

SUMMARY
Dopamine receptors, including D1- and D2-like receptors, are important therapeutic targets in a variety of neurological syndromes, as well as cardiovascular and kidney diseases. Here, we present five cryoelectron microscopy (cryo-EM) structures of the dopamine D1 receptor (DRD1) coupled to Gs heterotrimer in complex with three catechol-based agonists, a non-catechol agonist, and a positive allosteric modulator for endogenous dopamine. These structures revealed that a polar interaction network is essential for catecholamine-like agonist recognition, whereas specific motifs in the extended binding pocket were responsible for discriminating D1- from D2-like receptors. Moreover, allosteric binding at a distinct inner surface pocket improved the activity of DRD1 by stabilizing endogenous dopamine interaction at the orthosteric site. DRD1-Gs interface revealed key features that serve as determinants for G protein coupling. Together, our study provides a structural understanding of the ligand recognition, allosteric regulation, and G protein coupling mechanisms of DRD1.
Graphical Abstract
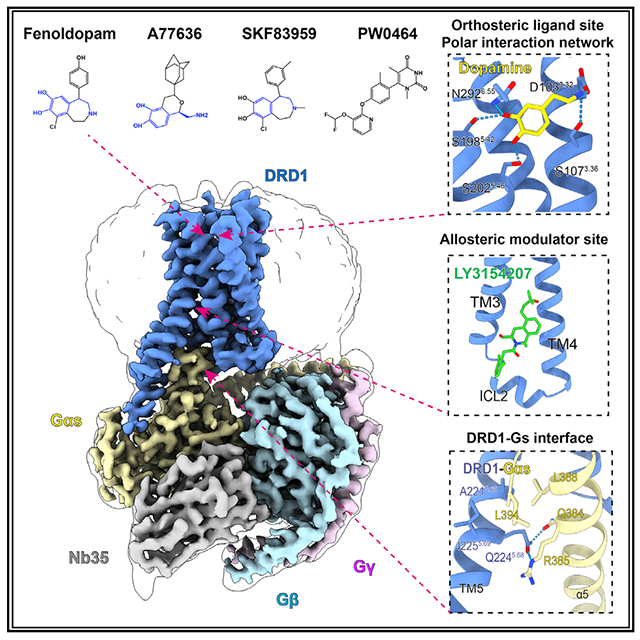
In Brief
The cryo-EM structures of DRD1 coupled to its G protein, in complex with important agonists, elucidates the mechanism of G-protein-coupled selectivity and will facilitate future drug discovery and design.
INTRODUCTION
Dopamine, also called a “feel good” hormone, enables animals not only to sense rewards but also to take actions to move toward them (Schultz, 2016). In addition to reward, dopamine also modulates multiple physiological processes, such as addiction, memory, metabolism, and hormone secretion, through two subfamilies of dopamine G-protein-coupled receptors (GPCRs) (Beaulieu and Gainetdinov, 2011; Kebabian and Calne, 1979; Missale et al., 1998). By activating Gs/Golf and stimulating cyclic AMP (cAMP) production, the D1-like receptors (DRD1 and DRD5) are essential regulators of reward, motor activity, and cognition in the central nervous system (CNS) and play beneficial roles in peripheral tissues including inhibiting inflammatory reactions and maintaining cardiovascular and kidney homeostasis (Beaulieu and Gainetdinov, 2011; Yan et al., 2015). By contrast, the D2-like receptors (DRD2, DRD3, and DRD4) couple to Gi/Go subunits, inhibiting cAMP accumulation and regulating a different set of functions (Missale et al., 1998; Spano et al., 1978). While these five dopamine receptors share high sequence homology and sense the same endogenous ligand, significant progress has been made in drug development to differentially affect these five dopamine receptor subtypes. Nonetheless, the mechanisms underlying agonist recognition, receptor activation, ligand selectivity, and G protein coupling selectivity between the dopamine receptor subfamilies remain cloudy.
To date, several structures of antagonists bound to D2-like receptors have revealed the mechanism of antagonist recognition (Chien et al., 2010; Wang et al., 2017, 2018a), offering an opportunity for understanding variable antagonist binding pockets involved in D2-like dopamine system signal inhibition. Recently, the cryoelectron microscopy (cyro-EM) structure of a selective agonist-bound DRD2-Gi complex in lipid membrane has been determined (Yin et al., 2020); however, the structural basis for ligand recognition by the D1-like subfamily is still lacking. There is an urgent need for highly selective agonists of DRD1 as antihypertensive drugs as well as for the treatment of Parkinson disease or kidney injury (Gurrell et al., 2018; Nichols et al., 1990; Noce et al., 2019). Structures of D1-like receptors, such as DRD1, will provide an avenue for finding selective agonists of these subfamily receptors. In this study, we determined near-atomic-resolution cryo-EM structures of activated DRD1 with a downstream Gs effector, either in complex with three catechol-containing agonists—the antihypertensive drug fenoldopam and the synthetic full agonists A77636 and SKF83959—or a non-catechol Gs-protein-biased agonist PW0464 (Wang et al., 2019) and a positive allosteric modulator (PAM) for endogenous agonist dopamine. Our structures reveal a conserved motif for dopaminergic receptor recognition with catecholamine agonists, a specific extended binding pocket (EBP) for ligand recognition, allosteric regulation, and the Gs coupling mechanisms of DRD1.
RESULTS
Cryo-EM structure of the DRD1-Gs complex
To obtain active structures of DRD1 in complex with Gs heterotrimer and catechol-containing agonists, either the antihypertensive drug fenoldopam, the full agonist A77636, or the full agonist SKF83959 was first used for complex assembly, because of their high binding affinities with the receptor (Figures S1A–S1C). The complex was formed on the membrane of insect cells in the presence of Nb35, which stabilizes the nucleotide-free complex by bridging the Gαs and Gβγ subunits (Rasmussen et al., 2011). The structures of DRD1-Gs-Nb35 in complex with fenoldopam, A77636, or SKF83959 were determined by single-particle cryo-EM with overall resolutions of 3.2, 3.5, and 3.3 Å, respectively (Figures 1A and 1B and S1D–S1F; Table S1).
Figure 1. Overall cryo-EM structures of the DRD1-Gs heterotrimer complexes.
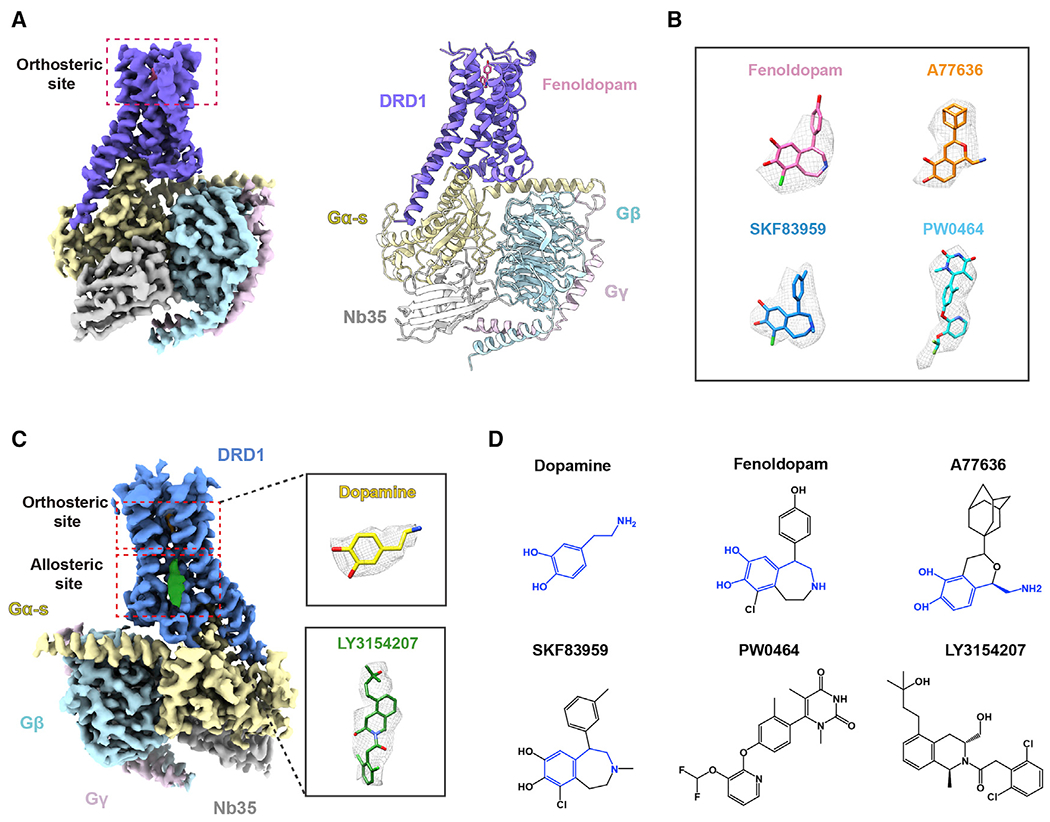
(A) The cryo-EM map (left) of the DRD1-Gs-Nb35 in complex with fenoldopam and the cartoon representations (right) of the complex structure are shown. Slate, DRD1; pink, fenoldopam; yellow, Gαs; cyan, Gβ; light pink, Gβ; and gray, Nb35. The cryo-EM density map colored according to different subunits is shown at 0.022 contour level.
(B) The cryo-EM density of four ligands presented as gray meshes allowed unambiguous identification of fenoldopam (pink), A77636 (orange), SKF83959 (blue), and PW0464 (cyan). The density maps of the agonists are depicted at contour level of 0.022, except for that of SKF83959 at 0.9.
(C) Cryo-EM map of the DRD1-Gs-Nb35 in complex with dopamine and positive allosteric modulator (PAM) LY3154207. The cryo-EM map contoured at the 0.022 level is colored according to different subunits. The densities of dopamine (yellow stick) and LY3154207 (green stick) are depicted as gray meshes shown in the inserted magnified panels.
(D) Two-dimensional representation of chemical structures of dopamine, fenoldopam, synthetic agonist A77636, SKF83959, non-catechol agonist PW0464, and PAM LY3154207. The common catecholamine pharmacophore is highlighted in blue.
The same method was then applied in obtaining the structures of DRD1-Gs-Nb35 complex bound with non-catechol agonist PW0464 at 3.1 Å resolution as well as concurrently bound with the endogenous dopamine and a PAM LY3154207 at 3.1 Å resolution (Figures 1B and 1C; Table S1). The high-quality cryo-EM maps allowed us to model the 7TM elements of the DRD1 receptor, the Gs heterotrimer, and Nb35; side chains of most residues are well defined in all components (Figure S1G). The above-mentioned ligands were unambiguously identified in the orthosteric pocket or the allosteric site of DRD1 (Figures 1D and S2A–S2F). The binding modes of the agonist in the cryo-EM density at the orthosteric site were further supported by molecular docking, which afforded a nearly identical pose at the top (Figure S2G).
Overall structures of active DRD1-Gs complexes with fenoldopam, A77636, and PW0464 were similar to dopamine-bound DRD1-Gs complex structures, with low root-mean-square deviation (RMSD) values of Cα ranging from 0.4 to 0.5, from 0.3 to 0.5, and from 0.4 to 0.6 Å for the receptor alone, the G proteins, and whole complex, respectively. However, the SKF83959-bound DRD1-Gs complexes exhibited larger differences compared with the other four structures, with RMSD values ranging from 2.1 to 2.3 Å for the whole complex.
Polar interaction motifs within the OBP
All DRD1 complex structures determined here exhibited a nearly identical orthosteric binding pocket (OBP), which is defined by TM3, TM5, TM6, and TM7 as well as the ECL2 region. Among the five agonists used in this study, dopamine is the endogenous agonist of DRD1 and belongs to the class of typical catecholamine transmitters. Anchored to the bottom of OBP in DRD1, dopamine engaged salt bridges with D1033.32 via its typical amine group and hydrogen bonds with S1985.42 and S2025.46 via its catechol hydroxyl groups, and it had hydrophobic contacts with I1043.33, F2886.51, and F2896.52 via its catechol ring (Figures 2A and S2H). The other four selective DRD1 agonists, including fenoldopam, A77636, SKF83959, and PW0464 (Conroy et al., 2015; Wang et al., 2019), formed extensive contacts with the hydrophobic OBP and were covered with a lid formed by ECL2 (Figure S2H). Importantly, the three agonists fenoldopam, SKF83959, and A77636 all contain a typical catecholamine group, which resembles that of the native agonist dopamine. In particular, catechol-based ligands bind to DRD1 via hydrogen bonds between the catechol hydroxyl groups and S1985.42, S1995.43, and S2025.46, as well as salt bridges between amine moiety and D1033.32 in TM3, exhibiting a catecholamine binding mode similar to that of the endogenous ligand dopamine (Figures 2B–2D). The non-catechol agonist PW0464 also forms polar network interactions with S1985.42, S2025.46, and D1033.32 in OBP (Figure 2E).
