

Abstract
Clear cell renal cell carcinoma (ccRCC) constitutes the most common renal cell carcinoma subtype and has long been recognized as an immunogenic cancer. As such, significant attention has been directed toward optimizing immune-checkpoints (IC)-based therapies. Despite proven benefits, a substantial number of patients remain unresponsive to treatment, suggesting that yet unreported, immunosuppressive mechanisms coexist within tumors and their microenvironment. Here, we comprehensively analyzed and ranked forty-four immune-checkpoints expressed in ccRCC on the basis of in‐depth analysis of RNAseq data collected from the TCGA database and advanced statistical methods designed to obtain the group of checkpoints that best discriminates tumor from healthy tissues. Immunohistochemistry and flow cytometry confirmed and enlarged the bioinformatics results. In particular, by using the recursive feature elimination method, we show that HLA-G, B7H3, PDL-1 and ILT2 are the most relevant genes that characterize ccRCC. Notably, ILT2 expression was detected for the first time on tumor cells. The levels of other ligand-receptor pairs such as CD70:CD27; 4-1BB:4-1BBL; CD40:CD40L; CD86:CTLA4; MHC-II:Lag3; CD200:CD200R; CD244:CD48 were also found highly expressed in tumors compared to adjacent non-tumor tissues. Collectively, our approach provides a comprehensible classification of forty-four IC expressed in ccRCC, some of which were never reported before to be co-expressed in ccRCC. In addition, the algorithms used allowed identifying the most relevant group that best discriminates tumor from healthy tissues. The data can potentially assist on the choice of valuable immune-therapy targets which hold potential for the development of more effective anti-tumor treatments.
Electronic supplementary material
The online version of this article (10.1007/s00262-020-02530-x) contains supplementary material, which is available to authorized users.
Keywords: HLA-G, Transcriptome, Immune-checkpoints, RNAseq, TCGA, ccRCC
Introduction
Clear cell renal cell carcinoma (ccRCC) is a renal tumor typically characterized by malignant epithelial cells with clear cytoplasm and a compact alveolar or acinar growth pattern interspersed with intricate, arborizing vasculature. Among kidney cancers, 70% are estimated to be ccRCC [1] constituting the most common renal cell carcinoma subtype. Patients cannot be treated by radiation or chemotherapy. Metastases at the diagnosis time are observed for 25–30% of the patients and less than 10% of these survive more than five years. Recurrence occurs in 20–30% of patients even after complete nephrectomy of primary tumors. Genetic evidence revealed that ccRCC originates from sequential losses of multiple tumor suppressor genes such as von Hippel-Lindau (VHL) gene. Other genes such as PBRM1 (Polybromo 1) and BAP1 (BRCA1-associated protein) have also been identified as renal cancer driver genes, mainly underexpressed in ccRCC [2]. These three genes are closely located on chromosome 3p. Remarkably, chromosome 3p loss occurs in more than 90% of sporadic ccRCCs [3].
ccRCC has long been recognized as an immunogenic cancer. As such, significant attention has been directed toward optimizing immune-checkpoints (IC)-based therapies. IC are defined as cell surface molecules that transduce positive or negative signals, into effector cells. When the same ligand interacts with an activating or an inhibitory receptor, inhibition is dominant and allows tumor cells to escape destruction by the immune system [4]. Therefore, blockade of inhibitory IC by antibody-based therapeutics constitutes a suitable strategy to restore an effective anti-tumor response [5, 6].
Clinical phase I/II trials have provided evidence that treatments with anti-CTLA-4 or PD-L1:PD-1 antibodies are capable of restoring suitable anti-tumor responses and that therapies using combinations of anti-CTLA-4 and PD-L1:PD-1 are more efficient than using each one individually [7–9]. Although these trials have helped to give a step forward in the treatment of some patients, others still remain unresponsive. Potential causes might be the complexity of IC pathways since several yet undescribed checkpoints, besides CTLA-4 and PD-1:PD-L1, might be expressed simultaneously and act concomitantly through non-overlapping, sometimes opposite, mechanisms. In addition, other causes of the inefficiency of the treatments might be the heterogeneity of expression within tumor cells or the toxicity due to their action on non-tumor tissues. Consequently, to achieve optimal specificity and limit potential side effects, IC targeted by immune-therapies must have restricted expression in normal tissues and high expression upon cellular transformation.
In this context, the search for novel IC targets has been intensified. Ongoing trials include antibody-based protocols against LAG-3, TIM-3 and VISTA [10] or agonist antibody-based protocols for co-stimulatory molecules ICOS, OX40 and 4-1BB [11]. Some of these molecules are evaluated in mono or combined therapies. However, the uncomplete list of their side effects and the lack of knowledge about their mechanisms of action prevent their immediate clinical utilization.
The advent of next-generation sequencing technologies, particularly applied to RNAseq, has demonstrated its usefulness in classifying patients with similar pathologies, screening genes involved in carcinogenesis and in identifying prognostic factors in cancer [12–14]. This technique also revealed inter- and intra-tumor heterogeneity which challenges the precise diagnosis and selection of reliable therapeutic targets [15]. The full exploration of large and growing public databases such as The Cancer Genome Atlas (TCGA) data portal (https://tcga-data.nci.nih.gov/tcga/tcgaHome2.jspa) which covers RNAseq transcriptome data for more than 10,000 samples of human cancers including over 30 different cancer types, holds the potential to help identify suitable therapeutic targets.
Many statistical tools were developed to analyze RNA-seq data [16]. Comparison of these different tools16 singled out DESeq2 as the most conservative [17]. However, several statistical challenges must still be faced in order to unravel molecular processes and interactions at stake for each cancer type.
Here, we provide a novel and comprehensible view on forty-four IC expressed in ccRCC and highlight the most relevant group of checkpoints that characterize ccRCC and the ones that best discriminate tumor from healthy tissues. The data can potentially assist on the choice of valuable immune-therapy targets which hold potential for the development and effectiveness of anti-tumor treatments.
Materials and methods
TCGA data collection and processing
RNA-sequencing profiles of ccRCC samples and their related clinical data obtained from The Cancer Genome Atlas (TCGA) data portal (https://tcga-data.nci.nih.gov/tcga/) were used to identify expressed IC. A set of 25 283 RNA-seq derived from the 539 patients with ccRCC was downloaded. These were normalized by the FPKM-UQ method that scales the number of reads of each gene compared to the total number of reads and the length of genes.
Statistical methods used for the differential analysis
The differential analysis was conducted on the RNA-seq data using DESeq2 [18] which is a statistical method implemented in an R software package. DESeq2 allows identifying differentially expressed genes using the standard comparison mode between two experimental conditions. A high variability between patients exists in RNA-seq data. Therefore, to take into account this variability and accurately analyze these data, it is mandatory to use a distribution that incorporates over-dispersion as does the negative binomial distribution. DESeq2 takes into account this variability by using Wald test based on a negative binomial distribution to test whether each model, tumor or healthy cells, differs significantly from zero. It models counts by a negative binomial distribution because counts are over-dispersed (the variance is higher than the mean), and the negative binomial distribution has two parameters called mean and dispersion which enable to model the mean–variance relationship. A scaling factor normalization procedure is performed to take into account the varying sequencing depths of the different samples. The generalized linear models (GLMs) with a logarithmic link are used to estimate coefficients that indicate the overall expression strength of the gene. The p-value adjustment for multiple tests was performed by the Benjamini–Höchberg procedure [19], which consists in controlling the false discovery rate (FDR). We considered differentially expressed genes those with an adjusted p value ≤ to 0.05.
