

Abstract
Vaccine-induced CD8 T cells directed to tumour-specific antigens are recognised as important components of protective and therapeutic immunity against tumours. Where tumour antigens have pathogenic potential or where immunogenic epitopes are lost from tumours, development of subunit vaccines consisting of multiple individual epitopes is an attractive alternative to immunising with whole tumour antigen. In the present study we investigate the efficacy of two DNA-based multiepitope (‘polytope’) vaccines containing murine (H-2b) and human (HLA-A*0201)–restricted epitopes of the E7 oncoprotein of human papillomavirus type 16, in eliciting tumour-protective cytotoxic T-lymphocyte (CTL) responses. We show that the first of these polytopes elicited powerful effector CTL responses (measured by IFN-γ ELISpot) and long-lived memory CTL responses (measured by functional CTL assay and tetramers) in immunised mice. The responses could be boosted by immunisation with a recombinant vaccinia virus expressing the polytope. Responses induced by immunisation with polytope DNA alone partially protected against infection with recombinant vaccinia virus expressing the polytope. Complete protection was afforded against challenge with an E7-expressing tumour, and reduced growth of nascent tumours was observed. A second polytope differing in the exact composition and order of CTL epitopes, and lacking an inserted endoplasmic reticulum targeting sequence and T-helper epitope, induced much poorer CTL responses and failed to protect against tumour challenge. These observations indicate the validity of a DNA polytope vaccine approach to human papillomavirus E7–associated carcinoma, and underscore the importance of design in polytope vaccine construction.
Keywords: Cytotoxic T lymphocyte, DNA vaccine, Human papillomavirus, Polytope, Tumour protection
Introduction
A number of squamous cell cancers are associated with human papillomaviruses (HPV). Mucoepitheliotropic HPV is detected in ca. 95% of cervical carcinomas, particularly the ‘high risk’ genotypes HPV 16 and 18. The oncogenic HPV E7 and E6 proteins are responsible for transforming cervical epithelial cells principally by abrogating the cell-cycle control functions of cellular retinoblastoma (RB) protein and p53, respectively [31, 33]. A corollary is that E7 and E6 proteins must persist for cells to remain transformed [43]. Thus, these viral proteins provide tumour-associated antigen (TAA) targets for immunotherapeutic strategies designed to eradicate HPV-associated neoplasia. CD8+ cytotoxic T lymphocytes (CTLs) are crucial components of protective immune responses to both murine [1] and human [30] tumours. CTLs kill tumour cells displaying epitopes derived from TAAs displayed on the tumour cell surface in conjunction with major histocompatibility complex (MHC) molecules. Using a number of experimental murine vaccines including peptides [13], plasmid DNA [20], viral vectors [23], recombinant fusion proteins [7] and chimeric virus-like particles (VLPs) [17], the CTL response to E7 protein has been shown to be an important determinant of HPV-associated disease outcome. While VLPs and viral vectors containing E7 are the most promising approaches which may be applicable to humans, putative disadvantages include the induction of neutralising antibodies against viral structural proteins in preimmune patients or following subsequent vaccination. The identification of human (and mouse) CTL epitopes in E7 protein restricted through MHC class I haplotypes (particularly HLA-A*0201) [12, 28] has provided the opportunity to develop epitope-based therapeutic vaccination approaches to HPV-associated tumours initially in HLA-transgenic mice [3, 10, 41] and subsequently in humans [17]. The outcome of E7-directed human clinical trials using peptide [4] or recombinant vaccinia viruses expressing E6–E7 [5] as immunogens has been equivocal, though CTL generation was seen in some patients. Thus, it is clear that more potent strategies for inducing CTLs, preferably against a broad array of E7 epitopes, are necessary for priming and boosting antitumour responses, particularly since peptide responses are likely to be short lived [37].
The use of recombinant DNA vaccines encoding individual CTL epitopes (usually eight to ten amino acids long) linked together in a linear ‘string-of-beads’ fashion has been a successful strategy for the simultaneous delivery of multiple CTL epitopes restricted through a number of class I haplotypes e.g. [35, 37]. The polytope approach has the added advantage of avoiding potential hazards associated with immunising with full-length antigens that are potentially oncogenic, such as HPV16 E7. Since it has been reported, at least for some polytope constructs, that each epitope within the construct may be individually immunogenic [35, 37], a polytope approach is attractive since a broad response directed simultaneously against multiple epitopes appears to be crucial for the development of effective vaccines against several diseases [19]. The multiple epitope approach is particularly applicable to cancer since expression of tumour-associated epitopes is highly variable, even within the same tumour mass [21].
