

Abstract
Background
Somatoform disorders are characterised by chronic, medically unexplained physical symptoms (MUPS). Although different medications are part of treatment routines for people with somatoform disorders in clinics and private practices, there exists no systematic review or meta‐analysis on the efficacy and tolerability of these medications. We aimed to synthesise to improve optimal treatment decisions.
Objectives
To assess the effects of pharmacological interventions for somatoform disorders (specifically somatisation disorder, undifferentiated somatoform disorder, somatoform autonomic dysfunction, and pain disorder) in adults.
Search methods
We searched the Cochrane Depression, Anxiety and Neurosis Review Group's Specialised Register (CCDANCTR) (to 17 January 2014). This register includes relevant randomised controlled trials (RCTs) from The Cochrane Library (all years), MEDLINE (1950 to date), EMBASE (1974 to date), and PsycINFO (1967 to date). To identify ongoing trials, we searched ClinicalTrials.gov, Current Controlled Trials metaRegister, the World Health Organization International Clinical Trials Registry Platform, and the Chinese Clinical Trials Registry. For grey literature, we searched ProQuest Dissertation & Theses Database, OpenGrey, and BIOSIS Previews. We handsearched conference proceedings and reference lists of potentially relevant papers and systematic reviews and contacted experts in the field.
Selection criteria
We selected RCTs or cluster RCTs of pharmacological interventions versus placebo, treatment as usual, another medication, or a combination of different medications for somatoform disorders in adults. We included people fulfilling standardised diagnostic criteria for somatisation disorder, undifferentiated somatoform disorder, somatoform autonomic dysfunction, or somatoform pain disorder.
Data collection and analysis
One review author and one research assistant independently extracted data and assessed risk of bias. Primary outcomes included the severity of MUPS on a continuous measure, and acceptability of treatment.
Main results
We included 26 RCTs (33 reports), with 2159 participants, in the review. They examined the efficacy of different types of antidepressants, the combination of an antidepressant and an antipsychotic, antipsychotics alone, or natural products (NPs). The duration of the studies ranged between two and 12 weeks.
One meta‐analysis of placebo‐controlled studies showed no clear evidence of a significant difference between tricyclic antidepressants (TCAs) and placebo for the outcome severity of MUPS (SMD ‐0.13; 95% CI ‐0.39 to 0.13; 2 studies, 239 participants; I2 = 2%; low‐quality evidence). For new‐generation antidepressants (NGAs), there was very low‐quality evidence showing they were effective in reducing the severity of MUPS (SMD ‐0.91; 95% CI ‐1.36 to ‐0.46; 3 studies, 243 participants; I2 = 63%). For NPs there was low‐quality evidence that they were effective in reducing the severity of MUPS (SMD ‐0.74; 95% CI ‐0.97 to ‐0.51; 2 studies, 322 participants; I2 = 0%).
One meta‐analysis showed no clear evidence of a difference between TCAs and NGAs for severity of MUPS (SMD ‐0.16; 95% CI ‐0.55 to 0.23; 3 studies, 177 participants; I2 = 42%; low‐quality evidence). There was also no difference between NGAs and other NGAs for severity of MUPS (SMD ‐0.16; 95% CI ‐0.45 to 0.14; 4 studies, 182 participants; I2 = 0%).
Finally, one meta‐analysis comparing selective serotonin reuptake inhibitors (SSRIs) with a combination of SSRIs and antipsychotics showed low‐quality evidence in favour of combined treatment for severity of MUPS (SMD 0.77; 95% CI 0.32 to 1.22; 2 studies, 107 participants; I2 = 23%).
Differences regarding the acceptability of the treatment (rate of all‐cause drop‐outs) were neither found between NGAs and placebo (RR 1.01, 95% CI 0.64 to 1.61; 2 studies, 163 participants; I2 = 0%; low‐quality evidence) or NPs and placebo (RR 0.85, 95% CI 0.40 to 1.78; 3 studies, 506 participants; I2 = 0%; low‐quality evidence); nor between TCAs and other medication (RR 1.48, 95% CI 0.59 to 3.72; 8 studies, 556 participants; I2 =14%; low‐quality evidence); nor between antidepressants and the combination of an antidepressant and an antipsychotic (RR 0.80, 95% CI 0.25 to 2.52; 2 studies, 118 participants; I2 = 0%; low‐quality evidence). Percental attrition rates due to adverse effects were high in all antidepressant treatments (0% to 32%), but low for NPs (0% to 1.7%).
The risk of bias was high in many domains across studies. Seventeen trials (65.4%) gave no information about random sequence generation and only two (7.7%) provided information about allocation concealment. Eighteen studies (69.2%) revealed a high or unclear risk in blinding participants and study personnel; 23 studies had high risk of bias relating to blinding assessors. For the comparison NGA versus placebo, there was relatively high imprecision and heterogeneity due to one outlier study. Although we identified 26 studies, each comparison only contained a few studies and small numbers of participants so the results were imprecise.
Authors' conclusions
The current review found very low‐quality evidence for NGAs and low‐quality evidence for NPs being effective in treating somatoform symptoms in adults when compared with placebo. There was some evidence that different classes of antidepressants did not differ in efficacy; however, this was limited and of low to very low quality. These results had serious shortcomings such as the high risk of bias, strong heterogeneity in the data, and small sample sizes. Furthermore, the significant effects of antidepressant treatment have to be balanced against the relatively high rates of adverse effects. Adverse effects produced by medication can have amplifying effects on symptom perceptions, particularly in people focusing on somatic symptoms without medical causes. We can only draw conclusions about short‐term efficacy of the pharmacological interventions because no trial included follow‐up assessments. For each of the comparisons where there were available data on acceptability rates (NGAs versus placebo, NPs versus placebo, TCAs versus other medication, and antidepressants versus a combination of an antidepressant and an antipsychotic), no clear differences between the intervention and comparator were found.
Future high‐quality research should be carried out to determine the effectiveness of medications other than antidepressants, to compare antidepressants more thoroughly, and to follow‐up participants over longer periods (the longest follow up was just 12 weeks). Another idea for future research would be to include other outcomes such as functional impairment or dysfunctional behaviours and cognitions as well as the classical outcomes such as symptom severity, depression, or anxiety.
Keywords: Adult; Humans; Middle Aged; Antidepressive Agents, Second‐Generation; Antidepressive Agents, Second‐Generation/therapeutic use; Antidepressive Agents, Tricyclic; Antidepressive Agents, Tricyclic/therapeutic use; Antipsychotic Agents; Antipsychotic Agents/therapeutic use; Randomized Controlled Trials as Topic; Selective Serotonin Reuptake Inhibitors; Selective Serotonin Reuptake Inhibitors/therapeutic use; Somatoform Disorders; Somatoform Disorders/drug therapy
Plain language summary
Medication as a treatment for long‐term medically unexplained physical symptoms (somatoform disorders): a review of the evidence
Who may be interested in this review?
‐ People with long‐term unexplained physical symptoms (somatoform disorders) and their family and friends.
‐ Professionals working with people with somatoform disorders.
‐ Professionals working in chronic pain services.
‐ General practitioners.
Why is this review important?
Around 6 in 100 people are affected by long‐term physical symptoms that have no clear medical cause (somatoform disorders). Symptoms can include pain, digestive problems, sexual or menstrual problems, breathing problems, and symptoms that mimic brain or nerve damage such as memory loss or sensory problems. Somatoform disorders often cause considerable distress and mean that people spend a lot of time consulting doctors and health professionals to try to find the cause of their symptoms and the correct treatment.
Guidelines for the treatment of somatoform disorders recommend that people receive talking therapies alongside medication. In current practice many people are treated 'off label' with medications that are intended for the treatment of anxiety, depression, and other mental health problems. However, it is unclear why medications such as antidepressants help to reduce the severity of medically unexplained physical symptoms.
What questions does this review aim to answer?
‐ What is the quality of current research on medication as a treatment for somatoform disorders?
‐ Is medication an effective treatment for physical symptoms in somatoform disorders compared with placebo (dummy pill)?
‐ Which types of medication are most effective?
‐ Are natural products such as St. John's wort an effective treatment for somatoform disorders compared with placebo?
‐ How well do people with somatoform disorders tolerate medication or natural products?
Which studies did we include in the review?
We searched databases to find all studies of medication for somatoform disorders published until January 2014. To be included in the review, studies had to compare medication with either placebo, usual treatment, another medication, or a combination of medication and include adults with a clear diagnosis of somatoform disorders. We included 26 studies in the review with 2159 participants aged between 18 and 77 years.
What does the evidence from the review tell us?
Although we identified 26 studies, each comparison only contained a few studies and a relatively small number of participants and so the findings must be interpreted with caution. We rated the quality of current research as low or very low and the risks of bias were high in many of the studies.
There was not sufficient evidence in order to make a statement about the efficacy of tricyclic antidepressants for the treatment of somatoform disorders.
New‐generation antidepressants were moderately effective treatments for physical symptoms, anxiety, and depression in somatoform disorders.
There was no difference found between the effectiveness of tricyclic antidepressants and new‐generation antidepressants for the treatment of physical symptoms. There was some evidence that a combination of antidepressants and antipsychotics was more effective than antidepressants alone.
Natural products, such as St. John's wort, significantly reduced the severity of physical symptoms compared with placebo.
High numbers of people dropped out of treatment due to side effects or lack of effects with antidepressant medication, and low numbers dropped out with natural products.
What should happen next?
The review authors suggest that future high‐quality research should be carried out to look at the effectiveness of medications other than antidepressants, to compare antidepressants more thoroughly and to follow up participants over longer periods (the longest follow‐up was just 12 weeks). The review authors also suggest that future research should measure changes in people's quality of life and daily functioning as well as physical symptoms and depression/anxiety symptoms.
Summary of findings
Summary of findings for the main comparison. Tricyclic antidepressants versus placebo for somatoform disorders in adults.
| Tricyclic antidepressants versus placebo for somatoform disorders in adults | ||||||
| Patient or population: somatoform disorders in adults Settings: outpatient setting Intervention: tricyclic antidepressants versus placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Tricyclic antidepressants versus placebo | |||||
| Severity/intensity of MUPS (post‐treatment score on self report scales) Different self report scales (SCL‐90‐R Somatisation Subscore, VAS)1 Follow‐up: 6‐12 weeks | ‐ | The mean severity/intensity of MUPS (post‐treatment score on self report scales) in the intervention groups was 0.13 standard deviations lower (0.39 lower to 0.13 higher) | ‐ | 239 (2 studies) | ⊕⊕⊝⊝ low2,3 | SMD ‐0.13 (95% CI ‐0.39 to 0.13) |
| Acceptability (all‐cause drop‐outs) | No data available | |||||
| Anxiety (post‐treatment score on self report and clinician‐rated scales) | No data available | |||||
| Depression (post‐treatment score on self report and clinician‐rated scales) | No data available | |||||
| Adverse effects (drop‐outs due to adverse effects) | No data available | |||||
| Treatment response (post‐treatment score on self report and clinician‐rated scales) | No data available | |||||
| Functional disability and quality of life (post‐treatment score on self report and clinician‐rated scales) | No data available | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; MUPS: medically unexplained physical symptoms; SCL: Symptom Checklist; SMD: standardised mean difference; VAS: visual analogue scale. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 SCL‐90‐R and VAS: high scale scores correspond to a negative outcome. 2 We considered the results to have a serious risk of bias and so we downgraded the quality of evidence by 1 point because none of the following criteria was met: a low risk of bias for sequence generation, allocation concealment, blinding of participants and assessors, incomplete outcome data, and selective outcome reporting. 3 We considered the results to be imprecise because the total population size was fewer than 400 and so we downgraded the quality of evidence by 1 point.
Summary of findings 2. New‐generation antidepressants versus placebo for somatoform disorders in adults.
| New‐generation antidepressants versus placebo for somatoform disorders in adults | ||||||
| Patient or population: somatoform disorders in adults Settings: outpatient setting Intervention: new‐generation antidepressants (SSRI, SNRI) versus placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | New‐generation antidepressants versus placebo | |||||
| Severity/intensity of MUPS (post‐treatment score on self report scales) Different self report scales (PHQ‐15, MOSPM)1 Follow‐up: 8‐12 weeks | ‐ | The mean severity/intensity of MUPS in the intervention groups was 0.91 standard deviations lower (1.36 to 0.46 lower) | ‐ | 243 (3 studies) | ⊕⊝⊝⊝ very low2,3,4 | SMD ‐0.91 (95% CI ‐1.36 to ‐0.46) |
| Acceptability (all‐cause drop‐outs)5 Follow‐up: mean 12 weeks | Study population | RR 1.01 (0.64 to 1.61) | 163 (2 studies) | ⊕⊕⊝⊝ low2,6 | ‐ | |
| 265 per 1000 | 268 per 1000 (170 to 427) | |||||
| Moderate | ||||||
| 193 per 1000 | 195 per 1000 (124 to 311) | |||||
| Anxiety (post‐treatment score on self report and clinician‐rated scales) Clinician‐rated scales (HARS)7 Follow‐up: mean 12 weeks | ‐ | The mean anxiety score in the intervention groups was 0.88 standard deviations lower (1.81 lower to 0.05 higher) | ‐ | 163 (2 studies) | ⊕⊝⊝⊝ very low2,3,4 | SMD ‐0.88 (95% CI ‐1.81 to 0.05) |
| Depression (post‐treatment score on self report and clinician‐rated scales) Different clinician‐rated scales (HDRS, MADRS)8 Follow‐up: mean 12 weeks | ‐ | The mean depression score in the intervention groups was 0.56 standard deviations lower (0.88 to 0.25 lower) | ‐ | 163 (2 studies) | ⊕⊝⊝⊝ very low2,3,4 | SMD ‐0.56 (95% CI ‐0.88 to ‐0.25) |
| Adverse effects (drop‐outs due to adverse effects)5 Follow‐up: mean 12 weeks | Study population | RR 2.26 (0.52 to 9.81) | 163 (2 studies) | ⊕⊕⊝⊝ low2,6 | ‐ | |
| 24 per 1000 | 54 per 1000 (13 to 236) | |||||
| Moderate | ||||||
| 18 per 1000 | 41 per 1000 (9 to 177) | |||||
| Treatment response (post‐treatment score on self report and clinician‐rated scales) Different self report and clinician‐rated scales (PHQ‐15, CGI ‐ Improvement Scale) Follow‐up: mean 12 weeks | Study population | RR 2 (0.9 to 4.43) | 163 (2 studies) | ⊕⊝⊝⊝ very low2,3,6 | ‐ | |
| 337 per 1000 | 675 per 1000 (304 to 1000) | |||||
| Moderate | ||||||
| 319 per 1000 | 638 per 1000 (287 to 1000) | |||||
| Functional disability and quality of life (post‐treatment score on self report scales) Different self report scales (SF‐36, SDS)9 Follow‐up: mean 12 weeks | ‐ | The mean functional disability score/quality of life score in the intervention groups was 0.52 standard deviations lower/higher (1 to 0.04 lower/higher) | ‐ | 163 (2 studies) | ⊕⊝⊝⊝ very low2,3,4 | SMD ‐0.52 (95% CI ‐1 to ‐0.04) |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CGI: Clinical Global Impression Scale; CI: confidence interval; HARS: Hamilton Anxiety Rating Scale; HDRS: Hamilton Depression Rating Scale; MADRS: Montgomery‐Åsberg Depression Rating Scale; MOSPM: Medical Outcomes Study Pain Measures; MUPS: medically unexplained physical symptoms; PHQ: Patient Health Questionnaire; RR: risk ratio; SDS: Sheehan Disability Scale; SF‐36: 36‐item Short Form; SMD: standardised mean difference; SNRI: serotonin norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 PHQ‐15 and MOSPM: high scale scores correspond to a negative outcome. 2 We considered the results to have a serious risk of bias and so we downgraded the quality of evidence by 1 point because none of the following criteria was met: a low risk of bias for sequence generation, allocation concealment, blinding of participants and assessors, incomplete outcome data, and selective outcome reporting. 3 We assumed that in 1 study, the SE instead of SD were reported (Muller 2008). Therefore, we re‐calculated the values of variance before we entered them into the meta‐analysis. The effect size of this study was still quite high in comparison to the other studies and could be considered as an outlier. A sensitivity analysis where we excluded this study did not change the pooled effect size significantly. Therefore, we considered the results to be inconsistent and so we downgraded the quality of evidence by 1 point. 4 We considered the results to be imprecise because the total population size was fewer than 400 and so we downgraded the quality of evidence by 1 point. 5 We calculated this rate as a proportion of the total number of randomised participants. 6 We considered the results to be imprecise because the total number of events was fewer than 300 and so we downgraded the quality of evidence by 1 point. 7 HARS: high scale scores correspond to a negative outcome. 8 HDRS and MADRS: high scale scores correspond to a negative outcome. 9 SF‐36: high scale scores correspond to a positive outcome and had to be re‐coded; SDS: high scale scores correspond to a negative outcome.
Summary of findings 3. Natural products versus placebo for somatoform disorders in adults.
| Natural products versus placebo for somatoform disorders in adults | ||||||
| Patient or population: somatoform disorders in adults Settings: outpatient setting Intervention: natural products versus placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Natural products versus placebo | |||||
| Severity/intensity of MUPS (post‐treatment score on self report scales) Different self report scales (SOMS‐7, SCL‐90‐R Somatisation Subscore)1 Follow‐up: mean 6 weeks | ‐ | The mean severity/intensity of MUPS in the intervention groups was 0.74 standard deviations lower (0.97 to 0.51 lower) | ‐ | 322 (2 studies) | ⊕⊕⊝⊝ low2,3 | SMD ‐0.74 (95% CI ‐0.97 to ‐0.51) |
| Acceptability (all‐cause drop‐outs4) Follow‐up: 2‐6 weeks | Study population | RR 0.85 (0.4 to 1.78) | 506 (3 studies) | ⊕⊕⊝⊝ low2,5 | ‐ | |
| 58 per 1000 | 50 per 1000 (23 to 104) | |||||
| Moderate | ||||||
| 68 per 1000 | 58 per 1000 (27 to 121) | |||||
| Anxiety (post‐treatment score on self report and clinician‐rated scales) Different self report and clinician‐rated scales (HARS, VAS)6 Follow‐up: 2‐6 weeks | ‐ | The mean anxiety score in the intervention groups was 0.83 standard deviations lower (1.13 to 0.52 lower) | ‐ | 321 (2 studies) | ⊕⊕⊝⊝ low2,3 | SMD ‐0.83 (95% CI ‐1.13 to ‐0.52) |
| Depression (post‐treatment score on self report and clinician‐rated scales) Different self report and clinician‐rated scales (HDRS, BDI)7 Follow‐up: 2‐6 weeks | ‐ | The mean depression score in the intervention groups was 0.64 standard deviations lower (0.87 to 0.41 lower) | ‐ | 321 (2 studies) | ⊕⊕⊝⊝ low2,3 | SMD ‐0.64 (95% CI ‐0.87 to ‐0.41) |
| Adverse effects (drop‐outs due to adverse effects4) Follow‐up: 2‐6 weeks | Study population | RR 0.54 (0.08 to 3.5) | 506 (3 studies) | ⊕⊕⊝⊝ low2,5 | ‐ | |
| 13 per 1000 | 7 per 1000 (1 to 47) | |||||
| Moderate | ||||||
| 16 per 1000 | 9 per 1000 (1 to 56) | |||||
| Treatment response (post‐treatment score on self report and clinician‐rated scales) Different self report and clinician‐rated scales (PHQ‐15, CGI ‐ Improvement Scale) Follow‐up: mean 12 weeks | Study population | RR 1.77 (1.34 to 2.34) | 324 (2 studies) | ⊕⊝⊝⊝ very low2,5,8 | ‐ | |
| 340 per 1000 | 601 per 1000 (455 to 794) | |||||
| Moderate | ||||||
| 352 per 1000 | 623 per 1000 (472 to 824) | |||||
| Functional disability and quality of life (post‐treatment score on self report scales) | No data available | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). BDI: Beck Depression Inventory; CGI: Clinical Global Impression Scale; CI: confidence interval; HARS: Hamilton Anxiety Rating Scale; HDRS: Hamilton Depression Rating Scale; MUPS: medically unexplained physical symptoms; PHQ: Patient Health Questionnaire; RR: risk ratio; SCL: Symptom Checklist; SMD: standardised mean difference; SOMS: Screening for Somatoform Symptoms; VAS: visual analogue scale. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 SOMS‐7 and SCL‐90‐R: high scale scores correspond to a negative outcome. 2 We considered the results to have a serious risk of bias and so we downgraded the quality of evidence by 1 point because none of the following criteria was met: a low risk of bias for sequence generation, allocation concealment, blinding of participants and assessors, incomplete outcome data, and selective outcome reporting. 3 We considered the results to be imprecise because the total population size was fewer than 400 and so we downgraded the quality of evidence by 1 point. 4 We calculated this rate as a proportion of the total number of randomised participants. 5 We considered the results to be imprecise because the total number of events was fewer than 300 and so we downgraded the quality of evidence by 1 point. 6 HARS and VAS: high scale scores correspond to a negative outcome. 7 HDRS and BDI: high scale scores correspond to a negative outcome. 8 We assumed that in 1 study, the SE instead of SD were reported (Muller 2008). Therefore, we re‐calculated the values of variance in SE before we entered them in the meta‐analysis. The effect size of this study was still quite high in comparison to the other studies and could be considered as an outlier. A sensitivity analysis where we excluded this study did not change the pooled effect size significantly. Therefore, we considered the results to be inconsistent and so we downgraded the quality of evidence by 1 point.
Summary of findings 4. Tricyclic antidepressants versus another medication for somatoform disorders in adults.
| Tricyclic antidepressants versus another medication for somatoform disorders in adults | ||||||
| Patient or population: somatoform disorders in adults Settings: outpatient and inpatient setting Intervention: tricyclic antidepressants versus another medication | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Tricyclic antidepressants versus another medication | |||||
| Severity/intensity of MUPS (post‐treatment score on self report scales) Different self report scales (VAS‐Pain, SCL‐90‐R Somatisation Subscore)1 Follow‐up: 6‐8 weeks | ‐ | The mean severity/intensity of MUPS in the intervention groups was 0.16 standard deviations lower (0.55 lower to 0.23 higher) | ‐ | 177 (3 studies) | ⊕⊕⊝⊝ low2,3 | SMD ‐0.16 (95% CI ‐0.55 to 0.23) |
| Acceptability (all‐cause drop‐outs4) Follow‐up: 4‐8 weeks | Study population | RR 1.48 (0.59 to 3.72) | 556 (8 studies) | ⊕⊕⊝⊝ low2,5 | ‐ | |
| 28 per 1000 | 41 per 1000 (16 to 103) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Anxiety (post‐treatment score on self report and clinician‐rated scales) Different self report and clinician‐rated scales (HARS, SCL‐90 Anxiety Subscore)6 Follow‐up: 6‐8 weeks | ‐ | The mean anxiety score in the intervention groups was 0.37 standard deviations higher (0.21 lower to 0.95 higher) | ‐ | 255 (4 studies) | ⊕⊝⊝⊝ very low2,3,7 | SMD 0.37 (95% CI ‐0.21 to 0.95) |
| Depression (post‐treatment score on self report and clinician‐rated scales) Different self report and clinician‐rated scales (VAS Sadness, HDRS, SCL‐90 Depression Subscore, ZDS)8 Follow‐up: 6‐8 weeks | ‐ | The mean depression score in the intervention groups was 0.17 standard deviations higher (0.07 lower to 0.4 higher) | ‐ | 395 (6 studies) | ⊕⊕⊝⊝ low2,3 | SMD 0.17 (95% CI ‐0.07 to 0.4) |
| Adverse effects (drop‐outs due to adverse effects4) Follow‐up: 4‐8 weeks | Study population | RR 2.37 (0.39 to 14.28) | 556 (8 studies) | ⊕⊕⊝⊝ low2,5 | ‐ | |
| 10 per 1000 | 25 per 1000 (4 to 148) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| Treatment response (post‐treatment score on self report and clinician‐rated scales) CGI ‐ Improvement Scale Follow‐up: mean 8 weeks | Study population | RR 0.93 (0.73 to 1.19) | 130 (2 studies) | ⊕⊕⊝⊝ low2,9 | ‐ | |
| 677 per 1000 | 630 per 1000 (494 to 806) | |||||
| Moderate | ||||||
| 681 per 1000 | 633 per 1000 (497 to 810) | |||||
| Functional disability and quality of life | No data available | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CGI: Clinical Global Impression Scale; CI: confidence interval; HARS: Hamilton Anxiety Rating Scale; HDRS: Hamilton Depression Rating Scale; MUPS: medically unexplained physical symptoms; RR: risk ratio; SCL: Symptom Checklist; SMD: standardised mean difference; VAS: Visual Analogue Scale; ZDS: Zung Depression Scale. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 VAS ‐ Pain and SCL‐90‐R: high scale scores correspond to a negative outcome. 2 We considered the results to have a serious risk of bias and so we downgraded the quality of evidence by 1 point because none of the following criteria was met: a low risk of bias for sequence generation, allocation concealment, blinding of participants and assessors, incomplete outcome data, and selective outcome reporting. 3 We considered the results to be imprecise because the total population size was fewer than 400 and so we downgraded the quality of evidence by 1 point. 4 We calculated this rate as a proportion of the total number of randomised participants. 5 We considered the results to be imprecise because the 95% CI around the pooled effect included both 1. no effect and 2. appreciable benefit or appreciable harm. Therefore, we downgraded the quality of evidence by 1 point. 6 HARS: high scale scores correspond to a negative outcome. 7 We considered the results to be inconsistent because the I2 value was large. Therefore, we downgraded the quality of evidence by 1 point. 8 VAS Sadness, HDRS, SCL‐90, and ZDS: high scale scores correspond to a negative outcome. 9 We considered the results to be imprecise because the total number of events was fewer than 300 and so we downgraded the quality of evidence by 1 point.
Summary of findings 5. Antidepressants versus a combination of medications for somatoform disorders in adults.
| Antidepressants versus a combination of medications for somatoform disorders in adults | ||||||
| Patient or population: somatoform disorders in adults Settings: outpatient setting Intervention: antidepressants versus a combination of antidepressant and antipsychotic | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Pharmacotherapy versus a combination of medications | |||||
| Severity/intensity of MUPS (post‐treatment score on self report scales) Different self report and clinician‐rated scales (SOMS‐7, SCL‐90 Somatisation Score)1 Follow‐up: 6‐8 weeks | ‐ | The mean severity/intensity of MUPS in the intervention groups was 0.77 standard deviations higher (0.32 to 1.22 higher) | ‐ | 107 (2 studies) | ⊕⊕⊝⊝ low2,3 | SMD 0.77 (95% CI 0.32 to 1.22) |
| Acceptability (all‐cause drop‐outs4) Follow‐up: 6‐8 weeks | Study population | RR 0.8 (0.25 to 2.52) | 118 (2 studies) | ⊕⊕⊝⊝ low2,5 | ‐ | |
| 190 per 1000 | 152 per 1000 (47 to 478) | |||||
| Moderate | ||||||
| 186 per 1000 | 149 per 1000 (47 to 469) | |||||
| Anxiety (post‐treatment score on self report and clinician‐rated scales) Different self report and clinician‐rated scales (HARS, SCL‐90 Anxiety Subscore)6 Follow‐up: 6‐8 weeks | ‐ | The mean anxiety in the intervention groups was 0.95 standard deviations higher (0.91 lower to 2.82 higher) | ‐ | 107 (2 studies) | ⊕⊝⊝⊝ very low2,3,7 | SMD 0.95 (95% CI ‐0.91 to 2.82) |
| Depression (post‐treatment score on self report and clinician‐rated scales) Different self report and clinician‐rated scales (HDRS, SCL‐90 depression subscore)8 Follow‐up: 6‐8 weeks | ‐ | The mean depression in the intervention groups was 0.58 standard deviations higher (0.33 lower to 1.48 higher) | ‐ | 107 (2 studies) | ⊕⊝⊝⊝ very low2,3,7 | SMD 0.58 (95% CI ‐0.33 to 1.48) |
| Adverse effects (drop‐outs due to adverse effects) | No data available | |||||
| Treatment response (post‐treatment score on self report and clinician‐rated scales) | No data available | |||||
| Functional disability and quality of life | No data available | |||||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HARS: Hamilton Anxiety Rating Scale; HDRS: Hamilton Depression Rating Scale; MUPS: medically unexplained physical symptoms; RR: risk ratio; SCL: Symptom Checklist; SMD: standardised mean difference; SOMS: Screening for Somatoform Symptoms. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 SOMS‐7 and SCL‐90 Somatisation Score: high scale scores correspond to a negative outcome. 2 We considered the results to have a serious risk of bias and so we downgraded the quality of evidence by 1 point because none of the following criteria was met: a low risk of bias for sequence generation, allocation concealment, blinding of participants and assessors, incomplete outcome data, and selective outcome reporting. 3 We considered the results to be imprecise because the total population size was fewer than 400 and so we downgraded the quality of evidence by 1 point. 4 We calculated this rate as a proportion of the total number of randomised participants. 5 We considered the results imprecise because the total number of events was fewer than 300 and so we downgraded the quality of evidence by 1 point. 6 HARS and SCL‐90: high scale scores correspond to a negative outcome. 7 We considered the results to be inconsistent because the I2 value was large. Therefore, we downgraded the quality of evidence by 1 point. 8 HDRS and SCL‐90 Depression Subscore: high scale scores correspond to a negative outcome.
Background
Description of the condition
Medically unexplained physical symptoms (MUPS) are somatic symptoms that cannot or have not been sufficiently explained by organic causes after a thorough physical examination (Sharpe 1995). The key feature of conditions known as 'somatoform disorders' is the presence of such MUPS with a chronic manifestation. Corresponding to the Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV; APA 1994), and the International Classification of Diseases (ICD‐10; WHO 1992), there are four somatoform diagnostic categories that include MUPS as their main indication:
somatisation disorder;
undifferentiated somatoform disorder;
somatoform autonomic dysfunction; and
pain disorder.
The current review is confined to these four categories. We did not consider other somatoform diagnoses because they either do not primarily focus on MUPS (hypochondriasis, body dysmorphic disorder), or the duration and chronic manifestation criteria are not specifically required (other somatoform disorders or somatoform disorders unspecified), or it is required that the physical symptoms are associated with a distressing event (conversion disorder).
Somatisation disorder depicts an extreme and chronic form of MUPS. To be diagnosed with this disorder, DSM‐IV (APA 1994) requires an overall number of eight unexplained physical symptoms, with a chronic manifestation relating to at least four different organ systems: pain, gastrointestinal, sexual, and pseudoneurological symptoms. Physical symptoms should begin before the age of 30 years and must lead to healthcare utilisation or significant impairment in important areas of functioning. Diagnostic criteria for somatisation according to the ICD‐10 are similar to those of DSM‐IV. The only differences are that ICD‐10 requires the duration of symptoms to be at least two years (independent of age at onset), that the lists of physical symptoms are structured in different ways, and that the person persistently refuses to accept that his or her symptoms are unexplained by a medical condition.
A less stringent form of somatisation disorder is undifferentiated somatoform disorder, where MUPS must have persisted for at least six months. The diagnostic category 'somatoform autonomic dysfunction' is only registered in the ICD‐10 and requires chronic symptoms especially related to the autonomic system. Finally, pain disorder involves the persistence of medically unexplained pain symptoms. In DSM‐5 (ww2.dsm‐5.org/) ‐ the new revision of the DSM (APA 2013) ‐ the diagnostic category 'somatic symptom disorder' has been proposed. Although this category includes a somewhat lesser emphasis on the "medically unexplained" criterion and requires three "positive" psychological criteria such as disproportionate or excessive thoughts, feelings, or behaviour, it covers the four mentioned somatoform diagnoses focusing on MUPS of DSM‐IV or ICD‐10.
Apart from the categories defined in DSM‐IV and ICD‐10, over the years further 'abridged' diagnostic labels have been developed because the classic criteria of the international classification systems are difficult to use for research purposes. Whereas for somatisation disorder the criteria are considered too stringent, for undifferentiated somatoform disorder the threshold level is considered too low. Therefore, diagnostic labels such as abridged somatisation disorder or the Somatic Symptom Index (SSI‐4,6; Escobar 1987), multisomatoform disorder (Kroenke 1997), polysymptomatic somatoform disorder (Rief 1999), or bodily distress syndrome (Fink 2010), were developed. All of these constructs have received varying amounts of attention in research on somatoform disorders.
One European study based on general populations and using a stepwise multi‐method approach, found the 12‐month prevalence for somatoform disorders in general (including somatisation disorder, undifferentiated somatoform disorder, pain disorder, and hypochondrias) to be approximately 6.3% (range 1.1% to 11%; Wittchen 2005; Wittchen 2011). Prevalence rates for the separate forms of somatoform disorders vary considerably. The 12‐month prevalence rate for people fulfilling the stringent criteria of somatisation disorder was found to be low (range 1.1% to 2.1%) among the European (Wittchen 2005), and American (Robins 1991), general populations. However, in contrast, the lifetime prevalence rates for the less stringent categories of somatoform disorders such as undifferentiated somatoform disorder, somatoform autonomic dysfunction, or pain disorder were much higher, ranging between 12% and 19% (Creed 2011; Fröhlich 2006; Grabe 2003; Jacobi 2004; Martin 2006; Meyer 2001; Robins 1991; Wittchen 1992).
Whereas female gender (e.g. Nimnuan 2001; Verhaak 2006), and low socioeconomic status (e.g. Jacobi 2004), seem to be clear risk factors for developing chronic MUPS, associations between MUPS and age seem to be more complex. MUPS appear in the general population more frequently in older than in younger people. However, studies on the epidemiology of somatoform diagnoses are different. For example, Leiknes 2007 showed that a peak of the prevalence of the multisomatoform disorder lies between 18 and 34 years. The highest co‐morbidity rates were found for anxiety disorders, affective disorders, and substance abuse (e.g. De Waal 2004; Fröhlich 2006; Kroenke 1997). Somatoform disorders in general are associated with excessive treatment and healthcare costs (Barsky 2005).
Aetiological theories of somatoform disorders and MUPS in general are varied (Rief 2007; Witthöft 2010). One of the most important concepts is that of somatosensory amplification (Barsky 1990). The authors assume that people with a tendency to experience somatic sensations as intense, noxious, and disturbing amplify benign somatic sensations by mis‐attributing them to serious illnesses and by focusing attention on them. Kirmayer and colleagues expanded this perceptional‐cognitive model by integrating social aspects (e.g. communication of distress with others, help‐seeking behaviour) (Kirmayer 1997). Another important model, focusing more on the perceptual process itself and its psychobiological correlates, is a signal‐filtering model of MUPS (Rief 2005). It emphasises the interaction of biological and psychological processes in the perception of MUPS. Finally, Ursin 1997 and Yunus 2007 have postulated mechanisms of central sensitisation for explaining MUPS. Central sensitisation describes a plastic response of an increased efficacy in synapses in specific brain areas ‐ especially in limbic structures ‐ as a consequence of repeated use (Ursin 1997).
Description of the intervention
In addition to psychological therapy approaches (Kleinstäuber 2011), pharmacological agents are also used to treat somatoform disorders. However, in contrast to psychological therapies, the mechanisms of action of pharmacotherapy in somatoform disorders are still partly unclear.
Based on the findings of research on chronic pain syndromes such as neuropathic pain (Saarto 2007), and fibromyalgia (Häuser 2009; O'Malley 2000), or other syndromes of MUPS such as irritable bowel syndrome (IBS) (Ford 2009; Jackson 2000; Jackson 2006a), or chronic fatigue syndrome (Pae 2009), antidepressants in particular have been used. Furthermore, findings from studies examining the effects of antidepressants on psychiatric co‐morbid conditions that are common in people with somatoform symptoms (e.g. depression or anxiety disorders), also support the administration of antidepressant drugs in people with somatoform disorders (Verdu 2008; Whitehead 2002).
Two groups of antidepressants are particularly relevant: tricyclic antidepressants (TCAs; e.g. amitriptyline, desipramine, trimipramine, doxepin, opipramol), and new‐generation antidepressants (NGAs). Typical NGAs that are used for treating somatoform symptoms are selective serotonin reuptake inhibitors (SSRIs; e.g. citalopram, escitalopram, sertraline, paroxetine, fluvoxamine, fluoxetine), serotonin and noradrenaline (norepinephrine) reuptake inhibitors (SNRIs; e.g. venlafaxine, duloxetine), or serotonin antagonist and reuptake inhibitors (SARI; e.g. trazodone) that operate as a serotonin receptor antagonist and by inhibiting the serotonin reuptake. Antiepileptic drugs are another group of pharmacological agents used for the treatment of somatoform disorders. Here there are also parallels to the research on chronic pain, where efficacy in pain relief of antiepileptics such as pregabalin (Moore 2009), or gabapentin (Moore 2011), has been demonstrated. For primarily pain‐dominated somatoform symptoms such as headache, the efficacy of antipsychotics (APs; e.g. olanzapine) has already been shown (Silberstein 2002). Finally, natural products (NPs) such as St. John's wort are also used in the treatment of MUPS.
In Germany, a guideline for the treatment of non‐specific functional or somatoform symptoms has been developed (AWMF 2012). The guideline recommends the use of different classes of antidepressants, particularly for severe syndromes dominated by pain symptoms and with or without co‐morbid depressive symptoms. For severe syndromes not determined by pain, the guideline recommends antidepressants only if there is co‐morbid depression. Furthermore, the guideline discourages the use of anxiolytic drugs (e.g. benzodiazepines), tranquillisers, or hypnotics, and APs when there is no co‐morbid symptomatology that justifies the prescription of such agents. The TCA opipramol has been officially approved in Germany for the medical treatment of somatoform disorders (www.rote‐liste.de). It should be taken into consideration that current guidelines in general do not recommend treating somatoform disorders with pharmacotherapy alone, but rather in combination with psychosocial interventions (e.g. AWMF 2012).
How the intervention might work
Antidepressants
The mechanisms of action of antidepressants on somatoform symptoms remain unclear. Once more, parallels to syndromes such as IBS or fibromyalgia can be drawn. In these syndromes, people have demonstrated increased prefrontal cortex activity with noxious stimulation. These are areas responsible for increased attention to a stimulus (Bonaz 2002; Drossman 2003). Furthermore, abnormal activity in brain areas involved with serotonin (5‐HT) and noradrenaline (norepinephrine; NE) have been observed in people with somatoform symptoms. In addition, 5‐HT and NE produce analgesic effects via inhibitory descending pain pathways (Jones 1991; Richardson 1990; Stahl 2002). Therefore, serotonin and NE could be involved in suppression of somatic symptoms at the level of the spinal cord. This could explain why people with IBS experience gastric and colonic distention as more painful than people without the syndrome (Naliboff 1997). The same can be observed in people with fibromyalgia: these people have lower thresholds when they experience pain from noxious stimulation (Montoya 2005; Petzke 2005). Therefore, antidepressant action may involve processing pain on a central as well as peripheral level. In addition, antidepressants may alter pathophysiological mechanisms involved in somatoform symptoms and could have direct effects on different organ systems. For example, TCAs may slow gastrointestinal transit due to anticholinergic effects. This can improve diarrhoea‐predominant IBS in particular (Gorard 1994). Additionally, especially in the treatment of fatigue with antidepressants, it is speculated that immunoregulatory effects could play an important role. Studies have demonstrated that an increased production of pro‐inflammatory cytokines may play a role in somatic symptoms such as anergy, sleeping disturbances, or psychomotor retardation (Maes 1999; Yirmiya 1996). Different studies demonstrated that the effects of antidepressants on such symptoms could be related to their negative immunoregulatory effects (Kubera 2001; Maes 2001). Finally, a mechanism of action could be that antidepressants reduce co‐morbid psychiatric conditions such as depressive disorders, anxiety disorders, and post‐traumatic stress (De Waal 2004). This can then influence symptom severity and functional impairment. For example, it could be demonstrated that anxiety and depression are associated with distinctive cognitive‐affective biases related to attention or encoding and recall processes. In turn, they can be critical for the cognitive processing of somatic changes. Suls 2012 reflected in their review that anxiety seems to be associated with an elevated report of momentary symptoms, whereas depression is related to an exaggerated recall of past symptoms.
Antiepileptic drugs
The mechanisms of action of antiepileptic drugs on MUPS are also unclear. In neuropathic pain, there is evidence that two antiepileptic drugs ‐ gabapentin and pregabalin ‐ bind calcium channels and modulate calcium influx (Urban 2005). Furthermore, they influence GABAergic neurotransmission (Gu 2002). Apart from antiepileptic effects, this mode of action can also produce analgesic, anxiolytic, and sedative effects. Pregabalin is more potent than gabapentin and is, therefore, administered at lower doses.
Antipsychotics
The use of APs in somatoform disorders is based on their analgesic effects (Nix 1998). The method by which APs reduce pain is still unclear. It is possible that the modes of action vary between different agents. The analgesic effect could be mediated by opioid mechanisms, serotonin antagonism (Schreiber 1999), or activity at alpha2‐adrenoreceptors (Silberstein 2002).
Natural products
Mechanisms of action are also unclear for NPs such as St. John's wort. Its administration in somatoform disorders is primarily based on diagnostic overlaps between depressive or anxiety and somatoform disorders (Linde 2009). Several studies on the efficacy of St. John's wort for mild depression demonstrated an additional positive effect on somatoform symptoms such as headache or gastrointestinal complaints (e.g. Sommer 1993; Woelk 2000). The effect of Hypericum extracts, and especially hyperforin and adhyperforin, may be mediated by their function as potent but non‐specific inhibitors of the synaptosomal reuptake of serotonin, noradrenaline, and dopamine (Butterweck 2003).
Why it is important to do this review
In addition to psychotherapeutic approaches, pharmacological agents are also often used in the treatment of somatoform disorders. Previously, the efficacy of these agents has been mainly researched in people with chronic pain where the use of specific medications has been judged critically. For example, the authors of another Cochrane review critically considered the application of APs for chronic painful conditions (Seidel 2008). They emphasised that the particularly strong extrapyramidal adverse effects and sedating effects have to be considered before they are prescribed. With few exceptions, there are no official indications (e.g. the TCA opipramol for treating somatoform disorders) of medication such as antidepressants, antiepileptic drugs, or APs for treating somatoform disorders. This 'off‐label' use of different pharmacological agents, that are in fact indicated for example for depressive or anxiety symptoms but not for somatic symptoms, is part of treatment routines for people with somatoform disorders or pain syndromes in clinics and private practices. One study demonstrated in a sample of people with headaches that 47% of the prescriptions met the criteria for off‐label use (Loder 2004). Although this 'off‐label' use is common practice (Di Franco 2010; Stone 2003), there exists no systematic review or meta‐analysis on the efficacy and tolerability of these medications.
Therefore, the intention of this meta‐analysis is to give an overview of: 1. the current status of research on the efficacy of pharmacological treatments for somatoform disorders, and 2. the acceptability of using medication to treat people with somatoform disorders. It will assist patients as well as providers in making optimal treatment decisions. Furthermore, it will highlight the shortcomings of previous research in pharmacotherapy for somatoform disorders and help to stimulate further research in this area. In this way, the current review adds to a portfolio of five Cochrane reviews covering somatoform disorders (the other four being Hoedeman 2010; Ipser 2009; Ruddy 2005; Thomson 2007).
Objectives
To assess the effects of pharmacological interventions for somatoform disorders (specifically somatisation disorder, undifferentiated somatoform disorder, somatoform autonomic dysfunction, and pain disorder) in adults.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) and cluster‐randomised controlled trials (CRCTs). We also included cross‐over trials, but used only data from the first randomisation period in the review. We excluded quasi‐randomised trials (e.g. allocation to the study group by day of the week). In the case that treatment outcome data were absent, we excluded the trial from meta‐analysis but included it as part of the narrative literature review.
Types of participants
Participant characteristics
Participants aged 18 to 65 years (where a trial has defined adults to include those older than 65 years but most participants were under 65 years of age we included the trial; however, we excluded any trial that focused on older adults or where the mean age of participants was greater than 65 years). We applied no restrictions on gender or culture.
Diagnosis
Participants had to meet the requirements for diagnosis of a somatoform disorder, based on chronic, multiple, MUPS, according to DSM‐III (APA 1980), DSM‐IV‐TR (APA 2000), ICD‐9 (WHO 1975), ICD‐10 (WHO 1992), the Chinese Classification of Mental Disorders (CCMD)‐III (Chinese Society of Psychiatry 2001), or the criteria of a somatic symptom disorder according to DSM‐5 (APA 2013). See Table 6 for an overview of all diagnostic categories of somatoform disorders and a clear indication of whether or not they were eligible for this review. A medical assessment of the physical symptoms was required to rule out the possibility that the physical symptoms and their intensity can be explained sufficiently by a medical condition.
1. Diagnostic categories of somatoform disorders.
| Diagnostic category | Eligible for the current review? | ||
| DSM‐IV | ICD‐10 | yes | no |
| Somatisation disorder (300.81) | Somatisation disorder (F45.0) | x | ‐ |
| Undifferentiated somatoform disorder (300.82) | Undifferentiated somatoform disorder (F45.1) | x | ‐ |
| ‐ | Somatoform autonomic dysfunction (F45.3) | x | ‐ |
| Pain disorder (307.8) | Persistent somatoform pain disorder (F45.4) | x | ‐ |
| Hypochondriasis (300.7) | Hypochondriacal disorder (F45.2) | ‐ | x |
| ‐ | Other somatoform disorders (F45.8) | ‐ | x |
| Somatoform disorders, unspecified (300.82) | Somatoform disorders, unspecified (F45.9) | ‐ | x |
| Body dysmorphic disorder (300.7) | Body dysmorphic disorder (F45.2) | ‐ | x |
| Conversion disorder (300.11) | Dissociative and conversion disorders (F44)* | ‐ | x |
DSM‐IV: Diagnostic and Statistical Manual of Mental Disorders; ICD‐10: International Classification of Diseases.
Note. *Conversion disorder is not classified as a somatoform disorder in ICD‐10 but is a separate diagnostic category.
Co‐morbidities
We included people with certain co‐morbidities, but the somatoform disorder had to be the primary diagnosis. We included studies that included participants with co‐morbid psychiatric disorders, with the exception of studies in which participants had a co‐morbid psychosis or dementia, which we excluded. We excluded studies that examined the efficacy of a pharmacotherapy in a group of participants diagnosed with only one specific functional syndrome (e.g. IBS, chronic fatigue syndrome, fibromyalgia).
Setting
We placed no restrictions on setting.
Types of interventions
Experimental interventions
Eligible studies included one or more of the following experimental interventions:
TCAs (e.g. amitriptyline);
NGAs, such as SSRIs (e.g. fluoxetine), SNRIs (e.g. venlafaxine), noradrenaline reuptake inhibitors (NRIs; e.g. reboxetine), tetracyclic antidepressants (TeCAs; e.g. maprotiline), noradrenergic and specific serotonergic antidepressants (NaSSAs; e.g. mirtazapine), SARIs (e.g. trazodone), and reversible inhibitors of monoamine oxidase type A (RIMAs; e.g. moclobemide)
any other antidepressants such as irreversible monoamine oxidase inhibitors (MAOIs; e.g. bupropion);
antiepileptics (e.g. pregabalin, gabapentin);
NPs (e.g. St. John's wort);
APs (e.g. paliperidone);
other pharmacological agents (e.g., benzodiazepines).
Comparator interventions
The following comparator interventions were accepted:
placebo;
treatment as usual;
another medication;
combination of medication.
Types of outcome measures
Primary outcomes
1. Severity/intensity of MUPS. If a validated self report scale was used, we considered this the primary outcome. Validated scales for the assessment of MUPS considered for this review were: Screening for Somatoform Symptoms (SOMS; Rief 2008), and Bradford Somatic Inventory (Mumford 1991). If no validated scales were available, we also accepted component subscales of validated standardised instruments for the assessment of general psychopathology or general health status, for example, the subscale 'Somatisation' of the Patient Health Questionnaire‐15 (PHQ‐15; Kroenke 2002), the subscale 'Somatisation' of the Symptom Checklist‐90‐R (SCL‐90‐R; Derogatis 1983), or the subscale 'Somatisation' of the Brief Symptom Inventory (BSI; Derogatis 1992). Where unvalidated or visual analogue self report scales (VAS) were used, we decided which scale most closely approximated MUPS. One of the review authors (WH) who is an expert in somatoform disorders and clinical diagnostics, but who was not directly involved in the process of study selection or data extraction and management, decided this, so that he could be blinded to the results. We examined clinician‐rated severity of MUPS separately and did not aggregate it with self report outcomes into one effect size index.
2. Acceptability. We considered the proportion of people who dropped out during the experimental as well as the comparator intervention. We calculated this rate as a proportion of the total number of randomised participants. In addition, we presented the acceptability rate as a risk ratio (RR) calculated from the total number of all randomised participants.
Secondary outcomes
3. Anxiety: using a. validated self report instruments (e.g. Beck Anxiety Inventory (BAI); Beck 1990), b. clinician‐rated instruments (e.g. Hamilton Anxiety Rating Scale (HARS); Hamilton 1959), or c. component subscales of validated standardised instruments for the assessment of general psychopathology or general health status (e.g. the subscale 'Anxiety' of the SCL‐90‐R; Derogatis 1983). Where unvalidated or self report VAS were used, we decided which scale most closely approximates anxiety. One of the review authors (WH) who is an expert in somatoform disorders and clinical diagnostics, but who was not directly involved in the process of study selection or data extraction and management, decided this, so that he could be blinded to the results.
4. Depression: using a. validated self report instruments (e.g. Beck Depression Inventory (BDI); Beck 1961), b. clinician‐rated instruments (e.g. Hamilton Depression Rating Scale (HDRS); Hamilton 1960), or c. component subscales of validated standardised instruments for the assessment of general psychopathology or general health (e.g. the subscale 'Depression' of the SCL‐90‐R; Derogatis 1983). Where unvalidated or self report VAS were used, we decided which scale most closely approximates depression. One of the review authors (WH) who is an expert in somatoform disorders and clinical diagnostics, but who was not directly involved in the process of study selection or data extraction and management, decided this, so that he could be blinded to the results.
5. Dysfunctional cognitions, emotions, or behaviours/participant‐rated: validated self report scales (e.g. the Whiteley Index (WI); Pilowsky 1967); Illness Attitude Scales (IAS); Kellner 1986); Scale for the Assessment of Illness Behavior (SAIB); Rief 2003); Cognitions About Body and Health Questionnaire (CABAH); Rief 1998)).
6. Adverse effects: when possible, we described the most common drug‐related adverse effects (defined as effects that occurred in at least 10% of people receiving medication) as well as significant differences in the rate of occurrence of drug‐related adverse events between medication and control groups, as part of the narrative literature review. We calculated the rate of participants who dropped out due to adverse effects during the experimental as well as the comparator intervention as a proportion of the total number of randomised participants. In addition, we presented the adverse effect‐related drop‐out rate as an RR calculated out of the total number of all randomised participants. As a limitation to interpreting these adverse effects, we noted that the inclusion of RCT or CRCT studies was not sufficient to gain information about the more rare or long‐term adverse outcomes.
7. Treatment response (responder versus non‐responder; with regard to the primary outcome 'severity/intensity of MUPS'); clinician‐rated Clinical Global Impression Scale (CGI) ‐ Improvement Scale (Guy 1976); we defined responders on this scale as those with a score of "1 = very much improved" or "2 = much improved". Alternatively, the number of participants who responded to the treatment according to the author's definition. We calculated response rates out of the total number of all randomised participants.
8. Functional disability and quality of life: a. validated clinician‐rated scales (e.g. Global Assessment of Functioning (GAF); APA 1994) or b. validated self report instruments (e.g. Sheehan Disability Scale (SDS); Sheehan 1983; 36‐item Short Form Questionnaire (SF‐36); Ware 1992).
Timing of outcome assessment
In the protocol, we had planned that the primary and secondary outcomes were classified as assessed: 1. post treatment, 2. within 12 months' post treatment, or 3. more than 12 months' post treatment. However, in all included studies data were only available for post treatment.
Search methods for identification of studies
The Cochrane Depression, Anxiety and Neurosis Review Group's Specialised Register (CCDANCTR)
The Cochrane Depression, Anxiety and Neurosis Group (CCDAN) maintain two clinical trials registers at their editorial base in Bristol, UK: a References Register and a Studies Register (ccdan.cochrane.org/specialised‐register). The CCDANCTR‐References Register contains over 36,000 reports of RCTs in depression, anxiety, and neurosis. Approximately 60% of these references have been tagged to individual, coded trials. The coded trials are held in the CCDANCTR‐Studies Register and records are linked between the two registers using unique Study ID tags. Coding of trials is based on the EU‐Psi coding manual, using a controlled vocabulary; please contact the CCDAN Trials Search Coordinator for further details. Reports of trials for inclusion in the Group's registers are collated from routine (weekly), generic searches of MEDLINE (1950 to date), EMBASE (1974 to date) and PsycINFO (1967 to date); quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and review‐specific searches of additional databases. Reports of trials are also sourced from international trials registers via the World Health Organization's (WHO) trials portal (the International Clinical Trials Registry Platform (ICTRP)), pharmaceutical companies, the handsearching of key journals, conference proceedings, and other (non‐Cochrane) systematic reviews and meta‐analyses.
Details of CCDAN's generic search strategies (used to identify RCTs) can be found on the Group's website.
Electronic searches
1. We searched the CCDANCTR‐Studies Register (to 17 January 2014) using the following terms: Condition = "Somatization Disorder" We screened records manually for pharmacological interventions.
2. We searched the CCDANCTR‐References Register for additional untagged references (to 17 January 2014), using a more sensitive set of free‐text terms: ("somatoform disorder*" or (somatoform and "autonomic dysfunction") or "somatic symptom disorder*" or somatization or somatisation or hysteri* or briquet or "pain disorder*" or polysymptom* or multisomatoform or somatizer* or (multiple and (MUPS or "medically unexplained" or "unexplained symptoms" or "physical symptoms" or "symptom diagnos*"))) We screened records manually for pharmacological interventions.
3. To ensure that we had missed no studies, we conducted complementary searches on the following bibliographic databases, using relevant subject headings (controlled vocabularies) and search syntax, appropriate to each resource: CENTRAL (all years, see Appendix 1), PsycINFO (all years, Appendix 2), and PSYNDEX (from 1977 onwards, see Appendix 3).
4. To identify ongoing trials, we searched the ClinicalTrials.gov register (clinicaltrials.gov/), the Current Controlled Trials metaRegister of Controlled Trials‐active registers (mRCT; www.controlled‐trials.com/mrct/), the WHO International Clinical Trials Registry Platform Search Portal (www.who.int/trialsearch), and the Chinese Clinical Trials Registry (www.chichtr.org/).
We applied no date or language restrictions to the searches.
Searching other resources
Grey literature
We searched the ProQuest Dissertation & Theses Database, OpenGrey (System for Information on Grey Literature in Europe launched by the Institute for Scientific and Technical Information INIST), and BIOSIS Previews for trials published in dissertations or theses, or other sources of grey literature.
Handsearching
We handsearched the proceedings of the following conferences since 2008 if available:
American Psychiatric Association (APA) Annual Meeting; World Congress of The International College of Neuro‐Psychopharmacology (CINP); European College of Neuropsychopharmacology (ECNP) Congress; International Congress of Behavioral Medicine (ICBM); European Conference on Psychosomatic Research (ECPR); Annual Meeting of the European Association for Consultation‐Liaison Psychiatry and Psychosomatics (EACLPP); and Congress of the German Association for Psychiatry, Psychotherapy, and Neurology (DGPPN).
Reference lists
We screened reference lists of all potentially relevant papers and of systematic reviews or meta‐analyses for further relevant studies. We identified systematic reviews or meta‐analyses using appropriate search filters in the above‐mentioned electronic databases (see Electronic searches).
Correspondence
We asked experts in the field of somatoform disorders, as well as authors who have published studies on pharmacotherapy or other therapies for MUPS, if they knew of any published or unpublished or ongoing trials meeting the criteria of the current review.
Data collection and analysis
Selection of studies
In a first step, two review authors (MK, MW) independently screened titles and abstracts of reports that were identified from the literature search. We discarded those studies that obviously did not fulfil the inclusion criteria at this stage of the screening process. We retrieved potentially relevant articles for full‐text assessment. In the next step, two review authors (MK, MW) independently assessed the main text of these retrieved trials for eligibility. We resolved disagreements by consensus, if necessary with the involvement of a third review author (WH). We had planned that studies for which additional information was required in order to determine their suitability for inclusion in the review would be listed in the 'Studies awaiting assessment' table in the Review Manager 5 software (RevMan 2012). One review author (MK) checked the reference lists of articles that were retrieved after the second stage of the selection process. The review authors were not blinded to the name(s) of the study author(s). We reported reasons for exclusion in the 'Characteristics of excluded studies' table.
We recorded all decisions that were made throughout the review process, along with the number of references and studies found and presented them in a PRISMA flow diagram (Moher 2009; Figure 1).
1.