Feature selection method
Supervised learning is a powerful tool for mathematical analysis of biological data. Here, we consider the following classification task: “how well can I predict if a tissue is tumoral or not with gene expression data?” In this section, our goal is to rank IC genes according to their powers of prediction: We want to find the smallest and the best subset of genes that realizes this classification task. To achieve this, we consider the recursive feature elimination (RFE) algorithm [20] with a linear-support vector machine (linear-SVM). This algorithm selects features by recursively considering smaller and smaller sets of features and assigns weights to the features directly linked to the coefficients of the linear model. A subset of the ranked genes is selected according to the SVM-IC [21]. The details of the method are explained below.
Dataset and tools
Python and scikit-learn were used [22]. The features are normalized (FPKM-UQ normalization) gene expressions of the differentially expressed genes (d = 41) in order to use well-known gene selection techniques for micro-array-based data [23]. The data are scaled with zero mean and variance one for better performance with the linear-SVM (see below for details of this algorithm).
Linear Support Vector Machine training
We focused on a binary classification model. We want to build a classifier f:
where is the gene expression data of a tissue sample and the target value is the type of the tissue: non-tumor adjacent or tumor.
Support vector machine with a linear kernel was chosen for the classification learning algorithm since the data are almost linearly separable. Also, linear decision functions capture very well the underlying distributions in micro-array classification tasks [23–25]. The goal is to find the optimal parameters of a classifier function that as the form:
We define the dataset as where is the gene expression data of the sample i and its label (non-tumor adjacent tissue or tumor tissue). For each gene , a weight is involved in the classification task. This weight is used for the ranking of genes. The linear-SVM learning algorithm can be defined as an optimization problem:
Recursive feature elimination (RFE) algorithm
The RFE method recursively deletes the genes that are the less important in the classification task. At an iteration , d minus k genes are left to rank. At each iteration, we train the linear SVM classifier used in the RFE method over the whole dataset. Each gene has a weight associated to this classifier in the decision making whether to distinguish if a tissue is tumoral or not. The square of this weight is the importance of a gene: It shows how the value of the gene expression is important in the decision making. The linear-SVM is trained over these genes, and the gene that has the least importance cj is deleted in the classification task (cj = Wj2).Let X be the matrix of the dataset (where i is the patient) so that (the vector of gene expression data of patient i) and the vector of expression data of gene j among all the patients. Let be the vector of the labels. The RFE algorithm is detailed below.
The set of ranked genes G is then obtained. We want to compare the classifiers trained on a subset of features in order to get the best subset of genes:
where is the most important gene and the least important one. To compare these classifiers, an information criterion is performed [21] which can be seen as an equivalent of Akaike's information criterion for linear-SVM. In the framework of the linear-SVM learning algorithm, one can define the SVM information criteria as: where the are the coefficients learned by the linear-SVM algorithm over and is the length of the subset. In this case, = k. The smaller this criterion is, the better the classifier is. So, we chose the best subset of features with: . This subset is the best compromise between classification performance and smallness of the subset in order to get the most significant genes.
Recovery of tumors from ccRCC patients
Tumors were obtained from patients who underwent a radical nephrectomy for ccRCC as first therapeutic intervention in the Urology Department of Saint-Louis Hospital (Paris, France). Renal tumors were classified as ccRCC by an experienced uropathologist according to the World Health Organization (WHO) classification of tumors of the kidney [26]. Adjacent non-tumor renal parenchyma was removed simultaneously. All patients participating in this study gave their informed written consent. The study was approved by the institutional review boards of Saint-Louis Hospital, Paris.
Immunohistochemistry (IHC)
Immunohistochemistry was performed for each pair of tumor and normal renal parenchyma on 4-μm-thick, formalin-fixed and paraffin-embedded or on snap-frozen tissue sections according to technical specificities of each antibody. The following murine antibodies were used: CD70, B7H4, HVEM, B7H5 (VISTA), CD40, CD163, HLA-G (clone 4H84) and PD-L1. Staining was performed on automated slide stainers from Roche (BenchMark ULTRA system, Tucson, AZ) using the OptiView DAB IHC Detection Kit (Roche), Cell Conditioning 1 (CC1) standard antigen retrieval, an antibody incubation time of 32 min at 37 °C, ultraWash procedure, counterstaining with Hematoxylin II for 4 min, and bluing reagent for 8 min. Isotype-matched immunoglobulins were used for negative controls simultaneously. The immunohistochemical analyses were finally realized by an uropathologist using a BX51 microscope (Olympus France S.A.S, Rungis).
Primary tumor cell culture
Tumor tissues were cultured in RPMI1640 supplemented with 20% fetal bovine serum, 20 μg/mL insulin, 10 μg/mL transferrin, 25 nM sodium selenite, 50 nM hydrocortisone, 1 ng/mL epidermal growth factor, 10 μM ethanolamine, 10 μM phosphorylethanolamine, 100 pM triiodothyronine, 2 mg/mL bovine serum albumin, 2 mM glutamine, 0.5 mM sodium pyruvate. After 21 days, medium was removed, tumor cells detached with EDTA 2 mM solution and analyzed by flow cytometry.
Flow cytometry analysis
Flow cytometry analyses were performed on tumor and tumor-infiltrating cells. Tumor-infiltrating cells were phenotyped immediately after isolation, whereas tumor cells were phenotyped after 3-week culture. This culture step was necessary to ensure sufficient cell numbers for flow cytometry analysis, even though some phenotype modifications by the in vitro culture might arise. Acquisition was performed on a MACSQuant 10 flow cytometer (Miltenyi Biotec), and analysis was performed using MACSQuantify (Miltenyi Biotec) and Flowjo softwares. Antibodies were CD45 VioGreen, CD3 PerCP, CD4 VioBright FITC, CD8 APC-Vio770, HLA-DR PerCP, CD28 PE-Vio770, CD137 PE-Vio770, B7-H3 VioBlue, ILT4 APC, CD70 APC, PD-L1 APC, CD137L PE from Miltenyi Biotec; HLA-G PE (clone MEM-G/09), ILT2 APC and ILT2PE (clone HP-F1) from eBioscience. PD-1 BV421 from Biolegend and CD27 PE, CD40 PE, CD86 FITC from Beckman Coulter.
Results
Landscape of IC expressed in ccRCC
To provide a global landscape of IC expressed in ccRCC patients, we analyzed the TCGA database which contains RNAseq information for 539 patients with ccRCC. To avoid selecting biased expression thresholds, we further focused on patients for whom data included tumor and adjacent non-tumor tissues. This information was available for 72 patients and is depicted in Table 1. A total of forty-four IC were chosen on the basis of a previously report [27].To test our methodology, three tumor suppressor genes expressed at very low levels in most ccRCC were added as controls: the von Hippel-Lindau (VHL), the Polybromo 1 (PBRM1) and the BRCA1-associated protein (BAP1).
Table 1.
