

Abstract
Lung cancer is the most common cause of cancer-related deaths globally. Although smoking-related lung cancers continue to account for the majority of diagnoses, smoking rates have been decreasing for several decades. Lung cancer in individuals who have never smoked (LCINS) is estimated to be the fifth most common cause of cancer-related deaths worldwide in 2023, preferentially occurring in women and Asian populations. As smoking rates continue to decline, understanding the aetiology and features of this disease, which necessitate unique diagnostic and treatment paradigms, will be imperative. New data have provided important insights into the molecular and genomic characteristics of LCINS, which are distinct from those of smoking-associated lung cancers and directly affect treatment decisions and outcomes. Herein, we review the emerging data regarding the aetiology and features of LCINS, particularly the genetic and environmental underpinnings of this disease as well as their implications for treatment. In addition, we outline the unique diagnostic and therapeutic paradigms of LCINS and discuss future directions in identifying individuals at high risk of this disease for potential screening efforts.
Introduction
Lung cancer is the leading cause of cancer-related death worldwide (see GLOBOCAN) and in every ethnic group in the USA1. Smoking-related lung cancers account for the majority of lung cancer diagnoses and continue to claim ~100,000 lives in the USA each year1; however, smoking rates have been decreasing for several decades. Although varying widely across the USA, cigarette smoking prevalence among adults reached an all-time low at 11.5% in 2021 (ref. 2), down by more than two-thirds (from ~42%) since the first Surgeon General’s Report linking cigarette smoking to lung cancer and heart disease in 1964 (ref. 3) (Fig. 1a) and is projected to fall further to 7.5% overall by 2065 (ref. 4). As smoking prevalence has declined, some reports indicate that the proportion of lung cancers occurring in individuals who have never smoked (LCINS) has increased, particularly among women and in younger age groups5. In 2023, >20,000 lung cancer-related deaths in the USA were projected to occur in people who have never smoked1, making LCINS the eighth leading cause of cancer-related mortality in the USA; data suggest that LCINS is currently the fifth most common cause of cancer-related deaths worldwide6 (Fig. 1b).
Fig. 1 |. Smoking rates and lung cancer-related deaths.
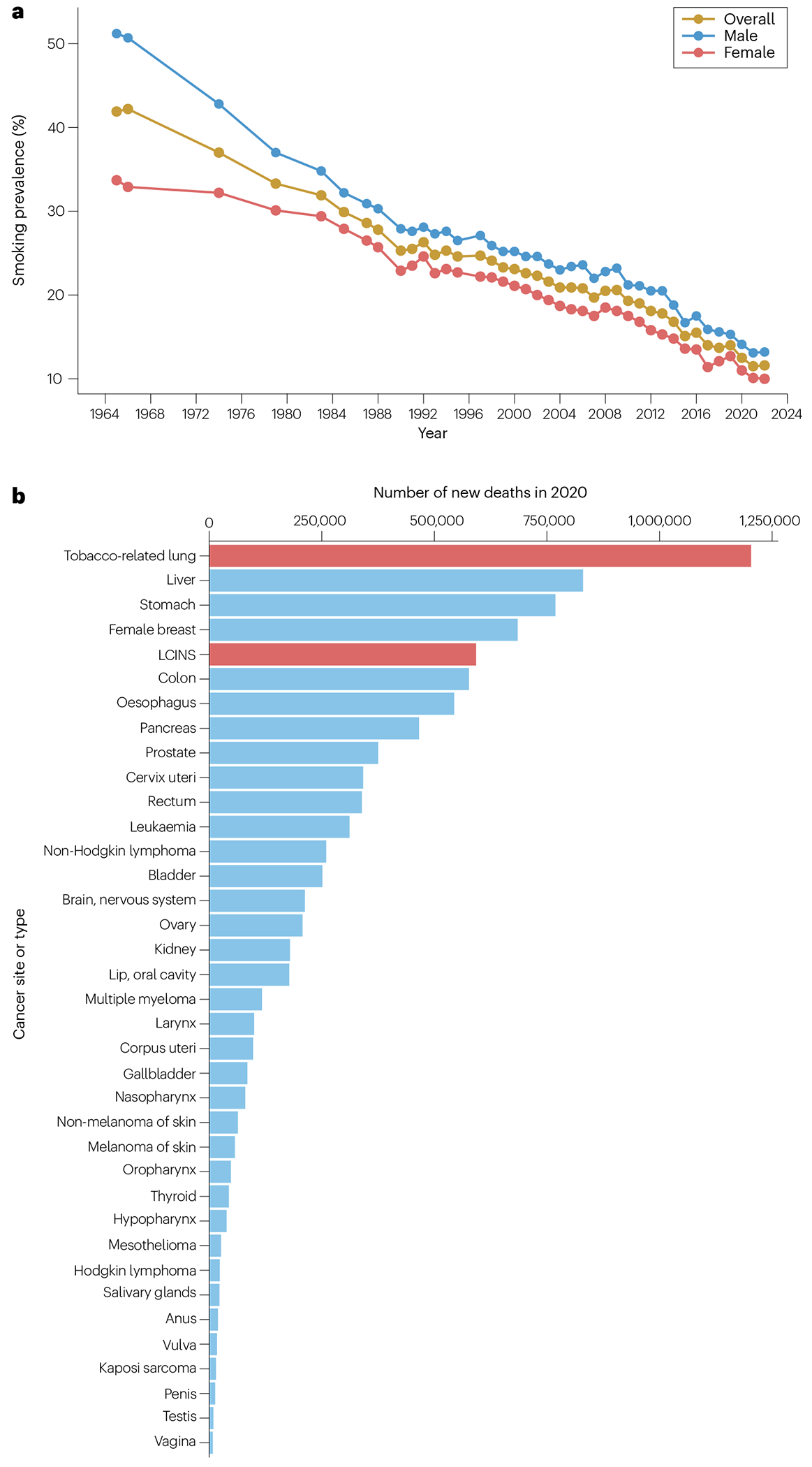
a, Smoking prevalence in the USA from 1965 to 2022 as the percentage of adults aged ≥18 years who reported currently smoking cigarettes, overall and by sex. The data are derived from the National Health Interview Survey (NHIS) 1965–2018 and 2019–2022 (refs. 2,3,385,386) and are not available for all years. b, Estimated number of global deaths attributed to 37 cancer types in 2020 (ref. 6). Smoking-related lung cancer and lung cancer in individuals who have never smoked (LCINS) are shown in red as the first and fifth leading cause of cancer deaths, respectively.
LCINS have histological and epidemiological distinctions from smoking-related lung cancers given that they are almost exclusively adenocarcinomas and most commonly occur in women and individuals of Asian ancestry7,8. Moreover, several studies have revealed that LCINS are also genomically and molecularly distinct from smoking-related lung cancers, highly enriched for targetable oncogenic alterations (such as EGFR mutations or ALK rearrangements as well as less common alterations), and thus often require different diagnostic and therapeutic strategies. Over the past decade, new data have emerged regarding the genetic risk of LCINS, conferred by both common and rare germline variants, as well as environmental risk factors and potential interactions between the two. Conceivably, LCINS could eventually become the most common form of lung cancer, necessitating a thorough understanding of its pathogenesis and risk factors. Herein, we review the epidemiological, clinical and genomic features of LCINS, along with data from studies examining both genetic and environmental risk factors, as well as diagnostic and treatment strategies.
Definitions
In an effort to mitigate the stigmatization of patients with lung cancer, the International Association for the Study of Lung Cancer released a language guide in 2021 that includes replacing the term ‘smoker’ with language such as ‘person who has smoked’, and ‘never-smoker’ with ‘person who has never smoked’9. Definitions of smoking status have varied; however, the Centers for Disease Control and Prevention and others have previously used the term ‘never-smoker’ (or sometimes ‘non-smoker’) to refer to individuals who have smoked <100 cigarettes in their lifetime, and LCINS is defined as lung cancer arising in such individuals. The previously used terms ‘former smoker’ or ‘ex-smoker’ are defined as people who have smoked >100 cigarettes in their lifetime but quit smoking ≥12 months prior to a lung cancer diagnosis. The term ‘long-term former smoker’ has been used to refer to those who have smoked >100 cigarettes in their lifetime but quit smoking ≥15 years prior to a lung cancer diagnosis, and individuals with ‘remote’ smoking histories include those who smoked infrequently and/or socially as adolescents and/or young adults but still smoked >100 cigarettes in their lifetime; both of these categories might share certain characteristics with those who have never smoked. A current smoking status refers to individuals who have smoked >100 cigarettes in their lifetime and who still report smoking every day or some days. Nevertheless, data from several studies suggest that smoking history should be quantified as a continuous rather than discrete variable, supported by findings such as the likelihood of a lung adenocarcinoma (LUAD) harbouring an EGFR mutation decreasing as number of pack-years increases10. As discussed below, the discovery of tumour mutational signatures corresponding to tobacco smoking might eventually help to delineate what constitutes a clinically significant level of exposure from a genomic perspective.
Epidemiology
Approximately two-thirds of LCINS cases occur in women, making women who have not smoked more than twice as likely to develop lung cancer than men who have not smoked5,11. The proportion of lung cancers attributable to tobacco smoking varies across countries, at >80% in men and women in the USA and the UK12–14, and 57.5% in men and 13% in women in China15. A never-smoking status is more frequent among female patients in Asia than those in other regions and, interestingly, >55% of female Asian patients and >30% of Hispanic female patients diagnosed with non-small-cell lung cancer (NSCLC) in the USA have never smoked16. However, even when compared specifically with non-smoking women in other geographical regions, lung cancer incidence rates have been observed to be higher among women in East Asia17, suggesting that genetic and/or environmental factors other than tobacco smoke exposure contribute to the global variation in the prevalence of LCINS.
The average age at LCINS diagnosis is similar to that reported for smoking-related lung cancers5 (median age at diagnosis of 67 years versus 65 years; P = 0.1)7, although younger patients with lung cancer are more likely to have never smoked. In a study including 121 patients <40 years of age at diagnosis and with a documented smoking history, 73% had never smoked; 90% of these patients had LUAD, >80% of which harboured a targetable oncogenic alteration18. Patients with ALK-rearranged NSCLC, which mostly occurs in individuals who have never smoked, are younger on average at diagnosis (median age at diagnosis 50–52 years)19–22, as are patients with other LUADs harbouring oncogenic fusions, for example, involving RET23,24, ROS1 (refs. 22,25–27) or NTRK1–3 (ref. 28).
In economically developed countries, smoking prevalence has decreased among all age groups (in the USA, most steeply amongst those <30 years old in the USA), reflected in an overall decreasing incidence of lung cancer1,29. Lung cancer incidence is decreasing twice as fast in men than in women, and perhaps four times as fast, based on an analysis of United States Cancer Statistics data30 examining annual percentage change in NSCLC incidence31,32 over 2001–2019 (J.L. et al., unpublished data), a difference that is only partially explained by differences in smoking trends33–35. Notably, the incidence of lung cancer is now higher in women than in men in both Hispanic and non-Hispanic white individuals born during or after the mid-1960s whereas, prior to this time, the incidence was higher in men than in women35. This pattern is also expected to reverse (that is, lung cancer incidence will become higher in women than in men) in the remaining birth cohorts by 2045 if current smoking trends continue4. Trends in NSCLC histology also reflect the decreasing smoking prevalence, with a relative increase in LUAD and decrease in lung squamous cell carcinoma (LSCC) over time36,37.
Precise national and global trends in LCINS epidemiology are unavailable owing to a lack of individual-level data on smoking status, which historically has not been collected in cancer registries or on death certificates. However, a retrospective study including >10,000 patients from three independent cancer centres revealed that the proportion of LCINS relative to total NSCLC diagnoses increased from 8.0% in 1990–1995 to 14.9% in 2011–2013 (P < 0.001)5. This trend was independent of sex, disease stage at diagnosis and ethnicity, and no statistically significant increase in the proportion of LCINS was observed among small-cell lung carcinoma (SCLC) or LSCC diagnoses5. However, additional and contemporary data are needed to determine true trends in LCINS epidemiology.
Features of LCINS
In addition to histological characteristics shared among LCINS, genomic and molecular features have been more recently characterized in LCINS as compared to lung cancers occurring in patients with a history of smoking. The findings underscore the distinct biology of LCINS, with important diagnostic and treatment implications (Table 1).
Table 1 |.
Somatic features reported for LCINS compared to tobacco-related lung cancers
| Feature | LCINS | Tobacco-related lung cancer |
|---|---|---|
| Histology | NSCLC; mostly LUAD1,2 | LUAD, LSCC or SCLC; strongest association with LSCC and SCLC334 |
| Targetable driver alterations | Present in 78–92% of LUADs3–5 | Present in 49.5% of LUADs (mainly KRAS mutations)7 |
| PD-L1 expression level | Low36–8,a | Highest with current smoking; PD-L1 staining intensity is positively correlated with pack-years of smoking history136 |
| TMB | 0–3mut/Mb (refs. 3–5,9–13) (median 1.1 mut/Mb) | Up to tenfold higher than in LCINS131, with a dose–response relationship between pack-years of smoking history and TMB132 |
| Genomic signatures | Devoid of tobacco-associated mutational signatures, including LCINS with SHS exposure3–5 | SBS signature 4b (mainly C>A transversions), attributable to misrepair of DNA damage40,135; less strongly associated with indel-based signature 3c and doublet-base substitution signature 2 (ref. 335); total number of SBS nearly fivefold higher in smoking-related versus non-smoking-related LUADs (mean 12.09 vs 2.65; P = 2.7 × 10−13); total number of indels higher in smoking-related versus non-smoking-related LUADs (mean 0.39 vs 0.14; P = 7.9 × 10−14)40 |
LCINS, Lung cancers in individuals who have never smoked; LUAD, lung adenocarcinoma; mut/Mb, mutations per megabase; NSCLC, non-small-cell lung cancer; SBS, single-base substitution; LSCC, lung squamous cell carcinoma; SCLC, small-cell lung cancer; SHS, secondhand smoke; TMB, tumour mutational burden.
Moderate to high levels of PD-L1 expression have been observed in LCINS with MET exon 14 mutations14,15.
Reliably detectable with targeted panel-based next-generation sequencing assays.
Histology
As noted above, LCINS are near-exclusively LUADs5,16 and largely driven by oncogenic alterations in key pro-survival signalling pathways. Although LUAD accounts for a substantial number of smoking-related lung cancers, both LSCC and SCLC are more strongly associated with smoking and typically arise in the larger, central airways that are more readily accessible to tobacco smoke. An estimated 6–8% of LSCCs and SCLCs occur in patients who have never smoked16, and the age-adjusted odds ratios (OR) for LUAD, LSCC and SCLC development in men who currently smoke are 21.9, 103.5 and 111.3, respectively38–40. An analysis of 11 cases of SCLC diagnosed in individuals with a history of former light smoking or never smoking revealed that most tumours (8 of 11; 73%) were of mixed histology or non-pulmonary origin41. Furthermore, driver mutations were detected in EGFR, NRAS, KRAS, BRCA1 and ATM, and one tumour had a TMPRSS2–ERG fusion41. These findings suggest that SCLC arising in those who have never smoked constitutes a distinct disease entity, requiring different therapeutic approaches than smoking-related SCLC.
LUADs occurring in the presence or absence of tobacco smoke exposure are largely similar in histological appearance and typically cannot be distinguished based on histological subtype or features when adjusted for pathological stage; differences in adverse prognostic features, such as lymphovascular invasion, visceral pleural invasion and spread of tumour through airways, have not been reported. However, cigarette smoking is strongly associated with the presence of a solid component within the tumour (prevalence of 53% versus 20% in stage I LUADs from individuals who had never smoked; multivariate HR 3.32, 95% CI 1.78–6.19; P < 0.001), and EGFR-mutant LUADs less frequently contain solid components than EGFR-wild-type LUADs (17% versus 51%; P = 0.033)42. Most studies have not characterized histology beyond LUAD and LSCC, although one study of 320 stage I LUADs revealed that those in patients with a never-smoking status are more frequently bronchioalveolar carcinoma (85% versus 58% in ever-smoking patients; P < 0.001) and more commonly have papillary components (81% versus 68%; P = 0.01)42. No histological differences in precursor lesions between never-smoking and ever-smoking patients have been reported, such that adenocarcinoma in situ in a never-smoking patient is indistinguishable from that occurring within the context of a field effect in a patient who has smoked; however, some evidence indicates that adenocarcinoma in situ and minimally invasive adenocarcinoma in patients with a history of smoking tend to be larger than those in patients who have never smoked43. Features including a signet ring cell morphology, cribriform formation and solid or acinar growth patterns have been associated with ALK-rearranged LUAD44–49; similar features have been identified in LUADs harbouring ROS1 or RET fusions50.
Imaging characteristics
Several studies have examined various radiographical features of molecular subtypes of LUAD and, in general, radiographical differences correlate with the molecular driver and not with smoking status. Fusion-positive LUADs often have striking solid components, corresponding to areas of invasiveness on histology51–53. In one study, ALK-rearranged LUADs tended to be centrally located, associated with large pleural effusions and lacking a pleural tail54. No characteristic imaging findings have been consistently identified in KRAS-mutant or EGFR-mutant LUADs, and conclusions have been confounded by differences in image acquisition, cancer stage and geographical location (that is, East Asian versus non-Asian cohorts)55–57. However, characteristic volumetric tumour response dynamics have been observed following treatment with EGFR tyrosine kinase inhibitors (TKIs), consisting of an initial marked decrease in tumour burden followed by a slower decrease until the point of maximal response58–62. The magnitude of the initial, and thus the maximal, response is predictive of survival duration in patients receiving EGFR TKIs58, and slower rates of tumour regrowth following maximal response are associated with longer overall survival (OS)63. Although not clinically useful at this time, radiomic machine learning algorithms using multimodal imaging features might eventually aid in determining the molecular driver present in an individual NSCLC57,64,65.
Patterns of metastatic disease
Certain patterns of metastatic spread also correlate with the molecular driver. Among patients with NSCLCs harbouring EGFR mutations or ALK rearrangements, 50–60% present with or eventually develop brain metastases, compared with 16–20% of unselected patients with NSCLC66–74. ROS1-rearranged LUADs are associated with roughly half this frequency of brain metastasis (~36%), although estimates vary widely owing to the relative rarity of this disease subtype22,25,75. With the advent of osimertinib – a third-generation, brain-penetrant EGFR TKI – the risk of developing brain metastasis while on treatment has been observed to be three times less than with earlier-generation TKIs76.
Oncogenic driver alterations
Constitutive activation of a growing number of oncogenes through mutation, rearrangement and/or amplification accounts for 78–92% of LCINS versus 49.5% of smoking-related LUADs7,40 (Fig. 2). The most common genetic alterations include mutations in EGFR, KRAS, HER2, MET and BRAF, rearrangements involving ALK, ROS1, RET and NTRK1–3, and MET amplification, and prevalences of these alterations vary according to age and genetic ancestry (for example, East Asian versus non-East Asian)77 as well as according to heterogeneity of the molecular testing assays used. Somatic sequencing studies include those at the whole-exome7 (Fig. 2a) and whole-genome78 (Fig. 2b) levels, as well as panel-based next-generation sequencing (NGS) data derived from the AACR Project GENIE cohort, which is the largest collective somatic sequencing study in patients with cancer and comprises clinical data from multiple institutions. A subset of the AACR Project GENIE cohort (Biopharma Collaborative NSCLC cohort79, n = 1,846) has been annotated for outcomes and clinicopathological variables, including smoking status, providing insights on the prevalence of oncogenic alterations in LUADs from patients who have never smoked (Fig. 2c). Rare molecular subtypes of lung cancer, most of which are more commonly found in LCINS, have been recently reviewed in this journal80.
Fig. 2 |. Prevalence of oncogenic somatic driver alterations in LCINS.
