

Abstract
The burden of human disease lies predominantly in polygenic, complex diseases. Since the early 2000s, genome-wide association studies (GWAS) have identified genetic variants and loci associated with complex traits. These have ranged from variants in coding sequences to mutations in regulatory regions, such as promoters and enhancers, as well as mutations affecting mediators of mRNA stability and other downstream regulators, such as 5’ and 3’ UTRs, long non-coding RNA (lncRNA), and miRNA. Recent research advances in genetics have utilized a combination of computational techniques, high-throughput in vitro and in vivo screening modalities, and precise genome editing to impute the function of diverse classes of genetic variants identified through GWAS. In this review, we highlight the vastness of genomic variants associated with disease and address recent advances in how genetic tools can be used to functionally characterize these diverse variants.
Keywords: GWAS variants, colocalization, CRISPR, chromatin capture, MPRA
Introduction
The burden of human diseases is dominated by complex, polygenic disease rather than monogenic disorders associated with single, highly penetrant genetic mutations. Since the mid-2000s, genome-wide association studies (GWAS) have been the preferred tool for identifying associations between genetic variants and complex traits [1]. While the association between genomic variation and phenotype has long been understood, GWA studies have greatly expanded the landscape of genomic variants associated with traits and disease [2]. The NHGRI-EBI GWAS Catalog has almost 6000 GWAS entries published since 2005 [3]. From these studies have emerged over 400,000 associations between genetic variants and human traits. These trait- and disease-associated variants lie in a variety of genomic locales, including coding and noncoding regions (Figure 1), and illuminate the complex biological pathways that regulate the formation of a functional protein product from a gene and how variant-mediated perturbations in relevant pathways can promote the pathogenesis of polygenic disorders (Figures 1–2).
Figure 1. Genome-wide association studies (GWAS) identify a diverse catalog of variants which affect gene expression and protein function across the central dogma.
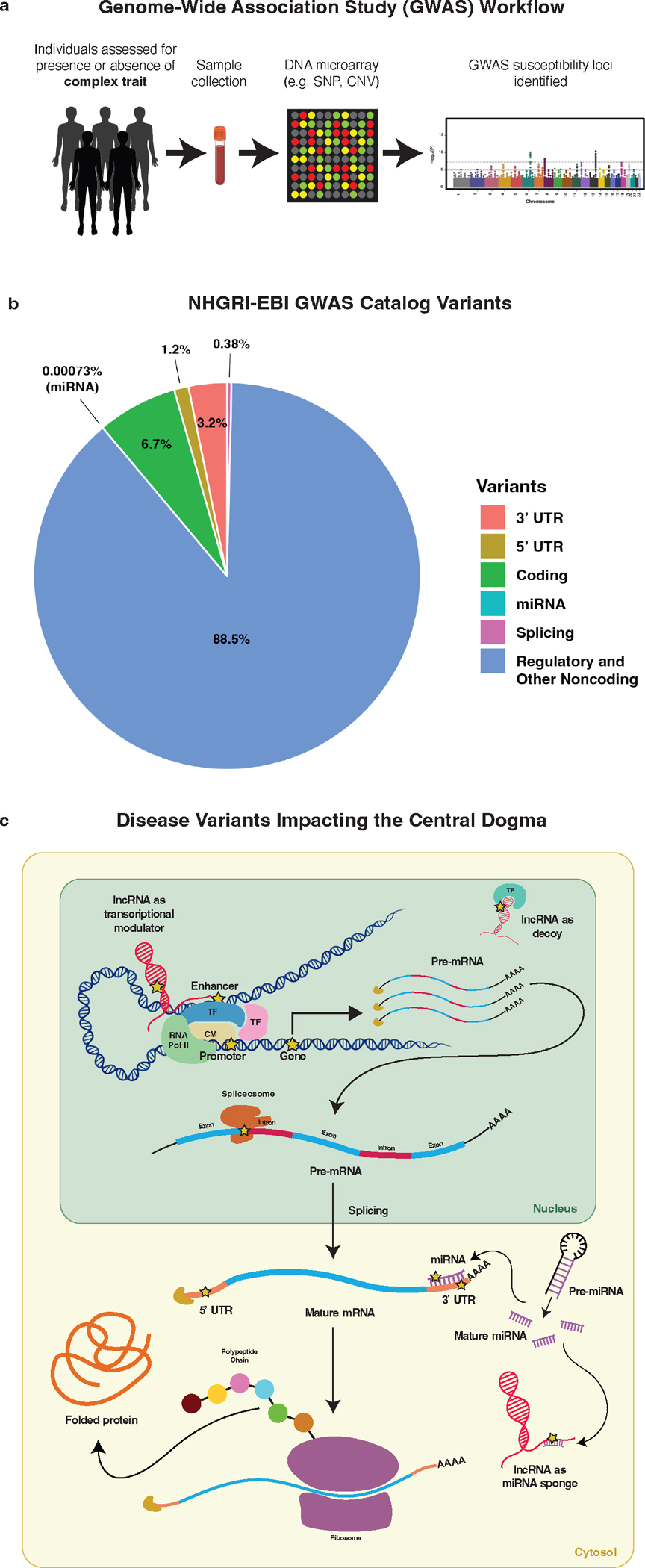
(a) Workflow for GWA studies. (b) Distribution of GWAS-identified variants across variant classes (based on the 2022 GWAS Catalog), highlighting large proportion of noncoding variants. “Regulatory and Other Noncoding” includes variants designated as regulatory, intronic, and other noncoding in the GWAS catalog. (c) Variants which are found to be associated with disease can disrupt gene expression and protein function through diverse mechanisms. Yellow stars highlight common genetic elements and mechanisms impacted by trait- and disease-associated variants. The diverse mechanisms highlight the need for diverse tools to functionally annotate variants. CM = chromatin modifier; TF = transcription factor.
Figure 2. Variants in various classes of genomic elements have diverse effects on gene expression and protein translation.
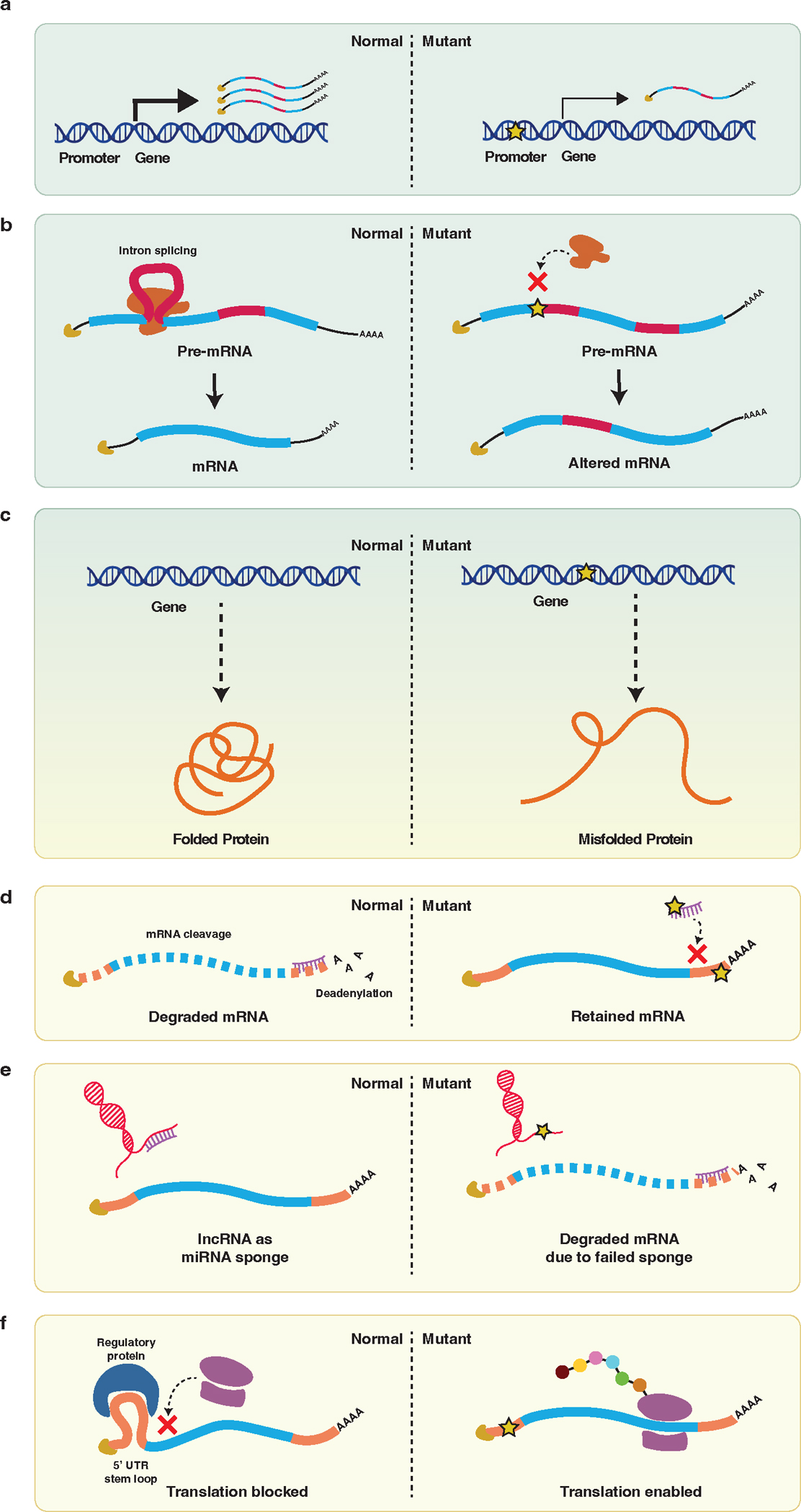
(a) Promoter and enhancer variants, as well as some types of lncRNA variants, disrupt the regulation of gene expression, decreasing transcription. (b) Splice site variants can result in abnormally spliced transcripts and the production of altered protein. (c) Coding variants can disrupt protein function by various mechanisms, including protein misfolding. (d) Variants in miRNA or 3’ UTR sequences can disrupt miRNA-mediated regulation of mRNA transcript abundance, resulting in mRNA retention. (e) Variants in some types of lncRNAs can disrupt the ability of lncRNAs to function as an miRNA sponge, resulting in mRNA degradation. (f) 5’ UTR variants can alter binding of regulatory proteins, resulting in altered translation.
An important limitation of the GWAS approach is that the findings are just that–associations. A GWAS hit does not specify the identity of the precise causal variant, nor does it reveal the causal trait-associated gene or the perturbed biological process [2]. Rather, GWAS only identify associations between traits and the lead or index SNPs present on the microarrays used in these studies. Thus, for GWAS hits to be translated into any meaningful clinical impact, the true functional variants need to be identified and their impact on gene expression and trait development interrogated. In this review, we will discuss efforts by researchers over the years to understand the diverse types of genomic variants associated with traits and disease by developing experimental techniques and tools tailored to the unique function and impact of the classes of variants being studied. We will focus particularly on recent developments in high-throughput screening methodologies and computational tools which have greatly facilitated ongoing efforts to functionally annotate and determine causal genes for the broad catalog of trait-associated genetic variants identified through GWAS.
Understanding the variants
Coding Variants
Long before the advent of the GWAS, the connection between coding variation and a disease or trait had been well understood, particularly for Mendelian disease. Since the early 2000s, however, greater efforts have been directed towards understanding the genes that drive complex traits and diseases. In their 2002 article, Glazier et al. provided a framework for identifying genes that drive complex traits which has come to reflect efforts since undertaken to uncover coding variants which drive complex disease [4]. They suggest first performing association studies to identify genomic intervals (i.e. loci) associated with a complex (or quantitative) trait–that is, its quantitative trait loci (QTLs). This should be followed by statistical fine mapping to reduce the size of the candidate genomic interval, including linkage disequilibrium (LD) studies to determine how genetic markers across the locus associate with the trait of interest. Because GWA studies typically identify lead or index SNPs associated with disease rather than the causal SNP itself, statistical fine mapping tools have been employed to attempt to nominate the true causal SNP (or SNPs) for downstream functional studies. Statistical fine mapping typically entails identifying the region of interest, exploring the region for its LD structure and the genes and SNPs contained in that region, partitioning the region independent regions which each affect the treat, and then employing statistical fine mapping methods (e.g. simple heuristic methods, penalized regression models, and Bayesian methods) to identify the SNP in the region most strongly associated with the trait or disease [5]. In their framework, Glazier et al. also note the importance of sequencing the region of interest to identify coding variants, which further improves the power of statistical mapping. Lastly, they stress the importance of performing functional assays to determine the role of a gene, especially as it relates to the trait, and the impact of a variant in that gene. These steps highlight the critical connection that must be made between QTL, variant, and gene in order to discover coding genetic variants which impact a trait. As researchers have endeavored over the past two decades to uncover genetic variants in coding regions which contribute to complex traits, varied approaches have been taken to connect coding variants and phenotype.
Researchers studying coding variation in complex traits prior to the development of GWAS commonly employed a gene → variant approach, rather than the locus → variant → gene approach outlined by Glazier et al. (2002) [4]. Studies often focused on the identification and characterization of single nucleotide variants/polymorphisms (SNVs/SNPs) and other genetic variants at gene loci already known to be involved in a particular trait or disease (Box 1). By contrast, GWAS is a locus-centric approach: GWAS identifies a susceptibility locus associated with a trait, and further work is done to identify the genes and causal variants driving the trait. Thus, a GWAS approach allows for the discovery of novel genes and pathways associated with a trait, thereby enabling insight into the complicated puzzle of genetically complex polygenic traits. It is also most aligned with the QTL-variant-gene triad which Glazier et al. propose for identifying genes that drive complex traits [4]. Studies which take this approach have often discovered and functionally characterized novel genes which may play a role in a trait. For example, Kim et al. (2017) used prior GWAS data on serum acylcarnitine levels to identify a susceptibility locus of interest [6]. Fine mapping of the locus determined acylcarnitine levels to be associated with variants in the SLC22A1 gene, which encodes a membrane transporter in the liver with no previously described role in acylcarnitine transport. Functional characterization of the gene and its human variants identified a novel role of SLC22A1 in mediating acylcarnitine efflux from the liver, and reduced protein levels and efflux activity resulting from genetic variants. In another study, Chang et al. (2018) performed exome-wide association analysis (similar to GWAS except for its exclusive focus on the human exome) of colorectal cancer cases and controls and identified a novel missense variant in TCF7L2, a TF which activates transcription of MYC, a proto-oncogene [7]. The authors show that the variant in TCF7L2 decreases its activity, leading to decreased MYC expression and lower risk for colorectal cancer.
Box 1. Pre-GWAS Era Characterization of Coding Variants.