Figure 2. Polar network in the orthosteric binding pocket (OBP) of DRD1.
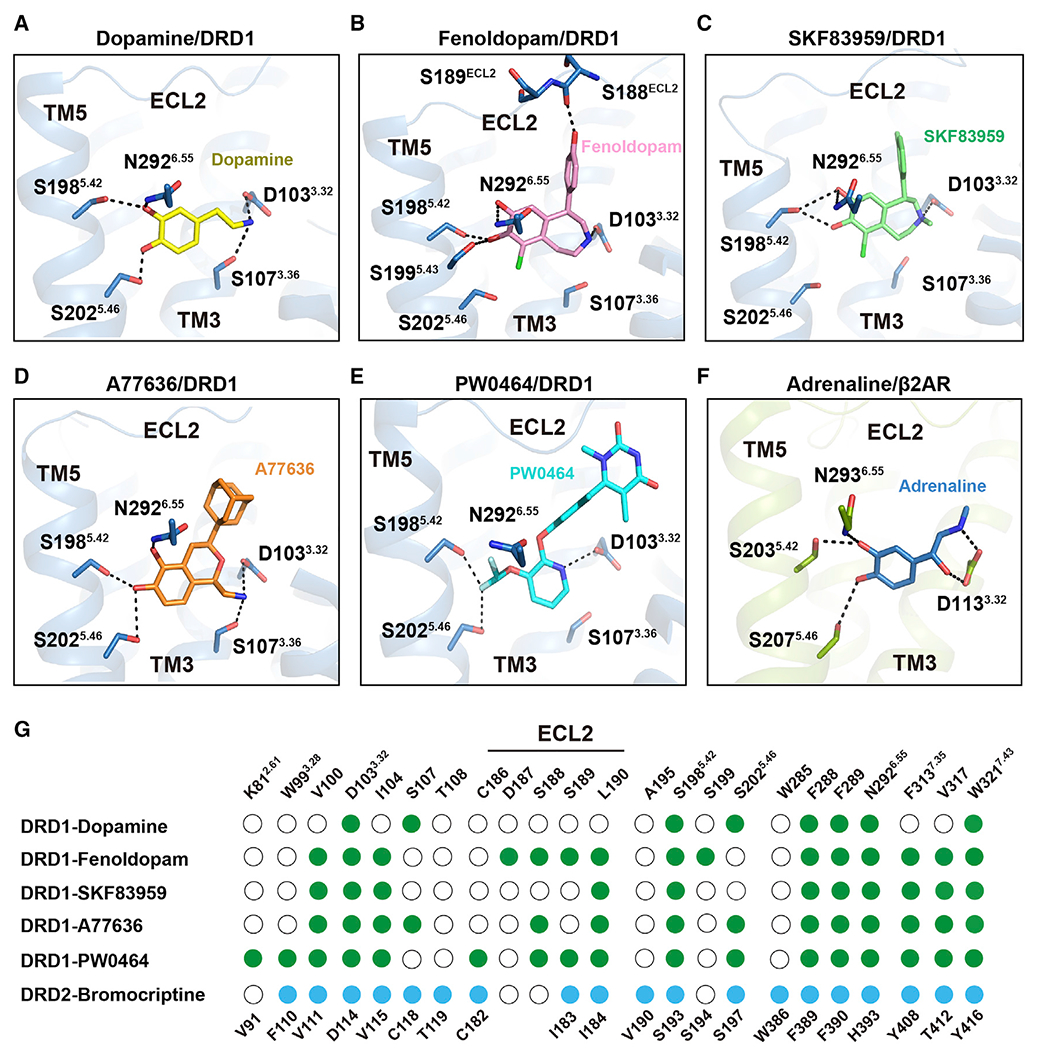
(A) Detailed interactions of dopamine (yellow) with DRD1 (sky blue); the hydroxyl groups of dopamine form potential polar interactions with the side chains of S1985.42, S2025.46, and N2926.55, and the amine group of dopamine forms salt bridges and hydrogen bond with D1033.32 and S1073.36 in DRD1, respectively. Polar interactions are highlighted as black dashed lines.
(B–E) Detailed interactions of fenoldopam (pink), SKF83959 (lime), A77636 (orange), and PW0464 (cyan) with DRD1 (sky blue). The polar interactions are indicated by black dashed lines.
(F) Detailed interactions of adrenaline (sky blue) with β2AR (limon) (PDB: 4LDO). The polar interactions are shown as black dashed lines.
(G) Comparison of agonist binding sites between DRD1 and DRD2. Residues in agonist-bound DRD1 or DRD2 that interact with ligands are indicated with green or blue dots, respectively. Residues that show no interaction with ligands are shown as white circles. Residue positions for DRD1 or DRD2 are indicated on the top or bottom of the scheme, respectively. Ballesteros-Weinstein numbers of D3.32-S5.42-S5.46, K2.61-W3.28-W7.43, or N6.55-F7.35 motif residues are shown as superscripts.
See also Figures S2 and S3 and Tables S2, S3 and S5.
Notably, the catechol-based agonists binding mode in DRD1 resembles the polar interaction network of the β2-adrenergic receptor (β2AR) with its endogenous agonist epinephrine (adrenaline) (Ring et al., 2013) Figure 2F). The polar interactions between the epinephrine and the S5.42 and S5.46 in β2AR have been proposed as key micro-switches essential for adrenergic receptor activation (Dror et al., 2011). Dopamine, nor-epinephrine, and epinephrine all share the same catecholamine pharmacophore (Masureel et al., 2018). Importantly, all dopamine receptors share S5.42 and S5.46 in TM5, which potentially engage with the catechol ring, as well as D3.32 in TM3 that interacts with the amine moiety (Masureel et al., 2018; Ring et al., 2013). Therefore, D3.32-S5.42-S5.46 is a key motif responsible for the recognition of agonists harboring catecholamine groups in dopamine receptors (Figure 2G). Consistently, alanine replacements of the D3.32-S5.42-S5.46 motif significantly impaired both ligand binding and cAMP accumulation in response to catechol-like agonists or native dopamine (Figures S3A and S3B; Tables S2–S5). These results are in agreement with those of previous pharmacological assays showing the importance of the motif for dopamine binding and functional activity (Fowler et al., 2012; Mansour et al., 1992). Therefore, the conserved polar interactions involving the D3.32-S5.42-S5.46 motif constitute one of the major agonist binding characteristics for all dopamine receptors. Interestingly, one of the dihydroxyl groups of the two agonists fenoldopam and SKF83959 was replaced by chloride in the antagonist SCH23390, which potentially lost the important hydrogen bonds engaged with S1985.42-S2025.46 in DRD1 (Figures S3C and S3D). The presence of dihydroxyl group in fenoldopam and SKF83959, but not in SCH23390, is putatively linked to their different activities.
Besides the D3.32-S5.42-S5.46 motif, subtle differences were observed in the polar network of OBP in these five EM structures. For example, S1985.42 was the only residue that forms polar interactions with the catechol hydroxyl groups of fenoldopam or SKF83959, whereas S1995.43 is not implicated in A77636 binding. Further functional assays and molecular dynamic simulation experiments suggest that the residue S1995.43 may participate in catecholamine-based agonist binding or stabilize the rotamer conformation of N2926.55 (Figures S3E–S3G).
Importantly, whereas all D1-like receptors and β-adrenergic receptors, with Gs coupling activity, contain an N6.55 in TM6 to contact the catechol meta-hydroxyl group, the D2-like receptors have an H6.55, and the Gi-coupled α2 adrenergic receptors bear a Y6.55 at the same position. On the opposite side of the catecholamine, the amine group is engaged in a hydrogen bond with S1073.36 in the dopamine-bound DRD1 structure (in addition to contacting D1033.32), which is conserved in D1-like receptors but replaced by C3.36 in D2-like receptors (Figures S3H and S3I). While sensing the same endogenous agonist dopamine, the distinct polar network motif of S3.36-N6.55 in D1-like receptors and C3.36-H6.55 in D2-like receptors differentiates these two types of receptors, and formation of these networks may trigger different allosteric paths leading to specific conformations on the cytoplasmic side to achieve selective G protein subtype engagement.
Non-catechol agonist binding mode of DRD1
Because of their poor penetration in CNS and rapid degradation metabolism, catechol-like agonists of DRD1 face great difficulty in treatment of CNS disorders (Zhang et al., 2009). A non-catechol-based agonist PW0464 was then developed to target to DRD1 with high affinity and selectivity, displaying a comparable cAMP signaling activity without detectable β-arrestin recruitment compared with A77636 (Figure S4A) (Wang et al., 2019). In our structure, PW0464 exhibited a different binding mode to catechol-based agonists, even though the ligand PW0464 was present at the same hydrophobic orthosteric site of DRD1 and the pocket residues of the receptor exhibited small RMSD of Cα compared with that of the DRD1-A77636 or DRD1-fenoldopam complexes (Figures S4B–S4D).
The non-catechol-based agonist PW0464 loses the typical dihydroxyphenyl catechol present in dopamine and catechol-containing ligands. However, PW0464 still forms potential hydrogen bonds with S1985.42 and S2025.46 in DRD1 via its fluorine atom (Sun et al., 2003; Zhou et al., 2009). Moreover, the residue D1033.32 formed both polar and charge-charge interactions with the nitrogen in the pyridine group (Figures 2E and 3A and 3B). A similar interaction has been proposed to stabilize the catecholamine-based ligand in the OBP of β2AR (Dror et al., 2011). In agreement with these observations, alanine mutagenesis of the D3.32-S5.42-S5.46 motif significantly impaired both ligand binding and cAMP activities of DRD1 in response to PW0464 (Figure 3C; Table S4).
Figure 3. Non-catechol agonist PW0464 binding mode of DRD1.
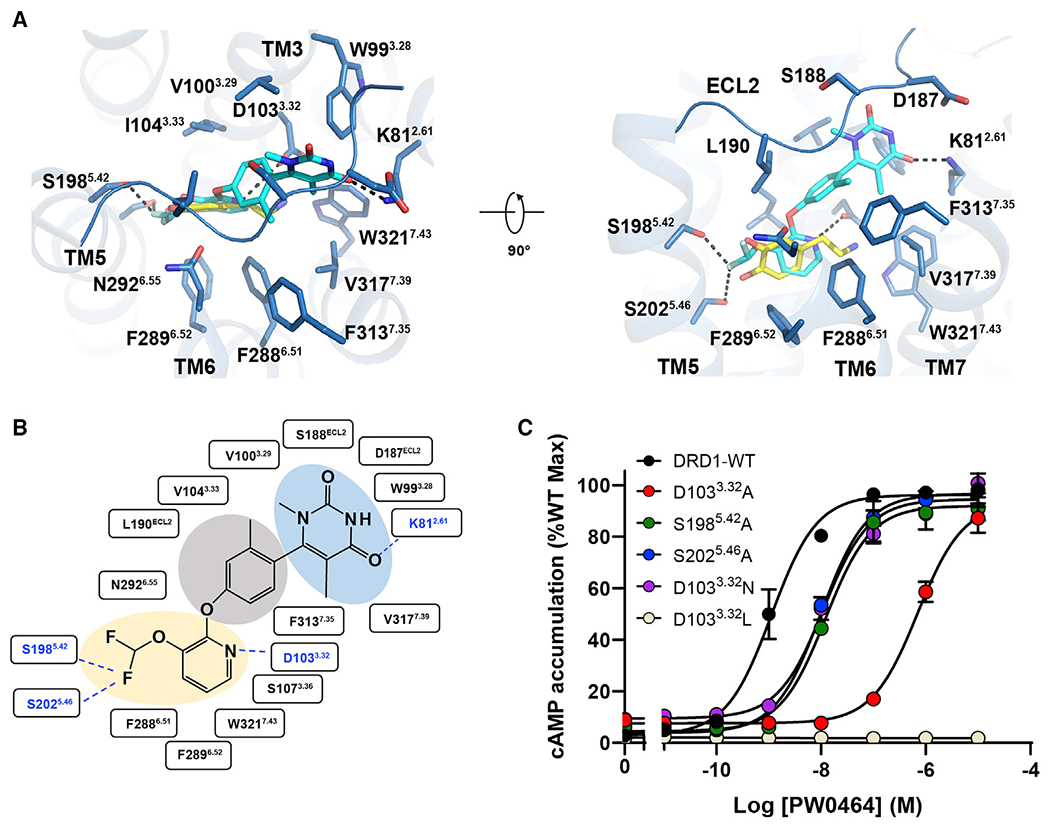
(A) Detailed interactions between PW0464 (cyan) and DRD1 (marine) shown in extracellular view (left panel) or side view (right panel). PW0464 occupies the same hydrophobic OBP as dopamine but exhibits a different binding mode. Polar interactions are highlighted with black dotted lines. The ligand PW0464 forms potential polar interactions with D3.32-S5.42-S5.46 motif via the pyridine nitrogen and difluoromethoxy group and retains the putative hydrogen bonding with N2926.55 and an additional direct interaction with K812.61.