PRISMA study flow diagram.
Data extraction and management
One review author (MK) and one research assistant independently conducted data extraction. We had previously prepared a data extraction form a priori and piloted it before use. The research assistant had training in completing the data extraction form. We assessed characteristics regarding the trial, participants, methods, intervention and outcome details, summary statistics, and associated commentaries. If necessary, we contacted authors of reports for clarification or additional information. We organised data using the most recent version of Review Manager 5 software (RevMan 2012). We resolved disagreements by consultation with another review author (MW or WH). We extracted the following information.
Characteristics of the trial: primary researcher, publication year, status of publication, language of publication, source of funding, study design, length of follow‐up.
Characteristics of participants: source of sample, sample size, gender, age, number of drop‐outs, nationality, applied diagnostic criteria, somatoform diagnosis, co‐morbidity, co‐morbid diagnoses, screening procedure (e.g. interview), screening instruments, inclusion and exclusion criteria, mean length of time since diagnosis of a somatoform disorder, previous treatments.
Characteristics of intervention: category of medication, medication, treatment setting, dose of medication, frequency of intake, mode of administration of medication, period over which the medication was administered, number of participants that dropped out due to adverse effects or inefficacy of treatment, most common drug‐related adverse effects, details of concurrent treatments (e.g. psychotherapy).
Details of methodology: number of centres involved; number of participants that were not included in the analyses (lost to follow‐up); whether blinding occurred for assessors, participants, or people who administered medication.
Outcome measures: primary and secondary outcome measures, summary statistics of continuous data (mean, standard deviation (SD)) and dichotomous data (number of responders), timing of outcome assessments, intention‐to‐treat (ITT) analysis (with last observation carried forward (LOCF)) or observed cases/completer analysis, other methods of estimating the outcome for participants who dropped out (e.g. mixed effect analyses).
As medication classes can all have different effects, where data allowed, we stratified the comparisons by medication class (see Types of interventions). Therefore, the following main comparisons were planned for each class of medication.
Pharmacotherapy versus placebo.
Pharmacotherapy versus usual treatment.
Pharmacotherapy versus another medication.
Pharmacotherapy versus a combination of medications.
Assessment of risk of bias in included studies
The first review author (MK) and a research assistant independently assessed the risk of bias within each included study. The assessment of risk of bias was based on a tool in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a), which included six categories. The following judgements had to be passed with consensus of the review author and the research assistant for each of these categories.
Random sequence generation. Was the method to generate the sequence of randomised allocation adequate to produce comparable groups?
Allocation concealment. Was the allocation concealed adequately so that intervention allocations could not have been foreseen in advance of or during enrolment?
Blinding. Was knowledge of the allocation of treatment of the participant and study personnel adequately prevented during the study? Was knowledge of the allocation of treatment of the outcome assessor(s) adequately prevented during the study? Were any measures applied to blind participants, personnel, and outcome assessors described. The assessment was made separately for each outcome domain.
Incomplete outcome data. Were incomplete outcome data adequately addressed? Was the completeness of outcome data described, including attrition and exclusions from analyses? If there were attritions and exclusions in the treatment and control group, were they reported, along with the reasons? If the review authors conducted any re‐inclusions in their analyses, we also reported this. We made the assessment separately for each outcome domain.
Selective reporting. Were reports of the study free of suggestion of selective outcome reporting?
Other sources of bias. Was the study apparently free of other sources that could produce risk of bias?
In order to assess risk of bias in the following specific types of study design, we passed the following additional judgements.
Multiple‐intervention studies. Were data presented for each of the groups to which participants were randomised?
Cross‐over trials. Was it clear that the order of receiving a treatment was randomised? Were unbiased data from the first treatment‐period available?
CRCTs. Were individuals recruited to the trial after the clusters had been randomised? Were methods of stratified or pair‐matched randomisations of clusters used? Were adequate statistical analyses (taking clustering into account) used?
We used the following scale to rate each of the categories: 'high' (high risk of bias), 'low' (low risk of bias), and 'unclear' (uncertain risk of bias).
We resolved disagreements regarding the ratings by consultation with a further review author (MW or WH). If necessary, we contacted study authors for further information.
Measures of treatment effect
Dichotomous data
For computing treatment effects based on dichotomous data, we used the pooled RR with 95% confidence interval (CI) for each comparison. We calculated the number needed to treat for an additional beneficial outcome (NNTB) for every class of pharmacological agent for which we found a statistically significant treatment effect. We interpreted the NNTB as the number of participants that needed to be treated for one to benefit compared with a control in a clinical trial. Therefore, we used the RR estimate and the control risk from the placebo group.
Continuous data
We collected the mean score, SD, and the number of participants at endpoint. Unfortunately, none of the included studies provided data for follow‐ups. We pooled these values for the single trials as follows: assuming that at least two studies using the same scale are available, we calculated the mean difference (MD) with a 95% CI between experimental and comparator intervention at endpoint and follow‐up. In case measures of an outcome domain varied across studies, we used the standardised mean difference (SMD) with 95% CI. We paid specific attention to the secondary outcome 'functional disability and quality of life' because the direction of corresponding scales can differ. Whereas an increase of scales of functional disability usually indicates deterioration, an increase of scales of quality of life often indicates improvement. The SMD method does not correct for such differences in the direction of the scale. In this case, we multiplied the mean values from one set of studies by ‐1 to ensure that all the scales pointed in the same direction.
Unit of analysis issues
Cluster‐randomised controlled trials
In order to avoid unit‐of‐analysis errors for trials in which incorrect statistical analyses were conducted, we performed approximate analyses based on inflating standard errors (SE). Before data were entered into Review Manager 5 for meta‐analytic calculations (RevMan 2012), we multiplied the SE of the effect estimate (from an analysis not taking into to account the clustering) by the square root of the so‐called design effect. The design effect is 1 + (M ‐ 1) ICC, where M is the mean cluster size and ICC is the intracluster correlation coefficient. We assumed a common design effect across intervention groups. If the ICC was not available in the published report, we used an external estimate obtained from similar studies or another external resource. It was planned that the meta‐analysis using the inflated variances would be performed using Review Manager 5 (RevMan 2012), and the generic inverse‐variance method.
Studies with multiple treatment groups
In trials comparing the efficacy of more than one medication for somatoform disorders, we considered three aspects.
If the different medications were of the same class of chemical agent (e.g. TCAs), we summarised the different experimental conditions into a single group that were compared with the control group. For continuous data, we pooled means and SDs across all of the treatment arms as a function of the number of participants in each arm. For dichotomous outcomes, we summed both the sample sizes and the numbers of people with events across groups.
If the different medications were of different classes of chemical agents (e.g. SSRIs versus TCAs), we included each pair‐wise comparison separately, but divided the 'shared group' into two or more groups (according to the number of intervention groups) with smaller sample size. For dichotomous outcomes, we divided up both the number of events and the total number of participants. For continuous outcomes, we divided up only the total number of participants and left the means and SDs unchanged. Although this method only partially overcomes the unit‐of‐analysis error, an advantage of this approach is that investigations of heterogeneity across intervention arms are possible.
In the case of data from trials employing multiple fixed doses of medication, we summarised the different experimental groups into one group corresponding to the pooling procedure of the aspect 2 above. We restricted the pooling of outcome data to those treatment arms that employed at least the minimum dose recommended by clinical guidelines or experts, in order to reduce the influence of data from arms that employ doses unlikely to have a clinical effect.
Cross‐over trials
We only included cross‐over trials in meta‐analytical calculations if it was possible to extract relevant data of the treatment and control group from the first treatment period.
Dealing with missing data
In the case of a missing continuous data summary, we preferred statistics based on mixed‐effects models, followed by statistics based on ITT analysis with LOCF and on observed cases. This is in accordance with the finding that mixed‐effects methods are more robust to bias than LOCF (Verbeke 2000). If SDs were not available directly, we attempted to calculate them from t, F, P, or CI values (Higgins 2011b), or SEs (Altman 1996).
In the case of missing dichotomous data, we applied ITT analysis, in which it was assumed that participants who dropped out after randomisation had a negative outcome. According to Gamble and Hollis, sensitivity analyses for dichotomous data were also conducted (Gamble 2005): we calculated best‐case/worst‐case scenarios for the clinical response outcome, in which it was assumed that drop‐outs in the active treatment group had positive outcomes and drop‐outs in the control group had negative outcomes (best‐case scenario), and that drop‐outs in the active treatment group had negative outcomes and drop‐outs in the control group had positive outcomes (worst‐case scenario).
If data were not in a suitable format or if relevant data were not available, we tried to contact trial authors to obtain further information. We reported reasons for missing data where they were provided in the published trials. Where we could not include data, we reported a qualitative summary of the results in the text of the review.
Assessment of heterogeneity
We first assessed statistical heterogeneity visually by inspecting forest plots of standardised mean effect sizes and of RR. Furthermore, we applied a Chi2 test to assess heterogeneity. The test has low power in general but especially when the sample size of the included studies is low or there are only a few included studies. Therefore, we used a P value of 0.10 to determine statistical significance in a conservative way. Another problem of the Chi2 statistic is that it indicates only significance or non‐significance but does not give any information about the level of heterogeneity. For this reason, we used the I2 statistic. The I2 statistic describes the percentage of variability in effect estimates that is due to heterogeneity rather than sampling error. We used conventions of interpretation that were defined by Higgins (Higgins 2011a). In the case of substantial levels (I2 = 50% to 90%) and considerable levels (I2 = 75% to 100%) of heterogeneity, we examined data by subgroup and sensitivity analyses (see Subgroup analysis and investigation of heterogeneity; Sensitivity analysis) for different aspects of clinical and methodological heterogeneity.
For the assessment and examination of clinical heterogeneity, we planned subgroup analyses (see Subgroup analysis and investigation of heterogeneity) for the following pre‐specified characteristics: class of medication, co‐morbidity, gender, source of funding for the trial, and source of outcome rating.
Assessment of reporting biases
In order to prevent publication bias, we made every attempt to include unpublished trials (e.g. by searching online trial registries or registries of unpublished doctoral theses). In order to assess for a publication bias, we implemented funnel plots (effect versus SE of the effect size) when a sufficient number of trials was available (according to recommendations of the Cochrane Handbook for Systematic Reviews of Interventions there should be at least 10 studies; Sterne 2011). For the analysis and the interpretation of the funnel plots, other reasons for asymmetry besides publication bias have to be considered (e.g. differences in methodological quality, true heterogeneity in intervention effects).
Data synthesis
In the case that two or more studies that were eligible for inclusion were found per comparison category (see Data extraction and management), and that these studies measured the same outcome construct, we performed a meta‐analysis of the results. One review author (MK) entered data into Review Manager 5 software (RevMan 2012). We obtained dichotomous and continuous treatment effects using a random‐effects model. However, we specified the fixed‐effect model as a sensitivity analysis in order to compare the results informally. Where heterogeneity analysis indicated significant heterogeneity, we performed subgroup analysis. We based the calculation of the mean effect size for each (sub)group as well as the 95% CI on the inverse‐variance method. This was reported for the post‐treatment assessment. If data were available, we also reported the summarised statistic for follow‐up assessment (less than 12 months post treatment or 12 months or more post treatment).
Subgroup analysis and investigation of heterogeneity
Although subgroup analyses have to be treated with caution, as they are rather hypothesis‐forming than hypothesis‐testing, we performed a priori defined analyses in order to explore whether methodological and clinical differences between the trials had systematically influenced the differences that were observed in the treatment outcomes. If sufficient data were available, we planned the following subgroup analyses for the main comparison 'pharmacotherapy versus placebo' (primary outcomes only).
1. Co‐morbidity. As known from other mental disorders, co‐morbid psychological problems can moderate the efficacy of a medication. Therefore, we planned to compare the effects of pharmacotherapy for people with somatoform disorders, with or without co‐morbid mental disorders.
2. Gender. Sex differences have been found in the absorption, metabolism, and excretion of many medications (Clayton 2005). Therefore, we planned to examine gender differences in the efficacy of pharmacotherapy for somatoform disorders.
3. Source of funding. Industrial funding of pharmacological trials can be associated with conflicts of interest for the trial conductors (e.g. when a new medication shall be placed on the market). Studies sponsored by pharmaceutical companies might be more likely to have outcomes favouring the sponsor than studies funded by other bodies (Heres 2006; Lexchin 2003). Therefore, we planned subgroup analyses in order to examine differences in the efficacy of medications between trials funded versus not funded by industry.
4. Source of outcome rating. Previous studies (e.g. Rief 2009), have demonstrated that results based on participant‐rated measures, or on clinician‐ratings, can differ considerably from each other. Therefore, we plan subgroup analyses relating to different sources of outcome rating.
Due to a lack of studies, we could only perform the subgroup analyses for co‐morbidity, gender, and source of outcome rating.
Sensitivity analysis
We conducted sensitivity analyses in order to determine whether conclusions were robust to decisions made during the review process (e.g. the inclusion or exclusion of specific studies or choice of the method of analysis). It should be verified that the results of the review do not depend on specific decisions that were made during the review process. As part of a sensitivity analysis, we compared effect sizes based on random‐effects and fixed‐effect analyses. If sufficient data were available (i.e. at least 10 studies), we planned to examine the following aspects in further sensitivity analyses for the primary outcomes.
Exclusion of studies with unclear allocation concealment.
Exclusion of studies with unclear methods of blinding of outcome assessors.
Exclusion of studies with unclear methods of sequence generation.
Exclusion of Chinese studies (because we did not run a comprehensive, up‐to‐date search of the Chinese biomedical literature and so the studies analysed in this review may be an incomplete list of relevant research from China and other Asian countries).
For continuous outcomes only: exclusion of results based on complete‐case analyses.
For dichotomous outcome of treatment response only: best‐case and worse‐case analyses (see also Dealing with missing data).
Exclusion of studies with a drop‐out higher than 20%.
With the exception of the best‐case and worst‐case analyses as well as the sensitivity analyses for the comparison of fixed‐effect and random‐effects models, we were unable to conduct any of the planned sensitivity analyses because there were not enough data available.
'Summary of findings' tables
In order to summarise the main findings of the review in a simple tabular format, we prepared 'Summary of findings' tables. The tables include a list of seven of the outcomes specified under Types of outcome measures (i.e. all outcomes apart from dysfunctional cognitions, emotions, or behaviours). The tables summarise effects based on a population of participants fulfilling inclusion criteria for this review (see Types of participants). For dichotomous outcomes (treatment response), we reported an assumed and corresponding absolute risk with 95% CI as well as an RR with 95% CI obtained from the meta‐analysis. For continuous outcomes (level of severity/intensity of MUPS, dysfunctional cognitions/emotions/behaviours, anxiety and depression, functional disability/quality of life), in the column of assumed and corresponding risk we present a difference in means or SMD with 95% CI. We use footnotes in order to specify the source or rationale for each assumed and corresponding risk. In order to assess the quality of body of evidence for each outcome, we used the GRADE approach (GRADEpro software; Schünemann 2011), providing a transparent procedure to classify the quality of evidence as 'high', 'moderate', 'low', and 'very low'. Judgements other than of 'high' quality are made transparent using footnotes or the Comments column in the 'Summary of findings' tables.
Results
Description of studies
See: Characteristics of included studies; Characteristics of excluded studies; Characteristics of ongoing studies.
Results of the search
The full literature search identified 26 RCTs (33 reports) (with 25 studies included in the final quantitative analysis). Figure 1 summarises each stage of the search process in the PRISMA study flow diagram.
Electronic search
The search of CCDANCTR‐Studies Register and CCDANCTR‐Reference Register (both searched 17 January 2014) yielded 157 references of potentially eligible studies. After de‐duplication, 149 references remained. Based on the screening of the title and abstracts, we excluded 94 references. Thus, we retrieved 55 full‐text papers for full inspection. Of these full‐text papers, we excluded 23 from further analyses. The subtotal of studies from the CCDANCTR was 25 studies included in the qualitative analyses and 24 studies in the quantitative analyses. Complementary searches in CENTRAL (10 April 2014), PSYNDEX (23 March 2014), and PsycINFO (10 April 2014) revealed 672 references of potentially eligible studies. After de‐duplication, 599 references remained. Based on the screening of title and abstract, we excluded 596 references. We retrieved three full‐text papers for full inspection. Of these full‐text references, we excluded two from further analyses and included one reference (Volz 2003), which was an erratum for one study (Volz 2002) that had already been identified with the search in the CCDANCTR‐Studies Register and CCDANCTR‐Reference Register.
It is important to note that the CCDANCTR retrieved many Chinese studies. While several of these were identified through routine searches of MEDLINE, EMBASE, and PsycINFO (used to inform the register), the provenance of some of the other Chinese studies in the CCDANCTR was less clear and probably dates back to an ad hoc search of Wang Fang Data (c/o The British Library) in 2007. While we ensured searches were run on sources other than the CCDANCTR, we did not run a comprehensive, up‐to‐date search of the Chinese biomedical literature and so the studies analysed in this review may be an incomplete list of relevant research from China and other Asian countries. However, an up‐to‐date search of the WHO ICTRP (see Electronic searches) retrieved no ongoing or unpublished studies from the Chinese Clinical Trial Registry (ChiCTR) for somatoform disorders or somatisation.
Handsearching of conference proceedings
Handsearch of proceedings of seven conferences (see Searching other resources) since 2008 revealed 10 references of potentially eligible studies. After de‐duplication, nine references remained. After the screening of titles and abstracts, we excluded eight references. The reference of one potentially eligible study remained (Agger 2014). We contacted the authors of this study in order to gain more information. The authors replied that the trial is still ongoing and will be finished in December 2014. For this reason, we included the study in the current review as an ongoing study after checking inclusion and exclusion criteria.
International trial registers
The search of international trial registers such as the ClinicalTrials.gov register (12 June 2014), the Current Controlled Trials metaRegister of Controlled Trials‐active registers (26 March 2014), the WHO ICTRP (26 March 2014), and the ChiCTR (15 August 2014) revealed protocols of 274 potentially eligible citations. After de‐duplication, 271 references remained. Screening titles and abstracts revealed five potentially relevant citations of which we excluded two after the screening of the full study protocol because they did not fulfil the intervention‐ or participant‐related criteria of the current review (Farnbach 2013; Liu 2011). Finally, three references remained (Fink 2011; Fink 2012a; Fink 2012b). These references are triplicate, international trial registration protocols for the ongoing study, which we identified from the handsearching of conference proceedings (Agger 2014).
Grey literature
The search of grey literature in web portals such as ProQuest Dissertation & Theses Database, OpenGrey, and BIOSIS Previews (all 26 March 2014) yielded 193 reference of potentially eligible studies and 113 after de‐duplication. However, after the screening of the titles and abstracts, we excluded all references for different reasons (see Figure 1).
Reference lists
The search of reference lists of 24 reviews or meta‐analyses (Allen 2002; Atkinson 1985; Decoutere 2011; Escobar 1996; Fallon 2004; Fishbain 1998; Gildenberg 1984; Grothe 2004; Jackson 2000; Jackson 2006a; Jackson 2006b; Kroenke 2007; Lynch 2001; Magni 1991; Marks 2009; McQuay 1995; O'Malley 1999; Price 2000; Prior 2013; Raine 2002; Satterthwaite 1990; Sharma 2013; Somashekar 2013; Sumathipala 2007) revealed six references of potentially eligible studies. After de‐duplication, five citations remained for titles and abstracts screening. Four references were entered in the full‐text inspection, which resulted in one additional eligible study (Eberhard 1988), after the exclusion of the other three references.
In addition to reviews and meta‐analyses, we also screened the reference lists of all potentially relevant trial reports. This search yielded 11 references of potentially relevant articles; this was reduced to eight citations after de‐duplication. However, after screening the titles and abstracts, none of these references fulfilled the inclusion criteria of the current review.
Correspondence and personal communication
We contacted 26 international experts in the field of somatoform disorders and 11 replied (response rate 42.3%). The correspondence with experts yielded one study that had already been identified with the handsearch of conference proceedings and was already included as an ongoing study in the current review (Agger 2014).
Included studies
We included 26 studies (2159 participants) in this review (see Characteristics of included studies).
Design
Of the 26 included RCTs, eight had a placebo‐controlled design where the medication was compared with placebo. Eighteen studies used a parallel‐group design. Most of these trials had two arms where the medication was compared with another medication. In one of these studies, there were three study arms (paroxetine versus "open paroxetine" versus amitriptyline; Kong 2004). One study used a combined placebo‐controlled, parallel‐group design (Melzer 2009). One study was a cross‐over trial (Zitman 1991). We included no CRCTs. In four of the 26 included studies, a placebo run‐in phase took place before the treatment started (Altamura 1991; Müller 2004; Volz 2000; Volz 2002). After this run‐in phase, placebo responders were excluded from the further trial.
Three studies included a one‐ or two‐week wash‐out phase before the study treatment started in order to discontinue other medications (Aragona 2005; Ouyang 2006; Zitman 1991). Of the included 26 studies, 15 were stated to be double‐blind.
One study included four study arms: amitriptyline plus psychotherapy, amitriptyline plus support, placebo plus psychotherapy, and placebo plus support (Pilowsky 1990). We only extracted data from the amitriptyline plus support and the placebo plus support groups. In fact, we originally defined in our inclusion criteria that no studies were included where pharmacotherapy was combined with a psychosocial intervention. However, in this study, the supportive interventions in comparison to the psychotherapeutic intervention was described by the trial authors as follows: "The 'support' included briefer and less frequent sessions, interaction with the clinician comprised a predominant focus on the physical symptoms themselves, and on the effects and side effects of medication prescribed during the treatment period. No attempt was made to broaden, or achieve, a re‐direction of the definition of the participant's problems, from the somatic to the interpersonal and intrapsychic domains. On those occasions where spontaneous disclosure of interpersonal and subjective personal problems were nonetheless revealed, there was sympathetic attention, encouragement and support, but not interpretation at a dynamic level. In particular, no attempt was made to link such personal experiences with the pain symptoms" (Pilowsky 1990, p. 6). We assumed that this type of support was comparable to a usual physician‐patient contact in the context of a study visit. For this reason, we did not exclude the study from the current review.
Twelve studies were trials with only one study centre (Aragona 2005; Huang 2012; Ju 2003; Luo 2009; Muller 2008; Ouyang 2006; Pilowsky 1990; Sanada 2010; Wang 2003; Yang 2006; Zhao 2006; Zitman 1991), three studies were multicentre trials (Eberhard 1988: six centres; Kroenke 2006: 19 centres; Melzer 2009: two centres), and for 12 studies no information was provided how many study centres were involved (Altamura 1991; Han 2008a; Han 2008b; Jiang 2005; Kong 2004; Li 2006; Müller 2004; Volz 2000; Volz 2002; Xu 2004; Ye 2006).
Sample sizes
The number of participants randomised to the relevant arms in the 26 trials ranged between 21 and 208 (median 69). For one of the 26 included studies, only the number of participants who complied with the study protocol was reported (Pilowsky 1990). The total sample size of included participants was 2159. The placebo‐controlled studies comprised 1031 participants and the parallel‐group trials comprised 1128 participants.
Setting
The included trials were conducted in numerous countries (China: 12, Germany: four, Italy: two, South Korea: two, Australia: one, Japan: one, the Netherlands: one, South Africa: one, Sweden: one, USA: one). There was a remarkably high number of Chinese studies (46.2%). Study participants were recruited in inpatient departments of general hospitals in eight studies (Huang 2012; Jiang 2005; Ju 2003; Ouyang 2006; Sanada 2010; Wang 2003; Yang 2006; Zhao 2006). It should be noted that all of these eight trials were conducted either in China (Huang 2012; Jiang 2005; Ju 2003; Ouyang 2006; Wang 2003; Yang 2006; Zhao 2006), or in Japan (Sanada 2010). In all remaining studies, either no information about the methods of recruitment were given (Han 2008a; Han 2008b; Kong 2004; Li 2006; Volz 2000; Xu 2004; Ye 2006), or participants were recruited in mixed settings such as outpatient centres (e.g. pain‐specialised centres or psychiatric consultation services); in departments of general, neurological, or psychiatric clinics; in general practitioner practices; or by other specialists in the community and other primary healthcare facilities (Altamura 1991; Aragona 2005; Eberhard 1988; Kroenke 2006; Luo 2009; Melzer 2009; Müller 2004; Muller 2008; Pilowsky 1990; Volz 2002; Zitman 1991). Six trials were conducted in an inpatient setting (Jiang 2005; Ju 2003; Ouyang 2006; Wang 2003; Yang 2006; Zhao 2006). They were all Chinese. The remaining studies took place in an outpatient setting or no information about the setting was provided.
Participants
Age
Most studies were limited to adults aged 18 to 65 years. In some studies, this age limit was slightly exceeded (Aragona 2005; Eberhard 1988; Kong 2004; Melzer 2009; Volz 2000; Wang 2003; Xu 2004; Yang 2006). The mean age of the included studies ranged between 37.04 and 53.64 years. The lower age limit was between 18 and 26 years whereas the higher age limit was between 51 and 77 years. For 17 studies, there was no information on the age range (see also Characteristics of included studies).
Proportion of men and women
In six studies, there were similar proportions of men and women. The proportion of women ranged between 43.3% and 58.8%, and the proportion of men ranged between 41.3% and 56.7%. Furthermore, there were 17 studies where the proportion of women was considerably higher than the proportion of men ranging between 60.0% and 90.2% for women and 9.8% and 40.0% for men. There was only one study with considerably more men (92.3%) than women (9.8%) (Sanada 2010). In almost all studies, the proportion of women and men was balanced between the trial conditions (Chi2 0.00 to 3.80; P value 0.116 to 1.000). Only in one study was there a statistically significant difference between the trial arms for the distribution of the gender (Müller 2004). In the treatment group, only slightly more women than men were included, whereas in the placebo group, considerably more women than men were included (Chi2 = 4.24; P value = 0.039).
Diagnosis and screening procedures
All included studies comprised participants diagnosed with a somatoform disorder according to explicit diagnostic criteria. Nine studies included people with somatoform/psychogenic pain disorder only (Aragona 2005; Eberhard 1988;Luo 2009;Ouyang 2006; Pilowsky 1990; Sanada 2010; Wang 2003; Xu 2004; Zitman 1991). Four studies included people with somatisation disorder, undifferentiated somatoform disorder, with somatoform autonomic dysfunction, or somatoform/psychogenic pain disorder (Jiang 2005; Kong 2004; Li 2006; Yang 2006). Three studies included people with somatisation disorder, undifferentiated somatoform disorder, or somatoform autonomic dysfunction (Huang 2012; Müller 2004; Volz 2000; Volz 2002). Three studies included people with a somatisation disorder only (Ju 2003; Ye 2006; Zhao 2006). Two studies included people with an undifferentiated somatoform disorder only (Han 2008a; Han 2008b). Two studies included people with a somatisation disorder or an undifferentiated somatoform disorder only (Altamura 1991; Melzer 2009). Two studies included people diagnosed with a multisomatoform disorder (Kroenke 2006; Muller 2008).
Nine studies applied diagnostic criteria of CCMD‐III (Jiang 2005; Ju 2003; Kong 2004; Li 2006; Ouyang 2006; Yang 2006; Ye 2006; Xu 2004; Zhao 2006). Seven studies applied the diagnostic criteria of ICD‐10 (Huang 2012; Luo 2009; Melzer 2009; Müller 2004; Volz 2000; Volz 2002; Wang 2003). Four studies used the diagnostic criteria of DSM‐III (Altamura 1991; Eberhard 1988; Pilowsky 1990; Zitman 1991). Four studies used the diagnostic criteria of DSM‐IV (Aragona 2005; Han 2008a; Han 2008b; Sanada 2010). Two studies used criteria of multisomatoform disorder that corresponded with the criteria of an undifferentiated somatoform disorder (Kroenke 2006; Muller 2008).
Most of the studies provided no information about the procedures for gaining the diagnosis (Altamura 1991; Huang 2012; Jiang 2005; Ju 2003; Kong 2004; Li 2006; Luo 2009; Melzer 2009; Ouyang 2006; Sanada 2010; Volz 2002; Wang 2003; Xu 2004; Yang 2006; Ye 2006; Zhao 2006). However, for these studies we assumed that a non‐structured, psychiatric interview was conducted because psychiatric diagnoses corresponding to explicit diagnostic criteria were made.
In two studies, a structured clinical interview (M.I.N.I. International Neuropsychiatric Interview; Sheehan 1998) was administered in order to make the diagnosis (Kroenke 2006; Muller 2008). Two studies applied diagnostic checklists (International Diagnostic Checklists (IDCL); Janca 1996) in order to make the diagnosis. In one study, a diagnostic assessment and clinical examination by a neurologist, a clinical assessment of the role of psychopathological components in the pain were conducted, as well as rating scales, checklists, and projective tests were available to be used by a clinical psychologist if psycho(patho)logical functioning had to be further explored (Aragona 2005). Trial authors of one study stated that participants had to have undergone a structured clinical interview in order to explore the type and severity of somatic symptoms as well as previous and existing co‐medication (Eberhard 1988). In two studies, the diagnosis of an undifferentiated somatoform disorder was evaluated according to DSM‐IV criteria by consensus between two board‐certified psychiatrists upon study entry (Han 2008a; Han 2008b). In one study, a diagnostic evaluation of participants over a two‐week period was conducted that also included an interview with the psychiatrist who explored the phenomenology of pain experience; evidence of psychiatric syndrome; aspects of the participant's development; interpersonal relationships and life stresses; participant's attitudes, beliefs, and expectation concerning pain and its management; and participant's affect (Pilowsky 1990). The probable diagnosis was discussed by the panel members. Finally, in one study, it was stated that a psychiatric interview was conducted to obtain other DSM‐III‐R diagnoses and to gather demographic data (Zitman 1991).
Ten studies used additional cut‐off scores on a measure of MUPS or depression (Huang 2012; Jiang 2005; Ju 2003; Kroenke 2006; Li 2006; Luo 2009; Müller 2004; Volz 2000; Volz 2002; Wang 2003).
Length of time since diagnosis of somatoform disorder
Thirteen studies provided information about the length of time since the diagnosis of somatoform disorder (Eberhard 1988; Han 2008a; Han 2008b; Huang 2012; Ju 2003; Li 2006; Luo 2009; Ouyang 2006; Wang 2003; Yang 2006; Ye 2006; Zhao 2006; Zitman 1991). The mean length of symptom duration in these studies ranged between 0.6 and 39.8 years (see also Characteristics of included studies).
Co‐morbidity
Twelve studies included information about the actual co‐morbid conditions of included participants (see Table 7). Of these studies, five excluded people with co‐morbid mental disorders. The remaining seven studies included people with different co‐morbid mental disorders. Mentioned co‐morbid conditions were major depression, dysthymia, atypical depression, anxiety disorders (panic disorder, agoraphobia, social anxiety disorder, generalised anxiety disorder, anxiety disorder not otherwise specified), hypochondriasis, nicotine abuse, and benzodiazepine abuse. Fourteen studies provided no information about actual co‐morbid conditions of the included participants (see Table 7).
2. Co‐morbid mental disorders or exclusion of co‐morbid mental conditions in trials comparing pharmacotherapy versus placebo or other medication (see also subgroup analyses 'Pharmacotherapy versus placebo (subgrouped by co‐morbidity of participants)').
| Trial ID | Experimental treatment group ‐ medication class | Control treatment group ‐ medication class | Co‐morbid mental disorders? Yes/no/ns |
Detailed information about co‐morbid diagnoses in participants or exclusion of participants with co‐morbid diagnoses (for studies that do not provide details about co‐morbid conditions) |
Detailed information about the exclusion of participants with co‐morbid diagnoses |
| Kroenke 2006 | SNRI | ‐ | Yes | Major depression, generalised anxiety disorder, social anxiety disorder | People with current/past history of mania, bipolar disorder, schizophrenia, or other psychotic disorder; history of serious or clinically unstable psychiatric condition; known or suspected alcohol or drug abuse within 6 months of screening |
| Luo 2009 | SSRI | ‐ | Yes | 38/80 participants had depression (17 item‐HDRS ≥ 17) | People with co‐exist depressive symptoms occurred prior to pain with HDRS ‐ Total Score ≥ 17 |
| Melzer 2009 | NP | ‐ | No | ‐ | People with historically known or clinical indication of a psychiatric disorder |
| Müller 2004 | NP | ‐ | ns | ‐ | People with co‐morbid depression, drug/alcohol abuse, schizophrenia, or schizo‐affective disorder |
| Muller 2008 | SSRI | ‐ | Yes | Dysthymia (27.5%); major depressive episode (2.0%); anxiety disorder (panic disorder/agoraphobia/social anxiety disorder/generalised anxiety disorder) (52.9%) | People with somatic symptoms that were judged to be secondary to a psychiatric disorder other than MDS; current or past psychotic disorder; significant suicidal risk |
| Pilowsky 1990 | TCA | ‐ | ns | ‐ | People with psychotic illness (including MDS), organic brain syndrome, or alcohol dependence |
| Volz 2000 | TCA | ‐ | No | ‐ | People with other significant Axis I diagnoses (e.g. panic disorder, major depressive disorder, substance abuse) |
| Volz 2002 | NP | ‐ | ns | ‐ | People with an additional diagnosis of depression, schizophrenia, schizo‐affective disorder, or dementia |
| Altamura 1991 | AP | AP | Yes | Dysthymia (n = 27), anxiety disorder NOS (n = 6) | ns |
| Aragona 2005 | SSRI | NRI | No | ‐ | People with a diagnosis of another mental disorder |
| Eberhard 1988 | TeCA | TCA | ns | ‐ | People with major depressive disorder, abuse of drugs, and other psychiatric illnesses |
| Han 2008b | SSRI | SSRI | ns | ‐ | People with history of or current (or both) psychotic disorders (such as schizophrenia, schizoaffective disorder, and bipolar disorder); current DSM Axis I disorders that could possibly account for the somatic symptoms (e.g. MDD, anxiety disorders, factitious disorder, malingering, or another somatoform disorder such as somatisation disorder); substance abuse of dependence in the previous 12 months; effect of co‐morbid psychiatric disorders on the effects of the antidepressants cannot be excluded because of the absence of a structured clinical interview, although participants were rigorously evaluated according to DSM‐IV criteria |
| Huang 2012 | SSRI + AP | SSRI | no | ‐ | People with a diagnosis of another mental disorder (e.g. panic disorder, MDD, or substance abuse) |
| Jiang 2005 | SNRI | TCA | ns | ‐ | People with drug dependence, severe psychosis, or paranoia |
| Ju 2003 | SNRI | TCA | ns | ‐ | People with psychotic symptoms, severe brain injury, or substance abuse |
| Kong 2004 | SSRI | TCA | ns | ‐ | ns |
| Li 2006 | SSRI + AP | SSRI | ns | ‐ | ns |
| Ouyang 2006 | NaSSA | TCA | Yes | 38/80 participants had depression (17 item‐HDRS ≥ 17). No information about other co‐morbidities in the sample was provided | People whose pain was caused by depression, anxiety, or schizophrenia |
| Sanada 2010 | SSRI | SNRI | ns | ‐ | ns |
| Wang 2003 | SARI | NSAID | Yes | Based on diagnostic criteria in CCMD‐3: depression (n = 46), dysthymia (n = 50), hypochondriasis (n = 20) | Severely depressed, suicidal people |
| Yang 2006 | SNRI | TCA | ns | ‐ | ns |
| Ye 2006 | NaSSA | TCA | ns | ns | People with any type of drug dependence |
| Xu 2004 | SNRI | TCA | ns | ‐ | ns |
| Zhao 2006 | NaSSA | TCA | no | ‐ | People with other mental disorders |
| Zitman 1991 | TCA + AP | TCA | yes | Atypical depression (n = 1), dysthymic disorder (n = 1), panic disorder (n = 1), nicotine abuse (n = 1), tea abuse (n = 1), benzodiazepine abuse (n = 1) | People with a serious psychiatric disease necessitating immediate treatment; or who were alcohol or illicit drug dependent |
Anxiety disorder NOS: anxiety disorder not otherwise specified; AP: antipsychotic; CCMD: Chinese Classification of Mental Disorders; SARI: serotonin antagonist and reuptake inhibitor; DSM: Diagnostic and Statistical Manual of Mental Disorders; HDRS: Hamilton Depression Rating Scale; ID: identification; ns: not specified; MDD: major depressive disorder; MDS: major depressive syndrome; n: number; NaSSA: noradrenergic specific serotonergic antidepressant; NP: natural product; NRI: noradrenaline reuptake inhibitor; NSAID: non‐steroidal anti‐inflammatory drug; SNRI: serotonin noradrenaline reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant; TeCA: tetracyclic antidepressant.
With the exception of six studies (see Table 7), studies provided information about the exclusion of specific mental conditions. In the trials that did not exclude participants with any form of co‐morbid mental condition, the common excluded conditions were: mania, bipolar disorder, schizophrenia or other psychotic disorder, schizo‐affective disorder, alcohol or drug abuse/dependence, dementia, severe depression including suicidal tendencies, and depressive symptoms occurring before the somatic symptoms started or were judged to be secondary to a psychiatric disorder other than the somatoform disorder.
Interventions
The 26 included studies primarily researched the efficacy of the following antidepressants for somatoform disorders in adults: TCAs, TeCAs, SSRIs, NRIs, SNRIs, NaSSAs, and SARIs.
Three studies examined the efficacy of NPs (butterbur root, valerian root, passionflower herb, lemon balm leaf‐Ze 185 4‐combination and valerian root, passionflower herb, lemon balm leaf‐Ze 185 3‐combination without butterbur: Melzer 2009; St. John's wort LI 160: Müller 2004; Volz 2002). One study examined the efficacy of APs (Altamura 1991), and three studies examined the combination treatment of an antidepressant and an AP (Huang 2012; Li 2006; Zitman 1991).
Maximum and daily mean dose, frequency of medication, and the mode of administration of the medication are described in detail in the Characteristics of included studies table. Fourteen studies implemented a flexible dosing scheme (Han 2008a; Han 2008b; Jiang 2005; Ju 2003; Kong 2004; Kroenke 2006; Li 2006; Muller 2008; Ouyang 2006; Pilowsky 1990; Sanada 2010; Xu 2004; Ye 2006; Zhao 2006), 11 studies implemented a fixed dosing scheme (Altamura 1991; Aragona 2005; Eberhard 1988; Huang 2012; Luo 2009; Melzer 2009; Müller 2004; Volz 2000; Volz 2002; Wang 2003; Zitman 1991), and one study provided no information about the dosing scheme (Yang 2006).
Comparators
We included eight studies comparing pharmacotherapy and placebo in the narrative literature but also in the meta‐analytical part of our review (Kroenke 2006; Luo 2009; Melzer 2009; Müller 2004; Muller 2008; Pilowsky 1990; Volz 2000; Volz 2002).
We included 15 studies comparing pharmacotherapy and another medication in the narrative literature review. Of these 15 studies, 14 could be included in the meta‐analytical part of our review (Aragona 2005; Eberhard 1988; Han 2008a; Han 2008b; Jiang 2005; Ju 2003; Kong 2004; Ouyang 2006; Sanada 2010; Wang 2003; Xu 2004; Yang 2006; Ye 2006; Zhao 2006). We excluded one study from the meta‐analytical calculations since means and SD were only provided in a graph in the trial report and according to the reply of the trial author were not available anymore (Altamura 1991).
In the following, we listed the studies according to the medication class and chemical agent. The chemical agent that was stated as experimental intervention in the trial report will also be seen as experimental intervention in the following listing. In four studies an SSRI was examined as experimental intervention (paroxetine: Kong 2004;Sanada 2010; citalopram: Aragona 2005; fluoxetine: Han 2008b). The SSRI was compared either with an NRI (reboxetine: Aragona 2005), with another SSRI (sertraline: Han 2008b), a TCA (amitriptyline: Kong 2004), or with an SNRI (milnacipran: Sanada 2010). In another four studies, the NaSSA, mirtazapine, was examined as experimental intervention (Han 2008a; Ouyang 2006; Ye 2006; Zhao 2006). The NaSSA was compared with either a TCA (amitriptyline: Ouyang 2006; Zhao 2006; clomipramine: Ye 2006), or an SNRI (venlafaxine: Han 2008a). In four studies, the SNRI, venlafaxine, was examined as experimental intervention (Jiang 2005; Ju 2003; Yang 2006; Xu 2004). In all four studies, the SNRI was compared with a TCA (doxepin: Ju 2003; amitriptyline: Jiang 2005; Yang 2006; Xu 2004). One study compared the TeCA, maprotiline, as experimental intervention with the TCA, clomipramine (Eberhard 1988). Another study compared the SARI, trazodone, as experimental intervention with the non‐steroidal anti‐inflammatory drug (NSAID) ibuprofen (Wang 2003). One study compared an AP, levosulpiride, with another AP, racemic sulpiride (Altamura 1991).
The efficacy of a combined treatment was examined in three trials. Two of these studies examined the combination of an SSRI and an AP with SSRI alone (citalopram plus paliperidone versus citalopram alone: Huang 2012; paroxetine plus quetiapine versus paroxetine alone: Li 2006). One study compared the efficacy of a combination of a TCA and an AP with the TCA only (amitriptyline plus flupentixol versus amitriptyline alone: Zitman 1991).
Treatment duration and study visits
All trials provided information about the duration of the treatment, which ranged between 14 and 84 days. Efficacy measures were collected throughout the treatment period and at completion of the trial. There were only two trials where follow‐ups were implemented after the completion of the treatment phase (months 3, 6, and 12 post treatment) (Eberhard 1988; Pilowsky 1990). However, in one of these studies, these follow‐up assessments did not take place in a standardised way (Eberhard 1988). The trial report stated that after the sixth week of study treatment the code for the individual participant could be broken and the person could continue the treatment in an open way. One of these trials mentioned that the follow‐up assessments were conducted by a blinded research assistant; however, the research report included no data on the follow‐up assessments (Pilowsky 1990).
Adverse effects and adverse effect‐specific drop‐outs
Detailed information about adverse effects for each trial are reported in the Characteristics of included studies table. It was difficult to give a summarising description of adverse effects, given the non‐standard way in which the adverse effects were reported.
Detailed information about the rate of drop‐outs due to adverse effects or inefficacy of the treatment are summarised in the Characteristics of included studies table. Four studies provided no information about drop‐outs due to adverse effects (Luo 2009; Pilowsky 1990; Zhao 2006; Zitman 1991).
Concomitant treatments
Detailed information about concomitant treatments of participants or about which concomitant treatments were allowed or excluded are summarised in Table 8.
3. Concomitant treatments or exclusion/permission of concomitant treatments in trials comparing pharmacotherapy versus placebo or other medication.
| Trial ID | Experimental treatment group ‐ medication class | Control treatment group ‐ medication class | Detailed information about the exclusion of or permission of concomitant treatments | Detailed information about concomitant treatments |
| Kroenke 2006 | SNRI | ‐ | ‐ Exclusion: use of triptans, psychoactive herbal medications, or any other psychoactive drugs or having a positive urine drug test at screening ‐ Permitted: zaleplon, zopiclone (1 dose nightly) as needed for insomnia for up to 6 nights during the 14 days immediately following randomisation and short‐term treatments for symptoms of allergies, colds, or influenza (without psychotropic effects) |
‐ Proportion of participants who took at least 1 concomitant medication in venlafaxine ER groups vs. placebo were 63.6% vs. 63.3%; for NSAIDs 10% vs. 13%; for paracetamol (acetaminophen) 7% vs. 9%; for COX‐2 inhibitors 8% vs. 3%; and for salicylates 4% vs. 4% |
| Luo 2009 | SSRI | ‐ | ‐ Exclusion: use of antidepressants for the treatment of pain or depression | ‐ |
| Melzer 2009 | NP | ‐ | ‐ Exclusion: psychotherapy, physiotherapy, acupuncture, or using psychoactive drugs including central stimulants and α‐/β‐blockers were excluded ‐ Permitted: in case of sleeping disorders, chloral hydrate was allowed up to 3 g/day |
‐ |
| Müller 2004 | NP | ‐ | ‐ Exclusion: use of psychotropic drugs 4 weeks before and during the study, concomitant psychotherapy, and concomitant treatment with phenprocoumon or cyclosporin (or both) | ‐ |
| Muller 2008 | SSRI | ‐ | ‐ Exclusion: use of psychotropics or cognitive‐behavioural therapy | ‐ |
| Pilowsky 1990 | TCA | ‐ | ‐ | ‐ |
| Volz 2000 | TCA | ‐ | ‐ | ‐ |
| Volz 2002 | NP | ‐ | ‐ Exclusion: concomitant treatment with psychopharmacological active compounds | ‐ |
| Altamura 1991 | AP | AP | ‐ | ‐ |
| Aragona 2005 | SSRI | NRI | ‐ Exclusion: psychotropic drugs endowed with an analgesic action (e.g. amitriptyline) ‐ Permitted: benzodiazepines at low doses for people with sleep disturbances ‐ Because people with other mental disorders were excluded, participants did not take any other type of psychotropic drugs |
‐ |
| Eberhard 1988 | TeCA | TCA | ‐ Exclusion: other central nervous system‐active drugs | ‐ |
| Han 2008a | NaSSA | ‐ | ‐ Exclusion: other psychotropic medications; use of prescription analgesics, muscle relaxants, and corticosteroids ‐ Permitted: hypnosedatives and benzodiazepines only for temporary control of insomnia or anxiety; concomitant medications such as non‐prescription paracetamol were allowed only on an as‐needed basis |
‐ Mirtazapine group: 46% lorazepam, 10% alprazolam ‐ Venlafaxine group: 32% lorazepam, 29% alprazolam |
| Han 2008b | SSRI | SSRI | ‐ Exclusion: other psychotropic medications; use of prescription analgesics, muscle relaxants, and corticosteroids ‐ Permitted: hypnosedatives and benzodiazepines only for temporary control of insomnia or anxiety; concomitant medications such as non‐prescription paracetamol were allowed only on an as‐needed basis |
‐ Fluoxetine group: 32.1% lorazepam, 7.1% alprazolam ‐ Sertraline group: 35.3% lorazepam, 17.6% alprazolam |
| Huang 2012 | SSRI + AP | SSRI | ‐ Exclusion: other psychotropic medications ‐ Permitted: benzodiazepines for insomnia, only for temporary control of the symptoms |
‐ |
| Jiang 2005 | SNRI | TCA | ‐ Permitted: treatment of adverse effects insomnia/anxiety with benzodiazepines and nausea with vitamin B6 | ‐ |
| Ju 2003 | SNRI | TCA | ‐ Exclusion: antidepressant and AP medication ‐ Permitted: sleep medication alprazolam (0.4‐0.8 mg/day) |
‐ |
| Kong 2004 | SSRI | TCA | ‐ Permitted: alprazolam of a maximum dose of 0.8 mg/day | ‐ |
| Li 2006 | SSRI + AP | SSRI | ‐ | ‐ |
| Ouyang 2006 | NaSSA | TCA | ‐ Exclusion: any other medication ‐ Permitted: exception of benzodiazepines for participants with sleep difficulties |
‐ |
| Sanada 2010 | SSRI | SNRI | ‐ | ‐ |
| Wang 2003 | DAS | NSAID | ‐ Exclusion: any other medication | ‐ |
| Xu 2004 | SNRI | TCA | ‐ | ‐ |
| Yang 2006 | SNRI | TCA | ‐ Permitted: alprazolam (0.4‐0.8 mg/day) for participants with sleep difficulties | ‐ |
| Ye 2006 | NaSSA | TCA | ‐ Permitted: zolpidem for participants with sleep difficulties | ‐ |
| Zhao 2006 | NaSSA | TCA | ‐ Exclusion: use of MAOI or other antidepressants during the first 2 weeks of treatment; use of other medication such as antidepressants, mood stabiliser, antipsychotic medication, or electroconvulsive therapy during the 8 treatment weeks ‐ Permitted: benzodiazepines were allowed for participants with insomnia. Benzhexol was allowed for participants with extrapyramidal adverse effects |
‐ |
| Zitman 1991 | TCA + AP | TCA | ‐ Permitted: benzodiazepines and non‐narcotic analgesics could be continued during the trial, but the participants were asked to keep the dose as low as possible | ‐ |
AP: antipsychotic; COX: cyclo‐oxygenase; DAS: ; ER: extended release; ID: identification; MAOI: monoamine oxidase inhibitors; NaSSA: noradrenergic specific serotonergic antidepressant; NP: natural product; NRI: noradrenaline reuptake inhibitor; NSAID: non‐steroidal anti‐inflammatory drug; SNRI: serotonin noradrenaline reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant; TeCA: tetracyclic antidepressant.
Outcomes
The most often assessed outcomes in the included studies of this review were the primary outcomes 'severity/intensity of MUPS' and 'depression'. Eighteen studies measured 'severity/intensity of MUPS' with several different self report scales (Aragona 2005; Eberhard 1988; Han 2008a; Han 2008b; Huang 2012; Kroenke 2006; Li 2006; Luo 2009; Melzack 1987; Müller 2004; Muller 2008; Pilowsky 1990; Sanada 2010; Ye 2006; Volz 2000; Volz 2002; Zhao 2006; Zitman 1991). Five studies used clinician‐rated scales (Huang 2012; Müller 2004; Muller 2008; Volz 2000; Volz 2002). 'Depression' was assessed in 20 included studies with self rating as well as clinician‐rated scales (Aragona 2005; Eberhard 1988; Han 2008a; Han 2008b; Huang 2012; Ju 2003; Kong 2004; Kroenke 2006; Li 2006; Luo 2009; Melzer 2009; Muller 2008; Ouyang 2006; Volz 2000; Volz 2002; Wang 2003; Yang 2006; Ye 2006; Zhao 2006; Zitman 1991). The remaining secondary outcomes were measured only in a small number of studies. 'Anxiety' was assessed with self rating as well as clinician‐rated scales in 11 studies (Huang 2012; Kong 2004; Kroenke 2006; Li 2006; Melzer 2009; Muller 2008; Volz 2000; Volz 2002; Yang 2006; Ye 2006; Zhao 2006). 'Treatment response' based on self rating as well as clinician‐rated scales was assessed in seven studies (Huang 2012; Müller 2004; Muller 2008; Volz 2000; Volz 2002; Ye 2006; Zhao 2006). 'Functional disability and quality of life' (measured in three studies: Kroenke 2006; Muller 2008; Pilowsky 1990) and 'dysfunctional cognitions, emotions, or behaviours' (measured in one study: Muller 2008) were assessed only in a small number of included studies. More information on the measures used can be found in the Characteristics of included studies table.
Regarding the outcome 'acceptability', 24 studies calculated the proportion of people who dropped out during the experimental as well as the comparator intervention. Table 9 displays the acceptability for the different medication classes, chemical agents, and comparisons. The acceptability could not be calculated for four studies for the following reasons. The trial by Luo 2009 stated that two participants quit the study due to side effects. However, it was not clear how these adverse effects related to drop‐outs were distributed between the treatment and placebo group. The trial by Zhao 2006 mentioned only the drop‐out rate for the total sample; however, the rate was not provided separately for control and treatment group. The trial by Zitman 1991 originally randomised 45 people; nine participants dropped out, resulting in a completer sample of 36 people. Afterwards, two people had to be excluded because of undetectable levels of amitriptyline and metabolites indicating non‐compliance resulting in a sample of 34 participants (amitriptyline alone group: 16 people, amitriptyline plus flupentixol group: 18 people). Unfortunately, it is unclear how many participants of the originally randomised sample were distributed among the amitriptyline alone group and the amitriptyline plus flupentixol group. Trial authors could only be partly contacted. Authors of the study by Luo 2009 did not reply to our request. For trial by Zhao 2006, no contact details of the authors were available (the article states only the affiliation of the authors and an online search for the authors' names was without result). The trial by Zitman 1991 was too old and we could not contact the author.
4. Attrition rate (acceptability: proportion of participants who dropped out during the experimental as well as the comparator intervention, calculated as a proportion of the total number of randomised participants).
| Trial ID | Experimental treatment | Placebo | Control treatment | ||||||||||
| Medication class |
Chemical agent |
Attrition | Total | % attrition | Attrition | Total | % attrition | Medication class | Chemical agent | Attrition | Total | % attrition | |
| Pilowsky 1990 | TCA | Amitriptyline | ns | 26 | 25.0 | ns | 24 | 31.0 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Kong 2004 | TCA | Amitriptyline | 0 | 15 | 0 | ‐ | ‐ | ‐ | SSRI | Paroxetinea | 0 | 45 | 0 |
| Jiang 2005 | TCA | Amitriptyline | 0 | 32 | 0 | ‐ | ‐ | ‐ | SNRI | Venlafaxine | 0 | 36 | 0 |
| Yang 2006 | TCA | Amitriptyline | 0 | 35 | 0 | ‐ | ‐ | ‐ | SNRI | Venlafaxine | 0 | 35 | 0 |
| Xu 2004 | TCA | Amitriptyline | 0 | 35 | 0 | ‐ | ‐ | ‐ | SNRI | Venlafaxine | 0 | 35 | 0 |
| Ouyang 2006 | TCA | Amitriptyline | 0 | 40 | 0 | ‐ | ‐ | ‐ | NaSSA | Mirtazapine | 0 | 40 | 0 |
| Zhao 2006 | TCA | Amitriptyline | ns | 30 | ne | ‐ | ‐ | ‐ | NaSSA | Mirtazapine | ns | 30 | ne |
| Zitman 1991 | TCA | Amitriptyline | ns | ns | ne | ‐ | ‐ | ‐ | TCA + AP | Amitriptyline + flupentixol | ns | ns | ne |
| Ye 2006 | TCA | Clomipramine | 2 | 35 | 5.7 | ‐ | ‐ | ‐ | NaSSA | Mirtazapine | 2 | 35 | 5.7 |
| Eberhard 1988 | TCA | Clomipramine | 13 | 40 | 32.5 | ‐ | ‐ | ‐ | TeCA | Maprotiline | 5 | 30 | 16.7 |
| Ju 2003 | TCA | Doxepin | 0 | 34 | 0 | ‐ | ‐ | ‐ | SNRI | Venlafaxine | 0 | 34 | 0 |
| Volz 2000 | TCA | Opipramol | 14 | 104 | 13.5 | 13 | 104 | 12.4 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Zitman 1991 | TCA + AP | Amitriptyline + flupentixol | ns | ns | ne | ‐ | ‐ | ‐ | TCA | Amitriptyline | ns | ns | ne |
| Kong 2004 | SSRI | Paroxetinea | 0 | 45 | 0 | ‐ | ‐ | ‐ | TCA | Amitriptyline | 0 | 15 | 0 |
| Li 2006 | SSRI | Paroxetine | 0 | 30 | 0 | ‐ | ‐ | ‐ | SSRI + AP | Paroxetine + quetiapine | 2 | 28 | 7.1 |
| Sanada 2010 | SSRI | Paroxetine | 3 | 11 | 27.3 | ‐ | ‐ | ‐ | SNRI | Milnacipran | 5 | 10 | 50.0 |
| Luo 2009 | SSRI | Fluoxetine | ns | 40 | ne | ns | 40 | ne | ‐ | ‐ | ‐ | ‐ | ‐ |
| Han 2008b | SSRI | Fluoxetine | 8 | 28 | 28.6 | ‐ | ‐ | ‐ | SSRI | Sertraline | 5 | 17 | 29.4 |
| Huang 2012 | SSRI | Citalopram | 9 | 30 | 30.0 | ‐ | ‐ | ‐ | SSRI + AP | Citalopram + paliperidone | 9 | 30 | 30.0 |
| Aragona 2005 | SSRI | Citalopram | 6 | 17 | 35.3 | ‐ | ‐ | ‐ | NRI | Reboxetine | 9 | 18 | 50.0 |
| Muller 2008 | SSRI | Escitalopram | 1 | 25 | 4.0 | 0 | 26 | 0 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Han 2008b | SSRI | Sertraline | 5 | 17 | 29.41 | ‐ | ‐ | ‐ | SSRI | Fluoxetine | 8 | 28 | 28.6 |
| Huang 2012 | SSRI + AP | Citalopram + paliperidone | 9 | 30 | 30.0 | ‐ | ‐ | ‐ | SSRI | Citalopram | 9 | 30 | 30.00 |
| Li 2006 | SSRI + AP | Paroxetine + quetiapine | 0 | 30 | 0 | ‐ | ‐ | ‐ | SSRI | Paroxetine | 2 | 28 | 7.1 |
| Kroenke 2006b | SNRI | Venlafaxine (extended release) | 21 | 55 | 38.2 | 22 | 57 | 38.6 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Jiang 2005 | SNRI | Venlafaxine | 0 | 36 | 0 | ‐ | ‐ | ‐ | TCA | Amitriptyline | 0 | 32 | 0 |
| Yang 2006 | SNRI | Venlafaxine | 0 | 35 | 0 | ‐ | ‐ | ‐ | TCA | Amitriptyline | 0 | 35 | 0 |
| Xu 2004 | SNRI | Venlafaxine | 0 | 35 | 0 | ‐ | ‐ | ‐ | TCA | Amitriptyline | 0 | 35 | 0 |
| Ju 2003 | SNRI | Venlafaxine | 0 | 34 | 0 | ‐ | ‐ | ‐ | TCA | Doxepin | 0 | 34 | 0 |
| Han 2008a | SNRI | Venlafaxine | 13 | 45 | 28.9 | ‐ | ‐ | ‐ | NaSSA | Mirtazapine | 11 | 50 | 22.0 |
| Sanada 2010 | SNRI | Milnacipran | 5 | 10 | 50.0 | ‐ | ‐ | ‐ | SSRI | Paroxetine | 3 | 11 | 27.3 |
| Ouyang 2006 | NaSSA | Mirtazapine | 0 | 40 | 0 | ‐ | ‐ | ‐ | TCA | Amitriptyline | 0 | 40 | 0 |
| Ye 2006 | NaSSA | Mirtazapine | 2 | 35 | 5.7 | ‐ | ‐ | ‐ | TCA | Clomipramine | 2 | 35 | 5.7 |
| Zhao 2006 | NaSSA | Mirtazapine | ns | 30 | ne | ‐ | ‐ | ‐ | TCA | Amitriptyline | ns | 30 | ne |
| Han 2008a | NaSSA | Mirtazapine | 11 | 50 | 22.0 | ‐ | ‐ | ‐ | SNRI | Venlafaxine | 13 | 45 | 28.9 |
| Eberhard 1988 | TeCA | Maprotiline | 5 | 30 | 16.7 | ‐ | ‐ | ‐ | TCA | Clomipramine | 13 | 40 | 32.5 |
| Aragona 2005 | NRI | Reboxetine | 9 | 18 | 50.0 | ‐ | ‐ | ‐ | SSRI | Citalopram | 6 | 17 | 35.29 |
| Wang 2003 | SARI | Traxodone | 0 | 70 | 0 | ‐ | ‐ | ‐ | NSAID | Ibuprofen | 0 | 70 | 0 |
| Altamura 1991 | AP | Levosulpride | 2 | 15 | 13.3 | ‐ | ‐ | ‐ | AP | Racemic sulpiride | 2 | 15 | 13.3 |
| Altamura 1991 | AP | Racemic sulpiride | 2 | 15 | 13.3 | ‐ | ‐ | ‐ | AP | Levosulpride | 2 | 15 | 13.3 |
| Müller 2004 | NP | St. John's wort LI 160 | 5 | 87 | 5.8 | 6 | 88 | 6.8 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Volz 2002d | NP | St. John's wort LI 160 | 0 | 75 | 0.0 | 2 | 74 | 2.7 | ‐ | ‐ | ‐ | ‐ | ‐ |
| Melzer 2009 | NP | Ze 185 3‐/4‐combinationc | 10 | 121 | 8.3 | 5 | 61 | 8.2 | ‐ | ‐ | ‐ | ‐ | ‐ |
AP: antipsychotic; ID: identification; NaSSA: noradrenergic specific serotonergic antidepressant; ns: not specified; ne: not estimable; NP: natural product; NRI: noradrenaline reuptake inhibitor, SARI: serotonin antagonist and reuptake inhibitor; SNRI: serotonin noradrenaline reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant; TeCA: tetracyclic antidepressant.
Total sample sizes and attrition rates refer to the total number of randomised participants.
a Trial included 3 arms: paroxetine, open paroxetine, and placebo; we combined the attrition rate of paroxetine and open paroxetine.
b 117 participants were originally randomised but information about sample sizes of treatment and control group were missing. However, study authors stated that in the intention‐to‐treat (ITT) population, they only included participants who had at least 1 post‐baseline efficacy evaluation yielding in an ITT sample of 112. No information was provided about how the 117 participants were distributed among treatment groups. Therefore, we calculated drop‐out rate based on the 112 sample.
c Trial included 3 arms: Ze 185 4‐combination, Ze 185 3‐combination, and placebo; we combined the attrition rate of the Ze 185 3‐combination and 4‐combination.
d Originally 151 participants were randomised. No participants were excluded after the placebo run‐in phase. However, study authors defined ITT population as a sample of all randomised participants with at least 1 assessment under trial medication. 2 participants had to be excluded from the analysis due to missing values for the primary efficacy variable after baseline. Therefore, the ITT sample included 149 participants. No information was provided how the original 151 participants were distributed among groups. Therefore, we calculated drop‐out rate based on the sample.
Excluded studies
After assessing the full‐texts, we excluded 30 studies (see Figure 1). We summarised the reasons for exclusion in the Characteristics of excluded studies table. The primary reason for exclusion (in 24 studies) was that the participants' diagnosis did not fit the inclusion criteria, especially in older studies that were conducted at a time where standardised diagnostic criteria had not been established, yet often old diagnostic concepts such as 'neurosis' or 'hysteria' were used to make a diagnosis. These definitions include a range of different diagnostic concepts such as generalised anxiety disorder, phobias, de‐personalisation disorder, dysthymic disorder, conversion disorder, obsessive‐compulsive disorder, or dissociative disorders, and so do not fulfil the strict diagnostic inclusion criteria of the current review. In addition, there were some studies with people diagnosed with 'psychosomatic disorders'. Furthermore, several studies included people only with very specific functional syndromes such as IBS (Davis 1988; Loldrup 1989; Pach 1976; Smouvelich 1996; Tanum 1996; Turkington 2002), which did not correspond with the inclusion criteria of the current review. One study comprised healthy participants (Lee 2012), or participants with reactive depression (Poinso 1988), and was therefore excluded. We excluded two studies because the criteria of treatment were not fulfilled. The study by Farnbach 2013 examined the efficacy of quetiapine fumarate as an adjunctive therapy to current pain treatment. However, the current pain treatment was not a standardised treatment. In the study by Xu 2006, the comparator intervention acupuncture did not correspond with the inclusion criteria of the current review. We excluded one study because it was just a case report (Kozian 2003). Another study implemented a cross‐over design (Onghena 1993); however, no data for the unbiased first study phase were provided. For this reason, this study could not contribute to this review, either in the narrative or the meta‐analytic sections. We excluded another study because it was not randomised (Altamura 2003). The status of randomisation in this study was not clear after screening the full‐text. Therefore, we contacted the authors of the trial who replied that the study was not randomised.
Ongoing studies
We identified one ongoing study (Agger 2014). The trial's registration documentation as well as the personal contacting of the principal investigators of this trial suggests that this study is planned to be completed in December 2014. The trial is being conducted in Denmark. In this randomised, placebo‐controlled, double‐blind study on the efficacy of the TCA, imipramine. The study will include participants diagnosed with the bodily distress syndrome (multi‐organ type) that is characterised by chronic MUPS (of a duration of at least two years) from at least three out of four symptoms that have moderate to severe impact on daily life. Included participants are aged 20 to 50 years. The primary outcome is CGI ‐ Improvement Scale and the secondary outcomes are functional level measured by the SF‐36 and symptom characterisation measured by VAS of the Functional Illness Checklist. The time points of evaluation are the following: participant‐rated improvement measured by the CGI ‐ Improvement Scale after 10 weeks of sufficient‐dosage study medication (minimum 25 mg/day). The SF‐36 will be administered before inclusion and after 10 weeks of sufficient‐dosage study medication. The VAS and the Functional Illness Checklist are completed before inclusion, after starting treatment, and after 10 weeks of sufficient‐dosage study medication. A sample size of 140 participants is planned with a study duration of 19 weeks for each participant. For detailed information see the Characteristics of ongoing studies table.
Studies awaiting classification
We identified no studies awaiting classification.
Risk of bias in included studies
For full details of risk of bias judgements for included studies see the Characteristics of included studies table. Figure 2 shows the proportion of studies with each of the judgements; the graph in Figure 3 shows all the judgements in a cross‐tabulation by trial.
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Random sequence generation
All studies were reported to be randomised trials. Only nine studies provided detailed information about the means of random sequence generation (Aragona 2005; Han 2008a; Han 2008b; Huang 2012; Li 2006; Melzer 2009; Müller 2004; Muller 2008; Pilowsky 1990). The studies used random number tables, a computer random number generator, and coin tossing as means of random sequence generation. One study used blocked randomisation (Müller 2004). We rated the nine studies as low risk of bias and 17 trials as unclear risk of bias for random sequence generation.
Allocation concealment
Two studies provided detailed information about allocation concealment (Huang 2012; Müller 2004). In both studies, the allocation concealment was conducted by a study‐independent institution. Huang 2012 used sequentially numbered, sealed envelopes. Müller 2004 applied sequentially numbered drug containers of identical appearance. We rated these two studies as low risk of bias and 24 trials as unclear risk of bias for allocation concealment.
Blinding
Blinding of study participants and personnel
Of the 26 included studies, 14 were stated to be double‐blind trials. Six of these 14 trials provided insufficient information about methods of blinding participants or study personnel (Altamura 1991; Aragona 2005; Kroenke 2006; Pilowsky 1990; Volz 2000; Volz 2002). The remaining eight studies described the methods of blinding participants and study personnel in detail (Eberhard 1988; Luo 2009; Melzer 2009; Müller 2004; Muller 2008; Wang 2003; Zhao 2006; Zitman 1991). The studies usually used medication that was of identical appearance as the strategy to blind participants and study personnel. In summary, we judged four studies to be at high risk of bias, 14 to be at unclear risk of bias, and eight studies to be at low risk of bias for blinding of participants and study personnel.
Blinding of outcome assessment
Most of the studies provided no information to determine whether the assessors were blinded. Three studies described their strategies in detail (Huang 2012; Müller 2004; Pilowsky 1990). We judged studies that stated that the trial was double‐blind without any information about blinding participants, study personnel, or assessors as at unclear risk of performance bias or detection bias. We judged two studies to be at high risk of bias, 21 studies to be at unclear risk of bias, and three studies to be at low risk of bias for blinding of assessors.
Incomplete outcome data
We judged studies to have a low risk of attrition bias when 1. they dealt adequately with missing values in the sense of conducting an ITT analysis and replacing missing values with adequate statistical methods (e.g. LOCF), 2. when the ITT sample corresponded to the number of originally randomised participants, and 3. when the number of drop‐outs and reasons for the drop‐out were balanced between the groups. We also judged a low risk of bias when there were no drop‐outs. In summary, we judged nine studies to be at high risk of attrition bias, four studies to be at unclear risk of bias, and 13 studies to be at low risk of bias. Seven studies had no drop‐outs (Jiang 2005; Ju 2003; Kong 2004; Ouyang 2006; Wang 2003; Xu 2004; Yang 2006). Interestingly, these studies were all Chinese trials and almost all of them took place in an inpatient setting. There were drop‐outs in the remaining 19 studies. Eleven of these 19 studies with drop‐outs conducted an ITT analysis (Aragona 2005; Han 2008a; Han 2008b; Huang 2012; Kroenke 2006; Luo 2009; Melzer 2009; Müller 2004; Muller 2008; Volz 2000; Volz 2002). All 11 studies replaced missing values with LOCF. However, in five of these 11 studies, the size of the ITT population did not correspond with the number of originally randomised participants (Aragona 2005; Han 2008a; Kroenke 2006; Müller 2004; Volz 2000). In these cases, the study authors included only participants who received at least one post‐baseline outcome assessment in the ITT sample. Six studies conducted statistical analyses only with the data of participants completing the trial and complying with the study protocol (Eberhard 1988; Li 2006; Pilowsky 1990; Sanada 2010; Ye 2006; Zitman 1991). Two of the 19 studies with drop‐outs provided no information about how the authors dealt with the missing values (Altamura 1991; Zhao 2006).
Selective reporting
The reporting bias was difficult to assess as none of the included trials had pre‐published study protocols. We judged studies as trials with low selective reporting bias ‐ even if there was no study protocol ‐ when they included a broad range of different outcomes, covered primary and secondary outcomes, as well as when these outcomes were assessed with validated measures. Furthermore, the outcomes had to be mentioned in the methods section of the trial report and findings of these outcomes had to be completely reported in the results section. Only three studies fulfilled these criteria (Muller 2008; Volz 2000; Volz 2002). We judged a further nine studies to be at unclear risk of reporting bias (Aragona 2005; Eberhard 1988; Huang 2012; Jiang 2005; Li 2006; Müller 2004; Ye 2006; Zhao 2006; Zitman 1991). They included a small range of primary and secondary outcomes. Because of the missing study protocols, it was not possible to make statements about the risk of reporting bias. Finally, we judged 14 studies to be at high reporting bias. Five of these trials only assessed treatment effects for secondary outcomes (e.g. depression, anxiety) whereas primary outcomes, such as severity of physical symptoms, were neglected (Ju 2003; Kong 2004; Melzer 2009; Ouyang 2006; Yang 2006). One study assessed only secondary outcomes with validated scales, whereas the primary outcome was assessed with a non‐validated clinician‐rated scale constructed by the trial authors (Wang 2003). One study administered clinician‐rated and non‐validated scales created by the trial authors (Xu 2004). Furthermore, three studies reporting no SD for post‐assessment values (Han 2008a; Han 2008b; Kroenke 2006). We contacted authors in order to gain these missing values, but they did not reply. One of these three studies did not report baseline values for all outcomes for which baseline‐to‐endpoint changes were indicated (Kroenke 2006). Therefore, post‐assessment values could not be calculated. We contacted the author requesting these missing values but he did not reply. Finally, one study only reported means and SD for the primary outcome in a graph and the numerical values were not available (Altamura 1991). The trial author informed us that the study was too old and that these missing data were no longer available. Another study only reported treatment effects for the primary outcome but not for the secondary outcome (depression) (Luo 2009). We contacted the trial author but we received no reply. One study assessed several validated scales but only at baseline (Pilowsky 1990). One study was available only as conference abstract in which means and SD were reported for one outcome (Sanada 2010). We contacted study authors but they did not reply to our request.
Other potential sources of bias
We judged 20 trials to be at low risk or unclear risk of other potential sources of bias. We judged six studies to be at high risk of other potential sources of bias (Altamura 1991; Müller 2004; Pilowsky 1990; Volz 2000; Volz 2002; Xu 2004). These relevant other potential sources of bias in the included studies are discussed in the following.
Pre‐randomisation intervention that could influence the effect of the subsequent randomised intervention
In two studies, there was a placebo run‐in phase before randomisation (Volz 2000; Volz 2002). In two studies, there was a placebo run‐in phase after the randomisation (Altamura 1991; Müller 2004). Three studies excluded placebo‐responders, participants exceeding a specific pre‐defined cut‐off score, after this run‐in phase (Altamura 1991; Müller 2004; Volz 2000). One trial offered treatments such as transcutaneous nerve stimulation, physiotherapy, regional nerve blocks, psychopharmacotherapy, and psychotherapy, for a finite period (Pilowsky 1990). It was then decided if the participant would be assigned to the study or not. Where it was thought that musculoligamentous or myofascial problems had not been adequately treated physiotherapeutically this was provided again before participants were entered into the trial. Furthermore, significant differences regarding demographics were identified between the group that was assigned to the study and the group that was not assigned to the study.
Baseline imbalance
Two studies had statistically significant differences between the study groups at baseline (Pilowsky 1990; Xu 2004).
Compliance
Twenty‐four trial reports did not describe any method for assessing compliance with an intervention. The remaining two trials assessed compliance by pill counts (Volz 2000; Volz 2002). The second study reported that compliance ranged from 85.7% to 110.1% (Volz 2002).
Multiple intervention trials
There were three multiple intervention trials (Kong 2004; Melzer 2009; Pilowsky 1990). All three trials presented the data separately for each group.
Cross‐over trials
We included one trial with cross‐over design (Zitman 1991). The trial randomised the order of receiving a treatment and presented unbiased data of the first treatment phase separately for the two study arms.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5
We conducted meta‐analyses for all comparisons planned in the review protocol with the exception of the comparison 'pharmacotherapy versus treatment as usual'. For this comparison, we found no trial that comprised usual treatment as control group. Furthermore, we were unable to assess at two of the three pre‐defined time points stated in our protocol (within 12 months' post treatment, or more than 12 months' post treatment) because in all included trials of this review data were only available for post treatment. As part of a sensitivity analysis, we compared effect sizes based on random‐effects and fixed‐effect analyses. In the following analyses, we reported results using the random‐effects model unless the results of the fixed‐effect analysis differed considerably, in which case we reported both. We classes a 'considerable' difference between results as a change in level of significance or where 95% CIs did not overlap.
Comparison 1: pharmacotherapy versus placebo
1. Tricyclic antidepressants versus placebo
Two studies (258 participants) provided data for comparing TCAs with placebo.
Primary outcomes
1.1 Severity/intensity of medically unexplained physical symptoms
There was no difference in the severity or intensity of MUPS between participants taking a TCA and those taking placebo (SMD ‐0.13; 95% CI ‐0.39 to 0.13; 2 studies, 239 participants). There was consistency in the data (I² = 2%) (Analysis 1.1; Figure 4).
1.1. Analysis.

