

Abstract
To develop an efficient antitumor immunotherapy, we have examined if dendritic cells (DCs) loaded with soluble antigens by electroporation present more antigens via the MHC (major histocompatibility complex) class I pathway, which mediate a cytotoxic T-cell response. DCs loaded with ovalbumin (OVA) by electroporation presented more MHC class I–restricted determinants compared with DCs pulsed with OVA. When electroporated DCs were pulsed with OVA for additional times, both MHC class I– and II–restricted presentation of OVA were increased compared with each single procedure, including electroporation or simple pulse. Immunization with DCs loaded with OVA by electroporation induced higher cytotoxicity of splenocytes to E.G7 cells, a clone of EL4 cells transfected with an OVA cDNA, than immunization with DCs pulsed with OVA. In the animal study, immunization with DCs loaded with OVA or tumor cell lysates by electroporation induced an effective antitumor immunity against tumor of E.G7 cells or Lewis lung carcinoma cells, respectively. In addition, immunization with DCs loaded with antigen by combination of electroporation and pulse, completely protected mice from tumor formation, and prolonged survival, in both tumor models. These results demonstrated that electroporation would be a useful way to enhance MHC class I–mediated antitumor immunity without functional deterioration, and that the combination of electroporation and pulse could be a simple and efficient antigen-loading method and consequently lead to induction of strong antitumor immunity.
Keywords: Dendritic cells, Cancer immunotherapy, Electroporation, Soluble antigens
Introduction
The ability of the immune system to recognize and attack tumors has been demonstrated unequivocally. Although both humoral and cellular effector arms of the immune response can contribute to tumor lysis, the latter appears to be responsible for tumor regression in the majority of cases through specific recognition of tumor-associated antigens (TAAs). Upon activation by dendritic cells (DCs), CD4+ helper T cells can cross-prime CD8+ cytotoxic T lymphocytes (CTLs) via IL-2 with IL-12 (and perhaps other cytokines) from the antigen-presenting cells (APCs). Antigen-specific CD8+ CTLs, if appropriately activated, can kill tumor cells [14]. Because DCs are the only APCs capable of initiating immune responses, and the ability of DCs to prime T cells capable of recognizing and killing tumor cells in an antigen-specific fashion has been demonstrated in various animal models [10, 19, 26, 44], many pilot clinical trials of DC-based cancer immunotherapy have been performed and yielded some promising results [2, 3, 20, 22, 30].
DCs are defined by their potency as APCs and distinction from other well-known, but less potent, APCs such as B cells and macrophages. Generally DCs being used in vaccine protocols have been derived from monocytes stimulated with IL-4 and GM-CSF or from monocyte precursors (CD34+ cells). Phenotypically immature DCs efficiently take up antigens through several mechanisms—including engulfment of apoptotic bodies, macropinocytosis and receptor-mediated endocytosis via mannose receptors and via CD32 and CD64 (these are Fc receptors) [7, 8, 38]. The captured antigens are delivered to MHC class II compartments, processed, and then directed to the cell surface as peptide-MHC complexes that can elicit CD4+ T-cell responses. Furthermore, it has been shown that DCs have the capacity to present exogenously introduced antigens on MHC I molecules via a mechanism that is proteasome dependent and TAP (transporter associated with antigen processing) dependent, a process referred to as “cross-presentation” [43]. Thus, DCs can stimulate both CD4+ and CD8+ T cells by specialized processing of exogenous antigens [41] and thereby induce strong antitumor immunity [11].
Although DCs are extremely efficient in taking up proteins, the processing of epitopes derived thereof into class I MHC requires high amounts of exogenous antigens and therefore appears to be rather inefficient, and the route of antigen internalization has been proposed to be critical for peptide-loading onto MHC class I molecules. Indeed, soluble proteins are poorly presented [23], whereas antigens introduced via receptor-mediated endocytosis and phagocytosis are presented efficiently by MHC class I molecules [1, 28, 32, 37].
The most commonly used and clinically approved DC-based vaccine is based on loading of empty MHC class I molecules with exogenous peptides. This is, however, limited by (1) peptide restriction to a given HLA type, (2) induction of CTL responses only, and (3) limitation of the induced responses to defined TAAs. In contrast to the peptide-based approach, unfractionated tumor material may provide both MHC class I and class II epitopes and does not require the identification of TAA [24]. Antigen presentation by both MHC class I and class II molecules leads to the diversification of immune responses and thereby induces strong antitumor immunity, and presentation of MHC class I antigens by DCs is an important pathway in priming CTL responses against tumor.