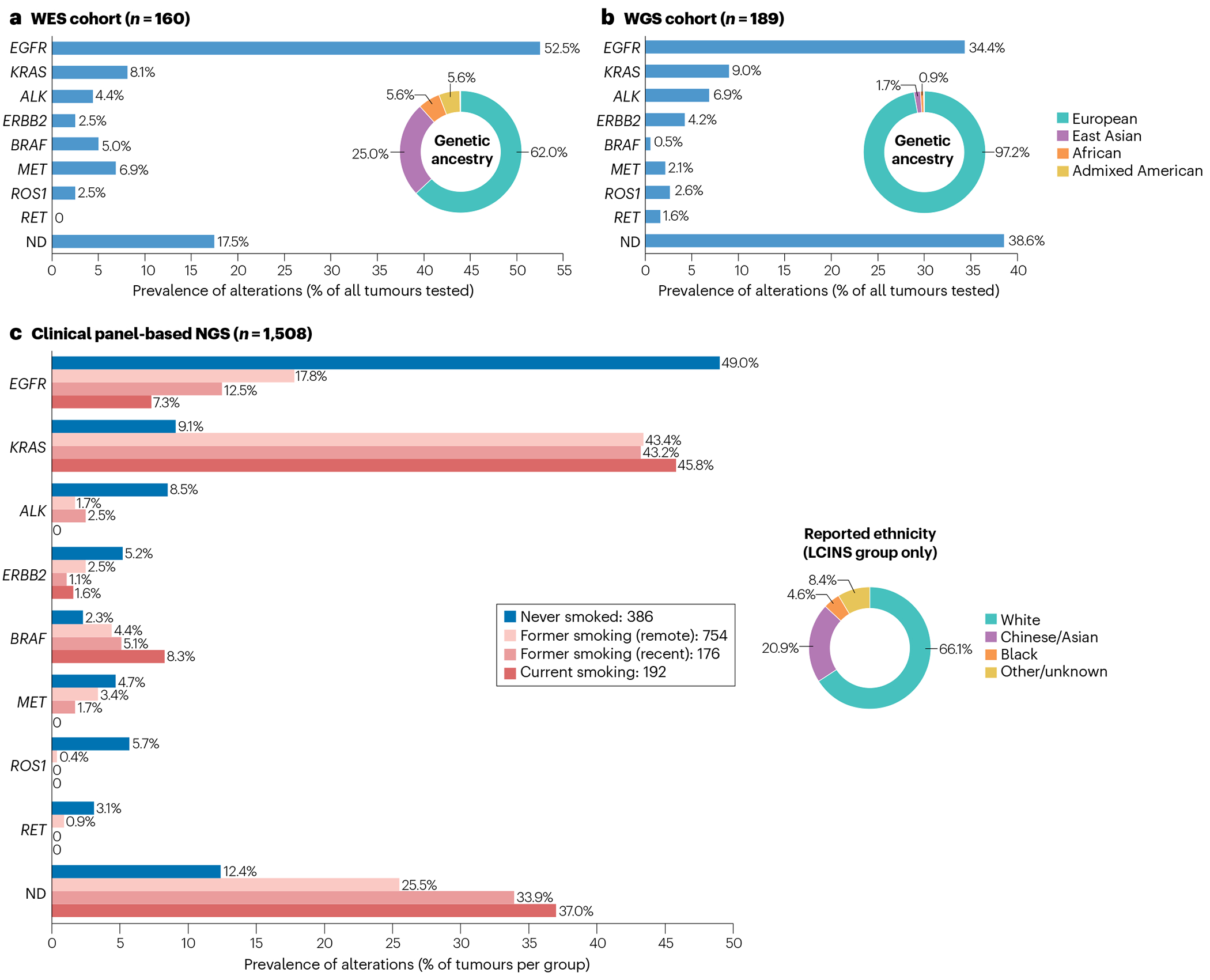
Data from somatic profiling studies of lung adenocarcinoma (LUAD) in individuals who have never smoked (LCINS). a, Whole-exome sequencing (WES) and RNA sequencing study7 of 160 samples, at an average depth of 20–30× for WES. The prevalence of each somatic driver alteration was calculated as a composite average using three individual cohorts included in the study, with values weighted proportionally to the number of samples in each cohort. Genetic ancestry was inferred by the authors based on germline sequencing data from matched peripheral blood. b, Whole-genome sequencing (WGS) study78 of 189 samples at an average depth of 85×. Genetic ancestry was inferred by the authors based on germline sequencing data from matched peripheral blood. c, Clinical panel-based next-generation sequencing (NGS) data from 1,508 LUADs, collected as part of the non-small-cell lung cancer (NSCLC) cohort of the AACR Project GENIE Biopharma Collaborative79, across four large academic cancer cancers in North America (Dana-Farber Cancer Institute, Memorial Sloan Kettering Cancer Center, Vanderbilt-Ingram Cancer Center, and Princess Margaret Cancer Centre-University Health Network). Clinical data available for this cohort include tumour histology, patient self-reported primary ethnicity and cigarette use at time of diagnosis: never smoked; quit smoking >1 year prior to diagnosis (‘former smoking (remote)’); quit smoking <1 year prior to diagnosis (‘former smoking (recent)’); and current smoking. Self-reported primary ethnicity is shown for samples with never-smoking status only. Included for analysis were somatic mutations (EGFR, KRAS, ERBB2, BRAF and MET) and gene fusions (ALK, ROS1 and RET) annotated as ‘putative drivers’ based on prior knowledge (OncoKB387,388) and statistical recurrence (Cancer Hotspots389,390). The GENIE Biopharma Collaborative Public release is available for download (https://www.synapse.org/#!Synapse:syn27056172/wiki/616601) and can be visualized and analysed using the cBioPortal interface (https://genie.cbioportal.org/study/summary?id=nsclc_public_genie_bpc). Note, wide ranges in the prevalence of particular alterations across studies probably reflect differences in cohort ascertainment (for example, 25.0% versus only 1.7% of patients were of East Asian ancestry in the WES and WGS studies, respectively) and heterogeneity of testing assays used. ND, none of the above alterations detected.
EGFR.
EGFR-mutant LUAD constitutes the largest proportion of LCINS, with rates as high as 60–74% in non-smoking East Asian women with lung cancer77. RNA sequencing (RNA-seq) and whole-exome sequencing (WES) data from a large cohort of never-smoking patients with LUAD (n = 160) found EGFR to be mutated in up to 52.5% of participants (versus ~10.4% of smoking-related LUADs)7. Canonical mutations in exons 19 and 21 (that is, exon 19 deletions and the L858R point mutation, respectively) account for the vast majority of EGFR alterations in LCINS (>85%), without a preponderance of one alteration relative to the other7, although the L858R mutation and exon 19 deletions seem to occur more frequently in older and younger patients, respectively18,81. EGFR mutations occur in a small fraction of smoking-related lung cancers, with the likelihood of EGFR mutation decreasing with increasing pack-years10. Regardless of pack-years smoked, the likelihood that a lung cancer will harbour an EGFR mutation increases with the number of smoke-free years prior to diagnosis, particularly in those who have stopped smoking for >25 years10,82.
Uncommon sensitizing and non-sensitizing EGFR mutations might be more likely to occur in smoking-related compared with non-smoking-related lung cancers83,84; however, ~60% of patients with uncommon EGFR exon 20 insertions have never smoked and most are women, in contrast to those with EGFR-wild-type cancers (but similar to those with classical sensitizing EGFR mutations)84–87. In a study of 102 NSCLC samples harbouring uncommon EGFR mutations, 85% of those with exon 18 alterations and 43% of those with exon 20 alterations occurred in patients with a history of smoking84. Rarely, oncogenic EGFR fusions occur, most commonly with RAD51 as the fusion partner, and reports of EGFR–RAD51 fusions in LUAD most often involve patients with a history of never or light (<5 pack-year) smoking, who are mostly young (<40 years of age)88–90. These fusions are targetable with several generations of EGFR TKIs and sustained clinical responses have been reported88–90.
ALK.
Rearrangement of ALK is the second most common oncogenic driver alteration in LCINS and is found in up to 14% of these cancers91, although this prevalence varies widely according to study design and population as well as between different age groups7,78,92. Overall, ALK rearrangements are found in ~5% of patients with NSCLC, with a roughly equal incidence in Asian and European populations93. Most patients with ALK rearrangements (70–80%) have never smoked19, and up to 23% of EGFR-wild-type NSCLCs occurring in individuals <50 years of age with a history of never or light smoking harbour ALK rearrangements94. Similar to other molecular subtypes of LCINS, ALK-rearranged lung cancers are almost exclusively adenocarcinomas, although very rare cases of ALK-rearranged LSCC have been reported95,96.
Other oncogenic fusions.
Novel gene fusions have been identified as oncogenic drivers in NSCLC using DNA-based and/or RNA-based NGS80. These include a variety of fusions involving ROS1, RET, NTRK1–3, FGFR1, FGFR3 or NRG1 as well as more recently identified CLIP1–LTK fusions97,98. ROS1 and RET fusions each occur in approximately 1–4% of all LUADs, with the prevalence of other fusions estimated at <1%; detection sensitivities might be increased when using both DNA-based and RNA-based sequencing panels, particularly for ROS1 and RET fusions80. Many of these fusions are enriched in LCINS as well as in younger patients and are targetable with various FDA-approved TKIs97,99,100.
KRAS.
KRAS mutations comprise the largest molecularly defined subset of LUAD owing to the large percentage of smoking-related LUADs harbouring mutant KRAS. Activating point mutations in KRAS most commonly occur at codon 12 (G12C, G12D, G12V and G12A) and less commonly at codons 13, 10 or 61. KRAS mutations are found in only 5–15% of lung cancers occurring in white never-smoking patients7,82,91,101, with the G12D variant being most common in this context (accounting for ~56% of KRAS mutations)82. By contrast, KRAS mutations are present in up to 47% of smoking-related LUADs7, in which the G12C variant predominates (accounting for ~41% of these mutations)82. Indeed, transversions leading to the KRAS G12C, G12V, G12A and G12R variants are part of a mutational signature associated with tobacco carcinogens82. Nevertheless, smoking has the potential to cause transition mutations, albeit at a relatively lower frequency, thus accounting for the limited occurrence of mutations such as KRAS G12D in smoking-related LUADs. Furthermore, transversion mutations are also associated with non-tobacco exposures (for example, reactive oxygen species102 and prior chemotherapy103), which might explain the occasional observation of KRAS G12C mutations in LCINS. A study comparing KRAS G12D-mutant versus non-G12D-mutant NSCLC revealed that KRASG12D-mutant tumours have lower PD-L1 expression, a lower tumour mutational burden (TMB), and lower intratumoural and total numbers of CD8+PD-1+ T cells. In keeping with these findings, KRASG12D-mutant disease was associated with a worse objective response rate (ORR; 15.8% versus 28.4%; P = 0.03), progression-free survival (HR 1.51, 95% CI 1.44–2.00; P = 0.003) and OS (HR 1.45, 95% CI 1.05–1.99; P = 0.02) when treated with anti-PD-(L)1 antibody monotherapy104. Moreover, stratification of KRASG12D-mutant tumours by smoking status revealed that PD-L1 expression and TMB were markedly lower in those with a history of light or never smoking (<10 pack-years), and these patients (n = 12) had an ORR of 0% with single-agent PD-(L)1 blockade.
MET alterations.
MET alterations are present in a small subset of NSCLCs and consist of mutations resulting in MET exon 14 skipping (METex14; estimated prevalence of 1–4%)80,105–108 and/or MET amplifications (prevalence of 1–6%, varying based on MET copy number)109–111. The percentage of patients with METex14-mutant NSCLC who have never smoked varies across studies (36–64%)108,112. One study found that METex14 mutations are significantly more likely to be identified in those who have never smoked compared to KRAS mutations (P < 0.001) but are more likely to be associated with a smoking history than EGFR mutations (P = 0.03)108. Patients with METex14-mutant NSCLC tend to be older at diagnosis (median age >70 years)108,112, whereas patients with MET amplifications are slightly younger (median age at diagnosis 60–64 years) and less often have a history of never smoking (7–34%)112–114. MET amplification also occurs as a mechanism of resistance to therapies targeting EGFR and ALK115–117. A high level of MET amplification (MET-to-CEP7 ratio of ≥5 on fluorescence in situ hybridization (FISH) or a ≥5–10-fold increase in MET copy number detected via NGS) has been used to distinguish tumours that are more likely to be MET-driven, based on the absence of co-occurring oncogenic drivers and response to MET-directed TKIs (highest in tumours with MET copy number ≥10)112,114,118; high-level MET amplification is considered an emerging biomarker in the National Comprehensive Cancer Network (NCCN) Guidelines for NSCLC. Case reports have identified the presence of MET fusions (specifically KIF5B–MET and STARD3NL–MET) in LUADs from patients with a history of never or light smoking, which were targetable with crizotinib (a TKI with activity against MET)119–121.
HER2.
Activating mutations in ERBB2 (also known as HER2) occur in 1–3% of NSCLCs and up to 5% of LUADs122–124, most commonly in patients who have never smoked and in women124,125. These mutations are typically small in-frame insertions in exon 20 (residues 770 to 783, most commonly the A775_G776insYVMA insertion or duplication) that result in constitutive HER2 kinase activity. Point mutations in the extracellular domain, transmembrane domain or kinase domain of HER2 have also been identified (for example, G660D, R678Q, E693K and Q709L)126,127. Notably, germline HER2G660D mutations have been identified in patients with familial lung cancer127. The relevance of HER2 amplification and/or overexpression in NSCLC is less clear, and efforts to target HER2 in patients with NSCLC have been focused mostly on those harbouring activating HER2 mutations, approximately 54–58% of which occur in patients who have never smoked128,129.
Unknown drivers.
Large-scale genomic analyses of LCINS show that a small percentage of these tumours have no detectable oncogenic driver alterations (Fig. 2). In one of the largest aggregated studies of LUADs from never-smoking patients7, samples deemed oncogene negative by WES had a lower mean tumour cellularity than those in which driver alterations were readily identified. Furthermore, among 13 tumours deemed oncogene negative based on standard-coverage WES, two were found to harbour METex14 mutations after additional deep WES (~400×)7. Exploratory WES and/or whole-genome sequencing (WGS) might uncover new variants in known driver oncogenes in regions not covered in current NGS panels, particularly in the case of fusions. Additionally, current variants of uncertain significance might be reclassified as oncogenic drivers (for example, the EGFR exon 18–25 kinase domain duplication130 and kinase domain A955R7 variants identified in 2021) in this population with a high pre-test probability of driver alterations. Moreover, ongoing genomic, epigenomic and proteomic analyses of LCINS are likely to elucidate new drivers and/or therapeutic targets.
Tumour mutational burden
LCINS generally have a markedly lower TMB (measured as non-synonymous mutations per megabase (mut/Mb)) in coding and non-coding regions compared to smoking-related lung cancers, with early studies suggesting as much as a tenfold difference131 and a dose–response relationship between TMB and pack-years132 (Table 1). Genomic profiling of >15,000 NSCLC samples has also revealed that KRAS and BRAF driver mutations, which are more commonly associated with a history of smoking, are associated with substantially higher TMB when compared to alterations in EGFR, ALK or ROS1 (refs. 133,134). Responses to immune-checkpoint inhibitors (ICIs) in patients with NSCLC correlate with a high TMB as well as with a high number of transversions as part of a smoking-related mutational signature135. Indeed, the lack of ICI response associated with LCINS, and with NSCLCs harbouring alterations in EGFR or ALK more generally, is thought to partly reflect their lower TMB.
PD-L1 expression
PD-L1 expression is typically low or absent in LCINS7,136 as well as in NSCLCs with non-KRAS oncogenic drivers94,137. Furthermore, PD-L1 positivity among KRAS-mutant lung cancers is lowest in patients who have never smoked, higher in those who formerly smoked and highest in those who currently smoke, with the intensity of staining positively correlating with pack-years of smoking history136. An exception is METex14-altered NSCLC, in which moderate to high levels of PD-L1 expression have been observed, although with a median TMB still substantially lower than that of unselected NSCLCs138. ICIs have been associated with low ORRs in patients with METex14-altered NSCLC (17–36% with ICI monotherapy139), and responses do not seem to correlate with PD-L1 expression or TMB138. ALK-rearranged and ROS1-rearranged NSCLCs are more frequently PD-L1 positive compared with EGFR-mutant NSCLCs94,137,140–142 (70.1% and 72.7%, respectively, versus 50.3% in those with classical EGFR mutations)142; however, PD-L1 positivity in these tumours is thought to reflect differential intrinsic oncogene-driven activation of downstream signalling effectors that transcriptionally upregulate PD-L1 expression (such as STAT3 and HIF1α)143,144 rather than reflecting true T cell-mediated immunogenicity through IFNγ signalling that correlates with a response to ICIs.
Genomic mutational signatures
Analysis of somatic alterations in lung cancers has enabled the identification of genomic mutational signatures that can emerge in tissues directly exposed to tobacco smoke. An analysis of WES or WGS data from 5,243 cancers of various types often associated with smoking demonstrated an increased burden of somatic mutations and several distinct mutational signatures in tumours from patients with a history of tobacco smoking, with total base substitutions nearly fivefold higher in LUADs from such patients compared with LUADs from patients who have never smoked40. The smoking-associated single-base substitution signature 4, or simply ‘signature 4’, consists mainly of C>A transversions with lesser contributions from other base substitutions40 and is very similar to the mutational signature that results from exposing cells to benzo[a]pyrene145, a carcinogen found in tobacco smoke. This signature can also be found in cells derived from the non-malignant bronchial epithelium in individuals without cancer but with a former or current smoking status and is not present in non-malignant and tumour tissues from people who have never smoked146. Interestingly, most lung cancers occurring in patients with reported passive exposure to tobacco smoke also do not contain these signatures7,78.
WES and WGS profiling of LCINS
Two large-scale genomic studies of LCINS were reported in 2021 (refs. 7,78). One study was a WGS analysis of 232 LCINS from patients of mostly European ancestry (97.4% European and 1.7% East Asian by inferred genetic ancestry) and described three different tumour subtypes according to somatic copy number alterations78. Approximately 60% of samples had alterations in EGFR, KRAS, ALK, MET, HER2, RET or ROS1. As expected, median TMB was sevenfold less than that of smoking-related lung cancers at 1.1 mut/Mb, and no tobacco smoking signatures were observed, even in cases with reported passive smoking exposure78. The second study was the previously discussed WES and RNA-seq analysis of 160 tumour and matched non-malignant tissue samples from never-smoking patients with LUAD, with a substantially higher percentage of East Asian patients (62% European and 25% East Asian by inferred genetic ancestry), and including 40 and 36 samples from The Cancer Genome Atlas (TCGA) and Clinical Proteomic Tumour Analysis Consortium cohorts, respectively7. As noted above, 78–92% of these LCINS harboured clinically actionable driver alterations, compared with 49.5% of smoking-related LUADs (P < 0.001). Only 6% of LCINS contained a smoking-related mutational signature potentially indicative of passive exposure to tobacco smoke, and the median TMB ranged from 1.25 to 2.93 mut/Mb across the internal and external (TCGA and Clinical Proteomic Tumour Analysis Consortium) cohorts. The immune landscape of these LUADs was examined using RNA-seq and consensus clustering of immune and stromal cell type markers. On the basis of PD-L1 and immune cell marker expression, three clusters were identified as relatively ‘immune cold’, ‘immune hot’ or intermediate tumours, with each cluster having a similar TMB and frequency of EGFR and KRAS mutations – suggesting the potential to identify subsets of LCINS that might be more likely to respond to immunotherapy.
Risk factors for LCINS
Genetic risk
Of the 20,000–40,000 LCINS diagnosed in the USA each year, second-hand smoke (SHS) and radon exposure are estimated to account for approximately 3,500 (ref. 13) and 2,900 cases147, respectively. In the remainder, few consistent environmental associations can be found, posing the question of underlying genetic predisposition. Overall lung cancer heritability has been estimated at 18%148 and might be even greater in those who have not smoked. Several large-scale genome-wide association studies (GWAS) have investigated common polymorphisms associated with lung cancer risk, mainly for smoking-associated cancers. Collectively, >50 loci mediating a small to moderate amount of lung cancer risk have been identified149. Additionally, data on rare germline pathogenic variants, for example, in DNA damage repair or tumour-suppressor genes, are emerging from somatic mutation profiling studies (WES and/or WGS analyses) that use matched non-malignant tissues for comparison150. The implications of germline variants for lung cancer therapy and familial genetic screening are largely unexplored. Indeed, a study testing a panel of 76–88 cancer predisposition genes to evaluate the prevalence of therapeutically actionable germline variants in almost 12,000 patients across >50 different cancer types did not include lung cancer151.
Family history of cancer can be used as a simplified surrogate for inherited susceptibility, particularly in the context of LCINS given that family members do not always share a history of tobacco smoking. This surrogacy is especially robust for rare, monogenic variants with large effect sizes, which are more likely to exhibit Mendelian patterns of inheritance within families than common variants with individually small effect sizes. A systematic review published in 2005 found that having a first-degree relative with lung cancer was associated with a twofold increased lung cancer risk; this was even greater when the first-degree relative was diagnosed at a young age and/or in individuals with multiple affected family members152. A subsequent study of epidemiological risk factors in people who had never smoked revealed that a family history of any cancer diagnosed before 50 years of age in a first-degree relative was a significant predictor of increased lung cancer risk (OR 1.87, 95% CI 1.13–3.10)153. Other proxies for genetic predisposition to lung cancer might include a lack of lifetime exposure to tobacco smoke and a young age at diagnosis, particularly for LCINS (which predominate lung cancer diagnoses among individuals <40 years of age)16,18. Nevertheless, such patients might not have a family history of lung cancer owing to rare pathogenic variants with incomplete penetrance or a polygenic risk architecture that obscures an inheritance pattern.
Insights from GWAS.
GWAS and candidate gene studies conducted over the past decade have identified common genetic variants, typically defined as single-nucleotide polymorphisms (SNPs) with a minor allele frequency of ≥1–5%, that are associated with lung cancer149. Common variant heritability is defined as the proportion of heritability that can be explained by common SNPs and has been estimated on an observed scale at 8.3% for lung cancer and 7.1% after removing genomic loci known to be associated with smoking behaviour154. Many studies using polygenic risk scores (PRS) to further estimate this attributable risk are under way. The GWAS have been focused mainly on smoking-related lung cancer (particularly a susceptibility locus at 15q25, corresponding to a cluster of nicotinic acetylcholine receptor subunit genes that mediate nicotine metabolism and smoking behaviour)155–158, with several additional studies of LUAD in non-smoking Asian women. Reported susceptibility loci generally have a low to moderate effect size (OR 1–2) and, for many loci, suspected causal genes have been identified. To avoid spurious associations owing to population stratification (that is, systematic differences in allele frequencies between subpopulations), GWAS are often restricted to individuals of a single ethnicity and/or ancestry. For this reason and because the landscape of NSCLC differs considerably between European and East Asian populations159, most GWAS of lung cancer have considered these populations separately. A comprehensive review of all GWAS in lung cancer is beyond the scope of this article. Instead, key GWAS that have identified loci contributing to the risk of LCINS are summarized (Table 2).
Table 2 |.
