

Abstract
The hypervirulent lineages of Klebsiella pneumoniae (HvKp) cause invasive infections such as Klebsiella-liver abscess. Invasive infection often occurs after initial colonization of the host gastrointestinal tract by HvKp. Over 80% of HvKp isolates belong to the clonal group 23 sublineage I that has acquired genomic islands (GIs) GIE492 and ICEKp10. Our analysis of 12 361 K. pneumoniae genomes revealed that GIs GIE492 and ICEKp10 are co-associated with the CG23-I and CG10118 HvKp lineages. GIE492 and ICEKp10 enable HvKp to make a functional bacteriocin microcin E492 (mccE492) and the genotoxin colibactin, respectively. We discovered that GIE492 and ICEKp10 play cooperative roles and enhance gastrointestinal colonization by HvKp. Colibactin is the primary driver of this effect, modifying gut microbiome diversity. Our in vitro assays demonstrate that colibactin and mccE492 kill or inhibit a range of Gram-negative Klebsiella species and Escherichia coli strains, including Gram-positive bacteria, sometimes cooperatively. Moreover, mccE492 and colibactin kill human anaerobic gut commensals that are similar to the taxa found altered by colibactin in the mouse intestines. Our findings suggest that GIs GIE492 and ICEKp10 enable HvKp to kill several commensal bacterial taxa during interspecies interactions in the gut. Thus, acquisition of GIE492 and ICEKp10 could enable better carriage in host populations and explain the dominance of the CG23-I HvKp lineage.
Keywords: hypervirulent, Klebsiella pneumoniae, colibactin, microcin, gut, commensal, colonization, genomic island
Graphical Abstract
Graphical Abstract.

Introduction
Klebsiella pneumoniae is a Gram-negative bacterium characterized as a nosocomial pathogen associated with pneumonia [1]. In contrast, hypervirulent K. pneumoniae (HvKp) is community acquired and the major cause of monomicrobial liver abscess in Asia [2, 3]. The dominant lineage of HvKp is clonal group 23-I (CG23-I), comprising mostly sequence type 23 (ST23) strains which cause ~80% of all Klebsiella-liver abscess (KLA) infections [4, 5]. Gastrointestinal carriage of HvKp can be high in Asian countries, ranging from 3% to 8% of patients with diarrhea in a Singaporean study to 21.1% of healthy individuals in a Korean study [6, 7]. ST23 HvKp has only been isolated from patients rather than environmental samples [6-9], and gut colonization is strongly associated with the development of KLA [10]. Therefore, factors favoring persistent gastrointestinal carriage of HvKp could benefit its dominance and spread.
Genomic islands (GIs) are horizontally acquired DNA elements integrated in bacterial chromosomes [11]. GIs contribute to the accessory genome of a bacterial species and can contain diverse genetic cargo such as antimicrobial resistance cassettes, metabolic operons, and virulence determinants [11-15]. Most K. pneumoniae chromosomes have at least four GIs and some strains possess up to 10 GIs, many encoding known or putative virulence factors [16]. Because GIs can confer beneficial phenotypes, they drive rapid evolution and the success of certain bacterial lineages within a species. Most HvKp strains possess a large virulence plasmid that encodes virulence factors [3, 17-19], as well as several GIs [16]. The GI E492 (GIE492) and integrative conjugative element Kp10 (ICEKp10) are tightly associated with the CG23-I lineage [4, 5, 16]. Currently there is a lack of studies addressing the evolutionary history and prevalence of GIE492 in the K. pneumoniae population. Moreover, the contribution of GIE492 and ICEKp10 to the pathogenesis of HvKp in the human host is poorly understood.
GIE492 contains the mce gene locus required for the synthesis and export of the bacteriocin microcin E492 (mccE492) [20]. MccE492 is a siderophore-microcin that kills other bacteria via a Trojan-horse mechanism [21]. MccE492 is a siderophore-microcin that enters the periplasm of susceptible prey via catechol siderophore receptors on the outer membrane [22]. Subsequently, mccE492 inserts into the inner membrane to cause cell death [21, 23, 24]. ICEKp10 carries the ybt locus encoding yersiniabactin siderophore and the clb locus enables the production of colibactin [25-27]. Colibactin is a small molecule alkylating genotoxin that causes double-stranded DNA breaks (DSBs). Gut carriage of clb-positive bacteria is associated with colorectal cancer [25, 28, 29]. Recent studies have shown that beyond its effect on the host, colibactin-producing Escherichia coli can utilize colibactin to kill other bacteria [30-32].
We profiled the evolution and prevalence of GIE492 in K. pneumoniae and found eight main variants of GIE492, of which a specific variant is co-associated with ICEKp10 in the hypervirulent CG23-I K. pneumoniae lineage. Furthermore, we discovered that GIE492 and ICEKp10 play cooperative roles during gastrointestinal colonization of SGH10, a representative clinical isolate of CG23-I [4]. Colibactin causes changes to the gut microbiome that benefit HvKp colonization. Our results support a model where GIE492 and ICEKp10 enable HvKp to compete effectively with other bacterial species in the ecological niche of the gut. Thus, we postulate that the acquisition of GIE492 and ICEKp10 is likely a contributing factor to the emergence ofje CG23-I as the dominant lineage of HvKp.
Materials and methods
Bacterial cultures
All bacterial strains used in this study are listed in Supplementary Table 1. Facultative anaerobes were revived from glycerol stocks on Lysogeny broth agar (ThermoFisher Scientific) and grown at 37°C under oxic conditions. Anaerobic bacteria were grown in Reinforced Clostridial Medium broth and agar (Sigma-Aldrich) under anoxic conditions at 37°C. Facultative anaerobes were grown at 37°C with shaking in Dulbecco’s modified Eagle’s medium (Life Technologies) with 10% fetal bovine serum (Singlab) unless otherwise specified.
Where necessary, 50 μg/ml kanamycin sulfate (Sigma-Aldrich), 10 μg/ml chloramphenicol (Sigma-Aldrich), 20 μg/ml tetracycline (Sigma-Aldrich), 10 μg/ml trimethoprim (Sigma-Aldrich), 10 μg/ml gentamicin sulfate (Sigma-Aldrich), and 50 μg/ml carbenicillin sodium salt (Sigma-Aldrich) were used in media.
Mouse experiments
Female C57BL/6 mice aged 7–8 weeks from InVivos were inoculated with 105 CFU of K. pneumoniae in 1 × Phosphate-buffered saline (PBS) via intraperitoneal injection to establish systemic infection. Mice were sacrificed at 30 hours post infection (hpi), and organ bacterial loads were enumerated by plating appropriate dilutions of organ homogenates on Klebsiella selective agar (KSA) supplemented with 50 μg/ml carbenicillin.
To establish gastrointestinal colonization, mice were gavaged with 2.5 mg of ampicillin (Sigma-Aldrich) in 100 μl 1 × PBS daily for 5 days. On the subsequent day, mice were infected with 5 × 106 colony forming units (CFU) of K. pneumoniae in 100 μl 1 × PBS via oral gavage. Fresh stools were collected from each mouse, and bacterial load was enumerated by plating on KSA.
To establish gut translocation, mice were gavaged with a cocktail of 2.5 mg ampicillin sodium salt, 0.2 mg of vancomycin hydrochloride, 1 mg of metronidazole, and 0.16 mg of colistin sulfate in 400 μl of 1 × PBS daily for 5 days. On the following day, mice were switched from normal drinking water to 15% Miralax (Bayer) in Milli-Q water. Mice were then orally gavaged with 109 CFU of K. pneumoniae 1 day later. Mice were weighed and scored for sickness daily. Mice reaching termination criteria or 20% weight loss were euthanized. Procedures were approved by the Institutional Animal Care and Use Committee at National University of Singapore (R18–0252).
