

Abstract
The bisphosphonates ((HO)2P(O)CR1R2P(O)(OH)2, BPs) were first shown to inhibit bone resorption in the 1960s, but it was not until 30 years later that a detailed molecular understanding of the relationship between their varied chemical structures and biological activity was elucidated. In the 1990s and 2000s, several potent bisphosphonates containing nitrogen in their R2 side chains (N-BPs) were approved for clinical use including alendronate, risedronate, ibandronate, and zoledronate. These are now mostly generic drugs and remain the leading therapies for several major bone-related diseases, including osteoporosis and skeletal-related events associated with bone metastases. The early development of chemistry in this area was largely empirical and only a few common structural features related to strong binding to calcium phosphate were clear. Attempts to further develop structure-activity relationships to explain more dramatic pharmacological differences in vivo at first appeared inconclusive, and evidence for mechanisms underlying cellular effects on osteoclasts and macrophages only emerged after many years of research. The breakthrough came when the intracellular actions on the osteoclast were first shown for the simpler bisphosphonates, via the in vivo formation of P-C-P derivatives of ATP. The synthesis and biological evaluation of a large number of nitrogen-containing bisphosphonates in the 1980s and 1990s led to the key discovery that the antiresorptive effects of these more complex analogs on osteoclasts result mostly from their potency as inhibitors of the enzyme farnesyl diphosphate synthase (FDPS/FPPS). This key branch-point enzyme in the mevalonate pathway of cholesterol biosynthesis is important for the generation of isoprenoid lipids that are utilized for the post-translational modification of small GTP-binding proteins essential for osteoclast function. Since then, it has become even more clear that the overall pharmacological effects of individual bisphosphonates on bone depend upon two key properties: the affinity for bone mineral and inhibitory effects on biochemical targets within bone cells, in particular FDPS. Detailed enzyme–ligand crystal structure analysis began in the early 2000s and advances in our understanding of the structure-activity relationships, based on interactions with this target within the mevalonate pathway and related enzymes in osteoclasts and other cells have continued to be the focus of research efforts to this day. In addition, while many members of the bisphosphonate drug class share common properties, now it is more clear that chemical modifications to create variations in these properties may allow customization of BPs for different uses.
Thus, as the appreciation for new potential opportunities with this drug class grows, new chemistry to allow ready access to an ever-widening variety of bisphosphonates continues to be developed. Potential new uses of the calcium phosphate binding mechanism of bisphosphonates for the targeting of other drugs to the skeleton, and effects discovered on other cellular targets, even at non-skeletal sites, continue to intrigue scientists in this research field.
Keywords: Bisphosphonates, Farnesyl diphosphate synthase (FDPS/FPPS), Osteoclasts, Bone resorption, Hydroxyapatite, Bone targeting
1. Introduction
It is now over 50 years since the biological effects of the bisphosphonates (BPs), originally called diphosphonates, were first reported in Science and Nature in 1969 [1].
The discovery and development of BPs as a major class of drugs for the treatment of bone diseases is a paradigm of a successful journey from ‘bench to bedside’. The study of BPs has become a vast field and more than 33,000 papers are currently listed on PubMed. In this article we will review the relationships between chemistry and pharmacological activity, building upon our publication from a decade ago [2]. We will also discuss advances in our current understanding of the structure-activity relationships with this drug class and look at what the future may hold.
The earliest uses of BPs were for bone scintigraphy and dental applications. The serendipitous discovery that BPs could inhibit bone resorption led initially to their first clinical approval for use in Paget’s disease. Later studies extended their use to the prevention of skeletal-related events in patients with bone metastases, and to reduce fractures in patients with osteoporosis. Currently, BPs continue to be the leading medicines used for such disorders in which bone resorption is excessive.
A major landmark event in the history of BPs was the elucidation of how bisphosphonates work. The pharmacological effects of BPs as inhibitors of bone resorption appear to depend upon two key properties; their affinity for bone minerals, and their inhibitory effects on osteoclasts. Despite many attempts, it was not until the later 1990s that their detailed molecular mechanisms of action were elucidated [3].
Bisphosphonates are internalized by osteoclasts and interfere with specific biochemical processes. Bisphosphonates can be classified into at least two groups with different molecular modes of action. The simpler non-nitrogen containing bisphosphonates (such as etidronate and clodronate, Fig. 1) can be metabolically incorporated into non-hydrolysable analogues of ATP, which interfere with ATP-dependent intracellular pathways. The more potent, nitrogen-containing bisphosphonates (including pamidronate, alendronate, risedronate, ibandronate, minodronate, and zoledronate, Fig. 1) are not metabolised in this way but inhibit key enzymes of the mevalonate/cholesterol biosynthetic pathway. The major enzyme target for bisphosphonates is farnesyl diphosphate synthase (FDPS), and the elucidation of the crystal structure for this enzyme revealed how BPs bind to and inhibit at the active site via their critical N atom. Inhibition of FDPS prevents the biosynthesis of isoprenoid compounds (notably farnesyl diphosphate and geranylgeranyl diphosphate) that are essential for the post-translational prenylation of small GTP-binding proteins (which are also GTPases) such as rab, ras, rho, and rac, which are critical for intracellular signaling events within osteoclasts. These mechanistic advances now offer medicinal chemists and biochemists a more rational basis to explain the wide variations in pharmacological activity of structurally different BPs. The nitrogen-containing BPs (N-BPs) approved by regulatory agencies have now largely become generic drugs, as key patents have expired. Nevertheless, the bisphosphonates are likely to remain major drugs for treating bone resorptive diseases [4,5]. Innovation to improve oral absorption and other routes of delivery of the bisphosphonate drug class continues to be a goal, as their highly hydrophilic nature and metal chelation properties result in limited passage through lipophilic membranes.
Fig. 1.
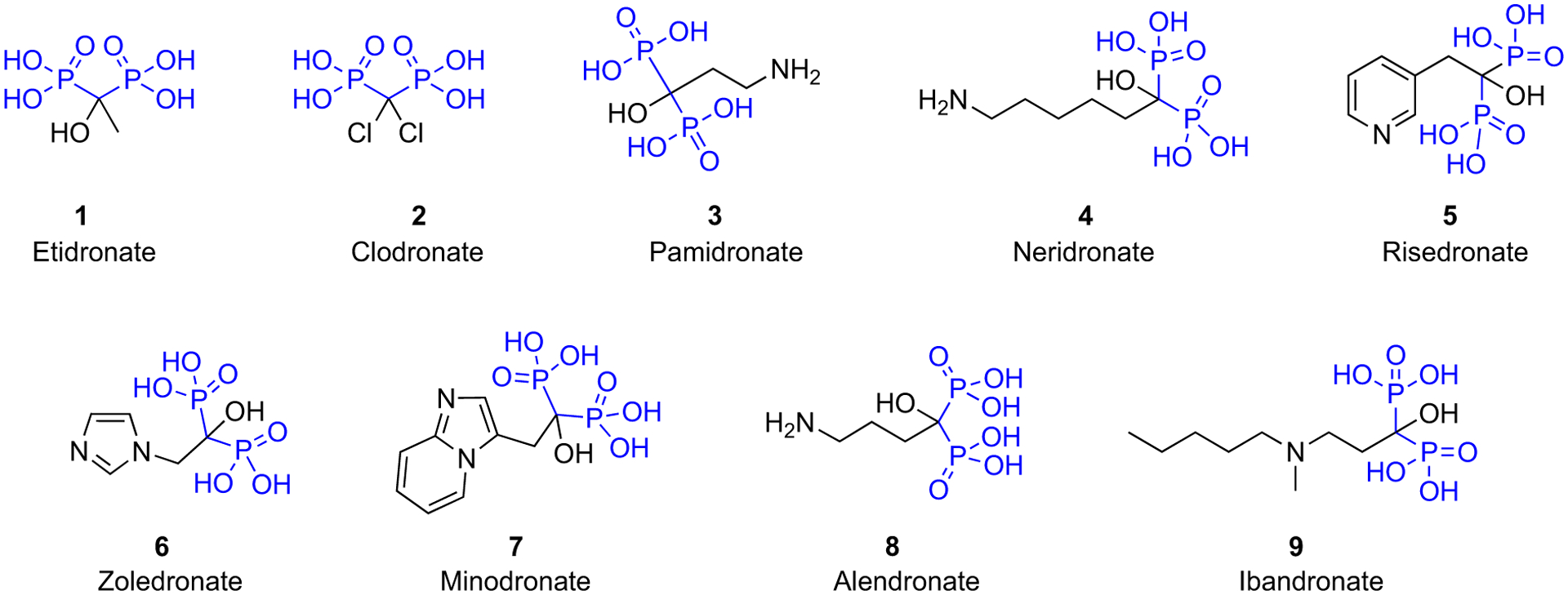
Clinically used bisphosphonates.
There are emerging opportunities for extending the use of BPs to other areas of medicine, and an encouraging amount of research in this field has continued during the past decade. In particular several recent studies indicate that BPs may be associated with exciting and hitherto unexpected clinical benefits outside the field of bone disease, e.g. in increasing lifespan, reducing cardiovascular disease, and the reduction of several cancers. The range of non-skeletal effects of BPs also includes the inhibition of several protozoan parasites, immunomodulatory effects via monocytes/macrophages and T cells, increasing longevity in animal progeria models, and enhancing human stem cell lifespan, DNA repair, and tissue regeneration. Another area of active research is the development of BP conjugates for selectively delivering other drugs to the bone micro-environment. Promising results have already been achieved with anti-myeloma agents, antimicrobials, and epigenetic modulators [6].
2. Advances in understanding of structure-activity relationships of bisphosphonates
2.1. Update on the chemistry of bisphosphonates
Significant advances have been made in the chemistry of bisphosphonates, including improved synthetic methods and the design of new bisphosphonate molecules, which have enhanced the understanding of the mechanism of action and access to customized analogs with potentially new applications. Several reviews have described the chemistry of the current leading BPs used globally in clinical therapy (clodronate (CLO), alendronate (ALE), pamidronate (PAM), risedronate (RIS), and zoledronate (ZOL)) (Fig. 1) [2]. More recently, additional reviews of the specific clinically important BPs have appeared, providing even more details of their discovery and commercialization history (CLO: [7]; ALE: [8]; PAM: [9]; RIS: [10]; ZOL: [11]; Etidronate (ETI): [12]; Minodronate (MIN): [13]). In addition, multiple reviews regarding other general chemistry advances have also been published in the last decade, covering the topic from various perspectives. For example, several comprehensive reviews of recent developments in the chemical synthesis of new bisphosphonates with potential utility in bone metabolism-related diseases include those by McKenna et al. [14] and other researchers [15–18].
The “classical” method for the preparation of 1-hydroxymethylene-bisphosphonates, by which the more recent clinically utilized bisphosphonates are synthesized, involves some variation of heating an appropriate carboxylic acid with phosphorous acid and phosphorus trichloride, followed by hydrolysis to obtain the product, and the reaction is usually slow, requiring one or more days to complete the process [2]. Thus, one of the major challenges in the synthesis of BPs is the efficiency of these methods in manufacturing BPs for pharmaceutical uses on a large scale. Therefore, more recently the focus of many researchers has been on developing efficient, cost-effective, and green methodologies that are scalable, reduce the reaction time, and eliminate the use of hazardous solvents. Insightful review articles have continued to be published that have focused on new synthetic routes of these pharmaceutically valuable compounds. In a review article by Barbosa et al. [17], these key synthetic procedures, along with their limitations have been discussed in more detail for the process chemistry of the hydroxy BPs currently used pharmaceutically.
As also discussed previously [2], in this “classical” synthetic method to prepare geminal hydroxy BPs, the intermediate reaction product from the phosphonylation step is not homogeneous and tends to solidify. Thus continuous efforts with different solvents, including methanesulfonic acid [19–21], sulfolane [22,23], and ionic liquids [23–25], have been tried to improve the process. Also, because of the slow reaction time of these procedures, microwave irradiation offers a promising remedy and has been shown to be effective in up to prep-scale syntheses of several well-known bisphosphonates, while generating comparable yields in shorter reaction time [16,26,27].
Other new synthetic methods have continued to appear, representing a burgeoning interest in bisphosphonate drugs and the concomitant desirability of expanding the structural scope of this class. In the past decade, a number of new methodologies have been developed that have given access to a wider library of BPs. Recently developed methodologies have granted chemists better control over the functionalization of BPs, as demonstrated in the following examples.
Substituting the α-hydroxy group for a different chemical moiety can dramatically change both enzyme inhibitory properties and mineral binding properties. Lawson, et al. [28] reported a new minodronate based bisphosphonate where the α-hydroxy group on the bisphosphonate bearing (geminal) carbon has been replaced with fluorine, 1-fluoro-2-(imidazo-[1,2 alpha]pyridin-3-yl)-ethyl-bisphosphonate, OX14 (10), through a useful fluorination sequence (Fig. 2). This resulted in the development of a strong inhibitor of FDPS, but with a lower binding affinity for bone mineral than most of the commonly studied bisphosphonates, as evidenced by both in vitro and in vivo experiments. This made OX14 (10) an interesting candidate for further investigation and potential development for its skeletal and nonskeletal benefits.
Fig. 2.
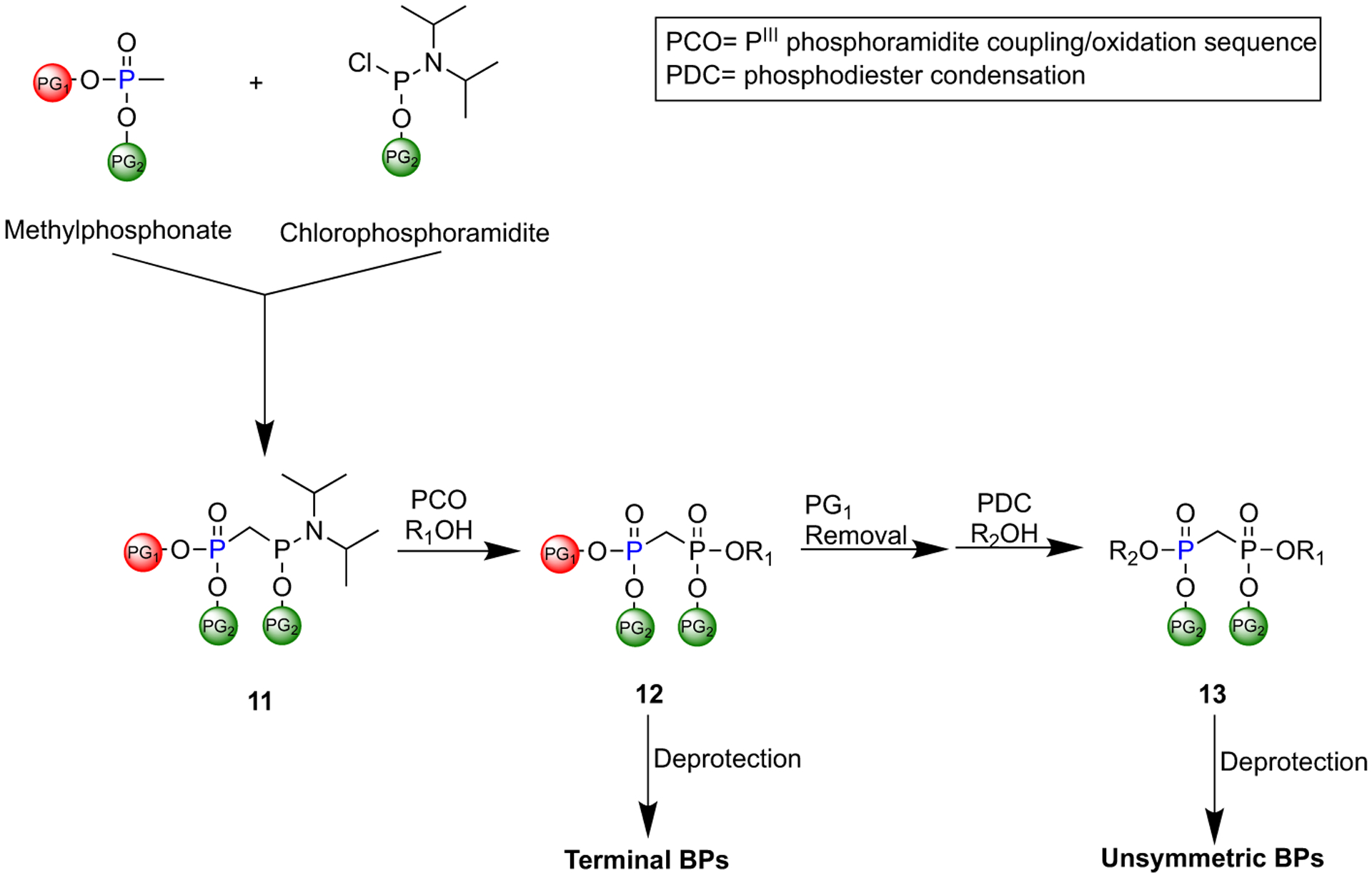
A novel protocol reported by Engelsma et al. for the synthesis of terminal and unsymmetric BPs.
Other substitutions for the alpha hydroxyl of minodronate led to much reduced inhibition of FDPS. Mazur, Ebetino and colleagues also prepared some closely related heterocycles to the imidazopyridine in OX14, which have also been found to be potent FDPS inhibitors such as OX139 (Table 1) [29]. Alkyl substituents added to risedronate (5) or deshydroxy risedronate (5a), such as in OX78 (5e), OX104 (5f), OX105 (5g), OX111 (5h), and OX112 (5i), led to interesting potency enhancements (unpublished), which could be anticipated as these were shown with molecular modelling to overlap well with the hydrophobic pyridyl ring of minodronate [30,31]. However, attempts to utilize fluorine substituents were less successful for the risedronate ring than with the minodronate ring system presumably because the binding of the geminal hydroxyl is relied on more greatly with less alternate hydrophobic interactions in the FDPS site as with minodronate [32]. Phosphonocarboxylate modifications of both series, where one phosphonate is replaced by one carboxylate (5d and 7d), led to analogs with little FDPS and no GGPPS inhibitory properties, but an emergence of inhibition of the RAB GGTase enzyme was observed [33–35]!
Table 1.
Structures of Minodronate and Risedronate analogues with altered substituents and bone mineral affinity leading to the development of OX14 (10).
| Name | Structure | Inhibition FDPS (IC50, nM) | Inhibition GGPPS (IC50, nM) | Mineral affinity Retention time (min) |
|---|---|---|---|---|
| Minodronate (7) |
|
1.9 | 869 | 10.33 |
| Deshydroxy Minodronate OX8 (7a) |
|
2.6 | >30,000 | 6.6 |
| OX9 (7b) |
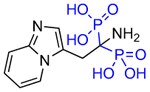
|
15.4 | >300,000 | 26.97 |
| OX14 (10) |
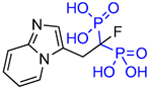
|
2.5 | 7345 | 6.17 |
| OX18 (7c) |
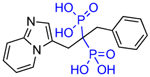
|
628 | 3100 | <3 |
| Minodronate phosphonocarboxylate (7d) |
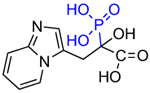
|
>60,000 | No inhibition | NA |
| OX139 (7e) |
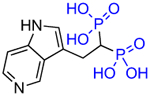
|
6.6 | 700,000 | 6.3 |
| Risedronate (5) |
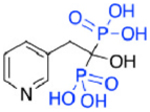
|
5.7 | 5675.4 | 9.97 |
| Deshydroxy Risedronate (NE58043) (5a) |
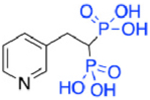
|
34.3 | ~2000 | 5.8 |
| OX11 (5b) |
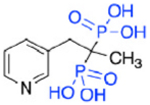
|
664 | NA | 4.6 |
| OX24 (5c) |
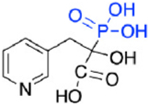
|
142 | >30,000 | 14.7 |
| Risedronate phosphonocarboxylate, NE10790 (5d) |
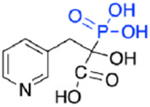
|
>250,000 | No inhibition | NA |
| OX78 (5e) |
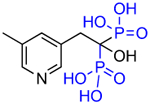
|
5 | - | 9.17 |
| OX104 (5f) |
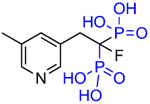
|
18 | ~327,000 | 5.9 |
| OX105 (5g) |
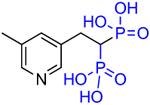
|
13 | >750,000 | 6.1 |
| OX111 (5h) |
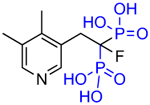
|
21 | 120,000 | 6.3 |
| OX112 (5i) |
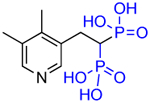
|
7.4 | >340,000 | 5.9 |
In 2017, Engelsma et al. reported a protocol that enables the controlled synthesis of mono- and diester methylene bisphosphonates [36]. They developed a new class of orthogonally protected phosphonylmethylphosphonate reagents (11) through treating a lithiated methylphosphonate diester with a chlorophosphoramidite (Fig. 2). Subsequently, 11 can be coupled to a variety of alcohols through a PIII phosphoramidite coupling/oxidation. Deprotection of 12 at this step would enable the preparation of terminally substituted BPs, while treating 12 with secondary alcohol through phosphodiester condensation would lead to a precursor of unsymmetric BPs (13). This protocol facilitates the introduction of BPs in structurally diverse pyrophosphate-containing biomolecules and is particularly advantageous for the development of BP analogues of natural products (ATP, NAD, FAD).
Egorov et al. have successfully developed a simple method for the synthesis of (geminal) α-hydroxy bisphosphonates (HBPs) (14) through a one-pot reaction of carboxylic acids and tris(trimethylsilyl) phosphite in the presence of catecholborane (Scheme 1) [37]. This method allows access to a larger library of HBPs, given that there are so many carboxylic acids commercially available. Moreover, this method is particularly useful for the synthesis of the highly useful nitrogen-containing hydroxy bisphosphonates, as it moves away from the additional steps often necessary for the protection/deprotection of the amino moieties.
Scheme 1.
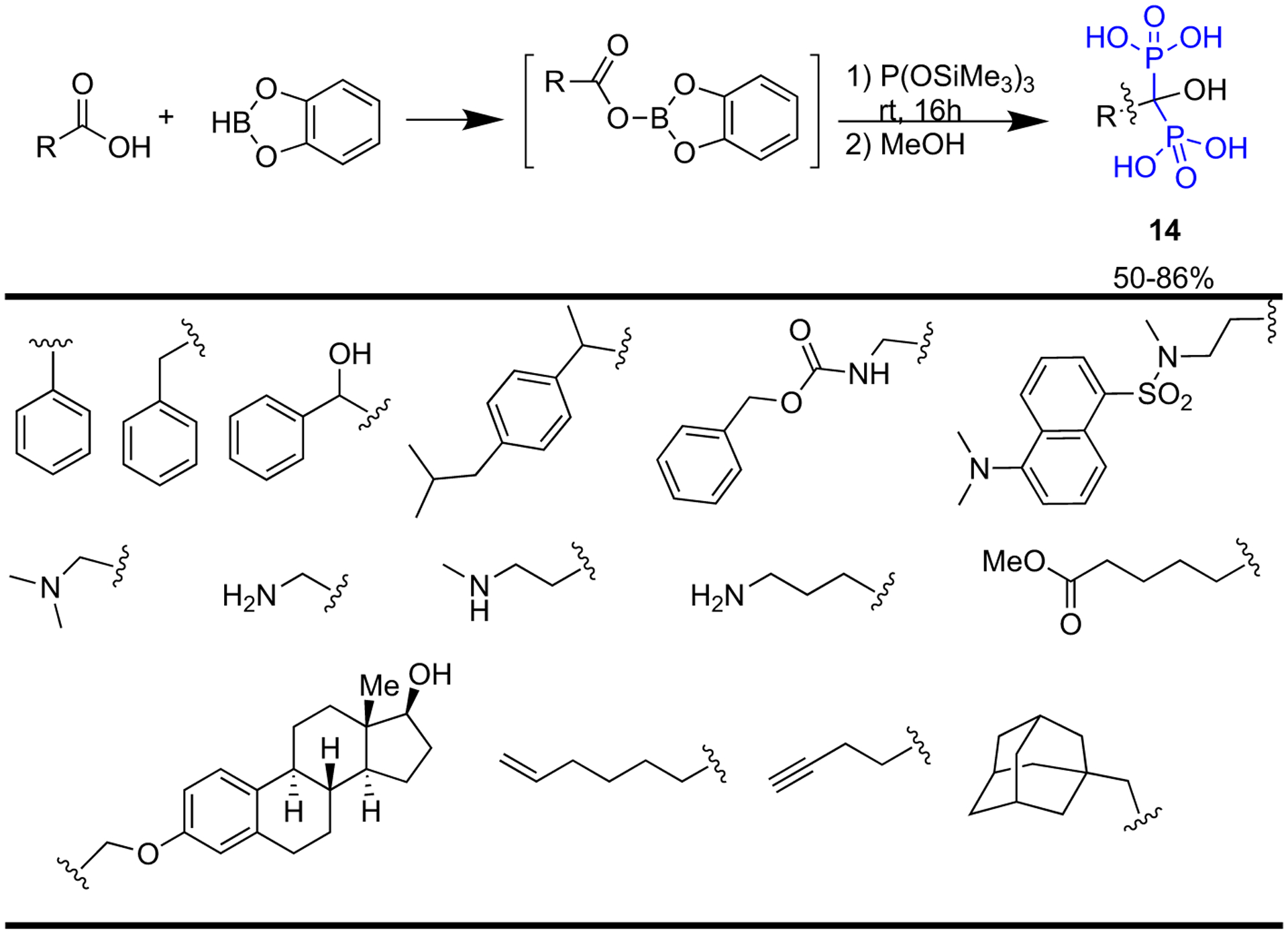
One-pot synthesis of α-hydroxy bisphosphonates from carboxylic acids.
Zhang et al. have developed an efficient methodology that creates structurally diverse aromatic bisphosphonates (15) through an Rh(III)-catalyzed directed methylene-diphosphonate carbenoid insertion into aromatic C—H bonds (Scheme 2). This one-step reaction combats the limitations of the traditional methods for the synthesis of aromatic bisphosphonates as they suffer from limited functional group diversity [38].
Scheme 2.
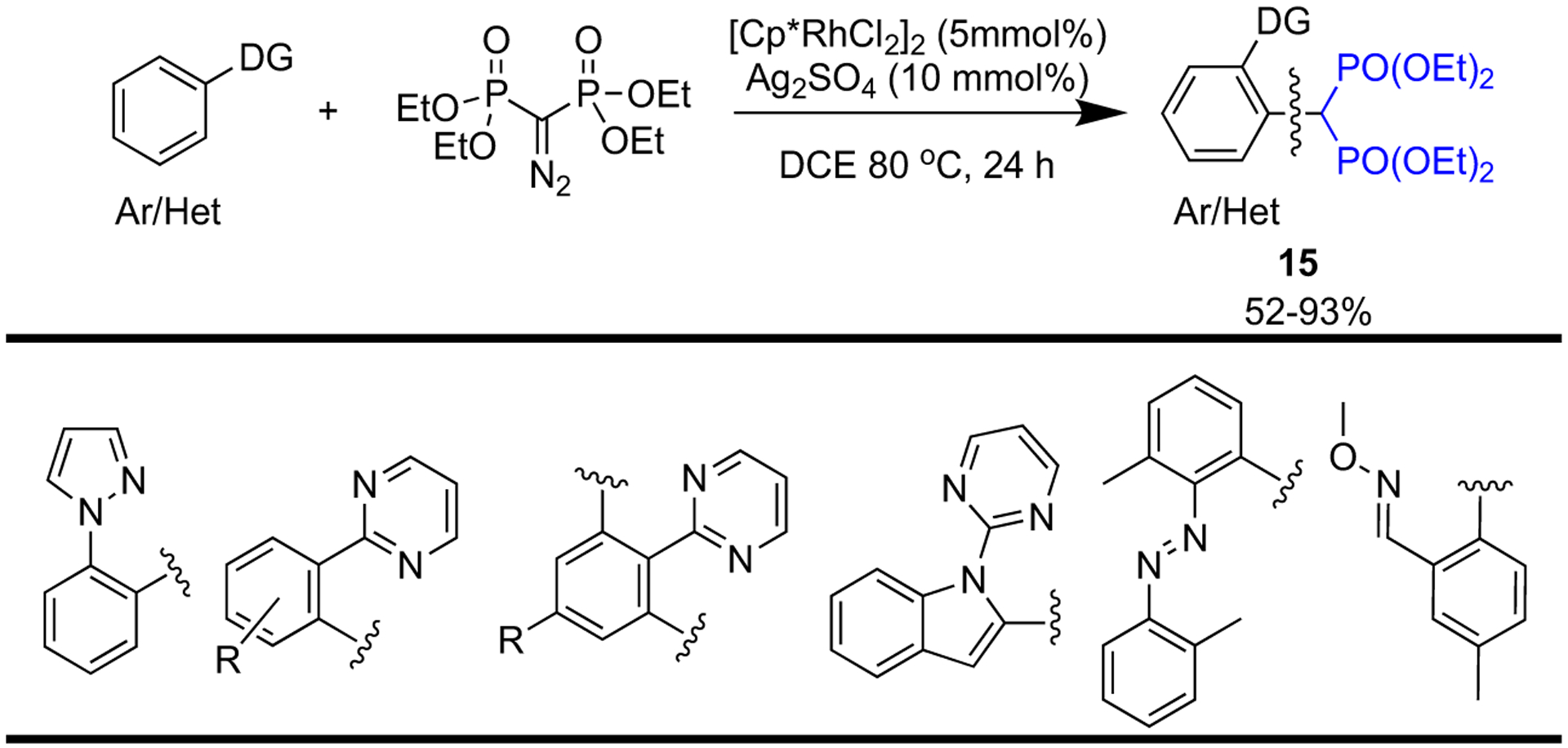
One-pot synthesis of aromatic bisphosphonates.
