

Abstract
Peptides derived from over-expressed p53 protein are presented by class I MHC molecules and may act as tumour-associated epitopes. Due to the diversity of p53 mutations, immunogenic peptides representing wild-type sequences are preferable as a basis for a broad-spectrum p53-targeting cancer vaccine. Our preclinical studies have shown that wild-type p53-derived HLA-A2–binding peptides are able to activate human T cells and that the generated effector T cells are cytotoxic to human HLA-A2+, p53+ tumour cells. In this phase I pilot study, the toxicity and efficacy of autologous dendritic cells (DCs) loaded with a cocktail of three wild-type and three modified p53 peptides are being analysed in six HLA-A2+ patients with progressive advanced breast cancer. Vaccinations were well tolerated and no toxicity was observed. Disease stabilisation was seen in two of six patients, one patient had a transient regression of a single lymph node and one had a mixed response. ELISpot analyses showed that the p53-peptide–loaded DCs were able to induce specific T-cell responses against modified and unmodified p53 peptides in three patients, including two of the patients with a possible clinical benefit from the treatment. In conclusion, the strategy for p53-DC vaccination seems safe and without toxicity. Furthermore, indications of both immunologic and clinical effect were found in heavily pretreated patients with advanced breast cancer. An independent clinical effect of repeated administration of DCs and IL-2 can not of course be excluded; further studies are necessary to answer these questions.
Keywords: Dendritic cells, Breast cancer, Vaccine, p53 peptides, Immunotherapy
Introduction
Dendritic cells (DCs) constitute a unique subset of extremely efficient antigen-presenting cells. They are potent stimulators of memory B and T cells and are able to activate resting or naïve T cells [3]. These features enable DCs to act as a natural adjuvant for cancer-specific vaccination [13, 23, 27]. Several antitumour vaccines have been constructed using peptide-pulsed DCs based on prior identification of cytotoxic T cell (CTL)–defined synthetic peptide epitopes and their presenting MHC class I molecules. DCs for antigen loading can be generated in clinical scale from blood leukapheresis products followed by in vitro manipulation and growth factor stimulation [12, 32].
The gene p53 is a tumour-suppressor gene and the normal protein is present in small amounts in the cell nucleus. Mutations in the p53 gene have frequently (>50%) been observed in a variety of human malignancies including about 30% of breast carcinomas resulting in a poor prognosis following conventional therapy [6, 7, 26]. Peptides derived from over-expressed p53 protein are presented by class I MHC molecules and may act as tumour-associated epitopes. Due to the diversity of p53 mutations, immunogenic peptides representing wild-type sequences are preferable as a basis for a broad-spectrum p53-targeting cancer vaccine [18, 20].
Our own preclinical studies have shown that wild-type p53-derived HLA-A2–binding peptides are able to activate human T cells and that the generated effector T cells are cytotoxic to human HLA-A2+, p53+ tumour cells [5, 24, 34]. In addition, single amino acid–modified p53 peptides can increase the HLA-A2-binding capacity and induction of p53-specific cytotoxic T cells [22]. In this phase I trial, safety and efficacy of autologous DCs loaded with a cocktail of three wild-type and three modified p53 peptides were analysed in six HLA-A2+ patients with progressive metastatic breast cancer. The patients are evaluated in a classical phase I/II design for toxicity, adverse events, clinical response, and induction of p53-specific T-cell immunity.
Methods
Patients and eligibility criteria
The main criteria for patient inclusion in the phase I trial were (1) histologically proven metastatic or locally advanced breast cancer, (2) progressive disease and no standard systemic treatment indicated, (3) at least one measurable lesion or osteolytic bone metastasis,(4) expression of the HLA-A2 allele, (5) WHO performance status 0–2, (6) life expectancy of more than 3 months. The main exclusion criteria included (1) evidence of brain metastasis, (2) use of immunosuppressive drugs such as steroid, (3) radiation therapy or chemotherapy within the prior 4 weeks, (4) significantly increased blood liver-enzyme level (>2.5 x upper normal limit), (5) other malignancies, or (6) pregnancy.
The study protocol was approved by the Institutional Ethical Committees, Copenhagen County, and the Danish Medicines Agency. Written consent was obtained from all patients at study entry.