ID and clinical information obtained from The Cancer Genome Atlas (TCGA) for the 72 patients for whom data included tumor and adjacent non-tumor tissues
| Patient ID | Case ID | Sex | Year of birth | Year of death | Tumor stage | Age at diagnosis (days) | Days to death | Days to last follow up |
|---|---|---|---|---|---|---|---|---|
| TCGA-CW-5581 | 2d0f6d4f-acb9-4b45-a69d-c9a3f68c3732 | Male | 1959 | – | Stage I | 16,195 | – | 2799 |
| TCGA-CZ-4865 | 305eaef4-4644-46e3-a696-d2e4a972f691 | Female | 1936 | 2006 | Stage I | 25,709 | 166 | – |
| TCGA-B0-5691 | 31a0dd95-8199-4d25-b40d-4b353644af46 | Female | 1936 | – | Stage I | 24,120 | – | 3431 |
| TCGA-CW-6088 | 4100b960-7963-4a1e-ba24-0a6526387e06 | Male | 1943 | – | Stage I | 22,020 | – | 3222 |
| TCGA-CW-6090 | 4dd51edf-05d1-445a-b43c-6fce29eb21d4 | Male | 1935 | – | Stage I | 24,909 | – | 2552 |
| TCGA-B0-5703 | 576ea0ef-3abb-479d-adb7-ba646cee344e | Male | 1936 | – | Stage I | 26,897 | – | 2246 |
| TCGA-CW-5589 | 58ef1a13-a549-4043-b66c-5327bfcbd2e6 | Male | 1952 | – | Stage I | 19,312 | – | 2378 |
| TCGA-B0-5705 | 62cf9546-b932-4a5e-bc9b-4103506d296d | Female | 1937 | – | Stage I | 23,746 | – | 4537 |
| TCGA-CJ-6030 | 6ab00314-5228-43ba-9880-9d5177b64c61 | Male | 1938 | 2009 | Stage I | 24,096 | 2299 | – |
| TCGA-B8-4619 | 78ec8bc9-e502-4f5b-b0f6-893,718,650,352 | Male | 1952 | – | Stage I | 21,206 | – | 523 |
| TCGA-B0-5690 | 794aeb92-205f-4c75-bb1a-0b2c6db99fea | Female | 1951 | – | Stage I | 19,685 | – | 3392 |
| TCGA-CZ-5986 | 883c37e2-c723-45d7-9e25-082d1bec34e2 | Male | 1945 | – | Stage I | 22,456 | – | 373 |
| TCGA-A3-3358 | 8a575e00-5dc5-416d-a8f5-2cdfb8e62c31 | Female | 1948 | – | Stage I | 21,087 | – | 1307 |
| TCGA-B8-5549 | 98dea82b-46f9-4b77-be8a-06669bcf731b | Male | 1957 | – | Stage I | 19,561 | – | 194 |
| TCGA-A3-3387 | 9dc7812b-c7a2-4de4-bf6d-4c7261384a62 | Male | 1957 | – | Stage I | 17,920 | – | 617 |
| TCGA-CZ-5982 | a43401ae-14cd-4f57-946b-9a9c073f2a47 | Female | 1946 | – | Stage I | 21,592 | – | 2439 |
| TCGA-B2-5641 | a682a2a3-dc53-4b7e-8929-07215737ec5e | Male | 1931 | – | Stage I | 28,953 | – | 656 |
| TCGA-B0-5699 | c814c26c-ee8e-4fd8-a3d3-441b302ead3b | Male | 1950 | – | Stage I | 19,647 | – | 3841 |
| TCGA-B0-5697 | dfd2c288-5054-4fc1-9cae-f437779dc2de | Male | 1957 | – | Stage I | 18,486 | – | 2630 |
| TCGA-B8-5552 | e58f2c7b-d1cc-48f3-a0c9-dcd2b27c37f6 | Female | 1969 | – | Stage I | 15,219 | – | 1046 |
| TCGA-CJ-5689 | e865d40a-9989-436c-8426-88cc84c863e8 | Male | 1915 | 2009 | Stage I | 32,872 | 1620 | – |
| TCGA-CZ-5984 | ea26d89f-2843-489b-8627-3d5ed0cabc2a | Male | 1954 | – | Stage I | 18,775 | – | 2067 |
| TCGA-B2-5636 | ed2e9354-5ee1-4fcf-92ec-a1b507818b91 | Male | 1931 | – | Stage I | 28,901 | – | 919 |
| TCGA-CJ-5672 | eee108d6-bb11-4369-b776-0f524afef6f3 | Male | 1920 | 2010 | Stage I | 30,900 | 2190 | – |
| TCGA-CZ-5988 | f2806652-1d7b-4c46-bb4e-ac7a2c96c68d | Male | 1968 | – | Stage I | 14,131 | – | 693 |
| TCGA-CZ-5989 | 13e25128-9be1-4f67-a43f-a8744a619203 | Male | 1946 | – | Stage II | 22,170 | – | 1905 |
| TCGA-B0-5706 | 3b2b492b-94af-4540-b283-3b9ef98d5b2f | Male | 1959 | – | Stage II | 16,442 | – | 3205 |
| TCGA-CZ-5456 | 467bf226-a646-4217-bce6-8d0f11c756eb | Male | 1949 | – | Stage II | 21,164 | – | 2422 |
| TCGA-CZ-5985 | 4c474c70-1d72-49e8-a8cc-a4145c5ab607 | Male | 1948 | – | Stage II | 21,242 | – | 1997 |
| TCGA-CZ-5453 | 577847b9-f9b6-4954-868f-3d5ab2b4f694 | Male | 1939 | – | Stage II | 24,640 | 2419 | 25 |
| TCGA-CZ-5469 | 6205c2d8-d431-4c4c-8ac6-ee407170e833 | Male | 1966 | 2009 | Stage II | 15,242 | 946 | – |
| TCGA-CZ-4864 | 7fc6b44d-ae66-409c-98e1-c1a518c33e95 | Male | 1916 | 2009 | Stage II | 31,557 | 2830 | 2830 |
| TCGA-CZ-5463 | 8e9e684c-20e5-48b1-9e40-970037fe959f | Male | 1931 | – | Stage II | 27,911 | – | 662 |
| TCGA-CZ-5470 | 99f59583-2728-4c1d-b98c-8fbc3bb0a819 | Female | 1936 | – | Stage II | 26,494 | – | 386 |
| TCGA-CZ-5451 | 9cdda9fa-d492-4902-afc1-eb2244e70c1b | Male | 1932 | – | Stage II | 27,312 | – | 1929 |
| TCGA-CZ-5452 | e9faa588-d45a-450a-81a2-949eb2834bc0 | Male | 1937 | – | Stage II | 25,486 | – | 1789 |
| TCGA-CW-5584 | 09c4ea05-928d-49b7-b7fb-30cff3481b14 | Male | 1929 | 2003 | Stage III | 27,352 | 164 | – |
| TCGA-B0-5694 | 11111b58-c7df-4291-ad8a-4baec9ff7d1f | Male | 1937 | 2009 | Stage III | 26,060 | 480 | – |
| TCGA-B0-5696 | 1d176c53-6cbb-4a39-9a6f-669b4f1cb575 | Male | 1938 | – | Stage III | 25,552 | – | 2609 |
| TCGA-CJ-5676 | 310c31bf-91ce-40b4-99e4-a10242296de9 | Male | 1957 | – | Stage III | 17,170 | – | 4067 |
| TCGA-CJ-5679 | 3fa6c93e-e7fe-402c-9526-c81411aa0920 | Male | 1930 | 2004 | Stage III | 26,858 | 679 | – |
| TCGA-CZ-5466 | 5722df9f-5631-476d-a11b-b3c1e9a40fbf | Male | 1940 | – | Stage III | 24,669 | – | 685 |
| TCGA-CZ-5465 | 5e248b21-e69d-4dee-a1e8-029ade80d0eb | Female | 1931 | – | Stage III | 27,838 | 2564 | 1446 |
| TCGA-CW-5587 | 73fc6ae6-7c5e-44de-a9a0-17292cbb01cc | Female | 1941 | – | Stage III | 22,693 | – | 2226 |
| TCGA-B0-5711 | 88c91a7b-5c41-4361-85d0-da759ab94204 | Male | 1954 | – | Stage III | 18,444 | – | 3989 |