Another highly valuable synthetic methodology that has been developed in recent years is for the synthesis of α-, β-, and γ-azido bisphosphonates. The introduction of the azido moiety has created a new and useful bisphosphonate synthon, as it can readily and efficiently engage in “click chemistry” [39,40]. The first examples of the synthesis of α-azido BPs (16, 17) were reported by Chamberlain et al. in 2011 (Scheme 3a) [41]. This is an example of an electrophilic azido transfer, where tosyl azide reacts with a carbanion to generate a triazene intermediate, which fragments into diazo and azido products. Chamberlain et al. were able to obtain the azido compound as the major product by controlling the reaction conditions. Later in 2019, an article by Chiminazzo et al. reported an optimized protocol affording (18, 19) β- and γ-azido bisphosphonates (Scheme 3b). Moreover, they have successfully conjugated these useful synthons to red-fluorescent diketopyrrolopyrrole by “click chemistry” to make a novel fluorescent probe for in vitro bone imaging [42].
Scheme 3.
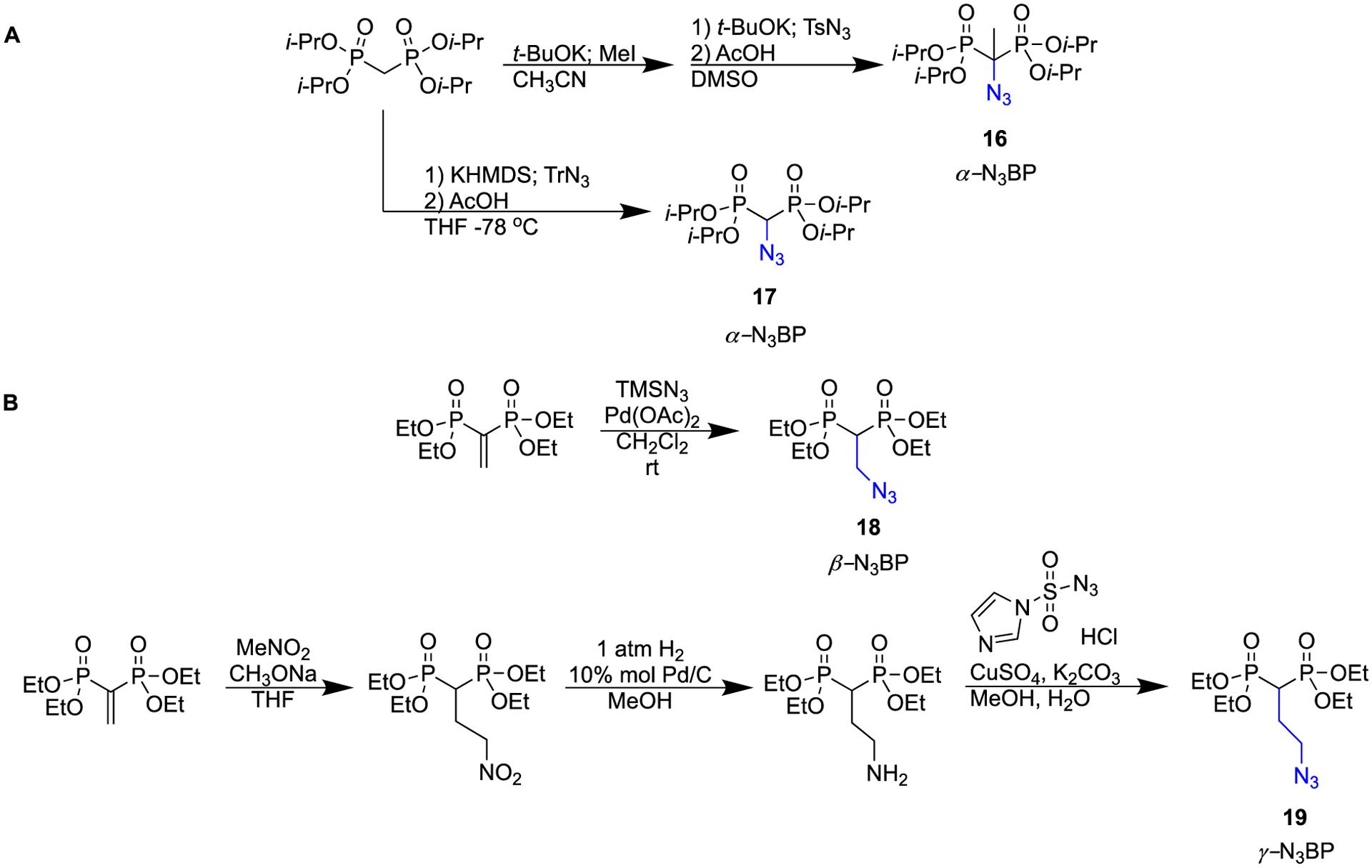
Synthetic routes to α-, β-, and γ-azido bisphosphonates.
New methodologies developed by Li et al. and Zhu et al. provide efficient access to a range of spiro bisphosphonates (Scheme 4) [43,44]. Through a 1,3-dipole cycloaddition reaction promoted by montmorillonite, Li et al. obtained spiro[indole-pyrrolizine], spiro[indole-indolizine], and spiro[indole-pyrrolidine] gem-bisphosphonates in good to excellent yields (Scheme 4a) [43]. Zhu et al. report a [3 + 2] cycloaddition reaction of azomethine imines with tetraethyl vinylidene BP catalyzed by CuI to furnish a series of dinitrogen-fused heterocycles coupled to geminal bisphosphonates (Scheme 4b) [44].
Scheme 4.
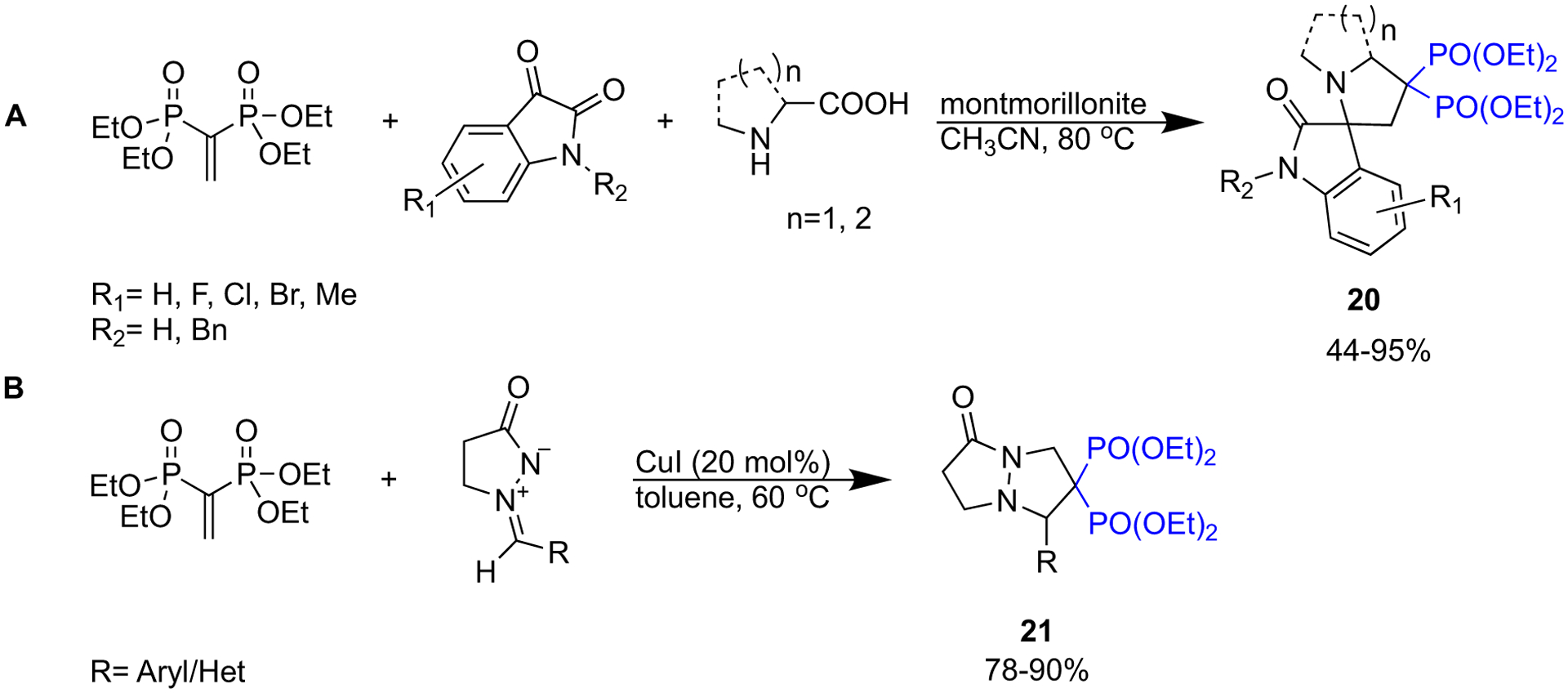
Synthesis of spiro bisphosphonates.
In the last decade, various “green” synthetic protocols have been developed to afford more facile access to α-substituted aminomethylene bisphosphonates (22). A straightforward synthesis of N-BPs consists of a one-pot reaction of diverse aryl/heteroaryl amines with dialkyl phosphite and triethyl orthoformate (Scheme 5a). Using conventional heating (oil bath) in these reactions would afford moderate yields of the corresponding α-amino bisphosphonates, and completion of the reaction generally takes a few hours [45,46]. Recently, by using microwave irradiation, eco-friendly protocols with comparable yields and shorter reaction time have been achieved (Scheme 5b) [26,47,48]. Employment of microwave irradiation in conjunction with the use of catalysts such as CuO nanoparticles [49], ZnO nanoparticles [50], and rGO-SO3H [51] have provided highly efficient green protocols that give access to a wide scope of α-amino bisphosphonates (Scheme 5c). Given the high demand for α-amino bisphosphonates in medicinal chemistry [52], such green and high yielding methodologies would facilitate the manufacturing of these pharmaceutically valuable motifs. Some recently synthesized α-amino bisphosphonates have been tested as antioxidant, antimicrobial, and anti-cancer drugs, and applications of such BPs will be discussed later in this review.
Scheme 5.
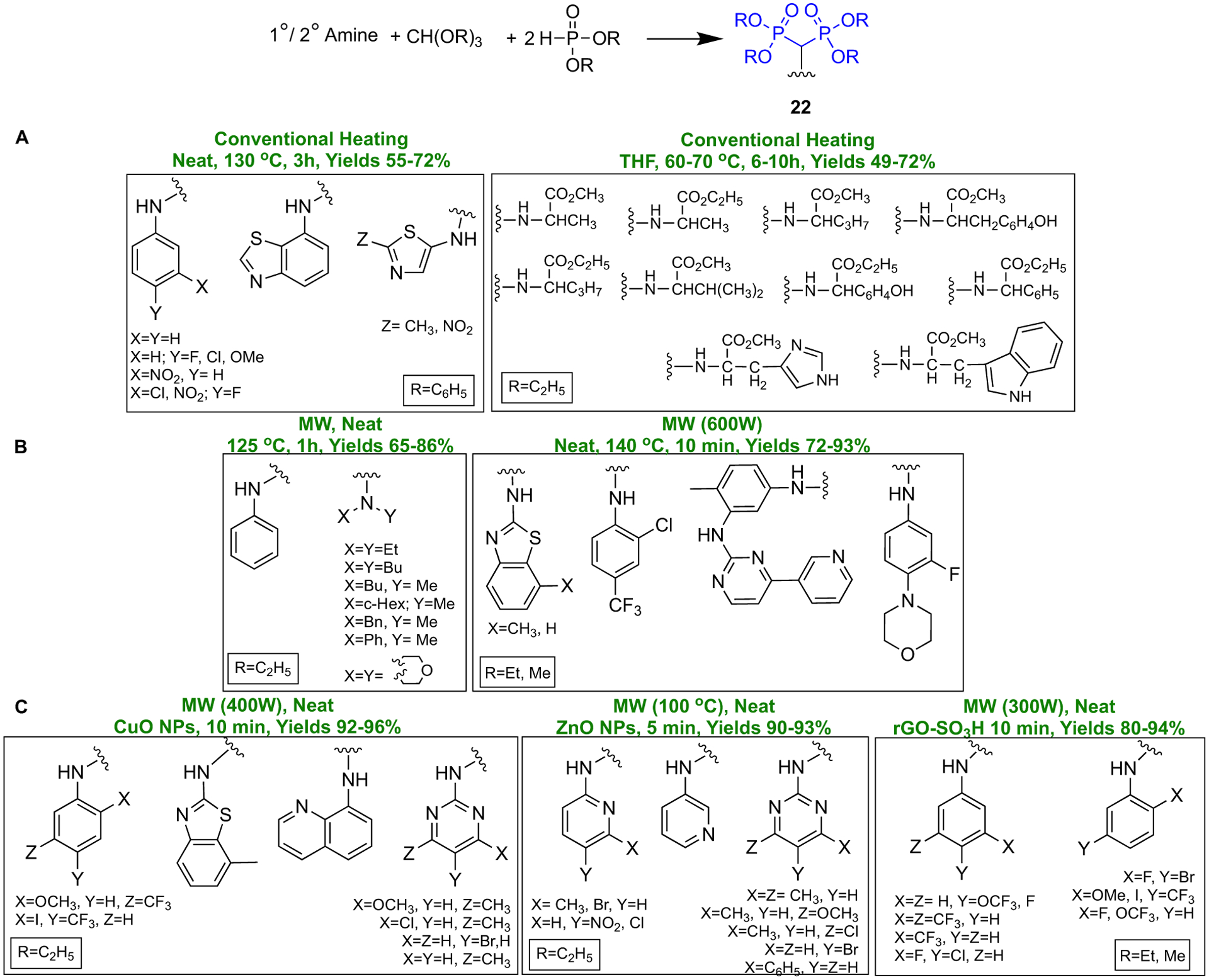
Recent methodologies to access α-amino bisphosphonates.
It is noteworthy that BPs are also used as synthons in the preparation of nucleotide analogs. When such BP analogues have different substituents at the bridging bisphosphonate carbon (as is the case with all the N-BP drugs), that carbon becomes chiral due to the asymmetric phosphonate groups in the nucleotide analogues. In a recent report, McKenna and collaborators have created a series of chiral synthons derived from differently substituted α-halo BPs which can be used to prepare individual stereoisomers of nucleotide BP analogues that showed non-equivalent interactions with an enzyme binding site, depending on the absolute configuration at the bridging carbon (Scheme 6) [53].
Scheme 6.
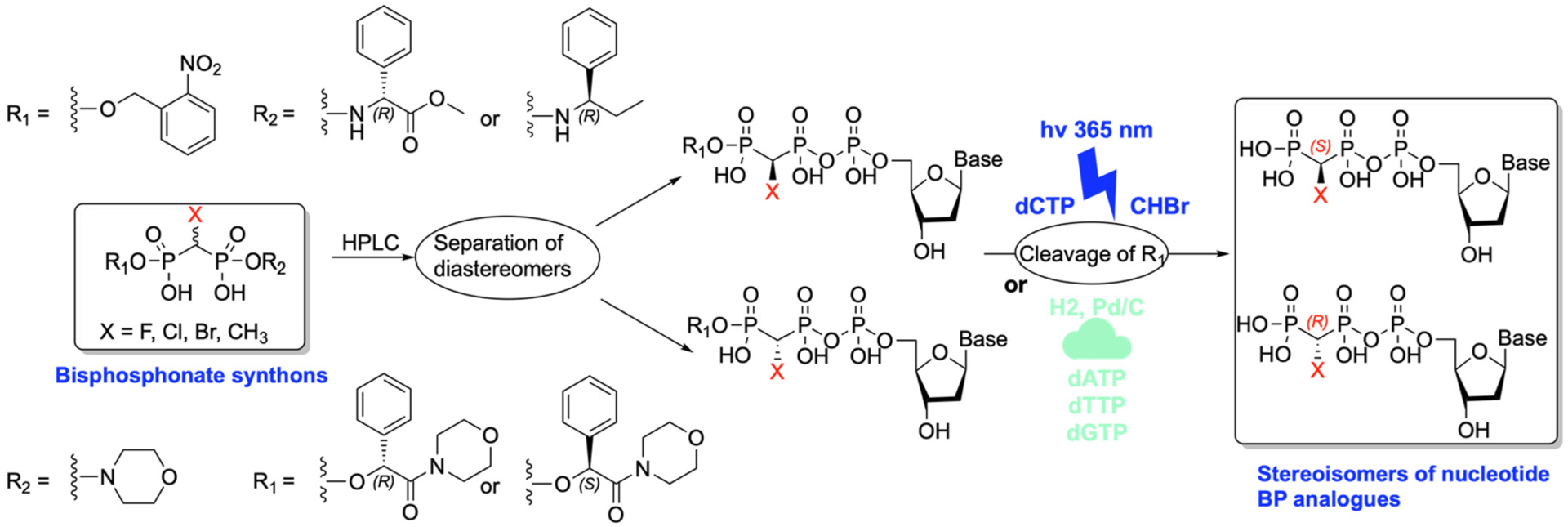
BPs used as synthons for preparation of nucleotide analogs.
2.2. Bisphosphonate esters and prodrugs
2.2.1. Bisphosphonate esters
Bisphosphonate esters are an interesting group of bisphosphonate derivatives, particularly those that have unique pharmacological properties of their own, that differ from the bone effects of classical bisphosphonates.
In the context of seeking potentially useful bisphosphonate derivatives, esters offer some novel properties. Being lipid soluble, they should have better absorption from the gut. Labile esters might also be used as prodrugs with improved bioavailability.
The compound called Apomine (not to be confused with ApomorphineR) is a 1,1-bisphosphonate ester (Fig. 3) which has been studied in considerable detail for its unique pharmacological properties.
Fig. 3.
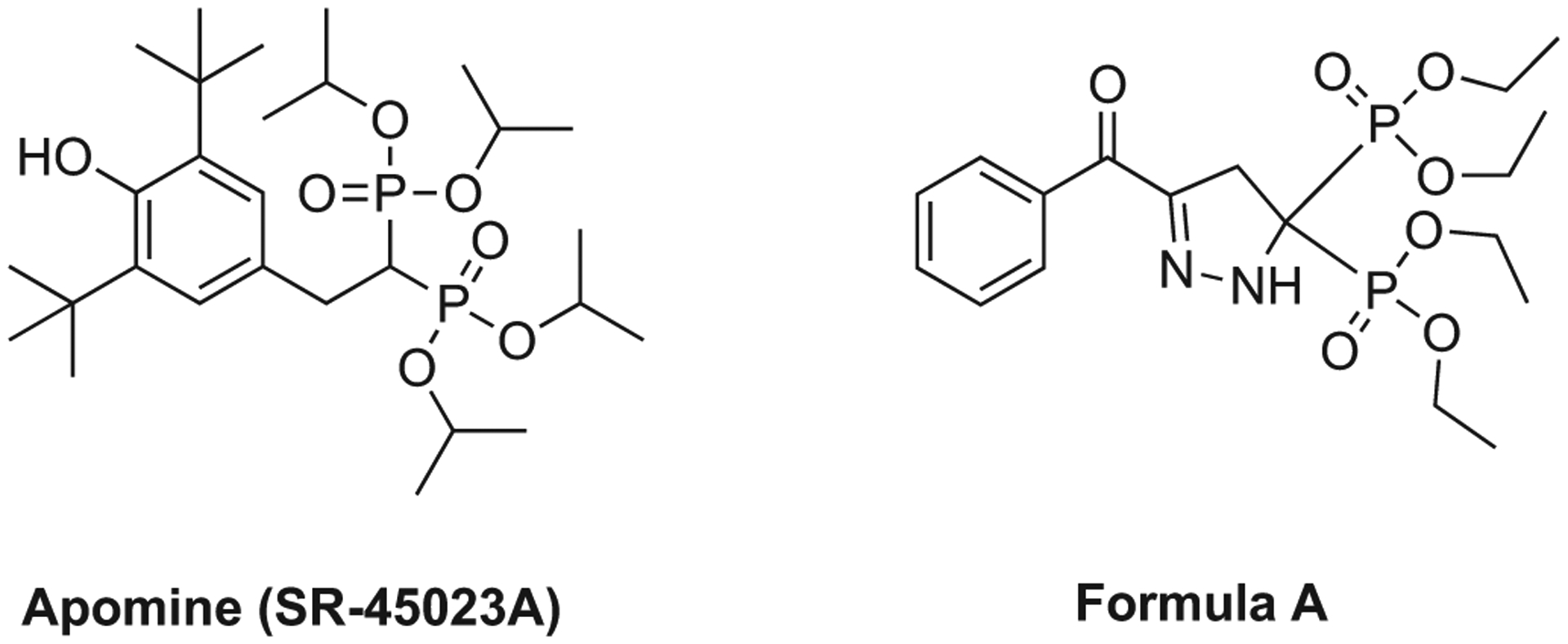
Examples of bisphosphonate esters: apomine (SR-45023A) and pyrazoline derived bisphosphonate ester (Formula A).
Apomine was one of several such compounds discovered by Eric Niesor and Craig Bentzen and colleagues at the Symphar SA company in Switzerland [54,55], and became the most intensively studied for its potential hypocholesterolemic [56–58] and antineoplastic activities [59].
The pioneering studies of this group showed that 1,1-bisphosphonate esters had the unexpected property of inducing hypocholesterolemia in experimental animals, an effect that was attributed to increasing the rate of degradation of HMGCoA reductase, a key enzyme in the cholesterol biosynthetic pathway and the well-known site of action of statin drugs. The mechanism underlying this hypocholesterolemic effect appears to be due to the ability of these esters to activate the orphan nuclear receptors FXR and LXR alpha. The farnesoid X activated receptor (FXR) is a member of the nuclear hormone superfamily implicated in cholesterol metabolism and bile acid transport. Natural endogenous agonists of FXR, like farnesol, as well as synthetic FXR agonists, like apomine, trigger differentiation, inhibit cell proliferation and are potent inducers of apoptosis. These actions may also contribute to their antineoplastic effects. These early studies with apomine (SR-45023A) on cholesterol metabolism led to it being studied by SmithKline Beecham as a novel cholesterol-lowering agent.
Apomine also underwent quite extensive development as an antineoplastic drug [59]. Preclinical studies showed activity against various tumor cell types, including osteosarcoma [60], myeloma [61,62], melanoma [63], and breast cancer [64]. Various clinical oncology studies were conducted, initially by Ilex Oncology based in San Antonio, which was later acquired by Genzyme Oncology in 2004. The clinical studies reached phase 2 exploration and included studies in refractory melanoma [65] and various solid tumors, including ovarian and prostate cancers. Despite several clinical studies and exploratory trials, apomine never reached the status of an approved drug however.
Pharmacologically, apomine is orally bioavailable but is a stable ester, so does not undergo cleavage and exhibit the typical bone effects of bisphosphonates. Pharmacokinetic studies were conducted in normal subjects as well as in cancer patients [66]. We might wonder whether the early promise apomine offered as a therapeutic with unique properties was thwarted by successive company mergers and acquisitions. Glaxo Wellcome merged with SmithKline Beecham in 2000 to become GlaxoSmithKline plc (GSK), and Sanofi acquired Genzyme in 2011. There are many examples of interesting drugs being victims of changing priorities when companies change hands and when key inventors are no longer able to champion their discoveries.
Nonetheless, the orphan nuclear receptors FXR and LXR alpha continue to be targets of interest for the discovery of new therapeutic agents. Bile acids and hydroxysterol intermediates are the respective natural ligands of these two structurally and functionally closely related receptors. Both FXR and LXR alpha are thought to play a major role in the control of cholesterol catabolism by regulating the expression of cholesterol 7-alpha-hydroxylase, the rate limiting enzyme of bile acid synthesis in the liver. Upon ligand binding, FXR regulates key genes involved in the metabolic processes of bile acid synthesis, transport and reabsorption and is also involved in the metabolism of carbohydrates and lipids. FXR is therefore considered to be a promising drug target for the therapy of bile acid-related liver diseases. With the approval of obeticholic acid (OCA) as the first small molecule to target FXR, several other small molecules are being evaluated in clinical trials. These newly emerging therapies based on agonists and antagonist of FXR are described in a recent review [67]. Interestingly, apomine is not specifically mentioned. However, apomine was recently identified as one of several compounds that might be repurposed for anti-viral activity against Covid infections [68].
As reviewed previously [69], bisphosphonate esters were also studied for their pharmacological effects useful in animal models of inflammation and arthritis. Nugent and coworkers reported studies to establish these effects with analogs related to pyrazoline (Formula A, Fig. 3), however, no compounds from this series ever advanced to clinical development [70]. It is uncertain whether these are in vivo prodrugs of the corresponding phosphonic acids.
2.2.2. Prodrugs
Bisphosphonates are notorious for their hydrophilic nature. This contributes to their excellent soft tissue safety as transport through many cell types and soft tissues is limited. Low oral absorption, and almost no absorption, reputedly due to an insoluble calcium salt formation “food effect” is also associated with orally administered bisphosphonates. While there are extensive studies on the phosphate and phosphonate prodrugs (reviewed in [71–73]), our focus here will be solely for studies of prodrugs of clinically used bisphosphonates. A group at P&G did demonstrate that addition of calcium chelators such as EDTA to oral formulations reduces the loss of absorption due to food intake prior to oral administration and the product Atelvia® was marketed as an any time formulation of risedronate that eliminated the need for fasting prior to weekly dosing of risedronate (Actonel®) [74]. Other researchers have investigated various ester, peptide (amide linkage) and anhydride prodrugs and formulations (for etidronate, clodronate, pamidronate and alendronate) for enhanced uptake after oral dosing and the studies have been reviewed extensively [75,76]. Those prodrug design strategies focus on masking the phosphonate groups as well as the –OH group on the bridging carbon and –NH2 group of the side chain substituent (Scheme 7). Webster et al. also evaluated the phosphonamidate prodrug of clodronate (compound 2a), and found the significant enhancement of in vitro anticancer activity of the prodrug compared to clodronate [77]. It should be noted that for the purpose of increasing oral bioavailability for bone disease treatment, BP prodrugs should have enough lability to be able to efficiently release the parent drugs in the bloodstream, but maintaining sufficient GI stability. In the case of BP prodrugs useful for extraskeletal diseases (e.g., cancer), it may be favorable for the prodrugs to have a somewhat slower cleavage in plasma to reduce the rate of sequestration on bone to allow greater distribution to other tissues. Additionally non-clinical studies have been reported demonstrating the potential utility of certain cyclic acetal esters of several nitrogen-containing bisphosphonates (e.g. risedronate ester (5j)). Although this series was not designed to enhance oral absorption in the fasted state, they appeared to be stable enough in the GI tract to dramatically reduce the food effect on orally administered bisphosphonates and then readily hydrolyze to the parent bisphosphonate (risedronate (5)) in the bloodstream (Scheme 8) [78].
Scheme 7.
