, Ramsey KH. 2005. Histopathologic changes related to fibrotic oviduct occlusion after genital tract infection of mice with Chlamydia muridarum. Sex Transm Dis 32:49–56. doi: 10.1097/01.olq.0000148299.14513.11 [DOI] [PubMed] [Google Scholar]
- 12. Sun X, Yang Z, Zhang H, Dai J, Chen J, Tang L, Rippentrop S, Xue M, Zhong G, Wu G. 2015. Chlamydia muridarum induction of glandular duct dilation in mice. Infect Immun 83:2327–2337. doi: 10.1128/IAI.00154-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Darville T, Hiltke TJ. 2010. Pathogenesis of genital tract disease due to Chlamydia trachomatis. J Infect Dis 201 Suppl 2:S114–25. doi: 10.1086/652397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Agrawal T, Vats V, Wallace PK, Singh A, Salhan S, Mittal A. 2009. Recruitment of myeloid and plasmacytoid dendritic cells in cervical mucosa during Chlamydia trachomatis infection. Clin Microbiol Infect 15:50–59. doi: 10.1111/j.1469-0691.2008.02113.x [DOI] [PubMed] [Google Scholar]
- 15. Peipert JF, Boardman L, Hogan JW, Sung J, Mayer KH. 1996. Laboratory evaluation of acute upper genital tract infection. Obstet Gynecol 87:730–736. doi: 10.1016/0029-7844(96)00040-3 [DOI] [PubMed] [Google Scholar]
- 16. Wiesenfeld HC, Heine RP, Krohn MA, Hillier SL, Amortegui AA, Nicolazzo M, Sweet RL. 2002. Association between elevated neutrophil defensin levels and endometritis. J Infect Dis 186:792–797. doi: 10.1086/342417 [DOI] [PubMed] [Google Scholar]
- 17. Ohman H, Tiitinen A, Halttunen M, Lehtinen M, Paavonen J, Surcel HM. 2009. Cytokine polymorphisms and severity of tubal damage in women with Chlamydia-associated infertility. J Infect Dis 199:1353–1359. doi: 10.1086/597620 [DOI] [PubMed] [Google Scholar]
- 18. Kimani J, Maclean IW, Bwayo JJ, MacDonald K, Oyugi J, Maitha GM, Peeling RW, Cheang M, Nagelkerke NJ, Plummer FA, Brunham RC. 1996. Risk factors for Chlamydia trachomatis pelvic inflammatory disease among sex workers in Nairobi, Kenya. J Infect Dis 173:1437–1444. doi: 10.1093/infdis/173.6.1437 [DOI] [PubMed] [Google Scholar]
- 19. McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, Zody MC, Hall JL, Brant SR, Cho JH, Duerr RH, Silverberg MS, Taylor KD, Rioux JD, Altshuler D, Daly MJ, Xavier RJ. 2008. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat Genet 40:1107–1112. doi: 10.1038/ng.215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kimura T, Watanabe E, Sakamoto T, Takasu O, Ikeda T, Ikeda K, Kotani J, Kitamura N, Sadahiro T, Tateishi Y, Shinozaki K, Oda S. 2014. Autophagy-related IRGM polymorphism is associated with mortality of patients with severe sepsis. PLoS One 9:e91522. doi: 10.1371/journal.pone.0091522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yao QM, Zhu YF, Wang W, Song ZY, Shao XQ, Li L, Song RH, An XF, Qin Q, Li Q, Zhang JA. 2018. Polymorphisms in autophagy-related gene IRGM are associated with susceptibility to autoimmune thyroid diseases. Biomed Res Int 2018:7959707. doi: 10.1155/2018/7959707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, Mari B, Barbry P, Mosnier J-F, Hébuterne X, Harel-Bellan A, Mograbi B, Darfeuille-Michaud A, Hofman P. 2011. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn's disease. Nat Genet 43:242–245. doi: 10.1038/ng.762 [DOI] [PubMed] [Google Scholar]
