

Summary
Horizontal basal cells (HBCs) mediate olfactory epithelium (OE) regeneration following severe tissue injury. The dynamism of the post-injury environment is well illustrated by in silico modeling of RNA sequencing data that demonstrate an evolving HBC transcriptome. Unfortunately, spatiotemporally dynamic processes occurring within HBCs in situ remain poorly understood. Here, we show that HBCs at 24 h post-OE injury spatially redistribute a constellation of proteins, which, in turn, helped to nominate Rac1 as a regulator of HBC differentiation during OE regeneration. Using our primary culture model to activate HBCs pharmacologically, we demonstrate that concurrent Rac1 inhibition attenuates HBC differentiation potential. This in vitro functional impairment manifested in vivo as decreased HBC differentiation into olfactory sensory neurons following HBC-specific Rac1 conditional knockout. Taken together, our data potentiate the design of hyposmia-alleviating therapies and highlight aspects of in situ HBC spatiotemporal dynamics that deserve further investigation.
Subject areas: sensory neuroscience, cell biology, Omics
Graphical abstract
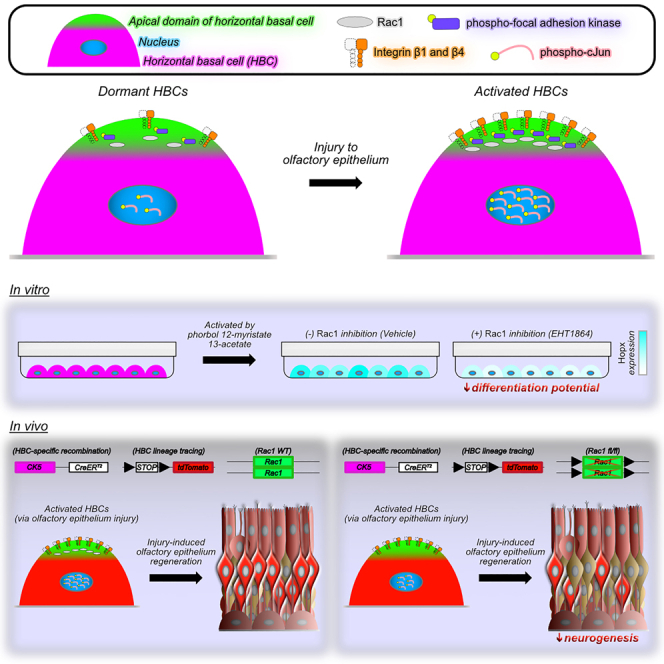
Highlights
-
•
Activated HBCs in vivo increase Rac1 within their apical domain following OE injury
-
•
Rac1-mediated signaling within activated HBCs in vitro is impaired by Rac1 inhibition
-
•
Differentiation potential of activated HBCs in vitro is attenuated by Rac1 inhibition
-
•
In vivo HBC neurogenesis following OE injury is impaired by HBC-specific Rac1 cKO
Sensory neuroscience; Cell biology; Omics
Introduction
Exposed to the external environment, the olfactory epithelium (OE) is a specialized neuroepithelium composed of sensory neurons and non-neuronal cell types that collectively facilitate transduction of volatile odorants. Critical to OE homeostasis, a population of reserve resident stem cells called horizontal basal cells (HBCs) reside atop the basal lamina. While typically dormant, HBCs activate and contribute to OE regeneration following severe tissue injury.1 Various mechanisms promote HBC-mediated OE repair, including those derived non-cell autonomously from the surrounding tissue environment. For example, targeted ablation of glial-like sustentacular (Sus) cells whose cell bodies form the OE-nasal cavity interface and extend processes basally that run adjacent to HBCs2 leads to HBC differentiation.3 This process following Sus cell destruction has been postulated to result from decreased cell-cell interactions between the Sus cell-expressed Notch ligand, Jagged1, and Notch1 receptor expressed by HBCs.3 Additionally, Sus cell destruction is thought to cause a localized reduction of retinoic acid, a cytokine synthesized by Sus cells that specifies HBC dormancy.4 Severe injury also elicits an inflammatory response that appears to regulate HBCs.5 Diminution of this response or impairing the ability of HBCs to respond to inflammatory cytokines negatively impacts HBC proliferation and subsequent OE regeneration.5 Moreover, discovery that HBC-specific ablation of cilia compromises OE regeneration further emphasizes the importance of extrinsic, environmental cues6 to HBC-mediated tissue repair.
Complementing these non-cell autonomous processes, ΔNp63 (p63) is a transcription factor that positively regulates HBC dormancy.7 Therefore, p63 downregulation is both necessary and sufficient to activate HBCs so that these resident stem cells can appropriately participate in OE regeneration.8 The critical relationship between p63 and HBC activation is underscored by HBC-specific p63 conditional knockout (cKO) that results in aberrant HBC differentiation within uninjured OE,7,8 and, conversely, p63 overexpression that attenuates basal cell differentiation following OE injury.8 Additionally, RNA sequencing technology has been a boon for computationally modeling HBCs to visualize their highly dynamic genomic landscape in silico following p63 downregulation.9,10 To date, however, little is known about the spatiotemporal dynamics exhibited by HBCs in situ soon after injury, and whether such dynamic processes involve a molecular component central to HBCs and their ability to contribute to OE regeneration.
To address this gap in knowledge, subcellular protein distribution within HBCs was assessed by dividing these stem cells into multiple intracellular domains. Comparing HBCs within uninjured and acutely injured OE revealed a constellation of differentially expressed proteins suggesting that Rac1 may influence HBCs as they mediate OE regeneration. Using our previously described primary HBC culture models11,12 to inform our in vivo investigation, the data in sum demonstrate that Rac1 positively regulates HBC neurogenesis. Furthermore, our work here establishes an in vitro model’s utility for elucidating mechanisms that govern HBC biology and provides the rationale for new avenues of exploration that may further enhance our understanding of HBCs.
Results
HBCs at 24 h post-injury (24 hpi) increase their apical expression of integrin β1 (Itgb1) and integrin β4 (Itgb4)
As p63 positively regulates HBC dormancy within the OE, its decline is sufficient to shift HBCs away from dormancy and serve as a marker of HBC activation.7,8 Relative to those within uninjured OE, cytokeratin 5+ (CK5+) HBCs in wild-type (WT) mice significantly downregulate p63 expression 24 h after intra-peritoneal injection with methimazole (MTZ) (Figures 1A–1E), an olfactotoxic agent known to induce severe OE injury that results in HBC-mediated regeneration of the tissue.1,13 Given that p63 can positively regulate expression of Itgb1 and Itgb4,14 it was therefore surprising that HBCs at 24 hpi did not decrease total Itgb1 (Figures S1A, S1B, S1E, S1F, and S1I) nor total Itgb4 relative to expression within uninjured OE (Figures S1K, S1L, S1O, S1P, and S1S) as defined by their respective fluorescence intensities within CK5+ HBCs. Because activated HBCs have been shown to undergo morphological changes suggestive of diminished contact with the basal lamina,8 it was also surprising that HBCs at 24 hpi do not decrease basal Itgb1 (Figures S1C, S1D, S1G, S1H, and S1J) nor basal Itgb4 (Figures S1M, S1N, S1Q, S1R, and S1T) relative to their counterparts within uninjured OE as defined by integrin fluorescence intensity within areas exclusively labeled by CK5 and the basal lamina constituent, Laminin. Notably, however, HBCs at 24 hpi increase apical domain expression of Itgb1 and Itgb4 by 91.2% (Figures 1F–1N) and 131.9% (Figures 1O–1W), respectively, relative to those within uninjured OE. For this analysis, the apical domain was defined as areas labeled by both CK5 and Ezrin (Figures 1H, 1I, 1L, 1M, 1Q, 1R, 1U, and 1V), the latter of which is a protein that regulates apical domain integrity by linking other apically localized proteins to the cytoskeleton.15 As such, these data demonstrate that, at 24 hpi, HBCs spatially modulate intracellular protein expression in part by enhancing apical expression of Itgb1 and Itgb4.
Figure 1.

HBCs at 24 hpi increase apical expression of Itgb1 and Itgb4
(A–D) Representative immunofluorescence images demonstrating that relative to HBCs within uninjured (uninj.) OE (A, B), HBCs at 24 hpi are activated as they demonstrate p63 downregulation (C, D).
(E) Quantification of nuclear p63 normalized (norm.) fluorescence (fluor.) density within CK5+ HBCs; each triangle represents a CK5+ Nuclear Object (Obj.) (blue outlines in B and D) used to quantify the encompassed p63 fluorescence.
(F–M) Representative immunofluorescence images demonstrating that HBCs at 24 hpi (J–M) increase apical Itgb1 (within green outlines, I and M) relative to HBCs within uninj. OE (F–I).
(N) Quantification of Itgb1 norm. fluor. density within HBC apical domains; each triangle denotes an analyzed region as represented in (I) and (M) (n = 19 regions across 3 mice).
(O–V) Representative immunofluorescence images demonstrating that HBCs at 24 hpi (S–V) increase apical Itgb4 (within green outlines, R and V) relative to HBCs within uninj. OE (O–R).
(W) Quantification of Itgb4 norm. fluor. density within HBC apical domains; each triangle denotes an analyzed region as represented in (R) and (V) (n = 17 regions across 3 mice). Unpaired t test (E, J), Mann-Whitney test (O). Uninj. used as baseline, error bars indicate mean ± SEM, ∗∗p < 0.01, ∗∗∗∗p < 0.0001 (E, N, W). Scale bar equals 10 μm (A).
