

Abstract
Rearranged during transfection (RET) rearrangements occur in 1% to 2% of lung adenocarcinomas as well as other malignancies and are now established targets for tyrosine kinase inhibitors. We developed three novel RET fusion–positive (RET+) patient–derived cancer cell lines, CUTO22 [kinesin 5B (KIF5B)–RET fusion], CUTO32 (KIF5B-RET fusion), and CUTO42 (echinoderm microtubule-associated protein-like 4–RET fusion), to study RET signaling and response to therapy. We confirmed each of our cell lines expresses the RET fusion protein and assessed their sensitivity to RET inhibitors. We found that the CUTO22 and CUTO42 cell lines were sensitive to multiple RET inhibitors, whereas the CUTO32 cell line was >10-fold more resistant to three RET inhibitors. We discovered that our RET+ cell lines had differential regulation of the mitogen-activated protein kinase and phosphoinositide 3-kinase/protein kinase B (AKT) pathways. After inhibition of RET, the CUTO42 cells had robust inhibition of phosphorylated AKT (pAKT), whereas CUTO22 and CUTO32 cells had sustained AKT activation. Next, we performed a drug screen, which revealed that the CUTO32 cells were sensitive (<1 nM IC50) to inhibition of two cell cycle–regulating proteins, polo-like kinase 1 and Aurora kinase A. Finally, we show that two of these cell lines, CUTO32 and CUTO42, successfully establish xenografted tumors in nude mice. We demonstrated that the RET inhibitor BLU-667 was effective at inhibiting tumor growth in CUTO42 tumors but had a much less profound effect in CUTO32 tumors, consistent with our in vitro experiments. These data highlight the utility of new RET+ models to elucidate differences in response to tyrosine kinase inhibitors and downstream signaling regulation. Our RET+ cell lines effectively recapitulate the interpatient heterogeneity observed in response to RET inhibitors and reveal opportunities for alternative or combination therapies.
SIGNIFICANCE STATEMENT
We have derived and characterized three novel rearranged during transfection (RET) fusion non-small cell lung cancer cell lines and demonstrated that they have differential responses to RET inhibition as well as regulation of downstream signaling, an area that has previously been limited by a lack of diverse cell line modes with endogenous RET fusions. These data offer important insight into regulation of response to RET tyrosine kinase inhibitors and other potential therapeutic targets.
Introduction
As understanding of the molecular landscape of non-small cell lung cancer (NSCLC) has grown, many small molecule drugs have been developed to inhibit aberrantly activated receptor tyrosine kinases. Oncogene-targeted therapies have been very successful in patients with NSCLC harboring anaplastic lymphoma kinase (ALK), BRAF, epidermal growth factor receptor (EGFR), ROS1, and neurotrophic tyrosine kinase receptor alterations (Shaw et al., 2014; Solomon et al., 2014; Peters et al., 2017; Planchard et al., 2017; Soria et al., 2018; Hong et al., 2019). Rearrangements involving the rearranged during transfection (RET) tyrosine kinase occur in approximately 1% to 2% of lung adenocarcinomas, are commonly found in papillary thyroid cancers, and more recently have been identified in breast and colorectal cancers (Le Rolle et al., 2015; Kato et al., 2017; Paratala et al., 2018). Notably, there have also been several reports of RET rearrangements arising as resistance mechanisms to EGFR and ALK inhibitors (McCoach et al., 2018b; Oxnard et al., 2018; Piotrowska et al., 2018; Schrock et al., 2018).
Typically, fusion kinases are activated by their 5′ partners by 1) providing an active promoter to enhance expression of the chimeric gene and 2) providing a dimerization domain that promotes constitutive, ligand-independent activation of the kinase. In lung cancer, kinesin 5B (KIF5B) is the most common 5′ partner linked to RET, occurring in ∼70% of RET rearrangements (Gautschi et al., 2017). Other fusion partners such as coiled coiled domain containing 6 (CCDC6), nuclear receptor coactivator 4, and tripartite motif containing 33 have also been identified (Takeuchi et al., 2012; Wang et al., 2012; Drilon et al., 2013). RET canonically activates several key pro-proliferative and prosurvival pathways, including mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K)/protein kinase B(AKT), and Janus kinase/signal transducer and activator of transcription (Mulligan, 2014). The growth factor receptor bound protein 2 adapter protein is recruited to RET Y1062 to activate son of sevenless and subsequently the MAPK pathway (Ohiwa et al., 1997). GRB2-associated-binding protein 1 also binds to the Y1062 site and activates the PI3K pathway (Hayashi et al., 2000). Mutation of this tyrosine residue has been shown to impair RET’s transforming ability (Asai et al., 1996).
Preclinical studies have shown that RET is amenable to small molecule inhibition; however, in early clinical trials, patients with NSCLC with RET rearrangements generally had poor responses to multikinase inhibitors, including cabozantinib, vandetanib, and ponatinib (Drilon et al., 2013; Falchook et al., 2016; Gautschi et al., 2017; Yoh et al., 2017). There has been great interest in developing more specific and potent RET inhibitors, several of which have had promising clinical trial results. Interestingly, one inhibitor, RXDX-105, yielded no successful responses in patients with the KIF5B-RET fusion but produced responses in ∼66% of patients with other 5′ fusion partners (Drilon et al., 2019). Two other novel RET inhibitors, BLU-667 (pralsetinib) and LOXO-292 (selpercatinib), have shown higher response rates, including in patients with KIF5B-RET rearrangements (Subbiah et al., 2018b,c; Gainor et al., 2019; Drilon et al., 2020). Selpercatinib and pralsetinib received approval from the Food and Drug Administration.
Despite overall improvements in response rates to RET inhibitors, there is still substantial variability in patient response, the mechanism for which is not well understood. Availability of RET fusion–positive (RET+) lung cancer models has been a limitation of prior studies and precluded a more detailed understanding of the mechanisms that regulate response or resistance to RET inhibitors. Here, we describe three novel, patient-derived cell lines, including two KIF5B-RET+ cell lines. In this study we sought to understand their response to RET inhibitors, regulation of downstream signaling, and potential sensitivities to other small molecule inhibitors. We used several unbiased screening approaches to address these questions and further explore RET fusion biology.
Materials and Methods
Cell Lines and Reagents.
All cells, except LC-2/Ad cells, were maintained in RPMI 1640 medium (Corning) supplemented with 10% FBS in a humidified 37°C incubator with 5% CO2. LC-2/Ad cells were maintained in 50% RPMI 1640 medium (Corning) and 50% Ham’s F-12 medium (Corning) with 10% FBS. The LC-2/Ad cell line was purchased from Sigma-Aldrich. All cell lines are tested for mycoplasma and short tandem repeat profiled every 6 months at the Molecular Biology Service Center at the Barbara Davis Center for Diabetes at the University of Colorado Anschutz Medical Campus. RXDX-105, ponatinib, trametinib, omipalisib, volasertib, and alisertib were purchased from Selleck Chemicals. BLU-667 and crizotinib were purchased from Chemietek.
The chemical names for compounds used are as follows:
RXDX-105: Urea, N-[3-[(6,7-dimethoxy-4-quinazolinyl)oxy]phenyl]-N-[5-(2,2,2-trifluoro-1,1-dimethylethyl)-3-isoxazolyl]-.
Ponatinib: 3-(2-(imidazo [1,2-b]pyridazin-3-yl)ethynyl)-4-methyl-N-(4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)benzamide.
Trametinib: N-(3-(3-cyclopropyl-5-(2-fluoro-4-iodophenylamino)-6,8-dimethyl-2,4,7-trioxo-3,4,6,7-tetrahydropyrido [4,3-day]pyrimidin-1(2H)-yl)phenyl)acetamide.
Omipalisib: 2,4-difluoro-N-(2-methoxy-5-(4-(pyridazin-4-yl)quinolin-6-yl)pyridin-3-yl)benzenesulfonamide.
Volasertib: N-((1r,4r)-4-(4-(cyclopropylmethyl)piperazin-1-yl)cyclohexyl)-4-((R)-7-ethyl-8-isopropyl-5-methyl6-oxo-5,6,7,8-tetrahydropteridin-2-ylamino)-3-methoxybenzamide.
Alisertib: Benzoic acid, 4-[[9-chloro-7-(2-fluoro-6-methoxyphenyl)-5H-pyrimido [5,4-day][2]benzazepin-2-yl]amino]-2-methoxy.
BLU-667: Cyclohexanecarboxamide, N-[(1S)-1-[6-(4-fluoro-1H-pyrazol-1-yl)-3-pyridinyl]ethyl]-1-methoxy4-[4-methyl-6-[(5-methyl-1H-pyrazol-3-yl)amino]-2-pyrimidinyl]-, cis.
Crizotinib: 3-((R)-1-(2,6-dichloro-3-fluorophenyl)ethoxy)-5-(1-(piperidin-4-yl)-1H-pyrazol-4-yl)pyridin-2-amine.