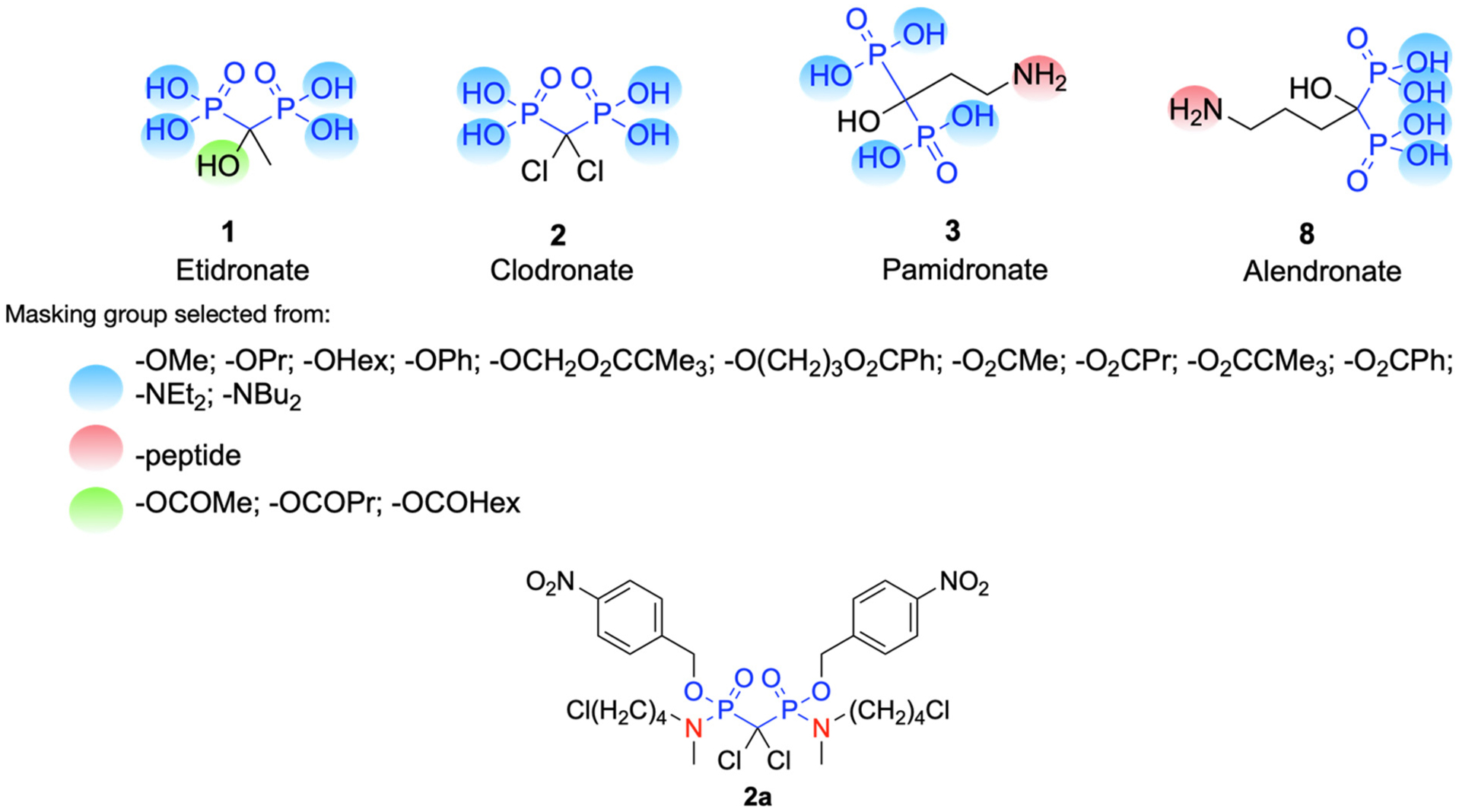
Prodrug design strategies for clinically used bisphosphonates: etidronate, clodronate, pamidronate and alendronate
Scheme 8.
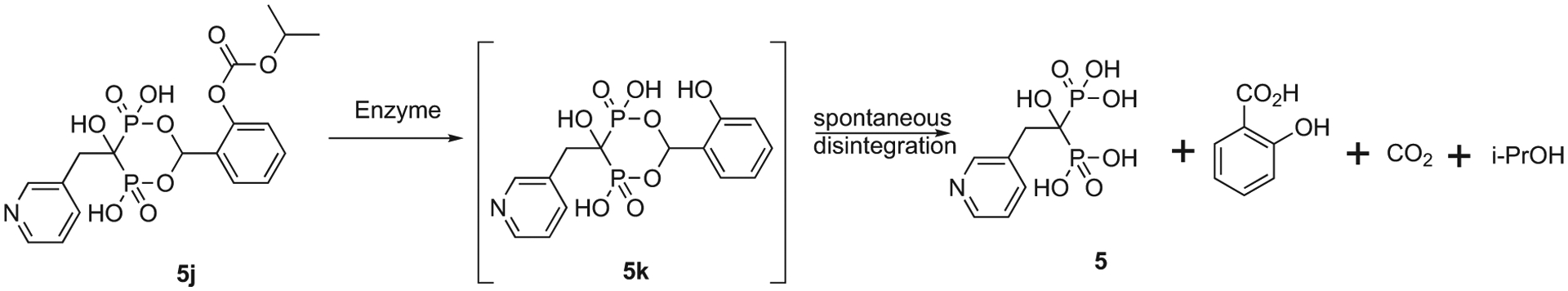
Enzymatic hydrolysis of relatively GI stable cyclic acetal ester of bisphosphonate (5j) releases tetracid drugs (5) in bloodstream.
2.3. Farnesyl diphosphate synthase (FDPS) inhibition: the key enzymatic target of nitrogen-containing bisphosphonates in osteoclasts – an interesting journey
The current understanding of the mechanisms of actions of BPs was not fully elucidated for almost thirty years after their first successful clinical use. The discovery of the major enzymatic target of N-BPs responsible for their antiresorptive activity has an interesting history and one not typical for modern drug development, where a clear identification of molecular targets and understanding of mechanisms of action is often required prior to regulatory approvals. The identification of the key enzymatic target, within the osteoclast, of these more recently developed, highly potent antiresorptive bisphosphonates (5–9) (Fig. 1) is perhaps the most important finding for this drug class since the discovery of the antiresorptive activity of BPs in the late 1960s. The key enzyme target, farnesyl diphosphate synthase (FDPS), was finally identified after the discovery of the involvement of the mevalonate pathway as a site of action of N-BPs. Followup studies using other research approaches, including protein crystallography, further illustrate the precise interactions between N-BPs and FDPS. The details of how bisphosphonates act within cells, and the details of this discovery history have been recently reviewed by Rogers et al. in the BP 50th anniversary celebratory issue of Bone [3]. The related in vitro assays based on inhibition of FDPS are now used as a semi-routine method to screen and evaluate the efficacy of newly synthesized BP molecules. Alongside the identification of FDPS, other enzymes in the mevalonate pathway, e.g., geranylgeranyl diphosphate synthase (GGPPS), were also found to be additional targets of some new BP molecules, and efforts to study in more detail the utility of the molecular interactions of BPs and FDPS or other key enzymes in the mevalonate pathway from different species have continued to emerge in the last decade.
Interestingly, the clinically used N-BPs all have weaker activity against geranylgeranyl diphosphate synthase (GGPPS) than FDPS. GGPPS is the enzyme beyond FDPS in the mevalonate pathway. When this weaker inhibition of GGPPS was first discovered, it was perhaps a rather surprising finding since it appears that it is the reduction in the levels of the metabolite GGPP, rather than FDP, that explains how BPs inhibit osteoclast function, which is the basis for their use in clinical disorders of bone resorption. This is because the GTP-ase signaling proteins prenylated by GGPP are more critical to osteoclast function than those prenylated by FDP. The probable explanation of this apparent paradox is that inhibition of FDPS reduces the levels of both metabolites, FDP and GGPP. In many experimental studies, the addition of GGPP reverses the biological actions of N-BPs, confirming that these are the key enzymes critical for the effects of N-BPs on osteoclast function.
In a recent article by Manaswiyoungkul and coworkers, the inhibitory potency of bisphosphonates towards human FDPS (hFDPS) and human GGPPS (hGGPPS) has been again extensively reviewed [79]. Selected recent examples that particularly shed light on the complexities of FDPS binding and inhibition are discussed in the sections below.
2.4. Insights from the interactions of bisphosphonates and FDPS via protein crystallography
It is now known that the BPs bind to the allylic (dimethylallyl diphosphate (DMADP) or geranyl diphosphate (GDP)) binding site of FDPS via three Mg2+ ions and a network of water molecules. The binding site contains two aspartate-rich motifs (103DDIMD107 and 243DDYLD247) which upon binding of BP produces a conformation change from open to partially closed. This creates a cavity for the binding of the homoallylic substrate i.e. isopentenyl pyrophosphate (IPP). Thus, the binding of IPP to FDPS rigidifies the C-terminal 350KRRK353 loop which causes the enzyme to develop a closed conformation to lock the BP and IPP inside the protein. Our team [2], and more recently, Park et al. [80] have provided further details of the binding of various BPs to FDPS.
Enzyme mutants have also been generated and analysed to further substantiate the key binding interactions required for optimal inhibition [81]. This includes investigation of the role of threonine 201 (Thr201) and tyrosine 204 (Tyr204) residues in substrate binding, catalysis, and inhibition by N-BPs, where kinetic and crystallographic studies of mutated FDPS proteins were employed. Mutants of Thr201 illustrated the importance of its methyl group in aiding the formation of the isopentenyl pyrophosphate (IPP) binding site, while Tyr204 mutations revealed the previously unknown role of this residue in both catalysis and IPP binding. The interaction between Thr201 and the side-chain nitrogen of N-BP was shown to be important for tight binding inhibition by zoledronate (ZOL) and risedronate (RIS), and RIS was shown to interact with the main-chain carbonyl of Lys200. The interaction of RIS with the phenyl ring of Tyr204 proved essential for the maintenance of the isomerized enzyme-inhibitor complex. Studies with conformationally restricted analogues of RIS reaffirmed the importance of Thr201 in the formation of hydrogen bonds with N-BPs. Thus new features of FDPS inhibition by N-BPs have recently been elucidated and additional unknown roles of the active site residues in catalysis and substrate binding were revealed in these studies [81].
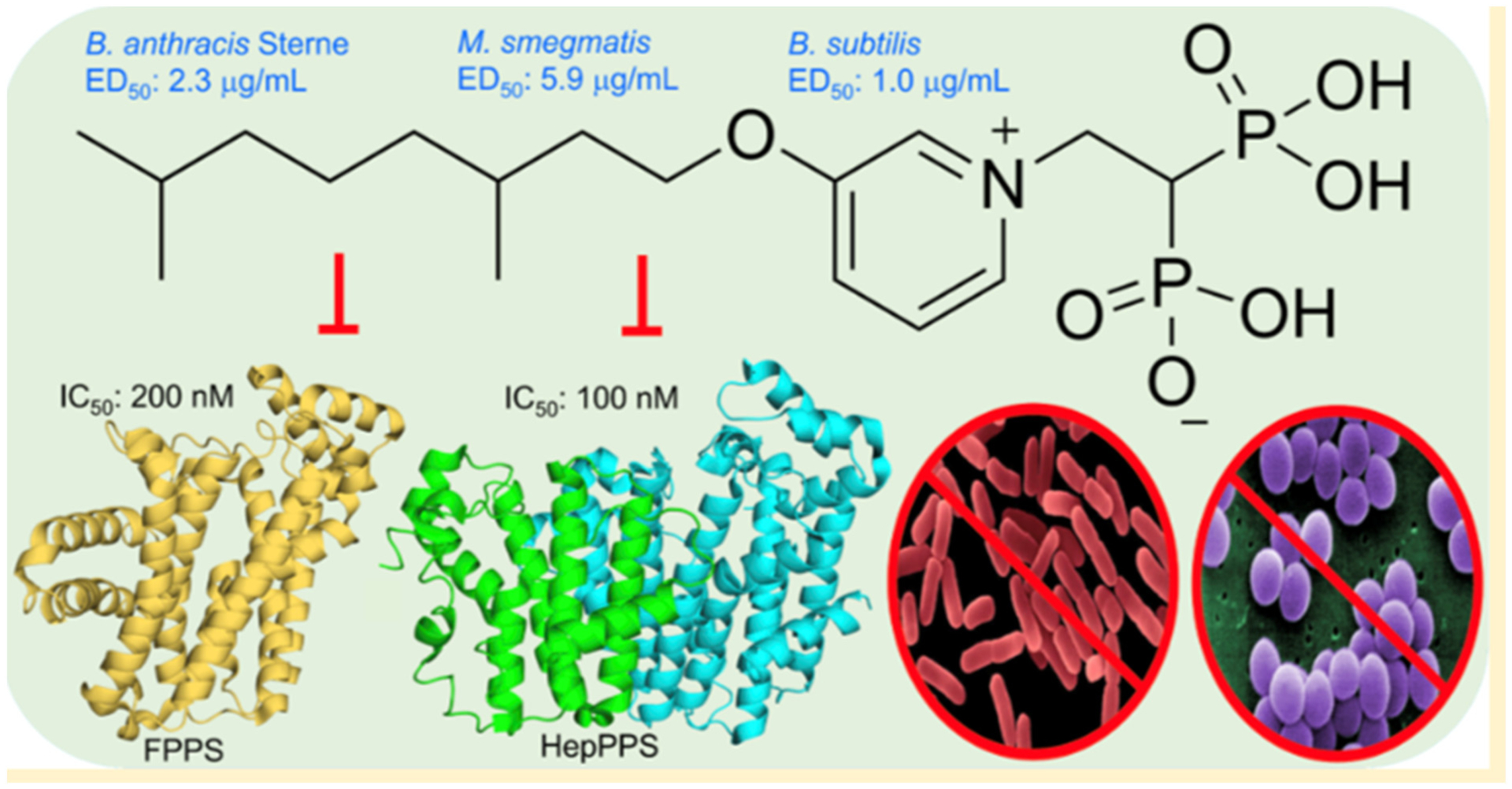
More work has continued to enhance the understanding of bisphosphonate binding characteristics of both FDPS and GGPPS. Notably, structure-activity studies from the Oldfield group, in related non-mammalian enzymes in particular, have led to the identification of additional bisphosphonates with exquisite sensitivity and specificity utilizing molecular modelling and crystallography techniques. The utility of this work has led to intriguing leads for parasitic diseases, anti-malarials [82], anti-tumor agents [83–85], and antibiotic research [86] (Fig. 4) (see detailed discussion in later sections in this manuscript).
Fig. 4.
Lipophilic antibacterial bisphosphonates that target the cell wall.
(Adapted from ref. [86]).
2.4.1. Recent studies of the binding of BPs to human FDPS
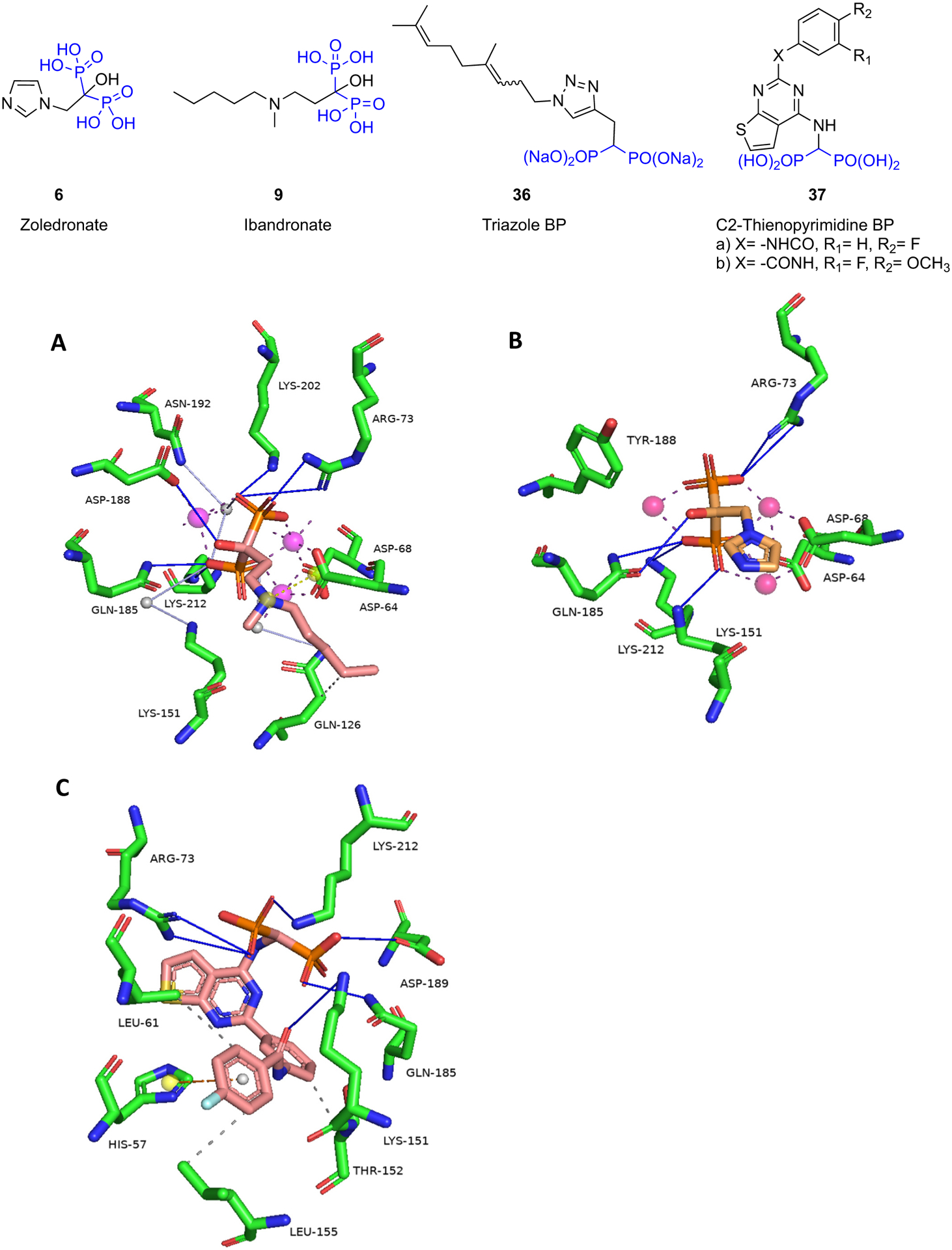
Yokoyama et al. [87] used neutron crystallography to further describe the protonation states and hydration structure of risedronate bound to FDPS. They obtained data for FDPS-RIS and FDPS-RIS-IPP complexes (Fig. 5A). In both X-ray structures, the pyridyl nitrogen was protonated. The pyridyl nitrogen formed a bifurcated H-bond with the side chain of Thr201 and the main chain carbonyl of Lys200. This main chain carbonyl of Lys200 also formed a CH–O bond with the 4-H of pyridine. The phosphonate groups of risedronate were completely deprotonated. Eight aspartic acid residues (Asp 103, 104, 107, 174, 243, 244, 247, and 261), Gln171, and Lys 266 interact indirectly with the phosphonate groups via water molecules and Mg2+ ions, while three basic amino acids Arg112, Lys200, and Lys257 interact directly with the phosphonate groups. The IPP was found to be fully deprotonated and interacting with basic residues and Mg2+ ions within the binding site. Neutron crystallography also revealed that the water molecules in the binding site are neither hydronium nor hydroxide ions. Hence, the neutralization of charge in the binding site upon the binding of RIS occurs via protonation-deprotonation of the adjacent amino acids. Thus, protonation of Asp264 enhances the binding of risedronate in FDPS. This work demonstrated that the pyrophosphate group of IPP was fully deprotonated and binding of IPP leads to the protonation of Glu93. Glu93 and Asp264 play an important role in stabilizing the net charge in the binding site to further enhance inhibitory potency. Also, it was shown that the hydroxyl group at the central bisphosphonate bearing carbon of risedronate enhances Mg2+ binding, further establishing its importance to enzyme binding, as well as binding to hydroxyapatite mineral surfaces.
Fig. 5.
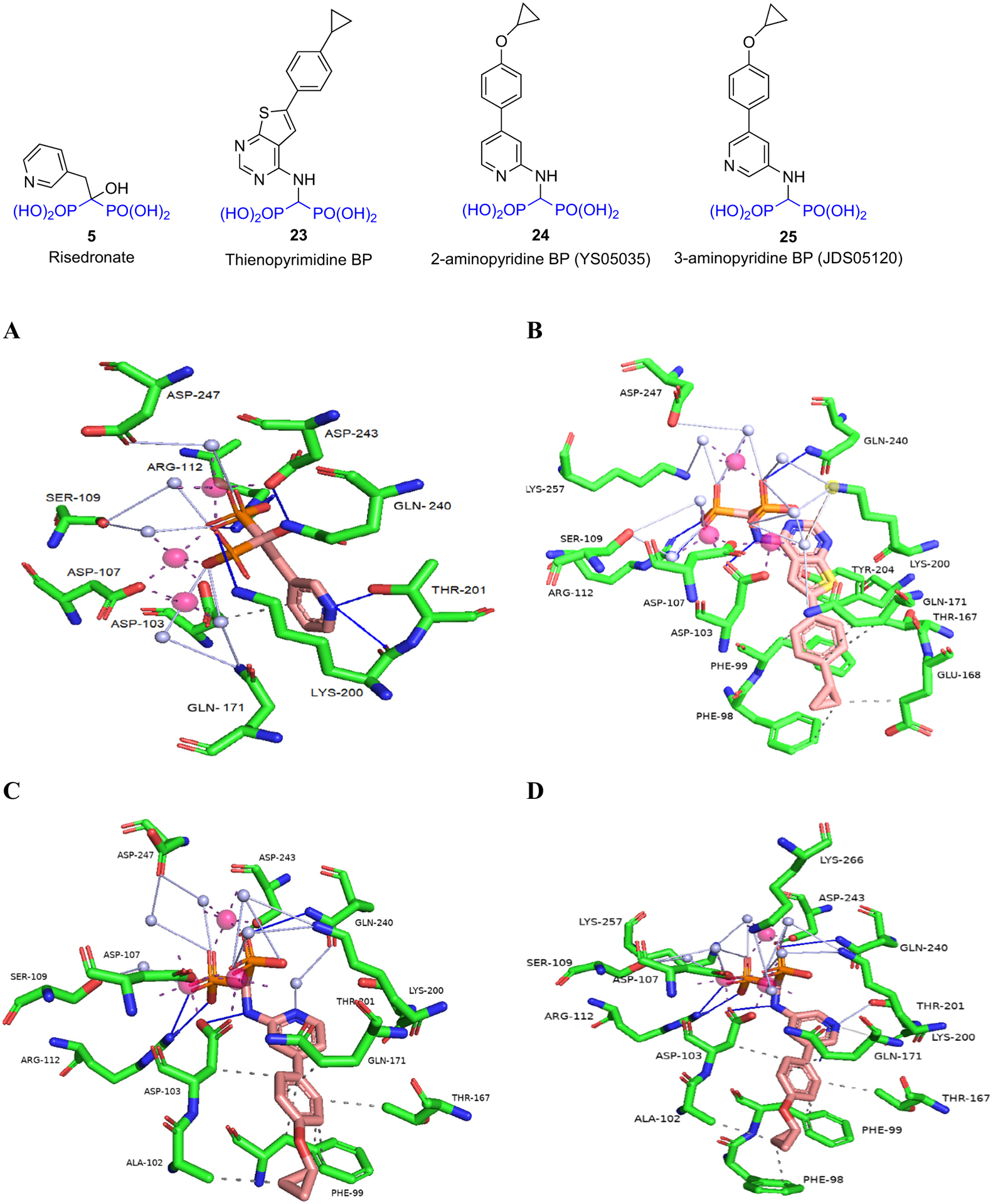
Binding of BPs to FDPS: A) Risedronate (5) (PDB ID 1YV5); B) Thienopyrimidine BP (23) (PDB ID 4L2X); C) 2-aminopyridine BP (24) (PDB ID 4PVX); D) 3-aminopyridine BP (25) (PDB ID 4NFI). Figure was generated using Protein-Ligand Interaction Profiler (PLIP) and Pymol. Code: blue line – hydrogen bond; grey line – water bridge; dashed line – hydrophobic interaction, white sphere – water molecule, pink sphere – Mg2+.
Leung et al. [88] synthesized a series of thienopyrimidine BPs which were potent inhibitors of hFDPS. An example from this series is 23 which was 8-fold less active than risedronate against hFDPS (IC50 39 nM vs 5.2 nM). The crystal structure of 23 - hFDPS shows that this thienopyrimidine BP can bind to the allylic (DMADP/GDP) binding portion of the enzyme in a similar orientation to that of risedronate (Fig. 5B). Thus, the phosphonate moieties of 23 can form similar interactions as those of risedronate. However, in comparison to the bifurcated H-bond formed by the pyridyl nitrogen with the side-chain hydroxyl of Thr210 and the main chain carbonyl of Lys200, the N3 of the thienopyrimidine ring only interacts with the side chain hydroxyl of Thr210, possibly reducing somewhat its overall enzyme binding/inhibition as in vitro studies suggest. The aromatic side chain of 23 extends into the lipophilic portion of the binding site containing two phenyl groups (Asp98 and Asp99), Leu100, and side-chain carbons of Gln171. Furthermore, the cyclopropyl ring extends towards the dimer interface of the homodimeric protein.
In addition to occupying the allylic (DMADP/GDP) binding site, 23 also occupies portions of the homoallylic (IPP) binding site, thus making it impossible for the IPP to bind concomitantly. Instead, the crystal structure of 23 – hFDPS contains inorganic pyrophosphate which functions similarly to IPP in closing the FDPS binding site, although no prenylation can then occur. Unlike risedronate, 23 lacks the Cα-hydroxy group which is known to increase the binding affinity of N-BPs to bone. Nevertheless, 23 had higher bone affinity than risedronate based on NMR studies. The higher bone affinity of 23 despite the lack of the Cα-OH group may result from less calcium salt solubility due to increased lipophilicity, or binding contributions from the other polar groups in the structure. Although no in vivo proof of concept studies are reported, the significance of this modification appears to allow improved cellular uptake as evidenced in their in vitro inhibition studies on myeloma cells. This could have important in vivo implications as good FDPS and bone binding characteristics are maintained.
The same group also designed a series of 2- and 3-aminopyridine BPs that showed inhibition of hFDPS comparable to risedronate [89]. A comparison of the binding modes of the 2-amino and 3-aminopyridine BPs showed that the pyridyl nitrogen of the 2-aminopyridine (24) interacts with Gln240 via a water molecule but it does not interact directly with other residues (Fig. 5C, D). In comparison, the pyridyl nitrogen of the 3-aminopyridine (25) forms a bifurcated H-bond with the side chain of Thr201 and main chain carbonyl of Lys200 reminiscent of the binding of risedronate pyridine. Nevertheless, both 2 and 3-aminopyridine analogs showed comparable activity against hFDPS (IC50 = 12 nM and 16 nM respectively). Thermodynamic characterization of the binding of the 2- and 3-aminopyridine BPs to hFDPS suggests that the enthalpic gain attained by the 3-aminopyridine BP (25) from the bifurcated H-bond formation is negated by entropic loss in the lipophilic portion of the molecule when compared to the 2-aminopyridine BP (24). Such findings will further fuel the discovery of analogs with strong FDPS binding, but with differing physiochemical properties that may offer leads towards analogs with improved nonskeletal benefits as well.
2.4.2. Binding of BPs to non-mammalian FDPS
In efforts to facilitate the development of new antiparasitic agents, Aripirala et al. [90] studied the binding of three pyridinium BP analogs to Leishmania major FDPS (LmFDPS) using X-ray and ITC. All three compounds are bound to the allylic (DMADP/GDP) binding site of LmFDPS (Fig. 6). Compounds 27 and 28 had similar binding modes to N-BPs. The binding of 27 and 28 with the LmFDPS allylic binding site involved three cations and IPP. In case of 26, there were two cations but no IPP. The authors suggest that the LmFDPS-26 complex might be an intermediary structure and it supports the ordered binding theory where the allylic sites on the homodimeric protein are occupied first leading to the formation of asymmetric monomers where one monomer was more favored for IPP binding. Binding of IPP to the first monomer leads to conformational change in the second monomer thus facilitating the binding of the second IPP. ITC studies of zoledronate and the three pyridinium BPs binding to LmFDPS suggest that the process was entropy-driven. Zoledronate and 26 bound more tightly to the enzyme than 27 which had a less favorable enthalpic component. Nevertheless, 27 still had better inhibitory activity than the other ligands which may be attributed to the more favorable entropic contribution arising from displacement of water by the phenyl ring from the hydrophobic region of the binding site.
Fig. 6.
