

Summary
This study examines the effectiveness of generative models in drug discovery, material science, and polymer science, aiming to overcome constraints associated with traditional inverse design methods relying on heuristic rules. Generative models generate synthetic data resembling real data, enabling deep learning model training without extensive labeled datasets. They prove valuable in creating virtual libraries of molecules for material science and facilitating drug discovery by generating molecules with specific properties. While generative adversarial networks (GANs) are explored for these purposes, mode collapse restricts their efficacy, limiting novel structure variability. To address this, we introduce a masked language model (LM) inspired by natural language processing. Although LMs alone can have inherent limitations, we propose a hybrid architecture combining LMs and GANs to efficiently generate new molecules, demonstrating superior performance over standalone masked LMs, particularly for smaller population sizes. This hybrid LM-GAN architecture enhances efficiency in optimizing properties and generating novel samples.
Keywords: masked language model, generative adversarial network, genetic algorithm, molecule design
Graphical abstract
Highlights
-
•
Uniting language models and GANs, LM-GAN elevates molecular design efficiency
-
•
Genetic algorithms address GAN limitations, ensuring structural diversity
-
•
LM-GAN outperforms in generating molecules with smaller population sizes
-
•
Consistently demonstrates superior performance in generating optimized molecules
The bigger picture
Recent advances in AI offer solutions to complex challenges in chemical and materials sciences, such as drug discovery and polymer synthesis. Traditional methods struggle due to the vast and intricate chemical compound space. AI, especially generative models, provides a promising alternative by creating synthetic data, reducing the need for extensive labeling. Challenges, however, persist, such as ensuring diverse structures. Improved methods could accelerate progress in fields such as drug discovery and materials design.
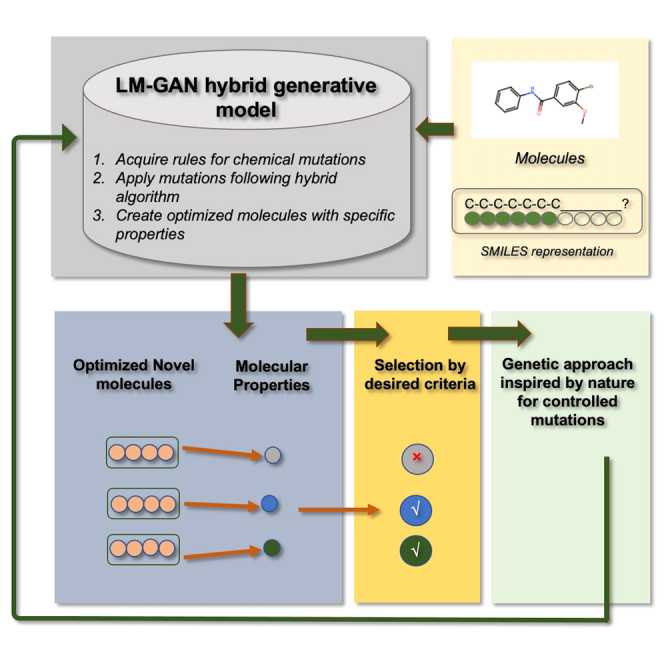
This study unveils a hybrid LM-GAN architecture, surmounting challenges such as mode collapse and dataset limitations. The research not only showcases enhanced efficiency in generating molecules but also addresses crucial issues related to population size and dataset requirements. From drug discovery to material synthesis, this novel AI algorithm promises transformative impacts and significant advancements for researchers seeking tailored molecules with diverse properties.
Introduction
Recent advances in artificial intelligence (AI) have laid the foundation to provide promising solutions for overcoming inverse design problems in the chemical and materials sciences, such as drug discovery, polymer science, and other condensed matter applications. Exploration of the vast chemical compound space for designing new drug candidates or customized molecules with desired functionality is challenging because of the high dimensionality of the associated chemical space, which makes searching for the appropriate novel compound difficult. Traditional inverse design approaches based on structural optimization methods typically rely on heuristic rules or domain-specific knowledge and might therefore encounter difficulties with novelty and generalizability. Although AI and machine learning (ML) techniques offer promise for overcoming this challenge, the need for massive labeled datasets to train these models remains.1 Gathering and annotating such datasets can be time-consuming, expensive, and sometimes not even feasible. Fortunately, recent advances in AI/ML, specifically concerning surrogate and generative models, have provided promising solutions by generating required synthetic data to overcome the need for generating massive labeled datasets using traditional, physics-based modeling and simulations.2
In this context, generative models have shown great potential in generating synthetic data that mimic the characteristics of real data.3 This ability has opened up new possibilities for training deep learning models without relying solely on large real or experimental labeled datasets. By leveraging generative models, researchers in material science, drug discovery, and polymer science can generate new molecules with desired properties, thus facilitating the exploration of novel chemicals and materials.4
Generative models play a pivotal role in the chemical and materials sciences, particularly in the generation of new molecules with tailored properties. In the chemical sciences, these models facilitate the creation of virtual libraries of molecules, allowing researchers to explore properties through simulations or experimental validations. This accelerates the discovery of novel compounds while reducing reliance on extensive labeled datasets.2 In drug discovery, generative models contribute by producing molecules with desired properties, such as high potency and low toxicity,5 expediting the process and lessening dependence on large labeled datasets. Similarly, in polymer science, these models aid in designing polymers with specific properties, mitigating the challenges posed by the need for massive labeled datasets. By training on existing databases, researchers can generate new polymer structures,6 advancing industries such as materials engineering, coatings, and energy storage.7 The use of generative models offers a promising solution in overcoming the requirement for extensive labeled datasets, revolutionizing the chemical and materials sciences and accelerating the discovery and development of innovative materials, drugs, and polymers.
In recent years, there has been a growing interest in developing data-driven generative models capable of predicting novel molecular structures with desired functionalities, especially those not reliant on labeled training data. Numerous approaches have emerged, focusing on effective generative models for molecular structure prediction.
A graph-based method has been designed to generate molecules with exact property values while ensuring the presence of desired scaffolds.8 A prominent category includes recurrent neural network (RNN)-based architectures, which have demonstrated considerable success in generating new molecules.9–14 For instance, Segler et al. utilized long short-term memory (LSTM) networks, showcasing the benefits of transfer learning and fine-tuning on smaller populations to achieve specific biological target activities.9 Previously, RNNs have been employed to generate molecules by considering given scaffolds.15 A comprehensive study by Arús-Pous et al. investigated the efficacy of different RNN models, such as LSTM and gated recurrent unit, on diverse data populations, ranging from 10,000 to 1 million molecules, using various simplified molecular input line entry system (SMILES) representations.10 Conditional adversarially regularized autoencoder16 and conditional RNNs9,17 are methods that sample molecules based on exact values. The work of Flam-Shepherd et al. extended the exploration of RNN-based language models (LMs) to learn complex chemical rules based on different molecular representations, such as SMILES or SELF-referencing embedded strings.11 Furthermore, the application of bidirectional encoder representations from transformers (BERT)-based large LMs demonstrated advantages when tested on benchmark models or datasets.18 Researchers, including Awale et al.,12 Zheng et al.,13 and Méndez-Lucio et al.,14 continued to enhance RNN models and introduced conditional generative adversarial networks (GANs) to address the challenges of generating diverse and novel molecular structures. However, despite their efficacy in exploring chemical space, GANs often face the issue of “mode collapse,” producing structures too similar to the training data.19
In response to these challenges, researchers have explored integrating genetic algorithm (GA)-based techniques into generative models to promote structural diversity by emulating the mutation and selection process in nature. GA-based methods have shown superiority over modern AI/ML techniques in generating novel molecules with desired properties.20,21 Previous investigations employed an adaptive training method influenced by GA, introducing valid and novel molecules through random and guided replacements in the training data.21,22 Despite the success of GA, there is a recognized need for a more efficient mutation operator to eliminate manual mutation rules and enable generalization, automation, and extension beyond single-atom mutation.