Pre-GWAS gene-centric studies often focused on the identification and characterization of SNPs and other genetic variants in genes already known to be involved in a particular trait or disease. For example, work by Blaisdell et al., (2002), Dai et al. (2001), and Zhang et al. (2001) identified novel SNPs in genes already known to play a role in drug metabolism, CYP2C19, CYP3A4, and PXR [210–212]. Similarly, Cargill et al. (1999) compiled a list of 106 genes encoding proteins thought to play a role in common diseases, such as type 2 diabetes, schizophrenia, and coronary artery diseases, and identified 392 coding-region SNPs which might influence these diseases [213]. Sharon et al. (2000) focused their studies on the olfactory receptor locus and identified 21 novel coding SNPs [214]. The SNPs identified in these studies are often further characterized to determine their effect on the genes of interest, using methods such as site-directed mutagenesis to generate plasmids expressing the mutant proteins and study variant effects on DNA binding affinity, catalytic activity, expression stability, hydroxylation activity and other functional assays relevant to the protein of interest [210–212]. What these studies have in common is their gene-centric approach, requiring a priori knowledge on the genes thought to play an important role the trait or disease. GWAS provides the advantage of nominating novel trait- and disease-associated loci and genes, and thus novel coding variants.
With methodological advances, the tools available to functionally characterize candidate genes identified in GWAS susceptibility loci and their coding variants have improved. In silico tools such as the Ensembl Variant Effect Predictor, PredictSNP, dbNSFP, Revel, and Envision have been employed to predict the deleterious effects of candidate coding variants on protein structure and function [8–14]. Furthermore, various functional assays have been carried out to characterize candidate genes and the effect of variants in in vitro and in vivo contexts. In a high throughput effort to functionally screen candidate genes, Chapuis et al. (2017) performed a genome-wide siRNA screen for genes which alter amyloid-β precursor protein metabolism and found that 8 of the top siRNA hits overlapped with genes located in GWAS-identified susceptibility loci for Alzheimer’s disease [15]. In vivo knockouts of candidate genes in mice and other animals are useful for interrogating the function of these genes in a whole organism [6,16]. Candidate variants may also be introduced into genes, using CRISPR/Cas9 homology-directed repair (HDR) or other tools for site-directed mutagenesis, and their effect on protein stability and function studied. Functional studies can also include reporter assays of the activity of variants of a protein known to be a transcription factor (TF) [7,9]. Other functional studies are particularly tailored to the protein of interest, such as studying the effects of SLC22A1 variants on carnitine transport [6], screening BRCA1 variants for their ability to repair DNA [17], or colony-formation assays to test the pathogenicity of TP53 mutations [9]. HDR may also be used in vivo to study gene variant effect, as was done by Zhu et al. (2017), who edited a human variant of the GWAS-identified STXBP5 gene into the corresponding mouse gene and found that the variant caused a decreased thrombotic phenotype in the mice [18].
In their 2019 study, Keller et al. provide an example of how to use the integrated QTL-SNP-gene approach to identify candidate genetic variants which drive disease [19]. Their study sought to identify genes associated with the modulation of insulin secretion in a non-diabetic state. They did so by isolating islet cells from non-diabetic mice fed a Western diet and measuring insulin secretion, while also conducting genetic screens of the mice and performing QTL analysis to identify loci associated with insulin secretion. These loci were integrated with SNP hits found in human GWAS for diabetes-related traits to identify genes and causal variants which may modulate insulin secretion and risk for type 2 diabetes. Three of these genes, Ptpn18, Hunk, and Zfp148, were further characterized in vivo, including mice with HDR-inactivated Ptpn18 which revealed the gene’s role in regulating insulin sensitivity. Because coding variants linked to pathogenic risk for specific polygenic disorders commonly affect protein function, they are particularly valuable in providing insight into disease pathogenesis.
Lastly, it is important to highlight the role of rare variants (MAF <1%) in advancing our understanding of genetic disease risk. It has been shown that GWAS variants, which are typically common (MAF >5%) with small effect sizes, do not completely explain disease heritability and risk [20]. Rare variants may explain some of this “missing heritability.” Rare coding variants in particular are more likely to be deleterious and have larger effect sizes, thus helping narrow down causal genes and allowing for better elucidation of disease mechanisms to improve therapeutic targeting. Recent rare variant-focused GWAS have shown how rare variants can identify associations for genes not previously known to play a role in disease. For example, in their analysis of very rare variants (MAF <0.1%) in the UK Biobank, Cirulli et al. identified novel associations between STAB1 and brain structure measurements [21]. Studies like these emphasize the importance of continuing to rare variant detection, as they will continue to play an important role in functional annotation of GWAS signals.
Variants That Affect Splicing
While coding variants are perhaps the best-characterized examples linking changes in protein sequence to impact protein function in polygenic disorders, an oft-underappreciated class of variants which also alter protein structure and function is comprised of those impacting pre-mRNA splicing, the process by which introns are removed and exons linked together to form mature mRNA. Splice site variants can generate mature mRNA transcripts containing introns or missing exons (Figure 2). Such mutations have been extensively investigated on a gene-by-gene basis, uncovering genetic variants causing alternative splicing in physiologically important genes/proteins such as CYP3A5 (drug metabolism) [22], tau protein (Alzheimer’s) [23], α-galactosidase A (Fabry disease) [24], and others. GWAS efforts have catalyzed further discovery of such variants that affect pre-mRNA splicing. However, while GWAS identifies associations between a SNP locus and a trait, it does not provide information on the effect of the variant, including if it impacts alternative splicing.
Thus, efforts have been undertaken to colocalize (i.e. overlap) the GWAS QTL map with a functional map of genomic loci–that is, determine which GWAS loci overlap with loci known to have some effect on gene expression [25]. This colocalization approach, used for several types of noncoding variants to be discussed in this article, is an extension of the approach outlined by Glazier et al. Once colocalization has identified the most promising functional SNP loci associated with a trait, fine mapping and functional characterization may proceed accordingly. In the case of splicing variants, researchers have worked to generate splicing QTL (sQTL) maps of various normal and abnormal (e.g. cancerous) tissue types [26–29]. sQTLs contain SNPs and other genomic variants which affect alternative splicing and thus change the transcripts produced from a gene. These sQTLs reside not only in canonical splicing sites at the exon-intron junction, but across the whole genome, including exons and 5’ and 3’ UTRs [30]. Such non-canonical splicing sites are referred to as cryptic splice sites. While variants in canonical splice sites lead to abnormal whole intron inclusion or exon skipping, cryptic splice variants, due to their location, cause partial inclusion or deletion of the intron or exon, respectively.
sQTL maps for specific tissue types can be colocalized with GWAS data for tissue-relevant diseases to identify potential causal splicing variants which may modulate disease risk. An example of this approach was illustrated by Takata et al. (2017), who began by analyzing prefrontal cortex RNA-Seq data matched with SNP genotyping for 206 individuals to identify sQTLs in the prefrontal cortex [30]. The authors followed this with enrichment analyses of the sQTL SNPs and the genomic loci found to be associated with disease through GWAS and found that the prefrontal cortex sQTL SNPs were most significantly enriched in schizophrenia-associated loci. Brotman et al. (2022) model a similar approach, beginning by sequencing and analyzing the adipose tissue of 426 individuals to identify sQTLs [26]. Colocalization analysis revealed gene sQTLs which overlapped with GWAS loci for several cardiometabolic traits, including lipid levels and BMI.
Noncoding Variants
Promoter / Enhancer Variants
While the classes of variants discussed thus far act primarily by changing the quality of the transcripts and proteins produced, there are several other variant classes, including those present in noncoding DNA, which can modulate the quantity of gene expression (Figure 2a). These noncoding variants make up more than 90% of all GWAS variants, consistent with their large proportion (>98%) of the human genome [3] (Figure 1b). Thus, characterization of noncoding variants and their target genes is important for understanding the diverse contributors to complex traits and diseases. An important group of noncoding variants resides in gene promoters and enhancers, noncoding regulatory elements in the genome which modulate gene expression. A promoter is a 100–1K bp long region of DNA located immediately upstream of the transcription start site (TSS) of a gene, to which proteins bind to initiate transcription of that gene. An enhancer is a 50–1.5K bp long region of DNA to which bind TFs that distally regulate transcription of a gene. Enhancers can be located up to 1 Mbp upstream or downstream the genes they regulate, and they can be found in intergenic or even intronic sequences, making their potential locations and reach expansive. What promoters and enhancers have in common is that they are bound by TFs which act to regulate transcription of their cis-target gene. Additionally, enhancers and promoters often form loops with each other to fine-tune gene expression. GWAS efforts have identified many noncoding variants which frequently overlap with putative promoter and enhancer regions as contributors to complex disease. Thus, in recent years, research on genetic variation has shifted towards understanding the consequences of these noncoding variants and the impacts they exert on their putative target genes and disease processes.
Variants in loci containing promoters and enhancers associated with a complex trait or disease (QTLs) can be nominated by GWAS. To confidently assign these noncoding loci to a dysregulated target gene, however, they must be colocalized with datasets that orthogonally connect the genomic regulatory region where the variant resides with a putative target gene. This is commonly undertaken using tissue-specific expression quantitative trait loci (eQTL) data, which, analogous to its sQTL counterpart, tracks associations between SNPs in a given genetic locus and changes in expression of genes within its genomic region. Noncoding variant linkage to putative target genes can also be assigned via DNA looping data, such as Hi-C and HiChIP, which identify physical contacts between putative enhancers and specific gene promoter, as well as via CRISPR studies, which can disrupt activity of a regulatory locus to identify its potential target gene(s). The latter is commonly initially achieved by CRISPR interference (CRISPRi) using catalytically inactive Cas9 fused to a mediator of gene silencing, which can impair function of enhancers and promoters, thereby nominating target genes as those whose expression levels are modulated by inhibition of a variant-containing regulatory locus. To identify and validate potential causal SNPs, fine mapping and prioritization of SNPs can also be coupled with reporter assays to test SNP activity compared to its non-risk control. Ultimately, the most likely causal GWAS-derived variant and its proposed target gene can be linked using gene editing tools like CRISPR HDR. Much debate in recent years has arisen in the field over how best to determine the target gene of a locus, particularly for long-range and often pleiotropic enhancers. The aforementioned tools and other methods which have been developed to improve locus target predictions will be discussed and compared in more detail in a later section.
Application of the colocalization approach to the characterization of GWAS noncoding variants associated with various complex traits and polygenic diseases has led to the identification of novel genes which may contribute to disease phenotypes. Gu et al. (2021) colocalized kidney disease and function GWAS with kidney QTL to identify MANBA as a novel gene whose low expression is associated with increased risk for chronic kidney disease [16]. They validated MANBA as a novel CKD-associated gene with phenome-wide association analysis of almost 41,000 patients, finding loss-of-function coding variants in MANBA associated with renal failure, as well as with a mouse knockout study, showing that Manba loss increased susceptibility to kidney fibrosis. Doke et al. (2021) and Gupta et al. (2017) similarly focused their studies one on particular locus [31,32]. Doke et al. (2021), similar to Gu et al. (2021), colocalized GWAS hits for eGFR (a measure of kidney function) with kidney eQTL data to nominate CASP9 as a novel kidney disease risk gene [31]. To identify the causal SNP, they analyzed kidney single nuclear assay for transposase-accessible chromatin sequencing (ATAC-Seq) data to identify variants located in open chromatin and defined a tubule-specific SNP in a locus which regulates CASP9 expression, as validated by gene knockout. Similar again to Gu et al., they used a mouse model to demonstrate the mechanism by which loss of Casp9 protects against renal disease and fibrosis. Gupta et al. (2017) performed similar studies, focusing on a locus associated with vascular disease which modulates EDN1 expression and using CRISPR HDR to validate potential causal SNPs [32].
With recent shifts to more high throughput approaches, however, other researchers have looked for ways to screen and prioritize multiple GWAS loci concurrently. Some have made use of massively parallel reporter assays (MPRAs), a pooled assay where an enhancer sequence modulates activity of a minimal promoter, which drives expression of a reporter. If reference and alternate sequences of an enhancer for a particular SNP are included, MPRA can be used to determine which GWAS-linked SNPs have differential functional activity. MPRAs have been used to screen SNPs in loci associated with diseases such as dementia [33,34], type 2 diabetes [35], melanoma [36], and chronic obstructive pulmonary disease [37]. MPRA-identified functional SNPs can then be further characterized using familiar techniques, including CRISPRi targeting SNP loci to identify responsive genes [33], eQTL co-localizations [33,35,36], HiChIP data linking SNP locus with candidate gene promoters [36], as well as testing of gene function using in vivo animal models [35].
Others have used alternative approaches to prioritize loci of interest, rooted in the predicted mechanistic impacts of variants in these loci. For example, after identifying 23,400 unique SNPs across 18 schizophrenia-associated loci identified through GWAS, Huo et al. (2019) used data from 30 ChIP-Seq experiments in brain tissue to derive TF DNA binding motifs and, from the 23,400 SNPs, identified 132 SNPs which were predicted to disrupt TF binding [38]. These SNPs were then co-localized with eQTL data to identify target genes and a subset validated with a luciferase reporter assay to test for reference versus alternate allele differential activity. Zhang et al. (2020), by contrast, focused not on TF binding site disruption, but rather on allele-specific open chromatin [39]. They began by identifying 20 people heterozygous for GWAS index SNPs at 70 schizophrenia susceptibility loci and made induced pluripotent stem cells (iPSCs) from these 20 individuals. These iPSCs were differentiated into various neuronal cell types, followed by ATAC-Seq and RNA-Seq to identify allele-specific open chromatin (ASoC) SNPs, which were significantly enriched in promoter and enhancer regions. Candidate target genes included genes nominated using colocalization with brain eQTLs and brain and neuronal Hi-C loops, as well as genes differentially expressed in CROP-Seq (single cell CRISPR) targeting ASoC sites. TF binding analysis identified a subset of ASoC SNPs predicted to disrupt a TF motif, matching the allelic direction of the allele-specific chromatin accessibility differences. CRISPR HDR-mediated introduction of a subset of ASoC SNPs validated the correct target genes of those nominated by eQTL, Hi-C, and CROP-Seq, as well as the mechanism by which SNPs act. They demonstrate that one schizophrenia-associated risk variant, rs2027349, is associated with allele-specific chromatin accessibility and TF footprinting. Zhao et al. (2021) took an alternative approach to prioritize loci, beginning by performing a GWAS of white matter microstructure, as measured by diffusion MRI (dMRI), followed by colocalization of dMRI GWAS data with GWAS data for various brain-related complex traits and diseases, such as schizophrenia, depression, stroke, and glioma [40]. MAGMA and FUMA, two prediction tools, were then used to nominate target genes and identified overlap between white matter microstructure-associated genes and those targeted by common nervous system drugs. This method of colocalizing GWAS loci for complex diseases/traits with GWAS loci linked to a physiological phenotype proposed to be associated with the disease helps unite candidate causal genes under one common physiological pathway.
Variants in 5’ and 3’ Untranslated Regions
5’ and 3’ untranslated regions (UTRs) are another frequent site of noncoding genetic variations. UTRs are regions present either immediately upstream (5’) or downstream (3’) of the mature coding mRNA and play an important role in regulating post-transcriptional gene expression. 5’ and 3’ UTRs are able to regulate translation through a wide array of mechanisms, and it is these mechanisms which may be impacted in disease-causing mutations. 5’ UTRs, for example, contain various regulatory components, including upstream open reading frames (uORFs) which, when translated, can inhibit expression of the downstream primary ORF, secondary structures (such as stem loops) which regulate translation initiation, and iron response elements (IREs) which modulate translation in response to iron levels [41]. 3’ UTRs also contain IREs, as well as miRNA response elements which bind an miRNA and lead to transcript degradation, AU-rich response elements (AREs) which bind to ARE binding proteins (ARE-BPs) to modulate translation, and sequences which regulate polyadenylation [42].