In the present study we investigated the capacity of two DNA-based polytope vaccines containing murine (H-2b) [12] and human (HLA-A*0201) [28] restricted epitopes of the HPV16 E7, to elicit protective E7-directed CTL responses. One of the polytope vaccines, encoding—in addition to the CTL epitopes—an endoplasmic reticulum (ER) signal sequence and a T-helper epitope, elicited multiple effector and long-lived memory CTL responses. These responses could be boosted by subsequent immunisation with recombinant vaccinia containing the polytope. This polytope vaccine afforded complete protection against challenge with an E7-expressing tumour, and partial protection against E7-expressing tumour in a therapeutic setting. The polytope vaccine also afforded E7-specific partial protection against challenge with recombinant vaccinia expressing the polytope. A second polytope vaccine, differing in composition and order of epitopes, and lacking the ER signal sequence and T-helper (Th) epitope, induced poor CTL responses and failed to protect against tumour challenge.
These data suggest the use of a DNA polytope approach to protect against E7-associated disease. They confirm that CTL responses restricted through multiple MHC haplotypes may be induced by polytope vaccination, and underscore the importance of polytope design in the construction of maximally effective vaccines.
Materials and methods
DNA polytopes
Artificial DNApoly1 and DNApoly2 polytope DNA constructs were derived essentially as described previously [38], and inserted into the eukaryotic expression plasmid pDNAVacc. DNApoly1 was constructed to encode an a polytope protein containing an ER (Ig κ-chain) signal sequence [19], the PADRE Th epitope [3] and a number of CTL epitopes including the HLA-A*0201–restricted epitopes E7/A2/1 and E7/A2/2 [28], the H-2Db–restricted epitope E7/Db of HPV16 E7 [12], and the HLA-A*0201–restricted epitope of influenza matrix protein, flu/A2 [8] (Fig. 1). DNApoly2 was constructed without the PADRE epitope or the ER signal sequence, and with the above epitopes arranged in a different linear order (Fig. 1). In addition, DNApoly2 encoded a H-2b-restricted epitope of ovalbumin. Authenticities of DNApoly1 and DNApoly2 were confirmed by DNA sequencing. STMPDV [37] is a DNA polytope encoding murine CTL epitopes unrelated to this study, and was used as a control DNA vaccine. Plasmid DNA for DNA vaccination was prepared using a Qiagen EndoFree plasmid kit.
Fig. 1.

Schematic presentation of domain structure of DNApoly1 and DNApoly2 vaccines, and amino acid sequences of encoded epitopes relevant to the present study. DNApoly1 contains, in addition to encoded epitopes, an endoplasmic reticulum (Ig κ-chain) signal sequence. The DNA polytopes minigenes were subcloned from bacterial expression plasmids into a pcDNA3-derived eukaryotic expression vector as described [37]
Mice
A2.1 Kb mice express a chimeric HLA class I molecule, A2.1 Kb, composed of the α-1 and α-2 domains of HLA-A*0201, and the α-3 transmembrane and cytoplasmic domains of H-2 Kb [42], on a predominantly C57Bl (H-2b) background. A2.1 Kb mice are capable of making CTL responses restricted through both HLA-A*0201 and H-2b class I molecules. HHD mice express a transgenic monochain class I molecule in which the C terminus of human β2-microglobulin is covalently linked to the N terminus of a chimeric heavy chain (α-1 and α-2 of HLA-A*0201, and α-3 of H-2Db) [12]. The H-2Db and mouse β2-microglobulin genes have been disrupted. Thus, HHD mice are capable of HLA-A*0201–restricted CTL responses, but not H-2–restricted CTL responses. Founder mice from both lines were derived by caesarean section, and all mice were housed under specific pathogen-free conditions. Genetic authenticity was confirmed at intervals by PCR for the transgene. Mice were used at 7–15 weeks of age, but within a given experiment were littermates or closely age- and sex-matched.
Peptides
Peptides representing defined HPV16 E7-, and influenza virus matrix protein-, and ovalbumin CTL epitopes (Fig. 1) were synthesised with free ends using 9-fluorenylmethoxycarbonyl (F-moc) chemistry and analysed by HPLC by Chiron Corporation (Melbourne, Australia). Peptide stocks were made at 10 mg/ml in dimethyl sulphoxide and diluted in tissue culture medium for assays.
Immunisation of mice and restimulation of splenocytes for CTLs
For DNA immunisations, anaesthetised mice were injected twice at 3-weekly intervals intradermally (i.d.) in the ear with 100 μg, or intramuscularly (i.m.) in each quadriceps with 50 μg, of purified plasmid DNA. Three weeks later, spleens were removed, and splenocytes were restimulated in vitro for 6 days in 24-well tissue-culture plates (5×106 cells per well) in the presence of 1 μg/ml cognate peptide. In some experiments, mice were ‘boosted’ by intraperitoneal immunisation with 107 plaque-forming units of recombinant vaccinia containing DNApoly1 before spleens were removed. In the case of HHD mice, restimulations were done by coculturing splenocytes with lipopolysaccharide-induced syngeneic lymphoblasts loaded with peptide at 10 μg/ml [14]. For peptide immunisations, mice were immunised subcutaneously (s.c.) at the tail base with 50 μg peptide + 0.25 μg tetanus toxoid (TT) as a source of Th epitopes + 10 μg Quil A adjuvant [15]. Ten days later, spleens were harvested, and splenocytes were restimulated as above.