(B) Diagram of ligand interactions between DRD1 and PW0464. The difluoromethoxy, pyridine (orange shade), phenoxy (gray shade), and pyrimidinedione groups (sky blue shade) are highlighted. Polar interactions are shown by the blue dashed lines.
(C) Effects of the D1033.32-S1985.42-S2025.46 mutants of DRD1 on PW0464-induced cAMP accumulation.
Notably, distinct from catechol-based agonists, PW0464 connected the pyridine head with a phenoxy group, which formed substantial hydrophobic interactions with I1043.33, L190ECL2, F2886.51, and F3137.35. The phenolic oxygen contacted with N2926.55 in DRD1. This observation is consistent with previous reports that the replacement of phenoxy with an aminopyridine or indoline ring nearly eliminated the activity of the compound (Gray et al., 2018; Hall et al., 2019). Pointing into the TM2 and ECL2 regions, the pyrimidinedione group of PW0464 had extensive contacts with the main chains of D187-S189 in ECL2, and side chains of W993.28 and V3177.39, and formed a hydrogen bond with K812.61 (Figure 3A).
The EBP of DRD1
All dopamine receptors contain an EBP, which helps to confer high ligand affinity and can allow for a high degree of receptor selectivity (Wang et al., 2017, 2018a). The well-resolved cryo-EM density maps in our structures permitted us to unambiguously assign the hydroxyphenyl group of fenoldopam, adamantane group of A77636, methylphenyl group of SKF83959, and pyrimidinedione group of PW0464 to a hydrophobic pocket created by the extracellular ends of TM2, TM3, TM6, and TM7 and covered by ECL2 (Figures S2A–S2F). This specific EBP of the DRD1 receptor is defined by F2886.51, N2926.55, and F3137.35 on one side, covered by S188 and L190 from ECL2, and surrounded by K812.61, W90ECL1, W993.28, and W3217.43 on the other side (Figure 4A). In the DRD1 structures bound with different agonists, the most conformational differences in the OBP of DRD1 were concentrated at the ECL2 region (Figures S5A and S5B), suggesting that the plasticity of the ECL2 contributes to the recognition of diverse ligands by DRD1. Importantly, several key motifs constituting the EBP commonly encircle these agonists, including the N2926.55-F3137.35 and the K812.61-W993.28-W3217.43 motifs (Figure 2G). These observations indicate that, despite assuming different conformations, certain conserved residues could be generally utilized by certain high-affinity agonist binding of the DRD1.
Figure 4. Comparison of extended binding pockets (EBPs) between DRD1 and D2-like receptors.
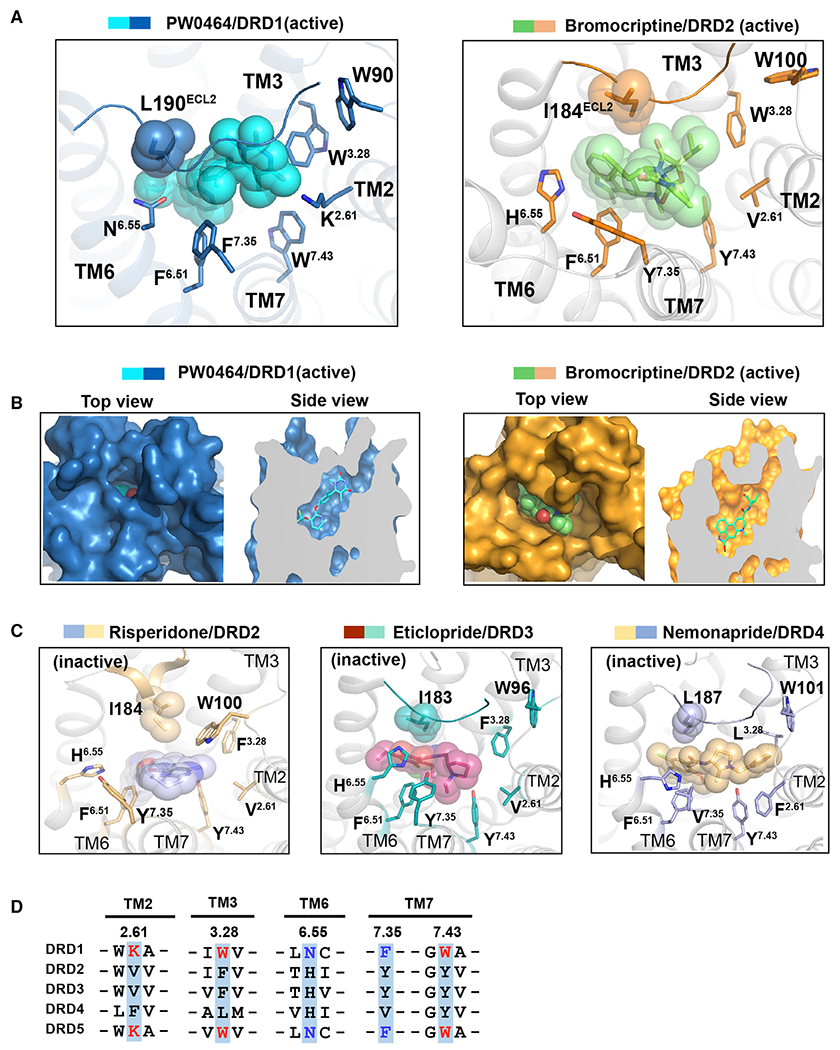
(A) Structural comparison of the EBP in PW0464 (cyan)-bound DRD1 (sky blue) structure with that in bromocriptine (pale green)-bound DRD2 (orange) (PDB: 6VMS). The ligands PW0464 and bromocriptine are represented as sticks/spheres by corresponding colors, and the side chains of key residues of EBP are shown in sticks for both receptors.
(B) Surface representation and cut away view of the ligand binding pocket for PW0464-bound DRD1 structure (left panel) and bromocriptine-bound DRD2 structure (right panel). The ligand PW0464 was covered by the ECL2 region of DRD1.
(C) Structural representation of the EBPs in antagonist-bound D2-like receptors. Risperidone (blue sphere)-bound DRD2 (light orange) (left, PDB: 6CM4), eticlopride (warm pink sphere)-bound DRD3 (teal) (middle, PDB: 3PBL), and nemonapride (light orange sphere)-bound DRD4 (light blue) (right, PDB: 5WIU).
(D) Sequence alignment of two critical motifs of EBP in five dopaminergic receptors located in TM2, TM3, TM6, and TM7. The K2.61-W3.28-W7.43 motifs of D1-like receptors are highlighted in red, whereas the N6.55-F7.35 pairs are highlighted in blue.
Compared with the recently solved DRD2-Gi structure, DRD1 displays substantial differences in ECL1, ECL2, TM6, and TM7, resulting in a distinct EBP. Notably, although assuming different conformations, the structural equivalent residues of the N2926.55-F3137.35 and the K812.61-W993.28-W3217.43 motifs were replaced by the V2.61-F3.28-Y7.43 and H6.55-Y7.35 pair in the bromocriptine-bound DRD2 structure, which also contacts the bound agonist bromocriptine (Figures 4A and S5C). The distinct shapes and sizes of the EBP of these two dopamine receptors are probably due to the different chemical properties of these positional and sequence equivalent residues (Figures 4B and S5D). Notably, the motifs of V/F2.61-F/L3.28-Y7.43 and H6.55-Y/V7.35 are shared in Gi-coupled D2-like receptors and have been experimentally demonstrated to contribute to ligand binding in previous studies (Figures 4C and S5E). For instance, the residue pair V2.61-F3.28 in inactive DRD3 was replaced by F2.61-L3.28 in inactive DRD4 (Figures 4C and 4D), resulting in a different EBP and enabling the discovery of a selective ligand for DRD4 (Wang et al., 2017).
Consistent with these observations and structural comparisons, the importance of these two motifs in DRD1 was confirmed in our mutagenesis studies. In particular, the K2.61 A mutation displayed approximately 50-fold and 70-fold decrease in PW0464 binding affinity and PW0464-induced signaling response, respectively, compared with the DRD1 wild type (Figures S5F and S5G). Similarly, replacing the N6.55-F7.35 pair in DRD1 with equivalent residues in DRD2 significantly reduced both the ligand binding and potency of agonist-induced cAMP accumulation compared with wild-type DRD1 (Figure S5H). Together, these distinct EBP features, especially the conserved K2.61-W3.28-W7.43 and N6.55-F7.35 motifs in DRD1, serve as important structural features for agonist recognition.
PAM binding site of DRD1
Unlike the conserved nature of orthosteric sites among the correlated GPCR subtypes, allosteric modulators are able to finely tune the activity of receptors with a high level of subtype selectivity, while still allowing for binding of endogenous ligands (Thal et al., 2018). LY3154207 is a DRD1 PAM with significant pharmacological advantages, wherein it markedly potentiates the cAMP activity of DRD1 in response to endogenous agonist dopamine (Hao et al., 2019).
The structure of DRD1-Gs in complex with both endogenous dopamine and PAM revealed that an EM density corresponding to LY3154207 localized at the membrane-embedded binding site created by intracellular loop 2 (ICL2), TM3, and TM4 (Figure 5A). The PAM binding pocket was found to be mainly composed of hydrophobic residues (Figure 5B). Because the EM density of the allosteric site was not detailed enough to unambiguously assign exact atom positions of the bound compound, the allosteric agonist LY3154207 could be modeled in two ways, in opposite directions. In the first model, the tertiary alcohol group of the PAM was projected onto TM3 and formed a hydrogen bond with C1153.44. The tetrahydroisoquinoline (THIQ) ring was involved in extensive hydrophobic interactions with V1193.48, W1233.52, and L1434.45. In addition, the dichlorophenyl group formed a π-cation interaction with the side chain of R130ICL2. This binding mode of LY3154207 was similar to previous computational modeling and supported by site mutagenesis experiments in both previous and present studies (Figure 5D) (Hao et al., 2019). In the other model, the aromatic group of the PAM was placed closer to the seven transmembrane (7TM) core of the receptor, which formed hydrophobic contacts with V1163.45. The hydrogen bond between PAM and C1153.44 in the first model was lost because the hydroxyl group was placed toward the outside region (Figure S6A). In our functional assays, the C1153.44 A mutant decreased the potency of PAM by 4.5-fold for dopamine-induced DRD1 activation. By contrast, the V1163.45 mutants showed no significant effects (Hao et al., 2019; Wang et al., 2018b). Therefore, our biochemical results and previous computational modeling preferred the first model that positioned the tertiary alcohol group of LY3154207 close to the receptor.
Figure 5. Binding of the PAM LY3154207 to DRD1.