HBCs at 24 hpi spatiotemporally synchronize intracellular focal adhesion components that suggest enhancement of Rac1-mediated signaling pathway activity
The cytoplasmic tails of both Itgb1 and Itgb4 can activate Rac1,16,17,18 a member of a family of small Rho GTPases19,20 that can mediate various stem cell functions.21,22,23,24 Concomitant with increased apical localization of Itgb1 and Itgb4 (Figures 1F–1W), HBCs at 24 hpi notably increase apical Rac1 by 81.6% relative to HBCs in uninjured OE (Figures 2A–2G). Furthermore, focal adhesion kinase phosphorylated at tyrosine 925 (pFAKY925), a critical intermediary that facilitates integrin-mediated Rac1 activation,25 is similarly increased by 91.7% in the apical domain of HBCs at 24 hpi when compared to the apical domain of HBCs prior to OE injury (Figures 2H–2N). Interestingly, the spatiotemporal synchronicity that HBCs demonstrate via enhanced apical localization of Itgb1 and Itgb4, pFAKY925, and Rac1 coincides with a greater than 8-fold increase in HBC expression of pcJun (Figures 2O–2U), a transcription factor subunit that can participate in regulating proliferation and tissue morphogenesis.26,27,28 As pcJun lies downstream of Rac1 activation,29 these data in sum suggest that HBCs at 24 hpi enhance Rac1-mediated signaling pathways.
Figure 2.

HBCs at 24 hpi spatiotemporally synchronize Rac1 with another intracellular focal adhesion component and a downstream effector of Rac1-mediated signaling
(A–F) Representative immunofluorescence images demonstrating that HBCs at 24 hpi (D–F) upregulate apical Rac1 (within green outlines, C and F) relative to HBCs within uninj. OE (A–C).
(G) Quantification of Rac1 norm. fluor. density within HBC apical domains; each triangle denotes an analyzed region as represented in (C) and (F) (n = 30 regions across 3 mice).
(H–M) Representative immunofluorescence images demonstrating that HBCs at 24 hpi (K–M) upregulate apical pFAKY925 (within green outlines, J and M) relative to HBCs within uninj. OE (H–J).
(N) Quantification of pFAKY925 norm. fluor. density within HBC apical domains; each triangle denotes an analyzed region as represented in (J) and (M) (n = 17 regions across 3 mice).
(O–T) Representative immunofluorescence images demonstrating that HBCs at 24 hpi (R–T) upregulate nuclear pcJun (within blue outlines, Q and T) relative to HBCs within uninj. OE (O–Q).
(U) Quantification of pcJun norm. fluor. density within HBC nuclei; each circle denotes a CK5+ Nuclear Obj. as represented in (Q) and (T) (n = 3 mice, 234 uninj. HBCs and 258 24 hpi HBCs). Uninj. used as baseline; error bars indicate mean ± SEM, Mann-Whitney test, ∗∗p < 0.01, ∗∗∗∗p < 0.0001 (G, N, U). Scale bar equals 10 μm (A).
HBC expression of Rac1 within the regenerating OE progressively wanes
At 2 and 3 days post-injury (dpi), distinct layers of cells begin to comprise the regenerating OE. Within these layers reside p63+ HBCs and p63- non-HBCs30 (Figure S2). Using the CK5 promoter to drive expression of tamoxifen-inducible Cre recombinase and lineage trace tdTomato+ (tdTom+) HBCs and their progeny (K5CreERT2; Rosa26R(tdTomato)), p63-/tdTom+ non-HBCs within these bi-genic mice at 2 dpi express 25.5% more Rac1 relative to p63+/tdTom+ HBCs (Figures S2A–S2E). At 3 dpi, Rac1 becomes increasingly enriched to p63-/tdTom+ non-HBCs (32.6%) (Figures S2F–S2J). These data demonstrate that Rac1 segregates away from HBCs as OE regeneration proceeds, thereby further suggesting that HBCs may only acutely utilize Rac1-mediated signaling after OE injury (Figures 1 and 2).
Primary HBCs activated in vitro demonstrate molecular sensitivity to Rac1 inhibition
To begin interrogating the potential relationship between HBC activation and Rac1 function, we leveraged our HBC culture system that enables transcriptional recapitulation of in vivo HBCs11 to treat primary HBCs with phorbol 12-myristate 13-acetate (PMA), a compound used to activate other stem cells in vitro.31,32 Importantly, in vitro rat HBCs treated for 12 h with 50 nM PMA emulate a myriad of transcriptomic patterns exhibited by in vivo HBCs through 24 hpi12 when they spatiotemporally synchronize a constellation of molecular components suggestive of Rac1-mediated signaling activity (Figures 1 and 2). Expectedly, 50 nM PMA treatment triggers HBC activation as evidenced by decreased p63 abundance when compared to vehicle-treated HBCs (Figures 3A and 3B). Mirroring the molecular characteristics exhibited by in vivo HBCs at 24 hpi (Figures 1A–1E and 2O–2U), p63 downregulation in vitro occurs concomitant with increased pcJun abundance (Figures 3A and 3D). To determine whether this increase in pcJun was a result of Rac1 activation, PMA-induced HBC activation was coupled with 50 μM EHT1864, an inhibitor of Rac1 activity.33 Indeed, PMA and EHT1864 treatment together attenuated pcJun abundance by 36.8% relative to PMA alone (Figures 3A and 3D). As Rac1 activation facilitates pcJun expression via upstream phosphorylation of cJun N-terminal kinase (JNK),34,35 the same treatment paradigm similarly decreased pJNK abundance by 37.1% relative to PMA alone (Figure S3), thus underscoring the involvement of Rac1-mediated signaling. Importantly, these effects were not due to any significant change in total Rac1 abundance (Figures 3A and 3C). Notably, the decrease in p63 abundance with concurrent PMA and EHT1864 treatment was not significantly different relative to PMA alone (Figure 3B). Collectively, these data demonstrate that primary HBCs enhance Rac1-mediated signaling following their activation in vitro, which in turn suggest that HBCs do so to regulate a currently undefined process independent of their activation.
Figure 3.

Primary HBCs activated in vitro enhance Rac1-mediated signaling
(A) Representative western blots depicting p63, Rac1, pcJun, total cJun, and GAPDH abundances in cultures of vehicle-treated (PMA-/EHT1864-), activated (PMA+/EHT1864-), activated and Rac1 inhibited (PMA+/EHT1864-), and Rac1 inhibited (PMA-/EHT1864+) HBCs.
(B–D) Quantification of densitometric measurements for p63 (B), Rac1 (C), and normalized pcJun per normalized total cJun (D) abundances (n = 3 independent trials), vehicle-treated used as baseline, error bars indicate mean +SEM, one-way ANOVA with post hoc Tukey’s multiple comparisons test, ∗<0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Rac1-mediated signaling influences primary HBC differentiation potential
As activated HBCs in culture exhibited molecular sensitivity to concurrent Rac1 inhibition (Figures 3 and S3), we examined how inhibiting Rac1 in PMA-activated in vitro rat HBCs might alter their function. Notably, in vivo HBCs become increasingly proliferative7 and upregulate Hopx, an early marker of HBC differentiation,10 shortly after their activation. Relative to HBCs treated with vehicle, PMA treatment alone more than doubled overall Hopx expression (Figures 4A, 4B, and 4E). When concurrently treated with PMA and EHT1864, HBC expression of Hopx was diminished by 43.8% relative to PMA alone (Figures 4B, 4C, and 4E). Surprisingly, in vitro HBCs attenuated Ki67 expression by nearly 25% following treatment with PMA in comparison to vehicle treatment (Figures 4F, 4G, and 4J). Treatment with both small molecules also resulted in a small, but statistically significant, decrease in overall Ki67 expression relative to PMA treatment alone (∼2.2%) (Figures 4G, 4H, and 4J). Together, these data demonstrate that inhibition of Rac1-mediated signaling functionally disrupts HBCs following their in vitro activation by PMA.
Figure 4.

Rac1 potentiates primary HBC differentiation in vitro
(A–D) Representative immunofluorescence images of Hopx expression in cultures of vehicle-treated (A), activated (B), activated and Rac1 inhibited (C), and Rac1 inhibited (D) HBCs.
(E) Quantification of norm. total Hopx fluor. density for conditions represented in (A) (4,927 CK5+ HBCs), (B) (8,065 CK5+ HBCs), (C) (7,235 CK5+ HBCs), and (D) (5,358 CK5+ HBCs) (n = 4 independent trials), each circle represents an analyzed CK5+ HBC.
(F–I) Representative immunofluorescence images of Ki67 expression in cultures of vehicle-treated (F), activated (G), activated and Rac1 inhibited (H), and Rac1 inhibited (I) HBCs.
(J) Quantification of norm. nuclear Ki67 fluor. density for conditions represented in (F) (4,716 CK5+ HBCs), (G) (7,325 CK5+ HBCs), (H) (7,297 CK5+ HBCs), and (I) (5,228 CK5+ HBCs) (n = 4 independent trials); each circle represents an analyzed DAPI+ area within a CK5+ HBC. Vehicle (PMA−/EHT1864-) used as baseline; error bars indicate mean ± SEM, Kruskal-Wallis test with post hoc Dunn’s multiple comparisons test, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 (E, J). Scale bar equals 50 μm (A).
In vivo HBCs at 24 hpi engage in Rac1-mediated signaling
Given that primary HBCs in vitro subjected to concurrent small molecule-mediated activation and Rac1 inhibition demonstrated diminished pcJun abundances (Figures 3A and 3D), we interrogated whether in vivo HBCs engage in Rac1-mediated signaling activity. To do so, we utilized tri-genic mice expressing tamoxifen-inducible Cre recombinase driven by the CK5 promoter to simultaneously conditionally knock out Rac1 (Figures S4 and S5) in an HBC-specific manner and identify HBCs and their progeny via a tdTom reporter (K5CreERT2; Rac1fl/fl; Rosa26R(tdTomato), hereafter referred to as Rac1 fl/fl). These Rac1 fl/fl mice were compared to Rac1 WT mice in which both Rac1 alleles are WT but otherwise similarly harbor CK5-driven Cre recombinase and the conditional tdTom reporter to identify HBCs and their derivatives. Following MTZ-induced OE injury, HBCs within Rac1 fl/fl OE at 24 hpi did not demonstrate a significant difference in nuclear p63 relative to their Rac1 WT counterparts (Figures 5A–5C, 5F–5H, and 5K). HBC-specific Rac1 cKO, however, decreased HBC nuclear pcJun by 44.5% (Figures 5D, 5E, 5I, 5J, and 5L). Taken together, the molecular consequences of in vivo Rac1 perturbation recapitulate those observed in vitro (Figures 3B and 3D) and demonstrate that Rac1 mediates signaling within in vivo HBCs following OE injury.