- 23. Mehto S, Jena KK, Nath P, Chauhan S, Kolapalli SP, Das SK, Sahoo PK, Jain A, Taylor GA, Chauhan S. 2019. The Crohn’s disease risk factor IRGM limits NLRP3 inflammasome activation by impeding its assembly and by mediating its selective autophagy. Mol Cell 73:429–445. doi: 10.1016/j.molcel.2018.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jena KK, Mehto S, Nath P, Chauhan NR, Sahu R, Dhar K, Das SK, Kolapalli SP, Murmu KC, Jain A, Krishna S, Sahoo BS, Chattopadhyay S, Rusten TE, Prasad P, Chauhan S, Chauhan S. 2020. Autoimmunity gene IRGM suppresses cGAS-STING and RIG-I-MAVS signaling to control interferon response. EMBO Rep 21:e50051. doi: 10.15252/embr.202050051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Singh SB, Ornatowski W, Vergne I, Naylor J, Delgado M, Roberts E, Ponpuak M, Master S, Pilli M, White E, Komatsu M, Deretic V. 2010. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol 12:1154–1165. doi: 10.1038/ncb2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pilla-Moffett D, Barber MF, Taylor GA, Coers J. 2016. Interferon-inducible GTPases in host resistance, inflammation and disease. J Mol Biol 428:3495–3513. doi: 10.1016/j.jmb.2016.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martens S, Howard J. 2006. The interferon-inducible GTPases. Annu Rev Cell Dev Biol 22:559–589. doi: 10.1146/annurev.cellbio.22.010305.104619 [DOI] [PubMed] [Google Scholar]
- 28. Ling YM, Shaw MH, Ayala C, Coppens I, Taylor GA, Ferguson DJP, Yap GS. 2006. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J Exp Med 203:2063–2071. doi: 10.1084/jem.20061318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Martens S, Parvanova I, Zerrahn J, Griffiths G, Schell G, Reichmann G, Howard JC. 2005. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog 1:e24. doi: 10.1371/journal.ppat.0010024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hunn JP, Koenen-Waisman S, Papic N, Schroeder N, Pawlowski N, Lange R, Kaiser F, Zerrahn J, Martens S, Howard JC. 2008. Regulatory interactions between IRG resistance GTPases in the cellular response to Toxoplasma gondii. EMBO J 27:2495–2509. doi: 10.1038/emboj.2008.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dockterman J, Coers J. 2022. How did we get here? Insights into mechanisms of immunity-related GTPase targeting to intracellular pathogens. Curr Opin Microbiol 69:102189. doi: 10.1016/j.mib.2022.102189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Haldar AK, Saka HA, Piro AS, Dunn JD, Henry SC, Taylor GA, Frickel EM, Valdivia RH, Coers J. 2013. IRG and GBP host resistance factors target aberrant, “non-self” vacuoles characterized by the missing of “self” IRGM proteins. PLoS Pathog 9:e1003414. doi: 10.1371/journal.ppat.1003414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Henry SC, Daniell XG, Burroughs AR, Indaram M, Howell DN, Coers J, Starnbach MN, Hunn JP, Howard JC, Feng CG, Sher A, Taylor GA. 2009. Balance of Irgm protein activities determines IFN-γ-induced host defense. J Leukoc Biol 85:877–885. doi: 10.1189/jlb.1008599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taylor GA. 2007. IRG proteins: key mediators of interferon-regulated host resistance to intracellular pathogens. Cell Microbiol 9:1099–1107. doi: 10.1111/j.1462-5822.2007.00916.x [DOI] [PubMed] [Google Scholar]