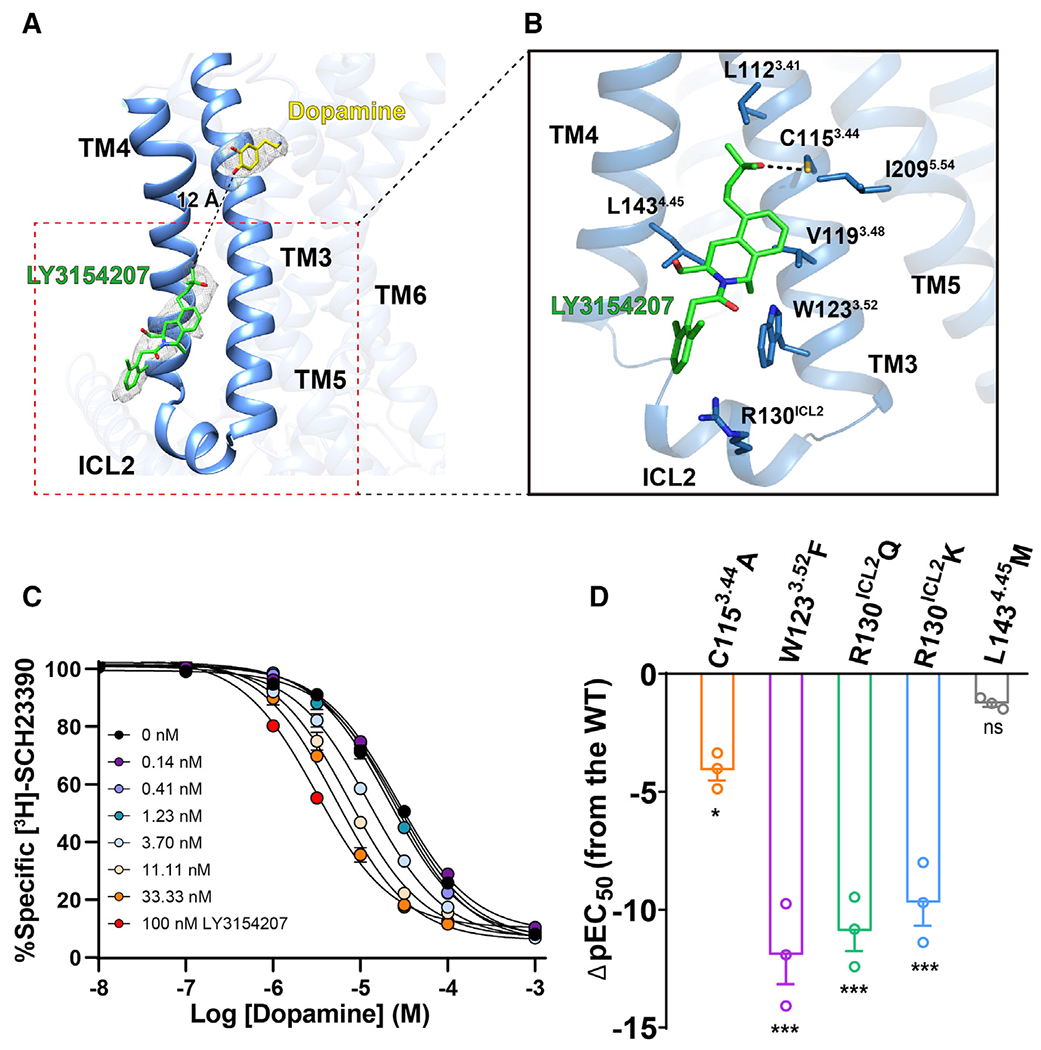
(A) Overall structure of DRD1 bound to dopamine (yellow stick) and LY3154207 (green stick). The structure reveals that an EM density (gray mesh, contoured at 0.022) corresponding to LY3154207 located at the membrane-embedded binding site was created by ICL2, TM3, and TM4, and the allosteric site in DRD1 is 12 Å away from the orthosteric site.
(B) Contact residues in dopamine-LY3154207-DRD1-Gs complex (sticks with sky-blue carbons) within 4 Å of the LY3154207 (green). Polar interaction is highlighted as a black dotted line.
(C) Competition binding curve of dopamine to wild-type (WT) DRD1 in the presence of different concentrations of LY3154207 (0.14–100 nM). LY3154207 enhanced the binding affinity of dopamine with DRD1. The Ki value (±SEM) of DRD1 for dopamine is 25.86 ± 1.9 μM, whereas the Ki value decreases to 3.16 ± 0.3 μM in the presence of 100 nM LY3154207. Data are presented as the mean ± SEM of three or more experiments run in triplicate.
(D) Dopamine-induced cAMP accumulation assay in the presence of 100 nM LY3154207. Bars represent differences in calculated potency of dopamine (ρEC50 [half maximal effective concentration]) for each mutation relative to WT of DRD1. *p < 0.01, ***p < 0.0001 (one-way analysis of variance [ANOVA] followed by the Dunnett’s test, compared with the response of WT).
See also Figure S6.
To understand the allosteric regulation mechanism of DRD1 by LY3154207, we performed assays with ligand binding at orthosteric site, cAMP activity, and mutagenesis substitutions (Figures S6B–S6H). Binding occurs between the allosteric modulator LY3154207 and the orthosteric endogenous agonist dopamine 12 Å away, and they are mainly connected by TM3 and TM6 residues (Figure 5A). Our radioligand binding assay indicated that the LY3154207 significantly enhanced the dopamine binding affinity with DRD1 by approximately 10-fold (Figures 5C and S6B), suggesting that allosteric modulation of the LY3154207 improved DRD1 activity by stabilizing the interaction of dopamine within the orthosteric site. Furthermore, once we compared the structures of PAM-bound DRD1 with other agonists-bound DRD1, the superposition reveals rotamer changes in side chains of W1233.52 and R130ICL2 (Figures S6I–S6K). Consistent with our observations, both mutations of W1233.52F and R130ICL2Q significantly diminished PAM activity on DRD1, due to loss of the potential interactions between receptor and PAM in our proposed model (Figures 5D, S6E, S6F, and S6H). It should be noted that an inverse substitution of equivalent Q130ICL2R in mouse DRD1 restored the allosteric activity of the LY3154207 to the same level compared with human DRD1 (Hao et al., 2019).
Taken together, allosteric binding of PAM LY3154207 stabilized the active conformation of TM3 end or ICL2 by direct contacts, thus positively increasing the dopamine-induced DRD1 activity through both increasing the dopamine binding affinity at the orthosteric site and stabilizing the ICL2 conformation for effective Gs coupling. Importantly, several allosteric modulators of DRD1 including LY3154207 have been progressed to phase 2 clinical studies. Our study thus may provide critical clues for development of effective DRD1 allosteric modulators to treat diseases.
Structural comparison of 7TM bundle of active DRD1 with that of DRD2
Superposition with the structure of bromocriptine-bound DRD2-Gi signaling complex with the five DRD1-Gs complex structures revealed an RMSD of Cα value ranging from 1.1 to 1.3 Å for receptor alone. The overall structure of the 7TM bundle of DRD1 in DRD1-Gs complexes was very similar to that of DRD2 in the bromocriptine-DRD2-Gi complex (Figure 6A), except for a larger separation between the cytoplasmic ends of TM3 and TM6 of DRD1 than that between those of DRD2.
Figure 6. Structural features of active DRD1 compared with that of active DRD2 receptor.
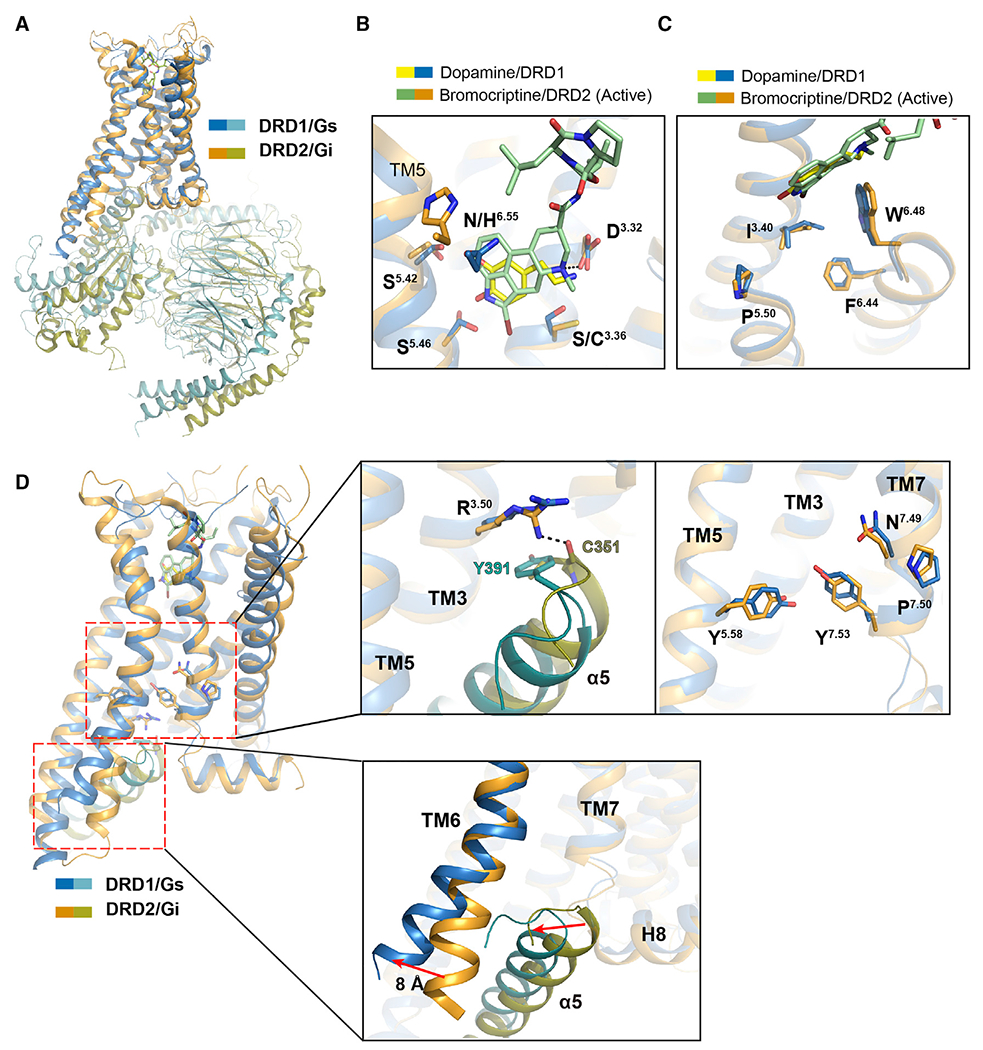
(A) Structural superposition of DRD1-Gs complex with DRD2-Gi complex when DRD1 and DRD2 were aligned. Sky blue, DRD1; teal, Gαs; orange, DRD2; and deep olive, Gi.
(B) Comparison of the OBP of dopamine (yellow) and PAM-bound DRD1 (sky blue) with that of bromocriptine (pale green)-bound DRD2 (orange). The amine group of bromocriptine is observed to form a polar interaction with D3.32 in DRD2, and the key residues are shown as sticks with corresponding colors.
(C) Structural comparison of the toggle switch W6.48 and P5.50-I3.40-F6.44 motif between DRD1 (sky blue) and DRD2 (orange).
(D) Relative orientation of TM5, TM6, and TM7 in DRD1 (sky blue) with Gαs (teal) coupling compared with that in DRD2 (orange) with Gαi (deep olive) coupling. Notable differences are the conformations of the D-R3.50-Y motif, N-P7.50-xx-Y7.53 motif and Y5.58 residue in DRD1 and DRD2. The orientations of TM6 and C-terminal α5 in Gαs or Gαi are highlighted according to receptor alignment.
In addition to similar motifs present in the OBPs of both DRD1 and DRD2 (Figure 6B), both of these two receptors sense agonists binding via the conserved toggle switch W6.48. Below the W6.48, a similar agonist-induced packing of the connector motif P5.50-I3.40-F6.44 was noticed (Figure 6C). At the cytoplasmic region, the residue R3.50 present in DRD1 and DRD2 displayed similar conformations in both receptors, forming hydrophobic interactions with Y391 of Gαs and hydrogen bonds with the main chain of C351 of Gαi in DRD1-Gs and DRD2-Gi structures, respectively. However, in spite of these common and important structural features for active GPCRs, there were significant differences found in the intracellular sides of the two active dopamine receptors. For example, the rotamers of the micro-switch N-P7.50-xx-Y7.53 in DRD1 showed rearrangements compared with those in DRD2. In particular, the side chain of Y7.53 was projected toward the receptor core and Y5.58 was inserted into the 7TM bundle, accompanied by a slight approach of the cytoplasmic ends of TM5 and TM7 of DRD1 compared with that of DRD2 (Figure 6D). The most striking difference between DRD1 and DRD2 was the further 8 Å displacement of TM6 outward in active DRD1 compared with those in active DRD2, resulting in a larger cytoplasmic cavity in DRD1 to accommodate the C-terminal α5 of Gαs.