| TCGA-CZ-4863 | 88e7ce26-5b3f-4e4e-89b7-f706063fc467 | Female | 1955 | – | Stage III | 18,730 | – | 1928 |
| TCGA-B0-5701 | 8f8a632d-7fbd-4c86-b909-afb7edb9ad28 | Male | 1942 | – | Stage III | 23,915 | – | 2461 |
| TCGA-CZ-5467 | b602a73b-809c-44c6-a787-d496705e7ae8 | Female | 1921 | 2007 | Stage III | 31,473 | 73 | – |
| TCGA-B0-5709 | bf768635-b809-4df4-a4e6-7495e66aa227 | Female | 1941 | – | Stage III | 22,843 | – | 3974 |
| TCGA-CZ-5458 | c4247ccb-8201-428e-a2e7-b5104f095588 | Male | 1963 | – | Stage III | 15,790 | – | 2789 |
| TCGA-B8-4620 | eda2b871-8a18-4854-92d8-d8d66fb127d8 | Female | 1940 | – | Stage III | 25,786 | – | 777 |
| TCGA-CZ-5457 | f00b7956-73a9-4e4b-85d2-60b3ba13f4a4 | Male | 1944 | – | Stage III | 22,763 | – | 2754 |
| TCGA-CJ-5681 | 2939c03a-6f3f-4f7b-b246-34361baadeb9 | Female | 1959 | 2004 | Stage IV | 16,121 | 552 | – |
| TCGA-CZ-5461 | 393abcf7-1155-4b32-85ca-cd44d259e4b4 | Male | 1954 | 2006 | Stage IV | 19,030 | 330 | 330 |
| TCGA-B0-4712 | 3f72d63f-ad48-4500-baf6-897b1d6dda7d | Male | 1930 | 2009 | Stage IV | 28,095 | 1337 | – |
| TCGA-CJ-6033 | 4edff57f-4b0e-4770-beac-590da7d7232c | Female | 1950 | 2004 | Stage IV | 19,919 | 224 | – |
| TCGA-B0-5712 | 514af471-31d2-43fa-88dc-8639a5e97181 | Female | 1936 | – | Stage IV | 24,988 | – | 2722 |
| TCGA-CJ-5680 | 5db69ebe-38db-4ef3-b825-674b4a6ddaee | Female | 1938 | 2005 | Stage IV | 23,778 | 768 | – |
| TCGA-CZ-5455 | 74749fe8-f5d0-4d0d-8c7a-e56eba6503b3 | Male | 1943 | 2007 | Stage IV | 23,150 | 561 | – |
| TCGA-CJ-5678 | 822cf6c1-dd65-4814-94b1-0c335208ad9b | Male | 1941 | 2004 | Stage IV | 22,944 | 574 | – |
| TCGA-CZ-5468 | 878d1caa-c4d8-4864-888c-310a0c1ee898 | Male | 1923 | 2007 | Stage IV | 30,729 | 59 | 59 |
| TCGA-CW-5580 | 88fc4bc4-32cf-4d92-8c29-20d920b8f719 | Female | 1930 | 2008 | Stage IV | 26,696 | 1964 | – |
| TCGA-B8-4622 | b125ff14-4fb4-4f90-8622-fed49bdfe954 | Male | 1953 | – | Stage IV | 21,135 | – | 1525 |
| TCGA-CW-6087 | bbdaa931-e922-49be-bfbf-fa0c2ae27d7a | Male | 1942 | 2003 | Stage IV | 22,438 | 41 | – |
| TCGA-CJ-5677 | d2664fde-ce3b-45e6-9a23-4a07980f7bac | Female | 1950 | 2006 | Stage IV | 19,849 | 782 | – |
| TCGA-B0-5402 | d7ab7ec0-3de7-4ffd-a5ac-f75579355b2a | Male | 1946 | – | Stage IV | 23,477 | – | 1290 |
| TCGA-CZ-5462 | e0127e51-43ba-4536-bc9d-004591f9c627 | Male | 1924 | 2007 | Stage IV | 30,659 | 311 | – |
| TCGA-B0-4700 | e33dff22-5bf1-4dd8-bed7-063cb555677c | Male | 1944 | 2009 | Stage IV | 22,034 | 1980 | – |
| TCGA-CW-5591 | f1ae0181-74f8-47fd-83be-83a7d01101cc | Male | 1948 | – | Stage IV | 20,712 | – | 2271 |
| TCGA-CZ-5454 | f2801b21-5444-4cc3-a642-c60c6d82cd3d | Male | 1943 | 2007 | Stage IV | 23,083 | 722 | – |
| TCGA-CZ-5987 | f5759059-c0e3-4f1a-af96-5c7197d3c33c | Male | 1946 | 2007 | Stage IV | 21,928 | 445 | – |
| TCGA-CW-5585 | 0487fc41-386c-4d76-9084-daf959bf5e98 | Male | 1952 | – | Stage IV | 18,898 | – | 2609 |
Expression ratios for tumor versus healthy adjacent tissues were first analyzed for each of the forty-four IC by bi-clustering genes for each of the 72 patients individually. The results are represented on a heatmap, a colored representation of a matrix of numbers that helps the visualization of gene expression data (Fig. 1). In this representation, each column represents a gene, each row a patient. The standardized expression levels are depicted by the color gradients. The results show perceptible differences between patients even though a global pattern emerges. Notably, the ratio of expression of tumor to adjacent non-tumors for B7H4 was found extremely low except for 2 out of 72 samples. The three control genes PBRM1, VHL and BAP1 that are mostly down-regulated in ccRCC were grouped together, and their low expressions in ccRCC samples were confirmed (Fig. 1).
Fig. 1.

Heatmap representation of the logarithm of the ratio of gene expression between tumor and non-tumor adjacent tissues. In this representation, each column represents a gene, and each row a patient. The standardized expression levels are depicted by the color gradient: green, low expression ratio in ccRCC samples; red, high expression ratio. Values in black represent a ratio equals to 0 (whose log is − ∞)
Statistically modulated IC in ccRCC
To establish statistically robust results, we chose to conduct a differential analysis to compare the expression levels of IC in ccRCC samples versus the adjacent non-tumor tissues using the DESeq2. A set of 1264 genes were found up-regulated and 1194 genes down-regulated, by considering a p-value of 0.05. The similar numbers of induced and repressed genes is consistent with an absence of methodological biases. From these, we extracted expression information for the forty-four IC simultaneously. The results obtained are represented by bar plots (Fig. 2). The x-axis represents the value of the log fold change and the y-axis the genes. A gradient of color is used in order to show the significance of genes in the study. The redder the bar, the more differentially expressed the gene.