Cells
EL4.A2 cells were derived by transfection of EL4 cells with A2.1 Kb plasmid encoding the chimeric MHC class I heavy chain (above), as decribed previously [9]. EL4.A2 cells are susceptible to specific CTL lysis through both H-2b and HLA-A*0201 restriction pathways. EL4S3− RobHHD cells [26], negative for endogenous MHC and transfected with the HHD construct, were maintained under G418 selection. TC-1 is an E7-expressing epithelial tumour cell line [23]. Cells were maintained in RPMI medium (Gibco) supplemented with 2 mM glutamine, 1 mM sodium pyruvate, 20 mM HEPES, 5×10−5 M β-mercaptoethanol, 100 IU/ml penicillin, 100 μg/ml streptomycin and 10% foetal bovine serum.
CTL assays
Cytotoxic T lymphocyte assays were conducted as previously described [9]. In summary, target cells (104 per well) sensitised at 37°C for 1 h with 1 μg/ml cognate or irrelevant peptide, or medium alone, or expressing the E7 gene, and labelled with 100 μCi chromium 51 (51Cr), were incubated with effector cells at various effector to target cell ratios in triplicate in 96-well microtitre plates. Negative controls included wells containing target cells but no effector cells (‘background’). Supernatants were harvested from CTL assays at 4 h, and 51Cr release quantified by gamma counting. Results are expressed as percentage cytotoxicity ± SD (51Cr release in experimental wells minus background / detergent-mediated total release minus background) × 100%.
ELISpot
Epitope-specific interferon gamma (IFN-γ)–secreting spleen cells were enumerated ex vivo by an enzyme-linked immunosorbent spot (ELISpot) assay with minimal CD8+ T-cell epitope peptides, essentially as described [22]. Microwell plates (MultiScreen-HA; Millipore, North Ryde, NSW, Australia) were coated with capture antibody (rat antimouse IFN-γ, clone RA-6A2; BD PharMingen, San Diego, CA, USA), blocked with DMEM 10% FBS, and washed. Serial dilutions of spleen cells, starting at 106/well were added, with 100 U/ml recombinant human interleukin 2 (Sigma, Castle Hill, NSW, Australia) and incubated with 10 μg/ml peptide or without peptide, for 18 h at 37°C. The cells were then lysed, the plates washed, and detection antibody (biotinylated antimouse IFN-γ, clone XMG 1.2; BD PharMingen) was added for 2 h at 37°C. After further washing, plates were developed using streptavidin-alkaline phosphatase (BD PharMingen) and BCIP-NBT substrate (Sigma FAST 5-bromo-4-chloro-3-indolylphosphate-nitroblue tetrazolium). IFN-γ spots were counted using an AID EliSpot reader system. Results were calculated as IFN-γ positive cells/106 spleen cells.
Recombinant vaccinia
A BamHI/SalI–excised DNApoly1 insert was cloned behind the vaccinia p7.5 promoter in the plasmid shuttle vector pBCB07 [37]. Construction of a TK− recombinant virus was then conducted using marker rescue recombination as described previously [35].
MHC class I tetramers and FACS staining
Phycoerythrin (PE)–conjugated HLA-A*0201 tetramers of epitopes E7/A2/2 and flu/A2 were constructed by the NIH Tetramer Facility (Atlanta, GA, USA). For staining, splenocytes or post in vitro restimulation cells were separated over Ficoll-Paque (Pharmacia, Uppsala), and 106 leukocytes costained with tetramer and FITC-antimouse CD8a (Sigma) at 4°C for 60 min, prior to washing, fixation in 1% paraformaldehyde and FACS analysis. Tetramer dilutions and staining conditions were optimised in preliminary experiments. Data sets included staining with ‘irrelevant’ MHC-matched tetramer, and staining of cognate tetramer on ‘irrelevant’ CD8+ T cells as negative controls. FACS plots record 60,000 events. Cells positive for both tetramer and CD8 staining are expressed as a percentage of total CD8+ive cells.
Tumour protection assays
To assay tumour prevention, groups of H-2b mice (ten per group) were immunised with 100 μg DNApoly1 or pSTMPDV DNA i.d., or 50 μg of E7 peptide + tetanus toxoid + Quil A s.c., before TC-1 cells (2×105 in 0.1 ml Hank’s buffered salt solution) were injected s.c. on the left flank using a 21-gauge needle. (The tumour dose was predetermined by titration experiments to discern a minimal dose giving rise to tumour in 100% of unimmunised mice). Tumour growth was monitored every 2 days, and mice were euthanised when tumour volume exceeded 1,000 mm3. Unimmunised mice received the same number of cells and served as a control. Data are represented as Kaplan-Meier curves of percentage tumour-free mice at given time points after tumour injection.