Patient-Derived Cell Lines.
Primary cell lines were derived from patient pleural fluid or needle core biopsies. For pleural fluid, the initial fluid underwent centrifugation followed by red blood cell lysis (ACK Lysis Buffer; KD Medical, Columbia, MD) to isolate nucleated cells. Adherent stromal cells were removed from the mixture by culturing the cells overnight on a tissue culture flask. The following day, the nonadherent cells were collected and subjected to a CD45 depletion step for the additional removal of lymphocytes and enrichment for tumor cells. This refined mixture of cells was then cultured in RPMI 1640 medium with 10% heat-inactivated fetal bovine serum (RPMI-10) until the establishment of the cell line. For needle core biopsy samples, the tissue was disaggregated using the “mechanical spill out method” to obtain tumor aggregates free of stromal components (Oie et al., 1996). Cell aggregates were plated onto 6 cm plates and cultured in RPMI-10. Once the tumor cells became the predominately established cell type in the culture plate, the culture was subjected to differential trypsinization to dislodge the remaining minor population of stromal cells. Cells were then maintained in RPMI-10 for the expansion of the cell line. Institution review board–approved informed consent was obtained from patients for the derivation of cancer cell lines. CUTO22 and CUTO32 cells were derived from pleural fluid; CUTO42 cells were derived from a core biopsy. All three patients were histologically confirmed to have lung adenocarcinoma. Comutations were determined from clinical sequencing panels from Foundation Medicine (CUTO22) and Guardant 360 ctDNA (CUTO32 and CUTO42).
Drug Screening.
Cells were seeded at 1000 cells per well in 384-well microtiter plates and treated after 24 hours with drug. Each library drug was tested in duplicate at 0.5 and 2.5 µM, respectively, in the presence and absence of 200 nM RXDX-105. Each treatment condition was performed in duplicate. Cell viability was measured by CellTiter-Glo Luminescent Cell Viability Assay (Promega) 72 hours after treatment.
RNA Sequencing.
Cells were treated with 100 nM RXDX-105 or vehicle (DMSO) for 24 hours, and then RNA was collected with the Qiagen RNeasy Mini Kit according to the manufacturer’s instructions. This was repeated in biologic triplicate. RNA quality was verified using a Tape Station 2200 (Agilent Technologies), and RNA concentration was measured using Qubit (Thermo Fisher Scientific). Library construction was performed using the Universal Plus mRNA Library Kit (NuGen Technologies), and sequencing was performed on the Illumina HiSEQ 4000 instrument using single-end reads (150 bp) by the University of Colorado Cancer Center Genomics and Microarray Core.
Illumina adapters were trimmed using BBDuk (sourceforge.net/projects/bbmap/) and reads <50 bp after trimming were discarded. Reads were aligned and quantified using STAR (version 2.6.0a) (Dobin et al., 2013) against the Ensembl human transcriptome [hg38.12 genome (release 95)]. Reads were normalized and differential expression was calculated using the limma R package (Ritchie et al., 2015). Gene set enrichment analysis was performed using the fgsea R package with 10,000 permutations (version 1.10.0; A. A. Sergushichev, preprint, DOI: https://doi.org/10.1101/060012) with hallmark gene sets from the Molecular Signatures Database (Liberzon et al., 2011). This RNA sequencing data have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus database and are accessible through accession number GSE168526.
Reverse Transcription Polymerase Chain Reaction.
RNA was extracted from cells using the Qiagen RNeasy Mini Kit according to the manufacturer’s instructions. cDNA was generated with the Invitrogen SuperScript IV First-Strand Synthesis System according to the manufacturer’s instructions using random hexamers. Polymerase chain reaction (PCR) amplification was performed using the following primer sequences and cycles: echinoderm microtubule-associated protein-like 4 (EML4) 5′-AAGCTCATGATGGCAGTGTG-3′ and RET 5′-CAGGCCCCATACAATTTGAT-3′, denatured at 98°C for 5 minutes; 40 cycles of 98°C for 30 seconds, 55°C for 30 seconds, and 72°C for 2 minutes. This procedure resulted in approximately 900 bp of PCR product. Sanger sequencing was performed by the Barbara Davis Center for Diabetes at the University of Colorado Anschutz Medical Campus.
Proliferation Assays.
Cells were seeded into 96-well plates at concentrations of 1000–4000 cells per well in RPMI 1640 medium (Invitrogen) with 10% FBS. After 24 hours, cells were treated with increasing concentrations of the indicated inhibitors and incubated for an additional 72 hours. The CellTiter 96 MTS assay (Promega) was then preformed according to the manufacturer’s instructions. Each assay was performed in triplicate with three biologic replicates. Control cells were treated with vehicle only (DMSO) and were used to normalize MTS data.
Immunoblotting.
Immunoblotting was performed as previously described (Davies et al., 2013). Briefly, cells were lysed in Pierce radioimmunoprecipitation assay buffer (Thermo Scientific) or T-PER (Thermo Scientific) supplemented with Halt protease and phosphatase inhibitor cocktail (Thermo Scientific). Proteins were quantified using the DC Protein Assay (Bio-Rad) according to the manufacturer’s instructions. Fifty micrograms of protein was loaded per sample. Protein Sample Loading Buffer (LI-COR) was added to lysates that were then separated on an SDS-PAGE gel. Proteins were then transferred to nitrocellulose and stained with specified primary antibodies followed by IR-Dye anti-mouse or anti-rabbit IgG (LI-COR). Membranes were imaged using the Odyssey Imager and Odyssey Image Studio Software (LI-COR). Antibodies were as follows, with dilutions indicated in brackets and product numbers indicated in parentheses: RET [1:1000] (D3D8R), pAKT S473 [1:2000] (D9E), AKT (40D4) [1:2000], phosphorylated extracellular signal-regulated kinase (ERK) 1/2 T202/Y204 [1:2000] (D13.14.4E), ERK1/2 [1:2000] (L34F12), phosphorylated MAPK/ERK kinase (MEK) 1/2 S217/221 [1:1000] (41G9), MEK1/2 [1:1000] (L38C12), phosphorylated hepatocyte growth factor receptor (MET) Y1234/1235 [1:1000] (D26), MET [1:1000] (L41G3), and c-MYC [1:1000] (D84C12) purchased from Cell Signaling Technology; phosphorylated RET (pRET) Y1062 [1:1000] (sc-20252) purchased from Santa Cruz Biotechnology; and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [1:5000] (6C5) and 4G10 platinum anti-phosphotyrosine [1:2500] (05-1050) were purchased from Millipore. GAPDH was included as a loading control in all immunoblotting experiments. Three biologic replicates of each experiment were performed.
Apoptosis Assay.
Cells were plated into 96-well plates at concentrations of 1000–4000 cells per well in RPMI 1640 medium (Corning) and incubated for 24 hours. Cells were then treated with the indicated inhibitors for an additional 24 hours. The Caspase-Glo 3/7 assay (Promega) was performed according to the manufacturer’s instructions. Each experiment was completed with three technical replicates and three biologic replicates.
In Vivo Xenografts and Animal Studies.
Female athymic nude mice were purchased from Envigo. At 5 to 6 weeks old, mice were anesthetized with 2%–4% isofluorane and injected subcutaneously with 2 × 106 cells for the CUTO32 cell line and 2.5 × 106 cells for the CUTO42 cell line per flank injection. Cells were resuspended in 50% RPMI 1640 medium and 50% Matrigel (Corning) prior to injection. Tumor size measurements were recorded via caliper twice per week, and the following formula was used to calculate tumor volume: (length × width2)/2. Once tumors reached volumes of approximately 150–200 mm3, 10 mice were randomly assigned to each treatment group. Treatment doses were selected based their use in the studies referenced below. Mice were treated with 60 mg/kg BLU-667 [in 10% DMSO, 20% 2-hydroxypropel beta-cyclodextrin (HPBCD), 10% solutol] (Subbiah et al., 2018b), 1 mg/kg trametinib (in water) (Hrustanovic et al., 2015), 1.5 mg/kg omipalisib (2.5% polyethylene glycol 400, 2.5% Tween 80) (Posch et al., 2013), 30 mg/kg alisertib (10% HPBCD, 1% sodium bicarbonate) (Mollaoglu et al., 2017), or control (10% DMSO, 20% HPBCD, 10% solutol). All treatments were administered once per day via oral gavage. Mice were treated for 24 days or until the tumors were too large or mice had other indications for sacrifice as per our Institutional Animal Care and Use Committee protocol. BLU-667, omipalisib, and alisertib were purchased from Chemitek; trametinib was from Novartis. Mouse weight data are displayed in Supplemental Fig. 7.
Statistical Analysis.