There are several sources of unidentified TAAs for antitumor immunotherapy, such as whole cell lysates, mRNA, eluted tumor peptides, apoptotic cells, and tumor cells fused with DCs [40]. Among these, the simplest approach to loading DCs with antigens in vitro is to pulse them with whole cell lysates [29]. However, development of more efficient antigen-loading methods may be required to improve the effectiveness of tumor cell lysates as an antigen source, since soluble antigens have been known to be poorly presented on MHC class I molecules via cross-priming.
Therefore, we have investigated if a direct introduction of ovalbumin (OVA) as a soluble antigen into the cytosol of DCs by electroporation leads to an increase in MHC class I–mediated presentation and consequently improves the effectiveness of soluble antigens as antigen sources.
Materials and methods
Cell lines and mouse strain
EL4 (a C57BL/6 T-cell lymphoma that expresses MHC class I but not class II determinants), E.G7 (a subline of EL4 cells transfected with cDNA of OVA), and T-T hybridomas, including DO11.10 (anti-OVA+I-Ad,b), which recognize OVA in association with both I-Ad and I-Ab [33, 42], and RF33.70 (anti-OVA+Kb) [34] cells, were cultured in RPMI 1640 supplemented with 10% heat-inactivated fetal calf serum (Life Technologies, Gaithersburg, MD), 2-mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. These cell lines were kindly provided by Dr Rock (Medical School, University of Massachusetts, MA). LL/2 Lewis lung carcinoma cells were maintained in DMEM supplemented with 10% heat-inactivated fetal calf serum, 2-mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Female wild-type C57BL/6 mice, 6–8 weeks of age, were obtained from Daehan Biolink (Chungbuk, Korea) and housed at the Animal Maintenance Facility of Pusan Medical Research Center. The animals were used for experiments at 7–9 weeks of age.
Generation of bone marrow–derived DCs
Bone marrow–derived DCs were generated as previously described [17], with minor modifications. Briefly, bone marrow cells flushed from tibias and femurs were depleted of erythrocytes by incubating in 0.9% ammonium chloride for 3 min at 37°C. The cells were washed in phosphate buffered saline, and cultured in complete medium (CM) consisting of OptiMEM (Life Technologies, Gaithersburg, MD) supplemented with 5% fetal bovine serum, recombinant mouse granulocyte/macrophage colony-stimulating factor (GM-CSF) 20 ng/ml (PeproTech, Rocky Hill, NJ), and recombinant mouse interleukin 4 (IL-4) 10 ng/ml (PeproTech, Rocky Hill, NJ) at 5×105 cells/ml. On the 3rd day in culture, the media and floating cells were removed and centrifuged for 5 min at 1,500 rpm. The cells were resuspended in CM and replaced in the original dishes. On day 6, nonadherent cells were harvested by gentle pipetting.
Antigen-loading into dendritic cells
OVA as antigen was loaded to DCs by electroporation, pulse, or combination of both. For electroporation, DCs (3.3×106 cells/500 μl of RPMI without serum and antibiotics) were incubated on ice for 10 min and mixed with OVA (4 mg/ml). Electroporation was carried out using Electro Cell Manipulator 2001 (BTX), which delivers a square wave pulse. Immediately after electroporation at various conditions, cuvettes were incubated on ice for an additional 10 min and then washed three times with cold PBS. For pulse, DCs (3.3×106 cells) were simply incubated in 4-ml CM containing OVA (4 mg/ml) at 37°C for 18 h. For the combination of both, the electroporated DCs were cultured with 4 mg/ml OVA in CM at 37°C for 18 h. When tumor cell lysates were used as antigen sources, 5×106 cells of DCs were treated and antigen concentration was maintained at 100 μg/ml during antigen-loading procedures.
Flow cytometric analysis
Stained cells were washed in PBS with 1% bovine serum albumin, resuspended in PBS with 1% glycerol, and analyzed with flow cytometry.
Immunization procedure
C57BL/6 mice were immunized subcutaneously with DCs (1×106 cells) in the right flank three times at 1-week intervals. Tumor cells were inoculated subcutaneously on the left flank 1 week after last immunization for a preventive immunity and 3 days before first immunization for a therapeutic immunity. All treated groups contained five mice. Tumor size was measured using calipers and calculated from the formula: Volume = (Width)2 × Length×0.52 [25].