For tumour therapy experiments, groups of H-2b mice (nine to ten per group) first received a s.c. injection of 1×105 TC-1 cells, in the flank. To evaluate the therapeutic efficacy of polytope immunisation, mice were immunised with 100 μg DNApoly1 or ‘irrelevant’ polytope DNA (STMPDV) three times on days 7, 14 and 21 after tumour inoculation. Tumour growth was monitored by calliper measurement in two perpendicular directions every 2 days, and volume of tumour was estimated by the formula: S2L, where S is the shorter dimension and L is the longer dimension of the tumour. Mice were euthanised when tumour volume exceeded 1,500 mm3, or earlier when mice showed signs of sickness. The significance of differences in rates of appearance of measurable tumour between DNApoly1- and STMPDV-immunised mice was calculated by the Wilcoxon two-tailed rank sum test.
Vaccinia protection challenge model
Groups of mice were immunised twice with 100 μg of pDNApoly1 or pSTMPDV DNA and 3 weeks later challenged intraperitoneally with 107 plaque-forming units (pfu) per mouse of recombinant vaccinia encoding DNApoly1. At day 4 postinfection, both ovaries were removed, washed and homogenised. A 100-μl aliquot of ovary suspension was then mixed with PBS containing trypsin (Sigma) at 1mg/ml, and incubated for 30 min at 37°C. The solution was briefly centrifuged at 10,000 g to remove debris, and 100 μl of the supernatant was mixed with 900 μl RPMI + 10% FCS medium. The vaccinia virus titres in the ovaries were then determined by plaque assay on confluent CV1 cells. The significance of the difference between the virus titres in the experimental and control groups was calculated by the Wilcoxon two-tailed rank sum test.
Results
Polyepitopes
Two polytope DNA vaccines, DNApoly1 and DNApoly2, were constructed as described (‘Materials and methods’). Both encoded the HPV 16 E7 H-2Db–restricted CTL epitope (E7/Db), and the HPV16 E7 HLA-A*0201–restricted CTL epitopes (E7/A2/1, E7/A2/2) (Fig. 1). They also encoded an HLA-A*0201–restricted influenza matrix CTL epitope (flu/A2) as a ‘control’ epitope, since flu/A2 has previously been shown to elicit powerful CTL responses in HLA-A2–transgenic (A2.1 Kb) mice when injected as a peptide immunogen [9]. In addition, DNApoly2 encoded a H-2b–restricted CTL epitope of ovalbumin (Ova/Kb). With a view to maximising immunogenicity, we included DNA encoding the ‘universal’ Th epitope PADRE (pan-DR epitope) [2] in DNApoly1 (Fig. 1). PADRE has been shown to have a high MHC-binding affinity to a wide range of mouse and human MHC class II haplotypes and to induce Th responses restricted through class II of both species. DNApoly1 was constructed to encode also an ER–translocating signal sequence (an Ig κ-chain signal sequence). This signal targets processing of proteins to the ER where they are degraded into peptides and loaded onto MHC class I molecules [4], a pathway important for CTL induction [39].
Immunogenicity of polytope DNA vaccines in HLA-A2–transgenic mice
In this study, we wished to evaluate the capacity of polytope vaccines encoding CTL epitopes of HPV16 E7 to elicit multiple CTL responses measured in vitro, and to confer E7-directed protection in vivo. First we assessed the capacity of DNApoly1 to induce CTL responses measured in vitro to E7 CTL epitopes (E7/Db, E7/A2/1, and E7/A2/2) and the flu/A2 CTL epitope. HLA-A2–transgenic (A2.1 Kb) mice immunised i.d. with DNApoly1 (Fig. 2a), but not mice immunised i.m. with DNApoly1 (Fig. 2b), displayed epitope-specific CTL responses to three of the four epitopes tested: namely, E7/Db, E7/A2/2, and flu/A2. The responses were approximately of the same magnitude as those elicited in peptide-immunised mice (Fig. 2c). Thus, strong responses were recorded for E7/Db and flu/A2 epitopes, and a lesser response recorded for E7/A2/2 epitope. We were unable to detect a DNApoly1-induced response to E7/A2/1 epitope (Fig. 2b), even though a response to this epitope was induced by immunisation with E7/A2/1 peptide (Fig. 2c).
Fig. 2.