All statistical analysis was performed with GraphPad Prism software. IC50 values were calculated with a nonlinear regression analysis log (inhibitor) versus response–variable slope (four parameters) and fit using least squares regression. Three regressions were generated from three biologic replicate experiments (each containing three technical replicates); the IC50 values were then calculated from interpolated x values for y = 0.5. Mean IC50 and S.D. were then calculated. Curves displayed in figures are derived from the pooled date of the same three biologic replicates using the same nonlinear regression models described above.
Results
Derivation and Characterization of RET+ Patient–Derived Cell Lines.
Currently, there are no commercially available KIF5B-RET+ cell lines and only one lung cancer cell line with a RET rearrangement. Therefore, to develop additional models to study RET signaling in the context of lung cancer, we established three patient-derived cell lines, CUTO22, CUTO32, and CUTO42. The CUTO22 and CUTO32 cell lines harbor KIF5B-RET fusions, whereas the CUTO42 cell line contains an EML4-RET rearrangement (Figs. 1, A and B). EML4 is the most common 5′ partner in ALK rearrangements in NSCLC; however, it is a rare RET 5′ partner (Camidge and Doebele, 2012). Each of these cell lines was derived from patients with NSCLC at the University of Colorado Cancer Center. Although the RET breakpoint was conserved at exon 12 in all three cell lines, the two KIF5B-RET+ cell lines had different KIF5B variants. The CUTO32 cells contain the more common KIF5B exon 15; RET exon 12 variant, and the CUTO22 cells contain the more rare KIF5B exon 23; RET exon 12 variant. Interestingly, the CUTO22 and CUTO32 cell lines grew in suspension, which is not typical for most non-small cell lung cancer cell lines; the CUTO42 cell line has a mixed phenotype with partially adherent cells (Fig. 1B; Supplemental Fig. 1A). We were able to derive an adherent subpopulation for the CUTO32 cell line, which was used for all subsequent experiments, but not for the CUTO22 cell line.
Fig. 1.

RET+ patient–derived cell lines. (A) Schematic describing RET rearrangements in cell lines derived from patients at University of Colorado Thoracic Oncology. (B) Table describing relevant patient characteristics, cell line growth behavior, and co-alterations detected in clinical sequencing assays. K23;R12 indicates KIF5B exon 23; RET exon 12. K15;R12 indicates KIF5B exon 15; RET exon 12. E19;R12 indicates EML4 exon 19; RET exon 12, C1;R12 indicates CCDC6 exon 1; RET exon 12. Adenomatous polyposis coli (APC), tryptophan-aspartic acid repeats (WD)
RET+ Cell Lines Exhibit Heterogeneous Responses to RET Inhibitors.
First, we wanted to determine the sensitivity of each cell line to several different RET inhibitors. In proliferation assays, the CUTO22 and CUTO42 cell lines were sensitive to the selective RET inhibitors RXDX-105 and BLU-667, whereas the CUTO32 cell line was markedly resistant to RXDX-105 and partially resistant to BLU-667. The CUTO22 and CUTO42 cell lines had <50 nM IC50 values forBLU-667, whereas the CUTO32 cell line’s IC50 value was >300 nM (Fig. 2A). The CUTO32 cell line was also resistant to the multikinase RET inhibitor ponatinib (Fig. 2A). The LC-2/Ad cell line, an NSCLC cell line with a CCDC6-RET fusion, has been previously characterized as sensitive to RET inhibition and served as a positive control (Matsubara et al., 2012; Nelson-Taylor et al., 2017). In vitro, the CUTO32 and CUTO42 cell lines recapitulated the RET inhibitor responses of the patients from whom they were derived. The CUTO22 cell line was sensitive to RET inhibitors; however, the patient these cells were derived from had a mixed response to RET inhibitors. Because the patient from whom the CUTO22 cell line was derived had a mixed response to a RET inhibitor, it is possible that the CUTO22 cell line is derived from a RET inhibitor–sensitive subpopulation of cells. We found that treatment of the CUTO22 and CUTO42, but not CUTO32, cell lines with ponatinib or RXDX-105 resulted in induction of apoptosis within 24 hours. Apoptosis was increased by at least 200% in all cell lines except for the CUTO32 cell line (Fig. 2B).
Fig. 2.
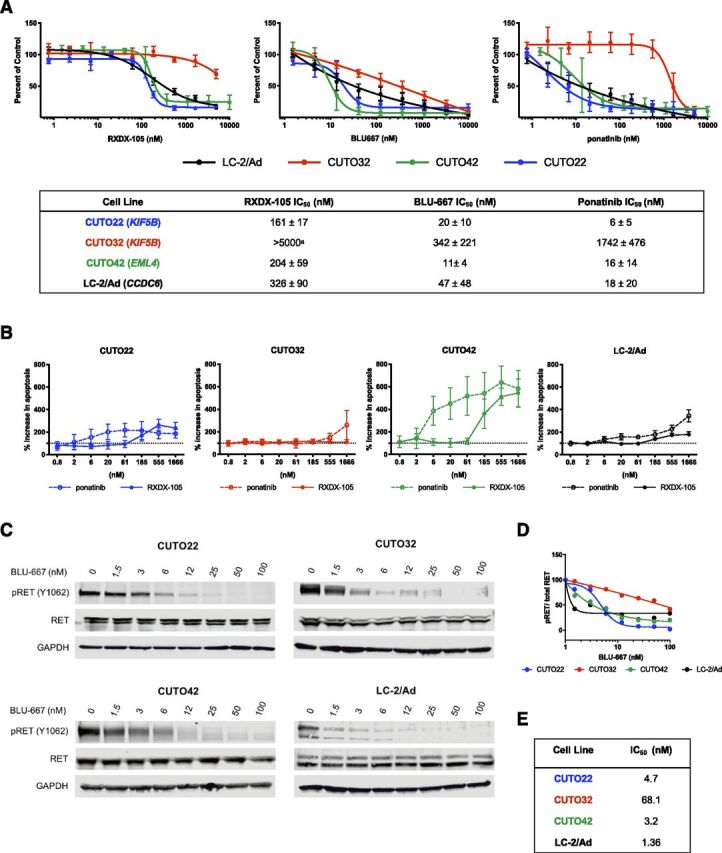
RET+ cell lines exhibit differential sensitivity to RET inhibitors. (A) MTS proliferation assays of CUTO22 (blue), CUTO32 (red), CUTO42 (green), and LC-2/Ad (black) cells treated with increasing concentrations of RXDX-105, BLU-667, or ponatinib for 72 hours. Error bars represent means ± S.D. for three replicate experiments. Lower table shows IC50 (nanomolar) values ± S.D. for each inhibitor and cell line. (B) Measurement of apoptosis induction with cleaved caspase 3/7 assay after treatment with increasing concentrations of ponatinib or RXDX-105 for 24 hours. Error bars represent means ± S.D. for three replicate experiments. (C) Western blot analysis of CUTO22, CUTO32, CUTO42, and LC-2/Ad cells treated with increasing concentrations of BLU-667 for 2 hours. pRET = 4G10 anti-phosphotyrosine in LC-2/Ad cells. (D) Quantification of dose-dependent inhibition of pRET with BLU-667. (E) IC50 values for pRET inhibition in immunoblot experiment.
RET Fusion Protein Is Expressed and Can Be Inhibited in a Dose-Dependent Manner by Tyrosine Kinase Inhibitors in RET+ Cell Lines.
We confirmed the expression of the RET fusion protein in each of our cell lines. The two KIF5B-RET+ cell lines expressed fusion proteins that were approximately 150 kDa (CUTO22) and 125 kDa (CUTO32); the EML4-RET fusion protein was also about 125 kDa (Supplemental Fig. 1B). Additionally, the presence of the RET fusion transcript was confirmed via RNA sequencing in the CUTO22 and CUTO32 cell lines and via reverse transcription PCR in the CUTO42 cell line (Supplemental Fig. 1C). Next, we wanted to assess RET inhibition in response to RET tyrosine kinase inhibitors (TKIs) in our cell lines. RET phosphorylation was successfully inhibited in all four cell lines after treatment with BLU-667. Notably, the KIF5B-RET+ fusions and non–KIF5B-RET fusions were inhibited at approximately equivalent concentrations of a given RET inhibitor; however, we noticed a small amount of residual pRET in the CUTO32 cell line (Figs. 2, C and E). We performed a time course experiment to determine if there was a difference in drug inhibition kinetics among different RET fusion proteins. We discovered that RET was inhibited within about 30 minutes in all four cell lines and was maintained up to 24 hours (Supplemental Figs. 2, A and B). Overall, these data suggest that differential responses to RET inhibitors are not likely to be due to differences in drug binding to the KIF5B-RET protein.
CUTO22 Cells Have Diminished AKT Inhibition After Treatment with RET Inhibitors.