Stool DNA extraction and Illumina sequencing of metagenomic libraries
DNA was extracted from frozen mouse stools using the QIAamp PowerFecal Pro DNA Kit (Qiagen). The purified DNA underwent NGS library construction steps using NEBNext Ultra II FS DNA Library Prep Kit (New England BioLabs) according to the manufacturer’s instructions. Post fragmentation, adaptor-ligation was performed using Illumina-compatible adaptors, diluted 10-fold as per kit’s recommendations prior to use. Postligation, samples were purified using Agencourt Ampure XP beads (Beckman Coulter) in a 7:10 beads-to-sample volume. Unique barcode indexes were added to each purified sample and amplified for 12 cycles under recommended kit conditions to achieve multiplexing within a batch of samples. Finally, each library sample was assessed for quality based on fragment size and concentration using the Agilent D1000 ScreenTape system and adjusted to identical concentrations by means of dilution and volume-adjusted pooling. The multiplexed sample pool was paired-end (2 × 151 bp) sequenced on the HiSeq X Ten system (Illumina). All sequencing was done in the Novogene sequencing facility at the Genome Institute of Singapore in accordance with standard Illumina sequencing protocols. An average of 14 893 204 raw reads, and an average of 7 446 602 paired reads were generated for each sample.
Statistical analysis
Data were plotted using Graphpad Prism 9.0 (La Jolla California USA, www.graphpad.com). Long read-sequencing data were plotted using R (v4.1.2; R Core Team 2021). Dunnett’s multiple comparison’s tests were used to compare means and standard deviations (SDs) in in vitro assays. For in vivo experiments, Dunnett’s multiple comparison’s test was used to compare geometric means relative to SGH10. A P value <.05 was considered statistically significant and denoted as *, and a P value <.01 is denoted as **. The Mantel–Cox test was performed to determine if there were significant differences in mortality between the groups. Microbiome analysis methods and K. pneumoniae phylogenomic analyses are described in Supplemental Methods. Bacterial mutant generation and bacterial prey-predator cocultures are also described in Supplemental Methods.
Results
Evolutionary history of GIE492 and its association with ICEKp10 in K. pneumoniae CG23
GIE492 was first characterized in the clonal group 35 (CG35) isolate K. pneumoniae RYC492 [20, 21, 33]. However, little is known about this GI in other lineages of K. pneumoniae and its potential roles in pathogenesis. We investigated the structure, distribution, and evolutionary history of GIE492 in K. pneumoniae and its relationship with CG23-I.
We produced a reference sequence for GIE492 by assembling the complete genome of K. pneumoniae RYC492 with Illumina and nanopore sequencing. Using this reference, we searched for GIE492 in 12 433 KpSC genomes from (1) the RefSeq database, (2) a multi-centre clinical trial studying KLA patients in Singapore (A-KLASS) [34], (3) a set of bloodstream infection isolates from Southeast Asia [35], (4) the CG23 genome set analyzed by Lam et al. [4], and (5) the Murray collection of bacterial isolates from the preantibiotic era. We identified 657 GIE492+ genomes in K. pneumoniae sensu stricto species (Supplementary Table 2) and discovered eight main GIE492 variants based on sequence comparison and allele calling for the 23 genes in GIE492 [20]. Six variants were similar in length to GIE492-III (first characterized in RYC492), and GIE492-II and GIE492-IV lacked a ~4.6-kbp internal region comprising the u1–u5 genes of unknown function (Fig. 1A). The remaining variants included up to four main variable alleles for some genes (Supplementary Data 1). The eight GIE492 variants were flanked by 100% conserved 20-bp direct repeats, carried the P4-like integrase-coding gene, and had the putative transfer origin (oriT). Other less frequent variants included insertion sequences (mainly ISKpn72 and ISKpn74), likely leading to variable deletions (Supplementary Fig. 1).
Figure 1.

Genomic structure and phylogenetic distribution of GIE492 in K. pneumoniae; (A) sequence alignment of GIE492 structural variants of K. pneumoniae (GIE492-I to GIE492-VIII), as well as GIE492 variants in K. michiganesis (GIE492-Kmi1 and 2) and E. coli (GIE492-Ec); (B) distribution of GIE492 and ICEKp variants in 588 GIE492+ K. pneumoniae genomes; a maximum-likelihood tree of the GIE492+ genomes was plotted based on core genome multilocus sequence alignment (cgMSA) of the scgMLSTv2 scheme; colors indicating GIE492-I and GIE492-II indicate their respective one-locus variants; ICEKp10 and ICEKp12 colored slots also include versions of these elements predicted as incomplete by Kleborate.
We also searched for GIE492 in a set of 82 206 Enterobacterales genomes from the RefSeq database. Other than in K. pneumoniae, GIE492 was only found in two E. coli (GIE492-Ec) and two Klebsiella michiganensis genomes (GIE492-Kmi1 and 2) (Fig. 1A). The latter carried a divergent GIE492 with more similarity to GIE492-II, with rearrangements and different gene content in the 3′ region, lacking the direct repeat (Fig. 1A). Thus, GIE492 is highly restricted to K. pneumoniae, although it may be transferred to other related species with relatively low frequency.
To investigate the phylogenetic relationships among the GIE492+ strains, sublineages (SLs; threshold: 190 allelic mismatches) and clonal groups (CGs; threshold: 43) were identified using cgMLST analysis. We also determined sequence type (ST) according to the classical seven-gene MLST scheme for better comparison with previous phylogroup definitions. SL23, SL405, SL35 (including RYC492), SL380, and SL152 are most prevalent, and are associated mainly with variants I, II, III, V, and VIII, respectively (Fig. 1B). This distribution suggests an early acquisition of this GI in different K. pneumoniae lineages, followed by vertical transmission within each lineage. The presence of the shortened variant GIE492-II in seven relatively distant lineages suggests an early occurrence of the putative deletion event leading to the shorter GIE492 variants (Fig. 1B, Supplementary Table 2). The oldest strains bearing GIE492 (found in the Murray collection), ERR230425 (1932, CG23), and GCA_022308495.1 (1940, CG35) possessed Variants I and III, respectively, further supporting the hypothesis that longer GIE492 variants are ancestral.
In terms of the co-occurrence of GIE492 and ICEKp variants in our dataset, a high proportion (82%) of GIE492+ genomes carried an ICEKp element (Supplementary Table 2). GIE492-I and ICEKp10 co-occur in the globally disseminated HvKp subclade CG23-I, corroborating earlier observations (Fig. 1B) [4]. CG23-I (described initially based on SNPs) mainly included isolates with LIN codes 0_0_429_0_4/5/6/8/9/44, and is also highly associated with the alleles ybt1, clb2, and the virulence plasmid KpVP-1. Moreover, GIE492-V and ICEKp10 also co-occurred in CG10118 (SL380), although few SL380 genomes were present in our dataset (Fig. 1B). Both SL23 and SL380 lineages are associated with KLA, although SL380 is a minor HvKp lineage [36, 37]. In all GIE492 + ICEKp10+ isolates, the colibactin and microcin immunity genes clbS and mceB were present (Supplementary Table 2). We did not find GIE492-ICEKp10-genomes that carried clbS or mceB (Supplementary Table 2).
GIs are unstable elements that may be easily lost after acquisition, but positive selection maintains GIs in bacterial lineages if their genetic cargo confers benefits. The co-occurrence of specific GIE492 variants and ICEKp10 in two hypervirulent lineages of K. pneumoniae suggests that two independent acquisitions of GIE492 occurred and were preserved in these hypervirulent clades. CG23-I is the dominant lineage of HvKp, and GIE492 and ICEKp10 may have contributed to its success.
SGH10 produces functional mccE492 and colibactin
SGH10 possesses GIE492-I and ICEKp10 [4]. The mce and clb genetic loci in SGH10 encode microcin E492 and colibactin, respectively (Fig. 2A). We created markerless deletion mutants of GIE492, ICEKp10, mceA, and clbP. Using an agar diffusion assay, we demonstrated that SGH10 makes a functional mccE492 that inhibits the growth of E. coli prey (Fig. 2B). The deletion mutants in GIE492 and mceA (microcin E492 precursor) did not kill E. coli (Fig. 2B). Because expressing excess mceA alone results in self-intoxication, we complemented SGH10ΔmceA by expressing both the microcin precursor and immunity protein (mceAB) under the control of the native promoter (Fig. 2C). The complemented strain killed E. coli prey, whereas SGH10ΔmceA expressing mceB alone or an empty vector did not (Fig. 2C).
Figure 2.