Comparison 1 Pharmacotherapy versus placebo, Outcome 1 Severity/intensity of medically unexplained physical symptoms (post‐treatment score on self report scales).
4.

Forest plot of comparison: 1 Pharmacotherapy versus placebo, outcome: 1.1 Severity/intensity of medically unexplained physical symptoms (post‐treatment score on self report scales).
1.2 Acceptability
The studies comparing TCAs with placebo found the rates of all‐cause drop‐outs to be 13.5% to 25.0% with TCAs versus 12.4% to 31.0% with placebo (Table 9). We identified one study comparing TCAs versus placebo for acceptability rate, which found no significant difference between groups (RR 1.08; 95% CI 0.53 to 2.18; 208 participants) (Analysis 1.2) (Volz 2000).
1.2. Analysis.

Comparison 1 Pharmacotherapy versus placebo, Outcome 2 Acceptability.
Secondary outcomes
1.3 Anxiety
We identified one study comparing TCAs versus placebo for anxiety, which found no significant difference in effect size between groups (SMD ‐0.01; 95% CI ‐0.29 to 0.26; 200 participants) (Analysis 1.3) (Volz 2000).
1.3. Analysis.
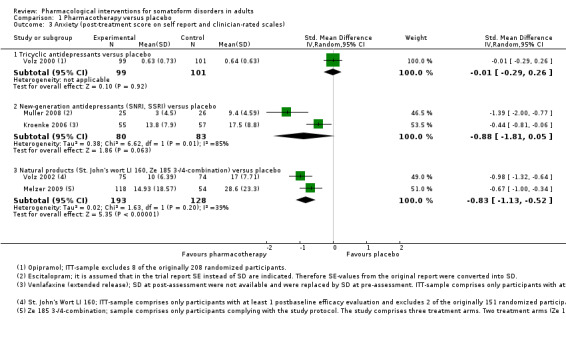
Comparison 1 Pharmacotherapy versus placebo, Outcome 3 Anxiety (post‐treatment score on self report and clinician‐rated scales).
1.4 Depression
We identified one study comparing TCAs versus placebo for depression, which indicated no significant difference between groups (SMD ‐0.27; 95% CI ‐0.55 to 0.01; 200 participants) (Analysis 1.4) (Volz 2000).
1.4. Analysis.

Comparison 1 Pharmacotherapy versus placebo, Outcome 4 Depression (post‐treatment score on self report and clinician‐rated scales).
1.5 Dysfunctional cognitions, emotions, and behaviours
We found no data comparing TCAs with placebo for dysfunctional cognitions, emotions, and behaviours.
1.6 Adverse effects
We identified one study comparing TCAs versus placebo for drop‐out rate due to adverse effects (Volz 2000). In both TCA and placebo arms, 2.9% of participants withdrew from treatment. There was no significant difference between groups (RR 1.00; 95% CI 0.21 to 4.84; 208 participants) (Analysis 1.6; see Characteristics of included studies table).
1.6. Analysis.

Comparison 1 Pharmacotherapy versus placebo, Outcome 6 Adverse effects.
1.7 Treatment response
We identified one study comparing TCAs versus placebo for treatment response, which found no significant difference between groups in the number of participants experiencing remission (RR 1.29; 95% CI 0.95 to 1.73; 208 participants; definition treatment response: 'very good' or at least 'good' treatment success on the CGI ‐ Improvement Scale) (Analysis 1.7) (Volz 2000).
1.7. Analysis.

Comparison 1 Pharmacotherapy versus placebo, Outcome 7 Treatment response (post‐treatment score on self report and clinician‐rated scales).
1.8 Functional disability and quality of life
We identified one study comparing TCAs versus placebo for functional disability and quality of life, which indicated no significant benefit of a TCA compared with placebo (SMD 0.01; 95% CI ‐0.58 to 0.60; 44 participants) (Analysis 1.8) (Pilowsky 1990).
1.8. Analysis.

Comparison 1 Pharmacotherapy versus placebo, Outcome 8 Functional disability and quality of life (post‐treatment score on self report scales).
2. New‐generation antidepressants versus placebo
Three studies (248 participants) provided data for comparing NGAs with placebo.
Primary outcomes
2.1 Severity/intensity of medically unexplained physical symptoms
Compared with placebo, taking an NGA (SNRI, SSRI) was considerably more effective in reducing the severity or intensity of MUPS (SMD ‐0.91; 95% CI ‐1.36 to ‐0.46; 3 studies, 243 participants; random‐effects model). Substantial heterogeneity was identified in the data (I² = 63%). This comparison included one study that for different outcomes and in comparison to other studies demonstrated obvious high effect sizes (Muller 2008). The values in the original trial report that were indicated as SD by the trial authors appeared to be very small and too homogeneous over the time points of assessment and between the different treatment groups. We assumed that in this trial report, SEs but not SDs were reported. Therefore, we used the common formula in order to convert SEs to SDs. However, the effect sizes still remained obviously high in comparison to the remaining studies. When we excluded this outlier study from analysis, the SMD was slightly decreased (SMD ‐0.74; 95% CI ‐1.17 to ‐0.31; 2 studies; 192 participants). Although the I2 statistic decreased, there was still a substantial level of heterogeneity (I2 = 52%) (Analysis 1.1).
2.2 Acceptability
The studies comparing NGAs (SNRI, SSRI) with placebo found the rates of all‐cause drop‐outs to be 4.0% to ‐38.2% with NGAs versus 0% to 38.6% with placebo (Table 9). There was no significant difference in the acceptability rate between NGAs (SNRI, SSRI) and placebo (RR 1.01; 95% CI 0.64 to 1.61; 2 studies, 163 participants). The I2 statistic of 0% indicated consistency in the data (Analysis 1.2).
Secondary outcomes
2.3 Anxiety
There was no significant difference between an NGA (SNRI, SSRI) and placebo for reducing anxiety (SMD ‐0.88; 95% CI ‐1.81 to 0.05; 2 studies; 163 participants) (Analysis 1.3). The I2 statistic indicated considerable heterogeneity in the data (I2 = 85%). However, it should be noted that this comparison included the outlier study by Muller 2008. The SMD decreased when calculated with a fixed‐effect model and became statistically significant in favour of NGA (SMD ‐0.69; 95% CI ‐1.02 to ‐0.37; I2 = 85%).
2.4 Depression
Results showed that NGAs (SNRI, SSRI) were significantly effective at reducing depressive symptoms compared with placebo (SMD ‐0.56; 95% CI ‐0.88 to ‐0.25; 2 studies; 163 participants). There was consistency in the data (I2 = 0%) (Analysis 1.4).
2.5 Dysfunctional cognitions, emotions, and behaviours
We identified one study comparing NGA (SSRI) versus placebo for dysfunctional cognitions, emotions, and behaviours (Muller 2008). The effect size indicated no significant difference between groups (SMD 0.26; 95% CI ‐0.29 to 0.81; 51 participants) (Analysis 1.5).
1.5. Analysis.

Comparison 1 Pharmacotherapy versus placebo, Outcome 5 Dysfunctional cognitions, emotions, and behaviours (post‐treatment score on self report scales).
2.6 Adverse effects
The drop‐out rate due to adverse effects ranged from 4.0% to 7.3% for the NGA group and from 0% to 3.5% for the placebo group (see Characteristics of included studies table). There was no significant difference between groups (RR 2.26; 95% CI 0.52 to 9.81; 2 studies; 163 participants). There was consistency in the data (I2 = 0%) (Analysis 1.6).
2.7 Treatment response
There was no difference in the percentage of participants who achieved remission between the NGA (SNRI, SSRI) and placebo groups (RR 2.00; 95% CI 0.90 to 4.43; 2 studies; 163 participants; definition treatment response: Kroenke 2006: PHQ‐15 less than 10; Muller 2008: CGI ‐ Improvement Scale 2 or less) (Analysis 1.7). There was considerable heterogeneity (I2 = 76%). However, it should be noted that this comparison included an outlier study (Muller 2008). The RR became significant in favour of NGA when calculated using a fixed‐effect model (RR 1.82; 95% CI 1.28 to 2.58; I2 = 76%).
2.8 Functional disability impairment and quality of life
NGA (SNRI, SSRI) was significantly more effective than placebo at reducing functional impairment or in increasing life quality (SMD ‐0.52; 95% CI ‐1.00 to ‐0.04; 2 studies; 163 participants). However, there was substantial heterogeneity (I2 = 51%) (Analysis 1.8).
3. Natural products versus placebo
Three studies (508 participants) provided data for comparing NPs with placebo. One of these studies used a combined placebo‐controlled, parallel‐group design comprising three study arms (Melzer 2009). In two of the three study arms, a specific NP combination was examined whereas in the third study arm a placebo was administered. For the following analyses, we combined the two combinations in one group.
Primary outcomes
3.1 Severity/intensity of medically unexplained physical symptoms
In comparison to placebo, NPs were significantly effective in reducing the severity of MUPS (SMD ‐0.74; 95% CI ‐0.97 to ‐0.51; 2 studies; 322 participants). There was high consistency in the data (I2 = 0%) (Analysis 1.1).
3.2 Acceptability
The studies comparing NPs with placebo found the rates of all‐cause drop‐outs to be 0% to 8.3% with NPs versus 7.0% to 8.2% with placebo (Table 9). There was no significant difference in the acceptability rate between the groups (RR 0.85; 95% CI 0.40 to 1.78; 3 studies; 506 participants). There was high consistency in the data (I2 = 0%) (Analysis 1.2).
Secondary outcomes
3.3 Anxiety
NPs were significantly more effective than placebo in reducing anxiety symptoms (SMD ‐0.83; 95% CI ‐1.13 to ‐0.52; 2 studies, 321 participants). There was moderate heterogeneity in the data (I2 = 39%) (Analysis 1.3).
3.4 Depression
NPs were significantly more effective than placebo in reducing depressive symptoms (SMD ‐0.64; 95% CI ‐0.87 to ‐0.41; 2 studies; 321 participants). There was consistency in the data (I2 = 0%) (Analysis 1.4).
3.5 Dysfunctional cognitions, emotions, and behaviours
We found no data comparing NPs with placebo for dysfunctional cognitions, emotions, and behaviours.
3.6 Adverse effects
The drop‐out rate due to adverse effects ranged from 0% to 1.7% with NPs and 0% to 2.7% with placebo (see Characteristics of included studies table). Three studies (506 participants) provided sufficient data for calculating an RR. There was no significant difference between groups (RR 0.54; 95% CI 0.08 to 3.50). There was consistency in the data (I2 = 0%) (Analysis 1.6).
3.7 Treatment response
Compared with the placebo group, there was a significantly higher percentage of participants who achieved remission in the NP group (RR 1.77; 95% CI 1.34 to 2.34; 2 studies; 324 participants; random‐effects model; definition treatment response: Müller 2004: CGI ‐ Improvement Scale 2 or less, Volz 2002: 'very much', 'much', or 'minimal' treatment success on the CGI ‐ Improvement Scale). There was low heterogeneity in the data (I2 = 24%) (Analysis 1.7). The NNTB was 4.
3.8 Functional disability and quality of life
We found no data comparing NPs with placebo for functional disability and quality of life.
Comparison 2: pharmacotherapy versus another medication
4. Tricyclic antidepressants versus another medication
Ten studies (616 participants) provided data comparing TCAs with another medication (exclusively NGAs). In one of these studies, there were three study arms (paroxetine versus 'open paroxetine' versus amitriptyline; Kong 2004). For the following analyses, we have combined the treatment arms paroxetine and 'open paroxetine' in one group.
Primary outcomes
4.1 Severity/intensity of medically unexplained physical symptoms
The results showed no difference between TCAs and NGAs (TeCA, NaSSA) in reducing the severity of MUPS (SMD ‐0.16; 95% CI ‐0.55 to 0.23; 3 studies; 177 participants; random‐effects model). There was moderate heterogeneity in the data (I2 = 42%) Analysis 2.1; Figure 5.
2.1. Analysis.

Comparison 2 Tricyclic antidepressants versus another medication, Outcome 1 Severity/intensity of medically unexplained physical symptoms (post‐treatment score on self report scales).
5.

Forest plot of comparison: 2 Tricyclic antidepressants versus new‐generation antidepressants, outcome: 2.1 Reduction of the level of severity/intensity of medically unexplained physical symptoms (post‐treatment score on self report scales).
4.2 Acceptability
Rates of attrition due to all causes for the comparison of TCAs versus NGAs (TeCA, NaSSA, SNRI, SSRI) were 0% to 32.4% for TCAs versus 0% to 16.7% with NGAs (Table 9). There was no significant difference in the acceptability rate between TCAs and NGAs (SNRI, TeCA, NaSSA) (RR 1.48; 95% CI 0.59 to 3.72; 8 studies, 556 participants). There was low heterogeneity in the data (I2 = 14%) (Analysis 2.2).
2.2. Analysis.

Comparison 2 Tricyclic antidepressants versus another medication, Outcome 2 Acceptability.
Secondary outcomes
4.3 Anxiety
There were no differences in levels of anxiety between TCAs and NGAs (NaSSA, SNRI, SSRI) (SMD 0.37; 95% CI ‐0.21 to 0.95; 4 studies; 255 participants) (Analysis 2.3). There was considerable heterogeneity in the data (I2 = 80%). The SMD changed slightly and became significant in favour of NGAs when we used a fixed‐effect model (SMD 0.36; 95% CI 0.10 to 0.62; I2 = 80%).
2.3. Analysis.

Comparison 2 Tricyclic antidepressants versus another medication, Outcome 3 Anxiety (post‐treatment score on self report and clinician‐rated scales).
4.4 Depression
There were no differences between TCAs and NGAs (NaSSA, SNRI, TeCA) for reducing depressive symptoms (SMD 0.17; 95% CI ‐0.07 to 0.40; 6 studies; 395 participants). There was low heterogeneity in the data (I2 = 29%) (Analysis 2.4).
2.4. Analysis.

Comparison 2 Tricyclic antidepressants versus another medication, Outcome 4 Depression (post‐treatment score on self report and clinician‐rated scales).
4.5 Dysfunctional cognitions, emotions, and behaviours
We found no data comparing TCAs with NGAs for dysfunctional cognitions, emotions, and behaviours.
4.6 Adverse effects
The drop‐out rate due to adverse effects ranged from 0% to 20.0% with TCAs versus 0% to 5.71% with NGAs (NaSSA, SNRI, SSRI, TeCA) (see Characteristics of included studies table). There was no significant difference between groups for the number of drop‐outs due to adverse effects (RR 2.37; 95% CI 0.39 to 14.28; 8 studies; 556 participants). There was moderate heterogeneity in the data (I2 = 40%) (Analysis 2.5).
2.5. Analysis.
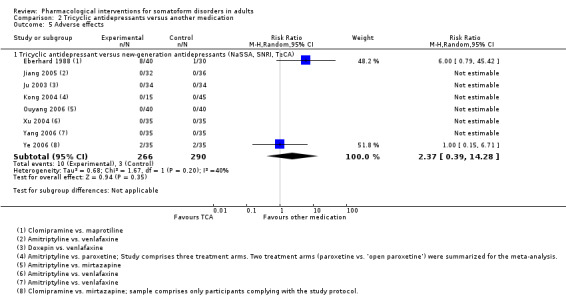
Comparison 2 Tricyclic antidepressants versus another medication, Outcome 5 Adverse effects.
4.7 Treatment response
Compared with the NGAs (NaSSA) group, there was no difference in the percentage of participants who achieved remission when taking a TCA (RR 0.93; 95% CI 0.73 to 1.19; 2 studies; 130 participants; definition treatment response: Ye 2006/Zhao 2006: 'full recovery' or 'significant improvement' on the CGI ‐ Improvement Scale). There was consistency in the data (I2 = 0%) (Analysis 2.6).
2.6. Analysis.