Figure 5.

In vivo HBCs at 24 hpi demonstrate diminished Rac1-mediated signaling as a consequence of HBC-specific Rac1 cKO
(A–L) Representative immunofluorescence images from Rac1 WT (A–C) and Rac1 fl/fl (F–H) OE demonstrate that HBC expression of nuclear p63 at 24 hpi (within cyan outlines, C and H) is not significantly affected by HBC-specific Rac1 cKO (quantified in K, each circle denotes a tdTom+ Nuclear Obj. as represented in C and H [n = 3 mice, 132 Rac1 WT HBCs and 124 Rac1 fl/fl HBCs]). Representative immunofluorescence images from Rac1 WT (A, D, E) and Rac1 fl/fl (F, I, J) OE demonstrate that nuclear pcJun at 24 hpi (within green outlines, E and J) is diminished by HBC-specific Rac1 cKO (quantified in L, each triangle denotes a tdTom+ Nuclear Obj. as represented in E and J [n = 3 mice, 132 Rac1 WT HBCs and 124 Rac1 fl/fl HBCs]). Rac1 WT used as baseline, error bars indicate mean ± SEM, Mann-Whitney test, ∗∗∗∗p < 0.0001 (K, L). Scale bar equals 10 μm (A).
Rac1 regulates the neurogenic potential of in vivo HBCs during OE regeneration
As in vivo HBCs utilize Rac1-mediated signaling pathways soon after HBC-mediated OE regeneration initiates, we examined the potential functional consequences of HBC-specific Rac1 cKO. Although Rac1 has been shown to organize integrin expression,36,37 Itgb1 (Figures S6A–S6M) and Itgb4 (Figures S6N–S6Z) localization to HBC intracellular domains was not affected within Rac1 fl/fl OE. Due to an inability in our hands to immunolabel Hopx within OE tissue sections, we were unable to correlate in vivo the dramatic downregulation of Hopx demonstrated by in vitro HBCs following their concurrent activation and Rac1 inhibition (Figures 4B, 4C, and 4E). However, because Hopx labels HBCs determined to differentiate,10 we interrogated the longer-term functional impact of Rac1 in vivo by assessing morphologically regenerated OE at 28 28 dpi in Rac1 WT and Rac1 fl/fl mice. Hopx-expressing HBCs primarily give rise to Sus cells and olfactory sensory neurons (OSNs),10 with the former identifiable by CK8 and interleukin-33 (IL33) located apically within the regenerated tissue10,38 while immature and mature OSNs are marked by PGP9.5.30,39,40,41,42 Following OE injury, HBC differentiation into CK8+/apical IL33+/tdTom+ Sus cells was unaffected (17.1 HBC-derived Sus cells per 100 μm Rac1 fl/fl OE vs. 18.3 HBC-derived Sus cells per 100 μm Rac1 WT OE) (Figures 6A–6K). Interestingly, however, HBC-specific Rac1 cKO attenuated HBC differentiation into PGP9.5+/tdTom+ OSNs at 28 dpi by nearly half (14.6 HBC-derived OSNs per 100 μm Rac1 fl/fl OE vs. 25.7 HBC-derived OSNs per 100 μm Rac1 WT OE) (Figures 6L–6V). In sum, these data demonstrate that Rac1 positively regulates the magnitude of HBC-derived neurogenesis during OE regeneration.
Figure 6.

In vivo HBC neurogenesis following OE injury is diminished by HBC-specific Rac1 cKO
(A–J) Representative immunofluorescence images demonstrating that HBC differentiation into CK8+/apical IL33+/tdTom+ Sus cells (magenta in D and I; magenta outlines in E and J) is not significantly altered at 28 dpi by HBC-specific Rac1 cKO (F–J) relative to Rac1 WT (A–E).
(K) Quantification of HBC-derived Sus cells found in regenerated OE at 28 dpi; each circle denotes an analyzed region as represented in (E) and (J) (n = 12 regions across 3 mice).
(L–U) Representative immunofluorescence images demonstrating that fewer PGP9.5+/tdTom+ OSNs (magenta in O and T; magenta outlines in P and U) arise from HBCs at 28 dpi following HBC-specific Rac1 cKO (Q–U) relative to Rac1 WT (L–P).
(V) Quantification of HBC-derived OSNs found in regenerated OE at 28 dpi; each circle denotes an analyzed region as represented in (P) and (U) (n = 15 regions across 3 mice). Error bars indicate mean ± SEM, Mann-Whitney test, ∗p < 0.05 (K, V). Scale bar equals 10 μm (A, D).
Discussion
While it has been known for more than a decade that HBCs can help repair the OE following tissue injury,1 the transcriptional and environmental mechanisms that enable HBC-mediated OE regeneration have only relatively recently been explored.5,6,8,30 However, it still remains unknown whether HBCs in situ exhibit processes that are spatiotemporally dynamic across subcellular domains soon after tissue injury and, if so, whether any of these early molecular players facilitates appropriate HBC contribution to OE reconstitution. We show here that HBCs reorganize Rac1 apically following OE injury. Subsequent to identifying this biological phenomenon, we interrogated these OE-resident stem cells in culture and in vivo. These investigations demonstrated for the first time that Rac1 regulates HBC differentiation potential in vitro and HBC neurogenesis during in vivo injury-induced OE regeneration.
The need to better understand HBC biology arises from the prevalence of olfactory deficits and their associated morbidities experienced by the aging population.43,44 OE biopsies from dysosmic patients demonstrate aneuronal regions42,45 that, despite the need for tissue repair, harbor persistently dormant HBCs.46,47 As such, one strategy to ameliorate olfactory dysfunction and its burdens includes artificially activating this endogenous HBC population. An alternate approach capitalizes on the ability to harvest, expand, activate,11 and re-introduce HBCs in an autologous manner. In either case, the mechanisms governing HBC differentiation need to be defined for therapeutic efficacy. Presently, inefficient HBC engraftment challenges the feasibility of cultured HBC transplantation,11,12 and this hindrance recapitulates a rodent model of stem cell infusion into the auditory cochlea.48 Their data in particular highlight the need to understand and regulate the differentiation of stem cells before transplantation, as evidenced by an inverse relationship between degree of cellular differentiation prior to infusion and cell survival following transplantation. Similar reasons may explain why HBCs, which upregulate the differentiation marker, Hopx, following PMA-mediated in vitro activation, exhibit low engraftment efficiency within injured OE. Notably, our in vitro data identify Rac1 as a positive regulator of HBC differentiation. Furthermore, concomitant PMA treatment with reversible inhibition of Rac1 attenuated Hopx without significantly affecting the decrease in p63 abundance that indicates HBC activation. Consequently, this present work offers a potential strategy to simultaneously improve HBC engraftment and maintain stem cell potency. Building from this framework, future investigations utilizing EHT1864 to optimize engraftment efficiency should remain mindful of the disparity between cellular and molecular processes. Although NIH 3T3 mouse fibroblasts attenuate lamellipodia formation after treatment with 5 μM EHT1864, BT-474 human breast cancer cells demonstrate an IC50 of 3.17 μM EHT1864 with respect to proliferation, and SH-SY5Y human neuroblastoma cells exhibit an IC50 of 5.44 μM in relation to Aβ plaque release, EHT1864 was required at much higher concentrations to inhibit Rac1 activity (50 μM, 50 μM, and >25 μM, respectively).33,49,50 This process-dependent variability is also exemplified by cell death. While significant increases in apoptotic OCI-AML3 human acute myeloid leukemia and bladder smooth muscle cells were observed after treatment with 30 μM and 50 μM EHT1864, respectively,51,52 rat HBCs in vehicle- and 50 μM EHT1864-treated cultures demonstrated similar morphological appearances (Figures 4A–4D, 4F, and 4I) and treated mouse platelets retained significant viability up to 200 μM EHT1864.53
The critical nature of reversible Rac1 inhibition for any potential HBC infusion therapy is underscored by HBC-specific Rac1 cKO and its attenuation of in vivo HBC neurogenesis following OE injury. The consequences of Rac1 cKO in the OE are consistent with Rac1’s role in promoting hippocampal neurogenesis.54,55,56 Although Rac1 has also been implicated in positively regulating glial differentiation,57 Rac1 cKO did not perturb HBC differentiation into Sus cells, which serve multiple glial-like functions within the OE such as regulating local metabolism, phagocytosis, and maintaining ion balance.58,59,60,61 Furthermore, Sus cells are thought to specify HBC dormancy via direct cell-cell interactions3 and contribute retinoic acid to the environmental milieu.4 Given that these support cells serve functions critical to tissue homeostasis, it would be worthwhile to interrogate whether Rac1 within HBCs operates in parallel with other redundant, sufficient-but-not-necessary mechanisms to ensure HBC differentiation into Sus cells. Interestingly, Rac1 localization to Sus cells in addition to HBCs (Figure S5) may further underscore the former cell type’s importance to overall OE integrity as this GTPase has been shown to promote tight junction formation.62,63 It remains to be seen whether Sus cells, which immediately abut the nasal cavity that is in direct contact with the external environment, employ Rac1 as a mechanism to maintain OE barrier function. From an evolutionary perspective, the need to restore barrier function may explain why Sus cells can differentiate directly from HBCs without the need for intermediary progenitors,10 which could represent possible failure points. Moreover, in light of unperturbed Sus cell differentiation following various HBC-specific manipulations30 (Figures 6A–6K), the question of whether a subset of HBCs is already determined during OE development to become Sus cells upon their activation deserves examination.
