

Abstract
Current immunotherapies have proven effective in strengthening anti-tumor immune responses, but constant opposing signals from tumor cells and the surrounding microenvironment eventually lead to immune escape. We hypothesized that in situ release of antigens and regulation of both the innate and adaptive arms of the immune system would provide a robust and long-term anti-tumor effect by creating immunological memory against tumors. To achieve this, we developed CARG-2020, a genetically modified virus-like vesicle (VLV) that is a self-amplifying RNA with oncolytic capacity and encodes immune regulatory genes. CARG-2020 carries three immune modulators: 1) the pleiotropic antitumor cytokine, IL12, in which the subunits (p35 and p40) are tethered together; 2) the extracellular domain (ECD) of the pro-tumor IL17RA, which serves as a dominant-negative antagonist; and 3) a shRNA targeting PD-L1. Using a mouse model of ovarian cancer, we demonstrated the oncolytic effect and immune-modulatory capacities of CARG-2020. By enhancing IL12 and blocking IL17 and PD-L1, CARG-2020 successfully reactivated immune surveillance by promoting M1, instead of M2, macrophage differentiation, inhibiting MDSC expansion and establishing a potent CD8+ T cell–mediated anti-tumoral response. Furthermore, we demonstrated that this therapeutic approach provided tumor-specific and long-term protection against the establishment of new tumors. Our results provide rationale for the further development of this platform as a therapeutic modality for ovarian cancer patients to enhance anti-tumor responses and prevent recurrence.
Keywords: onco-immunomodulators, cancer immune surveillance, immune escape, ovarian cancer recurrence
Introduction
The development of solid tumors requires modulation of the host immune system and successful escape from immune surveillance (1,2). This is accomplished by mechanisms that recruit and educate immune cells to support tumor growth and promote metastatic expansion (3–5). Immunotherapy, specifically immune checkpoint inhibitors, has transformed the treatment of several solid tumors and has demonstrated success in the modulation of the tumor microenvironment (TME), promotion of anti-tumor responses, and consequently regression of tumors (6–8). Unfortunately, not all tumor types have benefitted from this modality, and currently available immune modulators have shown limited success with recurrent disease (9,10).
Conventional treatment for solid tumors, such as ovarian cancer, involves chemotherapy that is administered prior to or after surgery (11). Although chemotherapy eradicates the majority of the tumor, undetectable micro-metastatic niches always persist (12). These chemoresistant cells are thought to drive relapse and are additionally known to create a unique microenvironment that modulates both the local and systemic immune system. Therefore, immune therapies that can successfully target these chemoresistant micro-metastases could increase the likelihood of permanent remission.
One of the well-known mechanisms that confers resistance to immune therapies is the establishment of a “cold” TME (13,14), wherein cytolytic T cells are actively prevented from infiltrating tumors and is attained by the recruitment of immune suppressor cells, such as myeloid-derived suppressor cells from the bone marrow (BM-MDSCs), M2 macrophages, and T regulatory cells (Tregs) (15). In contrast, “hot” tumors are characterized by presence of T cells, antigen-presenting cells (M1 macrophages and dendritic cells), and absence of immune suppressor cells, consequently demonstrating better response to immune therapy and ultimately better outcomes (16). A conceivable path to improve T-cell antitumor activity is to counteract immunosuppressive signals, such as those of BM-MDSCs. Indeed, studies have shown that poor clinical outcomes associate with an increase in tumor infiltration of immunosuppressive cells such as tumor-associated macrophages (TAMs) and BM-MDSCs. Another important characteristic of “cold” tumors is poor antigenicity, which further enhances the tolerogenic state of TME and impedes the success of anti-tumor vaccines. In situ vaccination (ISV) has been proposed as an efficient approach to remodel “cold” tumors into “hot” tumors while boosting anti-tumor T-cell responses (17). However, the selection of the most efficient antigen, which represents the heterogeneity of the tumor, has proven to be a major challenge (18–20). A promising strategy is to develop personalized vaccination by inducing cancer cell death and enhancing antigen presentation.
The use of oncolytic viruses has been proposed as an effective mechanism to induce the release of tumor antigens as a result of virus-induced cell death (19,21,22). Oncolytic viruses infect/replicate only in cancer cells, leading to cancer cell lysis, and their efficacy is amplified when used as an ISV by promoting cancer cells apoptosis, releasing high levels of tumor antigens, and evoking anti-tumor immune responses (23,24). However, although the oncolytic effect can enhance the release of tumor antigens, it is inefficient at modulating the components of immune system (23). It is plausible that the development of a more comprehensive immunotherapy strategy that engages multiple arms of the immune system and incorporates immune modulatory genes with the oncolytic effect can induce a more potent and persistent anti-tumor effect.
The objective of the study was to elucidate the efficacy of a novel therapeutic approach, CARG-2020, that can modulate immune responses while inducing cancer cells death. CARG-2020 is an armed oncolytic virus-like vesicle (VLV) vector with components from the alphavirus Semliki Forest Virus (SFV) and rhabdovirus vesicular stomatitis virus glycoprotein (VSV-G) and produces infectious, replication-competent, enveloped vesicles at high titers in vitro (25,26). VLVs are a capsid-free, self-replicating virus-like vaccine platform carrying positive-strand capped and polyadenylated RNA that encodes an in vitro-evolved SFV RNA-dependent RNA replicase and the VSV glycoprotein. VLVs can be engineered to express foreign antigens, proteins, or microRNAs (miRNA) that can modulate immune responses (27,28). The VLV platform replicates like a virus, but its only structural protein is the VSV glycoprotein (VSV-G), and unlike many other viral vectors, lacks pathogenicity (29,30). CARG-2020 is a membrane-encapsulated oncolytic VLV RNA replicon delivering three immunomodulators: IL12, dn-IL17RA (dominant-negative IL17 receptor A), and shRNA for PD-L1.
Previously, we evaluated a VLV and its potential impact on cancer cells and observed that it can function in the context of oncolytic virus therapy (24). In this present study, we tested the capacity of a VSV-based oncolytic virus, CARG-2020, to function as an ISV by inducing cancer cell death and to modulate both the innate and adaptive arms of the immune system through the expression of the three immune-modulatory molecules: IL12, IL17RA ECD (extracellular domain), and short hairpin RNA (shRNA) for PD-L1. Using human and mouse ovarian cancer models in both immunocompromised (human ovarian cancer cells) and immunocompetent mice (mice ovarian cancer cells), we demonstrated that CARG-2020 has potent oncolytic effect in cancer cells but not in normal cells and is well tolerated in vivo. In addition, we demonstrated that CARG-2020 can convert the “cold” ovarian TME to “hot” and provide specific long-term anti-tumor immunity capable of preventing tumor expansion and disease recurrence.
Methods and methods
Cell lines and culture conditions
Triple knockout (TKO) mouse ovarian cancer cells were kindly provided by Dr. Martin Matzuk (Baylor College of Medicine). These cells were obtained from spontaneously formed high-grade serous ovarian tumors in mice with conditional deletion of Dicer and PTEN and with p53 mutation (p53LSL-R172H/+Dicerflox/floxPtenflox/flox Amhr2cre/+ (31). mCherry fluorescence was stably expressed using a lentivirus as previously described (32) to allow monitoring of intraperitoneal (i.p.) tumors in real time. mCherry expression was confirmed by both microscopy and flow cytometry. TKO cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM)/F12 media (ThermoFisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (FBS) (ThermoFisher Scientific) and 1% penicillin-streptomycin (ThermoFisher Scientific). OCSC1-F2 cells were isolated in-house from tumors derived from ovarian cancer patients as previously described (24,33–38) and cultured in RPMI media(ThermoFisher Scientific) supplemented with 10% FBS, 1% penicillin-streptomycin, 1% sodium pyruvate, 1% HEPES, and 1% non-essential amino acids (ThermoFisher Scientific). BHK-21 cells (a kind gift from Dr. John Rose, Yale University) were cultured in DMEM supplemented with 5% FBS. Normal human endometrial stromal cells were established and cultured as previously described (39). B16 melanoma cells were a kind gift from Dr. Anthony Vella (University of Connecticut). All cell lines were grown at 37°C with 5% CO2. Cells were routinely tested for mycoplasma and authenticated once a year by STR profiling and used within 6 passages between experiments.
Generation of the recombinant CARG-2020 vector and particles
Previously described VLV double promoter (dp) vector (27) was used for cloning by linearizing with Asc I and Sbf I restriction enzymes (New England Biolabs (NEB), Ipswich, MA) in order to insert the VSV G glycoprotein fragment from New Jersey (NJ) serotype downstream of the second sub-genomic promoter (27). The resulting VLVdp VSV GNJ vector was then digested with BamH I and Pac I enzymes (NEB), and the resulting vector was used to insert cytokines and the shRNA downstream of the first sub-genomic promoter. In order to generate the CARG-2020 construct containing three transgenes, namely IL12 (IL12-p40 Accession #NP_001290173.1; IL12-p35 Accession #NP_001152896.2), IL17RA (Accession #NP_032385.1), and shRNA target sequences derived from PD-L1 (Accession #NM_021893.3), the full-length IL12 fragment (encompassing both fused subunits) and an IL17RA extracellular domain (ECD) containing a 3’ HA-tag and PD-L1-shRNA synthetic gene fragment incorporating three shRNA concatemers (Sh-1 :5’ATTTGCTGGCATTATATTCAC-3’; Sh-2: 5’-GCTGAAAGTCAATGCCCC ATA-3’, and Sh-3: 5’-CTGGACAAACAGTGACCACCA-3’) were amplified and infused into VLV dp - VSV GNJ between BamHI/PacI cloning sites. To clone GFP into the VLV vector, an insert was amplified from a VLV-GFP vector (kind gift from Dr. John Rose, Yale University) and cloned in VLV dp - VSV GNJ. Full-length GFP insert was infused into AscI and Sbfl digested VLV-NJ vector next to the second subgenomic promoter. All primers to amplify DNA fragments were designed according to NEBuilder instruction and infused into VLV dp - VSV GNJ vector using HiFi DNA Assembly Kit (NEB, E5520S). Sequences for these primers are in Supplementary Table S1. The recombinant clones were screened for positive inserts by DNA restriction digestion. The final positive constructs were further confirmed by DNA sequencing (GENEWIZ, NJ, USA).
The full-length IL12 fragment and IL17RA ECD HA tag/PD-L1-shRNA fragment driven by a sub-genomic promoter sequence were amplified by PCR and infused into VLV dp VSV-GNJ vector between BamHI/PacI cloning sites downstream of the first sub-genomic promoter. All primers to amplify DNA fragments were designed according to NEBuilder instruction, infused into the VLV backbone, screened for positive inserts, and the final positive constructs were confirmed by DNA sequencing as described above. Sequences for these primers are in Supplementary Table 1. VLVs were produced by transfecting BHK-21 cells with the VLV plasmid DNA, followed by collection of the master VLV stock. Propagation of the working stocks was performed by a single passage of the master stock in BHK-21 cells cultured in Opti-MEM™ Reduced Serum Medium (ThermoFisher, Waltham, MA, USA). Working stocks were concentrated using MacroSep® Advance 100K MWCO (Pall Laboratories, Port Washington, NY, USA) and titrated using plaque assays in BHK-21 cells.
Cell growth and cell death assays
3,000 TKO cells were seeded per well of a 96-well plate and allowed to reach ~80% confluency. Increasing doses of VLV-GFP or CARG-2020 (8, 16, 83, 166, and 833 PFU/cell) were added in a 200 μL total volume with added Celltox™ fluorescent dye (Promega, Madison, WI) at a 1:2,000 final dilution. Control wells received PBS at a dilution corresponding to the highest dose of VLV-GFP or CARG-2020. Plates were placed in Cytation5/Biospa (Agilent Technologies, Santa Clara, CA) for automatic imaging every 2 hours for a total of 90h. mCherry fluorescence was acquired using Texas Red filter cube (EX 586/15 nm, EM 647/57 nm). mCherry total sum area is the total masked area automatically detected by the Gen5 software using mCherry intensity threshold of 2000. Celltox fluorescence was acquired using GFP filter cube (EX 469/35 nm, EM 525/39 nm). Celltox total sum area was the total masked area automatically detected by the Gen5 software using green fluorescent intensity threshold of 7000.
Immunocompetent mouse model and treatment schedule
All the described experiments using mice were approved by Wayne State University Animal Care and Use Committee (IACUC 22–03-4474), and mice were housed at Wayne State University Division of Laboratory Animal Resources. 1×107 mCherry+ TKO mouse ovarian cancer cells were injected intraperitoneally (i.p.) in 6–8 week old female C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME). Tumor growth was monitored twice a week (typically every 3 to 4 days) by detecting mCherry fluorescence using Ami HT Imaging System (Spectral Instruments, Tucson, AZ). Tumor burden was quantified using mCherry fluorescent region of interest (ROI) using Aura Imaging Software (Spectral Instruments). Treatment commenced when the mCherry+ ROI reached 3.2 × 108 photons/sec (around 18 days post injection of cells, which is typically the beginning of the expansion phase; Fig. 1A). CARG-2020 or VLV-GFP was administered i.p. at 1×106 PFU/dose, given every three days for a total of three doses. Control mice received PBS. Animal weight and abdominal width by caliper measurement were monitored twice a week. Animals were sacrificed when the mCherry+ ROI reached 1 × 109 PFU/sec or when abdominal width reached or exceeded 3.4 cm.