The DRD1-Gs interface and G protein selectivity
The global architecture of the DRD1-Gs complex is similar to that of two previously reported class A GPCR-Gs complexes (Figure 7A) (García-Nafría et al., 2018; Rasmussen et al., 2011). The main interface between the receptor and Gs heterotrimer is composed of the insertion of the C-terminal α5 helix of Gαs into the 7TM bundle; the interaction between the ICL2 and the cytoplasmic end of TM5 of the receptor with the α4, α5 helix and β6 of Gαs; and multiple contacts between the ICL1 of the receptor and the Gβ subunit. Compared with the β2AR-Gs and A2AR-Gs interfaces, additional interactions were observed, such as the contacts between Q390 of the Gαs protein and T592.39 of DRD1 (Figures S7A–S7C). In addition, there were interactions between the extended TM5 of the DRD1 with α4-β6 of the Gαs protein, accompanied by a slight vertical orientation shift (approximately 1.4 Å) at the end of the α5 helix of Gαs (Figure 7A).
Figure 7. Comparison of receptor-G protein binding interface of DRD1-Gs complex with two previously determined class A GPCRs-Gs complexes (β2AR and A2AR) or DRD2-Gi complex.
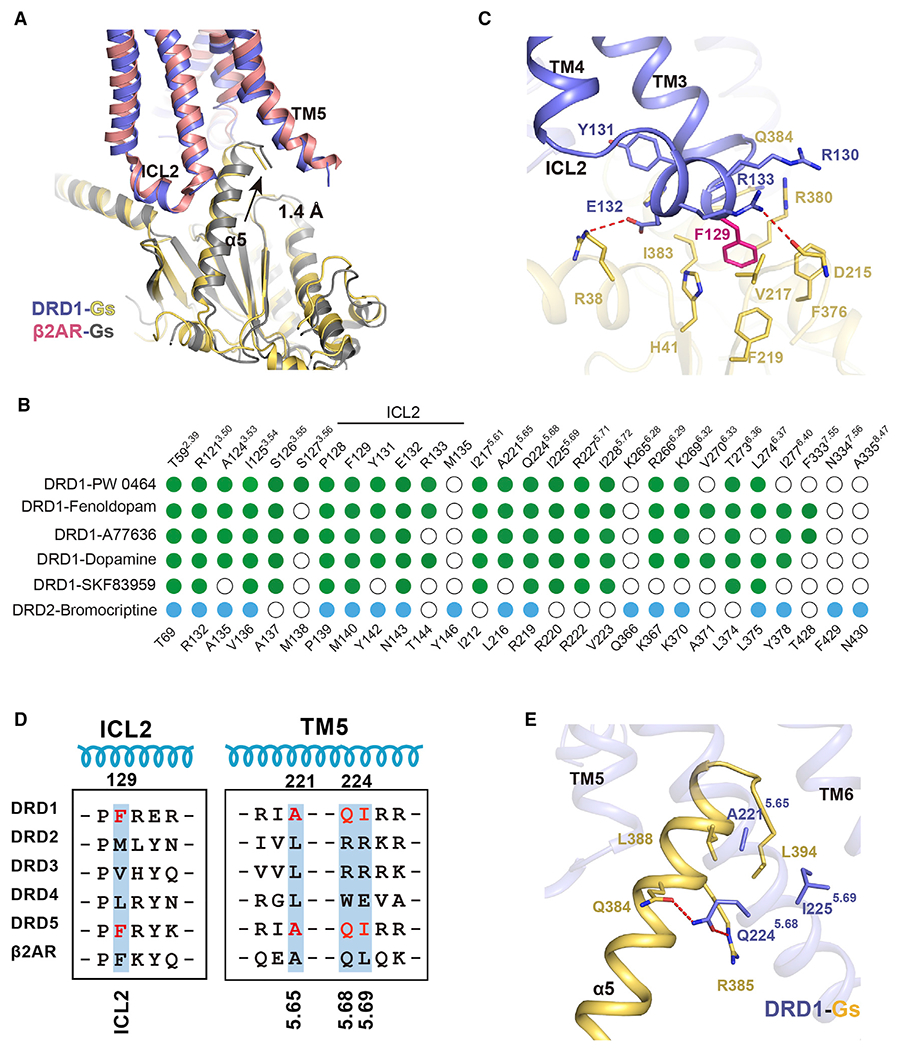
(A) Comparison of the interface of DRD1-Gs complex with that of β2AR-Gs complex when the TM3 helixes of DRD1 and DRD2 were aligned.
(B) Comparison of residues between agonist-bound DRD1 or DRD2 that contact their corresponding downstream G protein subtypes, the Gαs or Gαi, respectively. Residues in DRD1 that contact with Gαs or DRD2 that interact with Gαi are depicted as green or blue dots, respectively. Residues that show no interaction with G proteins are indicated as white circles. Residue positions with Ballesteros-Weinstein numbers shown as superscripts for DRD1 or DRD2 are indicated on the top or bottom of the scheme, respectively.
(C) Detailed interactions between ICL2 of DRD1 with Gαs. F129 (magenta stick) of DRD1 inserts into a hydrophobic pocket composed by Gαs. Additional polar interactions (highlighted by red dashed lines) strengthen the interaction between DRD1 and Gαs.
(D) Sequence alignment of ICL2 and TM5 of five dopaminergic receptors and the β2AR. The residues of F129, A221, Q224, and I225 in DRD1 and the equivalent residues in DRD5 are highlighted in red.
(E) Detailed interaction between α5 helix in Gαs (yellow) and DRD1 (blue). Polar interaction is highlighted in red dotted lines. The key A5.65xxQ5.68-I5.69 motif of DRD1 involved in G protein coupling is represented by the blue stick.
See also Figure S7.
Despite a high degree of sequence similarity, the two dopamine receptor subfamilies have distinct functions via distinct G protein subtype-dependent pathways; D1-like receptors couple to Gs, whereas D2-like receptors connect to Gi. Compared with the recently solved DRD2-Gi complex structure, the DRD1-Gs complex shared nine identical interactions but differed with 16 specific contacts (Figure 7B). Structural inspection of the interface between Gs and DRD1 revealed specific features that could contribute to G protein subtype coupling selectivity for these two subfamilies of receptors. First, the distinct contacts at ICL2 differed by four different contacts. In particular, the F129ICL2 of DRD1 sits in a hydrophobic pocket created by H41G.S1.02, V217G.S3.01, F219G.S3.03, F376G.H5.08, R380G.H5.12, and I383G.H5.15 from the β1 and β3 strands and the α5 helix of Gs (superscripts refer to the common Gα numbering system) (Flock et al., 2015). This interaction is further strengthened by additional contacts formed by P128ICL2, R130ICL2, E132ICL2, and R133ICL2 of DRD1 (Figure 7C). By contrast, the key DRD1-F129ICL2 was replaced by the structural equivalent residue M140ICL2 in DRD2, which only formed weak hydrophobic contacts with the structural equivalent L194G.S3.01 and I343G.H5.15 of Gi; but lost all other tight contacts (Figure S7D). Notably, the D1-like receptors, including DRD1 and DRD5, all have an FICL2 at this position, whereas D2-like receptors have MICL2, LICL2, or VICL2 (Figure 7D). In particular, previous studies have suggested that differences in the ICL2 region play a role in Gs versus Gi coupling selectivity in CB1 and CB2 receptors (Krishna Kumar et al., 2019); a bulky residue, such as phenylalanine (F139 in β2AR), is engaged in hydrophobic interactions with Gs and drives selective G protein coupling. Consistently, the F129LICL2 mutation in DRD1 displayed a notable 11-fold decrease in potency of cAMP response compared with that in the wild-type DRD1, and a smaller alanine substitution at F129ICL2 further impaired the ability to activate Gs, whereas the bulky phenylalanine substitution at the equivalent position of V/LICL2 in D2-like receptors significantly reduced Gi signaling (Figure S7E).
Furthermore, there is a conserved A5.65xxQ5.68-I5.69 motif in DRD1 and DRD5, which may serve as a key determinant for Gs coupling and is replaced with its structural equivalent L5.65xxR/W5.68-R/E5.69 in D2-like receptors (Figures 7D). In the DRD1-Gs complex structure, Q2245.68 forms hydrogen bonds with Q384G.H5.16 and R385G.H5.17 of Gαs (Figure 7E). By contrast, the corresponding residues of Gαi are I344G.H5.16 and K345G.H5.17, thus losing potential polar interactions with DRD1 and DRD5. The R2195.68 of DRD2 formed specific contacts with the I344G.HS.16 and its precedent acidic residue D341G.H5.13 of Gαi (Figures S7F and S7G). In addition, we found that small residues at the cytoplasmic end of TM5 in D1-like receptors, such as A2215.65 and I2255.69, allowed the accommodation of the bulkier five C-terminal residues in Gαs compared with those in Gαi. The residues L5.65 and R5.69/E5.69 in D2-like receptors (Figure S7F) were similarly oriented toward the five C-terminal residues but created a thinner crevice. Consistent with these observations, our functional assays showed that the replacement of any residue of the A5.65xxQ5.68-I5.69 motif by any residue of the L5.65xxR5.68-R5.69 motif in the DRD2 family caused a decrease in cAMP accumulation downstream of D1-like receptors in response to a panel of agonists. Similarly, substitutions of the residues of the L5.65xxR5.68 in D2-like receptors (DRD2, DRD3, and DRD4) by the equivalent residues in DRD1 also caused a reduction in Gi signaling (Figures S7H and S7I). Collectively, these results suggest that the combination of the conserved motif A5.65xxQ5.68-I5.69 in TM5 and FICL2 in DRD1 serve as key determinants of Gs coupling. The differences between the A5.65xxQ5.68-I5. 69 motif in DRD1 versus the L5.65xxR5.68 in D2-like receptors and the different hydrophobic residues in ICL2 might contribute to their G protein coupling selectivity.
Conclusions
In this study, DRD1-Gs complex structures provided a structural understanding of the agonist recognition, positive allosteric regulation, and coupling mechanism of DRD1 with Gs. In this issue of Cell, a companion study reported structures of DRD1 and DRD2 signaling complexes (Zhuang et al., 2021) Structural inspection of DRD1, compared with previously solved adrenergic receptor structures with endogenous ligands, and sequence alignment indicated that the D3.32-S5.42-S5.46 motif plays critical roles in the recognition of catecholamine agonists across dopaminergic and adrenergic receptor families. Moreover, the polar network motif of S3.36-N6.55 in the catecholamine binding pocket and the K2.61-W3.28-E7.43 motif in the EBP of DRD1, which are replaced by C3.36-H6.55 and F2.61-F/L3.28-Y7.43 in D2-like receptors, respectively, highlights the distinct ligand binding pocket properties between dopamine receptors, which could be exploited in the future development of selective ligands that uniquely modulate dopamine system function. In addition, a positive allosteric site of DRD1 was identified on the inner surface of the receptor created by TM3, ICL2, and TM4, which enabled selective and potent modulation of DRD1. Ligand binding and mutagenesis substitution experiments provided key insights into this allosteric modulation mechanism which may guide further clinical usage. Finally, we revealed that the conserved A5.65xxQ5.68-I5.69 motif and FICL2 are key determinants of the Gs coupling activity of DRD1.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead contact, Professor Zhenhua Shao (zhenhuashao@scu.edu.cn).
Materials availability
The plasmids generated from this study may be obtained directly from the lead contact.
Data and code availability
All data produced or analyzed in this study are included in the main text or the supplementary materials. The cryo-EM density maps and atomic coordinates have been deposited in the Electron Microscopy Data Bank (EMDB) and Protein Data Bank (PDB) under accession numbers EMD-30392 and 7CKW for Fenoldopam-DRD1 complex; EMD-30393 and 7CKX for A77636-DRD1 complex; EMD-30452 and 7CRH for A77636-DRD1 complex; EMD-30394 and 7CKY for PW0464-DRD1 complex; and EMD-30395 and 7CKZ for dopamine/LY3154207-DRD1 complex.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Spodoptera frugiperda (Sf9) cells were purchased from Expression system and were cultured in ESF921 medium (Expression system) at 27°C with 140 rpm. HEK293T cells were obtained from American Type Culture Collection (CRL-11268) and were grown in DEME medium (GIBCO, #11995) supplemented with 10% fetal bovine serum (FBS) (CELL-BOX, #SAG-01U-02), 1% penicillin/streptomycin (GIBCO, #15140122) at 37°C with 5% CO2.