As we demonstrated with Rac1 and HBC neurogenesis, 50 nM PMA can be used to activate HBCs in vitro as a starting point to elucidate mechanisms that govern HBC fate. In the present case, it is important to highlight that PMA treatment itself can lead to cJun phosphorylation via activation of protein kinase C,64,65 which thus may explain the still significant but not overly dramatic decrease in pcJun abundance with concurrent Rac1 inhibition (Figures 3A and 3D). To parse this potential in vitro confounder, HBC-specific Rac1 cKO in vivo demonstrated that activated HBCs at 24 hpi indeed express attenuated nuclear pcJun (Figures 5E–5J and 5L). This biological recapitulation is unsurprising as in vitro HBCs treated with 50 nM PMA for 12 h bear a striking transcriptomic resemblance to in vivo HBCs activated by OE injury.12 Firstly, decomposition of bulk RNA sequencing from PMA-treated in vitro HBCs demonstrates that the proportion of in vitro HBCs activated by 50 nM PMA proceeds in a temporally accurate manner when referenced against single-cell transcriptomes of in vivo HBCs from injured OE. Furthermore, when benchmarked against single-cell RNA sequencing of activated in vivo HBCs through 24 hpi,10 PMA-treated in vitro HBCs and their in vivo counterparts share core transcriptional elements as evidenced by significant overlap in the top 100 differentially expressed genes. These sets notably include enhancement of multiple wound response genes, upregulation of Hopx, and downregulation of p63,12 the latter two of which were again demonstrated (Figures 3A, 3B, 4A, 4B, and 4E). In addition to these extensive molecular data, PMA is known to activate Itgb1-mediated signaling pathways66 that can promote appropriate proliferation of mammary epithelial stem cells during mammary morphogenesis.67 This understanding in tandem with the in vivo data presented here following injury demonstrating that HBCs mobilize molecular machinery integral to Itgb1-mediated signaling suggests that further mechanistic interrogation of these pathways will bear fruit.
PMA treatment of HBCs, however, did not recapitulate the increase in Ki67 seen during in vivo HBC activation.7 Therefore, the decrease in Ki67 following PMA-induced HBC activation in vitro highlights the influence that in situ environmental cues may have on HBCs following OE injury. Given that skin wounding can prompt increased fibronectin deposition68 and skeletal muscle damage is followed by changes in laminin expression,69 the extracellular matrix (ECM) harbors promising candidate cues that are imperfectly recapitulated in the current culture model. Indeed, hematopoietic stem cell proliferation within bone marrow is dependent on interaction with tenascin-C,70 while collagen VI in skeletal muscle acts as an instructive cue for satellite cells to proliferate.71 In addition to positively regulating proliferation, chondroitin sulfate proteoglycans specify embryonic neural stem cell differentiation toward neuronal rather than glial fates.72 Moreover, stem cell function dependent on ECM interactions can be mediated by Itgb1.73,74 Therefore, our demonstration that in vivo HBCs spatiotemporally synchronize components of a signaling axis known to occur following Itgb1-ECM interactions75,76 furthers the need to inquire about the spatial localization of and role that ECM proteins may play in regulating HBC biology following OE injury. Indeed, data demonstrating that HBC-specific Rac1 cKO does not affect Itgb1 and Itgb4 expression at 24 hpi, including within the apical domain of HBCs (Figure S6), strengthen the case that Rac1 transduces signals initiated upstream by Itgb1 and/or Itgb4 interactions with currently unknown ECM components.
Limitations of the study
While the HBC cultures provided useful insights to motivate specific in vivo investigations, it is difficult to draw more definitive conclusions from these data given the use of small molecules had the relative biological simplicity inherent to in vitro systems. Due to technical limitations in our hands, we were unable to correlate in vitro Hopx expression to that in vivo before and after Rac1 perturbation. For this reason, we utilized a more indirect reference in vivo, namely HBC neuronal and Sus cell differentiation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse α-p63 | ATCC | Clone 4A4 |
| Mouse α-Ezrin | Developmental Studies Hybridoma Bank | CPTC-Ezrin-1 |
| Rabbit α-Laminin | Novus Biologicals | NB300-144 |
| Goat α-Itgb1 | R&D Systems | AF2405 |
| Rat α-Itgb4 | R&D Systems | MAB4054 |
| Chicken α-CK5 | BioLegend | Clone Poly9059 |
| Rabbit α-Rac1 | Proteintech | 24072-1-AP |
| Rabbit α-pFAKY925 | Cell Signaling Technology | 3284 |
| Rabbit α-pcJun | Cell Signaling Technology | 3270 |
| Rabbit α-PGP9.5 | Proteintech | 14730-1-AP |
| Rabbit α-ΔNp63 | BioLegend | 619002 |
| Rabbit α-cJun | Cell Signaling Technology | 9165 |
| Rabbit α-JNK | Cell Signaling Technology | 9252 |
| Rabbit α-pJNK | Cell Signaling Technology | 4668 |
| Mouse α-Rac1 | Proteintech | 66122-1-Ig |
| Mouse α-GAPDH | Cell Signaling Technology | 97166 |
| Chemicals, peptides, and recombinant proteins | ||
| Tamoxifen | Sigma-Aldrich | T5648 |
| Corn oil | Spectrum Chemical | CO136 |
| 4-hydroxytamoxifen | Hello Bio | HB6040 |
| Sunflower oil | Sigma-Aldrich | S5007 |
| Castor oil | Sigma-Aldrich | 259853 |
| Methimazole | Sigma-Aldrich | M8506 |
| EHT1864 | SellekChem | S7482 |
| Phorbol 12-myristate 13-acetate | SellekChem | S7791 |
| Critical commercial assays | ||
| RIPA Lysis and Extraction Buffer | Thermo Scientific | 89901 |
| Halt Protease and Phosphatase Inhibitor | Thermo Scientific | 78442 |
| Pierce BCA Protein Assay Kit | Thermo Scientific | 23227 |
| 4X Bolt LDS Sample Buffer | Invitrogen | B0007 |
| 10X Bolt Sample Reducing Agent | Invitrogen | B0009 |
| Bolt 4-12% Bis-Tris Plus Mini Protein Gel | Invitrogen | NW04120BOX |
| Broad Range Prestained Protein Marker | Proteintech | PL00002 |
| 20X Bolt MES SDS Running Buffer | Invitrogen | B0002 |
| PVDF/Filter Paper Sandwich (0.2 μm) | Invitrogen | LC2002 |
| 20X Bolt Transfer Buffer | Invitrogen | BT00061 |
| SuperSignal West Pico PLUS Chemiluminescent Substrate | Thermo Scientific | 34577 |
| Experimental models: Cell lines | ||
| Rat: primary HBCs | Taconic Biosciences | SD-M |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J; 129S1/SvlmJ F2 (wild-type) | This paper | N/A |
| Mouse: K5CreERT2 | Indra et al., 1999 | MGI:2177427 |
| Mouse: Rosa26R(tdTomato) | The Jackson Laboratory | 007909 |
| Mouse: Rac1tm1Djk/J | The Jackson Laboratory | 005550 |
| Software and algorithms | ||
| ImageJ | National Institutes of Health | https://imagej.net/ij/index.html |
| CellProfiler | Lamprecht et al., 2007 | https://cellprofiler.org/ |
| Affinity Designer | Serif Europe Ltd. | https://affinity.serif.com/en-us/designer/ |
| GraphPad Prism 10 | GraphPad Software | https://www.graphpad.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, James E. Schwob (jim.schwob@tufts.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Animals
All animals were maintained on ad libitum chow and water in a heat and humidity controlled, AALAC-accredited vivarium. All protocols for the use of vertebrate animal were approved by the Committee for the Humane Use of Animals at Tufts University School of Medicine. In vitro primary HBCs were harvested from male and female Sprague-Dawley rats (Taconic Biosciences, Cat. SD-M). WT mice were F2 generation mice from an initial cross of C57BL/6J (Jax Stock #000664) and 129S1/SvlmJ (Jax Stock #002448) mice performed in-house. K5CreERT2 mice, which enable HBC-specific inducible recombination, were generously provided by Dr. Pierre Chambon.77 Rosa26R(tdTomato) (Jax Stock #007909) mice were obtained from Jackson Labs. Rac1tm1Djk/J (Jax Stock #005550) mice were generously transferred from the Rejji Kuruvilla Lab, Johns Hopkins University. Bi-genic K5CreERT2; Rosa26R(tdTomato) (KT) mice enabling HBC-specific inducible lineage tracing were generated in-house. Tri-genic K5CreERT2; Rac1tm1Djk/ tm1Djk /J; Rosa26R(tdTomato) (Rac1 fl/fl) mice enabling HBC-specific inducible Rac1 KO and lineage tracing were generated in-house. K5CreERT2; Rac1WT/ WT /J; Rosa26R(tdTomato) mice (Rac1 WT) served as controls for genetic recombination experiments. All in vivo experiments used sex- and age-matched male and female mice that were 8-12 weeks old when initiated.
Method details
Tamoxifen induction of Cre-mediated recombination
Tamoxifen (Sigma-Aldrich, Cat. T5648) was dissolved in corn oil (Spectrum Chemical, Cat. CO136) at a stock concentration of 30 mg/mL. This tamoxifen stock was used to perform intra-peritoneal (IP) injections of KT mice at 150 mg/kg for two consecutive days prior to subsequent experimentation. 4-hydroxytamoxifen (Hello Bio, Cat. HB6040) was dissolved in 100% ethanol (Fisher Scientific, Cat. BP2818) at a stock concentration of 40 mg/mL. Prior to IP injection, this stock was emulsified in a mixture of 4 parts sunflower oil (Sigma-Aldrich, Cat. S5007) and 1 part castor oil (Sigma-Aldrich, Cat. 259853). Rac1 WT and Rac1 fl/fl mice each received 2.5mg 4-hydroxytamoxifen per day for five consecutive days prior to subsequent experimentation.