IL-2 production assay
T-T hybridoma cells (1×105 cells/200 μl) were stimulated with various numbers of DCs loaded with OVA by various procedures for 6 h. Supernatants was collected after centrifugation at 250 g for 10 min. The amounts of IL-2 were determined with an ELISA kit (Mouse IL-2 BD OptEIA ELISA Set; BD Biosciences, Mountain View, CA) in triplicate.
Cytotoxicity assay
Splenocytes (3×107 cells/5 ml) from immunized or control mice were restimulated by coculture with mitomycin C (10 μg/ml for 20 min)-treated target cells (3×106 cells) including EL4 and E.G7 cells for 5 days. After restimulation, target cells (2×104 cells/well) were cultured with restimulated splenocytes at various ratios in 96-well round-bottom plates (200 μl/well) for 6 h at 37°C. After centrifugation for 10 min at 250 g, 100 μl of supernatants from triplicate cultures was collected and lactate dehydrogenase (LDH) released from target cells was measured in vitro using a Cytotoxicity Detection Kit (Roche Molecular Biochemicals) according to manufacturer’s protocol. Briefly, cell-free supernatants were incubated in a separate 96-well plate with LDH substrate for 30 min before measuring absorbance using a microplate reader (μQuant; Bio-Tek Instruments, Winooski, VT) at 490 nm. The percentage of cytotoxicity was calculated according to the following formula: % Cytotoxicity = [(E−St−Se)/(M−St)]×100 (with E being the LDH release by effector-target coculture, St the spontaneous release by target cells, Se the spontaneous release by effector cells, and M the maximal release by target cells). Spontaneous release of effector and target cells was controlled by separate incubation of the respective populations. Maximal LDH release enzyme release was measured after lysis of the target cells with lysis solution (1% Triton × 100) in triplicate.
Statistical analysis
An unpaired Student’s t-test was performed to analyze the statistical significance. P<0.05 was considered significant.
Results
Antigen-loading into dendritic cells by electroporation
Both MHC class I– and II–mediated immunities are required for optimal antitumor immunotherapy, and tumor-specific CTL response can be considered as an endpoint to the success of immunotherapy. Therefore, to deliver more antigens into the cytosol of DCs and consequently increase MHC class I–mediated presentation of antigens, optimal conditions for electroporation were examined. Uptake of FITC-labeled OVA by DCs after electroporation at various conditions was monitored by flow cytometry (Fig. 1A). Amount of OVA transferred into DCs was primarily dependent on voltage level up to 750 V/cm. In addition, when DCs were pulsed twice sequentially, uptake of OVA was markedly increased compared with single pulse. However, cell viability was significantly decreased to approximately 55% at voltages higher than 750 V/cm, at which level, cell viability was approximately 75% to 80% (data not shown). Therefore, electroporation with two pulses at 750 V/cm for 2 ms was performed to load OVA as tumor antigen into DCs for the next studies. To determine if the procedure of electroporation can affect the phenotype of DCs, the surface makers were analyzed by flow cytometry 1 day after electroporation. The phenotype of the electroporated DCs was similar to that of the control DCs, showing that electroporation did not affect the phenotype of DCs (Fig. 1B).
Fig. 1A, B.

Flow cytometric analysis of OVA-loading into DCs by electroporation (A) and phenotype of DCs after electroporation (B). A DCs were electroporated in the presence of FITC-labeled OVA (2 mg/ml). Immediately after electroporation under various conditions, cuvettes were incubated on ice for an additional 10 min and then washed three times with cold PBS. As controls, unstained cells and mock-treated cells were included. Fluorescence was analyzed with FACSort (Becton-Dickinson, Mountain View, CA) and measured as mean fluorescence intensity. B One day after electroporation procedure at 750 V/cm, 2 ms, and 2 pulses, expression of surface markers in electroporated DCs (lower panels) was compared with that of untreated control DCs (upper panels). Fluorescence was analyzed with a Coulter Epics XL cytometer (Beckman Coulter, FL)
Enhancement of MHC class I–mediated presentation of OVA by electroporation
To verify the hypothesis that the direct introduction of soluble tumor antigens into the cytosol of DCs by electroporation may increase MHC class I–mediated presentation, IL-2 production was assayed after stimulation of OVA-specific and MHC-restricted T-T hybridomas with DCs, into which OVA was loaded by electroporation, pulse, or combination of both (Fig. 2). DCs loaded with OVA by electroporation stimulated the production of IL-2 from RF33.70 cells, a MHC class I–restricted T-T hybridoma, more than DCs loaded with OVA by pulse (Fig. 2A). On the other hand, DCs loaded with OVA by pulse markedly stimulated the production of IL-2 from DO11.10 cells, a MHC class II–restricted T-T hybridoma, while electroporated DCs stimulated IL-2 production weakly (Fig. 2B). In both experiments, when MHC-restricted T-T hybridomas were stimulated with DCs loaded with OVA by combination of electroporation and pulse, more IL-2 was produced from both MHC-restricted T-T hybridomas compared with each procedure (Fig. 2). These results indicated that the electroporation technique would be useful for loading more exogenous soluble antigens into MHC class I molecules than the conventional pulsing method.