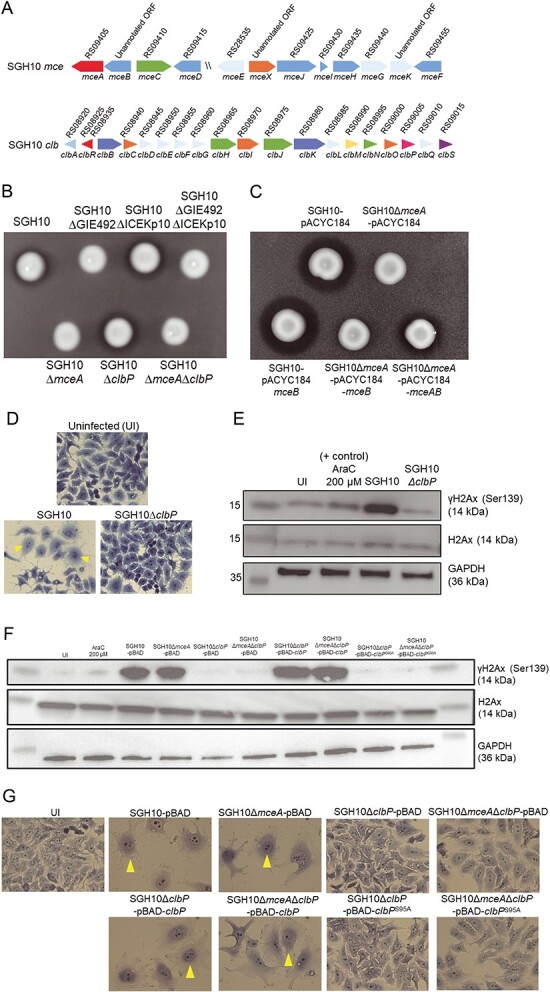
The clonal group-23 HvKp strain SGH10 produces active microcin E492 and colibactin; (A) genetic organization of the mce and clb loci in SGH10; mceB is an unannotated open reading frame (ORF) from base pairs 1 924 637–1 924 924, mceX is an unannotated ORF from base pairs 1 930 032–1 930 139, and mceK is an unannotated ORF from base pairs 1 935 889–1 936 298; (B) an agar diffusion assay was used to confirm if SGH10 produced a functional mccE492; SGH10 and mutant strains were spotted on a lawn of E. coli MG1655 prey; a growth inhibition halo can be observed around microcin producing strains; (C) the agar diffusion assay was used to demonstrate complementation of SGH10ΔmceA; SGH10 and SGH10ΔmceA containing a control plasmid pACYC184, pACYC184-mceB, or pACYC184-mceAB were spotted on a lawn of prey E. coli MG1655; strains producing mccE492 are surrounded by a halo of killed E. coli; (D) light micrographs of methylene blue-stained HepG2 cells infected with SGH10 and SGH10∆clbP at an MOI of 50:1 at 48 hpi; the cells were imaged at 20× magnification, and yellow arrows indicate megalocytotic cells with distended nuclei and cell bodies; (E) measurement of γH2Ax in Kp-infected cells; HepG2 cells were infected with the complemented SGH10 and mutant strains at an MOI of 50:1, and cytarabine (AraC) was used as a positive control; cell lysates were harvested at 8 hpi for western blotting to quantify γH2Ax (Ser139, phosphorylated at serine 139), and H2Ax and GAPDH were included as loading controls; (F) complementation of SGH10∆clbP. HepG2 cells was infected with SGH10, SGH10∆mceA, SGH10∆clbP, and SGH10∆mceA∆clbP carrying either the pmLBAD (pBAD) control vector or pBAD expressing either wildtype ClbP or ClbPS95A at an MOI of 50:1; 200 μM cytarabine (AraC) was used as a positive control for DNA damage and UI denotes uninfected cells; cell lysates were harvested at 8 hpi for western blotting to quantify γH2Ax, H2Ax, and GAPDH as a control; (G) at 48 hpi, HepG2 cells infected in the same manner as in (F) were fixed and stained with Giemsa and imaged at 20× magnification; megalocytotic cells are indicated with arrows.
To validate SGH10’s ability to make colibactin, HepG2 cells were infected with SGH10. Infected cells exhibited distended nuclei and cell bodies, characteristic of the megalocytotic phenotype induced by the genotoxic effect of colibactin (Fig. 2D) [38]. Conversely, cells infected with ∆clbP, where the enzyme responsible for maturation of colibactin has been deleted appeared similar in morphology to uninfected cells (Fig. 2D). γH2AX accumulation is a marker of double-stranded DNA breaks in mammalian cells [39]. We observed a strong γH2AX signal in HepG2 cells infected with SGH10 but not in cells infected with SGH10ΔclbP (Fig. 2E). Complementation of SGH10ΔclbP with wildtype clbP restored genotoxicity (Fig. 2F). Expression of ClbPS95A, a catalytically inactive ClbP, did not restore γH2AX accumulation or megalocytosis in SGH10ΔclbP (Fig. 2F and G). Thus, the genotoxic effect of SGH10 is dependent on the production of mature colibactin.
MccE492 and colibactin are important for gut colonization
Because GIE492 and ICEKp10 co-associate with the CG23-I lineage, we hypothesized that these GIs are important during disseminated infection. There were no defects in the ability of SGH10ΔGIE492 and SGH10ΔICEKp10 to infect the liver and spleen in a murine model of systemic infection, though there was a modest reduction in SGH10ΔICEKp10 in the lungs (Fig. 3A). These GIs are dispensable during systemic infection at the infectious dosage we used.
Figure 3.

The roles of mccE492 and colibactin during HvKp infection; a systemic infection was established via intraperitoneal injection of C57BL/6 mice with 105 CFU of SGH10, SGH10ΔGIE492, SGH10ΔICEKp10, and SGH10 ΔGIE492ΔICEKp10; bacterial loads were quantified in the liver, lungs, and spleen at 30 hpi; (B) mice were colonized with SGH10, SGH10ΔGIE492, SGH10ΔICEKp10, as well as C SGH10ΔmceA, SGH10ΔclbP and SGH10ΔmceAΔclbP; C57BL/6 mice were treated with ampicillin for 5 days before oral gavage with 5 x 106 CFU SGH10 and mutants; stool bacterial loads were quantified at appropriate intervals; mouse infection experiments were performed n = 2–4 times; the geometric mean and SD were plotted, and Dunnett’s multiple comparisons test was performed on log-transformed CFU values to determine differences in means; * denotes P < .05 and ** denotes P < .01.
We then examined whether GIE492 and ICEKp10 are important during gastrointestinal colonization using a murine model of infection that we previously established [40]. At Days 11 and 13, stool CFU of SGH10ΔICEKp10 was significantly lower than SGH10 (Fig. 3B). There were no differences in the stool CFU of SGH10ΔGIE492 and SGH10 (Fig. 3B). Hence, ICEKp10 but not GIE492 is important for gastrointestinal persistence.
ICEKp10 and GIE492 contain other genetic content besides mce and clb. We then created ∆mceA and ∆clbP to determine the specific roles of mccE492 and colibactin. SGH10∆mceA colonized as well as SGH10 (Fig. 3C). However, SGH10ΔclbP and SGH10ΔmceAΔclbP were attenuated in their ability to persist in the gut from Days 11 till 17 (Fig. 3C). Interestingly, stool bacterial loads of SGH10ΔmceAΔclbP were significantly lower than SGH10ΔclbP on Days 13 and 15 (Fig. 3C). Even on Days 19 and 20, bacterial loads of SGH10ΔmceAΔclbP were the lowest among all groups (Fig. 3C). Gut colonization appears to be colibactin-driven with mccE492 playing a supporting role by amplifying the effect of colibactin, as microcin alone did not affect colonization.
We developed a murine model of K. pneumoniae translocation dependent on an antibiotic cocktail and Miralax, a mild osmotic laxative that induces thinning of the gut mucosa [41] (Supplementary Fig. 2A). This treatment establishes high rates of HvKp translocation to the liver, lungs, and spleen (Supplementary Fig. 2B). We then determined whether colibactin is important in the context of gastrointestinal translocation (Fig. 4A). About 85.7% of the mice infected with SGH10 succumbed to infection, whereas SGH10ΔclbP killed only 35.7% of infected mice (Fig. 4B). There was a significant difference in stool bacterial loads on Day 8 (Fig. 4C), suggesting that attenuation of SGH10ΔclbP in this model may be due to transient differences in gut colonization. It is also possible that colibactin causes damage to the intestinal epithelium and increases translocation of SGH10. However, we observed no significant differences in weight as an indication of sickness, or stool lipocalin-2, a marker of inflammation (Fig. 4D and E).