Methimazole-induced OE injury
Methimazole (MTZ) (Sigma-Aldrich, Cat. M8506) was dissolved in 1X PBS (Gibco, Cat. 10010-023) at a stock concentration of 5 mg/mL. WT mice received an IP injection at 10μL MTZ stock/g mouse. WT mice used for uninjured OE controls received an IP injection at 10 uL 1X PBS vehicle/g mouse. KT mice received an IP injection at 10uL MTZ stock/g mouse 5 days after the initial tamoxifen injection. Rac1 WT and Rac1 fl/fl mice received an IP injection at 7.5μL MTZ stock/g mouse 21 days after the last 4-hydroxytamoxifen injection. This reduction in MTZ concentration was necessary as 10μL MTZ stock/g Rac1 WT or fl/fl mouse resulted in OE over-lesioning as evidenced by the absence of tdTom+ HBCs at 24 hpi.
Tissue processing
At indicated time points, mice were euthanized with an intra-muscular injection of ketamine (37.5mg/kg), xylazine (7.5mg/kg), and acepromazine (1.25mg/kg) triple cocktail. Following trans-cardial perfusion with 20mL PBS (Gibco Cat. 10010-023), mice were trans-cardially fixed with 40mL 1% PLP (1% paraformaldehyde, 0.1M monobasic and dibasic phosphates, 90mM lysine, and 0.1M meta-sodium periodate). After dissecting away the surrounding cranial structures, the isolated OE was post-fixed in 1% PLP under vacuum for 1.5hrs. Following post-fixation, the OE was briefly rinsed three times in 1X PBS to wash away residual fixative and then incubated in decalcification buffer (0.35M EDTA [Sigma-Aldrich, Cat. ED2SS] and 0.35M NaOH) overnight at 4°C. To cryopreserve the tissue, OE was equilibrated overnight in 30% sucrose (w/v in 1X PBS) at 4°C, embedded and frozen in OCT (Sakura, Cat. 4583), and then stored at −80°C until cryosectioning. A Leica cryostat was used to collect 10 μm coronal sections on positively-charged glass slides.
Immunofluorescent labeling
Specific antibodies and conditions to detect the various antigens can be found in Table S1. Of note, the Rac1 antibody utilized has been validated by knockdown experiments.78,79 All primary antibodies were diluted in Normal Donkey Block (NDB) (10% normal donkey serum v/v, 5% non-fat powdered milk w/v, 4% bovine serum albumin [BSA] w/v, and 0.1% Triton X-100 v/v in 1X PBS). Species-specific fluorophore- and biotin-conjugated donkey secondary antibodies (Jackson ImmunoResearch) were diluted in NDB at 1:150 and 1:100, respectively. Streptavidin-conjugated horseradish peroxidase (SA-HRP) was diluted in TNB (0.5% blocking reagent w/v [Akoya Biosciences, Cat. FP1020], 0.15M NaCl, and 0.1M Tris-HCl pH 7.5) at 1:400. All washes performed on day 1 and day 2 were in 1X PBS and 3X PBS, respectively, with those following antibody incubation performed 3x5min. Slides were dried on a slide warmer at ∼37°C for 45-60min. after application of rubber cement (Elmer’s) to isolate each section. Following a 10-15min. wash to remove OCT, sections used to detect antigens requiring antigen retrieval were incubated in antigen retrieval buffer (0.1M sodium citrate, pH 6) and placed in a pre-heated commercial steamer for 10min. After slides were washed for 10min., sections were permeabilized with 90% MeOH for 5min. Sections designated to detect antigens requiring tyramide signal amplication (TSA) were incubated for 5min. in 3% H2O2 diluted in MeOH in lieu of 90% MeOH. Slides were then washed 3x5min., and blocked for 1 hr. at room temperature with NDB. Following overnight primary antibody incubation at 4°C, sections were incubated in secondary antibodies for 1 hr. at room temperature in the dark. If antigen detection required TSA, appropriate sections were blocked for 30min. in TNB prior to incubation in SA-HRP for 1 hr. at room temperature in the dark. TSA was achieved by incubating appropriate sections for 15min. in a solution consisting of 98% TSA Buffer (0.1M boric acid pH 8.5, 0.003% H2O2), 1% of 10% BSA w/v in 1X PBS, and 1% FITC-tyramide. Nuclei were counterstained with DAPI. Sections were coverslipped in NPG (0.5% N-propyl gallate in 90% glycerol) mounting media, sealed with nail polish, and slides were stored at 4°C in the dark until imaging.
Primary HBC culture
Isolation, maintenance, and passaging of primary HBCs have been previously described in detail.11 PMA (Sellekchem, Cat. S7791) and EHT1864 (SellekChem, Cat. S7482) were dissolved in DMSO. For all experiments, HBCs were treated with 50 nM PMA and 50 μM EHT1864. DMSO concentration for all conditions was identical at all times. EHT1864 attenuates Rac1 activation in a dose-dependent manner and it was used at 50 μM as this concentration maximally inhibits Rac1.33 Passage 10 HBCs were used for all experiments and treated with the appropriate compounds for 12 hrs. prior to immediate lysis for western blot or fixation for immunocytochemistry. If determined to undergo Rac1 inhibition, HBCs were pre-incubated in 50 μM EHT1864 for 1 hr. HBCs cultured for western blot analyses were grown on 6-well plates coated with poly-D-lysine/laminin (Corning, Cat. 354595). HBCs cultured for immunocytochemistry were grown on 8-well culture slides coated with poly-D-lysine/laminin (Corning, Cat. 354688).
Western blot of primary HBCs
Primary HBCs were lysed by applying 200μL Lysis Buffer (RIPA buffer [Thermo Scientific, Cat. 89901] supplemented with a protease and phosphatase inhibitor cocktail [Thermo Scientific, Cat. 78442]) per well and freezing the 6-well plate at -80°C overnight. All subsequent steps were performed on ice. Immediately after thawing, an additional 100μL Lysis Buffer was applied to each well and mechanical cell lysis was performed with a cell scraper. The entire volume of lysate from each well was transferred to a pre-chilled 2mL microcentrifuge tube, and debris pelleted via centrifugation at maximum rpm for 10min. at 4°C. Supernatant from each experimental condition was transferred to a new pre-chilled 2mL microcentrifuge tube and total protein abundance was determined by BCA protein assay per manufacturer’s protocol (Thermo Scientific, Cat. 23227). Working protein stocks were prepared by diluting supernatant with 4X LDS Sample Buffer (Invitrogen, Cat. B0007), 10X Sample Reducing Agent (Invitrogen, Cat. B0009), and Lysis Buffer. These stocks were placed in a heating block set at 70°C for 10min and then stored at -80°C.
Protein from working stocks were loaded at ∼5μg in 4-12% Bis-Tris gels (Invitrogen, Cat. NW04120BOX) and bracketed by a protein ladder standard (Proteintech, Cat. PL00002). Gels were run per manufacturer’s protocol (Invitrogen, Cat. A25977) in MES SDS running buffer (Invitrogen, Cat. B0002). Protein was transferred to a 0.2 μm PVDF membrane (Invitrogen, Cat. LC2002) at 20V per manufacturer’s protocol (Invitrogen, Cat. BT00061).
Following protein transfer, membranes were rinsed in 1X TBST (Tris-buffered saline with 1% Tween 20) and blocked for 1 hr. at room temperature in Blocking Buffer (5% BSA w/v in 1X TBST). Primary antibodies against ΔNp63 (BioLegend, Cat. 619002; 1:500), cJun (Cell Signaling Technology, Cat. 9165; 1:1000), pcJun (Cell Signaling Technology, Cat. 3270; 1:1000), JNK (Cell Signaling Technology, Cat. 9252; 1:1000), pJNK (Cell Signaling Technology, Cat. 4668; 1:1000), Rac1 (Proteintech, Cat. 66122-1-Ig; 1:2000), and GAPDH (Cell Signaling Technology, Cat. 97166; 1:1000) were diluted in Blocking Buffer and incubated with membranes overnight at 4°C. The following day, membranes were washed 6x7min. in 1X TBST, incubated in the appropriate HRP-conjugated donkey secondary antibodies (Jackson ImmunoResearch) diluted 1:5000 in Blocking Buffer, and washed 6x5min in 1X TBST. Proteins were visualized in a gel dock system (Bio-Rad, Universal Hood II) after membranes were incubated in SuperSignal West Pico Plus Chemiluminescent substrate (Thermo Scientific, Cat. 34577). Bio-Rad Image Lab software was used to export TIFF files that were subsequently imported into ImageJ to perform densitometric analyses. Protein abundance was calculated by normalizing the densitometric value of the protein band to that of its paired GAPDH band. Given similar molecular weights between cJun, pcJun, JNK, pJNK, and GAPDH, membranes used to first detect either cJun, pcJun, JNK, or pJNK were dried overnight to deactivate HRP conjugated to donkey anti-rabbit antibodies. Following their reactivation in 100% methanol, membranes were then washed in 1X TBST and processed as described above following overnight primary antibody incubation to visualize GAPDH via HRP conjugated to donkey anti-mouse antibodies.
Immunocytochemistry
Primary HBCs were fixed with 10% buffered formalin (Fisher, Cat. SF100) for 15min. at room temperature, washed 3x5min in 1X PBS, then stored at 4°C in fresh 1X PBS until further processing. To note, all washes prior to primary antibody incubation and during day 2 were in 1X PBS and 3X PBS, respectively, with those following antibody incubation performed 3x5min. HBCs were subsequently permeabilized on ice with ice-cold 100% MeOH for 10min. and washed 3x5min. After 1 hr. of blocking in NDB at room temperature, HBCs were incubated overnight at 4°C in antibodies against Hopx (1:150; Proteintech, Cat. 11419-1-AP), Ki67 (1:75; BD Biosciences, Cat. 556003), and CK5 (1:400; BioLegend, Clone Poly9059) in NDB diluent. The next day, HBCs were incubated in the dark with species-specific fluorophore-conjugated donkey secondary antibodies (Jackson ImmunoResearch) at 1:250 in NDB diluent. Nuclei were counterstained with DAPI. Chamber walls of culture slides were removed per manufacturer’s protocol. Slides were coverslipped with NPG mounting media, sealed with nail polish, and stored at 4°C in the dark until imaging.