Immunisation of A2.1 Kb mice with DNApoly1 elicits epitope-specific memory CTL responses. Pooled splenocytes from mice (three per group) immunised twice with DNApoly1 DNA i.d. (a) or i.m. (b), or immunised once with an equimolar mix of epitope peptides (c), were restimulated with a single cognate peptide and reacted with EL4.A2 cells pulsed with the same peptide (open symbols) or without peptide (filled symbols), in a 51Cr-release cytotoxicity assay. Data points are means of triplicate tests ± standard deviation (SD). (Error bars, though plotted, do not appear, because SDs do not exceed symbol size). Data are from one of three experiments with similar results
We endeavoured to augment E7 HLA-restricted CTL responses by immunising HHD-A2–transgenic mice. HHD mice are constructed to be incapable of mounting CTL responses restricted through the H-2 locus [14]. We reasoned that elimination of a dominant E7/Db response might enhance E7 responses restricted through HLA-A*0201. We examined the A2-restricted effector CTL response in ex vivo splenocytes from DNApoly1-immunised HHD mice by IFN-γ ELISpot. Significant numbers of spots were recorded when ex vivo splenocytes were incubated with E7/A2/2 and flu/A2 peptide, but not with E7/A2/1 peptide or no peptide (Fig. 3a). We also examined memory CTL responses in splenocytes restimulated for a further 6 days in vitro with cognate peptide by 51Cr-release assay against peptide-pulsed EL4.A2 target cells. Splenocytes restimulated with E7/A2/2 or with flu/A2 specifically killed target cells coated with these peptides respectively (Fig. 3b). The relative amounts of cytotoxicity in each case mirrored that seen in peptide-immunised control mice (Fig. 3b). These data confirm the induction of an E7/A2/2-directed CTL response in A2-transgenic mice immunised with DNApoly1, but suggest that removal of a dominant E7/Db response did not enhance effector or memory CTL responses to HLA-A*0201–restricted E7 epitopes.
Fig. 3.
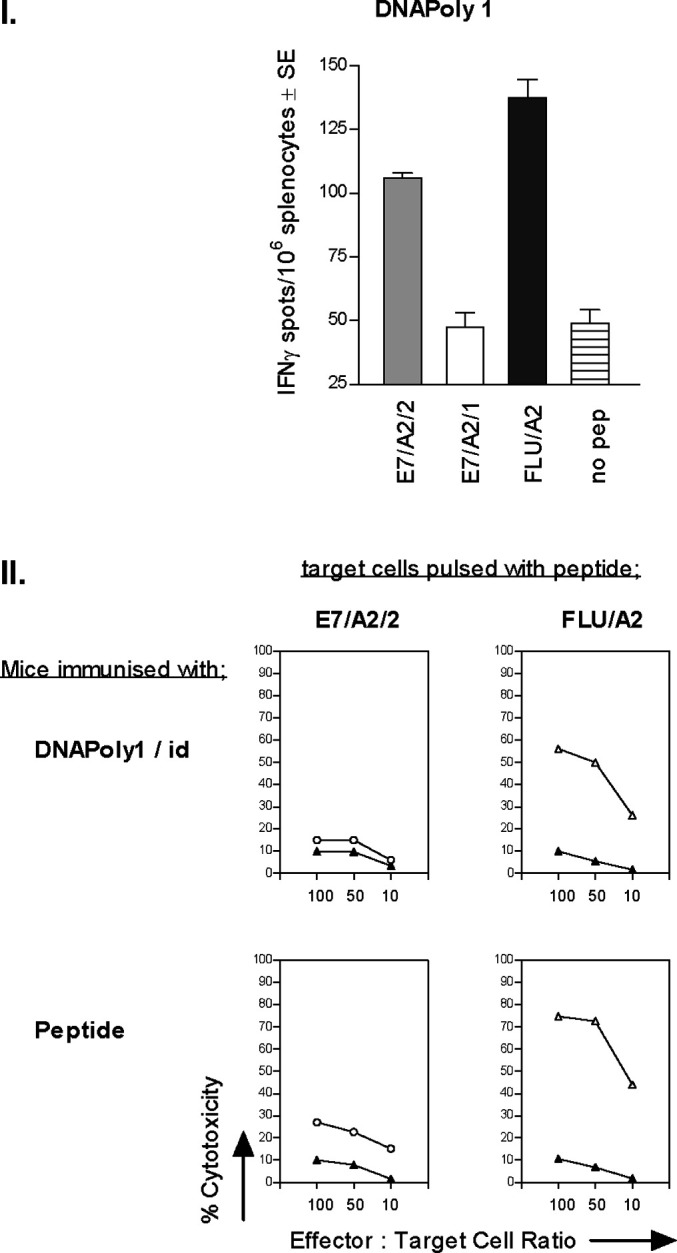
Immunisation of HHD mice with DNApoly1 DNA elicits epitope-specific effector and memory CTL responses: a Pooled ex vivo splenocytes from mice (three per group) immunised twice i.d. with DNApoly1 were incubated with or without E7/A2/2 or flu/A2, peptide as indicated, and IFN-γ–secreting cells were quantified by ELISpot assay. b Pooled splenocytes from mice (three per group) immunised i.d. twice with DNApoly1 DNA, or with an equimolar mix of epitope peptides, were restimulated with a single cognate peptide in vitro and reacted with EL4.A2 peptide pulsed target cells (open symbols), or without peptide (filled symbols) as indicated in a 51Cr-release cytotoxicity assay. Data points are means of triplicate tests ± SD. (Error bars, though plotted, do not appear, because SDs do not exceed symbol size). Data are from one of two experiments with similar results
DNApoly1-induced responses are boosted by recombinant vaccinia encoding the polytope
Boosting with virally encoded protein will often enhance the immune response generated by prior immunisation with DNA encoding that protein [27]. We therefore asked whether DNApoly1-immunised mice would mount augmented CTL responses when subsequently ‘boosted’ with recombinant vaccinia encoding the same polyepitope (Vacpoly1). Groups of A2.1 Kb mice were immunised twice with DNApoly1 either i.d. or i.m., or not immunised. The mice then received a ‘boost’ inoculation with Vacpoly1, 3 weeks later. Memory CTL responses measured by 51Cr-release assay, and the number of cognate CD8+ive T cells measured by specific MHC class I tetramer staining, were recorded. As indicated above, no measurable CTL response was seen in restimulated splenocytes from mice immunised i.m. with DNApoly1 alone (Fig. 2b). However, after boosting with Vacpoly1, easily measurable CTL responses were recorded against