Key genome-wide association studies identifying LCINS risk loci, stratified by population ancestry
| Study | Study cohorts: sample size (cases/controls) | Chromosomal region | Reference SNP cluster ID (rsID) | Associated genetic loci | OR (discovery; 95% CI) |
|---|---|---|---|---|---|
| European | |||||
| Li et al. (2010)160 | Mayo Clinic: 377/377 MDACC: 328/407 Harvard University: 92/161 UCLA: 91/439 |
13q31.3 | rs2352028 | GPC5 | 1.46 (1.26–1.70; P = 5.94 × 10−6) |
| Wang et al. (2010)338,a | Candidate gene study: 259/553 | 5p15.33 | rs4975616 | CLPTM1L–TERT | 0.69 (0.55–0.85; P = 7.95 × 10−4) |
| Sptiz et al. (2011)252,a | Pathway-based (inflammatory) association study: 451/508 | 12q13.13 | rs12809597 | ACVR1B | 0.72 (0.59–0.88; P = 0.0012) |
| Hung et al. (2019)339 | ILCCO: 3,636/6,295 | 5p15.33 | rs380286; rs31490; rs4975616 | CLPTM1L–TERT | rs380286: 0.77 (0.72–0.82; P=5.31×10−16) rs31490: 0.77 (0.72–0.82; P=4.32×10−16) rs4975616: 0.78 (0.73–0.83; P=1.04×10−14) |
| East Asian | |||||
| Hsiung et al. (2010)162,b | GELAC (Han Chinese): 584/585 GELAC (replication set): 610/560 CAMSCH: 287/287 SNU: 259/293 SWHS: 209/213 WHLCS: 207/207 KNUH: 121/119 KUMC: 95/87 GEL-S: 194/546 NJLCS: 203/203 |
5p15.33 | rs2736100 | CLPTM1L–TERT | 1.54 (1.41–1.68; P=2.60×10−20); 1.62 (1.40–1.87; P=8.51×10−11) when heterozygous; 2.54 (1.95–2.83; P=3.05×10−19) when homozygous |
| Hosgood et al. (2012)340,b | GELAC, CAMSCH, SNU, KUMC, KNUH, SWHS, GEL-S, SLCS, FLCS and TLCS: 3,467 (2,557 LUAD, 309 SCC)/3,787 in total | 3q28 | rs10937405; rs4488809 | TP63 | rs10937405: 0.80 (LUAD: 0.74–0.87; P=7.1×10−8); 0.82 (SCC: 0.67–0.99; P=0.037) rs4488809: 1.16 (LUAD: 1.08–1.24; P=7.4×10−5) |
| Shiraishi et al. (2012)341 | Japanese population study: 1,722/5,846 | 6p21.3 | rs3817963 | BTNL2 | 1.18 (1.12–1.24; P=2.7×10−10) |
| 17q24.3 | rs7216064 | BPTF | 1.20 (1.13–1.26; P=7.4×10−11) | ||
| Lan et al. (2012)163,b | FLCCA: 5,510/4,544 (from 14 studies) and 1,099/2,913 (replication set) | 10q25.2 | rs7086803 | VTI1A | 1.32 (1.24–1.41; P=5.04×10−17) |
| 6q22.2 | rs9387478 | ROS1, DCBLD1 | 0.85 (0.81–0.90; P=7.79×10−8) | ||
| 6p21.32 | rs2395185/rs28366298 | HLA class II region | 1.16 (1.09–1.23; P=2.60×10−6) | ||
| Ahn et al. (2012)171 | Korean population study: 446/497 (discovery set) and 434/1,000 (replication set) | 18p11.22 | rs11080466; rs11663246 | FAM38B (PIEZO2), APCDD1, NAPG | rs11080466: 0.61 (0.44–0.77; P=2.68×10−5) rs11663246: 0.60 (0.48–0.76; P=1.74×10−5) |
| Kim et al. (2013)175,b | Korean population study: 285/1,455 (discovery set), 294/495 (replication set 1) 546/733 (replication set 2) | 2p16.3 | rs10187911 | NRXN1 | 1.47 (1.22–1.78; P<0.001) |
| Wang et al. (2016)174,b | FLCCA: 6,877/6,277 (from 4 studies) and 5,878/7,046 (replication set) | 6p21.1 | rs7741164 | FOXP4, FOXP4–AS1 | 1.18 (1.10–1.26; P=2.05×10−6) |
| 9p21.3 | rs72658409 | CDKN2B, CDKN2B–AS1 | 0.75 (0.67–0.83; P=1.37×10−7) | ||
| 12q13.13 | rs116101143 | ACVR1B | 0.88 (0.83–0.93; P=2.21×10−6) | ||
| Shi et al. (2023)159 | FLCCA: 4,438/4,544 NJLCS: 1,923/3,544 NCC: 3,291/19,910 ACC: 1,471/2,564 |
2p23.3 | rs682888 | DTNB | 0.89 (0.86–0.93; P=4.94×10−10) |
| 3q22.2 | rs137884934 | PIK3CB | 0.81 (0.74–0.89; P=6.33×10−6) | ||
| 4p13 | rs117715768 | KCTD8 | 1.24 (1.14–1.34; P=4.48×10−7) | ||
| 4q32.1 | rs1373058 | PDGFC | 1.10 (1.05–1.15; P=8.55×10−6) | ||
| 6p12.1 | rs531557 | GCLC | 0.90 (0.87–0.94; P=7.73×10−7) | ||
| 7q31.33 | rs4268071 | GPR37 | 1.39 (1.25–1.54; P=7.27×10−10) | ||
| 10q26.13 | rs10901793 | FAM53B, METTL10 | 1.10 (1.06–1.14; P=3.14×10−7) | ||
| 11q12.2 | rs174559 | FADS1 | 0.91 (0.88–0.94; P=6.10×10−7) | ||
| 15q21.2 | rs764014 | RFX7 | 0.91 (0.88–0.95; P=5.75×10−7) | ||
| 15q21.3 | rs71467682 | FGF7, SECISBP2L | 0.91 (0.87–0.95; P=2.46×10−6) | ||
| 19p13.1 | rs116863980 | PALM | 1.31 (1.16–1.47; P=7.94×10−6) | ||
| East Asian and European | |||||
| Shi et al. (2023)159 | ILCCO multi-ancestry meta-analysis (similar effect sizes observed in both groups): 11,753/30,562 (East Asian) and 11,273/55,483 (European) | 2p11.2 | rs1130866 | SFTPB | 1.08 (P=1.56×10−8)c |
| 4q32.2 | rs2320614 | NAF1 | 1.08 (P=6.51×10−9)c | ||
| 16q23.3 | rs34638657 | MPHOSPH6 | 1.09 (P=2.19×10−9)c | ||
| 18q12.1 | rs638868 | GAREM1 | 1.08 (P=3.60×10−8)c | ||
The table lists the novel risk loci identified in each study. ACC, Aichi Cancer Center (Japan); CAMSCH, Chinese Academy of Medical Sciences Cancer Hospital; FLCCA, Female Lung Consortium in Asia; FLCS, Fudan Lung Cancer Study (China); GELAC, Genetic Epidemiological Study of Lung Adenocarcinoma (Taiwan); GEL-S, Genes and Environment in Lung Cancer, Singapore study; ILCCO, International Lung Cancer Consortium; KNUH, Kyungpook National University Hospital (South Korea); KUMC, Korea University Medical Center; LCINS, lung cancer in individuals who have never smoked; LUAD, lung adenocarcinoma; MDACC, MD Anderson Cancer Center; NCC, National Cancer Center of Japan Research Institute; NJLCS, Nanjing Lung Cancer Study (China); OR, odds ratio; SCC, squamous cell carcinoma; SLCS, Shenyang Lung Cancer Study (China); SNP, single-nucleotide polymorphism; SNU, Seoul National University (South Korea); SWHS, Shanghai Women’s Health Cohort Study (China); TLCS, Tianjin Lung Cancer Study (China); WHLCS, Wuhan Lung Cancer Study (China); UCLA, University of California Los Angeles (USA).
These studies are candidate gene and pathway-based analyses, which interrogate predefined biological pathways or gene sets; these are distinct from traditional genome-wide association studies and do not require genome-wide significance; therefore, the results should be interpreted with caution.
These studies were focused specifically on women.
No confidence intervals provided in summary statistics.
One of the first GWAS to assess the genetic risk of lung cancer in individuals who have never smoked was conducted in Europeans, and the findings published in 2010 identified a single locus at 13q31.3 where variants resulting in lower transcription of GPC5 were associated with increased susceptibility (OR 1.46, 95% CI 1.26–1.70; P = 5.94 × 10−6)160. GPC5 encodes glypican 5, a proteoglycan with poorly understood roles in physiology and tumorigenesis. Interestingly, GPC5 expression is reduced in LUAD but not in other histological subtypes of lung cancer compared with the surrounding non-malignant lung tissues160. The 5p15.33 locus containing TERT and CLPTM1L has been identified as a lung cancer susceptibility region in both smoking and non-smoking populations, including never-smoking women of East Asian descent161–163 (Table 2). Although specific pathogenetic mechanisms have not been elaborated, this locus is associated with the risk of lung, bladder, prostate and cervical cancers in both European and Asian populations162–164. Results of a meta-analysis of GWAS performed in two independent cohorts to identify variants associated with OS in never-smoking European individuals with NSCLC were reported in 2013 (ref. 165). Out of the top 25 SNPs (combined P < 1 × 10−6), 6 variants showed a genotype–expression association upon expression quantitative trait loci analysis, none of which were within genes previously found to be associated with overall lung cancer risk in people who have not smoked nor within genes associated with OS in patients with smoking-related lung cancer166–168.
GWAS of LCINS have been more extensive in patients of East Asian ancestry (Table 2), with risk variants at the aforementioned 5p15.33 locus recurrently identified in never-smoking women with LUAD162,169,170. Aside from this locus, the loci identified in never-smoking Asian women are generally distinct from those identified in European people with a history of smoking160,163,171. A LUAD susceptibility locus at 3q28 corresponding to TP63 was first identified in Japanese and Korean populations (OR 1.31; P = 7.26 × 10−12), with a trend towards higher ORs in women but no clear differences by smoking behaviour172, and was later confirmed specifically in never-smoking Asian women163. This locus was also subsequently associated with LUAD risk in European populations (OR 1.13; P = 7.22 × 10−10)173. A later study imputing data from four prior GWAS of lung cancer in never-smoking Asian women using the 1000 Genomes Project identified three new risk loci with small effect sizes at 6p21.1, 9p21.3 and 12q13.13, which map near FOXP4, CDKN2B and ACVR1B, respectively174 (Table 2). Subsequent studies in never-smoking Korean populations have identified additional LCINS susceptibility regions on chromosomes 2 and 18171,175 (Table 2).
Most recently, a large, two-stage GWAS of LUAD occurring in individuals of East Asian ancestry identified 12 novel susceptibility variants159 and identified novel alveolar lineage-specific candidate genes, including FADS1 and ELF5, via expression quantitative trait loci colocalization analyses. A multi-ancestry meta-analysis performed as part of the same study revealed four additional novel risk loci shared among East Asian and European individuals (Table 2), although the majority of associations identified in the East Asian cohorts did not extend to the European population. Historically, few studies have identified gene–environment interactions in lung cancer risk176. However, a PRS generated from the top 25 independent susceptibility variants that achieved genome-wide significance in the East Asian population stratified individuals in the highest risk quintile from those in the middle quintile (corresponding to average risk in the general population) to a greater extent in the non-smoking versus smoking population (OR 2.07 versus 1.80; PInteraction = 0.0058)159.
Taken together, the results from GWAS of LCINS are largely non-overlapping and have not converged on a set of robust associations. An analysis by the Transdisciplinary Research In Cancer of the Lung (TRICL) consortium using a custom SNP array restricted to rare variants in known cancer-associated genes identified a large effect association between germline ATM L2307F mutations and LUAD (OR 2.93–8.82 across discovery and replication cohorts)177. This effect seemed to be independent of smoking status, although never-smoking women carrying the L2307F variant were approximately four to seven times more likely to develop LUAD than non-carriers. Notably, this variant is much more prevalent in the Ashkenazi Jewish population (~4%) and might constitute a founder mutation. Variant-associated clinicopathological variables reaching high levels of statistical significance included never-smoking status and female sex but also adenocarcinoma histology and the presence of a somatic EGFR mutation177. Interestingly, never-smoking patients with lung cancers harbouring somatic EGFR mutations are more likely than those with EGFR-wild-type tumours to have a family history of lung cancer178,179. Moreover, a lung cancer genomics and ancestry analysis of admixed Latin American populations demonstrated that the frequency of somatic EGFR mutations varies by ethnicity, suggesting a germline component to EGFR mutation status180.
Polygenic risk scores.
PRS integrate the risk associated with GWAS-identified, disease-associated common SNPs present in the genome of an individual into weighted averages to produce a ‘personalized genetic susceptibility profile’ as a single measure of risk. Using data generated from previous GWAS conducted predominantly in people with a history of smoking, researchers developed PRS specific for 16 different cancer types; the investigators then compared the ability of the PRS versus self-reported family history of cancer in first-degree relatives to predict the risk of cancer within 5 years among 413,870 individuals included in the UK Biobank (with 22,755 incident cancer cases)181. Interestingly, lung cancer was unique in that family history was a significantly better predictor of risk than PRS, such that individuals with a positive family history and a low PRS had a higher 5-year risk of lung cancer than those with a high PRS and negative family history (0.54% versus 0.46%)181. This trend was not observed for several other common cancers, including prostate, breast and colorectal cancers. Assuming that affected relatives from different families did not share common exposures, this finding indicates that lung cancer heritability might be largely attributable to rare, high-effect alleles that mediate familial risk, although a possible alternative explanation is that a positive family history better predicted the risk in individuals who had not smoked given that the PRS was generated from GWAS of predominantly smoking-related NSCLCs.
Another lung cancer-specific PRS was generated by combining genetic and epidemiological data from approximately 13,000 patients and 10,000 control individuals and subsequently validated in almost 336,000 individuals included in the UK Biobank182. This PRS was generated based on a population in which >90% of patients had a history of smoking; therefore, a de novo model to predict the risk of LCINS was derived based on age, sex, body mass index, family and personal history of cancer, impaired lung function, and exposure to ambient air pollution and SHS, in addition to PRS182. Interestingly, including environmental exposures did not improve the predictive performance of the model for LCINS182, which is in contrast to the previously mentioned data from East Asian populations showing an interaction of PRS with smoking status159, probably because smoking is a stronger risk factor.
Ultimately, family history and PRS are not mutually exclusive but rather complementary factors that can provide different insights into the cancer risk of an individual. Indeed, combining family history and PRS has been shown to enhance risk assessment for breast and prostate cancers183,184, although a PRS for these malignancies has not yet been integrated into clinical practice. Whether PRS will have meaningful utility in predicting lung cancer risk remains unclear, and further efforts in this area will probably need to distinguish between smoking-related versus non-smoking-related lung cancers.
Insights from WES and WGS.
Susceptibility loci identified in GWAS account for only a small portion of the variation in lung cancer incidence, mainly because GWAS typically detect common variants with small to moderate effect sizes. Studies at the exome and genome levels are therefore needed to identify rare, large-effect/high-risk germline variants; only a handful of germline analyses in lung cancer have been conducted at these levels, either using data gleaned from matched non-malignant tissues in somatic tumour-profiling studies or as dedicated germline-directed efforts. The largest widely available datasets are derived from TCGA185 (containing WES data from ~580 LUADs and paired non-malignant tissues and WGS data from ~100 tumours) and the Hartwig Medical Foundation186,187. Additional datasets come from the Pan-Cancer Analysis of Whole Genomes (PCAWG) project188 as well as a WES study of LUAD in patients of East Asian ancestry189. In the Hartwig Medical Foundation cohort, which included 178 patients with lung cancer186, various nonsense, frameshift or splice site-altering germline variants were found in CHEK2 and Fanconi anaemia complementation group (FANC) genes (FANCI, FANCL, FANCM), involved in DNA repair, as well as DOCK8 and GJB2 (encoding dedicator of cytokinesis protein 8 and gap junction β2 protein, respectively). A germline WES study focused on SCLC or extrapulmonary small-cell carcinoma identified 42 deleterious germline variants across 35 cancer predisposition genes in 38 (44%) of 87 patients, although 90% had a current or former smoking status190.
WES and/or WGS studies had not distinguished between smoking and non-smoking individuals until 2021, when results emerged from the two formerly discussed somatic alteration profiling studies of never-smoking LUAD, which included data from matched non-malignant tissue analyses,7,78. In the WGS study78, 8 of 232 patients carried pathogenic germline variants (PGVs) in CYP21A2, which encodes the 21-hydroxylase enzyme involved in the synthesis of cortisol and aldosterone, 6 patients carried the same PGV in GLUD2, which encodes the mitochondrial enzyme glutamate dehydrogenase 2, and 5 patients had PGVs in the AR gene (encoding the androgen receptor); PGVs in BRCA1, ATM and RAD51 were each found in 2 or 3 patients. Interestingly, both CYP21A2 and AR are involved in hormone production and signalling, and might thus contribute to sex differences in the incidence of LCINS. In the WES study7, rare pathogenic or likely pathogenic (P/LP) mutations in known cancer predisposition genes were identified in patients who had never smoked; although the prevalence of P/LP mutations was similar to that in those who had smoked (6.9% and 6.4%, respectively), variants in BRCA1, BRCA2, FANCG, FANCM, HMBS, MSH6, NF1,POLD1, TMEM127 and WRN were exclusive to never-smoking individuals. Using a cancer-free control cohort derived from the Genome Aggregation Database (gnomAD), the never-smoking LUAD group was enriched for P/LP variants in FANCG (encoding a component of the Fanconi anaemia DNA repair complex) and TMEM127 (encoding a negative regulator of mTOR signalling).
Larger sample sizes along with functional validation will be needed to further implicate these germline variants in lung cancer pathogenesis, although these early studies in individuals who have never smoked have highlighted the presence of pathogenic alterations with large effect sizes in DNA repair-related genes. This finding is underscored by data from a broader study of common diseases showing that patients with a low common variant PRS are more likely to carry rare disease-specific pathogenic variants191, suggesting that these individuals could be prioritized for rare variant screening.
Lastly, in a study using WES data from participants in the UK Biobank and Mass General Brigham Biobank, the presence of clonal haematopoiesis was found to be associated with increased risk of lung cancer (meta-analysed OR 1.35, 95% CI 1.08–1.68)192, specifically for LUAD (OR 1.68, 95% CI 1.23–2.29) and LSCC (OR 1.59, 95% CI 1.51–1.68) but not for SCLC. Clonal haematopoiesis was also associated with a 36% increase in lung cancer risk among the UK Biobank participants after adjusting for major risk factors, including pack-years of smoking, age at sequencing, family history of lung cancer and lung cancer PRS192. Whether clonal haematopoiesis is a surrogate of currently unknown shared risk factors, including inherited genetic risk (for example, mediated by rare variants), or has a more causal role in lung cancer remains unclear.
Germline EGFR mutations in familial lung cancer.
Germline EGFR mutations have been identified in familial lung cancers193. Most common is the T790M mutation, which, in its somatic form, can be present at diagnosis or develop as a mechanism of resistance to earlier-generation EGFR TKIs. The germline EGFRT790M mutation was identified in 2005 among a family of European descent with several members across multiple generations developing LUAD193. Germline EGFRT790M has since been found in multiple unrelated kindreds, predominantly comprising white individuals in the USA, Europe and Australia, more frequently in never-smoking women, and in association with multiple primary lung lesions (either nodules or invasive adenocarcinomas)193–196. A cluster of families has been identified in the southeastern USA, suggesting a possible founder effect197. Notably, germline EGFRT790M has not been reported in patients of East Asian ethnicity with lung cancer despite the high prevalence of somatic EGFR mutations in this population. Germline EGFRT790M mutations have been estimated to occur in 0.5–1.0% of patients with NSCLC195,198 and in roughly 1 in 100,000 individuals in the general population194 (allele frequency of 9.9 × 10−6 in the gnomAD v4.0 database)199. Additional studies with larger sample sizes are needed to determine more precise frequencies of germline EGFRT790M mutations in patients with lung cancer as well as the magnitude of their effect on lung cancer risk.
Virtually all lung cancers that develop in the context of a germline EGFR mutation harbour a secondary somatic activating mutation in EGFR in cis193, most commonly L858R197,200. EGFRT790M carriers without known lung cancer are often found to have multiple groundglass nodules on CT imaging, suggestive of premalignant lesions that can develop into invasive adenocarcinoma over time194, and are likely to benefit from surveillance CT-based screening. EGFRT790M carriers do not seem to have an increased incidence of any other cancer type; the mechanism underlying LUAD specificity is unknown. Less common germline EGFR mutations include R776G/H201,202 and V769M in exon 20 (ref. 203) and V834L and V843I in exon 21, the latter identified specifically in Asian and Surinamese families204–206. Even rarer is the EGFR R831H germline variant reported in Chinese patients with NSCLC207,208, which was also described as co-segregating with prostate cancers harbouring somatic biallelic inactivation of CDK12 in two brothers within one family; prostate cancer cells derived from these patients were responsive to the EGFR TKI afatinib in vitro209. To our knowledge, EGFRR831H is the only germline EGFR mutation associated with a cancer type outside of the lung, although genotype–phenotype relationships among various germline EGFR mutations have not been extensively studied. Additional genetic or environmental modifiers might affect the lung cancer risk associated with germline EGFR mutations and account for phenotypic differences between individuals within families, despite carrying the same mutation.
Non-EGFR germline mutations in familial lung cancer.
Germline mutations in familial lung cancer pedigrees have also been found in HER2 (refs. 210-212), BRCA2, CHEK2 (ref. 173), MET213 and YAP1 (ref. 214), predominantly in never-smoking patients with LUAD and more commonly in female patients. The germline G660D mutation in the transmembrane domain of HER2 was initially reported in a Japanese family, identified in a female proband with a 1.2 pack-year smoking history and multifocal NSCLC210. Following lobectomy, pathology demonstrated that this woman had innumerable pre-invasive lesions; similar lesions were seen bilaterally on imaging in her 30-year-old daughter, also a HER2G660D carrier212. No additional somatic alterations were identified (including HER2 and EGFR), and the proband was treated with second-line afatinib following disease progression on chemotherapy, with a partial response in the lung and stable disease for ≥16 months in the bone211. Additionally, a candidate gene study using WGS in a never-smoking Taiwanese family with high frequency of LUAD identified a germline variant (R331W) in the transactivation domain of YAP1 (ref. 214), a transcriptional regulator in the Hippo signalling pathway that has been implicated in resistance of EGFR-mutant NSCLC to EGFR TKIs215,216. In a validation cohort derived from the Genetic Epidemiology Study of Lung Adenocarcinoma in Taiwan, the YAP1R331W allele frequency was 1.1% in patients with LUAD versus 0.18% in individuals without cancer, translating into an OR of 5.9 after adjusting for age, sex and smoking status214. All chest CT-screened YAP1R331W carriers had LUAD or groundglass lesions (40% and 60%, respectively)214. These studies support the existence of rare, highly penetrant PGVs that contribute to a subset of LCINS.
Hereditary cancer predisposition syndromes.
LUAD is a principal malignancy associated with several multi-organ cancer predisposition syndromes resulting from germline mutation of tumour-suppressor genes (for example, TP53, PTEN or LKB1)217. Several case reports have demonstrated oncogene-driven NSCLC in patients with Li–Fraumeni syndrome218–221. In one study, 21 (91%) of 23 NSCLC tumours in patients with Li–Fraumeni syndrome harboured an oncogenic alteration, 20 (87%) of which were EGFR mutations, most commonly exon 19 deletions220. EGFR-mutant LUAD has also been described in Cowden syndrome, caused by germline PTEN mutations, in case reports of younger patients with a history of light or never smoking222. An increased risk of lung cancer has been reported in survivors of hereditary retinoblastoma with germline RB1 mutations, with lung cancer diagnosis tending to occur before 40 years of age223, and there are case reports of lung cancers associated with Bloom224, Werner225 and Birt–Hogg–Dube syndromes217.
Environmental risk factors
Various environmental exposures have been implicated or hypothesized to contribute to the risk of LCINS (Table 3). An exhaustive discussion of these risk factors is beyond the scope of this Review; here, we focus on key exposures with established associations and/or those with contemporary evidence of an effect on lung cancer risk.
Table 3 |.