To address this gap, a masked LM inspired by natural language processing (NLP) has been implemented.21,23,24 This innovative approach constructs a vocabulary from common subsequences within a given population, tokenizing the training data for the LM to generate potential rearrangements or mutations. The masked LM provides a promising solution to challenges in manual mutation rules, aiming to enhance generalization and automation in the generation of novel molecular structures with desired properties.
Although LMs are effective generation tools that have proven successful in novel molecule generation, they are not without drawbacks and deficiencies. They lack common sense and rely solely on statistical patterns, which can lead to the generation of plausible, yet incorrect, outputs. These models might also perpetuate biases from the training data, overfit to specific datasets, and lack the ability to verify response accuracy. In addition, they are sensitive to changes in input, resulting in inconsistent outputs, and struggle with comprehending context, leading to confusion or repetitive results. Furthermore, LMs pose challenges in terms of interpretation, making it difficult to understand the reasoning behind their generated outputs.
In this work we go one step further to address some of the aforementioned issues. We study whether and how an efficient hybrid architecture can be built that can demonstrate higher efficacy in generating new molecules with desired properties by combining our previous works based on GA approaches with both LM and GAN as hybrid generative models. This new architecture combines advantages from both the LM and GANs while learning the commonly occurring sequences from the training dataset and being applied as an automated, generalized mutation operator for generating new molecules. Implementing a hybrid LM-GAN platform offers numerous advantages over using LMs or GANs separately. GANs enhance LMs by generating realistic and creative new samples, while LMs enhance GANs by producing informative and relevant samples. Hybrid models improve generation quality, enhance creativity and novelty, provide better control over sample generation, address data scarcity, capture contextual and structural information, and use adversarial training for improvement. However, careful consideration of training, model architecture, and optimization is necessary because of the increased complexity. Therefore, striking a balance between language modeling and GAN techniques is crucial for successful implementation.
While the integration of transformers with GANs has seen impressive success in computer vision and image generation, its application in molecule generation is noticeably restricted. Traditionally, GANs rely on convolutional neural networks (CNNs) for discriminator and generator networks, excelling in local relationships but struggling with global ones in feature spaces. Transformer networks, recognized for their proficiency in exploiting global relationships, when combined with GANs, recently have shown substantial improvements in computer vision. Dubey and Singh25 provide a thorough survey on GAN developments, examining the use of both CNNs and transformer networks as GAN components, across various computer vision applications. In addition to conducting a comprehensive comparative survey, the authors have emphasized the ongoing research trend in image and video synthesis, underscoring the significant impact of transformer-based GANs on the advancement of computer vision methods and applications. Although many transformer-based GANs primarily employ CNN-based discriminators, there are also endeavors to incorporate transformers into the discriminator component in certain models. In the pioneering work by Jiang et al.,26 who introduced TransGAN, a novel approach relying solely on transformers without convolutional components, they showcased TransGAN’s competitive performance in computer vision and image generation. Transformer-based GANs have proven successful in diverse image and computer vision, signaling the potential for future research in other domains, such as molecule generation, where the application of TransGAN or similar transformer-GAN combinations is still in its early stages.
In the realm of molecule generation, Transmol27 by Zhumagambetov et al. utilizes transformers exclusively for de novo molecular generation. Transmol outperforms baseline methods, including latentGAN, a GAN-based molecular generation strategy, showcasing superior internal diversity and a higher proportion of novel molecules. Another analogous contribution is MolGPT,28 which competes favorably in terms of validity, uniqueness, and novelty against various modern machine learning frameworks, including LatentGAN, in de novo molecular generation for drug design. While MolGPT exhibits lower novelty scores, it excels in managing long-term dependencies and SMILES grammar through attention mechanisms. This collective research highlights the promising yet underexplored potential of combining transformers with GANs in molecule generation. Despite encouraging results in computer vision and image generation, this integration has not been extensively applied in the domain of molecule generation. Nevertheless, within the specific context of molecule generation tasks, transformers have demonstrated success compared with alternative models, including deep learning generative models based on GANs. Ongoing research aims to enhance transformer-based models by integrating them with GANs in the context of molecule generation.
To test our hypothesis we performed a comparative study between LM and the new hybrid LM-GAN architecture (Figure 1) as a generative model for different and specific optimization tasks. To introduce diversity in the generated population, a simplified GA strategy was adopted. This GA algorithm is identical in both implementations in this study (i.e., standalone LM as well as LM-GAN). This involved survivor selection through random sampling across the population to choose parents, while mutation was used solely for generating new molecules. Ultimately, only unique molecules were kept in the population for the evolution of subsequent generations. The research addresses the limitations of LMs in molecule generation by proposing a hybrid architecture that combines genetic algorithm (GA) approaches with both LMs and GANs. The primary contribution lies in the development of an efficient hybrid LM-GAN platform that aims to overcome the deficiencies of standalone LMs. The hybrid model leverages the strengths of both LMs and GANs, utilizing the former to learn common sequences from the training dataset and serve as an automated mutation operator for molecule generation. This approach offers advantages over standalone LMs or GANs, enhancing generation quality, creativity, novelty, control over sample generation, addressing data scarcity, and capturing contextual and structural information. The research emphasizes the need for careful consideration in training, model architecture, and optimization to strike a balance between language modeling and GAN techniques for successful implementation.
Figure 1.

Novel generalizable hybrid LM-based GAN architecture for efficient new molecule generation with desired properties
This describes a new architecture that combines the benefits of both LMs and GANs to generate new molecules. Upper: in the LM-only architecture, the input molecules are provided as SMILES strings. A portion of each SMILES string is masked, and an LM model is used to predict mutations or alternatives to the original molecule. The newly generated molecules with mutated atoms are scored and selected based on a fitness function to create an optimized population of molecules. This process is repeated for multiple iterations to generate a final population. Lower: in the LM-GAN method, a GAN is implemented by defining generator and discriminator networks, designing a training loop, and using backpropagation for parameter updates. The generator randomly masks a portion of a SMILES representation and uses a pretrained LM to fill in the missing molecular structure, creating a new molecule. The discriminator, also a pretrained LM, classifies the original and generated SMILES strings as real or fake, respectively. The generator and discriminator LMs are iteratively trained to ensure that the masked LM generates molecules that can deceive the discriminator.