Generation of DCs
All procedures were performed according to good laboratory practice standards as approved by the Danish Medicines Agency. The patients underwent unmobilized leukapheresis using a continuous flow blood cell separator for isolation of large scale (>2×109) mononuclear cells. Remaining red blood cells were lysed with Ortho-mune lysing solution (provided by hospital pharmacy). Peripheral blood mononuclear cells (PBMCs) were washed, resuspended in culture medium (CM) (X-Vivo 15, 200-mM 2% l-glutamine, 1% autologous heat-inactivated plasma) at 7×106 cells/ml, and separated by 1 h adherence to plastic Nunclon dishes (Nunc, Biotech Line, Slangerup, Denmark). Nonadherent cells were removed, and adherent cells were subsequently cultured for 7 days in CM supplemented with 250 U/ml rh-IL-4 (CellGenix, Freiburg, Germany), and 1,000 U/ml GM-CSF (Leucomax, Schering Plough, Farum, Denmark). Cells were harvested at day 7 using a cell-scraper and cells with typical DC morphology were counted by light microscopy. Aliquots of 1×107 cells were frozen in 85% autologous serum, 10% DMSO (limited pool; BDH Laboratory Supplies, England) and 5% Glucosteril 40% (Fresenius, Albertslund, Denmark) using automated cryopreservation (Planer Freezing Unit; Planer, UK). Sterility controls of DCs were negative at all times.
DC phenotypic analysis
Aliquots of the cultured cells were subjected to phenotypic analysis at time of cryopreservation. The expressions of the cell surface antigens HLA-A2, HLA-DR, CD1a, CD11c, CD33, CD40, CD54 and CD86 were analysed. Antibodies against CD11c, CD33, HLA-DR (BD Bioscience, San José, CA), CD40, CD86, CD1a (PharMingen, San Diego, CA), CD54 (Immunotech, Ramcon, Denmark), HLA-A2 (One Lambda, Canoga Park, CA) were used together with relevant isotype controls. PE-Lineage (Lin) cocktail was prepared for simultaneous labelling of T cells, B cells and monocytes by antibodies against CD3, CD14 and CD19 (BD Bioscience, San José, CA). For four-colour analyses of DCs, cells were labelled simultaneously with lineage cocktail and three of the relevant DC cell surface markers. Flow cytometric analysis was performed on a FACS Calibur flow cytometer (BD Bioscience). Data were analysed using CellQuest software (BD Bioscience).
Pulsing of in vitro–generated DCs
On the day of vaccination one vial of 1×107 cells was thawed, washed twice, and resuspended in 500-µl RPMI. Cell viability >90% was invariably found. DCs were pulsed with 40 µg/ml of six HLA-A2-associated p53 peptides, and a pan-MHC class II peptide, PADRE [1], for 2 h at 37°C. After incubation, cells were washed twice, resuspended in 500-µl RPMI and transferred to a 0.5-ml insulin syringe for injection.
Treatment
Eligible patients were to receive a total of ten immunizations with at least 5×106 p53- and PADRE-peptide–pulsed autologous DCs. Vaccinations 1 to 4 were given weekly and vaccinations 5 to 10, biweekly. The vaccine was administered subcutaneously (s.c.) near the inguinal region on the same thigh every time. Concomitantly with each vaccination, 6 mIU/m2 interleukin 2 (Proleukin; Swedish Orphan, Denmark) was administered s.c.
Synthetic epitope peptides
HLA-A2–binding p53-derived peptides with high HLA-A2–binding affinities: p5365–73 RMPEAAPPV (R9 V) [5, 34], p53264–272 LLGRNSFEV (L9 V) [24, 34] and p53187–197 GLAPPQHLIRV (G11 V) [29, 35]. Position 2 anchor modified HLA-A2–binding p53-derived peptides [22]: p53149–157 SLPPPGTRV (S9Vm), p53139–147 KLCPVQLWV (K9Vm), p53103–111 YLGSYGFRL (Y9Lm). PADRE: aKXAAWTLKAAa. Purity of all peptides for clinical uses was >95%, these peptides were suspended at 1 mg/ml in RPMI, filtered through a 0.22-micron filter, and tested for sterility.
ELISpot control peptides: IMP58–66 GILGFVFTL from the influenza matrix protein, BMFL 1280–288 GLCTLVAML from the immediate-early lytic protein of Epstein–Barr virus and RTase 309–317 ILKEPVHGV from the reverse transcriptase of HIV-1 virus. All peptides were purchased from Schäfer N, Copenhagen, Denmark.