Environmental risk factors for LCINS
| Risk factor | Effect summary | Refs. |
|---|---|---|
| Radon | Second-leading environmental cause of lung cancer, after smoking, and estimated to account for ~12% of lung cancers in the USA annually; the association of radon with lung cancer was first noted in uranium miners in the 1980s228,229; numerous large, global studies have demonstrated an association between prolonged residential radon exposure and lung cancer in the general population; the risk is greatest when combined with smoking230,234,242 but applies to both smoking and non-smoking populations244; the strongest histological association is with SCLC, followed by LUAD245–247 | 226,228–239, 242–247,342 |
| SHS | SHS increases the risk of lung cancer development in individuals who do not smoke by 20–25% and is responsible for ~3,500 lung cancer deaths among non-smoking people in the USA annually1,13; other studies are limited but the reported effect sizes are moderate (OR range 1.2–2.1)153,253–256,343; no differences in the prevalence of various targetable alterations found in LCINS have been reported based on exposure to SHS258; tobacco-related mutational signatures have not been detected in SHS-related LCINS7,78 | 1,7,13,78,153,249–256,258,343 |
| Occupational carcinogens | Elements, such as sulfur, arsenic, beryllium, cadmium, chromium, nickel, plutonium and radon, fumes and particulate matter with a diameter of ≤10μm (PM10) or ≤2.5μm (PM2.5), diesel engine exhaust emissions, silica dust, X-rays, and γ-radiation have each been associated with an increased risk of LCINS281, as have bis(chloromethyl)ether, chloromethyl methyl ether (technical grade), coal-tar pitch and asbestos (all forms, including actinolite, amosite, anthophyllite, chrysotile, crocidolite and tremolite); asbestos is definitively associated with an increased risk of bronchogenic carcinoma and pleural mesothelioma285,286, with the risk being several-fold higher for lung cancer than for mesothelioma285,286, although for both, the risk is greatly multiplied when combined with cigarette smoking286,290–292,344; silica, diesel exhaust emissions and welding fumes increase the risk of lung cancer independently of smoking and co-exposures282–284 | 281–286,289–294,344,345 |
| Air pollution | Air pollution and fine particles (PM2.5) are classified as group 1 carcinogens by the WHO IARC268,310; WHO has estimated that ~6% of outdoor air pollution-related premature deaths in 2016 were due to lung cancer268; associations with lung cancer risk and mortality have been demonstrated in individuals who have smoked and those who have not (HR < 2)265,267,271,272, and air pollution-associated lung cancer risk is probably influenced by underlying genetic susceptibility265,278; PM2.5 levels positively correlated with rates of EGFR-mutant lung cancer in England, South Korea and Taiwan279 | 261–265,268,276–279,346 |
| Electronic cigarettes/vaping | The carcinogenic potential of electronic cigarette/vaping particles is not currently known and is probably dependent on the composition of aerosolized liquid, the device and user habits302–304; long-term follow-up studies are required to determine any lung cancer risk | 302–308,347–349 |
| Household use of solid fuels and high-temperature frying | Fumes and particulate matter generated from cooking and heating contain group 1 (carcinogenic) or group 2 (likely carcinogenic) substances as defined by the WHO IARC; dose–response relationships between reported exposure to cooking oil fumes and lung cancer risk have been demonstrated; in Chinese non-smoking women, the ORs are as high as 6.15 in the highest exposure groups (higher with deep frying versus stir-frying and in homes with poor ventilation)311–313; indoor burning of coal, wood and other biomass is associated with an increased risk of lung cancer with ORs <2, with greater levels of risk in women than in men315,316,350 | 310–316,350 |
| Diet | No consistent data exist that implicate dietary differences as being associated with lung cancer risk; primary chemoprevention studies focused on lung cancer have not demonstrated benefit from dietary supplements, although some studies have shown an association between β-carotene supplementation and a reduced risk of lung cancer in men who have smoked351–353 | 351–355 |
| HRT | Findings regarding the association between HRT and lung cancer risk are mixed; data from the Women’s Health Initiative trial demonstrated no statistically significant increase in the risk of lung cancer with either oestrogen–progestin or oestrogen-only HRT356–358; however, results of the Vitamins and Lifestyle study indicate an increased risk of lung cancer with oestrogen–progestin HRT after adjusting for smoking status (HR 1.48 if exposed to ≥10 years oestrogen plus progestin)359 | 78,356–364 |
| Prior and/or chronic lung disease | A history of lung disease (COPD, pneumonia, tuberculosis or interstitial lung disease) is associated with an increased risk of lung cancer365–370, although the associations are confounded by tobacco smoking and other environmental exposures; an elevated lung cancer risk associated with emphysema, pneumonia and tuberculosis has been observed consistently in white men367; the underlying mechanisms are presumed to be related to chronic inflammation and parenchymal lung injury; whether autoimmune diseases are associated with an increased risk of developing lung cancer in individuals who do not smoke remains unclear | 365–375 |
| Recreational and illicit drugs | Whether habitual smoking of marijuana or cocaine use increases the risk of lung cancer is unclear owing to confounding by tobacco smoking and/or insufficient follow-up time; opium has been classified as a group 1 carcinogen after smoking or ingestion of this drug was associated with the development of lung cancer among individuals in Golestan province, Iran (HR 2.2)376; the lung cancer risks associated with derivatives, such as heroin, morphine, codeine and fentanyl, have not been evaluated | 376–382 |
| Prior radiotherapy to the chest | Chest irradiation increases the risk of second primary lung cancer, with the greatest risk in combination with tobacco smoking; for example, a high risk of secondary lung cancer has been demonstrated in individuals who received prior radiotherapy for Hodgkin lymphoma (RR 2.6–7.0), particularly those treated after ≥45 years of age, with chemotherapy having an additive effect383; lung cancer risk increases over time up to 20–25 years after radiotherapy; similar observations have been reported in breast cancer survivors, with a tenfold higher lung cancer risk in patients who received radiation for invasive breast cancer versus those who did not384 | 383,384 |
COPD, chronic obstructive pulmonary disease; HRT, hormone replacement therapy; IARC, International Agency for Research on Cancer; LCINS, lung cancer in individuals who have never smoked; LUAD, lung adenocarcinoma; SCLC, small-cell lung cancer; SHS, secondhand smoke.
Radon.
Radon has been identified as the second-leading environmental cause of lung cancer (after tobacco smoking) and is estimated to contribute to ~21,000 lung cancer-related deaths annually in the USA, with roughly 2,900 of these deaths occurring in individuals who have never smoked147,226,227. The increased lung cancer incidence associated with radon was first noted in uranium miners in the 1980s228–230, and numerous large, international epidemiological case–control studies have also demonstrated an association between prolonged residential radon exposure and lung cancer in the general public231–239. These and additional studies led the US Environmental Protection Agency (EPA) to classify radon as a carcinogen owing to its causal association with lung cancer, and both the EPA and the National Radon Safety Board advocate radon screening and mitigation in homes across the USA240,241.
Radon exposure more than additively increases the lung cancer risk conferred by smoking234,242. In comparison with non-smoking individuals, lung cancer risk associated with radon exposure is eightfold to ninefold greater in individuals who also smoke, and >85% of radon-associated lung cancers occur in those who formerly smoked or currently smoke230,243, although the risk is still substantial in non-smoking groups244. Subsequent studies and meta-analyses have shown the strongest histological association of residential radon exposure with SCLC, followed by LUAD245–247. A population-based, case–control study performed among ~530 women with LCINS did not find an association between lung cancer and domestic levels of radon exposure, potentially owing to a relatively low level of exposure or differences in underlying genetic susceptibility. Interestingly, a meta-analysis has revealed a higher adjusted excess relative risk of lung cancer from residential radon exposure for men than for women among those who had never smoked (0.46 versus 0.09; P = 0.027)248.
Secondhand smoke.
SHS has long been studied as a potential causative factor in LCINS. SHS exposure is estimated to increase lung cancer risk by 20–25% in individuals who do not smoke and, in 2023, a projected 3,560 lung cancer-related deaths among non-smoking individuals in the USA were attributable to SHS1,13. In addition to the duration and intensity of exposure, genetic modifiers influencing carcinogen metabolism, DNA repair and inflammation all probably affect the lung cancer risk conferred by SHS249–252.
Many epidemiological studies performed in the 1990s to early 2000s established SHS as a risk factor for LCINS153,253–256, although the effect sizes are moderate. Most of these studies have compared individuals who have a spouse who smokes with those who do not. In a meta-analysis of 37 such studies (n = 4,600), the pooled relative risk of lung cancer in women who were exposed to SHS versus those who were not was 1.24 (95% CI 1.13–1.36; P < 0.001)253. A subsequent and much smaller case–control study including 280 patients with LCINS found a larger effect (OR 2.08, 95% CI 1.25–3.43 across both sexes) but lower than that associated with exposure to environmental dust (OR 2.43, 95% CI 1.53–3.88)153. Interestingly, a large prospective cohort study involving >75,000 women revealed a 13-fold higher incidence of lung cancer in individuals who currently smoke (HR 13.44, 95% CI 10.80–16.75) and fourfold higher incidence in those who formerly smoked (HR 4.20, 95% CI 3.48–5.08) compared with those who had never smoked, but no increase was observed among never-smoking women with passive smoke exposure (HR 0.88, 95% CI 0.52–1.49)257. Despite the limited number of lung cancers in never-smoking women with SHS exposure, a borderline significant trend towards an increased risk of lung cancer was observed in those who cohabitated for ≥30 years with someone who smoked (HR 1.61, 95% CI 1.00–2.58)257.
The molecular characteristics of SHS-associated lung cancers are similar to those of tumours in never-smoking patients, with no overall difference in rates of EGFR, ALK, KRAS, HER2, BRAF and PIK3CA alterations258. Although tobacco carcinogen metabolites have been found in the urine and blood of never-smoking individuals exposed to SHS253, large-scale genomic studies of LCINS have shown that reported SHS exposure often does not correlate with smoking-related somatic mutational signatures,7,78. Indeed, the small group in which such signatures were detected might consist of individuals with the highest SHS exposure and/or underlying genetic susceptibility to carcinogenesis. The WHO International Agency for Research in Cancer (IARC)259 and US National Institutes of Health260 have designated SHS as a human carcinogen, although SHS is generally accepted to confer only a modest risk of cancer and is not considered the sole causative factor for the majority of LCINS. Nonetheless, household exposure is probably more relevant than public exposure, and the continued declines in smoking rates as well as health regulations outlawing smoking in indoor public spaces will further mitigate the effects of SHS.
Air pollution.
Air quality can be measured by quantifying the amounts of ozone, particulate matter and chemical pollutants in ambient air. Particle pollution is a combination of small liquid and solid particles that can comprise dust, metals, soil, acids and organic chemicals. These small, inhaled particles emitted from vehicle exhaust, forest fires, coal-fired power plants and other industrial sources are thought to pass through the central airways and lodge in the more peripheral airways where LUADs typically form. The association between outdoor air pollution and lung cancer risk has been reported in several studies261–267, and both air pollution and particulate matter have been officially classified as group 1 carcinogens by the IARC265,268. Particulate matter with a diameter of ≤2.5 μm (PM2.5) can reach the alveoli and has been associated with heart disease and lung cancer263,266,267,269. According to the IARC, the largest and most important studies consist of combined analyses spanning 17 cohorts from 9 countries in Europe263 and a multi-cohort study among large cities in the USA266, reporting HRs of 1.18 and 1.55 per 5 μg/m3 of PM2.5 exposure for lung cancer and LUAD, respectively. Additionally, a large study in those who had never smoked showed a 15–27% increase in lung cancer mortality per 10 μg/m3 increase in PM2.5 concentrations267. In 2016, the IARC estimated that ~6% of outdoor air pollution-related premature deaths were attributable to lung cancer268. Unsurprisingly, risk is proportional to the extent of exposure270 and is greater than additive when combined with cigarette smoking (risk of lung cancer mortality 2.2 times greater than additive for joint exposure)271, although evidence indicates an increased risk even after accounting for smoking263,272 as well as when restricting the analysis to those who have never smoked267. The updated 2021 WHO global air quality guidelines273 recommend annual mean PM2.5 concentrations of ≤5 μg/m3, while the EPA primary standard274 is ≤12 μg/m3, with a proposal in January 2023 to revise this standard to 9.0–10.0 μg/m3 (ref. 275). Modelled annual mean PM2.5 concentrations for the USA276 and globally277 show the highest levels in portions of the Midwest, Southeast and California within the USA, and in northern Africa, India, China and Middle Eastern countries internationally (Fig. 3).
Fig. 3 |. Annual average PM2.5 concentrations across the world.
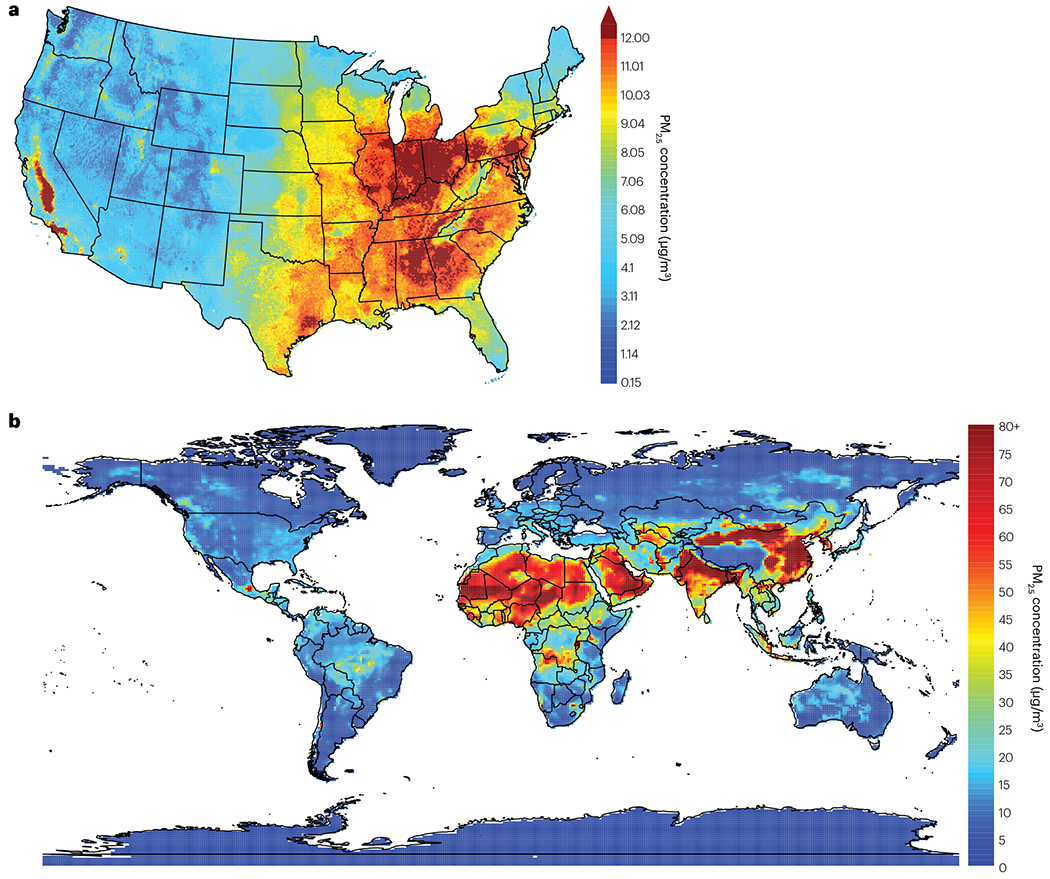
a, Mean annual average concentrations of particulate matter measuring ≤2.5 μm in diameter (PM2.5) across the contiguous United States, plotted in 1-km grids, spanning years 2000–2016 (ref. 276). The annual estimates are averages of daily predictions for each year in each grid cell. The current US Environmental Protection Agency annual average primary, health-based national ambient air quality standard for PM2.5 is ≤12.0 μg/m3, with a proposed revision to within 9.0–10.0 μg/m3 announced in January 2023 (ref. 275). b, Mean annual average PM2.5 concentrations across the globe, spanning years 2003–2022, according to European Centre for Medium-range Weather Forecasts Atmospheric Composition Reanalysis 4. Performed by the Copernicus Atmosphere Monitoring Service, the reanalysis combines data from modelling studies with observations from across the world into a globally complete dataset277. Annual estimates are averages of monthly predictions. The WHO air quality guidelines273 recommend annual mean PM2.5 concentrations of ≤5 μg/m3.
As with other environmental exposures, the lung cancer risk conferred by air pollution is probably influenced by underlying genetic susceptibility265,278 (Fig. 4a). In 2021, a study analysed SNP data from >450,000 individuals included in the UK Biobank combined with estimated particulate matter exposure265. After controlling for confounders, such as obesity and smoking, the investigators generated a PRS to model genetic risk and found an additive interaction between genetic susceptibility and air pollution, such that individuals with high genetic risk and high levels of exposure to air pollution were at greatest risk of developing lung cancer (PM2.5: HR 1.71, 95% CI 1.45–2.02) relative to those with low genetic risk and low air pollution exposure.
Fig. 4 |. Germline–somatic and gene–environment interactions in the pathogenesis of LCINS.
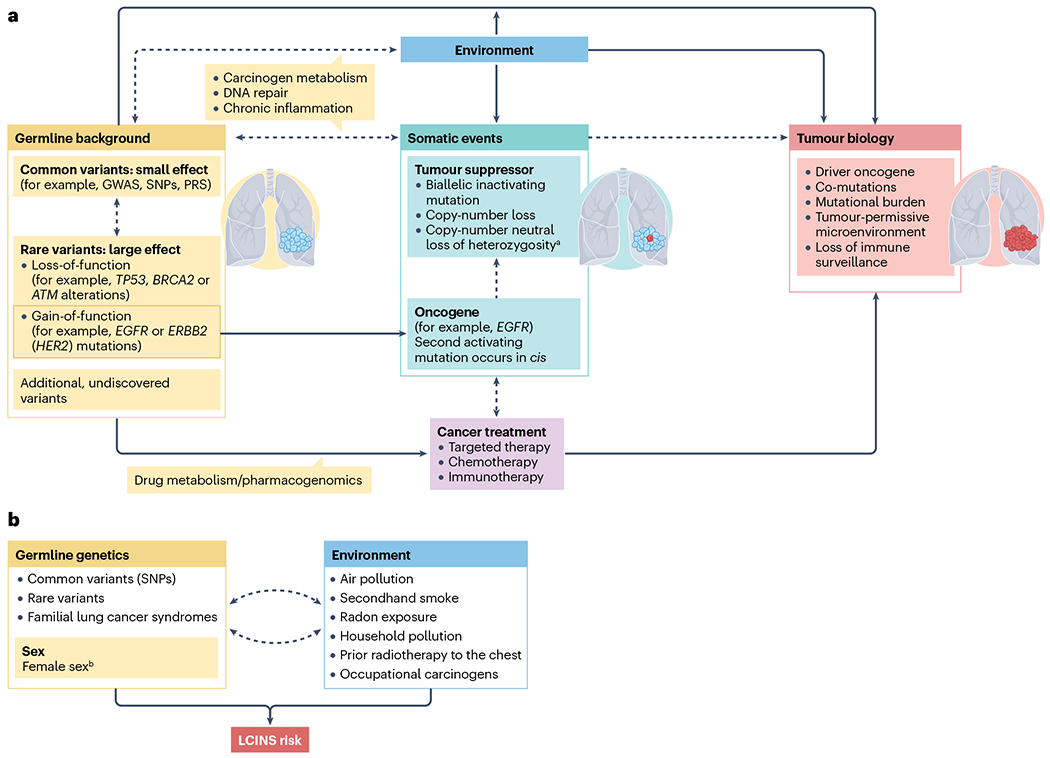
a, Model of germline–somatic interactions in lung cancer in individuals who have never smoked (LCINS). Dashed lines and arrows indicate interactions, whereas solid lines and arrows indicate direct processes. Germline variants and acquired somatic alterations can interact to promote cancer development and progression (potential mechanisms are detailed in the main text). Moreover, germline variants affecting carcinogen metabolism, DNA repair and inflammatory processes can interact with environmental exposures and thereby influence the acquisition of somatic alterations and tumour development, and polymorphisms in genes underlying drug metabolism can affect responses to therapy or the development of treatment-related toxicities (the concept of pharmacogenomics). b, Risk factors promoting LCINS include environmental exposures such as radon, secondhand smoke, outdoor (air) and indoor (household) pollution, occupational carcinogens, and prior radiotherapy to the chest. Patient-level risk factors include female sexb, Asian ethnicity and germline risk, the genetic architecture of which consists of common variants of small to moderate effect as well as rare and ultra-rare variants of large effect. Familial lung cancer syndromes are mediated by rare germline mutations in oncogenes such as EGFR. Interaction between genetics and environment can occur across any and/or multiple risk factors. GWAS, genome-wide association study; PRS, polygenic risk score; SNP, single-nucleotide polymorphism. aCopy number neutral loss of heterozygosity events can occur at loci harbouring either tumour-suppressor genes or oncogenes. bNote, environmental risks related to the socioeconomic and cultural aspects of female gender (such as historically greater exposure to household cooking fumes among women), and/or biological factors related to female sex, might contribute to the effect of female sex.
More recently, another study used UK Biobank data to examine exposure to PM2.5 in >400,000 individuals residing in the UK279. Increasing levels of PM2.5 exposure correlated with increased risk of lung cancer (HR 1.08; 95% CI 1.04–1.12); nominally significant (P < 0.05 and false discovery rate > 0.05) associations were also reported for mesothelioma (HR 1.11, 95% CI 1.00–1.24) and lip and oropharyngeal cancers (HR 1.10, 95% CI 1.01–1.19). Analyses of the interaction between PM2.5 exposure and ever-smoking status indicated that current or previous smoking and exposure to high levels of PM2.5 might act in combination to increase lung cancer risk (P = 0.049). Interestingly, PM2.5 levels correlated with the incidence of EGFR-mutant lung cancer in England as well as in South Korea and Taiwan, with the relative rates per 100,000 individuals increasing by 0.63 (P = 0.0028), 0.71 (P = 0.0091) and 1.82 (P = 4.01 × 10−6), respectively, with each additional 1 μg/m3 PM2.5 increment. Interestingly, DNA sequencing of non-malignant lung tissue revealed EGFR and KRAS mutations in 18% (54 out of 295) and 53% (43 out of 81) of samples, respectively, leading to the hypothesis that these mutations accumulate as part of the ageing process and that PM2.5 promotes subsequent tumour formation in the ‘at-risk epithelium’ harbouring these driver mutations279. Further studies are necessary to elucidate the degree of lung cancer risk conferred by PM2.5, particularly with regards to the amount and duration of exposure, as well as the direct mechanistic link between PM2.5 and EGFR and KRAS mutation and/or tumorigenesis driven by these alterations. Importantly, inequities in exposure to air pollution disproportionately affect lower socioeconomic status groups and non-white populations280 as well as those with less education or who live closer to major sources of pollution, necessitating a global effort to improve air quality for the most vulnerable populations.