Fig. 2A, B.

Presentation of exogenous soluble OVA on MHC class I and II molecules by DCs after loading OVA by electroporation (E), pulse (P), or combination of both (E+P). MHC class I–restricted (RF33.70) (A) or class II–restricted (DO11.10) (B) T-T hybridomas (1×105 cells/200 μl) were stimulated with various numbers of DCs loaded with OVA by various procedures for 6 h. The amounts of IL-2 in supernatants were determined with ELISA. Data reported as the mean ± SE of five mice per group
Enhancement of OVA-specific cytotoxicity of splenocytes by immunization with DCs, into which OVA was loaded by combination of electroporation and pulse
To determine whether immunization with DCs loaded with OVA by electroporation leads to an increased MHC class I–mediated cytotoxicity of splenocytes, cytotoxicity of splenocytes isolated from immunized mice was examined against OVA-expressing E.G7 cells (Fig. 3). Immunization with DCs loaded with OVA by electroporation induced higher splenocyte cytotoxicity than immunization with DCs loaded with OVA by pulse (Fig. 3A). In addition, consistent with the IL-2 production assay, MHC class I–mediated cytotoxicity of splenocytes was also increased more by combination of electroporation and pulse than by each single method. Meanwhile, when EL4 cells were used as target cells, overt cytotoxic effect of splenocytes was not observed in any case, indicating that tumor cell lysis was antigen specific, depending on expression of OVA by the target cells (Fig. 3B).
Fig. 3A, B.

MHC class I–mediated and OVA-specific cytotoxicity of splenocytes by immunization with DCs, into which OVA was loaded by electroporation (E), pulse (P), or both (E+P). Splenocytes (3×107 cells/5 ml) from immunized or control mice were restimulated by coculture with mitomycin C (10 μg/ml for 20 min)-treated target cells (3×106 cells) including E.G7 (A) and EL4 (B) cells for 5 days. After restimulation, target cells (2×104 cells/well) were cultured with splenocytes at various ratios for 6 h. Supernatants from triplicate cultures were collected, and activities of LDH were assayed with Cytotoxicity Detection Kit (Roche Molecular Biochemicals). Data reported as the mean cytotoxicity ± SE of five mice per group
Induction of strong antitumor immunity by immunization with DCs, into which OVA was loaded by combination of electroporation and pulse
To determine the ability of antigen-loaded DCs to induce a protective antitumor immunity in vivo, mice were subcutaneously immunized three times at 1-week intervals with DCs into which OVA was loaded by electroporation, pulse, or combination of both, and then challenged subcutaneously at a distant site with EL4 or E.G7 cells. When mice were immunized with DCs loaded with OVA by electroporation or pulse, tumor formations by E.G7 cells were delayed and survival rates were prolonged compared with control mice (Fig. 4A). Meanwhile, mice immunized with DCs loaded with OVA by combination of electroporation and pulse were completely protected from tumor formation, and survived for long time periods tumor-free (Fig. 4C).
Fig. 4A–D.

OVA-specific antitumor immunity induced by immunization with DCs, into which OVA was loaded by electroporation (E), pulse (P), or both (E+P). C57BL/6 mice were immunized subcutaneously with DCs (1×106 cells) in the right flank three times at 1-week intervals. Tumor cells (5×104 cells) including E.G7 (A and C) or EL4 (B and D) cells were inoculated subcutaneously on the left flank of immunized mice 1 week after last immunization. Tumor size (A and B) was assessed once a week. Data were reported as the mean tumor size ± SE of five mice per group. Survival (C and D) was recorded as the percentage of surviving mice. The number of tumor-bearing mice compared with the total number of mice in each group is indicated in A
However, tumor formations (Fig. 4B) and survival (Fig. 4D) after inoculation with EL4 cells were not affected by immunization with DCs loaded with OVA by electroporation, pulse, or combination of both, indicating that antitumor immunity is OVA-specific.
