

Abstract
We used fast absorbance spectroscopy to investigate in vivo binding dynamics and electron transfer between plastocyanin (pc) and photosystem I (PSI), and cytochrome (cyt) f oxidation kinetics in Chlamydomonas reinhardtii mutants in which either the binding or the release of pc from PSI was diminished. Under single flash-excitation conditions, electron flow between PSI and the cyt complex was not affected by a 5-fold lowering of the binding affinity of pc to PSI, as induced by a mutation replacing the tryptophan-651 of the PsaA subunit by a serine residue (PsaA-W651S). On the other hand, electron flow from PSI to the cyt b6f complex was very sensitive to a 2- to 3-fold decrease in the rate of pc release from PSI, obtained by replacing the glutamic acid residue 613 of the PsaB subunit with glutamine (PsaB-E613N). Thus, our data indicate that under these experimental conditions the release of oxidized pc limits electron transfer between cyt b6f complex and PSI in vivo.
Keywords: Chlamydomonas, photosynthesis, protein–protein interactions, systems biology
In vascular plants, most algae, and some cyanobacteria the type I copper protein plastocyanin (pc) is the lumen-localized carrier that shuttles electrons from the cytochrome (cyt) b6 f complex to photosystem I (PSI). pc can be replaced by cyt c6 in some cyanobacteria and green algae when the availability of copper is limiting (1–3). pc receives electrons from the cyt b6 f complex, which oxidizes a lipophilic, two-electron donor (plastoquinol). During this process, electron transfer is coupled to proton translocation across the membrane, which results in the establishment of an electrochemical proton gradient to be used for ATP synthesis (reviewed, e.g., in ref. 4). The cyt f subunit of this complex directly interacts with pc. This polypeptide is composed of a large and a small domain in which β structures are dominant (5). At the interface between the large and the small domain, a positively charged motif is observed, which comprises K58, K65, and K66 of the large domain and K188 and K189 of the small domain. This interface is believed to be important for electrostatic interaction with pc, allowing efficient electron transfer between the partners (6). However, its role has been questioned in vivo, where apparently other factors modulate the efficiency of the pc–cyt f interaction (7).
Reduced pc is oxidized by PSI, its electron being required for the reduction of the photo-oxidized primary donor P700+. The crystal structures of the cyanobacterial and plant PSIs suggest that the pc–PSI interaction is mediated by a flat luminal surface on the donor side of PSI (8, 9). This region is mainly composed of those loop regions that connect the transmembrane domains of the subunits PsaA and PsaB. These luminal loops, as well as the subunit PsaF, provide most of the structural elements present on this side. In vascular plants and algae, an additional extension of the luminal N-terminal domain of PsaF is observed, which is absent in cyanobacteria. This domain is highly positively charged and has been proposed to form an amphipathic helix where the basic side chains face the binding site of the soluble electron donors (10). This α-helical structure has been observed in the crystal structure of plant PSI, where its orientation might promote an interaction between K16 and K23 of the PsaF peptide and the conserved negatively charged residues present in pc (9). The involvement of these positively charged residues of PsaF in the binding of pc has been extensively studied by cross-linking, knock-out, and reverse genetics experiments (10–14). These studies have demonstrated the crucial role of these residues for proper binding, complex formation between pc and PSI, and fast electron transfer.
Besides PsaF, the existence of a second recognition motif that is essential for efficient binding and electron transfer between pc or cyt c6 and PSI has been demonstrated. This motif is provided by luminal loops i/j of PsaA and PsaB (15–17), where residues PsaA-W651 and PsaB-W627 constitute the key elements. They have their indole groups stacked at van der Waals distance (8) and are directly situated above P700 (8, 9). Previous in vitro studies of site-directed mutants of Chlamydomonas have shown that the removal of these tryptophan residues drastically decreases the affinity of pc for PSI, thus preventing the formation of an intermolecular complex between the two redox partners (15, 16). This lack of complex formation leads to a dramatic decrease of the second-order rate constant of electron transfer and to a significant increase of the dissociation constant as compared with the WT. It has been concluded therefore that binding of pc to the tryptophan dimer is critical for efficient binding and fast electron transfer in vitro (15, 16). An opposite effect has been observed in the case of glutamic acid at position 631, in loop j, of PsaB. Its substitution with an asparagine increased the PSI affinity for oxidized pc (15), reducing both the second-order rate constant for electron transfer and the dissociation constant for pc binding, when compared with the WT.