Occupational carcinogens.
In an extraction of data from the IARC monographs (spanning years 1971–2017), 20 different agents and their related compounds were found to be causally associated with lung cancer281 (Table 3). Several of these exposures (for example, silica, diesel exhaust and welding fumes) increase lung cancer risk independent of smoking status and co-exposures282–284. A large pooled analysis assessing the risk of lung cancer in men exposed to diesel exhaust fumes (with 16,901 cases and 20,965 controls) found an exposure–response relationship regardless of smoking history (OR 1.41, 95% CI 1.30–1.52 in never-smoking individuals at the highest exposure level), particularly for LSCC and SCLC (OR 1.38 for both cancer types at any exposure level, 95% CI 0.98–1.94 and 0.81–2.36, respectively)283. Similar exposure–response relationships were demonstrated for all histological subtypes of lung cancer using the same dataset to assess the risk associated with respirable crystalline silica282.
Exposure to asbestos is definitively associated with a high risk of both bronchogenic carcinoma and pleural mesothelioma285,286, with the risk being several-fold higher for lung cancer than mesothelioma. Nevertheless, approximately 80% of patients with pleural mesothelioma report asbestos exposure287, and 15–20% of all asbestos-related deaths in the USA result from mesothelioma288. Although these proportions are decreasing owing to asbestos abatement laws effected in the latter half of the twentieth century, they are likely to remain considerable for at least a decade owing to a long latency period to lung cancer development (15–35 years). Individuals employed in mining, construction, shipbuilding and firefighting as well as veterans exposed to military asbestos products continue to constitute the groups at highest risk. The risk of lung cancer is greatly multiplied when asbestos exposure is combined with cigarette smoking286,289–292 (OR 8.70 versus 1.70 in those exposed to asbestos who never smoked)292. In men with LUAD, an association has been found between occupational asbestos exposure and an increased prevalence of KRAS codon 12 mutations, independent of smoking status and age (adjusted OR 6.9, 95% CI 1.7–28.6)293. In addition to occupational exposure, lung cancer risk might be higher in individuals who live near environmental sources of asbestos (OR 1.48, 95% CI 1.18–1.86)294.
Electronic cigarettes and vaping.
Whether the use of electronic cigarettes (e-cigarettes) and similar devices is associated with an increased incidence or risk of lung cancer remains unclear owing to their relatively recent development and the tendency for users to also smoke tobacco. Nicotine exposure in the absence of tobacco smoking is not thought to increase cancer risk based on studies involving users of nicotine replacement therapy295,296, although these studies have had relatively short surveillance times (for example, 5–7 years). In fact, when used as tools for smoking cessation, nicotine replacement therapy has been correlated with a decrease in lung cancer risk and mortality297–300. Several thousand cases of severe vaping-related lung injury were reported in 2019 (ref. 301), thereafter known as e-cigarette or vaping product use-associated lung injury, largely owing to vitamin E acetate contamination of e-cigarette cartridges with either manufacturer-derived or illicit substances. However, the carcinogenic potential of e-cigarette vapours is less clear, and their acute and chronic effects probably depend on the composition of the liquid being aerosolized, the specific device used and user habits302–304. When heated, common nicotine solvents can yield byproducts, such as propylene oxide and acrolein, that are carcinogenic305–307. Depending on the aerosol, e-cigarette vapour can contain ultrafine particles (<0.3 μm in diameter) capable of reaching pulmonary alveolar regions, the distribution of which is also dependent on liquid solvent content (for example, vegetable glycerin-to-propylene glycol ratio) and inhalation practices303,308,309. Although similar in size to those found in tobacco and diesel engine smoke, the carcinogenic potential of vaping particles is unknown. Ultimately, long-term follow-up studies are needed to determine any association between e-cigarette use and lung cancer.
Household use of solid fuels and high-temperature frying.
Fumes and particulate matter generated from burning cooking oils and solid fuels used for heating (consisting of ‘smoky’ or bituminous coal and ‘biomass’ such as wood, charcoal and crop residue) contribute to indoor air pollution in homes across the world, particularly in developing nations and parts of Africa and Southeast Asia. These emissions contain known carcinogen substances, mainly polyaromatic hydrocarbons and aldehydes as well as particulate matter with a diameter of ≤0.25 μm, that have been classified as group 1 (carcinogenic) or group 2 (likely carcinogenic) by the IARC310 based on numerous studies worldwide. Traditional cooking in many Asian countries involves heating cooking oils to very high temperatures during the process of frying, which has historically exposed women to the resulting carcinogens to a greater extent than men. A dose–response relationship has been observed between reported exposure to cooking oil fumes and lung cancer risk, which is generally higher with deep frying versus stir-frying and in homes with poor ventilation311–313. With respect to indoor burning of coal, a large retrospective cohort study among smoky coal users in China (n = 27,310) found that the absolute risk of death from lung cancer before 70 years of age was 18% and 20% for men and women, respectively, compared with <0.5% for both sexes among smokeless coal users (n = 9,962), regardless of tobacco smoking status314. The lung cancer risk associated with indoor burning of wood and other biomass is similar to that attributed to coal smoke, with ORs of <2; the risk is greater in women than in men315–317, which might reflect a greater amount of time spent in the household by women and might therefore be associated with the social construct of female gender rather than being biologically related to female sex. Household pollutants are predominantly associated with LUAD, although cases of LSCC and SCLC have been reported. Whether an underlying genetic susceptibility contributes to the lung cancer risk associated with indoor pollutants remains unclear.
Germline–somatic–environment interactions
Cancers arise in the context of a complex interplay between germline and somatic genomes and the environment (Fig. 4a). With regard to lung cancer, the architecture of germline risk includes many small-effect common variants and larger-effect rare and ultra-rare variants. Established rare risk variants include loss-of-function mutations in tumour-suppressor genes as well as gain-of-function alterations in oncogenes, the latter of which underlie familial lung cancer syndromes associated with germline mutations in EGFR and HER2. Interactions between inherited genetic variants and the acquisition of somatic alterations as well as environmental exposures can affect the risk and management of LCINS (Fig. 4), underscoring the importance of studies evaluating matched tumour and non-malignant tissue samples. An example of such an interaction comes from the previously discussed study in which germline ATML2307F mutations were found to be associated with LUAD arising in individuals with a history of light versus heavy smoking, later age of onset, and broad and local somatic features (that is, EGFR mutation and biallelic ATM inactivation, respectively)177.
Potential mechanisms for this interaction include (1) cooperation between germline and somatic variants in different pathways to promote a clonal advantage318, (2) germline variants acting downstream of somatic variants to modulate clonal advantage (and vice versa), (3) germline variants promoting the formation of larger-scale somatic events such as chromosomal abnormalities177, and (4) germline variants (for example, in DNA repair pathways) contributing to clonal advantage, agnostic to the nature of the somatic mutation driving the clone. Treatments for lung cancer typically target somatic alterations irrespective of germline genetic background, although a study in patients with SCLC demonstrated that PGVs in DNA repair genes can influence recurrence-free survival and response to agents targeting defective DNA repair190. Considering ICI treatment and toxicity, a germline PRS enriched for variants regulating gene expression in macrophages and dendritic cells has been shown to predict the nature of the tumour immune microenvironment and ICI response318. Additionally, a germline PRS for hypothyroidism is predictive of thyroid-specific immune-related adverse events in patients with NSCLC319. Moreover, a cross-cancer GWAS among patients receiving ICIs identified three common variants that are associated with all-grade immune-related adverse events, the most consistently replicated of which was an intronic variant in IL7 that is predictive of increased lymphocyte stability after ICI initiation as well as improved OS320. Thus, underlying germline variation can affect both tumour evolution and the development of targetable somatic alterations, and consequently influence treatment response and clinical outcomes.
Diagnostic and management considerations
Given the high likelihood of targetable somatic alterations, several diagnostic and management principles can be applied to LCINS (Fig. 5). The NCCN Guidelines for NSCLC321 recommend testing for mutations or fusions involving the following 11 oncogenes: EGFR, KRAS, ALK, ROS1, RET, HER2, N7RK1–3, BRAF and MET (exon 14 skipping), with high-level MET amplification as an emerging target. Testing methods vary, although panel-based NGS is preferable to maximize target coverage. Blood-based NGS might not detect driver alterations owing to insufficient shedding of tumour DNA; therefore, samples cannot be deemed oncogene negative based on liquid biopsy results alone. Most fusions (for example, those involving ALK, ROS1 or RET) are detectable with break-apart FISH assays or hybrid capture-based NGS, and DNA-based NGS has been used to identify novel fusion partners not detectable with FISH and reverse transcription PCR80. However, genomic breakpoints for rarer fusions (for example, involving NTRK1–3 or NRG1 as well as some ROS1 fusions) can occur within long intronic sequences that are not adequately covered in targeted DNA-based panels. For this reason, RNA-based NGS has been used, and can also enable the identification of fusions when low tumour purity limits DNA-based detection owing to the high expression of fusion oncogenes. Broader sequencing efforts, such as WES, can reveal therapeutic targets, particularly in the case of novel fusions or other genomic alterations in regions with low coverage in standard NGS panels.
Fig. 5 |. Molecular diagnostic algorithm for advanced-stage LCINS.
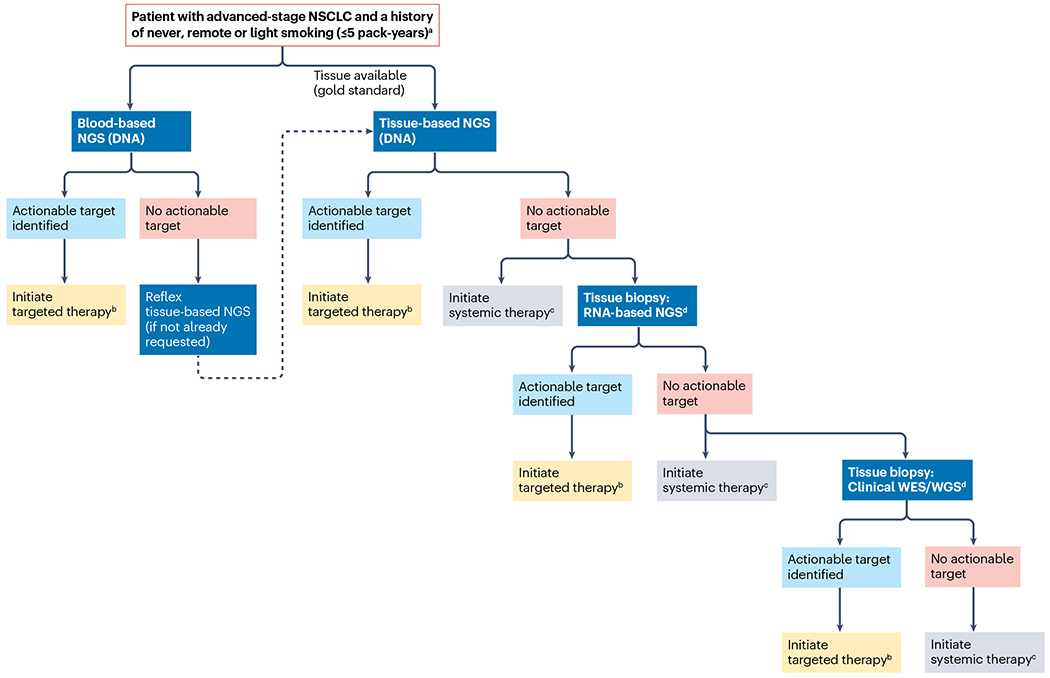
The algorithm outlines a sequential approach to evaluating the presence of targetable oncogenic driver alterations in lung cancers in individuals who have never smoked (LCINS) or who have a limited smoking history, which are almost exclusively non-small-cell lung cancer (NSCLC) of adenocarcinoma histology. The National Comprehensive Cancer Network (NCCN) Guidelines321 recommend that molecular analysis of advanced-stage NSCLC should include assays to identify actionable alterations in EGFR, KRAS, ALK, ROS1, RET, ERBB2 (also known as HER2), NTRK1–3, BRAF (V600E) and MET exon 14, for which FDA-approved targeted therapies are available. Emerging therapeutic biomarkers include high-level MET amplification, with the current NCCN Guidelines (version 5.2023) noting that thresholds constituting high-level MET amplification are evolving; when next-generation sequencing (NGS) is used, a MET copy number of ≥10 is consistent with high-level MET amplification and is associated with clinical benefit from MET tyrosine kinase inhibitors. Although NGS is the preferred analytical modality, molecular profiling is defined as broad testing that identifies all relevant clinically actionable biomarkers in either a single assay or a combination of a limited number of assays and, ideally, also identifies emerging biomarkers. WES, whole-exome sequencing; WGS, whole-genome sequencing. aSimultaneous blood-based and tissue-based NGS testing recommended, with ideal turnaround times of 7–10 days and 2–3 weeks, respectively. bIf a matched targeted therapy is not approved in the first-line setting, may consider clinical trials offering first-line targeted therapy versus standard-of-care treatment (for example, NCT05048797 for HER2-mutant NSCLC). cClinical decision-making should include consideration of clinicopathological variables such as smoking history, histology, PD-L1 positivity and tumour mutational burden, noting that the majority of LCINS are associated with poor response to immunotherapy. dIf available, to maximize detection of fusion proteins and emerging targets.
The management of LCINS is largely driven by genomic findings as outlined in NCCN guidelines for NSCLC321. Discussion of genotype-directed therapy is beyond the scope of this Review but, for patients with advanced-stage disease, targeted therapy is generally preferable when available321. Newer strategies involving TKI treatment to the point of maximal response followed by local consolidative treatment are under investigation322–325 as are upfront combinations of targeted agents with chemotherapy in patients with advanced-stage disease326. For patients with central nervous system metastases at diagnosis but without severe mass effects and in whom a targetable alteration is found for which a central nervous system-penetrant TKI is available, potential therapeutic strategies include TKI treatment prior to radiotherapy or surgical resection of brain lesions given the high intracranial efficacy of these drugs in clinical trials; however, comparisons with upfront radiotherapy and/or surgery have not yet been made. Ongoing or completed clinical trials have also incorporated targeted therapies in the neoadjuvant and adjuvant settings327–330. Indeed, adjuvant osimertinib is now approved for patients with completely resected stage IB–IIIA EGFR-mutant NSCLC, with a final analysis of the phase III ADAURA trial showing 5-year OS of 88% with osimertinib versus 78% with placebo (HR 0.49, 95% CI 0.33–0.73)328,331.
In individuals with an elevated germline risk of cancer, screening has an important role in disease prevention. Low-dose CT (LDCT) screening has been shown to reduce lung cancer mortality332,333 but is only approved for individuals ≥50 years of age with a current or recent heavy smoking status. Future efforts in the LCINS space include identifying a population with elevated germline and/or exposure-mediated risk to prioritize for LDCT screening (for example, individuals carrying large-effect rare variants associated with familial EGFR-mutant lung cancer), with studies already under way (such as NCT05587439 and NCT05265429). These individuals might benefit from germline genetic testing and carriers of pathogenic variants might also benefit from LDCT screening as part of future changes to clinical care.
Conclusions
LCINS is an evolving and complex disease with several risk factors and open questions. With smoking rates declining, LCINS might eventually predominate lung cancer diagnoses. Investigations with specific attention to smoking history are therefore paramount both for determining the global and national trends in LCINS epidemiology, and establishing registries with detailed exposure histories. Advances have been made in understanding LCINS biology at the somatic exome and genome levels, yet the germline contribution remains largely unexplored and large-scale sequencing studies in diverse populations are needed to define germline genetic risk. Furthermore, our understanding of environmental carcinogens continues to evolve, and future directions for research should include the mechanisms driving carcinogenesis mediated by non-tobacco-related exposures, such as environmental pollution, and their interaction with underlying germline variation. The biological distinctions between LCINS and smoking-related lung cancers necessitate a unique approach to diagnosis and treatment, with integration of environmental exposures and both germline and somatic genetics to deliver precision oncology strategies for the treatment and prevention of this increasingly important disease.
Key points.
The global incidence of lung cancer is decreasing in parallel with declining smoking rates in developed countries; however, the incidence of lung cancer in individuals who have never smoked (LCINS) is stable or increasing.
LCINS is the eighth leading cause of cancer-related mortality in the USA and the fifth most common cause of cancer-related deaths worldwide.
LCINS has histological and epidemiological distinctions from smoking-related lung cancers, occurring almost exclusively as adenocarcinomas and most commonly in women and individuals of Asian ancestry.
LCINS are highly enriched for targetable oncogenic alterations, have low tumour mutational burden and low rates of PD-L1 positivity, and lack mutational signatures, even in patients who report passive, secondhand smoke exposure.
LCINS development probably involves interactions between genetic risk, mediated by common and rare germline variants, and environmental exposures, including air pollution and particulate matter, with potential opportunities for broader lung cancer screening.
In the era of precision oncology, the biological underpinnings of LCINS necessitate unique diagnostic and treatment paradigms and warrant consideration of this disease as an important and distinct clinical entity.
Acknowledgements
The authors gratefully acknowledge M. Makarem, R. Collins, I. Odintsov and B. Johnson (at the Dana-Farber Cancer Institute) for their careful review of the manuscript and/or thoughtful discussions as well as Z. Wei and A. Tramontano (at the Dana-Farber Cancer Institute) and A. Shtein and Y. Wei (at the Harvard School of Public Health) for their assistance with data gathering and visualization.
Footnotes
Competing interests
P.A.J. declares consultancy roles for Abbvie, Accutar Biotech, Allorion Therapeutics, AstraZeneca, Bayer, Biocartis, Boehringer Ingelheim, Chugai Pharmaceuticals, Daiichi Sankyo, DualityBio, Eisai, Eli Lilly, Frontier Medicines, Hongyun Biotechnology, Merus, Mirati Therapeutics, Monte Rosa, Novartis, Nuvalent, Pfizer, Roche/Genentech, Sanofi, Scorpion Therapeutics, SFJ Pharmaceuticals, Silicon Therapeutics, Syndax, Takeda Oncology, Transcenta and Voronoi; stock ownership in Gatekeeper Pharmaceuticals; research funding from AstraZenenca, Boehringer Ingelheim, Daiichi Sankyo, Eli Lilly, PUMA Biotechnology, Revolution Medicines and Takeda Oncology; and post-marketing royalties from Dana-Farber Cancer Institute-owned intellectual property relating to EGFR mutations that has been licensed to Lab Corp. The other authors declare no competing interests.
Related links
Cancer Hotspots: https://www.cancerhotspots.org/#/home
ClinicalTrials.gov: https://clinicaltrials.gov/
GLOBOCAN: https://gco.iarc.fr/today/home
National Comprehensive Cancer Network (NCCN) Guidelines for NSCLC: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1450
OncoKB: https://www.oncokb.org/
References
- 1.Siegel RL, Miller KD, Wagle NS & Jemal A Cancer statistics, 2023. CA Cancer J. Clin 73, 17–48 (2023). [DOI] [PubMed] [Google Scholar]
- 2.Cornelius ME et al. Tobacco product use among adults - United States, 2021. MMWR Morb. Mortal. Wkly Rep 72, 475–483 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.The American Lung Association. Overall Tobacco Trends. Analysis of Centers for Disease Control and Prevention, National Center for Health Statistics. National Health Interview Survey 1965-2018. lung.org, https://www.lung.org/research/trends-in-lung-disease/tobacco-trends-brief/overall-tobacco-trends (2019). [Google Scholar]
- 4.Jeon J et al. Smoking and lung cancer mortality in the United States from 2015 to 2065: a comparative modeling approach. Ann. Intern. Med 169, 684–693 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pelosof L et al. Proportion of never-smoker non-small cell lung cancer patients at three diverse institutions. J. Natl Cancer Inst 109, djw295 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sung H et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin 71, 209–249 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Devarakonda S et al. Genomic profiling of lung adenocarcinoma in never-smokers. J. Clin. Oncol 39, 3747–3758 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ha SY et al. Lung cancer in never-smoker Asian females is driven by oncogenic mutations, most often involving EGFR. Oncotarget 6, 5465–5474 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.International Association for the Study of Lung Cancer. IASLC Language Guide https://www.iaslc.org/IASLCLanguageGuide (2021).
- 10.Pham D et al. Use of cigarette-smoking history to estimate the likelihood of mutations in epidermal growth factor receptor gene exons 19 and 21 in lung adenocarcinomas. J. Clin. Oncol 24, 1700–1704 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Thu KL et al. Lung adenocarcinoma of never smokers and smokers harbor differential regions of genetic alteration and exhibit different levels of genomic instability. PLoS ONE 7, e33003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown KF et al. The fraction of cancer attributable to modifiable risk factors in England, Wales, Scotland, Northern Ireland, and the United Kingdom in 2015. Br. J. Cancer 118, 1130–1141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Islami F et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J. Clin 68, 31–54 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Parkin DM 2. Tobacco-attributable cancer burden in the UK in 2010. Br. J. Cancer 105, S6–S13 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Islami F et al. Cancer deaths and cases attributable to lifestyle factors and infections in China, 2013. Ann. Oncol 28, 2567–2574 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Siegel DA, Fedewa SA, Henley SJ, Pollack LA & Jemal A Proportion of never smokers among men and women with lung cancer in 7 US States. JAMA Oncol. 7, 302–304 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thun MJ et al. Lung cancer occurrence in never-smokers: an analysis of 13 cohorts and 22 cancer registry studies. PLoS Med. 5, e185 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gitlitz BJ et al. The genomics of young lung cancer: comprehensive tissue genomic analysis in patients under 40 with lung cancer. JTO Clin. Res. Rep 2, 100194 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaw AT et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J. Clin. Oncol 27, 4247–4253 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong DW et al. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer 115, 1723–1733 (2009). [DOI] [PubMed] [Google Scholar]
- 21.Kim H et al. A comprehensive comparative analysis of the histomorphological features of ALK-rearranged lung adenocarcinoma based on driver oncogene mutations: frequent expression of epithelial-mesenchymal transition markers than other genotype. PLoS ONE 8, e76999 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gainor JF et al. Patterns of metastatic spread and mechanisms of resistance to crizotinib in ROS1-positive non-small-cell lung cancer. JCO Precis. Oncol 2017, PO.17.00063 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin C, Wang S, Xie W, Chang J & Gan Y The RET fusion gene and its correlation with demographic and clinicopathological features of non-small cell lung cancer: a meta-analysis. Cancer Biol. Ther 16, 1019–1028 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hess LM, Han Y, Zhu YE, Bhandari NR & Sireci A Characteristics and outcomes of patients with RET-fusion positive non-small lung cancer in real-world practice in the United States. BMC Cancer 21, 28 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mazieres J et al. Crizotinib therapy for advanced lung adenocarcinoma and a ROS1 rearrangement: results from the EUROS1 cohort. J. Clin. Oncol 33, 992–999 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Park S et al. Characteristics and outcome of ROS1-positive non-small cell lung cancer patients in routine clinical practice. J. Thorac. Oncol 13, 1373–1382 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Chen YF et al. Clinical and the prognostic characteristics of lung adenocarcinoma patients with ROS1 fusion in comparison with other driver mutations in East Asian populations. J. Thorac. Oncol 9, 1171–1179 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Farago AF et al. Clinicopathologic features of non-small-cell lung cancer harboring an NTRK gene fusion.JCO Precis. Oncol 2018, PO.18.00037 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leiter A, Veluswamy RR & Wisnivesky JP The global burden of lung cancer: current status and future trends. Nat. Rev. Clin. Oncol 20, 624–639 (2023). [DOI] [PubMed] [Google Scholar]
- 30.United States Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute. National Program of Cancer Registries and Surveillance, Epidemiology and End Results Program SEER*Stat Database: NPCR and SEER Incidence — U.S Cancer Statistics Public Use Research Database, 2021 Submission (2001-2019) www.cdc.gov/cancer/uscs/public-use (2022). [Google Scholar]
- 31.Walters KA, Li Y, Tiwari RC & Zou Z A weighted-least-squares estimation approach to comparing trends in age-adjusted cancer rates across overlapping regions. J. Data Sci 8, 631–644 (2011). [PMC free article] [PubMed] [Google Scholar]
- 32.National Cancer Institute Surveillance Research Program. SEER*Stat software version 8.4.2. — August 14, 2023 https://seer.cancer.gov/seerstat/ (2023).