In the pre-GWAS era, mutations in 5’ and 3’ UTR were already shown to play important roles in disease, particularly Mendelian disorders, by disrupting the aforementioned mechanisms of transcriptional regulation (Figure 2d, f) (Box 2). The advent of GWAS has led to the discovery of additional 5’ and 3’ UTR variants which contribute to the pathophysiology of complex diseases. However, similar to other variants, and particularly noncoding variants, the challenge lies in functionally annotating the variants associated with disease. As with other variant classes, part of functional annotation requires linking the variant to a target gene which is mediating its effects. In some cases, UTRs have been linked to genes using eQTL data, as, similar to promoters and enhancers, they can affect transcript abundance through, for example, miRNA-mediated transcript degradation [43,44]. However, eQTL analysis is insufficient for linking UTR variance with target genes, given that UTR mutations affect not only transcript levels, but also the rate of translation, alternative polyadenylation, and other processes not captured by expression analysis. Thus, others have performed alternative analyses to predict the effects of loci variation on genes, using readouts which capture additional functions of UTRs. For example, Li et al. (2021) used RNA sequencing data generated for GTEx (an eQTL database) to identify tissue-specific 3′UTR alternative polyadenylation (APA) quantitative trait loci (3′aQTLs)—that is, loci associated with the generation of different polyadenylation sites on an mRNA, resulting in 3’ UTRs of varying length with differing susceptibilities to translational regulation [45]. They find that these loci are significantly enriched in 3’ UTRs. From this atlas of 3’aQTLs, CLIP-Seq was performed to identify RNA-binding protein (RBP) binding sites interrupted by 3’aQTL SNPs. 3’aQTLs were found to differ significantly from both eQTLs and sQTLs in their genomic element enrichments, highlighting the importance of mechanism-specific loci-gene associations. Others have described similar prediction and identification of APA-specific QTLs [46,47]. In another effort, Wei et al. (2022) employed PIVar, an algorithm that predicts variants which cause posttranscriptional impairment of a gene (piSNVs), to identify 3’-UTR piSNVs [48]. They identified piSNVs that overlapped with GWAS SNPs to identify the potential GWAS causal gene and, similar to Li et al. (2021), used CLIP-Seq to predict RBP binding site disruption by 3’-UTR piSNVs. Protein QTL (pQTL) analysis is another downstream alternative to eQTLs, measuring variant-dependent differences in protein abundance. Given the ability for UTRs to modulate protein abundance by modulating translation, pQTL analysis can valuable tool for identifying variants that impact translation, though the study of UTR pQTLs has been limited [49–51].
Box 2. Pre-GWAS Era Characterization of UTR Variants.
In the pre-GWAS era, mutations in 5’ and 3’ UTRs were already shown to play important roles in disease, particular Mendelian disorders, by disrupting mechanisms of transcript regulation. For example, myotonic dystrophy, a genetic disorder which causes progressive muscle wasting and weakness, is caused by expanded CTG trinucleotide repeats in the 3’ UTR of the DMPK gene [215]. These repeats are recognized by an RNA-binding protein (RBP), resulting in transcripts being retained in cell nuclei and not being translated [215–217]. Relatedly, a 3’ UTR variant in the FMR1 gene which causes Fragile X syndrome does so by disrupting binding of an RBP, resulting in loss of FMR1 translation [218]. In mantle cell lymphoma, the t(11;14)(q13;q32) translocation which drives the disease leads to deletions at the 3’ end of the CCND1 gene [215,219]. These deletions disrupt AREs which normally reduce the lifespan of the CCND1 mRNA, resulting in less mRNA degradation and more CCND1 translation. Mutations in one of the 5’ UTR uORFs of thrombopoietin (THPO), a hormone that regulates platelet production, have been shown to cause loss of uORF-mediated repression, resulting in increased THPO translation and hereditary thrombocythemia (a clotting disorder caused by excess platelets) [220,221]. Similarly, mutations in the 5’ UTR of the L-ferritin, an iron storage protein, disrupt the IRE and impair the interaction between the IRE and iron-regulatory proteins (IRPs) [220,222]. This leads to increased L-ferritin translation, causing hereditary hyperferritinemia/cataract syndrome, a disorder characterized by abnormally high blood ferritin levels. Additional 5’ UTR variants with alter the efficiency of translation efficiency of various genes, including BRCA1 (breast cancer) and MYC (multiple myeloma), have been identified [220,223–225].
Functional annotation of a variant entails not just linking the GWAS-associated locus with a candidate target gene, but also identifying and studying the causal variant in that locus. We previously described the use of MPRAs to screen enhancer sequences and their SNPs for differential activity by linking the enhancer to a minimal promoter which drives expression of a reporter. These traditional MPRA methods have helped some researchers identify functional UTR variants with differential activity [52,53]. However, similar to the limitations of using eQTLs for UTRs, the MPRA assay, which solely measures a sequence’s ability to increase transcript abundance, can miss diverse mechanisms whereby UTRs and their variants can function in disease. Griesemer et al. (2021) addressed this challenge by creating MPRAu, an MPRA designed to specifically test 3’ UTR sequences and variants [54]. Using MPRAu, Griesemer et al. (2021) identify and characterize two functional 3’ UTR SNPs—one which interrupts the 3’ UTR miRNA binding site of TRIM14 and another which changes PILRB transcript levels through an unknown mechanism. Other UTR MPRA adaptations have included measuring the impact of APA variants in 3’ UTRs and studying how 5’ UTR sequences affect ribosomal loading [55,56].
Noncoding RNA Variants
Unlike coding genes which get translated into protein, noncoding RNA (ncRNA) genes are a special class of genes which, as the name implies, do not get translated but rather get transcribed and can carry out their regulatory functions as RNA molecules. There are several types of ncRNAs which carry out a wide array of functions. These include long ncRNAs (lncRNAs) and microRNAs (miRNAs), both of which have been shown to be altered in both Mendelian and complex diseases.
Much like UTRs, ncRNAs carry out diverse, complex functions which are important for understanding the functional impact of variants in these RNA genes. LncRNAs in particular carry out a variety of functions, including acting as genome guides for regulatory factors, decoys for regulatory factors to prevent them from acting on a gene, sponges for miRNAs to block their inhibitory functions, precursors for miRNAs, and scaffolds for chromatin looping [57]. miRNAs work by binding to an mRNA transcript, usually in the 3’ UTR, and inhibiting its translation by inducing mRNA cleavage, speeding up deadenylation, or blocking ribosome-mediated translation [58]. In addition to playing important and previously underappreciated roles in regulating gene expression, ncRNAs are also widely expressed and often located in regions previously thought to be “junk”. It has been estimated that lncRNAs, for example, make up 68% of expressed genes, with disease-associated SNPs overlapping with 7% of lncRNAs [59].
Given that over 80% of GWAS-discovered variants are predicted to lie in intronic and other noncoding regions, it is important to functionally annotate ncRNA variants to understand their role in complex disease (Figure 2d, e). However, efforts to functionally annotate disease-associated ncRNA variants have lagged in comparison to other variant classes thus far. Most of the work looking at GWAS-identified ncRNA variants has studied variants which lie in ncRNA gene promoters and enhancers, affecting ncRNA expression levels, rather than the RNA sequence itself. For example, Wu et al. (2020), co-localized GWAS for steroid-associated phenotypes with eQTLs across several tissues to validate the role of lincNORS, a long intergenic ncRNA (lincRNA), in sterol homeostasis [60]. They identified SNPs in the promoter of the lincRNA which are associated with steroid-linked traits (e.g. age of menarche, age of first facial hair, and others), and used a luciferase reporter assay to validate the effect of the SNPs on promoter activity. GWAS SNPs affecting the expression of a lncRNA, often by interrupting TF binding, have been identified and characterized in several other diseases, including colorectal cancer [61], prostate cancer [62], and renal cell carcinoma [63]. While these SNPs do not impact lncRNA sequence, they are nonetheless useful for identifying disease-relevant lncRNAs and the pathways they regulate. By contrast, others have identified SNPs in lncRNAs genes which impact the sequence of the RNA and thus its function. Feng et al. (2020), for example, identified a lncRNA SNP in a GWAS susceptibility locus associated with non-small cell lung cancer and found that overexpression of one allele caused reduced proliferation of cancer cells [64]. To understand the mechanism of action of the SNP, they used lncRNASNP, a bioinformatic tool which predicts a SNP’s ability to create/disrupt an miRNA binding site, and they find that the SNP in the lncRNA LOC146880 lies in a putative miRNA binding site for miR-539–5p. They validate this finding with a luciferase assay, showing differential miRNA binding to the reference and alternate allele. Further mechanistic studies reveal that the low risk allele results in increased miR-539–5p binding to LOC146880, preventing phosphorylation of ENO1, which is important for activating downstream cancer-linked genes.
The landscape of miRNA research has similarly identified SNPs which impact miRNA expression as well as those which impact their function by disrupting binding through mutations in miRNAs themselves or miRNA response elements (primarily 3’ UTRs). As an example of the former, Nikpay et al. (2019) used miRNA-Seq data to identify miRNA-eQTLs (miQTLs) in blood—that is, regulatory eQTLs containing SNPs which explain variations in miRNA levels [65]. These miQTLs were then co-localized with GWAS data for cardiometabolic traits to identify trait-associated SNPs which might act through altered miRNA levels. Larson et al. (2022) similarly focused on diseases impacted by miRNA regulation by performing a transcriptome wide association study to find associations between miRNA expression levels and prostate cancer, and integrating this with GWAS data to predict prostate cancer-associated SNPs which regulate levels of top candidate miRNAs [66]. Others have identified SNPs which affect not miRNA levels, but the processing, stability, and function of the miRNA itself [67–69]. Ghanbari et al. (2017) identified SNPs associated with open-angle glaucoma which lie in miRNA genes and used the Vienna RNAfold algorithm to predict which SNPs affect the stem-loop structure of precursor miRNAs (pre-miRNAs) [67,70]. Using this method, they identified two glaucoma-associated variants, one which was predicted to affect miRNA secondary structure and another predicted to reduce the interaction between the miRNA and its target genes. The authors then used TargetScan and miRDB, two computational tools, to predict target genes of the candidate miRNAs and performed luciferase assays (with target 3’ UTR sequences linked to the reporter) to validate the miRNA SNP effects [67,71,72]. Mens et al. (2020) used a multi-omics approach to find miRNAs associated with disease [68]. They used GWAS data to find variants in miRNA genes associated with cardiometabolic traits and then layered this with miRNA CpG site data (epigenomic) and miRNA expression data (transcriptomic) to nominate candidate miRNAs altered in cardiometabolic traits.
As with other noncoding variant classes, some of the key challenges in functionally annotating SNPs affecting noncoding RNAs includes understanding the mechanism of the impact, identifying the target gene(s), and validating the effect of the SNP on the gene and proposed disease-relevant pathway. While some novel associations between noncoding RNAs and disease have been identified, the limited depth of research in this area, and particularly the limited use of high-throughput functional screens to annotate SNPs and noncoding RNA genes, suggests that there is much to still be discovered connecting noncoding RNA variants with complex disease.
Copy Number Variants
While a majority of GWA studies have focused on identifying single base pair SNPs associated with disease, there is another class of variants which have been shown to be linked to disease: structural variants. Structural variants are those which, rather than a single nucleotide change, involve larger alterations to the structure of a chromosome. Copy number variants (CNVs) are a type of structural variants which in particular make up a large proportion of genomic variants and have been the target of GWAS as they are thought to play an important role in complex disease [73,74]. CNVs are duplications, deletions, and other alterations of genomic segments between 50 bases and 5 megabases [75]. There are several reasons for studying CNV associations with complex disease alongside SNPs. For example, while there are more individual SNPs than CNVs in the genome, CNVs account for 1.2% of genomic variation from the human reference sequence, whereas SNPs account for 0.1%, highlighting the potentially important role of CNVs in driving human phenotypic variation in traits and disease [76–78]. Additionally, while many GWAS-identified CNV loci overlap with SNP loci for the same trait, CNV-specific GWAS has also identified several novel disease susceptibility loci [73,77,79]. Furthermore, in susceptibility loci that contain both a SNP and a CNV for a trait, identifying and functionally annotating both is important as the presence of a CNV can distort the haplotype and therefore the significance of a co-localized SNP [80].
CNV GWAS have identified loci containing these genomic alterations which associate with complex traits and disease, including autoimmune diseases [73,81], schizophrenia and other psychiatric illnesses [81,82], hematological traits [79], anthropometric traits [79,83], cardiometabolic traits [73,81], and others. As previously noted, the loci identified are often co-localized with SNP loci identified in previous trait-matched GWAS to support the role of the CNV locus in the phenotype. Of particular interest, however, are those studies which identify novel loci associated with the trait, highlighting the unique contribution of CNVs to complex traits and disease. For example, Macé et al. (2017) discovered five novel associations between CNV loci and anthropometric traits, while Marshall et al. (2017) identified novel CNV loci associated with schizophrenia. Marshall et al. (2017) took a further look at these loci, performing pathway analysis of genes located in the loci, and found an enrichment of genes associated with synaptic function and mouse neurobehavioral phenotypes. Nonetheless, critically missing from studies of disease-associated CNVs thus far has been extensive characterization and annotation of the CNV locus. Given that CNVs are structural alterations which can impact multiple genes and other regions with potential regulatory function, an important next step, as has been done with SNPs, is identifying the causal genomic segments driving the phenotype. While these efforts have been limited, Collins et al. (2022) have begun to make headway on this goal, using associations between rare CNVs and several disorders to construct a machine learning model which predicts the dosage sensitivity (i.e. haploinsufficiency or triplosensitivity) of individual genes in connection to disease [84]. Hujoel et al. (2022) similarly developed a computational approach, HI-CNV, to advance our understanding of how rare CNVs affect complex traits by taking advantage of haplotype sharing in human biobanks [85]. Future work will require making uses of advances in in silico and experimental tools to further and fully characterize CNV loci associated with traits and disease.
Understanding the tools
Efforts to functionally annotate genomic variants connected to disease through GWAS have led to the development of a variety of tools to predict variant effects. These have included computational in silico prediction, high-throughput variant screens, and, for noncoding variants in particular, putative target gene prediction.
Computational/In Silico Tools
Due to their minimal upfront costs, computational tools have long been used to perform in silico functional annotation and characterization of genomic variants (Table 1). A plethora of tools to predict the functional effect of missense and nonsynonymous variants in the coding sequence of a gene have been developed, including PredictSNP [8], Ensembl Variant Effect Predictor [14], SIFT (Sorting Tolerant from Intolerant) [86], PolyPhen-2 (Polymorphism Phenotyping v2) [87], Protein Variation Effect Analyzer (PROVEAN) [88], SNPs&GO [89], and others [90–96]. Other tools have gone further by performing structural analysis of the effect of SNPs, such as surface accessibility with NetSurfP-2.0 [97], secondary structure with SOPMA [98], protein stability with I-mutant 3.0 [99], and others [100–102]. These tools have aided in the characterization of coding SNPs in various disease-associated genes, including the androgen receptor gene [103], CYP2U1 gene [104], RETN gene [10], PPT1 gene [105], BRCA2 (in male breast cancer) [106], and PNMT gene (in neurodegenerative disorders) [107]. Structural characterization of coding SNPs in the BRCA2 gene associated with male breast cancer even led to the discovery of a therapeutic hit with potential anticancer properties [106].