Figure 1. Characterization of immune response during ovarian tumor progression.
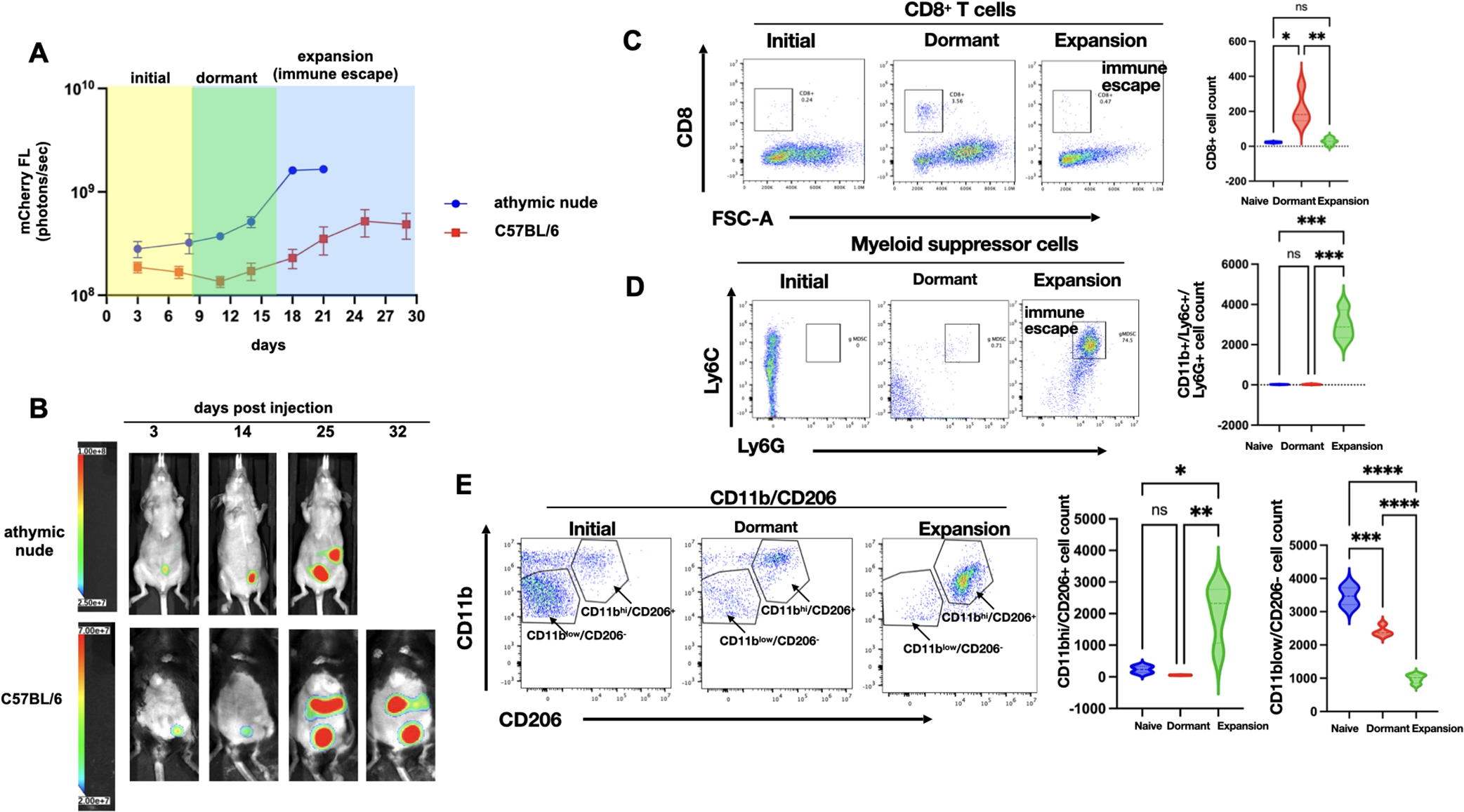
mCherry+ TKO mouse ovarian cancer cells were injected i.p. in C57BL/6 immunocompetent mice or immunocompromised athymic nude mice. A. Tumor growth was quantified using mCherry ROI fluorescence. Note difference in tumor kinetics between mouse strains with the dormant phase observed only in C57BL/6 mice; data shown are Mean ± SEM for representative experimental cohort; B. Representative images of live animal imaging showing progression of i.p. tumor burden; C. Peritoneal lavage from TKO-bearing C57BL/6 mice from each phase of tumor progression (n=6) was stained with anti-CD8 and analyzed by flow cytometry; D. Peritoneal lavage from TKO-bearing C57BL/6 mice from each phase of tumor progression (n=6) was stained with anti-CD11b, anti-Ly6C, and anti-Ly6G and analyzed by flow cytometry; E. Peritoneal lavage from TKO-bearing C57BL/6 mice from each phase of tumor progression (n=6) was stained with anti-CD11b and CD206 and analyzed by flow cytometry. Representative dot plots are shown. Gating strategy is shown in Supp. Fig. 1. Violin plots show Mean ± SEM and One-way ANOVA was used for statistical analysis. p value <0.05 was considered significant.
Immune-incompetent model and treatment schedule
5×106 mCherry+ TKO mouse ovarian cancer cells were injected i.p. in 6–8 week old female athymic nude mice (Envigo, Indianapolis, IN). Tumor growth was monitored and quantified as above. Treatment commenced when the mCherry+ ROI reached 3.2 × 108 PFU/sec (around day 11 post injection of cells). CARG-2020 was administered i.p. at 1×107 PFU/dose, given every three days for a total of three doses. Control animals received PBS. Animal weight and abdominal width by caliper measurement were monitored twice a week as specified above. Animals were sacrificed when the mCherry+ ROI reached 1 × 109 PFU/sec or when abdominal width reached or exceeded 3.5 cm.
Re-challenge experiments
TKO tumor-bearing C57BL/6 mice were established and treated with CARG-2020 as described above. Fifty days after the 1st dose of CARG-2020, disease-free mice were re-challenged with either 1×107 mCherry+ TKO mouse ovarian cancer cells or with 1×106 B16 mouse melanoma cells injected i.p. Tumor progression was monitored by imaging as described above (for TKO cells) or measurement of abdominal width using calipers (for B16 cells). Age-matched C57BL/6 mice were used as control.
Adoptive cell transfer
TKO tumor-bearing C57BL/6 mice were established and treated with CARG-2020 as described above. Spleens from disease-free CARG-2020-treated mice were collected 35 days after the 3rd dose of CARG-2020. Control mice received PBS. Spleens from control mice were collected when mCherry ROI reached 1×109 photons/sec. Splenocytes were obtained by homogenizing spleens using the back of 1 mL syringe. Splenocytes were pelleted, and red blood cells were lysed by resuspending cell pellets in 9 mL double distilled water, followed immediately by the addition of 1 mL 10x PBS. Samples were centrifuged at 1500 rpm for 5 min. to collect cell pellets, and pellets were resuspended in PBS to obtain cell count. 5×105 splenocytes in 200 μL PBS were injected i.p. to recipient mice. Recipient mice were C57BL/6 mice in dormant phase of disease.
Collection and preparation of peritoneal cells
For all the mice (C57BL/6 and athymic nude) treated with CARG-2020, VLV-GFP, and the corresponding PBS Controls, peritoneal cells were collected from ascites-free mice by i.p. injection of sterile PBS, followed by fluid aspiration into heparin-containing tubes. Peritoneal fluid from ascites-containing mice was collected by peritoneal tap into heparin-containing tubes. Cell pellets were obtained from peritoneal fluid by centrifugation at 1500 rpm for 5 min. Red blood cells were lysed by resuspending cell pellets in 9 mL double distilled water, followed immediately by the addition of 1 mL 10x PBS. Samples were centrifuged at 1500 rpm for 5 min. to collect cell pellets, and pellets were resuspended in FACS buffer (PBS + 1% bovine serum albumin + 0.05% sodium azide) to obtain cell counts.
Immunephenotyping by FACS analysis
Extracellular staining was performed on 1×106 peritoneal cells per 100 μL volume using the following antibodies: APC-conjugated CD8 (clone 53–6.7; 1:100); Superbright 785-conjugated CD11b (clone M1/70; 1:100); APCFire 750-conjugated Ly6C (clone HK1.4 1:50), Pacific Blue-conjugated Ly6G (clone 1A8; 1:50) , APC-conjugated CD206 (clone C068X2; 1:100), Brilliant Violet 605-conjugated MHC-II (clone M5/114,15.2; 1:50), Superbright 785-conjugated CD44 (clone lm7; 1:50), and FITC-conjugated CD62L (clone MEL-14; 1:100). Staining was performed for 30 minutes. Stained cells were then washed in FACS buffer and resuspended in 4% paraformaldehyde in PBS until acquisition. Flow cytometry was carried out with electronic gates set on >200k FSC-A and single cells. A minimum of 5 × 104 events was collected per sample, and data were analyzed with FlowJo software (FlowJo LLC). Gating strategies are shown in Supplementary Figure S1.
In vitro treatment with CARG-2020
7×104 TKO cells were plated in 60mm dishes in 3 mL growth media. 48h later, cells were treated with 1×105 PFU/mL of CARG-2020. Cell pellets and cell-free conditioned media were collected after 24h.
Measurement of secreted IL12
TKO cells were treated as above, and cell-free conditioned media were obtained from treated cells, as well as PBS controls. Undiluted conditioned media were used to quantify IL12-p40 and IL12-p70 using Bioplex Pro™ mouse cytokine IL12 (p70) paired antibodies and Bioplex Pro™ mouse cytokine IL12 (p70) paired antibodies using xMAP technology (Bioplex System, Biorad, Hercules, CA).
RNA extraction, cDNA synthesis, and quantitative (q)PCR
Total RNA was extracted from freshly isolated cell pellets using RNAeasy Mini Kit (Qiagen, Austin, TX) according to the manufacturer’s instructions and with DNase treatment. RNA was quantified, and purity assessed using an Epoch Microplate Spectrophotometer (Agilent, Santa Clara, CA). cDNA was synthesized using the iScript cDNA kit (Bio-Rad, Hercules, CA) from 1 microgram of total RNA according to manufacturer’s instructions. qPCR was performed using SYBR Green Supermix (Bio-Rad, Hercules, CA), with 1:10 dilution of cDNA in a final volume of 10 μL according to manufacturer’s instructions. qPCR was run on CFX96TM PCR detection system (Bio-Rad, Hercules, CA) using the following thermocycling parameters: initial denaturation step of 2 minutes at 95°C; primer annealing step for 30 seconds at 55°C and elongation step for 60 seconds at 74°C for 30 cycles; and final extension step for 5 minutes at 74°C. Primers used are shown in Supplementary Table S2. A “No RT” control was used as negative control. Relative expression was calculated using the comparative ΔΔCT method with Ppia as the housekeeping gene and the Control no treatment group as the reference. All reactions were performed in triplicates.
Western blot analysis
Whole cell protein lysates were isolated by resuspending cell pellets in 1X Cell lysis buffer (Cell Signaling Techonologies, Danvers, MA) with added cOmplete™ Protease Inhibitor Cocktail (Millipore Sigma, Burlington, MA) and centrifugation for 20 minutes at 1,500 rpm. 20 μg of protein lysate was electrophoresed on 12% SDS-polyacrylamide gels made in-house then transferred to PVDF membranes (EMD Millipore, Burlington, MA). After blocking with 5% milk, membranes were probed with the following primary antibodies at 4°C overnight: ß-Actin (Millipore # MAB1501); IL12 (R&D Systems # MAB1570–500); HA tag (for IL17RA; Invitrogen # 71–5500); PD-L1 (Bio X Cell # BE0101). Blots are then placed in secondary antibodies for 1 hour at room temperature. The blots were developed using enhanced chemiluminescence and imaged using GE ImageQuant LAS 500 chemiluminescence (Cytiva Life Sciences, Marlborough, MA).
Statistical analysis
Unpaired two-tailed Student t-tests, assuming Gaussian distribution, or one-way analysis of variance (ANOVA) with Dunnett’s multiple comparisons were used for comparison between different groups. P values of 0.05 or less were considered statistically significant. Statistical analysis was performed, and all data were graphed, using GraphPad Prism v9.3.1 (San Diego, CA; RRID:SCR_002798). Data are presented as mean ±SEM.
Data availability statement
All data are present in the main paper and/or the supplementary materials and are available upon reasonable request to the corresponding author.
Results
Carcinomatosis associates with an immunosuppressive peritoneal microenvironment
To characterize the temporal regulation of the immune response during the process of carcinomatosis formation, we utilized a previously reported syngeneic mouse model of ovarian cancer established using mCherry-expressing (mChery+) TKO mouse ovarian cancer cells injected i.p. in C57BL/6 immunocompetent mice (5,24). TKO mouse ovarian cancer cells were isolated from spontaneously formed high-grade serous ovarian cancer from a conditional Dicer-PTEN KO mouse with a p53 mutation (p53LSLR172H/+Dicerflox/floxPten flox/flox Amhr2cre/+). Three days after inoculation, mCherry+ TKO cancer cells could be detected in the peritoneal cavity. This initial mCherry signal gradually dissipated 5–10 days post-injection without any treatment intervention and remained below background until about day 21, when the mCherry signal returned and continued to increase, resulting in animals developing substantial i.p. carcinomatosis and ascites by day 30 (Figure 1A–B). Thus, in this model, we observed three distinct phases of tumor progression: 1) Initial phase (yellow box); 2) Dormant phase (green box) potentially associated with immune surveillance; and 3) Expansion/Immune Escape phase (blue box) due to a brake on immune surveillance (Supp. Fig. S2).