METHOD DETAILS
Constructs
The human wild-type DRD1 gene was cloned into pFastBac1 with an N-terminal FLAG tag (DYKDDDDA). To facilitate expression and purification, the DRD1 signal peptide was substituted with that of hemagglutinin (HA). Human DNGαs, human Gβ1 with an N-terminal 6 × Histidine tag and human Gγ2 were also sub-cloned into pFastBac1 vector. The same constructs were used for the expression and functional assays in this study.
Expression of DRD1, Gαs/Gβγ heterotrimer
Recombinant baculovirus was generated using the Bac-to-Bac Baculovirus Expression System (Invitrogen). Briefly, FuGENE HD transfection reagent (Promega) was used to prepare baculovirus. Sf9 suspension cells were seeded in ESF921 medium at a density of 3 × 106 cells/ml and infected with DRD1 baculovirus. After 48h incubation at 27°C, shaking at 110 rpm, cells were collected by centrifugation, flash-frozen in liquid nitrogen and stored at −80°C. Gs heterotrimer was co-expressed via infection with virus of Gs and Gβ1γ2. Infected cells were also cultured at 27°C, 110 rpm for 48 h. The harvested cells were collected by centrifugation and the cell pellets were stored at −80°C.
DRD1-Gs complex formation and purification
Sf9 cell pellets infected with DRD1 and Gs heterotrimer were resuspended in lysis buffer (20 mM HEPES, pH 7.4, 100 mM NaCl, 3 mM MgCl2, 5 mM CaCl2, 2.5 mg/ml leupeptin and 0.2 mg/ml benzamidine). The DRD1-Gs complex was prepared in membranes, and the mixture was incubated for 2 h at room temperature by adding either 10 μM fenoldopam, A77636 and SKF83959, 10 mg/ml Nb35, and 25 mU/ml Apyrase. The complex was then solubilized in a buffer containing 20 mM HEPES, pH 7.4, 100 mM NaCl, 3 mM MgCl2, 5 mM CaCl2, 2.5 mg/ml leupeptin, 0.2 mg/ml benzamidine, 0.5% (w/v) lauryl maltose neopentylglycol (LMNG, Anatrace), 0.1% (w/v) cholesteryl hemisuccinate TRIS salt (CHS) for 3 h at 4°C. The supernatant was collected by centrifugation at 25,000 rpm for 30 min, and the solubilized complex was incubated with M1 anti-FLAG resin for 2 h at 4°C. The complex was immobilized on Flag-M1 resin, loaded on a Flag-M1 column and washed with 20 column volumes of 20 mM HEPES, pH 7.4, 100 mM NaCl, 3 mM MgCl2, 5 mM CaCl2, 0.01% (w/v) LMNG, 0.002% (w/v) CHS, 10 μM ligand, 2.5 mg/ml leupeptin, 0.2 mg/ml benzamidine. The DRD1-Gs complex was eluted with 20 mM HEPES, pH 7.4, 100 mM NaCl, 0.01% (w/v) LMNG, 0.002% (w/v) CHS, 10 μM ligand, 5 mM EGTA and 0.2 mg/ml FLAG peptide. The complex was collected and concentrated, then loaded onto a Superdex 6 Increase 10/300 GL column (GE Healthcare) with buffer containing 20 mM HEPES, pH 7.4, 100 mM NaCl, 0.00075% (w/v) LMNG, 0.00025% GDN, 0.0002% (w/v) CHS, 10 μM ligand. The complex fractions were concentrated with a 100 kDa MWCO Millipore concentrator for electron microscopy. The DRD1-Gs-Nb35 in complex with PW0464, or dopamine and LY3154207 were prepared with similar procedure as fenoldopam bound DRD1 complex. All samples were concentrated over 5mg/ml for making cryo grid.
Cryo-grid preparation and EM data collection
Forcryo-EM grid preparation, 3.5 μL aliquots of concentrated ligands bound DRD1-Gs-Nb35 complexes were loaded onto glow-discharged holey carbon grids (Quantifoil Au R1.2/1.3, 300 mesh). Grids were blotted for 3.0 s and plunge-frozen in liquid ethane cooled by liquid nitrogen using a Vitrobot Mark IV (Thermo Fisher) at 4°C and with 100% humidity. Grids were then transferred to a Titan Krios electron microscope (Thermo Fisher) operating at 300 kV and equipped with spherical aberration (Cs) image corrector. Micrographs were recorded using a K2 Summit counting camera (Gatan Company) in super-resolution mode with a nominal magnification of 22,500 × , resulting in a physical pixel size of 1 Å. Movie stacks were obtained with a defocus range of −1.0 to −2.0 μm (Table S1) using SerialEM with a set of customized scripts enabling automated low-dose image acquisition. Each movie stack was recorded for a total of 8 s with 0.2 s exposure per frame and exposure dose set to 8 electrons per pixel per second. For micrographs recording of SKF83959 bound DRD1 complex, the magnification is 105,000 × and pixel size is 0.85 Å, movie stacks were obtained with a defocus range of −1.0 to −2.5 μm (Table S1).
Image processing and 3D reconstructions
For the fenoldopam-DRD1-Gs-Nb35 complex, a total 2316 movie stacks were collected and all 40 frames in each stack were summed and motion-corrected using MotionCorr2 (Roh et al., 2017) (Figure S1), with twofold binned to a pixel size of 1Å per pixel and dose weighting was performed. Each micrograph was manually inspected to remove bad pictures that were contaminated by crystalline ice or other forms of visible contamination, and CTF parameters were estimated by GCTF (Zhang, 2016). After sorting, micrographs with maximum estimated resolution beyond 4.0 Å were discarded. A total of 2,386,124 particles were auto-picked by Gautomatch (Zhang, 2016) for the fenoldopam-DRD1-Gs-Nb35 sample with a box size of 216 pixels. Particle picking was performed using low-pass filtered templates to 40 Å to limit reference bias. Particles extracted from the dataset were downscaled 4 times and subjected to reference-free 2D classification to remove false picks and obvious junk classes leaving behind approximately 1,581,300 particles. The map generated in cryoSPARC (Punjani et al., 2017) low passed filtered to 60 Å was used as an initial reference model for maximum-likelihood-based three-dimensional classifications. The total good 1,581,300 particles were 3D classified with 6 classes in RELION3.0 (Zivanov et al., 2018). Two good classes (887,222 particles) were selected for another round of 3D classification. A dataset of 539,327 particles were subjected to 3D auto-refinement, resulting in an initial 3.3 Å density map. Further Bayesian polishing of these particle projections was performed in RELION3.0, followed by another round of auto-refinement, which generated a final 3.2 Å map determined by gold standard Fourier shell correlation using the 0.143 criterion. The density maps were corrected for the modulation transfer function (MTF) of the K2 summit direct detector and then sharpened by applying a temperature factor that was estimated using post-processing in RELION3.0. Local resolution estimation was performed in RELION3.0 using the unfiltered half map.
For A77636-DRD1-Gs-Nb35 complex, a total 3509 movie stacks were collected (Data S1A). 720,719 particles were extracted for further 3D classification and we obtained four different conformational subclasses. 391,771 particle projections of the best class were further applied for final homogeneous refinement in RELION3.0 and obtained the best density map with global l resolution of 3.5 Å.
For SKF83959-DRD1-Gs-Nb35 complex, a total 3610 movie stacks were collected (Data S1D). 140,000 particle projections (38.9%) of the best class were further applied for final homogeneous refinement in RELION3.0 and obtained the best density map with global resolution of 3.3 Å.
For the PW0464-DRD1-Gs-Nb35 complex, a total of 4,690 movie stacks were collected (Data S1B). 720,786 particles were extracted for further 3D classification and we obtained four different conformational subclasses. 672,890 particle projections of the best class were further applied for final homogeneous refinement in RELION3.0 and obtained the best density map with global resolution of 3.1 Å.
For dopamine-LY3154207-DRD1-Gs-Nb35 complex (Data S1C), a total of 4,475 movie stacks were collected. 1,206,885 particles were extracted for further 3D classification and obtained four different conformational subclasses. 584,166 particle projections of the best class were further applied for final homogeneous refinement in RELION3.0 and obtained the best density map with global resolution of 3.1 Å.
Model building and structure refinement
The crystal structure of β2 receptor with Gs protein and NB35 complex (PDB code: 3SN6) (Rasmussen et al., 2011) was used as initial model for model rebuilding and refinement against EM density map. The model was docked into the EM density map using UCSF Chimera (Pettersen et al., 2004), followed by manual adjustment and rebuilding in COOT (Emsley and Cowtan, 2004). Real space refinement was carried out in Phenix programs (Adams et al., 2010). The model statistics was validated using MolProbity (Williams et al., 2018). The final structures showed a good model geometry, and the detailed refinement statistics are provided in Table S1. The extent of any model overfitting during refinement was measured by refining the final model against one of the half-maps and by comparing the resulting map versus model FSC curves with the two half-maps and the full model. Structural figures were prepared in PyMoL (https://pymol.org/2/) and UCSF Chimera.
Radioligand binding assay
Binding assays were performed using wild-type or mutant human DRD1 transfected into HEK293 cell. Briefly, wild-type or mutant DRD1 was transfected into HEK293 cells using Lipofectamine 2000 (Thermo Fisher) according to the manufacturer’s instructions, and cultured at 37°C with 5% CO2 for 48 h. Cells were then harvested with a cell scraper and dounced to homogenize in ice-cold lysis buffer containing 50 mM HEPES, pH 7.4, 2 mM EDTA. Cell membranes were collected by centrifugation at 25,000 g for 45 min and homogenized in ice-cold buffer containing 75 mM Tris pH 7.4, 1 mM EDTA, and 12.5 mM MgCl2. The total membrane surface protein expression was determined by Bradford protein assay. For saturation binding assay, membranes (8~10 μg) were incubated for 45 min at 30°C while shaking with 0.075-5 nM [3H]-SCH23390 (Perkin Elmer). Non-specific binding was determined in the presence of 2 μM A77636. Kd value of the wild-type DRD1 with [3H]-SCH23390 is 0.28 ± 0.03 nM.
For competition assays, membranes (~8 μg) were incubated for 45 min at 30°C shaking with 0.5 nM [3H]-SCH23390 (PerkinElmer) and an increasing concentration of competing ligand concentrations in binding buffer (50 mM HEPES, pH 7.4, 10 mM MgCl2, 1 mM EDTA, 0.1% protease-free BSA, 0.01% ascorbic acid). In case of the dopamine competition assay in the presence of an allosteric regulator, different concentrations of LY3154207 (from 0.14 nM to 100 nM) were added. Cells were filtered over GF/C glass microfiber filters (Whatman) pre-soaked in ice-cold binding buffer supplemented 0.5% polyethylenimine using a 96-well filtermate harvester (Perkin Elmer) and rinsed 3 times with 2 mL ice-cold assay buffer to remove any free [3H]-SCH23390. Afterward, filters were dried and subjected to liquid scintillation counting on a MicroBeta TriLux scintillation counter (Perkin Elmer). All data were analyzed by GraphPad Prism 7 (GraphPad Software Inc.). Each measurement was repeated in at least three independent experiments, each in triplicate.
cAMP functional assays
The coding sequence of wild-type human DRD1 and DRD2 was sub-cloned into the expression vector pcDNA3.1+ with a HA signal sequence followed by a Flag tag at the N terminus. Point mutations used in our study were generated using Q5 site-Directed Mutagenesis kit (NEB). The constructs were expressed in HEK293 cells using jetPRIME transfection reagent (Polyplus transfection, France) according to the manufacturer’s instruction. Cells were harvested 48 h post-transfection. Cell surface expression level of receptors were determined by flow cytometry with an anti-Flag M2-FITC antibody (Sigma, F4049).
cAMP accumulation level was measured using a cAMP-Gs kit (Cisbio Bioassays, 62AM4PEB) or GloSensor cAMP assay (Promega) for DRD1/DRD5. In brief, for cAMP accumulation assays of DRD1/DRD5 activated by agonists, the harvested cells were seeded into 384-well plates and stimulated with the corresponding agonists and incubated for 1 h at 37°C with 5% CO2. After that, cAMP Eu Cryptate conjugate and anti-cAMP-d2 were added to 384-wells plates and incubated for 1 h at room temperature. Finally, plates were read on BIOTEK Cytation3 reader with excitation at 320 nm, emission at 620 nm and 665 nm. In addition, increase of cAMP level by positive allosteric modulator LY3154207 were measured in the presence and absence of EC20 concentration of dopamine (10 nM). The allosteric regulation efficacy of LY3154207 on dopamine action on DRD1 was performed using different concentrations of LY3154207 from 0.14 nM to 100 nM. For GloSensor cAMP assay, HEK293 cells transfected with wild-type or mutant DRD1 and Glosensor plasmid were plated into 96-well plates and cultured for 48 h at 37°C with 5% CO2. Cells were stimulated with serum-free media containing 5% v/v dilution of the GloSensor cAMP reagent stock solution (Promega). Afterward, a range of concentrations of ligands were added, and the luminescence signal were counted on an EnVision multimode plate reader (Perkin Elmer). cAMP level was calculated according to a standard dose-response curve. EC50 were calculated using nonlinear regression (curve fit) using GraphPad Prism 7 (GraphPad Software). To validate our results, each measurement was repeated in at least three independent experiments, each in triplicate.