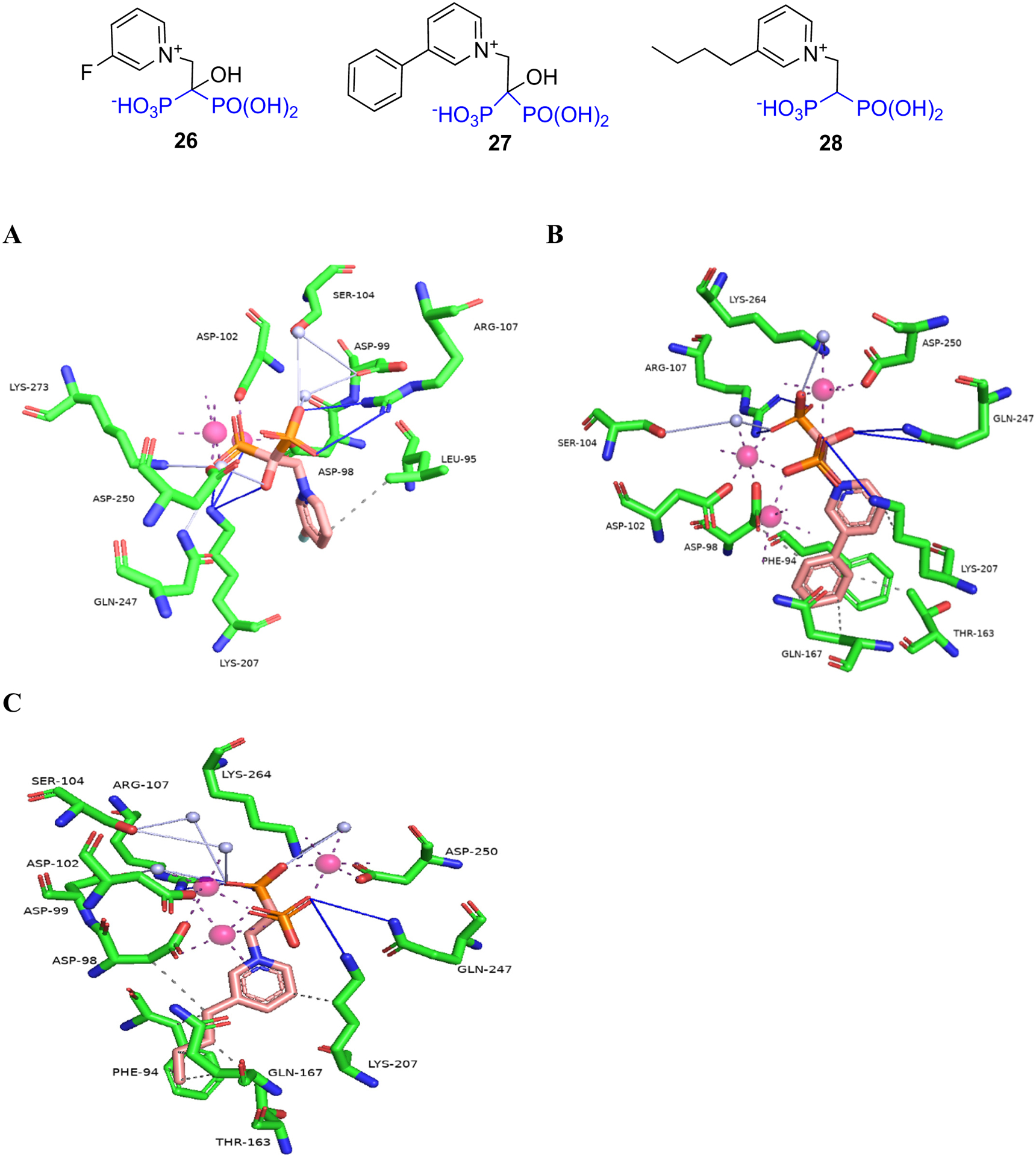
Binding of pyridinium BPs to Leishmania major FDPS: A) 26 (PDB ID 4K10); B) 27 (PDB ID 4JZB); C) 28 (PDB ID 4JZX).
Yang et al. [91] synthesized and tested a series of lipophilic BPs as potential antiparasitics for Trypanosomes. The series consisted of pyridinium, sulfonium, aminomethylene BPs, as well as N-alkyl imidazolium analogs of zoledronate and dehydoxyzoledronate. These were screened for biological activities in T. brucei FDPS enzyme inhibition, T. brucei cell growth inhibition, and also human cell (HEK293T) cytotoxicity assay in order to determine their selectivity index (SI = HEK293TCC50/T. bruceiEC50). Compounds with chain lengths of zero to seven carbons (C0-C7) showed comparable inhibitory activities against TbFDPS. The activity of the compounds decreased if the chain length was more than nine carbons (C9) which most likely was due to steric hindrance. Among the pyridiniums, sulfoniums, and aminomethylenes, the best compound was 29 (BPH-597) (TbFDPS IC50 = 0.25 ± 0.29 μM, SI = 4.3) which contained a six-carbon C6 chain at the 3-position of the pyridine ring. Among the N-alkyl imidazolium series, the dehydroxyzoledronate analog 30b (BPH-1326) (TbFDPS IC50 = 0.56 ± 0.13 μM, SI = 62) containing a seven-carbon chain and zoledronate analog 31b (BPH-1238) (TbFDPS IC50 = 1.26 ± 0.6 μM, SI = 293) containing a nine-carbon chain displayed the best overall activities. X-ray studies showed that the binding mode of these compounds is similar to the N-BPs. They bound to the allylic (DMADP/GDP) binding site of TbFDPS via three Mg2+ ions and a network of water molecules (Fig. 7). Thermodynamic studies using ITC showed that the binding is strongly driven by entropy. The addition of the seven-carbon chain to dehydroxyzoledronate 30a (BPH-301) led to an increase in the entropic (ΔS) value (53 cal mol−1 K−1 for 30b vs 41 cal mol−1 K−1 for 30a). This was a favorable entropic contribution as 30b displayed better inhibitory activity against TbFDPS than 30a (IC50 = 0.74 ± 0.7 μM, SI = 0.3). 30b was 4-fold less active than zoledronate (IC50 = 0.14 ± 0.057 μM, SI = 0.5) against the TbFDPS but had better T. brucei cell inhibitory activity (EC50 = 1 ± 0.0078 μM vs 14 ± 1.5 μM) and displayed a better selectivity index.
Fig. 7.
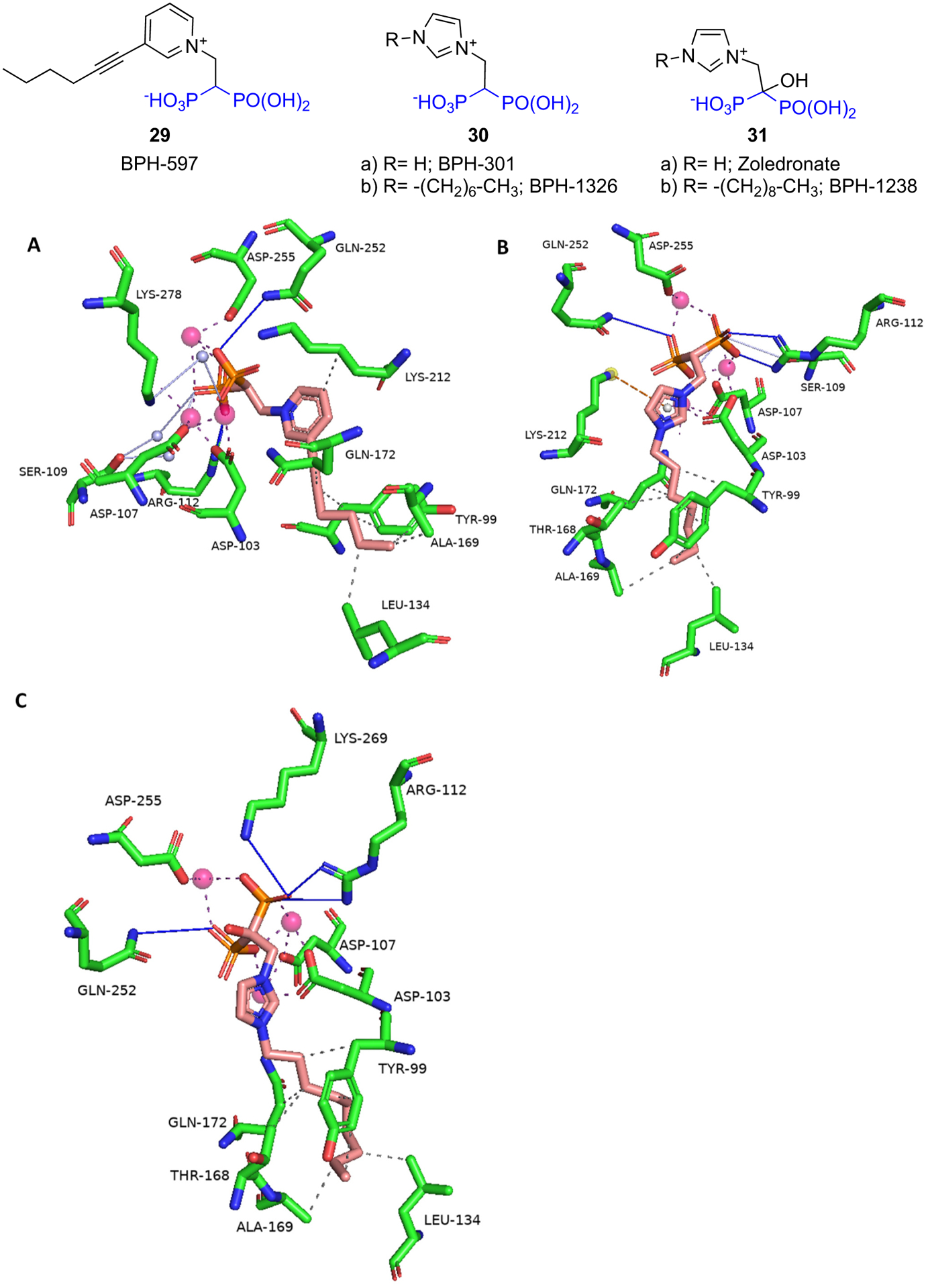
Binding of BPs to Trypanosoma brucei FDPS: A) 29 (BPH-597) (PDB ID 5AEL); B) 30b (BPH-1326) (PDB ID 5AHU); C) 31b (BPH-1238) (PDB ID 5AFX).
2.4.3. Binding of lipophilic BPs to other non-mammalian enzymes
Malwal et al. [86] designed lipophilic BPs that inhibit bacterial growth by targeting bacterial cell wall and quinone biosynthesis (Fig. 4). These BPs target the long-chain prenyltransferase enzyme octaprenyl diphosphate synthase of E. coli (EcOPPS) and the corresponding heptaprenyl diphosphate synthase of S. aureus (SaHepPPS). In addition, these BPs also target the FDPS enzyme due to the structural similarity of the active sites of these enzymes. Initial screening of a diverse set of compounds identified the long alkyl chain pyridinium analog 32 (BPH-715) as a potent inhibitor of both SaHepPPS (IC50 = 110 nM) and EcOPPS (IC50 = ~6 nM). 32 was previously reported to be a potent inhibitor of tumor cell growth with activity against both FDPS (IC50 = 100 nM) and GGPPS (IC50 = 280 nM). When screened against a panel of gram-positive and gram-negative bacteria, 32 was only active against gram-positive bacteria. Thus, 32 displayed good activity against S. aureus (ED50 = 10 ± 0.68 μg/mL) but it had weak or no activity (ED50 > 21 μg/mL) against E. coli. When compared with other structures in their set, compounds containing an aromatic group at the end of the lipophilic chain were generally more active against gram-negative bacteria. For example, compound 33 (BPH-1625) had quite good activity against gram-negative bacteria (ED50 = ~2 μg/mL) but it had weak activity against gram-positive bacteria (ED50 > 30 μg/mL). Such information will be valuable when developing selective inhibitors against gram-positive and gram-negative bacteria. SAR study of 32 led to compound 34 (BPH-728), which had comparable activity to 32 against SaHepPPS (IC50 = 100 nM), EcOPPS (IC50 = 12 nM) and human FDPS (IC50 = 230 nM).
The authors were unable to obtain an X-ray structure of their most active inhibitors bound to SaHepPPS or EcOPPS. However, they were able to obtain the structure of a weaker inhibitor 35 (BPH-629, IC50 = 330 μM) bound to EcOPPS (Fig. 8). Compound 35 bound to the allylic (farnesyl diphosphate, FDP) binding site of EcOPPS and coordinated with only one Mg2+ ion. Arg93 and Asp88 directly interacted with one of the phosphate groups while Asp84 interacted with the second phosphate via Mg2+ ion. The phenyl ring formed hydrophobic interactions with Thr80 while the tricyclic ring formed hydrophobic interactions with side-chains of Leu110, Ile142, and Ala143, and the side-chain carbons of Glu146 and Asp113. Compound 34 is expected to bind in a similar fashion as 35.
Fig. 8.
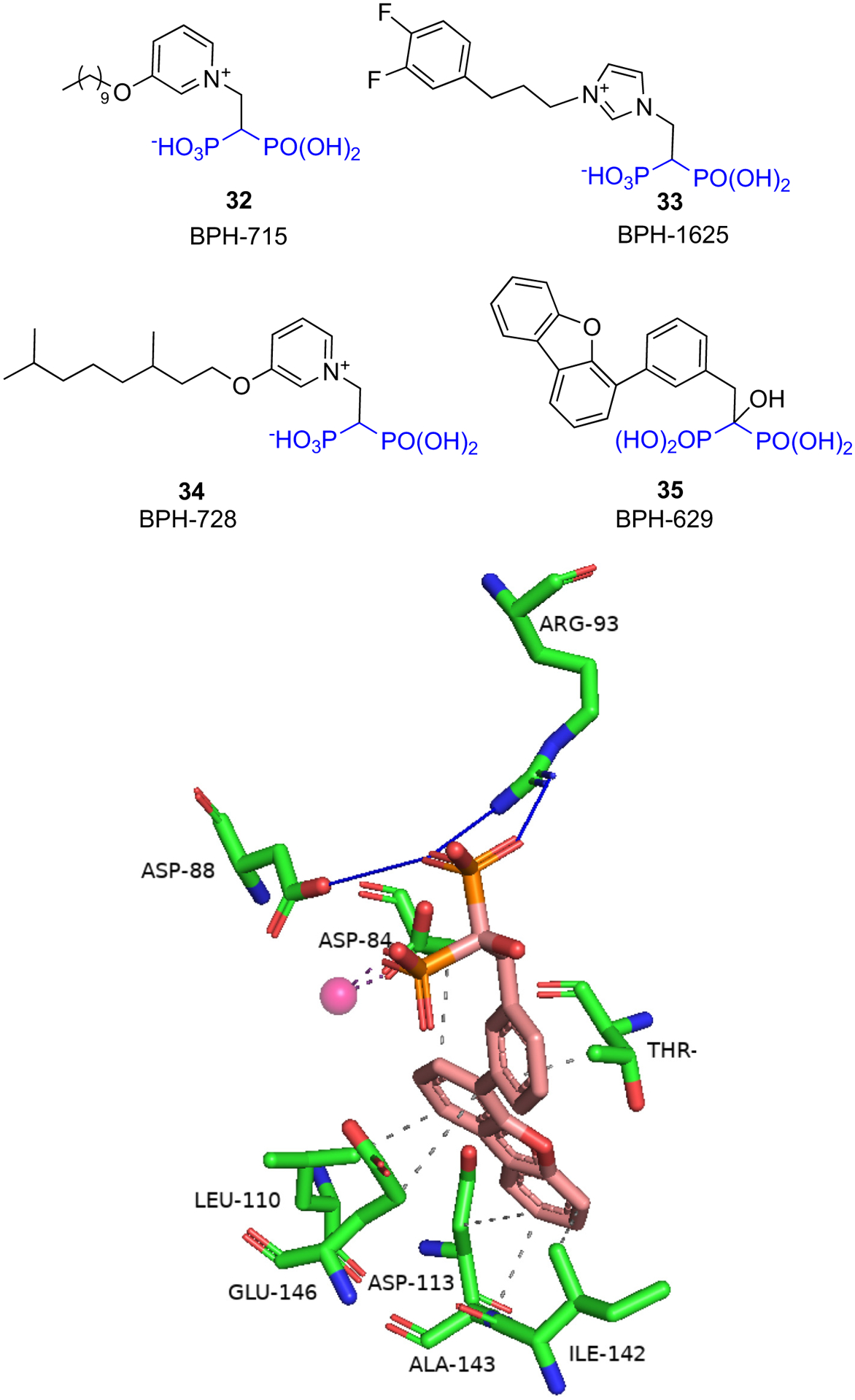
Binding of 35 to E. coli OPPS (PDB ID 5ZLF).
2.5. Further studies of interactions between bisphosphonates and GGPPS via protein crystallography
As noted earlier, the clinically used N-BPs have weaker activity against geranylgeranyl diphosphate synthase (GGPPS) than FDPS. GGPPS is the enzyme succeeding FDPS in the mevalonate pathway.
GGPPS catalyzes the formation of geranylgeranyl pyrophosphate (GGPP, C20) by condensing farnesyl diphosphate (FDP, C15) and isopentenyl pyrophosphate (IPP, C5). The catalytic site of GGPPS contains two aspartate-rich motifs similar to FDPS and the catalytic mechanism of the two enzymes is expected to be similar. The catalytic site of GGPPS is larger than FDPS in order to accommodate the larger GGPP product. Unlike the human FDPS enzyme which exists as a homodimer, the human GGPPS (hGGPPS) is a complex hexameric propeller-blade shaped structure consisting of three homodimers which makes it difficult to crystallize and study hGGPPS – inhibitor interactions. Most of the current knowledge regarding GGPPS – BP interactions comes from yeast GGPPS (yGGPPS) – BP X-ray structures. In comparison to hGGPPS, the yGGPPS exists as a homodimer and the BPs bind to the allylic binding site via Mg2+ ions and water molecules. Unlike FDPS where three Mg2+ are required to bind to BP, the number of Mg2+ ions in the binding site of yGGPPS - BP X-ray structures varies from zero to three. Recently, the first structure of hGGPPS complexed with BP (ibandronate) was reported [92]. Ibandronate (9) bound the allylic (FDP) binding site of GGPPS with the bisphosphonate moiety interacting with the aspartate-rich regions (64DDIED68 and 188DDYAN192) via three Mg2+ ions and a network of water molecules (Fig. 9A). The three Mg2+ ions were found to be crucial for hGGPPS – ibandronate interaction using ITC experiments and computational studies. Based on this finding, the authors reanalyzed several of the yGGPPS – BP X-ray structures and were able to assign three Mg2+ ions in the binding site which improved the overall quality of the X-ray structures. The X-ray structure of hGGPPS – ibandronate (9) will be a valuable tool for finding new GGPPS inhibitors using computational methods.
Fig. 9.
A) X-ray of hGGPPS – ibandronate (9) (PDB ID 6R4V); B) X-ray of hGGPPS (D118Y) – zoledronate (6) (6G31); C) X-ray of hGGPPS (Y246D) – 37a. Note: yellow sphere represents a charge center.
The same research group also reported the structure of mutant GGPPS-D118Y in complex with zoledronate (6) [93] (Fig. 9B). The D118Y mutation in GGPPS has been associated with increased susceptibility to BP-induced atypical fractures [94,95]. The GGPPS-D188Y mutant was ~4-fold less active than the wild-type GGPPS and had a reduced affinity for zoledronate. X-ray structure of the GGPPS-D188Y – zoledronate complex shows the displacement of Tyr188 compared to the apo structure to accommodate zoledronate (6) and one Mg2+ ion. Nevertheless, zoledronate (6) significantly reduced the activity of GGPPS-D188Y at physiologically relevant concentrations. It is noteworthy that while the residual activity of the wild-type GGPPS after zoledronate inhibition may be sufficient to carry out critical bone functions, inhibition of the GGPPS-D118Y mutant which already has low enzymatic activity may result in impairment in bone remodeling. Since this mutation was found in a family of three sisters with osteoporosis [94,95], it is still unclear whether it has a causal and wider relationship to the occurrence of atypical fractures associated with BP administration. Nonetheless these findings indicate a potentially interesting new direction of BP drug development by exploiting novel BPs that target hFDPS, but are less active on hGGPPS than the ones currently available, with the aim of reducing the side-effects of BPs used to treat bone diseases.
Inhibitors of hGGPPS may have potential use in the treatment of different cancers such as colorectal cancer [83], metastatic prostate cancer [84], and lung adenocarcinomas [85]. The majority of the BP-based inhibitors of GGPPS are characterized by one or two lipophilic chains in their structure. One of the most potent and selective hGGPPS inhibitors is a triazole-based BP (36) which has IC50 of 45 nM and is >600-fold selective for hGGPPS vs hFDPS [96].
Another example is the C2-thienopyrimidine BP compound 37a which has IC50 of 42 nM and is >24-fold more selective for hGGPPS vs hFDPS. The C2-thienopyrimidine BPs block the proliferation of multiple myeloma (MM) cells by inhibiting protein prenylation. In order to study the binding of 37a to hGGPPS, the authors generated an easily crystallizable version of hGGPPS via Y246D mutation in the protein (Fig. 9C). The hGGPPS – Y246D mutant was dimeric in nature and was catalytically active. A low resolution hGGPPS(Y246D) – 37a complex showed that the compound bound to the allylic (FDP) binding site with the p-fluorophenyl extending into the hydrophobic cavity that would accommodate the farnesyl group of FDP. The low resolution of the X-ray structure hampered the assignment of the Mg2+ ions, however the requirement for Mg2+ ions was confirmed by differential scanning fluorimetry thermal shift assays. Another C2-thienopyrimidine BP from the series, compound 37b which has an IC50 of 86 nM and is ~16-fold more selective for hGGPPS vs hFDPS, displayed antimyeloma effects in a MM disease mouse model thus validating hGGPPS as therapeutic target for MM.
Furthermore, as discussed in our previous review [2], the BP-based inhibitors of GGPPS were exemplified by the compound digeranyl bisphosphonate 38 (DGBP, Fig. 10A), synthesized by the Wiemer group in 2007. Interestingly, this compound was crystallized with yeast GGPPS by the Oldfield group as part of their crystallographic studies of BP inhibitors of this enzyme [97]. As can be seen (Fig. 10B), these compounds inhabit a binding site with a distinctive V-shape that accommodates both of the geranyl lipophilic chains with the BP head group still positioned to interact with 2 Mg2+ ions. This allows considerable interaction space to be modeled for further synthetic efforts around these structures with the aim of improving selectivity and affinity [98–100]. More recently, Zhou et al. [101] have demonstrated this through the synthesis of an isoprenoid bisphosphonate ether series where the most potent compound O-citronellylgeranyl bisphosphonate (OCGBP, 39, Fig. 10C) contained an ether based on an (S)-(+)-citronellyl motif replacing one of the geranyl groups of DGBP. This substitution increased the affinity for the enzyme by 2.6 fold compared to the parent (210 nM for DGBP vs. 82 nM for OCGBP).
Fig. 10.
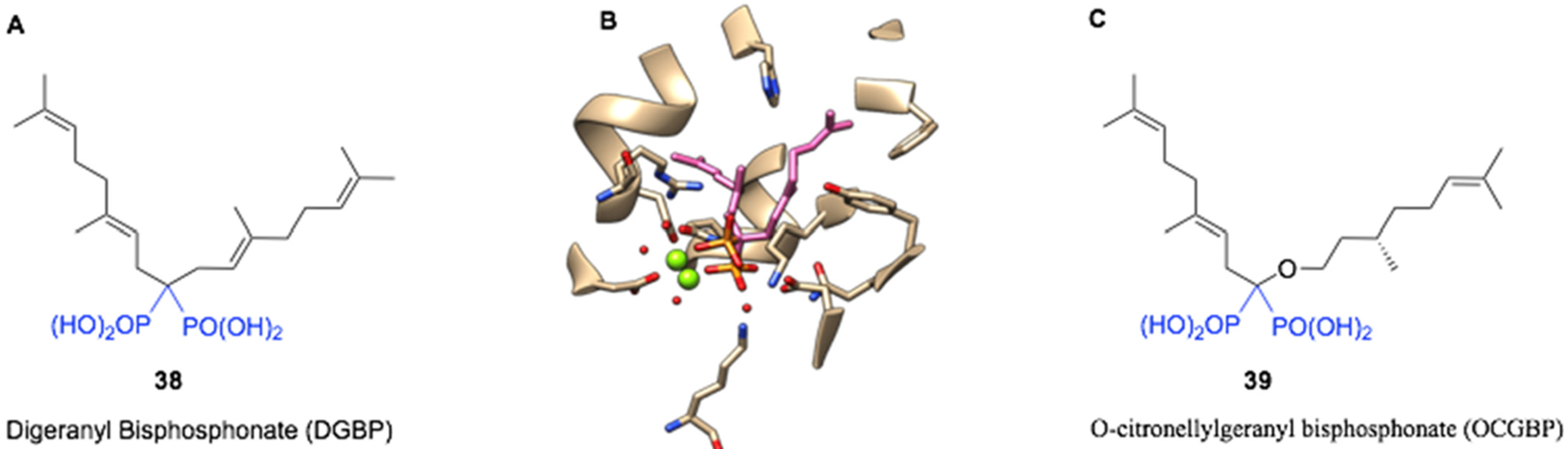
A: Structure of Digeranyl bisphosphonate (DGBP), B: Crystal structure of DGBP bound to S. cerevisiae GGPPS (pdb file 2z4w), and C: The more potent O-citronellylgeranyl bisphosphonate (OCGBP).
2.6. Study of interactions between bisphosphonates and squalene synthase (SQS) via protein crystallography
The SQS enzyme catalyzes the condensation of two FDP (C15) molecules to produce squalene (C30) which is crucial for the synthesis of sterols such as cholesterol and ergosterol. Inhibitors of SQS may be of therapeutic value in the treatment of hyperlipidemia, cancer, neurode-generative disorders, and as antimicrobial agents [102]. N-BPs such as ibandronate and incadronate were reported to inhibit SQS from rat microsomes, inhibit sterol biosynthesis in mouse macrophage J774 cells, and inhibit cholesterol biosynthesis in the liver in rats [103,104]. In another study, lipophilic BPs were shown to inhibit rat microsomal SQS, inhibit cholesterol biosynthesis in rats, and lower plasma cholesterol levels in rats and hamsters [105]. More recently, the Oldfield group tested diverse compounds including N-alkyl imidazolium analogs of zoledronate and deshydroxyzoledronate as inhibitors of Trypanosoma cruzi SQS (TcSQS) for the treatment of Chagas disease [106]. The N-alkyl portion of the BP analogs varied from one to fifteen carbons. Zoledronate, deshydroxyzoledronate, and shorter alkyl chains analogs (C1–C5) were inactive against TcSQS (IC50 > 10 μM). The activity of the longer chain analogs increased with chain length with the C9–C13 analogs showing the highest activity (IC50 < 0.1 μM). The corresponding zoledronate and deshydroxyzoledronate analogs had comparable activity. The most active compounds were the C10 zoledronate analog 41a (BP-1237, IC50 = 0.018 μM) and C11 deshydroxyzoledronate analog 41b (BP-1328, IC50 = 0.01 μM). The active analogs were generally more selective for TcSQS vs human SQS (HsSQS), TcFDPS, and hFDPS. For example, 41b was 11-fold more selective for TcSQS vs HsSQS, 39-fold more selective vs TcFDPS, and 28-fold more selective vs hFDPS. 41b also inhibited the growth of T. cruzi amastigotes (ED50 = 4 ± 1.3 μM) and displayed low cytotoxicity against vero cells (ED50 ~ 1000 μM). The authors also determined the X-ray structures of TcSQS and HsSQS complexed with representative BPs and compared these to the X-ray of the enzymes complexed with the substrate-like inhibitor FSPP (S-thiolo-farnesyldiphosphate, 40). The SQS – FSPP X-ray shows two FSPP molecules in the active site – one mimicking the allylic FDP and the other mimicking the homoallylic FDP (Fig. 11). The X-ray of TcSQS – 41a shows the compound bound to the homoallylic binding site (Fig. 12). The bisphosphonate group interacts with the side-chains of Ser42, Ser44, Tyr68, and Arg68 and the main chains of Ser44 and Phe45. The lipophilic tail extends into a hydrophobic pocket formed by Gly172, Leu175, Met199, Gly200, and Leu203 in a similar fashion as the farnesyl group of FSPP, and the terminal methyl of 41a and FSPP end in the same position. The C15-deshydroxyzoledronate analog 41c (BPH-1344) (TcSQS IC50 = 0.14 μM) shows a similar binding mode as 41a except that the alkyl chain bends at the initial carbons to occupy portions of the allylic binding site (Fig. 12). The terminal methyl of 41c (BPH-1344) reaches the same position as the terminal methyl of 41a and FSPP. These BPs bind to HsSQS in the same manner as TcSQS. The authors identified certain differences in close proximity of the hydrophobic pocket in the allylic binding site, for example Tyr179 and Ser283 in TcSQS are substituted by Phe187 and Cys289 respectively in HsSQS, that can be exploited to develop more selective inhibitors of TcSQS.