In pc–PSI complex from spinach, a value of 10 has been estimated for the electron transfer equilibrium constant, based on redox titrations (18). This value is lower than expected, due to a positive shift of the midpoint potential of pc CuII/CuI couple upon binding to PSI. As a consequence, oxidized pc is less tightly bound to PSI than is reduced pc. The lower driving force for electron transfer is compensated by a faster dissociation of the oxidized product, leading to an optimized turnover of PSI. According to a kinetic model developed by Drepper and coworkers (18), the fast release of oxidized pc from PSI is crucial for optimum electron transfer between the cyt b6 f complex and PSI.
To challenge this hypothesis in vivo, we have studied different Chlamydomonas strains with a modified oxidizing side of PSI. By comparing the kinetic properties of mutants in which either the kon or the koff for pc binding to PSI was affected, we conclude that unbinding of oxidized pc from PSI represents the limiting step of electron transfer between cyt b6 f complex and PSI in vivo.
Materials and Methods
Strains and Culture Conditions. Chlamydomonas WT (from strain 137 C), 6a+, PsaA-W651S, and PsaB-E613N strains (hereafter called W651S and E613N, respectively) were grown at 24°C in acetate-supplemented medium (19) under ≈6 μE·m–2·s–1 of continuous white light [1 einstein (E) = 1 mol of photons].
Isolation of PSI Complexes. Isolation of PSI particles was performed as described in ref. 20. Chlorophyll (Chl) concentrations were determined according to Porra et al. (21).
NADP+ Photoreduction Measurements. PSI-dependent NADP+ photoreduction was measured under donor side limiting conditions. Measurements were performed under saturating illumination provided by a 150-W halogen lamp. The reaction mixture contained PSI at a concentration of 5 μg of Chl per ml, 20 mM N-tris(hydroxymethyl)methylglycine–KOH (pH 7.4), 0.05% β-dodecyl maltoside, 3 mM MgCl2, 2 mM ascorbate, 0.06 mM dichloroindophenol, 0.5 μM ferredoxin–NADP+ reductase from spinach (Sigma), 4 μM ferredoxin, 0.5 mM NADP+, and pc at the indicated concentrations. NADPH generation was monitored by absorption changes at 340 nm. pc and ferredoxin were isolated from Chlamydomonas cells according to (20, 22) respectively.
Spectroscopic Measurements. For spectroscopic measurements, cells were harvested during exponential growth (≈2 × 106 cells per ml) and resuspended in minimum high salt medium (19), with addition of 20% (wt/vol) Ficoll to prevent cell sedimentation. Measurements were realized on intact cells, at a Chl concentration of ≈70 μg·ml–1. Spectroscopic analysis was performed at room temperature, using home-made pump and probe spectrophotometers. Three different setups were used: Fast cyt f oxidation kinetics (Fig. 2) were measured with a light-emitting diode (LED)-based spectrophotometer, as described elsewhere (24). Actinic flashes were provided by a dye laser at 690 nm (10-ns duration), whereas measuring flashes were provided by a white LED (Luxeon, Lumileds, San Jose, CA) filtered with interferential filters (10-nm full width at one-half maximum). In this case, flash duration was 10 μs. The fast P700+ reduction phase (Fig. 3) was measured with a second setup, having a time resolution of 10 ns (23, 24). Actinic flashes were provided by a dye laser at 690 nm, whereas detecting flashes were provided by an optical parametric oscillator pumped by a neodymium:yttrium/aluminum garnet laser. For multiple turnover experiments and steady-state electron flow measurements (Fig. 4), a xenonlamp-based spectrophotometer was used (22). Actinic flashes were provided by a xenon flashlamp (giving saturating light pulses, 5-μs duration at one-half height) filtered through a Schott RG695 filter. Detecting flashes were provided by a second xenon flashlamp (3-μs duration at one-half height) filtered through a monochromator (Jobin Yvon, Longjumeau, France). Continuous light was provided by a diode laser [SDL-LFI 3225, Wavelength Electronics (Bozeman, MT), providing 500 mW, emission peak at 690 nm] screened with neutral filters to deliver ≈1,000 μE·m–2·s–1 (70–80 photons per photosystem per s) to the samples.
Fig. 2.