Table 1.
Computational/in silico tools for annotation and characterization of genomic variants
| Variant Type | Tool | Description | Citation |
|---|---|---|---|
| Coding | Ensembl | Predicting effect of variant/amino acid substitution | [14] |
| Meta-SNP | [92] | ||
| PANTHER | [93] | ||
| PolyPhen-2 | [87] | ||
| PredictSNP 2.0 | [8,90] | ||
| PROVEAN | [88] | ||
| SIFT | [86] | ||
| SNPs&GO | [89] | ||
| SNPsnap | [94] | ||
| SuSPect | [96] | ||
| UMD-Predictor | [95] | ||
| NetSurf2.0 | Surface accessibility effects | [97] | |
| SOPMA | Secondary structure effects | [98] | |
| I-mutant-3.0 | Protein stability effects | [99] | |
| ConSurf | Evolutionary conservation | [100] | |
| HOPE | 3D structure effects | [101] | |
| SWISS-MODEL | Homology modeling | [102] | |
| Promoter/Enhancer | MotifBreakR | TFBS disruption | [117] |
| RegulomeDB | Regulatory DNA variant prediction | [108] | |
| Basenji | Deep learning | [123] | |
| DeepSea | [121] | ||
| COLOC | Colocalization | [124] | |
| eCaviar | [25] | ||
| FUSION | [127] | ||
| MetaXcan | [125,126] | ||
| Splicing | GeneSplicer | Splicing site and variant effect prediction | [131] |
| Human Splicing Finder | [130] | ||
| NetGene2 | [132,133] | ||
| RegSNPs-intron | [128] | ||
| SpliceAI | [129] | ||
| 5’/3’ UTRs | ExUTR | 3’ UTR sequence prediction | [111] |
| RegulomeDB | UTR variant effect prediction | [108] | |
| UTRannotator | 5’ UTR variant effect prediction | [112] | |
| UTRscan | UTR sequence prediction | [109,110] | |
| miRNA/lncRNA | CPSS 2.0 | miRNA and lncRNA sequence prediction | [116] |
| MicroSNiPer | miRNA variant effect prediction | [115] | |
| miR2GO | [114] | ||
| SubmiRine | [113] |
Computational tools have also been developed which can predict the effects of a wide array of noncoding variants associated with disease. For example, SNPs which are predicted to lie in the 5’ or 3’ UTR of a gene can be further analyzed for disruption of a TF binding site (TFBS) or miRNA seeding region. RegulomeDB is one tool which can perform such functional analysis, integrating data from public datasets like ENCODE with information on DNase hypersensitivity site (chromatin accessibility), ChIP-Seq and TF binding sites, and other DNA regulatory elements [108]. UTRScan is a tool which similarly predicts the functional effects of SNPs, with a specific focus on 5’ and 3’ UTR elements, and was used by Elkhattabi et al. (2019) to identify SNPs associated with obesity and insulin resistance which disrupt the polyadenylation signal in the 3’ UTR of the RETN gene [10,109,110]. Additional tools for identifying UTRs and their variants include ExUTR, which predicts predicts 3’ UTR sequences from RNA-Seq data, and UTRannotator, a 5’ UTR variant effect prediction tool [111,112]. Furthermore, several tools for miRNA variant effect prediction have been developed, including SubmiRine [113], miR2GO [114], and others [115,116]. However, tools for predicting the effects of variants in lncRNAs have lagged behind and present an opportunity for future computational tool development. Other types of noncoding SNPs, such as those predicted to lie in promoter and enhancer regions, can be similarly computationally annotated using tools which predict disruption of TF binding sites or other epigenetic alterations. For example, motifbreakR is an R/bioconductor package which uses TF binding motif information from public datasets to predict whether a variant will break the motif [117]. It has proved useful in predicting the TF binding disruption of SNPs in regulatory elements associated with Parkinson’s disease [118], colorectal cancer [119], ovarian cancer [120], and several other diseases. However, a key limitation of motifbreakR is that it requires a priori knowledge of TF binding motif sequences, thus limiting the analysis to motifs which have already been discovered.
Alternatively, deep learning tools like DeepSEA and Basenji can be trained on tissue-specific regulatory features and used to predict the epigenetic state of a novel DNA sequence and the chromatin effects of variants [121–123]. Other tools have been identified to co-localize GWAS SNPs with tissue-specific eQTLs to nominate disease-associated genes, including COLOC [124], eCaviar [25], MetaXcan [125,126], and FUSION [127]. Computational tools have also been developed which predict how SNPs can affect splicing. For example, Lin et al. (2019) developed RegSNPs-intron, a tool for predicting the effect of intronic SNPs on splicing and protein structure [128]. Jaganathan et al. (2019) developed SpliceAI, a tool for predicting splice junctions (particularly cryptic splice sites) from a given pre-mRNA sequence, allowing it to effectively predict how variants affect splicing [129]. Other splice site prediction computational tools include Human Splicing Factor [130], GeneSplicer [131], NetGene2 [132,133], and MLCsplice [134]. One crucial limitation of in silico prediction tools is the quality of the data from which the tools are built, as they are often developed from public epigenomic and transcriptomic datasets like ENCODE with wide varieties of data quality, thus limiting their predictive power. Improved standards for data quality, such as high signal-to-noise ratio and high replicate correlation, will be important moving forward to address this issue [123]. Furthermore, given the known cell type-specificity of genomic regulation, it will be important to collect more single-cell sequencing data for better cell type resolution, and thus better cell type-specific regulation and variant prediction.
High Throughput Functional Reporter Assays
While computational tools are useful for nominating the most likely causal variants, the function of a SNP must ultimately be experimentally validated to help make accurate predictions about the SNP’s effect in human disease. Given the high volume of candidate SNPs which emerge from GWAS, high throughput screening assays have been developed by researchers to screen thousands of disease-associated genomic variants for some functional effect. Such assays have been particularly employed in the study of variants in noncoding regions, such as promoters, enhancers, introns, and UTRs, with some downstream reporter serving as a readout for differential activity (Figure 3a).
Figure 3. Overview of high-throughput screening tools used to test variant function.
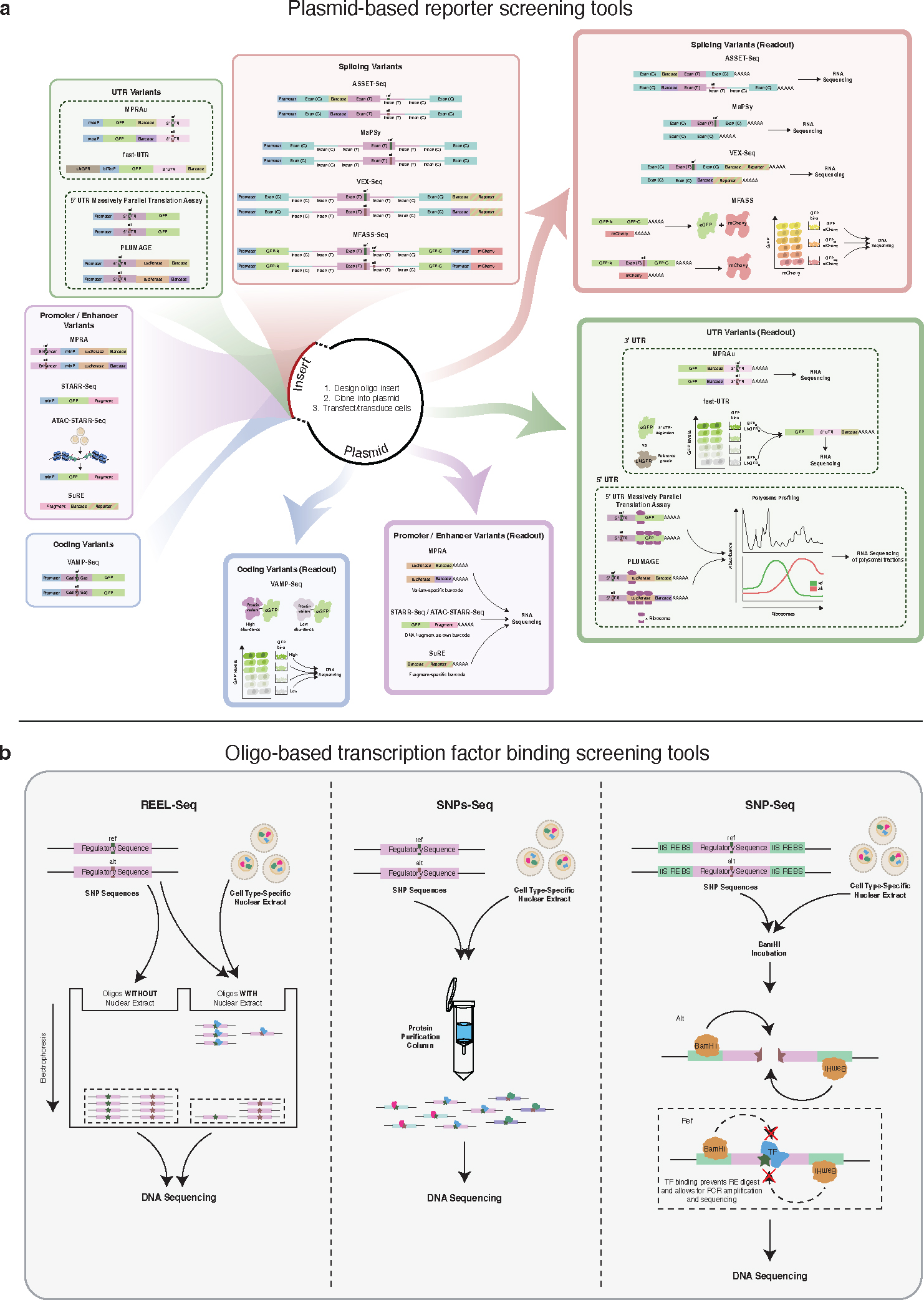
(a) Plasmid-based reporter screening tools have been adapted for various types of variants, with readouts specifically tailored to the variant effects on function. This includes MPRA (enhancer), MPRAu (3’ UTR), VAMP-Seq (coding), and ASSET-Seq (splicing). T = test; C = constant. (b) Oligo-based transcription factor binding screening tools used to test ability of variant to disrupt TF binding. REEL-Seq relies on differential migration oligos bound vs unbound to TF. SNPs-Seq uses protein purification columns to enrich for oligo SNPs bound to TFs. SNP-Seq uses restriction enzyme digest to test the ability of a variant to disrupt TF binding.
One such assay is the massively parallel reporter assay (MPRA), first developed to study the activity of transcriptional regulatory elements by having the candidate sequences control a minimal promoter which drives expression of a reporter and element-specific tag [135]. Because of the use of the minimal promoter, MPRA is most useful for identifying sequences which increase baseline gene expression, rather than those which decrease it. MPRA was later adapted to study how variants in enhancer regions affect regulatory activity by introducing reference and alternative alleles into the enhancer sequences [136]. This approach has been used widely to screen GWAS-identified disease-associated SNPs for regulatory function, covering diseases like neurodegenerative diseases [33], type 2 diabetes [35], melanoma [36], COPD [37], and several others. Integration of MPRA profiles with eQTL data through colocalization allows predicted causal variants to be linked to gene expression, as shown by Abell et al. (2022) [137]. Kircher et al. (2019) adapted MPRA in a different manner, by performing saturation mutagenesis of 20 disease-associated promoters and enhancers and cloning the tagged sequences into a reporter construct, such that the different tested sequences drove expression of a luciferase reporter and their unique tags [138]. This adapted MPRA protocol is particularly useful for investigating enhancers and promoters commonly associated with disease, as it generates single nucleotide resolution information about the effect of variants on regulatory function and can thus be integrated with GWAS SNP data to nominate functional SNPs.
Other parallel reporter assays have been developed and adapted to study the functional effects of SNPs in regulatory elements. STARR-Seq is a reporter assay where, rather than driving expression of a barcoded reporter from upstream the promoter, the enhancer is located downstream the promoter and adjacent to the reporter open reading frame, such that the enhancer transcribes itself and the strength of an enhancer is reflected by its transcript abundance [139]. More recently, STARR-Seq has been adapted into the method HiDRA, a combination of experimental regulatory element testing (ATAC-STARR-Seq) and computational modeling to identify driver regulatory nucleotides (SHARPR-RE) [140]. ATAC-STARR-Seq involves selecting and amplifying accessible regions of fragmented whole genome and cloning these fragments (enriched for regulatory elements) into a STARR-Seq plasmid construct, such that each DNA fragment serves as its own barcode. Overlap between various fragments allowed for the development of SHARPR-RE, a machine learning model which can predict regulatory driver nucleotides and look for overlap with disease-associated genetic variants. Adapting HiDRA to cell types of interest could lead to discovery of unique driver nucleotides located in key regulatory elements which can be colocalized with GWAS SNP data to nominate most likely causal variants. Another reporter technology, SuRE (survey of regulatory elements) similarly relies on fragmented whole genome which gets inserted into a promoter-less barcoded plasmid, thus testing the ability of the fragment to initiate transcription [141]. van Arensbergen et al. (2019) performed SuRE with genomes from four individuals of different ethnic backgrounds to effectively test 5.9 million SNPs for their impact on transcription, which were then linked to cell type-relevant GWAS traits and diseases [142].
While the aforementioned parallel reporter assays test the downstream effect of a regulatory SNP–that is, increased or decreased expression–another class of high throughput assays tests the upstream event of differential TF binding (Figure 3b). Several assays have been developed which involve incubation of synthesized oligos containing regulatory elements and their SNPs with nuclear extract from a disease-relevant cell type of interest and then assaying SNPs for differential TF binding. REEL-Seq uses electrophoresis mobility shift assay to isolate and sequence oligos not bound to TFs and identify SNPs affecting TF binding affinity [143]. SNPs-Seq, by contrast, passes incubated oligos through a protein purification column to isolate and sequence oligos that are bound to TFs to identify SNPs [144]. SNP-Seq, another method, takes advantage of sequence-independent cleavage by Type IIS restriction enzymes (REs) and situates the regulatory element containing the SNP at the cleavage site of the RE, such that after incubation with nuclear extract, SNPs that bind to a regulatory protein are protected from RE cleavage and can be PCR amplified and analyzed [145]. Another oligo-based TF binding tool is SNP-SELEX, a high-throughput plate-based assay which combines SNP-containing oligos with recombinantly expressed TFs to screen millions of unique TF-DNA combinations to analyze TF binding specificity and how disease-linked SNPs disrupt TF affinity for DNA [146]. A recently developed non-high throughout assay, PROBER, complements these approaches by providing an unbiased approach to identify TFs and associated DNA-associated proteins that differentially bind variants of interest in living cells [147]. The PROBER assay is applicable to diverse cell types and enables quantitation of regulatory SNPs that act as TF binding quantitative trait loci (bQTLs).