Induction of therapeutic antitumor immunity against lung cancer cells by immunization with DCs, into which tumor cell lysates were loaded by combination of electroporation and pulse
Since we observed that antigen-loading by combination of electroporation and pulse led to induction of an effective antitumor immunity in an OVA model system, we examined therapeutic antitumor immunity against Lewis lung carcinoma after immunization with DCs into which tumor cell lysates were loaded by electroporation, pulse, or combination of both (Fig. 5). Tumor sizes were evaluated after 1 month of tumor inoculation. Although immunization with DCs loaded with total cell lysates by electroporation or pulse showed effective antitumor immunity, immunization with DCs loaded by combination of both resulted in no tumor formation. These results indicate that electroporation can be a generally applicable and effective antigen-loading method, and the combined method of electroporation and pulse is more effective than either single method, including electroporation and pulse.
Fig. 5.
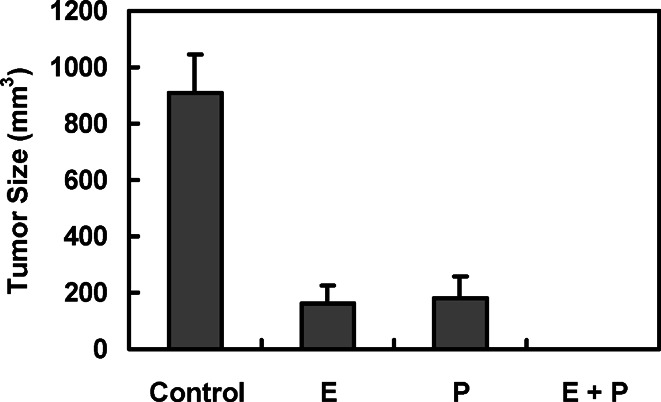
Induction of therapeutic antitumor immunity against Lewis lung carcinoma cells by immunization with DCs, into which total tumor cell lysates were loaded by electroporation (E), pulse (P), or combination of both (E+P). A solid tumor was established on the left flank by injecting LL/2 cells (1×105 cells). After 3 days, mice were immunized with antigen-loaded DCs (1×106 cells) three times at 1-week intervals. Tumor sizes were assessed 1 month after tumor inoculation. Data reported as the mean tumor size ± SE of five mice per group
Discussion
DCs are the most potent APCs identified thus far and are crucial for priming the immune response. An increasing number of studies have demonstrated that immunization with DC-based vaccines is capable of inducing a specific CTL and antitumor immune response. For example, DCs pulsed with MHC class I–restricted peptide induced antigen-specific CTL-mediated antitumor immunity in a number of experimental systems and human trials [4, 11, 27]. In addition, DCs were transduced with tumor antigen genes to elicit a protective and therapeutic antitumor immunity [5, 39]. However, these approaches require identification of tumor-specific antigens for individual tumors and the demonstration of their recognition by CTLs, a process that is difficult and tedious. Therefore, antigens that can be used to pulse DCs for immunotherapy are very limited at the present time [31, 36]. Although there are various sources of unidentified tumour antigens, due to its simplicity an interesting alternative may be the unfractionated tumor cell lysates, which induce effective antitumor immunity when pulsed into DCs [9]. In the present study, it was demonstrated that electroporation could improve the presentation of soluble OVA antigen on MHC class I molecules, and consequently the effectiveness of soluble antigens as an antigen source for DC-based cancer immunotherapy.
Electroporation has been widely used to deliver DNA, mRNA, drugs, and peptide vaccines for cancer treatment [12, 13]. In the present study, when DCs were electroporated twice at 750 V/cm for 2 ms, the electroporation efficiency was markedly improved without a change in the expression of surface markers and serious loss of viability. It was shown that proteins of up to 230 kDa were taken up by Chinese hamster ovary fibroblasts exposed to electroporation under conditions generally similar to those used to mediate DNA transfection [16]. Recently, a unique membrane transport pathway linking the lumen of endocytic compartments and the cytosol was identified in DCs [35]. This endosome-to-cytosol transport is restricted to DCs, specific to internalized antigens, and selective for the size (3–20 kDa) of the transported molecules. Therefore, it might be possible to deliver much larger tumor antigens into the cytoplasm of DCs by electroporation.