The comparative study between a state-of-the-art LM and the proposed LM-GAN hybrid architecture reveals several noteworthy findings. The hybrid LM-GAN architecture demonstrates superior performance in predicting a higher fraction of acceptable molecules with improved target properties compared with the standalone LM. The incorporation of GA-based mutation strategies helps mitigate mode-collapse issues commonly found in GANs. The research also highlights that the hybrid LM-GAN performs better with smaller population sizes, addressing the challenge of requiring large amounts of data for model training. In addition, the hybrid model consistently outperforms the standalone LM in terms of efficiency when computing the ratio of accepted molecules to generated novel molecules with desired optimized molecular properties across various population sizes. The implications of this novel AI algorithm extend to diverse domains, including drug discovery, polymer synthesis, and materials design.
Results
LM vs. LM-GAN
Experimental design
As part of our study we selected our evaluation metrics to be based on two different functions, namely different population size and different masking scheme for mutations to be propagated into the molecule population. The initial population size was evaluated within a range from 5k to 50k, and the mutation rates were modified from 10% to 100%. The evaluated metrics were synthesizability, drug likeliness, solubility, number of atoms, number of novel generated molecules, number of accepted molecules, fitness function (composed of harmonic mean of synthesizability and drug likeliness), and the efficiency of the generative model as determined by computing the ratio between accepted to novel molecules. An experiment was designed where 50% of the samples are permitted to be mutated (i.e., to be sent to the generator). Each run for each of the methods, mutation rates, and population size is performed for 50 generations five times, independently from each other, to estimate the statistical error.
Fitness function
We first discuss the synthesizability score. The method we follow was proposed by Ertl and Schuffenhauer29 as applied in several of our previous works.21,22,23,24 Here, synthetic accessibility is computed by an empirical technique via combining molecular complexity with molecular fragment data that were collected by processing a large set of chemical structures that were already synthesized. This accessibility is then tested and validated with “ease of synthesis” ranks that were collected by domain experts. In other words, this technique thus behaves like a surrogate model trained, tested, and validated on historical synthesizability data. The agreement is generally acceptable. In our implementation, low synthesizability is indicated by scores trending to zero, while high synthesizability is reflected in scores trending to one. We also estimated the drug likeliness and solubility of the molecules following the methods proposed by29,30,31 as also implemented in our previous works.21,22,23,24 Once these metrics are computed, the fitness function is estimated by taking the harmonic mean of synthesizability and drug likeliness. In Figure 2 we show the fitness score, both as a function of the initial population size and of the mutation rate. We see that the performance of LM and LM-GAN are close to each other, with LM slightly and consistently performing better compared with LM-GAN, especially for larger population sizes (>20k) or with higher mutation rates (>0.5).
Figure 4.

Plot showing the number of accepted molecules included in the generated dataset
The results indicate that, with smaller population sizes and lower mutation rates, the LM-GAN method performs slightly better. However, as the population size and mutation rate increase, the performance of the standalone LM method gradually improves and surpasses that of the LM-GAN method.
Novel molecules
In Figure 3 we have plotted the comparison between these two methods in terms of their ability to generate novel molecules not contained in the original dataset. Straightforward LM is clearly producing more novel molecules. In this case we also see a trend of an initial increase in the number of novel molecules up to a mutation rate of 50% and then gradually decreasing. For the hybrid LM-GAN, the number of novel molecules is considerably smaller and decreases gradually with increasing mutation rate.
Figure 3.

The comparison plot illustrates the ability of two methods to generate novel molecules not present in the original dataset
The LM method clearly outperforms the LM-GAN method in terms of producing a higher number of novel molecules. Specifically, in the case of the LM method, there is an initial increase in the number of novel molecules up to a mutation rate of 50%, followed by a gradual decrease. In contrast, the LM-GAN method generates considerably fewer novel molecules, and this number decreases gradually as the mutation rate increases.
Accepted molecules
In Figure 4 we show the number of accepted molecules that were included into the generated dataset. Not all of the generated, novel molecules were accepted because only those with higher fitness scores were eventually merged into the new population, except at the beginning when novel molecules with positive fitness scores would be selected until the population size reaches a maximum; after that, molecules with higher scores are accepted during survivor selection. We see that, for smaller population size (<20k) and lower mutation rates, the hybrid LM-GAN performs slightly better with decreasing population size, while for a higher mutation rate the pure LM performance gradually improves as the population size becomes larger than 20k.
Efficiency
Finally, we estimate the efficiency of both LM and LM-GAN techniques in terms of their efficiency in generating new valid molecules with desired fitness scores. To determine their efficiency, we compute the fraction of novel molecules that were accepted into the population of newly generated molecules. As mentioned previously, we included only those novel molecules with higher fitness scores than their parent molecules. Therefore, in Figure 5, we plotted this ratio for the two different techniques for all different population sizes and mutation rates. It is clear that the hybrid LM-GAN consistently performs better (almost twice as much) compared with the simple LM. This finding is independent of population size, and we note that for the hybrid LM-GAN there is a tendency to deliver “better” molecules at higher masking rates than the straightforward LM. This is because the number of novel molecules generated by the LM-GAN is significantly lower compared with pure LM because of the action of the discriminator GAN network.
Figure 5.

Plot showing the efficiency values of two different techniques across various population sizes and mutation rates
The LM-GAN technique consistently outperforms the LM-only technique, with an efficiency value nearly twice as high. The population size does not appear to have a significant effect on the efficiency values, but there is a slight trend indicating that LM-GAN performs slightly better at higher masking rates.
LM pretraining on different population sizes
To eliminate any uncertainty due to the population size on which the LM was prepretrained, we performed our evaluation rungs using two different LM models trained on two different populations—1 million for one and 3 billion for the other—while keeping other parameters unchanged. We did not see any difference in outcome between these two models in fitness function, novel molecules, accepted molecules, or efficiency estimation. These results are plotted in Figures S1–S4.
Population distribution
Here, we compare the distribution of number density of different metrics (i.e., fitness function, synthesizability, drug likeness, and solubility) between LM and LM-GAN alongside the initial distribution for four different population sizes to show how much these two methods (i.e., LM and LM-GAN) have been able to improve the different metrics. The different metrics depict different behavior in each population. For example, in terms of synthesizability (Figure 6), LM-GAN performs better for smaller population sizes up to 10k compared with LM both in number density and scoring values. A similar trend is observed in drug likeliness (Figure 7). In the case of solubility (Figure 8), we do not see much difference between LM and LM-GAN up to 10–20k population sizes; however, in larger populations, the simple LM dominates. Finally, when we compare the final fitness function (Figure 9), we observe similar behaviors where LM-GAN performs better in terms of scoring values up to 10k, after which we observe the opposite trend.
Figure 6.