To measure cAMP inhibition by D2-like receptors (DRD2, DRD3 and DRD4) activating, cell-based luciferase activity assay was performed by pGL3-CRE-luciferase reporter systems (Promega), as described in a previous study (Wu et al., 2019) . Briefly, cells expressing wild-type and mutant DRD2 were treated with dopamine diluted in 2 μM forskolin stimulating buffer. After 6 h incubation, 1 × Passive Lysis Buffer was added to the 96-well plate to measure luciferase assay in cell lysates as determined by the luciferase assay reagent kit (Promega). The luciferase activities in agonist treatment groups were first calculated as ratios to that of control group and normalized by wild-type receptor. To validate our function results, all experiments were repeated at least three times independently, each in triplicate.
β-arrestin recruitment assay
To measure β-arrestin recruitment by DRD1 upon PW0464 or A77636 activation, intracellular bioluminescence resonance energy transfer (BRET) assay was performed as previously described (Yang et al., 2018, 2020). Briefly, Flag-DRD1-WT-eYFP, β-arrestin-2-Rluc plasmids were co-transfected into HEK293 cells using PEI and cultured at 37°C with 5% CO2 for 24 h. Afterward, the transfected cells were seeded (50,000 cells per well) into clear-bottom 96-well plate and cultured at 37°C for another 24 h. The cells were washed 3 times with 200 μL PBS and incubated with increasing concentrations of PW0464, A77636 or vehicle for 10 min. BRET signal was determined, as the ratio of the light emitted by YFP (530 nm) and Rluc (485 nm), after addition of 5 μM of luciferase substrate coelenterazine 400a (Cayman) using a Mithras LB940 microplate reader (Berthold Technologies).
Molecule docking
The GLIDE program (version 5.6, Schrödinger, LLC, New York, NY, 2010) (Friesner et al., 2006; Halgren et al., 2004) was used for docking studies. Prior to docking, ligands were prepared using “Ligand Prep” module. For GLIDE docking, complex structures of DRD1 was preprocessed according to the protein preparation procedure recommended and docking site was defined in terms of ligands. All docking calculations were run in the “Standard Precision” (SP) mode (Friesner et al., 2004) with default values for all parameters. Five poses per prepared ligand were saved for each docking calculation. The best-docked structure was chosen using the Glide Score (Jorgensen et al., 1996).
Molecular dynamics (MD) simulation
Prior to running MD simulations, the receptor-ligand complex model was further prepared in Protein Preparation Wizard in Maestro (Schrödinger) to fix the missing side chains, add hydrogen atoms and cap the receptor chain termini while the residues D70 and D120 were protonated in the active-state simulations, all other titratable residues were left in their dominate protonation state at pH 7.0. The structures were inserted into a pre-equilibrated POPC bilayer and solvated with 0.15 M NaCl in explicitly water using dabble (Betz, 2018).
Parameters for the MD simulations were generated using the CHARMM-GUI web interface (Jo et al., 2008; Lee et al., 2016; Wu et al., 2014) using the CHARMM36 forcefield (Huang and MacKerell, 2013) with CGenFF (Vanommeslaeghe et al., 2010) for ligands. Simulations were performed CUDA enabled version of PMEMD in AMBER16 (Le Grand et al., 2013). The system was heated from 0 to 100 K in the NVT ensemble using the Langevin thermostat over 12.5 ps with harmonic restraints of 10.0 kcal mol−1 Å−2 on the non-hydrogen atoms of lipid, protein, and ligand. Then the system was heated to 310 K over 125 ps in the NPT ensemble with semi-isotropic pressure coupling and a pressure of 1 bar. Further equilibration was performed at 310K with harmonic restraints on the protein and ligand starting at 5.0 kcal mol−1 Å−2 and reduced by 1.0 kcal mol−1 Å−2 in a stepwise fashion every 2 ns, for a total of 10 ns of additional restrained equilibration. Multiple independent simulations were initialized from the final snapshot of the restrained equilibration. These simulations were conducted in the NPT ensemble at 310K and 1 bar, using a Langevin thermostat and Monte Carlo barostat under periodic boundary conditions. A time step of 2.0 fs with hydrogen mass repartitioning was used, bond lengths to hydrogen atoms were constrained using SHAKE (Ryckaert et al., 1977). Non-bonded interactions were cut off at 9.0 Å, and long-range electrostatic interactions were computed using the particle mesh Ewald method. The simulation frames were written every 100 ps. Root-mean-square deviation (RMSD) was calculated using the CPPTRAJ module (Roe and Cheatham, 2013) to monitor the fluctuation of the protein and ligand during the simulations.
QUANTIFICATION AND STATISTICAL ANALYSIS
The resolutions of all cryo-EM maps were estimated with the gold-standard Fourier shell correlation 0.143 criterion indicated in figure legends. The detailed can be found in Figure S1, supplemental information Data S1, legends and method.
Statistical analysis of radioligand binding assays, cAMP function assays, BRET assays and receptor expression level assays were processed using Prism 7.0 (GraphPad). Data in figures and tables are presented as the mean ± standard error of the mean (SEM) of three independent experiments performed in triplicate indicated in figure and table legends. The “n” value in tables means the numbers of independent experiment. The details can be found in method and Figures 5C, 5D, S3A, S3B, S3G, S3I, S4A, S5F–S5H, S6B–S6H, S7E, S7H, and S7I, and Tables S2–S5 and legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli cells DH10Bac | Invitrogen | 10361012 |
| Chemicals, peptides, and recombinant proteins | ||
| Benzamidine | Sigma-Aldrich | Cat# B6506 |
| Leupeptin | Sigma-Aldrich | Cat# L2884 |
| Lauryl Maltose Neopentyl Glycol (LMNG) | Anatrace | Cat# NG310 |
| Glyco-diosgenin (GDN) | Anatrace | Cat# GDN101 |
| Cholesterol hemisucinate TRIS Salt (CHS) | Sigma-Aldrich | Cat# C6512 |
| Sodium Cholate | Sigma-Aldrich | Cat# C1254 |
| TCEP | Thermo Fisher Scientific | Cat# 77720 |
| Apyrase | NEB | Cat# M0398S |
| Ethylenediamine Tetraacetic Acid (EDTA) | Thermo Fisher Scientific | Cat# S311-500 |
| Monoclonal ANTI-FLAG M2-FITC antibody | Sigma- Aldrich | Cat# F4049 |
| Fenoldopam | Med Chem Express | Cat# HY-B0735A |
| A77636 | TOCRIS | Cat# 1701 |
| Dopamine | Selleck | Cat# S2529 |
| SKF83959 | TOCRIS | Cat# 2074 |
| PW0464 | This paper | N/A |
| LY3154207 | This paper | N/A |
| [3H]-SCH23390 | PerkinElmer | NET930250UC |
| Critical commercial assays | ||
| HTRF Dynamic cAMP kit | Cisbio | Cat# 62AM4PEB |
| Q5 site directed mutagenesis kit | NEB | Cat# E0554S |
| cAMP Glosensor Assay | Promega | Cat# E1291 |
| Deposited data | ||
| Fenoldopam-DRD1-Gs EM map and coordinate | This paper | EMDB: EMD-30392 PDB: 7CKW |
| A77636-DRD1-Gs EM map and coordinate | This paper | EMDB: EMD-30393 PDB: 7CKX |
| PW0464-DRD1-Gs EM map and coordinate | This paper | EMDB: EMD-30394 PDB: 7CKY |
| Dopamine-PAM-DRD1-Gs EM map and coordinate | This paper | EMDB: EMD-30395 PDB: 7CKZ |
| SKF83959-DRD1-Gs EM map and coordinate | This paper | EMDB: EMD-30452 PDB: 7CRH |
| Experimental models: cell lines | ||
| Insect cell line Sf9 | Expression Systems | N/A |
| HEK293 cell | ATCC | N/A |
| Primers for site-direct mutagenesis | ||
| DRD1-S198A_forward: catctcaGCCtctgtaataagcttttacatccctg | This paper | N/A |
| DRD1-S198A_reverse: attacagaGGCtgagatggcatatgtcctgctga | This paper | N/A |
| DRD1-S202A_forward: CtgtaataGCCttttacatccctgtggccatcatg | This paper | N/A |
| DRD1-S202A_reverse: tgtaaaaGGCtattacagaggatgagatggcatat | This paper | N/A |
| DRD1-D103A_forward: cctttGCCatcatgtgctccactgcatccatc | This paper | N/A |
| DRD1-D103A_reverse: agcacatgatGGCaaaggccacccagatgttacagaag | This paper | N/A |
| Recombinant DNA | ||
| pFastBac1-HA-FLAG-DRD1-His*10 | This paper | N/A |
| pcDNA3.1-HA-FLAG-DRD1 (WT and mutants) | This paper | N/A |
| pFastBac1-Gαs | This paper | N/A |
| pFastBac1Dual-Gβγ | This paper | N/A |
| Software and algorithms | ||
| SerialEM | Mastronarde DN | https://bio3d.colorado.edu/SerialEM/ |
| MotionCor2 | (Zheng et al., 2017) | https://emcore.ucsf.edu/ucsf-software |
| Gctf | Zhang, 2016 | https://www.mrc-lmb.cam.ac.uk/kzhang/Gctf/ |
| Gautomatch | Zhang, 2016 | https://www.mrc-lmb.cam.ac.uk/kzhang/Gautomatch |
| Relion3.0 | (Zivanov et al., 2018) | https://www2.mrc-lmb.cam.ac.uk/relion |
| CHIMERA | Pettersen et al., 2004 | http://www.cgl.ucsf.edu/chimera |
| COOT | (Emsley and Cowtan, 2004) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix | Adams et al., 2010 | https://www.phenix-online.org |
| PyMOL | Schrodinger | https://www.pymol.org/2/ |
| Prism v.7.0 | GraphPad Software | https://www.graphpad.com |
| Other | ||
| Superdex 200 Increase column 10/300 | GE healthcare | Cat# 28990944 |
| BIOTEK Cytation3 reader | BIO-TEK | N/A |
| ESF921 cell culture medium | Expression Systems | Cat# 96-001 |
| DMEM basic cell culture media | GIBCO | Cat# 11995 |
| FBS (fetal bovine serum) | Cell Box | Cat# SAG-01U-02 |
| Penicillin-Streptomycin | GIBCO | Cat# 15140122 |
| Trypsin 0.5% EDTA | GIBCO | Cat# 25300054 |
| 9 cm2 Cell Culture Dish | Corning | Cat# 430166 |
| 6-well Cell Culture Dish | Corning | Cat# 430166 |
| 96-well Cell Culture Dish | Corning | Cat# 3599 |
| 96-well white microplate assay plates | Corning | Cat# 3917 |
Highlights.