To determine if the same kinetics would be observed in immunocompromised mice, we injected the same number of mCherry+ TKO cells into athymic nude mice. In this model, we did not observe a decrease in mCherry signal corresponding to the Dormant phase observed in C57BL/6 mice. In the athymic nude mice, the disease continuously progressed, with generalized carcinomatosis around day 20–25 (Fig. 1A–B). These findings demonstrate that the presence of a functional immune system impacts the kinetics of tumor growth and suggests that it is necessary for the early control of cancer cell implantation and continued disease progression.
To test the hypothesis that the Dormant phase observed in immunocompetent C57BL/6 mice associated with an active anti-tumor immune response, we characterized and compared the i.p. immune phenotype between naïve mice (not injected with TKO cancer cells) and mice in the Dormant phase and Expansion phase using peritoneal lavage. We first assessed the CD8+ T-cell population and observed transient, but significant, expansion of CD8+ T cells in the Dormant phase, which was lost in the Expansion phase (Fig. 1C), confirming the presence of active immune surveillance during the Dormant phase.
Next, we evaluated the components of the innate immune response. We first focused on MDSCs because they are known to blunt protective anti-tumor immunity (40,41). We observed an inverse correlation between the presence of MDSCs and CD8+ T cells. MDSCs were not observed in the peritoneal lavage of naïve and Dormant phase mice. Instead, a significant expansion of CD11b+Ly6C+Ly6G+ granulocytic MDSCs (gMDSCs) was observed in the Expansion phase (Fig. 1D). Because M2 macrophages constitute the predominant cell type in neoplastic tissues contributing to angiogenesis and cancer metastasis, we tested for the presence of CD11b+CD206+ M2 macrophages in the peritoneal cavity. Naïve mice were characterized by the presence of a dominant population of CD11blowCD206− macrophages (Fig. 1E). In contrast, Dormant and Expansion phases were characterized by a dominant population of CD11bhighCD206+ M2 macrophages. The percentage of CD11bhighCD206+ M2 macrophages was significantly higher in the Expansion phase compared to naïve mice and mice in Dormant phase (Fig. 1E).
Taken together, these data demonstrate that although there is an initial anti-tumor response during the Dormant phase, the Expansion phase is characterized by the loss of CD8+ T cells and the expansion of immunosuppressive Ly6C+Ly6G+ gMDSCs and CD11bhighCD206+ M2 macrophages, which together can facilitate tumor growth and carcinomatosis (Supp. Fig. S2). We postulate that inhibiting the expansion of MDSCs and CD11b+CD206+ M2 macrophages will aid in the promotion of CD8+ T-cell expansion to consequently prevent tumor progression.
In vitro activity of CARG-2020
To test our hypothesis that combined modulation of the innate and adaptive immune system can successfully prevent tumor progression and protect from tumor recurrence, we engineered CARG-2020, a DNA plasmid based on SFV-VSV chimeric viruses, in which the native SFV glycoprotein was replaced with the VSV glycoprotein sequence (Supp. Fig. S3A). CARG-2020 carries the expression sequence for three immune modulators: IL12, which is expected to amplify the anti-cancer response by promoting T-cell proliferation; IL17RA ECD, which is expected to antagonize IL17 signaling and block tumor-promoting inflammation; and shRNA-PD-L1, which is expected to inhibit the PD-L1/PD-1 immune checkpoint pathway (Supp. Fig. S3A). We first tested whether CARG-2020 could infect ovarian cancer cells and express its cargo in vitro. Thus, TKO mouse ovarian cancer cells were exposed to CARG-2020 for 24h, and Il12 mRNA expression was quantified by qPCR. We observed a significant increase in Il12 mRNA expression upon treatment with CARG-2020 compared to Control [no] treatment (Supp. Fig. S3B). Next, we determined if the mRNA expression was translated into protein by quantifying secreted IL12 in cell culture supernatants. Both the p40 and p70 IL12 subunits were significantly higher in CARG-2020- infected cells compared to the vector Control (Supp. Fig. S3B). Western blot analysis showed protein expression of IL12, as well as IL17RA ECD, only in the CARG-2020-infected cells, but not in vector Control cells (Supp. Fig. S3C). Because the action of the IL17RA ECD is mediated by its binding to circulating IL17, we examined the presence of secreted IL17RA ECD in the supernatant of cells treated with CARG-2020 by western blot analysis. IL17R ECD was detected only in supernatants of cells treated with CARG-2020 and not supernatants from cells treated with VLV-GFP control (Supp. Fig. S3D). Finally, we tested the expression of shRNA-PD-L1 and its impact on PD-L1 expression. Cells were treated with CARG-2020, and PD-L1 expression was determined by western blot analysis in the cell lysate at 0-, 6- and 16-hours post infection. PD-L1 expression decreased in a time dependent manner in CARG-2020-treated cells but not in cells treated with vector control (Supp. Fig. S3E). These findings were confirmed by flow cytometry, which showed a decrease on PD-L1 MFI in the CARG-2020-treated cells, but not with VLV-IL12 or VLV-IL17 ECD (Supp. Fig. S3F)
We next evaluated the oncolytic effect of CARG-2020. Thus, mCherry+ TKO mouse ovarian cancer cells were exposed to increasing concentrations of CARG-2020 or VLV-GFP, and the effect on cell growth and viability were monitored by real-time cell imaging. Control [no] treatment cells received PBS. Both CARG-2020 and VLV-GFP induced a dose-dependent decrease in cell growth as measured by quantifying the mCherry region of interest (ROI) (Supp. Fig. S4A). To determine whether the decrease in cell growth was due to cell death, we also quantified the number of cells positive for the Celltox dye (Celltox+). We observed a dose-dependent increase in Celltox+ cells in both CARG-2020- and VLV-GFP-treated cultures (Supp. Fig. S4A) compared to Control. The increase in Celltox+ cells peaked in the first 24 hours with CARG-2020 treatment and around 40 hours with VLV-GFP treatment (Supp. Fig. S4A). Supplementary Figure S4B shows representative mCherry and Celltox images of cultures.
To further validate that the cytotoxic effect was not limited to mouse cancer cells, we treated human ovarian cancer cells, clone OCSC1-F2, with CARG-2020 and VLV-GFP. We selected the minimal cytotoxic concentration observed in the mouse cells for the human ovarian cancer cells and normal immortalized human endometrial stromal cells. 8 and 16 PFU/cell induce cell death in both mouse and human cancer cells but not in the normal human stromal cells (Supp. Fig. S4C). As we recently reported, VLV vectors have no cytotoxic effect on normal human cell lines (24), and we confirmed these findings by exposing normal human endometrial stromal cells to similar doses of CARG-2020. In these cultures, CARG-2020 did not demonstrate cytotoxic activity (Supp. Fig. S4C). Together, these results demonstrate that the VLV vector is oncolytic and that CARG-2020 can induce expression of its cargo in vitro.
To define whether the cell death observed following CARG-2020 was intrinsic to the VLV and not to the cargo, e.g. IL12, we evaluated the expression of pro-apoptotic genes, Trail and Fas, in mouse ovarian cancer cells exposed to VLV-GFP, which lacks any of the immune modulatory genes. Exposure to VLV-GFP induced the expression of Trail and Fas in a time dependent manner. We further validated these findings in vivo by determining Trail and Fas expression in tumors exposed to VLV-GFP. 24h after the administration of the second dose, tumor samples were collected and examined for Trail and Fas mRNA expression by qPCR. We observed a significant increase on the expression of these two apoptotic genes in tumors from animals treated with VLV-GFP (Supp. Fig S5B). These findings support our premise that the VLV component of CARG-2020 can induce apoptosis of tumor cells independently of the immune-modulatory factors.
In vivo efficacy of CARG-2020
Having demonstrated an in vitro oncolytic effect and successful expression of the immune-modulatory factors carried by CARG-2020, we next evaluated its activity in vivo. Thus, we established TKO tumors i.p. in C57/BL6 mice and began treatment with CARG-2020, VLV-GFP, or PBS Control in the beginning of the Expansion phase as detailed in the Materials and Methods section. The time of the first dose was designated as day 0 (Fig. 2A). Three i.p. doses of 1×106 PFU per dose were given every 72h, and mice were imaged to follow disease progression. Mice in the CARG-2020-treated group demonstrated complete tumor regression with no detectable mCherry signal by day 14 (Fig. 2B–C). These mice remained disease-free when followed up to 60 days (Fig. 2B–C). In contrast, tumor progression and carcinomatosis was observed in mice in the VLV-GFP and PBS Control groups. Kaplan-Meier survival analysis showed significantly longer overall survival in mice treated with CARG-2020 compared to mice treated with VLV-GFP and PBS (Fig. 2D). The difference in the in vivo activity between CARG-2020 and VLV-GFP was interesting given that CARG-2020 and VLV-GFP were both cytotoxic in vitro. These findings suggest that the efficacy observed in CARG-2020-treated mice is secondary to the immune-modulators carried by CARG-2020.
Figure 2. CARG-2020 causes tumor regression and significantly improves overall survival.
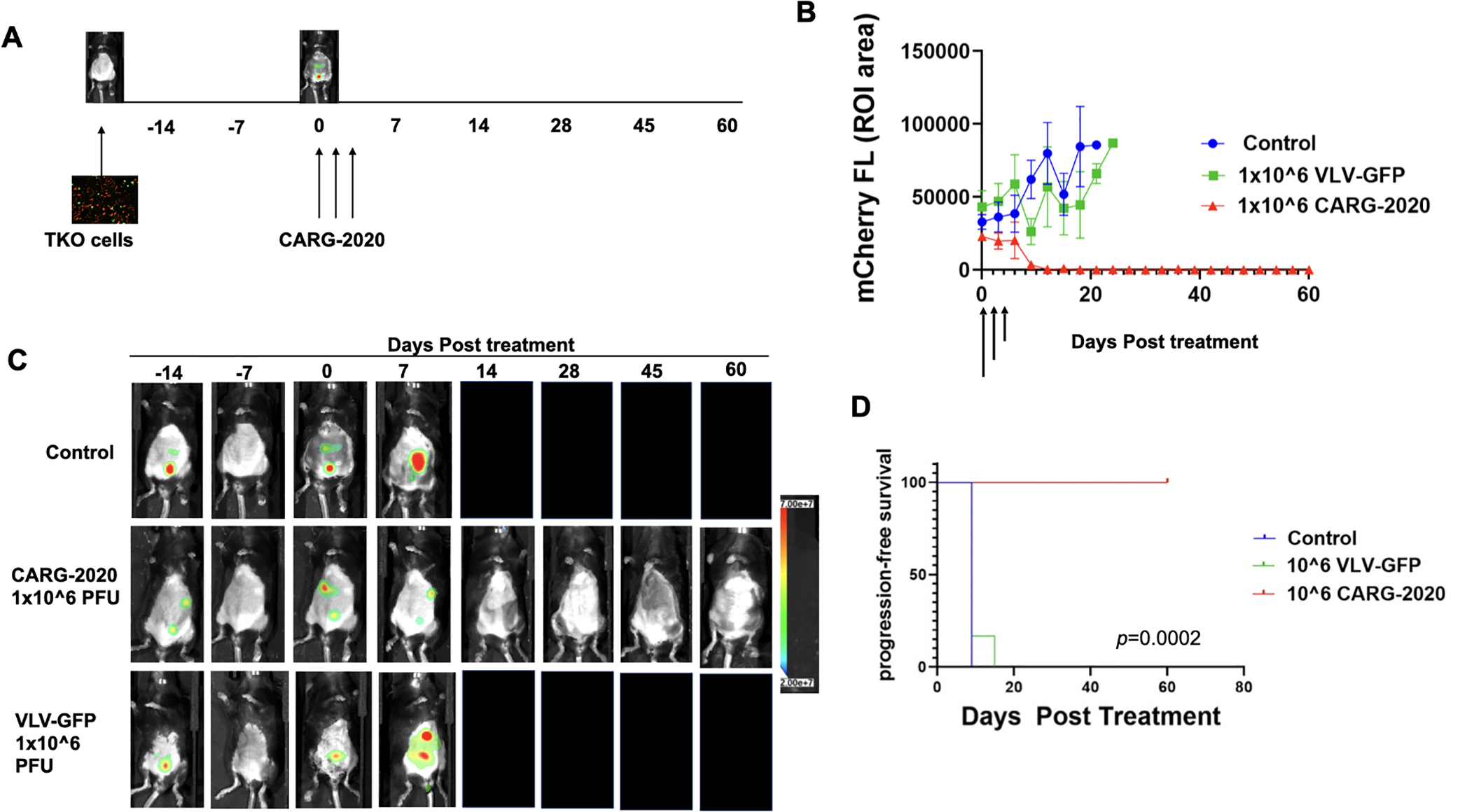
A. Diagram of experimental design. 1×107 mCherry+ TKO mouse ovarian cancer cells were injected i.p. in C57BL/6 mice. Mice were randomized into treatment groups and treatment began when mCherry ROI fluorescence reached 3.2×108 PFU/cell (designated as day 0). Treatment was 1×106 PFU of CARG-2020 or VLV-GFP given i.p. every 3 days for a total of 3 doses (arrows show days of treatment). Control no treatment mice received PBS (n=6 per group); B. Tumor growth was measured by mCherry ROI fluorescence (arrows show day of treatment); data show Mean ± SEM C. Representative images showing tumor regression only in CARG-2020 treated group; D. overall survival was defined as day mCherry ROI area reached 1×109 PFU/cell and calculated using Kaplan Meier analysis.