E7/Db- and flu/A2-coated target cells (Fig. 4b). To quantify more accurately the response, we stained flu/A2-restimulated splenocytes with flu tetramer, in parallel. The number of CD8+ive splenocytes bearing flu/A2-specific T-cell receptors increased after Vacpoly1 boosting in mice which had received DNApoly1 i.m. (0.6–34.5%; Fig. 4b). Immunisation with DNApoly1 alone i.d. elicits powerful E7/Db and flu/A2 CTL memory responses, which was not improved upon by boosting with Vacpoly1, when measured by 51Cr-release assay (cf. Figs. 2a and 4a). However, an augmentation in the numbers of cognate epitope-specific T cells after VacPoly1 boosting was demonstrated in these mice by staining with flu/A2 tetramer (7.6–18.3%; Fig. 4a). (Note that although a Vacpoly1 inoculation of mice which have not been previously immunised with DNApoly1 induces a CTL response to E7/Db and flu/A2 [Fig. 4c], the response is relatively small compared with Vacpoly1-boosted DNApoly1-immunised mice, when measured by specific 51Cr-release or by staining with flu tetramer [2.2%]).
Fig. 4.

Cytotoxic T-lymphocyte responses in DNApoly1-immunised mice are ‘boosted’ by subsequent inoculation with vaccinia virus encoding the polytope (Vacpoly1). Pooled restimulated splenocytes from A2.1 Kb mice (three per group) immunised twice with DNApoly1 i.d. (a), DNApoly1 i.m. (b), or unimmunised (c), were ‘boosted’ with VacPoly1, 3 weeks after the second immunisation. Spleens were harvested 2 weeks after ‘boost’, and CTL responses measured in specifically restimulated splenocytes by 51Cr-release cytotoxicity assay, and by staining with flu/A2-specific PE-tetramer. (d) 51Cr-release cytotoxicity and tetramer staining using pooled splenocytes from mice (three per group) immunised once with peptide mix + tetanus toxoid + Quil A, 10 days prior to in vitro restimulation. In 51Cr-release assays, open symbols represent EL4.A2 target cells pulsed with peptide; filled symbols represent unpulsed EL4.A2 target cells. Data points are means of triplicate tests ± SD. (Error bars, though plotted, do not appear, because SDs do not exceed symbol size). Data are from one of two experiments with similar results
Together, the data in Figs. 2 and 4 indicate that boosting with vaccinia encoding the DNA polytope enhances epitope-specific T-cell responses in mice previously immunised with DNApoly1.
CTL response induced by DNApoly1 immunisation is long-lived
We asked whether the CTL response induced by DNApoly1 immunisation persisted for a long period. Splenocytes from mice harvested 16 weeks after immunisation with DNApoly1 were restimulated in vitro, and reacted with cognate peptide-pulsed target cells in a 51Cr-release cytotoxicity assay, or reacted with specific tetramer. In mice immunised i.d. with DNApoly1, the flu/A2-directed cytotoxicity response measured at 16 weeks postimmunisation had not diminished compared with the response measured at 6 weeks (cf. Figs. 2a and5). Indeed the response remained as high as that recorded in mice recently immunised (10 days previously) with a vast molar excess of flu/A2 peptide (Fig. 5c). In agreement with this finding, flu/A2-specific tetramer binding at 16 weeks (5.7%) had barely diminished compared with that at 6 weeks (7.6%) (cf. Figs. 4a and 5a). The E7/Db-specific CTL response fell somewhat at 16 weeks compared with 6 weeks postimmunisation (cf. Figs. 2a and 5a). Together, these data indicate that intradermal immunisation with DNApoly1 elicits long-lived memory CTL responses, which, for at least one epitope (flu/A2), remain undiminished at 16 weeks. When the intramuscular route was used for immunisation with DNApoly1 no CTL responses to flu/A2 (measured by 51Cr-release or by tetramer) or to E7/Db (measured by 51Cr-release) were seen at 16 weeks (Fig. 5b) consistent with our findings at 6 weeks postimmunisation (Fig. 2b).