Parallel reporter assays have also been adapted for screening UTRs and their variants. Griesemer et al. (2021) developed the massively parallel reporter assay for 3’ UTRs (MPRAu), where a moderately strong promoter controls the expression of a reporter gene and a library of 3’ UTRs [54]. Using MPRAu, they screened thousands of GWAS 3’ UTR variants and identified several potentially functional causal variants across various traits and diseases, including prostate cancer, triglycerides, and coffee consumption. Zhao et al. (2014) developed fast-UTR, another parallel reporter assay which uses a bidirectional promoter to drive constant expression of a reference protein in one direction and 3’ UTR-dependent expression of eGFP in the other direction [148]. Cells are sorted via flow cytometry into GFP-high and GFP-low populations to identify UTR segments that increase or decrease reporter production. While not originally developed to screen UTR variants, fast-UTR could easily be adapted to screen SNP or indel variants in 3’ UTR sequences of interest. Sample et al. (2019) adapted MPRA for screening 5’ UTR variants, designing a vector where a strong promoter drove expression of a 5’ UTR and eGFP reporter, which was then transfected into cells which subsequently underwent polysome profiling to identify 5’ UTR variants that affect ribosomal loading [56]. Lim et al. (2021) took a similar approach when developing PLUMAGE, a high-throughput 5’ UTR screening tool where a promoter drives expression of variable 5’ UTR sequences and a luciferase reporter gene [149].
Assays which test the effects of variants on splicing have also been developed, all of which in some way test for the inclusion or exclusion of an exon of interest. ASSET-Seq was developed to test whether intronic SNPs of interest affect splicing [128]. The test exon and intron (containing the SNP) are inserted into a plasmid between two universal exons downstream a unique barcode and the resulting transcripts from transfected cells are sequenced to determine SNP effects on splicing variant abundance. MaPSy and VEX-Seq are two alternative assays which, by contrast, test exonic SNPs and, similarly, involve inserting the test exon with mutations of interest between two universal exons and comparing variant abundance [150,151]. MFASS is another high-throughput splicing assay where the test exon and intron are inserted within the GFP coding sequence, such that skipping of the middle exon results in reconstitution of GFP expression and cells can be sorted into GFP-high and GFP-low populations to identify SNPs which drive exon skipping and inclusion, respectively [152]. Fewer massively parallel reporter assays have been developed for screening coding variants, but one such assay is VAMP-Seq. VAMP-Seq is a fluorescence-reporter system developed to assay the effects of thousands of coding variants on protein stability by linking the coding sequence to a GFP reporter, applied to study the pathogenicity of poorly characterized PTEN and TPMT variants [153].
One important limitation of these parallel reporter assays is that they separate the genetic event being tested (splicing, transcription regulation, etc.) from its native context, thus limiting the biological validity of the findings. To truly validate the effect of a coding or noncoding variant on biological pathways, the genomic element and its variant must be assessed in its native genomic context.
Genomic Element Characterization
An important aspect of functional annotation of a GWAS-identified variant is characterizing the genomic element affected by variant, including coding genes, noncoding RNA genes, and regulatory elements, in their native genomic context to predict the disease-relevant biological effects of the variant. For coding and noncoding genes, this entails using tools which can perturb expression and function of the genes to determine downstream effects and identify associated pathways. For regulatory elements, particularly those like enhancers and distal promoters which act at a distance from the target gene, it is important to first identify the genes linked to the regulatory elements of interest in order to accurately predict the effect of the variant in disease. Importantly, it is critical to not only assess the element in its native genomic context, but also in a system reflective of the native biological context and disease of interest, as the activity of genes and proteins, and their regulation, is often highly dependent on cell/tissue type and state.
The tool most commonly employed to study the function of a candidate risk gene is CRISPR-Cas9, a gene editing technology that uses a Cas protein coupled with a guide RNA (gRNA) to introduce double-stranded breaks into a sequence of interest, effectively blocking gene activity [154] (Figure 4a). Single-cell methods like Perturb-Seq [155,156] and CROP-Seq [157] have been developed which allow for screening of a large pool of gRNAs, coupled with single-cell RNA-Seq to determine the effects of individual gRNAs on RNA expression, thus linking genes of interest with potential biological pathways. While most of these single-cell CRISPR studies are performed in cell culture, Jin et al. (2020) developed this technology further by performing Perturb-Seq in vivo in the developing mouse neocortex to screen autism risk genes in their biological context [158]. Alternative read-outs to RNA expression can be used–for example, Rubin et al. (2019) developed Perturb-ATAC, coupling CRISPR screening with single-cell ATAC-Seq, which they used to screen TFs, chromatin modifying factors, and noncoding RNAs for effects on chromatin accessibility [159]. CRISPR-Cas9 has also been modified into tools like CRISPR interference (CRISPRi) and CRISPRa activation (CRISPRa), which provide alternative ways to test the function of a gene without generating DNA breaks (Figure 4a). CRISPRi uses a catalytically dead Cas9 (dCas9) which is targeted to the sequence of interest using a gRNA and blocks transcription [160]. CRISPRa similarly uses dCas9, but it is fused to transcriptional activators such that, when targeted to the gene of interest, it can activate transcription [161]. Both CRISPRi and CRISPRa have been used to screen large gene sets for the consequences of inhibition and activation, respectively, and are useful tools for evaluating the function of genes nominated by GWAS [155,162–164]. Other tools for silencing gene expression which have been used to screen GWAS-identified genes include short hairpin RNAs (shRNAs), used by Nandakumar et al. (2019) to study genes linked to blood traits, and small interfering RNAs (siRNAs), used by Chapuis et al. (2017) to screen genes in 19 Alzheimer’s susceptibility loci [15,165] (Figure 4a). Both are forms of RNA interference (RNAi) which, unlike CRISPR tools which target DNA, bind and target mRNA for degradation and can be designed to target a gene transcript of interest.
Figure 4. Overview of tools to characterize diverse types of genomic loci.
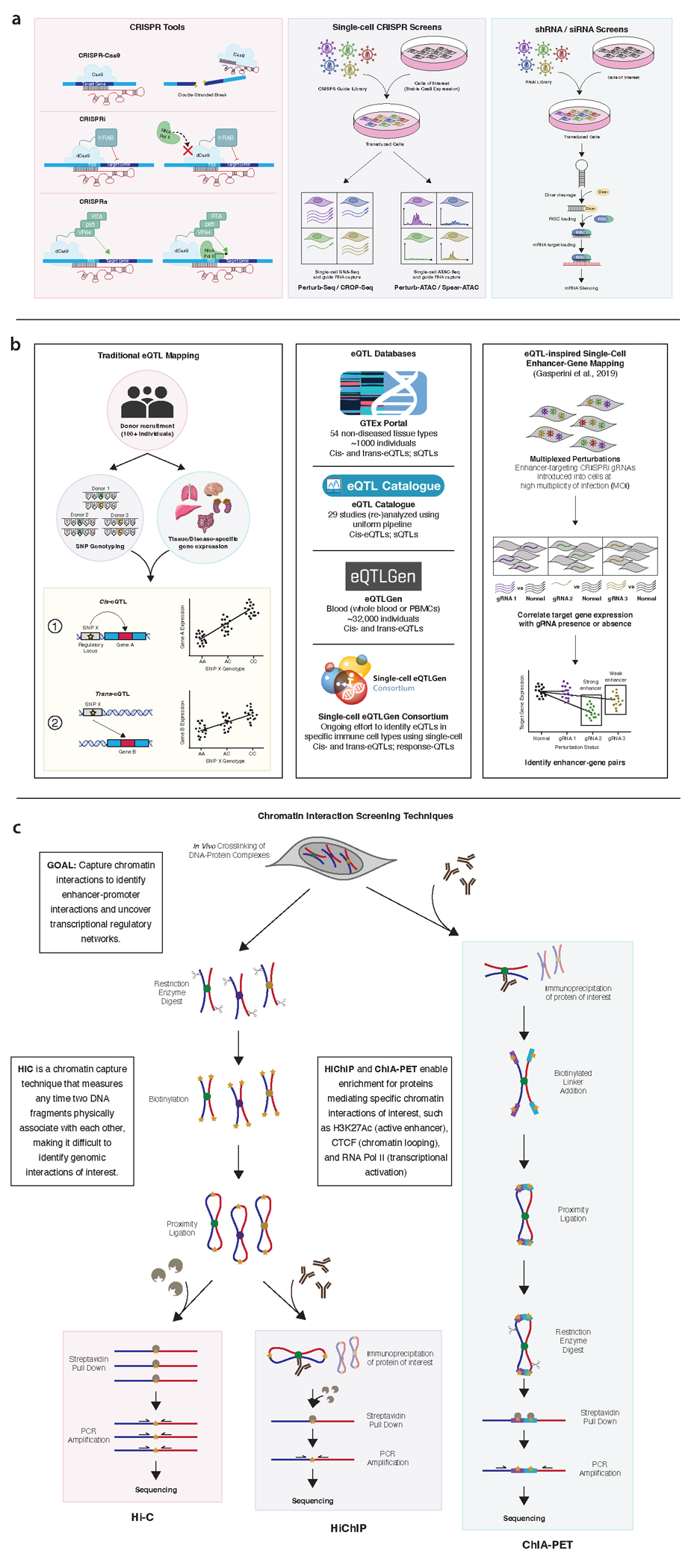
(a) CRISPR tools, including CRISPRi and CRISPRa, have been used to characterize coding and noncoding regions. CRISPR screens like Perturb-Seq can be leveraged to study the effect of a CRISPR perturbation on gene expression in a high-throughput, single-cell manner. shRNA and siRNA screens can similarly be used to test for the effect of gene silencing, this time at the RNA level, on a phenotype. (b) Colocalization of GWAS susceptibility loci with eQTLs is a common strategy for identifying target genes for non-coding variants/loci. Traditional eQTL mapping uses hundreds of individuals to identify associations between a variant and a change in gene expression. eQTL databases include eQTLGen and GTEx. Single-cell eQTL mapping using high MOI CRISPR perturbation can bypass the need for hundreds of human samples. (c) Hi-C, HiChIP, and ChIA-PET are chromatin capture techniques used to identify physical contacts between DNA to predict enhancer-promoter interactions. HiChIP and ChIA-PET allow for capture of specific proteins to enrich for enhancer-promoter loops.
As has already been discussed, a critical component of the functional annotation of SNPs in regulatory regions involves predicting the genes that these regions are associated with, as this will determine the downstream disease-relevant impacts of the variant. While regulatory elements are often presumed to act on the nearest gene, many elements, such as enhancers or distal promoters, can act on genes from a distance and can be located up to 1 Mbp from their target genes [166]. Furthermore, target genes are often context-specific and so a great challenge in this work is identifying the precise regulatory networks that are active in the tissue and cell state of interest [167]. Nonetheless, a plethora of tools and analysis techniques have been developed to link regulatory elements with genes. One commonly used tool which we have already discussed is eQTL mapping, which identifies genomic loci that explain a portion of variation in mRNAs, based on associations between SNPs in these loci and gene expression differences [168,169] (Figure 4b). These eQTLs can be co-localized with GWAS susceptibility loci to identify genes whose expression is most likely affected by loci [25,170]. Recent eQTL mapping work has shifted to single-cell eQTL mapping to uncover cell type-specific gene regulatory networks lost during bulk RNA-Seq [171–173]. Importantly, eQTL mapping requires hundreds of samples at a minimum from different donors in order to identify SNPs and have the power necessary to detect eQTLs [174]. Thus, rather than performing their own eQTL analyses, many researchers often use databases like eQTLGen and GTEx which have identified SNP eQTLs in blood (eQTLGen) and other diverse tissue types (GTEx) [175–177] (Figure 4b). Other forms of QTLs, such as sQTLs, miQTLs, and pQTLs, are also cataloged in databases such as these to capture the diverse effects of variants. However, the limitation of these databases is that the tissue states captured in their data may not be trait- or disease-concordant. One alternative approach has been to adapt the eQTL approach to a single cell level. Gasperini et al. (2019) developed an eQTL-inspired framework for identifying enhancer-gene pairs in a single cell line by using multiplexed CRISPRi enhancer perturbations coupled with single cell RNA-Seq [178] (Figure 4b). Rather than requiring hundreds or thousands of people with tissue-level transcriptomes and SNP variants between them, this approach only required thousands of cells with single cell-level transcriptomes and multiple CRISPRi perturbations (i.e. variants) per cell. It is an approach that can be adapted to identifying disease context-specific enhancer-gene pairs for GWAS-identified regulatory loci.
Alternative non-eQTL tools have also been developed which can match regulatory regions with putative target genes (Figure 4c). Hi-C is a chromatin capture technique which involves crosslinking two DNA fragments that are physically associated, ligating them together, and sequencing to identify interacting genomic fragments [179]. Hi-ChIP is a derivative of Hi-C which pairs the technique with chromatin immunoprecipitation (ChIP) to enrich for genomic interactions occurring near a regulatory protein of interest [180]. H3K27Ac HiChIP in particular has been used to capture enhancer-promoter loops to identify genes linked to enhancers of interest, as H3K27Ac is an epigenetic marker associated with active enhancers [181]. This technique is similar to ChIA-PET, another protein-centered chromatin capture technique which can enrich for CTCF- and cohesin-associated chromatin loops, although HiChIP has been shown to require much lower cell input with greater read output [181–183]. These methods have been able to identify links between regulatory regions and genes that are not revealed by eQTL mapping [184,185]. NEAT-Seq is an alternative protein-centered approach developed by Chen et al. (2022) that entails simultaneous profiling of enhancer-associated TFs (using antibody-derived tags), open chromatin peaks (ATAC-Seq) and gene expression (RNA-Seq) to interrogate gene regulatory mechanisms [186]. The authors showed that the technique is capable of linking SNPs in TF motifs to putative target gene expression, highlighting its potential use for GWAS target gene nomination. In a reversal of the previously outlined methods, if a putative target gene of interest has been identified, CRISPRi-FlowFISH and activity-by-contact (ABC) modeling has been shown to accurately predict enhancer-gene connections [187]. Developed by Fulco et al. (2019), CRISPRi-FlowFISH entails performing CRISPRi with gRNAs against enhancer regions of interest, using RNA fluorescence in situ hybridization (FISH) to label single cells based on expression of the gene of interest, sorting the labeled cells into bins based on RNA abundance using fluorescence-activated cell sorting (FACS), and sequencing each bin to identify guides enriched in low or high target gene expressing populations. Naturally, a key limitation of this technique is that it requires a priori knowledge on the putative target gene to link it to the appropriate enhancers.
Variant Effect Characterization
By far the most rigorous method for functionally annotating a GWAS-identified variant (SNP, insertion, deletion, CNV, etc.) involves introducing that precise variant into its native genomic context and studying the functional effects in a relevant cell and tissue context. This approach is particularly powerful when the variant is compared to its non-disease associated counterpart allele in an otherwise genetically identical isogenic context which allows isolation of any impacts to the variant in question. This approach is laborious but recent advances in the efficiency of gene editing have facilitated its application to human organoid tissue to study acquired somatic SNVs in collagen genes, which established a role for common COL11A1 gene mutations as functionally active promoters of neoplastic invasion [188]. While the tools described up to this point can all predict the effect of a variant, only such precise gene editing with isogenic controls can definitively determine the disease-relevant biological perturbations resulting from a genetic variant and thus this approach represents the gold standard for disease-linked variant characterization.