Comparison 2 Tricyclic antidepressants versus another medication, Outcome 6 Treatment response (post‐treatment score on self report and clinician‐rated scales).
4.8 Functional disability and quality of life
We found no data comparing TCAs with NGAs for functional disability and quality of life.
5. New‐generation antidepressants versus another medication
Primary outcomes
Five studies (336 participants) provided data for comparing NGAs with another medication (TCAs, NGAs, NSAIDs). In the following analyses, the experimental condition of included studies was the trial arm that was defined as the experimental arm in the trial report.
5.1 Severity/intensity of medically unexplained physical symptoms
See results for the comparison TCA versus NGAs in section 4 above (tricyclic antidepressants versus another medication) and Analysis 2.1.
There were no significant differences for reducing the severity of MUPS between NGAs (NaSSA, SSRI) and another NGA (NRI, SNRI, SSRI) (SMD ‐0.16; 95% CI ‐0.45 to 0.14; 4 studies; 182 participants). There was consistency in the data (I2 = 0%) (Analysis 3.1; Figure 6.
3.1. Analysis.

Comparison 3 New‐generation antidepressants (SARI, NaSSA, SSRI) versus another medication, Outcome 1 Severity/intensity of medically unexplained physical symptoms (post‐treatment score on self report scales).
6.

Forest plot of comparison: 3 New‐generation antidepressants (serotonin antagonist and reuptake inhibitors (SARI), noradrenergic and specific serotonergic antidepressant (NaSSA), selective serotonin reuptake inhibitor (SSRI)) versus other new‐generation antidepressants, outcome: 3.1 Reduction of the level of severity/intensity of medically unexplained physical symptoms (post‐treatment score on self report scales).
5.2 Acceptability
Rates of attrition rates comparing NGAs with another medication were:
NGAs versus TCAs: see results for the comparison TCAs versus NGAs in section 4 above (tricyclic antidepressants versus another medication) (Analysis 2.2);
NGAs (NaSSA, SSRI) versus another NGAs (NRI, SNRI): 22.0% to 35.3% with NGAs (NaSSA, SSRI) versus 28.9% to 50.0% with NGAs (NRI, SNRI);
NGA (SARI) versus NSAID (ibuprofen): 0% with NGA (SARI) versus 0% with NSAID.
See also Table 9. Information about the RR comparing NGAs versus TCAs for acceptability is given in section 4 above (tricyclic antidepressants versus another medication) (Analysis 2.2). Four studies contributed sufficient data to compare the acceptability rate between NGAs (NaSSA, SSRI) and another NGAs (NRI, SNRI, SSRI), which found no significant difference between groups (RR 0.92; 95% CI 0.60 to 1.40; 4 studies; 182 participants). There was consistency in the data (I2 = 0%) (Analysis 3.2).
3.2. Analysis.

Comparison 3 New‐generation antidepressants (SARI, NaSSA, SSRI) versus another medication, Outcome 2 Acceptability.
Secondary outcomes
5.3 Anxiety
See results for the comparison NGAs versus TCAs in section 4 above (tricyclic antidepressants versus another medication) (Analysis 2.3). We found no data comparing the efficacy of NGAs with forms of medication other than TCAs.
5.4 Depression
See results for the comparison NGAs versus TCAs in section 4 above (tricyclic antidepressants versus another medication) (Analysis 2.4).
We identified one study comparing an NGA (SARI) with an NSAID, which demonstrated that SARI was significantly more effective in reducing depressive symptoms compared with NSAID (SMD ‐3.87; 95% CI ‐4.44 to ‐3.31; 140 participants) (Analysis 3.3) (Wang 2003).
3.3. Analysis.

Comparison 3 New‐generation antidepressants (SARI, NaSSA, SSRI) versus another medication, Outcome 3 Depression (post‐treatment score on self report and clinician‐rated scales).
Three studies compared an NGA (NaSSA, SSRI) with another NGA (NRI, SNRI, SSRI). There was no significant difference between groups (SMD 0.41; 95% CI 0 to 0.82; 3 studies; 169 participants) (Analysis 3.3). There was moderate heterogeneity in the data (I2 = 36%). The SMD and its CI changed only slightly when we used a fixed‐effect model but became significant in favour of the other NGA (NRI, SNRI, SSRI) (SMD 0.46; 95% CI 0.15 to 0.77; I2 = 36%).
5.5 Dysfunctional cognitions, emotions, and behaviours
We found no data comparing an NGA (NaSSA, SSRI) with another NGA (NRI, SNRI, SSRI) for dysfunctional cognitions, emotions, and behaviours.
5.6 Adverse effects
Rates of attrition due to adverse effects of the treatment were:
NGAs (NaSSA, SNRI, SSRI, TeCA) versus TCAs: see results for the comparison TCAs versus NGAs in section 4 above (tricyclic antidepressants versus another medication) (Analysis 2.5);
NGA (NaSSA, SSRI) versus another NGA (NRI, SNRI, SSRI): 6.0% to 27.27% with NGA (NaSSA, SSRI) versus 0% to 22.22% with NGA (NRI, SNRI, SSRI);
NGA (SARI) versus NSAID: 0% with NGA (SARI) versus 0% with NSAID.
See also the Characteristics of included studies table for detailed information about adverse effects in the single studies. Four studies (196 participants) provided sufficient data to compare the attrition rates due to adverse events between NGAs (NaSSA, SSRI) and other NGAs (NRI, SNRI, SSRI). The effect size indicated no significant difference between groups (RR 0.84; 95% CI 0.35 to 2.03). There was consistency in the data (I2 = 0%) (Analysis 3.4).
3.4. Analysis.
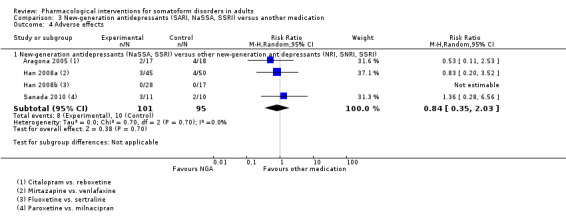
Comparison 3 New‐generation antidepressants (SARI, NaSSA, SSRI) versus another medication, Outcome 4 Adverse effects.
5.7 Treatment response
See results for the comparison NGAs versus TCAs in section 4 above (tricyclic antidepressants versus another medication) (Analysis 2.6).
5.8 Functional disability and quality of life
We found no data comparing NGAs with another medication for functional disability and quality of life.
Comparison 3: pharmacotherapy versus a combination of medications
The literature search identified three studies (163 participants) where a single antidepressant treatment was compared with a combined treatment of an antidepressant and an AP. In two of these studies, the AP was combined with an SSRI (Huang 2012; Li 2006), and in the other study, a TCA was combined an AP (Zitman 1991).
Primary outcomes
6.1 Severity/intensity of medically unexplained physical symptoms
We identified one study comparing a TCA alone versus TCA plus AP, which found no significant difference between groups (SMD 0.26; 95% CI ‐0.42 to 0.94; 34 participants) (Zitman 1991). We found two studies comparing SSRI alone versus SSRI plus AP. SSRI plus AP had a significantly higher effect than SSRI alone (SMD 0.77; 95% CI 0.32 to 1.22; 2 studies, 107 participants). There was a low level of heterogeneity in the data (I2 = 23%) (Analysis 4.1).
4.1. Analysis.

Comparison 4 Pharmacotherapy versus a combination of medications, Outcome 1 Severity/intensity of medically unexplained physical symptoms (post‐treatment score on self report scales).
6.2 Acceptability
The studies comparing an antidepressant alone with antidepressant plus AP found attrition rates of:
SSRI alone versus SSRI plus AP: 7.1% to 30.0% with SSRI alone versus 0% to 30.0% with SSRI plus AP;
TCA alone versus TCA plus AP: was not calculated due to missing data.
See also Table 9. There was no significant difference in the acceptability rate between SSRI alone and SSRI plus AP (RR 0.80; 95% CI 0.25 to 2.52; 2 studies, 118 participants). There was low heterogeneity in the data (I2 = 16%) (Analysis 4.2).
4.2. Analysis.

Comparison 4 Pharmacotherapy versus a combination of medications, Outcome 2 Acceptability.
Secondary outcomes
6.3 Anxiety
There was no significant difference between an SSRI plus AP and SSRI alone (SMD 0.95; 95% CI ‐0.91 to 2.82; 2 studies, 107 participants) (Analysis 4.3). There was considerable heterogeneity in the data (I2 = 95%). The effect size was significant when calculated using a fixed‐effect model (SMD 0.73; 95% CI 0.32 to 1.15; I2 = 95%).
4.3. Analysis.

Comparison 4 Pharmacotherapy versus a combination of medications, Outcome 3 Anxiety (post‐treatment score on self report and clinician‐rated scales).
6.4 Depression
We identified one study comparing a TCA alone versus TCA plus AP for depression (Zitman 1991). There was a significant benefit of TCA plus AP compared with TCA alone (SMD 0.79; 95% CI 0.09 to 1.49; 34 participants) (Analysis 4.4). There was no significant difference between SSRI plus AP and SSRI alone (SMD 0.58; 95% CI ‐0.33 to 1.48; 2 studies, 107 participants) (Analysis 4.4). There was considerable heterogeneity in the data (I2 = 81%). The effect size did not change considerably when we used a fixed‐effect model, but did reach a significant level in favour of SSRI plus AP (SMD 0.53; 95% CI 0.14 to 0.93).
4.4. Analysis.

Comparison 4 Pharmacotherapy versus a combination of medications, Outcome 4 Depression (post‐treatment score on self report and clinician‐rated scales).
6.5 Dysfunctional cognitions, emotions, and behaviours
We found no data comparing pharmacotherapy versus a combination of medications for dysfunctional cognitions, emotions, and behaviours.
6.6 Adverse effects
Drop‐out rates due to adverse effects were 0% to 10.0% with SSRI alone versus 0% to 16.7% with SSRI plus AP. In order to calculate a RR, only the study authors of Huang 2012 provided data. The study by Li 2006 had no drop‐outs in either group and, therefore, could not be used for RR calculations. For the study by Huang 2012, there was no significant difference in drop‐outs related to adverse effects between SSRI plus AP and SSRI alone (RR 0.60; 95% CI 0.16 to 2.29; 60 participants). For the study by Zitman 1991 comparing TCA plus AP with TCA alone, we could not calculate an RR value because of missing data. We could not contact the authors of the study, as the study was too old.
6.7 Treatment response
We identified one study comparing SSRI plus AP with SSRI alone, which found no significant difference between groups for treatment response (RR 1.70; 95% CI 0.94 to 3.08; 60 participants; definition of treatment response: 50% reduction on the SOMS‐7; Analysis 4.6) (Huang 2012).
4.6. Analysis.

Comparison 4 Pharmacotherapy versus a combination of medications, Outcome 6 Treatment response (post‐treatment score on self report and clinician‐rated scales).
6.8 Functional disability and life quality
We found no data comparing pharmacotherapy versus a combination of medications for functional disability and life quality.
Subgroup analyses
We conducted all subgroup analyses that were originally planned in the review protocol with the only exception of the analysis 'gender'. All included studies had both men and women in their samples.
Subgroup analysis 1: co‐morbidity (based on pharmacotherapy versus placebo)
Table 7 gives an overview of the studies where information on co‐morbid mental disorders was provided.
1.1 Severity/intensity of medically unexplained physical symptoms
In the included studies with participants having co‐morbid mental disorders there was a significant effect in favour of pharmacotherapy (SMD ‐0.91; 95% CI ‐1.36 to ‐0.46; 3 studies; 243 participants; random‐effects model). However, there was substantial heterogeneity in this subgroup (I2 = 63%). A test for subgroup differences could not be applied because we identified only one study in the subgroup of no co‐morbid mental disorder that found no significant difference between pharmacotherapy and placebo (SMD ‐0.07; 95% CI ‐0.35 to 0.21; 200 participants) (Analysis 5.1) (Volz 2000).
5.1. Analysis.

Comparison 5 Subgroup analysis 1: Co‐morbidity (based on the comparison pharmacotherapy versus placebo), Outcome 1 Severity/intensity of medically unexplained physical symptoms (post‐treatment score on self report scales).
1.2 Acceptability
The studies comparing pharmacotherapy with placebo in participants with co‐morbid mental disorders found all‐cause attrition rates to be 4.0% to 38.2% with pharmacotherapy versus 0% to 38.6% with placebo. The studies in participants with no co‐morbid mental disorders found all‐cause attrition rates to be 8.1% to 13.5% with pharmacotherapy versus 8.2% to 12.4% with placebo (Table 9). There were no significant differences on acceptability rates between pharmacotherapy and placebo for participants with or without co‐morbid mental disorders (with co‐morbid mental disorders: RR 1.01; 95% CI 0.64 to 1.61, 2 studies; 163 participants; with no co‐morbid mental disorders: RR 1.05; 95% CI 0.59 to 1.89; 2 studies; 390 participants). There was consistency in the data in both groups (with co‐morbidity mental disorders: I2 = 0%; with no co‐morbidity mental disorders: I2 = 0%). The test of differences between subgroups indicated no significant effect (I2 = 0%, Chi2 = 0.01, degrees of freedom (df) = 1, P value = 0.920) (Analysis 5.2).
5.2. Analysis.

Comparison 5 Subgroup analysis 1: Co‐morbidity (based on the comparison pharmacotherapy versus placebo), Outcome 2 Acceptability.
Subgroup analysis 2: source of funding (based on pharmacotherapy versus placebo)
The Characteristics of included studies table provided information about the funding of the included studies or if study authors were involved in pharmaceutical industry.
2.1 Severity/intensity of medically unexplained physical symptoms
In the subgroup comprising studies with funding by pharmaceutical industry there was a significant effect to reducing the severity and intensity of MUPS in favour of pharmacotherapy (with funding: SMD ‐0.64; 95% CI ‐1.02 to ‐0.27; 5 studies; 685 participants; random‐effects model; with no funding SMD ‐0.75; 95% CI ‐1.29 to ‐0.21; 2 studies; 119 participants; random‐effects model). In the subgroup with funding, there was considerable heterogeneity in the data (I2 = 81%), whereas in the group with no funding, there was moderate heterogeneity in the data (I2 = 48%). The test of subgroup differences revealed no significant effect (I2 = 0%, Chi2 = 0.10, df = 1, P value = 0.760) (Analysis 6.1).
6.1. Analysis.
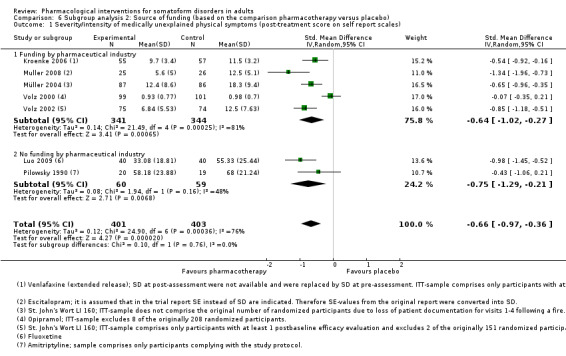
Comparison 6 Subgroup analysis 2: Source of funding (based on the comparison pharmacotherapy versus placebo), Outcome 1 Severity/intensity of medically unexplained physical symptoms (post‐treatment score on self report scales).
2.2 Acceptability
The studies with funding from pharmaceutical industry reported the attrition rate to be 0% to 38.2% with pharmacotherapy versus 0% to 38.6% with placebo. There was only one study with no financial support where we could calculate the attrition rate for every study arm (Pilowsky 1990). In this study, the attrition rate was 25.0% for pharmacotherapy and 31.0% for placebo (Table 9). In studies with funding by the pharmaceutical industry, there was no significant difference in the acceptability rate between pharmacotherapy and placebo (RR 0.99; 95% CI 0.70 to 1.40; 6 studies; 877 participants). There was consistency in the data (I2 = 0%) (Analysis 6.2). We could not calculate subgroup differences of acceptability because we identified no placebo‐controlled study with no financial support from pharmaceutical industry.
6.2. Analysis.

Comparison 6 Subgroup analysis 2: Source of funding (based on the comparison pharmacotherapy versus placebo), Outcome 2 Acceptability.
Subgroup analysis 3: source of outcome rating (based on pharmacotherapy versus placebo)
3.1 Severity/intensity of medically unexplained physical symptoms
In the subgroup comprising self report scales, there was a significant effect size in favour of pharmacotherapy compared with placebo (SMD ‐0.66; 95% CI ‐0.97 to ‐0.36; 7 studies; 804 participants; random‐effects model). There was considerable heterogeneity in the data (I2 = 76%). In the subgroup comprising clinician‐rated scales, there was a high, significant effect size in favour of pharmacotherapy (SMD ‐0.91; 95% CI ‐1.42 to ‐0.40; 4 studies; 573 participants; random‐effects model). There was considerable heterogeneity in the data (I2 = 87%). The test of subgroup differences revealed no significant difference between data based on self report and clinician‐rated scales (I2 = 0%, Chi2 = 0.66, df = 1, P value = 0.420) (Analysis 7.1).
7.1. Analysis.

Comparison 7 Subgroup analysis 3: Source outcome rating (based on the comparison pharmacotherapy versus placebo), Outcome 1 Severity/intensity of medically unexplained physical symptoms (post‐treatment score).
3.2 Acceptability
This subgroup analysis could only be conducted for outcomes that were assessed by self report or clinician‐rated measures but not for the acceptability rate.
Sensitivity analyses
For reasons of clarity and comprehensibility most of the sensitivity analyses were reported in the sections of 'Comparison 1: pharmacotherapy versus placebo', 'Comparison 2: pharmacotherapy versus another medication', and 'Comparison 3: pharmacotherapy versus a combination of medications'.
Best‐case/worst‐case analysis (based on outcome 'treatment response')
Comparison: pharmacotherapy versus placebo
In order to deal with missing data in analyses of dichotomous outcomes we conducted best‐case/worst‐case scenarios. For the comparison TCA versus placebo, there were not enough studies available for conducting this analysis.
NGA versus placebo: the first analysis that was based on complete cases showed that there was no significant difference between NGA and placebo (RR 2.02; 95% CI 0.82 to 4.97; 2 studies; 119 participants; random‐effects model). There was considerable heterogeneity in the data (I2 = 84%). The second analysis was based on a best‐case scenario, which means that drop‐outs in the active treatment group were assumed to have positive outcomes and drop‐outs in the control group were assumed to have negative outcomes. Results showed that NGA was significantly more effective than placebo (RR 2.59; 95% CI 1.90 to 3.53; 2 studies; 163 participants; random‐effects model). There was high consistency in the data (I2 = 0%). The third analysis was based on a worst‐case scenario, which means that drop‐outs in the active treatment group had negative outcomes and drop‐outs in the control group had positive outcomes. Results showed that there was no significant difference between NGA and placebo (RR 1.41; 95% CI 0.30 to 6.63; 2 studies; 163 participants; random‐effects model). There was considerable heterogeneity in the data (I2 = 95%) (Analysis 8.1). In summary, the complete cases as well as the worst‐cases scenario demonstrated no differences between NGAs and placebos whereas the best‐case scenario did.
8.1. Analysis.

Comparison 8 Best‐case/worst‐case analysis (based on comparison 'new‐generation antidepressants versus placebo'), Outcome 1 Treatment response (post‐treatment score on self report and clinician‐rated scales).
NP versus placebo: there was a statistically significant effect in favour of NP for the complete‐case analysis (RR 1.75; 95% CI 1.29 to 2.36; 2 studies; 311 participants; random‐effects model), the best‐case analysis (RR 1.92; 95% CI 1.26 to 2.94; 2 studies; 324 participants; random‐effects model), and the worst‐case analysis (RR 1.57; 95% CI 1.27 to 1.93; 2 studies; 324 participants; random‐effects model). In the complete‐case analysis there was moderate heterogeneity (I2 = 33%), in the best‐case analysis there was substantial heterogeneity (I2 = 63%), and in the worst‐case consistency there was consistency in the data (I2 = 0%) (Analysis 9.1).
9.1. Analysis.

Comparison 9 Best‐case/worst‐case analysis (based on comparison 'natural products versus placebo'), Outcome 1 Treatment response (post‐treatment score on self report and clinician‐rated scales).
Comparison: pharmacotherapy versus other medication
There were not enough data available for comparing pharmacotherapy and placebo in a sensitivity analysis.
Comparison: pharmacotherapy versus combination of medication
There were not enough data available for comparing pharmacotherapy and placebo in a sensitivity analysis.
Reporting bias
Originally, we planned to assess for a publication bias by implementing funnel plots. However, creating funnel plots would only make sense for the medication versus placebo comparisons. It would be difficult to interpret studies where a medication was compared with another medication. For the comparison of medication versus placebo we included only eight studies for meta‐analytical calculations, which is below the number of trials recommended by the Cochrane Handbook for Systematic Reviews of Interventions (there should be at least 10 studies) (Sterne 2011). However, in order to prevent publication bias, we made every attempt to include unpublished trials (e.g. by searching online trial registries or registries of unpublished doctoral theses).
Studies that did not contribute data that allowed them to be included in the meta‐analysis
The following study could only contribute to the narrative literature part of the review. For detailed information about study, see the Characteristics of included studies table.
Altamura 1991
This randomised, parallel‐group, double‐blind, fixed‐dose trial included two arms comparing the efficacy of two APs ‐ levosulpiride and racemic sulpiride in 30 participants with somatisation disorder or undifferentiated somatoform disorder (Altamura 1991). We could not include the study in our meta‐analysis because all results presented graphically. We contacted the trial authors but they replied that the study was too old and that they no longer had access to the data. Thus, we have summarised the most important findings of this study narratively. The severity of MUPS was assessed with the somatisation subscore of the SCL‐90. There were no significant differences between groups. Starting in the treatment week two, there was a significant decrease of somatic symptoms with every further week until the end of treatment in both groups. Depressive symptoms (depression subscore of the SCL‐90) started to decrease significantly from the second treatment week until the end of treatment. With anxiety (using HARS), the sulpiride group obtained a significant decrease of symptoms in week two whereas the racemic sulpiride group revealed a decrease of symptoms in week one. In both groups, this reduction of anxiety remained stable until the end of treatment. In both treatment arms, there was a drop‐out rate of 13.3%. Extrapyramidal and anticholinergic adverse effects were more frequent with racemic sulpiride.
Discussion
Summary of main results
See also a summary of the main results for the main comparison pharmacotherapy versus placebo in Table 1; Table 2; and Table 3; and for the comparisons of 'TCAs versus another medication' (Table 4) and 'pharmacotherapy versus a combination of medications' (Table 5).
Comparison 1: pharmacotherapy versus placebo
Tricyclic antidepressants versus placebo
Evidence from two studies in 239 people contributing data to the primary outcomes of this review showed that the TCAs given for between six and 12 weeks did not reduce the severity of MUPS more than placebo. There were not enough data available to conduct meta‐analyses for our secondary outcomes. The attrition rates due to adverse effects and the rates of all‐case drop‐outs in these studies were comparable between medication and placebo group. In summary, the evidence for the efficacy of a TCA in comparison to a placebo is low quality. Statements about efficacy are only possible for the severity of MUPS, but not secondary outcomes.
New‐generation antidepressants versus placebo
Results from three studies with 243 people showed that the severity of MUPS could be reduced significantly by treatment with NGAs for between eight and 12 weeks compared with placebo. Evidence from two studies with 163 people demonstrated that administering NGAs for 12 weeks significantly reduced depressive symptoms and functional disability, while increasing quality of life. For anxiety symptoms and treatment response, there were no differences between NGAs and placebo. In summary, the impact of NGAs compared with placebo was inconsistent across outcomes at post‐treatment. For an adequate interpretation, it has to be noted that the results for NGAs had high levels of heterogeneity. Furthermore, the analyses included one study with outlier values (Muller 2008). The exclusion of this study in a sensitivity analysis did not change the SMD considerably. It still remained significant in favour of an impact of NGAs on the severity of MUPS. Results did not considerably change under further sensitivity analyses. However, best‐case/worst‐case analysis did cast some doubt over the results. The attrition rates in these studies were comparable between NGAs and placebo groups, as well as the number of drop‐outs due to adverse effects of treatment.
Natural products versus placebo
Two studies with 322 people showed that treatment with an NP over six weeks significantly reduced the severity of MUPS and led to a significantly higher number of participants with symptom remission. Evidence from two studies with 321 participants contributing data to the secondary outcomes showed that NPs given for two to six weeks significantly reduced anxiety and depressive symptoms. Results did not change considerably under sensitivity analyses and were confirmed by the best‐case/worst‐case analysis. For functional disability and quality of life, we did not find enough data to conduct a meta‐analysis. Attrition rates due to all causes and due to adverse effects of treatment were quite low and similar in NP and placebo groups.
Comparison 2: pharmacotherapy versus another medication
Tricyclic antidepressants versus another medication
There were only enough studies available for the comparison between TCAs and NGAs. Results were based on 10 studies with 616 participants contributing data partly to primary and secondary outcomes of this review. The central finding was that TCAs administered over four to 17 weeks were as effective as administering NGAs at reducing the severity of MUPS, at decreasing depressive and anxiety symptoms, and in symptom remission. Results did not change considerably under sensitivity analyses. The attrition rates due to all causes and due to adverse effects were about three times as high with TCA compared with NGA. However, due to the considerable levels of heterogeneity in the data this difference between the study conditions was not statistically significant.
New‐generation antidepressants versus another medication
In addition to the studies comparing NGAs with TCAs (see summary of the findings of this analysis in section 4 above (tricyclic antidepressants versus another medication), we found four studies with 196 participants comparing the efficacy of one NGA with another NGA. Evidence from this analysis showed that different NGAs taken over eight to 12 weeks were comparably effective at reducing the severity of MUPS depressive symptoms. The attrition rates due to all causes and due to adverse effects were similar for the different types of NGAs.
Comparison 3: pharmacotherapy versus a combination of medications
For the comparison of a TCA alone versus a TCA plus AP, we found only one study (Zitman 1991). However, for the comparison of SSRIs alone versus SSRIs plus AP over six to eight weeks, evidence was based on two studies with 118 people contributing data partly to the primary and secondary outcomes of this review. The findings showed that the combined treatment was more successful in reducing the intensity of MUPS and depressive symptoms than single pharmacotherapy. Both treatments were similarly effective at decreasing anxiety. The attrition rates due to all causes and due to adverse effects were comparable in both treatment groups.
Subgroup analyses
Subgroup analyses for co‐morbid mental disorders of included participants and the funding of trials by pharmaceutical companies did not reveal any significant effects. However, results have to be interpreted with caution due to high heterogeneity. For severity of MUPS, analyses clearly demonstrated a considerably higher effect when the assessment was done with clinician‐rated scales than with self report scales.
Overall completeness and applicability of evidence
The objective of the current review was to assess the effects of pharmacological interventions for somatoform disorders (specifically somatisation disorder, undifferentiated somatoform disorder, somatoform autonomic dysfunction, and pain disorder) in adults. We performed a thorough literature search of different electronic databases and many other resources such as conference proceedings, international trial registers, grey unpublished literature, and reference lists, which resulted in 26 studies that could be included in this review. In comparison to other existing reviews on this topic, this number of eligible studies was quite high (e.g. Kroenke 2007; O'Malley 1999; Sumathipala 2007). Due to the connection of the CCDANCTR database to many Asian journals to which other common databases have no access, we had the opportunity to screen studies that have never been mentioned in existing reviews. However, it has to be emphasised that the provenance of some of the other Chinese studies in the CCDANCTR was less clear and probably dated back to an opportunistic search of Wang Fang Data (c/o The British Library) in 2007. While we ensured searches were run on sources other than the CCDANCTR, we did not run a comprehensive, up‐to‐date search of the Chinese biomedical literature and so the studies analysed in this review may be an incomplete list of relevant research from China and other Asian countries. However, we retrieved a considerable number of studies in order to address our questions.
Our review has several limitations. The studies that we identified focused primarily on antidepressants and our objectives focused on pharmacological interventions in general. A critical issue was that maybe our results were of 'artificial' nature because we focused very specifically on studies with participants with somatoform disorders characterised by chronic MUPS. However, somatoform disorders are conceptually overlapping with functional somatic syndromes (Wessely 1999), which we excluded from the current review. The results of the current review should be interpreted as part of a portfolio of five Cochrane reviews covering somatoform disorders (Hoedeman 2010; Ipser 2009; Ruddy 2005; Thomson 2007), as well as Cochrane reviews focusing on different functional syndromes (e.g. Huertas‐Ceballos 2014; Moore 2012). A further problem of this review was dealing with missing data for meta‐analytic calculations. Although we tried to contact study authors in order to be provided with missing data, we had to exclude one study from the meta‐analytic part of our review because data were no longer available (Altamura 1991). Unfortunately, this study examined a medication class that had to be neglected in the current review due to a lack of studies, that is, APs. Another problem of the current review was that there were not enough studies to assess the reporting bias with funnel plots. Creating funnel plots would only make sense for the medication versus placebo comparisons. Bringing together the studies where a medication was compared with another medication would be very difficult to interpret. For the medication versus placebo comparisons, we could only include eight studies for meta‐analytical calculations. This is below the number of trials recommended by the Cochrane Handbook for Systematic Reviews of Interventions, which states that there should be at least 10 studies (Sterne 2011). We could report adverse effects of the different medication classes in the treatment of somatoform disorders only in a narrative way in the Characteristics of included studies table. Adverse effects were reported in an unstandardised way in all reports. Therefore, it was not possible to code them in a standardised way in order to include them in our meta‐analytical calculations. We have to emphasise again that the inclusion of RCT and CRCT studies is not sufficient to gain information about the more rare or long‐term adverse events. A further consequence of the lack of a sufficient number of studies was that we could not conduct many of our pre‐planned subgroup and sensitivity analyses. These analyses ‐ and potentially additional interesting subgroup analyses (e.g. according to the duration of symptoms) ‐ can hopefully be considered in future updates of this review.
Finally, it should be noted that none of the included studies examined the efficacy of pharmacotherapy over the long term. This means that the trial reports comprised no follow‐up data but only that of post‐treatment assessments. Furthermore, the pharmacological interventions in the included studies had a maximum duration of 12 weeks and can therefore be declared as short‐term treatments. This is particularly since included participants had the MUPS over a long period and it could be considered as a chronic population. It is questionable whether short‐term interventions can really provide a realistic estimation of the efficacy of pharmacotherapy in the sample of people with somatoform disorders.
Quality of the evidence
According to the first quality criterion risk of bias defined by the guidelines by GRADE (Guyatt 2008), Figure 2 and Figure 3 show that for different types of biases, most of our included studies show an unclear or high risk of bias. Although many studies are classified as 'randomised' and 'double‐blind', detailed descriptions of random sequence generation; allocation concealment; blinding of participants and study personnel, and outcome assessment were missing. For example, even if a study described how participants and study personnel were blinded with regard to the study condition, there was no study that attempted to blind participants with regard to adverse effects. Because there was only a small number of studies showing low risk of bias, it was often not possible to conduct sensitivity analyses in order to check for the biases.
Another important quality problem of the included studies was with attrition bias and the way missing data were dealt with. There were several studies that did not define the ITT sample as a sample of originally randomised participants. Usually only participants with at least one post‐baseline efficacy evaluation were included in this population. This procedure can lead to an overestimation of effects. We checked this problem with sensitivity analyses that revealed no impact.
For reporting bias, there was another problem. Although several studies implemented a broad range of different outcomes in the form of self report and clinician‐rated scales, there were also some studies that assessed outcomes that we classified as secondary in our review. An important outcome such as the severity of physical symptoms was neglected in such studies. In addition, there were several studies with a low variety in the form of assessment ‐ self report versus clinician‐rated scales. In our subgroup analyses, we could demonstrate that clinician‐rated scales can lead to higher effect sizes than self report scales. However, we had to be careful with interpreting these results because tests of subgroup differences were not significant and there was high heterogeneity in the data of the subgroups and there are also other studies that could demonstrate this effect (Lambert 1994; Lambert 1986; Rief 2009). Furthermore, the study protocols were not available as publications in an international trial register for any of the included studies. This increases the risk of reporting bias. In addition, reporting bias could not be checked with funnel plots because there was not a sufficient number of studies. One specific problem that appeared in the context of reporting bias was that three trial reports did not report SDs for the post assessment (Han 2008a; Han 2008b; Kroenke 2006). After contacting trial authors and receiving no reply, we replaced these missing SD with SD from the pre‐assessment. We are aware that this procedure can produce bias in the results ‐ in both the form of underestimation or overestimation of the effects. However, it is discussed as a common procedure in the evaluation of therapeutic outcome (Bergin 1971), and appeared to us to be a better alternative than excluding these studies. We used sensitivity analyses to determine the impact of a possible bias but could not identify one.
There are still some other sources of risk of bias in our included studies that have to be considered. For example, there were four studies that implemented a placebo run‐in phase before they started with the treatment. This design feature severely endangers accurate estimates of the placebo response rate (Fournier 2010). Because early placebo responders were removed from the trial before they can contribute data, the true rate of placebo response may be underestimated in trials that use this feature. We checked the impact of this pre‐intervention before the study treatment started in sensitivity analyses, which revealed no considerable impact. Furthermore, compliance of participants in taking their medication was reported only in one trial. Without this compliance check, trial authors cannot ensure that participants complied with the treatment protocol, which in turn can produce biases, especially in pharmacological interventions with considerable adverse effects. Although all included studies were randomised trials there was a small number of studies where significant baseline differences between the study arms in the outcomes appeared. Finally, the funding of studies by the pharmacological industry or the involvement of study authors in pharmacological companies can have an influence on the results. We checked for this bias using sensitivity analyses and could not find any influence for our included studies. However, heterogeneity in the data was very high in the funded studies compared with the non‐funded studies.
With regard to the GRADE criterion 'inconsistency' it must be noted that under the included placebo‐controlled trials there was one study that demonstrated outlier values for almost all outcomes (Muller 2008). Sensitivity analyses showed that the exclusion of this study reduced the level of heterogeneity in the data. The values in the original trial report that were indicated as SD by the trial authors appeared to be very small and too homogenous over the time points of assessment and between the different treatment groups. We assumed that in this trial reported SE and not SD. Therefore, we used the common formula in order to convert SEs to SDs. However, the effect sizes still remained obviously high compared with the remaining studies.
For the GRADE criterion 'imprecision', we had to downgrade the ratings in several cases. The central limitation was that the total (cumulative) sample size was too small or that the 95% CI around the pooled effect included both no effect and appreciable benefit or appreciable harm. Although we identified 26 studies, each comparison only contained a few studies and had a relatively small number of participants.
For the GRADE criterion 'publication bias', it should be emphasised for our review in general that we neglected literature search of biomedical electronic databases of Asian countries. The implications of this limitation are difficult to gauge. Due to a lack of data, we could not conduct sensitivity analyses where Asian studies were excluded.
The criteria of quality defined by GRADE 'indirectness' was less of a problem in our included studies. Finally, it has to be noted that several of the included studies were performed before the publication and implementation of current quality criteria for conducting and reporting RCTs (Moher 2001).
Potential biases in the review process
Although this review has several strengths, such as a pre‐published protocol, an experienced librarian who performed thorough searches, the collection of data from various sources, two review authors to select studies/data extract/assess risk of bias, and a third review author to resolve disputes, there were also post hoc decisions in the review process that could have had a bias effect. The management of antidepressant classes was one. For example, it would be logical to consider SSRIs as an NGA separately from the other NGAs. This is because SSRIs build an important medication class that is often used off‐label for treating somatoform pain. However, for the medication versus placebo comparisons, there were not enough studies available to calculate analyses separately for SSRIs. Therefore, we decided to combine SSRI studies and trials on other NGAs for all meta‐analytic calculations.
Another problem appeared with the high number of included Asian studies (40.0%). Research on mental and somatic disorders in various cultures shows cross‐national differences occurring in somatic distress. Although the patterns of these differences seem not to follow clear cultural lines, the role of culture cannot be excluded (Gureje 2004). Research on cultural aspects shows that there are differences in experiencing, presenting, and coping with physical symptoms between Asian and Western cultures. For example, one study demonstrated that Chinese outpatients reported more somatic symptoms on spontaneous problem report and structured clinical interview compared with Euro‐Canadians, who in turn reported more psychological symptoms (Ryder 2008).
It appears that other factors are also associated with the cultural background of the studies. For example, whereas an outpatient setting was chosen in almost all studies that were conducted in Western cultures, an inpatient setting was used only in the Asian studies. This factor maybe confounded with the attrition rate. In an inpatient setting, the barrier to withdraw study participation is maybe higher than in outpatient settings. Interestingly, there were several studies with no drop‐outs in each study arm and these studies were all Asian studies. Furthermore, although a high proportion of our included studies was of Asian origin, we cannot claim that this review is a complete list of relevant research from China and other Asian countries because, while we ensured searches were run on sources other than the CCDANCTR, we did not run a comprehensive, up‐to‐date search of the Chinese biomedical literature and so the studies analysed in this review may be an incomplete list of relevant research from China and other Asian countries. We also have to emphasise that we had no restrictions regarding language for including studies. This means that of the included studies, we had to translate one Italian and eight Chinese articles. There were three translators who supported us. All of them were native speakers in the language in which the article was written and lived for a longer time in a country with English as principal language. Furthermore, they were all graduate clinical psychologists. However, they were not specialist in pharmacotherapy.
Another post hoc decision in the review process that could have a bias effect was the combination of study arms with different NP combinations (Melzer 2009), or with the same medication administered in different settings (Kong 2004). We did this combination of study arms in order to prevent a unit‐of‐analysis error.
Agreements and disagreements with other studies or reviews
We could not find an existing meta‐analysis; however, we identified four literature reviews that were very close to the topic of our review (Fallon 2004; Kroenke 2007; Somashekar 2013; Sumathipala 2007). Fallon 2004 identified only one open‐label study on the pharmacological treatment of people with a variety of somatoform disorders. The authors concluded that there was a research gap regarding pharmacological interventions for people with primarily somatic cluster of somatoform disorders. Kroenke 2007 included four RCTs on the pharmacological treatment of multiple MUPS (antidepressants and NPs). The authors found that three of the four pharmacological trials demonstrated efficacy of the medication. The authors concluded that preliminary but not conclusive evidence existed for antidepressant treatment of MUPS. Somashekar 2013 examined the efficacy of antidepressants in somatoform and related disorders. The authors concluded that there was evidence that antidepressants with both serotonergic and noradrenergic activity were more effective than SSRIs for somatoform pain disorder. Furthermore, they stated that although TCAs, especially amitriptyline, have been commonly used in clinical practice, it is not systematically evaluated specifically in somatic symptoms disorders. Sumathipala 2007 described the results of the review by O'Malley 1999 (see below). Sumathipala 2007 concluded that there was evidence for the effectiveness of antidepressants available for different subgroups of somatoform disorders, but that there was not much evidence on other medications.
We found one meta‐analysis and two other reviews that focused on specific functional syndromes. These publications also have important parallels with our review. The review and meta‐analysis by O'Malley 1999 examined the efficacy of antidepressant therapy on six symptom syndromes: headache, fibromyalgia, functional gastrointestinal syndromes, idiopathic pain, tinnitus, and chronic fatigue. They judged the quality of the included studies as fair. They identified studies focusing on TCAs, anti‐serotonin antidepressants, SSRIs, or multiple agents. They quoted an SMD of 0.87 (95% CI 0.59 to 1.14). Unfortunately, it is unclear to which outcome this SMD refers. The authors found high drop‐out rates (40% of the included studies had drop‐out rates greater than 20%). Jackson 2006a examined the efficacy of antidepressant therapy in 11 somatic syndromes (IBS, chronic back pain, headache, fibromyalgia, chronic fatigue syndrome, tinnitus, menopausal symptoms, chronic facial pain, non‐cardiac chest pain, interstitial cystitis, and chronic pelvic pain). The authors concluded that there was good RCT evidence of benefit from antidepressant therapy for headaches, fibromyalgia, and IBS. The evidence of improvement with antidepressant therapy was rather weak for back pain and chronic fatigue syndrome. The authors criticise many studies of somatic syndromes that have used subtherapeutic doses of antidepressants for relatively short durations, making it less likely that the benefit is entirely due to antidepressant properties of these drugs. The systematic review by Raine 2002 primarily focused on mental healthcare interventions for people with common somatic symptoms in general (chronic fatigue syndrome, IBS, chronic back pain). They identified 18 trials that investigated the efficacy of TCA, TeCA, anxiolytic plus TCA, SSRIs, and MAOIs. The authors concluded that antidepressants seem to be effective in IBS but ineffective in chronic fatigue syndrome.
In summary, these reviews and the one meta‐analysis suggested that particularly antidepressants might be effective in the treatment of MUPS. They emphasise that there is not much research on the efficacy of medications other than antidepressants. Furthermore, the quality of the studies was judged as moderate. Drop‐out rates were high. Therefore, the results of these reviews were consistent with our findings.
Authors' conclusions
Implications for practice.
The results of this review demonstrated only low or very low quality evidence for the efficacy of pharmacological interventions for somatoform disorders in adults. This evidence is limited and refers only to two specific classes of pharmacological interventions: new‐generation antidepressants (NGAs) and natural products (NPs). Placebo‐based comparisons demonstrated consistent efficacy only for NPs for diverse primary and secondary outcomes. For NGAs, the evidence was very inconsistent across outcomes. Findings showed no difference between TCAs and placebo for the severity of MUPS but there was not enough evidence to assess any other outcomes. A small number of studies compared different medications for somatoform disorders. The findings of these studies suggested that NGAs seemed to be as effective as TCAs; however, the evidence was of low to very low quality. Furthermore, the results showed that different NGAs seem to be similarly effective for diverse outcomes. Finally, meta‐analyses on studies comparing selective serotonin‐reuptake inhibitors (SSRIs) with combined treatments of SSRIs and antipsychotics suggest that there could be a benefit of a combination of medications only for reducing the severity of MUPS and depression. For other outcomes, such as anxiety symptoms, no effects of the study arm were identified. Here again the evidence was of low or very low quality.
Overall, these results have to be considered with caution given several shortcomings associated with the included studies, such as the imprecision of the data (due to the small numbers of studies and sample sizes contributing to each comparison), the bias possibly produced by cultural factors and the inclusion of a high number of Asian studies, or the lack of studies with follow‐up assessments (see also Table 1Table 2; Table 3; Table 4; and Table 5). Furthermore, the significant effects of antidepressant treatment have to be balanced against the relatively high rates of adverse effects. For people experiencing somatic symptoms without medical causes, adverse effects of medication can have amplifying effects on symptom perceptions. In addition, drop‐out rates as indicators of a low treatment acceptability were quite high for the different antidepressants compared with NPs.
Implications for research.
This review revealed that the evidence base is lacking in studies that examine the efficacy of medication class (such as antipsychotics) other than antidepressants or NPs for somatoform disorders in adults. Therefore, research on other medication classes in the treatment of somatoform disorders and chronic medically unexplained physical symptoms (MUPS) should be facilitated. Furthermore, there were some implications particularly regarding antidepressant trials. There was not enough evidence or the existing evidence was not of good enough quality to support the common off‐label use of antidepressants ‐ especially TCAs ‐ in the treatment of somatoform disorders and MUPS. This review only partly allowed conclusions to be made about differences in the efficacy between different chemical agents based on included randomised controlled trials (RCTs) where different medications were compared. However, with this low number of studies per medication class, and the sometimes high heterogeneity in the data, the findings from our review have to be interpreted with caution. Replication studies comparing different antidepressant agents ‐ NGAs but also TCAs ‐ would be desirable and should ensure that they address the elements of bias that we found for existing antidepressant trials. Furthermore, an important limitation of the included studies was the lack of examining the long‐term effects of pharmacological interventions. This would be very important especially for the comparability with other interventions for somatoform disorders for which long‐term stability of the effects has already been confirmed (e.g. cognitive‐behavioural therapy; Kleinstäuber 2011).
For several outcomes, only a small number studies was available (e.g. functional impairment or cognitive/emotional/behavioural symptoms). Therefore, another idea for future research would be to include other outcomes such as functional impairment or dysfunctional behaviours and cognitions as well as the classical outcomes such as symptom severity, depression, and anxiety. This is also important for the comparison of pharmacological interventions with other interventions. In this context, it would be also important to push forward research of the impact of different modes of measures on the results in studies on the efficacy of pharmacotherapy for somatoform disorders. An important research question could be, for example, if clinician ratings overestimate improvement. In addition, more research is needed on the mechanisms of efficacy in the different medication classes. Research shows that somatoform disorders have a high co‐morbidity with other mental disorders such as depression or anxiety disorders. This, and the fact that medications that were originally developed for the treatment of such co‐morbid disorders are administered 'off‐label' to treat somatoform syndromes, could motivate research on connections between MUPS and co‐morbid mental disorders. Such research could provide important information about the efficacy mechanisms of 'off‐label' pharmacotherapy in somatoform disorders. Typical examples could be immunoregulatory mechanisms that could play a role in somatoform disorders as well as in depression. Furthermore, distinctive cognitive‐affective biases related to attention or encoding and recall processes could be important connections between anxiety or depression and somatoform disorders.
Our review and other previous studies (Rief 2009) could demonstrate that there seems to be differences between clinician‐rated and self report measures. In addition, Lambert 1994 and Lambert 1986 demonstrated that not only the mode of assessment for itself but in interaction with the blinding of the assessor has an important impact on effect sizes. This important aspect could unfortunately not be considered in this review because the number studies with blinded assessors was too small. Finally, in future research guidelines for conducting and reporting high‐quality RCTs should be followed more strictly (Guyatt 2008; Moher 2001). Interesting research questions for further studies could be to compare the efficacy of different dosages of one chemical agent. It would be also interesting to investigate if the benefit that people with somatoform disorders experience in a pharmacological treatment is dependent on specific participant variables.
Acknowledgements
Special thanks to CCDAN's Trials Search Coordinator for assistance in developing search strategies for our review; to Kim Jones for proofreading; to Yoko Tsui, Li Ping Claire Su, Sean Woodland, and Norio Watanabe for translations; to Changlin Ai and Zheng Gang Bai for providing us with important information about included Chinese articles and Chinese classifications of mental disorders; to Maruschka Heil for double coding the studies; and to Harm van Marwijk for supporting us with managing the antidepressant classes. Furthermore, we thank all experts in somatoform disorders who provided us with information about ongoing trials, who supported our literature search, or who provided us with missing data (Carlo Altamura, Massimiliano Aragona, Brian Fallon, Harald Freyberger, Fumiyuki Goto, Peter Henningsen, Jeffrey L. Jackson, Thomas Müller, Russell Noyes, Patrick Onghena, Judith Rosmalen, Andreas Schröder, and Amarendra N. Singh).
Funding for this review was provided by the National Institute for Health Research (NIHR) Cochrane Incentive Award Scheme 2014.
CRG Funding Acknowledgement:
The National Institute for Health Research (NIHR) is the largest single funder of the Cochrane Depression, Anxiety and Neurosis Review Group.
Disclaimer:
the views and opinions expressed therein are those of the authors and do not necessarily reflect those of the NIHR, NHS, or the Department of Health.
Appendices
Appendix 1. Cochrane Central Register of Controlled Trials (CENTRAL) search strategy
CENTRAL search terms (10 April 2014):
(somatization OR somatisation OR somatoform OR (pain NEXT disorder) OR MUPS OR medically unexplained OR unexplained symptoms) AND (randomized)
Appendix 2. PsycINFO Ovid search strategy
Ovid PsycINFO search terms (2 May 2014): [RCT Filter] 1. treatment effectiveness evaluation.sh. 2. clinical trials.sh. 3. mental health program evaluation.sh. 4. placebo.sh. 5. placebo*.ti,ab. 6. randomly.ab. 7. randomi#ed.ti,ab. 8. trial.ti,ab. 9. ((singl* or doubl* or trebl* or tripl*) adj3 (blind* or mask* or dummy)).mp. 10. (control* adj3 (trial* or study or studies or group*)).ti,ab. 11. factorial*.ti,ab. 12. allocat*.ti,ab. 13. assign*.ti,ab. 14. volunteer*.ti,ab. 15. (crossover* or cross over*).ti,ab. 16. "2000".md. 17. ((waitlist* or wait* list* or treatment as usual or TAU) adj3 (control or group or versus)).ab. 18. or/1‐17 19. exp somatization/ 20. hysteria/ 21. (somatiz* or somatis* or hysteri* or briquet or polysymptom* or poly symptom* or multisomat* or multi somat*).mp. 22. (multiple and (medically unexplained or unexplained symptoms or physical symptoms or symptom diagnos*)).mp. 23. MUPS.ti,ab. 24. or/19‐23 25. 18 and 24 26. exp Pharmacology/ 27. exp Drugs/ 28. exp Drug Therapy/ 29. "3340".cc. 30. (drug* or pharma* or psychopharma* or psycho pharma* or psychotropic* or psycho tropic* or antipsychotic* or anti psychotic* or antiepileptic* or anti epileptic* or neuroleptic* or anticonvuls* or anti conculs* or antiemetic* or anti emetic* or hypnotic*).mp. 31. (antidepress* or anti depress* or MAOI* or monoamine oxidase inhibit* or ((serotonin or norepinephrine or noradrenaline or nor epinephrine or nor adrenaline or neurotransmitt* or dopamine*) and (uptake or reuptake or reuptake)) or noradrenerg* or antiadrenergic or anti adrenergic or SSRI* or SNRI* or TCA* or tricyclic* or tetracyclic* or heterocyclic*).mp. 32. (anxiolytic* or ((anti anxiety or antianxiety) adj1 (drug* or agent*)) or tranquili#er* or sedative*).mp. 33. exp "Medicinal Herbs and Plants"/ 34. exp Dietary Supplements/ 35. (nutraceutical* or herbal).mp. 36. or/26‐35 37. 25 and 36
Key: "2000".md. = Methodology: 'Treatment Outcome/Clinical Trial' "3340".cc. = Concept Code: 'Clinical Psychopharmacology'
Appendix 3. PSYNDEX search
PSYNDEX search terms (23 March 2014):
(somatisier* OR somatoform* OR schmerzstoerung OR medizinisch unerklaert* OR medizinisch ungeklaert* OR koerperbeschwerden OR koerpersymptome) AND randomisiert*
Data and analyses
Comparison 1. Pharmacotherapy versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Severity/intensity of medically unexplained physical symptoms (post‐treatment score on self report scales) | 7 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Tricyclic antidepressants versus placebo | 2 | 239 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.13 [‐0.39, 0.13] |
| 1.2 New‐generation antidepressants (serotonin noradrenaline reuptake inhibitor (SNRI), selective serotonin reuptake inhibitor (SSRI)) versus placebo | 3 | 243 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.91 [‐1.36, ‐0.46] |
| 1.3 Natural products (St. John's wort LI 160) versus placebo | 2 | 322 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.74 [‐0.97, ‐0.51] |
| 2 Acceptability | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Tricyclic antidepressant versus placebo | 1 | 208 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.53, 2.18] |
| 2.2 New‐generation antidepressants (SNRI, SSRI) versus placebo | 2 | 163 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.64, 1.61] |
| 2.3 Natural products (St. John's wort LI 160, Ze 185 3‐/4‐combination) versus placebo | 3 | 506 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.40, 1.78] |
| 3 Anxiety (post‐treatment score on self report and clinician‐rated scales) | 5 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Tricyclic antidepressants versus placebo | 1 | 200 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.29, 0.26] |
| 3.2 New‐generation antidepressants (SNRI, SSRI) versus placebo | 2 | 163 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.88 [‐1.81, 0.05] |
| 3.3 Natural products (St. John's wort LI 160, Ze 185 3‐/4‐combination) versus placebo | 2 | 321 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.83 [‐1.13, ‐0.52] |
| 4 Depression (post‐treatment score on self report and clinician‐rated scales) | 5 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Tricyclic antidepressant versus placebo | 1 | 200 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.27 [‐0.55, 0.01] |
| 4.2 New‐generation antidepressants (SSRI, SNRI) versus placebo | 2 | 163 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.56 [‐0.88, ‐0.25] |
| 4.3 Natural products (St. John's wort, Ze 185) versus placebo | 2 | 321 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.64 [‐0.87, ‐0.41] |
| 5 Dysfunctional cognitions, emotions, and behaviours (post‐treatment score on self report scales) | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 New‐generation antidepressants (SSRI) versus placebo | 1 | 51 | Std. Mean Difference (IV, Random, 95% CI) | 0.26 [‐0.29, 0.81] |
| 6 Adverse effects | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Tricyclic antidepressant versus placebo | 1 | 208 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.21, 4.84] |
| 6.2 New‐generation antidepressants (SNRI, SSRI) versus placebo | 2 | 163 | Risk Ratio (M‐H, Random, 95% CI) | 2.26 [0.52, 9.81] |
| 6.3 Natural products (St. John's wort LI 160, Ze 185 3‐/4‐combination) versus placebo | 3 | 506 | Risk Ratio (M‐H, Random, 95% CI) | 0.54 [0.08, 3.50] |
| 7 Treatment response (post‐treatment score on self report and clinician‐rated scales) | 5 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Tricyclic antidepressant versus placebo | 1 | 208 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.95, 1.73] |
| 7.2 New‐generation antidepressants (SNRI, SSRI) versus placebo | 2 | 163 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.90, 4.43] |
| 7.3 Natural products (St. John's wort LI 160) versus placebo | 2 | 324 | Risk Ratio (M‐H, Random, 95% CI) | 1.77 [1.34, 2.34] |
| 8 Functional disability and quality of life (post‐treatment score on self report scales) | 3 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8.1 Tricyclic antidepressant versus placebo | 1 | 44 | Std. Mean Difference (IV, Random, 95% CI) | 0.01 [‐0.58, 0.60] |
| 8.2 New‐generation antidepressants (SNRI, SSRI) versus placebo | 2 | 163 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.52 [1.00, ‐0.04] |
Comparison 2. Tricyclic antidepressants versus another medication.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Severity/intensity of medically unexplained physical symptoms (post‐treatment score on self report scales) | 3 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Tricyclic antidepressants versus new‐generation antidepressants (noradrenergic and specific serotonergic antidepressant (NaSSA), tetracyclic antidepressant (TeCA)) | 3 | 177 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.16 [‐0.55, 0.23] |
| 2 Acceptability | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Tricyclic antidepressants versus new‐generation antidepressants (NaSSA, serotonin noradrenaline reuptake inhibitor (SNRI), TeCA) | 8 | 556 | Risk Ratio (M‐H, Random, 95% CI) | 1.48 [0.59, 3.72] |
| 3 Anxiety (post‐treatment score on self report and clinician‐rated scales) | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Tricyclic antidepressants versus new‐generation antidepressants (NaSSA, SNRI, SSRI) | 4 | 255 | Std. Mean Difference (IV, Random, 95% CI) | 0.37 [‐0.21, 0.95] |
| 4 Depression (post‐treatment score on self report and clinician‐rated scales) | 6 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Tricyclic antidepressants versus new‐generation antidepressants (NaSSA, SNRI, TeCA) | 6 | 395 | Std. Mean Difference (IV, Random, 95% CI) | 0.17 [‐0.07, 0.40] |
| 5 Adverse effects | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Tricyclic antidepressant versus new‐generation antidepressants (NaSSA, SNRI, TeCA) | 8 | 556 | Risk Ratio (M‐H, Random, 95% CI) | 2.37 [0.39, 14.28] |
| 6 Treatment response (post‐treatment score on self report and clinician‐rated scales) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Tricyclic antidepressant versus new‐generation antidepressants (NaSSA) | 2 | 130 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.73, 1.19] |
Comparison 3. New‐generation antidepressants (SARI, NaSSA, SSRI) versus another medication.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Severity/intensity of medically unexplained physical symptoms (post‐treatment score on self report scales) | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 New‐generation antidepressants (noradrenergic and specific serotonergic antidepressant (NaSSA), selective serotonin reuptake inhibitor (SSRI)) versus other new‐generation antidepressants (noradrenaline reuptake inhibitor (NRI), serotonin noradrenaline reuptake inhibitor (SNRI), SSRI) | 4 | 182 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.16 [‐0.45, 0.14] |
| 2 Acceptability | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 New‐generation antidepressants (NaSSA, SSRI) versus other new‐generation antidepressants (NRI, SNRI, SSRI) | 4 | 196 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.60, 1.40] |
| 3 Depression (post‐treatment score on self report and clinician‐rated scales) | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 New‐generation antidepressants (NaSSA, SSRI) versus other new‐generation antidepressants (NRI, SNRI, SSRI) | 3 | 169 | Std. Mean Difference (IV, Random, 95% CI) | 0.41 [0.00, 0.82] |
| 3.2 New‐generation antidepressant (serotonin antagonist and reuptake inhibitors (SARI)) versus non‐steroidal anti‐inflammatory drugs | 1 | 140 | Std. Mean Difference (IV, Random, 95% CI) | ‐3.87 [‐4.44, ‐3.31] |
| 4 Adverse effects | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 New‐generation antidepressants (NaSSA, SSRI) versus other new‐generation antidepressants (NRI, SNRI, SSRI) | 4 | 196 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.35, 2.03] |
Comparison 4. Pharmacotherapy versus a combination of medications.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Severity/intensity of medically unexplained physical symptoms (post‐treatment score on self report scales) | 3 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Tricyclic antidepressant versus tricyclic antidepressant + antipsychotic | 1 | 34 | Std. Mean Difference (IV, Random, 95% CI) | 0.26 [‐0.42, 0.94] |
| 1.2 Selective serotonin reuptake inhibitor versus selective serotonin reuptake inhibitor + antipsychotic | 2 | 107 | Std. Mean Difference (IV, Random, 95% CI) | 0.77 [0.32, 1.22] |
| 2 Acceptability | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Selective serotonin reuptake inhibitor versus selective serotonin reuptake inhibitor + antipsychotic | 2 | 118 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.25, 2.52] |
| 3 Anxiety (post‐treatment score on self report and clinician‐rated scales) | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 Selective serotonin reuptake inhibitor versus selective serotonin reuptake inhibitor + antipsychotic | 2 | 107 | Std. Mean Difference (IV, Random, 95% CI) | 0.95 [‐0.91, 2.82] |
| 4 Depression (post‐treatment score on self report and clinician‐rated scales) | 3 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Tricyclic antidepressant versus tricyclic antidepressant + antipsychotic | 1 | 34 | Std. Mean Difference (IV, Random, 95% CI) | 0.79 [0.09, 1.49] |
| 4.2 Selective serotonin reuptake inhibitor versus selective serotonin reuptake inhibitor + antipsychotic | 2 | 107 | Std. Mean Difference (IV, Random, 95% CI) | 0.58 [‐0.33, 1.48] |
| 5 Adverse effects | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Selective serotonin reuptake inhibitor versus selective serotonin reuptake inhibitor + antipsychotic | 2 | 118 | Risk Ratio (M‐H, Random, 95% CI) | 0.6 [0.16, 2.29] |
| 6 Treatment response (post‐treatment score on self report and clinician‐rated scales) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Selective serotonin reuptake inhibitor versus selective serotonin reuptake inhibitor + antipsychotic | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 1.7 [0.94, 3.08] |
4.5. Analysis.