Clinical monitoring
Patients receiving one vaccination or more were evaluable for toxicity and adverse events, and patients receiving four vaccinations or more were evaluable for tumour response. With the exception of patients with osteolytic bone metastasis, all patients underwent response evaluation according to the RECIST criteria [17, 30] at baseline, after the sixth (8 weeks from trial entry) and after the tenth (16 weeks from trial entry) DC vaccination. Adverse events were graded according to the NCI common toxicity criteria.
Immunologic monitoring
Heparinized blood samples were collected preimmunization and postimmunization after the fourth, sixth and tenth vaccination. PBMCs were separated by centrifugation on a Lymphoprep (NYCOMED, Norway) density gradient using standard procedures. Aliquots of PBMCs were frozen in RPMI with 10% AB-serum and 10% DMSO.
ELISpot assay
The IFN-γ ELISpot assay was used to quantify p53 peptide-specific CTLs during the vaccination trial. In advance, PBMCs (2×105/well) were cultured in X-Vivo 15 medium (BioWhittaker, Wokingham, England) containing 2% inactivated human AB-serum and 10 µM of relevant p53-derived peptide in 96-well microtiter plates (Nunc, Roskilde, Denmark). Wells with no peptide or mock peptide were added as negative controls, and wells containing HLA-A2–binding peptides from the influenza matrix protein or Epstein–Barr virus were included as positive controls. RhIL-2 (Proleukin; Chiron, The Netherlands) 300 U/ml was added on day 2. On day 10, cells were harvested directly to 96-well ELISpot plates (Multiscreen, MAHAS4510; Millipore, Molsheim, France) precoated with 7.5 µg/ml antihuman IFN-γ (M-700A, Endogen; Pierce Biotechnology, Rockford, IL, USA). Fresh peptides were added and after a 24-culture period plates were washed several times. Biotinylated antihuman IFN-γ (M-701B, Endogen; Pierce Biotechnology, Rockford, IL, USA), 0.75 µg/ml, was then added prior to visualization with HRP-streptavidin (DAKO, Copenhagen, Denmark) and substrate. The number of spot-forming cells was determined with computer-assisted image analysis software (KS ELISpot, Zeiss, Munich, Germany).
Results
Patient characteristics
A total of nine patients were enrolled; however, two (patients 003 and 004) were excluded prior to the first treatment and one (patient 006) after second vaccination, either due to rapid disease progression and death, or due to demonstration of brain metastases and treatment with steroid.
The characteristics of treated patients are summarized in Table 1. All patients were HLA-A2–positive and in 3/6 patients primary tumour samples were stained positive (>50% positive cells) for p53 by immunohistochemistry. Four of six patients had at least one measurable tumour lesion, the remaining two had osteolytic bone metastases. Prior treatment included chemotherapy, endocrine therapy, and radiation therapy. Four patients had performance status (PS) 0, one had PS 1, and one had PS 2.
Table 1.
Patient and treatment characteristics. TAM tamoxifen; EPI epirubicin; LN lymph nodes; RT radiotherapy; FEC 5-fluorouracil, epirubicin, cyclophosphamide; TAX paclitaxel; CMF cyclophosphamide, methotrexate, 5-fluorouracil; GEM gemcitabine
| Patient no. | Age (years) | Sites of metastases | Previous therapy | p53 expression (%) | No. of vaccinations | Clinical outcome after 6 vaccinations | Immune response (+/–) | Survival after first vaccination (months) |
|---|---|---|---|---|---|---|---|---|
| 001 | 46 | Bones | TAM, EPI | 80 | 10 | Stable disease | + | +24 |
| 002 | 65 | Liver, LN | TAM, EPI, Anastazol | 50 | 8 | Progression | – | 7 |
| 005 | 49 | Bones, LN | RT, FEC, TAX | 0 | 8 | Mixed response | (+) | 17 |
| 007 | 51 | Lung, pleura, bones | RT, CMF, TAM, TAX/GEM | 1 | 7 | Progression | + | 41/2 |
| 008 | 41 | LN | RT, FEC, TAX | 95 | 10 | Unconfirmed regression | + | +15 |
| 009 | 62 | Bones | CMF, EPI, LET, TAM | 2 | 10 | Stable disease | – | +14 |
Of the six patients recruited, three completed the planned 10 immunizations. Three patients discontinued the treatment after 7–8 immunizations because of progressive disease. Two patients received additional immunizations biweekly in continuation of the study due to disease stabilization.