Figure 4.

The role of colibactin in bacterial translocation; (A) a murine model of HvKp translocation was used to determine the lethality of SGH10 or SGH10ΔclbP; 1 × PBS was used as a control; (B) the mice were scored daily for signs of sickness and culled when they reached termination criteria; a survival curve of mortality induced by SGH10 and SGH10ΔclbP was plotted; The Mantel–Cox test was performed to determine if there were significant differences in mortality between the groups; (C) stool bacterial-loads of SGH10 and SGH10ΔclbP infected mice; (D) weight change (%) relative to the initial weight of the mice during the experiment was plotted; mice were culled when they met termination criteria or if weight loss was greater than 20%; (E) stool lipocalin-2 was measured by ELISA; the geometric mean and SD are plotted for (C and E), and mouse experiments were performed n = 2 times; Dunnett’s multiple comparisons test was performed on log-transformed values to determine differences in means; * denotes P < .05, and ** denotes P < .01.
Colibactin-driven changes in the gut microbiome
We hypothesized that mccE492 and colibactin cause changes in the gut microbiome that benefit K. pneumoniae colonization. To determine whether gut bacterial taxa were affected, shotgun metagenomic sequencing was conducted on samples from the experiment in Fig. 3C. Stools were collected before the microbiome was perturbed with ampicillin (Day 0), and when the greatest difference in the abundance of K. pneumoniae was observed between groups (Days 11 and 13). We observed a colibactin-associated reduction of α-diversity in the gut microbiome and not with other mutants on Days 11 (Richness) and 13 (Simpson) (Fig. 5A). Subsequently, we plotted β-diversity (Bray–Curtis distance) on a PCoA plot (Fig. 5B). The points at Day 0 cluster more tightly (Fig. 5B), but after ampicillin treatment and colonization with K. pneumoniae, there is a shift that differs depending on bacterial mutants (Fig. 5C). Permutational multivariate ANOVA (PERMANOVA) analysis was performed on the Bray–Curtis distances between samples from different groups and indicated that there were significant differences in gut microbiome composition of SGH10 with SGH10∆clbP on both Days 11 and 13 (R2 = 9.14, P = .011; R2 = 6.09, P = .026), as well as between SGH10 and SGH10∆mceA∆clbP infected mice, but only on Day 11 (R2 = 8.78, P = .023) (Fig. 5C). We did not observe significant differences in microbiome composition upon infection with SGH10ΔmceA compared to SGH10, and smaller R2 values were observed for SGH10ΔmceA (Fig. 5C), indicating that the primary driver of changes in the gut microbiome is colibactin.
Figure 5.

Colibactin-dependent alteration of the murine gut microbiome; (A) differences in α-diversity between mice colonized with SGH10 or mutants; richness, Shannon and Simpson index values were plotted and linear mixed models that adjust for batch were used to determine if there were significant differences between groups; the P value of SGH10 vs. SGH10ΔclbP at D11 and D13 is .012 and .027 respectively; (B) PCoA (principal Co-ordinates analysis) was performed on Bray–Curtis distance data as a measure of β-diversity, and PCoA1–PCoA2 were plotted for all groups; centroids of each group are indicated with black dots and connected with arrows; (C) PCoA1–PCoA2 plots of Bray–Curtis distance data (β-diversity) were individually plotted by group; ellipses which represent 90% confidence interval are drawn around each timepoint and arrows are plotted connecting the centroids of each timepoint within groups; PERMANOVA analysis of the Bray–Curtis data determined that there were significant differences in β-diversity between SGH10 vs. SGH10ΔclbP and SGH10 vs. SGH10ΔmceAΔclbP at D11; R2 and P values are listed in Supplementary Table 3; the R2 value represents the percentage of variance in the data explained by the distinctions between groups when comparing each mutant to the wild type; a higher R2 indicates a more pronounced impact of the group differences on the dissimilarity observed among samples; the alpha value cutoff was set to 0.05; (D) differential abundance analysis was conducted using MaAsLin2, comparing SGH10 vs. SGH10ΔclbP and SGH10 vs SGH10ΔmceAΔclbP at D11 and D13; colored cells marked with “•” correspond to FDR-adjusted P value <.1; taxa which are positively or negatively abundant relative to SGH10 are plotted, and P values are listed in Supplementary Table 4; the Oscillibacter, Lachnospiraceae, Eubacterium, Dorea, Acetatifactor taxa belong to the Clostridiales order, whereas Muribaculae belongs to the Bacteroidales order and Parvibacter belongs to the Eggerthellales order. Klebsiella variicola and Escherichia belong to the Enterobacterales order, E. faecalis to the Lactobacillales order, and Acutalibacter muris to the Eubacteriales.
We plotted relative abundance of microbial taxa in the groups at the Family level but did not observe significant differences at this taxonomic rank (Supplementary Fig. 3). Subsequently, MaAsLin2 was used to conduct differential abundance analysis at different time points. In mice colonized with SGH10∆clbP, multiple taxa of Oscillibacter, Lachnospiraceae (14-2), Christensenella (CAG-552), and Muribaculaceae (CAG-873) were enriched relative to mice colonized with SGH10 (Fig. 5D, Supplementary Fig. 4). Conversely, there was a reduction in the abundance of Enterobacterales, K. pneumoniae, Enterococcus faecalis, and E. coli in mice colonized with SGH10∆clbP relative to mice colonized with SGH10 (Fig. 5D, Supplementary Fig. 4, Supplementary Table 4). Although PERMANOVA analysis showed that the differences in microbiome composition were insignificant between SGH10 and SGH10∆mceA∆clbP colonized mice on Day 13 (Fig. 5C), we observed enrichment in one Oscillibacter and one Acetatifactor taxon relative to wildtype (Fig. 5D). The colibactin-induced changes in gut microbiome composition may be due to colibactin-mediated inhibition of these taxa.
K. pneumoniae kills other bacteria via microcin- and colibactin-dependent mechanisms
Although mccE492 is not required for gut colonization, it is beneficial for HvKp to express both mccE492 and colibactin in the gut. E. coli was killed by SGH10 under oxic and anoxic conditions but it did not affect K. pneumoniae growth (Fig. 6A, Supplementary Fig. 5). Deletion of both GIE492 and ICEKp10 restored survival of E. coli (Fig. 6A). When competed with SGH10ΔmceA or SGH10ΔclbP, more E. coli survived relative to SGH10, showing that mccE492 and colibactin kill E. coli through independent pathways (Fig. 6A). Additionally, more E. coli survived when competed with SGH10ΔmceAΔclbP than with SGH10ΔmceA or SGH10ΔclbP under oxic conditions (Fig. 6A). Although the GIs may contain other factors that enable SGH10 to kill E. coli or scavenge nutrients such as iron, these results suggest that mccE492 and colibactin cooperate to enhance the effect of colibactin.
Figure 6.

HvKp can utilize mccE492 and colibactin to kill other bacteria; (A) survival of E. coli MG1655 (Ec) when competed with K. pneumoniae on solid media for 24 h; (B) sensitivity of anaerobic bacteria to killing by mccE492 and colibactin; we competed D. longicatena (Dl), (C) Lachnospiraceae 24 430 (Lachno), (D) O. acetigenes (Oa), (E) C. difficile (Cd), (F) B. adolescentis (BA), (G) B. longum (BL), (H) B. thetaiotaomicron, and (I) B. uniformis with SGH10 and mutants deficient in the synthesis of colibactin, mccE492, or both under anoxic conditions; Dunnett’s multiple comparisons test was performed on CFU values to determine differences in means; * denotes P < .05 and ** denotes P < .01.
Complementation of SGH10ΔclbP and SGH10ΔmceAΔclbP with wildtype ClbP but not the catalytically inactive ClbPS95A resulted in increased killing of E. coli (Supplementary Fig. 6A), confirming the effects due to active colibactin. To demonstrate the specificity of killing, we expressed their cognate immunity proteins MceB and ClbS in E. coli. E. coli carrying control vectors was susceptible to killing by mccE492 and colibactin (Supplementary Fig. 6B). When E. coli expressing mceB was competed with SGH10 and SGH10ΔclbP, no difference in survival was observed due to protection against microcin. However, colibactin-specific killing could be observed when E. coli expressing mceB were competed with SGH10ΔmceA or SGH10ΔmceAΔclbP (Supplementary Fig. 6B). When clbS was expressed in E. coli, only microcin-dependent killing was observed. When both immunity proteins were expressed, there were no differences in survival when competed with SGH10 or all mutants, indicating that the immunity proteins could protect E. coli against both forms of killing (Supplementary Fig. 6B). In these experiments, E. coli did not affect the growth of K. pneumoniae (Supplementary Fig. 6C and D).