In vivo cyt f oxidation kinetics in WT and mutant Chlamydomonas cells. Cells were harvested during exponential growth (≈2 × 106 cells per ml) and resuspended in minimum high salt medium (19), with addition of 20% (wt/vol) Ficoll to prevent cell sedimentation. Kinetics were obtained by deconvolution of absorption changes at 545, 554, and 573 nm after excitation with a laser flash, as described in Materials and Methods. Traces were normalized to the maximum amplitude of the cyt f oxidation signal; r.u., relative units. The duration of the actinic laser flash and of the detecting light-emitting diode flash was 10 ns and 10 μs, respectively.
Fig. 3.

Estimation of the fast P700+ reduction phase in vivo, reflecting predocking of pc to PSI. Signals represent the decay-associated spectrum assigned to rereduction of P700+ in the blue region, as measured by pump-probe spectroscopy in vivo. Signals were normalized to the amplitude of the signal at 430 nm, measured 900 ns after illumination. Experimental conditions were the same as in Fig. 2.
Fig. 4.

Number of PSI charge separations induced by an actinic xenon flash after a saturating laser flash given at time 0. Data were normalized to the amplitude of the signal induced by the laser flash, taken as 100% of PSI charge separation. Other conditions were as in Fig. 2. Data were calculated from the extent of the electrochromic signal, which is linearly related to the amplitude of light-driven charge separation. Hydroxylamine and 3-(3,4-dichlorophenyl)-1,1-dimethylurea were added to inhibit PSII photochemistry. Traces represent the difference between the absorption at 515 and 545 nm.
cyt f redox changes were calculated as the difference between the absorption at 554 nm and a baseline drawn between 545 and 573 nm (25) and were corrected for the contribution of the electrochromic signal [5% of the signal observed at 515 nm (25)].
Results
NADP Photoreduction in E613N PSI Particles. Previous studies on Chlamydomonas mutants with a modified oxidizing or reducing side of PSI (22) have shown that electron transfer from pc to P700+ limits light-driven electron transfer to NADP+. In particular, PSI donor side mutants showing a diminished pc binding to PSI also exhibited lower NADP+ photoreduction rates. In the E613N mutant, the release of pc+ from PSI is slowed down (15, 22). To test whether this mutation was also able to affect electron transfer through PSI, we measured the NADP+ photoreduction rate as a function of pc concentration with PSI particles isolated from mutant and WT cells (Fig. 1A). Compared with the WT, the NADP+ photoreduction measured in the E613N mutant PSI particles showed a lower apparent Km value, consistent with the higher pc affinity for PSI, and a reduced maximal rate, consistent with the electron transfer through the PSI complex being slowed down (Fig. 1 A). Importantly, these differences were not due to a lower PSI content of mutant particles. Indeed, the PSI amount in E613N particles was only slightly reduced (15%) compared with the WT, as indicated by SDS/PAGE fractionation and immunoblotting analysis using anti-PsaD antibodies (Fig. 1B). It appears therefore that mutations that decrease either the kon or the koff for pc binding to PSI similarly affect the rate of NADP+ reduction in vitro.
Fig. 1.

In vitro characterization of PSI activity and content in mutant strain PsaB-E613N. (A) NADP+ photoreduction measurements showing that electron transfer through PSI is limited by mutation PsaB-E613N. The SDs of the NADP+ photoreduction rates were calculated from a linear regression of the various time points at a given pc concentration. Measurements were performed with isolated PSI particles at 5 μg of Chl per ml, as described in Materials and Methods.(B) Quantification of PSI amounts in thylakoids isolated from the WT and E613N strains, upon SDS/PAGE fractionation and immunoblotting analysis using anti-PsaD antibodies.
pc-Mediated cyt f Oxidation in Chlamydomonas pc-Docking Side Mutants. We then tested the impact of mutations affecting either the kon or the koff for pc–PSI binding on electron flow between PSI and the cyt b6 f complex in vivo. To this aim, we performed a detailed spectroscopic kinetic analysis of the W651S strain, where the Kd is increased by a factor of 5, owing to a decrease of kon (16), and of the E613N mutant, where the Kd is decreased by a factor of 3 because of a lower koff value (15). As a control, we measured WT and the 6a+ strains. In the latter strain pc binding is strongly impaired due to removal of the PsaF subunit (11). Measurements were performed in the presence of the plastoquinol oxidation inhibitor stigmatellin (26), to inhibit cyt f reduction by plastoquinol, and thereby allow the measure of the full extent of the oxidation kinetics (see, e.g., ref. 27). The ionophore carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) was also added to increase the rate of cyt f oxidation, as shown (27). Laser illumination was used to avoid the occurrence of PSI double turnovers, which would otherwise complicate the kinetic analysis.