- 35. Collazo CM, Yap GS, Sempowski GD, Lusby KC, Tessarollo L, Vande Woude GF, Sher A, Taylor GA. 2001. Inactivation of Lrg-47 and Irg-47 reveals a family of interferon γ-inducible genes with essential, pathogen-specific roles in resistance to infection. J Exp Med 194:181–188. doi: 10.1084/jem.194.2.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Springer HM, Schramm M, Taylor GA, Howard JC. 2013. Irgm1 (LRG-47), a regulator of cell-autonomous immunity, does not localize to mycobacterial or listerial phagosomes in IFN-γ-induced mouse cells. J Immunol 191:1765–1774. doi: 10.4049/jimmunol.1300641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Coers J, Gondek DC, Olive AJ, Rohlfing A, Taylor GA, Starnbach MN. 2011. Compensatory T cell responses in IRG-deficient mice prevent sustained Chlamydia trachomatis infections. PLoS Pathog 7:e1001346. doi: 10.1371/journal.ppat.1001346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pradipta A, Sasai M, Motani K, Ma JS, Lee Y, Kosako H, Yamamoto M. 2021. Cell-autonomous Toxoplasma killing program requires Irgm2 but not its microbe vacuolar localization. Life Sci Alliance 4:e202000960. doi: 10.26508/lsa.202000960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dockterman J, Fee BE, Taylor GA, Coers J. 2021. Murine Irgm paralogs regulate nonredundant functions to execute host defense to Toxoplasma gondii. Infect Immun 89:e0020221. doi: 10.1128/IAI.00202-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Intemann CD, Thye T, Niemann S, Browne ENL, Amanua Chinbuah M, Enimil A, Gyapong J, Osei I, Owusu-Dabo E, Helm S, Rüsch-Gerdes S, Horstmann RD, Meyer CG. 2009. Autophagy gene variant IRGM -261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog 5:e1000577. doi: 10.1371/journal.ppat.1000577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Singh SB, Davis AS, Taylor GA, Deretic V. 2006. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313:1438–1441. doi: 10.1126/science.1129577 [DOI] [PubMed] [Google Scholar]
- 42. Liu B, Gulati AS, Cantillana V, Henry SC, Schmidt EA, Daniell X, Grossniklaus E, Schoenborn AA, Sartor RB, Taylor GA. 2013. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am J Physiol Gastrointest Liver Physiol 305:G573–G584. doi: 10.1152/ajpgi.00071.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Azzam KM, Madenspacher JH, Cain DW, Lai L, Gowdy KM, Rai P, Janardhan K, Clayton N, Cunningham W, Jensen H, Patel PS, Kearney JF, Taylor GA, Fessler MB. 2017. Irgm1 coordinately regulates autoimmunity and host defense at select mucosal surfaces. JCI Insight 2:e91914. doi: 10.1172/jci.insight.91914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taylor GA, Huang HI, Fee BE, Youssef N, Jewell ML, Cantillana V, Schoenborn AA, Rogala AR, Buckley AF, Feng CG, Vallance BA, Gulati AS, Hammer GE. 2020. Irgm1-deficiency leads to myeloid dysfunction in colon lamina propria and susceptibility to the intestinal pathogen Citrobacter rodentium. PLoS Pathog 16:e1008553. doi: 10.1371/journal.ppat.1008553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bafica A, Feng CG, Santiago HC, Aliberti J, Cheever A, Thomas KE, Taylor GA, Vogel SN, Sher A. 2007. The IFN-inducible GTPase LRG47 (Irgm1) negatively regulates TLR4-triggered proinflammatory cytokine production and prevents endotoxemia. J Immunol 179:5514–5522. doi: 10.4049/jimmunol.179.8.5514 [DOI] [PubMed] [Google Scholar]
- 46. Schmidt EA, Fee BE, Henry SC, Nichols AG, Shinohara ML, Rathmell JC, MacIver NJ, Coers J, Ilkayeva OR, Koves TR, Taylor GA. 2017. Metabolic alterations contribute to enhanced inflammatory cytokine production in Irgm1-deficient macrophages. J Biol Chem 292:4651–4662. doi: 10.1074/jbc.M116.770735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rai P, Janardhan KS, Meacham J, Madenspacher JH, Lin WC, Karmaus PWF, Martinez J, Li QZ, Yan M, Zeng J, Grinstaff MW, Shirihai OS, Taylor GA, Fessler MB. 2021. IRGM1 links mitochondrial quality control to autoimmunity. Nat Immunol 22:312–321. doi: 10.1038/s41590-020-00859-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Feng CG, Collazo-Custodio CM, Eckhaus M, Hieny S, Belkaid Y, Elkins K, Jankovic D, Taylor GA, Sher A. 2004. Mice deficient in LRG-47 display increased susceptibility to mycobacterial infection associated with the induction of lymphopenia. J Immunol 172:1163–1168. doi: 10.4049/jimmunol.172.2.1163 [DOI] [PubMed] [Google Scholar]