ClbP has also been shown to be important in the export of microcins in E. coli Nissle [42]. To rule out the possibility that our microcin and colibactin mutants possess defects in other pathways, we assessed the amounts of mccE492 of SGH10 and SGH10ΔclbP (Supplementary Fig. 6E). There were no differences in mccE492 activity between the strains, and so mccE492 export is independent of ClbP.
We then competed HvKp with obligate anaerobes known to colonize the human gut. Intriguingly, we found that the human gut obligate anaerobes Dorea longicatena, Lachnospiraceae 24430, and Oscillibacter acetigenes were susceptible to the effects of colibactin (Fig. 6B–D). These human gut isolates correspond to the taxa from the Clostridiales order that were depleted in mice colonized with colibactin-producing SGH10 in Fig. 5C. Other important human gut anaerobes such as Clostridioides difficile, Bifidobacterium adolescentis, and Bifidobacterium longum were also sensitive to mccE492 and colibactin (Fig. 6E–G).
Additionally, we competed HvKp with a panel of Klebsiella isolates that were clb-mce- and clbS-mceB-. We have observed differences in the susceptibility of E. coli and Klebsiella species to mccE492 and colibactin during oxic or anoxic conditions (Figs 6A and 7A, Supplementary Figs 7 and 8). Some strains were sensitive to mccE492 under oxic conditions but these strains were not killed by mccE492 under anoxic conditions. Some strains insensitive to colibactin in oxic conditions were susceptible to colibactin during anaerobic growth. Only K. pneumoniae NUH28 and NUH56 were susceptible to both mccE492 and colibactin during anoxic conditions (Figs 7A and 8C). In these experiments, the growth of K. pneumoniae was not affected by co-culture with other bacteria (Supplementary Figs 9 and 10).
Figure 7.

Effect of mccE492 and colibactin on other bacteria; (A) susceptibility of bacteria to mccE492 and colibactin; these bacteria did not possess immunity proteins to colibactin or mccE492; first column (yellow) denotes susceptibility to mccE492, second column (blue) denotes susceptibility to colibactin, and third column (green) denotes that a cooperative killing effect of both molecules is observed; unshaded boxes indicate insensitivity to these molecules; bacteria CFU are plotted in Fig. 6, and Supplementary Fig. 7,8; for all datasets described in this figure, mean ± SD are plotted (n = 3–4), and Dunnett’s multiple comparisons test was performed on CFU values to determine differences in means; * denotes P < .05 and ** denotes P < .01; (B) an in vivo predator–prey experiment was conducted in the murine model of gastrointestinal colonization with K. pneumoniae (Kp) NUH56 as prey; C57BL6/J mice were infected with 105 CFU of Kp NUH56 by oral gavage after ampicillin treatment for 5 days; 8 h later, the mice were gavaged with 5 x 105 CFU of SGH10ΔlacZ, SGH10ΔmceAΔlacZ, SGH10ΔclbPΔlacZ, or SGH10ΔmceAΔclbPΔlacZ; (C) the bacterial loads of NUH56 or D the SGH10 ΔlacZ mutants in stool were quantified; the geometric mean and SD are plotted (n = 1), and Dunnett’s multiple comparisons test was performed on log-transformed CFU values to determine differences in means; * denotes P < .05.
Figure 8.

Mechanism of mccE492 and colibactin mediated killing of other bacteria; (A) outer membrane perturbation by mccE492 was measured by NPN uptake in E. coli MG1655; (B) colistin was used as a positive control for outer membrane perturbation; NPN assays were performed n = 3 times and values are expressed as mean ± SD; (C) the TUNEL assay was used to quantify DNA damage of E. coli MG1655 co-incubated with SGH10 and mutants on solid media for 4 h under oxic and (D anoxic conditions; 50 μg/ml ciprofloxacin (Cip) was used as a positive control for DNA damage; (E) Propidium iodide (PI) uptake was measured in BA and (F) BL treated with mccE492 for 30 min; a positive control (+ control) for PI uptake was generated by treating BA and BL with lysozyme, mutanolysin, and isopropanol; (G) TUNEL assays were conducted to measure DNA damage in BA and H BL co-incubated with SGH10 and mutants for 4 h under anoxic conditions; 1% H2O2 is used as a positive control for DNA damage, and mean fluorescence intensity (MFI) values were quantified; in this figure, mean ± SD are plotted (n = 3–4), and Dunnett’s multiple comparisons test was performed to determine differences in means; * denotes P < .05.
Our screen revealed that some Gram-positive bacteria such as C. difficile, B. adolescentis, and B. longum were susceptible to both colibactin and mccE492 (Figs 6E–G and 7A). The major Gram-negative human gut species Bacteroides thetaiotaomicron was resistant K. pneumoniae to both (Figs 6F and 7A). Bacteroides uniformis was susceptible only to colibactin (Figs 6I and 7A). Co-culture with anaerobes did not affect the growth of K. pneumoniae (Supplementary Fig. 11) In E. coli BW25113, K. pneumoniae NUH11 and NUH56, and B. adolescentis, the effect of microcin and colibactin together is greater than the effect of either factor alone (Fig. 6A, Supplementary Figs 7H and 8C).
K. pneumoniae does not use mccE492 and colibactin to compete with closely related isolates
To determine whether mccE492 and colibactin in K. pneumoniae are important for competition with closely related bacterial prey, NUH56 was used as prey during in vivo competition (Fig. 7B) as it is a mce-clb-ST23 lineage strain that was susceptible to killing by colibactin and mccE492 (Fig. 7A). We did not observe statistically significant differences in the colonization of the NUH56 prey in competition with SGH10 or its various mutants (Fig. 7C).
We observed that expression of colibactin benefited SGH10 colonization even though it did not kill NUH56. Stool carriage of SGH10ΔclbPΔlacZ and SGH10ΔmceAΔclbPΔlacZ was significantly reduced compared to SGH10ΔlacZ on Day 11 (Fig. 7D). On Day 13, stool carriage of SGH10ΔmceAΔclbPΔlacZ but not SGH10ΔclbPΔlacZ was reduced relative to SGH10 (Fig. 7D). SGH10ΔmceAΔlacZ colonized the gut as well as SGH10ΔlacZ (Fig. 7D). Stool bacterial loads of NUH56 were ~2 logs lower in mice colonized with SGH10ΔclbPΔlacZ and SGH10ΔmceAΔclbPΔlacZ as compared to mice colonized with SGH10ΔlacZ and SGH10ΔmceAΔlacZ on Day 13 (Fig. 7D). The expression of colibactin or colibactin and mccE492 is important for SGH10 to colonize even when a competing K. pneumoniae strain is present. These results support a model where the crucial targets of microcin and colibactin are not competitors closely related to HvKp such as other susceptible K. pneumoniae, but other members of the microbiome that affect colonization resistance. In fact, colibactin and mccE492 seem to even benefit the colonization of other K. pneumoniae strain such as NUH56 that do not produce these factors themselves.
Effect of mccE492 and colibactin on bacterial prey
MccE492 is a pore-forming bacteriocin that exhibits activity against Gram-negatives [23, 24]. Using 1-N-phenylnaphthylamine (NPN) uptake as an output for outer membrane permeability, we demonstrated that purified mccE492 can perturb the outer membrane of E. coli in a dose-dependent manner (Fig. 8A and B). Moreover, purified mccE492 has a dose-dependent effect on the survival of the Gram-positives B. adolescentis, B. longum, and C. difficile (Supplementary Fig. 12).
Employing the TUNEL assay to examine DNA damage, we found that SGH10 induced a significant increase in TUNEL-positive E. coli under oxic and anoxic conditions (Fig. 8C and D, Supplementary Fig. 13). No significant increases in TUNEL-positive E. coli were observed when they were competed with SGH10ΔmceA, SGH10ΔclbP, or SGH10ΔmceAΔclbP (Fig. 8C and D, Supplementary Fig. 13).