Comparison 4 Pharmacotherapy versus a combination of medications, Outcome 5 Adverse effects.
Comparison 5. Subgroup analysis 1: Co‐morbidity (based on the comparison pharmacotherapy versus placebo).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Severity/intensity of medically unexplained physical symptoms (post‐treatment score on self report scales) | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Co‐morbid mental disorders | 3 | 243 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.91 [‐1.36, ‐0.46] |
| 1.2 No co‐morbid mental disorders | 1 | 200 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.35, 0.21] |
| 2 Acceptability | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Co‐morbid mental disorder | 2 | 163 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.64, 1.61] |
| 2.2 No co‐morbid mental disorder | 2 | 390 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.59, 1.89] |
Comparison 6. Subgroup analysis 2: Source of funding (based on the comparison pharmacotherapy versus placebo).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Severity/intensity of medically unexplained physical symptoms (post‐treatment score on self report scales) | 7 | 804 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.66 [‐0.97, ‐0.36] |
| 1.1 Funding by pharmaceutical industry | 5 | 685 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.64 [‐1.02, ‐0.27] |
| 1.2 No funding by pharmaceutical industry | 2 | 119 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.75 [‐1.29, ‐0.21] |
| 2 Acceptability | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Funding by pharmaceutical industry | 6 | 877 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.70, 1.40] |
Comparison 7. Subgroup analysis 3: Source outcome rating (based on the comparison pharmacotherapy versus placebo).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Severity/intensity of medically unexplained physical symptoms (post‐treatment score) | 7 | 1377 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.75 [‐1.00, ‐0.49] |
| 1.1 Self report scales | 7 | 804 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.66 [‐0.97, ‐0.36] |
| 1.2 Clinician‐rated scales | 4 | 573 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.91 [‐1.42, ‐0.40] |
Comparison 8. Best‐case/worst‐case analysis (based on comparison 'new‐generation antidepressants versus placebo').
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Treatment response (post‐treatment score on self report and clinician‐rated scales) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Complete‐case analysis | 2 | 119 | Risk Ratio (M‐H, Random, 95% CI) | 2.02 [0.82, 4.97] |
| 1.2 Best‐case analysis | 2 | 163 | Risk Ratio (M‐H, Random, 95% CI) | 2.59 [1.90, 3.53] |
| 1.3 Worst‐case analysis | 2 | 163 | Risk Ratio (M‐H, Random, 95% CI) | 1.41 [0.30, 6.63] |
Comparison 9. Best‐case/worst‐case analysis (based on comparison 'natural products versus placebo').
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Treatment response (post‐treatment score on self report and clinician‐rated scales) | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Complete‐case analysis | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 1.75 [1.29, 2.36] |
| 1.2 Best‐case analysis | 2 | 324 | Risk Ratio (M‐H, Random, 95% CI) | 1.92 [1.26, 2.94] |
| 1.3 Worst‐case analysis | 2 | 324 | Risk Ratio (M‐H, Random, 95% CI) | 1.57 [1.27, 1.93] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Altamura 1991.
| Methods | Randomised, parallel‐group, double‐blind, fixed‐dose triala,f | |
| Participants |
n(randomised/ITT) = 30 Diagnosis: somatisation disorder, undifferentiated somatoform disorder (DSM‐III‐R) Inclusion criteria: no information provided Exclusion criteria: no information provided Age: mean (± SD) 49.20 ± 9.64 years, range 18‐77 years; sex: 43.3% women; mean length of time since diagnosis of a somatoform disorder: ns; country: Italy; setting: no information provided |
|
| Interventions | Levosulpiride (n = 15; AP; mean dose = 150 mg/day, max dose = 150 mg/day; no information regarding mode/frequency of administration provided) Racemic sulpiride (n = 15; AP; mean dose= 300 mg/day, max dose = 300 mg/day; no information regarding mode/frequency of administration provided) Treatment duration: 21 days |
|
| Outcomes |
Primary outcome: Scale for Somatoform Disorders (Lipman 1969) ‐ Total Score, Somatisation Score (self rated) Secondary outcomes: Scale for Somatoform Disorders ‐ Depression Score (self rated): baseline, week 1, 2, 3, 4, 5 HARS ‐ Total Score (clinician‐rated): baseline, week 1, 2, 3, 4, 5 Simpson & Angus Scale (extrapyramidal adverse effects): baseline, week 1, 2, 3, 4, 5 Anticholinergic Side Effects Check List: baseline, week 1, 2, 3, 4, 5 All‐cause drop‐outs: n = 2 (levosulpiride), n = 2 (racemic sulpiride) Drop‐outs due to adverse effects: n = 1 (levosulpiride), n = 1 (racemic sulpiride) Reported drug‐related adverse effects: Levosulpiride: anticholinergic adverse effects (n = 1, 6.6%) Racemic sulpiride: extrapyramidal adverse effects (n = 2, 13.3%), anticholinergic adverse effects (n = 3, 20.0%) No information about the total rate of participants with drug‐related adverse events provided. There were no significant differences between groups regarding severity of anticholinergic adverse effects |
|
| Notes |
Funding: no information provided Ethics approval: no information provided Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Authors states that participants were randomly assigned to 1 of 2 treatment groups (p. 26); no information provided about the random sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No information about method of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Participants "were treated under double blind conditions" (p. 25); insufficient information provided about who was and was not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about method of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Drop‐out rate in total sample: 13.3%; although reasons for missing outcome data were partly likely to be related to true outcome, missing outcome data were balanced in number across intervention with similar reasons for missing data across groups; however, no information was provided about how missing values were replaced; furthermore, no information was provided about how many of the randomised participants (placebo‐responder) were excluded from the trial after the 1‐week placebo run‐in phase before the treatment started |
| Selective reporting (reporting bias) | High risk | No protocol available; generally accepted outcomes were used. Means and SD only presented in a graph, but not as concrete values in a table or text |
| Other bias | High risk | "After one week of placebo treatment, not responders were treated under double blind conditions" (p. 25), "A placebo week followed the treatment" (p. 25), participants were randomised and passed a 1‐week placebo run‐in phase before they started the treatment, only placebo‐non‐responders (improvement < 20% on clinical rating scales) were included to the study. This exclusion of placebo‐responders before the treatment started could have led to a change of the magnitude of the effect estimation |
Aragona 2005.
| Methods | Randomised, parallel‐group, double‐blind, fixed‐dose trial | |
| Participants |
n(randomised)
= 35; n(ITT) = 29 Diagnosis: pain disorder associated with psychological factors (DSM‐IV‐TR) Inclusion criteria: presence of psychological factors that might have influenced the onset or the clinical course (or both) of pain; passing the 2 steps of diagnostic procedure; aged > 18 years Exclusion criteria: pregnancy, medical conditions of clinical importance, direct organic explanation of symptoms, a diagnosis of another mental disorder, use of psychotropic drugs endowed with an analgesic effect (e.g. amitriptyline) and anti‐inflammatory medications (were suspended in 1‐week washout phase before randomisation) Ageb: mean (± SD) 53.64 ± 15.79 years, range 18‐77 years; sexb: 72.4% women; mean length of time since diagnosis of a somatoform disorder: ns; country: Italy; setting: outpatient |
|
| Interventions | Citalopram (n = 17; SSRI; mean dose = 40 mg/day, max dose = 40 mg/day; 1 tablet twice/day) Reboxetine (n = 18; SNRI; mean dose = 8 mg/day, max dose = 8 mg/day; 1 tablet twice/day) Treatment duration: 56 days |
|
| Outcomes |
Primary outcome: McGill Pain Questionnaire ‐ Present Pain Intensity, Pain Rating Index (self rated): baseline, week 2, 4, 8 Secondary outcomes: Zung Self‐Rating Depression Scale (self rated): baseline, week 2, 4, 8 Treatment Emergent Symptom Scale: baseline, week 2, 4, 8 All‐cause drop‐outs: n = 6 (citalopram), n = 9 (reboxetine) Drop‐outs due to adverse effects: n = 2 (citalopram), n = 4 (reboxetine) Reported drug‐related adverse effects: Citalopram: dry mouth (n = 2, 11.7%); somnolence (n = 6, 35.3%); insomnia (n = 4, 23.5%); tremor (n = 5, 29.4%); tachycardia (n = 2, 11.7%); increased motor activity (n = 5, 29.4%); nausea (n = 2, 11.7%); blurred vision (n = 2, 11.7%); dizziness (n = 1, 5.8%); diarrhoea (n = 2, 11.7%) Reboxetine: dry mouth (n = 9, 50.0%); somnolence (n = 4, 22.2%); insomnia (n = 6, 33.3%); tremor (n = 4, 22.2%); tachycardia (n = 7, 38.8%); increased motor activity (n = 2, 11.1%); nausea (n = 3, 16.6%); blurred vision (n = 3, 16.6%); dizziness (n = 4, 22.2%); diarrhoea (n = 1, 5.5%); sweating (n = 2, 11.1%) 100% of people in both groups reported adverse effects. Both treatments were similarly tolerated |
|
| Notes |
Funding: no information provided Ethics approval: obtained Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomly assigned (by random tables)" (p. 35) |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Under double‐blind conditions" (p. 35); insufficient information provided about who was and was not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided; only self rating scales were used; however, it was not clear who was and was not blinded in this trial |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Drop‐out rate in total sample: 42.9%; only participants who received at least 1 outcome assessment were included in the ITT sample: originally, 35 participants were randomised (citalopram: n = 17; reboxetine: n = 18), 6 participants abandoned treatment before the first outcome assessment and were excluded from ITT sample (n = 29) that was used for statistical analyses; although reasons for missing outcome data were partly likely to be related to true outcome, missing outcome data were balanced in number across interventions with similar reasons for missing data across groups; LOCF was used to replace missing values for statistical analyses (p. 35) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available; generally accepted outcomes were used |
| Other bias | Low risk | No significant differences were found in the baseline demographic or clinical characteristics between groups; participants who had previously used psychotropic/analgesic drugs were similarly distributed in the 2 groups |
Eberhard 1988.
| Methods | Randomised, parallel‐group, double‐blind, fixed‐dose trial | |
| Participants |
n(randomised/ITT) = 70 Diagnosis: idiopathic pain syndrome according to the definition by Williams 1982 (comparable to psychogenic pain disorder of the DSM‐III‐TR) Inclusion criteria: no further inclusion criteria Exclusion criteria: severe somatic disease, MDD (DSM‐III‐TR), other psychiatric illnesses, clinically relevant contraindications towards the use of antidepressive agents, antidepressive treatment for the previous 3 months, intake of other CNS active drug, abuse of drugs Age: mean (± SD) 50.29 ± 12.60 years, range 21‐73 years; sex: 72.9% women; mean (± SD) length of time since diagnosis of a somatoform disorder: 60.0 ± 67.11 months; country: Sweden; setting: outpatient |
|
| Interventions | Maprotiline (n = 30; TCA; mean dose = 100 mg/day, max dose = 150 mg/day; 1 tablet twice/day) Clomipramine (n = 40; TeCA; mean dose = 97.2 mg/day, max dose = 150 mg/day; 1 tablet twice/day) Treatment duration: 42 days |
|
| Outcomes |
Primary outcomes: 2 VAS on pain and on bodily discomfort (Johansson 1982) (self rating): week 2, 6, month 3, 6, 12 Secondary outcomes: Depressive SCL (clinician‐rated): baseline, week 6, month 3, 6, 12 4 VAS (Johansson 1982)on sadness, inner tension, concentration difficulties, memory disturbances (self rated): week 2, 6, month 3, 6, 12 Global Assessment of degree of disease and improvement (clinician‐rated): baseline, week 6, month 3, 6, 12 Side effect checklist: baseline, week 6 Spontaneously reported side effects: baseline, week 1, 2, 4, 6 All‐cause drop‐outs: n = 5 (maprotiline), n = 13 (clomipramine) Drop‐outs due to adverse effects: n = 1 (maprotiline), n = 8 (clomipramine) Reported drug‐related adverse effects: Maprotiline: tremor (n = 3, 10.0%), tachycardia (n = 3, 10.0%), headache (n = 1, 3.3%), dry mouth (n = 7, 60.0%), sweating (n = 7, 23.3%), micturition disturbances (n = 4, 13.3%), constipation (n = 5, 16.7%), asthenia (n = 2, 6.7%), dizziness (n = 2, 6.7%), orthosatism (n = 4, 13.3%), sedation (n = 1, 3.3%) Clomipramine: tremor (n = 12, 30.0%), tachycardia (n = 2, 5.0%), headache (n = 2, 5.0%), dry mouth (n = 20, 50.0%), sweating (n = 12, 30.0%), micturition disturbances (n = 4, 10.0%), constipation (n = 10, 25.0%), asthenia (n = 7, 17.5%), dizziness (n = 2, 5.0%), orthosatism (n = 3, 7.5%), sedation (n = 1, 2.5%), vertigo (n = 4, 10%), gastritis (n = 1, 2.5%) No information about the total rate of participants with drug‐related adverse events was provided or if both treatment groups differed in the number of adverse effects |
|
| Notes |
Funding: medication in both study groups was provided by the Ciba‐Geigy AG Ethics approval: obtained Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomized, double‐blind multicenter study" (p. 27); no information provided about the random sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Randomized, double‐blind multicenter study" (p. 27); "Both drugs were given in 25‐mg tablets of identical shape, taste, and color" (p. 29) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Drop‐out rate in total sample: 25.7%; reasons for missing outcome data were partly likely to be related to true outcome. Furthermore, missing outcome data were not balanced in number across interventions. More participants dropped out of the clomipramine than of the maprotiline group because of adverse effects; missing values were not replaced, statistical analyses were based only on the sample of participants who complied with the protocol; "patients did not complete at least 2 weeks of treatment […] were not included in the efficacy analysis" (p. 32). However, it was not clear how many participants were excluded for this reason |
| Selective reporting (reporting bias) | Unclear risk | No protocol available; generally accepted outcomes were used |
| Other bias | Low risk | Clients who were recruited in a pain clinic or in a psychiatric clinic were compared with the rest of the participants who were recruited from general practitioner practices "with regard to sex, age, duration of illness, and to treatment outcome and no special trends were found" (p. 27). "The two treatment groups were compared as concerns age, sex, and duration of symptoms. No significant differences emerged" (p. 28) |
Han 2008a.
| Methods | Randomised, parallel‐group, flexible‐dose trial | |
| Participants |
n(randomised) = 95; n(ITT) = 77 Diagnosis: undifferentiated somatoform disorder (DSM‐IV) Inclusion criteria: aged ≥ 18 years; somatic symptoms almost every day for ≥ 6 months, not taking any active prescription medications to control somatic symptoms (non‐prescription medications, e.g. paracetamol (acetaminophen) 2 g/day or ibuprofen 1.2 g/day were allowed), women of reproductive age had to agree to use adequate contraception Exclusion criteria: history of (or current, or both) psychotic disorders (such as schizophrenia, schizoaffective disorder, and bipolar disorder); current DSM Axis I disorders that could possibly account for the somatic symptoms (e.g. MDD, anxiety disorders, factitious disorder, malingering or another somatoform disorder such as somatisation disorder), substance abuse or dependence in the previous 12 months, history of hypersensitivity to venlafaxine or mirtazapine, people currently treated with any psychotropic medication; participation in any clinical trials in the previous 30 days; people involved in workers' compensation, disability or related litigation, breast‐feeding or pregnant women Age: mean (± SD) 45.20 ± 12.59 years, range ns; sex: 61.1% women; mean (± SD) length of time since diagnosis of a somatoform disorder: 32.5 ± 22.5 months; country: South Korea; setting: no information provided, but outpatient setting was assumed due to affiliations of study authors |
|
| Interventions | Mirtazapine (n = 50; NaSSA; mean dose = 31.70 mg/day, max dose = 60 mg/day; no information regarding mode/frequency of administration provided) Venlafaxine (n = 45; SNRI; mean dose = 105.50 mg/day, max dose = 225 mg/day; no information regarding mode/frequency of administration provided) Treatment duration: 84 days |
|
| Outcomes |
Primary outcome: PHQ‐15 (self rated): baseline, week 1, 2, 4, 8, 12 Secondary outcomes: Beck Depression Inventory (self rated): baseline, week 1, 2, 4, 8, 12 12‐item General Health Questionnaire (self rated): baseline, week 1, 2, 4, 8, 12 Treatment Emergent Events‐General Inquiry: baseline, week 1, 2, 4, 8, 12 All‐cause drop‐outs: n = 11 (mirtazapine), n = 13 (venlafaxine) Drop‐outs due to adverse effects: n = 3 (mirtazapine), n = 4 (venlafaxine) Reported drug‐related adverse effects: Mirtazapine: nausea (n = 2, 4.0%), vomiting (n = 1, 2.0%), somnolence (n = 4, 8.0%), dry mouth (n = 5, 10.0%), anorexia (n = 1, 2.0%), yawning (n = 3, 6.0%), sweating (n = 2, 4.0%), dizziness (n = 3, 6.0%), headache (n = 1, 2.0%) Venlafaxine: nausea (n = 4, 8.9%), vomiting (n = 2, 4.4%), somnolence (n = 1, 2.2%), dry mouth (n = 6, 13.3%), anorexia (n = 2, 4.4%), sweating (n = 1, 2.2%), insomnia (n = 2, 4.4%), dizziness (n = 2, 4.4%), headache (n = 1, 2.2%) Both treatments were well tolerated. No information about the total rate of participants with drug‐related adverse events was provided |
|
| Notes |
Funding: Dr Han: research support from Korea Research Foundation Grant (MOEHRD) (KRF‐2007‐013‐E00033), Korea University Neuropsychiatric Alumni Grant, member of speaker's bureaux of GlaxoSmithKline Korea, Janssen Pharmaceuticals Korea, and Otsuka Korea
Dr. Pae: received research grants from GlaxoSmithKline Korea, GlaxoSmithKline, AstraZeneca Korea, Janssen Pharmaceuticals Korea, Eli Lilly and Company Korea, Korean Research Foundation, Otsuka Korea, Wyeth Korea, and the Korean Institute of Science and Technology Evaluation and Planning; received honoraria from/is on the speaker's bureaux of GlaxoSmithKline Korea, Lundbeck Korea, AstraZeneca Korea, Janssen Pharmaceuticals Korea, Eli Lilly and Company Korea, McNeil Consumer and Speciality Inc. And Otsuka Korea
Dr Patkar: consultant for Bristol‐Myers Squibb, GlaxoSmithKline, and Reckitt Benckiser; is on the speaker's bureaux of Bristol‐Myers Squibb, GlaxoSmithKline, and Reckitt Benckiser; received research support from the National Institutes of Health, Astra Zeneca, Bristol Myers Squibb, Forest Laboratories Inc., GlaxoSmithKline, Janssen, McNeil Consumer and Speciality Inc., Organon, Jazz Pharmaceuticals and Pfizer
Dr Masand: consultant for Bristol‐Myers Squibb Company, Cephalon Inc., Eli Lilly and Company, Forest Laboratories Inc., GlaxoSmithKline, i3CME, Janssen Pharmaceuticals, Jazz Pharmaceuticals, Organon, Pfizer Inc., Targacept Inc., and Wyeth Pharmaceuticals; is on speaker's bureaux of Astra Zeneca, Bristol Myers Squibb Company, Forest Laboratories Inc., GlaxoSmithKline, Janssen Pharmaceuticals, Pfizer Inc., and Wyeth Pharmaceuticals; received research support from Astra Zeneca, Bristol Myers Squibb Company, Cephalon Inc., Eli Lilly and Company, Forest Laboratories Inc., GlaxoSmithKline, Ortho McNeil Pharmaceutical Inc., Janssen Pharmaceuticals, and Wyeth Pharmaceuticals Ethics approval: obtained Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Computer‐generated randomization code" (p. 253) |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Observer bias should be considered" (p. 259); because the study report included no information about blinding procedures it is assumed that neither participants nor study personnel were blinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "Observer bias should be considered" (p. 259); because the study report includes no information about blinding procedures it is assumed that outcome assessment was not blinded; only self report scales were used; however, it is not clear if participants were blinded to the treatment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Drop‐out rate in total sample: 25.2%; only participants who received at least 1 dose of a study medication and had at least 1 post‐baseline visit assessment were included in the ITT sample: originally, 95 participants were randomised (citalopram: n = 17; reboxetine: n = 18), 6 participants abandoned treatment before the first outcome assessment and were excluded from ITT sample (n = 7) that was used for statistical analyses; although reasons for missing outcome data were partly likely to be related to true outcome, missing outcome data were balanced in number across intervention with similar reasons for missing data across groups; LOCF was applied for replacing missing values (p. 254) |
| Selective reporting (reporting bias) | High risk | SD for outcomes at post‐assessment were not provided. No protocol available; generally accepted outcomes were used |
| Other bias | Low risk | No significant differences were found in the baseline demographic or clinical characteristics between groups |
Han 2008b.
| Methods | Randomised, parallel‐group, flexible‐dose trial | |
| Participants |
n(randomised/ITT) = 45 Diagnosis: undifferentiated somatoform disorder (DSM‐IV) Inclusion criteria: aged ≥ 18 years, somatic symptoms almost every day for ≥ 6 months, not taking any active prescription medications to control their somatic symptoms (non‐prescription medications, e.g. paracetamol (acetaminophen) 2 g/day or ibuprofen 1.2 g/day were allowed), women of reproductive age had to agree to use adequate contraception Exclusion criteria: history of (or current, or both) psychotic disorders (such as schizophrenia, schizoaffective disorder, and bipolar disorder); current DSM Axis I disorders that could possibly account for the somatic symptoms (e.g. MDD, anxiety disorders, factitious disorder, malingering or another somatoform disorder such as somatisation disorder); substance abuse or dependence in the previous 12 months; history of hypersensitivity to venlafaxine or mirtazapine; people currently treated with any psychotropic medication; participation in any clinical trials in the previous 30 days; people involved in workers' compensation, disability, or related litigation; breast‐feeding or pregnant women Age: mean (± SD) 37.64 ± 11.90 years, range ns; sex: 57.8% women; mean (± SD) length of time since diagnosis of a somatoform disorder: 38.9 ± 27.5 months; country: South Korea; setting: no information provided, but outpatient setting was assumed due to affiliations of study authors |
|
| Interventions | Fluoxetine (n = 28; SSRI; mean dose = 36.10 mg/day, max dose = 60 mg/day; no information regarding mode/frequency of administration provided) Sertraline (n = 17; SSRI; mean dose = 167.70 mg/day, max dose = 350 mg/day; no information regarding mode/frequency of administration provided) Treatment duration: 84 days |
|
| Outcomes |
Primary outcome: PHQ‐15 (self rated): baseline, week 1, 2, 4, 8, 12 Secondary outcomes: Beck Depression Inventory (self rated): baseline, week 1, 2, 4, 8, 12 12‐item General Health Questionnaire (self rated): baseline, week 1, 2, 4, 8, 12 Treatment Emergent Events‐General Inquiry: baseline, week 1, 2, 4, 8, 12 All‐cause drop‐outs: n = 8 (fluoxetine), n = 5 (sertraline) Drop‐outs due to adverse effects: n = 0 (fluoxetine), n = 0 (sertraline) Reported drug‐related adverse effects: Fluoxetine: nausea (n = 5, 17.9%), vomiting (n = 2, 7.1%), somnolence (n = 1, 3.6%), dry mouth (n = 3, 10.7%), anorexia (n = 3, 10.7%), sweating (n = 1, 3.6%), insomnia (n = 2, 7.1%), constipation (n = 1, 3.6%), dizziness (n = 1, 3.6%) Sertraline: nausea (n = 3, 17.6%), vomiting (n = 1, 5.9%), somnolence (n = 3, 17.6%), dry mouth (n = 2, 11.8%), anorexia (n = 1, 5.9%), constipation (n = 2, 11.8%), dizziness (n = 2, 11.8%) Both treatments were well tolerated. No information about the total rate of participants with drug‐related adverse events was provided |
|
| Notes |
Funding: Dr Han: research support from Korea Research Foundation Grant (MOEHRD) (KRF‐2007‐013‐E00033), Korea University Neuropsychiatric Alumni Grant
Dr. Pae: received research grants from GlaxoSmithKline Korea, GlaxoSmithKline, AstraZeneca Korea, Janssen Pharmaceuticals Korea, Eli Lilly and Company Korea, Korean Research Foundation, Otsuka Korea, Wyeth Korea, and the Korean Institute of Science and Technology Evaluation and Planning; received honoraria from/is on the speaker's bureaux of GlaxoSmithKline Korea, Lundbeck Korea, AstraZeneca Korea, Janssen Pharmaceuticals Korea, Eli Lilly and Company Korea, McNeil Consumer and Speciality Inc. And Otsuka Korea
Dr Patkar: consultant for Bristol‐Myers Squibb, GlaxoSmithKline, and Reckitt Benckiser; is on the speaker's bureaux of Bristol‐Myers Squibb, GlaxoSmithKline, and Reckitt Benckiser; received research support from the National Institutes of Health, Astra Zeneca, Bristol Myers Squibb, Forest Laboratories Inc., GlaxoSmithKline, Janssen, McNeil Consumer and Speciality Inc., Organon, Jazz Pharmaceuticals, and Pfizer
Dr Masand: consultant for Bristol‐Myers Squibb Company, Cephalon Inc., Eli Lilly and Company, Forest Laboratories Inc., GlaxoSmithKline, i3CME, Janssen Pharmaceuticals, Jazz Pharmaceuticals, Organon, Pfizer Inc., Targacept Inc. and Wyeth Pharmaceuticals; is on speaker's bureaux of Astra Zeneca, Bristol Myers Squibb Company, Forest Laboratories Inc., GlaxoSmithKline, Janssen Pharmaceuticals, Pfizer Inc., and Wyeth Pharmaceuticals; received research support from Astra Zeneca, Bristol Myers Squibb Company, Cephalon Inc., Eli Lilly and Company, Forest Laboratories Inc., GlaxoSmithKline, Ortho McNeil Pharmaceutical Inc., Janssen Pharmaceuticals, and Wyeth Pharmaceuticals Ethics approval: obtained Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Computer‐generated randomization code" (p. 438) |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Observer bias should be considered" (p. 442); because the study report included no information about blinding procedures it was assumed that neither participants nor study personnel were blinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | "Observer bias should be considered" (p. 259); because the study report included no information about blinding procedures it is assumed that outcome assessment was not blinded; only self report scales were used; however, it was not clear if participants were blinded to the treatment |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Drop‐out rate in total sample: 28.9%; only participants who received at least 1 dose of a study medication and had at least 1 post‐baseline visit assessment were included in the ITT sample: originally, 45 participants were randomised (fluoxetine: n = 28; sertraline: n = 17), because all participants returned for at least 1 post‐baseline follow‐up visit in both treatment groups yielding an ITT sample of n = 45; although reasons for missing outcome data were partly likely to be related to true outcome, missing outcome data were balanced in number across intervention with similar reasons for missing data across groups; LOCF was applied for replacing missing values (p. 439) |
| Selective reporting (reporting bias) | High risk | SD for outcomes at post‐assessment were not provided; no protocol available; generally accepted outcomes were used |
| Other bias | Low risk | No significant differences were found in the baseline demographic or clinical characteristics between groups |
Huang 2012.
| Methods | Randomised, parallel‐group, fixed‐dose trial | |
| Participants |
n(randomised/ITT) = 60 Diagnosis: somatisation disorder (F45.0), undifferentiated somatoform disorder (F45.1), SAD (F45.3) (ICD‐10) Inclusion criteria: duration of the disorder > 6 months, somatic subscore of HARS ≥ 12 and ≥ 5 points higher than the Psychic Subscore of the HARS, the SOMS‐7 score had to be ≥ 12 Exclusion criteria: 17‐item HDRS score ≥ 17, had other mental disorders (e.g. panic disorder, MDD, or substance abuse), exhibited a severe and unstable physical illness (e.g. epilepsy, severe renal or hepatic impairment, or cancer), pregnant or breast‐feeding women Age: mean (± SD) 44.40 ± 9.75 years, range ns; sex: 65.0% women; mean (± SD) length of time since diagnosis of a somatoform disorder: 30.9 ± 18.1 months; country: China; setting: no information provided, but outpatient setting was assumed due to affiliations of study authors |
|
| Interventions | Citalopram + paliperidone (n = 30; SSRI + AP; mean dose citalopram = 20 mg/day, mean dose paliperidone = 3 mg/day, max dose citalopram = 20 mg/day, max dose paliperidone = 3 mg/day; no mode/frequency of administration provided) Citalopram (n = 30; SSRI; mean dose = 20 mg/day, max dose = 20 mg/day; no information regarding mode/frequency of administration provided) Treatment duration: 42 days |
|
| Outcomes |
Primary outcome: SOMS‐7 (self rated): baseline, week 2, 4, 6 Secondary outcomes: HARS ‐ Total Score, Somatisation Score (clinician‐rated): baseline, week 2, 4, 6 HDRS (clinician‐rated): baseline, week 2, 4, 6 Response rate (50% reduction in SOMS‐7 score; SOMS‐7 score < 12): week 6 Treatment Emergent Symptom Scale: baseline, week 2, 4, 6 All‐cause drop‐outs: n = 9 (citalopram + paliperidone), n = 9 (citalopram) Drop‐outs due to adverse effects: n = 3 (citalopram), n = 5 (citalopram + paliperidone) Reported drug‐related adverse effects: Citalopram + paliperidone: drowsiness (n = 2, 6.7%), dry mouth (n = 6, 20.0%), somnolence (n = 4, 13.3%), constipation (n = 3, 10.0%), nasal obstruction (n = 1, 3.3%), sweating (n = 2, 3.3%), nausea or vomiting (n = 2, 6.7%), dizziness (n = 3, 10.0%), headache (n = 2, 6.7%), elevated prolactin (n = 1, 3.3%) Citalopram: drowsiness (n = 1, 3.3%), dry mouth (n = 4, 13.3%), somnolence (n = 1, 3.3%), constipation (n = 1, 3.3%), sweating (n = 1, 3.3%), nausea or vomiting (n = 2, 6.7%), dizziness (n = 1, 3.3%), headache (n = 1, 3.3%) 86.7% of the citalopram + paliperidone group and 40.0% of the citalopram group experienced adverse effects. Both groups did not differ statistically significant regarding the adverse effects |
|
| Notes |
Funding: no information provided. Authors state that there are no conflicts of interest Ethics approval: obtained Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The allocation of each medication was on the basis of a computer‐generated randomization code" (p. 152) |
| Allocation concealment (selection bias) | Low risk | "Each code was sealed in an envelope saved in the Clinical Research Center. In addition, another researcher who did not know the code helped with the process. Only when a patient fulfilled the inclusion criteria and was enrolled in the study was the corresponding coded envelope opened and the code of the group given" (p. 152) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "This is not a double‐blind study, but only a randomized study. But in order to ensure the reliability and validity of research results, we carried out the study in accordance with the requirements of a double‐blind study as much as possible. [...] Therefore, the patients were completely randomly assigned to receive citalopram plus paliperidone treatment or receive citalopram treatment alone." "Only when a patient met the inclusion criteria and was enrolled in the study was the corresponding coded envelope opened and the code of the group given." (p. 152); no means of blinding participants/study personnel after randomisation and opening the envelope were described; it was assumed that according to the statement of the trial authors the current trial was not double‐blinded and that, therefore, there was a high risk regarding blinding participants/study personnel after randomisation |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "The rater was blinded to the kind of treatment patients received" (p. 151); "The data management personnel and statistical analyst participating this study were not aware of the kind of treatment patients received" (p. 153) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Drop‐out rate in total sample: 30.0%; although reasons for missing outcome data were partly likely to be related to true outcome, missing outcome data were balanced in number across intervention with similar reasons for missing data across groups; missing values were replaced by LOCF (p. 153) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available; generally accepted outcomes were used |
| Other bias | Low risk | "No statistically significant difference was observed between the two groups at the baseline" (p. 153); "We chose a moderate dose of 20 mg of citalopram as the FDA [Food and Drug Administration] and European Medicines Agency have recently advised against using 40 mg of citalopram because of concerns of cardiotoxicity (08/24/2011 ‐ Drug Safety Communication3 ‐ FDA http://www.fda.gov/Safety/ MedWatch/SafetyInformation/SafetyAlertsforhumanmedicalproducts/ucm269481.htm)" (p.157) |
Jiang 2005.
| Methods | Randomised, parallel‐group, flexible‐dose trial | |
| Participants |
n(randomised/ITT) = 68 Diagnosis:somatoform disorder (CCMD‐III) Inclusion criteria: SCL‐90 Total Score ≥ 3, CGI ‐ Severity Scale ≥ 3, aged 18‐58 years, time since diagnosis of somatoform disorder > 6 months Exclusion criteria: drug dependence, severe psychosis, paranoia, organic brain disease, physical illness, pregnant or breastfeeding women, epilepsy Age: mean (± SD) 37.04 ± 10.40 years, range ns; sex: 67.7% women; mean (± SD) length of time since diagnosis of a somatoform disorder: 168.49 ± 97.92 months; country: China; setting: inpatient |
|
| Interventions | Venlafaxine (n = 36; SNRI; mean dose = 105.0 mg/day, max dose = 200 mg/day; no information regarding mode/frequency of administration provided) Amitriptyline (n = 32; TCA; mean dose = 168.0 mg/day, max dose = 200 mg/day; no information regarding mode/frequency of administration provided) Treatment duration: 42 days |
|
| Outcomes |
Primary outcome: No primary outcome was assessed Secondary outcomes: CGI ‐ Severity Scale (clinician‐rated): baseline, week 1, 2, 4, 6 SCL‐90 (self rated): baseline, week 1, 2, 4, 6 Effectiveness rate (no further information about this measure provided): week 6 Treatment Emergent Symptom Scale: week 1, 2, 6 All‐cause drop‐outs: n = 0 (venlafaxine), n = 0 (amitriptyline) Drop‐outs due to adverse effects: n = 0 (venlafaxine), n = 0 (amitriptyline) Reported drug‐related adverse effects: Venlafaxine: nausea (n = 20; 55.6%), dizziness (n = 10, 27.8%), anxiety (n = 7, 19.4%), insomnia (n = 5, 13.9%) Amitriptyline: dry mouth (n = 21, 65.6%), constipation (n = 17, 53.1%), blurred vision (n = 14, 43.8%), dizziness (n = 12, 37.5%), tremor (n = 9, 28.1%), sweating (n = 8, 25.0%), micturition disturbances (n = 1, 3.1%), abnormalities on electrocardiograph (n = 2, 5.6%). In the amitriptyline group, statistically significantly more participants experienced adverse effects compared with the venlafaxine group in treatment weeks 1, 2, and 6. No information about the total rate of participants with drug‐related adverse events was provided |
|
| Notes |
Funding: no information provided Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients [...] were randomly assigned to receive venlafaxine or amitriptyline" (p. 1177); no information provided about the random sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information about methods of blinding participants/study personnel provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the study and there were no losses to follow‐up and no treatment withdrawals |
| Selective reporting (reporting bias) | Unclear risk | No protocol available; generally accepted outcomes were used |
| Other bias | Low risk | There were no significant differences in gender, age, and duration of symptoms between groups (p. 1177) |
Ju 2003.
| Methods | Randomised, parallel‐group, flexible‐dose trial | |
| Participants |
n(randomised/ITT) = 68 Diagnosis: somatisation disorder (CCMD‐III) Inclusion criteria: inpatient of Wutaishan Hospital of Yangzhou (Jiangsu, China) between May 2001 and January 2002; HARS ≥ 14 Exclusion criteria: psychotic symptoms, severe brain injury, substance abuse, pregnant and breast‐feeding women Age: mean (± SD) 41.75 ± 9.57 years, range ns; sex: 77.9% women; mean (± SD) length of time since diagnosis of a somatoform disorder: 154.8 ± 82.3 months; country: China; setting: inpatient |
|
| Interventions | Venlafaxine (n = 34; SNRI; mean dose: ns, max dose = 150 mg/day; no information regarding mode/frequency of administration provided) Doxepin (n = 34; TCA; mean dose: ns, max dose = 150 mg/day; no information regarding mode/frequency of administration provided) Treatment duration: 42 days |
|
| Outcomes |
Primary outcome: No primary outcome was assessed Secondary outcomes: HDRS ‐ Total Score, responder rate (clinician‐rated): baseline, week 1, 2, 4, 6 Treatment Emergent Symptom Scale: week 1, 2, 4, 6 All‐cause drop‐outs: n = 0 (venlafaxine), n = 0 (doxepin) Drop‐outs due to adverse effects: n = 0 (venlafaxine), n = 0 (doxepin) Reported drug‐related adverse effects: Venlafaxine: headache (n = 5, 14.7%), dry mouth (n = 5, 14.7%), insomnia (n = 3, 8.8%) Doxepin: fatigue, headache, dry mouth, constipation (no information about the number of participants with each adverse effect provided), micturition disturbances (n = 1, 2.9%), irregularities on electrocardiograph (n = 3, 8.8%) In the doxepin group, statistically significantly more participants experienced adverse effects compared with the venlafaxine group. 29.4% of the venlafaxine group and 82.4% of the doxepin group experienced adverse effects |
|
| Notes |
Funding: no information provided Ethics approval: no information provided Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomly assigned to venlafaxine group and doxepin group" (p. 590); no information provided about the random sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information about methods of blinding participants/study personnel provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the study and there were no losses to follow‐up and no treatment withdrawals |
| Selective reporting (reporting bias) | High risk | No protocol available; only 2 outcomes (depression, adverse effects) were assessed, important common primary outcomes (such as severity of MUPS) were neglected |
| Other bias | Low risk | No differences were found between groups regarding age and symptom duration at baseline (p. 590) |
Kong 2004.
| Methods | Randomised, parallel‐group, flexible‐dose trial | |
| Participants |
n(randomised/ITT) = 60 Diagnosis:somatoform disorder (CCMD‐III) Inclusion criteria: aged 24‐71 years Exclusion criteria: somatic disorder that explains somatoform symptoms, people who used or stopped using a specific medication (anti‐anxiety drugs, antidepressants, AP medication) for ≥ 2 weeks before treatment started Age: mean (± SD) 48.50 ± 10.92 years, range 23‐70 years; sex: 48.3% women; mean length of time since diagnosis of a somatoform disorder: ns; country: China; setting: ns |
|
| Interventions | Paroxetine (n = 15; SNRI; mean dose: ns, max dose = 50 mg/day; twice/day; no information regarding mode of administration provided) Amitriptyline (n = 15; TCA; mean dose: ns, max dose = 150 mg/day; 3 times/day; no information regarding mode administration provided) Open paroxetine (n = 30; no information regarding mean dose, max dose, and frequency/mode of administration provided) Treatment duration: 42 days |
|
| Outcomes |
Primary outcome: No primary outcome was assessed Secondary outcomes: HDRS ‐ Total Score; anxiety, de‐realisation, retardation, weight, sleep disturbance score; responder rate (clinician‐rated): baseline, week 1, 2, 4, 6 HARS (clinician‐rated): baseline, week 1, 2, 4, 6 CGI ‐ Severity Scale (clinician‐rated): baseline, week 1, 2, 4, 6 Treatment Emergent Symptom Scale: baseline, week 1, 2, 4, 6 All‐cause drop‐outs: n = 0 (paroxetine), n = 0 (amitriptyline), n = 0 (open paroxetine) Drop‐outs due to adverse effects: n = 0 (paroxetine), n = 0 (amitriptyline), n = 0 (open paroxetine) Reported drug‐related adverse effects: Paroxetine: dry mouth (n = 4, 26.6%), constipation (n = 2, 13.3%), micturition disturbances (n = 2, 13.3%), headache + nausea (n = 3, 20.0%), blurred vision (n = 2, 13.3%), headache (n = 4, 26.6%), insomnia (n = 1, 6.6%), increased blood pressure (n = 1, 6.6%), increased motor activity (n = 3, 20.0%) Amitriptyline: dry mouth (n = 6, 40.0%), constipation (n = 5, 33.3%), headache + nausea (n = 4, 26.6%), blurred vision (n = 7, 46.6%), headache (n = 5, 33.3%), insomnia (n = 4, 26.6%), hypersomnia (n = 7, 46.6%), increased blood pressure (n = 4, 26.6%) Open paroxetine: dry mouth (n = 8, 26.6%), constipation (n = 3, 10.0%), micturition disturbances (n = 1, 3.3%), headache + nausea (n = 5, 16.6%), blurred vision (n = 3, 10.0%), headache (n = 5, 16.6%), insomnia (n = 4, 13.3%), hypersomnia (n = 2, 6.6%), increased blood pressure (n = 3, 10.0%), increased motor activity (n = 4, 13.3%) No information about the total rate of participants with drug‐related adverse events was provided or if both treatment groups differed in the number of adverse effects |
|
| Notes |
Funding: medication in all 3 study groups was provided by the Zhejiang Huahai Pharmaceutical Co., Ltd Ethics approval: no information provided Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients […] were randomly assigned" (p. 733); no information provided about the random sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Differences in the frequency of which medication was administered between the groups are described (p. 733), therefore, it is assumed that no blinding of participants/study personnel was given |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the study and there were no losses to follow‐up and no treatment withdrawals. |
| Selective reporting (reporting bias) | High risk | No protocol available; generally accepted outcomes were used; however, important common primary outcomes (such as severity of MUPS) were neglected |
| Other bias | Low risk | In this multiple‐intervention trial data were presented separately for each of the groups to which participants were randomised |
Kroenke 2006.
| Methods | Randomised, placebo‐controlled, double‐blind, flexible‐dose trial | |
| Participants |
n(randomised) = 117, n(ITT) = 112 Diagnosis: MSD (≥ 3 medically unexplained symptoms with a frequency or severity beyond that expected for a known medical condition, with > 1 of these symptoms present for at least 6 months); symptoms were defined as medically unexplained when they were, in the opinion of the evaluating physician, 1. idiopathic, and 2. a symptom characteristic of a known medical disorder but reported at a frequency or severity considered out of proportion to that expected for the known medical condition; the MSD corresponded to the criteria of an undifferentiated somatoform disorder according to the DSM‐IV or ICD‐10) Inclusion criteria: ≥ 18 years; DSM‐IV diagnostic criteria for MDD, GAD, SAD, or a combination were fulfilled; met clinical criteria for MSD measured by the PHQ‐15; HDRS‐17 Total Score ≥ 14 and a HARS Total Score of ≥ 12; ≤ 25% decrease in the HDRS‐17 Total Score or HARS Total Score from screening to randomisation Exclusion criteria: history of inability to tolerate/failure to respond to ≥ 2 antidepressants of sufficient dose/duration of administration for the treatment of symptoms present in the current illness (depressive or anxiety); current/past history of mania, bipolar disorder, schizophrenia, or other psychotic disorder; history of seizure disorder other than childhood febrile seizure; evidence of serious or clinically unstable medical illness or psychiatric condition; previous intolerance or hypersensitivity to (extended‐release) venlafaxine; use of any non‐psychopharmacological drug with psychotropic effects within 7 days of study randomisation, an MAOI of fluoxetine within 30 days of screening, or electroconvulsive therapy within 3 months of screening; chronic use of analgesics containing opiates for > 6 months or for ≥ consecutive weeks prior screening; known of suspected alcohol or drug abuse within 6 months of screening; use of triptans, psychoactive herbal medications, or any other psychoactive drugs; or a positive urine drug test at screening; pregnant or breastfeeding women or women who expected becoming pregnant during the course of study or were sexually active and were not using contraception Ageb: mean (± SD) 47.00 ± 12.06 years, range ns; sexb: 80.4% women; mean length of time since diagnosis of a somatoform disorderb: ns; country: USA; setting: outpatient |
|
| Interventions | Venlafaxine (n = 55b; SNRI; mean dose = 177 mg/day, max dose = 225 mg/day; 1 tablet once/day) Placebo (n = 57b) Treatment duration: 84 days |
|
| Outcomes |
Primary outcome: PHQ‐15 ‐ Total Score, responder rate (PHQ‐15 score < 10) (self rated): baseline, week 4, 8, 12 Secondary outcomes: HDRS‐17 (clinician‐rated): baseline, week 2, 4, 8, 12 HARS (clinician‐rated): baseline, week 2, 4, 8, 12 CGI ‐ Severity Scale (clinician‐rated): baseline, week 2, 4, 8, 12 CGI ‐ Improvement Scale (clinician‐rated): week 2, 4, 8, 12 McGill Quality of Life Questionnaire Physical Symptoms Scale (self rated): baseline, week 12 Medical Outcomes Study Short‐Form 36‐Item Questionnaire ‐ physical health, mental health, bodily pain (self rated): baseline, week 12 Medical Outcomes Study Concentration Scale (self rated): baseline, week 12 Adverse effects reported by participants: baseline, week 1, 2, 4, 8, 12, post‐taper All‐cause drop‐outsb: n = 21 (venlafaxine extended release), n = 22 (placebo) Drop‐outs due to adverse effectsb: n = 4 (venlafaxine extended release), n = 10 (placebo) Reported drug‐related adverse effectsb: Venlafaxine: nausea (n = 16, 29.1%), headache (n = 13, 23.6%), fatigue (n = 8, 14.5%), dizziness (n = 6, 10.9%), constipation (n = 6, 10.9%), tremor (n = 6, 10.9%), back pain (n = 5, 9.1%), contusion (n = 4, 7.3%), decreased appetite (n = 3, 5.5%), hypoaesthesia (n = 3, 5.5%), upper abdominal pain (n = 3, 5.5%), yawning (n = 3, 5.5%), nasopharyngitis (n = 2, 3.6%), migraine (n = 2, 3.6%), urinary tract infection (n = 2, 3.6%) Placebo: nausea (n = 9, 15.8%), headache (n = 7, 12.3%), fatigue (n = 1, 1.8%), dizziness (n = 3, 5.3%), constipation (n = 3, 5.3%), back pain (n = 1, 1.8%), decreased appetite (n = 1, 1.8%), nasopharyngitis (n = 1, 1.8%), migraine (n = 1, 1.8%), urinary tract infection (n = 1, 1.8%) No information about the total rate of participants with drug‐related adverse events was provided. There was a somewhat higher incidence of adverse effects in the venlafaxine compared with placebo |