Fig. 11.
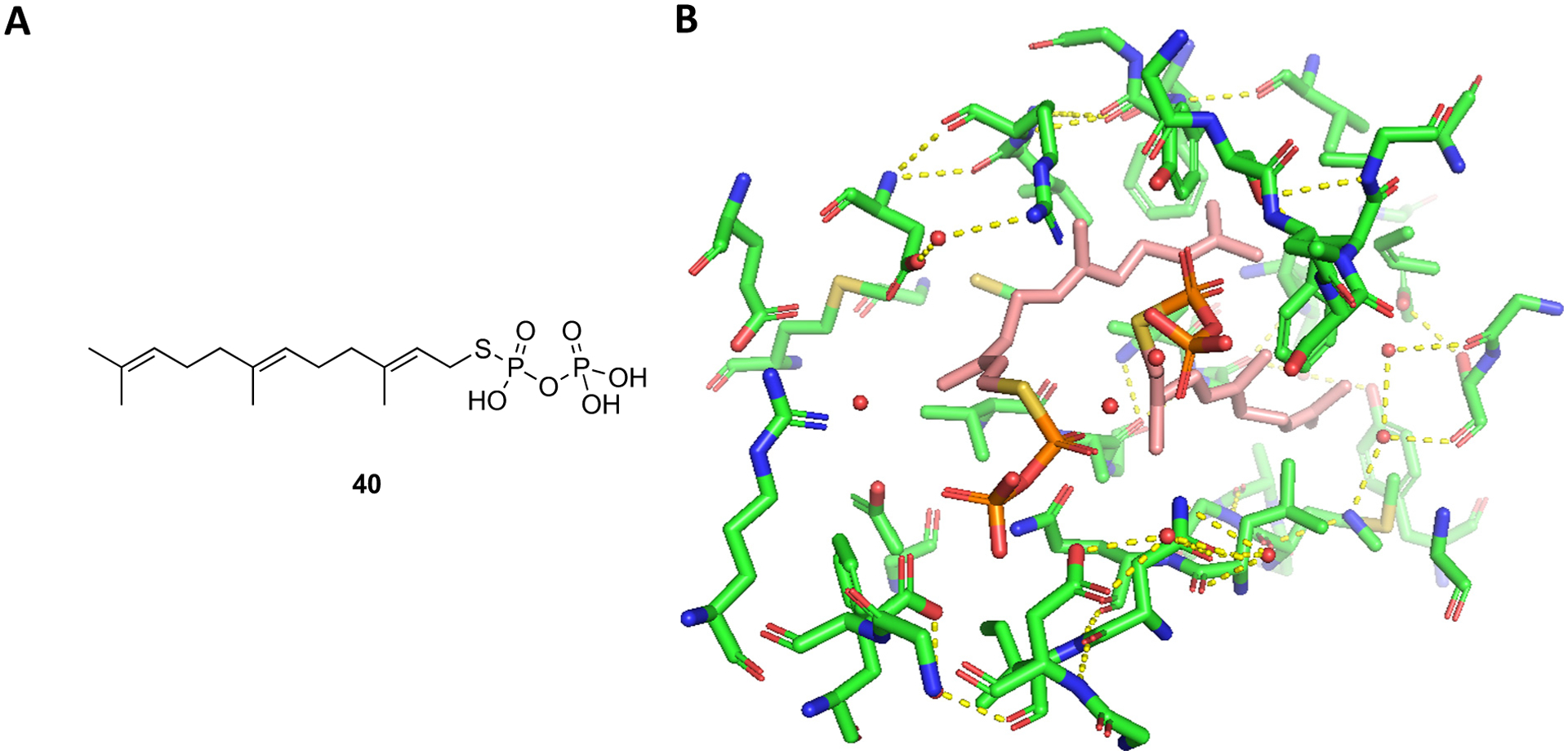
A) Structure of FSPP (40); B) Two molecules of FSPP bound to the active site of TcSQS (PDB ID 3WCA).
Fig. 12.
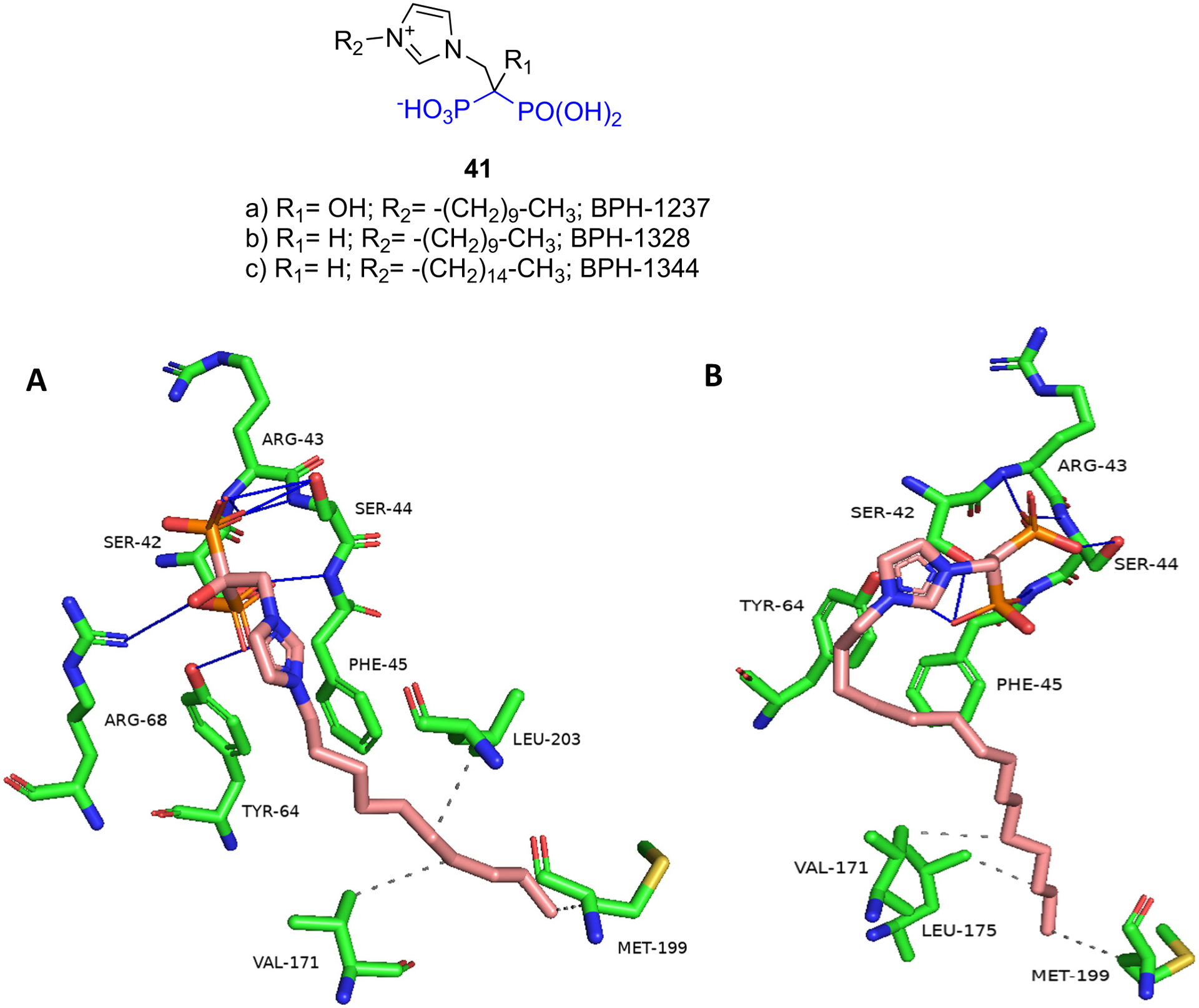
A) X-ray of TcSQS – 41a (BPH-1237) (PDB ID 3WCB); B) X-ray of TcSQS – 41c (BPH-1344) (3WCG).
2.7. Advances in the study of differing bone affinities among bisphosphonates and predicting structure-activity relationships: combining data from HAP mineral binding and inhibition (IC50) of FDPS predicts pharmacological potency
The bone binding property of BPs is an integral part of their biological activity and it significantly influences their skeletal uptake and retention, bone distribution, and potential recycling. A variety of in vitro methods have been utilized over the years to study the bone affinity of BPs as reviewed in detail in our 2011 paper [2]. The results of the in vitro studies were significantly affected by assay conditions such as pH, buffer concentration, and methodology, which has produced some apparent inconsistencies in the rank ordering of the bone binding affinities of BPs. Nevertheless, alendronate, zoledronate, and pamidronate typically rank as high-affinity N-BPs and risedronate and minodronate as somewhat lower affinity N-BPs [107]. In an additional study of the bone binding rank order of BPs in vivo, Lundy et al. [108] performed a direct comparison of the skeletal binding of BPs including fluorescently labeled BPs by simultaneously administering the BPs to rats and measuring their urinary excretion using LC-MS. Approximately 30–50% of the administered dose of the highest-affinity BPs and up to 95% of the administered dose of very low-affinity BPs were found in urine within 24 h (Fig. 13). Approximately 90% of the amount excreted within 24 h was excreted within the first 4 h. The rank order for bone binding of the BPs from this analysis was: Alendronate > Etidronate ~ Zoledronate > Neridronate > Risedronate > Minodronate > FAM-RIS ~ Clodronate > Ibandronate > OX14 ≫ NE-10790. Generally there was an inverse relationship between urinary excretion and affinity to HAP, indicating that binding of BPs to HAP strongly influences their in vivo distribution and excretion profiles [28,108]. While there are some discrepancies among typically described bone affinity measurement methods (e.g., for etidronate, ibandronate), it is still not known how much protein binding might be a confounder as well as the balance between kinetic binding vs thermodynamic retention in this in vivo analysis [5].
Fig. 13.
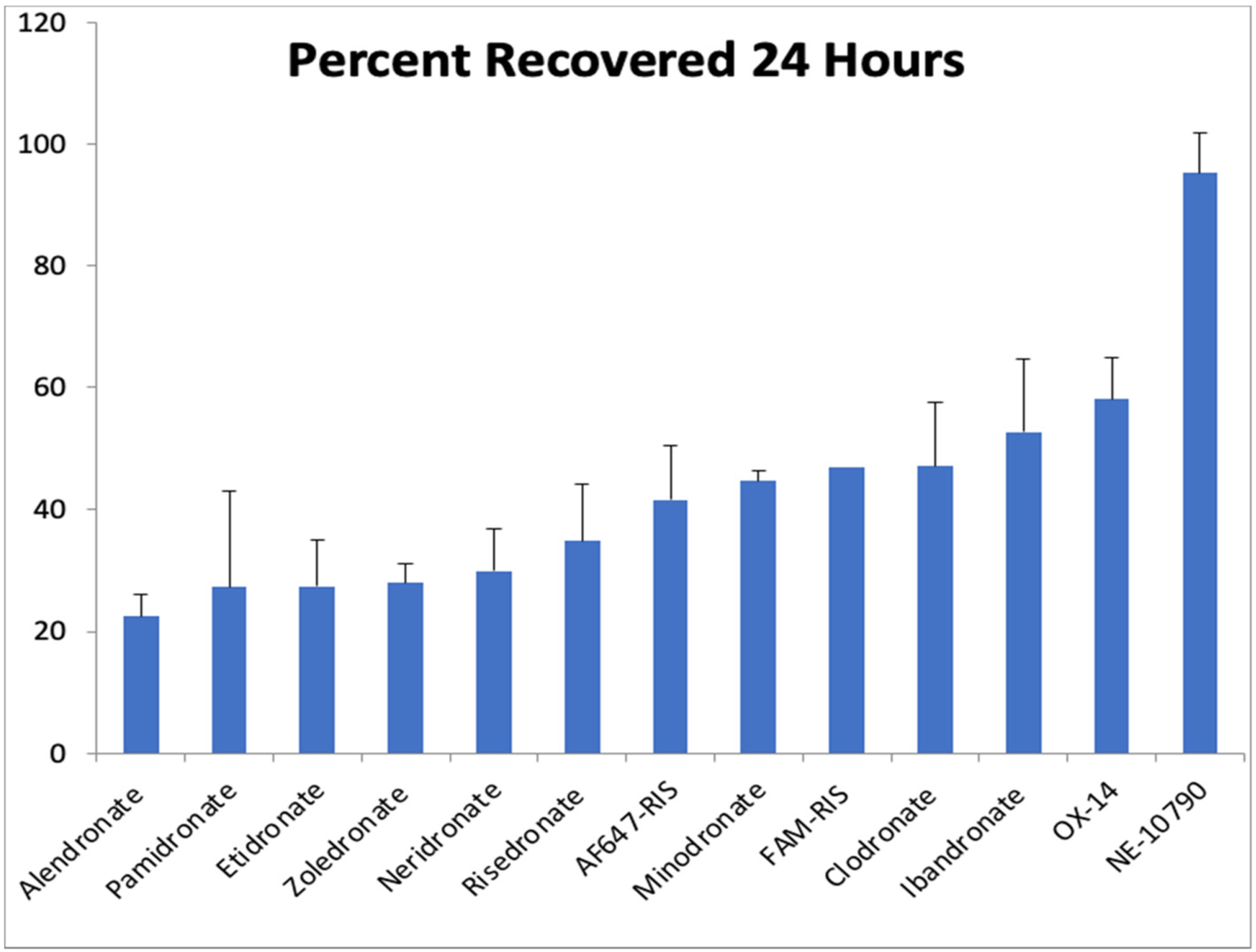
Percentage urinary excretion of bisphosphonates collected 24 h after i.v. injection (multiple compounds were administered in the same injection) (adapted from ref. [108]). Higher affinity compounds have a lower recovery in the urine. AF647-RIS (compound 52a6 in Fig. 17) & FAM-RIS (compound 52a1 in Fig. 17) are fluorescently labeled bisphosphonates [109,110].
As discussed earlier in this review, the two key properties of BPs: FDPS inhibitory activity and bone binding affinity, play the major role in determining the overall antiresorptive efficacy of BPs in vivo. Thus, the predictive potential of the in vivo efficacy of a novel BP molecule from in vitro assays has been addressed [108]. Similar to previous modelling, that was based on the combination of the IC50 of FDPS inhibition and the measure of bone affinity obtained from the HAP affinity column as discussed in our 2011 review [2,111], the relationship between predicted D20 and experimental D20 (D20 is the dose calculated from a dose-response study necessary to change BMD (assessed using DEXA) by 20% vs control animals in the growing rat model) was reanalyzed by Mark Lundy and Lin Fei using a multiple regression model with additional bisphosphonate analogs. By using the binding affinity data obtained from the HAP affinity column assay and FDPS inhibition data (IC50) to calculate the predicted D20, the striking correlation to the in vivo efficacy (experimental D20) was reinforced (r2 = 0.90) (Fig. 14) [108]. This result further demonstrates the prediction utility of such correlation modelling and that these two parameters (FDPS inhibition and bone affinity) are the major drivers of the pharmacological potency of bisphosphonates in vivo. This better understanding may therefore enable more efficient screening of new analogs for their desired in vivo efficacy profile.
Fig. 14.
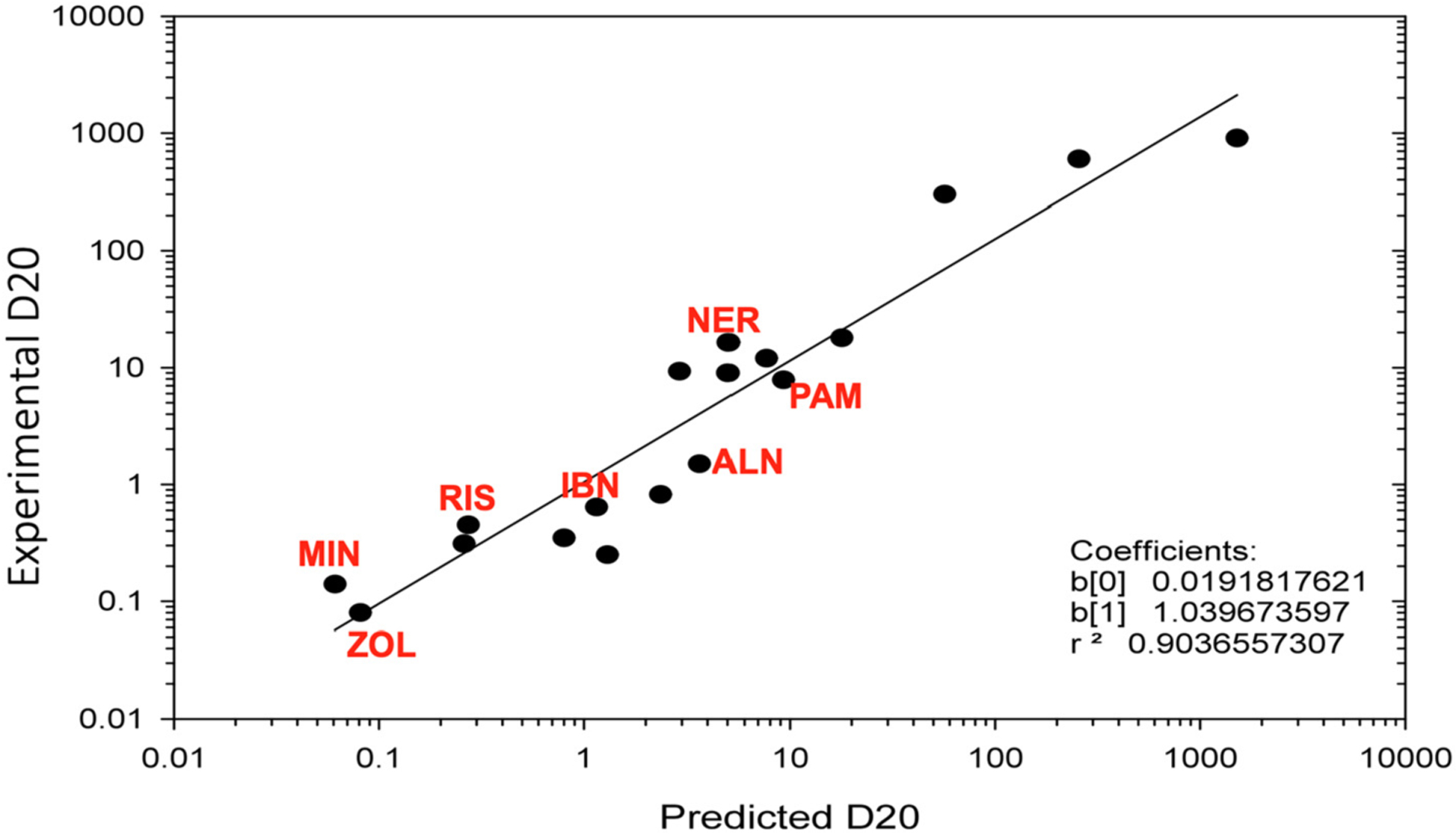
Correlation model of experimental D20 and predicted D20 (predicted D20 (pD20) is calculated from both in vitro FDPS inhibition and bone affinity data, via formula log(pD20) = −0.801 + 1.2605 * log (FDPS IC50 (nM)) − 0.0656 * Affinity − 0.01841 * log(FDPS IC50 (nM)) * Affinity). This model allows a very accurate estimate of the anti-resorptive in vivo efficacy (experimental D20), enabling rapid screening of new analogs based on in vitro data. Each dot represents a bisphosphonate analog, and all clinically used bisphosphonates are annotated in red: PAM – pamidronate (3), NER – neridronate (4), RIS – risedronate (5), ZOL – zoledronate (6), MIN – minodronate (7), ALN – alendronate (8), IBN – ibandronate (9).
(Figure adapted from ref. [108]).
3. New directions in bisphosphonate research
3.1. New uses of bisphosphonates and their relation to FDPS or other mechanisms
Now with many years of patient experience with bisphosphonates, and multiple prospective and epidemiologic clinical analyses, along with the advanced understanding of the mechanism of action of the bisphosphonates [3], opportunities for their use in new directions are receiving increased attention, both in skeletal diseases and even in non-skeletal fields. This includes immunomodulatory, anti-viral, and anti-tumor activities, and effects on protozoan parasites. For example, further studies have recently shown that N-BPs suppress the spread of various cancer cells in vitro [50,51,112,113]. However, it is not clear that the relatively high concentrations used in these studies are attainable in vivo. Others have discussed their ability to activate certain T-cell which may be related to an “acute phase response” sometimes observed on initial clinical dosing with iv bisphosphonates [114,115].
Promising nonclinical results with newly synthesized α-amino bisphosphonates raise hopes for the development of novel bisphosphonates with useful antioxidant, antimicrobial, and anti-cancer properties. Sudileti et al. have studied the anticancer effects of series of fluoro-substituted α-amino bisphosphonates against the human breast cancer cell lines (MCF-7) using the MTT assay method [51]. They found compounds 42(a–f) (Fig. 15) have shown greater antiproliferative effects in vitro as compared to the anticancer drug exemestane. In a different study, Lacbay et al. [116] have presented α-amino bisphosphonate 37a (Fig. 15) as a new class of inhibitors of hGGPPS and have reported the antimyeloma effects of 37a both in vitro and in multiple myeloma (MM) disease mouse models, as discussed in the section of “BP-GGPPS interaction” above. The study of antimicrobial activity of a series of α-amino bisphosphonates by Tellamekala et al. [49] have shown that compounds 43c, 44(a–b), and 45 displayed superior anti-fungal activity to that of voriconazole. Similarly, compounds 43(a–d) exhibited excellent antioxidant activity compared to that of ascorbic acid (Fig. 15). Moreover, recent computation docking studies conducted by Shaik et al. have predicted that compounds 46(a–b) (Fig. 15) have stronger inhibitory activity on human DNA ligase enzyme than that of the antibiotic ciprofloxacin [48].
Fig. 15.
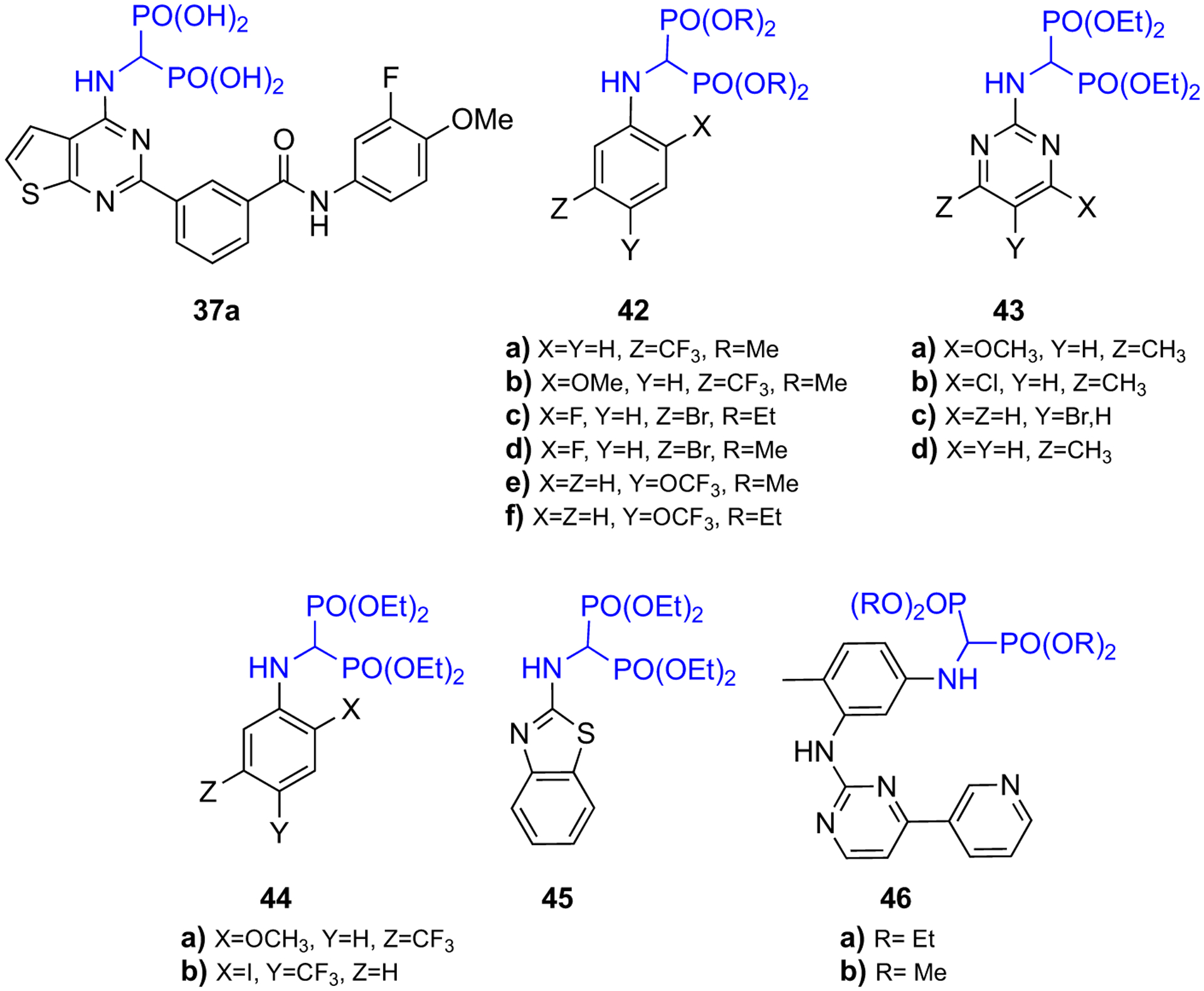
Structures of bioactive α-amino bisphosphonates.
In the past decade, a series of lipophilic bisphosphonates was synthesized, among which some of the triazole-based ones have shown exceptional inhibitory activities towards enzymes in the mevalonate pathway (Fig. 16). Assembly of triazole-based bisphosphonates with lipophilic tails can be obtained through a click chemistry reaction between an azide and an acetylene. In a report by Zhou et al. [117], compound 47 has shown potency towards inhibiting GGTase II with an IC50 of 0.1 mM, while 48 is a potent and specific inhibitor of GGPPS with an IC50 = 0.38 μM [118]. In 2015, Wills and coworkers reported that compound 36 (a 3:1 mixture of E and Z olefin isomers) shows an excellent inhibitory effect against GGPPS with an IC50 = 45 nM, which thus far is the most potent GGPPS inhibitor reported [96]. Later, in 2018 Matthiesen et al. modified the structure of 36 by incorporating a methyl group at the α-carbon to obtain 49a (Z isomer) and 50a (E isomer) [119]. This modification has slightly reduced the GGPPS inhibitory of 49a (IC50 = 85 nM) and 50a (IC50 = 125 nM); however, α-methylation allowed the preparation of POM-prodrugs (49b and 50b), which exhibited a 10-fold increase in cellular activity compared to the corresponding salt.
Fig. 16.