Dendritic cell preparations
Data regarding in vitro DC yield are given in Table 2. The collected number of PBMCs after leukapheresis ranged from 2.6×109 to 7.5×109 cells with a median harvest of 10.0% monocytes. After 7 days of culture, a median 18.7% of the harvested population were defined as large dendritic-like cells by morphology.
Table 2.
Dendritic cell yield from apheresis products
| Median | Range | |
|---|---|---|
| Cell harvest (x109) | 7.1 | 2.6–7.5 |
| Monocyte fraction (%) | 10.0 | 7.0–15.1 |
| DC yielda (x107) | 13.1 | 6.7–26.0 |
| DC % of MNCa | 18.7 | 15.6–43.4 |
| DCs per vaccinea (x106) | 10 | 7–20 |
aNumber of cells with morphology characteristic for DCs was counted by light microscopy
Flow cytometry analysis was carried out. The gating strategy and expression level of relevant surface markers presented in Fig. 1 are representative for the generated DC preparations. DCs were defined as large granular cells not expressing CD3, CD14 and CD19 lineage markers. The phenotype of these cells was characteristic for intermediate mature DCs with a pronounced expression of MHC class I including HLA-A2, MHC class II and CD11c, and a moderate to high expression of the adhesion and costimulatory molecules CD54, CD86 and CD40. Based on flow cytometry (Fig. 1), on average 37.8% of the harvested cells were gated as DCs with 24.8% of these being lineage negative (Table 3). Approximately 50% of the lineage-negative cells expressed CD86, and 41% were postitive for CD1a. CD40, CD54 and MHC molecules were expressed by the vast majority of cells.
Fig. 1a–h.

Flow cytometric phenotyping of in vitro–generated DCs from patient 008. a Gating on forward-side scatter including all potential DCs (R2). b DCs are further gated by low or negative staining with a lineage cocktail including mAbs for CD3, CD14 and CD19 (R3). c–h Fluorescence intensities of gated lineage-negative cells stained with mAbs for relevant DC surface markers
Table 3.
Dendritic cell surface marker expression (percentages)
| Patient no. | DCa | Lin− cellsb | DRc | CD86c | CD40c | CD54c | HLA-A2c | CD1ac | CD11cc | CD33c |
|---|---|---|---|---|---|---|---|---|---|---|
| 001 | 53.86 | 28.45 | 96.31 | 73.79 | 51.64 | 25.29 | 98.3 | 7.04 | 96.00 | 39.53 |
| 002 | 28.97 | 54.48 | 99.74 | 1.20 | 96.41 | 86.35 | 99.93 | 82.00 | 98.96 | 0.28 |
| 005 | 27.14 | 30.23 | 92.75 | 12.94 | 96.48 | 99.01 | 99.87 | 87.2 | 97.23 | 90.00 |
| 007 | 30.05 | 13.28 | 87.11 | 53.55 | 89.83 | 95.08 | 93.36 | 15.95 | 90.07 | 70.11 |
| 008 | 38.37 | 17.4 | 88.66 | 84.31 | 83.07 | 92.42 | 92.70 | 11.83 | 93.07 | 84.27 |
| 009 | 48.37 | 5.12 | 81.42 | 70.01 | 76.37 | 78.88 | 88.55 | 42.49 | 83.03 | 61.21 |
| Mean | 37.79 | 24.83 | 91.00 | 49.30 | 82.30 | 79.51 | 95.45 | 41.09 | 93.06 | 57.57 |
aDC gate: large granular cells gated on FSC SSC dot plot
bLin−: cells negative for labelling with PE-lineage cocktail (CD3, CD14, CD19)
cPercentage of DC-gated, Lin− cells positive for the surface marker
Safety and toxicity
The vaccine was well tolerated. None of the patients developed irritation at the site of injection. No allergic or autoimmune reactions were observed. No haematologic, hepatic, pulmonary or renal toxicities were observed (Fig. 2). The most common side effect was mild to moderate local reaction at the site of Proleukin injection. All patients experienced mild flu-like symptoms lasting 12–24 h after injection of Proleukin. One patient had significant joint pain, which disappeared after a 50% reduction of the Proleukin dose.
Fig. 2.