Next, we investigated two canonical downstream pathways of RET, the MAPK pathway and the PI3K/AKT pathway, in our three RET inhibitor–sensitive cell lines. Remarkably, we observed differential signaling responses upon RET inhibition in each of our cell lines. The CUTO42 cells showed dramatic inhibition of pAKT after treatment with RET inhibitors, whereas the CUTO22 cells maintained pAKT (Figs. 3, A and B). We also noticed differences in inhibition of phosphorylated ERK (pERK) between different inhibitors. In the CUTO22, CUTO42, and LC-2/Ad cells we observed more complete inhibition of pERK with BLU-667 than either ponatinib or RXDX-105 (Figs. 3, A–C). Because we observed that our RET+ cell lines showed different downstream signaling after treatment with RET inhibitors, we wanted to assess their dependence on the RAS/MAPK and PI3K/AKT pathways. We treated the CUTO22, CUTO42, and LC-2/Ad cell lines with the MEK inhibitor trametinib and found that the CUTO22 cells were sensitive to trametinib (Fig. 4A). Surprisingly, the CUTO42 and LC-2/Ad cells were resistant to MEK inhibition despite being sensitive to RET inhibitors (Fig. 4A). The CUTO22 and LC-2/Ad cells were more than five times more sensitive to trametinib than CUTO42 cells. Additionally, we found that treatment with 15 nM trametinib did not greatly enhance sensitivity to RXDX-105 in the CUTO42 cell line but did enhance RET inhibitor sensitivity in the CUTO22 cell line. Addition of trametinib to RXDX-105 had moderate effects on the LC-2/Ad cell line (Fig. 4B). We used the pan-PI3K/mechanistic target of rapamycin (mTOR) inhibitor omipalisib to assess our RET+ cell lines’ dependence on the PI3K pathway. We observed an inverse pattern with omipalisib treatment, where the CUTO42 and LC-2/Ad cells were more sensitive to omipalisib than trametinib, whereas the CUTO22 cells were more sensitive to trametinib than omipalisib (Fig. 4A). The CUTO22 cells were approximately five times more resistant to omipalisib than the CUTO42 or LC-2/Ad cells. Treatment with 20 nM omipalisib enhanced sensitivity to RXDX-105 in the CUTO42 and LC-2/Ad cells but not in the CUTO22 cells. We confirmed via Western blot that our trametinib and omipalisib treatments inhibited their intended targets as indicated by inhibition of pERK and pAKT, respectively (Fig. 4, D and E). We observed crosstalk between the MAPK and PI3K/AKT pathways during these treatments where the CUTO22 cells had a decrease in pERK after AKT inhibition (Fig. 4D). The RET inhibitor–resistant CUTO32 cells did not have substantial inhibition of either pERK or pAKT despite successful RET inhibition (Supplemental Fig. 3A). Additionally, these cells were resistant to trametinib and moderately resistant to omipalisib. Neither inhibitor sensitized the CUTO32 cells to RET inhibition (Supplemental Figs. 3, B–D).
Fig. 3.
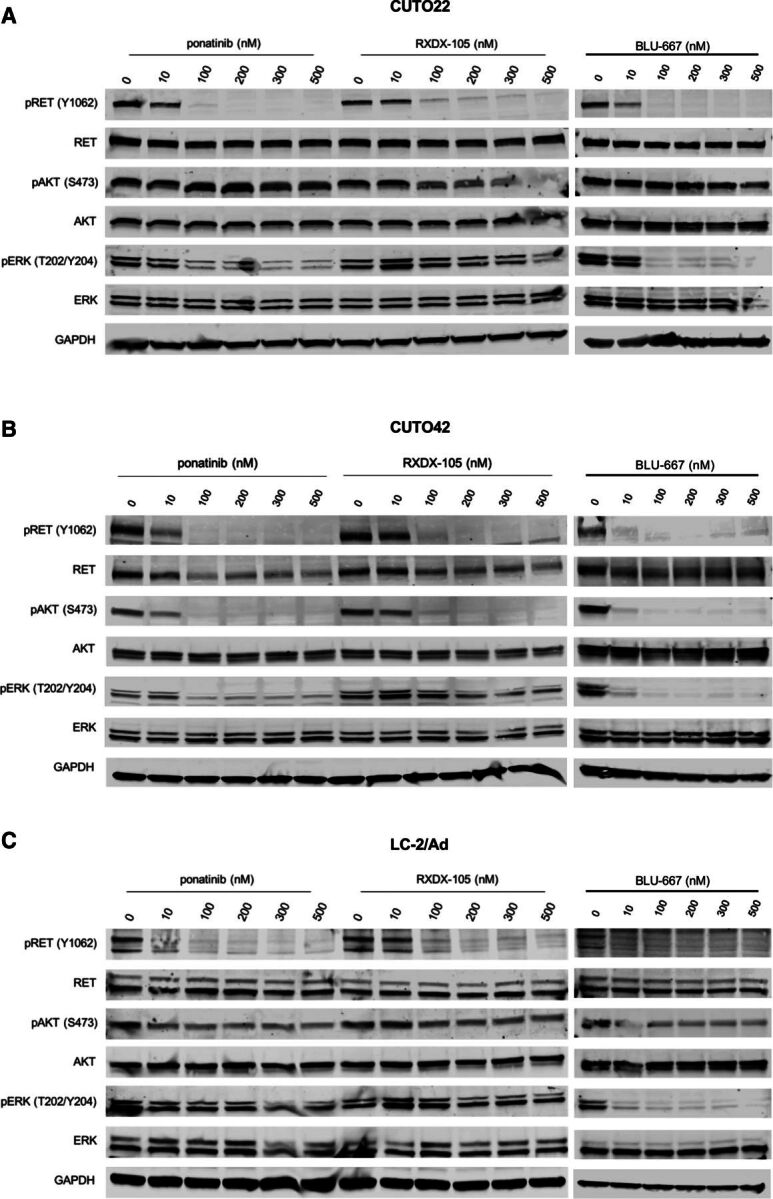
RET inhibitors successfully decrease KIF5B-RET activation but show variable MAPK and AKT inhibition. Western blot analysis of RET inhibition and downstream signaling in CUTO22 (A), CUTO42 (B), and LC-2/Ad (C) cells treated with increasing concentrations of ponatinib, RXDX-105, or BLU-667 for 2 hours. pRET = 4G10 anti-phosphotyrosine for LC-2/Ad.
Fig 4.
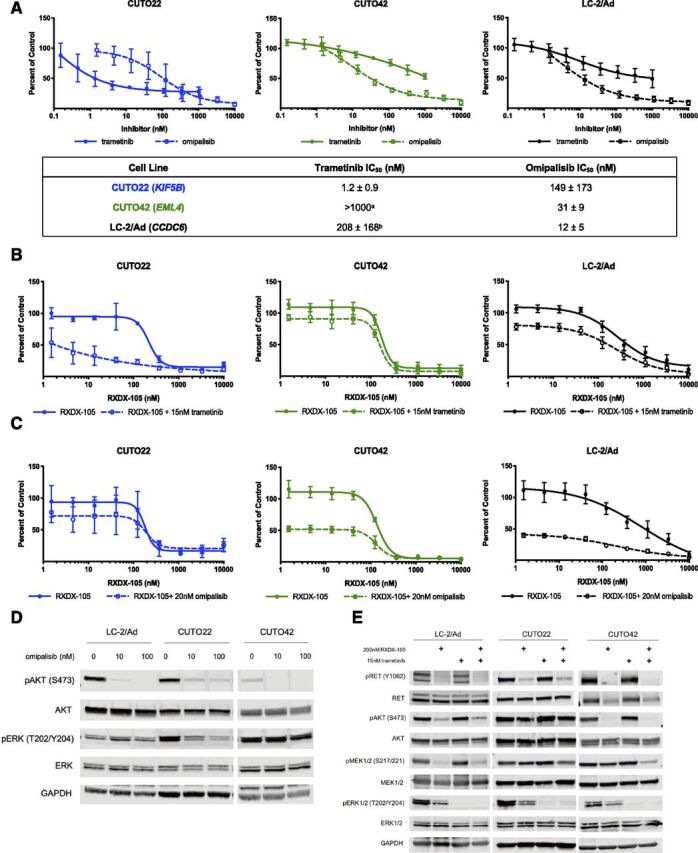
RET+ cells have differential responses to MEK and PI3K inhibition. (A) MTS proliferation assays in CUTO22, CUTO42, and LC-2/Ad cells treated with increasing concentrations of trametinib or omipalisib. Lower table describes IC50 values ± S.D. Error bars represent means ± S.D. for three replicate experiments. MTS proliferations assays in CUTO22, CUTO32, CUTO42, or LC-2/Ad cells treated with increasing concentrations of RXDX-105 alone or in combination with 15 nM trametinib (B) or 20 nM omipalisib (C). Error bars represent means ± S.D. for three biologic replicate experiments. (D) Western blot analysis of cells treated with 10 or 100 nM omipalisib for 2 hours. (E) Western blot analysis of RET+ cell lines treated with RXDX-105, trametinib, or both for 2 hours. aIC50 not calculated because cells did not reach 50% inhibition of proliferation. bIC50 calculated from N = 2 replicates because third replicate did not reach 50% of proliferation.