While a wide array genome editing tools has historically been used to introduce genetic variation in situ, including zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), CRISPR-based tools have become the favorite gene editing tool and several iterations of this technology have been developed to improve the accuracy of gene editing (Figure 5). CRISPR-Cas9 was first adapted for precise genome editing by taking advantage of the homology-directed repair (HDR) pathway by introducing a DNA template (containing the mutation) into cells along with the CRISPR-Cas9 construct. This DNA sequence can serve as a repair template after CRISPR-mediated cutting to introduce the mutation into the cell’s genome [189,190]. CRISPR HDR has been used to study the effects of both coding and noncoding variants, as well as CNVs, in vitro and in vivo [37,191–193]. There are, however, several drawbacks to CRISPR HDR which limit its use, particularly in a more high throughput manner. When the Cas9 protein generates double-stranded breaks (DSBs), both HDR and non-homologous end-joining (NHEJ) pathways compete to repair the breaks, resulting in a high frequency of insertions and deletions (indels) near the target site and limiting the precision of the edits [189]. Furthermore, HDR is restricted to G2 and S phases of the cell cycle and it is thus not efficient in non-dividing cells [194]. Alternatives to CRISPR HDR have been developed which aim to improve the efficiency of genetic editing.
Figure 5. CRISPR-based precision gene editing tools.
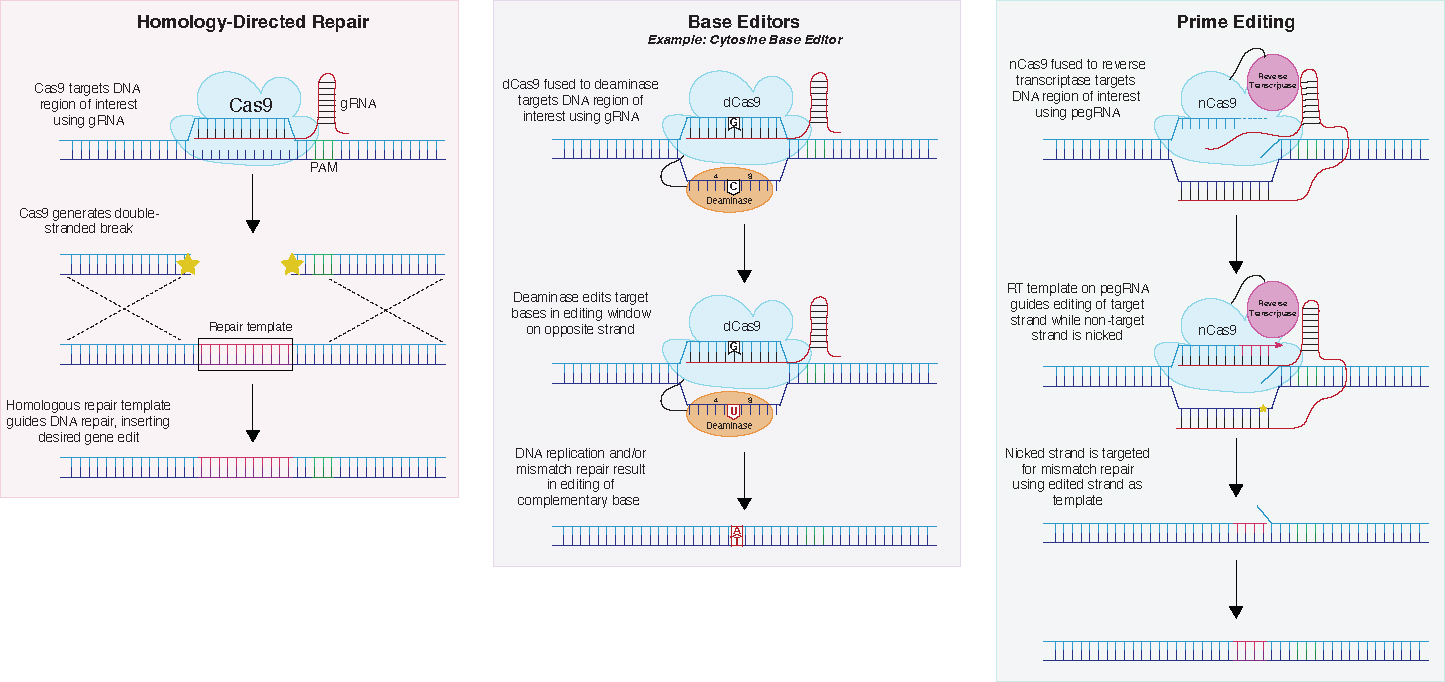
Homology-directed repair (HDR), base editing, and prime editing are all tools for introducing a precise genetic edit of a variant (SNP, indel, or CNV) at its native genomic context. HDR involves the generation of double-stranded breaks using CRISPR-Cas9, followed by homology-directed repair using a supplied repair template with homology to the DNA region of interest. Base editors use a dCas9 fused to a cytidine deaminase, adenosine deaminase, or uracil DNA glycosylase which can introduce a single base change. Prime editing uses nCas9 and reverse transcriptase to insert a precise edit included in the pegRNA template.
One such alternative is base editors, which use dCas9 and a gRNA fused to a cytidine deaminase (C to T), an adenosine deaminase (A to G), or a uracil DNA glycosylase (C to G) to generate the appropriate edit to the bases in its 5 bp activity window [195–197]. Unlike CRISPR HDR, base editing does not generate DSBs and thus indels are not found in edited cells. However, like any technology, base editors have limitations. If, for example, there are two of the same target base within the activity window, both will get edited, generating unwanted bystander edits [198]. Additionally, base editors can sometimes generate unwanted edits even outside the activity window. Furthermore, the scope of edits is limited to those for which base editors have been developed, with about 30% of pathogenic mutations not covered [199]. Nonetheless, base editors have increasingly been used to introduce disease-associated point mutations of interest [196,200–202]. Furthermore, Hanna et al. (2021) recently demonstrated the ability to introduce variants into their endogenous loci in a massively parallel manner using base editors, thus opening the door for high throughput endogenous variant screens [203].
Prime editors are a novel gene editing tool which aim to overcome some of the limitations of HDR and base editors. They use a Cas9 nickase (which generates single-stranded nicks instead of double-stranded breaks) fused to a prime editing gRNA (pegRNA) and a reverse transcriptase [204]. The pegRNA guides the construct to the target sequence, Cas9 nicks one strand, binding and template-guided repair occur, and finally, the non-edited strand is nicked so that it is targeted for repair which will match the edited strand. Prime editors can be used to insert not only point mutations, but also up to 44bp insertions and 80 bp deletions. Like base editors, prime editors do not generate DSBs, resulting in fewer indels. Additionally, while gRNA target sequences for both CRISPR HDR and base editing must be located within approximately 15 nucleotides of protospacer adjacent motif (PAM), a short DNA recognition sequence used by CRISPR proteins, the PAM sequence of prime editors can be over 30 nucleotides from the edit site, thus expanding the scope of targetable regions. Finally, although still a relatively new technology, prime editors have been shown to generate higher efficiency edits than both HDR and base editors, supporting further exploration and optimization of this tool [204,205].
Once genomic edits have been introduced, familiar tools can be used to explore the effect of the variant on gene expression, protein stability, molecular pathways, and other downstream effects. For example, RNA-Seq and ATAC-Seq can be used to explore variant effects on gene expression through mRNA levels and chromatin accessibility at transcription start sites and enhancer loci [39]. ChIP-Seq might be used to explore how the variant impacts TF binding [7]. In vivo editing can demonstrate how the variant affects organism function and biological pathways [206–209]. Understanding the mechanism of the variant’s effect and how that is connected to its associated disease can deliver the ultimate holy grail of GWAS: a variant whose resulting deficit can be corrected to either alleviate or completely eliminate disease.
Concluding Remarks and Future Perspectives
GWAS efforts have broadly expanded our appreciation for the diverse types of genomic elements whose variation can contribute to the pathogenesis of polygenic disease. Far from limited to coding genes, disease-associated variants can lie in promoters, enhancers, long noncoding RNAs, and other genomic elements which can perturb gene expression and molecular pathways through diverse mechanisms. As a result, a diverse array of tools, from computational to high throughput to gene editing, have been developed to identify and interrogate these diverse element-specific mechanisms, extending insight from GWAS-identified susceptibility loci to functional variant to putative causal gene and how its function is related to the trait or disease (Figure 6). The great task of the coming years will be to continue to improve upon and apply these tools to uncover the wide array of variants which in some way contribute to a trait- or disease-causing pathway.
Figure 6. Flowchart showing how to go from GWAS to disease/trait-relevant function.
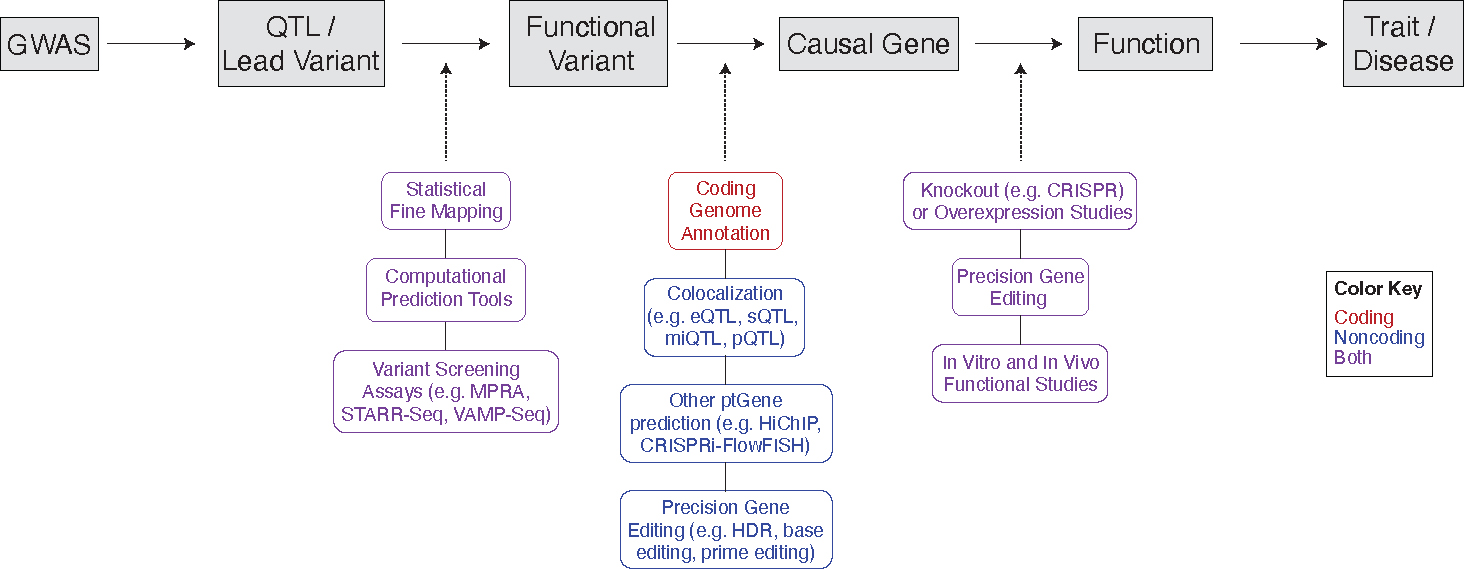
A diverse array of computational, high-throughput screening, gene editing, and other tools can be used to annotate coding and/or noncoding variants. In all cases, the goal is to identify the causal gene driving GWAS disease heritability and characterize the disease- or trait-relevant function to provide better understanding of disease and guide future therapeutics.
There are several feasible next steps in GWAS functional annotation with the tools we currently have, as well as important next directions in tool development (see Outstanding Questions Box). Characterization of noncoding variants, including introns, long noncoding RNAs, UTRs, and others, has lagged behind that of coding variants, despite making up over 90% of GWAS-identified variants. While the effects of noncoding variants are often less clear due to complex regulatory mechanisms, we now have tools which aid us further in untangling this web. As in silico prediction tools play an important role in noncoding variant functional annotation in particular, improved data quality for better modeling is critical. Tools like Basenji, DeepSEA, and other predictive
Outstanding Questions Box [separate doc].
While the importance of coding variation disease is appreciated, how can understanding the genes and pathways linked to disease through noncoding variation help us understand and treat disease?
How can genomic tools, including high-throughput screening modalities, be adapted to study the functional effects of copy number variation in disease?
How well do high-throughput reporter screens like MPRA and STARR-Seq capture the activity of a variant in its native locus?
How can the different enhancer-gene pairing tools, from eQTL mapping to HiChIP, best be integrated to nominate the most likely target gene for an enhancer in the trait- or disease-relevant context?
What depth of specificity is necessary in eQTL mapping (e.g. whole tissue versus single-cell sequencing) to capture true locus/SNP-gene relationships that are relevant to GWAS traits/diseases?
How can precision gene editing tools be optimized for high throughput variant editing tools to allow for massive screening of GWAS variants for function in their native genomic context?
Additionally, as the importance of CNVs in disease heritability is now increasingly appreciated, additional efforts should be directed at understanding the mechanisms of CNV disease-associated perturbations. Furthermore, further tool development will be important to improve the accuracy of GWAS functional annotation results. For example, there is currently no consensus on which enhancer-gene pairing strategy is the most accurate, which makes definitively connecting an enhancer variant to a causal gene and disease pathway difficult. Additionally, the efficiency of precision editing tools must be improved to be more widely applied to screening variants in their endogenous loci, the current gold standard for predicting variant effects. Relatedly, further development of high-throughput variant editing screens would improve our ability to screen large numbers of variants with high confidence. Lastly, as diverse contributors to complex diseases continue to be discovered and functionally characterized, it will be important to explore how to best target the multiple genes and interwoven pathways found to be causal in the disease to develop the most effective treatments, as is the ultimate goal of the GWAS.