Kinetics of cyt f oxidation turned out to be differently affected by mutations at the PSI donor side (Fig. 2). In the WT, the half-time of cyt f oxidation was 130 μs (±15 μs), in agreement with previous values obtained in green algae (see, e.g., ref. 27). A lag in cyt f oxidation was also observed, which was in the order of tens of microseconds and consistent with previous data (27). The W651S mutation was essentially without consequences on the rate of cyt f oxidation, the half-time of this reaction being 140 μs(±18 μs). An almost identical phenotype was observed in the symmetrical mutant (PsaB-W627F, not shown), suggesting that the removal of one of the two tryptophan residues, which are required for efficient binding of pc to PSI in vitro, has no impact on cyt f oxidation kinetics in vivo. On the other hand, the half-time of cyt f oxidation was increased to 300 μs (±30 μs) in the E613N strain, suggesting that the slowing down of pc release from PSI limits the overall rate of cyt f oxidation in vivo. In the case of the PsaF-deficient mutant 6a+, where the second-order electron transfer rate constant is ≈100 times slower as compared with WT, the half-time of cyt f oxidation was largely slowed down (600 ± 30 μs), and a significant lag in the oxidation kinetics was observed (hundreds of microseconds), in agreement with previous observations (11).
With the only exception of the 6a+ strain, the total amounts of cyt f oxidized by the laser flash were very similar. This finding suggests that neither the redox potential of pc nor its relative abundance in the thylakoid membranes was modified in the various PSI mutant strains characterized here.
pc–PSI Interaction in Chlamydomonas pc-Docking Side Mutants. The different kinetics of cyt f oxidation reported above might reflect the different ability of mutant PSI complexes to bind pc. Indeed, the release of oxidized pc from PSI is thought to represent the limiting step of electron transfer between PSI and the cyt b6 f complex (18). To test this hypothesis, we measured the ability of PSI to form a pc prebound complex in the dark. We thus measured the amplitude of the 4-μs component of the decay-associated spectra of P700+. This phase reflects electron transfer between pc and PSI in this intermolecular complex (27). Therefore, its amplitude gives an estimation of the amount of pc that is predocked to PSI. Fig. 3 shows the results of such measurements in the strains analyzed above.
We first checked that the spectral features associated with this component corresponded to a P700+ – P700 spectrum, as previously measured in detergent-solubilized PSI particles. With the only exception of the 6a+ strain, this turned out to be the case (see, e.g., ref. 28 in the case of Chlamydomonas particles). We found that the amplitude of this component was very similar in the WT and in the E613N and W651S mutants. This result suggests that in vivo the same amount of pc was predocked to PSI in these mutants. On the other hand, not only the spectral features but also the amplitude of the signal were largely modified in the 6a+ mutant (Fig. 3). This finding confirms the existence of a PSI–pc complex in the E613N mutant, as well as its disappearance upon the removal of the PsaF subunit, in agreement with previous reports (11, 20). On the other hand, the result obtained in the case of the W651S mutant indicates that increasing the PSI–pc dissociation constant by a factor of 5 does not prevent an efficient binding of pc to PSI in vivo.
pc Exchanges at the pc-Docking Site. Although the data of Fig. 3 indicate that pc is prefixed to PSI in the E613N and W651S mutants in the dark, they do not provide any dynamic information on the binding of pc to PSI. Indeed, these experiments were performed on cells that had been dark adapted for a long time (>10 min) before a single turnover flash-driven experiment was performed.
To gain information on the pc-PSI binding dynamics in vivo, we repeated a double-flash experiment already performed to characterize the consequences of the PsaF deletion in Chlamydomonas (11). This experiment is based on the consideration that the recovery of photoactive PSI centers is limited by the rate of P700+ rereduction, the reoxidation of PSI acceptors being a faster process (29, 30).