Fig. 5.

Immunisation with DNApoly1 induces long-term memory. Pooled splenocytes from A2.1 Kb mice (three per group) immunised twice with DNApoly1 i.d. (a), DNApoly1 i.m. (b), were restimulated in vitro 16 weeks after the second immunisation, and CTL response measured by 51Cr-release cytotoxicity assay against EL4-A2 target cells, and by staining of effector splenocytes with flu/A2 PE-tetramer. (c) CTL response measured by 51Cr-release cytotoxicity assay using pooled splenocytes from ‘control’ mice (three per group) immunised once with peptide mix + tetanus toxoid + Quil A, 10 days prior to in vitro restimulation. In 51Cr-release assays, open symbols represent EL4.A2 target cells pulsed with cognate peptide, filled symbols represent unpulsed EL4.A2 target cells. Data points are means of triplicate tests ± SD. (Error bars, though plotted, do not appear, because SDs do not exceed symbol size). Data are from one of two experiments with similar results
Poor response to epitopes encoded by DNApoly2
We investigated the immunogenicity of a second DNA polytope vaccine, DNApoly2 (Fig. 1b). DNApoly2 differs from DNApoly1 in not having an ER signal sequence or PADRE Th epitope, and in the arrangement of epitopes (Fig. 1). A2.1 Kb mice were immunised with DNApoly2, and CTL responses to encoded epitopes E7/Db, flu/A2 and OVA/Kb measured in restimulated splenocytes by 51Cr-release assay on peptide pulsed target cells. No CTL responses against either E7/Db or flu/A2 were recorded (Fig. 6a). However, a specific CTL response directed to OVA/Kb epitope was observed (Fig. 6a) which approximated in magnitude that seen in OVA peptide immunised mice (Fig. 6c), though it was not as great as that seen in mice immunised with a control DNA polytope, pSTMPDV, which also encodes the OVA/Kb epitope. These findings demonstrate that though immunogenic (for OVA/Kb), DNApoly2 was less effective than DNApoly1 at inducing CTL memory responses to E7/Db and flu/A2 epitopes encoded by both vaccines.
Fig. 6.

Immunisation with DNApoly2 fails to induce E7/Db or flu/A2-directed immune responses. Pooled splenocytes from A2.1 Kb mice (three per group) immunised twice i.d. with DNApoly2 DNA (a) STMPDV DNA (b) or once with peptide mix + tetanus toxoid + Quil A (c), were restimulated with individual peptides and reacted with EL4.A2 cells pulsed with the cognate peptide (open symbols) or without peptide (filled symbols) in 51Cr-release cytotoxicity assay. Data points are means of triplicate tests ± SD. (Error bars, though plotted, do not appear, because SDs do not exceed symbol size). Data are from one of two experiments with similar results
Immunisation with DNApoly1 induces in vivo protection against proliferation of recombinant vaccinia encoding E7 and flu epitopes
We investigated whether CTL responses induced by immunisation with DNApoly1 correlated with in vivo protection. Mice were immunised twice with DNApoly1 or with ‘irrelevant’ DNA polytope STMPDV, and 3 weeks after the second immunisation, challenged intraperitoneally with Vacpoly1. Four days later, mice were sacrificed, ovaries removed and homogenised, and the amount of vaccinia virus they contained determined by plaque assay. In a parallel group of immunised mice, spleens were removed on the day of vaccinia challenge and assessed for CTL in order to confirm a concomitant epitope-specific T-cell response. Mice immunised with DNApoly1 and challenged with Vacpoly1 had significantly lower titres of Vacpoly1 recorded in the ovary, compared with mice immunised with the ‘irrelevant’ polytope STMPDV identically challenged with Vacpoly1 (p<0.001) (Fig. 7a). This specific protection correlated with immunisation-induced E7/Db- and flu/A2-directed CTL responses present at the time of Vacpoly1 challenge (Fig. 7b). These data indicate that DNApoly1 immunogen elicits protective immune responses in vivo. It also demonstrates that the protective effect correlates with concomitant CTL responses measured in vitro to epitopes encoded by DNApoly1 and the recombinant vaccinia used for challenge.
Fig. 7.