Fig. 2.

Categorization of forty-four differentially expressed IC. Numbers on the x-axis represent the value of the logarithm fold change. If this value is negative, then the gene is underexpressed, and if this one is positive, then the gene is overexpressed. The higher the value is, the more the gene is affected by the condition (tumor). To represent differentially expressed genes, we used a color gradient. More red the bar is, more the IC is considered as differentially expressed
Moreover, the genes on the y-axis are ranked according to their adjusted p-values. The lower the adjusted p-value, the lower the position of the associated gene on the axis, and the more significant the differential expression. If the value of the log fold change is negative, the gene is underexpressed, and if it is positive, the gene is overexpressed. Values representing the mean of normalized counts for all samples, the log2 FoldChange, the p values and the adjusted p-values of the test are represented in supplementary Table 1.
Notably, among the most significantly differentially overexpressed genes in tumor cells we found those encoding ligand-receptor pairs such as: CD70:CD27; HLA-G:ILTs; 4-1BB:4:1BBL, CD40:CD40L; CD86:CTLA4; MHC-II:Lag3; CD200:CD200R; CD244:CD48. In contrast, adjacent non-tumor cells express significantly higher levels of B7-H4 compared to tumor cells. As expected, the expression levels of the three control genes: PBRM1, VHL and BAP1, were lower in tumors than in adjacent non-tumor tissues, which further confirms the robustness of our statistical analysis.
Localization of representative IC within tumor compartments.
To deepen the results of the TCGA analysis, we aimed at identifying more precisely the nature of the IC-expressing cells. To this end, we performed IHC analysis on different samples derived from a new cohort of ccRCC patients that underwent a radical nephrectomy at the Urology Department of Saint-Louis Hospital (Paris, France).
Tumor and normal renal parenchyma of ten patients were assessed simultaneously. We focused on B7-H4, the most significantly down-regulated gene and three up-regulated genes in tumor: CD70, CD40 and B7-H5 (VISTA). When samples were analyzed with the antibody directed against B7H4, strong labeling of the normal renal parenchyma of all renal tubular epithelial cells was observed in all cases. On the other hand, a very heterogeneous labeling was observed within and between ccRCC samples. In the majority of ccRCC tumors, no labeling was noted in both tumor and tumor-infiltrating inflammatory cells. In the other cases, a slight labeling was noted in tumor cells and/or tumor-infiltrating inflammatory cells. This confirms further the downregulation of B7H4 highlighted in the RNAseq analysis. A representative example is illustrated in Fig. 3. When antibodies directed against the three up-regulated genes CD70, CD40 and B7-H5 (VISTA) were used, tumors were strongly stained when compared to normal renal parenchyma which further confirms the transcriptome results. Nevertheless, the supplementary information given by the morphologic analysis concerns the nature of cells that highly express these checkpoints. Indeed, IHC results showed that tumor cells themselves expressed high levels of CD70 whereas only rare parietal cells of Bowman’s capsule, some interstitial fibrous areas and glomerular capillary vessels were labeled with anti-CD70 in normal renal parenchyma. In sharp contrast, CD40 and B7-H5 (VISTA) antibodies intensely labeled numerous inflammatory cells in contact with tumor cells such as CD163-labeled macrophages representative stains are illustrated in Fig. 3.
Fig. 3.

IHC illustration of the localization of differentially expressed IC within tumor compartments of a representative ccRCC patient. Protein products of the selected genes CD70, B7H4, CD40, HVEM, B7H5 and CD163 were assessed in normal renal tissue and tumor tissue (H&E and immunoperoxidase stains are also shown)
Altogether, IHC results confirmed and extended those obtained by the analysis of RNAseq data showing for the first time the down-regulation of B7H4 and overexpression of CD70, CD40 and B7-H5 (VISTA) in ccRCC.
The ID of each patient included in the TCGA database (Table 1) allows to gain further insight into IHC results which are digitalized and made available on https://cancer.digitalslidearchive.org.
Most relevant IC expressed in tumors of ccRCC patients
Our survey of IC expressed in ccRCC reveals the overexpression of 38 IC at different levels. To find out which of them are the most important representatives of this type of cancer, we considered the recursive feature elimination (RFE) algorithm with a linear-SVM. The model eliminates the least important features, one by one iteratively until all features in the dataset are exhausted. Features are then ranked according to when they were eliminated. As such, it is a robust method for finding the best performing subset of features and owns the advantage that it can be applied to the whole dataset without fixing a threshold beforehand.
The RFE algorithm was therefore applied to the 539 patients included in the TCGA database followed by the linear-SVM model. The results, named SVMIC, revealed that the optimal number of features is 7. The accuracy is visible on the plot represented in Fig. 4a and was estimated by tenfold cross-validation over the whole dataset. The dataset was separated in ten parts, for each part we trained the model on the nine others and computed the accuracy on the singled-out part. Finally, we took the mean of the ten computed accuracies. The output of the whole algorithm is the ranked subset of the following genes: 'HLA-G' 'HVEM' 'PD-L1′ 'B7-H3′ 'ILT2′ 'CD40′ 'B7-H5′. The importance of each gene, defined as the square of its weight (see Materials and Methods for more details), shows how the value of the gene expression is important in the decision making. It is depicted by a graphical representation in Fig. 4b. The x-axis represents the selected genes, and the y-axis the importance of the genes in the linear-SVM model. Although HLA-G has been identified as the most significant gene, the graph shows that its bar plot is smaller. This would mean that HLA-G shares information with one or several of the other selected genes. This interesting feature should be the aim of future work.
Fig. 4.