Plot comparing the distribution of number density for synthesizability between the LM (blue) and LM-GAN (orange) methods
The initial distribution is shown in green. The goal is to assess the improvement achieved by these methods. For synthesizability, LM-GAN exhibits better performance in smaller populations (up to 10,000) compared with LM, both in terms of number density and scoring values. The plot illustrates how the LM and LM-GAN methods have improved the different metrics, with varying effects depending on the specific matrix and population size.
Figure 7.

Plot comparing the distribution of number density for the drug likeness matrix between the LM (blue) and LM-GAN (orange) methods
The initial distribution is represented by the color green. The purpose of the plot is to evaluate the improvement achieved by these methods. In terms of drug likeness, the LM-GAN method performs better than LM in smaller populations (up to 10,000), as indicated by both the number density and scoring values. The plot demonstrates how the LM and LM-GAN methods have improved the drug likeness matrix, with the effect varying depending on the specific matrix and population size.
Figure 8.

Plot comparing the distribution of number density of solubility between LM (blue) and LM-GAN (orange), as well as the initial distribution (green)
The plot is shown for four different populations to demonstrate the improvement achieved by the LM and LM-GAN methods. The behavior of the different metrics varies across the populations. For solubility, there is not much difference between LM and LM-GAN up to populations of 10–20k. However, in larger populations, the LM method outperforms LM-GAN.
Figure 9.

Plot comparing the distribution of number density for the fitness function between LM (blue) and LM-GAN (orange) methods, along with the initial distribution (green)
The plot analyzes four different populations to assess the extent of improvement achieved by these two methods (LM and LM-GAN) across various metrics. The behavior of the different metrics varies depending on the population size. In terms of the fitness function, LM-GAN outperforms LM in scoring values up to 10,000. However, beyond this point, there is an opposite trend observed, indicating a shift in performance between the two methods.
Discussion
Sequence-based models for molecular design
In this work the used LM models take molecular sequence (SMILES representation for molecular fingerprints) as input data to generate new molecules based on scoring values or desired properties. Recently, sequence-based approaches have been used in contrast to time-consuming feature engineering or curation that strongly depend on molecular properties and fingerprints at local or global scales.32 On the other hand, classical techniques such as modeling and simulation use 3D structures to estimate interactions with target proteins, but they are restricted to small numbers of molecules because of the computational complexity of physics-based evaluations. This causes a high resource demand for very long periods of time, and at the same time the acquired information is not transferable in most cases. Therefore, a sequence-based method not only offers simplicity in terms of input to the model training but also is able to train and predict desired molecular structure without requiring 3D information and has been shown to perform in a favorable manner compared with the traditional techniques that require manual feature characterization.33,34,35
Mutation for molecule generation
In our works, the token-based mutation of molecular structure is key for generating new molecule designs that have desired properties. This mutation technique works differently from traditional GANs22,30 or variational autoencoders.36,37 This is because, rather than constructing entirely new molecules from the latent space by introducing specific noise into the distribution, we employ a controlled mechanism. This mechanism selects particular subsequences of molecules and introduces mutations into these selections. The mutation technique preserves specific structural components of the original molecule while systematically exploring the chemical space. This exploration is contingent upon various control parameters, such as mutation rates, population replacement strategies, or selection criteria based on scoring methodologies. Evidently, such a mutation strategy has multiple benefits. First, mutation is applied to a sampled population so each generation of a new molecule population dataset affects the training process, leading to providing solutions to the mode collapse problem. Second, the vast chemical space could be explored38 in a controlled way by selecting the mutation rate and frequency. Last, because mutations in every generation of evolution or scoring criteria can be manually examined or tuned, the interpretability of the mutation strategy is higher than for latency space-based generative models.
Figure 2 illustrates the relationship between the fitness score and the population size of the initial molecular dataset, as well as the mutation rate. Comparing the performance of LM and LM-GAN, we observe that they are similar, with LM consistently slightly outperforming LM-GAN. This difference is particularly evident in larger populations (20k) or with higher mutation rates (0.5).
Figure 2.

The fitness score is analyzed in relation to the population of the initial molecular dataset and the mutation rate
The performance of the LM and LM-GAN is similar, with the LM consistently performing slightly better than the LM-GAN. This difference becomes more pronounced, particularly in larger populations (20k) or with higher mutation rates (0.5).
A higher masking rate in LMs, requiring the model to predict more missing tokens, enhances its proficiency in comprehending context, making predictions based on surrounding words, and improving its ability to handle incomplete or partially observed text. However, in the case of LMs integrated into GANs, higher masking rates might not yield the same advantages. GANs are specifically designed to capture the underlying data distribution and generate realistic samples. The LM component within the GAN focuses on generating text that adheres to both learned language patterns and the data distribution captured by the GAN. Higher masking rates can disrupt the coherency of the generated text since the LM has less information available to guide the generation process. Producing coherent and meaningful text becomes more challenging for the LM when a significant portion of the input is masked.
Therefore, finding the right balance in the masking rate is crucial within the context of GANs. A masking rate that is too low might not provide sufficient training signal for the LM to learn meaningful language patterns. Conversely, a masking rate that is too high might hinder the coherency and quality of the generated text. The optimal masking rate in a GAN depends on the specific task, dataset, and interplay between the LM and GAN components.
Therefore, although higher masking rates can benefit standalone LMs, LMs integrated into GANs require careful consideration of the masking rate to maintain a balance between language coherency and the data distribution captured by the GAN.
Combined LM and GAN approach
Large language models, such as GPT, utilize the transformer architecture as their core foundation, enabling excellence in tasks such as language translation, text summarization, and conversational agents. This proficiency is attained through pretraining on extensive text data, leveraging the transformative role of the transformer architecture as the fundamental framework that enhances their language processing capabilities. In terms of generative models, the GANs and the transformers are two powerful ML generative models. They have been used separately in various diverse domain applications, although primarily for NLP and image generation. However, as we have shown here in our novel implementation for molecular structure generation, when used together they can produce even better results in a number of cases, including designing new and improved molecules with improved target properties. There are several reasons why this specific combination of GANs and transformers is able to produce better results. First, the combined approach can deliver improved generative capabilities. This is because GANs are generally designed to generate new data that closely resemble the training data, while transformers have the ability to model complex dependencies in the heterogeneous data taking into account the context and subsequently generate realistic outputs from that. Thus, together, these two models can deliver more accurate and diverse outputs than when compared individually. Second, coupled implementations have better control over the generated output. GANs are generally conditioned on specific input data, while the use of transformers can be tuned to certain criteria or characteristics of the generated output, such as specific properties. The next advantage is efficient training where the training of GANs and the transformers are performed separately, but then they can be fine-tuned together with the pretrained model depending on the specific requirements, thus substantially reducing the need for extensive training every single time. Last, a joint implementation of GANs and transformers enables the generative model to learn the high-level and low-level features, as well as the long-range and short-range relation in the data. This, in turn, results in a more efficient and accurate prediction of material behavior and properties. In summary, the joint approach of GANs and transformers can provide advantages for more efficient molecular and materials design with desired properties, with the condition that the quality of the outcome will still depend on initial data quality and its respective diversity.