|
| Notes |
Funding: Wyeth Research, Collegeville, PA. Drs. Benattia and Graepel and Mr. Musgnung are employees of Wyeth; Dr. Kroenke has received grant/research support from Wyeth, Eli Lilly, and Pfizer and has received honoraria from Wyeth, Pfizer, Eli Lilly, and Astra Zeneca. Dr. Messina has received grant/research support from Vista Medical Research Inc. Dr. Graepel is a major stock shareholder of Wyeth. Ethics approval: obtained Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomized, double‐blind, placebo‐controlled study" (p. 74); no information provided about the random sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Randomized, double‐blind, placebo‐controlled study" (p. 74); insufficient information provided about who was and was not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Drop‐out rate in total sample: 41.0%; only participants who received at least 1 post‐baseline efficacy evaluation were included in the ITT sample: originally, 117 participants were randomised (information about sample sizes of treatment and control group are missing), 5 participants abandoned treatment before the first outcome assessment and were excluded from ITT sample (n = 112) that was used for statistical analyses; although reasons for missing outcome data were partly likely to be related to true outcome, missing outcome data were balanced in number across intervention with similar reasons for missing data across groups; LOCF was applied for replacing missing values (p. 75) |
| Selective reporting (reporting bias) | High risk | SD of post‐assessment not provided; baseline values were not reported for all outcomes for which baseline‐to‐endpoint changes were reported; no protocol available; generally accepted outcomes were used |
| Other bias | Low risk | "The proportion of patients in each group who took at least 1 concomitant medication was almost identical" (p. 74); "Patients who could not tolerate at least 75mg/d at any time during the study were withdrawn from the study" (p. 74); "Baseline characteristics were similar between groups" (p. 75); used dose range in the current study was "within the dose range recommended for venlafaxine ER's [extended release] U.S. Food and Drug Administration (FDA‐) approved indications for MDD, GAD, and SAD" (p. 78) |
Li 2006.
| Methods | Randomised, parallel‐group, flexible‐dose trial | |
| Participants |
n(randomised/ITT) = 58 Diagnosis: somatoform disorder (CCMD‐III) Inclusion criteria: SCL‐90 Somatisation Subscale ≥ 3; HDRS Total Score ≥ 20; if participants also had other mental problems, somatic complaints had to appear before them; somatoform symptoms could not be explained by a somatic disorder Exclusion criteria: ns Age: mean (± SD) 41.73 ± 12.34 years, range 20‐68 years; sex: 60.3% women; mean (± SD) length of time since diagnosis of a somatoform disorder: 84.60 ± 59.2 months; country: China; setting: no information provided, but inpatient setting was assumed due to affiliations of study authors |
|
| Interventions | Paroxetine + quetiapine (n = 30; SSRI + AP; mean dose paroxetine = 26.7 mg/day, mean dose quetiapine = 403.3 mg/day, max dose paroxetine = 40 mg/day, max dose quetiapine = 600 mg/day; no information about mode/frequency of administration provided) Paroxetine (n = 28; SSRI; mean dose = 27.30 mg/day, max dose = 40 mg/day; no information about mode/frequency of administration provided) Treatment duration: 56 days |
|
| Outcomes |
Primary outcome: SCL‐90 ‐ Somatisation Subscore (self rated): baseline, week 2, 8 Secondary outcomes: SCL‐90 ‐ Depression Subscore, Anxiety Subscore (self rated): baseline, week 2, 8 HDRS (clinician‐rated) ‐ Total Score, responder rate: baseline, week 2, 8 Treatment Emergent Symptom Scale: no information about time points of measurement provided Drop‐outs due to adverse effects: n = 0 (paroxetine + quetiapine), n = 0 (paroxetine) Reported drug‐related adverse effects: n = 0 (paroxetine + quetiapine), n = 2 (paroxetine) Reported drug‐related adverse effects: Paroxetine + quetiapine: fatigue (26.7%), nausea (23.3%), dry mouth (16.7%) Paroxetine: nausea (26.9%), dizziness (23.1%), dry mouth (19.2%) No information about the total rate of participants with drug‐related adverse events was provided or if treatment groups differed in the number of adverse effects |
|
| Notes |
Funding: no information provided Ethics approval: no information provided Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly assigned to 1 study groups by coin tossing, if it was the coin's head participants were assigned to paroxetine + quetiapine group, if it was the coin's tail it was the paroxetine group (p. 598) |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information about methods of blinding participants/study personnel assessment provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There were only 2 drop‐outs in the paroxetine group (drop‐out rate: 3.4%); missing values were not replaced; statistical analyses are based only on the sample of participants who complied with the protocol |
| Selective reporting (reporting bias) | Unclear risk | No protocol available. Generally accepted outcomes were used |
| Other bias | Unclear risk | No information is provided |
Luo 2009.
| Methods | Randomised, placebo‐controlled, double‐blind, fixed‐dose trial | |
| Participants |
n(randomised/ITT) = 80 Diagnosis: persistent somatoform pain disorder (ICD‐10) Inclusion criteria: aged 18‐65 years; pain existed ≥ 6 months Exclusion criteria: co‐exist depressive symptoms occurred prior to pain with HDRS‐17 ≥ 17; positive family history of pain disorder or depressive episodes; severe and unstable physical illnesses; use of antidepressants for the treatment of pain or depression; pregnancy Age: mean (± SD) 40.96 ± 12.69 years, range ns; sex: 57.5% women; mean (± SD) length of time since diagnosis of a somatoform disorder: 21.02 ± 19.02 months; country: China; setting: outpatient |
|
| Interventions | Fluoxetine (n = 40; SSRI; mean dose = 20 mg/day, max dose = 10 mg/day; mode of administration: tablet; no information about frequency of administration provided) Placebo (n = 40) Treatment duration: 56 days |
|
| Outcomes |
Primary outcome: Medical Outcomes Study Pain Measures (self rated): baseline, week 2, 4, 8 Secondary outcomes: HDRS‐17 (clinician‐rated): baseline, week 2, 4, 8 Treatment Emergent Symptom Scale: baseline, week 2, 4, 8 Drop‐outs due to adverse effects: it was stated that 2 participants quit the study due to adverse effects. However, it is not unclear how these adverse effect‐related drop‐out were distributed between groups Reported drug‐related adverse effects: Fluoxetine: drowsiness (n = 1, 2.5%), dry mouth (n = 2, 5.0%), constipation (n = 1, 2.5%), sweating (n = 1, 2.5%), nausea or vomiting (n = 10, 25.0%) Placebo: dry mouth (n = 2, 5.0%), nasal obstruction (n = 1, 2.5%), sweating (n = 1, 2.5%), nausea or vomiting (n = 4, 10.0%), dizziness (n = 1, 2.5%) 35.0% of the fluoxetine group and 22.5% of the placebo group reported adverse effects. There was no statistically significant difference between groups regarding the adverse effects |
|
| Notes |
Funding: Shanghai Science and Technology Committee Ethics approval: no information provided Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "All patients were randomly allocated to either treatment group or control group" (p. 1523); no information provided about the random sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Double‐blind, placebo‐controlled, […] study" (p. 1523); "placebo capsules […] having the same color, weight, shape and taste like the fluoxetine capsules" (p. 1523) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information was provided for incomplete outcome data; it was only stated that "intention‐to‐treat (ITT) analysis was performed and last observation carry forward (LOCF) was used for missing values" (p. 1524), thus it can be concluded that there were no losses to follow‐up; the only information about drop‐outs stated that "2 patients quit the study due to" adverse effects (p. 1524); however, it is not clear how these 2 patients were distributed among the fluoxetine and placebo groups |
| Selective reporting (reporting bias) | High risk | No protocol available; generally accepted outcomes were used; however, treatment effects were only reported for the primary outcome and not for HDRS‐17 (depression) |
| Other bias | Low risk | "No statistically significant difference was observed between the fluoxetine and placebo group at the baseline" (p. 1523) |
Melzer 2009.
| Methods | Combined randomised, placebo‐controlled, parallel‐group, double‐blind, fixed‐dose trial | |
| Participants |
n(randomised/ITT) = 182 Diagnosis: somatisation disorder (F45.0), undifferentiated somatoform disorder (F45.1) (ICD‐10) Inclusion criteria: aged ≥ 18 years; symptom duration ≥ 6 months Exclusion criteria: pregnancy or lactation; known allergies to trial medication; alcohol, drug, or medication abuse; concomitant participation in another clinical trial or < 4 weeks ago; historically known or clinical indication of: inflammatory or non‐inflammatory neurological disease, endocrinological disorders, inflammatory bowel disease, psychiatric disorders, rheumatic diseases, bronchial asthma, infectious diseases, serious conditions such as cancer/uncontrolled hypertension/heart failure/endocarditis, myocarditis/primary cardiomyopathies or severe cardiac dysrhythmias/severe hepatic diseases/renal insufficiency (creatinine > 3 mg/dL) or anaemia (haemoglobin < 9 g/dL)/severe nutritional disorders or vitamin deficiency illnesses Age: mean (± SD) 41.64 ± 14.79 years, range ns; sex: 57.5% women; mean length of time since diagnosis of a somatoform disorder: ns; nationality: Germany; country: no information provided, but outpatient setting was assumed due to information about recruitment source |
|
| Interventions | Ze 185 4‐combination: butterbur root, valerian root, passionflower herb, lemon balm leaf (n = 53; NP; mean dose Ze 185 4‐combination = 330 mg/day, max dose Ze 185 4‐combination = 330 mg/day; 1 tablet 3 times/day) Ze 185 3‐combination: root, valerian root, passionflower herb, lemon balm leaf (n = 58; NP; mean dose Ze 185 3‐combination = 330 mg/day, max dose Ze 185 3‐combination = 330 mg/day; 1 tablet 3 times/day) Placebo (n = 61) Treatment duration: 14 days |
|
| Outcomes |
Primary outcome: Pain diary (self rated): days 1‐14 Secondary outcomes: VAS anxiety (Rickels 1994) (self rated): baseline, day 14 Beck Depression Inventory (self rated): baseline, day 14 CGI ‐ Severity Scale (clinician‐rated): baseline, day 14 Participant's rating of treatment efficacy: day 14 Treatment responder (50% improvement on VAS Anxiety or Beck Depression Inventory, self rated): day 14 All‐cause drop‐outs: n = 4 (Ze 185 4‐combination), n = 6 (Ze 185 3‐combination), n = 5 (placebo) Drop‐outs due to adverse effects: n = 0 (Ze 185 4‐combination), n = 2 (Ze 185 3‐combination), n = 1 (placebo) Reported drug‐related adverse effects: Ze 185 4‐combination: vomiting (n = 1, 1.6%), flatulence (n = 1, 1.6%) Ze 185 3‐combination: nausea (n = 2, 3.4%), constipation (n = 1, 1.7%) Placebo: nausea (n = 1, 1.6%) 3.2% of the Ze 185 4‐combination group, 5.2% of the Ze 185 3‐combination, and 1.6% of the placebo group reported adverse effects. The distribution of adverse events did not differ significantly between groups |
|
| Notes |
Funding: study medication was supplied by Zeller AG (sponsor). Every investigator received a grant from the sponsor. The sponsor covered the costs for monitoring and data management (contract research organisation) as well as for the auditing (Chair of Medical Informatics, University of Giessen, Germany) Ethics approval: obtained Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The allocation to the respective treatment group was carried out based on a computer‐generated randomization list" (p. 1305); "Stratification was carried out using the computer program STATXACT" (p. 1305) |
| Allocation concealment (selection bias) | Unclear risk | "Patients were assigned a patient number and the corresponding numbered medication box in the order of their admission into the trial" (p. 1305); insufficient information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Tablets of all three treatment groups were identical in color, odor, and consistency" (p.1304); "The double‐blind nature of the trial was guaranteed by adherence to standard procedures and audits" (p. 1305) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Drop‐out rate in total sample: 8.2%; "The ITT population included all randomized patients who used treatment at least once and had a postbaseline value for comparison" (p. 1305), however, it was unclear if and how many participants were not included to the ITT sample for this reason, in Table 2 (p. 1305). The difference between the sample of randomised participants and the ITT sample was explained only by justifications such as withdrawal of consent or adverse events; although reasons for missing outcome data were partly likely to be related to true outcome, missing outcome data were balanced in number across intervention with similar reasons for missing data across groups; LOCF was used to replace missing values (p. 1305) |
| Selective reporting (reporting bias) | High risk | No protocol available; treatment effects are only reported for the secondary outcomes such as depression and anxiety; important primary outcomes such as severity of physical symptoms were not assessed |
| Other bias | Low risk | At baseline, there were no significant differences between groups regarding demographic or clinical characteristics (p. 1305); "At the end of the trial, 99% of patients had taken at least 75% of the medication" (p. 1305); in this multiple‐intervention trial data were presented separately for each of the groups to which participants were randomised |
Muller 2008.
| Methods | Randomised, placebo‐controlled, double‐blind, flexible‐dose trial | |
| Participants |
n(randomised/ITT) = 51 Diagnosis: MSD (bothersome medically unexplained ≥ 3 symptoms within the past month, together with a history of ≥ 1 somatoform symptoms for at ≥ 2 years; the MSD corresponded to the criteria of an undifferentiated somatoform disorder according to the DSM‐IV or ICD‐10) Inclusion criteria: ns Exclusion criteria: people with somatic symptoms that were judged to be secondary to a psychiatric disorder other than MSD, current or past psychotic disorder, significant suicidal risk, any serious unstable medical illness (as assessed by medical history, physical examination, and standard laboratory investigations), pregnancy or breastfeeding, use of recent or concomitant psychotropics, concurrent cognitive‐behavioural therapy Age: mean (± SD) 39.64 ± 9.75 years, range 18‐65 years; sex: 57.5% women; mean length of time since diagnosis of a somatoform disorder: ns; country: South Africa; setting: no information provided, but outpatient setting was assumed due to information about recruitment source |
|
| Interventions | Escitalopram (n = 25; SSRI; mean dose = 14.4 mg/day, max dose = 20 mg/day; no information about mode/frequency of administration provided) Placebo (n = 26) Treatment duration: 84 days |
|
| Outcomes |
Primary outcomes: PHQ (self rated): baseline, week 2, 4, 6, 8, 12 PHQ ‐ responder rate (PHQ score < 10) (self rated): week 2, 4, 6, 8, 12 Visual Analogue Pain Rating Scale (Katz 1999) (self rated): baseline, week 4, 8, 12 Secondary outcomes: CGI ‐ Improvement Scale (clinician‐rated): week 2, 4, 6, 8, 12 CGI ‐ Severity Scale (clinician‐rated): baseline, week 2, 4, 6, 8, 12 CGI ‐ Severity Scale ‐ remission rate (clinician‐rated): week 2, 4, 6, 8, 12 HARS ‐ Total, Psychic, Somatic Score (clinician‐rated): baseline, week 4, 8, 12 Montgomery‐Åsberg Depression Rating Scale (self rated): baseline, week 4, 8, 12 Scale for the Assessment of Illness Behaviour (self rated): baseline, week 12 Sheehan Disability Scale (self rated): baseline, week 4, 8, 12 All‐cause drop‐outs: n = 1 (escitalopram), n = 0 (placebo) Drop‐outs due to adverse effects: n = 1 (escitalopram), n = 0 (placebo) Reported drug‐related adverse effects: Escitalopram: Week 0‐2: headache (n = 11, 44%), nausea (n = 10, 40%), abdominal discomfort (n = 3, 12%), insomnia (n = 3, 12%), yawning (n = 3, 12%), nasopharyngitis (n = 2, 8%), diarrhoea (n = 2, 8%), tremor/anxiety (n = 2, 8%), drowsiness (n = 2, 8%), dry mouth (n = 2, 8%) Week 2‐12: headache (n = 9, 36%), nasopharyngitis (n = 4, 16%), diarrhoea (n = 3, 12%), nausea (n = 3, 12%), insomnia (n = 3, 12%), urinary tract infection (n = 2, 8%), staphylococcal skin infection (n = 1, 4%) Placebo: Week 0‐2: headache (n = 7; 27%), nausea (n = 6; 23%), abdominal discomfort (n = 2; 8%), insomnia (n = 1; 4%), yawning (n = 1; 4%), nasopharyngitis (n = 4; 15%), diarrhoea (n = 3; 12%), tremor/anxiety (n = 1; 4%), drowsiness (n = 2; 8%), dry mouth (n = 1; 4%) Week 2‐12: headache (n = 11; 42%), nasopharyngitis (n = 8; 31%), diarrhoea (n = 6; 23%), nausea (n = 5; 19%), staphylococcal skin infection (n = 2; 8%); abdominal discomfort (n = 4; 15%); dizziness (n = 2; 8%) No information about the total rate of participants with drug‐related adverse events was provided or if both treatment groups differed in the number of adverse effects |
|
| Notes |
Funding: H. Lundbeck A/S; at the time this study was conducted, Professor Stein, Professor Seedat, and Dr. Muller were funded by the Medical Research Council of South Africa Ethics approval: obtained Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients were randomly assigned (via computer‐generated randomization lists)" (p. 44) |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Escitalopram and placebo medication were identical in appearance" (p. 44) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Drop‐out rate in total sample: 2.0%; reasons for missing outcome data were very unlikely to be related to true outcome (there was only 1 participant withdrawn due to adverse events in the escitalopram group); missing outcome data were balanced in number across groups with similar reasons for missing data across groups; LOCF was be used to replace missing values (p. 45); trial authors stated that ITT included "all randomized patients with at least one valid post‐baseline efficacy measure" (p. 45). However, there was no difference in the number of randomised participants and the ITT sample. Therefore, it was concluded that all randomised participants had at least 1 valid post‐baseline efficacy measure |
| Selective reporting (reporting bias) | Low risk | No protocol available; generally accepted outcomes were used and data were reported for a broad variety of primary and secondary outcomes (self report and clinician‐rated scales) |
| Other bias | Low risk | "The trial consisted of a 2‐week screening phase for medication washout (if required) and for obtaining results of selected laboratory investigations" (p. 44); "no significant difference existed in the extent of comorbidity across the medication and placebo groups" (p. 45) |
Müller 2004.
| Methods | Randomised, placebo‐controlled, double‐blind fixed‐dose triala | |
| Participants |
n(randomised) = 175, n(ITT) = 173 Diagnosis: somatisation disorder (F45.0), undifferentiated somatoform disorder (F45.1), persistent somatoform pain disorder (F45.3) (ICD‐10) Inclusion criteria: aged 18‐65 years; baseline HARS ‐ Somatisation Score ≥ 12; baseline HARS ‐ Psychic Subscore ≥ 5 and < HARS ‐ Somatisation Score; baseline HDRS ‐ Total Score ≤ 12; baseline SOMS‐2 score ≥ 4 (men) and ≥ 6 (women); SOMS‐7 score 12‐30; standardised good clinical practice criteria for study participation Exclusion criteria: people exhibiting a decrease in SOMS‐7 score of 6 during the placebo run‐in phase ("placebo responders"), current diagnoses of major depression, drug and alcohol abuse, epilepsy, organic mental disorder, any other serious unstable acute or chronic medical condition, current or anamnestic diagnosis of schizophrenia or schizoaffective disorder, use of psychotropic drugs 4 weeks before and during the study, concurrent psychotherapy, increased suicidal risk, need for concomitant treatment with phenprocoumon or cyclosporin (or both) Ageb: mean (± SD) 47.65 ± 11.35 years, range ns; sexb: 57.5% women; mean length of time since diagnosis of a somatoform disorder: ns; country: Germany; setting: outpatient |
|
| Interventions | St. John's wort (n = 87b; NP; mean dose = 600 mg/day, max dose = 600 mg/day; 1 tablet twice/day) Placebo (n = 86b) Treatment duration: 42 days |
|
| Outcomes |
Primary outcomes: SOMS‐7 (self rated): baseline, week 2, 4, 6 SCL‐90‐R ‐ Somatisation Score (self rated): baseline, week 2, 4, 6 Secondary outcomes: HARS ‐ Psychic Anxiety Score, Somatisation Score (clinician‐rated): baseline, week 2, 4, 6 CGI ‐ Severity Scale (clinician‐rated): baseline, week 2, 4, 6 CGI ‐ Improvement Scale (clinician‐rated): baseline, week 2, 4, 6 Responder rate (decrease of ≥ 50% of the SOMS‐7 and 'very much better'/'much better' rating on the CGI ‐ Improvement Scale): week 6 Adverse effects were assessed in an open question fashion: week 2, 4, 6 Tolerability was assessed in analogy to the CGI 'therapeutic risks' (1 = no adverse effect, 2 = impairment not significant, 3 = significant impairment, 4 = risks outweighing therapeutic effect): week 6 All‐cause drop‐outs: n = 5 (St. John's wort), n = 6 (placebo) Drop‐outs due to adverse effects: n = 0 (St. John's wort), n = 0 (placebo) Reported drug‐related adverse effects: St. John's wort: nightmares (n = 1, 1.1%) Placebo: no adverse effects Tolerability and safety of St. John's wort treatment was comparable to that of placebo. 1.1% of the St. John's wort and 0% of the placebo group reported adverse effects |
|
| Notes |
Funding: Medical Services, Berlin, Germany (Marcus Mannel); Lichtwer Pharma GmbH, Berlin, Germany (Harald Murck); data analysis and experimental design, Gauting, Germany (Volker W. Rahlfs); Study medications were provided by Lichtwer Pharma AG (Berlin, Germany) Ethics approval: obtained Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomized, placebo‐controlled […] trial" (p. 539); "Patients [...] were randomly assigned in a 1:1 ratio to receive either SJW [St. John's wort] or placebo" (p. 539); "Computer‐aided randomization and preparation of a coding list (Rancode; Wiedey, Konstanz, Germany) was performed in blocks of six" (p. 539) |
| Allocation concealment (selection bias) | Low risk | "Randomization […] was performed […] by the independent Quality Assurance Unit of the sponsor" (p. 539); "Blistered study medication and medication containers were labeled sequentially according to the coding list by the manufacturing department of the sponsor and provided to the investigators in blocks of six" (p. 539) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Double‐blind trial" (p. 539); "placebo tablets identical in shape, size, taste, and color were administered" (p. 541); "Tolerability ratings and adverse events of SJW LI 160 were indistinguishable from corresponding placebo figures. Therefore, both investigators and patients could not have been unblinded by recognizing a specific side effect pattern of SJW LI 160, which further validates the study results" (p. 543) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All investigators, personnel of contracted partners and of the sponsor who were actively involved in the trial were blinded to group assignment until the code was broken at the end of the trial. Success of blinding was not been evaluated systematically in this trial, because experience from previous trials with identical formulations has revealed good results (p. 539) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Drop‐out rate in total sample: 7.5%; 175 participants were originally randomised (St. John's wort: n = 87, placebo: n = 88); however, due to loss of participant documentation for visits 1‐4 following a fire the ITT population comprises only 173 participants (St. John's wort: n = 87, placebo: n = 86); reasons for missing outcome data were very unlikely to be related to true outcome (no participant was withdrawn due to adverse events in either groups), missing outcome data were balanced in number with similar reasons for missing data across groups; "Results in the PP [treated per protocol] population were nearly identical, thereby corroborating the ITT results" (p. 542); "Missing values were handled conservatively according to the Last Value Carried Forward (LVCF)" (p. 541) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available; generally accepted outcomes were used |
| Other bias | High risk | "Patients exhibiting a decrease in SOMS‐7 score 6 during the placebo run‐in phase ("placebo responders") were not to be included in the trial" (p. 539) this placebo run‐in phase was single‐blind, randomisation was conducted just after the placebo run‐in phase, 9 participants were excluded from the trial after this placebo run‐in phase; "Baseline demographics and clinical characteristics did not differ significantly between groups, except for small‐sized differences regarding sex ratio and CGI subscore "severity" " (p. 539); "It further remains open whether 600 mg of SJW [St. John's wort] daily is the optimal dose and if higher dosages may result in increased efficacy" (p. 545) |
Ouyang 2006.
| Methods | Randomised, parallel‐group, flexible‐dose trial | |
| Participants |
n(randomised/ITT) = 80 Diagnosis: somatoform pain disorder (CCMD‐III) Inclusion criteria: ns Exclusion criteria: people whose pain was caused by depression, anxiety, schizophrenia, lung or physical illness Age: mean (± SD) 37.85 ± 9.71 years, range ns; sex: 58.8% women; mean (± SD) length of time since diagnosis of a somatoform disorder: 39.8 ± 7.10 months; country: China; setting: inpatient |
|
| Interventions | Mirtazapine (n = 40; NaSSA; mean dose: ns, max dose = 275 mg/day; medication was administered twice/day; no information regarding mode of administration provided) Amitriptyline (n = 40; TCA; mean dose: ns, max dose: ns; medication was administered twice/day; no information regarding mode of administration provided) Treatment duration: 56 days |
|
| Outcomes |
Primary outcome: No primary outcome was assessed Secondary outcomes: HDRS ‐ Total Score, responder rate (clinician‐rated): baseline, week 1, 2, 4, 6, 8 Treatment Emergent Symptom Scale: baseline, week 1, 2, 4, 6, 8 All‐cause drop‐outs: n = 0 (mirtazapine), n = 0 (amitriptyline) Drop‐outs due to adverse effects: n = 0 (mirtazapine), n = 0 (amitriptyline) Reported drug‐related adverse effects: Mirtazapine: dry mouth, constipation, micturition disturbances, palpitation, sexual difficulty, electrocardiograph irregularities Amitriptyline: dizziness, hypersomnia, increased appetite, weight gain, limb swallow, hypertension In week 1 in the mirtazapine group, the Treatment Emergent Symptom Scale score was significantly higher than in amitriptyline group; in week 2, 4, 6, and 8 the score was significantly higher in amitriptyline compared with mirtazapine. No information about the total rate of participants with drug‐related adverse events was provided |
|
| Notes |
Funding: no information provided Ethics approval: no information provided Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Trial authors stated that participants were randomly assigned to the 2 groups (p. 560); no information provided about random sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information about methods of blinding participants/study personnel provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the study and there were no losses to follow‐up and no treatment withdrawals |
| Selective reporting (reporting bias) | High risk | No protocol available; generally accepted outcomes were used; however, treatment effects were only reported for the secondary outcomes such as depression, adverse effects, and general symptom improvement; important primary outcomes such as severity of physical symptoms were not assessed |
| Other bias | Low risk | There was no significant difference between groups regarding demographic variables; 1 week before treatment started participants were instructed to wash‐out other medication (p. 560); groups did not significantly differ in HDRS scores before treatment (p. 560) |
Pilowsky 1990.
| Methods | Combined randomised, placebo‐controlled, parallel‐group, double‐blind, flexible‐dose trialc | |
| Participants |
n(complete cases) = 50 Diagnosis: somatoform pain disorder (DSM‐III‐R) Inclusion criteria: pain for ≥ 1 month that was not responding adequately to appropriate treatment; absence of objective evidence for the presence of any significant organic disease sufficient to explain the presence or severity of the pain experience and degree of disability; impairment of functioning by at least 25% taking into account biological, personal, social, occupational, and recreational aspects; absence of a psychotic illness (including major depressive syndrome), organic brain syndrome, or alcohol dependence; adequate comprehension of English; absence of any physical disorder contraindicating the use of a TCA Exclusion criteria: major depression Age: ns; sex: ns; mean length of time since diagnosis of a somatoform disorder: ns; country: Australia; setting: outpatient |
|
| Interventions | Amitriptyline + support (n: ns; TCA; mean dose: ns, max dose: ns; medication was administered as tablets; no information regarding frequency of administration provided) Placebo (n: ns) Treatment duration: 84 days |
|
| Outcomes |
Primary outcomes (all primary outcome measures were assessed at baseline, week 4, 8, 12, month 3, 6, 12) Global pain assessment over the last week (location, intensity, character, distribution, chronicity, tune relationship) (self rated) McGill Pain Questionnaire (self rated) Pain diary (VAS over 2 weeks) Secondary outcomes (all secondary outcome measures were assessed at baseline, week 4, 8, 12, month 3, 6, 12): VAS‐productivity (self rated) Sickness Impact Profile (self rated) Activity diary (VAS over 2 weeks) (self rated) Illness Behaviour Assessment Schedule (self rated) Illness Behaviour Questionnaire (self rated) Affective Status ‐ Clinical assessment on 4‐point scales (self rated) Zung Depression Questionnaire (self rated) Levine‐Pilowsky Depression Questionnaire (self rated) Spielberger State‐Trait Anxiety Questionnaire (self rated) Semi‐structured interview where participants had to judge their progress concerning 3 'global areas': well‐being, pain, activity VAS on mean intensity of pain, amount of pain, and degree of impairment or 'productivity' (self rated) All‐cause drop‐outs/drop‐outs due to adverse effects: no information provided Reported drug‐related adverse effects: no information provided |
|
| Notes |
Funding: financial support for the study by National Health and Medical Research Council Ethics approval: obtained Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were "allocated to 1 of 4 treatment groups with the use of a table of random numbers" (p. 6) |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "At the end of therapy, i.e., after 12 weeks, the therapist broke the code to establish whether the patient had been on the active or inert preparation and wrote a summary of the treatment and progress with a recommendation concerning further management" (p. 6); insufficient information about blinding of participants provided |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Patients were followed up […] by one of the research assistants who had not carried out the baseline measures and who was also blind to the drug treatment and, as far as possible, to the non‐drug treatment" (p. 7) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | In this study, the following 4 groups were examined: amitriptyline + psychotherapy (n = 26); amitriptyline + support (n = 26); placebo tablet + psychotherapy (n = 26); placebo tablet + support (n = 24). In the context of the current review, only results for amitriptyline + support vs. placebo tablet + support were considered, originally, 129 participants were randomised to the 4 study groups, 102 participants completed the trial, drop‐out rates for the single groups were given only as percentage (amitriptyline + support group: 25%, placebo + support group: 31%). Because ITT sample size in the single groups was not mentioned, percentages cannot be converted in absolute values. Trial authors only state that "There was no significant difference in dropout numbers between the 4 groups" (p. 9); no information was provided about how drop‐outs were distributed between the 4 study groups, therefore, no drop‐out rate can be estimated; statistical analyses were based only on the sample of participants who complied with the protocol |
| Selective reporting (reporting bias) | High risk | Study authors only evaluated outcome measures based on 3 ratings of the participants of improvement in well‐being, pain, activity, as well as the following 3 continuous variables (VAS): mean intensity of pain, amount of pain, and degree of impairment or 'productivity'; scores on validated scales such as McGill Pain Questionnaire, Pain diary, Zung Depression Scale, or Spielberger State‐Trait Anxiety Questionnaire were only assessed at baseline but were not used to evaluate the therapy efficacy, these variables were only used as predictors of therapy efficacy (p. 7); follow‐up month 3, 6, and 12 post treatment was reported in the study report but no follow‐up data were provided; no protocol available |
| Other bias | High risk | "Patients were offered treatments like transcutaneous nerve stimulation, physiotherapy, regional nerve blocks, to psychopharmacotherapy and psychotherapy, in each case, the treatment is offered for a finite period of time after which the patient returns to the panel for a review. Where it was thought that musculoligamentous or myofascial problems had not been adequately treated physiotherapeutically this was provided before patients were entered into the trial, even if considered suitable" (p. 5); participants who were admitted to the study were compared with people who were not: "Those admitted were more likely to be women, more highly educated, more likely to be employed, less likely to be on an invalidity pension and younger" (p. 8); "marked baseline differences" between the [study] groups regarding specific dependent variables were identified (p. 11); in this multiple‐intervention trial data were presented separately for each of the groups to which participants were randomised |
Sanada 2010.
| Methods | Randomised, parallel‐group, flexible‐dose triale | |
| Participants |
n(randomised/ITT) = 21 Diagnosis: pain disorder (DSM‐IV‐TR) Inclusion criteria: ns Exclusion criteria: ns Aged: mean (± SD) 48.17 ± 14.26 years, range ns; sexd: 58.8% women; mean length of time since diagnosis of a somatoform disorder: ns; country: Japan; setting: outpatient |
|
| Interventions | Paroxetine (n = 11; SSRI; mean dose: ns, max dose = 40 mg/day; no information about mode/frequency of administration provided) Milnacipran (n = 10; SNRI; mean dose: ns, max dose = 150 mg/day; no information about mode/frequency of administration provided) Treatment duration: 56 days |
|
| Outcomes |
Primary outcome: Short‐Form McGill Pain Questionnaire ‐ Total Pain Rating Index, Present Pain Intensity, VAS (self rated): baseline, week 4, 8 Secondary outcomes: HDRS (clinician‐rated): baseline, week 4, 8 Zung Self‐Rating Depression Scale (self rated): baseline, week 4, 8 All‐cause drop‐outs: n = 3 (paroxetine), n = 5 (milnacipran) Drop‐outs due to adverse effects: n = 3 (paroxetine), n = 2 (milnacipran) Reported drug‐related adverse effects: no information provided |
|
| Notes |
Funding: no information provided Ethics approval: no information provided Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomly assigned" (p. S543); no information provided about the random sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information about methods of blinding participants/study personnel provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Drop‐out rate in total sample: 38.1%; although reasons for missing outcome data were partly likely to be related to true outcome, missing outcome data were balanced in number across intervention with similar reasons for missing data across groups; however, sample size was very small and the drop‐out rate was very high, therefore, the chance of a bias produced by incomplete data was high; statistical analyses were based only on the sample of participants who complied with the protocol |
| Selective reporting (reporting bias) | High risk | No protocol available; generally accepted outcomes were used; for this trial only a conference abstract was available in which data were only reported completely for 1 outcome (severity of pain) |
| Other bias | Unclear risk | No information was provided |
Volz 2000.
| Methods | Randomised, placebo‐controlled, double‐blind fixed‐dose triala | |
| Participants |
n(randomised) = 208; n(ITT) = 200 Diagnosis: somatisation disorder (F45.0), undifferentiated somatoform disorder (F45.1), persistent somatoform pain disorder (F45.3) (ICD‐10) Inclusion criteria: aged 18‐76 years, no significant actual or history of other Axis I diagnoses (e.g. panic disorder, MDD, schizophrenia, substance abuse), no relevant concomitant diseases (e.g. epilepsy, severe renal or hepatic impairment, cancer), HARS ‐ Somatisation Score ≥ 12 and ≥ 5 points than HARS ‐ Psychic Score; HDRS ≤ 24 Exclusion criteria: pregnant or breastfeeding women, contraindication to the use of the study medication Ageb: mean (± SD) 45.61 ± 13.00 years, range ns; sexb: 63.5% women; mean length of time since diagnosis of a somatoform disorder: ns; country: Germany; setting: ns |
|
| Interventions | Opipramol (n = 104; TCA; mean dose = 200 mg/day, max dose = 200 mg/day; 1 capsule twice/day) Placebo (n = 104) Treatment duration: 42 days |
|
| Outcomes |
Primary outcomes: SCL‐90‐R ‐ Somatic Subscore (self rated): day ‐7, 0, 7, 14, 28, 42 HARS ‐ Somatic Score (clinician‐rated): day ‐7, 0, 7, 14, 28, 42 Secondary outcomes: HARS ‐ Total Score, Psychic Score (clinician‐rated): day ‐7, 0, 7, 14, 28, 42 HDRS (clinician‐rated): day ‐7, 0, 7, 14, 28, 42 SCL‐90‐R ‐ Total Score, Anxiety Subscore (self rated): day ‐7, 0, 7, 14, 28, 42 CGI ‐ Improvement Scale (clinician‐rated): day ‐7, 0, 7, 14, 28, 42 Adverse events: day 7, 14, 28, 42 All‐cause drop‐outs: n = 14 (opipramol), n = 13 (placebo) Drop‐outs due to adverse effects: n = 3 (opipramol), n = 3 (placebo) Reported drug‐related adverse effects: Opipramol:single adverse effects with a frequency of ≥5%: tiredness (n = 9, 9.1%), dizziness (n = 5, 5.1%), nausea (n = 3, 3.0%), back pain (n = 5, 5.1%); sum of adverse effects in 1 category with a frequency of ≥5%: body as whole (n = 12, 12.1%), central or peripheral nervous system (n = 7, 7.1%), gastrointestinal system (n = 6, 6.1%), skeletomuscular system (n = 14, 14.1%) Placebo:single adverse effects with a frequency of ≥5%: tiredness (n = 2, 2.0%), dizziness (n = 8, 7.9%), gastroenteritis (n = 6, 5.9%), nausea (n = 6, 5.9%), back pain (n = 3, 3.0%); sum of adverse effects in 1 category with a frequency of ≥5%: body as whole (n = 4, 4.0%), central or peripheral nervous system (n = 13, 12.9%), gastrointestinal system (n = 21, 20.8%), skeletomuscular system (n = 7, 6.9%) 37.0% of the opipramol group and 38.0% of the placebo group reported adverse effects. No information if treatment groups differed in the number of adverse effects was provided |
|
| Notes |
Funding: Novartis Pharma GmbH, Nürnberg, Germany, was involved in the trial (Klaus Dieter Stoll is employee of Novartis Pharma GmbH); however, it was not stated how Ethics approval: obtained Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomized, placebo‐controlled trial" (p. 214); no information provided about the random sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Double‐bind treatment phase" (p. 212); no sufficient information provided about who was and was not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Drop‐out rate in total sample: 13.0%; originally, 208 participants were randomised (opipramol group: n = 104, placebo group: n = 104), according to the trial authors' definition of the ITT sample (no further information about this definition provided) 8 participants were excluded from ITT sample (n = 200) that was used for statistical analyses; although reasons for missing outcome data were partly likely to be related to true outcome, missing outcome data were balanced in number across intervention with similar reasons for missing data across groups; "The last observation was carried forward (LOCF)" (p. 213) was used to replace missing values |
| Selective reporting (reporting bias) | Low risk | No protocol available; generally accepted outcomes were used and data were reported for a broad variety of primary and secondary outcomes |
| Other bias | High risk | "Compliance was assessed by immediate pill‐count at visits as well as by the results of opipramol plasma concentration measurements integrated into the data set when the whole monitoring procedures were terminated" (p. 212); "There were no relevant differences between the treatment groups" for initial symptom severity (p. 214); "[…] patients underwent a 7‐day, single blind, washout period with four placebo capsules per day […]. After this 7‐day period, placebo responders (HAMA [HARS]‐SOM decrease of more than 4 points) had to be excluded" (p. 212); the randomisation took place after the placebo run‐in phase; however, it is unclear how many participants were excluded after this placebo run‐in phase |
Volz 2002.
| Methods | Randomised, placebo‐controlled, double‐blind fixed‐dose triala | |
| Participants |
n(randomised) = 151; n(ITT) = 149 Diagnosis: somatisation disorder (F45.0), undifferentiated somatoform disorder (F45.1), persistent somatoform pain disorder (F45.3) (ICD‐10) Inclusion criteria: aged 18‐65 years; HARS ‐ Somatic Scale ≥ 12 and ≥ 5 points than HARS ‐ Psychic Scale; HDRS ≤ 24 Exclusion criteria: additional diagnosis of depression, schizophrenia, schizo‐affective disorder, dementia; pregnancy; nursing mothers; concomitant treatment with psychopharmacological active compounds and relevant physical diseases (i.e. severe cardiac, renal or hepatic dysfunction or disease, underlying malignant disease, bone‐marrow depression or dyscrasia, clinically manifest liver dysfunction, renal insufficiency [serum creatinine > 2.0 mg/dL], non‐compensated cardiac insufficiency, epilepsy, or organic brain disease), and laboratory value deviations (i.e. > 10% deviation from the upper/lower normal ranges of erythrocytes, haemoglobin, and haematocrit, leukocytopenia (< 2500 cells/mm3), granulocytopenia (< 1500 cells/mm3), thrombocytopenia (< 100,000 cells/mm3), GOT > 90 IU/L, bilirubin > 1.5 mg/dL) Ageb: mean (± SD) 47.74 ± 12.00 years, range ns; sexb: 62.0% women; mean length of time since diagnosis of a somatoform disorder: ns; country: Germany; setting: outpatient |
|
| Interventions | St. John's wort extract LI160 (n = 75; NP; mean dose = 600 mg/day, max dose = 600 mg/day; 1 capsules twice/day) Placebo (n = 74) Treatment duration: 42 days |
|
| Outcomes |
Primary outcomes: SCL‐90‐R ‐ Somatic Subscore (self rated): day ‐7, 0, 14, 28, 42 HARS ‐ Somatic Score (clinician‐rated): day ‐7, 0, 14, 28, 42 Secondary outcomes: HARS ‐ Total Score, Psychic Score (clinician‐rated): day ‐7, 0, 14, 28, 42 HDRS (clinician‐rated): day ‐7, 0, 14, 28, 42 SCL‐90‐R ‐ Total Score, Anxiety Subscore (self rated): day ‐7, 0, 14, 28, 42 CGI ‐ Improvement Scale (clinician‐rated): 0, 14, 28, 42 CGI ‐ Severity Scale (clinician‐rated): 0, 14, 28, 42 Adverse events: day ‐7, 0, 14, 28, 42 All‐cause drop‐outs: n = 14 (St. John's wort extract), n = 13 (placebo) Drop‐outs due to adverse effects: n = 0 (St. John's wort extract), n = 2 (placebo) Reported drug‐related adverse effects: St. John's wort extract: abdominal pain (n = 1, 1.3%), arthritis (n = 1, 1.3%), arrhythmia (n = 1, 1.3%), bronchitis (n = 2, 2.7%), cystitis (n = 1, 1.3%), headache (n = 2, 2.7%), neuralgia (n = 1, 1.3%) Placebo: back pain (n = 3, 3.9%), gastroenteritis (n = 1, 1.3%), influenza‐like symptoms (n = 1, 1.3%) 10.7% of the St. John's wort extract group and 5.4% of the placebo group reported adverse effects. No information if treatment groups differed in the number of adverse effects was provided. No statistically significant differences between groups were found regarding the physicians' and participants' ratings of the tolerability of treatments |
|
| Notes |
Funding: Lichtwer Pharma AG, Berlin, Germany, was involved in the trial (Hans Murck is employee of Lichtwer Pharma AG); however, it was not stated how Ethics approval: obtained Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomised treatment phase" (p. 295); no information provided about random sequence |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "Double blind, randomised treatment phase" (p. 295); insufficient information provided about who was and was not blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Drop‐out rate in total sample: 17.9%; originally, 151 participants were randomised; only participants who received at least 1 outcome assessment were included in the ITT sample: no information was provided how the original 151 participants were distributed between the 2 groups, 2 participants abandoned treatment before the first outcome assessment and were excluded from ITT sample (n = 149) that was used for statistical analyses; "During the double‐blind treatment phase, no participant was excluded in the LI160 group, two drop‐outs occurred in the placebo group [one due to a serious adverse event (apoplexy), the other due to an adverse event (swelling and burning underneath the eyes)] (p. 296); reasons for missing outcome data were very unlikely to be related to true outcome (only 2 participants withdrew due to adverse events in the placebo group); "The last observation was carried forward" (p. 296) was used to replace missing values |