CARG-2020 induces specific anti-tumor immune memory
Given that both CARG-2020 and VLV-GFP demonstrated comparable cytotoxic activity in vitro (Supp. Fig. S3), we hypothesized that the superior in vivo activity of CARG-2020 may due to its ability to modulate the immune response. To elucidate these mechanisms, we treated a new set of mice with CARG-2020 and monitored their response. As observed previously (Fig. 2B), CARG-2020-treated mice again showed complete regression and remained disease-free for up to 55 days post-treatment (Fig. 3A). At this time, to determine the establishment of immunological memory and the capacity to prevent recurrent disease, we re-challenged CARG-2020-treated mice with i.p. injection of mCherry+ TKO cells. After the re-challenge, we observed a transient mCherry signal, but this lasted for only 7 days and failed to form tumors (Fig. 3A). The mice remained disease-free for up to 5 weeks post the 1st re-challenge. Thus, we re-challenged for the second time with another i.p. injection of mCherry+ TKO cells. Similarly, we observed a transient mCherry signal, but mice remained disease-free for up to 40 days after the 2nd re-challenge (Fig. 3A).
Figure 3. CARG-2020 provides specific immunological memory.
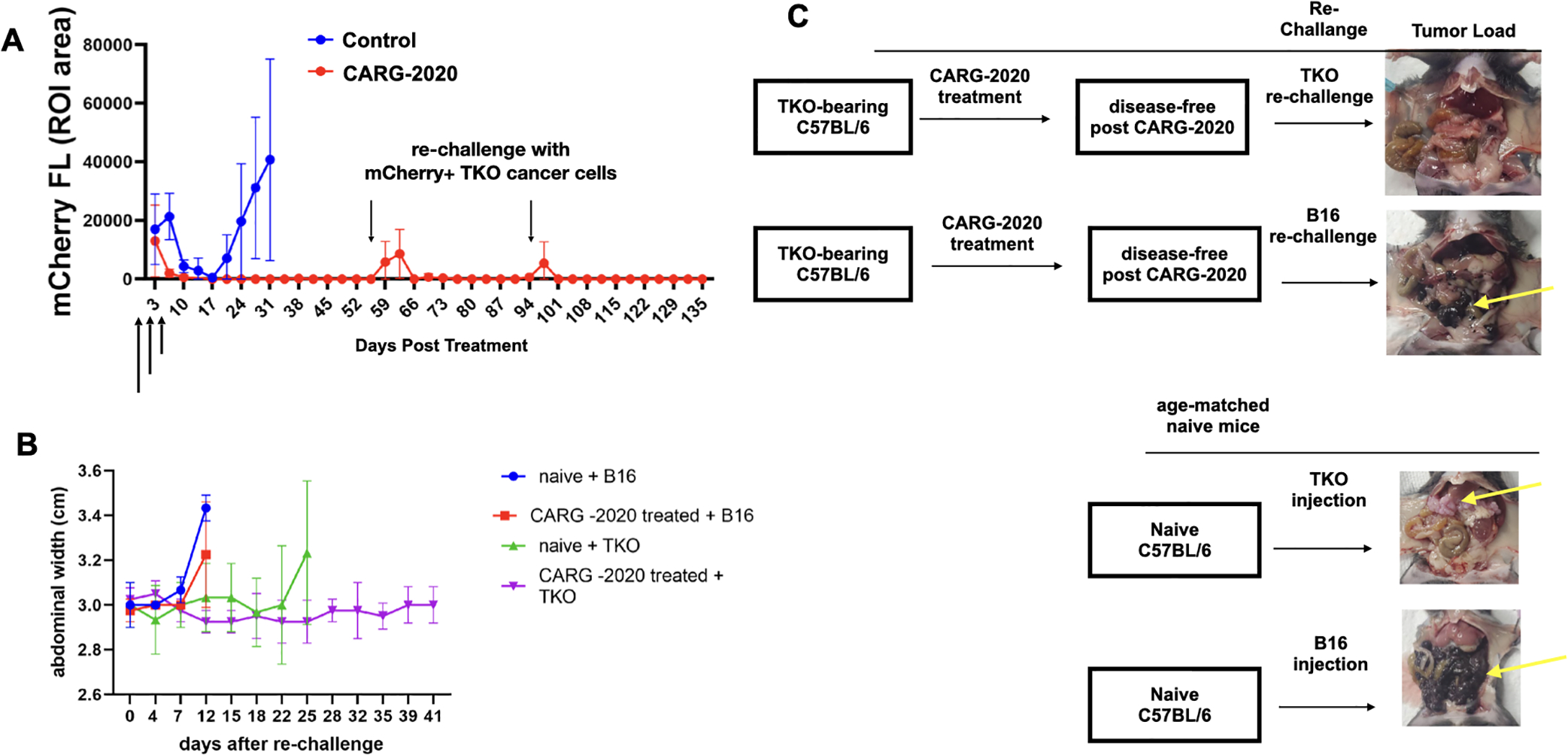
A. 1×107 mCherry+ TKO mouse ovarian cancer cells were injected i.p. in C57BL/6 mice. Mice were randomized into PBS control and CARG-2020 groups (n=6) when mCherry ROI fluorescence reached 3.2×108 PFU/cell (designated as day 0). Treatment was given i.p. in three doses of 1×106 PFU CARG-2020 (arrows). Note disappearance of mCherry signal in CARG-2020 treatment group up to day 55, at which point mice were re-challenged with i.p. injection of 1×107 mCherry+ TKO mouse ovarian cancer cells. Mice were followed for tumor formation and another re-challenge of TKO cells were given on day 94; B. To test for specificity, experiment in A was repeated with the addition of a group re-challenged with B16 mouse melanoma cells (n=5). Ascites formation was used as surrogate for i.p. tumor growth and monitored by measuring abdominal width. Naïve (age-matched, no tumors, no treatment) mice served as controls for i.p. growth of cancer cells; C. Representative necropsy images showing that CARG-2020 treatment of TKO-bearing mice protected from re-challenge with TKO ovarian cancer cells but not B16 melanoma cells. Yellow arrows point to i.p. tumors.
To determine if this was a specific immunological memory against TKO mouse ovarian cancer cells, we established TKO i.p. tumors in another set of mice, administered three doses of CARG-2020, and re-challenged mice at day 55 as before. However, instead of re-challenging with TKO ovarian cancer cells, the mice were re-challenged with B16 mouse melanoma cells. B16 melanoma cells promptly formed i.p. tumors, demonstrating that previous treatment of TKO-bearing mice with CARG-2020 protected only against TKO cells (Fig. 3B–C). Age-matched naïve mice were used during the re-challenge experiments as control for the cancer cell growth in vivo. Taken together these findings demonstrate that CARG-2020 can induce a long-term and specific immune memory.
CARG-2020-induced immune memory is transferable
To demonstrate and characterize the immunological response induced by CARG-2020, we performed adoptive transfer of splenocytes (Fig. 4A). mCherry+ TKO i.p. tumors were established in C57BL/6 mice followed by treatment with CARG-2020 as stated. Similar to data shown above (Fig. 2B, Fig. 3A), treated mice demonstrated no quantifiable mCherry signal after three doses of CARG-2020 (Fig. 4B). At day 35, we collected spleens from disease-free mice, isolated splenocytes, and injected 5 million splenocytes i.p. into another set of TKO-bearing mice in the Dormant phase of the disease (Fig. 4A). In this transfer experiment, the control groups (PBS or VLV-GFP) received splenocytes from TKO-bearing mice that never received CARG-2020 and hence had measurable disease. Splenocytes from CARG-2020-treated mice induced tumor regression and prevented the progression of recipient mice to the Expansion phase of the disease. This translated to significant improvement in overall survival compared to Control mice (Fig. 4C). These results further demonstrate that in addition to its oncolytic activity, CARG-2020 can induce a transferable immunological memory capable of promoting anti-tumor immune responses potent enough to induce complete disease regression and significantly improve overall survival.
Figure 4. CARG-2020-induced immune memory is transferable.
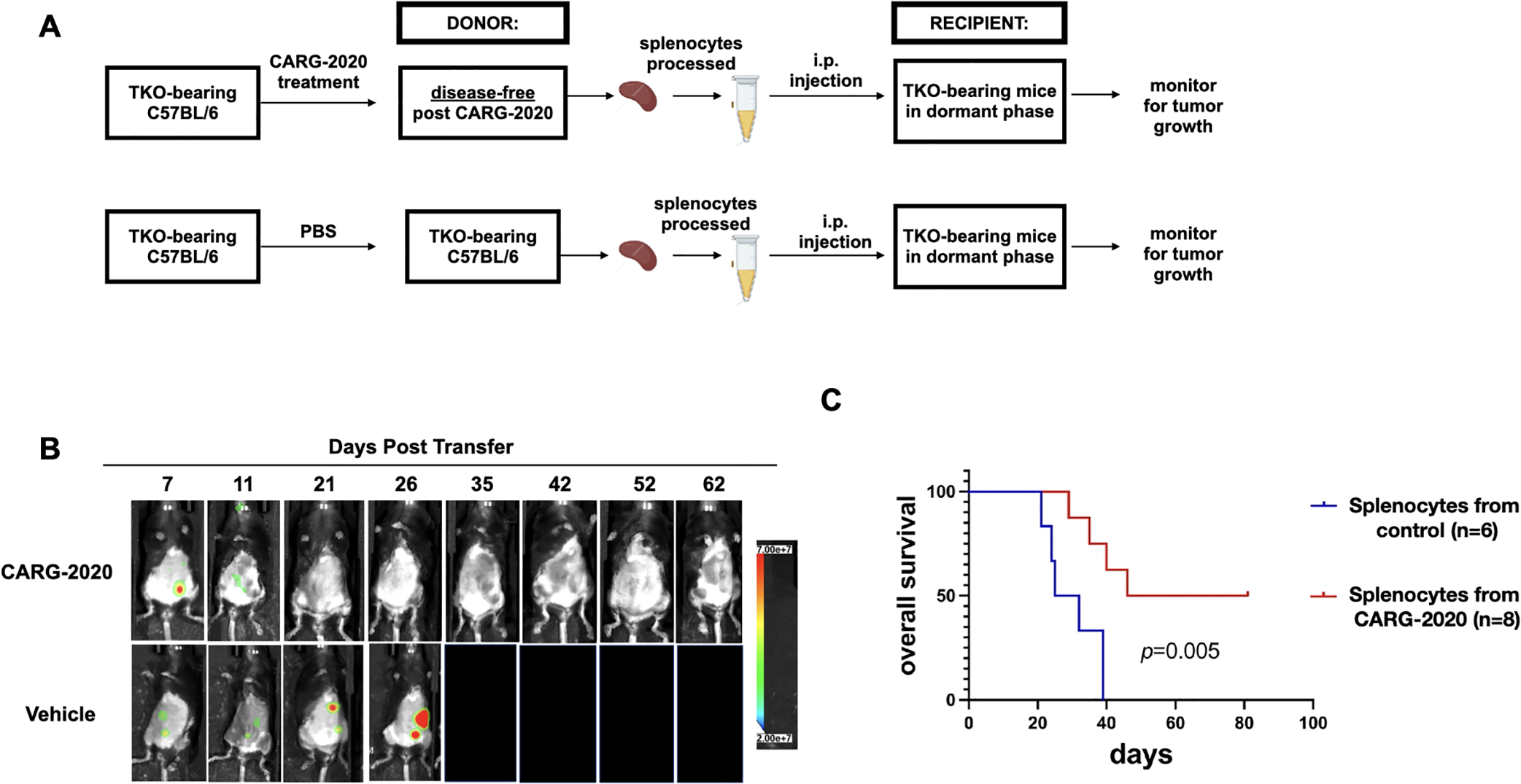
A. Diagram of spleen transfer study design. mCherry+ TKO mouse ovarian cancer cells were injected i.p. in C57BL/6 mice. CARG-2020 donor mice were treated with CARG-2020 (3 doses of 1×106 PFU CARG-2020) when mCherry ROI fluorescence reached 3.2×108 PFU/sec. Splenocytes were isolated from CARG-2020 donor mice 35 days after the start of treatment when mice were disease-free. Splenocytes were injected i.p. in another set of TKO-bearing mice in Dormant phase of the disease. Control recipients received splenocytes isolated from non-treated tumor-bearing donor mice. Non-treated tumor-bearing donor mice received PBS and spleens were isolated when mCherry ROI area reached 1×109 photons/sec; B. Representative images of Recipient mice showing mCherry+ i.p. tumor burden. C. overall survival was defined as day mCherry ROI area reached 1×109 PFU/sec or abdominal width reached 3.4 cm and calculated using Kaplan Meier analysis.