Laboratory values. Detection of haemoglobin, amorph nucleated cells (ANCs), thrombocytes, white blood cells (WBCs), LDH and creatinine
Immune response
All patients had blood samples eligible for analysing p53-peptide–specific T-cell response. Two of the patients (005 and 007) had preexisting T-cell reactivity against some of the p53 peptides. After four or six vaccinations, four patients (001, 005, 007 and 008) showed increased specific T-cell responses against one or more modified peptides (S9Vm, K9Vm and Y9Lm), while three patients (005, 007 and 008) showed increased response against one or two unmodified p53 peptides (R9 V, L9 V and G11 V) (Fig. 3). In patients with reactivity against the modified p53 peptides, reactivity was also shown against the corresponding unmodified variants (Fig. 4). In three patients (001, 007 and 008) with vaccine-induced reactivity, ELISpot analyses showed a subsequent decrease in T-cell response at late time intervals. We suggest that this decrease might reflect entry of p53-specific T cells from the blood stream into the tumour site. In two patients, no T-cell response to p53 peptides was demonstrated.
Fig. 3.

Frequency of p53-peptide–specific CTLs in PBMCs from breast cancer patients during the vaccination trial. Reactivity was tested in all six patients, data represent the four patients with reactivity in the ELISpot assay. Peptides are p1 p5365–73, p2 p53264–272, p3 p53187–197, p4 p53149–157 (modified), p5 p53139–147 (modified), p6 p53103–111 (modified), and the vehicle is included as a negative control. PBMCs were set up in duplicate wells and the graph depicts the average number of spot-forming cells (SFC) / 200,000 PBMCs in the IFN-γ ELISpot assay at different times
Fig. 4.
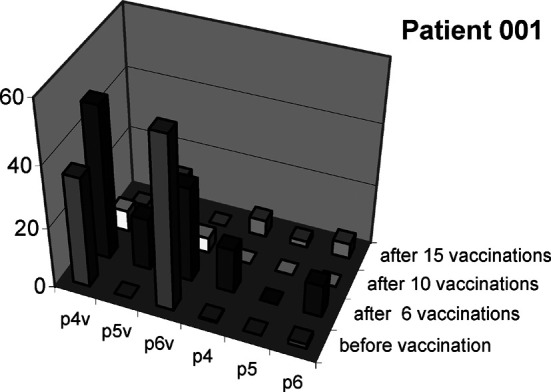
CTL reactivity against modified peptides is associated with reactivity against the corresponding wild-type peptides. In patients with CTLs specific for the three modified p53 peptides, reactivity was tested against the corresponding wild-type peptides in the ELISpot assay. Peptides are p4v p53149–157, p5v p53139–147, p6v p53103–111, p4 p53149–157 (modified), p5 p53139–147 (modified), p6 p53103–111 (modified). PBMCs were set up in duplicate wells and the graph depicts the average number of spot forming cells (SFC) / 200,000 PBMCs in the IFN-γ ELISpot assay with data for vehicle control subtracted. Data on wild-type and modified peptides, respectively, are from two individual experiments with PBMCs from patient 001
We also attempted to measure the frequency of p53-specific CTLs from the PBMCs collected prior to expansion by FACS analyses. By loading MHC class I dimers (DimerX; BD Bioscience, San José, CA) we identified a population of EBV-specific CTLs (positive control) on approximately 0.23% of the PBMCs. However, we were unable to identify p53-specific CTLs by this method in samples that had shown high reactivity in the ELISpot assay (data not shown).
Clinical response
Stable disease for more than 6 months was attained in two patients (001 and 009): both had progressive bone metastases at study entry (Table 1). Furthermore, patient 001 had spontaneous normalization of the haemoglobin level after four vaccinations (Fig. 2). And after six vaccinations, the PSPA score was reduced from the initial 4, to 1 due to cessation of bone pain and reduced requirement of analgetics. Both patients are still alive +24/+14 months after the first vaccination. T-cell reactivity against two modified peptides was induced in patient 001, who also had a high primary expression of p53. In patient 008 with a supraclavicular lymph node metastasis, temporary lymph node regression and replacement with cicatricial tissue was demonstrated by MRI scan after six immunizations (Fig. 5). However, a rescan after 5 weeks showed new lesions on the neck as well as regrowth of the primary lesion. This patient is, however, still alive +15 months. The patient had a high p53 expression level and developed T-cell responses against three of the p53 peptides. Patient 005 had a mixed response with partial regression of an axillary lesion, but appearance of new skin metastases. Two patients (002 and 007), one with multiple liver metastases and one with multiple lung and bone metastases had progressive disease.