| Selective reporting (reporting bias) | Low risk | No protocol available; generally accepted outcomes were used and data were reported for a broad variety of primary and secondary outcomes |
| Other bias | High risk | "Patient population underwent a 7‐day, single blind placebo run‐in period, receiving one placebo capsule in the morning and one in the evening. At the end of this period, placebo responders, defined as those patients exhibiting a HDRS‐SOM decrease of more than four points, were excluded from the further trial" (p. 295), single‐blind placebo run‐in phase was conducted before the randomisation; however, no participant had to be excluded in this placebo run‐in phase; "Compliance was assessed by immediate pill count at visits. The compliance ranged from 85.7 to 110.1% and was therefore well in the predefined range of 75‐125%" (p. 295); for initial symptom severity "there were no relevant differences between the treatment groups" (p. 296) |
Wang 2003.
| Methods | Randomised, parallel‐group, double‐blind, fixed‐dose trial | |
| Participants |
n(randomised/ITT) = 140 Diagnosis: persistent somatoform pain disorder (ICD‐10) Inclusion criteria: HDRS score > 17; pain continued over the last 6 months at least 5 days/week and 30 minutes/day Exclusion criteria: depressed, suicidal, and chronically ill people Age: mean (± SD) 44.50 ± 21.16 years, range ns; sex: 47.1% women; mean (± SD) length of time since diagnosis of a somatoform disorder: 7.2 ± 5.3 months; country: China; setting: inpatient |
|
| Interventions | Trazodone (n = 70; SARI; mean dose = 100 mg/day, max dose = 100 mg/day; 1 capsule twice/day) Ibuprofen (n = 70; NSAID; mean dose = 400 mg/day, max dose = 400 mg/day; 1 capsule twice/day) Treatment duration: 28 days |
|
| Outcomes |
Primary outcome: Effectiveness rate concerning reduction of pain (defined by authors, no further information about that measure provided) (clinician‐rated): week 4 Secondary outcomes: HDRS (clinician‐rated): baseline, week 4 Treatment Emergent Symptom Scale: week 4 All‐cause drop‐outs: n = 0 (trazodone), n = 0 (ibuprofen) Drop‐outs due to adverse effects: n = 0 (trazodone), n = 0 (ibuprofen) Reported drug‐related adverse effects: Trazodone: hypersomnia, nausea, poor appetite, lethargy Ibuprofen: reduced white blood cells (n = 3), allergy (n = 1), stomach irritation (n = 10) No information about the total rate of participants with drug‐related adverse events was provided. The groups differed significantly with regard to the Treatment Emergent Symptom Scale ‐ Total Score |
|
| Notes |
Funding: no information was provided Ethics approval: no information provided Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomly divided into two groups" (p. 70); no information provided about random sequence |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "The clinical effects and side‐effects were double‐blind evaluated" (p. 70); trial authors stated that the trial pharmacist put the trazodone and the placebo in similar capsules, both participants and physicians did not know the treatment condition until the end of the treatment (p. 71) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the study and there were no losses to follow‐up and no treatment withdrawals |
| Selective reporting (reporting bias) | High risk | No protocol available; generally accepted outcomes were used; however, treatment effects were assessed with validated scales only for secondary outcomes such as depression and adverse effects; important primary outcome was assessed with non‐validated clinician‐rated scale that was constructed by the study authors |
| Other bias | Low risk | No significant differences between groups regarding gender, age, duration of symptoms, and diagnosis (p. 71); there were no significant differences between groups in the HDRS at baseline (p. 71) |
Xu 2004.
| Methods | Randomised, parallel‐group, flexible‐dose trial | |
| Participants |
n(randomised/ITT) = 70 Diagnosis: somatoform pain disorder (CCMD‐III) Inclusion criteria: pain symptom presentation of ≥ 6 months; the pain that the participants experience from onset and over the course of disorder was related to the emotional stability or to psychosocial problems Exclusion criteria: pain explained by a somatic disorder Age: mean 52 years, range 26‐73 years; sex: 60.0% women; mean length of time since diagnosis of a somatoform disorder: 10.65 months; country: China; setting: outpatient |
|
| Interventions | Venlafaxine (n = 35; SNRI; mean dose = 132.4 mg/day, max dose = 150 mg/day; 1 capsule twice/day) Amitriptyline (n = 35; TCA; mean dose = 127.3 mg/day, max dose = 150 mg/day; 2 capsules twice/day) Treatment duration: 28 days |
|
| Outcomes |
Primary outcome: No primary outcome was assessed Secondary outcomes: Treatment effectiveness score based on rating of degree of pain, frequency how often they felt the pain, improvement of the activity daily living quality (no further information about that measure provided) (clinician‐rated): baseline, day 4, week 1, 2, 4 Treatment Emergent Symptom Scale: day 4, week 1, 2, 4 All‐cause drop‐outs: n = 0 (venlafaxine), n = 0 (amitriptyline) Drop‐outs due to adverse effects: n = 0 (venlafaxine), n = 2 (amitriptyline) Reported drug‐related adverse effects: Venlafaxine: dry mouth (n = 4, 11.4%), constipation (n = 5, 14.3%), blurred vision (n = 4, 11.4%), increased heart rate (n = 2, 5.7%), hypersomnia (n = 3, 8.6%), headache (n = 2, 5.7%), dizziness (n = 3, 8.6%), nausea (n = 5, 14.3%) Amitriptyline: dry mouth (n = 20, 57.1%), constipation (n = 18, 51.4%), blurred vision (n = 17, 48.6%), increased heart rate (n = 15, 42.9%), hypersomnia (n = 21, 60.0%), headache (n = 5, 14.3%), dizziness (n = 7, 20.0%), nausea (n = 7, 20.0%). With the exception of dizziness and nausea, all mentioned adverse effects appeared significantly more often in the amitriptyline compared with venlafaxine. No information about the total rate of participants with drug‐related adverse events was provided |
|
| Notes |
Funding: Southwest Pharmaceutical Co., Ltd., Chengdu, Sichuan (venlafaxine); Hunan Dongting Pharmaceutical Co., Deshan, Changde City, Hunan (amitriptyline) Ethics approval: no information provided |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "70 […] outpatients […] were randomly divided" (p. 397); no information provided about random sequence |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information about methods of blinding participants and study personnel provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the study and there were no losses to follow‐up and no treatment withdrawals |
| Selective reporting (reporting bias) | High risk | No protocol available; generally accepted outcomes were used; however, only clinician‐rated and no participant self rated measures were used. Furthermore, only non‐validated scales defined by the trial authors were administered |
| Other bias | High risk | Significant differences between groups regarding sex, age, duration of symptoms, occupational status, and pain regions at baseline were identified (p. 398) |
Yang 2006.
| Methods | Randomised, parallel‐group trial (unclear if this study was a fixed‐ or flexible‐dose trial) | |
| Participants |
n(randomised/ITT) = 70 Diagnosis: somatoform disorder (CCMD‐III) Inclusion criteria: inpatients of the Kangning Hospital of Chaoyang City Exclusion criteria: no information provided Age: mean 41.60 years, range 20‐72 years; sex: 70.0% women; mean length of time since diagnosis of a somatoform disorder: 3‐144 months; 61.4% were 12‐36 months; country: China; setting: inpatient |
|
| Interventions | Venlafaxine (n = 35; SNRI; mean dose: ns, max dose: ns; no information about mode/frequency of administration provided) Amitriptyline (n = 35; TCA; mean dose = 127.3 mg/day, max dose = 150 mg/day; no information about mode/frequency of administration provided) Treatment duration: 42 days |
|
| Outcomes |
Primary outcome: No primary outcome was assessed Secondary outcomes: HDRS ‐ Total Score, responder rate (clinician‐rated): baseline, week 1, 2, 4, 6 HARS (clinician‐rated): baseline, week 1, 2, 4, 6 No information provided how adverse effects were assessed All‐cause drop‐outs: n = 0 (venlafaxine), n = 0 (amitriptyline) Drop‐outs due to adverse effects: n = 0 (venlafaxine), n = 2 (amitriptyline) Reported drug‐related adverse effects: Venlafaxine: poor digestion (n = 12, 23.3%), insomnia (n = 3, 8.6%), headache (n = 3, 8.6%), sweating (n = 5, 14.3%), hypertension (n = 2, 5.7%) Amitriptyline: dry mouth (n = 10, 28.6%), nervousness/alertness (n = 10, 28.6%), constipation (n = 10, 28.6%), increased heart rate (n = 3, 8.6%), blurred vision (n = 6, 17.1%), whole body did not feel well (n = 3, 8.6%) No information about the total rate of participants with drug‐related adverse events was provided or if both treatment groups differed in the number of adverse effects |
|
| Notes |
Ethics approval: no information provided Funding: no information provided Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "70 patients […] were randomly assigned" (p. 262); no information provided about random sequence |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information about methods of blinding participants and study personnel provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants completed the study and there were no losses to follow‐up and no treatment withdrawals |
| Selective reporting (reporting bias) | High risk | No protocol available; generally accepted outcomes were used; however, treatment effects were only reported for the secondary outcomes such as depression, anxiety, and adverse effects; important primary outcomes such as severity of physical symptoms were not assessed |
| Other bias | Low risk | There was no significant difference between groups in age, gender, duration of symptoms, or HARS and HDRS scores at baseline (p. 262) |
Ye 2006.
| Methods | Randomised, parallel‐group, flexible‐dose trial | |
| Participants |
n(randomised/ITT) = 70 Diagnosis: somatisation disorder (CCMD‐III) Inclusion criteria: aged 18‐65 years Exclusion criteria: people with diabetes; history of any liver, lung, kidney, or related diseases; people with any type of drug dependence; pregnant or breastfeeding women; people who used MAOIs and other antidepressants in the 2 week before treatment started Age: mean (± SD) 43.50 ± 10.0 years, range ns; sex: 72.9% women; mean (± SD) length of time since diagnosis of a somatoform disorder: 7.5 ± 5.7 months; country: China; setting: ns |
|
| Interventions | Mirtazapine (n = 35; NaSSA; mean dose: ns, max dose = 45 mg/day; medication was administered as pills; no information about frequency of administration provided) Clomipramine (n = 35; TCA; mean dose: ns, max dose = 150 mg/day; medication was administered as pills; no information about frequency of administration provided) Treatment duration: 56 days |
|
| Outcomes |
Primary outcome: SCL‐90‐Somatisation Subscore (self rated): baseline, week 2, 4, 8 Secondary outcomes: SCL‐90 ‐ Interpersonal Sensitivity, Obsessive‐compulsive, Depression, Anxiety, Hostility, Phobic Anxiety, Paranoid Ideation, Psychoticism Subscore (self rated): baseline, week 2, 4, 8 CGI ‐ Improvement Scale (clinician‐rated): week 8 Treatment Emergent Symptom Scale: week 2, 4, 8 All‐cause drop‐outs: n = 2 (mirtazapine), n = 3 (clomipramine) Drop‐outs due to adverse effects: n = 2 (mirtazapine), n = 2 (clomipramine) Reported drug‐related adverse effects: Mirtazapine: dry mouth (n = 4, 11.4%), constipation (n = 3, 8.6%), increased heart rate (n = 1, 2.9%), blurred vision (n = 1, 2.9%), headache (n = 4, 11.4%), fatigue (n = 3, 8.6%), increased body weight (n = 6, 17.1%) Clomipramine: dry mouth (n = 17, 48.6%), constipation (n = 9, 25.7%), nausea, (n = 7, 20.0%), loss of appetite (n = 6, 17.1%), increased heart rate (n = 13, 37.1%), hypotension (n = 6, 17.1%), blurred vision (n = 18, 51.4%), tremor (n = 7, 20%), micturition disturbances (n = 9, 25.7%), headache (n = 6, 17.1%), fatigue (n = 5, 14.3%), increased body weight (n = 9, 25.7%) Dry mouth, constipation, increased heart rate, and blurred vision appeared statistically more often in the mirtazapine group than in the clomipramine group. Dry mouth, constipation, nausea, loss of appetite, increased heart rate, hypotension, blurred vision, tremor, and micturition disturbances appeared statistically more often in the clomipramine group than in the mirtazapine group. No information about the total rate of participants with drug‐related adverse events was provided |
|
| Notes |
Funding: no information provided Ethics approval: no information provided Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Seventy patients […] were randomly divided into two groups" (p. 14); no information provided about random sequence |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information about methods of blinding participants and study personnel provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Drop‐out rate in total sample: 7.1%; although reasons for missing outcome data were partly likely to be related to true outcome, missing outcome data were balanced in number with similar reasons for missing data across groups; missing values were not replaced, statistical analyses were based only on the sample of participants who complied with the protocol |
| Selective reporting (reporting bias) | Unclear risk | No protocol available; generally accepted outcomes were used |
| Other bias | Low risk | There were no statistical significant differences between groups regarding sex, age, and mean duration of somatoform symptoms (p. 14) |
Zhao 2006.
| Methods | Randomised, parallel‐group, double‐blind, flexible‐dose trial | |
| Participants |
n(randomised/ITT) = 60 Diagnosis: somatisation disorder (CCMD‐III) Inclusion criteria: no further information provided Exclusion criteria: other mental disorders, history of physical illnesses, drug dependence, pregnant or breastfeeding (or both) women; taking MAOI or other antidepressants during the first 2 weeks of treatment Age: mean (± SD) 38.15 ± 7.58 years, range 25‐51 years; sex: 68.3% women; mean (± SD) length of time since diagnosis of a somatoform disorder: 61.8 ± 39 months; country: China; setting: inpatient |
|
| Interventions | Mirtazapine (n = 30; NaSSA; mean dose = 40 mg/day, max dose = 45 mg/day; medication was administered as capsules; no information about frequency of administration provided) Amitriptyline (n = 30; TCA; mean dose = 116.3 mg/day, max dose = 200 mg/day; medication was administered as capsules; no information about frequency of administration provided) Treatment duration: 56 days |
|
| Outcomes |
Primary outcome: SCL‐90 ‐ Somatisation Subscore (self rated): baseline, week 2, 4, 8 Secondary outcomes: SCL‐90 ‐ Interpersonal Sensitivity, Obsessive‐compulsive, Depression, Anxiety, Hostility, Phobic Anxiety, Paranoid Ideation, Psychoticism Subscore (self rated): baseline, week 2, 4, 8 CGI ‐ Improvement Scale: week 8 Treatment Emergent Symptom Scale: week 2, 4, 8 All‐cause drop‐outs: 7 participants dropped out; however, it was unclear how they were distributed among groups Drop‐outs due to adverse effects: no information provided Reported drug‐related adverse effects: Mirtazapine: weight gain (n = 14, 46.7%), hypersomnia (n = 3, 10.0%), hyperventilation (n = 4, 13.3%), hypotension (n = 2, 6.7%), blurred vision (n = 1, 3.3%), headache (n = 8, 26.7%), constipation (n = 10, 33.3%), nausea (n = 6, 20.0%) Amitriptyline: weight gain (n = 2, 6.7%), hypersomnia (n = 15, 50.0%), hyperventilation (n = 15, 50.0%), hypotension (n = 12, 40.0%), blurred vision (n = 17, 56.7%), micturition disturbances (n = 7, 23.3%), headache (n = 7, 23.3%), constipation (n = 12, 40.0%), nausea (n = 7, 23.3%) Weight gain appeared statistically more often in the mirtazapine group than in the amitriptyline group. Hypersomnia, hyperventilation, hypotension, blurred vision, micturition disturbances, headache, constipation, and nausea appeared statistically more often in the clomipramine group than in the mirtazapine group. No information about the total rate of participants with drug‐related adverse events was provided |
|
| Notes |
Funding: no information provided Ethics approval: no information provided Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "60 patients […] were randomly divided" (p. 175); no information provided about random sequence |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Trial authors stated that it was a double‐blind study (p. 175); both medications were administered in identical capsules (p. 175) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 7 drop‐outs (drop‐out rate in total sample: 11.7%) but no information about the reason for drop‐out and how drop‐outs were distributed among the 2 trial arms was provided; no information about how and if missing values were replaced in the statistical analyses was provided; in the result tables the number of originally randomised participants was depicted, therefore, it can be concluded that an ITT analysis with replaced missing values was conducted |
| Selective reporting (reporting bias) | Unclear risk | No protocol available; generally accepted outcomes were used |
| Other bias | Low risk | There were no significant differences in demographics and the duration of the symptoms between groups at baseline (p. 175) |
Zitman 1991.
| Methods | Randomised, parallel‐group, cross‐over, double‐blind, fixed‐dose trial | |
| Participants |
n(randomised/ITT) = 45 Diagnosis: psychogenic pain disorder (307.80) (DSM‐III‐R) Inclusion criteria: aged 30‐60 years Exclusion criteria: serious psychiatric disease necessitating immediate treatment; alcohol or illicit drug dependence; renal, hepatic, or cardiac disorders; epilepsy; glaucoma; hypertension; prostate dysfunction; intake of medication consisting of enzyme‐inducing drugs; use of antidepressants or neuroleptic drugs (or both) during the previous year Aged: mean (± SD) 41.20 ± 11.00 years, range ns; sexd: 61.1% women; mean (± SD) length of time since diagnosis of a somatoform disordere : 113.4 ± 111.6 months; country: The Netherlands; setting: outpatient |
|
| Interventions | Amitriptyline + flupentixol (n = 16d; TCA + AP; mean dose amitriptyline = 75.0 mg/day, mean dose flupentixol = 3.0 mg/day, max dose amitriptyline = 75 mg/day, max dose flupentixol = 3 mg/day; medication was administered as capsules; no information about frequency of administration provided) Amitriptyline alone (n = 18d; TCA; mean dose = 116.3 mg/day, max dose = 200 mg/day; medication was administered as capsules; no information about frequency of administration provided) Treatment duration: 35 days |
|
| Outcomes |
Primary outcomes: Participant's estimation of the amount of pain felt at that time as a percentage of the pain on day 1 (self rated): first day of week 3, 5, 8, 10, 12, 15, 17 Pain diaries (numerical scale for rating pain intensity and the trouble the pain caused) (self rating): baseline, week 5, 7, 9, 12, 14, 16 Secondary outcomes: Zung Depression Scale (self rated): first day of week 1, 3, 5, 8, 10, 12, 15, 17 HDRS (clinician‐rated): first day of week 1, 3, 5, 8, 10, 12, 15, 17 Adverse effects registration: combined checklist for subjective adverse effects and withdrawal symptoms developed by the study authors, clinical screening for EPS, Abnormal Involuntary Movement Scale: first day of week 1, 3, 5, 8, 10, 12, 15, 17 All‐cause drop‐outs: 9 participants dropped out; however, it was not clear how they were distributed among groups Drop‐outs due to adverse effects: no information provided Reported drug‐related adverse effects: It was only mentioned that the adverse effect dry mouth became significantly more serious during the treatment in the amitriptyline as well as in the amitriptyline + flupentixol groups. Withdrawal symptoms of antidepressants and neuroleptics were included in the checklist, but no statistically significant increase in the severity of these symptoms was observed. Regarding the extrapyramidal side effects scored on the Abnormal Involuntary Movement Scale and the EPS, there was no statistically significant difference between groups. The mean scores were very low. On the Abnormal Involuntary Movement Scale (range 0‐42) they were 0.13‐0.67, and on the EPS all participants scored 0 |
|
| Notes |
Funding: H. Lundbeck A/S, Copenhagen supplied the medication and financial support for the trial Ethics approval: obtained Underlined outcome measures and time points were considered in the meta‐analytical part of the review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomized [...] crossover trial" (p. 26); "there were 2 treatment schemes to which the patients were randomly assigned" (p. 26); no information provided about random sequence |
| Allocation concealment (selection bias) | Unclear risk | No information about methods of allocation concealment provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Double‐blind crossover trial" (p. 26); "Four types of tablets were used: 25mg AT [amitriptyline], 1 mg FP [flupentixol], placebo tablets looking identical to AT tablets (ATpi) and placebo tablets looking identical to FP tablets (FPpl)" (p. 26) |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information about methods of blinding outcome assessment provided |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Reasons for missing outcome data and distribution of drop‐outs were not provided. For this reason, we could not judge if missing outcome data were balanced in number across intervention with similar reasons for missing data across groups. Total sample size was very small and the total drop‐out rate was relatively high (20.0%). Therefore, the chance of a bias produced by incomplete data was high. Missing values were not replaced, statistical analyses were based only on the sample of participants who complied with the protocol (p. 27) |
| Selective reporting (reporting bias) | Unclear risk | No protocol available; generally accepted outcomes were used |
| Other bias | Low risk | "The 2 groups to which the patients had been randomly assigned did not show statistically significant differences with respect to demographic variables (sex, age, marital status, level of education) or to pain variables (localization and duration of pain, baseline pain intensity and pain trouble)" (p. 27); "is questioned if a higher dose of flupentixol [would have] have yielded better analgesic effects" (p. 29); in this cross‐over trial, the order receiving a treatment was randomised: "there were 2 treatment schemes to which the patients were randomly assigned" (p. 26), in addition, the means/SD for pain intensity/burden and depression in Table 1 and 3 were given separately for group 1 and 2 for day 15/29/50/64/78/99/113 |
AP: antipsychotic; CCMD: Chinese Classification of Mental Disorders; CGI: Clinical Global Impression Scale; DSM: Diagnostic and Statistical Manual of Mental Disorders; EPS: Extrapyramidal Side Effects; GAD: generalised anxiety disorder; HARS: Hamilton Anxiety Rating Scale; HDRS: Hamilton Depression Rating Scale; ICD: International Classification of Diseases; ITT: intention to treat; LOCF: last observation carried forward; MAOI: monoamine oxidase inhibitor; max: maximum; MDD: major depressive disorder; MSD: multisomatoform disorder; MUPS: medically unexplained physical symptoms; n: number; NaSSA: noradrenergic and specific serotonergic antidepressant; NP: natural product; ns: not specified; NSAID: non‐steroidal anti‐inflammatory drug; PHQ: Patient Health Questionnaire; SAD: somatoform autonomic dysfunction; SCL: Symptom Checklist; SD: standard deviation; SNRI: serotonin noradrenaline reuptake inhibitor; SOMS: Screening for Somatoform Symptoms; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant; TeCA: tetracyclic antidepressant; VAS: visual analogue scale.
Notes: percentages, numbers, and mean values/SDs regarding age, sex, length of time since the diagnosis of somatoform disorder, dose, drop‐outs, and adverse effects relate to the originally randomised sample. Deviations from this rule are indicated with a footnote.
aStudy design included a single‐blind placebo run‐in phase after which 'placebo‐responders' were excluded from the further trial.
bValues refer to the ITT sample that was not the same as the originally randomised sample. Usually participants abandoned before the first outcome assessment or before they received at least 1 dose of a study medication were excluded from the ITT sample.
cThe trial originally involved 4 study groups: group A: amitriptyline + psychotherapy, group B: amitriptyline + support, group C: placebo + psychotherapy, group D: placebo + support. The following study‐related information referred only to the comparison between group B and D.
dMean values/SDs and percentages relate to the completer‐sample.
eFor this study, only a conference abstract was available. Authors were contacted in order to obtain more information about the study; however, we received no reply.
fThe study was included in the narrative but not in the meta‐analytical part of the review.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Altamura 2003 | No randomisation (we contacted the authors because the article included no information about randomisation) |
| Ballbe 1970 | People with hysteria and anxiety neurosis (focus on conversion symptoms) |
| Bratfos 1967 | People with neurosis |
| Cui 2004 | People with hysteria |
| Davis 1988 | People with a specific functional syndromes (non‐ulcer dyspepsia) |
| Farnbach 2013 | Efficacy of quetiapine fumarate as a adjunctive therapy to current pain treatment of participants was examined |
| Fukuda 1971 | People with psychosomatic symptoms (primary diagnoses: depression, anxiety, somatic disorders) |
| Hasegawa 1977 | People psychosomatic disease and neurosis |
| Holdevici 1995 | People with symptoms of anxious neurosis and somatoform disorders, no information provided how participants were diagnosed and how long participants' symptoms had to persist |
| Kozian 2003 | Case report |
| Lee 2012 | Anti‐stress effect of Korean red ginseng in a general population with various stress‐related somatic symptoms was examined |
| Liu 2011 | People fulfilling CCMD‐III criteria of a depressive disorder with a co‐morbid somatic diagnosis were included |
| Loldrup 1989 | 45.1% of the participants had tension headache (not classified as somatoform disorder) |
| Onghena 1993 | Cross‐over design applied but no data for the first study phase available |
| Pach 1976 | People with functional physical symptoms in combination with anxious, depressive mood; functional physical symptoms included predominantly different heart sensations, circulation problems or tension headache |
| Poinso 1988 | People with reactive depression |
| Raich 1966 | Anxious somatising neurotic medical clinic participants; no information provided how participants were diagnosed and how long participants' symptoms had to persisted |
| Smith 1971 | People with psychoneurotic anxiety with/without depressive symptoms |
| Smouvelich 1996 | People with cardioneurotic disease with an anxiety disorder (agoraphobia, generalised anxiety disorder, panic disorder) and a somatoform disorder (somatisation disorder, hypochondriasis) |
| Tanum 1996 | People with specific functional syndromes (irritable bowel syndrome, non‐ulcer dyspepsia) |
| Tsutsui 1984 | People with a disorder in the field of internal medicine (psychosomatic medicine), geriatric medicine (focus on various sleeping problems with the frequency of symptoms of ≥ 4 times/week) |
| Tsutsui 1985 | People with a disorder in the field of internal medicine (psychosomatic medicine), geriatric medicine (focus on various sleeping problems with the frequency of symptoms of ≥ 4 times/week) |
| Tsutsui 1986 | People with a disorder in the field of internal medicine (psychosomatic medicine), geriatric medicine (focus on various sleeping problems with the frequency of symptoms of 4 times or more per week) |
| Tsutsui 1987 | People with a disorder in the field of internal medicine (psychosomatic medicine), geriatric medicine (focus on various sleeping problems with the frequency of symptoms of ≥ 4 times/week) |
| Tsutsui 1992 | People with a disorder in the field of internal medicine (psychosomatic medicine) and geriatric medicine (focus on insomnia) |
| Turkington 2002 | People with 1 specific functional syndrome (prostatodynia) |
| Xu 2006 | Interventions: paroxetine vs. acupuncture (no placebo or other medication group) |
| Zang 1991 | People fulfilling diagnostic criteria of neurosis defined by the Chinese Neuropsychiatric Committee in October 1985 (these criteria do not correspond to the inclusion criteria of the review regarding the diagnosis) |
| Zhao 1989 | People with neurosis (included anxiety, phobia, somatisation, and compulsion) and depression |
| Zitman 1990 | People with chronic pain of various origins (only 35.9% fulfilled criteria of psychogenic pain disorder) |
CCMD: Chinese Classification of Mental Disorders.
Characteristics of ongoing studies [ordered by study ID]
Agger 2014.
| Trial name or title | Imipramine Treatment for Patients with Multi‐Organ Bodily Distress Syndrome (STreSS‐3) |
| Methods | Randomised, placebo‐controlled, double‐blind, flexible‐dose trial |
| Participants | n = 140 Diagnosis: bodily distress syndrome multi‐organ type with symptoms for more than 3 of 4 symptom categories (resembles somatisation disorder in ICD‐10) a more specific set of diagnostic criteria Inclusion criteria: moderate or severe impact on daily life; symptoms lasting for at least 2 years; aged 20‐50 years; born in Denmark or have Danish parents; participant understands, speaks, writes, and reads Danish Exclusion criteria: presence of other physical of psychiatric condition; continuous antidepressant treatment because of moderate or severe depression; other severe psychiatric disorder that demands treatment or suicidality; lifetime diagnosis of psychoses, mania, or depression with psychotic symptoms (International Classification of Diseases‐10: F20‐29, F30‐31, F32.3, F33.3); abuse of alcohol, narcotics, or drugs; pregnancy, breastfeeding, or current pregnancy wish; fertile women must use effective contraception; treatment with all pain‐modulating drugs, e.g. all analgesics, antidepressants, antiepileptics, and other types of medication with pain‐relieving properties must be discontinued at least 2 weeks before the treatment phase; imipramine treatment in sufficient dosage within the last year, i.e. 25 mg daily continuously for at least 8 weeks; allergy to study medication or excipients in study medication; people with previous myocardial infarction, congestive heart failure, signs of conduction defects or abnormalities on electrocardiograph, narrow‐angle glaucoma, porphyria, inherited galactose intolerance, epilepsy, hepatic insufficiency, and severe renal impairment; simultaneous use of: antipsychotics, oral anticoagulants, diuretics, sympathomimetics and central nervous system‐stimulating drugs (amphetamine‐like drugs), all serotonergic drugs, e.g. selective serotonin reuptake inhibitor, serotonin noradrenaline reuptake inhibitor, and tricyclic antidepressant, the dietary supplement hypericum perforatum, non‐selective, irreversible, or selective reversible monoamine oxidase inhibitors, triptans, tramadol, pethidine and tryptophan, cimetidine, quinidine, clonidine, fluconazole (antimycotics), clindamycin, clarithromycin, erythromycin, droperidol, levodopa, mefloquine, phenytoin, barbiturates, carbamazepine, bupropion, celecoxib, cinacalcet, duloxetine, fluphenazine, fluoxetine, gefitinib, moclobemide, paroxetine, sertraline, terbinafine, yohimbine, fluvoxamine, ciprofloxacin, or enoxacin |
| Interventions | Imipramine (n = 70, 25‐75 mg/day, medication administered as tablets) Placebo (n = 70) Treatment duration: 133 days (19 weeks) |
| Outcomes |
Primary outcomes: CGI ‐ Improvement Scale (clinician‐rated): week 13 Participant‐rated improvement of health since the beginning of the study: week 13 Secondary outcomes: Medical Outcomes Study Short‐Form 36‐Item Questionnaire (participant‐rated): week 1, 13 Visual Analogue Scale for pain and worst symptom (participant‐rated): week 1, 13 Symptom Checklist (participant‐rated): week 1, 3, 13 Functional Illness Checklist: week 1, 3, 13 World Health Organization Disability Assessment Schedule II (interview): week 1, 13 |
| Starting date | January 2012 (estimated study completion date: December 2014) |
| Contact information | Per K Fink, Dr. med; telephone: +4578464310; email: per.fink@aarhus.rm.dk Johanne L Agger, M.D.; telephone: +4578464344; email: johanne.agger@aarhus.rm.dk Research Clinic for Functional Disorders Recruiting Aarhus, Denmark, 8000 |
| Notes | Funding: Aarhus University Hospital |
n: number.
Differences between protocol and review
1) For literature search the following search terms were added: "somatoform disorder*", somatoform and "autonomic dysfunction", "somatic symptom disorder*","pain disorder".
Rationale: the former search strategy did not include these important terms although they are part of the title of the review and the inclusion criteria. Therefore, it was necessary to add them.
2) The syntax for the PsycINFO search was adapted.
Rationale: according to the changes in the search strategy (see the first difference between protocol and review), the syntax for the PsycINFO search had to be adapted.
3) We made changes to the searches of electronic databases: instead of the search in PubMed and EMBASE, we conducted an additional search in CENTRAL.
Rationale: we decided to run one search in CENTRAL because this database included all records from PubMed, indexed as publication type=RCT as well as records from EMBASE, obtained from a Cochrane RCT screening project.
4) In the section 'Types of interventions', we added the comparator intervention 'combination of medications'. Correspondingly, we added the comparison 'medication versus a combination of medications' to the section 'Data extraction and management'.
Rationale: the literature search obtained three studies where the efficacy of a combination of medications was compared with a single medication. In order to avoid excluding these studies and because they also fit the objectives of our review, we decided to add another comparison.
5) In the section 'Types of participants' under 'Participants characteristics', we modified the age criterion. We added the following sentence: "where a trial has defined adults to include those older than 65 years but most participants were under 65 years of age we included the trial; however, we excluded any trial that focused on older adults or where the mean age of participants was greater than 65 years'.
Rationale: we modified the inclusion criteria of people regarding age in order to prevent the exclusion of good‐quality studies only because they slightly exceed our originally defined age range.
6) In the section 'Types of participants' under 'Diagnosis', we deleted the originally required structured clinical interview for making the diagnoses.
Rationale: with screening the full texts of the potentially eligible studies for this review it became clear that requiring a structured clinical interview was too strict and inappropriate a criterion. There were several studies where a unstructured psychiatric interview was conducted in order to determine the diagnosis(es) of a participant.
7) In the section 'Types of participants' under 'Diagnosis', we added the DSM‐5 diagnosis 'somatic symptom disorder' and the Chinese Classification of Mental Disorders (CCMD‐III).
Rationale: the DSM‐5 was published in 2014 shortly after the protocol of this review had been published. In the protocol, we had already announced that the diagnostic criteria would be adapted as soon as the DSM‐5 was published. The criteria of somatoform disorders in the CCMD‐III correspond to the criteria of somatoform disorders in DSM‐IV or ‐III and in ICD‐10 or ‐9.
8) In the section 'Types of outcome measures', we made the following changes:
under 'Primary outcomes', we noted that the acceptability rate was also presented as risk ratio.
Rationale: the calculation of the risk ratio gives the opportunity to compare the treatment and comparator group for the number of drop‐outs.
under 'Secondary outcomes', we divided the originally combined outcome 'depression and anxiety' into two separate outcomes.
Rationale: during coding the characteristics and statistical values of the included studies we realised that in many studies qualitatively equivalent measures of depression and anxiety were available (e.g. the Hamilton Depression Rating Scale (HDRS) and the Hamilton Anxiety Rating Scale (HARS)). Therefore, it was impossible to decide which of these measures should be included. Furthermore, we decided against excluding one or both outcomes from the review because anxiety as well as depressive symptoms are import co‐morbid complaints in people with somatoform disorders.
under 'Secondary outcomes', we added that for the outcomes anxiety and depression the subscales of validated questionnaires assessing general psychopathology and VAS were acceptable.
Rationale: subscales of validated questionnaires assessing general psychopathology or VAS were accepted measures for the 'severity/intensity of MUPS'. We decided afterwards to accept them as eligible measures of depression and anxiety.
under 'Secondary outcomes', we noted that the drop‐out rate due to adverse effects was also presented as an RR.
Rationale: the calculation of the RR gives the opportunity to compare the treatment and comparator group for number of drop‐outs due to adverse effects.
11) In the section 'Types of outcome measures' under 'Secondary outcomes' we clarified that the treatment response referred to the primary outcome 'severity of MUPS'.
Rationale: during the coding of the statistical values of the included studies it became clear that the section about treatment response as outcome was not formulated clearly enough. Therefore, we tried to clarify this section.
12) In the section 'Subgroup analysis and investigation of heterogeneity', we changed the following aspects: Subgroup analyses were conducted only for the primary outcomes
Rationale: subgroup analyses have to be treated with caution, as they are hypothesis‐forming rather than hypothesis‐testing and should be reduced to a reasonable minimum.
13) In the section 'Sensitivity analysis', we changed the following analysis:
we changed exclusion of results based on mixed‐effects models or on intention‐to‐treat approach (last observation carried forward (LOCF)) into exclusion of results based on complete‐case analyses
Rationale: we changed this sensitivity analysis because we realised that a large number of studies dealt with missing values by applying the LOCF approach and only in a small number of trials conducted complete‐case analyses.
We added further sensitivity analyses:
exclusion of Chinese studies.
Rationale: the CCDANCTR retrieved many Chinese studies. While several of these were identified through routine searches of MEDLINE, EMBASE, and PsycINFO (used to inform the register), the provenance of some of the other Chinese studies in the CCDANCTR was less clear and probably dated back to an opportunistic search of Wang Fang Data (c/o The British Library) in 2007. Therefore, we decided to add a sensitivity analysis of excluding studies that were conducted in China. Unfortunately, none of the comparisons had enough studies available in order to conduct this sensitivity analysis.
We excluded the following sensitivity analyses that were originally proposed in the protocol from the review:
exclusion of CRCTs;
exclusion of CRCTs for which intracluster correlation coefficients were used;
for dichotomous outcomes only: exclusion of results based on available/observed cases.
Rationale: we excluded these sensitivity analyses because no or insufficient studies were available.
Sensitivity analyses were conducted only when at least 10 studies were available and only for the primary outcomes.
Rationale: results of sensitivity analyses with a very small number of studies are difficult to interpret. Furthermore, sensitivity analyses have to be treated with caution, as they are hypothesis‐forming rather than hypothesis‐testing and should be reduced to a reasonable minimum.
14) In the section 'Summary of findings tables', we deleted the sentence on reporting Summary of findings tables only for the medication versus placebo comparisons.
Rationale: several peer reviewers suggested that a 'Summary of findings' table for the tricyclic antidepressants versus new‐generation antidepressants and antidepressants alone versus antidepressants plus antipsychotic comparison should be added.
Contributions of authors
Preparation of the protocol:
Maria Kleinstäuber: developed and drafted the protocol.
Michael Witthöft, Andrés Steffanowski, Michael Lambert, Günter Meinhardt, Klaus Lieb, Wolfgang Hiller: supervised preparation of the protocol and gave feedback on the draft version of the protocol.
Preparation of the review:
Maria Kleinstäuber and Michael Witthöft independently reviewed titles and abstracts in the first step and screened full‐texts regarding eligibility of studies for the review in the second step.
Maria Kleinstäuber and Michael Witthöft independently appraised the quality of the included studies.
Maria Kleinstäuber and a research assistant trained in the data extraction procedure independently extracted data of the included studies.
Andrés Steffanowski, Wolfgang Hiller, Harm van Marwijk, and Michael Lambert supervised the preparation of the review process and arbitrated in the case of disagreements.
Sources of support
Internal sources
Johannes Gutenberg‐University of Mainz, Germany.
External sources
-
National Institute for Health Research, UK.
Funding for this review was provided by the NIHR Cochrane Incentive Award Scheme 2014
Declarations of interest
Maria Kleinstäuber: none known.
Michael Witthöft: none known.
Andrés Steffanowski: none known.
Harm van Marwijk: none known.
Wolfgang Hiller: none known.
Michael Lambert: none known.
New
References
References to studies included in this review
Altamura 1991 {published data only (unpublished sought but not used)}
- Altamura AC, Mauri MC, Regazzetti G, Coppola MT. L‐sulpiride in the treatment of somatoform disturbances: a double‐blind study with racemic sulpiride [L‐sulpiride nel trattamento dei disturbi somatoformi: uno studio in doppio cieco vs sulpiride racema]. Minerva Psichiatrica 1991;32(1):25‐9. [PubMed] [Google Scholar]
Aragona 2005 {published data only}
- Aragona M, Bancheri L, Perinelli D, Tarsitani L. Randomized double‐blind comparison of serotonergic (citalopram) versus noradrenergic (reboxetine) reuptake inhibitors in outpatients with somatoform, DSM‐IV‐TR pain disorder. European Journal of Pain 2005;9(1):33‐8. [DOI] [PubMed] [Google Scholar]
Eberhard 1988 {published data only}
- Eberhard G, Vonknorring L, Nilsson HL, Sundequist U, Bjorling G, Linder H, et al. A double‐blind randomized study of clomipramine versus maprotiline in patients with idiopathic pain syndromes. Neuropsychobiology 1988;19(1):25‐34. [DOI] [PubMed] [Google Scholar]
Han 2008a {published data only (unpublished sought but not used)}
- Han C, Pae CU, Lee BH, Ko YH, Masand PS, Patkar AA, et al. Venlafaxine versus mirtazapine in the treatment of undifferentiated somatoform disorder: a 12‐week prospective, open‐label, randomized, parallel‐group trial. Clinical Drug Investigation 2008;28(4):251‐61. [DOI] [PubMed] [Google Scholar]
Han 2008b {published data only (unpublished sought but not used)}
- Han C, Pae CU, Lee BH, Ko YH, Masand PS, Patkar AA, et al. Fluoxetine versus sertraline in the treatment of patients with undifferentiated somatoform disorder: a randomized, open‐label, 12‐week, parallel‐group trial. Progress in Neuro‐Psychopharmacology and Biological Psychiatry 2008;32(2):437‐44. [DOI] [PubMed] [Google Scholar]
Huang 2012 {published data only}
- Huang M, Luo B, Hu J, Wei N, Chen L, Wang S, et al. Combination of citalopram plus paliperidone is better than citalopram alone in the treatment of somatoform disorder: results of a 6‐week randomized study. International Clinical Psychopharmacology 2012;27(3):151‐8. [DOI] [PubMed] [Google Scholar]
Jiang 2005 {published data only}
- Jiang X, Yang K, Hu W, Zheng JG. A clinical effective analysis of venlafaxine in treatment of somatoform disorders. Chinese General Practice 2005;8(14):1177‐8. [Google Scholar]
Ju 2003 {published data only}
- Ju G. A study of venlafaxine in the treatment of somatization disorder. Medical Journal of Chinese People's Health 2003;15(10):590‐4. [Google Scholar]
Kong 2004 {published data only}
- Kong LS, Li J, Zhang X. Comparison study of domestic paroxetine and amitriptyline in treatment of somatoform disorders randomized double‐blind study. Medical Journal of Chinese People's Health 2004;10(12):733‐4, 742. [Google Scholar]
Kroenke 2006 {published data only (unpublished sought but not used)}
- Kroenke K, Benattia I, Musgnung J, Graepel J, Stahl S. Short‐term treatment of depressed and anxious primary care patients with multiple, unexplained somatic symptoms: a pilot study. Neuropsychopharmacology 2004;29(Suppl 1):S210. [Google Scholar]
- Kroenke K, Messina N, Benattia I, Graepel J, Musgnung J. Venlafaxine extended release in the short‐term treatment of depressed and anxious primary care patients with multisomatoform disorder. Journal of Clinical Psychiatry 2006;67(1):72‐80. [DOI] [PubMed] [Google Scholar]
Li 2006 {published data only}
- Li G, Jin W. Comparison study of paroxetine and paroxetine combined with quetiapine in treatment of somatoform disorder. Chinese Journal of Behavioral Medical Science 2006;15(7):598‐9. [Google Scholar]
Luo 2009 {published data only (unpublished sought but not used)}
- Luo YL, Zhang MY, Wu WY, Li CB, Lu Z, Li QW. A randomized double‐blind clinical trial on analgesic efficacy of fluoxetine for persistent somatoform pain disorder. Progress in Neuro‐Psychopharmacology and Biological Psychiatry 2009;33(8):1522‐5. [DOI] [PubMed] [Google Scholar]
Melzer 2009 {published data only}
- Kraft K. Additional benefit of Petasites hybridus (butterbur) in a four‐combination for somatoform disorders. Focus on Alternative and Complementary Therapies 2010;15(1):25‐6. [Google Scholar]