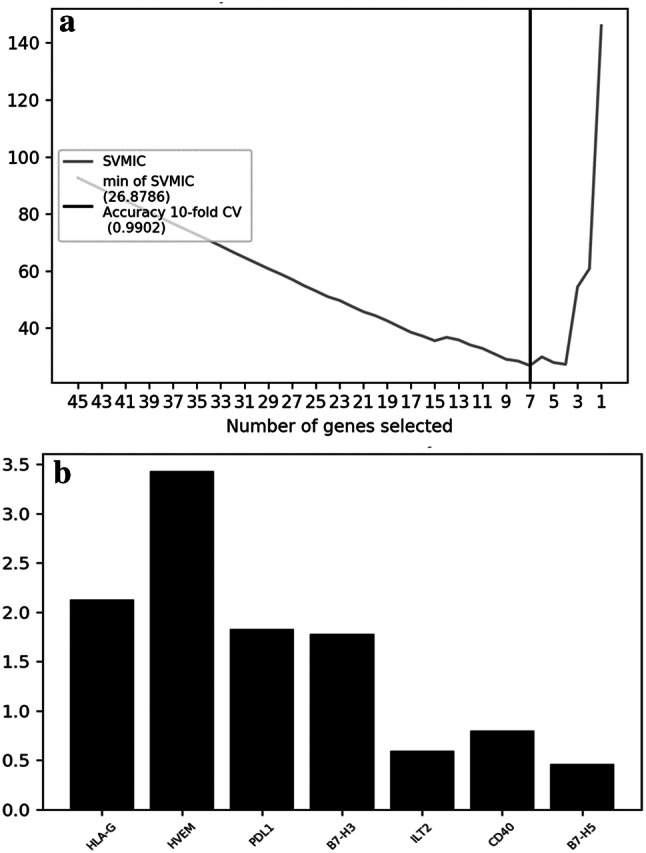
A. Plot of the SVM information criteria as a function of the subsets generated by the RFE algorithm. x-axis: number of selected genes; y-axis: cross-validations score (number of correct classifications). B. Barplot showing the importance of each of the seven features selected. The x-axis represents the selected genes and the y-axis the importance of the genes in the linear-SVM model
Checkpoints expression in tumor cells
To validate the RFE algorithm results, we analyzed ten additional tumors derived from ccRCC patients. The results obtained by immunohistochemistry for six representative samples are illustrated in Fig. 5. A constant labeling of tumor cells and more rarely tumor-infiltrating inflammatory cells was noted in all ccRCC tumors with the antibody directed against HVEM. Nevertheless, this antibody also labeled renal tubular epithelial cells of the normal renal parenchyma. Immunostaining with HLA-G and PD-L1 antibodies revealed high and heterogeneous inter- and intra-tumor labeling. In sharp contrast, no staining was observed in normal renal parenchyma.
Fig. 5.

IHC illustration of the localization of the three more important selected IC obtained by the linear-SVM model
To better quantify the cell populations that express the IC, we analyzed tumor cells individually by flow cytometry. We focus on the most significant checkpoints revealed by the RFE algorithm: HLA-G:ILT2 and HLA-G:ILT4, 4-1BBL:4-1BB (CD137L:CD137), PD-L1:PD-1, and B7-H3, to which we added CD70, one of the most overexpressed IC in ccRCC. Thus, ten additional ccRCC tumors were more thoroughly analyzed (Fig. 6a) and a representative example, expressing the majority of the IC is shown in Fig. 6b. Consistent with the results of the RFE model, PD-L1, HLA-G and CD137L were expressed in all tumors studied. CD70 and B7-H3 were found in 6/7 and 5/8 tumors studied, respectively (Fig. 6c). There was a high inter-individual variability with respect to the proportion of tumor cells expressing a given checkpoint. For instance, the proportion of tumor cells expressing PD-L1 varied from 27% in patient #1 to 100% in patient #3. Similarly, this range was 17–74% for 4-1BB (CD137L) and 5–97% for HLA-G (Fig. 6c). Notably, ILT2 expression was observed in tumor cells from six out of ten patients, and particularly significant in three of them (16%, 20% and 30%). Similarly, ILT4 expression was also observed in tumor cells from six out of ten patients, and particularly significant (from 28 to 79%). This is noteworthy since ILT2 and ILT4 are usually considered as being expressed only by leukocytes [28]. All the values are represented in Fig. 6c. These results are thus consistent with those derived from the RNAseq analysis and the RFE model and reveal for the first time the expression of ILT2 on tumor cells. In addition, a very important conclusion that follows from culturing individual tumors is that a particular tumor expresses several IC simultaneously, and this has to be taken into account for optimizing future IC-based future therapies, since the treatment with a single checkpoint does not seem to be efficient enough.
Fig. 6.

Expression of selected IC by tumor cells. a Cell surface expression of the indicated IC by tumor cells from ten patients. b Illustration of cell surface profiles of all the IC expressed in tumor of patient 7. c Percentage of cell surface IC expression for the ten ccRCC patients studied
Discussion
Immune-checkpoint blockade by antibodies that target specific ligand–receptor interactions has emerged as one of the most promising therapeutic options for patients with ccRCC. Despite the success of these therapies, a considerable proportion of patients remain unresponsive to treatment suggesting that multiple non-redundant immunosuppressive mechanisms coexist within tumor and its microenvironment.
In this study, we comprehensibly analyzed forty-four IC expressed in tumors derived from patients with ccRCC in terms of gene and protein expression differences between tumor and non-tumor-adjacent tissues and ranked them according to their relevances to characterize ccRCC. Among the ligand-receptor pairs identified in this study, some are involved in inhibitory pathways, whereas others are implicated in stimulatory pathways and might be expressed in the same tumor cells or the microenvironment.
One of the most highly differentially expressed RNA in ccRCC is CD70, a type II integral membrane protein that belongs to the tumor necrosis factor superfamily. This result was confirmed at the protein level by IHC analysis and is consistent with previous report [29] adding arguments in favor of the robustness of our analysis.
Our TCGA survey also revealed high expression of CD40 in ccRCC samples compared to adjacent non-tumor tissues. Using IHC, we have confirmed this and further show that CD40 is expressed in the abundant CD163-labeled macrophage population surrounding tumor cells. Clinical trials using CD40 agonists are still ongoing even though its administration is associated with particular toxicities [30]. Another ligand-receptor pair found highly overexpressed in ccRCC samples is 4-1BB (CD137) and 4-1BBL (CD137L). These are co-stimulatory molecules that are involved in multiple steps during the progression of inflammation and hematopoiesis. High expression of 4-1BB has also been noted on a number of cancer cell lines such as liver or leukemia cell lines [31]. Clinical trials with two agonist antibodies, urelumab and utomilumab, are ongoing. Despite initial signs of efficacy, clinical development of urelumab has been hampered by inflammatory liver toxicity [32]. To our knowledge, the presence of high levels of 4-1BBL in ccRCC is reported here for the first time. The presence of co-stimulatory molecules would provide a “second” signal that activates T-cell and promote anti-tumor response following the blockage of the negative IC [33]. In agreement with this, blockade of CTLA-4 (inhibitory IC) together with engagement of ICOS (stimulatory IC) enhanced anti-tumor responses and significantly improved tumor rejection [34].
Several members of the B7 family were also found modulated in ccRCC. A hallmark of the B7 family is the capability to co-stimulate or co-inhibit T cell responses in the presence of peptide/MHC complex-mediated TCR signaling [35]. The B7 co-stimulatory ligands are important for full activation of naïve T cells in the lymphoid organs. In contrast, B7 co-inhibitory ligands are crucial for the termination of over activated T cell response and maintenance of self-tolerance [36]. The analysis of the TCGA data highlighted members of the B7 family that are differentially modulated in ccRCC, mainly B7H3, B7-H4 and B7H5/VISTA.
The levels of B7H4 were highly decreased in ccRCC. This correlates with results of our protein expression analysis performed by IHC on our cohort of ccRCC patients but sharply contrast with studies performed in other cancer types such as pancreatic or hepatocellular carcinoma, in which the overexpression of the B7-H4 protein constitute a negative prognostic marker for patient’s survival [37]. This emphasizes the need to assess the expression of each IC in the different cancer types.