Fig. 5a, b.
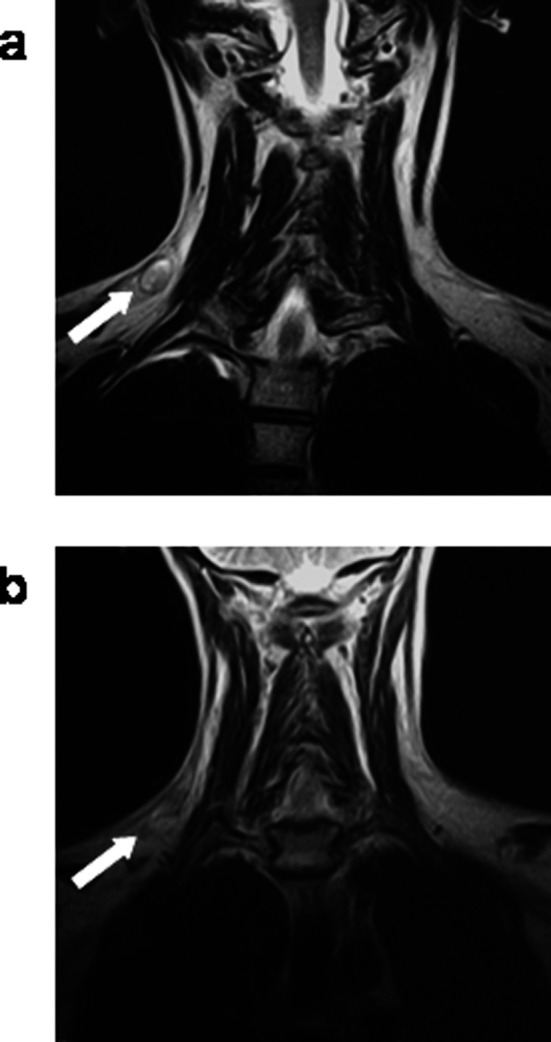
MRI scan of a supraclavicular lymph node metastasis (patient 008) at baseline (a) and after six immunizations (b) showing tumour regression
Discussion
The main observation of the present study is that vaccinations with p53-peptide–pulsed autologous DCs are safe and not associated with significant toxicity and that these cells are capable of expanding p53-specific peripheral blood T cells. In addition, our data suggest that immunizations with p53-peptide–pulsed autologous DCs may be able to induce antitumour activity.
The procedures for generation of DCs from leukapheresis-separated PBMCs used in the present work resemble those used by other authors [9, 21]. Approximately 40% of our DC preparations contain large granular cells and more than 90% of the cells are DR- and CD11c-positive, i.e. a phenotype typical for intermediate-to-mature DCs [3]. Ongoing studies on our DC preparations attempt to characterize their cytokine profile and their capacity for cross-presentation of naïve and recall antigens.
The role of DCs for the induction of strong antigen-specific immune responses in humans is unclear. Vaccinations with DCs pulsed with peptides derived from melanoma-associated antigenic peptides were previously shown to activate specific T cells and correlate with disease stabilization and regression of metastatic melanoma lesions [4, 31]. In the present work, we found that four to six vaccinations in half of the patients led to p53-peptide–specific T-cell responses in the peripheral blood, as measured by IFN-γ–secreting CD8+ T cells in ELISpot assays. However, these p53-specific CTLs could not be identified by direct fluorescence staining with relevant MHC class I dimers on freshly obtained T cells although a low, but significant, fraction of the T cells stained for a dominant EBV-peptide epitope. Thus, these data suggest that in order to quantify their frequency, peptide-specific T cells have to be expanded as done in the ELISpot assay, which recorded T-cell reactivity against both unmodified and modified peptides. At late time intervals after vaccination, p53-peptide–specific T cells in peripheral blood appeared to decline to background levels. Since repeated IL-2 treatments have been shown to shift tumour-specific T cells from the blood to the tumour site [2], our data might suggest that p53-peptide–specific T cells, as time passes, will localize at the tumour site. On the contrary, induction of tolerance due to repeated injections of the antigen can not be ruled out; further studies will be needed to clarify this [10].