CARG-2020 prevents the shift towards a pro-tumor immunologic milieu and sustains a successful anti-tumor immune response
So far, our results have shown that CARG-2020 can promote a transferrable and specific immune memory capable of inducing complete disease regression and significant improvement of survival. To fully characterize the immunological changes, we investigated the immune profile of CARG-2020-treated disease-free mice using peritoneal lavage. We observed a significant decrease in CD11b+Ly6C+Ly6G+ pro-tumor gMDSCs in CARG-2020-treated mice compared to Control (Fig. 5A). Similarly, CD11bhighCD206+ M2 pro-tumor macrophages were also significantly decreased in mice treated with CARG-2020 (Fig. 5B). In CARG-2020-treated mice, CD11blowCD206− macrophages were the dominant population (Fig. 5B). Further analysis of the macrophage population showed that CARG-2020 promoted the expansion of CD11b+MHCII+ M1 anti-tumor macrophages (Fig. 5C).
Figure 5. CARG-2020 modulates the innate and adaptive arms immune system.
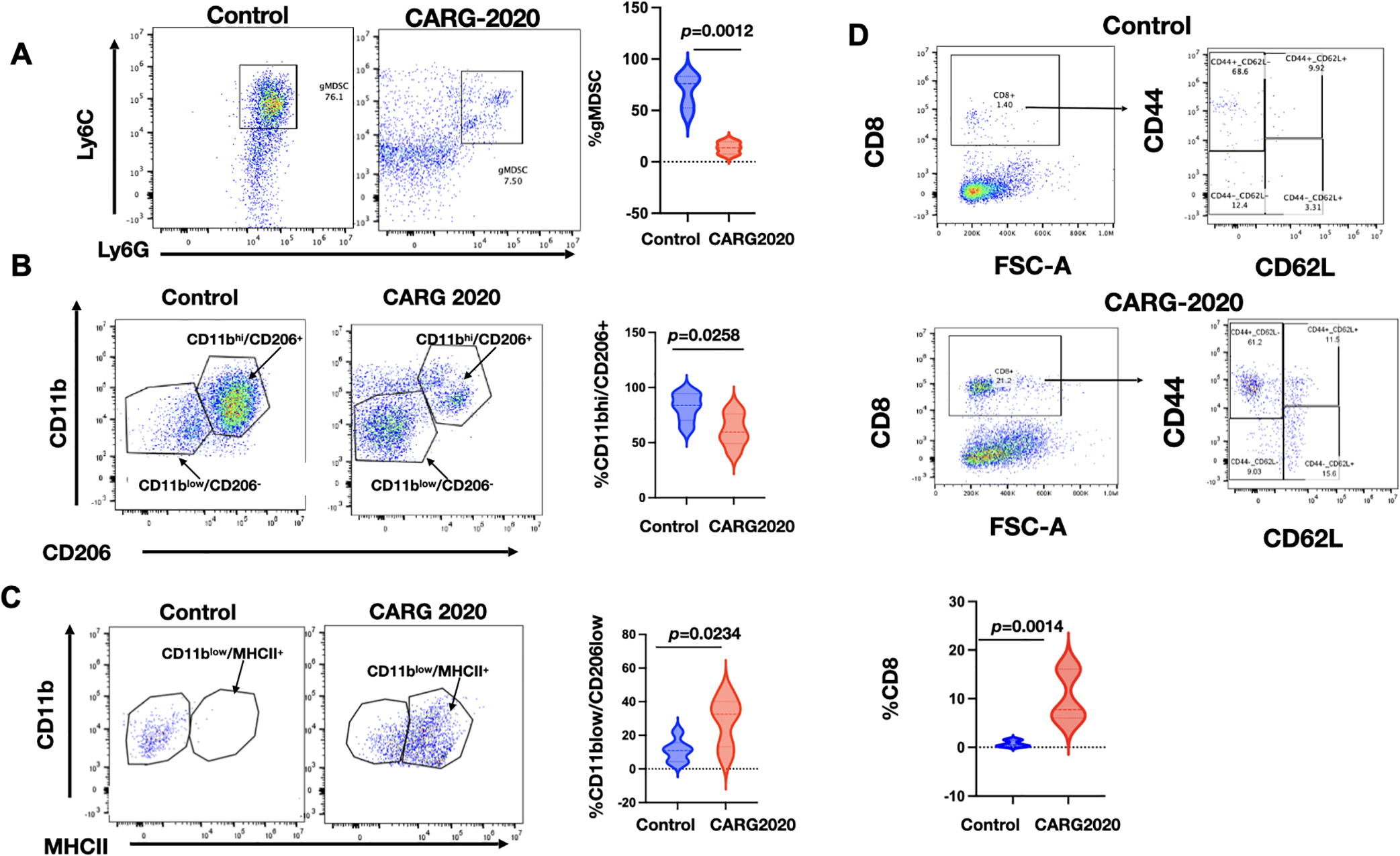
Peritoneal lavage from PBS Control and CARG-2020-treated mice (n=6) were analyzed by flow cytometry for: A. granulocytic myeloid suppressor cells (CD11b+/Ly6Clo/Ly6G+). B. macrophages using CD11b and CD206 and; C. antigen presentation using CD11b and MHCII; and D. CD8 memory T cells using CD8, CD44, and CD62L. Representative dot plots are shown. Gating strategy is shown in Supp. Fig. S1. Data are presented as Mean ± SEM and unpaired two-tailed Student t-test was used for statistical analysis.
Finally, we characterized the adaptive immune response and determined the levels of splenic CD8+ T cells. We observed significantly higher percentage of CD8+ T cells in mice treated with CARG-2020 compared to Control (Fig. 5D). Further characterization of the expanded CD8+ T-cell population showed that they were CD8+CD44+CD62L− effector memory T cells (Fig. 5D). These data demonstrate that CARG-2020 can prevent the expansion of immunosuppressive immune cells and successfully sustain an effective cytolytic and memory T-cell response.
CARG2020 anti-tumoral effect requires adaptive immune activation
Because our data showed that CARG-2020 could modulate both the innate and adaptive arms of the immune system, we next determined if both were essential for CARG-2020’s anti-tumor activity. To test this hypothesis, we used athymic nude mice, which specifically lack T cells but have an intact innate immune system. As shown above, the tumor kinetics of TKO cells in athymic nude mice did not include a Dormant phase as was observed in immune competent C57BL/6 mice (Fig. 1A). We injected 5×106 mCherry+ TKO mouse ovarian cancer cells i.p. and initiated treatment 10 days post injection. At this time, mice were randomized into Control and CARG-2020 groups. CARG-2020 was administered i.p. at 1×106 PFU per dose for a total of 3 doses (Fig. 6A). Animal imaging showed an initial and transient decline in tumor growth in CARG-2020-treated mice, but all mice eventually progressed (Fig. 6B). Flow cytometry analysis of peritoneal lavage showed no difference in the percentage of CD11b+CD206+ M2 macrophages (Fig. 6C). However, we observed a significant increase in the percentage of CD11b+MHCII+ M1 macrophages in mice treated with CARG-2020 (Fig. 6D–E). Thus, even though CARG-2020 was successful in promoting the expansion of CD11b+MHCII+ M1 macrophages, this did not translate to an effective anti-tumor response in mice lacking T cells. These results demonstrate that modulation of both the innate and adaptive arms of the immune system is necessary for successful control of tumor growth.
Figure 6. Adaptive immune system is required for CARG-2020 anti-tumor activity.
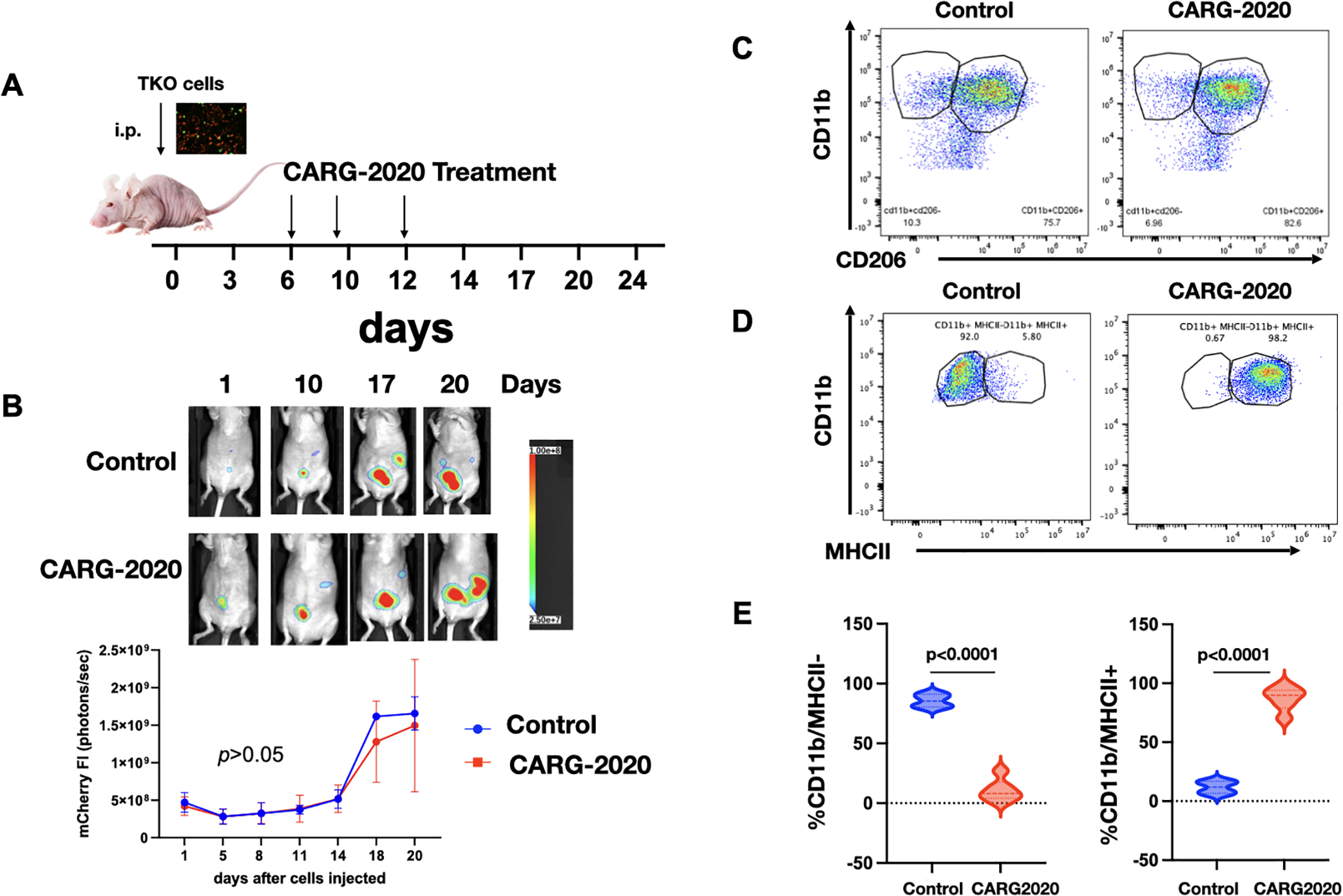
A. Diagram of study design. TKO mouse ovarian cancer cells were injected i.p. in athymic nude mice (day 0). Mice were randomized into PBS control and CARG-2020 treatment groups on day 6 (n=5). Treatment was administered i.p. at 1×106 PFU per dose for 3 doses (arrows); B. top panel, representative live animal imaging; bottom panel, quantification of tumor burden using mCherry ROI fluorescence area; C. Representative dot plots of peritoneal lavage analyzed for CD11b and CD206; D. Representative dot plots of peritoneal lavage analyzed for CD11b and MCHII; E. Quantification of D. Gating strategy is shown in Supp. Fig. S1. Data are presented as Mean ± SEM and unpaired two-tailed Student t-test was used for statistical analysis.
Characterization of CARG-2020’s individual components
Although there is abundant information related to the anti-tumoral effect of each of the modulatory factors carried by CARG-2020, we evaluated whether the “whole is greater than its parts” by treating tumor-bearing animals with VLV-IL12, VLV-IL17R ECD, or VLV-shRNA PDL1. We observed that VLV-IL12 alone was able to increase overall survival similar to CARG-2020 (Supp. Fig. S6A). This was not the case for VLV-IL17R ECD and VLV-shRNA PDL-1 (Supp. Fig. S6A). Although treatment with IL12 alone represents an ideal approach for tumor immunotherapy due to its ability to activate cytotoxic T lymphocytes (42–48), it has modest effects in clinical trials (49,50). Indeed, when we compared the immunological changes induced by CARG-2020 and VLV-IL12, we observed major differences in the regulation of M1/M2 macrophages and MDSCs. CARG-2020, in addition to inhibiting tumor growth, promoted the generation of M1 macrophages and decreased CD11b+Ly6C+Ly6G+ pro-tumor gMDSCs. In contrast, treatment with VLV-IL12 did not induce these immunological changes (Sup. Fig. S6B–C). These findings demonstrate that unlike CARG-2020, VLV-IL12 is not sufficient to modulate the innate immune response.