It was shown that electroporated B cells not only presented exogenous OVA to CD8+ MHC class I–restricted T cells but also stimulated CD4+ MHC class II–restricted T cells [18]. However, in the present study the DCs loaded with OVA by electroporation presented exogenous OVA primarily on MHC class I molecules and consequently stimulated a MHC class I–restricted immune response, but not efficiently on MHC class II molecules. This difference may originate from the different antigen-presenting cells used or the procedure of electroporation. Here, DCs were incubated on ice for 10 min immediately before and after electroporation and then washed three times with cold PBS to remove the opportunity to engulf soluble OVA as much as possible. When the electroporated DCs were pulsed with OVA for additional times, both MHC class I– and II–restricted presentation of OVA was increased compared with each single procedure, including electroporation or pulse. These results suggest that antigen delivery to DCs by electroporation provides a simple and efficient way to increase antitumor immunity without interference with the uptake and subsequent processing of antigen during additional pulses, and the combination of electroporation and pulse for antigen-loading leads to induction of strong antitumor immunity.
Although cross-presentation can be activated developmentally by a subset of maturation stimuli in DCs, such as CD40 ligation and disruption of cell-cell contacts [6], it seems likely that electroporation can improve the presentation of exogenous antigens to MHC class I molecules by delivering exogenous antigens directly into the cytoplasm via disruption of the lipid bilayer of the cell surface membrane [16, 21]. These exogenous antigens would be processed similarly to endogenously derived antigens, which are enzymatically digested into peptides, mainly by cytosolic proteases called proteasomes, and are then transported by TAP molecules into the endoplasmic reticulum (ER). In the ER lumen, peptides bind to MHC class I molecules, which are subsequently transported via the Golgi apparatus to the cell surface [15].
From recent clinical trials, patients treated with tumor cell lysates–loaded DCs showed better response rates compared with patients treated with peptide-loaded DCs [30], suggesting that tumor cell lysates are a good source of tumor antigens for a polyvalent antitumor vaccine. In the present study, immunization with DCs, into which tumor cell lysates were loaded by combination of electroporation and pulse, resulted in induction of an effective therapeutic antitumor immunity against Lewis lung carcinoma cells.
Taken together, our results suggest that electroporation can be combined with the conventional pulsing method to load more tumor antigens into the MHC class I pathway of dendritic cells and consequently improve the efficacy of tumor cell lysates as tumor antigens.
Acknowledgements
This work was supported by grant no. R01-2000-00119 from the Basic Research Program of the Korea Science & Engineering Foundation.
Abbreviations
- DCs
dendritic cells
- MHC
major histocompatibility complex
- OVA
ovalbumin
- TAA
tumor-associated antigen
- CTL
cytotoxic T lymphocyte
- LDH
lactate dehydrogenase
References
- 1.Bachmann Eur J Immunol. 1996;26:2595. doi: 10.1002/eji.1830261109. [DOI] [PubMed] [Google Scholar]
- 2.Banchereau Annu Rev Immunol. 2000;18:767. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 3.Brossart Exp Hematol. 2001;29:1247. doi: 10.1016/S0301-472X(01)00730-5. [DOI] [PubMed] [Google Scholar]
- 4.Celluzzi J Exp Med. 1996;183:283. doi: 10.1084/jem.183.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De J Immunol. 1999;162:144. [Google Scholar]
- 6.Delamarre J Exp Med. 2003;198:111. doi: 10.1084/jem.20021542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fanger J Immunol. 1996;157:541. [PubMed] [Google Scholar]