Population size and distribution for molecule generation
The impact of population size in generating customized novel molecules using different algorithms is distinct. When considering the population size of the molecule data, the choice between using an LM or an LM implemented on a GAN (LM-GAN) depends on the specific circumstances. With smaller population sizes, LM-GAN is undoubtedly the better choice. However, as the population size increases (typically exceeding 20k), the advantage of using a GAN diminishes when examining individual metrics.
When evaluating the efficacy of these two different approaches in terms of accepted molecules, LM-GAN is significantly better compared with LM. We surmise that this is a consequence of the general observation that the GAN approach works generally better with smaller training data, while LM-like generative models require considerably larger datasets. Therefore, the hybrid LM-GAN reported here provides a viable pathway to generate novel structures when there is not a large amount of data available to work with.
Population and molecule size
Handling of dataset sizes differs between LMs and GANs. Generally, LMs tend to excel with larger datasets, while GANs can be effective even with smaller ones.
LMs derive benefits from larger datasets because of their reliance on statistical patterns and their ability to learn the underlying language structure. With a substantial dataset, the model gains access to a wide range of language patterns, vocabulary, and context. This enables the LM to generalize better, capture diverse linguistic nuances, and generate text that is more coherent and contextually appropriate. The abundance of data facilitates the learning of statistical regularities and dependencies within the language, leading to improved performance.
In contrast, GANs, particularly in tasks such as image generation, can work well with smaller datasets. GANs are specifically designed to learn the data distribution and produce realistic samples. In some cases, having a smaller dataset can be advantageous as GANs can better capture the unique characteristics of the available data. With a limited dataset, GANs can focus on learning the intricate details, textures, and specific patterns that are distinctive to the data. This results in more accurate and faithful generation of new samples. Furthermore, GANs can be prone to overfitting when trained on larger datasets, which might hinder their ability to capture fine-grained details and generate realistic samples.
However, it is important to note that these observations are general tendencies and not absolute rules. The performance of both LMs and GANs can still vary depending on the specific task, dataset quality, and model architecture. Dataset size is just one of many factors that can influence the performance of these models.
In terms of molecule size, i.e., number of atoms in a molecule, as detailed in the molecular structure data within the experimental procedures, our study initiated with an initial population from GDB9, consisting of molecules with up to nine heavy atoms (CONF), excluding hydrogen. The application of both LM-based models’ generative capabilities subsequently resulted in an approximately 2-fold increase in the average molecule size in the subsequent generations of newly generated molecules. Specifically, the average molecule size increased from 8.8 atoms in the starting population to 17.6 atoms when utilizing the LM as the generative model, while, in contrast, employing the LM-GAN as the generative model yielded a further increase to an average molecule size of 19.2 atoms. The LM-GAN also favored heavier atoms, as shown in Figure S5. Furthermore, the topological accessible surface was increased in the LM-GAN compared with the LM only approach, along with the maximum partial charge and the number of valence electrons. Twenty molecules from the population of 5k molecules generated using the LM-GAN and LM only are shown in Figure S7.
Population distribution
The behavior of LMs and LMs integrated into GANs differs concerning the population distribution of generated samples.
In a standalone LM, the generated samples are typically representative of the language patterns and statistical properties present in the training data. The LM learns to produce text that aligns with the underlying data distribution on which it was trained. Consequently, the population distribution of generated samples from a standalone LM tends to resemble the distribution of the training data. The LM prioritizes generating coherent, fluent text that is statistically similar to the training data, while maintaining the learned language patterns.
On the other hand, when an LM is integrated into a GAN, the population distribution of generated samples can be influenced by both the LM component and the GAN component. GANs aim to capture the overall data distribution and generate samples that are realistic and indistinguishable from real data. The GAN component introduces an additional factor that shapes the population distribution of generated samples.
The interaction between the LM and the GAN in a GAN-based LM can result in a population distribution of generated samples that differs from that of a standalone LM. The GAN component might prioritize generating samples that align with the overall data distribution captured by the GAN, which might diverge from the distribution of the training data. This can lead to generated samples exhibiting realistic characteristics and conforming to the data distribution captured by the GAN but deviating from the precise language patterns and statistical properties of the training data. We demonstrate this difference in Figure S6, which shows the distribution of 5k molecules after 50 generations for a small population size (5k molecules). In this chemical space, we see distinct differences in the population diversity. The increased diversity of the LM-GAN-generated molecules is consistent with the chemical features shown in Figure S5.
In conclusion, although a standalone LM primarily focuses on generating text that aligns with the language patterns and statistical properties of the training data, an LM integrated into a GAN takes into account both the learned language patterns and the overall data distribution captured by the GAN. This interplay can result in differences in the population distribution of generated samples, with GAN-based LMs potentially producing samples that are realistic and that conform to the GAN’s data distribution but might deviate from the exact language patterns observed in the training data.
Conclusions
In this work we present a comparative study on the efficacy of two generative molecular structure models, namely a state-of-the art LM on one hand, and a novel hybrid architecture where the same LM is deployed on a GAN platform (i.e., LM-GAN). The objective of performing various specific or user-defined optimizations tasks was accomplished in both models by taking advantage of nature-inspired GA-based mutations strategy. The obtained results have implications that are multidimensional. First, we show that the hybrid LM-GAN architecture predicts a higher fraction of acceptable molecules from the set of newly generated structures with improved target properties for any dataset size than the standalone LM, while the use of the GA addresses the problem of mode-collapse commonly found in GANs. Second, the results show that, with smaller population sizes, the hybrid LM-GAN performs better in generating desired molecules both in terms of improved optimized properties and larger number of atoms. This trend reverses when the population size increases. This addresses another important issue of requiring a large amount of data in model training for generating new samples because the LM-GAN is able to work with comparatively smaller population sizes. Third, the LM method performs better when counting generation of more numbers of novel molecules. But the most impactful observation is when estimating the efficiency of respective algorithms (i.e., when computing the ratio of the number of accepted molecules to the number of generated novel molecules with desired optimized molecular properties, LM-GAN performance is consistently better compared with LM among all population sizes of the molecule dataset). This novel AI algorithm will enable researchers to design new molecules with desired properties with different sizes of available training datasets in a wide range of domains from drug discovery to synthesizing novel polymers to building new materials.
Experimental procedures
Resource availability
Lead contact
Further information and requests for resources and data should be directed to and will be fulfilled by the lead contact, Debsindhu Bhowmik (bhowmikd@ornl.gov).
Materials availability
The materials employed in this study are openly accessible and outlined in the following molecular structure data section. For requests regarding the newly generated molecular structures and properties data, please find published data at Zenodo39 or contact the lead author.