C. difficile, B. adolescentis (BA), and B. longum (BL) were sensitive to colibactin and mccE492 (Figs 6E–G and 7A). MccE492 has not been reported to have an effect against Gram-positives. However, it is possible that unmodified mccE492 can perturb their membranes. Using a propidium iodide (PI) uptake assay, we demonstrated that treatment with mccE492 increased permeability in BA and BL (Fig. 8F and G, Supplementary Fig. 14). Although PI is not a specific assay for membrane perturbation, the NPN assay does not work with Gram-positives because the layer of peptidoglycan that shields the cytoplasmic membrane of Gram-positives is not hydrophobic, and NPN fluoresces in a hydrophobic environment such as in the Gram-negative outer membranes. These results show that mccE492 causes perturbation of membranes in BA and BL.
Additionally, we observed that SGH10 induced a significant increase in DNA damage in BA and BL (Fig. 8G and H, Supplementary Fig. 15). K. pneumoniae-induced DNA damage is dependent on both microcin and colibactin in both BA and BL (Fig. 8G and H). MccE492 appears to perturb the membrane of susceptible Gram-negative and Gram-positive bacteria. However, the genotoxic effect of colibactin on prey was observed only when both mccE492 and colibactin were expressed.
Colibactin can potentiate the death of susceptible bacteria by prophage induction [31]. We utilized the Δ9 strain of E. coli BW25113 that has been cured of all prophages [43] and found that it was still sensitive to colibactin, demonstrating that prophage induction is not necessary for colibactin-mediated killing of E. coli (Supplementary Fig. 16). Thus, K. pneumoniae-induced DNA damage was sufficient to kill E. coli without prophage induction. K. pneumoniae was unaffected by the presence of E. coli in these assays (Supplementary Fig. 16).
Discussion
In this study, we demonstrate that GIs GIE492 and ICEKp10 are co-associated with the CG23-I and CG10118 HvKp lineages. The products of GIE492 and ICEKp10 play cooperative roles during gastrointestinal colonization of HvKp. Epistatic interactions such as co-selection can occur between genetic loci that possess complementary functions. ICEKp10 integration is linked to clonal expansion of the CG23-I subclade within ST23 [4], further supporting a model where a beneficial relationship exists between these two GIs. Indeed, we also observe convergent evolution in CG10118, another hypervirulent lineage distant from CG23 that has acquired ICEKp10, GIE492, and the Klebsiella virulence plasmid.
There is a strong, colibactin-driven benefit of expressing mccE492 and colibactin during colonization. Colibactin is not essential during systemic infection, which contrasts with previous work reporting that a clbA deletion mutant in the HvKp strain KP 1084 was attenuated in virulence [44]. This phenotype is likely due to pleiotropic effects of deleting ClbA on siderophore maturation [45]. Gastrointestinal colonization precedes translocation and development of disseminated infection. SGH10∆clbP was attenuated in lethality in our murine model of HvKp translocation, likely because it colonizes poorly, although colibactin-induced tissue damage could also enhance bacterial adhesion and translocation.
Many K. pneumoniae lineages have acquired GIE492, implying a benefit of producing mccE492. However, mccE492 is dispensable for HvKp SGH10 to colonize the murine gut. MccE492 could be more important in the context of competition in the environment rather than in the mammalian gut. Indeed, mccE492 has a strong effect on E. coli during oxic but not anoxic conditions. MccE492 alone did not cause significant shifts in the composition of murine gut microbiota, and mccE492 seems to play a supportive role to colibactin during colonization of K. pneumoniae.
Colibactin likely benefits gastrointestinal colonization because it alters the gut microbiome. In a murine model involving maternal to fetal transmission of colibactin-producing E. coli, colibactin was shown to be important for intraspecies competition with closely related Enterobacterales and Firmicutes [46]. In our model, colibactin-producing K. pneumoniae caused changes in the gut microbiome of adult mice, inhibiting several gut commensal taxa belonging to the Clostridiales order (Lachnospiraceae and Christensenellaceae) and Muribaculaceae (Bacteroidales). Lachnospiraceae are important in short chain fatty acid (SCFA) and bile acid production, and Christensenellaceae are associated with host metabolic health [47-49]. Inhibition of these taxa by colibactin could reshape the metabolic landscape of the gut to benefit K. pneumoniae colonization. We observed colibactin-dependent killing of D. longicatena, Lachnospiraceae 24430, and O. acetigenes, human gut commensal species that corresponded to the taxa inhibited in mice. These results corroborated our murine experiments. The human commensal isolates used have also been reported to produce SCFAs and modify host metabolism, further supporting the idea that inhibition of bacterial taxa by colibactin could affect metabolism in the gut [50-52].
Besides the above species, inhibition of human commensal taxa belonging to Clostridiales (O. acetigenes, Lachnospiraceae 24430) and Bacteroidales (B. uniformis) order further bolsters the possibility that HvKp could be in competition with commensal species in the gut. SGH10 did not use colibactin and mccE492 to kill a closely related susceptible prey strain NUH56 in the gut. These factors may only provide a slight competitive advantage against closely related Enterobacterales bacteria perhaps because of spatial segregation. Our results strongly suggest that colibactin has a greater effect on gut commensals. The benefit for HvKp SGH10 inhibiting NUH56 is likely outweighed by the advantage of SGH10 killing gut commensals that compete with NUH56, overall benefiting both K. pneumoniae strains. The main target of colibactin appears to be gut commensal species in closer proximity to the colonizing strain, and the specificity of colibactin is not as narrow as initially imagined.
We made the unexpected discovery that Gram-positive anaerobes were susceptible to mccE492 because mccE492 is thought to be restricted in activity to related Gram-negative bacteria expressing catechol siderophore receptors. Perhaps, mccE492 can insert into the membrane of Gram-positives through a siderophore-independent mechanism. MccE492 can insert into artificial liposomes and lipid bilayers and form pores [21]. We observed a cooperative killing effect of colibactin and mccE492 in a small number of bacteria. Moreover, K. pneumoniae-induced DNA damage in E. coli, B. adolescentis, and B. longum is dependent on the presence of mccE492 and colibactin. The mechanism by which colibactin enters mammalian or bacterial cells is still unknown. We hypothesize that during cooperative killing of prey, mccE492 disrupts the cellular envelope and enhances uptake of colibactin. Alternatively, mccE492 could cause DNA damage in bacterial prey as it has been shown to induce apoptosis in Hela cells [53]. Moreover, these observations are supported by in vivo gastrointestinal colonization where mccE492 alone is dispensable for colonization but enhances the beneficial effect of colibactin.
In summary, we describe a co-association between GIE492 and ICEKp10 in two HvKp lineages. The combination of colibactin and mccE492 is beneficial during colonization of HvKp in the gastrointestinal tract. Colibactin and mccE492 enable HvKp to kill bacterial gut commensals to gain a foothold in the crowded endogenous microbiome space. Our results suggest that the benefits that GIE492 and ICEKp10 confer during colonization have contributed to the dominance of the CG23-I lineage.
Supplementary Material
Acknowledgements
We thank Dr Jeanette Teo (National University Hospital) for sharing clinical isolates of Klebsiella oxytoca.
Contributor Information
Yi Han Tan, Infectious Diseases Translational Research Programme, Yong Loo Lin School of Medicine, National University of Singapore, 5 Science Drive 2, MD4, Level 2, Singapore 117545, Republic of Singapore; Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, MD7, 8 Medical Drive, Singapore 117596, Republic of Singapore; Genome Institute of Singapore (GIS), Agency for Science, Technology and Research (A*STAR), Singapore 138672, Republic of Singapore.
Patricio Arros, Grupo de Microbiología Integrativa, Laboratorio de Biología Estructural y Molecular BEM, Facultad de Ciencias, Departamento de Biología, Universidad de Chile, Las Palmeras 3425 Ñuñoa, Santiago, Chile.
Camilo Berríos-Pastén, Grupo de Microbiología Integrativa, Laboratorio de Biología Estructural y Molecular BEM, Facultad de Ciencias, Departamento de Biología, Universidad de Chile, Las Palmeras 3425 Ñuñoa, Santiago, Chile.
Indrik Wijaya, Genome Institute of Singapore (GIS), Agency for Science, Technology and Research (A*STAR), Singapore 138672, Republic of Singapore.