Drug Screen Reveals Unique Vulnerabilities in Cell Cycle Regulation in CUTO32 Cells.
We performed a drug screen with the CUTO22, CUTO32, and LC-2/Ad cell lines with and without 200 nM RXDX-105 treatment to determine possible therapeutic vulnerabilities in the RET inhibitor–resistant CUTO32 cell line. This high-throughput screen included approximately 300 compounds designed to inhibit a wide variety of protein targets at one higher concentration (2500 nM) and one lower concentration (500 nM) (Fig. 5A). We found that the CUTO32 cells were sensitive to three polo-like kinase 1 (PLK1) inhibitors, volasertib, BI2536, and rigosertib. The CUTO32 cells were also sensitive to several Aurora kinase inhibitors, alisertib, AT9283, and SNS-314 (Supplemental Fig. 4). It was notable that the CUTO22 cell line was resistant to most of the PLK1 and Aurora kinase inhibitors, suggesting that these cell lines have differential dependencies on these pathways.
Fig. 5.
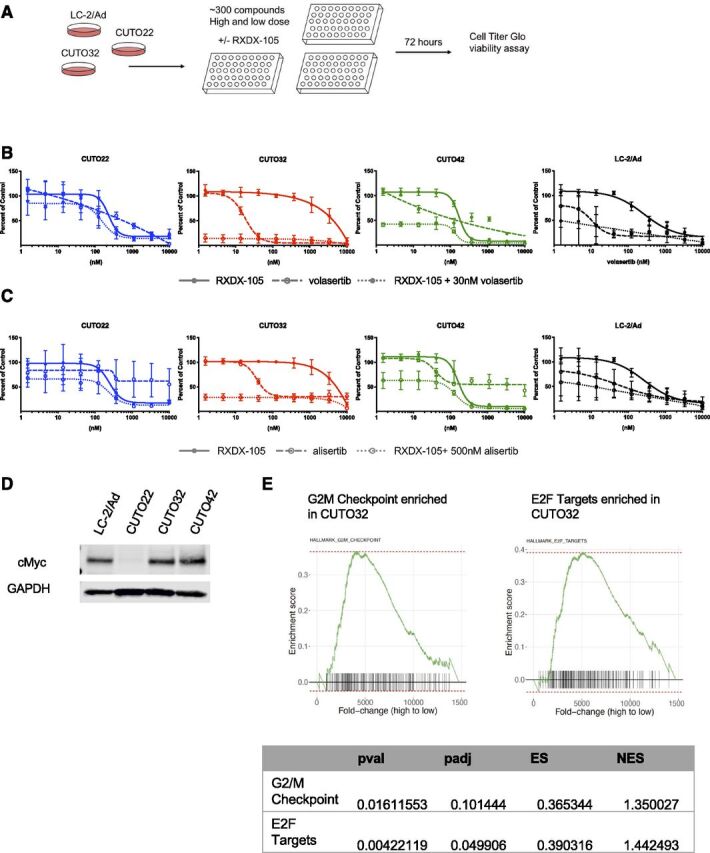
Drug screening reveals unique vulnerabilities in cell cycle regulation. (A) Schematic describing drug screening strategy. CUTO22, CUTO32, and LC-2/Ad cells were treated with approximately 300 compounds at two fixed concentrations of inhibitors with or without 200 nM RXDX-105. Cell viability was assayed after 72 hours using Cell Titer Glo. (B) MTS proliferation assays of CUTO22, CUTO32, CUTO42, and LC-2/Ad cells treated with RXDX-105, volasertib, or RXDX-105 combined with 30 nM volasertib for 72 hours. Error bars represent means ± S.D. for three replicate experiments. (C) MTS proliferation assays of CUTO22, CUTO32, CUTO42, and LC-2/Ad cells treated with RXDX-105, alisertib, or RXDX-105 combined with 500 nM alisertib for 72 hours. Error bars represent means ± S.D. for three biologic replicate experiments. (D) Immunoblot analysis for MYC expression in CUTO22, CUTO32, CUTO42, and LC-2/Ad cells. (E) Gene set enrichment analysis of CUTO32 (untreated) compared with CUTO22 (untreated) cells shows an enrichment of genes associated with G2/M transition and E2F targets in CUTO32 cells. Adjusted p-value (padj), p-value (pval), enrichment score (ES), normalized enrichment score (NES).
CUTO32 Cells Are Dependent on G2/M Cell Cycle–Regulating Proteins.
We performed proliferation assays on CUTO22, CUTO32, CUTO42, and LC-2/Ad cells treated with volasertib, RXDX-105, or RXDX-105 with the addition of 30 nM volasertib. Our results corroborated the drug screen data and showed that the CUTO32 cells were the most sensitive to PLK1 inhibition with volasertib. The LC-2/Ad and CUTO42 cell lines had moderate sensitivity (Fig. 5B). Treatment with alisertib mirrored the results with volasertib and showed that the CUTO32 cells were most sensitive to Aurora kinase inhibition (Fig. 5C). The CUTO22 cell line was not sensitive to either volasertib or alisertib. It has recently been shown in small cell lung cancer that high MYC expression correlated with sensitivity to Aurora kinase inhibitors (Mollaoglu et al., 2017). With immunoblot analysis, we found that the CUTO22 cell line lacked MYC expression but that the CUTO32, CUTO42, and LC-2/Ad cells, which all had moderate to high sensitivity to alisertib, expressed MYC (Fig. 5D). Further support for these conclusions is found in our RNA sequencing data where we found that genes associated with the G2/M checkpoint were enriched in the CUTO32 cells compared with CUTO22 cells in gene set enrichment analysis. E2/F targets, which promote the G1/S transition, were also enriched in CUTO32 cells (Fig. 5E).
Co-Inhibition of MET Does Not Enhance Sensitivity to RET Inhibition.
Our RNA sequencing data showed high expression of MET, which has been shown to promote resistance to targeted therapies (Camidge et al., 2014). MET expression was high in all of our cell lines but was highest in the RET inhibitor–resistant CUTO32 cell line (Supplemental Fig. 5). All four of our cell lines expressed MET in Western blots, but MET was only phosphorylated in the CUTO32 and LC-2/Ad cells. Inhibition of MET with 500 nM crizotinib, in combination with RXDX-105, however, did not sensitize the CUTO32 cells to RET inhibition (Supplemental Figs. 6, A and B).
RET Inhibitor Treatment Inhibits Tumor Growth in CUTO42 Xenografts More Effectively Than CUTO32 Xenografts.
Finally, we sought to evaluate the efficacy of RET, PI3K, MEK, and Aurora kinase inhibitors in vivo. We subcutaneously implanted CUTO32 and CUTO42 cells into the flanks of nude mice. Once tumors were established, we began daily oral dosing with BLU-667, trametinib, alisertib, or omipalisib (Fig. 6A). We were unable to establish xenografted tumors with the LC-2/Ad or CUTO22 cell lines. We found that the CUTO32 tumors showed a decrease in tumor growth rate but did not show stabilization of tumor growth or regression when treated with BLU-667 (Fig. 6B). Consistent with our in vitro data, the CUTO42 tumors rapidly regressed with BLU-667 treatment and had sustained tumor growth inhibition (Fig. 6B). Alisertib was not effective at reducing tumor growth rate in either the CUTO32 or CUTO42 tumors. Omipalisib was more effective at reducing tumor growth in the CUTO42 tumors than the CUTO32 tumors, and trametinib resulted in a modest delay in tumor growth in both models (Fig. 6B).
Fig. 6.