- 33.Harris JE Cigarette smoking among successive birth cohorts of men and women in the United States during 1900-80. J. Natl Cancer Inst 71, 473–479 (1983). [PubMed] [Google Scholar]
- 34.Jemal A, Ma J, Rosenberg PS, Siegel R & Anderson WF Increasing lung cancer death rates among young women in southern and midwestern States. J. Clin. Oncol 30, 2739–2744 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jemal A et al. Higher lung cancer incidence in young women than young men in the United States. N. Engl. J. Med 378, 1999–2009 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health. The Health Consequences of Smoking — 50 Years of Progress: A Report of the Surgeon General (Centers for Disease Control and Prevention (US), 2014). [PubMed] [Google Scholar]
- 37.Lewis DR, Check DP, Caporaso NE, Travis WD & Devesa SS US lung cancer trends by histologic type. Cancer 120, 2883–2892 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pesch B et al. Cigarette smoking and lung cancer — relative risk estimates for the major histological types from a pooled analysis of case-control studies. Int. J. Cancer 131, 1210–1219 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Agudo A et al. Impact of cigarette smoking on cancer risk in the European prospective investigation into cancer and nutrition study. J. Clin. Oncol 30, 4550–4557 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Alexandrov LB et al. Mutational signatures associated with tobacco smoking in human cancer. Science 354, 618–622 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ogino A et al. Genomic and pathological heterogeneity in clinically diagnosed small cell lung cancer in never/light smokers identifies therapeutically targetable alterations. Mol. Oncol 15, 27–42 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maeda R et al. Influence of cigarette smoking on histological subtypes of stage I lung adenocarcinoma. J. Thorac. Oncol 6, 743–750 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Sugawara H et al. Adenocarcinoma in situ and minimally invasive adenocarcinoma in lungs of smokers: image feature differences from those in lungs of non-smokers. BMC Med. Imaging 21, 172 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Inamura K et al. EML4-ALK lung cancers are characterized by rare other mutations, a TTF-1 cell lineage, an acinar histology, and young onset. Mod. Pathol 22, 508–515 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Rodig SJ et al. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the Western population. Clin. Cancer Res 15, 5216–5223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoshida A et al. Frequent ALK rearrangement and TTF-1/p63 co-expression in lung adenocarcinoma with signet-ring cell component. Lung Cancer 72, 309–315 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Yoshida A et al. Comprehensive histologic analysis of ALK-rearranged lung carcinomas. Am. J. Surg. Pathol 35, 1226–1234 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Popat S et al. ALK translocation is associated with ALK immunoreactivity and extensive signet-ring morphology in primary lung adenocarcinoma. Lung Cancer 75, 300–305 (2012). [DOI] [PubMed] [Google Scholar]
- 49.Nishino M et al. Histologic and cytomorphologic features of ALK-rearranged lung adenocarcinomas. Mod. Pathol 25, 1462–1472 (2012). [DOI] [PubMed] [Google Scholar]
- 50.Pan Y et al. ALK, ROS1 and RET fusions in 1139 lung adenocarcinomas: a comprehensive study of common and fusion pattern-specific clinicopathologic, histologic and cytologic features. Lung Cancer 84, 121–126 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Nakada T et al. Imaging characteristics in ALK fusion-positive lung adenocarcinomas by using HRCT. Ann. Thorac. Cardiovasc. Surg 21, 102–108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoon HJ et al. Decoding tumor phenotypes for ALK, ROS1, and RET fusions in lung adenocarcinoma using a radiomics approach. Medicine 94, e1753 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Han X et al. CT features associated with EGFR mutations and ALK positivity in patients with multiple primary lung adenocarcinomas. Cancer Imaging 20, 51 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamamoto S et al. ALK molecular phenotype in non-small cell lung cancer: CT radiogenomic characterization. Radiology 272, 568–576 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Glynn C, Zakowski MF & Ginsberg MS Are there imaging characteristics associated with epidermal growth factor receptor and KRAS mutations in patients with adenocarcinoma of the lung with bronchioloalveolar features? J. Thorac. Oncol 5, 344–348 (2010). [DOI] [PubMed] [Google Scholar]
- 56.Pinheiro G et al. Identifying relationships between imaging phenotypes and lung cancer-related mutation status: EGFR and KRAS. Sci. Rep 10, 3625 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rossi G et al. Radiomic detection of EGFR mutations in NSCLC. Cancer Res. 81, 724–731 (2021). [DOI] [PubMed] [Google Scholar]
- 58.Nishino M et al. Tumor volume decrease at 8 weeks is associated with longer survival in EGFR-mutant advanced non-small-cell lung cancer patients treated with EGFR TKI. J. Thorac. Oncol 8, 1059–1068 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nishino M et al. Volumetric tumor growth in advanced non-small cell lung cancer patients with EGFR mutations during EGFR-tyrosine kinase inhibitor therapy: developing criteria to continue therapy beyond RECIST progression. Cancer 119, 3761–3768 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nishino M et al. Volumetric tumor response and progression in EGFR-mutant NSCLC patients treated with erlotinib or gefitinib. Acad. Radiol 23, 329–336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nishino M Tumor response assessment for precision cancer therapy: response evaluation criteria in solid tumors and beyond. Am. Soc. Clin. Oncol. Educ. Book 38, 1019–1029 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Park H, Sholl LM, Hatabu H, Awad MM & Nishino M Imaging of precision therapy for lung cancer: current state of the art. Radiology 293, 15–29 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nishino M et al. Tumor growth rate after nadir is associated with survival in patients with egfr-mutant non-small-cell lung cancer treated with epidermal growth factor receptor tyrosine kinase inhibitor. JCOPrecis. Oncol 5, 1603–1610 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shiri I et al. Next-generation radiogenomics sequencing for prediction of EGFR and KRAS mutation status in NSCLC patients using multimodal imaging and machine learning algorithms. Mol. Imaging Biol 22, 1132–1148 (2020). [DOI] [PubMed] [Google Scholar]
- 65.Wang G et al. Radiomics signature of brain metastasis: prediction of EGFR mutation status. Eur. Radiol 31, 4538–4547 (2021). [DOI] [PubMed] [Google Scholar]
- 66.Schuette W Treatment of brain metastases from lung cancer: chemotherapy. Lung Cancer 45, S253–257 (2004). [DOI] [PubMed] [Google Scholar]
- 67.Heon S et al. Development of central nervous system metastases in patients with advanced non-small cell lung cancer and somatic EGFR mutations treated with gefitinib or erlotinib. Clin. Cancer Res 16, 5873–5882 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Costa DB et al. Clinical experience with crizotinib in patients with advanced ALK-rearranged non-small-cell lung cancer and brain metastases. J. Clin. Oncol 33, 1881–1888 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang I, Zaorsky NG, Palmer JD, Mehra R & Lu B Targeting brain metastases in ALK-rearranged non-small-cell lung cancer. Lancet Oncol. 16, e510–521 (2015). [DOI] [PubMed] [Google Scholar]
- 70.Johung KL et al. Extended survival and prognostic factors for patients with ALK-rearranged non-small-cell lung cancer and brain metastasis. J. Clin. Oncol 34, 123–129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Soria JC et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet 389, 917–929 (2017). [DOI] [PubMed] [Google Scholar]
- 72.Peters S et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N. Engl. J. Med 377, 829–838 (2017). [DOI] [PubMed] [Google Scholar]
- 73.Shin DY et al. EGFR mutation and brain metastasis in pulmonary adenocarcinomas. J. Thorac. Oncol 9, 195–199 (2014). [DOI] [PubMed] [Google Scholar]
- 74.Rangachari D et al. Brain metastases in patients with EGFR-mutated or ALK-rearranged non-small-cell lung cancers. Lung Cancer 88, 108–111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Patil T et al. The incidence of brain metastases in stage IV ROS1-rearranged non-small cell lung cancer and rate of central nervous system progression on crizotinib. J. Thorac. Oncol 13, 1717–1726 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reungwetwattana T et al. CNS response to osimertinib versus standard epidermal growth factor receptor tyrosine kinase inhibitors in patients with untreated EGFR-mutated advanced non-small-cell lung cancer. J. Clin. Oncol 10.1200/JCO.2018.78.3118 (2018). [DOI] [PubMed] [Google Scholar]
- 77.Zhou F & Zhou C Lung cancer in never smokers-the East Asian experience. Transl. Lung Cancer Res 7, 450–463 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang T et al. Genomic and evolutionary classification of lung cancer in never smokers. Nat. Genet 53, 1348–1359 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Choudhury NJ et al. The GENIE BPC NSCLC cohort: a real-world repository integrating standardized clinical and genomic data for 1,846 patients with non-small cell lung cancer. Clin. Cancer Res 29, 3418–3428 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Harada G, Yang SR, Cocco E & Drilon A Rare molecular subtypes of lung cancer. Nat. Rev. Clin. Oncol 20, 229–249 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee B, Lee T, Lee SH, Choi YL & Han J Clinicopathologic characteristics of EGFR, KRAS, and ALK alterations in 6,595 lung cancers. Oncotarget 7, 23874–23884 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dogan S et al. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin. Cancer Res 18, 6169–6177 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee YJ et al. Dose effect of cigarette smoking on frequency and spectrum of epidermal growth factor receptor gene mutations in Korean patients with non-small cell lung cancer. J. Cancer Res. Clin. Oncol 136, 1937–1944 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Beau-Faller M et al. Rare EGFR exon 18 and exon 20 mutations in non-small-cell lung cancer on 10 117 patients: a multicentre observational study by the French ERMETIC-IFCT network. Ann. Oncol 25, 126–131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Oxnard GR et al. Natural history and molecular characteristics of lung cancers harboring EGFR exon 20 insertions. J. Thorac. Oncol 8, 179–184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Arcila ME et al. EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol. Cancer Ther 12, 220–229 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Burnett H et al. Epidemiological and clinical burden of EGFR exon 20 insertion in advanced non-small cell lung cancer: a systematic literature review. PLoS ONE 16, e0247620 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Konduri K et al. EGFR fusions as novel therapeutic targets in lung cancer. Cancer Discov. 6, 601–611 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhu YC et al. EGFR-RAD51 fusion variant in lung adenocarcinoma and response to erlotinib: a case report. Lung Cancer 115, 131–134 (2018). [DOI] [PubMed] [Google Scholar]
- 90.Guan Y et al. Effectiveness of EGFR-TKIs in a patient with lung adenocarcinoma harboring an EGFR-RAD51 fusion. Oncologist 24, 1027–1030 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yamaguchi N et al. Smoking status and self-reported race affect the frequency of clinically relevant oncogenic alterations in non-small-cell lung cancers at a United States-based academic medical practice. Lung Cancer 82, 31–37 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Couraud S et al. BioCAST/IFCT-1002: epidemiological and molecular features of lung cancer in never-smokers. Eur. Respir. J 45, 1403–1414 (2015). [DOI] [PubMed] [Google Scholar]
- 93.Solomon B, Varella-Garcia M & Camidge DR ALK gene rearrangements: a new therapeutic target in a molecularly defined subset of non-small cell lung cancer. J. Thorac. Oncol 4, 1450–1454 (2009). [DOI] [PubMed] [Google Scholar]
- 94.Chang GC et al. ALK variants, PD-L1 expression, and their association with outcomes in ALK-positive NSCLC patients. Sci. Rep 10, 21063 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Inamura K et al. EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. J. Thorac. Oncol 3, 13–17 (2008). [DOI] [PubMed] [Google Scholar]
- 96.Boland JM et al. Anaplastic lymphoma kinase immunoreactivity correlates with ALK gene rearrangement and transcriptional up-regulation in non-small cell lung carcinomas. Hum. Pathol 40, 1152–1158 (2009). [DOI] [PubMed] [Google Scholar]
- 97.Izumi H et al. The CLIP1-LTK fusion is an oncogenic driver in non-small-cell lung cancer. Nature 600, 319–323 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cooper AJ, Sequist LV, Johnson TW & Lin JJ LTK fusions: a new target emerges in non-small cell lung cancer. Cancer Cell 40, 23–25 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cadranel J et al. Therapeutic potential of afatinib in NRG1 fusion-driven solid tumors: a case series. Oncologist 26, 7–16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu SV, Minasi LAE, Herpers M & Frohn C Efficacy of afatinib in patients with advanced/metastatic solid tumors harboring NRG1 gene fusions: a novel, prospective real-world outcomes study based on single-patient protocol data. J. Clin. Oncol 40, TPS3180 (2022). [Google Scholar]
- 101.Riely GJ et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin. Cancer Res 14, 5731–5734 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kucab JE et al. A compendium of mutational signatures of environmental agents. Cell 177, 821–836.e16 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pich O et al. The mutational footprints of cancer therapies. Nat. Genet 51, 1732–1740 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ricciuti B et al. Dissecting the clinicopathologic, genomic, and immunophenotypic correlates of KRAS(G12D)-mutated non-small-cell lung cancer. Ann. Oncol 33, 1029–1040 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Frampton GM et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 5, 850–859 (2015). [DOI] [PubMed] [Google Scholar]
- 106.Schrock AB et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J. Thorac. Oncol 11, 1493–1502 (2016). [DOI] [PubMed] [Google Scholar]
- 107.Tong JH et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin. Cancer Res 22, 3048–3056 (2016). [DOI] [PubMed] [Google Scholar]
- 108.Awad MM et al. MET exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent met genomic amplification and c-Met overexpression. J. Clin. Oncol 34, 721–730 (2016). [DOI] [PubMed] [Google Scholar]
- 109.Schildhaus HU et al. MET amplification status in therapy-naive adeno- and squamous cell carcinomas of the lung. Clin. Cancer Res 21, 907–915 (2015). [DOI] [PubMed] [Google Scholar]
- 110.Okuda K, Sasaki H, Yukiue H, Yano M & Fujii Y Met gene copy number predicts the prognosis for completely resected non-small cell lung cancer. Cancer Sci. 99, 2280–2285 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Onozato R et al. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J. Thorac. Oncol 4, 5–11 (2009). [DOI] [PubMed] [Google Scholar]
- 112.Wolf J et al. Capmatinib in MET exon 14-mutated or MET-amplified non-small-cell lung cancer. N. Engl. J. Med 383, 944–957 (2020). [DOI] [PubMed] [Google Scholar]
- 113.Noonan SA et al. Identifying the appropriate FISH criteria for defining MET copy number-driven lung adenocarcinoma through oncogene overlap analysis. J. Thorac. Oncol 11, 1293–1304 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Le X et al. Tepotinib in patients with non-small cell lung cancer with high-level MET amplification detected by liquid biopsy: VISION Cohort B. Cell Rep. Med 4, 101280 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yu HA et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res 19, 2240–2247 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chabon JJ et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat. Commun 7, 11815 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Dagogo-Jack I et al. MET alterations are a recurring and actionable resistance mechanism in ALK-positive lung cancer. Clin. Cancer Res 26, 2535–2545 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kron A et al. Genetic heterogeneity of MET-aberrant NSCLC and its impact on the outcome of immunotherapy. J. Thorac. Oncol 16, 572–582 (2021). [DOI] [PubMed] [Google Scholar]
- 119.Plenker D et al. Structural alterations of MET trigger response to MET kinase inhibition in lung adenocarcinoma patients. Clin. Cancer Res 24, 1337–1343 (2018). [DOI] [PubMed] [Google Scholar]
- 120.Cho JH et al. KIF5B-MET gene rearrangement with robust antitumor activity in response to crizotinib in lung adenocarcinoma. J. Thorac. Oncol 13, e29–e31 (2018). [DOI] [PubMed] [Google Scholar]
- 121.Liu LF, Deng JY, Lizaso A, Lin J & Sun S Effective response to crizotinib of concurrent KIF5B-MET and MET-CDR2-rearranged non-small cell lung cancer: a case report. World J. Clin. Cases 10, 2529–2536 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stephens P et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature 431, 525–526 (2004). [DOI] [PubMed] [Google Scholar]
- 123.Barlesi F et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 387, 1415–1426 (2016). [DOI] [PubMed] [Google Scholar]
- 124.Arcila ME et al. Prevalence, clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin. Cancer Res 18, 4910–4918 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhao J & Xia Y Targeting HER2 alterations in non-small-cell lung cancer: a comprehensive review. JCO Precis. Oncol 4, 411–425 (2020). [DOI] [PubMed] [Google Scholar]
- 126.Ou SI et al. HER2 transmembrane domain (TMD) mutations (V659/G660) that stabilize homo- and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J. Thorac. Oncol 12, 446–457 (2017). [DOI] [PubMed] [Google Scholar]
- 127.Pahuja KB et al. Actionable activating oncogenic ERBB2/HER2 transmembrane and juxtamembrane domain mutations. Cancer Cell 34, 792–806.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Li BT et al. Trastuzumab deruxtecan in HER2-mutant non-small-cell lung cancer. N. Engl. J. Med 386, 241–251 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Goto K. et al. Trastuzumab deruxtecan in patients with HER2-mutant metastatic non-small-cell lung cancer: primary results from the randomized, phase II DESTINY-Lung02 trial. J. Clin. Oncol 41, 4852–4863 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Du Z. et al. Structure-function analysis of oncogenic EGFR kinase domain duplication reveals insights into activation and a potential approach for therapeutic targeting. Nat. Commun 12, 1382 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Govindan R. et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 150, 1121–1134 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang X. et al. Association between smoking history and tumor mutation burden in advanced non-small cell lung cancer. Cancer Res. 81, 2566–2573 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Schrock AB et al. Pulmonary sarcomatoid carcinomas commonly harbor either potentially targetable genomic alterations or high tumor mutational burden as observed by comprehensive genomic profiling. J. Thorac. Oncol 12, 932–942 (2017). [DOI] [PubMed] [Google Scholar]
- 134.Skoulidis F. et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 8, 822–835 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rizvi NA et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Calles A. et al. Expression of PD-1 and its ligands, PD-L1 and PD-L2, in smokers and never smokers with KRAS-mutant lung cancer. J. Thorac. Oncol 10, 1726–1735 (2015). [DOI] [PubMed] [Google Scholar]
- 137.Tseng JS et al. Characteristics and predictive value of PD-L1 status in real-world non-small cell lung cancer patients. J. Immunother 41, 292–299 (2018). [DOI] [PubMed] [Google Scholar]
- 138.Sabari JK et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann. Oncol 29, 2085–2091 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Otano I, Ucero AC, Zugazagoitia J & Paz-Ares L At the crossroads of immunotherapy for oncogene-addicted subsets of NSCLC. Nat. Rev. Clin. Oncol 20, 143–159 (2023). [DOI] [PubMed] [Google Scholar]
- 140.Evans M et al. The clinicopathological and molecular associations of PD-L1 expression in non-small cell lung cancer: analysis of a series of 10,005 cases tested with the 22c3 assay. Pathol. Oncol. Res 26, 79–89 (2020). [DOI] [PubMed] [Google Scholar]
- 141.Lee J et al. PD-L1 expression in ROS1-rearranged non-small cell lung cancer: a study using simultaneous genotypic screening of EGFR, ALK, and ROS1. Thorac. Cancer 10, 103–110 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Negrao MV et al. Oncogene-specific differences in tumor mutational burden, PD-L1 expression, and outcomes from immunotherapy in non-small cell lung cancer. J. Immunother. Cancer 9, e002891 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Koh J et al. EML4-ALK enhances programmed cell death-ligand 1 expression in pulmonary adenocarcinoma via hypoxia-inducible factor (HIF)-1α and STAT3. Oncoimmunology 5, e1108514 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Shen J et al. PD-L1 expression is associated with ALK positivity and STAT3 activation, but not outcome in patients with systemic anaplastic large cell lymphoma. Mod. Pathol 33, 324–333 (2020). [DOI] [PubMed] [Google Scholar]
- 145.Nik-Zainal S et al. The genome as a record of environmental exposure. Mutagenesis 30, 763–770 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yoshida K et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature 578, 266–272 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kim SH, Hwang WJ, Cho JS & Kang DR Attributable risk of lung cancer deaths due to indoor radon exposure. Ann. Occup. Environ. Med 28, 8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mucci LA et al. Familial risk and heritability of cancer among twins in Nordic countries. JAMA 315, 68–76 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bosse Y & Amos CI A decade of GWAS results in lung cancer. Cancer Epidemiol. Biomark. Prev 27, 363–379 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mukherjee S et al. Germline pathogenic variants impact clinicopathology of advanced lung cancer. Cancer Epidemiol. Biomark. Prev 31, 1450–1459 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Stadler ZK et al. Therapeutic implications of germline testing in patients with advanced cancers. J. Clin. Oncol 39, 2698–2709 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Matakidou A, Eisen T & Houlston RS Systematic review of the relationship between family history and lung cancer risk. Br. J. Cancer 93, 825–833 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gorlova OY et al. Never smokers and lung cancer risk: a case-control study of epidemiological factors. Int. J. Cancer 118, 1798–1804 (2006). [DOI] [PubMed] [Google Scholar]
- 154.Byun J et al. The shared genetic architectures between lung cancer and multiple polygenic phenotypes in genome-wide association studies. Cancer Epidemiol. Biomark. Prev 30, 1156–1164 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Amos CI et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat. Genet 40, 616–622 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hung RJ et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 452, 633–637 (2008). [DOI] [PubMed] [Google Scholar]
- 157.Thorgeirsson TE et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature 452, 638–642 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Liu P et al. Familial aggregation of common sequence variants on 15q24-25.1 in lung cancer. J. Natl Cancer Inst 100, 1326–1330 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Shi J et al. Genome-wide association study of lung adenocarcinoma in East Asia and comparison with a European population. Nat. Commun 14, 3043 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Li Y et al. Genetic variants and risk of lung cancer in never smokers: a genome-wide association study. Lancet Oncol 11, 321–330 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.McKay JD et al. Lung cancer susceptibility locus at 5p15.33. Nat. Genet 40, 1404–1406 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hsiung CA et al. The 5p15.33 locus is associated with risk of lung adenocarcinoma in never-smoking females in Asia. PLoS Genet 6, e1001051 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lan Q et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat. Genet 44, 1330–1335 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Rafnar T et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat. Genet 41, 221–227 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wu X. et al. Genome-wide association study of genetic predictors of overall survival for non-small cell lung cancer in never smokers. Cancer Res 73, 4028–4038 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Huang YT et al. Genome-wide analysis of survival in early-stage non-small-cell lung cancer. J. Clin. Oncol 27, 2660–2667 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wu X et al. Genome-wide association study of survival in non-small cell lung cancer patients receiving platinum-based chemotherapy. J. Natl Cancer Inst 103, 817–825 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sato Y et al. Genome-wide association study on overall survival of advanced non-small cell lung cancer patients treated with carboplatin and paclitaxel. J. Thorac. Oncol 6, 132–138 (2011). [DOI] [PubMed] [Google Scholar]
- 169.Yoon KA et al. A genome-wide association study reveals susceptibility variants for non-small cell lung cancer in the Korean population. Hum. Mol. Genet 19, 4948–4954 (2010). [DOI] [PubMed] [Google Scholar]
- 170.Hymowitz N, Ploshnick A, Laemle L & Brezenoff H Effects of repeated administration of soman on schedule-controlled behavior and brain in the rat. Neurotoxicol. Teratol 12, 47–56 (1990). [DOI] [PubMed] [Google Scholar]
- 171.Ahn MJ et al. The 18p11.22 locus is associated with never smoker non-small cell lung cancer susceptibility in Korean populations. Hum. Genet 131, 365–372 (2012). [DOI] [PubMed] [Google Scholar]
- 172.Miki D et al. Variation in TP63 is associated with lung adenocarcinoma susceptibility in Japanese and Korean populations. Nat. Genet 42, 893–896 (2010). [DOI] [PubMed] [Google Scholar]
- 173.Wang Y et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat. Genet 46, 736–741 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang Z et al. Meta-analysis of genome-wide association studies identifies multiple lung cancer susceptibility loci in never-smoking Asian women. Hum. Mol. Genet 25, 620–629 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kim JH et al. Genome-wide association study of lung cancer in Korean non-smoking women. J. Korean Med. Sci 28, 840–847 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Hosgood HD III et al. Interactions between household air pollution and GWAS-identified lung cancer susceptibility markers in the Female Lung Cancer Consortium in Asia (FLCCA). Hum. Genet 134, 333–341 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ji X et al. Protein-altering germline mutations implicate novel genes related to lung cancer development. Nat. Commun 11, 2220 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gaughan EM, Cryer SK, Yeap BY, Jackman DM & Costa DB Family history of lung cancer in never smokers with non-small-cell lung cancer and its association with tumors harboring EGFR mutations. Lung Cancer 79, 193–197 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Cheng YI et al. Potential genetic modifiers for somatic EGFR mutation in lung cancer: a meta-analysis and literature review. BMC Cancer 19, 1068 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Carrot-Zhang J et al. Genetic ancestry contributes to somatic mutations in lung cancers from admixed Latin American populations. Cancer Discov. 11, 591–598 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kachuri L et al. Pan-cancer analysis demonstrates that integrating polygenic risk scores with modifiable risk factors improves risk prediction. Nat. Commun 11, 6084 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hung RJ et al. Assessing lung cancer absolute risk trajectory based on a polygenic risk model. Cancer Res. 81, 1607–1615 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kapoor PM et al. Combined associations of a polygenic risk score and classical risk factors with breast cancer risk. J. Natl Cancer Inst 113, 329–337 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.van den Broek JJ et al. Personalizing breast cancer screening based on polygenic risk and family history. J. Natl Cancer Inst 113, 434–442 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hoadley KA et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Priestley P et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210–216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hartwig Medical Foundation. Hartwig Data Catalogue https://www.hartwigmedicalfoundation.nl/en/data/database/ (2023).