- Melzer J. Results of a randomized, placebo‐controlled pharmaco‐clinical study with a combined herbal preparation for the treatment of patients with somatoform disorders [Ergebnisse einer randomisierten, placebokontrollierten pharmakoklinischen Studie mit einem pflanzlichen Kombinationspreparat zur Behandlung von Patienten mit somatoformen Störungen]. Schweizerische Zeitschrift für Ganzheitsmedizin 2012;24(5):284‐6. [Google Scholar]
- Melzer J, Schrade E, Brattström A, Schellenberg R, Saller R. Herbal drug combination with and without butterbur (Ze 185) for the treatment of patients with somatization disorders: randomized, placebo‐controlled pharmaco‐clinical trial [Pflanzliches Kombinationspräparat mit und ohne Pestwurz (Ze 185) für die Behandlung von Patienten mit Somatisierungsstörungen: Eine randomisiert, placebokontrollierte pharmako‐klinische Studie]. Deutsche Gesellschaft für Psychiatrie, Psychotherapie und Neurologie (DGPPN) Congress; 2009 Nov 25‐28; Berlin, Germany. 2009.
- Melzer J, Schrader E, Brattström A, Schellenberg R, Saller R. Fixed herbal drug combination with and without butterbur (Ze 185) for the treatment of patients with somatoform disorders: randomized, placebo‐controlled pharmaco‐clinical trial. Phytotherapy Research 2009;23(9):1303‐8. [DOI] [PubMed] [Google Scholar]
Müller 2004 {published data only}
- Müller T, Mannel M, Murck H, Rahlfs VW. Treatment of somatoform disorders with St. John's wort: a randomized, double‐blind and placebo‐controlled trial. Psychosomatic Medicine 2004;66(4):538‐47. [DOI] [PubMed] [Google Scholar]
Muller 2008 {published data only}
- Müller JE, Wentzel I, Koen L, Niehaus DJ, Seedat S, Stein DJ. Escitalopram in the treatment of multisomatoform disorder: a double‐blind, placebo‐controlled trial. International Clinical Psychopharmacology 2008;23(1):43‐8. [DOI] [PubMed] [Google Scholar]
Ouyang 2006 {published data only}
- Ouyang H, Liu Y. Comparison of mirtazapine and amitriptyline in the treatment of somatoform disorders. Chinese Journal of Health Psychology 2006;14(5):560‐1. [Google Scholar]
Pilowsky 1990 {published data only}
- Pilowsky I, Barrow CG. A controlled study of psychotherapy and amitriptyline used individually and in combination in the treatment of chronic intractable 'psychogenic' pain. Pain 1990;40(1):3‐19. [DOI] [PubMed] [Google Scholar]
Sanada 2010 {published data only (unpublished sought but not used)}
- Sanada K, Mimura M, Noda Y, Hida M, Nakahara M, Tsukurimichi A, et al. Comparison of effect of paroxetine and milnacipran for outpatients with pain disorder conference abstract. European Neuropsychopharmacology 2010;20 Suppl 3:S543. [Google Scholar]
Volz 2000 {published data only}
- Möller HJ, Volz HP, Stoll KD. Psychopharmacotherapy of somatoform disorders: effects of opipramol on symptoms of somatization, anxiety, and depression. Acta Neuropsychiatrica 2003;15(4):217‐26. [DOI] [PubMed] [Google Scholar]
- Volz HP, Möller HJ, Reimann I, Stoll KD. Opipramol for the treatment of somatoform disorders results from a placebo‐controlled trial. European Neuropsychopharmacology 2000;10(3):211‐7. [DOI] [PubMed] [Google Scholar]
Volz 2002 {published data only}
- Anonymus. St. John's wort effective for somatoform disorders independent of depressive symptoms. Brown University Psychopharmacology Update 2003;14(2):3. [Google Scholar]
- Murck H, Moller HJ, Volz HP. St. John's wort extract (LI 160) in somatoform disorders ‐ results of a placebo‐controlled trial. European Neuropsychopharmacology 2002;12(Suppl 3):S342. [DOI] [PubMed] [Google Scholar]
- Volz HP, Murck H, Kasper S, Moller HJ. St John's wort extract (LI 160) in somatoform disorders: results of a placebo‐controlled trial. Erratum [Johanniskrautextrakt (LI 160) bei somatoformen Stoerungen: Ergebnisse einer plazebokontrollierten Studie. Erratum]. Psychopharmacology 2003;167(3):333. [Google Scholar]
- Volz HP, Murck H, Kasper S, Möller HJ. St John's wort extract (li 160) in somatoform disorders: results of a placebo‐controlled trial [Johanniskrautextrakt (LI 160) bei somatoformen Stoerungen: Ergebnisse einer plazebokontrollierten Studie]. Psychopharmacology 2002;164(3):294‐300. [DOI] [PubMed] [Google Scholar]
Wang 2003 {published data only}
- Wang YX, Ren ZH, Wang SJ. A double‐blind study on trazodone in the treatment of persistent somatoform pain disorder. Chinese Journal of Pain Medicine 2003;9(2):70‐2. [Google Scholar]
Xu 2004 {published data only}
- Xu J, Li S. Venlafaxine in the treatment of 35 cases of persistent somatoform pain disorder. Herald of Medicine 2004;23(1):397‐99. [Google Scholar]
Yang 2006 {published data only}
- Yang Z, Lu X, Liu J, Zhang WB. A control study of venlafaxine in the treatment of somatoform disorder. Journal of Clinical Psychosomatic Diseases 2006;12(4):262‐3. [Google Scholar]
Ye 2006 {published data only}
- Ye M, Xie Z, Wang J, Zheng W, Jiang D. Mirtazapine in treatment of somatization disorder. Clinical Medicine 2006;16(6):14‐5. [Google Scholar]
Zhao 2006 {published data only}
- Zhao H. A controlled study of mirtazapine and amitriptyline in somatization. Evaluation and Analysis of Drug‐Use in Hospitals of China 2006;6(3):175‐6. [Google Scholar]
Zitman 1991 {published data only}
- Zitman FG, Linssen ACG, Edelbroek PM, Kempen GMJ. Does addition of low‐dose flupenthixol enhance the analgesic effects of low‐dose amitriptyline in somatoform pain disorder?. Pain 1991;47(1):25‐30. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Altamura 2003 {published and unpublished data}
- Altamura AC, Di RA, Ermentini A, Guaraldi GP, Invernizzi G, Rudas N, et al. Levosulpiride in somatoform disorders: a double‐blind, placebo‐controlled cross‐over study. International Journal of Psychiatry in Clinical Practice 2003;7(3):155‐9. [DOI] [PubMed] [Google Scholar]
Ballbe 1970 {published data only}
- Ballbe R, Jocano H. Action of WY 3498 in hysteria and anxiety neuroses [Acción del WY 3498 en neurosis histéricas o ansiosas.]. Prensa medica argentina 1970;58(19):946‐50. [PubMed] [Google Scholar]
Bratfos 1967 {published data only}
- Bratfos O, Lingjaerde O, Salvesen C. Symptomatic relief of neurotic symptoms with tybamate: a double‐blind, placebo‐controlled study. Acta Psychiatrica Scandinavica 1967;43(3):282‐5. [DOI] [PubMed] [Google Scholar]
Cui 2004 {published data only}
- Cui GM, Wang XF, Liu JX. A controlled study of the treatment of electric acupuncture and low dose perphenazine on hysteria. Chinese Journal of the Practical Chinese with Modern Medicine 2004;4(19):2928‐9. [Google Scholar]
Davis 1988 {published data only}
- Davis RH, Clench MH, Mathias JR. Effects of domperidone in patients with chronic unexplained upper gastrointestinal symptoms: a double‐blind, placebo‐controlled study. Digestive Diseases and Sciences 1988;33(12):1505‐11. [DOI] [PubMed] [Google Scholar]
Farnbach 2013 {published data only}
- Farnbach P. A double‐blind, randomised, placebo‐controlled, flexible‐dose of 50 mg/day to 400 mg/day, phase IIIb study of the efficacy and safety of quetiapine fumarate (Seroquel XR) as an add‐on therapy in patients with chronic somatoform pain disorder. apps.who.int/trialsearch/Trial2.aspx?TrialID=ACTRN12610000603011 (accessed 24 October 2014).
Fukuda 1971 {published data only}
- Fukuda I, Kamide H, Higuchi H, Fujiwara K, Nakamura Y, Hayashi M. Comparative evaluation of the effect on the psychosomatic symptoms between medazepam (s‐804) and diazepam by means of double blind method [Comparative evaluation of the effect on the psychosomatic symptoms between medazepam (s‐804) and diazepam by means of double blind method]. Modern Medicine 1971;26(8):1564‐7. [PubMed] [Google Scholar]
Hasegawa 1977 {published data only}
- Hasegawa N, Namiki M, Kawakami K, Sasaki D, Suzuki J, Ueno K, Mizushima N, Tanaka M, Kitanaka I, Ogawa C. The clinical efficacy of id‐540, a psychotropic drug, for psychosomatic disease and neurosis: double‐blind comparative study. Rinsyotokenkyu 1977;54(6):2075‐85. [Google Scholar]
Holdevici 1995 {published data only}
- Holdevici I, Truia I, Girlasu O. The suggestive action of some 'placebo' medications in the balancing of patients with somatoform disorders. Revue Roumaine de Psychologie 1995;39(2):129‐38. [Google Scholar]
Kozian 2003 {published data only}
- Kozian R. Olanzapin in the therapy of a somatoform disorder [Olanzapin in der Therapie einer somatoformen Störung]. Fortbildung und Diskussion 2003;30:450‐7. [PubMed] [Google Scholar]
Lee 2012 {published data only}
- Lee JH, Kim B, Kang EH, Yu BH. Anti‐stress effect of Korean red ginseng (Panax ginseng C.A. Mayer) in healthy individuals with medically unexplained stress‐related somatic symptoms: a randomized, double‐blind, and placebo controlled clinical study. Proceedings of the 15th Pacific Rim College of Psychiatrists Scientific Meeting; 2012 Oct 25‐27; Seoul, Korea. 2012.
Liu 2011 {published data only}
- Liu Y, Liu Z. Short‐term deanxit in combination with sertraline treats somatic disease accompanied with depressive disorder. Study protocol (ChiCTR‐TRC‐11001732). www.chictr.org/en/proj/show.aspx?proj=2080 (accessed 24 October 2014).
Loldrup 1989 {published data only}
- Loldrup D, Langemarck M, Hansen H, Olesen J, Bech P. Clomipramine and mianserin in chronic idiopathic pain syndrome: a placebo controlled study. Psychopharmacology 1989;99(1):1‐7. [DOI] [PubMed] [Google Scholar]
Onghena 1993 {published data only}
- Onghena P, Cuyper H, Houdenhove B, Verstraeten D. Mianserin and chronic pain: a double‐blind placebo‐controlled process and outcome study. Acta Psychiatrica Scandinavica 1993;88(3):198‐204. [DOI] [PubMed] [Google Scholar]
Pach 1976 {published data only}
- Pach J, Waniek W. A comparison of the tranquilizing effect of fluspirilene and diazepam [Vergleichende Untersuchung zum Tranquilizereffekt von Fluspirilene und Diazepam]. Pharmakopsychiatrie, Neuro‐Psychopharmakologie 1976;9(2):61‐6. [DOI] [PubMed] [Google Scholar]
Poinso 1988 {published data only}
- Poinso Y, Gouvernet J, Sambuc R. A multicentre double‐blind trial of sulpiride versus toloxatone in patients with reactive depressions and somatization receiving non‐specialized care [Etude multicentrique en double insu sulpiride versus toloxatone dans les depressions reactionnelles avec somatisations en medecine generale]. Semaine des hopitaux 1988;64(17):1201‐5. [Google Scholar]
Raich 1966 {published data only}
- Raich WA, Rickels K, Raab E. A double‐blind evaluation of fenfluramine in anxious somatizing neurotic medical clinic patients. [1966]. Current Therapeutic Research, Clinical and Experimenta 1966;8(1):31‐3. [PubMed] [Google Scholar]
Smith 1971 {published data only}
- Smith ME. A controlled comparative study of doxepin and chlordiazepoxide in psychoneurotic anxiety. Journal of Clinical Pharmacology & New Drugs 1971;11(2):152‐6. [DOI] [PubMed] [Google Scholar]
Smouvelich 1996 {published data only}
- Smoulevich A, Tkhostov A, Ivanov S, Kolutskaya E. A study of the efficiency of therapy of cardioneurotic patients. Xth World Congress of Psychiatry, Madrid, Spain, 1996.
Tanum 1996 {published data only}
- Tanum L, Malt UF. A new pharmacologic treatment of functional gastrointestinal disorder: a double‐blind placebo‐controlled study with mianserin. Scandinavian Journal of Gastroenterology 1996;31(4):318‐25. [DOI] [PubMed] [Google Scholar]
Tsutsui 1984 {published data only}
- Tsutsui S, Katsunuma H, Hirai S, Katsura K, Suematsu H, Kikuchi N, et al. The clinical efficacy of brotizolam (we941), a hypnotic, by the double‐blind method in the field of internal medicine (psychosomatic medicine), geriatric medicine. Journal of Clinical and Experimental Medicine, Medicine in Progress 1984;131(6):412‐27. [Google Scholar]
Tsutsui 1985 {published data only}
- Tsutsui S, Katsura T, Kono T, Oosita A, Saito T, Namba T. The clinical evaluation of efficacy and safety of brotizolam (we 941), a hypnotic, in the fields of internal medicine and psychosomatic medicine: A comparative study by the double‐blind method. Medical Consultation & New Remedies 1985;22(1):41‐56. [Google Scholar]
Tsutsui 1986 {published data only}
- Tsutsui S, Katsura T, Kono T, Matsuzaki H, Yamamoto H, Yamaoka M, et al. The clinical evaluation of lormetazepam, a new hypnotic, in the field of internal medicine, psychosomatic medicine: a double‐blind comparative study with haloxazolam. Japanese Journal of Clinical and Experimental medicine 1986;63(3):1627‐64. [Google Scholar]
Tsutsui 1987 {published data only}
- Tsutsui S, Katsura T, Kono T, Katsura K, Namba T, Kawano T. Multi‐center double‐blind clinical trial on 450191‐s, a new hypnotic, in the field of psychosomatic medicine. Clinical Report 1987;1(8):3683‐99. [Google Scholar]
Tsutsui 1992 {published data only}
- Tsutsui S, Katsura T, Kono T, Ogawa C, Tanaka M. Dose‐finding study of sch161 in the field of psychosomatic medicine: double‐blind comparative study with placebo. Journal of Clinical Therapeutics and Medicine 1992;8(2):335‐56. [Google Scholar]
Turkington 2002 {published data only}
- Turkington D, Grant JB, Ferrier IN, Rao NS, Linsley KR, Young AH. A randomized controlled trial of fluvoxamine in prostatodynia, a male somatoform pain disorder. Journal of Clinical Psychiatry 2002;63(9):778‐81. [DOI] [PubMed] [Google Scholar]
Xu 2006 {published data only}
- Xu Y, Fu C. A comparative study of paroxetine versus acupuncture‐therapy in treatment of somatization disorder. Journal of Practical Medical Techniques 2006;13(13):2302‐3. [Google Scholar]
Zang 1991 {published data only}
- Zang DX. A self body double blind clinical study of L‐tryptophan and placebo in treated neurosis. Chinese Journal of Neurology and Psychiatry 1991;24(2):77‐80. [PubMed] [Google Scholar]
Zhao 1989 {published data only}
- Zhao H, Chang ZZ, Cao ML, Zang DX. A self‐body double‐blind trial of L‐tryptophan in neurosis and depression. Acta Academiae Medicinae Hubei 1989;10(3):256‐9. [Google Scholar]
Zitman 1990 {published data only}
- Zitman FG, Linssen ACG, Edelbroek PM, Stijnen T. Low‐dose amitriptyline in chronic pain: the gain is modest. Pain 1990;42(1):35‐42. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
Agger 2014 {published and unpublished data}
- Agger J, Schroder A, Fink P, Gormsen L, Jensen TS. Pharmacological treatment of multi‐organ bodily distress syndrome: a double‐blind, placebo‐controlled trial of the effects of imipramine (STreSS‐3). Journal of Psychosomatic Research 2013;75(2):196. [Google Scholar]
- Fink PK. Treatment of multi‐organ bodily distress syndrome. A double‐blinded placebo controlled trial of the effect of imipramine (STreSS‐3) ‐ Stress‐3. Study protocol. apps.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2011‐004294‐87‐DK (accessed 24 October 2014).
- Fink PK, Agger JL. Imipramine treatment for patients with multi‐organ bodily distress syndrome (STreSS‐3). Study protocol. clinicaltrials.gov/show/NCT01518634 (accessed 24 October 2014).
- Fink PK, Agger JL. Imipramine treatment for patients with multi‐organ bodily distress syndrome. Study protocol. www.controlled‐trials.com/mrct/trial/2272077/ (accessed 24 October 2014).
Additional references
Allen 2002
- Allen LA, Escobar JI, Lehrer PM, Gara MA, Woolfolk RL. Psychosocial treatments for multiple unexplained physical symptoms: a review of literature. Psychosomatic Medicine 2002;64(6):393‐50. [DOI] [PubMed] [Google Scholar]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313:1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 1980
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM III). 3rd Edition. Washington, DC: American Psychiatric Press, 1980. [Google Scholar]
APA 1994
- American Psychiatric Association. Diagnostic and Statistical Manual for Mental Disorders (DSM IV). 4th Edition. Washington, DC: American Psychiatric Press, 1994. [Google Scholar]
APA 2000
- American Psychiatric Association. Diagnostic and Statistical Manual for Mental Disorders (DSM‐IV‐TR). 4th edition, text revision. Washington, DC: American Psychiatric Press, 2000. [Google Scholar]
APA 2013
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM III).. Diagnostic and Statistical Manual of Mental Disorders. Fifth edition. DSM‐5. Washington, DC: American Psychiatric Press, 2013. [Google Scholar]
Atkinson 1985
- Atkinson JH, Kremer EF, Garfin SR. Psychopharmacological agents in the treatment of pain. Journal of Bone and Joint Surgery ‐ American Volume 1985;67A(2):337‐42. [PubMed] [Google Scholar]
AWMF 2012
- Arbeitsgemeinschaft der Wissenschaftlichen Medizinischen Fachgesellschaften. Guideline for the treatment of patients with non‐specific, functional and somatoform physical complaints [Leitlinie zum Umgang mit Patienten mit nicht‐spezifischen, funktionellen und somatoformen Körperbeschwerden]. AWMF 2012.
Barsky 1990
- Barsky AJ, Wyshak GL. Hypochondriasis and somatosensory amplification. British Journal of Psychiatry 1990;157:404‐9. [DOI] [PubMed] [Google Scholar]
Barsky 2005
- Barsky AJ, Orav EJ, Bates DW. Somatization increases medical utilization and costs independent of psychiatric and medical comorbidity. Archives of General Psychiatry 2005;62:903‐10. [DOI] [PubMed] [Google Scholar]
Beck 1961
- Beck AT, Erbaugh J, Ward CH, Mock J, Mendelsohn M. An inventory for measuring depression. Archives of General Psychiatry 1961;4:561‐71. [DOI] [PubMed] [Google Scholar]
Beck 1990
- Beck AT, Steer RA. Beck Anxiety Inventory Manual. San Antonio, TX: The Psychological Corporation, Harcourt Brace, 1990. [Google Scholar]
Bergin 1971
- Bergin AE. The evaluation of therapeutic outcomes. Handbook of Psychotherapy and Behavior Change. 1. New York: Wiley, 1971:217‐70. [Google Scholar]
Bonaz 2002
- Bonaz B, Baciu M, Papillon E, Bost R, Gueddah N, Bas JF, et al. Central processing of rectal pain in patients with irritable bowel syndrome: an fMRI study. American Journal of Gastroenterology 2002;97:654‐61. [DOI] [PubMed] [Google Scholar]
Butterweck 2003
- Butterweck V. Mechanism of action of St John's wort in depression ‐ what is known?. CNS Drugs 2003;17:539‐62. [DOI] [PubMed] [Google Scholar]
Chinese Society of Psychiatry 2001
- Chinese Society of Psychiatry. The Chinese Classification and Diagnostic Criteria of Mental Disorders Version 3 (CCMD‐3). Jinan: Chinese Society of Psychiatry, 2001. [Google Scholar]
Clayton 2005
- Clayton AH. Gender differences in clinical psychopharmacology. Journal of Clinical Psychiatry 2005;66:1191. [DOI] [PubMed] [Google Scholar]
Creed 2011
- Creed F, Barsky AJ, Leiknes KA. Epidemiology: prevalence, causes and consequences. In: Creed F, Henningsen P, Fink P editor(s). Medically Unexplained Symptoms, Somatisation and Bodily Distress. New York: Cambridge University Press, 2011. [Google Scholar]
De Waal 2004
- Waal MW, Arnold IA, Eekhof JA, Hemert AM. Somatoform disorders in general practice: prevalence, functional impairment and comorbidity with anxiety and depressive disorders. British Journal of Psychiatry 2004;184:470‐6. [DOI] [PubMed] [Google Scholar]
Decoutere 2011
- Decoutere L, Eede F, Moorkens G, Sabbe BG. Antipsychotic agents in the treatment of somatoform disorders: a review [Gebruik van antipsychotica bij somatoformestoornissen; een literatuuroverzicht]. Tijdschrift voor Psychiatrie 2011;53(3):163‐73. [PubMed] [Google Scholar]
Derogatis 1983
- Derogatis LR. The SCL‐90‐R Manual. Scoring and Administration Procedures for the SCL‐90‐R. Baltimore, MA: John Hopkins University School of Medicine, 1983. [Google Scholar]
Derogatis 1992
- Derogatis LR. The Brief Symptom Inventory (BSI): Administration, Scoring, and Procedures Manual II. Baltimore, MD: Clinical Psychometric Research, 1992. [Google Scholar]
Di Franco 2010
- Franco M, Iannuccelli C, Atzeni F, Cazzola M, Salaffi F, Valesini G, et al. Pharmacological treatment of fibromyalgia. Clinical and Experimental Rheumatology 2010;28(6):S110‐6. [PubMed] [Google Scholar]
Drossman 2003
- Drossman DA, Toner BB, Whitehead WE, Diamant NE, Dalton CB, Duncan S, et al. Cognitive‐behavioral therapy versus education and desipramine versus placebo for moderate to severe functional bowel disorders. Gastroenterology 2003;125:19‐31. [DOI] [PubMed] [Google Scholar]
Escobar 1987
- Escobar JI, Burnam MA, Karno M, Forsythe A, Golding JM. Somatization in the community. Archives of General Psychiatry 1987;44:713‐18. [DOI] [PubMed] [Google Scholar]
Escobar 1996
- Escobar JI. Pharmacological treatment of somatization/hypochondriasis. Psychopharmacology Bulletin 1996;32:589‐96. [PubMed] [Google Scholar]
Fallon 2004
- Fallon BA. Pharmacotherapy of somatoform disorders. Journal of Psychosomatic Research 2004;56(4):455‐60. [DOI] [PubMed] [Google Scholar]
Fink 2010
- Fink P, Schröder A. One single diagnosis, bodily distress syndrome, succeeded to capture 10 diagnostic categories of functional somatic syndromes and somatoform disorders. Journal of Psychosomatic Research 2010;68:415‐26. [DOI] [PubMed] [Google Scholar]
Fink 2011
- Fink PK. Treatment of multi‐organ bodily distress syndrome. A double‐blinded placebo controlled trial of the effect of imipramine (STreSS‐3) ‐ Stress‐3. Study protocol. apps.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2011‐004294‐87‐DK (accessed 24 October 2014).
Fink 2012a
- Fink PK, Agger JL. Imipramine treatment for patients with multi‐organ bodily distress syndrome (Stress‐3). Study protocol. clinicaltrials.gov/show/NCT01518634 (accessed 24 October 2014).
Fink 2012b
- Fink PK, Agger JL. Imipramine treatment for patients with multi‐organ bodily distress syndrome. Study protocol. www.controlled‐trials.com/mrct/trial/2272077/ (accessed 24 October 2014).
Fishbain 1998
- Fishbain DA, Cutler RB, Rosomoff HL, Steele‐Rosomoff RS. Do antidepressants have an analgesic effect in psychogenic pain and somatoform pain disorder? A meta‐analysis. Psychosomatic Medicine 1998;60(4):503‐9. [DOI] [PubMed] [Google Scholar]
Ford 2009
- Ford AC, Talley NJ, Schoenfeld PS, Quigley EM, Moayyedi P. Efficacy of antidepressants and psychological therapies in irritable bowel syndrome: systematic review and meta‐analysis. Gut 2009;58:367‐78. [DOI] [PubMed] [Google Scholar]
Fournier 2010
- Fournier JC, DeRubeis RR, Hollon SD, Dimidjian S, Amsterdam JD, Shelton RC, et al. Antidepressant drug effects and depression severity: a patient‐level meta‐analysis. Journal of the American Medical Association 2010;303(1):47‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fröhlich 2006
- Fröhlich C, Jacobi F, Wittchen HU. DSM‐IV pain disorder in the general population ‐ an exploration of the structure and threshold of medically unexplained pain symptoms. European Archives of Psychiatry and Clinical Neuroscience 2006;256:187‐96. [DOI] [PubMed] [Google Scholar]
Gamble 2005
- Gamble C, Hollis S. Uncertainty method improved on best‐worst case analysis in a binary meta‐analysis. Journal of Clinical Epidemiology 2005;58:579‐88. [DOI] [PubMed] [Google Scholar]
Gildenberg 1984
- Gildenberg PL. Management of chronic pain. Applied Neurophysiology 1984;47(4‐6):157‐70. [DOI] [PubMed] [Google Scholar]
Gorard 1994
- Gorard DA, Libby GW, Farthing MJG. Influence of antidepressants on whole gut and orocecal transit times in health and irritable‐bowel‐syndrome. Alimentary Pharmacology & Therapeutics 1994;8:159‐66. [DOI] [PubMed] [Google Scholar]
Grabe 2003
- Grabe HJ, Meyer C, Hapke U, Rumpf HJ, Freyberger HJ, Dilling H, et al. Specific somatoform disorder in the general population. Psychosomatics 2003;44:304‐11. [DOI] [PubMed] [Google Scholar]
Grothe 2004
- Grothe DR, Scheckner B, Albano D. Treatment of pain syndromes with venlafaxine. Pharmacotherapy 2004;24(5):621‐9. [DOI] [PubMed] [Google Scholar]
Gu 2002
- Gu YP, Huang LYM. Gabapentin potentiates N‐methyl‐D‐aspartate receptor mediated currents in rat GABAergic dorsal horn neurons. Neuroscience Letters 2002;324:177‐80. [DOI] [PubMed] [Google Scholar]
Gureje 2004
- Gureje O. What can we learn from a cross‐national study of somatic distress?. Journal of Psychosomatic Research 2004;56(4):409‐12. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy W. Clinical Global Impressions. ECDEU Assessment Manual for Psychopharmacology, Revised. Rockville, MD: National Institute of Mental Health, 1976:218‐22. [Google Scholar]
Guyatt 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck‐Ytter Y, Alonso‐Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336:924‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hamilton 1959
- Hamilton M. The assessment of anxiety states by rating. British Journal of Psychiatry 1959;32:50‐5. [DOI] [PubMed] [Google Scholar]
Hamilton 1960
- Hamilton M. A rating scale for depression. Journal of Neurology Neurosurgery and Psychiatry 1960;23:56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Heres 2006
- Heres S, Davis J, Maino K, Jetzinger E, Kissling W, Leucht S. Why olanzapine beats risperidone, risperidone beats quetiapine, and quetiapine beats olanzapine: an exploratory analysis of head‐to‐head comparison studies of second‐generation antipsychotics. American Journal of Psychiatry 2006;163(2):185‐94. [DOI] [PubMed] [Google Scholar]
Higgins 2011a
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane.org.
Higgins 2011b
- Higgins JPT, Deeks JJ. Chapter 7: Selecting studies and collecting data. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hoedeman 2010
- Hoedeman R, Blankenstein NAH, Feltz‐Cornelis CM, Krol B, Groothoff JJW. Consultation letters for medically unexplained physical symptoms in primary care. Cochrane Database of Systematic Reviews 2010, Issue 12. [DOI: 10.1002/14651858.CD006524.pub2] [DOI] [PubMed] [Google Scholar]
Huertas‐Ceballos 2014
- Huertas‐Ceballos AA, Logan S, Bennett C, Macarthur C, Martin AE. Pharmacological interventions for recurrent abdominal pain (RAP) and irritable bowel syndrome (IBS) in childhood. Cochrane Database of Systematic Reviews 2014, Issue 2. [DOI: 10.1002/14651858.CD003017.pub3] [DOI] [PubMed] [Google Scholar]
Häuser 2009
- Häuser W, Bernardy K, Uceyler N, Sommer C. Treatment of fibromyalgia syndrome with antidepressants: a meta‐analysis. Journal of the American Medical Association 2009;301:198‐209. [DOI] [PubMed] [Google Scholar]
Ipser 2009
- Ipser JC, Stein DJ. Pharmacotherapy and psychotherapy for body dysmorphic disorder. Cochrane Database of Systematic Reviews 2009, Issue 1. [DOI: 10.1002/14651858.CD005332.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jackson 2000
- Jackson JL, O'Malley PG, Tomkins G, Balden E, Santoro J, Kroenke K. Treatment of functional gastrointestinal disorders with antidepressant medications: a meta‐analysis. American Journal of Medicine 2000;108:65‐72. [DOI] [PubMed] [Google Scholar]
Jackson 2006a
- Jackson JL, O'Malley PG, Kroenke K. Antidepressants and cognitive‐behavioral therapy for symptom syndromes. CNS Spectrums 2006;11:212‐22. [DOI] [PubMed] [Google Scholar]
Jackson 2006b
- Jackson JL, Kroenke K. Managing somatization: medically unexplained should not mean medically ignored. Journal of General Internal Medicine 2006;21(7):797‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jacobi 2004
- Jacobi F, Wittchen HU, Holting C, Hofler M, Pfister H, Muller N, et al. Prevalence, co‐morbidity and correlates of mental disorders in the general population: results from the German Health Interview and Examination Survey (GHS). Psychological Medicine 2004;34:597‐611. [DOI] [PubMed] [Google Scholar]
Janca 1996
- Janca A, Hiller W. CD‐10 Checklists ‐ a tool for clinicians' use of the ICD‐10 classification of mental and behavioral disorders. Comprehensive Psychiatry 1996;37:180‐7. [DOI] [PubMed] [Google Scholar]
Johansson 1982
- Johansson F. Patients with Chronic Pain. A Clinical, Clinical‐pharmacological and Biochemical Study [medical dissertation]. Umeå, Sweden: Umeå University, 1982. [Google Scholar]
Jones 1991
- Jones SL. Descending noradrenergic influences on pain. Progress in Brain Research 1991;88:381‐94. [DOI] [PubMed] [Google Scholar]
Katz 1999
- Katz J, Melzack R. Measurement of pain. Surgical Clinics of North America 1999;79:231‐52. [DOI] [PubMed] [Google Scholar]
Kellner 1986
- Kellner R. Somatization and Hypochondriasis. New York: Praeger‐Greenwood Publishers, 1986. [Google Scholar]
Kirmayer 1997
- Kirmayer LJ, Looper KJ, Taillefer, S. Somatoform disorders. In: Turner SM, Hersen M editor(s). Adult Psychopathology and Diagnosis. 4th Edition. New York: John Wiley, 1997:420‐75. [Google Scholar]
Kleinstäuber 2011
- Kleinstäuber M, Witthöft M, Hiller W. Efficacy of short‐term psychotherapy for multiple medically unexplained physical symptoms: a meta‐analysis. Clinical Psychology Review 2011;31(1):146‐60. [DOI] [PubMed] [Google Scholar]
Kroenke 1997
- Kroenke K, Spitzer RL, deGruy RV, Hahn SR, Linzer M, Williams JB. Multisomatoform disorder. An alternative to undifferentiated somatoform disorder for the somatizing patient in primary care. Archives of General Psychiatry 1997;54:352‐8. [DOI] [PubMed] [Google Scholar]
Kroenke 2002
- Kroenke K, Spitzer RL, Williams JBW. The PHQ‐15: validity of a new measure for evaluating the severity of somatic symptoms. Psychosomatic Medicine 2002;64:258‐66. [DOI] [PubMed] [Google Scholar]
Kroenke 2007
- Kroenke K. Efficacy of treatment for somatoform disorders: a review of randomized controlled trials. Psychosomatic Medicine 2007;69(9):881‐8. [DOI] [PubMed] [Google Scholar]
Kubera 2001
- Kubera N, Lin AH, Kenis G, Bosmans E, Bockstaele D, Maes M. Anti‐inflammatory effects of antidepressants through suppression of the interferon‐gamma/interleukin‐10 production ratio. Journal of Clinical Psychopharmacology 2001;21:199‐206. [DOI] [PubMed] [Google Scholar]
Lambert 1986
- Lambert MJ, Hatch DR, Kingston MD, Edwards BC. Zung, Beck, and Hamilton Rating Scales as measures of treatment outcome: a meta‐analytic comparison. Journal of Consulting and Clinical Psychology 1986;54(1):54‐9. [DOI] [PubMed] [Google Scholar]
Lambert 1994
- Lambert MJ, Hill CE. Assessing psychotherapy outcomes and process. In: Bergin AE, Garfield SL editor(s). Handbook of Psychotherapy and Behavior Change. 4th Edition. New York: Wiley, 1994:72‐113. [Google Scholar]
Leiknes 2007
- Leiknes KA, Finset A, Moum T, Sandanger I. Course and predictors of medically unexplained pain symptoms in the general population. Journal of Psychosomatic Research 2007;62(2):119‐28. [DOI] [PubMed] [Google Scholar]
Lexchin 2003
- Lexchin J, Bero LA, Djulbegovic B, Clark O. Pharmaceutical industry sponsorship and research outcome and quality: systematic review. BMJ 2003;326(7400):1167‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Linde 2009
- Linde K. St. John's wort ‐ an overview. Forschende Komplementärmedizin 2009;16:146‐55. [DOI] [PubMed] [Google Scholar]
Lipman 1969
- Lipman RS, Chase C, Rickles K, Covi L, Derogatis LR, Uhlenhuth EH. Factors of symptom distress. Doctor ratings of anxious neurotic outpatients. Archives of General Psychiatry 1969;21:328‐31. [DOI] [PubMed] [Google Scholar]
Loder 2004
- Loder EW, Biondi DM. Off‐label prescribing of drugs in specialty headache practice. Headache 2004;44(7):636‐41. [DOI] [PubMed] [Google Scholar]
Lynch 2001
- Lynch ME. Antidepressants as analgesics: a review of randomized controlled trials. Journal of Psychiatry & Neuroscience 2001;26(1):30‐6. [PMC free article] [PubMed] [Google Scholar]
Maes 1999
- Maes M. Major depression and activation of the inflammatory response system. Cytokines, Stress, and Depression 1999;461:25‐46. [DOI] [PubMed] [Google Scholar]
Maes 2001
- Maes M. The immunoregulatory effects of antidepressants. Human Psychopharmacology ‐ Clinical and Experimental 2001;16:95‐103. [DOI] [PubMed] [Google Scholar]
Magni 1991
- Magni G. The use of antidepressants in the treatment of chronic pain: a review of the current evidence. Drugs 1991;42(5):730‐48. [DOI] [PubMed] [Google Scholar]
Marks 2009
- Marks DM, Shah MJ, Patkar AA, Masand PS, Park GY, Pae CU. Serotonin‐norepinephrine reuptake inhibitors for pain control: premise and promise. Current Neuropharmacology 2009;7(4):331‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Martin 2006
- Martin A, Jacobi F. Features of hypochondriasis and illness worry in the general population in Germany. Psychosomatic Medicine 2006;68:770‐7. [DOI] [PubMed] [Google Scholar]
McQuay 1995
- Mcquay H, Carroll D, Jadad AR, Wiffen P, Moore A. Anticonvulsant drugs for management of pain: a systematic review. BMJ 1995;311(7012):1047‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Melzack 1987
- Melzack R. The short‐form McGill Pain Questionnaire. Pain 1987;30:191‐7. [DOI] [PubMed] [Google Scholar]
Meyer 2001
- Meyer C, Rumpf HJ, Hapke U, John U. Prevalence of DSM‐IV psychiatric disorders including nicotine dependence in the general population: results from the Northern German TACOS study. Neurology Psychiatry and Brain Research 2001;9:75‐80. [Google Scholar]
Moher 2001
- Moher D, Schulz KF, Altman DG. The CONSORT statement: revised recommendations for improving the quality of reports of parallel group randomized trials. BMC Medical Research Methodology 2001;1:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. PLoS Medicine 2009;6(7):e1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
Montoya 2005
- Montoya P, Pauli P, Batra A, Wiedemann G. Altered processing of pain‐related information in patients with fibromyalgia. European Journal of Pain 2005;9:293‐303. [DOI] [PubMed] [Google Scholar]
Moore 2009
- Moore RA, Straube S, Wiffen PJ, Derry S, McQuay HJ. Pregabalin for acute and chronic pain in adults. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD007076.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2011
- Moore RA, Wiffen PJ, Derry S, McQuay HJ. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2011, Issue 3. [DOI: 10.1002/14651858.CD007938.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2012
- Moore RA, Derry S, Aldington D, Cole P, Wiffen PJ. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2012, Issue 12. [DOI: 10.1002/14651858.CD008242.pub2] [DOI] [PubMed] [Google Scholar]
Mumford 1991
- Mumford DB, Bavington JT, Bhatnagar KS, Hussain Y, Mirza S, Naraghi MM. The Bradford Somatic Inventory ‐ a multiethnic inventory of somatic symptoms reported by anxious and depressed patients in Britain and the Indo‐Pakistan subcontinent. British Journal of Psychiatry 1991;158:379‐86. [DOI] [PubMed] [Google Scholar]
Naliboff 1997
- Naliboff BD, Munakata J, Fullerton S, Gracely RH, Kodner A, Harraf F, et al. Evidence for two distinct perceptual alterations in irritable bowel syndrome. Gut 1997;41:505‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Nimnuan 2001
- Nimnuan C, Hotopf M, Wessely S. Medically unexplained symptoms: an epidemiological study in seven specialities. Journal of Psychosomatic Research 2001;51:361‐7. [DOI] [PubMed] [Google Scholar]
Nix 1998
- Nix WA. What is certain in pain therapy? The analgesic potency of neuroleptics in the treatment of chronic pain. A meta‐analysis [Was ist gesichert in der Schmerztherapie? Haben Neuroleptika eine analgetische Potenz? Eine Metaanalyse]. Schmerz 1998;12:30‐8. [DOI] [PubMed] [Google Scholar]
O'Malley 1999
- O'Malley PG, Jackson JL, Santoro J, Tomkins G, Balden E, Kroenke K. Antidepressant therapy for unexplained medical symptoms and syndromes. Journal of Family Practice 1999;48(12):980‐90. [PubMed] [Google Scholar]
O'Malley 2000
- O'Malley PG, Balden E, Tomkins G, Santoro J, Kroenke K, Jackson JL. Treatment of fibromyalgia with antidepressants: a meta‐analysis. Journal of General Internal Medicine 2000;15:659‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pae 2009
- Pae CU, Marks DM, Patkar AA, Masand PS, Luyten P, Serretti A. Pharmacological treatment of chronic fatigue syndrome: focusing on the role of antidepressants. Expert Opinion on Pharmacotherapy 2009;10:1561‐70. [DOI] [PubMed] [Google Scholar]
Petzke 2005
- Petzke F, Harris RE, Williams DA, Clauw DJ, Gracely RH. Differences in unpleasantness induced by experimental pressure pain between patients with fibromyalgia and healthy controls. European Journal of Pain 2005;9:325‐35. [DOI] [PubMed] [Google Scholar]
Pilowsky 1967
- Pilowsky I. Dimensions of hypochondriasis. British Journal of Psychiatry 1967;113:89‐93. [DOI] [PubMed] [Google Scholar]
Price 2000
- Price J. Review: antidepressants are effective for clinical unexplained symptoms and syndromes. Evidence Based Mental Health 2000;3:84. [Google Scholar]
Prior 2013
- Prior KN, Bond MJ. Somatic symptom disorders and illness behaviour: current perspectives. International Review of Psychiatry 2013;25(1):5‐18. [DOI] [PubMed] [Google Scholar]
Raine 2002
- Raine R, Haines A, Sensky T, Hutchings A, Larkin K, Black N. Systematic review of mental health interventions for patients with common somatic symptoms: can research evidence from secondary care be extrapolated to primary care?. BMJ 2002;325(7372):1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan 2012 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.2. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2012.
Richardson 1990
- Richardson BP. Serotonin and nociception. Annals of the New York Academy of Sciences 1990;600:511‐20. [DOI] [PubMed] [Google Scholar]
Rickels 1994
- Rickels K, Noyes RJ, Robinson DS, Schweizer E, Uhlenhut EH. Evaluating drug treatments of generalized anxiety disorder and adjustment disorders with anxious mood. In: Prien RF, Robinson DS editor(s). Clinical Evaluation of Psychotropic Drugs: Principles and Guidelines. New York: Raven Press, 1994:373‐410. [Google Scholar]
Rief 1998
- Rief W, Hiller W, Margraf J. Cognitive aspects of hypochondriasis and the somatization syndrome. Journal of Abnormal Psychology 1998;107:587‐95. [DOI] [PubMed] [Google Scholar]
Rief 1999
- Rief W, Hiller W. Toward empirically based criteria for the classification of somatoform disorders. Journal of Psychosomatic Research 1999;46:507‐18. [DOI] [PubMed] [Google Scholar]
Rief 2003
- Rief W, Ihle D, Pilger F. A new approach to assess illness behaviour. Journal of Psychosomatic Research 2003;54:405‐14. [DOI] [PubMed] [Google Scholar]
Rief 2005
- Rief W, Barsk AJ. Psychobiological perspectives on somatoform disorders. Psychoneuroendocrinology 2005;30:996‐1002. [DOI] [PubMed] [Google Scholar]
Rief 2007
- Rief W, Broadbent E. Explaining medically unexplained physical symptoms ‐ models and mechanisms. Clinical Psychology Review 2007;27:821‐41. [DOI] [PubMed] [Google Scholar]
Rief 2008
- Rief W, Hiller W. SOMS Screening for Somatoform Disorders [SOMS Das Screening für Somatoforme Störungen. Manual zum Fragebogen]. 4th Edition. Bern, Switzerland: Huber, 2008. [Google Scholar]
Rief 2009
- Rief W, Nestoriuc Y, Weiss S, Welzel E, Barsky AJ, Hofmann SG. Meta‐analysis of the placebo response in antidepressant trials. Journal of Affective Disorders 2009;118:1‐8. [DOI] [PubMed] [Google Scholar]
Robins 1991
- Robins LN, Regier DA. Psychiatric Disorders in America: The Epidemiological Catchment Area Study. New York: The Free Press, 1991. [Google Scholar]
Ruddy 2005
- Ruddy R, House A. Psychosocial interventions for conversion disorder. Cochrane Database of Systematic Reviews 2005, Issue 4. [DOI: 10.1002/14651858.CD005331.pub2] [DOI] [PubMed] [Google Scholar]
Ryder 2008
- Ryder AG, Yang J, Zhu XZ, Yao SQ, Yi JY, Heine SJ, et al. The cultural shaping of depression: somatic symptoms in China, psychological symptoms in North America?. Journal of Abnormal Psychology 2008;117(2):300‐13. [DOI] [PubMed] [Google Scholar]
Saarto 2007
- Saarto T, Wiffen PJ. Antidepressants for neuropathic pain. Cochrane Database of Systematic Reviews 2007, Issue 4. [DOI: 10.1002/14651858.CD005454.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Satterthwaite 1990
- Satterthwaite JR, Tollison CD, Kriegel ML. The use of tricyclic antidepressants for the treatment of intractable pain. Comprehensive Therapy 1990;16(4):10‐5. [PubMed] [Google Scholar]
Schreiber 1999
- Schreiber S, Getslev V, Backer MM, Weizman R, Pick CG. The atypical neuroleptics clozapine and olanzapine differ regarding their antinociceptive mechanisms and potency. Pharmacology Biochemistry and Behavior 1999;64:75‐80. [DOI] [PubMed] [Google Scholar]
Schünemann 2011
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Seidel 2008
- Seidel S, Aigner M, Ossege M, Pernicka E, Wildner B, Sycha T. Antipsychotics for acute and chronic pain in adults. Cochrane Database of Systematic Reviews 2008, Issue 4. [DOI: 10.1002/14651858.CD004844.pub2] [DOI] [PubMed] [Google Scholar]
Sharma 2013
- Sharma MP, Manjula M. Behavioural and psychological management of somatic symptom disorders: an overview. International Review of Psychiatry 2013;25(1):116‐24. [DOI] [PubMed] [Google Scholar]
Sharpe 1995
- Sharpe M, Mayou R, Bass C. Concepts, theories and terminology. In: Mayou R, Bass C, Sharpe M editor(s). Treatment of Functional Somatic Symptoms. Oxford: Oxford University Press, 1995:3‐16. [Google Scholar]
Sheehan 1983
- Sheehan DV. The Anxiety Disease. New York: Scribner, 1983. [Google Scholar]
Sheehan 1998
- Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E, et al. The Mini‐International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM‐IV and ICD‐10. Journal of Clinical Psychiatry 1998;59:22‐33. [PubMed] [Google Scholar]
Silberstein 2002
- Silberstein SD, Peres MFP, Hopkins MM, Shechter AL, Young WB, Rozen TD. Olanzapine in the treatment of refractory migraine and chronic daily headache. Headache 2002;42:515‐8. [DOI] [PubMed] [Google Scholar]
Somashekar 2013
- Somashekar B, Jainer A, Wuntakal B. Psychopharmacotherapy of somatic symptoms disorders. Review. International Review of Psychiatry 2013;25(1):107‐15. [DOI] [PubMed] [Google Scholar]
Sommer 1993
- Sommer H, Harrer G. Placebo‐controlled double‐blind study examining the effectiveness of an Hypericum preparation in 105 patients with depressions. Nervenheilkunde 1993;12:274‐7. [DOI] [PubMed] [Google Scholar]
Stahl 2002
- Stahl SM. The psychopharmacology of painful physical symptoms in depression. Journal of Clinical Psychiatry 2002;63:382‐3. [DOI] [PubMed] [Google Scholar]
Sterne 2011
- Sterne JAC, Egger M, Moher D. Chapter 10: Addressing reporting bias. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Stone 2003
- Stone KJ, Viera AJ, Parman CL. Off‐label applications for SSRIs. American Family Physician 2003;68(3):498‐504. [PubMed] [Google Scholar]
Suls 2012
- Suls J, Howren MB. Understanding the physical‐symptom experience: the distinctive contributions of anxiety and depression. Current Directions in Psychological Science 2012;21(2):129‐34. [Google Scholar]
Sumathipala 2007
- Sumathipala A. What is the evidence for the efficacy of treatments for somatoform disorders? A critical review of previous intervention studies. Psychosomatic Medicine 2007;69(9):889‐900. [DOI] [PubMed] [Google Scholar]
Thomson 2007
- Thomson AB, Page LA. Psychotherapies for hypochondriasis. Cochrane Database of Systematic Reviews 2007, Issue 4. [DOI: 10.1002/14651858.CD006520.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Urban 2005
- Urban MO, Ren K, Park KT, Campbell B, Anker N, Stearns B, et al. Comparison of the antinociceptive profiles of gabapentin and 3‐methylgabapentin in rat models of acute and persistent pain: implications for mechanism of action. Journal of Pharmacology and Experimental Therapeutics 2005;31:1209‐16. [DOI] [PubMed] [Google Scholar]
Ursin 1997
- Ursin H. Sensitization, somatization, and subjective health complaints. International Journal of Behavioral Medicine 1997;4:105‐16. [DOI] [PubMed] [Google Scholar]
Verbeke 2000
- Verbeke M, Molenberghs G. Linear Mixed Models for Longitudinal Data. New York: Springer, 2000. [Google Scholar]
Verdu 2008
- Verdu B, Decosterd I, Buclin T, Stiefel F, Berney A. Antidepressants for the treatment of chronic pain. Drugs 2008;68:2611‐32. [DOI] [PubMed] [Google Scholar]
Verhaak 2006
- Verhaak PF, Meijer SA, Visser AP, Wolters G. Persistent presentation of medically unexplained symptoms in general practice. Family Practice 2006;23:414‐20. [DOI] [PubMed] [Google Scholar]
Volz 2003
- Volz HP, Murck H, Kasper S, Moller HJ. St John's wort extract (LI 160) in somatoform disorders: results of a placebo‐controlled trial. Erratum [Johanniskrautextrakt (LI 160) bei somatoformen Stoerungen: Ergebnisse einer plazebokontrollierten Studie. Erratum]. Psychopharmacology 2003;167(3):333. [Google Scholar]
Ware 1992
- Ware JE, Sherbourne CD. The MOS 36‐Item Short‐Form Health Survey (SF‐36) .1. Conceptual‐framework and item selection. Medical Care 1992;30:473‐83. [PubMed] [Google Scholar]
Wessely 1999
- Wessely S, Nimnuan C, Sharpe M. Functional somatic syndromes: one or many?. Lancet 1999;354:936‐9. [DOI] [PubMed] [Google Scholar]
Whitehead 2002
- Whitehead WE, Palsson O, Jones KR. Systematic review of the comorbidity of irritable bowel syndrome with other disorders: what are the causes and implications?. Gastroenterology 2002;122:1140‐56. [DOI] [PubMed] [Google Scholar]
WHO 1975
- World Health Organization. International Classification of Diseases. 9th revision (ICD‐9). Geneva, Switzerland: World Health Organization, 1975. [Google Scholar]
WHO 1992
- World Health Organization. International Statistical Classification of Diseases. 10th Revision (ICD‐10). Geneva, Switzerland: World Health Organization, 1992. [Google Scholar]
Williams 1982
- Williams JBW, Spitzer RL. Idiopathic pain disorder: a critique of pain‐prone disorder and a proposal for a revision of the DSM‐III category psychogenic pain disorder. Journal of Nervous and Mental Disease 1982;170:415‐9. [PubMed] [Google Scholar]
Wittchen 1992
- Wittchen HU, Essau CA, Vonzerssen D, Krieg JC, Zaudig M. Lifetime and 6‐month prevalence of mental disorders in the Munich follow‐up‐study. European Archives of Psychiatry and Clinical Neuroscience 1992;241:247‐58. [DOI] [PubMed] [Google Scholar]
Wittchen 2005
- Wittchen HU, Jacobi F. Size and burden of mental disorders in Europe ‐ a critical review and appraisal of 27 studies. European Neuropsychopharmacology 2005;15:357‐76. [DOI] [PubMed] [Google Scholar]
Wittchen 2011
- Wittchen HU, Jacobi F, Rehm J, Gustavsson A, Svensson M, Jonsson B, et al. The size and burden of mental disorders and other disorders of the brain in Europe 2010. European Neuropsychopharmacology 2011;21:655‐79. [DOI] [PubMed] [Google Scholar]
Witthöft 2010
- Witthöft M, Hiller W. Psychological approaches to origins and treatments of somatoform disorders. Annual Review of Clinical Psychology 2010;6:257‐83. [DOI] [PubMed] [Google Scholar]
Woelk 2000
- Woelk H. Comparison of St John's wort and imipramine for treating depression: randomised controlled trial. BMJ 2000;321:536‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yirmiya 1996
- Yirmiya R. Endotoxin produces a depressive‐like episode in rats. Brain Research 1996;711(1‐2):163‐74. [DOI] [PubMed] [Google Scholar]
Yunus 2007
- Yunus MB. Fibromyalgia and overlapping disorders: the unifying concept of central sensitivity syndromes. Seminars in Arthritis and Rheumatism 2007;36:339‐56. [DOI] [PubMed] [Google Scholar]