Data and code availability
Source code for this work is published and can be found at Zenodo.40
LM-based GAN architecture
The primary focus of this study is to investigate, within the GAN paradigm,41 LMs trained on text-based molecular representations (SMILES) both as generator and discriminator networks. For this purpose we compute the boost in efficiency in generating valid molecules while simultaneously demonstrating a comparative study on fitness optimization. As in our previous work, we use a masked LM21,23,24 for our automated mutation operator driven by a GA. In Figure 1 we have shown the major components of the two architectures used in this study. In the LM-only architecture, the input population of molecules is provided as a SMILES string format. A part of this SMILES string representing each of the molecules is then masked, and subsequently an LM model is used to predict mutation or alternative to the original input molecule. This newly generated molecule with mutated atom(s) is then scored and selected based on a specific fitness function to generate a new dataset consisting of a population of optimized molecules. This process of introducing a mutation and the novel way of selection is repeated for multiple iterations to generate the final molecule population. In our hybrid LM-GAN method, to implement a GAN, the generator and discriminator networks are defined, a training loop is designed, and backpropagation is used for parameter updates. This involves initializing the networks, specifying loss functions, creating optimizers, and implementing a training loop that alternates between updating the generator and discriminator. In this GAN paradigm, as shown in Figure 1 (lower), the generator creates a new molecule by randomly masking a portion of a SMILES molecular representation and using a pretrained LM to fill in the missing molecular structure. The discriminator network is a (pretrained) LM that classifies the original or generated SMILES strings as real or fake, respectively. One might think of training the discriminator as a fine-tuning task for the LM and of training the generator as a masked language modeling task. The generator and discriminator LMs are iteratively trained such that the masked LM generates molecules that can fool the discriminator.
Implementing an LM on a GAN platform entails several differences compared with using a GAN in isolation. First, the architecture incorporates both the GAN components (generator and discriminator) and the LM, integrating the LM into the generator. This integration allows the generator to generate context and sequence based on learned language patterns.
The training process combines adversarial training and language modeling. Adversarial training involves the generator and discriminator competing to enhance the quality of generated samples, while the LM learns statistical patterns in the training data. This joint training empowers the generator to produce samples that align with both the GANs data distribution and the language patterns learned by the LM.
In a GAN, the objective function includes the generator’s loss for promoting realistic text generation and the discriminator’s loss for distinguishing real and generated sequences. Integrating an LM introduces an additional language modeling loss, encouraging the generator to generate sequences that are aligned with the language patterns captured by the LM. By combining these distinct loss components, the training process optimizes overall sequence generation quality, considering realism and pattern coherence.
The integration of an LM within a GAN platform enhances sequence generation quality compared with using a standalone GAN. LMs excel at capturing language patterns and generating coherent sequences, while GANs effectively capture data distribution and generate realistic ones. Leveraging the strengths of both approaches enables higher-quality sample generation that exhibits both realistic characteristics and adherence to language patterns. The result is an LM-based generator that produces realistic molecules for a broad scope of potential tasks.
GA
As mentioned previously and following our previous works,22,23,24,25 we have adopted in both LM- and LM-GAN-based generative models a simplified GA strategy to introduce diversity in the generated population. To this end, we have implemented an algorithm that follows a survivor selection. We deploy random sampling uniformly applied over the population to select μ parents, and mutation was used only to generate new molecules as described in our earlier work.21 An initial population ranging in size from 5k to 50kmolecules was used for comparing different scenarios, and mutated molecules were created by taking the top 5 predictions of the model for a given set of applied masks. The validity and uniqueness of the generated molecules were evaluated using functionalities in the rdkit42 library to convert SMILES representation into a canonical form. In the end, only unique molecules were retained in the population for subsequent structure evolution.
Mask generation and fitness calculations
The use of various masking ratios in an LM holds great significance for multiple reasons.21 It enhances the model’s capability to handle incomplete or noisy data by leveraging contextual cues to predict missing tokens. The incorporation of diverse masking ratios fosters generalization, empowering the model to effectively handle different levels of missing information, thereby improving its real-world performance. Moreover, it facilitates transfer learning by capturing distinct contextual dependencies across different ratios. These masking ratios also serve as a means of data augmentation, effectively mitigating overfitting and bolstering generalization on unseen data. The flexibility to adjust the masking ratio allows the model to cater to specific task requirements, whether it involves inferring missing words or generating coherent text. Therefore, in this comparative study we used different mutation rates for mask generation, or, in other words, we used a series of various probabilities for which a given token in a molecular sequence would be masked. These mutation rates range from 0.10 to 1.00 with a gap of 0.10. Furthermore, these mutations were deployed in three different ways: replacement, insertion, and deletion. At least one mask was applied on each molecule with a randomly chosen masking location. As for the replacement strategy, the token location was replaced with the mask; for insertion, a mask was inserted on the token location and, for deletion token, location was deleted following the mask. For insertion or deletion, any remaining locations followed replacement strategy.
The optimized fitness property was determined by taking the harmonic mean of multiple molecular metrics. As an example for metric and , the resultant fitness function F will be = . We took the drug likeness, synthesizability, and solubility as individual metrics for quantitative estimation of optimized fitness function as done in many earlier works on molecular optimization.22,29,30,31
Molecular structure data
For this study, we chose an initial population derived from a subset of molecules sourced from the QM9 dataset,43 referred to as GDB9 in this investigation. The QM9 dataset is recognized as the gold standard for ML predictions in various chemical properties, given its calculated geometric, energetic, electronic, and thermodynamic properties for 134k stable small organic molecules composed of CHONF elements. Notably, the QM9 dataset is grounded in the exploration of the chemical space. GDB9 molecules constitute a subset of 133,885 species within the GDB-17 chemical universe, encompassing 166 billion organic molecules with up to 9 heavy atoms (CONF), excluding hydrogen. This forms a comprehensive and diverse foundation for our comparative analysis. Importantly, our approach utilizes only the SMILES structure as the starting point to generate new molecules in subsequent iterations.
LM training
The pretraining phase of the LM involves two key stages: tokenization and mask prediction. In the tokenization process, a vocabulary is constructed based on frequently occurring subsequences found in the SMILES representation of molecules. SMILES sequences are split using punctuation, following the default setting of the BERT WordPiece tokenizer in the Hugging Face transformers library.44 This results in the creation of a vocabulary for the entire dataset, comprising molecules, as mentioned earlier. The vocabulary size is set to 32,768. Simultaneously, in the mask prediction stage, PyTorch and Hugging Face Transformers, along with DeepSpeed for distributed training,45 are employed. The transformer architecture utilized for the molecule LM is BERT based, boasting approximately 109 million learnable parameters. The pretraining process adopts data parallelism, where each GPU is trained with the model on distinct data. DeepSpeed’s fused LAMB optimizer is used for efficient training at scale on a dataset consisting of 3 billion molecules (i.e., the first 6,000 partitions of the complete molecule dataset). The pretraining is executed on the Summit supercomputer using 1,000 nodes, each equipped with 6 Nvidia 16 GB V100 GPUs. Each GPU is assigned a partition of the dataset, and a batch size of 80 molecules with 3 gradient accumulation steps per GPU is utilized, resulting in a global batch size of 1.44 million. Despite the need for a large number of GPUs for training, once the LM is pretrained it can be employed with just one GPU for fine-tuning or downstream tasks on smaller datasets. The pretraining phase spans 7 epochs, taking approximately 2.5 h, with model validation conducted using mask prediction on a hold-out set of molecules. The best validation accuracy is achieved in the final epoch, and the resulting model weights are frozen for subsequent LM mutations.