Another member of the B7 family, CD276/B7-H3, was found overexpressed at mRNA and protein levels. Given its immunomodulatory capacities and its overexpression on both, cancer cells and tumor-infiltrating blood vessels [38, 39], CD276/B7-H3 may be considered a more general target in cancer’s treatment. However, its therapeutic use awaits more information due to its dual activity, either as stimulatory effect on the proliferation of both CD4+ and CD8+-T cells or as co-inhibitory molecule. Moreover, since B7-H3 is broadly expressed, especially in peripheral healthy tissues, B7-H3′s blockade by specific antibodies might be associated with severe adverse effects. In addition, B7H3 seems to share a yet unidentified receptor with B7H4. Considering that levels of B7H3 and B7H4 are highly increased and decreased respectively in ccRCC, it would be necessary to determine which of the two will work and in what circumstances.
This may also be the case of B7H5/VISTA, which is a molecule with dual activity. It behaves as a stimulatory ligand for antigen presenting cells (APCs) causing immune activation and as a negative ligand for T-cells, suppressing activation and proliferation [40]. In this study, the mRNA and protein levels of this molecule were found increased in ccRCC samples. In particular, IHC clearly showed that B7H5/VISTA is expressed on abundant macrophage surrounding tumor cells. It is likely that given the multiple binding partners of these molecules and their bidirectionality with regard to signaling, their molecular mechanisms of action have to be clarified more thoroughly before therapeutic application.
The recursive method pointed out HLA-G and PD-L1 as highly relevant IC expressed in tumor cells of ccRCC patients. These confirmed recent reports show high expression of HLA-G in ccRCC which may coexist with PD-L1 [41, 42]. This was also demonstrated here by in vitro culture of tumor cells derived from ccRCC patients. If PD-L1 is already used for therapeutic purposes, HLA-G still is not. We have previously made the proof of concept that anti-HLA-G antibodies can be used in immunotherapy protocols [43]. In addition, HLA-G offers a significant therapeutic window for targeted therapy since: (1) HLA-G has a restricted expression in normal tissues, (2) HLA-G has broad immune-inhibitory functions through binding to its receptors ILT2 and ILT4 present on immune cells. Indeed, HLA-G/ILTs can block all stages of an immune response, from APC activation and effector priming to the function of fully activated CTLs, NK or B cells [44]. Therefore, developing strategies to block this pathway would offer the advantage of exerting extended effects with less prominent systemic toxicities. We propose that simultaneously engaging PD-L1 and HLA-G blockade would constitute a promising new therapeutic opportunity. Moreover, the demonstration that ILT4 [41, 45] and for the first time in this study, ILT2, are expressed not only by tumor infiltrating cells but also by CD45-negative tumor cells themselves, adds supplementary interest of this checkpoint as a therapeutic target.
In conclusion, the approach adopted in this study, which couples transcriptome stringent data analyses and protein measurement by IHC and flow cytometry, highlighted key IC, some of which were never reported before to be expressed in ccRCC. In particular, the ranking of these checkpoints and the demonstration that each tumor express several IC simultaneously emphasize the importance that exists to expand the potential therapeutic targets to treat ccRCC. Targeting HLA-G/ILT in HLA-G-positive tumors, either concomitantly or in case of non-responsiveness to anti-PD1/PD-L1, would be one strategy to be tested. The remaining challenges include the understanding of fundamental mechanisms of IC action in pathological conditions and the identification of optimal combinations of IC for ccRCC patient’s benefit.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors are particularly grateful to Dr. Nathalie Rouas-Freiss for very instructive discussions and critical reading of the manuscript. We are also thankful to Dr. Marcela Garcia and Santiago Miriuka for their enlightened suggestions on tumor culture procedures. We kindly appreciate the experimental assistance of Alix Jacquier on cytometric assays and Jerome Delmotte on immunochemistry. The results shown are based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcha.
Abbreviations
- ccRCC
Clear cell renal cell carcinoma
- HLA-G
Human leukocyte antigen-G
- IC
Immune-checkpoints
- ILT2
Immunoglobulin-like transcript 2 (LILRB1)
- ILT4
Immunoglobulin-like transcript 4 (LILRB2)
- RFEM
Recursive feature elimination method
- SVM
Support vector machine
- TCGA
The Cancer Genome Atlas
Authors contributions
DTLR was involved in conceptualization, formal analysis, writing original draft, supervision; MS helped in methodology, software, formal analysis, writing original draft; MB contributed to methodology, software, formal analysis, writing original draft; JV performed experiments, formal analysis and diagnosis; MBP contributed to methodology, performed experiments; MD performed experiments and analysis; FB performed experiments; JLM contributed to methodology, formal analysis, writing original draft; FD provided samples; S.L. contributed to formal analysis, writing original draft; PHC helped in formal analysis, methodological supervision, writing-original draft, funding; EDC was involved in research design, funding.
Funding
This work was funded by the Commissariat à l’Energie Atomique et aux Energies Alternatives (CEA) and Ecole CentraleSupélec, Université Paris-Saclay.
Compliance with ethical standards
Conflict of interest
The authors declare no potential conflicts of interest.
Ethical approval
The study was approved by the institutional ethics committee of Saint-Louis Hospital, Paris.
Informed consent
Patients provided written informed consent before sampling.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Mathilde Sautreuil and Mahmoud Bentriou have contributed equally to this work.
References
- 1.Znaor A, Lortet-Tieulent J, Laversanne M, Jemal A, Bray F. International variations and trends in renal cell carcinoma incidence and mortality. Eur Urol. 2015;67:519–530. doi: 10.1016/j.eururo.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Brugarolas J. PBRM1 and BAP1 as novel targets for renal cell carcinoma. Cancer J. 2013;19:324–332. doi: 10.1097/PPO.0b013e3182a102d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsieh JJ, Le VH, Oyama T, Ricketts CJ, Ho TH, Cheng EH. Chromosome 3p loss-orchestrated VHL, HIF, and epigenetic deregulation in clear cell renal cell carcinoma. J Clin Oncol. 2018;36:JCO2018792549. doi: 10.1200/JCO.2018.79.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levi-Schaffer F, Mandelboim O. Inhibitory and coactivating receptors recognising the same ligand: immune homeostasis exploited by pathogens and tumours. Trends Immunol. 2018;39:112–122. doi: 10.1016/j.it.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kruger S, Ilmer M, Kobold S, Cadilha BL, Endres S, Ormanns S, et al. Advances in cancer immunotherapy 2019—latest trends. J Exp Clin Cancer Res. 2019;38:268–278. doi: 10.1186/s13046-019-1266-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rotte A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J Exp Clin Cancer Res. 2019;1(381):255. doi: 10.1186/s13046-019-1259-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lalani AA, McGregor BA, Albiges L, Choueiri TK, Motzer R, Powles T, et al. Systemic treatment of metastatic clear cell renal cell carcinoma in 2018: current paradigms, use of immunotherapy, and future directions. Eur Urol. 2019;1:100–110. doi: 10.1016/j.eururo.2018.10.010. [DOI] [PubMed] [Google Scholar]