- 188.ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Chen J et al. Genomic landscape of lung adenocarcinoma in East Asians. Nat. Genet 52, 177–186 (2020). [DOI] [PubMed] [Google Scholar]
- 190.Tlemsani C et al. Whole-exome sequencing reveals germline-mutated small cell lung cancer subtype with favorable response to DNA repair-targeted therapies. Sci. Transl. Med 13, eabc7488 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lu T et al. Individuals with common diseases but with a low polygenic risk score could be prioritized for rare variant screening. Genet. Med 23, 508–515 (2021). [DOI] [PubMed] [Google Scholar]
- 192.Tian R et al. Clonal hematopoiesis and risk of incident lung cancer. J. Clin. Oncol 41, 1423–1433 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bell DW et al. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat. Genet 37, 1315–1316 (2005). [DOI] [PubMed] [Google Scholar]
- 194.Gazdar A et al. Hereditary lung cancer syndrome targets never smokers with germline EGFR gene T790M mutations. J. Thorac. Oncol 9, 456–463 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Thomas A et al. Concurrent molecular alterations in tumors with germ line epidermal growth factor receptor T790M mutations. Clin. Lung Cancer 14, 452–456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Yu HA et al. Germline EGFR T790M mutation found in multiple members of a familial cohort. J. Thorac. Oncol 9, 554–558 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Oxnard G. et al. OA 06.02 final report of the INHERIT EGFR Study — 33 unrelated kindreds carrying germline EGFR mutations. J. Thorac. Oncol 12, S1758 (2017). [Google Scholar]
- 198.Girard N. et al. Analysis of genetic variants in never-smokers with lung cancer facilitated by an Internet-based blood collection protocol: a preliminary report. Clin. Cancer Res 16, 755–763 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Chen S. et al. A genome-wide mutational constraint map quantified from variation in 76,156 human genomes. Nature 625, 92–100 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Berry DK et al. Clinical cohort analysis of germline EGFR T790M demonstrates penetrance across ethnicities and races, sexes, and ages. JCO Precis. Oncol 4, PO.19.00297 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Sovich JL et al. Lung adenocarcinoma associated with germline EGFR R776H variant: a case report and review of the literature. JCO Precis. Oncol 6, e2100559 (2022). [DOI] [PubMed] [Google Scholar]
- 202.Guo T. et al. Two cases of non-small cell lung cancer patients with somatic or germline EGFR R776H mutation. Lung Cancer 161, 94–97 (2021). [DOI] [PubMed] [Google Scholar]
- 203.Hellmann MD et al. Identification and functional characterization of EGFR V769M, a novel germline variant associated with multiple lung adenocarcinomas. JCO Precis. Oncol 1, PO.16.00019 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ikeda K, Nomori H, Mori T, Sasaki J & Kobayashi T Novel germline mutation: EGFR V843I in patient with multiple lung adenocarcinomas and family members with lung cancer. Ann. Thorac. Surg 85, 1430–1432 (2008). [DOI] [PubMed] [Google Scholar]
- 205.Ohtsuka K. et al. Familial lung adenocarcinoma caused by the EGFR V843I germ-line mutation. J. Clin. Oncol 29, e191–2 (2011). [DOI] [PubMed] [Google Scholar]
- 206.van der Leest C. et al. Novel EGFR V834L germline mutation associated with familial lung adenocarcinoma. JCO Precis. Oncol 2, PO.17.00266 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lu S. et al. EGFR and ERBB2 germline mutations in Chinese lung cancer patients and their roles in genetic susceptibility to cancer. J. Thorac. Oncol 14, 732–736 (2019). [DOI] [PubMed] [Google Scholar]
- 208.Lin X. et al. Identification of the unique clinical and genetic features of Chinese lung cancer patients with EGFR germline mutations in a large-scale retrospective study. Front. Oncol 11, 774156 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Qian K. et al. A novel germline EGFR variant p.R831H causes predisposition to familial CDK12-mutant prostate cancer with tandem duplicator phenotype. Oncogene 39, 6871–6878 (2020). [DOI] [PubMed] [Google Scholar]
- 210.Yamamoto H. et al. Novel germline mutation in the transmembrane domain of HER2 in familial lung adenocarcinomas. J. Natl Cancer Inst 106, djt338 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Yamamoto H. et al. Therapeutic potential of afatinib for cancers with ERBB2 (HER2) transmembrane domain mutations G660D and V659E. Oncologist 23, 150–154 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Yamamoto H, Yatabe Y & Toyooka S Inherited lung cancer syndromes targeting never smokers. Transl. Lung Cancer Res 7, 498–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Tode N. et al. Exome sequencing deciphers a germline MET mutation in familial epidermal growth factor receptor-mutant lung cancer. Cancer Sci. 108, 1263–1270 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Chen HY et al. R331W missense mutation of oncogene yap1 is a germline risk allele for lung adenocarcinoma with medical actionability. J. Clin. Oncol 33, 2303–2310 (2015). [DOI] [PubMed] [Google Scholar]
- 215.Chaib I. et al. Co-activation of STAT3 and YES-associated protein 1 (YAP1) pathway in EGFR-mutant NSCLC. J. Natl Cancer Inst 109, djx014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kurppa KJ et al. Treatment-induced tumor dormancy through YAP-mediated transcriptional reprogramming of the apoptotic pathway. Cancer Cell 37, 104–122.e12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Furuya M. et al. Pulmonary neoplasms in patients with Birt-Hogg-Dube syndrome: histopathological features and genetic and somatic events. PLoS ONE 11, e0151476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Jia Y. et al. Successful treatment of a patient with Li-Fraumeni syndrome and metastatic lung adenocarcinoma harboring synchronous EGFR L858R and ERBB2 extracellular domain S310F mutations with the pan-HER inhibitor afatinib. Cancer Biol. Ther 15, 970–974 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Michalarea V. et al. EGFR-mutated lung cancer in Li-Fraumeni syndrome. Lung Cancer 85, 485–487 (2014). [DOI] [PubMed] [Google Scholar]
- 220.Mezquita L. et al. High prevalence of somatic oncogenic driver alterations in patients with NSCLC and Li-fraumeni syndrome. J. Thorac. Oncol 15, 1232–1239 (2020). [DOI] [PubMed] [Google Scholar]
- 221.Kerrigan K. et al. Lung cancer in Li-fraumeni syndrome. JCO Precis. Oncol 5, PO.20.00468 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Boespflug A. et al. Primary lung adenocarcinoma occurring in a PTEN related syndrome (Cowden’s disease): routine EGFR sequencing also highlights two rare somatic mutations S768I and V769L. Lung Cancer 79, 318–320 (2013). [DOI] [PubMed] [Google Scholar]
- 223.Kleinerman RA et al. Hereditary retinoblastoma and risk of lung cancer. J. Natl Cancer Inst 92, 2037–2039 (2000). [DOI] [PubMed] [Google Scholar]
- 224.Takemiya M, Shiraishi S, Teramoto T & Miki Y Bloom’s syndrome with porokeratosis of Mibelli and multiple cancers of the skin, lung and colon. Clin. Genet 31, 35–44 (1987). [DOI] [PubMed] [Google Scholar]
- 225.Yamanaka A, Hirai T, Ohtake Y & Kitagawa M Lung cancer associated with Werner’s syndrome: a case report and review of the literature. Jpn J. Clin. Oncol 27, 415–418 (1997). [DOI] [PubMed] [Google Scholar]
- 226.Zeeb H, Shannoun F & World Health Organization. WHO Handbook on Indoor Radon: A Public Health Perspective (WHO, 2009). [PubMed] [Google Scholar]
- 227.National Research Council (US) Committee on Health Risks of Exposure to Radon (BEIR VI). Health Effects of Exposure to Radon: BEIR VI (National Academies Press, 1999). [PubMed] [Google Scholar]
- 228.Lubin JH et al. Lung cancer in radon-exposed miners and estimation of risk from indoor exposure. J. Natl Cancer Inst 87, 817–827 (1995). [DOI] [PubMed] [Google Scholar]
- 229.National Research Council (US) Committee on the Biological Effects of Ionizing Radiations. Health Risks of Radon and Other Internally Deposited Alpha-Emitters: Beir IV (National Academies Press, 1988). [PubMed] [Google Scholar]
- 230.Lubin JH & Boice JD Jr Lung cancer risk from residential radon: meta-analysis of eight epidemiologic studies. J. Natl Cancer Inst 89, 49–57 (1997). [DOI] [PubMed] [Google Scholar]
- 231.Lubin JH et al. Estimating lung cancer mortality from residential radon using data for low exposures of miners. Radiat. Res 147, 126–134 (1997). [PubMed] [Google Scholar]
- 232.Krewski D. et al. A combined analysis of North American case-control studies of residential radon and lung cancer. J. Toxicol. Environ. Health A 69, 533–597 (2006). [DOI] [PubMed] [Google Scholar]
- 233.Zhang ZL et al. Residential radon and lung cancer risk: an updated meta-analysis of case-control studies. Asian Pac. J. Cancer Prev 13, 2459–2465 (2012). [DOI] [PubMed] [Google Scholar]
- 234.Darby S. et al. Radon in homes and risk of lung cancer: collaborative analysis of individual data from 13 European case-control studies. BMJ 330, 223 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Krewski D. et al. Residential radon and risk of lung cancer: a combined analysis of 7 North American case-control studies. Epidemiology 16, 137–145 (2005). [DOI] [PubMed] [Google Scholar]
- 236.Lubin JH et al. Risk of lung cancer and residential radon in China: pooled results of two studies. Int. J. Cancer 109, 132–137 (2004). [DOI] [PubMed] [Google Scholar]
- 237.Garzillo C, Pugliese M, Loffredo F & Quarto M Indoor radon exposure and lung cancer risk: a meta-analysis of case-control studies. Transl. Cancer Res 6, S934–S943 (2017). [Google Scholar]
- 238.Turner MC et al. Radon and lung cancer in the American Cancer Society cohort. Cancer Epidemiol. Biomark. Prev 20, 438–448 (2011). [DOI] [PubMed] [Google Scholar]
- 239.Ngoc LTN, Park D & Lee YC Human health impacts of residential radon exposure: updated systematic review and meta-analysis of case-control studies. Int. J. Environ. Res. Public Health 20, 97 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.United States Environmental Protection Agency. A Citizen’s Guide to Radon: The Guide to Protecting Yourself and Your Family from Radon https://www.epa.gov/sites/default/files/2016-12/documents/2016_a_citizens_guide_to_radon.pdf (2012). [Google Scholar]
- 241.United States Environmental Protection Agency (EPA). The National Radon Action Plan – A Strategy for Saving Lives. epa.gov, https://www.epa.gov/radon/national-radon-action-plan-strategy-saving-lives (2023). [Google Scholar]
- 242.Lee ME, Lichtenstein E, Andrews JA, Glasgow RE & Hampson SE Radon-smoking synergy: a population-based behavioral risk reduction approach. Prev. Med 29, 222–227 (1999). [DOI] [PubMed] [Google Scholar]
- 243.United States Environmental Protection Agency (EPA). Assessment of Risks from Radon in Homes (EPA Report No. 402-R-03-003) (Office of Radiation and Indoor Air, 2003). [Google Scholar]
- 244.Roscoe RJ, Steenland K, Halperin WE, Beaumont JJ & Waxweiler RJ Lung cancer mortality among nonsmoking uranium miners exposed to radon daughters. JAMA 262, 629–633 (1989). [PubMed] [Google Scholar]
- 245.Li C. et al. Residential radon and histological types of lung cancer: a meta-analysis of case-control studies. Int. J. Environ. Res. Public Health 17, 1457 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Yao SX et al. Exposure to radon progeny, tobacco use and lung cancer in a case-control study in southern China. Radiat. Res 138, 326–336 (1994). [PubMed] [Google Scholar]
- 247.Rodriguez-Martinez A, Torres-Duran M, Barros-Dios JM & Ruano-Ravina A Residential radon and small cell lung cancer. A systematic review. Cancer Lett. 426, 57–62 (2018). [DOI] [PubMed] [Google Scholar]
- 248.Cheng ES et al. Systematic review and meta-analysis of residential radon and lung cancer in never-smokers. Eur. Respir. Rev 30, 200230 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Bennett WP et al. Environmental tobacco smoke, genetic susceptibility, and risk of lung cancer in never-smoking women. J. Natl Cancer Inst 91, 2009–2014 (1999). [DOI] [PubMed] [Google Scholar]
- 250.Kiyohara C. et al. Risk modification by CYP1A1 and GSTM1 polymorphisms in the association of environmental tobacco smoke and lung cancer: a case-control study in Japanese nonsmoking women. Int. J. Cancer 107, 139–144 (2003). [DOI] [PubMed] [Google Scholar]
- 251.Gorlova OY et al. DNA repair capacity and lung cancer risk in never smokers. Cancer Epidemiol. Biomark. Prev 17, 1322–1328 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Spitz MR et al. Variants in inflammation genes are implicated in risk of lung cancer in never smokers exposed to second-hand smoke. Cancer Discov. 1, 420–429 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Hackshaw AK, Law MR & Wald NJ The accumulated evidence on lung cancer and environmental tobacco smoke. BMJ 315, 980–988 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Lee CH et al. Lifetime environmental exposure to tobacco smoke and primary lung cancer of non-smoking Taiwanese women. Int. J. Epidemiol 29, 224–231 (2000). [DOI] [PubMed] [Google Scholar]
- 255.Zhong L, Goldberg MS, Parent ME & Hanley JA Exposure to environmental tobacco smoke and the risk of lung cancer: a meta-analysis. Lung Cancer 27, 3–18 (2000). [DOI] [PubMed] [Google Scholar]
- 256.Johnson KC, Hu J, Mao Y & Canadian Cancer Registries Epidemiology Research Group. Lifetime residential and workplace exposure to environmental tobacco smoke and lung cancer in never-smoking women, Canada 1994-97. Int. J. Cancer 93, 902–906 (2001). [DOI] [PubMed] [Google Scholar]
- 257.Wang A. et al. Active and passive smoking in relation to lung cancer incidence in the Women’s Health Initiative prospective cohort study. J. Clin. Oncol 31, 1504 (2013). 1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Couraud S. et al. No impact of passive smoke on the somatic profile of lung cancers in never-smokers. Eur. Respir. J 45, 1415–1425 (2015). [DOI] [PubMed] [Google Scholar]
- 259.International Agency for Research on Cancer, World Health Organization. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans: Tobacco Smoke and Involuntary Smoking Vol. 83 (IARC, WHO, 2004). [PMC free article] [PubMed] [Google Scholar]
- 260.Office of the Surgeon General, Office on Smoking and Health (US). The Health Consequences of Smoking: A Report of the Surgeon General (Centers for Disease Control and Prevention, 2004). [Google Scholar]
- 261.Olsson AC et al. Exposure to diesel motor exhaust and lung cancer risk in a pooled analysis from case-control studies in Europe and Canada. Am. J. Respir. Crit. Care Med 183, 941–948 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Silverman DT et al. The diesel exhaust in miners study: a nested case-control study of lung cancer and diesel exhaust. J. Natl Cancer Inst 104, 855–868 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Raaschou-Nielsen O. et al. Air pollution and lung cancer incidence in 17 European cohorts: prospective analyses from the European Study of Cohorts for Air Pollution Effects (ESCAPE). Lancet Oncol. 14, 813–822 (2013). [DOI] [PubMed] [Google Scholar]
- 264.GBD 2019 Respiratory Tract Cancers Collaborators Global, regional, and national burden of respiratory tract cancers and associated risk factors from 1990 to 2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Respir. Med 9, 1030–1049 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Huang Y. et al. Air pollution, genetic factors, and the risk of lung cancer: a prospective study in the UK biobank. Am. J. Respir. Crit. Care Med 204, 817–825 (2021). [DOI] [PubMed] [Google Scholar]
- 266.Krewski D. et al. Extended follow-up and spatial analysis of the American Cancer Society study linking particulate air pollution and mortality. Res. Rep. Health Eff. Inst 140, 5–114 (2009). [PubMed] [Google Scholar]
- 267.Turner MC et al. Long-term ambient fine particulate matter air pollution and lung cancer in a large cohort of never-smokers. Am. J. Respir. Crit. Care Med 184, 1374–1381 (2011). [DOI] [PubMed] [Google Scholar]
- 268.International Agency for Research on Cancer, World Health Organization. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans: Outdoor Air Pollution Vol. 109 (IARC, WHO, 2016). [PMC free article] [PubMed] [Google Scholar]
- 269.Brook RD et al. Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation 121, 2331–2378 (2010). [DOI] [PubMed] [Google Scholar]
- 270.Cohen AJ et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015. Lancet 389, 1907–1918 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Turner MC et al. Interactions between cigarette smoking and fine particulate matter in the Risk of Lung Cancer Mortality in Cancer Prevention Study II. Am. J. Epidemiol 180, 1145–1149 (2014). [DOI] [PubMed] [Google Scholar]
- 272.Myers R. et al. High-ambient air pollution exposure among never smokers versus ever smokers with lung cancer. J. Thorac. Oncol 16, 1850–1858 (2021). [DOI] [PubMed] [Google Scholar]
- 273.Hoffmann B. et al. WHO Air Quality Guidelines 2021 — aiming for healthier air for all: a joint statement by medical, public health, scientific societies and patient representative organisations. Int. J. Public Health 66, 1604465 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Shi L. et al. Low-concentration PM2.5 and mortality: estimating acute and chronic effects in a population-based study. Environ. Health Perspect 124, 46–52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.United States Environmental Protection Agency. National Ambient Air Quality Standards (NAAQS) for PM https://www.epa.gov/pm-pollution/national-ambient-air-quality-standards-naaqs-pm (2023).