Wilson H W Chu, National Public Health Laboratory, National Centre for Infectious Diseases, 16 Jln Tan Tock Seng, Singapore 308442, Republic of Singapore.
Yahua Chen, Infectious Diseases Translational Research Programme, Yong Loo Lin School of Medicine, National University of Singapore, 5 Science Drive 2, MD4, Level 2, Singapore 117545, Republic of Singapore; Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, MD7, 8 Medical Drive, Singapore 117596, Republic of Singapore.
Guoxiang Cheam, Infectious Diseases Translational Research Programme, Yong Loo Lin School of Medicine, National University of Singapore, 5 Science Drive 2, MD4, Level 2, Singapore 117545, Republic of Singapore; Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, MD7, 8 Medical Drive, Singapore 117596, Republic of Singapore.
Ahmad Nazri Mohamed Naim, Genome Institute of Singapore (GIS), Agency for Science, Technology and Research (A*STAR), Singapore 138672, Republic of Singapore.
Andrés E Marcoleta, Grupo de Microbiología Integrativa, Laboratorio de Biología Estructural y Molecular BEM, Facultad de Ciencias, Departamento de Biología, Universidad de Chile, Las Palmeras 3425 Ñuñoa, Santiago, Chile.
Aarthi Ravikrishnan, Genome Institute of Singapore (GIS), Agency for Science, Technology and Research (A*STAR), Singapore 138672, Republic of Singapore.
Niranjan Nagarajan, Infectious Diseases Translational Research Programme, Yong Loo Lin School of Medicine, National University of Singapore, 5 Science Drive 2, MD4, Level 2, Singapore 117545, Republic of Singapore; Genome Institute of Singapore (GIS), Agency for Science, Technology and Research (A*STAR), Singapore 138672, Republic of Singapore.
Rosalba Lagos, Grupo de Microbiología Integrativa, Laboratorio de Biología Estructural y Molecular BEM, Facultad de Ciencias, Departamento de Biología, Universidad de Chile, Las Palmeras 3425 Ñuñoa, Santiago, Chile.
Yunn-Hwen Gan, Infectious Diseases Translational Research Programme, Yong Loo Lin School of Medicine, National University of Singapore, 5 Science Drive 2, MD4, Level 2, Singapore 117545, Republic of Singapore; Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, MD7, 8 Medical Drive, Singapore 117596, Republic of Singapore.
Conflicts of interest
We declare that the authors do not possess any competing financial interests in relation to the work published.
Funding
We acknowledge funding support from A*STAR, National Medical Research Council, Singapore (SC18/23-739915-OOE). YH TAN was supported by the NUS Research Scholarship (Ministry of Education, Singapore). P Arros was supported by the Maria Ghilardi Venegas Foundation (Chile). A Marcoleta was supported by the grant FONDECYT 1221193 (Agencia Nacional de Investigación y Desarrollo, ANID, Chile). C Berríos-Pastén was supported by the ANID doctoral scholarship (Chile). Computational resources were provided by the supercomputing infrastructure of Soroban (SATREPS MACH – JPM/JSA1705) at Centro de Modelación y Computación Científica at Universidad de La Frontera, Chile. The work is funded by National Research Foundation (NRFMOSTID21-0005) and National University of Singapore (Reimagine Research Fund Type 1) to YH Gan.
Data availability
The complete genome sequence of K. pneumoniae RYC492 was deposited under the Bioproject accession PRJNA179092. K. pneumoniae genome assemblies from the A-KLASS study have been deposited in GenBank under BioProject PRJNA956314. The complete sequences of GIE492 variants and mce alleles have been made available in Supplementary Data 1. Supplemental methods are available in Supplementary Information.
References
- 1. Belk WP. Pulmonary infections by Friedländer’s bacillus. J Infect Dis 1926;38:115–26. 10.1093/infdis/38.2.115 [DOI] [Google Scholar]
- 2. Liu YC, Cheng DL, Lin CL. Klebsiella pneumoniae liver abscess associated with septic endophthalmitis. Arch Intern Med 1986;10, 146:1913. 10.1001/archinte.1986.00360220057011 [DOI] [PubMed] [Google Scholar]
- 3. Shon AS, Bajwa RPS, Russo TA. Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: a new and dangerous breed. Virulence 2013;4:107–18. 10.4161/viru.22718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lam MMC, Wyres KL, Duchêne S et al. Population genomics of hypervirulent Klebsiella pneumoniae clonal-group 23 reveals early emergence and rapid global dissemination. Nat Commun 2018;9:2703. 10.1038/s41467-018-05114-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Struve C, Roe CC, Stegger M et al. Mapping the evolution of hypervirulent Klebsiella pneumoniae. mBio 2015;6:e00630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chung DR, Lee H, Park MH et al. Fecal carriage of serotype K1 Klebsiella pneumoniae ST23 strains closely related to liver abscess isolates in Koreans living in Korea. Eur J Clin Microbiol Infect Dis 2012;31:481–6. 10.1007/s10096-011-1334-7 [DOI] [PubMed] [Google Scholar]
- 7. Koh TH, Lee V, Chng J et al. Hypervirulent Klebsiella pneumoniae carriage in polyclinic attendees and national servicemen presenting with diarrhoea. Ann Acad Med Singap 2021;50:90–1. 10.47102/annals-acadmedsg.2020323 [DOI] [PubMed] [Google Scholar]
- 8. Fung CP, Lin YT, Lin JC et al. Klebsiella pneumoniae in gastrointestinal tract and pyogenic liver abscess. Emerg Infect Dis 2012;18:1322–5. http://wwwnc.cdc.gov/eid/article/18/8/11-1053_article.htm [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin YT, Siu LK, Lin JC et al. Seroepidemiology of Klebsiella pneumoniae colonizing the intestinal tract of healthy Chinese and overseas Chinese adults in Asian countries. BMC Microbiol 2012;12:13. 10.1186/1471-2180-12-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim JH, Jeong Y, Lee CK et al. Characteristics of Klebsiella pneumoniae isolates from stool samples of patients with liver abscess caused by hypervirulent K. pneumoniae. J Korean Med Sci 2020;35:e18. 10.3346/jkms.2020.35.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gal-Mor O, Finlay BB. Pathogenicity islands: a molecular toolbox for bacterial virulence. Cell Microbiol 2006;8:1707–19. 10.1111/j.1462-5822.2006.00794.x [DOI] [PubMed] [Google Scholar]
- 12. Botelho J, Mourão J, Roberts AP et al. Comprehensive genome data analysis establishes a triple whammy of carbapenemases, ICEs and multiple clinically relevant bacteria. Microb Genom 2020;6, 6:mgen000424. 10.1099/mgen.0.0004246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Botelho J, Schulenburg H. The role of integrative and conjugative elements in antibiotic resistance evolution. Trends Microbiol 2021;29:8–18. 10.1016/j.tim.2020.05.011 [DOI] [PubMed] [Google Scholar]
- 14. Regmi A, Boyd EF. Carbohydrate metabolic systems present on genomic islands are lost and gained in Vibrio parahaemolyticus. BMC Microbiol 2019;19:112. 10.1186/s12866-019-1487-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McDaniel TK, Jarvis KG, Donnenberg MS et al. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc Natl Acad Sci U S A 1995;92:1664–8. 10.1073/pnas.92.5.1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Berríos-Pastén C, Acevedo R, Arros P et al. Properties of genes encoding transfer RNAs as integration sites for genomic islands and prophages in Klebsiella pneumoniae. Genomics 2020. Nov cited 2022 Aug 10. 10.1101/2020.11.02.365908 [DOI] [Google Scholar]
- 17. Nassif X, Fournier JM, Arondel J et al. Mucoid phenotype of Klebsiella pneumoniae is a plasmid-encoded virulence factor. Infect Immun 1989;57:546–52. 10.1128/iai.57.2.546-552.1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Russo TA, Olson R, MacDonald U et al. Aerobactin mediates virulence and accounts for increased siderophore production under iron-iimiting conditions by hypervirulent (hypermucoviscous) Klebsiella pneumoniae. Infect Immun 2014;82:2356–67. 10.1128/IAI.01667-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vornhagen J, Bassis CM, Ramakrishnan S et al. A plasmid locus associated with Klebsiella clinical infections encodes a microbiome-dependent gut fitness factor. PLoS Pathog 2021;17:e1009537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marcoleta B-PC, Nuñez G, Monasterio O et al. Klebsiella pneumoniae asparagine tDNAs are integration hotspots for different genomic islands encoding microcin E492 production determinants and other putative virulence factors present in hypervirulent strains. Front Microbiol 2016;7:849. 10.3389/fmicb.2016.00849/abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lagos R, Baeza M, Corsini G et al. Structure, organization and characterization of the gene cluster involved in the production of microcin E492, a channel-forming bacteriocin. Mol Microbiol 2001;42:229–43. 10.1046/j.1365-2958.2001.02630.x [DOI] [PubMed] [Google Scholar]