- 9.George S, Rini BI, Hammers HJ. Emerging role of combination immunotherapy in the first-line treatment of advanced renal cell carcinoma: a review. JAMA Oncol. 2019;5:411–421. doi: 10.1001/jamaoncol.2018.4604. [DOI] [PubMed] [Google Scholar]
- 10.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 11.Marin-Acevedo JA, Soyano AE, Dholaria B, Knutson KL, Lou Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J Hematol Oncol. 2018;11:8. doi: 10.1186/s13045-017-0552-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015;21:1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The immune landscape of cancer. Immunity. 2018;48(812–30):e14. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rover LK, Gevensleben H, Dietrich J, Bootz F, Landsberg J, Goltz D, et al. PD-1 (PDCD1) promoter methylation is a prognostic factor in patients with diffuse lower-grade gliomas harboring isocitrate dehydrogenase (IDH) mutations. EBioMedicine. 2018;28:97–104. doi: 10.1016/j.ebiom.2018.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez JI, Angulo JC. Pathological bases and clinical impact of intratumor heterogeneity in clear cell renal cell carcinoma. Curr Urol Rep. 2018;19:3. doi: 10.1007/s11934-018-0754-7. [DOI] [PubMed] [Google Scholar]
- 16.Soneson C, Delorenzi M. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics. 2013;14:91. doi: 10.1186/1471-2105-14-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reiner A, Yekutieli D, Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003;19:368–375. doi: 10.1093/bioinformatics/btf877. [DOI] [PubMed] [Google Scholar]
- 20.Guyon I, Weston J, Barnhill S, Vapnik V. Gene selection for cancer classification using support vector machines. Mach Learn. 2002;46(1):389–422. doi: 10.1023/A:1012487302797. [DOI] [Google Scholar]
- 21.Claeskens G, Croux C, Van Kerckhoven J. An information criterion for variable selection in support vector machines. J Mach Learn Res. 2008;9:541–558. [Google Scholar]
- 22.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay E. Scikit-learn: Machine learning in Python. J Mach Learn Res. 2011;12:2825–2830. [Google Scholar]
- 23.Statnikov A, Wang L, Aliferis CF. A comprehensive comparison of random forests and support vector machines for microarray-based cancer classification. BMC Bioinformatics. 2008;9:319. doi: 10.1186/1471-2105-9-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dudoit S, Fridlyand J, Speed TP. Comparison of discrimination methods for the classification of tumors using gene expression data. J Am Stat Assoc. 2002;97(457):77–87. doi: 10.1198/016214502753479248. [DOI] [Google Scholar]
- 25.Dupuy A, Simon RM. Critical review of published microarray studies for cancer outcome and guidelines on statistical analysis and reporting. J Natl Cancer Inst. 2007;99:147–157. doi: 10.1093/jnci/djk018. [DOI] [PubMed] [Google Scholar]
- 26.Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM. The 2016 WHO classification of tumours of the urinary system and male genital organs-part A: renal, penile, and testicular tumours. Eur Urol. 2016;70:93–105. doi: 10.1016/j.eururo.2016.02.029. [DOI] [PubMed] [Google Scholar]
- 27.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Desgrandchamps F, LeMaoult J, Goujon A, Riviere A, Rivero-Juarez A, Djouadou M, et al. Prediction of non-muscle-invasive bladder cancer recurrence by measurement of checkpoint HLAG's receptor ILT2 on peripheral CD8(+) T cells. Oncotarget. 2018;9:33160–33169. doi: 10.18632/oncotarget.26036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Junker K, Hindermann W, von Eggeling F, Diegmann J, Haessler K, Schubert J. CD70: a new tumor specific biomarker for renal cell carcinoma. The Journal of urology. 2005;173:2150–2153. doi: 10.1097/01.ju.0000158121.49085.ba. [DOI] [PubMed] [Google Scholar]
- 30.Hassan SB, Sorensen JF, Olsen BN, Pedersen AE. Anti-CD40-mediated cancer immunotherapy: an update of recent and ongoing clinical trials. Immunopharmacol Immunotoxicol. 2014;36:96–104. doi: 10.3109/08923973.2014.890626. [DOI] [PubMed] [Google Scholar]
- 31.Vinay DS, Kwon BS. Immunotherapy of cancer with 4-1BB. Mol Cancer Therapeutics. 2012;11:1062–1070. doi: 10.1158/1535-7163.MCT-11-0677. [DOI] [PubMed] [Google Scholar]
- 32.Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood. 2018;131:49–57. doi: 10.1182/blood-2017-06-741041. [DOI] [PubMed] [Google Scholar]
- 33.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan X, Quezada SA, Sepulveda MA, Sharma P, Allison JP. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J Exp Med. 2014;211:715–725. doi: 10.1084/jem.20130590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen YP, Zhang J, Wang YQ, Liu N, He QM, Yang XJ, et al. The immune molecular landscape of the B7 and TNFR immunoregulatory ligand-receptor families in head and neck cancer: a comprehensive overview and the immunotherapeutic implications. Oncoimmunology. 2017;6:e1288329. doi: 10.1080/2162402X.2017.1288329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jung K, Choi I. Emerging co-signaling networks in T cell immune regulation. Immune Netw. 2013;13:184–193. doi: 10.4110/in.2013.13.5.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Podojil JR, Miller SD. Potential targeting of B7-H4 for the treatment of cancer. Immunol Rev. 2017;276:40–51. doi: 10.1111/imr.12530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castellanos JR, Purvis IJ, Labak CM, Guda MR, Tsung AJ, Velpula KK, et al. B7-H3 role in the immune landscape of cancer. Am J Clin Exp Immunol. 2017;6:66–75. [PMC free article] [PubMed] [Google Scholar]
- 39.Seaman S, Zhu Z, Saha S, Zhang XM, Yang MY, Hilton MB, et al. Eradication of tumors through simultaneous ablation of CD276/B7-H3-positive tumor cells and tumor vasculature. Cancer Cell. 2017;31:501–515. doi: 10.1016/j.ccell.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O'Connell S, et al. VISTA is an immune checkpoint molecule for human T cells. Cancer research. 2014;74:1924–1932. doi: 10.1158/0008-5472.CAN-13-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rouas-Freiss N, LeMaoult J, Verine J, Tronik-Le Roux D, Culine S, Hennequin C, et al. Intratumor heterogeneity of immune checkpoints in primary renal cell cancer: Focus on HLA-G/ILT2/ILT4. Oncoimmunology. 2017;6:e1342023. doi: 10.1080/2162402X.2017.1342023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tronik-Le Roux D, Renard J, Verine J, Renault V, Tubacher E, LeMaoult J, et al. Novel landscape of HLA-G isoforms expressed in clear cell renal cell carcinoma patients. Mol Oncol. 2017;11:1561–1578. doi: 10.1002/1878-0261.12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Agaugue S, Carosella ED, Rouas-Freiss N. Role of HLA-G in tumor escape through expansion of myeloid-derived suppressor cells and cytokinic balance in favor of Th2 versus Th1/Th17. Blood. 2011;117:7021–7031. doi: 10.1182/blood-2010-07-294389. [DOI] [PubMed] [Google Scholar]
- 44.Carosella ED, Rouas-Freiss N, Tronik-Le Roux D, Moreau P, LeMaoult J. HLA-G: an immune checkpoint molecule. Adv Immunol. 2015;127:33–144. doi: 10.1016/bs.ai.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 45.Zhang P, Guo X, Li J, Yu S, Wang L, Jiang G, et al. Immunoglobulin-like transcript 4 promotes tumor progression and metastasis and up-regulates VEGF-C expression via ERK signaling pathway in non-small cell lung cancer. Oncotarget. 2015;6:13550–13563. doi: 10.18632/oncotarget.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.