- 49. Feng Carl G, Zheng L, Jankovic D, Báfica A, Cannons JL, Watford WT, Chaussabel D, Hieny S, Caspar P, Schwartzberg PL, Lenardo MJ, Sher A. 2008. The immunity-related GTPase Irgm1 promotes the expansion of activated CD4+ T cell populations by preventing interferon-γ-induced cell death. Nat Immunol 9:1279–1287. doi: 10.1038/ni.1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rai P, Sharpe M, Ganta CK, Baker PJ, Mayer-Barber KD, Fee BE, Taylor GA, Fessler MB. 2023. IRGM1 supports host defense against intracellular bacteria through suppression of type I interferon in mice. J Clin Invest 133:e171982. doi: 10.1172/JCI171982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Eren E, Planès R, Bagayoko S, Bordignon P, Chaoui K, Hessel A, Santoni K, Pinilla M, Lagrange B, Burlet‐Schiltz O, Howard JC, Henry T, Yamamoto M, Meunier E. 2020. Irgm2 and Gate-16 cooperatively dampen Gram-negative bacteria-induced caspase-11 response. EMBO Rep 21:e50829. doi: 10.15252/embr.202050829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Finethy R, Dockterman J, Kutsch M, Orench-Rivera N, Wallace GD, Piro AS, Luoma S, Haldar AK, Hwang S, Martinez J, Kuehn MJ, Taylor GA, Coers J. 2020. Dynamin-related Irgm proteins modulate LPS-induced caspase-11 activation and septic shock. EMBO Rep 21:e50830. doi: 10.15252/embr.202050830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wilburn KM, Meade RK, Heckenberg EM, Dockterman J, Coers J, Sassetti CM, Olive AJ, Smith CM. 2023. Differential requirement for IRGM proteins during tuberculosis infection in mice. Infect Immun 91:e0051022. doi: 10.1128/iai.00510-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lijek RS, Helble JD, Olive AJ, Seiger KW, Starnbach MN. 2018. Pathology after Chlamydia trachomatis infection is driven by nonprotective immune cells that are distinct from protective populations. Proc Natl Acad Sci U S A 115:2216–2221. doi: 10.1073/pnas.1711356115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bougnères L, Helft J, Tiwari S, Vargas P, Chang BH-J, Chan L, Campisi L, Lauvau G, Hugues S, Kumar P, Kamphorst AO, Dumenil A-ML, Nussenzweig M, MacMicking JD, Amigorena S, Guermonprez P. 2009. A role for lipid bodies in the cross-presentation of phagocytosed antigens by MHC class I in dendritic cells. Immunity 31:232–244. doi: 10.1016/j.immuni.2009.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Guo J, McQuillan JA, Yau B, Tullo GS, Long CA, Bertolino P, Roediger B, Weninger W, Taylor GA, Hunt NH, Ball HJ, Mitchell AJ. 2015. IRGM3 contributes to immunopathology and is required for differentiation of antigen-specific effector CD8+ T cells in experimental cerebral malaria. Infect Immun 83:1406–1417. doi: 10.1128/IAI.02701-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sturdevant GL, Caldwell HD. 2014. Innate immunity is sufficient for the clearance of Chlamydia trachomatis from the female mouse genital tract. Pathog Dis 72:70–73. doi: 10.1111/2049-632X.12164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mosser