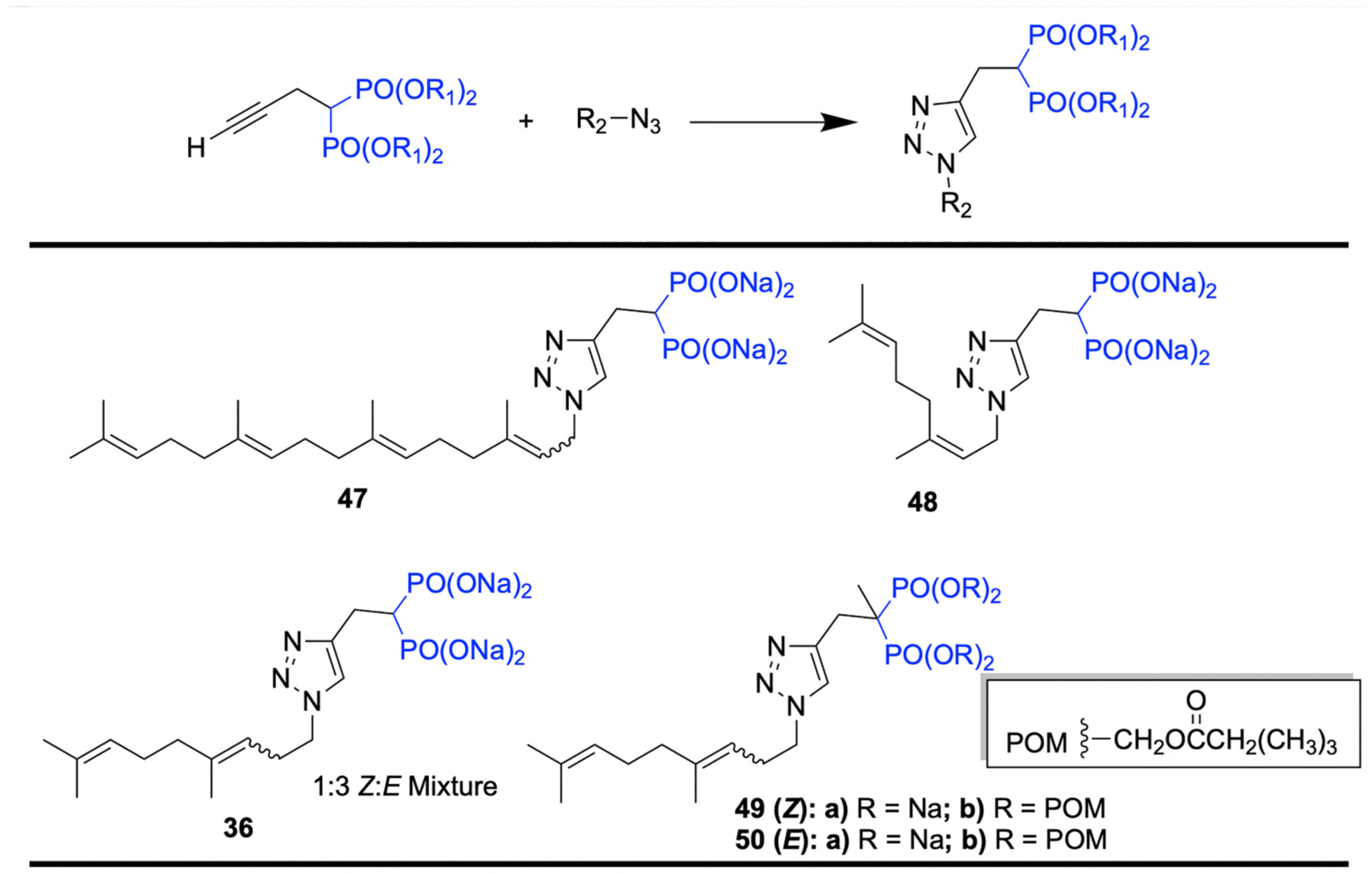
Triazole based lipophilic bisphosphonates.
Other observations suggesting potential nonskeletal effects with the current clinically used bisphosphonates have also appeared in the literature in recent years. An exciting recent report discusses the evidence for the mesenchymal stem cell protective effects of the bisphosphonates and their ability to block DNA damage and enhance stem cell repair after irradiation or other damage [120]. Inhibition of FDPS within these cells appears to be an important step leading to these effects. Examples of enhanced wound healing and perhaps even anti-fibrotic activity have been demonstrated in models of ulcerative colitis [121], lung fibrosis [122], and lifespan and cardiac benefits [123]. The finding that inhibition of GGPPS is a potential treatment for pulmonary fibrosis has recently been followed with a report showing that simply increasing the flux of metabolites through the pathway of isoprenoid synthesis leads to increased macrophage-mediated fibrosis [124]. This result points to the use of GGPPS inhibitors in fibrotic diseases in general where this type of macrophage dysregulation mediates the pathology. Thus, advances in the chemistry of bisphosphonates and our advanced mechanistic understanding of enzyme targets may yield future designer bisphosphonates in these fields, perhaps with lower bone affinity and higher targeting to other tissue compartments.
Furthermore, there are also many new opportunities for improved studies and new data for bisphosphonates in the veterinary field, as reports of their use in pets, horses, and other domestic animals are increasing [125,126]. Surprisingly little work has been done to develop bisphosphonate drugs for uses in this field. Additional work is clearly needed for generating optimized veterinary products.
3.2. Fluorescent BP analogs and applications
Over the past two decades, many more fluorescent bisphosphonates have been designed and made available as probes for mechanistic studies and as diagnostic research tools [2]. The McKenna lab introduced a facile “magic linker” strategy to create the first fluorescent conjugates of a modern N-BP drug, RIS bisphosphonate as imaging tools [110]. Since then, additional fluorescent bisphosphonates were prepared, expanding the “toolkit” to cover all clinically used N-heterocyclic BPs (ZOL, MIN) and related analogues, as well as a wider spectroscopic window from visible to near-infrared range (Fig. 17) [109]. These probes have been used in various imaging studies in skeletal-related fields [127–130], and further expanded in non-skeletal fields in recent years, including dental research [131–136], otology [137–139], drug distribution studies [140,141], cancer research [142,143], nephrology [144] and brain calcification studies [145].
Fig. 17.
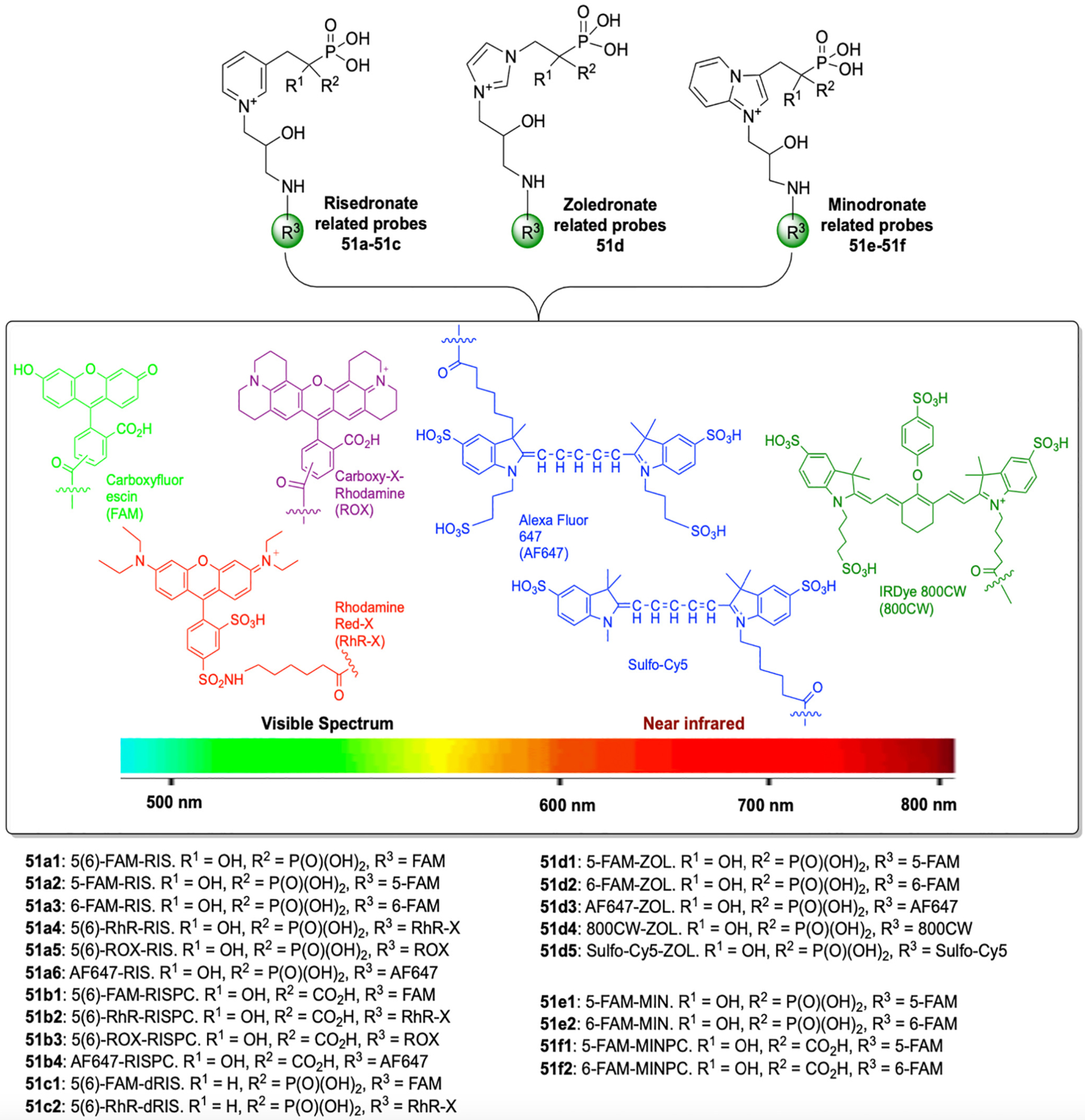
Fluorescent bisphosphonate imaging probe “toolkit” derived from clinically used N-heterocyclic BPs (RIS, ZOL, MIN). These probes cover a wider spectroscopic window from visible to near-infrared range, allowing various imaging applications both in vitro and in vivo.
(Adapted from ref. [109]).
Another noteworthy highlight concerning the use of fluorescent bisphosphonates is the contribution they offer to a more in-depth understanding of how structure-activity relationships among bisphosphonates influence their skeletal distribution. This may lead to new avenues of therapeutic exploration, for example in gaining access to osteocytes within the canalicular network in bone [128,129]. Of course fluorescent BPs may not only have diagnostic potential but also combined therapeutic/diagnostic value as well. For example, McKenna and collaborators have exploited the fact that one bisphosphonate can displace another on the bone surface, which has led to the development of an “antidote” procedure [131]. By utilizing relatively inactive (low potency) bisphosphonates (e.g., some fluorescent bisphosphonates) that maintain their bone binding affinity, it has been shown that this strategy may aid the prevention and treatment of related symptoms of zoledronate associated osteonecrosis of the jaw in a rodent model [136]. In addition, a recent significant study by McDonald, et al. reported a novel cellular program for osteoclast reuse through their fission, transport, and reassembly process (osteoclast-osteomorph recycling) [146]. This finding redefines the understanding of osteoclast physiology and also points to new actionable targets for novel therapeutic development [147]. Fluorescent bisphosphonates have played an indispensable role in this project as imaging probes, which enable direct observation of the process via intravital imaging technique.
These fluorescent bisphosphonates are “always on” in the sense that their fluorescence persists wherever they may bind. The complementary concept of an imaging bisphosphonate whose fluorescence is “off” until triggered by a specific enzyme targeted in situ was recently demonstrated by McKenna and collaborators, who synthesized a molecule consisting of a Förster internally-quenched dye pair linked by a cathepsin K peptide substrate attached to a bisphosphonate, to detect in situ bone resorption by osteoclasts, which secrete proteolytic cathepsin K to break down the collagen matrix in the bone. This “osteoadsorptive fluorogenic sentinel” or OFS probe adsorbed on hydroxyapatite generated a localized fluorescent signal when activated by a resorbing osteoclast. Its exciting diagnostic potential was shown by its ability to sensitively image multiple myeloma cells metastasized to a nascent bone tumor site in a murine model [148].
3.3. Targeting drugs to the bone with bisphosphonates
Ideally, drugs should act directly upon their intended targets, selectively in the relevant tissues, in order to achieve optimal efficacy with minimal side-effects. There are several ways to pursue this in drug design, but this goal is not always straightforward. With statins, for example, selective liver uptake is known to be a desirable property, but off-target effects on muscle can lead to side effects. Key principles underlying the development of selective bone-targeted bisphosphonate – drug conjugates continue to evolve [6,149], and more recent successes with in vivo “Target and Release” strategies offer an exciting tissue-selective drug design approach [150].
The risk of off-target toxicities with non-selective drug classes can also hamper the drug discovery process, particularly for early in vivo evaluation of a drug class. In the case of drugs acting on or within the bone, the bisphosphonates offer an obvious and unique opportunity for selective drug delivery. In theory, selectivity from targeting bone with bisphosphonates could be achieved through their conjugation with many different relevant drug classes. In practice, the resulting bone-targeted drugs may offer important new therapies for many unmet medical needs. While this concept is not new, there are significant challenges with this drug design approach and it has achieved only limited clinical success. The permanent covalent linking of two drug classes generally can significantly diminish the activity of each component in drug conjugates. Efficient and effective release mechanisms are likely to be needed in most cases as precise local concentrations of released drug need to be generated which complicates the pharmacokinetics of releasable BP-drug conjugates. These hurdles have led to a slower than desired evolution of such a bone targeting strategy in drug design [151].
Only recently, advances in chemistry have enabled the identification of bisphosphonate linkers that provide adequate stability in the bloodstream, and rapid enough break-down and release of pharmacologically active agents at the bone surface in relevant concentrations to affect the desired biochemical targets. These new conjugates are being designed to release active drugs in the vicinity of cells near the bone surface in a “depot” fashion. The repertoire of bisphosphonate conjugates of various known and experimental drug classes includes steroids [152], prostaglandins [153], antibiotics [154,155], and several anticancer drug classes [156]. Such an approach may also enhance the likelihood of success in new drug design by offering more rapid in vivo validation of new drug classes and their targets for further optimization of drug leads. Therefore, it is likely that further research on the development of bone-targeted drugs for a variety of bone-related diseases will offer potential new therapeutic opportunities. In particular, potential new treatments of multiple myeloma and other bone metastatic cancers, osteomyelitis, arthritis, fracture repair, and osteoporosis are currently being pursued. Several recent examples have built upon historic attempts of drug development with this strategy, and exemplify many of these principles with new “proof of concept” in vivo data. In particular, linkages between the bisphosphonate and the active drug have been optimized for adequate stability in the circulation, but with useful lability at the bone surface to yield effective local concentrations of the drug. Furthermore, pharmacologically less active bisphosphonates have been utilized recently, in order to minimize confounding activities, concerns about differing relative dose levels for each component, and to avoid any un-toward effects of the bisphosphonate. Thus, the bisphosphonate is primarily used as a carrier in some of the recent examples of in vivo “proof of concept” studies described below.
The development of a bone-targeted drug for use in multiple myeloma (MM) would represent a potential medical breakthrough for the treatment and management of MM, and also, perhaps, for other haematopoietic malignancies or primary bone tumors. First, the future drug would augment existing drug treatments by addressing simultaneously the detrimental hallmarks of these tumor types, namely infiltration of bone tissue by cancer cells and induction of excessive bone resorption, which in turn leads to increased skeletal complications, including bone destruction, fractures, and pain. Second, a tissue-targeted strategy will lower systemic drug levels, resulting in reduced adverse side effects, an important factor that in clinical practice often limits efficient treatments. Third, the ability to selectively and directly modulate and reshape the tumor microenvironment (including bone tissue, osteoclasts, osteoblasts, and immune cells), addresses the challenges in MM therapy, where relapse and development of therapy resistance are thought to be critically dependent on the complex changes occurring in the cancer microenvironment.
A team at Rochester recently pursued a bone targeting approach for the treatment of multiple myeloma with the design of a bisphosphonate linked bortezomib analog via a linker that appears to have significant bloodstream stability, but which steadily releases bortezomib at the bone surface due to acid instability of the boronate ester linkage (Fig. 18, BP-Btz, compound 52) [157]. Notably, a dramatic reduction in soft tissue toxicities were observed relative to equivalent doses of bortezomib. This approach has also been evaluated as a new bone formation stimulant with exciting results in animal models [158].
Fig. 18.
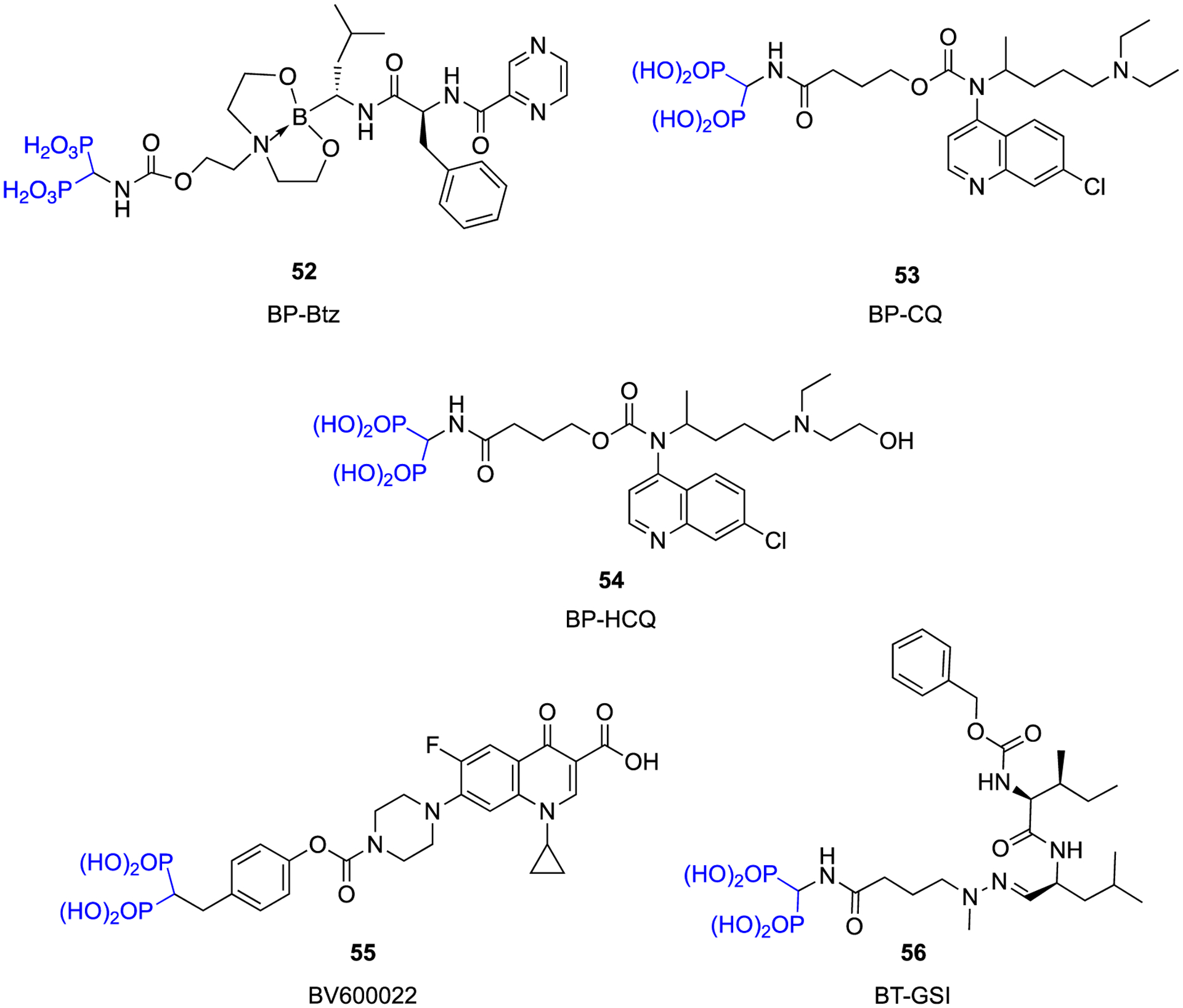
Novel conjugates designed based on a “target and release” concept.
The Rochester team has also optimized a carbamate linkage to a low activity bisphosphonate to chloroquine (Fig. 18, BP-CQ, compound 53) and hydroxychloroquine (Fig. 18, BP-HCQ, compound 54) [150,159]. In vitro, these conjugates prevented osteoclast formation whereas a stable amide-linked conjugate was nearly inactive in this respect. Enhanced in vivo efficacy was also observed with the conjugates when compared to chloroquine/hydroxchloroquine.
Collaborating BioVinc/USC research teams recently designed, synthesized, and evaluated a novel bone-targeting antimicrobial agent comprising a pharmacologically “inert” bisphosphonate (BP) moiety that was conjugated – through metabolically hydrolyzable aryl carbamate linkers – to a fluoroquinolone (FQ) antibiotic, ciprofloxacin, via another “target and release” chemical strategy (Fig. 18, BV600022, compound 55) [160]. These linkers are relatively stable in the bloodstream, while the antibiotic is delivered to metabolically active bone surfaces, but are cleaved presumably by the enzymatic environment produced by both the bone-resorbing cell, the osteoclast, and at the bacterial cell bone surface interface. FQ antibiotics remain a mainstay for the treatment of bone infections in adults, particularly first-line oral monotherapy with agents like ciprofloxacin, levofloxacin, and moxifloxacin. As noted above, a pharmacologically inactive BP was used as a bone targeting carrier for this purpose. Also, it was further demonstrated that BPs are particularly ideal for targeting anti-infective agents to the bone surfaces where biofilm infections occur, not only because they have a high affinity for hydroxyapatite (HA) and selectively bind to bone, but also because they preferentially accumulate at sites of active bone disease or infection, where the highest turnover is taking place [161].
BPs also penetrate the canalicular network to osteocytes and osteocytic lacunae where no blood flow exists and where S. aureus organisms, in particular, are known to embed [162,163]. Accordingly, the demonstrated efficacy of using a novel bone-targeted BP-FQ conjugate for the treatment of osteomyelitis biofilms is a particularly good demonstration of a new therapeutic opportunity for such “custom designed” bisphosphonates. Thus, in vitro antimicrobial susceptibility testing revealed an effective bactericidal profile and sustained release of the parent antibiotic over time. Efficacy and safety were demonstrated in an animal model of jawbone periprosthetic osteomyelitis, where a single dose of 10 mg/kg (15.6 μmol/kg) conjugate reduced the bacterial load by 99% and demonstrated nearly an order of magnitude greater activity than the parent antibiotic alone given in multiple doses (total dose: 30 mg/kg, 90.6 μmol/kg) [160].
Another academic research team has recently reported studies of a bisphosphonate-targeted gamma-secretase inhibitor analog GSI-XII (Fig. 18, BT-GSI, compound 56). This “target and release” approach was designed to release this Notch inhibitor near osteoclasts where the acid sensitive linker cleaves at the bone surface, and it has been shown to bring enhanced in vivo activity to this ubiquitous and relatively toxic series of Notch inhibitors. In vivo efficacy was demonstrated in a mouse model of multiple myeloma and no gastrointestinal (GI) side effects typically associated with GSI-XII alone were observed [164,165].
4. Conclusions
Studies of the structure-activity relationships of bisphosphonates over many years have led to a better understanding of their unique properties and how they work. Continued efforts in synthetic chemistry have provided additional methods for more efficient or “greener” approaches to produce new BPs for scientific and potential clinical use. It has also enlarged the structural repertoire of BP molecules, enabling further research and understanding of their mechanism of action, as well as expanding their uses in more fields. The identification of the major enzymatic target, FDPS, for the nitrogen-containing BPs, was probably the most remarkable landmark achievement along the way. Further efforts to elucidate the interactions between BPs and FDPS, and other important enzymes in the mevalonate pathway (GGPPS, SQS) via crystallography, and new HAP crystal physical chemical interactions have augmented our knowledge of the structure-activity relationships, and also indicated potential new uses of BPs in other disease areas.
With the advanced understanding of these mechanisms of action, we now have a clearer consensus about the two key properties responsible for their antiresorptive efficacy (bone affinity and cellular effects). We can now more clearly explain their clinical features, and can rationally design novel BPs with desired pharmacological properties. New therapeutic areas that could potentially benefit from this optimized and rational drug design include bone-related and other cancers, inflammatory and arthritic conditions, viral infections, and the intriguing possibility of altering age related health span, by extending longevity and reducing multimorbidity.
Another area of growing importance and research attention is the ability of conjugated bisphosphonates to act as carriers to target other drug classes to the bone, due to their exceptional selectivity for bone as the target organ. Although this concept is not new, additional clinical success remains to be accomplished with this approach. A classical application that has long enjoyed clinical success is the technetium-BP bone scanning using SPECT. Recent advances in conjugating BPs with fluorescent dyes have proven to be invaluable research tools for elucidating BP drug pharmacology and hold promise for eventual development as clinical imaging probes. It is encouraging that significant work has been done in the last decade and is receiving increasing interest. A recent emphasis on conjugates that provide a “target and release” of active drugs at bone surfaces is offering unique and exciting treatment possibilities for several medical problems where current therapies could be significantly improved, such as bone infections and bone metastatic disease. Success with these endeavours would open a new era of broadening applications of new medicines from the extensive range of molecular structures based on BP molecules.
As is evident from this review, chemistry continues to play a backbone role for BP related research by providing important molecular design and new pharmacological leads. The integrated expertise of biochemical, preclinical, clinical, and pharmacology researchers in the musculoskeletal and related fields should enable a bright future for this unique class of compounds.
In conclusion, we anticipate that the fascinating scientific journey that began 50 years ago will continue well into the future.
Acknowledgements
The authors wish to dedicate this article to the memory of Professor Robert K. Boeckman who sadly passed away while this manuscript was being prepared. He made outstanding contributions to synthetic chemistry throughout his long career. He played an important role in generating novel bisphosphonates and in the bone targeted drug field. FHE & SS are supported by small business grants from the NIH including the NIDCR (R42-DE025789 and R44-DE025524), NIAMS (R43-AR073727), NCI (R41-CA235996) and NIAID (R44-AI125060). PPS is supported by grant (R42-DE025789) from the NIH (NIDCR). JED was supported by the NIHR Biomedical Research Centre, Oxford, UK. We also wish to acknowledge the many friends and colleagues who have contributed to this work over the years.
Crystallographic data were obtained from PDB bank and crystallographic figures were generated using Protein-Ligand Interaction Profiler (PLIP) and Pymol.
Footnotes
CRediT authorship contribution statement
Conceptualization: Frank H. Ebetino, Shuting Sun; Writing - Original draft preparation: Frank H. Ebetino, Shuting Sun, Philip Cherian, Sahar Roshandel; Writing - Reviewing and Editing: all listed authors; Visualization: Philip Cherian, Sahar Roshandel, and Shuting Sun prepared all figures; Supervision: Frank H. Ebetino, Shuting Sun; Funding acquisition: Frank H. Ebetino, Shuting Sun.