Experiments were conducted by firing a laser flash to oxidize P700. The amount of PSI centers with reduced P700 was then detected by measuring PSI activity upon firing a second flash, using a xenon source capable of inducing ≈40% of double photochemical acts, at discrete times (Fig. 4). The extent of PSI active centers was probed by measuring the electrochromic shift of carotenoids at 515 nm, which gives a linear response to the amplitude of the charge separation generated by PSI photochemistry (31). Hydroxylamine and 3-(3,4-dichlorophenyl)-1,1-dimethylurea were added to inhibit PSII photochemistry (32). Because P700 is fully oxidized by the laser flash, the xenon flash can only induce charge separation in PSI centers where P700+ has been rereduced by prefixed pc in the time elapsing between the two illuminations according to the following reactions:
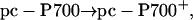 |
[1] |
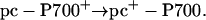 |
[2] |
Thus, the fast recovery of PSI activity (<20 μs) observed in Fig. 4 reflects the reduction of P700+ in PSI complexes with prebound pc. The similar amplitude of this phase observed in WT, the E613N, and the W651S strains, which is equal to the number of charge separations induced by the laser flash, confirms that ≈100% of PSI complexes are able to prefix a pc molecule in these strains, in agreement with the data of Fig. 3. Furthermore, the large inhibition observed in the case of the 6a+ mutant confirms the inability of its PSI to fix pc in the dark (11), again in agreement with the data of Fig. 3.
The xenon flash used in this experiment is able to induce double photoreactions. On the other hand, these reactions can occur only in complexes where both PSI and the prefixed pc are rereduced in the time between the laser and the xenon illumination. Indeed, illumination of centers in the pc–P700 state can produce double photoreactions, provided that illumination lasts longer than the time required for the accomplishment of reaction 1. On the other hand, illumination of pc+–P700 can generate only a pc+–P700+ state, i.e., does not result in any double photochemical acts.
Accordingly, the second phase (in the millisecond time range) of PSI recovery observed in Fig. 4 could be associated with the generation of the reduced pc–P700 complex, in line with the following scheme:
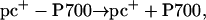 |
[3] |
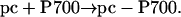 |
[4] |
The large inhibition of the slow phase of PSI recovery observed in the 6a+ mutant, where the rate of pc binding to PSI is slowed (11), suggests that this phase is controlled by reaction 4, in agreement with previous suggestions (11). On the other hand, this phase was also decelerated in the E613 mutant, where the release of oxidized pc is slowed down, but was almost unchanged in the W651S mutant. This result suggests that the recovery of PSI active photocenters is also kinetically controlled by the release of oxidized pc from PSI (i.e., by reaction 3), in agreement with previous suggestions (ref. 18; see also ref. 33 for similar conclusions in the case of Rhodobacter sphaeroides).
Discussion
Previous modeling of electron transfer at the donor side of PSI has suggested that the release of oxidized pc from PSI might represent the kinetic limiting step in electron transfer between cyt b6 f complex and PSI (18). In this article, we have tested this hypothesis in vivo, addressing the consequences of changing the rate constants kon for pc binding and koff for pc+ release on the efficiency of pc prefixation to PSI and on the rate of cyt f oxidation.
Decreasing kon for pc: The 6a+ and W651S Phenotype. Although the in vitro data clearly show that the W651S substitution decreases the PSI affinity for pc (16), this mutation is apparently silent in vivo, where it does not affect the pc exchanges at the PSI donor side or the generation of a pc–PSI complex in the dark. On the other hand, both parameters are largely inhibited by the removal of the PsaF subunit in the 6a+ mutant. This finding suggests that the luminal pc concentration in vivo might be sufficiently high to compensate for the moderate decrease in PSI affinity for pc induced by the W651S mutation, but not the drastic impact on pc binding as observed in the 6a+ strain (16). This interpretation implies that the pc concentration in Chlamydomonas thylakoids is significantly >500 μM, which represents the dissociation constant for pc and PSI electron transfer complex formation in the W651S mutant. This high concentration is likely due to the very strict regulation of the lumen dimension in vivo (see, e.g., ref. 34), which results in optimization of pc concentration.
In light of this very high pc concentration, the existence of a strong binding affinity between PSI and pc in vivo, mediated by both electrostatic and hydrophobic contacts, is somewhat surprising. One possible rationale for maintaining such an elevated affinity could be the necessity of preventing photodamage to the photosynthetic apparatus, which is otherwise observed upon impairment of electron transfer between pc and PSI in vivo. It has been indeed reported that illumination of the PsaF-deficient mutants 3bF and 6a+ or of the E613N strain with saturating light results in an elevated lipid peroxidation and a rapid photoinhibition of PSII (22). In our conditions, sustained illumination of these strains clearly induced a decrease in the rate of cyt f reduction by PSII, as expected in the case of photoinhibition (not shown). The mechanisms underlying the relationship between impairment of electron transfer from pc to f to PSI and occurrence of PSII photoinhibition are poorly understood. Clearly, this light sensitivity cannot be ascribed to overreduction of the PSI electron acceptors in the mutants, because electron flow to NADPH is reduced in both strains (ref. 22 and Fig. 1). Nevertheless, the optimization of pc binding to PSI clearly provides a benefit to the cell in terms of photoprotection. This benefit is possibly the reason for keeping several “parallel” mechanisms, all of which contribute to the overall optimization of the pc–PSI interaction.