Acknowledgments
This research used resources of the Oak Ridge Leadership Computing Facility at Oak Ridge National Laboratory (ORNL), which is supported by the Office of Science of the US Department of Energy (DOE) under contract no. DE-AC05-00OR22725. This research was funded by the AI Initiative, as part of the Laboratory Directed Research and Development Program of ORNL, managed by UT-Battelle LLC for DOE; the Exascale Computing Project (17-SC-20-SC), a collaborative effort of the DOE Office of Science and the National Nuclear Security Administration.
This manuscript has been authored by UT-Battelle LLC under contract no. DE-AC05-00OR22725 with the US Department of Energy (DOE). The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of the manuscript, or allow others to do so, for US government purposes. DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (http://energy.gov/downloads/doe-public-access-plan).
Author contributions
Conceptualization, D.B.; methodology, D.B. and J.G.; investigation, D.B., Z.F., and J.G.; writing – original draft, D.B.; writing – review & editing, D.B., P.Z., Z.F., S.I., and J.G.; funding acquisition, D.B., J.G., and S.I.; resources, D.B. and J.G.
Declaration of interests
The authors declare no competing interests.
Published: April 12, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.patter.2024.100947.
Supplemental information
References
- 1.EDITORIAL For chemists, the ai revolution has yet to happen. Nature. 2023;617:438. doi: 10.1038/d41586-023-01612-x. [DOI] [PubMed] [Google Scholar]
- 2.Anstine D.M., Isayev O. Generative models as an emerging paradigm in the chemical sciences. J. Am. Chem. Soc. 2023;145:8736–8750. doi: 10.1021/jacs.2c13467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bond-Taylor S., Leach A., Long Y., Willcocks C.G. Deep generative modelling: A comparative review of vaes, gans, normalizing flows, energy-based and autoregressive models. IEEE Trans. Pattern Anal. Mach. Intell. 2022;44:7327–7347. doi: 10.1109/TPAMI.2021.3116668. [DOI] [PubMed] [Google Scholar]
- 4.Yao Z., Lum Y., Johnston A., Mejia-Mendoza L.M., Zhou X., Wen Y., Aspuru-Guzik A., Sargent E.H., Seh Z.W. Machine learning for a sustainable energy future. Nat. Rev. Mater. 2023;8:202–215. doi: 10.1038/s41578-022-00490-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bilodeau C., Jin W., Jaakkola T., Barzilay R., Jensen K.F. Generative models for molecular discovery: Recent advances and challenges. WIREs Comput. Mol. Sci. 2022;12 doi: 10.1002/wcms.1608. [DOI] [Google Scholar]
- 6.Zhang P., Kearney L., Bhowmik D., Fox Z., Naskar A.K., Gounley J. Transferring a molecular foundation model for polymer property predictions. J. Chem. Inf. Model. 2023;63:7689–7698. doi: 10.1021/acs.jcim.3c01650. [DOI] [PubMed] [Google Scholar]
- 7.Sattari K., Xie Y., Lin J. Data-driven algorithms for inverse design of polymers. Soft Matter. 2021;17:7607–7622. doi: 10.1039/D1SM00725D. [DOI] [PubMed] [Google Scholar]
- 8.Lim J., Hwang S.-Y., Moon S., Kim S., Kim W.Y. Scaffold-based molecular design with a graph generative model. Chem. Sci. 2019;11:1153–1164. doi: 10.1039/C9SC04503A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Segler M.H.S., Kogej T., Tyrchan C., Waller M.P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 2018;4:120–131. doi: 10.1021/acscentsci.7b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arús-Pous J., Johansson S.V., Prykhodko O., Bjerrum E.J., Tyrchan C., Reymond J.-L., Reymond J.-L., Chen H., Engkvist O. Randomized smiles strings improve the quality of molecular generative models. J. Cheminf. 2019;11 doi: 10.1186/s13321-019-0393-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flam-Shepherd D., Zhu K., Aspuru-Guzik A. Language models can learn complex molecular distributions. Nat. Commun. 2022;13:3293–3310. doi: 10.1038/s41467-022-30839-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Awale M., Sirockin F., Stiefl N., Reymond J.-L. Drug analogs from fragment-based long short-term memory generative neural networks. J. Chem. Inf. Model. 2019;59:1347–1356. doi: 10.1021/acs.jcim.8b00902. [DOI] [PubMed] [Google Scholar]
- 13.Zheng S., Yan X., Gu Q., Yang Y., Du Y., Lu Y., Xu J. Qbmg: quasi-biogenic molecule generator with deep recurrent neural network. J. Cheminf. 2019;11 doi: 10.1186/s13321-019-0328-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Méndez-Lucio O., Baillif B., Clevert D.-A., Rouquié D., Wichard J. De novo generation of hit-like molecules from gene expression signatures using artificial intelligence. Nat. Commun. 2020;11 doi: 10.1038/s41467-019-13807-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aruś-Pous A.J., Patronov A. Smiles-based deep generative scaffold decorator for de-novo drug design. J. Cheminf. 2020;12:1–18. doi: 10.1186/s13321-020-00441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong S.H., Ryu S., Lim J., Kim W.Y. Molecular generative model based on an adversarially regularized autoencoder. J. Chem. Inf. Model. 2020;60:29–36. doi: 10.1021/acs.jcim.9b00694. [DOI] [PubMed] [Google Scholar]
- 17.Kotsias P.C., Arús-Pous J., Aruś-Pous, Engkvist O., Tyrchan C., Bjerrum E.J. Direct steering of de novo molecular generation with descriptor conditional recurrent neural networks. Nat. Mach. Intell. 2020;2:254–265. doi: 10.1038/s42256-020-0174-5. [DOI] [Google Scholar]
- 18.Fabian B., Edlich T., Gaspar H., Segler M.H.S., Meyers J., Fiscato M., Ahmed M. Molecular representation learning with language models and domain-relevant auxiliary tasks. arXiv. 2020 doi: 10.48550/arXiv.2011.13230. Preprint at. [DOI] [Google Scholar]
- 19.Lin Z., Khetan A., Fanti G.C., Oh S. Pacgan: The power of two samples in generative adversarial networks. arXiv. 2017;1 doi: 10.48550/arXiv.1712.04086. Preprint at. [DOI] [Google Scholar]
- 20.Eiben A.E., Smith J.E. 2nd ed. Springer-Verlag GmbH Germany: Springer Publishing Company; 2015. Introduction to Evolutionary Computing. [DOI] [Google Scholar]
- 21.Blanchard A.E., Shekar M.C., Gao S., Gounley J., Lyngaas I., Glaser J., Bhowmik D. Automating Genetic Algorithm Mutations for Molecules Using a Masked Language Model. IEEE Trans. Evol. Comput. 2022;26:793–799. doi: 10.1109/TEVC.2022.3144045. [DOI] [Google Scholar]