References
- [1].Russell RG, Bisphosphonates: the first 40 years, Bone 49 (1) (2011) 2–19, 10.1016/j.bone.2011.04.022. [DOI] [PubMed] [Google Scholar]
- [2].Ebetino FH, Hogan AM, Sun S, Tsoumpra MK, Duan X, Triffitt JT, Kwaasi AA, Dunford JE, Barnett BL, Oppermann U, Lundy MW, Boyde A, Kashemirov BA, McKenna CE, Russell RG, The relationship between the chemistry and biological activity of the bisphosphonates, Bone 49 (1) (2011) 20–33, 10.1016/j.bone.2011.03.774. [DOI] [PubMed] [Google Scholar]
- [3].Rogers MJ, Mönkkönen J, Munoz MA, Molecular mechanisms of action of bisphosphonates and new insights into their effects outside the skeleton, Bone 139 (2020), 115493, 10.1016/j.bone.2020.115493. [DOI] [PubMed] [Google Scholar]
- [4].Cremers S, Drake MT, Ebetino FH, Bilezikian JP, Russell RGG, Pharmacology of bisphosphonates, Br. J. Clin. Pharmacol 85 (6) (2019) 1052–1062, 10.1111/bcp.13867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Russell RG, Watts NB, Ebetino FH, Rogers MJ, Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy, Osteoporos. Int 19 (6) (2008) 733–759, 10.1007/s00198-007-0540-8. [DOI] [PubMed] [Google Scholar]
- [6].Sun S, Tao J, Sedghizadeh PP, Cherian P, Junka AF, Sodagar E, Xing L, Boeckman RK Jr., Srinivasan V, Yao Z, Boyce BF, Lipe B, Neighbors JD, Russell RGG, McKenna CE, Ebetino FH, Bisphosphonates for delivering drugs to bone, Br. J. Pharmacol 178 (9) (2021) 2008–2025, 10.1111/bph.15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].McCloskey E, Paterson AH, Powles T, Kanis JA, Clodronate, Bone 143 (2021), 115715, 10.1016/j.bone.2020.115715. [DOI] [PubMed] [Google Scholar]
- [8].Cummings SR, Santora AC, Black DM, Russell RGG, History of alendronate, Bone 137 (2020), 115411, 10.1016/j.bone.2020.115411. [DOI] [PubMed] [Google Scholar]
- [9].Papapoulos SE, Pamidronate: A model compound of the pharmacology of nitrogen-containing bisphosphonates; A Leiden historical perspective, Bone 134 (2020), 115244, 10.1016/j.bone.2020.115244. [DOI] [PubMed] [Google Scholar]
- [10].McClung MR, Ebetino FH, History of risedronate, Bone 137 (2020), 115407, 10.1016/j.bone.2020.115407. [DOI] [PubMed] [Google Scholar]
- [11].Reid IR, Green JR, Lyles KW, Reid DM, Trechsel U, Hosking DJ, Black DM, Cummings SR, Russell RGG, Eriksen EF, Zoledronate, Bone 137 (2020), 115390, 10.1016/j.bone.2020.115390. [DOI] [PubMed] [Google Scholar]
- [12].Watts NB, Chesnut CH 3rd, Genant HK, Harris ST, Jackson RD, Licata AA, Miller PD, Mysiw WJ, Richmond B, Valent D, History of etidronate, Bone 134 (2020), 115222, 10.1016/j.bone.2020.115222. [DOI] [PubMed] [Google Scholar]
- [13].Matsumoto T, Endo I, Minodronate, Bone 137 (2020), 115432, 10.1016/j.bone.2020.115432. [DOI] [PubMed] [Google Scholar]
- [14].McKenna CE, Haratipour P, Duro MVV, Ebetino FH, Chemistry of bisphosphonates, in: Zaidi M (Ed.), Encyclopedia of Bone Biology, Academic Press, Oxford, 2020, pp. 551–564, 10.1016/B978-0-12-801238-3.11260-7. [DOI] [Google Scholar]
- [15].Holstein SA, A patent review of bisphosphonates in treating bone disease, Expert Opin. Ther. Pat 29 (5) (2019) 315–325, 10.1080/13543776.2019.1608180. [DOI] [PubMed] [Google Scholar]
- [16].Romanenko VD, α-heteroatom-substituted gem-bisphosphonates: advances in the synthesis and prospects for biomedical application, Curr. Org. Chem 23 (5) (2019) 530–615, 10.2174/1385272823666190401141844. [DOI] [Google Scholar]
- [17].Barbosa JS, Braga SS, Almeida Paz FA, Empowering the medicinal applications of bisphosphonates by unveiling their synthesis details, Molecules 25 (12) (2020) 2821, 10.3390/molecules25122821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kuznik A, Pazdzierniok-Holewa A, Jewula P, Kuznik N, Bisphosphonates-much more than only drugs for bone diseases, Eur. J. Pharmacol 866 (2020), 172773, 10.1016/j.ejphar.2019.172773. [DOI] [PubMed] [Google Scholar]
- [19].Keglevich G, Grün A, Aradi K, Garadnay S, Greiner I, Optimized synthesis of N-heterocyclic dronic acids; closing a black-box era, Tetrahedron Lett. 52 (21) (2011) 2744–2746, 10.1016/j.tetlet.2011.03.093. [DOI] [Google Scholar]
- [20].Kovacs R, Nagy D, Grün A, Balogh G, Garadnay S, Greiner I, Keglevich G, Optimized synthesis of etidronate, Lett. Drug Des. Discovery 10 (8) (2013) 733–737, 10.2174/15701808113109990026. [DOI] [Google Scholar]
- [21].Grun A, Kovacs R, Garadnay S, Greiner I, Keglevich G, The synthesis of risedronic acid and alendronate applying phosphorus oxychloride and phosphorous acid in methanesulfonic acid, Lett. Drug Des. Discovery 12 (4) (2015) 253–258, 10.2174/1570180811666141010220427. [DOI] [Google Scholar]
- [22].Kovács R, Grün A, Garadnay S, Greiner I, Keglevich G, “Greener” synthesis of bisphosphonic/dronic acid derivatives, Green Processes Synth. 3 (2) (2014) 111–116, 10.1515/gps-2013-0107. [DOI] [Google Scholar]
- [23].Nagy DI, Grün A, Lévay K, Garadnay S, Greiner I, Keglevich G, Efficient syntheses of zoledronic acid as an active ingredient of a drug against osteoporosis, Synth. Commun 48 (6) (2018) 663–671, 10.1080/00397911.2017.1410894. [DOI] [Google Scholar]
- [24].Nagy DI, Grun A, Pavela O, Garadnay S, Greiner I, Keglevich G, Efficient synthesis of ibandronate in the presence of an ionic liquid, Lett. Drug Des. Discovery 15 (7) (2018) 713–720, 10.2174/1570180814666171027160324. [DOI] [Google Scholar]
- [25].Nagy DI, Grün A, Sinkovicz J, Garadnay S, Greiner I, Keglevich G, A study on the synthesis of risedronic acid: the role of an ionic liquid additive, Lett. Drug Des. Discovery 16 (3) (2019) 238–244, 10.2174/1570180815666180626122630. [DOI] [Google Scholar]
- [26].Mustafa DA, Kashemirov BA, McKenna CE, Microwave-assisted synthesis of nitrogen-containing 1-hydroxymethylenebisphosphonate drugs, Tetrahedron Lett. 52 (18) (2011) 2285–2287, 10.1016/j.tetlet.2011.02.058. [DOI] [Google Scholar]
- [27].Lenin R, Raju RM, Rao DVNS, Ray UK, Microwave-assisted efficient synthesis of bisphosphonate libraries: a useful procedure for the preparation of bisphosphonates containing nitrogen and sulfur, Med. Chem. Res 22 (4) (2013) 1624–1629, 10.1007/s00044-012-0153-4. [DOI] [Google Scholar]
- [28].Lawson MA, Ebetino FH, Mazur A, Chantry AD, Paton-Hough J, Evans HR, Lath D, Tsoumpra MK, Lundy MW, Dobson RL, Quijano M, Kwaasi AA, Dunford JE, Duan X, Triffitt JT, Jeans G, Russell RGG, The pharmacological profile of a novel highly potent bisphosphonate, OX14 (1-Fluoro-2-(Imidazo-[1,2-alpha]Pyridin-3-yl)-ethyl-Bisphosphonate), J. Bone Miner. Res 32 (9) (2017) 1860–1869, 10.1002/jbmr.3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ebetino FH, Mazur A, Lundy MW, Russell RG, 5-Azaindole bisphosphonates, in: United States Patent (Ed.), USPTO, 2012. US Patent # 8,252,774. [Google Scholar]
- [30].Ebetino FH, Rozé CN, McKenna CE, Barnett BL, Dunford JE, Russell RGG, Mieling GE, Rogers MJ, Molecular interactions of nitrogen-containing bisphosphonates within farnesyl diphosphate synthase, J. Organomet. Chem 690 (10) (2005) 2679–2687, 10.1016/j.jorganchem.2005.03.005. [DOI] [Google Scholar]
- [31].Zhang Y, Cao R, Yin F, Hudock MP, Guo RT, Krysiak K, Mukherjee S, Gao YG, Robinson H, Song Y, No JH, Bergan K, Leon A, Cass L, Goddard A, Chang TK, Lin FY, Van Beek E, Papapoulos S, Wang AH, Kubo T, Ochi M, Mukkamala D, Oldfield E, Lipophilic bisphosphonates as dual farnesyl/geranylgeranyl diphosphate synthase inhibitors: an X-ray and NMR investigation, J. Am. Chem. Soc 131 (14) (2009) 5153–5162, 10.1021/ja808285e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Marma MS, Xia Z, Stewart C, Coxon F, Dunford JE, Baron R, Kashemirov BA, Ebetino FH, Triffitt JT, Russell RG, McKenna CE, Synthesis and biological evaluation of alpha-halogenated bisphosphonate and phosphonocarboxylate analogues of risedronate, J. Med. Chem 50 (24) (2007) 5967–5975, 10.1021/jm0702884. [DOI] [PubMed] [Google Scholar]
- [33].Coxon FP, Helfrich MH, Larijani B, Muzylak M, Dunford JE, Marshall D, McKinnon AD, Nesbitt SA, Horton MA, Seabra MC, Ebetino FH, Rogers MJ, Identification of a novel phosphonocarboxylate inhibitor of Rab geranylgeranyl transferase that specifically prevents Rab prenylation in osteoclasts and macrophages, J. Biol. Chem 276 (51) (2001) 48213–48222, 10.1074/jbc.M106473200. [DOI] [PubMed] [Google Scholar]
- [34].Baron RA, Tavare R, Figueiredo AC, Blazewska KM, Kashemirov BA, McKenna CE, Ebetino FH, Taylor A, Rogers MJ, Coxon FP, Seabra MC, Phosphonocarboxylates inhibit the second geranylgeranyl addition by Rab geranylgeranyl transferase, J. Biol. Chem 284 (11) (2009) 6861–6868, 10.1074/jbc.M806952200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Blazewska KM, Ni F, Haiges R, Kashemirov BA, Coxon FP, Stewart CA, Baron R, Rogers MJ, Seabra MC, Ebetino FH, McKenna CE, Synthesis, stereochemistry and SAR of a series of minodronate analogues as RGGT inhibitors, Eur. J. Med. Chem 46 (10) (2011) 4820–4826, 10.1016/j.ejmech.2011.04.063. [DOI] [PubMed] [Google Scholar]
- [36].Engelsma SB, Meeuwenoord NJ, Overkleeft HS, van der Marel GA, Filippov DV, Combined phosphoramidite-phosphodiester reagents for the synthesis of methylene bisphosphonates, Angew. Chem 129 (11) (2017) 3001–3005, 10.1002/ange.201611878. [DOI] [PubMed] [Google Scholar]
- [37].Egorov M, Aoun S, Padrines M, Redini F, Heymann D, Lebreton J, Mathé-Allainmat M, A one-pot synthesis of 1-Hydroxy-1,1-bis(phosphonic acid)s starting from the corresponding carboxylic acids, Eur. J. Org. Chem 2011 (35) (2011) 7148–7154, 10.1002/ejoc.201101094. [DOI] [Google Scholar]
- [38].Zhang C, Pu Y-C, Yu Z-J, Wu C-Y, Brem J, McDonough MA, Schofield CJ, Li G-B, Wu Y, Rh(III)-catalyzed directed C-H carbenoid coupling reveals aromatic bisphosphonates inhibiting metallo- and serine-β-lactamases, Org. Chem. Front 5 (8) (2018) 1288–1292, 10.1039/C8QO00009C. [DOI] [Google Scholar]
- [39].Kolb HC, Finn MG, Sharpless KB, Click chemistry: diverse chemical function from a few good reactions, Angew. Chem. Int. Ed. Engl 40 (11) (2001) 2004–2021, . [DOI] [PubMed] [Google Scholar]
- [40].Hein CD, Liu XM, Wang D, Click chemistry, a powerful tool for pharmaceutical sciences, Pharm. Res 25 (10) (2008) 2216–2230, 10.1007/s11095-008-9616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chamberlain BT, Upton TG, Kashemirov BA, McKenna CE, α-azido bisphosphonates: synthesis and nucleotide analogues, J. Org. Chem 76 (12) (2011) 5132–5136, 10.1021/jo200045a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chiminazzo A, Borsato G, Favero A, Fabbro C, McKenna CE, Dalle Carbonare LG, Valenti MT, Fabris F, Scarso A, Diketopyrrolopyrrole bisphosphonate conjugate: a new fluorescent probe for in vitro bone imaging, Chem. Eur. J 25 (14) (2019) 3617–3626, 10.1002/chem.201805436. [DOI] [PubMed] [Google Scholar]
- [43].Li G, Wu M, Liu F, Jiang J, One-pot, highly regioselective 1,3-dipole cycloaddition promoted by montmorillonite for the synthesis of Spiro[indole-pyrrolizine], Spiro[indole-indolizine], and Spiro[indole-pyrrolidine] gem-bisphosphonates, Synthesis 47 (23) (2015) 3783–3796, 10.1055/s-0035-1560463. [DOI] [Google Scholar]
- [44].Zhu Z, Wang Q, Kong D, Huang T, Wu M, CuI-catalyzed [3+2] cycloaddition of hindered vinylidenebisphosphonates (VBP) with azomethine imines for highly regioselective access to dinitrogen-heterobicycle-containing bisphosphonates, Synthesis 50 (13) (2018) 2601–2607, 10.1055/s-0036-1591555. [DOI] [Google Scholar]
- [45].Balakrishna A, Narayana Reddy MV, Rao PV, Kumar MA, Kumar BS, Nayak SK, Reddy CS, Synthesis and bio-activity evaluation of tetraphenyl (phenylamino) methylene bisphosphonates as antioxidant agents and as potent inhibitors of osteoclasts in vitro, Eur. J. Med. Chem 46 (5) (2011) 1798–1802, 10.1016/j.ejmech.2011.02.038. [DOI] [PubMed] [Google Scholar]
- [46].Reddy CB, Reddy NB, Raju CN, Design, synthesis, characterization of geminal bisphosphonates and bioactivity evaluation, in: Proceedings of International Conference on Recent Trends in Mechanical and Materials Engineering: ICRTMME 2019, 2020, p. 040041. [Google Scholar]
- [47].Bálint E, Tajti Á, Dzielak A, Hägele G, Keglevich G, Microwave-assisted synthesis of (aminomethylene)bisphosphine oxides and (aminomethylene) bisphosphonates by a three-component condensation, Beilstein J. Org. Chem 12 (2016) 1493–1502, 10.3762/bjoc.12.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Shaik MS, Nadiveedhi MR, Gundluru M, Katike U, Obulam VSR, Cirandur SR, Efficient catalyst free green synthesis and in vitro antimicrobial, antioxidant and molecular docking studies of α-substituted aromatic/heteroaromatic aminomethylene bisphosphonates, Synth. Commun (2020) 1–18, 10.1080/00397911.2020.1853778. [DOI] [Google Scholar]
- [49].Tellamekala S, Gundluru M, Sudileti M, Sarva S, Putta CRK, Cirandur SR, Green one-pot synthesis of N-bisphosphonates as antimicrobial and antioxidant agents, Monatsh. Chem 151 (2) (2020) 251–260, 10.1007/s00706-020-02551-3. [DOI] [Google Scholar]
- [50].Mohan G, Santhisudha S, Reddy NM, Sreekanth T, Murali S, Reddy CS, Nano ZnO catalyzed green synthesis and cytotoxic assay of pyridinyl and pyrimidinyl bisphosphonates, Monatsh. Chem 148 (10) (2017) 1843–1851, 10.1007/s00706-017-2000-2. [DOI] [Google Scholar]
- [51].Sudileti M, Nagaripati S, Gundluru M, Chintha V, Aita S, Wudayagiri R, Chamarthi N, Cirandur SR, rGO-SO3H catalysed green synthesis of fluoro-substituted aminomethylene bisphosphonates and their anticancer, molecular docking studies, ChemistrySelect 4 (44) (2019) 13006–13011, 10.1002/slct.201903191. [DOI] [Google Scholar]
- [52].Mucha A, Kafarski P, Berlicki Ł, Remarkable potential of the α-Aminophosphonate/Phosphinate structural motif in medicinal chemistry, J. Med. Chem 54 (17) (2011) 5955–5980, 10.1021/jm200587f. [DOI] [PubMed] [Google Scholar]
- [53].Haratipour P, Minard C, Nakhjiri M, Negahbani A, Chamberlain BT, Osuna J, Upton TG, Zhao M, Kashemirov BA, McKenna CE, Completing the beta, gamma-CXY-dNTP stereochemical probe toolkit: synthetic access to the dCTP diastereomers and (31)P and (19)F NMR correlations with absolute configurations, J. Org. Chem 85 (22) (2020) 14592–14609, 10.1021/acs.joc.0c01204. [DOI] [PubMed] [Google Scholar]
- [54].Niesor EJ, Flach J, Lopes-Antoni I, Perez A, Bentzen CL, The nuclear receptors FXR and LXRalpha: potential targets for the development of drugs affecting lipid metabolism and neoplastic diseases, Curr. Pharm. Des 7 (4) (2001) 231–259, 10.2174/1381612013398185. [DOI] [PubMed] [Google Scholar]
- [55].Flach J, Antoni I, Villemin P, Bentzen CL, Niesor EJ, The mevalonate/isoprenoid pathway inhibitor apomine (SR-45023A) is antiproliferative and induces apoptosis similar to farnesol, Biochem. Biophys. Res. Commun 270 (1) (2000) 240–246, 10.1006/bbrc.2000.2421. [DOI] [PubMed] [Google Scholar]
- [56].Roitelman J, Masson D, Avner R, Ammon-Zufferey C, Perez A, Guyon-Gellin Y, Bentzen CL, Niesor EJ, Apomine, a novel hypocholesterolemic agent, accelerates degradation of 3-hydroxy-3-methylglutaryl-coenzyme a reductase and stimulates low density lipoprotein receptor activity, J. Biol. Chem 279 (8) (2004) 6465–6473, 10.1074/jbc.M308094200. [DOI] [PubMed] [Google Scholar]
- [57].Jackson B, Gee AN, Guyon-Gellin Y, Niesor E, Bentzen CL, Kerns WD, Suckling KE, Hypocholesterolaemic and antiatherosclerotic effects of tetra-iso-propyl 2-(3,5-di-tert-butyl-4-hydroxyphenyl)ethyl-1,1-diphosphonate (SR-9223i), Arzneimittelforschung 50 (4) (2000) 380–386, 10.1055/s-0031-1300217. [DOI] [PubMed] [Google Scholar]
- [58].Toyota Y, Yoshioka H, Sagimori I, Hashimoto Y, Ohgane K, Bisphosphonate esters interact with HMG-CoA reductase membrane domain to induce its degradation, Bioorg. Med. Chem 28 (14) (2020), 115576, 10.1016/j.bmc.2020.115576. [DOI] [PubMed] [Google Scholar]
- [59].Alberts DS, Hallum AV 3rd, Stratton-Custis M, Garcia DJ, Gleason-Guzman M, Salmon SE, Santabarbara P, Niesor EJ, Floret S, Bentzen CL, Phase I pharmacokinetic trial and correlative in vitro phase II tumor kinetic study of apomine (SR-45023A), a novel oral biphosphonate anticancer drug, Clin. Cancer Res 7 (5) (2001) 1246–1250. [PubMed] [Google Scholar]
- [60].Moriceau G, Roelofs AJ, Brion R, Redini F, Ebetion FH, Rogers MJ, Heymann D, Synergistic inhibitory effect of apomine and lovastatin on osteosarcoma cell growth, Cancer 118 (3) (2012) 750–760, 10.1002/cncr.26336. [DOI] [PubMed] [Google Scholar]
- [61].Edwards CM, Mueller G, Roelofs AJ, Chantry A, Perry M, Russell RG, Van Camp B, Guyon-Gellin Y, Niesor EJ, Bentzen CL, Vanderkerken K, Croucher PI, Apomine, an inhibitor of HMG-CoA-reductase, promotes apoptosis of myeloma cells in vitro and is associated with a modulation of myeloma in vivo, Int. J. Cancer 120 (8) (2007) 1657–1663, 10.1002/ijc.22478. [DOI] [PubMed] [Google Scholar]
- [62].Roelofs AJ, Edwards CM, Russell RG, Ebetino FH, Rogers MJ, Hulley PA, Apomine enhances the antitumor effects of lovastatin on myeloma cells by down-regulating 3-hydroxy-3-methylglutaryl-coenzyme a reductase, J. Pharmacol. Exp. Ther 322 (1) (2007) 228–235, 10.1124/jpet.106.116467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Pourpak A, Dorr RT, Meyers RO, Powell MB, Stratton SP, Cytotoxic activity of apomine is due to a novel membrane-mediated cytolytic mechanism independent of apoptosis in the A375 human melanoma cell line, Investig. New Drugs 25 (2) (2007) 107–114, 10.1007/s10637-006-9015-6. [DOI] [PubMed] [Google Scholar]
- [64].Lowe LC, Senaratne SG, Colston KW, Induction of apoptosis in breast cancer cells by apomine is mediated by caspase and p38 mitogen activated protein kinase activation, Biochem. Biophys. Res. Commun 329 (2) (2005) 772–779, 10.1016/j.bbrc.2005.02.032. [DOI] [PubMed] [Google Scholar]
- [65].Lewis KD, Thompson JA, Weber JS, Robinson WA, O’Day S, Lutzky J, Legha SS, Floret S, Ruvuna F, Gonzalez R, A phase II open-label trial of apomine (SR-45023A) in patients with refractory melanoma, Investig. New Drugs 24 (1) (2006) 89–94, 10.1007/s10637-005-4544-y. [DOI] [PubMed] [Google Scholar]
- [66].Bonate PL, Floret S, Bentzen C, Population pharmacokinetics of APOMINE: a meta-analysis in cancer patients and healthy males, Br. J. Clin. Pharmacol 58 (2) (2004) 142–155, 10.1111/j.1365-2125.2004.02111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Jiang L, Zhang H, Xiao D, Wei H, Chen Y, Farnesoid X receptor (FXR): structures and ligands, Comput. Struct. Biotechnol. J 19 (2021) 2148–2159, 10.1016/j.csbj.2021.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Tejera E, Munteanu CR, López-Cortés A, Cabrera-Andrade A, Pérez-Castillo Y, Drugs repurposing using QSAR, docking and molecular dynamics for possible inhibitors of the SARS-CoV-2 M(pro) protease, Molecules 25 (21) (2020), 10.3390/molecules25215172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ebetino FH, Dansereau SM, in: Bisphosphonate Antiresorptive Structure Activity Relationships, Bisphosphonate on Bones, Elsevier Science, 1995, pp. 139–153. [Google Scholar]
- [70].Nugent RA, Murphy M, Schlachter ST, Dunn CJ, Smith RJ, Staite ND, Galinet LA, Shields SK, Aspar DG, Richard KA, Rohloff NA, Pyrazoline bisphosphonate esters as novel antiinflammatory and antiarthritic agents, J. Med. Chem 36 (1) (1993) 134–139, 10.1021/jm00053a017. [DOI] [PubMed] [Google Scholar]
- [71].Pradere U, Garnier-Amblard EC, Coats SJ, Amblard F, Schinazi RF, Synthesis of nucleoside phosphate and phosphonate prodrugs, Chem. Rev 114 (18) (2014) 9154–9218, 10.1021/cr5002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Wiemer AJ, Wiemer DF, Prodrugs of phosphonates and phosphates: crossing the membrane barrier, Top. Curr. Chem 360 (2015) 115–160, 10.1007/128_2014_561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Heidel KM, Dowd CS, Phosphonate prodrugs: an overview and recent advances, Future Med. Chem 11 (13) (2019) 1625–1643, 10.4155/fmc-2018-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].McClung MR, Miller PD, Brown JP, Zanchetta J, Bolognese MA, Benhamou CL, Balske A, Burgio DE, Sarley J, McCullough LK, Recker RR, Efficacy and safety of a novel delayed-release risedronate 35 mg once-a-week tablet, Osteoporos. Int 23 (1) (2012) 267–276, 10.1007/s00198-011-1791-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ezra A, Hoffman A, Breuer E, Alferiev IS, Monkkonen J, El Hanany-Rozen N, Weiss G, Stepensky D, Gati I, Cohen H, Tormalehto S, Amidon GL, Golomb G, A peptide prodrug approach for improving bisphosphonate oral absorption, J. Med. Chem 43 (20) (2000) 3641–3652, 10.1021/jm980645y. [DOI] [PubMed] [Google Scholar]
- [76].Vepsalainen JJ, Bisphosphonate prodrugs, Curr. Med. Chem 9 (12) (2002) 1201–1208, 10.2174/0929867023369998. [DOI] [PubMed] [Google Scholar]
- [77].Webster MR, Zhao M, Rudek MA, Hann CL, Freel Meyers CL, Bisphosphonamidate clodronate prodrug exhibits potent anticancer activity in non-small-cell lung cancer cells, J. Med. Chem 54 (19) (2011) 6647–6656, 10.1021/jm200521a. US Patent # 8,252,774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Pavlov VA, Ebetino FH, Lundy MW, Joly J, Dobson RL, Russell RGG, Mazur AW, Developments in the synthesis of new functionalized bisphosphonate drug candidates such as cyclic prodrugs, Phosphorus Sulfur Silicon Relat. Elem 191 (11–12) (2016) 1504–1508, 10.1080/10426507.2016.1212342. [DOI] [Google Scholar]
- [79].Manaswiyoungkul P, de Araujo ED, Gunning PT, Targeting prenylation inhibition through the mevalonate pathway, RSC Med. Chem 11 (1) (2020) 51–71, 10.1039/C9MD00442D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Park J, Pandya VR, Ezekiel SJ, Berghuis AM, Phosphonate and bisphosphonate inhibitors of farnesyl pyrophosphate synthases: a structure-guided perspective, Front. Chem 8 (2020), 612728, 10.3389/fchem.2020.612728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Tsoumpra MK, Muniz JR, Barnett BL, Kwaasi AA, Pilka ES, Kavanagh KL, Evdokimov A, Walter RL, Von Delft F, Ebetino FH, Oppermann U, Russell RGG, Dunford JE, The inhibition of human farnesyl pyrophosphate synthase by nitrogen-containing bisphosphonates. Elucidating the role of active site threonine 201 and tyrosine 204 residues using enzyme mutants, Bone 81 (2015) 478–486, 10.1016/j.bone.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].No JH, de Macedo Dossin F, Zhang Y, Liu YL, Zhu W, Feng X, Yoo JA, Lee E, Wang K, Hui R, Freitas-Junior LH, Oldfield E, Lipophilic analogs of zoledronate and risedronate inhibit Plasmodium geranylgeranyl diphosphate synthase (GGPPS) and exhibit potent antimalarial activity, Proc. Natl. Acad. Sci. U. S. A 109 (11) (2012) 4058–4063, 10.1073/pnas.1118215109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Baranyi M, Rittler D, Molnar E, Shirasawa S, Jalsovszky I, Varga IK, Hegedus L, Nemeth A, Dank M, Aigner C, Tovari J, Timar J, Hegedus B, Garay T, Next generation lipophilic bisphosphonate shows antitumor effect in colorectal cancer in vitro and in vivo, Pathol. Oncol. Res 26 (3) (2020) 1957–1969, 10.1007/s12253-019-00789-9. [DOI] [PubMed] [Google Scholar]
- [84].Reilly JE, Neighbors JD, Hohl RJ, Targeting protein geranylgeranylation slows tumor development in a murine model of prostate cancer metastasis, Cancer Biol. Ther 18 (11) (2017) 872–882, 10.1080/15384047.2016.1219817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Xia Y, Liu YL, Xie Y, Zhu W, Guerra F, Shen S, Yeddula N, Fischer W, Low W, Zhou X, Zhang Y, Oldfield E, Verma IM, A combination therapy for KRAS-driven lung adenocarcinomas using lipophilic bisphosphonates and rapamycin, Sci. Transl. Med 6 (263) (2014) 263ra161, 10.1126/scitranslmed.3010382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Malwal SR, Chen L, Hicks H, Qu F, Liu W, Shillo A, Law WX, Zhang J, Chandnani N, Han X, Zheng Y, Chen CC, Guo RT, AbdelKhalek A, Seleem MN, Oldfield E, Discovery of lipophilic bisphosphonates that target bacterial Cell Wall and quinone biosynthesis, J. Med. Chem 62 (5) (2019) 2564–2581, 10.1021/acs.jmedchem.8b01878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Yokoyama T, Mizuguchi M, Ostermann A, Kusaka K, Niimura N, Schrader TE, Tanaka I, Protonation state and hydration of bisphosphonate bound to farnesyl pyrophosphate synthase, J. Med. Chem 58 (18) (2015) 7549–7556, 10.1021/acs.jmedchem.5b01147. [DOI] [PubMed] [Google Scholar]
- [88].Leung CY, Park J, De Schutter JW, Sebag M, Berghuis AM, Tsantrizos YS, Thienopyrimidine bisphosphonate (ThPBP) inhibitors of the human farnesyl pyrophosphate synthase: optimization and characterization of the mode of inhibition, J. Med. Chem 56 (20) (2013) 7939–7950, 10.1021/jm400946f. [DOI] [PubMed] [Google Scholar]