Imaging
All sections and cells were imaged on a Zeiss LSM800 confocal microscope. Zeiss Zen 2.6 software was used to set imaging parameters to eliminate spectral overlap between channels, which included capturing fluorescence in multi-track mode, adjusting the variable secondary dichroics, and enabling appropriate emission filters. For proteins quantified by relative fluorescence intensity, all excitation and emission capture settings were identical between control and experimental conditions. Imaging OE included capturing multiple z-planes stepped at 2 μm intervals to sample from a minimum of ∼5 cellular cross-sections. First and last z-planes were established when the maximum fluorescence intensity of the channel to be quantified fell within lowest quartile of a 16bit display range. Because primary HBCs can pile on top of each another in culture, imaging HBC islands in vitro involved capturing the z-plane in which CK5+ HBCs appeared most confluent.
Image analyses
ImageJ was used to process all uncompressed, 16bit tiff files obtained from confocal imaging. Z-planes were sum projected to maximize bit depth of captured fluorescence. The ImageJ Rolling ball background subtraction function was applied to every channel. CellProfiler image analysis software80 was used to quantify total fluorescence intensity. Images of channels in which captured fluorescence was intended to facilitate cellular domain demarcation rather than for intensity quantification (namely Laminin, Ezrin, and CK5) were converted to 16bit so that CellProfiler could process those images, identify relevant cellular landmarks, and quantify the area that those landmarks occupied if applicable. The basal domain of HBCs was determined within CellProfiler and defined as Laminin+ objects that overlapped with CK5+ objects. The apical domain of HBCs was similarly determined within CellProfiler and defined as Ezrin+ objects that overlapped with CK5+ objects without encroaching on areas occupied by Laminin+ objects. The differing appearance of CK5+/ Ezrin+ objects between Figures 1 and 2 are a result of mutually exclusive tissue processing requirements of Itgb1, Itgb4, Rac1, pFAKY925, and pcJun prior to primary antibody incubation. Specifically, antigen retrieval was necessary to immunolabel Rac1, pFAKY925, and pcJun, and enabled Ezrin detection via donkey anti-mouse IgG conjugated fluorophores. However, antigen retrieval had to be avoided in order to retain Itgb1 and Itgb4 antigenicity, which also then necessitated Ezrin detection via streptavidin-conjugated fluorophores. Moreover, antigen retrieval significantly compromised Laminin antigenicity, and as such, limited the ability to objectively demarcate HBC basal domains. Consequently, some CK5+/ Ezrin+ objects in Figure 2 appear sub-apical as images were sum projected for reasons stated above. For experiments quantifying relative fluorescence, the average fluorescence density of the control condition was used as the denominator to calculate normalized fluorescence densities. For analyses at 28 dpi, images acquired from the tdTom channel were used to morphologically identify and measure the length of the basal lamina using the freehand tool within ImageJ. To negate the potential bias associated with manual counting, CellProfiler was utilized to identify individual OSN objects via segmentation by Otsu thresholding of PGP9.5 fluorescence that co-localized with DAPI+ nuclei (channel not shown for illustrative clarity). These PGP9.5+ objects were counted as tdTom+ if at least 25% of their area co-localized with tdTom+/CK8- fluorescence as determined by CellProfiler.
Data presentation
Only brightness and contrast were adjusted for each representative image within ImageJ or Affinity Designer, and these parameters were applied globally to the image. Graphs were generated using GraphPad Prism Version 10. All figures were constructed in Affinity Designer.
Quantification and statistical analysis
All statistical analyses were performed with GraphPad Prism Version 10. For single mean comparisons, data sets demonstrating normal and non-normal distributions were compared with unpaired t-test and Mann-Whitney test, respectively. For multiple mean comparisons, data sets demonstrating normal and non-normal distributions were compared with one-way ANOVA with post-hoc Tukey’s multiple comparisons test and Kruskal-Wallis test with post-hoc Dunn’s multiple comparisons test, respectively. A p-value < 0.05 was considered statistically significant (∗<0.05, ∗∗p<0.01, ∗∗∗p<0.001, ∗∗∗∗p<0.0001). Error bars indicate mean ± SEM.
Acknowledgments
We would like to express immense gratitude to Po Kwok-Tse for her tremendous technical assistance. This work was supported by the NIH, USA: F30 DC018450 (J.D.L.) and R01 DC017869 (J.E.S.).
Author contributions
Conceptualization, J.D.L., C.M.B.-C., B.H.B., and J.E.S.; methodology, J.D.L.; formal analysis, J.D.L.; investigation, J.D.L., C.M.B-C., B.H.B., and C.A.H.; resources, J.E.S. and J.D.L.; writing – original draft, J.D.L.; writing – review and editing, J.D.L., C.M.B.-C., B.H.B., and J.E.S.; visualization, J.D.L.; funding acquisition, J.E.S. and J.D.L.
Declaration of interests
J.E.S. is a co-founder of Rhino Therapeutics, Inc.
Published: March 27, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109600.
Supplemental information
References
- 1.Leung C.T., Coulombe P.A., Reed R.R. Contribution of olfactory neural stem cells to tissue maintenance and regeneration. Nat. Neurosci. 2007;10:720–726. doi: 10.1038/nn1882. [DOI] [PubMed] [Google Scholar]
- 2.Holbrook E.H., Szumowski K.E., Schwob J.E. An immunochemical, ultrastructural, and developmental characterization of the horizontal basal cells of rat olfactory epithelium. J. Comp. Neurol. 1995;363:129–146. doi: 10.1002/cne.903630111. [DOI] [PubMed] [Google Scholar]
- 3.Herrick D.B., Lin B., Peterson J., Schnittke N., Schwob J.E. Notch1 maintains dormancy of olfactory horizontal basal cells, a reserve neural stem cell. Proc. Natl. Acad. Sci. USA. 2017;114:E5589–E5598. doi: 10.1073/pnas.1701333114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Håglin S., Berghard A., Bohm S. Increased Retinoic Acid Catabolism in Olfactory Sensory Neurons Activates Dormant Tissue-Specific Stem Cells and Accelerates Age-Related Metaplasia. J. Neurosci. 2020;40:4116–4129. doi: 10.1523/JNEUROSCI.2468-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen M., Reed R.R., Lane A.P. Acute inflammation regulates neuroregeneration through the NF-kappaB pathway in olfactory epithelium. Proc. Natl. Acad. Sci. USA. 2017;114:8089–8094. doi: 10.1073/pnas.1620664114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joiner A.M., Green W.W., McIntyre J.C., Allen B.L., Schwob J.E., Martens J.R. Primary Cilia on Horizontal Basal Cells Regulate Regeneration of the Olfactory Epithelium. J. Neurosci. 2015;35:13761–13772. doi: 10.1523/JNEUROSCI.1708-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fletcher R.B., Prasol M.S., Estrada J., Baudhuin A., Vranizan K., Choi Y.G., Ngai J. p63 regulates olfactory stem cell self-renewal and differentiation. Neuron. 2011;72:748–759. doi: 10.1016/j.neuron.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schnittke N., Herrick D.B., Lin B., Peterson J., Coleman J.H., Packard A.I., Jang W., Schwob J.E. Transcription factor p63 controls the reserve status but not the stemness of horizontal basal cells in the olfactory epithelium. Proc. Natl. Acad. Sci. USA. 2015;112:E5068–E5077. doi: 10.1073/pnas.1512272112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fletcher R.B., Das D., Gadye L., Street K.N., Baudhuin A., Wagner A., Cole M.B., Flores Q., Choi Y.G., Yosef N., et al. Deconstructing Olfactory Stem Cell Trajectories at Single-Cell Resolution. Cell Stem Cell. 2017;20:817–830.e8. doi: 10.1016/j.stem.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gadye L., Das D., Sanchez M.A., Street K., Baudhuin A., Wagner A., Cole M.B., Choi Y.G., Yosef N., Purdom E., et al. Injury Activates Transient Olfactory Stem Cell States with Diverse Lineage Capacities. Cell Stem Cell. 2017;21:775–790.e9. doi: 10.1016/j.stem.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peterson J., Lin B., Barrios-Camacho C.M., Herrick D.B., Holbrook E.H., Jang W., Coleman J.H., Schwob J.E. Activating a Reserve Neural Stem Cell Population In Vitro Enables Engraftment and Multipotency after Transplantation. Stem Cell Rep. 2019;12:680–695. doi: 10.1016/j.stemcr.2019.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barrios-Camacho C.M., Zunitch M.J., Louie J.D., Jang W., Schwob J.E. An in vitro model of acute horizontal basal cell activation reveals dynamic gene regulatory networks underlying the acute activation phase. bioRxiv. 2023;2 doi: 10.1101/2023.12.14.568855. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Genter M.B., Deamer N.J., Blake B.L., Wesley D.S., Levi P.E. Olfactory toxicity of methimazole: dose-response and structure-activity studies and characterization of flavin-containing monooxygenase activity in the Long-Evans rat olfactory mucosa. Toxicol. Pathol. 1995;23:477–486. doi: 10.1177/019262339502300404. [DOI] [PubMed] [Google Scholar]