Efficacy of human CARG-2020 against human ovarian cancer cells in athymic nude mice
To determine the efficacy of CARG-2020 in human cancer cells in vivo, we used a previously reported animal model consisting of mCherry OCSC-F2 human ovarian cancer cells injected i.p. into athymic nude mice (2,37,51). Animals received three injections of CARG-2020. Tumor growth was monitored twice a week, and mCherry fluorescent ROI was plotted against time in days (Supp. Fig. S7). An initial decrease in tumor growth was observed in the CARG-2020-treated group, which was statistically significant by day 20 (p=0.0461) compared to Control group. However, this effect was transient, and tumor recurrence and growth was observed (Supp. Fig. S7A). Survival analysis did not show statistically significant differences between the groups (Supp. Fig. S7B). Taken together with the in vitro cytotoxicity data, these results demonstrate that CARG-2020 is cytolytic to human ovarian cancer cells but requires adaptive immunity to provide long-term anti-tumor protection.
Discussion
The development of intrinsic or acquired resistance is a major hurdle in the success of immune therapy in cancers, especially ovarian cancer. Resistance is typically acquired through several mechanisms targeting both the innate and adaptive arms of the immune system. Here we demonstrated that CARG-2020, an oncolytic vector containing multiple immune modulatory factors, could induce a successful anti-tumoral immune response against ovarian tumors by modifying both the innate and adaptative immune response and consequently conferring long-term protection against recurrent disease.
Dormant disseminated tumor cells (DTCs) are thought to be the seed of carcinomatosis and the origin of metastatic tumors. Upon seeding, DTCs create a supportive niche that can sustain their dormant state and allow their escape from immune surveillance. These mechanisms involve decreasing cancer cell antigenicity, recruitment of MDSCs, M2 macrophages, and Tregs, and expression of checkpoints such as PD-L1. Thus, a successful anti-tumor immunotherapy should have the capacity to reverse this state of tolerance and should be able to target most of the components of this complex network.
We have developed a disseminated intra-peritoneal ovarian cancer syngeneic mouse model. In this model, we observed a dormant phase, wherein majority of the cancer cells disappear either because they undergo cell death or are eliminated by the host immune system. However, a small number of cells not detected by imaging survive. The presence of these cells in our animal model is supported by the development of recurrent disease (expansion phase), which is observed after 10 to 15 days. The immunological milieu also changes in each of these phases. Most notable is the development of a tolerogenic condition that allows the expansion of the tumors. The main characteristics that we observed in our model during the expansion phase were: 1) suppression of CD8+ T cells; 2) recruitment of Ly6C+Ly6G+ gMDSCs and polarization of macrophages towards an M2 phenotype. Our model resembles many of the findings reported in patients with ovarian cancer. It has been demonstrated in vivo that T cells can halt ovarian carcinoma progression (52,53). However, antitumor immunity against established tumors is often insufficient to significantly impact tumor growth and clinical outcomes. In part, this is because ovarian cancers (and likely all solid tumors) suppress anti-tumor immunity by recruiting immunosuppressive stromal myeloid leukocytes, Tregs, and M2 macrophages (4,53). These cells not only have the capacity to blunt protective anti-tumor immunity (40,52–54), but they are crucial for both the generation and maintenance of tumor vasculature as well as the promotion of metastasis (5,55). Suppressive myeloid cells establish a pro-tumor microenvironment through multiple mechanisms including the expression of PD-L1, production of arginase, secretion of Galectin-1, and up-regulation of tolerogenic butyrophilins, all of which are mechanisms that can block anti-tumor immune responses (4,52,56).
In this study we tested the efficacy of a novel oncolytic virus carrying immune modulatory factors targeted to “open up” the TME to T-cell control, independent of tumor chemoresistance. VLVs are hybrid vectors based on an evolved Semliki Forest virus (SFV) RNA replicon and the envelope glycoprotein (G) from vesicular stomatitis virus (VSV) (28,30). VLVs can also express immune regulatory proteins, dominant negatives, and miRNAs and shRNAs. CARG-2020 was engineered to express IL12, antagonist to IL17RA, and shRNA for PDL-1. IL12 has been demonstrated to regulate both innate and adaptive immunities in cancer (57).
IL12 polarizes T cells into a type 1 helper T (Th1) effector cell phenotype. Additionally, IL12 programs effector T cells for optimal generation of effector memory T cells and T follicular helper cells (58). This cytokine was proposed as a potential agent in cancer immunotherapy due to its impressive antitumor effects in various animal models (59–61); unfortunately, the majority of clinical trials involving treatment with recombinant IL12 failed to show sustained antitumor responses (62,63) due to the presence of a strong anti-tumoral environment and its association with toxic side-effects when administrated systemically (58,64–66). We postulated that IL12 used in combination with other immune modulatory factors and delivered within the tumor microenvironment will minimize toxicity and provide a better anti-tumor effect (67). In this study, we confirmed that IL12 alone had anti-tumoral effects but was not able to regulate the innate immune system. IL12 acts predominantly on lymphocytes such as T and NK cells, but its function is inhibited by factors produced in the TME, mainly by macrophages and cancer cells (67). Thus, to enhance IL12 function, it is necessary to modulate the TME and abrogate factors that can inhibit IL12. To achieve this objective, we introduced two additional immune modulatory factors that could reverse the status of the TME: inhibition of IL17 and PD-L1.
IL17, a potent proinflammatory cytokine, has been linked to inflammation and wound healing, but has also been shown to contribute to the formation, growth, and metastasis of solid tumors (68,69). Increasing evidence demonstrates that the pro-tumorigenic mechanism of IL17 includes the promotion of an immunosuppressive endogenous microenvironment. In this regard, IL17 works through two main mechanisms: 1) recruitment of immunosuppressive myeloid cells either by direct secretion by TH17 cells or indirectly by cancer cells; and 2) enhancement of cancer cell survival and promotion of EMT and angiogenesis. The presence of IL17+ Th cells and high levels of circulating IL17 has been described in patients with ovarian cancer compared to healthy controls (70). Furthermore, there is strong evidence suggesting the role of IL17 in ovarian cancer metastasis (71,72). We reasoned that VLVs encoding the dominant-negative receptor form (dnIL-17RA) could disrupt the essential IL17 signaling pathway. Therefore, we incorporated into VLVs the ECD of IL17RA lacking the intracellular domain as a decoy. Because the IL17 receptor family consists of five different receptors (IL17RA, B, C, D, and E) and IL17RA is the common subunit for all the other receptors, we generated a potent dominant-negative receptor. This mutant receptor allows the assembly of heterodimers which are competent to compete with wild-type receptors for some rate-limiting step in IL17 signaling pathway (e.g. IL17 ligands). In this way, the ability to induce the production of inflammatory mediators in cooperation with a wide range of ligands in the TME is abrogated, and the favorable environment needed for the tumor progression is diminished.
Although immune checkpoint blockade is arguably the most effective current cancer therapy approach (73) , the overall response rate in most solid tumors is only around 20% (8,74). This efficacy is limited to patients with “hot” tumors, thereby warranting an effective approach to transform “cold” tumors. Oncolytic viruses are known to modulate the TME and convert cold tumors into hot tumors (75,76). CARG-2020 can turn a cold tumor into a hot tumor, while avoiding the systemic toxicity of IL12. Intriguingly, the remodeling the TME combined with the co-expression of the three transgenes prolonged mice survival and prevented tumor recurrence.
Conceptually, the three different genetic approaches we adopted in this study to generate CARG-2020 complemented each other. First, by genetically linking p35 and p40 with a flexible linker, we enforced co-expression of the tethered subunits, thus favoring the antitumor signaling properties of IL12 through the JAK-STAT pathway. Second, by designing an IL17 receptor A extracellular domain (IL17RA ECD) lacking the intracellular domain, the expressed IL17RA ECD (dnIL-17RA) does not inhibit dimerization but results in a nonfunctional dimer. Thus, IL17 downstream signaling cannot foster a favorable environment conducive for tumor progression because of lack of ability to induce the production of inflammatory mediators in cooperation with a wide range of ligands in the TME. Third, PD-L1 is expressed by diverse cell types in the TME, and high PD-L1 expression dampens antitumor immunity. In addition, the PD-1/PD-L1 axis exerts a crucial role in regulating Treg development in TME. It is likely that shRNA-mediated inhibition of PD-L1 leads to the reversal of T-cell exhaustion and thereby contributes to the augmentation of anti-tumor immunity. Theoretically, the suppression of PD-L1 using a gene silencing approach remains by far a better option, as a single interfering gene fragment is able to “switch off” protein synthesis. Because shRNA post-transcriptionally inhibits PD-L1 expression, we predict that this method should prove more efficient in turning off PD-L1 expression, resulting in low protein production than small molecules or antibody blockades. Given their common roles in limiting anti-cancer immunity and promoting therapy resistance, simultaneous inhibition of IL17 and immune checkpoints may generate synergistic anti-cancer efficacy in cancer treatment. Thus, it is plausible that each genetic component used in CARG-2020 alone is necessary but not sufficient to prevent tumor recurrence. Thus, the use of VLV as an oncolytic agent in conjunction with genetic approaches is a promising single-agent immunotherapeutic for preventing cancer recurrence.
In our model of ovarian cancer, we observed the recruitment of immunosuppressive myeloid cells during tumor expansion, and hypothesized that inhibition of IL17 could inhibit their recruitment and together with IL12 could allow the infiltration of CD8+ T cells. Indeed, treatment with CARG-2020 associates with a significant decrease in the presence of CD11b+Ly6C+Ly6G+ myeloid suppressor cells and expansion of CD8+ T cells. In addition to these changes, we also observed a significant change in macrophage differentiation.
Macrophages can be phenotypically polarized by surrounding micro-environment to mount specific functional programs. M2 macrophages are a subset also known as tumor-associated macrophages (TAMS) and constitute a major cell component in neoplastic tissues that contribute to angiogenesis and cancer metastasis. They are characterized by the expression of surface markers such as CD206 and the secretion of cytokines such as IL10, CCL1, Light, and IL6. IL17 is one of the main regulators of macrophage polarization and is responsible for the promotion of M2 macrophage differentiation (77,78). Although it was originally considered that IL17 was produced by the T helper cell subset TH17, numerous studies have demonstrated a wide range of innate immune cells as sources of IL17 (77,79). IL17 promotes recruitment of macrophages into the tumor microenvironment and induces their polarization towards an M2 phenotype (78,80). In this study we focused on M2 macrophages together with MDSCs because they are potent inhibitors CD8+ T-cell activation and thus of anti-tumoral immune responses. Inhibition of both immunosuppressive cell types has been a long-time goal of immunotherapy. CARG-2020, through delivery of immunomodulators, has successfully achieved this goal. However, we cannot ignore the potential contribution of other cell types, e.g. Tregs, neutrophils, γδT cells, etc. Ongoing studies are evaluating the potential contribution of these cell types in the response to CARG-2020 treatment.
Due to a high tolerogenic condition and low antigenicity, ovarian tumors are considered “cold” tumors. In order to enhance the release of antigens and reverse the “cold” status, we used the in situ vaccination approach (ISV) by promoting cell death of cancer cells together with the inhibition of M2 macrophages and MDSCs. The outcome was a specific anti-tumoral response that promotes an efficient establishment of memory, the vaccination effect, a protective response to subsequent challenges (Fig. 7). The treatment with CARG-2020 did not induce the generation of neutralizing antibodies and was able to enhance the expression of Fas and TRAIL. Fas is a proapoptotic receptor that when expressed renders cancer cells more susceptible to its ligand (FASL), which is also expressed by cancer cells as well as by immune cells (81,82). Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) plays an important role in apoptosis and tumor immunosurveillance (83,84). TRAIL selectively induces apoptosis in cancer cells by binding to DR4 and DR5. Increased expression of TRAIL in cancer cells has been associated with increase apoptosis in response to cellular and tissue stress (85–87).
Figure 7. Proposed mechanism of action of CARG-2020.