Decreasing the koff for pc+: The E613N Phenotype. At variance with the W651S mutant, the E613N mutation has clear consequences on the rate of cyt f oxidation and on the efficiency of pc exchanges at the PSI docking site in vivo. The slowing down of cyt f oxidation rate observed in vivo (a factor of ≈2) is very similar to the estimated decrease of the koff for release of oxidized pc from PSI in the E613N strain (15). This observation suggests that changes in koff parameter have a higher impact on the overall electron transfer between PSI and the cyt b6 f complex than variations of the kon in vivo.
Sommer et al. (15) have proposed that the presence of negatively charged glutamic acid residue in PsaB at position 613 is required for fast release of oxidized pc. According to Drepper and coworkers (18), this reaction step is crucial for optimization of the PSI–cyt b6 f connection. Our data are in line with these conclusions and demonstrate to our knowledge for the first time the validity of these hypotheses in vivo, under physiological conditions.
Electron Transfer Under Single-Turnover and Continuous-Light Conditions. In our experiments, the pc, cyt f, and PSI pools appear to be close to their thermodynamic equilibrium because a saturating flash illumination results in the oxidation of ≈35% of the total amount of cyt f. This value is in good agreement with the value of 0.29, which can be estimated on the basis of the redox midpoints potentials of P700, of free pc, and of cyt f (475 mV, 360 mV, and 330 mV, respectively), and of their relative concentration in the thylakoids [3 pc and 0.6 cyt b6 f/PSI (27, 35)]. The slight difference between measured and calculated values likely reflects the higher redox midpoint potential (Em) of the pc molecule that is bound by PSI. Indeed, a value of 0.35 can be calculated if the same computation is made assuming that two pc molecules have an Em of 360 mV, whereas the Em of the third one is increased by ≈60 mV, as expected in the case of pc binding to PSI via PsaF (18). In agreement with this hypothesis, the amplitude of cyt f oxidation measured is reduced in the PsaF-deficient 6a+ strain (Fig. 2), where pc fixation to PSI is prevented.
On the other hand, a very low apparent equilibrium constant (Kapp) between cyt b6 f and PSI [lower than the one calculated on the basis of the Em value (Keq)] has been observed in vascular plant chloroplasts upon continuous light illumination (36, 37). When tested in continuous light, a low Kapp between PSI and the cyt f was also observed in Chlamydomonas (not shown). This difference between Kapp and Keq has been recently interpreted in terms of the existence of pc diffusion domains, having different stoichiometries of PSI, pc and cyt b6 f complexes. These domains have been tentatively identified as the different chloroplast compartments previously identified by Albertsson (38), namely the grana stacks, the end and margin membranes, and the stroma lamellae. Within this model, the rationale for the discrepancy between Kapp and Keq is the following: Owing to uneven complex stoichiometry, P700 can be oxidized by light in some domains [the ones with a high P700/(cyt f + pc) ratio], well before the cyt f oxidation in other domains, where the P700/(cyt f + pc) ratio is lower. This results in the simultaneous detection of cyt f+ and P700+, when the average redox state of the different domains is measured in the thylakoid membranes, leading to the estimation of a low Kapp. See Lavergne and Joliot (39) for a more detailed discussion in the case of a similar model and Joliot and Joliot (40) for a recent application of this model to interpret the occurrence of cyclic electron flow in plant leaves. Nevertheless, it is of note that diffusion of pc from the distant stroma lamella to the grana core in the light has been demonstrated (41). It is unclear, however, whether the long-distance movement of pc is fast enough to support a sustained electron transfer (42).