- 22.Blanchard A.E., Stanley C., Bhowmik D. Using GANs with adaptive training data to search for new molecules. J. Cheminf. 2021;13 doi: 10.1186/s13321-021-00494-3. 14–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blanchard A.E., Zhang P., Bhowmik D., Mehta K., Gounley J., Reeve S.T., Irle S., Pasini M.L. In: Accelerating Science and Engineering Discoveries Through Integrated Research Infrastructure for Experiment, Big Data, Modeling and Simulation. SMC. Doug K., Al G., Pophale S., Liu H., Parete-Koon S., editors. Vol. 1690. Communications in Computer and Information Science; 2022. Computational workflow for accelerated molecular design using quantum chemical simulations and deep learning models; pp. 3–19. Springer Nature Switzerland. [DOI] [Google Scholar]
- 24.Blanchard A.E., Gounley J., Bhowmik D., Chandra Shekar M., Lyngaas I., Gao S., Yin J., Tsaris A., Wang F., Glaser J. Language Models for the Prediction of SARS-CoV-2 Inhibitors. Int. J. High Perform. Comput. Appl. 2022;36:587–602. doi: 10.1177/1094342022112180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubey S.R., Singh S.K. Transformer-based generative adversarial networks in computer vision: A comprehensive survey. arXiv. 2023 doi: 10.48550/arXiv.2302.08641. Preprint at. [DOI] [Google Scholar]
- 26.Jiang Y., Chang S., Wang Z. Transgan: Two pure transformers can make one strong gan, and that can scale up. arXiv. 2021 doi: 10.48550/arXiv.2102.07074. Preprint at. [DOI] [Google Scholar]
- 27.Zhumagambetov R., Molnár F., Peshkov V.A., Fazli S. Transmol: repurposing a language model for molecular generation. RSC Adv. 2021;11:25921–25932. doi: 10.1039/D1RA03086H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bagal V., Aggarwal R., Vinod P.K., Priyakumar U.D. Molgpt: Molecular generation using a transformer-decoder model. J. Chem. Inf. Model. 2022;62:2064–2076. doi: 10.1021/acs.jcim.1c00600. [DOI] [PubMed] [Google Scholar]
- 29.Ertl P., Schuffenhauer A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminf. 2009;1:8–11. doi: 10.1186/1758-2946-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Cao N., Kipf T. MolGAN: An implicit generative model for small molecular graphs. arXiv. 2018 doi: 10.48550/arXiv.1805.11973. Preprint at. [DOI] [Google Scholar]
- 31.Bickerton G.R., Paolini G.V., Besnard J., Muresan S., Hopkins A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012;4:90–98. doi: 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minnich A.J., McLoughlin K., Tse M., Deng J., Weber A., Murad N., Madej B.D., Ramsundar B., Rush T., Calad-Thomson S., et al. AMPL: A Data-Driven Modeling Pipeline for Drug Discovery. J. Chem. Inf. Model. 2020;60:1955–1968. doi: 10.1021/acs.jcim.9b01053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang S., Guo Y., Wang Y., Sun H., Huang J. Smiles-Bert: Large scale unsupervised pre-training for molecular property prediction. ACM-BCB. 2019;2019:429–436. doi: 10.1145/3307339.3342186. [DOI] [Google Scholar]
- 34.Xue D., Zhang H., Xiao D., Gong Y., Chuai G., Sun Y., Tian H., Wu H., Li Y., Liu Q. X-MOL: large-scale pre-training for molecular understanding and diverse molecular analysis. bioRxiv. 2020 doi: 10.1101/2020.12.23.424259. Preprint at. [DOI] [PubMed] [Google Scholar]
- 35.Kim H., Lee J., Ahn S., Lee J.R. A merged molecular representation learning for molecular properties prediction with a web-based service. Sci. Rep. 2021;11 doi: 10.1038/s41598-021-90259-7. 11028–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gómez-Bombarelli R., Wei J.N., Duvenaud D., Hernández-Lobato J.M., Sánchez-Lengeling B., Sheberla D., Aguilera-Iparraguirre J., Hirzel T.D., Adams R.P., Aspuru-Guzik A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018;4:268–276. doi: 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobs S.A., Moon T., McLoughlin K., Jones D., Hysom D., Ahn D.H., Gyllenhaal J., Watson P., Lightstone F.C., Allen J.E., et al. Enabling rapid COVID-19 small molecule drug design through scalable deep learning of generative models. Int. J. High Perform. Comput. Appl. 2021;35:469–482. doi: 10.1177/10943420211010930. [DOI] [Google Scholar]
- 38.Virshup A.M., Contreras-García J., Wipf P., Yang W., Beratan D.N. Stochastic voyages into uncharted chemical space produce a representative library of all possible drug-like compounds. J. Am. Chem. Soc. 2013;135:7296–7303. doi: 10.1021/ja401184g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhowmik D., Zhang P., Fox Z., Irle S., Gounley J. 2023. molecular data for Genetic Algorithm Mutations for Molecules with a Hybrid Language Model-Based GAN Architecture. Zenodo. URL: [DOI] [Google Scholar]
- 40.Bhowmik D., Zhang P., Fox Z., Irle S., Gounley J. 2023. Genetic Algorithm Mutations for Molecules with a Hybrid Language Model-Based GAN Architecture. Zenodo. URL: [DOI] [Google Scholar]
- 41.Goodfellow I.J., Pouget-Abadie J., Mirza M., Xu B., Warde-Farley D., Ozair S., Courville A., Bengio Y. Generative adversarial networks. arXiv. 2014 doi: 10.48550/arXiv.1406.2661. Preprint at. [DOI] [Google Scholar]
- 42.Landrum G., Tosco P., Kelley B., Ric, Cosgrove D., sriniker, gedeck, Vianello R., NadineSchneider, Kawashima E., Jones G., N D., Dalke A., et al. 2023. jasondbiggs, and strets123. RDKit: Open-source cheminformatics. Zenodo. URL: [DOI] [Google Scholar]
- 43.Ramakrishnan R., Dral P.O., Rupp M., Von Lilienfeld O.A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data. 2014;1:140022–140027. doi: 10.1038/sdata.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolf T., Debut L., Sanh V., Chaumond J., Delangue C., Moi A., Cistac P., Rault T., Louf R., Funtowicz M., et al. Proceedings of the 2020 Conference on Empirical Methods in Natural Language Processing: System Demonstrations. 2020. Transformers: State-of-the-art natural language processing; pp. 38–45. [DOI] [Google Scholar]
- 45.Rajbhandari S., Rasley J., Ruwase O., He Y. Zero: Memory optimizations toward training trillion parameter models. Int. Conf. High Perform. Comput. Netw. Storage Anal. 2020:1–24. doi: 10.1109/SC41405.2020.00024. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source code for this work is published and can be found at Zenodo.40