- [89].Park J, Rodionov D, De Schutter JW, Lin YS, Tsantrizos YS, Berghuis AM, Crystallographic and thermodynamic characterization of phenylaminopyridine bisphosphonates binding to human farnesyl pyrophosphate synthase, PLoS ONE 12 (10) (2017), e0186447, 10.1371/journal.pone.0186447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Aripirala S, Gonzalez-Pacanowska D, Oldfield E, Kaiser M, Amzel LM, Gabelli SB, Structural and thermodynamic basis of the inhibition of leishmania major farnesyl diphosphate synthase by nitrogen-containing bisphosphonates, Acta Crystallogr. D Biol. Crystallogr 70 (Pt 3) (2014) 802–810, 10.1107/S1399004713033221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Yang G, Zhu W, Kim K, Byun SY, Choi G, Wang K, Cha JS, Cho HS, Oldfield E, No JH, In vitro and in vivo investigation of the inhibition of trypanosoma brucei cell growth by lipophilic bisphosphonates, Antimicrob. Agents Chemother 59 (12) (2015) 7530–7539, 10.1128/AAC.01873-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Lisnyansky M, Yariv E, Segal O, Marom M, Loewenstein A, Ben-Tal N, Giladi M, Haitin Y, Metal coordination is crucial for geranylgeranyl diphosphate synthase-bisphosphonate interactions: a crystallographic and computational analysis, Mol. Pharmacol 96 (5) (2019) 580–588, 10.1124/mol.119.117499. [DOI] [PubMed] [Google Scholar]
- [93].Lisnyansky M, Kapelushnik N, Ben-Bassat A, Marom M, Loewenstein A, Khananshvili D, Giladi M, Haitin Y, Reduced activity of geranylgeranyl diphosphate synthase mutant is involved in bisphosphonate-induced atypical fractures, Mol. Pharmacol 94 (6) (2018) 1391–1400, 10.1124/mol.118.113670. [DOI] [PubMed] [Google Scholar]
- [94].Roca-Ayats N, Balcells S, Garcia-Giralt N, Falco-Mascaro M, Martinez-Gil N, Abril JF, Urreizti R, Dopazo J, Quesada-Gomez JM, Nogues X, Mellibovsky L, Prieto-Alhambra D, Dunford JE, Javaid MK, Russell RG, Grinberg D, Diez-Perez A, GGPS1 mutation and atypical femoral fractures with bisphosphonates, N. Engl. J. Med 376 (18) (2017) 1794–1795, 10.1056/NEJMc1612804. [DOI] [PubMed] [Google Scholar]
- [95].Roca-Ayats N, Ng PY, Garcia-Giralt N, Falco-Mascaro M, Cozar M, Abril JF, Quesada Gomez JM, Prieto-Alhambra D, Nogues X, Dunford JE, Russell RG, Baron R, Grinberg D, Balcells S, Diez-Perez A, Functional characterization of a GGPPS variant identified in atypical femoral fracture patients and delineation of the role of GGPPS in bone-relevant cell types, J. Bone Miner. Res 33 (12) (2018) 2091–2098, 10.1002/jbmr.3580. [DOI] [PubMed] [Google Scholar]
- [96].Wills VS, Allen C, Holstein SA, Wiemer DF, Potent triazole bisphosphonate inhibitor of geranylgeranyl diphosphate synthase, ACS Med. Chem. Lett 6 (12) (2015) 1195–1198, 10.1021/acsmedchemlett.5b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Hudock MP, Zhang Y, Guo RT, Cao R, No JH, Liang PH, Ko TP, Chang TH, Chang SC, Song Y, Axelson J, Kumar A, Wang AH, Oldfield E, K.M. C. C, Inhibition of geranylgeranyl diphosphate synthase by bisphosphonates: a crystallographic and computational investigation, J. Med. Chem 51 (18) (2008) 5594–5607, 10.1021/jm800325y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Shull LW, Wiemer DF, Copper-mediated displacements of allylic THP ethers on a bisphosphonate template, J. Organomet. Chem 690 (10) (2005) 2521–2530, 10.1016/j.jorganchem.2004.10.013. [DOI] [Google Scholar]
- [99].Shull LW, Wiemer AJ, Hohl RJ, Wiemer DF, Synthesis and biological activity of isoprenoid bisphosphonates, Bioorg. Med. Chem 14 (12) (2006) 4130–4136, 10.1016/j.bmc.2006.02.010. [DOI] [PubMed] [Google Scholar]
- [100].Wiemer AJ, Tong H, Swanson KM, Hohl RJ, Digeranyl bisphosphonate inhibits geranylgeranyl pyrophosphate synthase, Biochem. Biophys. Res. Commun 353 (4) (2007) 921–925, 10.1016/j.bbrc.2006.12.094. [DOI] [PubMed] [Google Scholar]
- [101].Zhou X, Reilly JE, Loerch KA, Hohl RJ, Wiemer DF, Synthesis of isoprenoid bisphosphonate ethers through C-P bond formations: potential inhibitors of geranylgeranyl diphosphate synthase, Beilstein J. Org. Chem 10 (2014) 1645–1650, 10.3762/bjoc.10.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Kourounakis AP, Bavavea E, New applications of squalene synthase inhibitors: membrane cholesterol as a therapeutic target, Arch. Pharm 353 (9) (2020), e2000085, 10.1002/ardp.202000085. [DOI] [PubMed] [Google Scholar]
- [103].Amin D, Cornell SA, Gustafson SK, Needle SJ, Ullrich JW, Bilder GE, Perrone MH, Bisphosphonates used for the treatment of bone disorders inhibit squalene synthase and cholesterol biosynthesis, J. Lipid Res 33 (11) (1992) 1657–1663. [PubMed] [Google Scholar]
- [104].Amin D, Cornell SA, Perrone MH, Bilder GE, 1-Hydroxy-3-(methylpentylamino)-propylidene-1,1-bisphosphonic acid as a potent inhibitor of squalene synthase, Arzneimittelforschung 46 (8) (1996) 759–762. [PubMed] [Google Scholar]
- [105].Ciosek CP Jr., Magnin DR, Harrity TW, Logan JV, Dickson JK Jr., Gordon EM, Hamilton KA, Jolibois KG, Kunselman LK, Lawrence RM, et al. , Lipophilic 1,1-bisphosphonates are potent squalene synthase inhibitors and orally active cholesterol lowering agents in vivo, J. Biol. Chem 268 (33) (1993) 24832–24837. [PubMed] [Google Scholar]
- [106].Shang N, Li Q, Ko TP, Chan HC, Li J, Zheng Y, Huang CH, Ren F, Chen CC, Zhu Z, Galizzi M, Li ZH, Rodrigues-Poveda CA, Gonzalez-Pacanowska D, Veiga-Santos P, de Carvalho TM, de Souza W, Urbina JA, Wang AH, Docampo R, Li K, Liu YL, Oldfield E, Guo RT, Squalene synthase as a target for chagas disease therapeutics, PLoS Pathog. 10 (5) (2014), e1004114, 10.1371/journal.ppat.1004114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Cole LE, Vargo-Gogola T, Roeder RK, Targeted delivery to bone and mineral deposits using bisphosphonate ligands, Adv. Drug Deliv. Rev 99 (Pt A) (2016) 12–27, 10.1016/j.addr.2015.10.005. [DOI] [PubMed] [Google Scholar]
- [108].Lundy MW, Sun S, Duan X, McKenna CE, Jeans G, Dobson RL, Quijano M, Triffitt JT, Russell G, Ebetino FH, Skeletal retention and urinary excretion of nitrogen-containing bisphosphonates including fluorescently-labeled bisphosphonates in rats, J. Bone Miner. Res 29 (2014). S262–S262. [Google Scholar]
- [109].Sun S, Blazewska KM, Kadina AP, Kashemirov BA, Duan X, Triffitt JT, Dunford JE, Russell RG, Ebetino FH, Roelofs AJ, Coxon FP, Lundy MW, McKenna CE, Fluorescent bisphosphonate and carboxyphosphonate probes: a versatile imaging toolkit for applications in bone biology and biomedicine, Bioconjug. Chem 27 (2) (2016) 329–340, 10.1021/acs.bioconjchem.5b00369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Kashemirov BA, Bala JL, Chen X, Ebetino FH, Xia Z, Russell RG, Coxon FP, Roelofs AJ, Rogers MJ, McKenna CE, Fluorescently labeled risedronate and related analogues: “magic linker” synthesis, Bioconjug. Chem 19 (12) (2008) 2308–2310, 10.1021/bc800369c. [DOI] [PubMed] [Google Scholar]
- [111].Lundy MW, Ebetino FH, Fei L, Xia Z, Pozzi M, Trokhan D, Dunford JE, Russell RGG, Bisphosphonate affinity to hydroxyapatite influences in vivo efficacy, J. Bone Miner. Res 22 (S1) (2007), s443. [Google Scholar]
- [112].Rennert G, Rennert HS, Pinchev M, Lavie O, The effect of bisphosphonates on the risk of endometrial and ovarian malignancies, Gynecol. Oncol 133 (2) (2014) 309–313, 10.1016/j.ygyno.2014.02.014. [DOI] [PubMed] [Google Scholar]
- [113].Garay T, Kenessey I, Molnár É, Juhász É, Réti A, László V, Rózsás A, Dobos J, Döme B, Berger W, Klepetko W, Tóvári J, Tímár J, Hegedűs B, Prenylation inhibition-induced cell death in melanoma: reduced sensitivity in BRAF Mutant/PTEN wild-type melanoma cells, PLoS ONE 10 (2) (2015), e0117021, 10.1371/journal.pone.0117021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Roelofs AJ, Ebetino FH, Reszka AA, Russell RGG, Rogers MJ, Chapter 81 - bisphosphonates: mechanisms of action, in: Bilezikian JP, Raisz LG, Martin TJ (Eds.), Principles of Bone Biology, Third Edition, Academic Press, San Diego, 2008, pp. 1737–1767, 10.1016/B978-0-12-373884-4.00095-1. [DOI] [Google Scholar]
- [115].Cremers S, Drake MT, Ebetino FH, Rogers MJ, Bilezikian JP, Russell RGG, Clinical and translational pharmacology of bisphosphonates, in: Bilezikian JP, Martin TJ, Clemens TL, Rosen CJ (Eds.), Principles of Bone Biology, Academic Press, 2020, pp. 1671–1687, 10.1016/b978-0-12-814841-9.00072-5. [DOI] [Google Scholar]
- [116].Lacbay CM, Waller DD, Park J, Gómez Palou M, Vincent F, Huang XF, Ta V, Berghuis AM, Sebag M, Tsantrizos YS, Unraveling the prenylation–cancer paradox in multiple myeloma with novel geranylgeranyl pyrophosphate synthase (GGPPS) inhibitors, J. Med. Chem 61 (15) (2018) 6904–6917, 10.1021/acs.jmedchem.8b00886. [DOI] [PubMed] [Google Scholar]
- [117].Zhou X, Hartman SV, Born EJ, Smits JP, Holstein SA, Wiemer DF, Triazole-based inhibitors of geranylgeranyltransferase II, Bioorg. Med. Chem. Lett 23 (3) (2013) 764–766, 10.1016/j.bmcl.2012.11.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Zhou X, Ferree SD, Wills VS, Born EJ, Tong H, Wiemer DF, Holstein SA, Geranyl and neryl triazole bisphosphonates as inhibitors of geranylgeranyl diphosphate synthase, Bioorg. Med. Chem 22 (9) (2014) 2791–2798, 10.1016/j.bmc.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Matthiesen RA, Varney ML, Xu PC, Rier AS, Wiemer DF, Holstein SA, Alpha-methylation enhances the potency of isoprenoid triazole bisphosphonates as geranylgeranyl diphosphate synthase inhibitors, Bioorg. Med. Chem 26 (2) (2018) 376–385, 10.1016/j.bmc.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Misra J, Mohanty ST, Madan S, Fernandes JA, Hal Ebetino F, Russell RG, Bellantuono I, Zoledronate attenuates accumulation of DNA damage in mesenchymal stem cells and protects their function, Stem Cells 34 (3) (2016) 756–767, 10.1002/stem.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Tsoumpra MK, Lehr HA, Ebetino FH, Neighbors JD, Russell RGG, Ferrari-Lacraz S, Ferrari SL, Orally administered bisphosphonates alleviate colonic inflammation and bone loss in a mouse model of acute colitis, J. Bone Miner. Res 29 (S1) (2014) 404. [Google Scholar]
- [122].Osborn-Heaford HL, Murthy S, Gu L, Larson-Casey JL, Ryan AJ, Shi L, Glogauer M, Neighbors JD, Hohl R, Carter AB, Targeting the isoprenoid pathway to abrogate progression of pulmonary fibrosis, Free Radic. Biol. Med 86 (2015) 47–56, 10.1016/j.freeradbiomed.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Center JR, Lyles KW, Bliuc D, Bisphosphonates and lifespan, Bone 141 (2020), 115566, 10.1016/j.bone.2020.115566. [DOI] [PubMed] [Google Scholar]
- [124].Larson-Casey JL, Vaid M, Gu L, He C, Cai GQ, Ding Q, Davis D, Berryhill TF, Wilson LS, Barnes S, Neighbors JD, Hohl RJ, Zimmerman KA, Yoder BK, Longhini ALF, Hanumanthu VS, Surolia R, Antony VB, Carter AB, Increased flux through the mevalonate pathway mediates fibrotic repair without injury, J. Clin. Invest 129 (11) (2019) 4962–4978, 10.1172/JCI127959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Mitchell A, Watts AE, Ebetino FH, Suva LJ, Bisphosphonate use in the horse: what is good and what is not? BMC Vet. Res 15 (1) (2019) 211, 10.1186/s12917-019-1966-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Suva LJ, Cooper A, Watts AE, Ebetino FH, Price J, Gaddy D, Bisphosphonates in veterinary medicine: the new horizon for use, Bone 142 (2021), 115711, 10.1016/j.bone.2020.115711. [DOI] [PubMed] [Google Scholar]
- [127].Roelofs AJ, Coxon FP, Ebetino FH, Lundy MW, Henneman ZJ, Nancollas GH, Sun S, Blazewska KM, Bala JL, Kashemirov BA, Khalid AB, McKenna CE, Rogers MJ, Fluorescent risedronate analogues reveal bisphosphonate uptake by bone marrow monocytes and localization around osteocytes in vivo, J. Bone Miner. Res 25 (3) (2010) 606–616, 10.1359/jbmr.091009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Roelofs AJ, Stewart CA, Sun S, Blazewska KM, Kashemirov BA, McKenna CE, Russell RG, Rogers MJ, Lundy MW, Ebetino FH, Coxon FP, Influence of bone affinity on the skeletal distribution of fluorescently labeled bisphosphonates in vivo, J. Bone Miner. Res 27 (4) (2012) 835–847, 10.1002/jbmr.1543. [DOI] [PubMed] [Google Scholar]
- [129].Turek J, Ebetino FH, Lundy MW, Sun S, Kashemirov BA, McKenna CE, Gallant MA, Plotkin LI, Bellido T, Duan X, Triffitt JT, Russell RG, Burr DB, Allen MR, Bisphosphonate binding affinity affects drug distribution in both intracortical and trabecular bone of rabbits, Calcif. Tissue Int 90 (3) (2012) 202–210, 10.1007/s00223-012-9570-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Cohn Yakubovich D, Eliav U, Yalon E, Schary Y, Sheyn D, Cook-Wiens G, Sun S, McKenna CE, Lev S, Binshtok AM, Pelled G, Navon G, Gazit D, Gazit Z, Teriparatide attenuates scarring around murine cranial bone allograft via modulation of angiogenesis, Bone 97 (2017) 192–200, 10.1016/j.bone.2017.01.020. [DOI] [PubMed] [Google Scholar]
- [131].Hokugo A, Sun S, Park S, McKenna CE, Nishimura I, Equilibrium-dependent bisphosphonate interaction with crystalline bone mineral explains anti-resorptive pharmacokinetics and prevalence of osteonecrosis of the jaw in rats, Bone 53 (1) (2013) 59–68, 10.1016/j.bone.2012.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Vermeer JA, Jansen ID, Marthi M, Coxon FP, McKenna CE, Sun S, de Vries TJ, Everts V, Jaw bone marrow-derived osteoclast precursors internalize more bisphosphonate than long-bone marrow precursors, Bone 57 (1) (2013) 242–251, 10.1016/j.bone.2013.08.007. [DOI] [PubMed] [Google Scholar]
- [133].Bae S, Sun S, Aghaloo T, Oh JE, McKenna CE, Kang MK, Shin KH, Tetradis S, Park NH, Kim RH, Development of oral osteomucosal tissue constructs in vitro and localization of fluorescently-labeled bisphosphonates to hard and soft tissue, Int. J. Mol. Med 34 (2) (2014) 559–563, 10.3892/ijmm.2014.1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Cheong S, Sun S, Kang B, Bezouglaia O, Elashoff D, McKenna CE, Aghaloo TL, Tetradis S, Bisphosphonate uptake in areas of tooth extraction or periapical disease, J. Oral Maxillofac. Surg 72 (12) (2014) 2461–2468, 10.1016/j.joms.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Zhu W, Xu R, Du J, Fu Y, Li S, Zhang P, Liu L, Jiang H, Zoledronic acid promotes TLR-4-mediated M1 macrophage polarization in bisphosphonate-related osteonecrosis of the jaw, FASEB J. 33 (4) (2019) 5208–5219, 10.1096/fj.201801791RR. [DOI] [PubMed] [Google Scholar]
- [136].Hokugo A, Kanayama K, Sun S, Morinaga K, Sun Y, Wu Q, Sasaki H, Okawa H, Evans C, Ebetino FH, Lundy MW, Sadrerafi K, McKenna CE, Nishimura I, Rescue bisphosphonate treatment of alveolar bone improves extraction socket healing and reduces osteonecrosis in zoledronate-treated mice, Bone 123 (2019) 115–128, 10.1016/j.bone.2019.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Kang WS, Sun S, Nguyen K, Kashemirov B, McKenna CE, Hacking SA, Quesnel AM, Sewell WF, McKenna MJ, Jung DH, Non-ototoxic local delivery of bisphosphonate to the mammalian cochlea, Otol. Neurotol 36 (6) (2015) 953–960, 10.1097/MAO.0000000000000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Seist R, Tong M, Landegger LD, Vasilijic S, Hyakusoku H, Katsumi S, McKenna CE, Edge ASB, Stankovic KM, Regeneration of Cochlear synapses by systemic administration of a bisphosphonate, Front. Mol. Neurosci 13 (2020) 87, 10.3389/fnmol.2020.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Kempfle JS, Duro MV, Zhang A, Amador CD, Kuang R, Lu R, Kashemirov BA, Edge AS, McKenna CE, Jung DH, A novel small molecule Neurotrophin-3 analogue promotes inner ear neurite outgrowth and synaptogenesis in vitro, Front. Cell. Neurosci 15 (2021), 666706, 10.3389/fncel.2021.666706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Li X, Naguib YW, Cui Z, In vivo distribution of zoledronic acid in a bisphosphonate-metal complex-based nanoparticle formulation synthesized by a reverse microemulsion method, Int. J. Pharm 526 (1–2) (2017) 69–76, 10.1016/j.ijpharm.2017.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Li X, Valdes SA, Alzhrani RF, Hufnagel S, Hursting SD, Cui Z, Zoledronic acid-containing nanoparticles with minimum premature release show enhanced activity against extraskeletal tumor, ACS Appl. Mater. Interfaces 11 (7) (2019) 7311–7319, 10.1021/acsami.8b16588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Tseng HC, Kanayama K, Kaur K, Park SH, Park S, Kozlowska A, Sun S, McKenna CE, Nishimura I, Jewett A, Bisphosphonate-induced differential modulation of immune cell function in gingiva and bone marrow in vivo: role in osteoclast-mediated NK cell activation, Oncotarget 6 (24) (2015) 20002–20025, 10.18632/oncotarget.4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Junankar S, Shay G, Jurczyluk J, Ali N, Down J, Pocock N, Parker A, Nguyen A, Sun S, Kashemirov B, McKenna CE, Croucher PI, Swarbrick A, Weilbaecher K, Phan TG, Rogers MJ, Real-time intravital imaging establishes tumor-associated macrophages as the extraskeletal target of bisphosphonate action in cancer, Cancer Discov. 5 (1) (2015) 35–42, 10.1158/2159-8290.CD-14-0621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Verhulst A, Sun S, McKenna CE, D’Haese PC, Endocytotic uptake of zoledronic acid by tubular cells may explain its renal effects in cancer patients receiving high doses of the compound, PLoS ONE 10 (3) (2015), e0121861, 10.1371/journal.pone.0121861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Zarb Y, Weber-Stadlbauer U, Kirschenbaum D, Kindler DR, Richetto J, Keller D, Rademakers R, Dickson DW, Pasch A, Byzova T, Nahar K, Voigt FF, Helmchen F, Boss A, Aguzzi A, Klohs J, Keller A, Ossified blood vessels in primary familial brain calcification elicit a neurotoxic astrocyte response, Brain 142 (4) (2019) 885–902, 10.1093/brain/awz032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].McDonald MM, Khoo WH, Ng PY, Xiao Y, Zamerli J, Thatcher P, Kyaw W, Pathmanandavel K, Grootveld AK, Moran I, Butt D, Nguyen A, Corr A, Warren S, Biro M, Butterfield NC, Guilfoyle SE, Komla-Ebri D, Dack MRG, Dewhurst HF, Logan JG, Li Y, Mohanty ST, Byrne N, Terry RL, Simic MK, Chai R, Quinn JMW, Youlten SE, Pettitt JA, Abi-Hanna D, Jain R, Weninger W, Lundberg M, Sun S, Ebetino FH, Timpson P, Lee WM, Baldock PA, Rogers MJ, Brink R, Williams GR, Bassett JHD, Kemp JP, Pavlos NJ, Croucher PI, Phan TG, Osteoclasts recycle via osteomorphs during RANKL-stimulated bone resorption, Cell 184 (5) (2021) 1330–1347, 10.1016/j.cell.2021.02.002, e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Iqbal J, Zaidi M, Bone resorption goes green, Cell 184 (5) (2021) 1137–1139, 10.1016/j.cell.2021.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Richard ET, Morinaga K, Zheng Y, Sundberg O, Hokugo A, Hui K, Zhou Y, Sasaki H, Kashemirov BA, Nishimura I, McKenna CE, Design and synthesis of cathepsin-K-activated osteoadsorptive fluorogenic sentinel (OFS) probes for detecting early osteoclastic bone resorption in a multiple myeloma mouse model, Bioconjug. Chem 32 (5) (2021) 916–927, 10.1021/acs.bioconjchem.1c00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Farrell KB, Karpeisky A, Thamm DH, Zinnen S, Bisphosphonate conjugation for bone specific drug targeting, Bone Rep. 9 (2018) 47–60, 10.1016/j.bonr.2018.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Xing L, Ebetino FH, Boeckman RK Jr., Srinivasan V, Tao J, Sawyer TK, Li J, Yao Z, Boyce BF, Targeting anti-cancer agents to bone using bisphosphonates, Bone 138 (2020), 115492, 10.1016/j.bone.2020.115492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Zhang S, Gangal G, Uludag H, ‘Magic bullets’ for bone diseases: progress in rational design of bone-seeking medicinal agents, Chem. Soc. Rev 36 (3) (2007) 507–531, 10.1039/b512310k. [DOI] [PubMed] [Google Scholar]
- [152].Morioka M, Kamizono A, Takikawa H, Mori A, Ueno H, Kadowaki S, Nakao Y, Kato K, Umezawa K, Design, synthesis, and biological evaluation of novel estradiol-bisphosphonate conjugates as bone-specific estrogens, Bioorg. Med. Chem 18 (3) (2010) 1143–1148, 10.1016/j.bmc.2009.12.041. [DOI] [PubMed] [Google Scholar]
- [153].Young RN, Grynpas MD, Targeting therapeutics to bone by conjugation with bisphosphonates, Curr. Opin. Pharmacol 40 (2018) 87–94, 10.1016/j.coph.2018.03.010. [DOI] [PubMed] [Google Scholar]
- [154].Houghton TJ, Tanaka KS, Kang T, Dietrich E, Lafontaine Y, Delorme D, Ferreira SS, Viens F, Arhin FF, Sarmiento I, Lehoux D, Fadhil I, Laquerre K, Liu J, Ostiguy V, Poirier H, Moeck G, Parr TR Jr., Far AR, Linking bisphosphonates to the free amino groups in fluoroquinolones: preparation of osteotropic prodrugs for the prevention of osteomyelitis, J. Med. Chem 51 (21) (2008) 6955–6969, 10.1021/jm801007z. [DOI] [PubMed] [Google Scholar]
- [155].Tanaka KS, Dietrich E, Ciblat S, Metayer C, Arhin FF, Sarmiento I, Moeck G, Parr TR Jr., Far AR, Synthesis and in vitro evaluation of bisphosphonated glycopeptide prodrugs for the treatment of osteomyelitis, Bioorg. Med. Chem. Lett 20 (4) (2010) 1355–1359, 10.1016/j.bmcl.2010.01.006. [DOI] [PubMed] [Google Scholar]
- [156].Zinnen SP, Karpeisky A, Von Hoff DD, Plekhova L, Alexandrov A, First-in-human phase I study of MBC-11, a novel bone-targeted cytarabine-etidronate conjugate in patients with cancer-induced bone disease, Oncologist 24 (3) (2019) 303–e102, 10.1634/theoncologist.2018-0707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Wang H, Xiao L, Tao J, Srinivasan V, Boyce BF, Ebetino FH, Oyajobi BO, Boeckman RK Jr., Xing L, Synthesis of a bone-targeted bortezomib with in vivo anti-myeloma effects in mice, Pharmaceutics 10 (3) (2018), 10.3390/pharmaceutics10030154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Wang H, Zhang H, Srinivasan V, Tao J, Sun W, Lin X, Wu T, Boyce BF, Ebetino FH, Boeckman RK Jr., Xing L, Targeting bortezomib to bone increases its bone anabolic activity and reduces systemic adverse effects in mice, J. Bone Miner. Res 35 (2) (2020) 343–356, 10.1002/jbmr.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Yao Z, Hou XD, Lei W, Xiao L, Ebetino FH, Boeckman RK Jr., Boyce BF, Bone-targeted chloroquine inhibits osteoclastogenesis and bone resorption more effectively than chloroquine, J. Bone Miner. Res 31 (S1) (2016) S225. [Google Scholar]
- [160].Sedghizadeh PP, Sun S, Junka AF, Richard E, Sadrerafi K, Mahabady S, Bakhshalian N, Tjokro N, Bartoszewicz M, Oleksy M, Szymczyk P, Lundy MW, Neighbors JD, Russell RG, McKenna CE, Ebetino FH, Design, synthesis, and antimicrobial evaluation of a novel bone-targeting bisphosphonate-ciprofloxacin conjugate for the treatment of osteomyelitis biofilms, J. Med. Chem 60 (6) (2017) 2326–2343, 10.1021/acs.jmedchem.6b01615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Adjei-Sowah E, Peng Y, Weeks J, Jonason JH, de Mesy Bentley KL, Masters E, Morita Y, Muthukrishnan G, Cherian P, Hu XE, McKenna CE, Ebetino FH, Sun S, Schwarz EM, Xie C, Development of bisphosphonate-conjugated antibiotics to overcome pharmacodynamic limitations of local therapy: initial results with carbamate linked sitafloxacin and tedizolid, Antibiotics (Basel) 10 (6) (2021), 10.3390/antibiotics10060732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].de Mesy Bentley KL, Trombetta R, Nishitani K, Bello-Irizarry SN, Ninomiya M, Zhang L, Chung HL, McGrath JL, Daiss JL, Awad HA, Kates SL, Schwarz EM, Evidence of Staphylococcus Aureus deformation, proliferation, and migration in Canaliculi of live cortical bone in murine models of osteomyelitis, J. Bone Miner. Res 32 (5) (2017) 985–990, 10.1002/jbmr.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Junka A, Szymczyk P, Ziolkowski G, Karuga-Kuzniewska E, Smutnicka D, Bil-Lula I, Bartoszewicz M, Mahabady S, Sedghizadeh PP, Bad to the bone: on in vitro and ex vivo microbial biofilm ability to directly destroy colonized bone surfaces without participation of host immunity or osteoclastogenesis, PLoS ONE 12 (1) (2017), e0169565, 10.1371/journal.pone.0169565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Amorim T, Ferrari AJ, Sabol HM, Anderson J, Cregor M, Sweet M, Srinivasan V, Ebetino FH, Boeckman RK Jr., Roodman GD, Bellido T, Delgado-Calle J, Low doses of the bone-targeted notch inhibitor BT-GSI exhibit higher anti-myeloma activity and preserve bone compared to unconjugated GSI or zolendronic acid, J. Bone Miner. Res 35 (S1) (2020) S101. [Google Scholar]
- [165].Sabol HM, Ferrari AJ, Adhikari M, Amorim T, McAndrews K, Anderson J, Vigolo M, Lehal R, Cregor M, Khan S, Cuevas PL, Helms JA, Kurihara N, Srinivasan V, Ebetino FH, Boeckman RK, Roodman GD, Bellido T, Delgado-Calle J, Targeting notch inhibitors to the myeloma bone marrow niche decreases tumor growth and bone destruction without gut toxicity, Cancer Res. (2021), 10.1158/0008-5472.CAN-21-0524. [DOI] [PMC free article] [PubMed] [Google Scholar]