- 8.Fanger J Immunol. 1997;158:3090. [PubMed] [Google Scholar]
- 9.Fields Proc Natl Acad Sci U S A. 1998;95:9482. doi: 10.1073/pnas.95.16.9482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flamand Eur J Immunol. 1994;24:605. doi: 10.1002/eji.1830240317. [DOI] [PubMed] [Google Scholar]
- 11.Fong Annu Rev Immunol. 2000;18:245. doi: 10.1146/annurev.immunol.18.1.245. [DOI] [PubMed] [Google Scholar]
- 12.Gehl Acta Physiol Scand. 2003;177:437. doi: 10.1046/j.1365-201X.2003.01093.x. [DOI] [PubMed] [Google Scholar]
- 13.Hui Technol Cancer Res Treat. 2002;1:373. doi: 10.1177/153303460200100508. [DOI] [PubMed] [Google Scholar]
- 14.Hurwitz Curr Opin Immunol. 2000;12:589. doi: 10.1016/S0952-7915(00)00147-3. [DOI] [PubMed] [Google Scholar]
- 15.Kloetzel Nat Rev Mol Cell Biol. 2001;2:179. doi: 10.1038/35056572. [DOI] [PubMed] [Google Scholar]
- 16.Lambert Biochem Cell Biol. 1990;68:729. doi: 10.1139/o90-105. [DOI] [PubMed] [Google Scholar]
- 17.Lambert J Immunother. 2001;24:232. doi: 10.1097/00002371-200105000-00006. [DOI] [Google Scholar]
- 18.Li J Leukoc Biol. 1994;56:616. doi: 10.1002/jlb.56.5.616. [DOI] [PubMed] [Google Scholar]
- 19.Mayordomo Nat Med. 1995;1:1297. doi: 10.1038/nm1295-1297. [DOI] [PubMed] [Google Scholar]
- 20.McIlroy D, Gregoire M (2003) Optimizing dendritic cell–based anticancer immunotherapy: maturation state does have clinical impact. Cancer Immunol Immunother (in press) [DOI] [PMC free article] [PubMed]
- 21.Mir Exp Cell Res. 1988;175:15. doi: 10.1016/0014-4827(88)90251-0. [DOI] [PubMed] [Google Scholar]
- 22.Nestle Oncogene. 2000;19:6673. doi: 10.1038/sj.onc.1204095. [DOI] [PubMed] [Google Scholar]
- 23.Norbury Eur J Immunol. 1997;27:280. doi: 10.1002/eji.1830270141. [DOI] [PubMed] [Google Scholar]
- 24.Nouri-Shirazi Immunol Lett. 2000;74:5. doi: 10.1016/s0165-2478(00)00243-1. [DOI] [PubMed] [Google Scholar]
- 25.O’Reilly Nat Med. 1996;2:689. doi: 10.1038/nm0696-689. [DOI] [PubMed] [Google Scholar]
- 26.Paglia J Exp Med. 1996;183:317. doi: 10.1084/jem.183.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Porgador J Immunol. 1996;156:2918. [PubMed] [Google Scholar]
- 28.Regnault J Exp Med. 1999;189:371. doi: 10.1084/jem.189.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reid Br J Haematol. 2001;112:874. doi: 10.1046/j.1365-2141.2001.02626.x. [DOI] [PubMed] [Google Scholar]
- 30.Reinhard Br J Cancer. 2002;86:1529. doi: 10.1038/sj.bjc.6600316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Renkvist Cancer Immunol Immunother. 2001;50:3. doi: 10.1007/s002620000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rescigno Proc Natl Acad Sci U S A. 1998;95:5229. doi: 10.1073/pnas.95.9.5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rock J Exp Med. 1983;157:1618. doi: 10.1084/jem.157.5.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rock J Immunol. 1990;145:804. [PubMed] [Google Scholar]
- 35.Rodriguez Nat Cell Biol. 1999;1:362. doi: 10.1038/14058. [DOI] [PubMed] [Google Scholar]
- 36.Rosenberg Immunity. 1999;10:281. doi: 10.1016/s1074-7613(00)80028-x. [DOI] [PubMed] [Google Scholar]
- 37.Rovere J Immunol. 1998;161:4467. [PubMed] [Google Scholar]
- 38.Sallusto J Exp Med. 1995;182:389. doi: 10.1084/jem.182.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Specht J Exp Med. 1997;186:1213. doi: 10.1084/jem.186.8.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tarte Leukemia. 1999;13:653. [Google Scholar]
- 41.Thery Curr Opin Immunol. 2001;13:45. doi: 10.1016/S0952-7915(00)00180-1. [DOI] [PubMed] [Google Scholar]
- 42.White J Immunol. 1983;130:1033. [PubMed] [Google Scholar]
- 43.Yewdell Adv Immunol. 1999;73:1. doi: 10.1016/s0065-2776(08)60785-3. [DOI] [PubMed] [Google Scholar]
- 44.Zitvogel J Exp Med. 1996;183:87. doi: 10.1084/jem.183.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]