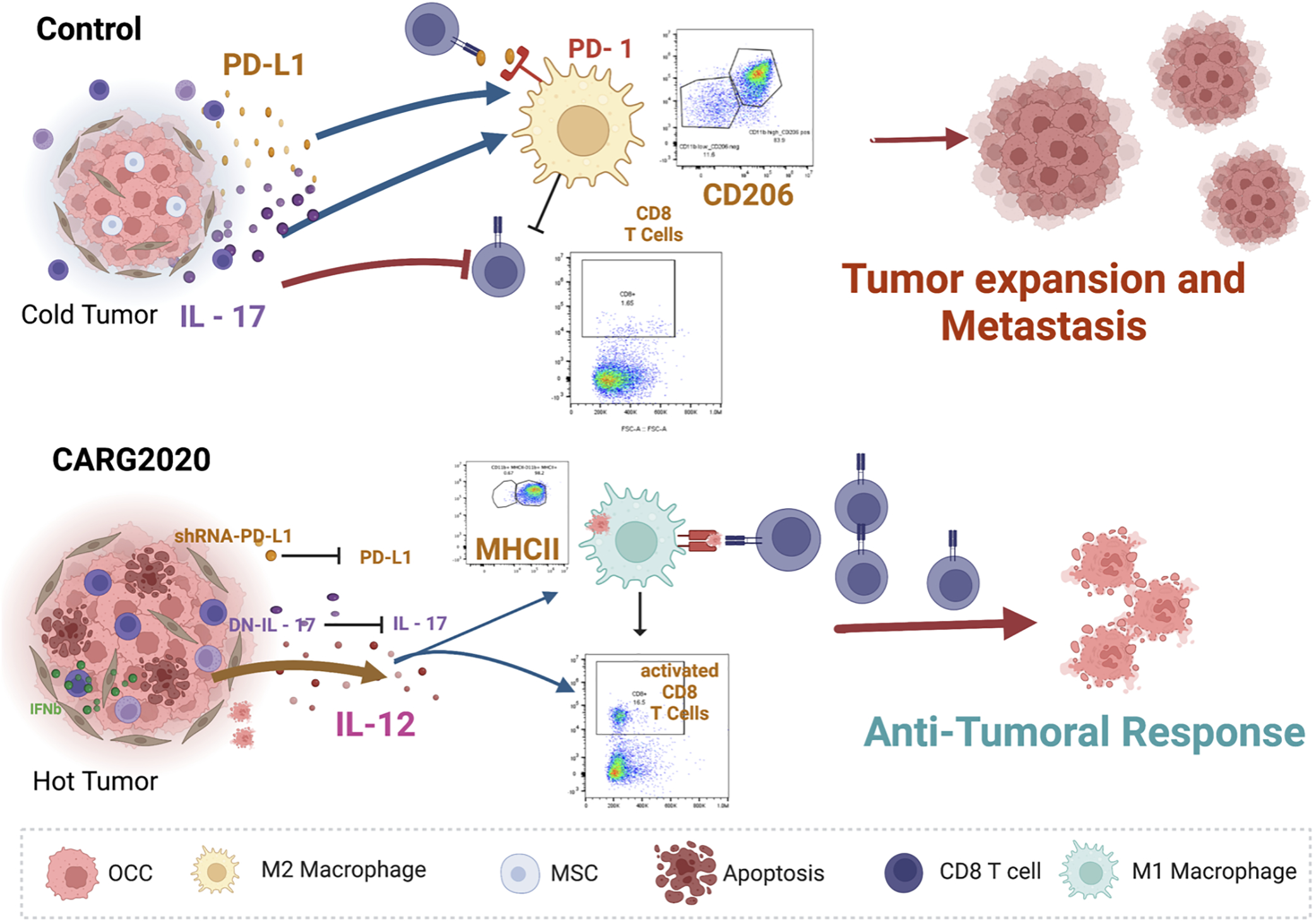
top panel, Control no treatment ovarian tumors are “cold” and express/secrete IL17 and PD-L1 leading to differentiation of pro-tumor M2 macrophages, which inhibit the expansion of cytotoxic CD8+ T cells; bottom panel, CARG-2020, by expressing shRNA for PD-L1 and dominant negative for IL17, in addition to promoting the secretion of IL12, shifts the environment to a “hot” tumor and promotes the differentiation of antigen presenting cells and consequently expansion of cytotoxic CD8+ T cells.
In advance-stage disease, tumors establish a strong immunosuppressive microenvironment (iTME) and reversing it is a challenge. Indeed, the efficacy of CARG-2020 is most optimal when given at early stages of the disease, before the full establishment of an iTME. In summary, we described a novel therapeutic approach that induced in situ vaccination by promoting cancer cell apoptosis, inhibition of MDSCs, polarization of M2 macrophages into M1 antigen presenting cells, leading to an efficient anti-tumoral immune response that conferred protection against potential recurrence. By using the combination of well-established immune modulatory factors, we successfully demonstrated anti-tumor responses and long-term protection. We postulate that the combination of oncolysis and immune modulation of the innate immune response is the most efficient way to activate long-term protection by the adaptive immune system that will be effective in the prevention of relapse.
Supplementary Material
Synopsis:
The virus-like-vesicle, CARG-2020, which combines oncolytic activity with immune-modulatory effects, is demonstrated to remodel the tumor microenvironment. CARG-2020 used as a therapy induces complete tumor regression, generates immune memory, and protects against future tumor recurrence.
Acknowledgment
This research was funded in part by the Janet Burros Memorial Foundation, the National Institute of Health, National Institute of Diabetes and Digestive and Kidney award (NIH NIDDK R44DK113858), and National Institute of Allergy and Infectious Diseases award (NIH NIAID R43AI149798) to V.N.
References
- 1.Salmon H, Remark R, Gnjatic S, Merad M. Host tissue determinants of tumour immunity. Nat Rev Cancer 2019;19(4):215–27 doi 10.1038/s41568-019-0125-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang-Hartwich Y, Gurrea-Soteras M, Sumi N, Joo WD, Holmberg JC, Craveiro V, et al. Ovulation and extra-ovarian origin of ovarian cancer. Scientific reports 2014;4:6116 doi 10.1038/srep06116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci 2012;125(Pt 23):5591–6 doi 10.1242/jcs.116392. [DOI] [PubMed] [Google Scholar]
- 4.Alvero AB, Montagna MK, Craveiro V, Liu L, Mor G. Distinct subpopulations of epithelial ovarian cancer cells can differentially induce macrophages and T regulatory cells toward a pro-tumor phenotype. Am J Reprod Immunol 2012;67(3):256–65 doi 10.1111/j.1600-0897.2011.01068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvero AB, Hanlon D, Pitruzzello M, Filler R, Robinson E, Sobolev O, et al. Transimmunization restores immune surveillance and prevents recurrence in a syngeneic mouse model of ovarian cancer. Oncoimmunology 2020;9(1):1758869 doi 10.1080/2162402x.2020.1758869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018;359(6382):1350–5 doi 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366(26):2455–65 doi 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Science translational medicine 2016;8(328):328rv4 doi 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371(23):2189–99 doi 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348(6230):124–8 doi 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartz PE, Chambers JT, Kohorn EI, Chambers SK, Weitzman H, Voynick IM, et al. Tamoxifen in combination with cytotoxic chemotherapy in advanced epithelial ovarian cancer. A prospective randomized trial. Cancer 1989;63(6):1074–8. [DOI] [PubMed] [Google Scholar]
- 12.Reid A, Ercolano E, Schwartz P, McCorkle R. The management of anxiety and knowledge of serum CA-125 after an ovarian cancer diagnosis. Clin J Oncol Nurs 2011;15(6):625–32 doi 10.1188/11.CJON.625-632. [DOI] [PubMed] [Google Scholar]
- 13.Dale B, Cheng M, Park KS, Kaniskan H, Xiong Y, Jin J. Advancing targeted protein degradation for cancer therapy. Nat Rev Cancer 2021;21(10):638–54 doi 10.1038/s41568-021-00365-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J, Wu J, Han J. Analysis of Tumor Microenvironment Heterogeneity among Breast Cancer Subtypes to Identify Subtype-Specific Signatures. Genes (Basel) 2022;14(1) doi 10.3390/genes14010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Frontiers in immunology 2019;10 doi 10.3389/fimmu.2019.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sevenich L Turning “Cold” Into “Hot” Tumors—Opportunities and Challenges for Radio-Immunotherapy Against Primary and Metastatic Brain Cancers. Frontiers in Oncology 2019;9 doi 10.3389/fonc.2019.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin MJ, Svensson-Arvelund J, Lubitz GS, Marabelle A, Melero I, Brown BD, et al. Cancer vaccines: the next immunotherapy frontier. Nat Cancer 2022;3(8):911–26 doi 10.1038/s43018-022-00418-6. [DOI] [PubMed] [Google Scholar]
- 18.Saha C, Bojdo J, Dunne NJ, Duary RK, Buckley N, McCarthy HO. Nucleic acid vaccination strategies for ovarian cancer. Front Bioeng Biotechnol 2022;10:953887 doi 10.3389/fbioe.2022.953887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lundstrom K Therapeutic Applications for Oncolytic Self-Replicating RNA Viruses. Int J Mol Sci 2022;23(24) doi 10.3390/ijms232415622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu X, Bian C, Zhao X, Yi T. Efficacy evaluation of multi-immunotherapy in ovarian cancer: From bench to bed. Frontiers in immunology 2022;13:1034903 doi 10.3389/fimmu.2022.1034903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haines BB, Denslow A, Grzesik P, Lee JS, Farkaly T, Hewett J, et al. ONCR-177, an Oncolytic HSV-1 Designed to Potently Activate Systemic Antitumor Immunity. Cancer Immunol Res 2021;9(3):291–308 doi 10.1158/2326-6066.Cir-20-0609. [DOI] [PubMed] [Google Scholar]
- 22.Andtbacka RHI, Curti B, Daniels GA, Hallmeyer S, Whitman ED, Lutzky J, et al. Clinical Responses of Oncolytic Coxsackievirus A21 (V937) in Patients With Unresectable Melanoma. J Clin Oncol 2021;39(34):3829–38 doi 10.1200/jco.20.03246. [DOI] [PubMed] [Google Scholar]
- 23.Geoffroy K, Bourgeois-Daigneault M-C. The pros and cons of interferons for oncolytic virotherapy. Cytokine & Growth Factor Reviews 2020;56:49–58 doi 10.1016/j.cytogfr.2020.07.002. [DOI] [PubMed] [Google Scholar]
- 24.van den Pol AN, Zhang X, Lima E, Pitruzzello M, Albayrak N, Alvero A, et al. Lassa-VSV chimeric virus targets and destroys human and mouse ovarian cancer by direct oncolytic action and by initiating an anti-tumor response. Virology 2021;555:44–55 doi 10.1016/j.virol.2020.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rolls MM, Webster P, Balba NH, Rose JK. Novel infectious particles generated by expression of the vesicular stomatitis virus glycoprotein from a self-replicating RNA. Cell 1994;79(3):497–506 doi 10.1016/0092-8674(94)90258-5. [DOI] [PubMed] [Google Scholar]
- 26.Rose NF, Publicover J, Chattopadhyay A, Rose JK. Hybrid alphavirus-rhabdovirus propagating replicon particles are versatile and potent vaccine vectors. Proc Natl Acad Sci U S A 2008;105(15):5839–43 doi 10.1073/pnas.0800280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chiale C, Yarovinsky TO, Mason SW, Madina BR, Menon M, Krady MM, et al. Modified Alphavirus-Vesiculovirus Hybrid Vaccine Vectors for Homologous Prime-Boost Immunotherapy of Chronic Hepatitis B. Vaccines (Basel) 2020;8(2) doi 10.3390/vaccines8020279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yarovinsky TO, Mason SW, Menon M, Krady MM, Haslip M, Madina BR, et al. Virus-like Vesicles Expressing Multiple Antigens for Immunotherapy of Chronic Hepatitis B. iScience 2019;21:391–402 doi 10.1016/j.isci.2019.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong MT, Chen SS. Emerging roles of interferon-stimulated genes in the innate immune response to hepatitis C virus infection. Cell Mol Immunol 2016;13(1):11–35 doi 10.1038/cmi.2014.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynolds TD, Buonocore L, Rose NF, Rose JK, Robek MD. Virus-Like Vesicle-Based Therapeutic Vaccine Vectors for Chronic Hepatitis B Virus Infection. J Virol 2015;89(20):10407–15 doi 10.1128/jvi.01184-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim J, Coffey DM, Ma L, Matzuk MM. The ovary is an alternative site of origin for high-grade serous ovarian cancer in mice. Endocrinology 2015;156(6):1975–81 doi 10.1210/en.2014-1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Craveiro V, Yang-Hartwich Y, Holmberg JC, Joo WD, Sumi NJ, Pizzonia J, et al. Phenotypic modifications in ovarian cancer stem cells following Paclitaxel treatment. Cancer Med 2013;2(6):751–62 doi 10.1002/cam4.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tedja R, Roberts CM, Alvero AB, Cardenas C, Yang-Hartwich Y, Spadinger S, et al. Protein kinase Cα-mediated phosphorylation of Twist1 at Ser-144 prevents Twist1 ubiquitination and stabilizes it. J Biol Chem 2019;294(13):5082–93 doi 10.1074/jbc.RA118.005921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cardenas C, Montagna MK, Pitruzzello M, Lima E, Mor G, Alvero AB. Adipocyte microenvironment promotes Bclxl expression and confers chemoresistance in ovarian cancer cells. Apoptosis 2017;22(4):558–69 doi 10.1007/s10495-016-1339-x. [DOI] [PubMed] [Google Scholar]
- 35.Alvero AB, Kim D, Lima E, Sumi NJ, Lee JS, Cardenas C, et al. Novel approach for the detection of intraperitoneal micrometastasis using an ovarian cancer mouse model. Scientific reports 2017;7:40989 doi 10.1038/srep40989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alvero AB, Heaton A, Lima E, Pitruzzello M, Sumi N, Yang-Hartwich Y, et al. TRX-E-002–1 Induces c-Jun-Dependent Apoptosis in Ovarian Cancer Stem Cells and Prevents Recurrence In Vivo. Mol Cancer Ther 2016;15(6):1279–90 doi 10.1158/1535-7163.mct-16-0005. [DOI] [PubMed] [Google Scholar]
- 37.Sumi NJ, Lima E, Pizzonia J, Orton SP, Craveiro V, Joo W, et al. Murine model for non-invasive imaging to detect and monitor ovarian cancer recurrence. Journal of visualized experiments : JoVE 2014(93):e51815 doi 10.3791/51815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alvero AB, Montagna MK, Sumi NJ, Joo WD, Graham E, Mor G. Multiple blocks in the engagement of oxidative phosphorylation in putative ovarian cancer stem cells: implication for maintenance therapy with glycolysis inhibitors. Oncotarget 2014;5(18):8703–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krikun G, Mor G, Alvero A, Guller S, Schatz F, Sapi E, et al. A novel immortalized human endometrial stromal cell line with normal progestational response. Endocrinology 2004;145(5):2291–6 doi 10.1210/en.2003-1606. [DOI] [PubMed] [Google Scholar]
- 40.Balint K, Conejo-Garcia JR, Buckanovich R, Coukos G. Role of vascular leukocytes in ovarian cancer neovascularization. Adv Exp Med Biol 2008;622:273–80 doi 10.1007/978-0-387-68969-2_22. [DOI] [PubMed] [Google Scholar]
- 41.Coukos G, Conejo-Garcia JR, Buckanovich R, Benencia F. Vascular leukocytes: a population with angiogenic and immunossuppressive properties highly represented in ovarian cancer. Adv Exp Med Biol 2007;590:185–93 doi 10.1007/978-0-387-34814-8_13. [DOI] [PubMed] [Google Scholar]