- 276.Di Q. et al. Daily and Annual PM2.5 Concentrations for the Contiguous United States, 1-km Grids, v1 (2000–2016) (NASA Socioeconomic Data and Applications Center, 2021). [Google Scholar]
- 277.Inness A. et al. The CAMS reanalysis of atmospheric composition. Atmos. Chem. Phys 19, 3515–3556 (2019). [Google Scholar]
- 278.Christiani DC Ambient air pollution and lung cancer: nature and nurture. Am. J. Respir. Crit. Care Med 204, 752–753 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Hill W. et al. Lung adenocarcinoma promotion by air pollutants. Nature 616, 159–167 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Jbaily A. et al. Air pollution exposure disparities across US population and income groups. Nature 601, 228–233 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Loomis D, Guha N, Hall AL & Straif K Identifying occupational carcinogens: an update from the IARC Monographs. Occup. Environ. Med 75, 593–603 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Ge C. et al. Respirable crystalline silica exposure, smoking, and lung cancer subtype risks. a pooled analysis of case-control studies. Am. J. Respir. Crit. Care Med 202, 412–421 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Ge C. et al. Diesel engine exhaust exposure, smoking, and lung cancer subtype risks. a pooled exposure-response analysis of 14 case-control studies. Am. J. Respir. Crit. Care Med 202, 402–411 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Honaryar MK et al. Welding fumes and lung cancer: a meta-analysis of case-control and cohort studies. Occup. Environ. Med 76, 422–431 (2019). [DOI] [PubMed] [Google Scholar]
- 285.Banks DE et al. American College of Chest Physicians consensus statement on the respiratory health effects of asbestos. Results of a Delphi study. Chest 135, 1619–1627 (2009). [DOI] [PubMed] [Google Scholar]
- 286.Markowitz SB, Levin SM, Miller A & Morabia A Asbestos, asbestosis, smoking, and lung cancer. New findings from the North American insulator cohort. Am. J. Respir. Crit. Care Med 188, 90–96 (2013). [DOI] [PubMed] [Google Scholar]
- 287.Bridda A, Padoan I, Mencarelli R & Frego M Peritoneal mesothelioma: a review. MedGenMed 9, 32 (2007). [PMC free article] [PubMed] [Google Scholar]
- 288.Mazurek JM, Syamlal G, Wood JM, Hendricks SA & Weston A Malignant mesothelioma mortality — United States, 1999-2015. MMWR Morb. Mortal. Wkly Rep 66, 214–218 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Selikoff IJ, Hammond EC & Churg J Asbestos exposure, smoking, and neoplasia. JAMA 204, 106–112 (1968). [PubMed] [Google Scholar]
- 290.Lee PN Relation between exposure to asbestos and smoking jointly and the risk of lung cancer. Occup. Environ. Med 58, 145–153 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Liddell FD The interaction of asbestos and smoking in lung cancer. Ann. Occup. Hyg 45, 341–356 (2001). [PubMed] [Google Scholar]
- 292.Ngamwong Y. et al. Additive synergism between asbestos and smoking in lung cancer risk: a systematic review and meta-analysis. PLoS ONE 10, e0135798 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Nelson HH et al. k-ras mutation and occupational asbestos exposure in lung adenocarcinoma: asbestos-related cancer without asbestosis. Cancer Res. 59, 4570–4573 (1999). [PubMed] [Google Scholar]
- 294.Kwak K, Kang D & Paek D Environmental exposure to asbestos and the risk of lung cancer: a systematic review and meta-analysis. Occup. Environ. Med 79, 207–214 (2022). [DOI] [PubMed] [Google Scholar]
- 295.Murray RP, Connett JE & Zapawa LM Does nicotine replacement therapy cause cancer? Evidence from the Lung Health Study. Nicotine Tob. Res 11, 1076–1082 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Lee PN & Fariss MW A systematic review of possible serious adverse health effects of nicotine replacement therapy. Arch. Toxicol 91, 1565–1594 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Sheikh M, Mukeriya A, Shangina O, Brennan P & Zaridze D Postdiagnosis smoking cessation and reduced risk for lung cancer progression and mortality: a prospective cohort study. Ann. Intern. Med 174, 1232–1239 (2021). [DOI] [PubMed] [Google Scholar]
- 298.Fares AF et al. Association between duration of smoking abstinence before non-small-cell lung cancer diagnosis and survival: a retrospective, pooled analysis of cohort studies. Lancet Public Health 8, e691–e700 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Caini S. et al. Quitting smoking at or around diagnosis improves the overall survival of lung cancer patients: a systematic review and meta-analysis. J. Thorac. Oncol 17, 623–636 (2022). [DOI] [PubMed] [Google Scholar]
- 300.Godtfredsen NS, Prescott E & Osler M Effect of smoking reduction on lung cancer risk. JAMA 294, 1505–1510 (2005). [DOI] [PubMed] [Google Scholar]
- 301.Christiani DC Vaping-induced acute lung injury. N. Engl. J. Med 382, 960–962 (2020). [DOI] [PubMed] [Google Scholar]
- 302.Kosmider L. et al. Carbonyl compounds in electronic cigarette vapors: effects of nicotine solvent and battery output voltage. Nicotine Tob. Res 16, 1319–1326 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Mulder HA et al. The effect of electronic cigarette user modifications and e-liquid adulteration on the particle size profile of an aerosolized product. Sci. Rep 9, 10221 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Lalo H, Leclerc L, Sorin J & Pourchez J Aerosol droplet-size distribution and airborne nicotine portioning in particle and gas phases emitted by electronic cigarettes. Sci. Rep 10, 21707 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Goniewicz ML et al. Levels of selected carcinogens and toxicants in vapour from electronic cigarettes. Tob. Control 23, 133–139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Jensen RP, Luo W, Pankow JF, Strongin RM & Peyton DH Hidden formaldehyde in e-cigarette aerosols. N. Engl. J. Med 372, 392–394 (2015). [DOI] [PubMed] [Google Scholar]
- 307.Laino T. et al. Mechanisms of propylene glycol and triacetin pyrolysis. J. Phys. Chem. A 116, 4602–4609 (2012). [DOI] [PubMed] [Google Scholar]
- 308.Ingebrethsen BJ, Cole SK & Alderman SL Electronic cigarette aerosol particle size distribution measurements. Inhal. Toxicol 24, 976–984 (2012). [DOI] [PubMed] [Google Scholar]
- 309.Eversole A. et al. E-cigarette solvent ratio and device power influence ambient air particulate matter. Tob. Regul. Sci 7, 177–183 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.International Agency for Research on Cancer, World Health Organization. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans: Household Use of Solid Fuels and High-temperature Frying Vol. 95 (IARC, WHO, 2010). [PMC free article] [PubMed] [Google Scholar]
- 311.Jia PL et al. The risk of lung cancer among cooking adults: a meta-analysis of 23 observational studies. J. Cancer Res. Clin. Oncol 144, 229–240 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Xue Y, Jiang Y, Jin S & Li Y Association between cooking oil fume exposure and lung cancer among Chinese nonsmoking women: a meta-analysis. Onco Targets Ther. 9, 2987–2992 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Yu IT, Chiu YL, Au JS, Wong TW & Tang JL Dose-response relationship between cooking fumes exposures and lung cancer among Chinese nonsmoking women. Cancer Res. 66, 4961–4967 (2006). [DOI] [PubMed] [Google Scholar]
- 314.Barone-Adesi F. et al. Risk of lung cancer associated with domestic use of coal in Xuanwei, China: retrospective cohort study. BMJ 345, e5414 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Kurmi OP, Arya PH, Lam KB, Sorahan T & Ayres JG Lung cancer risk and solid fuel smoke exposure: a systematic review and meta-analysis. Eur. Respir. J 40, 1228–1237 (2012). [DOI] [PubMed] [Google Scholar]
- 316.Naeher LP et al. Woodsmoke health effects: a review. Inhal. Toxicol 19, 67–106 (2007). [DOI] [PubMed] [Google Scholar]
- 317.Mehta SS et al. Indoor wood-burning from stoves and fireplaces and incident lung cancer among Sister Study participants. Environ. Int 178, 108128 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Pagadala M. et al. Germline modifiers of the tumor immune microenvironment implicate drivers of cancer risk and immunotherapy response. Nat. Commun 14, 2744 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Luo J. et al. Immunotherapy-mediated thyroid dysfunction: genetic risk and impact on outcomes with PD-1 blockade in non-small cell lung cancer. Clin. Cancer Res 27, 5131–5140 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Groha S. et al. Germline variants associated with toxicity to immune checkpoint blockade. Nat. Med 28, 2584–2591 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.National Comprehensive Cancer Network. Non-Small Cell Lung Cancer (Version 5.2023), https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf (accessed 4 August 2023).
- 322.Anderson MD Cancer Center & National Cancer Institute. Local consolidative therapy and brigatinib in treating patients with stage IV or recurrent non-small cell lung cancer. ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT03707938 (2018). [Google Scholar]
- 323.Anderson MD Cancer Center, National Cancer Institute & National Comprehensive Cancer Network. Osimertinib, surgery, and radiation therapy in treating patients with stage IIIB or IV non-small cell lung cancer with EGFR mutations, NORTHSTAR study. ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT03410043 (2018). [DOI] [PubMed]
- 324.Elamin YY et al. LocaL consoLidation therapy (LCT) after first Line tyrosine kinase inhibitor (TKI) for patients with EGFR mutant metastatic non-small-cell lung cancer (NSCLC). Clin. Lung Cancer 20, 43–47 (2019). [DOI] [PubMed] [Google Scholar]
- 325.Zeng Y. et al. The value of local consolidative therapy in osimertinib-treated non-small cell lung cancer with oligo-residual disease. Radiat. Oncol 15, 207 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Planchard D. et al. Osimertinib with or without chemotherapy in EGFR-mutated advanced NSCLC. N. Engl. J. Med 10.1056/NEJMoa2306434 (2023). [DOI] [PubMed] [Google Scholar]
- 327.Solomon BJ et al. ALINA: a phase III study of alectinib versus chemotherapy as adjuvant therapy in patients with stage IB-IIIA anaplastic lymphoma kinase-positive (ALK+) non-small cell lung cancer (NSCLC). J. Clin. Oncol 37, TPS8569 (2019). [Google Scholar]
- 328.Herbst RS et al. Adjuvant osimertinib for resected EGFR-mutated stage IB-IIIA non-small-cell lung cancer: updated results from the phase III randomized ADAURA trial. J. Clin. Oncol 41, 1830–1840 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Urisman A. et al. Phase II trial of neoadjuvant osimertinib for surgically resectable EGFR-mutated non-small cell lung cancer. J. Clin. Oncol 41, 8508–8508 (2023). [Google Scholar]
- 330.Solomon BJ et al. LBA2 ALINA: efficacy and safety of adjuvant alectinib versus chemotherapy in patients with early-stage ALK+ non-small cell lung cancer (NSCLC). Ann. Oncol 34, S1295–S1296 (2023). [Google Scholar]
- 331.Tsuboi M. et al. Overall survival with osimertinib in resected EGFR-mutated NSCLC. N. Engl. J. Med 389, 137–147 (2023). [DOI] [PubMed] [Google Scholar]
- 332.National Lung Screening Trial Research Teamet al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N. Engl. J. Med 365, 395–409 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.de Koning HJ et al. Reduced lung-cancer mortality with volume CT screening in a randomized trial. N. Engl. J. Med 382, 503–513 (2020). [DOI] [PubMed] [Google Scholar]
- 334.Khuder SA Effect of cigarette smoking on major histological types of lung cancer: a meta-analysis. Lung Cancer 31, 139–148 (2001). [DOI] [PubMed] [Google Scholar]
- 335.Alexandrov LB et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Ernst SM et al. Tobacco smoking-related mutational signatures in classifying smoking-associated and nonsmoking-associated NSCLC. J. Thorac. Oncol 18, 487–498 (2023). [DOI] [PubMed] [Google Scholar]
- 337.Lindsay CR et al. Abstract 6463: persistence of smoking mutational signatures in the non-small cell lung cancer genome. Cancer Res. 83, 6463–6463 (2023). [Google Scholar]
- 338.Wang Y, Broderick P, Matakidou A, Eisen T & Houlston RS Role of 5p15.33 (TERT-CLPTM1L), 6p21.33 and 15q25.1 (CHRNA5-CHRNA3) variation and lung cancer risk in never-smokers. Carcinogenesis 31, 234–238 (2010). [DOI] [PubMed] [Google Scholar]
- 339.Hung RJ et al. Lung cancer risk in never-smokers of European descent is associated with genetic variation in the 5(p)15.33 TERT-CLPTM1Ll region. J. Thorac. Oncol 14, 1360–1369 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Hosgood HD III et al. Genetic variant in TP63 on locus 3q28 is associated with risk of lung adenocarcinoma among never-smoking females in Asia. Hum. Genet 131, 1197–1203 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Shiraishi K. et al. A genome-wide association study identifies two new susceptibility loci for lung adenocarcinoma in the Japanese population. Nat. Genet 44, 900–903 (2012). [DOI] [PubMed] [Google Scholar]
- 342.Yngveson A. et al. p53 Mutations in lung cancer associated with residential radon exposure. Cancer Epidemiol. Biomark. Prev 8, 433–438 (1999). [PubMed] [Google Scholar]
- 343.Peres J. No clear link between passive smoking and lung cancer. J. Natl Cancer Inst 105, 1844–1846 (2013). [DOI] [PubMed] [Google Scholar]
- 344.Hammond EC, Selikoff IJ & Seidman H Asbestos exposure, cigarette smoking and death rates. Ann. N. Y. Acad. Sci 330, 473–490 (1979). [DOI] [PubMed] [Google Scholar]
- 345.Sichletidis L. et al. Mortality from occupational exposure to relatively pure chrysotile: a 39-year study. Respiration 78, 63–68 (2009). [DOI] [PubMed] [Google Scholar]
- 346.Shapiro MZ et al. Cancer in general responders participating in World Trade Center Health Programs, 2003-2013. JNCI Cancer Spectr. 4, pkz090 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Ward AM, Yaman R & Ebbert JO Electronic nicotine delivery system design and aerosol toxicants: a systematic review. PLoS ONE 15, e0234189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Goniewicz ML et al. Comparison of nicotine and toxicant exposure in users of electronic cigarettes and combustible cigarettes. JAMA Netw. Open 1, e185937 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Shahab L. et al. Nicotine, carcinogen, and toxin exposure in long-term E-cigarette and nicotine replacement therapy users: a cross-sectional study. Ann. Intern. Med 166, 390–400 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Barcenas CH et al. Wood dust exposure and the association with lung cancer risk. Am. J. Ind. Med 47, 349–357 (2005). [DOI] [PubMed] [Google Scholar]
- 351.Tanvetyanon T & Bepler G Beta-carotene in multivitamins and the possible risk of lung cancer among smokers versus former smokers: a meta-analysis and evaluation of national brands. Cancer 113, 150–157 (2008). [DOI] [PubMed] [Google Scholar]
- 352.Omenn GS et al. Chemoprevention of lung cancer: the beta-Carotene and Retinol Efficacy Trial (CARET) in high-risk smokers and asbestos-exposed workers. IARC Sci. Publ 136, 67–85 (1996). [PubMed] [Google Scholar]
- 353.Alpha-Tocopherol, Beta Carotene Cancer Prevention Study Group. The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. N. Engl. J. Med 330, 1029–1035 (1994). [DOI] [PubMed] [Google Scholar]
- 354.Brasky TM, White E & Chen CL Long-term, supplemental, one-carbon metabolism-related vitamin B use in relation to lung cancer risk in the vitamins and lifestyle (VITAL) cohort. J. Clin. Oncol 35, 3440–3448 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Hennekens CH et al. Lack of effect of long-term supplementation with beta carotene on the incidence of malignant neoplasms and cardiovascular disease. N. Engl. J. Med 334, 1145–1149 (1996). [DOI] [PubMed] [Google Scholar]
- 356.Chlebowski RT et al. Lung cancer among postmenopausal women treated with estrogen alone in the women’s health initiative randomized trial. J. Natl Cancer Inst 102, 1413–1421 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Chlebowski RT et al. Oestrogen plus progestin and lung cancer in postmenopausal women (Women’s Health Initiative trial): a post-hoc analysis of a randomised controlled trial. Lancet 374, 1243–1251 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Heiss G. et al. Health risks and benefits 3 years after stopping randomized treatment with estrogen and progestin. JAMA 299, 1036–1045 (2008). [DOI] [PubMed] [Google Scholar]
- 359.Slatore CG, Chien JW, Au DH, Satia JA & White E Lung cancer and hormone replacement therapy: association in the vitamins and lifestyle study. J. Clin. Oncol 28, 1540–1546 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Pesatori AC et al. Hormone use and risk for lung cancer: a pooled analysis from the International Lung Cancer Consortium (ILCCO). Br. J. Cancer 109, 1954–1964 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Seow A, Koh WP, Wang R, Lee HP & Yu MC Reproductive variables, soy intake, and lung cancer risk among nonsmoking women in the Singapore Chinese Health Study. Cancer Epidemiol. Biomark. Prev 18, 821–827 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Weiss JM et al. Menstrual and reproductive factors in association with lung cancer in female lifetime nonsmokers. Am. J. Epidemiol 168, 1319–1325 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Ganti AK, Sahmoun AE, Panwalkar AW, Tendulkar KK & Potti A Hormone replacement therapy is associated with decreased survival in women with lung cancer. J. Clin. Oncol 24, 59–63 (2006). [DOI] [PubMed] [Google Scholar]
- 364.Liu Y, Inoue M, Sobue T & Tsugane S Reproductive factors, hormone use and the risk of lung cancer among middle-aged never-smoking Japanese women: a large-scale population-based cohort study. Int. J. Cancer 117, 662–666 (2005). [DOI] [PubMed] [Google Scholar]
- 365.Hopkins RJ et al. Reduced expiratory flow rate among heavy smokers increases lung cancer risk. results from the National Lung Screening Trial — American College of Radiology Imaging Network Cohort. Ann. Am. Thorac. Soc 14, 392–402 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Denholm R. et al. Is previous respiratory disease a risk factor for lung cancer? Am. J. Respir. Crit. Care Med 190, 549–559 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Brenner DR et al. Previous lung diseases and lung cancer risk: a pooled analysis from the International Lung Cancer Consortium. Am. J. Epidemiol 176, 573–585 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Brenner DR, McLaughlin JR & Hung RJ Previous lung diseases and lung cancer risk: a systematic review and meta-analysis. PLoS ONE 6, e17479 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Shiels MS, Albanes D, Virtamo J & Engels EA Increased risk of lung cancer in men with tuberculosis in the alpha-tocopherol, beta-carotene cancer prevention study. Cancer Epidemiol. Biomark. Prev 20, 672–678 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Yu YH et al. Increased lung cancer risk among patients with pulmonary tuberculosis: a population cohort study. J. Thorac. Oncol 6, 32–37 (2011). [DOI] [PubMed] [Google Scholar]
- 371.Jacob S. et al. Lung cancer survival in patients with autoimmune disease. JAMA Netw. Open 3, e2029917 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Liu X. et al. Clinicopathological features of lung cancer in patients with rheumatoid arthritis. J. Thorac. Dis 10, 3965–3972 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Naccache JM et al. Lung cancer and interstitial lung disease: a literature review. J. Thorac. Dis 10, 3829–3844 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Onishi A, Sugiyama D, Kumagai S & Morinobu A Cancer incidence in systemic sclerosis: meta-analysis of population-based cohort studies. Arthritis Rheum. 65, 1913–1921 (2013). [DOI] [PubMed] [Google Scholar]
- 375.Siemes C. et al. C-reactive protein levels, variation in the C-reactive protein gene, and cancer risk: the Rotterdam Study. J. Clin. Oncol 24, 5216–5222 (2006). [DOI] [PubMed] [Google Scholar]
- 376.Sheikh M. et al. Opium use and subsequent incidence of cancer: results from the Golestan Cohort Study. Lancet Glob. Health 8, e649–e660 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.IARC Monographs Vol 126 Group. Carcinogenicity of opium consumption. Lancet Oncol. 21, 1407–1408 (2020). [DOI] [PubMed] [Google Scholar]
- 378.Rahimi-Movaghar A. et al. Pharmacological therapies for management of opium withdrawal. Cochrane Database Syst. Rev 6, CD007522 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Callaghan RC, Allebeck P & Sidorchuk A Marijuana use and risk of lung cancer: a 40-year cohort study. Cancer Causes Control. 24, 1811–1820 (2013). [DOI] [PubMed] [Google Scholar]
- 380.Tashkin DP Effects of marijuana smoking on the lung. Ann. Am. Thorac. Soc 10, 239–247 (2013). [DOI] [PubMed] [Google Scholar]
- 381.Sarafian TA, Magallanes JA, Shau H, Tashkin D & Roth MD Oxidative stress produced by marijuana smoke. An adverse effect enhanced by cannabinoids. Am. J. Respir. Cell Mol. Biol 20, 1286–1293 (1999). [DOI] [PubMed] [Google Scholar]
- 382.Barsky SH, Roth MD, Kleerup EC, Simmons M & Tashkin DP Histopathologic and molecular alterations in bronchial epithelium in habitual smokers of marijuana, cocaine, and/or tobacco. J. Natl Cancer Inst 90, 1198–1205 (1998). [DOI] [PubMed] [Google Scholar]
- 383.Lorigan P, Radford J, Howell A & Thatcher N Lung cancer after treatment for Hodgkin’s lymphoma: a systematic review. Lancet Oncol. 6, 773–779 (2005). [DOI] [PubMed] [Google Scholar]
- 384.Huang YJ et al. Radiation therapy for invasive breast cancer increases the risk of second primary lung cancer: a nationwide population-based cohort analysis. J. Thorac. Oncol 12, 782–790 (2017). [DOI] [PubMed] [Google Scholar]
- 385.Cornelius ME, Loretan CG, Wang TW, Jamal A & Homa DM Tobacco product use among adults — United States, 2020. MMWR Morb. Mortal. Wkly Rep 71, 397–405 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Cornelius ME, Wang TW, Jamal A, Loretan CG & Neff LJ Tobacco product use among adults — United States, 2019. MMWR Morb. Mortal. Wkly Rep 69, 1736–1742 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Suehnholz SP et al. Quantifying the expanding landscape of clinical actionability for patients with cancer. Cancer Discov. 10.1158/2159-8290.CD-23-0467 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Chakravarty D. et al. OncoKB: a precision oncoLogy knowledge base. JCO Precis. Oncol 2017, PO.17.00011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Chang MT et al. Accelerating discovery of functional mutant alleles in cancer. Cancer Discov. 8, 174–183 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Chang MT et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol 34, 155–163 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]