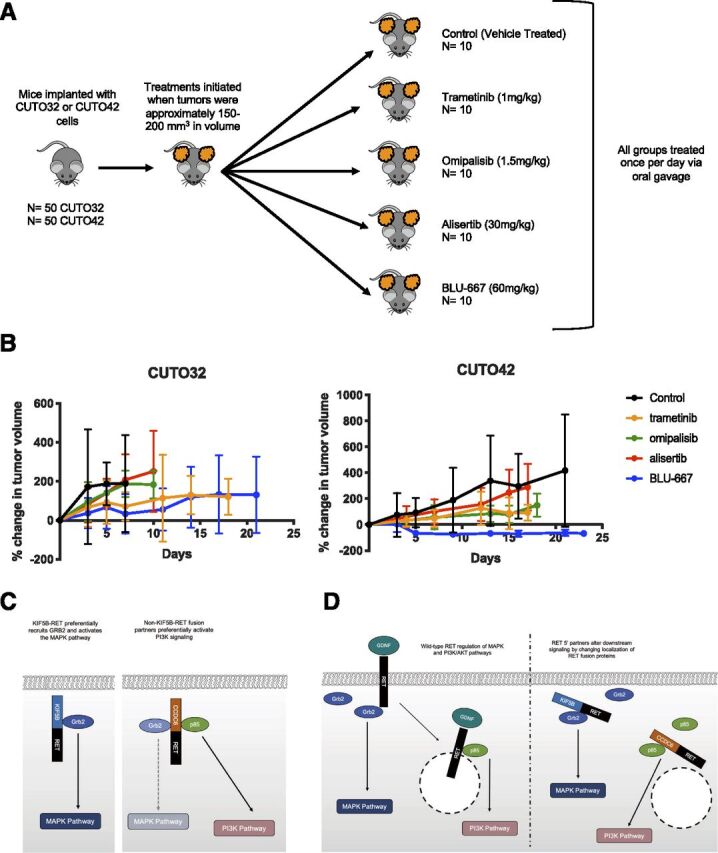
RET inhibitor treatment inhibits tumor growth in CUTO42 xenografts more effectively than CUTO32 xenografts. (A) CUTO32 or CUTO42 cells were subcutaneously implanted in the flanks of nude mice. Once tumors developed, mice were treated once per day with 60 mg/kg BLU-667, 1 mg/kg trametinib, 1.5 mg/kg omipalisib, 30 mg/kg alisertib, or control (vehicle) via daily oral gavage. N = 10 mice per treatment group. (B) Graphs of percent change in tumor growth (relative to starting tumor volume at initiation of treatment). Error bars represent ±S.D. (C) Summary figure describing potential for differential recruitment of adapter proteins in KIF5B-RET+ and non–KIF5B-RET+ cells. (D) Potential differential subcellular localization of RET fusion proteins. Glial cell line-derived neurotrophic factor (GDNF), growth factor receptor bound protein 2(Grb2).
Discussion
A lack of RET+ models has previously been a major limitation in studying RET signaling and response to targeted therapies in lung cancer. In this study we have derived and characterized several novel RET+ cell lines that have revealed differential signaling dynamics, despite harboring the same driver oncogene. To our knowledge, this is the first example of a patient-derived NSCLC cell line with EML4-RET fusion. These cell lines offer the unique ability to study the signaling mechanisms of endogenous RET fusion proteins with different fusion partners.
Previous studies have postulated that KIF5B-RET may be more difficult to inhibit because the KIF5B promoter results in higher fusion kinase expression than the promoters associated with other 5′ partners, which consequently might require higher doses of drugs to inhibit (Drilon et al., 2019). In our RNA sequencing data, RET was most highly expressed in the CUTO22 cells. The CUTO32 and LC-2/Ad cells had similar RET expression levels (Supplemental Fig. 5). These data suggest that there may be other regulation of RET expression beyond the 5′ fusion promoter. Our Western blot and time course data suggest that pRET is inhibited at approximately equal concentrations in the KIF5B-RET+ and non–KIF5B-RET+ cell lines. Furthermore, we did not notice major differences in RET or pRET expression in Western blots. It is worth noting that the CUTO32 and CUTO42 cell lines each contain different variants of the KIF5B-RET fusion, which could potentially contribute to their difference in RET inhibitor sensitivity.
There has been growing interest in understanding fusion kinase biology and how it is affected by the 5′ partner. Historically, different 5′ partners have been largely considered equivalent in clinical decision making; however, evidence has been accumulating that they may affect drug responses and resistance. It has been shown that different ALK variants, which contain differing lengths of the 5′ EML4 gene, have a significantly different frequency and spectrum of acquired resistance mutations (Lin et al., 2018). Although the mechanism of this remains unknown, it suggests that each ALK variant has a unique structure or propensity to develop resistance through a particular mechanism.
Here, our data suggest that KIF5B-RET+ cell lines may be more dependent on the RAS/MAPK pathway and lack inhibition of pAKT after RET inhibition. It has been previously shown that PI3K/AKT pathway activation is necessary for RET to transform cells (Segouffin-Cariou and Billaud, 2000). It has also been shown that activation of AKT by RET does not require complete RET internalization after ligand binding but that activation of ERK does (Richardson et al., 2006). Based on these previous studies, there could be several mechanisms that regulate this differential signaling. First, it is possible that different 5′ partners lead to differential recruitment and activation of adapter proteins and downstream pathways (Fig. 6C). A Drosophila model has shown that KIF5B-RET, specifically, can recruit other kinases through its kinesin domain, which further enhances its downstream signaling activation (Das and Cagan, 2017). Second, it is possible that the RET 5′ partner could alter subcellular localization of the RET fusion protein, which can subsequently alter its downstream signaling (Fig. 6D). In ROS1 fusions it has been demonstrated that Syndecan-4-ROS1 fusions localized to endosomes, whereas CD74-ROS1 fusions localized to the endoplasmic reticulum (Neel et al., 2019). Future experiments analyzing RET subcellular localization as well as RET binding partners in our cell lines will be informative in determining the mechanism underlying the differential pathway regulation we observed. One of the limitations of this study is the small number of KIF5B-RET and non–KIF5B-RET cell lines included; therefore, these conclusions may not be generalizable to all KIF5B-RET fusions. Our data do, however, show that there are different signaling patterns and dependencies present in RET+ patients.
Unfortunately, acquired resistance to TKIs is nearly universal, and therefore it will be important to understand mechanisms of resistance to RET inhibitors. Currently, there are only a few studies describing mechanisms of resistance to RET inhibitors. Kinase domain mutations that block drug binding will likely be a common mechanism of resistance, as they have been well documented in resistance to other TKIs (McCoach et al., 2018a,b). BLU-667 and LOXO-292 have activity against the two RET gatekeeper mutations, V804M and V804L; however, it is possible that patients could develop other mutations that prevent drug binding. Our laboratory has previously shown that reactivation of RAS/MAPK signaling is capable of driving resistance to ponatinib in RET+ cells (Nelson-Taylor et al., 2017). The study described here suggests that the PI3K/AKT pathway may be another possible avenue of resistance, particularly in KIF5B-RET+ patients. These studies also demonstrate that combined inhibition of RET and AKT may be an effective upfront combination therapy in in some RET+ patients. Consistent with this strategy, a recent clinical trial demonstrated effectiveness of a combination of vandetanib, which has low activity as monotherapy (Falchook et al., 2016; Yoh et al., 2017), and the mTOR inhibitor, everolimus (Subbiah et al., 2018a). Importantly, our data also suggest that not all RET+ patients would derive benefit from MEK inhibitors, despite the MAPK pathway being a key pro-proliferative pathway downstream of RET. Work is ongoing to develop RET inhibitor–resistant derivations of the CUTO22 and CUTO42 cell lines to assess mechanisms of resistance to RET inhibitors.
MET amplification and/or activation is a well characterized mechanism of resistance to TKIs in lung cancer (Bean et al., 2007; Engelman et al., 2007). All four of the RET+ cell lines used in this study robustly express MET at the RNA and protein level; however, MET was only phosphorylated in the LC-2/Ad and CUTO32 cell lines. Although inhibiting MET did not further sensitize our cells to RET inhibitors, the high MET expression suggests that these cells may be primed to use MET signaling as a mechanism of resistance to RET inhibitors. Indeed, a recent case report demonstrates MET gene amplification as a mechanism of resistance to selpercatinib in a patient with RET fusion–positive NSCLC (Zhu et al., 2020).
We identified that the CUTO32 cell line was sensitive (<1 nM IC50) to inhibition of two cell cycle–related proteins, PLK1 and Aurora kinase. PLK1 serves several important roles during the cell cycle in centrosome formation, maintaining genome stability and promoting entry into M phase (Strebhardt and Ullrich, 2006). PLK1 expression is elevated in a number of cancers and is associated with poor prognosis as well as resistance to several chemotherapy drugs (Gutteridge et al., 2016). Aurora kinases occupy a similar role in promoting the M phase of the cell cycle and regulate proper chromosome segregation (Tang et al., 2017). Aurora kinase activity has also been shown to promote cell survival and subsequent resistance to the EGFR inhibitor osimertinib (Shah et al., 2019). Although establishing biomarkers of predicted response to cell cycle inhibitors may be challenging, these inhibitors may be a potential option for patients who are initially resistant to RET inhibitors or eventually develop resistance. We found that MYC expression corresponded with response to Aurora kinase inhibitors. Interestingly, another study demonstrated a relationship between RET expression and a MYC transcriptional signature, which could be inhibited with cabozantinib ((Hayashi et al., 2020)Hayashi et al., 2020).
The role of co-alterations in tumor suppressors in RET rearranged cancers is not fully understood. According to clinical testing of patients’ tumors, the CUTO22 cell line had neurofibromin 1 (NF1) and Adenomatous polyposis coli mutations, the CUTO32 cells had tumor protein 53 (TP53) and NF1 mutations, and the CUTO42 cells had a NOTCH1 mutation. In general, TP53 mutations confer a worse prognosis in EGFR-mutated lung cancers, and the role of NF1 mutations in lung cancer remains uncertain (Redig et al., 2016; Labbé et al., 2017). These mutations did not correlate with decreased expression compared with our other cell lines in our RNA sequencing data, making it unclear to us if they are serving a functional role in our cell lines (Supplemental Fig. 5).