In agreement with previous work [4, 31], two patients (001 and 008) showed some relation between T-cell reactivity and clinical course with respect to disease stabilization, normalization of haemoglobin levels, and regression of a lymph node metastasis. Two of the three patients (005 and 007) with progressive disease or only a mixed response showed reactivity toward the p53 epitopes prior to vaccination. The lack of clinical responses in these patients might suggest that the tumour cells had already evaded anti-p53 immune reactivity prior to vaccination.
Because wild-type p53 is ubiquitously expressed, including in the thymus, immunologic tolerance by thymic deletion might restrict the natural CTL repertoire that can be exploited for p53-specific immunotherapy of cancer. Murine studies have revealed that tolerance markedly restricts the p53-specific CTL repertoire in p53+/+ mice [29], while experiments in HLA-A2-transgenic mice have shown that low avidity CTLs against an A2-binding murine p53-derived peptide was not deleted, indicating the persistence of a part of this repertoire. In addition, multiple groups have found evidence for the presence of CTLs against HLA-A2–binding p53 peptides in humans [11, 14, 24, 28].
Although the responses against modified peptides with improved HLA class I binding are directed against non-self-peptides per se, our experiments showed that the responding CTLs cross-react with the three corresponding unmodified peptides. A similar cross-reactivity of CTLs against modified and unmodified p53 peptides was observed in HLA-A2-transgenic mice immunized with anchor-modified p53 peptides [22]. These results have important implications for the generation of antitumour reactivity against self-epitopes. They indicate that cancer vaccines based on anchor-modified peptides with high affinity for HLA class I are capable of expanding CTL responses against the corresponding wild-type peptide epitopes.
Clinical responses to cancer vaccination with regression and/or stabilization of generalized disease have been observed in different vaccine trials. Regression of metastases were often associated with measurable immune responses to the vaccine [1, 4, 8, 16, 36], and tumour progression was often associated with low levels or absence of tumour MHC class I [15, 19]. In the present study, three of six patients experienced changes in the course of the disease in association with the vaccination treatment, suggesting that vaccine-induced immunity may have therapeutic effects in some patients with defined tumour characteristics. However, this small phase I study brings no direct proof of clinical effect, only indications of a possible connection with vaccination. The study focused primarily on toxicity associated with the use of modified self-peptides in cancer vaccination therapy and only secondarily on induced immunologic and tumour response. Furthermore, the possibility that clinical effect is related to unpulsed DCs and IL-2 treatment alone, rather than p53-specific reactivity can of course not be excluded. However, it is not possible within the limitations of a phase I study to address this issue properly.
A correlation between p53 protein expression and clinical effect could not be clarified in this small series. The p53 expression level was determined by immunohistochemical staining of tissue samples from the primary tumour, which might not reflect the expression level in metastatic lesions. Furthermore, it was recently demonstrated that p53-restricted CTL-mediated killing is not necessarily dependent on a high p53 protein level in the tumour cells [33].
Tumour cells might lose antigens and therefore display a reduced susceptibility to vaccine-induced immunity in the course of the vaccinations. Multiepitope vaccine strategies as well as ways to stabilize antigen expression of tumours in vivo should be considered to prevent tumour escape of antigen-loss variants. In addition, tumour-associated factors such as the absence of costimulatory molecules, secretion of immunosuppressive cytokines, and interference with T-cell-receptor signal transduction may limit the effectiveness of vaccine-induced immune responses. The prospective analysis of these factors in future clinical vaccine trials will help to determine their role as prognostic factors for immunotherapy of cancer [25].
Patients included in the trial had metastatic breast cancer often with a high tumour burden and were all heavily pretreated; as a consequence, significant tumour responses may not be obtainable despite activation of the immune system. However, the clinical results obtained here encourage further clinical studies at an earlier stage of the disease.
Acknowledgements
This work was supported by grants from The Danish Cancer Research Foundation, The Danish Cancer Society, The Michaelsen Foundation and The Aase og Ejnar Danielsen’s Foundation.