- 42.Manetti R, Parronchi P, Giudizi MG, Piccinni MP, Maggi E, Trinchieri G. Natural killer cell stimulatory factor (interleukin 12 [IL-12]) induces T helper type 1 (Th1)-specific immune responses and inhibits the development of IL-4-producing Th cells. J Exp Med 1993;177 doi 10.1084/jem.177.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nizzoli G, Krietsch J, Weick A, Steinfelder S, Facciotti F, Gruarin P, et al. Human CD1c+ dendritic cells secrete high levels of IL-12 and potently prime cytotoxic T-cell responses. Blood 2013;122(6):932–42 doi 10.1182/blood-2013-04-495424. [DOI] [PubMed] [Google Scholar]
- 44.Lee SM, Suen Y, Qian J, Knoppel E, Cairo MS. The regulation and biological activity of interleukin 12. Leuk Lymphoma 1998;29(5–6):427–38 doi 10.3109/10428199809050903. [DOI] [PubMed] [Google Scholar]
- 45.Asselin-Paturel C, Pardoux C, Gay F, Chouaib S. Failure of TGF beta1 and IL-12 to regulate human FasL and mTNF alloreactive cytotoxic T-cell pathways. Tissue Antigens 1998;51(3):242–9. [DOI] [PubMed] [Google Scholar]
- 46.Vile RG, Castleden S, Marshall J, Camplejohn R, Upton C, Chong H. Generation of an anti-tumour immune response in a non-immunogenic tumour: HSVtk killing in vivo stimulates a mononuclear cell infiltrate and a Th1-like profile of intratumoural cytokine expression. Int J Cancer 1997;71(2):267–74 doi . [DOI] [PubMed] [Google Scholar]
- 47.Tsutsui T, Mu J, Ogawa M, Yu W, Suda T, Nagaga S, et al. Administration Of Il-12 Induces a Cd3(+)Cd4(−)Cd8(−)B220(+) Lymphoid Population Capable Of Eliciting Cytolysis Against Fas-Positive Tumor Cells. J Immunol 1997;159(6):2599–605. [PubMed] [Google Scholar]
- 48.Lasek W, Zagożdżon R, Jakobisiak M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol Immunother 2014;63(5):419–35 doi 10.1007/s00262-014-1523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang ZZ, Grote DM, Ziesmer SC, Niki T, Hirashima M, Novak AJ, et al. IL-12 upregulates TIM-3 expression and induces T cell exhaustion in patients with follicular B cell non-Hodgkin lymphoma. J Clin Invest 2012;122(4):1271–82 doi 10.1172/jci59806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Strauss J, Deville JL, Sznol M, Ravaud A, Maruzzo M, Pachynski RK, et al. First-in-human phase Ib trial of M9241 (NHS-IL12) plus avelumab in patients with advanced solid tumors, including dose expansion in patients with advanced urothelial carcinoma. J Immunother Cancer 2023;11(5) doi 10.1136/jitc-2022-005813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Craveiro V, Yang-Hartwich Y, Holmberg JC, Sumi NJ, Pizzonia J, Griffin B, et al. Phenotypic modifications in ovarian cancer stem cells following Paclitaxel treatment. Cancer Medicine 2013;2(6):751–62 doi 10.1002/cam4.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015;161(7):1527–38 doi 10.1016/j.cell.2015.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nesbeth Y, Scarlett U, Cubillos-Ruiz J, Martinez D, Engle X, Turk MJ, et al. CCL5-mediated endogenous antitumor immunity elicited by adoptively transferred lymphocytes and dendritic cell depletion. Cancer Res 2009;69(15):6331–8 doi 10.1158/0008-5472.CAN-08-4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huarte E, Cubillos-Ruiz JR, Nesbeth YC, Scarlett UK, Martinez DG, Buckanovich RJ, et al. Depletion of dendritic cells delays ovarian cancer progression by boosting antitumor immunity. Cancer Res 2008;68(18):7684–91 doi 10.1158/0008-5472.CAN-08-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Santoiemma PP, Powell DJ Jr., Tumor infiltrating lymphocytes in ovarian cancer. Cancer Biol Ther 2015;16(6):807–20 doi 10.1080/15384047.2015.1040960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huarte E, Cubillos-Ruiz JR, Nesbeth YC, Scarlett UK, Martinez DG, Buckanovich RJ, et al. Depletion of dendritic cells delays ovarian cancer progression by boosting antitumor immunity. Cancer Research 2008;68(18):7684–91 doi 0008–5472.CAN-08–1167 [pii] 10.1158/0008-5472.CAN-08-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, vom Berg J, et al. New insights into IL-12-mediated tumor suppression. Cell Death & Differentiation 2015;22(2):237–46 doi 10.1038/cdd.2014.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu S, Cao X. Interleukin-17 and its expanding biological functions. Cellular & Molecular Immunology 2010;7(3):164–74 doi 10.1038/cmi.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Indrová M, Bieblová J, Jandlová T, Vonka V, Pajtasz-Piasecka E, Reinis M. Chemotherapy, IL-12 gene therapy and combined adjuvant therapy of HPV 16-associated MHC class I-proficient and -deficient tumours. Int J Oncol 2006;28(1):253–9. [PubMed] [Google Scholar]
- 60.Adris S, Chuluyan E, Bravo A, Berenstein M, Klein S, Jasnis M, et al. Mice vaccination with interleukin 12-transduced colon cancer cells potentiates rejection of syngeneic non-organ-related tumor cells. Cancer Res 2000;60(23):6696–703. [PubMed] [Google Scholar]
- 61.Afonso LC, Scharton TM, Vieira LQ, Wysocka M, Trinchieri G, Scott P. The adjuvant effect of interleukin-12 in a vaccine against Leishmania major. Science 1994;263(5144):235–7 doi 10.1126/science.7904381. [DOI] [PubMed] [Google Scholar]
- 62.Asselin-Paturel C, Megherat S, Vergnon I, Echchakir H, Dorothée G, Blesson S, et al. Differential effect of high doses versus low doses of interleukin-12 on the adoptive transfer of human specific cytotoxic T lymphocyte in autologous lung tumors engrafted into severe combined immunodeficiency disease-nonobese diabetic mice: relation with interleukin-10 induction. Cancer 2001;91(1):113–22. [PubMed] [Google Scholar]
- 63.Atkins MB, Robertson MJ, Gordon M, Lotze MT, DeCoste M, DuBois JS, et al. Phase I evaluation of intravenous recombinant human interleukin 12 in patients with advanced malignancies. Clin Cancer Res 1997;3(3):409–17. [PubMed] [Google Scholar]
- 64.Gerosa F, Paganin C, Peritt D, Paiola F, Scupoli MT, Aste-Amezaga M, et al. Interleukin-12 primes human CD4 and CD8 T cell clones for high production of both interferon-gamma and interleukin-10. J Exp Med 1996;183(6):2559–69 doi 10.1084/jem.183.6.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paganin C, Gerosa F, Peritt D, Paiola F, Scupoli MT, Aste-Amezaga M, et al. Effect of interleukin-12 on the cytokine profile of human CD4 and CD8 T-cell clones. Ann N Y Acad Sci 1996;795:382–3 doi 10.1111/j.1749-6632.1996.tb52699.x. [DOI] [PubMed] [Google Scholar]
- 66.Portielje JE, Gratama JW, van Ojik HH, Stoter G, Kruit WH. IL-12: a promising adjuvant for cancer vaccination. Cancer Immunol Immunother 2003;52(3):133–44 doi 10.1007/s00262-002-0356-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheng EM, Tsarovsky NW, Sondel PM, Rakhmilevich AL. Interleukin-12 as an in situ cancer vaccine component: a review. Cancer Immunology, Immunotherapy 2022;71(9):2057–65 doi 10.1007/s00262-022-03144-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moreira AP, Cavassani KA, Ismailoglu UB, Hullinger R, Dunleavy MP, Knight DA, et al. The protective role of TLR6 in a mouse model of asthma is mediated by IL-23 and IL-17A. The Journal of clinical investigation 2011;121(11):4420–32 doi 10.1172/JCI44999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li Y, Cao ZY, Sun B, Wang GY, Fu Z, Liu YM, et al. Effects of IL-17A on the occurrence of lung adenocarcinoma. Cancer Biology & Therapy 2011;12(7):610–6 doi 10.4161/cbt.12.7.16302. [DOI] [PubMed] [Google Scholar]
- 70.Guo N, Zhang J. Interleukin-17 promotes the development of ovarian cancer through upregulation of MTA1 expression. Am J Cancer Res 2022;12(12):5646–56. [PMC free article] [PubMed] [Google Scholar]
- 71.Jackson AM, Mulcahy LA, Porte J, Franks HA, El Refaee M, Wang Q, et al. Role of mitogen-activated protein kinase and PI3K pathways in the regulation of IL-12-family cytokines in dendritic cells and the generation of T H-responses. Eur Cytokine Netw 2010;21(4):319–28 doi 10.1684/ecn.2010.0219. [DOI] [PubMed] [Google Scholar]
- 72.Zhu X, Mulcahy LA, Mohammed RA, Lee AH, Franks HA, Kilpatrick L, et al. IL-17 expression by breast-cancer-associated macrophages: IL-17 promotes invasiveness of breast cancer cell lines. Breast Cancer Res 2008;10(6):R95 doi 10.1186/bcr2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363(8):711–23 doi 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366(26):2443–54 doi 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Garofalo M, Pancer KW, Wieczorek M, Staniszewska M, Salmaso S, Caliceti P, et al. From Immunosuppression to Immunomodulation - Turning Cold Tumours into Hot. J Cancer 2022;13(9):2884–92 doi 10.7150/jca.71992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gujar S, Pol JG, Kroemer G. Heating it up: Oncolytic viruses make tumors ‘hot’ and suitable for checkpoint blockade immunotherapies. Oncoimmunology 2018;7(8):e1442169 doi 10.1080/2162402X.2018.1442169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kuen DS, Kim BS, Chung Y. IL-17-Producing Cells in Tumor Immunity: Friends or Foes? Immune Netw 2020;20(1):e6 doi 10.4110/in.2020.20.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shen J, Sun X, Pan B, Cao S, Cao J, Che D, et al. IL-17 induces macrophages to M2-like phenotype via NF-κB. Cancer Manag Res 2018;10:4217–28 doi 10.2147/cmar.S174899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen C, Itakura E, Nelson GM, Sheng M, Laurent P, Fenk LA, et al. IL-17 is a neuromodulator of Caenorhabditis elegans sensory responses. Nature 2017;542(7639):43–8 doi 10.1038/nature20818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Song X, Qian Y. The activation and regulation of IL-17 receptor mediated signaling. Cytokine 2013;62(2):175–82 doi 10.1016/j.cyto.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 81.Abrahams V, Straszewski S, Kamsteeg M, Hanczaruk B, Schwartz P, Rutherford T, et al. Epithelial Ovarian Cancer secrete funcitonal Fas Ligand. Cancer Res 2003;63:5573–81. [PubMed] [Google Scholar]
- 82.Abrahams VM, Kamsteeg M, Mor G. The Fas/Fas ligand system and cancer: immune privilege and apoptosis. Mol Biotechnol 2003;25(1):19–30 doi 10.1385/mb:25:1:19. [DOI] [PubMed] [Google Scholar]
- 83.Pimentel JM, Zhou JY, Wu GS. The Role of TRAIL in Apoptosis and Immunosurveillance in Cancer. Cancers (Basel) 2023;15(10) doi 10.3390/cancers15102752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pimentel JM, Zhou JY, Wu GS. Regulation of programmed death ligand 1 (PD-L1) expression by TNF-related apoptosis-inducing ligand (TRAIL) in triple-negative breast cancer cells. Mol Carcinog 2023;62(2):135–44 doi 10.1002/mc.23471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cardoso Alves L, Corazza N, Micheau O, Krebs P. The multifaceted role of TRAIL signaling in cancer and immunity. The FEBS journal 2021;288(19):5530–54 doi 10.1111/febs.15637. [DOI] [PubMed] [Google Scholar]
- 86.Singh D, Tewari M, Singh S, Narayan G. Revisiting the role of TRAIL/TRAIL-R in cancer biology and therapy. Future Oncol 2021;17(5):581–96 doi 10.2217/fon-2020-0727. [DOI] [PubMed] [Google Scholar]
- 87.von Karstedt S, Montinaro A, Walczak H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat Rev Cancer 2017;17(6):352–66 doi 10.1038/nrc.2017.28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are present in the main paper and/or the supplementary materials and are available upon reasonable request to the corresponding author.