Our in vivo experiments recapitulated our in vitro RET inhibitor sensitivity data; the CUTO42 xenograft tumors regressed with BLU-667 treatment, and our CUTO32 tumors showed a reduction in rate of tumor growth, compared with control mice, but continued to grow steadily during BLU-667 treatment. A recent study by Hayashi et al. (2021) also investigated cell signaling in several novel RET+ patient–derived cell lines/patient-derived xenograft models. Interestingly, their study also showed differential sensitivity to RET inhibitors in vivo. They found that one KIF5B-RET+ patient-derived xenograft tumor was resistant to RXDX-105 but sensitive to cabozantinib. Although we predicted that the Aurora kinase inhibitor, alisertib, would result in decreased tumor growth in the CUTO32 cell line, it failed to do so as a single-agent treatment. Previous studies have similarly shown potent sensitivity to alisertib in vitro but only slight effects in vivo. One study showed that the effect of alisertib was enhanced by the addition of chemotherapy, suggesting that Aurora kinase inhibition may be most effective in combination with RET inhibitors or other therapies (Mollaoglu et al., 2017). We expected the PI3K/mTOR inhibitor omipalisib to inhibit tumor growth in the CUTO42 tumors but it did not do so. In the CUTO42 cells, treatment with a combination of a RET inhibitor and omipalisib was most effective at inhibiting cell proliferation in vitro; therefore, perhaps combination treatment is necessary for greater reduction in tumor growth in vivo.
Overall, these data demonstrate that RET+ cancers have heterogeneous biology, despite harboring the same oncogene driver, and highlight the importance of gaining a detailed understanding of fusion kinases’ signaling networks. Collectively, these studies shed light on the complex nature of fusion oncogene signaling and provide insight on the basis for differential response to RET inhibitors.
Acknowledgments
We would like to thank the University of Colorado Cancer Center (UCCC) Bioinformatics and Biostatistics Shared Resource, the UCCC Genomics and Microarray Shared Resource, and the UCCC Protein Production/MoAB/Tissue Culture Shared Resource for their help and expertise.
Abbreviations
- AKT
protein kinase B
- ALK
anaplastic lymphoma kinase
- CCDC6
coiled coiled domain containing 6
- EGFR
epidermal growth factor receptor
- EML4
echinoderm microtubule-associated protein-like 4
- ERK
extracellular signal-regulated kinase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HPBCD
2-hydroxypropel beta-cyclodextrin
- KIF5B
kinesin 5B
- MAPK
mitogen-activated protein kinase
- MEK
MAPK/ERK kinase
- MET
hepatocyte growth factor receptor
- NF1
neurofibromin 1
- NSCLC
non-small cell lung cancer
- pAKT
phosphorylated AKT
- PCR
polymerase chain reaction
- pERK
phosphorylated ERK
- PI3K
phosphoinositide 3-kinase
- PLK1
polo-like kinase 1
- pRET
phosphorylated RET
- RET
rearranged during transfection
- TKI
tyrosine kinase inhibitor
- TP53
tumor protein 53
Authorship Contributions
Participated in research design: Schubert, Le, Doebele.
Conducted experiments: Schubert, Le, Estrada-Bernal, Doak, Kinose.
Contributed new reagents or analytic tools: Le, Goodspeed, Rix, Tan.
Performed data analysis: Yoo, Ferrara, Goodspeed, Rix, Tan.
Wrote or contributed to the writing of the manuscript: Schubert, Doebele.
Footnotes
This work was funded by National Institutes of Health National Cancer Institute Lung Cancer SPORE Pilot Grant [P50-CA058187], National Institutes of Health National Research Service Awards [F31-1F31CA228320-01 and T32-T32CA190216-01A1], the Colorado Clinical and Translational Sciences Institute [CCTSI TL1 5TL1TR001081], and National Institutes of Health National Cancer Institute [P30-CA046934].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Asai N, Murakami H, Iwashita T, Takahashi M (1996) A mutation at tyrosine 1062 in MEN2A-Ret and MEN2B-Ret impairs their transforming activity and association with shc adaptor proteins. J Biol Chem 271:17644–17649. [DOI] [PubMed] [Google Scholar]
- Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, et al. (2007) MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA 104:20932–20937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camidge DR, Doebele RC (2012) Treating ALK-positive lung cancer--early successes and future challenges. Nat Rev Clin Oncol 9:268–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camidge DR, Pao W, Sequist LV (2014) Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol 11:473–481. [DOI] [PubMed] [Google Scholar]
- Das TK, Cagan RL (2017) KIF5B-RET oncoprotein signals through a multi-kinase signaling hub. Cell Rep 20:2368–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KD, Mahale S, Astling DP, Aisner DL, Le AT, Hinz TK, Vaishnavi A, Bunn PA Jr, Heasley LE, Tan AC, et al. (2013) Resistance to ROS1 inhibition mediated by EGFR pathway activation in non-small cell lung cancer. PLoS One 8:e82236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A, Fu S, Patel MR, Fakih M, Wang D, Olszanski AJ, Morgensztern D, Liu SV, Cho BC, Bazhenova L, et al. (2019) A phase I/Ib trial of the VEGFR-sparing multikinase RET inhibitor RXDX-105. Cancer Discov 9:384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A, Oxnard GR, Tan DSW, Loong HHF, Johnson M, Gainor J, McCoach CE, Gautschi O, Besse B, Cho BC, et al. (2020) Efficacy of selpercatinib in RET fusion-positive non-small-cell lung cancer. N Engl J Med 383:813–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drilon A, Wang L, Hasanovic A, Suehara Y, Lipson D, Stephens P, Ross J, Miller V, Ginsberg M, Zakowski MF, et al. (2013) Response to Cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov 3:630–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316:1039–1043. [DOI] [PubMed] [Google Scholar]
- Falchook GS, Ordóñez NG, Bastida CC, Stephens PJ, Miller VA, Gaido L, Jackson T, Karp DD (2016) Effect of the RET inhibitor vandetanib in a patient with RET fusion-positive metastatic non-small-cell lung cancer. J Clin Oncol 34:e141–e144. [DOI] [PubMed] [Google Scholar]
- Gainor JF, Lee DH, Curigliano G, Doebele RC, Kim D-W, Baik CS, Tan DS-W, Lopes G, Gadgeel SM, Cassier PA, et al. (2019) Clinical activity and tolerability of BLU-667, a highly potent and selective RET inhibitor, in patients (pts) with advanced RET-fusion+ non-small cell lung cancer (NSCLC). J Clin Oncol 37(suppl):9008. [Google Scholar]
- Gautschi O, Milia J, Filleron T, Wolf J, Carbone DP, Owen D, Camidge R, Narayanan V, Doebele RC, Besse B, et al. (2017) Targeting RET in patients with RET-rearranged lung cancers: results from the global, multicenter RET registry. J Clin Oncol 35:1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutteridge REA, Ndiaye MA, Liu X, Ahmad N (2016) Plk1 inhibitors in cancer therapy: from laboratory to clinics. Mol Cancer Ther 15:1427–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Ichihara M, Iwashita T, Murakami H, Shimono Y, Kawai K, Kurokawa K, Murakumo Y, Imai T, Funahashi H, et al. (2000) Characterization of intracellular signals via tyrosine 1062 in RET activated by glial cell line-derived neurotrophic factor. Oncogene 19:4469–4475. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Odintsov I, Smith RS, Ishizawa K, Liu AJW, Delasos L, Kurzatkowski C, Tai H, Gladstone E, Vojnic M, et al. (2020) RET inhibition in novel patient-derived models of RET-fusion positive lung adenocarcinoma reveals a role for MYC upregulation. Dis Model Mech 14:dmm047779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DS, Bauer TM, Lee JJ, Dowlati A, Brose MS, Farago AF, Taylor M, Shaw AT, Montez S, Meric-Bernstam F, et al. (2019) Larotrectinib in adult patients with solid tumours: a multi-centre, open-label, phase I dose-escalation study. Ann Oncol 30:325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrustanovic G, Olivas V, Pazarentzos E, Tulpule A, Asthana S, Blakely CM, Okimoto RA, Lin L, Neel DS, Sabnis A, et al. (2015) RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat Med 21:1038–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Subbiah V, Marchlik E, Elkin SK, Carter JL, Kurzrock R (2017) RET aberrations in diverse cancers: next-generation sequencing of 4,871 patients. Clin Cancer Res 23:1988–1997. [DOI] [PubMed] [Google Scholar]