To explain the apparent discrepancy between the results obtained under single-turnover flash and continuous light, we propose that the cyt b6 f/pc stoichiometry might be very similar in all of the domains considered in the model described above. On the other hand, the PSI/pc and PSI/cyt b6 f ratios would remain variable, as required by the observation of a low Kapp in continuous light. In this case, no deviations between Kapp and Keq are expected upon excitation by a single-turnover flash. Indeed, the positive charge generated by PSI is rapidly injected into its donor pool, because of the high Em difference between P700 and pc. In this case, redox equilibration occurs only between the pc and cyt f pools, and therefore heterogeneous distribution of the electron carriers can be neglected. On the contrary, in the case of continuous light, partition of multiple charges between P700 and its donor pool takes place. In this case, the heterogeneous PSI/(cyt b6 f + pc) stoichiometry becomes relevant, and deviations between Kapp and Keq are expected, as discussed above.
A corollary of this hypothesis is that a calculation of the pc/cyt f equilibration curve should give identical Kapp and Keq values also in the experiments performed in continuous light. In agreement with this conclusion, we could estimate that the apparent equilibrium constant between pc and cyt f that can be calculated from data of refs. 36 and 37 is very close to the Keq value that is predicted from standard Em values (not shown).
Acknowledgments
Discussions with Drs. Friedel Drepper (University of Freiburg, Freiburg, Germany) and Helmut Kirchhoff (University of Münster, Münster, Germany) are gratefully acknowledged. This work was supported by funds of the University of Pennsylvania (to M.H.) and of the Centre National de la Recherche Scientifique (to G.F.).
Author contributions: G.F. and M.H. designed research; G.F. and F.S. performed research; G.F., F.S., and M.H. analyzed data; and G.F. and M.H. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: cyt, cytochrome; pc, plastocyanin, PS, photosystem; Chl, chlorophyll.
References
- 1.Wood, P. M. (1978) Eur. J. Biochem. 87, 9–19. [DOI] [PubMed] [Google Scholar]
- 2.Ho, K. K. & Krogmann, D. W. (1984) Biochim. Biophys. Acta 766, 310–316. [Google Scholar]
- 3.Merchant, S. & Bogorad, L. (1986) Mol. Cell. Biol. 6, 462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hope, A. B. (1993) Biochim. Biophys. Acta 1143, 1–22. [DOI] [PubMed] [Google Scholar]
- 5.Martinez, S. E., Huang, D., Szczepaniak, A., Cramer, W. A. & Smith, J. L. (1994) Structure 2, 95–105. [DOI] [PubMed] [Google Scholar]
- 6.Soriano, G. M., Ponamarev, M. V., Tae, G. S. & Cramer, W. A. (1996) Biochemistry 35, 14590–14598. [DOI] [PubMed] [Google Scholar]
- 7.Soriano, G. M., Ponamarev, M. V., Piskorowski, R. A. & Cramer, W. A. (1998) Biochemistry 37, 15120–15128. [DOI] [PubMed] [Google Scholar]
- 8.Jordan, P., Fromme, P., Witt, H. T., Klukas, O., Saenger, W. & Krauss, N. (2001) Nature 411, 909–917. [DOI] [PubMed] [Google Scholar]
- 9.Ben-Shem, A., Frolow, F. & Nelson, N. (2003) Nature 426, 630–635. [DOI] [PubMed] [Google Scholar]
- 10.Hippler, M., Reichert, J., Sutter, M., Zak, E., Altschmied, L., Schröer, U., Herrmann, R. G. & Haehnel, W. (1996) EMBO J. 15, 6374–6384. [PMC free article] [PubMed] [Google Scholar]
- 11.Farah, J., Rappaport, F., Choquet, Y., Joliot, P. & Rochaix, J. D. (1995) EMBO J. 14, 4976–4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hippler, M., Drepper, F., Haehnel, W. & Rochaix, J. D. (1998) Proc. Natl. Acad. Sci. USA 95, 7339–7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hippler, M., Drepper, F., Rochaix, J. D. & Muhlenhoff, U. (1999) J. Biol. Chem. 274, 4180–4188. [DOI] [PubMed] [Google Scholar]