- 14.Carroll D.K., Carroll J.S., Leong C.O., Cheng F., Brown M., Mills A.A., Brugge J.S., Ellisen L.W. p63 regulates an adhesion programme and cell survival in epithelial cells. Nat. Cell Biol. 2006;8:551–561. doi: 10.1038/ncb1420. [DOI] [PubMed] [Google Scholar]
- 15.Casaletto J.B., Saotome I., Curto M., McClatchey A.I. Ezrin-mediated apical integrity is required for intestinal homeostasis. Proc. Natl. Acad. Sci. USA. 2011;108:11924–11929. doi: 10.1073/pnas.1103418108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berrier A.L., Martinez R., Bokoch G.M., LaFlamme S.E. The integrin beta tail is required and sufficient to regulate adhesion signaling to Rac1. J. Cell Sci. 2002;115:4285–4291. doi: 10.1242/jcs.00109. [DOI] [PubMed] [Google Scholar]
- 17.Cruz-Monserrate Z., O'Connor K.L. Integrin alpha 6 beta 4 promotes migration, invasion through Tiam1 upregulation, and subsequent Rac activation. Neoplasia. 2008;10:408–417. doi: 10.1593/neo.07868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russell A.J., Fincher E.F., Millman L., Smith R., Vela V., Waterman E.A., Dey C.N., Guide S., Weaver V.M., Marinkovich M.P. Alpha 6 beta 4 integrin regulates keratinocyte chemotaxis through differential GTPase activation and antagonism of alpha 3 beta 1 integrin. J. Cell Sci. 2003;116:3543–3556. doi: 10.1242/jcs.00663. [DOI] [PubMed] [Google Scholar]
- 19.Didsbury J., Weber R.F., Bokoch G.M., Evans T., Snyderman R. rac, a novel ras-related family of proteins that are botulinum toxin substrates. J. Biol. Chem. 1989;264:16378–16382. [PubMed] [Google Scholar]
- 20.ten Klooster J.P., Hordijk P.L. Targeting and localized signalling by small GTPases. Biol. Cell. 2007;99:1–12. doi: 10.1042/BC20060071. [DOI] [PubMed] [Google Scholar]
- 21.Benitah S.A., Frye M., Glogauer M., Watt F.M. Stem cell depletion through epidermal deletion of Rac1. Science. 2005;309:933–935. doi: 10.1126/science.1113579. [DOI] [PubMed] [Google Scholar]
- 22.Castilho R.M., Squarize C.H., Leelahavanichkul K., Zheng Y., Bugge T., Gutkind J.S. Rac1 is required for epithelial stem cell function during dermal and oral mucosal wound healing but not for tissue homeostasis in mice. PLoS One. 2010;5 doi: 10.1371/journal.pone.0010503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Myant K.B., Scopelliti A., Haque S., Vidal M., Sansom O.J., Cordero J.B. Rac1 drives intestinal stem cell proliferation and regeneration. Cell Cycle. 2013;12:2973–2977. doi: 10.4161/cc.26031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olabi S., Ucar A., Brennan K., Streuli C.H. Integrin-Rac signalling for mammary epithelial stem cell self-renewal. Breast Cancer Res. 2018;20:128. doi: 10.1186/s13058-018-1048-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deramaudt T.B., Dujardin D., Hamadi A., Noulet F., Kolli K., De Mey J., Takeda K., Rondé P. FAK phosphorylation at Tyr-925 regulates cross-talk between focal adhesion turnover and cell protrusion. Mol. Biol. Cell. 2011;22:964–975. doi: 10.1091/mbc.E10-08-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bohmann D., Bos T.J., Admon A., Nishimura T., Vogt P.K., Tjian R. Human proto-oncogene c-jun encodes a DNA binding protein with structural and functional properties of transcription factor AP-1. Science. 1987;238:1386–1392. doi: 10.1126/science.2825349. [DOI] [PubMed] [Google Scholar]
- 27.Rauscher F.J., 3rd, Voulalas P.J., Franza B.R., Jr., Curran T. Fos and Jun bind cooperatively to the AP-1 site: reconstitution in vitro. Genes Dev. 1988;2:1687–1699. doi: 10.1101/gad.2.12b.1687. [DOI] [PubMed] [Google Scholar]
- 28.Shaulian E., Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- 29.Han S.I., Oh S.Y., Woo S.H., Kim K.H., Kim J.H., Kim H.D., Kang H.S. Implication of a small GTPase Rac1 in the activation of c-Jun N-terminal kinase and heat shock factor in response to heat shock. J. Biol. Chem. 2001;276:1889–1895. doi: 10.1074/jbc.M006042200. [DOI] [PubMed] [Google Scholar]
- 30.Louie J.D., Bromberg B.H., Zunitch M.J., Schwob J.E. Horizontal basal cells self-govern their neurogenic potential during injury-induced regeneration of the olfactory epithelium. Development. 2023;150 doi: 10.1242/dev.201552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seo H.H., Lee C.Y., Lee J., Lim S., Choi E., Park J.C., Lee S., Hwang K.C. The role of nuclear factor of activated T cells during phorbol myristate acetate-induced cardiac differentiation of mesenchymal stem cells. Stem Cell Res. Ther. 2016;7:90. doi: 10.1186/s13287-016-0348-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song J.K., Lee C.H., Hwang S.M., Joo B.S., Lee S.Y., Jung J.S. Effect of phorbol 12-myristate 13-acetate on the differentiation of adipose-derived stromal cells from different subcutaneous adipose tissue depots. Korean J. Physiol. Pharmacol. 2014;18:289–296. doi: 10.4196/kjpp.2014.18.4.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shutes A., Onesto C., Picard V., Leblond B., Schweighoffer F., Der C.J. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J. Biol. Chem. 2007;282:35666–35678. doi: 10.1074/jbc.M703571200. [DOI] [PubMed] [Google Scholar]
- 34.Teramoto H., Coso O.A., Miyata H., Igishi T., Miki T., Gutkind J.S. Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway. A role for mixed lineage kinase 3/protein-tyrosine kinase 1, a novel member of the mixed lineage kinase family. J. Biol. Chem. 1996;271:27225–27228. doi: 10.1074/jbc.271.44.27225. [DOI] [PubMed] [Google Scholar]
- 35.Smeal T., Binetruy B., Mercola D., Grover-Bardwick A., Heidecker G., Rapp U.R., Karin M. Oncoprotein-mediated signalling cascade stimulates c-Jun activity by phosphorylation of serines 63 and 73. Mol. Cell Biol. 1992;12:3507–3513. doi: 10.1128/mcb.12.8.3507-3513.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mohri T., Adachi Y., Ikehara S., Hioki K., Tokunaga R., Taketani S. Activated Rac1 selectively up-regulates the expression of integrin alpha6beta4 and induces cell adhesion and membrane ruffles of nonadherent colon cancer Colo201 cells. Exp. Cell Res. 1999;253:533–540. doi: 10.1006/excr.1999.4720. [DOI] [PubMed] [Google Scholar]
- 37.Kiosses W.B., Shattil S.J., Pampori N., Schwartz M.A. Rac recruits high-affinity integrin alphavbeta3 to lamellipodia in endothelial cell migration. Nat. Cell Biol. 2001;3:316–320. doi: 10.1038/35060120. [DOI] [PubMed] [Google Scholar]
- 38.Maurya D.K., Henriques T., Marini M., Pedemonte N., Galietta L.J.V., Rock J.R., Harfe B.D., Menini A. Development of the Olfactory Epithelium and Nasal Glands in TMEM16A-/- and TMEM16A+/+ Mice. PLoS One. 2015;10 doi: 10.1371/journal.pone.0129171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zunitch M.J., Fisch A.S., Lin B., Barrios-Camacho C.M., Faquin W.C., Tachie-Baffour Y., Louie J.D., Jang W., Curry W.T., Gray S.T., et al. Molecular Evidence for Olfactory Neuroblastoma as a Tumor of Malignant Globose Basal Cells. Mod. Pathol. 2023;36 doi: 10.1016/j.modpat.2023.100122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishijima H., Zunitch M.J., Yoshida M., Kondo K., Yamasoba T., Schwob J.E., Holbrook E.H. Rapid fluorescent vital imaging of olfactory epithelium. iScience. 2022;25 doi: 10.1016/j.isci.2022.104222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gavid M., Coulomb L., Thomas J., Aouimeur I., Verhoeven P., Mentek M., Dumollard J.M., Forest F., Prades J.M., Thuret G., et al. Technique of flat-mount immunostaining for mapping the olfactory epithelium and counting the olfactory sensory neurons. PLoS One. 2023;18 doi: 10.1371/journal.pone.0280497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holbrook E.H., Wu E., Curry W.T., Lin D.T., Schwob J.E. Immunohistochemical characterization of human olfactory tissue. Laryngoscope. 2011;121:1687–1701. doi: 10.1002/lary.21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doty R.L., Shaman P., Applebaum S.L., Giberson R., Siksorski L., Rosenberg L. Smell identification ability: changes with age. Science. 1984;226:1441–1443. doi: 10.1126/science.6505700. [DOI] [PubMed] [Google Scholar]
- 44.Doty R.L. Clinical studies of olfaction. Chem. Senses. 2005;30(Suppl 1):i207–i209. doi: 10.1093/chemse/bjh187. [DOI] [PubMed] [Google Scholar]
- 45.Fitzek M., Patel P.K., Solomon P.D., Lin B., Hummel T., Schwob J.E., Holbrook E.H. Integrated age-related immunohistological changes occur in human olfactory epithelium and olfactory bulb. J. Comp. Neurol. 2022;530:2154–2175. doi: 10.1002/cne.25325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Child K.M., Herrick D.B., Schwob J.E., Holbrook E.H., Jang W. The Neuroregenerative Capacity of Olfactory Stem Cells Is Not Limitless: Implications for Aging. J. Neurosci. 2018;38:6806–6824. doi: 10.1523/JNEUROSCI.3261-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwob J.E., Jang W., Holbrook E.H., Lin B., Herrick D.B., Peterson J.N., Hewitt Coleman J. Stem and progenitor cells of the mammalian olfactory epithelium: Taking poietic license. J. Comp. Neurol. 2017;525:1034–1054. doi: 10.1002/cne.24105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hildebrand M.S., Dahl H.H.M., Hardman J., Coleman B., Shepherd R.K., de Silva M.G. Survival of partially differentiated mouse embryonic stem cells in the scala media of the guinea pig cochlea. J. Assoc. Res. Otolaryngol. 2005;6:341–354. doi: 10.1007/s10162-005-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hampsch R.A., Shee K., Bates D., Lewis L.D., Désiré L., Leblond B., Demidenko E., Stefan K., Huang Y.H., Miller T.W. Therapeutic sensitivity to Rac GTPase inhibition requires consequential suppression of mTORC1, AKT, and MEK signaling in breast cancer. Oncotarget. 2017;8:21806–21817. doi: 10.18632/oncotarget.15586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Désiré L., Bourdin J., Loiseau N., Peillon H., Picard V., De Oliveira C., Bachelot F., Leblond B., Taverne T., Beausoleil E., et al. RAC1 inhibition targets amyloid precursor protein processing by gamma-secretase and decreases Abeta production in vitro and in vivo. J. Biol. Chem. 2005;280:37516–37525. doi: 10.1074/jbc.M507913200. [DOI] [PubMed] [Google Scholar]