- Labbé C, Cabanero M, Korpanty GJ, Tomasini P, Doherty MK, Mascaux C, Jao K, Pitcher B, Wang R, Pintilie M, et al. (2017) Prognostic and predictive effects of TP53 co-mutation in patients with EGFR-mutated non-small cell lung cancer (NSCLC). Lung Cancer 111:23–29. [DOI] [PubMed] [Google Scholar]
- Le Rolle AF, Klempner SJ, Garrett CR, Seery T, Sanford EM, Balasubramanian S, Ross JS, Stephens PJ, Miller VA, Ali SM, et al. (2015) Identification and characterization of RET fusions in advanced colorectal cancer. Oncotarget 6:28929–28937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP (2011) Molecular signatures database (MSigDB) 3.0. Bioinformatics 27:1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JJ, Zhu VW, Yoda S, Yeap BY, Schrock AB, Dagogo-Jack I, Jessop NA, Jiang GY, Le LP, Gowen K, et al. (2018) Impact of EML4-ALK variant on resistance mechanisms and clinical outcomes in ALK-positive lung cancer. J Clin Oncol 36:1199–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara D, Kanai Y, Ishikawa S, Ohara S, Yoshimoto T, Sakatani T, Oguni S, Tamura T, Kataoka H, Endo S, et al. (2012) Identification of CCDC6-RET fusion in the human lung adenocarcinoma cell line, LC-2/ad. J Thorac Oncol 7:1872–1876. [DOI] [PubMed] [Google Scholar]
- McCoach CE, Blakely CM, Banks KC, Levy B, Chue BM, Raymond VM, Le AT, Lee CE, Diaz J, Waqar SN, et al. (2018a) Clinical utility of cell-free DNA for the detection of ALK fusions and genomic mechanisms of ALK inhibitor resistance in non-small cell lung cancer. Clin Cancer Res 24:2758–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoach CE, Le AT, Gowan K, Jones K, Schubert L, Doak A, Estrada-Bernal A, Davies KD, Merrick DT, Bunn PA Jr, et al. (2018b) Resistance mechanisms to targeted therapies in ROS1+ and ALK+ non-small cell lung cancer. Clin Cancer Res 24:3334–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollaoglu G, Guthrie MR, Böhm S, Brägelmann J, Can I, Ballieu PM, Marx A, George J, Heinen C, Chalishazar MD, et al. (2017) MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell 31:270–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan LM (2014) RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer 14:173–186. [DOI] [PubMed] [Google Scholar]
- Neel DS, Allegakoen DV, Olivas V, Mayekar MK, Hemmati G, Chatterjee N, Blakely CM, McCoach CE, Rotow JK, Le A, et al. (2019) Differential subcellular localization regulates oncogenic signaling by ROS1 kinase fusion proteins. Cancer Res 79:546–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson-Taylor SK, Le AT, Yoo M, Schubert L, Mishall KM, Doak A, Varella-Garcia M, Tan AC, Doebele RC (2017) Resistance to RET-inhibition in RET-rearranged NSCLC is mediated by reactivation of RAS/MAPK signaling. Mol Cancer Ther 16:1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohiwa M, Murakami H, Iwashita T, Asai N, Iwata Y, Imai T, Funahashi H, Takagi H, Takahashi M (1997) Characterization of Ret-Shc-Grb2 complex induced by GDNF, MEN 2A, and MEN 2B mutations. Biochem Biophys Res Commun 237:747–751. [DOI] [PubMed] [Google Scholar]
- Oie HK, Russell EK, Carney DN, Gazdar AF (1996) Cell culture methods for the establishment of the NCI series of lung cancer cell lines. J Cell Biochem Suppl 24:24–31. [DOI] [PubMed] [Google Scholar]
- Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, Feeney N, Sholl LM, Dahlberg SE, Redig AJ, et al. (2018) Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol 4:1527–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paratala BS, Chung JH, Williams CB, Yilmazel B, Petrosky W, Williams K, Schrock AB, Gay LM, Lee E, Dolfi SC, et al. (2018) RET rearrangements are actionable alterations in breast cancer. Nat Commun 9:4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim DW, Ou SI, Pérol M, Dziadziuszko R, Rosell R, et al. ALEX Trial Investigators (2017) Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N Engl J Med 377:829–838. [DOI] [PubMed] [Google Scholar]
- Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, Marcoux N, Banwait MK, Digumarthy SR, Su W, et al. (2018) Landscape of acquired resistance to osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion. Cancer Discov 8:1529–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planchard D, Smit EF, Groen HJM, Mazieres J, Besse B, Helland Å, Giannone V, D’Amelio AM Jr, Zhang P, Mookerjee B, et al. (2017) Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: an open-label, phase 2 trial. Lancet Oncol 18:1307–1316. [DOI] [PubMed] [Google Scholar]
- Posch C, Moslehi H, Feeney L, Green GA, Ebaee A, Feichtenschlager V, Chong K, Peng L, Dimon MT, Phillips T, et al. (2013) Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc Natl Acad Sci USA 110:4015–4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redig AJ, Capelletti M, Dahlberg SE, Sholl LM, Mach S, Fontes C, Shi Y, Chalasani P, Jänne PA (2016) Clinical and molecular characteristics of NF1-mutant lung cancer. Clin Cancer Res 22:3148–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson DS, Lai AZ, Mulligan LM (2006) RET ligand-induced internalization and its consequences for downstream signaling. Oncogene 25:3206–3211. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrock AB, Zhu VW, Hsieh WS, Madison R, Creelan B, Silberberg J, Costin D, Bharne A, Bonta I, Bosemani T, et al. (2018) Receptor tyrosine kinase fusions and BRAF kinase fusions are rare but actionable resistance mechanisms to EGFR tyrosine kinase inhibitors. J Thorac Oncol 13:1312–1323. [DOI] [PubMed] [Google Scholar]
- Segouffin-Cariou C, Billaud M (2000) Transforming ability of MEN2A-RET requires activation of the phosphatidylinositol 3-kinase/AKT signaling pathway. J Biol Chem 275:3568–3576. [DOI] [PubMed] [Google Scholar]
- Shah KN, Bhatt R, Rotow J, Rohrberg J, Olivas V, Wang VE, Hemmati G, Martins MM, Maynard A, Kuhn J, et al. (2019) Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat Med 25:111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, Riely GJ, Varella-Garcia M, Shapiro GI, Costa DB, et al. (2014) Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med 371:1963–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T, et al. PROFILE 1014 Investigators (2014) First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med 371:2167–2177. [DOI] [PubMed] [Google Scholar]
- Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura F, Nogami N, Kurata T, et al. FLAURA Investigators (2018) Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med 378:113–125. [DOI] [PubMed] [Google Scholar]
- Strebhardt K, Ullrich A (2006) Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer 6:321–330. [DOI] [PubMed] [Google Scholar]
- Subbiah V, Cascone T, Hess KR, Subbiah IM, Nelson S, Morikawa N, Nilsson MB, Bhatt T, Ali S, William WN, et al. (2018a) Multi-kinase RET inhibitor vandetanib combined with mTOR inhibitor everolimus in patients with RET rearranged non-small cell lung cancer. J Clin Oncol 36(suppl):9035. [Google Scholar]
- Subbiah V, Gainor JF, Rahal R, Brubaker JD, Kim JL, Maynard M, Hu W, Cao Q, Sheets MP, Wilson D, et al. (2018b) Precision targeted therapy with BLU-667 for RET-driven cancers. Cancer Discov 8:836–849. [DOI] [PubMed] [Google Scholar]
- Subbiah V, Velcheti V, Tuch BB, Ebata K, Busaidy NL, Cabanillas ME, Wirth LJ, Stock S, Smith S, Lauriault V, et al. (2018c) Selective RET kinase inhibition for patients with RET-altered cancers. Ann Oncol 29:1869–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, Asaka R, Hamanaka W, Ninomiya H, Uehara H, et al. (2012) RET, ROS1 and ALK fusions in lung cancer. Nat Med 18:378–381. [DOI] [PubMed] [Google Scholar]
- Tang A, Gao K, Chu L, Zhang R, Yang J, Zheng J (2017) Aurora kinases: novel therapy targets in cancers. Oncotarget 8:23937–23954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Hu H, Pan Y, Li Y, Ye T, Li C, Luo X, Wang L, Li H, Zhang Y, et al. (2012) RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol 30:4352–4359. [DOI] [PubMed] [Google Scholar]
- Yoh K, Seto T, Satouchi M, Nishio M, Yamamoto N, Murakami H, Nogami N, Matsumoto S, Kohno T, Tsuta K, et al. (2017) Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): an open-label, multicentre phase 2 trial. Lancet Respir Med 5:42–50. [DOI] [PubMed] [Google Scholar]
- Zhu VW, Madison R, Schrock AB, Ou SI (2020) Emergence of high level of MET amplification as off-target resistance to selpercatinib treatment in KIF5B-RET NSCLC. J Thorac Oncol 15:e124–e127. [DOI] [PubMed] [Google Scholar]