- 14.Haldrup, A., Simpson, D. J. & Scheller, H. V. (2000) J. Biol. Chem. 275, 31211–31218. [DOI] [PubMed] [Google Scholar]
- 15.Sommer, F., Drepper, F. & Hippler, M. (2002) J. Biol. Chem. 277, 6573–6581. [DOI] [PubMed] [Google Scholar]
- 16.Sommer, F., Drepper, F., Haehnel, W. & Hippler, M. (2004) J. Biol. Chem. 279, 20009–20017. [DOI] [PubMed] [Google Scholar]
- 17.Sun, J., Xu, W., Hervas, M., Navarro, J. A., Rosa, M. A. & Chitnis, P. R. (1999) J. Biol. Chem. 274, 19048–19054. [DOI] [PubMed] [Google Scholar]
- 18.Drepper, F., Hippler, M., Nitschke, W. & Haehnel, W. (1996) Biochemistry 35, 1282–1295. [DOI] [PubMed] [Google Scholar]
- 19.Harris, E. H. (1989) The Chlamydomonas Sourcebook: A Comprehensive Guide to Biology and Laboratory Use (Academic, San Diego). [DOI] [PubMed]
- 20.Hippler, M., Drepper, F., Farah, J. & Rochaix, J. D. (1997) Biochemistry 36, 6343–6349. [DOI] [PubMed] [Google Scholar]
- 21.Porra, R. J., Thompson, W. A. & Kriedemann, P. E. (1989) Biochim. Biophys. Acta 975, 384–394. [Google Scholar]
- 22.Sommer, F., Hippler, M., Biehler, K., Fischer, N. & Rochaix, J. D. (2003) Plant Cell Environ. 26, 1881–1892. [Google Scholar]
- 23.Joliot, P. & Joliot, A. (2002) Proc. Natl. Acad. Sci. USA 99, 10209–10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Béal, D., Rappaport, F. & Joliot, P. (1999) Rev. Sci. Instrum. 70, 202–207. [Google Scholar]
- 25.Finazzi, G., Furia, A., Barbagallo, R. P. & Forti, G. (1999) Biochim. Biophys. Acta 1413, 117–129. [DOI] [PubMed] [Google Scholar]
- 26.Frank, K. & Trebst, A. (1995) Photochem. Photobiol. 61, 2–9. [DOI] [PubMed] [Google Scholar]
- 27.Delosme, R. (1991) Photosynth. Res. 29, 45–54. [DOI] [PubMed] [Google Scholar]
- 28.Li, Y., Lucas, M. G., Konovalova, T., Abbott, B., MacMillan, F., Petrenko, A., Sivakumar, V., Wang, R., Hastings, G., Gu, F., et al. (2004) Biochemistry 43, 12634–12647. [DOI] [PubMed] [Google Scholar]
- 29.Thurnauer, M. T. & Norris, J. R. (1980) Chem. Phys. Lett. 76, 557–564. [Google Scholar]
- 30.Thurnauer, M. T., Rutherford, A. W. & Norris, J. R. (1982) Biochim. Biophys. Acta 593, 384–399. [Google Scholar]
- 31.Junge, W. & Witt, H. T. (1968) Z. Naturforsch. 24, 1038–1041. [Google Scholar]
- 32.Bennoun, P. (1970) Biochim. Biophys. Acta 216, 357–363. [DOI] [PubMed] [Google Scholar]
- 33.Gerencser, L., Laczko, G. & Maroti, P. (1999) Biochemistry 38, 16866–16875. [DOI] [PubMed] [Google Scholar]
- 34.Cruz, J. A., Salbilla, B. A., Kanazawa, A. & Kramer, D. M. (2001) Plant Physiol. 127, 1167–1179. [PMC free article] [PubMed] [Google Scholar]
- 35.Pierre, Y., Breyton, C., Kramer, D. & Popot, J. L. (1995) J. Biol. Chem. 270, 29342–29349. [DOI] [PubMed] [Google Scholar]
- 36.Joliot, P. & Joliot, A. (1984) Biochim. Biophys. Acta 765, 219–226. [Google Scholar]
- 37.Kirchhoff, H., Schottler, M. A., Maurer, J. & Weis, E. (2004) Biochim. Biophys. Acta 1659, 63–72. [DOI] [PubMed] [Google Scholar]
- 38.Albertsson, P. (2001) Trends Plant Sci. 6, 349–358. [DOI] [PubMed] [Google Scholar]
- 39.Lavergne, J. & Joliot, P. (1991) Trends Biochem. Sci. 16, 129–134. [DOI] [PubMed] [Google Scholar]
- 40.Joliot, P. & Joliot, A. (2005) Proc. Natl. Acad. Sci. USA 102, 4913–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haehnel, W., Ratajczak, R. & Robenek, H. (1989) J. Cell Biol. 108, 1397–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haehnel, W. (1984) Ann. Rev. Plant Physiol. 35, 659–693. [Google Scholar]