- 22. Destoumieux-Garzón D, Peduzzi J, Thomas X et al. Parasitism of iron-siderophore receptors of Escherichia coli by the siderophore-peptide microcin E492m and its unmodified counterpart. Biometals 2006;19:181–91. 10.1007/s10534-005-4452-9 [DOI] [PubMed] [Google Scholar]
- 23. Baquero F, Lanza VF, Baquero MR et al. Microcins in Enterobacteriaceae: peptide antimicrobials in the eco-active intestinal chemosphere. Front Microbiol 2019;10:2261. 10.3389/fmicb.2019.02261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Biéler S, Silva F, Belin D. The polypeptide core of Microcin E492 stably associates with the mannose permease and interferes with mannose metabolism. Res Microbiol 2010;161:706–10. 10.1016/j.resmic.2010.07.003 [DOI] [PubMed] [Google Scholar]
- 25. Lai YC, Lin AC, Chiang MK et al. Genotoxic Klebsiella pneumoniae in Taiwan. PLoS One 2014;9:9. 10.1371/journal.pone.0096292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lam MMC, Wyres KL, Judd LM et al. Tracking key virulence loci encoding aerobactin and salmochelin siderophore synthesis in Klebsiella pneumoniae. Genomics 2018;10:77. 10.1186/s13073-018-0587-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wami H, Wallenstein A, Sauer D et al. Insights into evolution and coexistence of the colibactin- and yersiniabactin secondary metabolite determinants in enterobacterial populations. Microb Genom 2021;7:000577. 10.1099/mgen.0.000577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Buc E, Dubois D, Sauvanet P et al. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. Battista JR, editor. PLoS One 2013;8:e56964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dubinsky V, Dotan I, Gophna U. Carriage of colibactin-producing bacteria and colorectal cancer risk. Trends Microbiol 2020;28:874–6. 10.1016/j.tim.2020.05.015 [DOI] [PubMed] [Google Scholar]
- 30. Chen BH, Liu R, Jung IJ et al. A commensal-encoded genotoxin drives restriction of Vibrio cholerae colonization and host gut microbiome remodeling. Proc Natl Acad Sci U S A 2022;119:e2121180119. 10.1073/pnas.2121180119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Silpe JE, Wong JWH, Owen SV et al. The bacterial toxin colibactin triggers prophage induction. Nature 2022;603:315–20. 10.1038/s41586-022-04444-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wong JJ, Ho FK, Choo PY et al. Escherichia coli BarA-UvrY regulates the pks island and kills Staphylococci via the genotoxin colibactin during interspecies competition. PLoS Pathog 2022;18:e1010766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. De Lorenzo V, Martinez JL, Asensio C. Microcin-mediated interactions between Klebsiella pneumoniae and Escherichia coli strains. Microbiology 1984;130:391–400. 10.1099/00221287-130-2-391 [DOI] [PubMed] [Google Scholar]
- 34. Molton JS, Chan M, Kalimuddin S et al. Oral vs intravenous antibiotics for patients with Klebsiella pneumoniae liver abscess: a randomized, controlled noninferiority study. Clin Infect Dis 2020;71:952–9. 10.1093/cid/ciz881 [DOI] [PubMed] [Google Scholar]
- 35. Wyres KL, Nguyen TNT, Lam MMC et al. Genomic surveillance for hypervirulence and multi-drug resistance in invasive Klebsiella pneumoniae from South and Southeast Asia. Genome Med 2020;12:11. 10.1186/s13073-019-0706-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li Y, Li Z, Qian S et al. A fatal case of liver abscess caused by hypervirulent Klebsiella pneumoniae in a diabetic adolescent: a clinical and laboratory study. Pediatr Invest 2021;5:118–24. 10.1002/ped4.12238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Palmieri M, Wyres KL, Mirande C et al. Genomic evolution and local epidemiology of Klebsiella pneumoniae from a major hospital in Beijing, China, over a 15 year period: dissemination of known and novel high-risk clones. Microb Genom 2019;7:000520. 10.1099/mgen.0.0005207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nougayrède JP, Homburg S, Taieb F et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 2006;313:848–51. 10.1126/science.1127059 [DOI] [PubMed] [Google Scholar]
- 39. Kuo LJ, Yang LX. γ-H2AX – a novel biomarker for DNA double-strand breaks. In Vivo 2008;22:305–9. [PubMed] [Google Scholar]
- 40. Tan YH, Chen Y, Chu WHW et al. Cell envelope defects of different capsule-null mutants in K1 hypervirulent Klebsiella pneumoniae can affect bacterial pathogenesis. Mol Microbiol 2020;113:889–905. 10.1111/mmi.14447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tropini C, Moss EL, Merrill BD et al. Transient osmotic perturbation causes long-term alteration to the gut microbiota. Cell 2018;173:1742–1754.e17. 10.1016/j.cell.2018.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Massip C, Branchu P, Bossuet-Greif N et al. Deciphering the interplay between the genotoxic and probiotic activities of Escherichia coli Nissle 1917. PLoS Pathog 2019;15:e1008029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang X, Kim Y, Ma Q et al. Cryptic prophages help bacteria cope with adverse environments. Nat Commun 2010;1:147. 10.1038/ncomms1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lu MC, Chen YT, Chiang MK et al. Colibactin contributes to the hypervirulence of pks+ K1 CC23 Klebsiella pneumoniae in mouse meningitis infections. Front Cell Infect Microbiol 2017;7:103. 10.3389/fcimb.2017.00103/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Martin P, Marcq I, Magistro G et al. Interplay between siderophores and colibactin genotoxin biosynthetic pathways in Escherichia coli. PLoS Pathog 2013;9:e1003437. 10.1371/journal.ppat.1003437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tronnet S, Floch P, Lucarelli L et al. The genotoxin colibactin shapes gut microbiota in mice. mSphere 2020;5:11. 10.1128/mSphere.00589-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fusco W, Lorenzo MB, Cintoni M et al. Short-chain fatty-acid-producing bacteria: key components of the human gut microbiota. Nutrients 2023;15:2211. 10.3390/nu15092211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Krautkramer KA, Fan J, Bäckhed F. Gut microbial metabolites as multi-kingdom intermediates. Nat Rev Microbiol 2021;19:77–94. 10.1038/s41579-020-0438-4 [DOI] [PubMed] [Google Scholar]
- 49. Waters JL, Ley RE. The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. BMC Biol 2019;17:83. 10.1186/s12915-019-0699-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yang J, Li Y, Wen Z et al. Oscillospira - a candidate for the next-generation probiotics. Gut Microbes 2021;13:1987783. 10.1080/19490976.2021.1987783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Abdugheni R, Wang W, Wang Y et al. Metabolite profiling of human‐originated Lachnospiraceae at the strain level. iMeta 2022;1:e58. 10.1002/imt2.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Prudêncio APA, Fonseca DC, Machado NM et al. Red meat intake, indole-3-acetate, and Dorea longicatena together affect insulin resistance after gastric bypass. Nutrients 2023;15:1185. 10.3390/nu15051185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hetz C, Bono MR, Barros LF et al. Microcin E492, a channel-forming bacteriocin from Klebsiella pneumoniae induces apoptosis in some human cell lines. Proc Natl Acad Sci U S A 2002;99:2696–701. 10.1073/pnas.052709699 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The complete genome sequence of K. pneumoniae RYC492 was deposited under the Bioproject accession PRJNA179092. K. pneumoniae genome assemblies from the A-KLASS study have been deposited in GenBank under BioProject PRJNA956314. The complete sequences of GIE492 variants and mce alleles have been made available in Supplementary Data 1. Supplemental methods are available in Supplementary Information.