Structures of DRD1-Gs in complex with catechol-based and non-catechol agonists
Key polar interaction network is involved in DRD1 activation
Specific features for agonists interaction and Gs coupling of DRD1 are determined
Structure of DRD1-Gs complexed with dopamine and positive allosteric modulator
Structural basis of allosteric regulation of DRD1 by LY3154207 is investigated
ACKNOWLEDGMENTS
Cryo-EM data were collected at the cryo-EM facility of the National Center for Protein Science in Shanghai. We thank Sichuan University West China Cryo-EM Center and Duyu High Performance Computing Center and the Instrumental Analysis Center of Xi’an Jiaotong University for cryo-EM sample preparation. We thank Professor Yajun Wang, Dr. Jiannan Zhang, and Can Lu from the College of Life Sciences Sichuan University for their help on cAMP measurement. This study was supported by the Science and Technology Department of Sichuan Province (2020YJ0208 to Z.S.), the National Key R&D Program of China (2019YFA0508800 to Z.S. and P.X.), the National Science Fund for Excellent Young Scholars (81822008 to Xiao Yu and 11922410 to Lei Zhang), the National Science Fund for Distinguished Young Scholars (81825022 to J.-P.S.), the National Natural Science Foundation of China (31972916 to Z.S. and 31971195 to P.X.), Shandong University Youth Interdisciplinary Science Innovation Group (2020QNQT002 to Xiao Yu), and the National Institutes of Health (R35GM136387 to D.M.R.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2021.01.028.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, and Gainetdinov RR (2011). The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev 63, 182–217. [DOI] [PubMed] [Google Scholar]
- Betz R. (2018). Dabble 10.5281/zenodo.836914. [DOI] [Google Scholar]
- Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, and Stevens RC (2010). Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 330, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conroy JL, Free RB, and Sibley DR (2015). Identification of G protein-biased agonists that fail to recruit β-arrestin or promote internalization of the D1 dopamine receptor. ACS Chem. Neurosci 6, 681–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, Borhani DW, and Shaw DE (2011). Activation mechanism of the β2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 108, 18684–18689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Flock T, Ravarani CNJ, Sun D, Venkatakrishnan AJ, Kayikci M, Tate CG, Veprintsev DB, and Babu MM (2015). Universal allosteric mechanism for Ga activation by GPCRs. Nature 524, 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler JC, Bhattacharya S, Urban JD, Vaidehi N, and Mailman RB (2012). Receptor conformations involved in dopamine D(2L) receptor functional selectivity induced by selected transmembrane-5 serine mutations. Mol. Pharmacol 81, 820–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, et al. (2004). Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, and Mainz DT (2006). Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- García-Nafría J, Lee Y, Bai X, Carpenter B, and Tate CG (2018). Cryo-EM structure of the adenosine A2A receptor coupled to an engineered hetero-trimeric G protein. eLife 7, e35946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray DL, Allen JA, Mente S, O’Connor RE, DeMarco GJ, Efremov I, Tierney P, Volfson D, Davoren J, Guilmette E, et al. (2018). Impaired β-arrestin recruitment and reduced desensitization by non-catechol agonists of the D1 dopamine receptor. Nat. Commun 9, 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurrell R, Duvvuri S, Sun P, and DeMartinis N (2018). A Phase I Study of the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of the Novel Dopamine D1 Receptor Partial Agonist, PF-06669571, in Subjects with Idiopathic Parkinson’s Disease. Clin. Drug Investig 38, 509–517. [DOI] [PubMed] [Google Scholar]
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, and Banks JL (2004). Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- Hall A, Provins L, and Valade A (2019). Novel Strategies To Activate the Dopamine D1 Receptor: Recent Advances in Orthosteric Agonism and Positive Allosteric Modulation. J. Med. Chem 62, 128–140. [DOI] [PubMed] [Google Scholar]
- Hao J, Beck JP, Schaus JM, Krushinski JH, Chen Q, Beadle CD, Vidal P, Reinhard MR, Dressman BA, Massey SM, et al. (2019). Synthesis and Pharmacological Characterization of2-(2,6-Dichlorophenyl)-1-((1S,3R)-5-(3-hydroxy-3-methylbutyl)-3-(hydroxymethyl)-1-methyl-3,4-dihydroisoquinolin-2(1H)-yl)ethan-1-one (LY3154207), a Potent, Subtype Selective, and Orally Available Positive Allosteric Modulator of the Human Dopamine D1 Receptor. J. Med. Chem 62, 8711–8732. [DOI] [PubMed] [Google Scholar]
- Huang J, and MacKerell AD Jr. (2013). CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem 34, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S, Kim T, Iyer VG, and Im W (2008). CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, Maxwell DS, and TiradoRives J (1996). Developmentand testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc 118, 11225–11236. [Google Scholar]
- Kebabian JW, and Calne DB (1979). Multiple receptors for dopamine. Nature 277, 93–96. [DOI] [PubMed] [Google Scholar]
- Krishna Kumar K, Shalev-Benami M, Robertson MJ, Hu H, Banister SD, Hollingsworth SA, Latorraca NR, Kato HE, Hilger D, Maeda S, et al. (2019). Structure of a Signaling Cannabinoid Receptor 1-G Protein Complex. Cell 176, 448–458.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Cheng X, Swails JM, Yeom MS, Eastman PK, Lemkul JA, Wei S, Buckner J, Jeong JC, Qi Y, et al. (2016). CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput 12, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grand S, Gotz AW, and Walker RC (2013). SPFP: Speed without compromise–A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun 184, 374–380. [Google Scholar]
- Mansour A, Meng F, Meador-Woodruff JH, Taylor LP, Civelli O, and Akil H (1992). Site-directed mutagenesis of the human dopamine D2 receptor. Eur. J. Pharmacol 227, 205–214. [DOI] [PubMed] [Google Scholar]
- Masureel M, Zou Y, Picard LP,van der Westhuizen E, Mahoney JP, Rodrigues JPGLM, Mildorf TJ, Dror RO, Shaw DE, Bouvier M, et al. (2018). Structural insights into binding specificity, efficacy and bias of a β2AR partial agonist. Nat. Chem. Biol 14, 1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missale C, Nash SR, Robinson SW, Jaber M, and Caron MG (1998). Dopamine receptors: from structure to function. Physiol. Rev 78, 189–225. [DOI] [PubMed] [Google Scholar]
- Nichols AJ, Ruffolo RR Jr., and Brooks DP (1990). The pharmacology of fenoldopam. Am. J. Hypertens 3, 116S–119S. [DOI] [PubMed] [Google Scholar]
- Noce A, Marrone G, Rovella V, Busca A, Gola C, Ferrannini M, and Di Daniele N (2019). Fenoldopam Mesylate: A Narrative Review of Its Use in Acute Kidney Injury. Curr. Pharm. Biotechnol 20, 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryo-SPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. (2011). Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, and Kobilka BK (2013). Adrenaline-activated structure of β2-adrenoceptor stabilized by an engineered nanobody. Nature 502, 575–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe DR, and Cheatham TE 3rd. (2013). PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- Roh SH, Hryc CF, Jeong HH, Fei X, Jakana J, Lorimer GH, and Chiu W (2017). Subunit conformational variation within individual GroEL oligomers resolved by Cryo-EM. Proc. Natl. Acad. Sci. USA 114, 8259–8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryckaert J-P, Ciccotti G, and Berendsen H (1977). Numerical-Integration of Cartesian Equations of Motion of a System with Constraints – Molecular-Dynamics of N-Alkanes. J. Comput. Phys 23, 327–341. [Google Scholar]
- Schultz W. (2016). Dopamine reward prediction-error signalling: a two-component response. Nat. Rev. Neurosci 17, 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spano PF, Govoni S, and Trabucchi M (1978). Studies on the pharmacological properties of dopamine receptors in various areas of the central nervous system. Adv. Biochem. Psychopharmacol 19, 155–165. [PubMed] [Google Scholar]
- Sun JP, Fedorov AA, Lee SY, Guo XL, Shen K, Lawrence DS, Almo SC, and Zhang ZY (2003). Crystal structure of PTP1B complexed with a potent and selective bidentate inhibitor. J. Biol. Chem 278, 12406–12414. [DOI] [PubMed] [Google Scholar]
- Thal DM, Glukhova A, Sexton PM, and Christopoulos A (2018). Structural insights into G-protein-coupled receptor allostery. Nature 559, 45–53. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, and Mackerell AD Jr. (2010). CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wacker D, Levit A, Che T, Betz RM, McCorvy JD, Venkatakrishnan AJ, Huang XP, Dror RO, Shoichet BK, and Roth BL (2017). D4 dopamine receptor high-resolution structures enable the discovery of selective agonists. Science 358, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Che T, Levit A, Shoichet BK, Wacker D, and Roth BL (2018a). Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 555, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Heinz BA, Qian YW, Carter JH, Gadski RA, Beavers LS, Little SP,Yang CR, Beck JP, Hao J, et al. (2018b). Intracellular Binding Site for a Positive Allosteric Modulator of the Dopamine D1 Receptor. Mol. Pharmacol 94, 1232–1245. [DOI] [PubMed] [Google Scholar]
- Wang P, Felsing DE, Chen H, Raval SR, Allen JA, and Zhou J (2019). Synthesis and Pharmacological Evaluation of Noncatechol G Protein Biased and Unbiased Dopamine D1 Receptor Agonists. ACS Med. Chem. Lett 10, 792–799. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, Verma V, Keedy DA, Hintze BJ, Chen VB, et al. (2018). MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu EL, Cheng X, Jo S, Rui H, Song KC, Dávila-Contreras EM, Qi Y, Lee J, Monje-Galvan V, Venable RM, et al. (2014). CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem 35, 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Lv C, Wan Y, Li X, Zhang J, Li J, and Wang Y (2019). Arginine vasotocin (AVT)/mesotocin (MT) receptors in chickens: Evidence for the possible involvement of AVT-AVPR1 signaling in the regulation of oviposition and pituitary prolactin expression. Gen. Comp. Endocrinol 281, 91–104. [DOI] [PubMed] [Google Scholar]
- Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, and Zhou R (2015). Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160, 62–73. [DOI] [PubMed] [Google Scholar]
- Yang F, Xiao P, Qu CX, Liu Q, Wang LY, Liu ZX, He QT, Liu C, Xu JY, Li RR, et al. (2018). Allosteric mechanisms underlie GPCR signaling to SH3-domain proteins through arrestin. Nat. Chem. Biol 14, 876–886. [DOI] [PubMed] [Google Scholar]
- Yang F, Mao C, Guo L, Lin J, Ming Q, Xiao P, Wu X, Shen Q, Guo S, Shen DD, et al. (2020). Structural basis of GPBAR activation and bile acid recognition. Nature 587, 499–504. [DOI] [PubMed] [Google Scholar]
- Yin J, Chen KM, Clark MJ, Hijazi M, Kumari P, Bai XC, Sunahara RK, Barth P, and Rosenbaum DM (2020). Structure of a D2 dopamine receptor-G-protein complex in a lipid membrane. Nature 584, 125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K. (2016). Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Xiong B, Zhen X, and Zhang A (2009). Dopamine D1 receptor ligands: where are we now and where are we going. Med. Res. Rev 29, 272–294. [DOI] [PubMed] [Google Scholar]
- Zheng S, Palovcak E, Armache J, Verba K, Cheng Y, and Agard D (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Zou J, Tian F, and Shang Z (2009). Fluorine bonding–how does it work in protein-ligand interactions? J. Chem. Inf. Model 49, 2344–2355. [DOI] [PubMed] [Google Scholar]
- Zhuang Youwen, Xu Peiyu, Mao Chunyou, Wang Lei, Krumm Brian, Zhou X. Edward, Huang Sijie, Liu Heng, and Cheng Xi (2021). Structrural insight into the human D1 and D2 dopamine receptor signaling complexes. Cell 184 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJH, Lindahl E, and Scheres SHW (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data produced or analyzed in this study are included in the main text or the supplementary materials. The cryo-EM density maps and atomic coordinates have been deposited in the Electron Microscopy Data Bank (EMDB) and Protein Data Bank (PDB) under accession numbers EMD-30392 and 7CKW for Fenoldopam-DRD1 complex; EMD-30393 and 7CKX for A77636-DRD1 complex; EMD-30452 and 7CRH for A77636-DRD1 complex; EMD-30394 and 7CKY for PW0464-DRD1 complex; and EMD-30395 and 7CKZ for dopamine/LY3154207-DRD1 complex.