DM, Zhang X. 2008. Activation of murine macrophages. Curr Protoc Immunol 83:14 doi: 10.1002/0471142735.im1402s83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rajaram K, Nelson DE. 2015. Chlamydia muridarum infection of macrophages elicits bactericidal nitric oxide production via reactive oxygen species and cathepsin B. Infect Immun 83:3164–3175. doi: 10.1128/IAI.00382-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nagarajan UM, Tripathy M, Kollipara A, Allen J 4th, Goodwin A, Whittimore J, Wyrick PB, Rank RG. 2018. Differential signaling pathways are initiated in macrophages during infection depending on the intracellular fate of Chlamydia spp. Immunol Cell Biol 96:246–256. doi: 10.1111/imcb.1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Naglak EK, Morrison SG, Morrison RP. 2017. Neutrophils are central to antibody-mediated protection against genital Chlamydia. Infect Immun 85:e00409-17. doi: 10.1128/IAI.00409-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ajayi TA, Innes CL, Grimm SA, Rai P, Finethy R, Coers J, Wang X, Bell DA, McGrath JA, Schurman SH, Fessler MB. 2019. Crohn's disease IRGM risk alleles are associated with altered gene expression in human tissues. Am J Physiol Gastrointest Liver Physiol 316:G95–G105. doi: 10.1152/ajpgi.00196.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tiwari S, Choi HP, Matsuzawa T, Pypaert M, MacMicking JD. 2009. Targeting of the GTPase Irgm1 to the phagosomal membrane via PtdIns(3,4)P2 and PtdIns(3,4,5)P3 promotes immunity to mycobacteria. Nat Immunol 10:907–917. doi: 10.1038/ni.1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. MacMicking JD, Taylor GA, McKinney JD. 2003. Immune control of tuberculosis by IFN-γ-inducible LRG-47. Science 302:654–659. doi: 10.1126/science.1088063 [DOI] [PubMed] [Google Scholar]
- 65. Henry SC, Schmidt EA, Fessler MB, Taylor GA. 2014. Palmitoylation of the immunity related GTPase, Irgm1: impact on membrane localization and ability to promote mitochondrial fission. PLoS One 9:e95021. doi: 10.1371/journal.pone.0095021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Haldar AK, Piro AS, Finethy R, Espenschied ST, Brown HE, Giebel AM, Frickel EM, Nelson DE, Coers J. 2016. Chlamydia trachomatis is resistant to inclusion ubiquitination and associated host defense in gamma interferon-primed human epithelial cells. mBio 7:e01417-16. doi: 10.1128/mBio.01417-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chang JT. 2020. Pathophysiology of inflammatory bowel diseases. N Engl J Med 383:2652–2664. doi: 10.1056/NEJMra2002697 [DOI] [PubMed] [Google Scholar]
- 68. Wang Y, Kahane S, Cutcliffe LT, Skilton RJ, Lambden PR, Clarke IN. 2011. Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog 7:e1002258. doi: 10.1371/journal.ppat.1002258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Darville T, Andrews CW, Laffoon KK, Shymasani W, Kishen LR, Rank RG. 1997. Mouse strain-dependent variation in the course and outcome of chlamydial genital tract infection is associated with differences in host response. Infect Immun 65:3065–3073. doi: 10.1128/iai.65.8.3065-3073.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Increased inflammation, tissue distortion, and pathology in Irgm- and Rag1-deficient C. muridarum-infected mice.
Data Availability Statement
The numerical values of all quantified data depicted in data panels in this manuscript are openly available in the Digital Repositories at Duke at https://doi.org/10.7924/r4r214s79