References
- 1.Visscher PM et al. (2012) Five Years of GWAS Discovery. Am. J. Hum. Genet. 90, 7–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Visscher PM et al. (2017) 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 101, 5–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buniello A et al. (2019) The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glazier AM et al. (2002) Finding Genes That Underlie Complex Traits. Science 298, 2345–2349 [DOI] [PubMed] [Google Scholar]
- 5.Schaid DJ et al. (2018) From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 19, 491–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim HI et al. (2017) Fine Mapping and Functional Analysis Reveal a Role of SLC22A1 in Acylcarnitine Transport. Am. J. Hum. Genet. 101, 489–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang J et al. (2018) A Rare Missense Variant in TCF7L2 Associates with Colorectal Cancer Risk by Interacting with a GWAS-Identified Regulatory Variant in the MYC Enhancer. Cancer Res. 78, 5164–5172 [DOI] [PubMed] [Google Scholar]
- 8.Bendl J et al. (2014) PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. PLOS Comput. Biol. 10, e1003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doffe F et al. (2021) Identification and functional characterization of new missense SNPs in the coding region of the TP53 gene. Cell Death Differ. 28, 1477–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elkhattabi L et al. (2019) In Silico Analysis of Coding/Noncoding SNPs of Human RETN Gene and Characterization of Their Impact on Resistin Stability and Structure. J. Diabetes Res. 2019, e4951627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gray VE et al. (2018) Quantitative Missense Variant Effect Prediction Using Large-Scale Mutagenesis Data. Cell Syst. 6, 116–124.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ioannidis NM et al. (2016) REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 99, 877–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X et al. (2020) dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 12, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McLaren W et al. (2016) The Ensembl Variant Effect Predictor. Genome Biol. 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chapuis J et al. (2017) Genome-wide, high-content siRNA screening identifies the Alzheimer’s genetic risk factor FERMT2 as a major modulator of APP metabolism. Acta Neuropathol. (Berl.) 133, 955–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu X et al. (2021) Kidney disease genetic risk variants alter lysosomal beta-mannosidase (MANBA) expression and disease severity. Sci. Transl. Med. DOI: 10.1126/scitranslmed.aaz1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Starita LM et al. (2018) A Multiplex Homology-Directed DNA Repair Assay Reveals the Impact of More Than 1,000 BRCA1 Missense Substitution Variants on Protein Function. Am. J. Hum. Genet. 103, 498–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu QM et al. (2017) Novel Thrombotic Function of a Human SNP in STXBP5 Revealed by CRISPR/Cas9 Gene Editing in Mice. Arterioscler. Thromb. Vasc. Biol. 37, 264–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keller MP et al. (2019) Gene loci associated with insulin secretion in islets from nondiabetic mice. J. Clin. Invest. 129, 4419–4432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibson G (2012) Rare and common variants: twenty arguments. Nat. Rev. Genet. 13, 135–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cirulli ET et al. (2020) Genome-wide rare variant analysis for thousands of phenotypes in over 70,000 exomes from two cohorts. Nat. Commun. 11, 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuehl P et al. (2001) Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet. 27, 383–391 [DOI] [PubMed] [Google Scholar]
- 23.Varani L et al. (1999) Structure of tau exon 10 splicing regulatory element RNA and destabilization by mutations of frontotemporal dementia and parkinsonism linked to chromosome 17. Proc. Natl. Acad. Sci. 96, 8229–8234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishii S et al. (2002) Alternative Splicing in the α-Galactosidase A Gene: Increased Exon Inclusion Results in the Fabry Cardiac Phenotype. Am. J. Hum. Genet. 70, 994–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hormozdiari F et al. (2016) Colocalization of GWAS and eQTL Signals Detects Target Genes. Am. J. Hum. Genet. 99, 1245–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brotman SM et al. (2022) Subcutaneous adipose tissue splice quantitative trait loci reveal differences in isoform usage associated with cardiometabolic traits. Am. J. Hum. Genet. 109, 66–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garrido-Martín D et al. (2021) Identification and analysis of splicing quantitative trait loci across multiple tissues in the human genome. Nat. Commun. 12, 727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian J et al. (2019) CancerSplicingQTL: a database for genome-wide identification of splicing QTLs in human cancer. Nucleic Acids Res. 47, D909–D916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y et al. (2020) Regional Variation of Splicing QTLs in Human Brain. Am. J. Hum. Genet. 107, 196–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takata A et al. (2017) Genome-wide identification of splicing QTLs in the human brain and their enrichment among schizophrenia-associated loci. Nat. Commun. 8, 14519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doke T et al. (2021) Genome-wide association studies identify the role of caspase-9 in kidney disease. Sci. Adv. 7, eabi8051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta RM et al. (2017) A Genetic Variant Associated with Five Vascular Diseases Is a Distal Regulator of Endothelin-1 Gene Expression. Cell 170, 522–533.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper YA et al. (2022) Functional regulatory variants implicate distinct transcriptional networks in dementia. Science 377, eabi8654. [DOI] [PubMed] [Google Scholar]
- 34.Myint L et al. (2020) A screen of 1,049 schizophrenia and 30 Alzheimer’s-associated variants for regulatory potential. Am. J. Med. Genet. B Neuropsychiatr. Genet. 183, 61–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khetan S et al. (2021) Functional characterization of T2D-associated SNP effects on baseline and ER stress-responsive β cell transcriptional activation. Nat. Commun. 12, 5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi J et al. (2020) Massively parallel reporter assays of melanoma risk variants identify MX2 as a gene promoting melanoma. Nat. Commun. 11, 2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castaldi PJ et al. (2019) Identification of Functional Variants in the FAM13A Chronic Obstructive Pulmonary Disease Genome-Wide Association Study Locus by Massively Parallel Reporter Assays. Am. J. Respir. Crit. Care Med. 199, 52–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huo Y et al. (2019) Functional genomics reveal gene regulatory mechanisms underlying schizophrenia risk. Nat. Commun. 10, 670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang S et al. (2020) Allele-specific open chromatin in human iPSC neurons elucidates functional disease variants. Science 369, 561–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao B et al. (2021) Common genetic variation influencing human white matter microstructure. Science 372, eabf3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leppek K et al. (2018) Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 19, 158–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mayr C (2019) What Are 3′ UTRs Doing? Cold Spring Harb. Perspect. Biol. 11, a034728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flynn ED et al. (2022) Transcription factor regulation of eQTL activity across individuals and tissues. PLOS Genet. 18, e1009719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levran O et al. (2019) A 3’ UTR SNP rs885863, a cis-eQTL for the circadian gene VIPR2 and lincRNA 689, is associated with opioid addiction. PLOS ONE 14, e0224399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li L et al. (2021) An atlas of alternative polyadenylation quantitative trait loci contributing to complex trait and disease heritability. Nat. Genet. 53, 994–1005 [DOI] [PubMed] [Google Scholar]
- 46.Mariella E et al. (2019) The Length of the Expressed 3′ UTR Is an Intermediate Molecular Phenotype Linking Genetic Variants to Complex Diseases. Front. Genet. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shulman ED and Elkon R (2020) Systematic identification of functional SNPs interrupting 3’UTR polyadenylation signals. PLOS Genet. 16, e1008977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei W et al. (2022) Comprehensive characterization of posttranscriptional impairmentrelated 3′-UTR mutations in 2413 whole genomes of cancer patients. Npj Genomic Med. 7, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emilsson V et al. (2022) Coding and regulatory variants are associated with serum protein levels and disease. Nat. Commun. 13, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He B et al. (2020) Genome-wide pQTL analysis of protein expression regulatory networks in the human liver. BMC Biol. 18, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yao C et al. (2018) Genome-wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat. Commun. 9, 3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Klein JC et al. (2019) Functional testing of thousands of osteoarthritis-associated variants for regulatory activity. Nat. Commun. 10, 2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schrode N et al. (2019) Synergistic effects of common schizophrenia risk variants. Nat. Genet. 51, 1475–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Griesemer D et al. (2021) Genome-wide functional screen of 3′UTR variants uncovers causal variants for human disease and evolution. Cell 184, 5247–5260.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bogard N et al. (2019) A Deep Neural Network for Predicting and Engineering Alternative Polyadenylation. Cell 178, 91–106.e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sample PJ et al. (2019) Human 5′ UTR design and variant effect prediction from a massively parallel translation assay. Nat. Biotechnol. 37, 803–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sweta S et al. (2019) Importance of Long Non-coding RNAs in the Development and Disease of Skeletal Muscle and Cardiovascular Lineages. Front. Cell Dev. Biol. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.O’Brien J et al. (2018) Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iyer MK et al. (2015) The Landscape of Long Noncoding RNAs in the Human Transcriptome. Nat. Genet. 47, 199–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu X et al. (2020) Regulation of cellular sterol homeostasis by the oxygen responsive noncoding RNA lincNORS. Nat. Commun. 11, 4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tian J et al. (2020) Risk SNP-Mediated Enhancer–Promoter Interaction Drives Colorectal Cancer through Both FADS2 and AP002754.2. Cancer Res. 80, 1804–1818 [DOI] [PubMed] [Google Scholar]
- 62.Hua JT et al. (2018) Risk SNP-Mediated Promoter-Enhancer Switching Drives Prostate Cancer through lncRNA PCAT19. Cell 174, 564–575.e18 [DOI] [PubMed] [Google Scholar]
- 63.Wang J et al. (2021) SNP-mediated lncRNA-ENTPD3-AS1 upregulation suppresses renal cell carcinoma via miR-155/HIF-1α signaling. Cell Death Dis. 12, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Feng T et al. (2020) A SNP-mediated lncRNA (LOC146880) and microRNA (miR-539–5p) interaction and its potential impact on the NSCLC risk. J. Exp. Clin. Cancer Res. 39, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nikpay M et al. (2019) Genome-wide identification of circulating-miRNA expression quantitative trait loci reveals the role of several miRNAs in the regulation of cardiometabolic phenotypes. Cardiovasc. Res. 115, 1629–1645 [DOI] [PubMed] [Google Scholar]
- 66.Larson NB et al. (2022) A microRNA Transcriptome-wide Association Study of Prostate Cancer Risk. Front. Genet. 13, 836841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ghanbari M et al. (2017) A Genome-Wide Scan for MicroRNA-Related Genetic Variants Associated With Primary Open-Angle Glaucoma. Invest. Ophthalmol. Vis. Sci. 58, 5368–5377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mens MMJ et al. (2020) Multi-Omics Analysis Reveals MicroRNAs Associated With Cardiometabolic Traits. Front. Genet. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rhead B et al. (2019) miRNA contributions to pediatric-onset multiple sclerosis inferred from GWAS. Ann. Clin. Transl. Neurol. 6, 1053–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lorenz R et al. (2011) ViennaRNA Package 2.0. Algorithms Mol. Biol. 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McGeary SE et al. (2019) The biochemical basis of microRNA targeting efficacy. Science 366, eaav1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wong N and Wang X (2015) miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 43, D146–D152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Craddock N et al. (2010) Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 464, 713–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McCarroll SA (2008) Extending genome-wide association studies to copy-number variation. Hum. Mol. Genet. 17, R135–R142 [DOI] [PubMed] [Google Scholar]
- 75.Carvalho CMB and Lupski JR (2016) Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 17, 224–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.MacDonald JR et al. (2014) The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 42, D986–D992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pang AW et al. (2010) Towards a comprehensive structural variation map of an individual human genome. Genome Biol. 11, R52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sherry ST et al. (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Auwerx C et al. (2022) The individual and global impact of copy-number variants on complex human traits. Am. J. Hum. Genet. 109, 647–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu J et al. (2018) The coexistence of copy number variations (CNVs) and single nucleotide polymorphisms (SNPs) at a locus can result in distorted calculations of the significance in associating SNPs to disease. Hum. Genet. 137, 553–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li YR et al. (2020) Rare copy number variants in over 100,000 European ancestry subjects reveal multiple disease associations. Nat. Commun. 11, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marshall CR et al. (2017) Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 49, 27–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Macé A et al. (2017) CNV-association meta-analysis in 191,161 European adults reveals new loci associated with anthropometric traits. Nat. Commun. 8, 744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Collins RL et al. (2022) A cross-disorder dosage sensitivity map of the human genome. Cell 185, 3041–3055.e25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hujoel MLA et al. (2022) Influences of rare copy-number variation on human complex traits. Cell 185, 4233–4248.e27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vaser R et al. (2016) SIFT missense predictions for genomes. Nat. Protoc. 11, 1–9 [DOI] [PubMed] [Google Scholar]
- 87.Adzhubei IA et al. (2010) A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Choi Y (2012) A fast computation of pairwise sequence alignment scores between a protein and a set of single-locus variants of another protein. in Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine, New York, NY, USA, pp. 414–417 [Google Scholar]
- 89.Calabrese R et al. (2009) Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 30, 1237–1244 [DOI] [PubMed] [Google Scholar]
- 90.Bendl J et al. (2016) PredictSNP2: A Unified Platform for Accurately Evaluating SNP Effects by Exploiting the Different Characteristics of Variants in Distinct Genomic Regions. PLoS Comput. Biol. 12, e1004962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Capriotti E et al. (2006) Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 22, 2729–2734 [DOI] [PubMed] [Google Scholar]
- 92.Capriotti E et al. (2013) Collective judgment predicts disease-associated single nucleotide variants. BMC Genomics 14, S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mi H et al. (2019) PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pers TH et al. (2015) SNPsnap: a Web-based tool for identification and annotation of matched SNPs. Bioinformatics 31, 418–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Salgado D et al. (2016) UMD-Predictor: A High-Throughput Sequencing Compliant System for Pathogenicity Prediction of any Human cDNA Substitution. Hum. Mutat. 37, 439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yates CM et al. (2014) SuSPect: Enhanced Prediction of Single Amino Acid Variant (SAV) Phenotype Using Network Features. J. Mol. Biol. 426, 2692–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Klausen MS et al. (2019) NetSurfP-2.0: Improved prediction of protein structural features by integrated deep learning. Proteins Struct. Funct. Bioinforma. 87, 520–527 [DOI] [PubMed] [Google Scholar]
- 98.Geourjon C and Deléage G (1995) SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics 11, 681–684 [DOI] [PubMed] [Google Scholar]
- 99.Capriotti E et al. (2005) I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 33, W306–W310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ashkenazy H et al. (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Venselaar H et al. (2010) Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics 11, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Waterhouse A et al. (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Adiba M et al. (2021) In silico characterization of coding and non-coding SNPs of the androgen receptor gene. Inform. Med. Unlocked 24, 100556 [Google Scholar]
- 104.Akhtar A et al. (2021) In silico computation of functional SNPs of CYP2U1 protein leading to hereditary spastic paraplegia. Inform. Med. Unlocked 24, 100610 [Google Scholar]
- 105.Thirumal Kumar D et al. (2022) Chapter Three - Computational and structural investigation of Palmitoyl-Protein Thioesterase 1 (PPT1) protein causing Neuronal Ceroid Lipofuscinoses (NCL). In Advances in Protein Chemistry and Structural Biology 132 (Donev R, ed), pp. 89–109, Academic Press; [DOI] [PubMed] [Google Scholar]
- 106.Shinde SD et al. (2022) Computational Biology of BRCA2 in Male Breast Cancer, through Prediction of Probable nsSNPs, and Hit Identification. ACS Omega 7, 30447–30461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Saxena S et al. (2022) In-silico analysis of deleterious single nucleotide polymorphisms of PNMT gene. Mol. Simul. 0, 1–15 [Google Scholar]
- 108.Boyle AP et al. (2012) Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 22, 1790–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pesole G and Liuni S (1999) Internet resources for the functional analysis of 5′ and 3′ untranslated regions of eukaryotic mRNAs. Trends Genet. 15, 378. [DOI] [PubMed] [Google Scholar]