References
- 1.Alexander Immunol Res. 1998;18:79. doi: 10.1007/BF02788751. [DOI] [PubMed] [Google Scholar]
- 2.Andersen MH, Gehl J, Reker S, Pedersen LO, Becker JC, Geertsen P, Straten PT (2003) Dynamic changes of specific T cell responses to melanoma correlate with IL-2 administration. Semin Cancer Biol (in press) [DOI] [PubMed]
- 3.Banchereau Annu Rev Immunol. 2000;18:767. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 4.Banchereau Cancer Res. 2001;61:6451. [PubMed] [Google Scholar]
- 5.Barfoed Scand J Immunol. 2000;51:128. doi: 10.1046/j.1365-3083.2000.00668.x. [DOI] [PubMed] [Google Scholar]
- 6.Chang J Clin Oncol. 1995;13:1009. doi: 10.1200/JCO.1995.13.4.1009. [DOI] [PubMed] [Google Scholar]
- 7.Chariyalertsak J Med Assoc Thai. 1998;81:698. [PubMed] [Google Scholar]
- 8.Coulie Proc Natl Acad Sci USA. 2001;98:10290. [Google Scholar]
- 9.De J Immunother. 2002;25:429. doi: 10.1097/00002371-200209000-00007. [DOI] [PubMed] [Google Scholar]
- 10.Dhodapkar Blood. 2002;100:174. doi: 10.1182/blood.V100.1.174. [DOI] [PubMed] [Google Scholar]
- 11.Eura Clin Cancer Res. 2000;6:979. [Google Scholar]
- 12.Feuerstein J Immunol Methods. 2000;245:15. doi: 10.1016/S0022-1759(00)00269-6. [DOI] [PubMed] [Google Scholar]
- 13.Fong Annu Rev Immunol. 2000;18:245. doi: 10.1146/annurev.immunol.18.1.245. [DOI] [PubMed] [Google Scholar]
- 14.Gnjatic J Immunol. 1998;160:328. [PubMed] [Google Scholar]
- 15.Jager Int J Cancer. 1997;71:142. doi: 10.1002/(SICI)1097-0215(19970410)71:2<142::AID-IJC3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 16.Jager Proc Natl Acad Sci USA. 2000;97:12198. [Google Scholar]
- 17.James J Natl Cancer Inst. 1999;91:523. doi: 10.1093/jnci/91.6.523. [DOI] [PubMed] [Google Scholar]
- 18.Lutzker Clin Cancer Res. 2001;7:2. [Google Scholar]
- 19.Mendez Tissue Antigens. 2001;57:508. doi: 10.1034/j.1399-0039.2001.057006508.x. [DOI] [PubMed] [Google Scholar]
- 20.Nikitina Clin Cancer Res. 2001;7:127. [PubMed] [Google Scholar]
- 21.O’Rourke Cancer Immunol Immunother. 2003;52:387. doi: 10.1007/s00262-003-0375-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petersen Scand J Immunol. 2001;53:357. doi: 10.1046/j.1365-3083.2001.00887.x. [DOI] [PubMed] [Google Scholar]
- 23.Reid Br J Haematol. 2001;112:874. doi: 10.1046/j.1365-2141.2001.02626.x. [DOI] [PubMed] [Google Scholar]
- 24.Ropke Proc Natl Acad Sci USA. 1996;93:14704. doi: 10.1073/pnas.93.25.14704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenberg Nature. 2001;411:380. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 26.Soussi Ann N Y Acad Sci. 2000;910:121. doi: 10.1111/j.1749-6632.2000.tb06705.x. [DOI] [PubMed] [Google Scholar]
- 27.Svane APMIS. 2003;111:818. doi: 10.1034/j.1600-0463.2003.11107813.x. [DOI] [PubMed] [Google Scholar]
- 28.Theobald Proc Natl Acad Sci USA. 1995;92:11993. [Google Scholar]
- 29.Theobald J Exp Med. 1997;185:833. doi: 10.1084/jem.185.5.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Therasse J Natl Cancer Inst. 2000;92:205. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 31.Thurner J Exp Med. 1999;190:1669. doi: 10.1084/jem.190.11.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thurner J Immunol Methods. 1999;223:1. doi: 10.1016/s0022-1759(98)00208-7. [DOI] [PubMed] [Google Scholar]
- 33.Vierboom Cancer Res. 2000;60:5508. [PubMed] [Google Scholar]
- 34.Wurtzen Int J Cancer. 2002;99:568. doi: 10.1002/ijc.10375. [DOI] [PubMed] [Google Scholar]
- 35.Wurtzen Int J Cancer. 2001;93:855. doi: 10.1002/ijc.1417. [DOI] [PubMed] [Google Scholar]
- 36.Zwaveling Cancer Res. 2002;62:6187. [PubMed] [Google Scholar]