- 51.Ramos D.F.V., Mancuso R.I., Contieri B., Duarte A., Paiva L., de Melo Carrilho J., Saad S.T.O., Lazarini M. Rac GTPases in acute myeloid leukemia cells: Expression profile and biological effects of pharmacological inhibition. Toxicol. Appl. Pharmacol. 2022;442 doi: 10.1016/j.taap.2022.115990. [DOI] [PubMed] [Google Scholar]
- 52.Wang R., Yu Q., Wang X., Li B., Ciotkowska A., Rutz B., Wang Y., Stief C.G., Hennenberg M. Rac1 silencing, NSC23766 and EHT1864 reduce growth and actin organization of bladder smooth muscle cells. Life Sci. 2020;261 doi: 10.1016/j.lfs.2020.118468. [DOI] [PubMed] [Google Scholar]
- 53.Dütting S., Heidenreich J., Cherpokova D., Amin E., Zhang S.C., Ahmadian M.R., Brakebusch C., Nieswandt B. Critical off-target effects of the widely used Rac1 inhibitors NSC23766 and EHT1864 in mouse platelets. J. Thromb. Haemost. 2015;13:827–838. doi: 10.1111/jth.12861. [DOI] [PubMed] [Google Scholar]
- 54.Corbetta S., Gualdoni S., Ciceri G., Monari M., Zuccaro E., Tybulewicz V.L.J., de Curtis I. Essential role of Rac1 and Rac3 GTPases in neuronal development. FASEB J. 2009;23:1347–1357. doi: 10.1096/fj.08-121574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haditsch U., Anderson M.P., Freewoman J., Cord B., Babu H., Brakebusch C., Palmer T.D. Neuronal Rac1 is required for learning-evoked neurogenesis. J. Neurosci. 2013;33:12229–12241. doi: 10.1523/JNEUROSCI.2939-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vadodaria K.C., Brakebusch C., Suter U., Jessberger S. Stage-specific functions of the small Rho GTPases Cdc42 and Rac1 for adult hippocampal neurogenesis. J. Neurosci. 2013;33:1179–1189. doi: 10.1523/JNEUROSCI.2103-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liang X., Draghi N.A., Resh M.D. Signaling from integrins to Fyn to Rho family GTPases regulates morphologic differentiation of oligodendrocytes. J. Neurosci. 2004;24:7140–7149. doi: 10.1523/JNEUROSCI.5319-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Breipohl W., Laugwitz H.J., Bornfeld N. Topological relations between the dendrites of olfactory sensory cells and sustentacular cells in different vertebrates. An ultrastructural study. J. Anat. 1974;117:89–94. [PMC free article] [PubMed] [Google Scholar]
- 59.Dahl A.R., Hadley W.M., Hahn F.F., Benson J.M., McClellan R.O. Cytochrome P-450-dependent monooxygenases in olfactory epithelium of dogs: possible role in tumorigenicity. Science. 1982;216:57–59. doi: 10.1126/science.7063870. [DOI] [PubMed] [Google Scholar]
- 60.Rafols J.A., Getchell T.V. Morphological relations between the receptor neurons, sustentacular cells and Schwann cells in the olfactory mucosa of the salamander. Anat. Rec. 1983;206:87–101. doi: 10.1002/ar.1092060111. [DOI] [PubMed] [Google Scholar]
- 61.Suzuki Y., Takeda M., Farbman A.I. Supporting cells as phagocytes in the olfactory epithelium after bulbectomy. J. Comp. Neurol. 1996;376:509–517. doi: 10.1002/(SICI)1096-9861(19961223)376:4<509::AID-CNE1>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 62.Mertens A.E.E., Rygiel T.P., Olivo C., van der Kammen R., Collard J.G. The Rac activator Tiam1 controls tight junction biogenesis in keratinocytes through binding to and activation of the Par polarity complex. J. Cell Biol. 2005;170:1029–1037. doi: 10.1083/jcb.200502129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guillemot L., Guerrera D., Spadaro D., Tapia R., Jond L., Citi S. MgcRacGAP interacts with cingulin and paracingulin to regulate Rac1 activation and development of the tight junction barrier during epithelial junction assembly. Mol. Biol. Cell. 2014;25:1995–2005. doi: 10.1091/mbc.E13-11-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Castagna M., Takai Y., Kaibuchi K., Sano K., Kikkawa U., Nishizuka Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J. Biol. Chem. 1982;257:7847–7851. [PubMed] [Google Scholar]
- 65.Adler V., Franklin C.C., Kraft A.S. Phorbol esters stimulate the phosphorylation of c-Jun but not v-Jun: regulation by the N-terminal delta domain. Proc. Natl. Acad. Sci. USA. 1992;89:5341–5345. doi: 10.1073/pnas.89.12.5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hellweg C.E., Arenz A., Bogner S., Schmitz C., Baumstark-Khan C. Activation of nuclear factor kappa B by different agents: influence of culture conditions in a cell-based assay. Ann. N. Y. Acad. Sci. 2006;1091:191–204. doi: 10.1196/annals.1378.066. [DOI] [PubMed] [Google Scholar]
- 67.Taddei I., Deugnier M.A., Faraldo M.M., Petit V., Bouvard D., Medina D., Fässler R., Thiery J.P., Glukhova M.A. Beta1 integrin deletion from the basal compartment of the mammary epithelium affects stem cells. Nat. Cell Biol. 2008;10:716–722. doi: 10.1038/ncb1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.To W.S., Midwood K.S. Plasma and cellular fibronectin: distinct and independent functions during tissue repair. Fibrogenesis Tissue Repair. 2011;4:21. doi: 10.1186/1755-1536-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rayagiri S.S., Ranaldi D., Raven A., Mohamad Azhar N.I.F., Lefebvre O., Zammit P.S., Borycki A.G. Basal lamina remodeling at the skeletal muscle stem cell niche mediates stem cell self-renewal. Nat. Commun. 2018;9:1075. doi: 10.1038/s41467-018-03425-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakamura-Ishizu A., Okuno Y., Omatsu Y., Okabe K., Morimoto J., Uede T., Nagasawa T., Suda T., Kubota Y. Extracellular matrix protein tenascin-C is required in the bone marrow microenvironment primed for hematopoietic regeneration. Blood. 2012;119:5429–5437. doi: 10.1182/blood-2011-11-393645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Urciuolo A., Quarta M., Morbidoni V., Gattazzo F., Molon S., Grumati P., Montemurro F., Tedesco F.S., Blaauw B., Cossu G., et al. Collagen VI regulates satellite cell self-renewal and muscle regeneration. Nat. Commun. 2013;4:1964. doi: 10.1038/ncomms2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sirko S., von Holst A., Wizenmann A., Götz M., Faissner A. Chondroitin sulfate glycosaminoglycans control proliferation, radial glia cell differentiation and neurogenesis in neural stem/progenitor cells. Development. 2007;134:2727–2738. doi: 10.1242/dev.02871. [DOI] [PubMed] [Google Scholar]
- 73.Brizzi M.F., Tarone G., Defilippi P. Extracellular matrix, integrins, and growth factors as tailors of the stem cell niche. Curr. Opin. Cell Biol. 2012;24:645–651. doi: 10.1016/j.ceb.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 74.Shen Q., Wang Y., Kokovay E., Lin G., Chuang S.M., Goderie S.K., Roysam B., Temple S. Adult SVZ stem cells lie in a vascular niche: a quantitative analysis of niche cell-cell interactions. Cell Stem Cell. 2008;3:289–300. doi: 10.1016/j.stem.2008.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lawson C.D., Burridge K. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases. 2014;5 doi: 10.4161/sgtp.27958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yu W., Datta A., Leroy P., O'Brien L.E., Mak G., Jou T.S., Matlin K.S., Mostov K.E., Zegers M.M.P. Beta1-integrin orients epithelial polarity via Rac1 and laminin. Mol. Biol. Cell. 2005;16:433–445. doi: 10.1091/mbc.e04-05-0435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Indra A.K., Warot X., Brocard J., Bornert J.M., Xiao J.H., Chambon P., Metzger D. Temporally-controlled site-specific mutagenesis in the basal layer of the epidermis: comparison of the recombinase activity of the tamoxifen-inducible Cre-ER(T) and Cre-ER(T2) recombinases. Nucleic Acids Res. 1999;27:4324–4327. doi: 10.1093/nar/27.22.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xiong D., Hu W., Han X., Cai Y. Rhein Inhibited Ferroptosis and EMT to Attenuate Diabetic Nephropathy by Regulating the Rac1/NOX1/beta-Catenin Axis. Front. Biosci. 2023;28:100. doi: 10.31083/j.fbl2805100. [DOI] [PubMed] [Google Scholar]
- 79.Su Y., Huang H., Luo T., Zheng Y., Fan J., Ren H., Tang M., Niu Z., Wang C., Wang Y., et al. Cell-in-cell structure mediates in-cell killing suppressed by CD44. Cell Discov. 2022;8:35. doi: 10.1038/s41421-022-00387-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lamprecht M.R., Sabatini D.M., Carpenter A.E. CellProfiler: free, versatile software for automated biological image analysis. Biotechniques. 2007;42:71–75. doi: 10.2144/000112257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.