- 110.Pesole G et al. (2002) UTRdb and UTRsite: specialized databases of sequences and functional elements of 5′ and 3′ untranslated regions of eukaryotic mRNAs. Update 2002. Nucleic Acids Res. 30, 335–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Huang Z and Teeling EC (2017) ExUTR: a novel pipeline for large-scale prediction of 3′-UTR sequences from NGS data. BMC Genomics 18, 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang X et al. (2021) Annotating high-impact 5′untranslated region variants with the UTRannotator. Bioinformatics 37, 1171–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Maxwell EK et al. (2015) SubmiRine: assessing variants in microRNA targets using clinical genomic data sets. Nucleic Acids Res. 43, 3886–3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bhattacharya A and Cui Y (2015) miR2GO: comparative functional analysis for microRNAs. Bioinformatics 31, 2403–2405 [DOI] [PubMed] [Google Scholar]
- 115.Barenboim M et al. (2010) MicroSNiPer: a web tool for prediction of SNP effects on putative microRNA targets. Hum. Mutat. 31, 1223–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wan C et al. (2017) CPSS 2.0: a computational platform update for the analysis of small RNA sequencing data. Bioinformatics 33, 3289–3291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Coetzee SG et al. (2015) motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics 31, 3847–3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schilder BM and Raj T (2022) Fine-mapping of Parkinson’s disease susceptibility loci identifies putative causal variants. Hum. Mol. Genet. 31, 888–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Leberfarb EY et al. (2020) Potential regulatory SNPs in the ATXN7L3B and KRT15 genes are associated with gender-specific colorectal cancer risk. Pers. Med. 17, 43–54 [DOI] [PubMed] [Google Scholar]
- 120.Jones MR et al. (2020) Ovarian Cancer Risk Variants Are Enriched in Histotype-Specific Enhancers and Disrupt Transcription Factor Binding Sites. Am. J. Hum. Genet. 107, 622–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhou J and Troyanskaya OG (2015) Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 12, 931–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhou J et al. (2018) Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet. 50, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kelley DR et al. (2018) Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res. 28, 739–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wallace C (2021) A more accurate method for colocalisation analysis allowing for multiple causal variants. PLOS Genet. 17, e1009440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Barbeira AN et al. (2018) Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 9, 1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Barbeira AN et al. (2019) Integrating predicted transcriptome from multiple tissues improves association detection. PLOS Genet. 15, e1007889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Grishin D and Gusev A (2022) Allelic imbalance of chromatin accessibility in cancer identifies candidate causal risk variants and their mechanisms. Nat. Genet. 54, 837–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lin H et al. (2019) RegSNPs-intron: a computational framework for predicting pathogenic impact of intronic single nucleotide variants. Genome Biol. 20, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jaganathan K et al. (2019) Predicting Splicing from Primary Sequence with Deep Learning. Cell 176, 535–548.e24 [DOI] [PubMed] [Google Scholar]
- 130.Desmet F-O et al. (2009) Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37, e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pertea M et al. (2001) GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res. 29, 1185–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brunak S et al. (1991) Prediction of human mRNA donor and acceptor sites from the DNA sequence. J. Mol. Biol. 220, 49–65 [DOI] [PubMed] [Google Scholar]
- 133.Hebsgaard SM et al. (1996) Splice Site Prediction in Arabidopsis Thaliana Pre-mRNA by Combining Local and Global Sequence Information. Nucleic Acids Res. 24, 3439–3452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Liu H et al. (2022) Performance evaluation of computational methods for splice-disrupting variants and improving the performance using the machine learning-based framework. Brief. Bioinform. 23, bbac334. [DOI] [PubMed] [Google Scholar]
- 135.Melnikov A et al. (2012) Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 30, 271–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tewhey R et al. (2016) Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 165, 1519–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Abell NS et al. (2022) Multiple causal variants underlie genetic associations in humans. Science 375, 1247–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kircher M et al. (2019) Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat. Commun. 10, 3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Arnold CD et al. (2013) Genome-Wide Quantitative Enhancer Activity Maps Identified by STARR-seq. Science 339, 1074–1077 [DOI] [PubMed] [Google Scholar]
- 140.Wang X et al. (2018) High-resolution genome-wide functional dissection of transcriptional regulatory regions and nucleotides in human. Nat. Commun. 9, 5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.van Arensbergen J et al. (2017) Genome-wide mapping of autonomous promoter activity in human cells. Nat. Biotechnol. 35, 145–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.van Arensbergen J et al. (2019) High-throughput identification of human SNPs affecting regulatory element activity. Nat. Genet. 51, 1160–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhao Y et al. (2020) A sequential methodology for the rapid identification and characterization of breast cancer-associated functional SNPs. Nat. Commun. 11, 3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang P et al. (2018) High-throughput screening of prostate cancer risk loci by single nucleotide polymorphisms sequencing. Nat. Commun. 9, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Li G et al. (2018) High-throughput identification of noncoding functional SNPs via type IIS enzyme restriction. Nat. Genet. 50, 1180–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yan J et al. (2021) Systematic analysis of binding of transcription factors to noncoding variants. Nature 591, 147–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mondal S et al. (2022) PROBER identifies proteins associated with programmable sequence-specific DNA in living cells. Nat. Methods 19, 959–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhao W et al. (2014) Massively parallel functional annotation of 3′ untranslated regions. Nat. Biotechnol. 32, 387–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lim Y et al. (2021) Multiplexed functional genomic analysis of 5’ untranslated region mutations across the spectrum of prostate cancer. Nat. Commun. 12, 4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Adamson SI et al. (2018) Vex-seq: high-throughput identification of the impact of genetic variation on pre-mRNA splicing efficiency. Genome Biol. 19, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Soemedi R et al. (2017) Pathogenic variants that alter protein code often disrupt splicing. Nat. Genet. 49, 848–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cheung R et al. (2019) A Multiplexed Assay for Exon Recognition Reveals that an Unappreciated Fraction of Rare Genetic Variants Cause Large-Effect Splicing Disruptions. Mol. Cell 73, 183–194.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Matreyek KA et al. (2018) Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat. Genet. 50, 874–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jinek M et al. (2012) A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 337, 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Adamson B et al. (2016) A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167, 1867–1882.e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Dixit A et al. (2016) Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Datlinger P et al. (2017) Pooled CRISPR screening with single-cell transcriptome read-out. Nat. Methods 14, 297–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jin X et al. (2020) In vivo Perturb-Seq reveals neuronal and glial abnormalities associated with autism risk genes. Science 370, eaaz6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rubin AJ et al. (2019) Coupled Single-Cell CRISPR Screening and Epigenomic Profiling Reveals Causal Gene Regulatory Networks. Cell 176, 361–376.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Larson MH et al. (2013) CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 8, 2180–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Perez-Pinera P et al. (2013) RNA-guided gene activation by CRISPR-Cas9–based transcription factors. Nat. Methods 10, 973–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Horlbeck MA et al. (2016) Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife 5, e19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Joung J et al. (2017) Genome-scale Activation Screen Identifies a LncRNA Locus that Regulates a Gene Neighborhood. Nature 548, 343–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Liu SJ et al. (2017) CRISPRi-based genome-scale identification of functional long non-coding RNA loci in human cells. Science 355, aah7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nandakumar SK et al. (2019) Gene-centric functional dissection of human genetic variation uncovers regulators of hematopoiesis. eLife 8, e44080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chepelev I et al. (2012) Characterization of genome-wide enhancer-promoter interactions reveals co-expression of interacting genes and modes of higher order chromatin organization. Cell Res. 22, 490–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Nott A et al. (2019) Brain cell type–specific enhancer–promoter interactome maps and disease-risk association. Science 366, 1134–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mackay TFC et al. (2009) The genetics of quantitative traits: challenges and prospects. Nat. Rev. Genet. 10, 565–577 [DOI] [PubMed] [Google Scholar]
- 169.Michaelson JJ et al. (2010) Data-driven assessment of eQTL mapping methods. BMC Genomics 11, 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wu Y et al. (2019) Colocalization of GWAS and eQTL signals at loci with multiple signals identifies additional candidate genes for body fat distribution. Hum. Mol. Genet. 28, 4161–4172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Perez RK et al. (2022) Single-cell RNA-seq reveals cell type–specific molecular and genetic associations to lupus. Science 376, eabf1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.van der Wijst M et al. (2020) The single-cell eQTLGen consortium. eLife 9, e52155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yazar S et al. (2022) Single-cell eQTL mapping identifies cell type–specific genetic control of autoimmune disease. Science 376, eabf3041. [DOI] [PubMed] [Google Scholar]
- 174.Schliekelman P (2008) Statistical Power of Expression Quantitative Trait Loci for Mapping of Complex Trait Loci in Natural Populations. Genetics 178, 2201–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.GTEx Consortium (2017) Genetic effects on gene expression across human tissues. Nature 550, 204–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.GTEx Consortium (2020) The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Võsa U et al. (2021) Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 53, 1300–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gasperini M et al. (2019) A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 176, 377–390.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lieberman-Aiden E et al. (2009) Comprehensive mapping of long range interactions reveals folding principles of the human genome. Science 326, 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mumbach MR et al. (2016) HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mumbach MR et al. (2017) Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat. Genet. 49, 1602–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Fullwood MJ et al. (2009) An oestrogen-receptor-α-bound human chromatin interactome. Nature 462, 58–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tang Z et al. (2015) CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell 163, 1611–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Giambartolomei C et al. (2021) H3K27ac HiChIP in prostate cell lines identifies risk genes for prostate cancer susceptibility. Am. J. Hum. Genet. 108, 2284–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wang W et al. (2021) Functional Interrogation of Enhancer Connectome Prioritizes Candidate Target Genes at Ovarian Cancer Susceptibility Loci. Front. Genet. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Chen AF et al. (2022) NEAT-seq: simultaneous profiling of intra-nuclear proteins, chromatin accessibility and gene expression in single cells. Nat. Methods 19, 547–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fulco CP et al. (2019) Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lee CS et al. (2021) Mutant collagen COL11A1 enhances cancerous invasion. Oncogene 40, 6299–6307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Lin S et al. (2014) Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 3, e04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ran FA et al. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lee MN et al. (2014) Common Genetic Variants Modulate Pathogen-Sensing Responses in Human Dendritic Cells. Science 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Long C et al. (2014) Prevention of muscular dystrophy in mice by CRISPR/Cas9–mediated editing of germline DNA. Science 345, 1184–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Soldner F et al. (2016) Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 533, 95–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Cox DBT et al. (2015) Therapeutic genome editing: prospects and challenges. Nat. Med. 21, 121–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Gaudelli NM et al. (2017) Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Komor AC et al. (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kurt IC et al. (2021) CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 39, 41–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Antoniou P et al. (2021) Base and Prime Editing Technologies for Blood Disorders. Front. Genome Ed. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Rees HA and Liu DR (2018) Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 19, 770–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hu Y et al. (2021) Whole-genome sequencing association analysis of quantitative red blood cell phenotypes: The NHLBI TOPMed program. Am. J. Hum. Genet. 108, 874–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Yuan J et al. (2018) Genetic Modulation of RNA Splicing with a CRISPR-Guided Cytidine Deaminase. Mol. Cell 72, 380–394.e7 [DOI] [PubMed] [Google Scholar]
- 202.Zeng Y et al. (2018) Correction of the Marfan Syndrome Pathogenic FBN1 Mutation by Base Editing in Human Cells and Heterozygous Embryos. Mol. Ther. 26, 2631–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Hanna RE et al. (2021) Massively parallel assessment of human variants with base editor screens. Cell 184, 1064–1080.e20 [DOI] [PubMed] [Google Scholar]
- 204.Anzalone AV et al. (2019) Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gao P et al. (2021) Prime editing in mice reveals the essentiality of a single base in driving tissue-specific gene expression. Genome Biol. 22, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Böck D et al. (2022) In vivo prime editing of a metabolic liver disease in mice. Sci. Transl. Med. 14, eabl9238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Cai Y et al. (2019) In vivo genome editing rescues photoreceptor degeneration via a Cas9/RecA-mediated homology-directed repair pathway. Sci. Adv. 5, eaav3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Koblan LW et al. (2021) In vivo base editing rescues Hutchinson–Gilford progeria syndrome in mice. Nature 589, 608–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Xu L et al. (2021) Efficient precise in vivo base editing in adult dystrophic mice. Nat. Commun. 12, 3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Blaisdell J et al. (2002) Identification and functional characterization of new potentially defective alleles of human CYP2C19. Pharmacogenet. Genomics 12, 703–711 [DOI] [PubMed] [Google Scholar]
- 211.Dai D et al. (2001) Identification of Variants of CYP3A4 and Characterization of Their Abilities to Metabolize Testosterone and Chlorpyrifos. J. Pharmacol. Exp. Ther. 299, 825–831 [PubMed] [Google Scholar]
- 212.Zhang J et al. (2001) The human pregnane X receptor: genomic structure and identification and functional characterization of natural allelic variants. Pharmacogenet. Genomics 11, 555–572 [DOI] [PubMed] [Google Scholar]
- 213.Cargill M et al. (1999) Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat. Genet. 22, 231–238 [DOI] [PubMed] [Google Scholar]
- 214.Sharon D et al. (2000) Identification and characterization of coding single-nucleotide polymorphisms within a human olfactory receptor gene cluster. Gene 260, 87–94 [DOI] [PubMed] [Google Scholar]
- 215.Conne B et al. (2000) The 3′ untranslated region of messenger RNA: A molecular ‘hotspot’ for pathology? Nat. Med. 6, 637–641 [DOI] [PubMed] [Google Scholar]
- 216.Davis BM et al. (1997) Expansion of a CUG trinucleotide repeat in the 3′ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc. Natl. Acad. Sci. 94, 7388–7393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Lu X et al. (1999) Cardiac Elav-type RNA-binding Protein (ETR-3) Binds to RNA CUG Repeats Expanded in Myotonic Dystrophy. Hum. Mol. Genet. 8, 53–60 [DOI] [PubMed] [Google Scholar]
- 218.Suhl JA et al. (2015) A 3′ untranslated region variant in FMR1 eliminates neuronal activity-dependent translation of FMRP by disrupting binding of the RNA-binding protein HuR. Proc. Natl. Acad. Sci. U. S. A. 112, E6553–E6561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Rimokh R et al. (1994) Rearrangement of CCND1 (BCL1/PRAD1) 3’ untranslated region in mantle- cell lymphomas and t(11q13)-associated leukemias. Blood 83, 3689–3696 [PubMed] [Google Scholar]
- 220.Chatterjee S and Pal JK (2009) Role of 5′- and 3′-untranslated regions of mRNAs in human diseases. Biol. Cell 101, 251–262 [DOI] [PubMed] [Google Scholar]
- 221.Kondo T et al. (1998) Familial Essential Thrombocythemia Associated With One-Base Deletion in the 5′-Untranslated Region of the Thrombopoietin Gene. Blood 92, 1091–1096 [PubMed] [Google Scholar]
- 222.Allerson CR et al. (1999) Clinical Severity and Thermodynamic Effects of Iron-responsive Element Mutations in Hereditary Hyperferritinemia-Cataract Syndrome*. J. Biol. Chem. 274, 26439–26447 [DOI] [PubMed] [Google Scholar]
- 223.Chappell SA et al. (2000) A mutation in the c-myc-IRES leads to enhanced internal ribosome entry in multiple myeloma: A novel mechanism of oncogene de-regulation. Oncogene 19, 4437–4440 [DOI] [PubMed] [Google Scholar]
- 224.Evans JR et al. (2003) Members of the poly (rC) binding protein family stimulate the activity of the c-myc internal ribosome entry segment in vitro and in vivo. Oncogene 22, 8012–8020 [DOI] [PubMed] [Google Scholar]
- 225.Signori E et al. (2001) A somatic mutation in the 5’UTR of BRCA1 gene in sporadic breast cancer causes down-modulation of translation efficiency. Oncogene 20, 4596–4600 [DOI] [PubMed] [Google Scholar]