Recombinant vaccinia virus challenge and CTL response following polytope DNA immunisation. a Mice (three per group) were immunised once with pDNApoly1 or with pSTMPDV DNA (control), and then challenged intraperitoneally with 107 pfu of vaccinia recombinant for DNApoly1(Vacpoly1). Vaccinia virus titres in the ovaries were measured 4 days later. Histogram bars represent the means of individual mice ± SD. b Pooled restimulated splenocytes from a parallel group of DNApoly1-immunised mice (three per group) identical to those subject to Vacpoly1 challenge were reacted with EL4.A2 cells pulsed with the cognate peptide (open symbols) or without peptide (filled symbols) in 51Cr-cytotoxicity assay. Data points are means of triplicate tests ± SD. (Error bars, though plotted, do not appear, because SDs do not exceed symbol size.)
Tumour prevention
We asked whether immunisation with DNApoly1 would protect against growth of a challenge inoculum of E7-expressing tumour. Groups of mice were immunised twice i.d., either at the tail base, or in the ear pinna, with DNApoly1, with ‘irrelevant’ STMPDV, or were left unimmunised. Two weeks after the second immunisation, the mice received an inoculum of E7-expressing TC-1 tumour cells, and tumour growth was monitored over the ensuing weeks. All unimmunised and irrelevantly immunised mice had acquired tumour by day 14 (Fig. 8). In contrast, tumour growth was abolished or delayed in mice immunised with DNApoly1. One hundred percent (10/10) of mice immunised with DNApoly1 at the tail base and 80% (8/10) immunised with DNApoly1 in the ear pinna remained tumour free at 14 days (Fig. 8). Protection was durable; 100% and 70% of mice immunised with DNApoly1 in the tail or ear pinna, respectively, remained free of tumour at 100 days after tumour challenge.
Fig. 8.

Tumour growth in immunised mice. Growth of E7-expressing tumour (TC-1) in H-2b mice (ten per group) immunised twice on the days indicated with 100 μg DNApoly1 i.d. in the ear pinna or at the base of tail, or with 100 μg pSTMPDV DNA i.d. at the base of the tail. Control groups of mice (ten per group) were immunised with 30 μg E7/Db peptide + tetanus toxoid + Quil A adjuvant, or were unimmunised. Mice were immunised 2 and 4 weeks before challenge with 2×105 TC-1 cells (s.c.) on day 0. Results are expressed as tumour-free mice (%) at the indicated time points. Data are from one of three experiments with similar results
Tumour therapy
We investigated whether growth of a preexisting tumour challenge would be controlled by subsequent immunisation with DNApoly1. Groups of mice were inoculated with TC-1 tumour cells, and 7, 14 and 21 days later immunised with DNApoly i.d. in the ear pinna or tail base, or with ‘irrelevant’ STMPDV DNA. Mice were monitored for tumour growth every 2 days, and tumour measured. Quantifiable tumours first appeared in irrelevantly immunised mice on day 14, and 100% (9/9) of these mice had developed tumour by day 19 (Fig. 9a). In contrast, significantly fewer mice displayed tumours and at later points in those groups immunised with DNApoly1 (p<0.005) (Fig. 9a). As well as delayed appearance of quantifiable tumour, retardation of tumour growth was evident in some individual DNApoly1-immunised mice when progressive tumour volumes were measured (Fig. 9b).
Fig. 9.

Therapy against early TC-1 tumours. Mice (ten per group) received 1×105 TC-1 cells s.c. on day 0. Mice subsequently received 100 μg DNA vaccine (pDNApoly i.d. in the ear pinna or tail base, or irrelevant pSTMPDV i.d. in the ear pinna) three times on days 7, 14 and 21 (a). Tumour-free mice (%) at the indicated time-points. (b) Onset and growth of tumours in evaluable individual mice, by group
Discussion
Polytope vaccines against a number of diseases have been shown to induce multiple protective CD8+ive CTL responses in experimental systems (reviewed in [20]), and some polytopes are in clinical trials [18, 34]. A preferred strategy is to induce a broad CTL response directed against several CTL epitopes simultaneously, which confers advantages where individual epitopes may be lost or mutated or where the target antigen is oncogenic (and therefore cannot be used as an immunogen). Inclusion of epitopes restricted through a range of MHC class I haplotypes allows wide coverage of a MHC polymorphic population. Epitopes from diverse antigens may be combined in a single polytope construct.
HLA-transgenic mice are accepted as a relevant model for the induction of human MHC class I–restricted CTL responses, since there is sufficient similarity in murine and human T-cell repertoires for murine CD8+ T cells to recognise the same HLA-restricted epitopes as those recognised by human CTLs which bear the corresponding HLA haplotype [10, 14]. Thus HLA-transgenic mice provide an animal model for preclinical evaluation of immunoprotection against tumours and other diseases.
In this study we show that immunisation of HLA-A2–transgenic mice with a DNA polytope vaccine, DNApoly1, containing murine (H-2b)- and human (HLA-A*0201)–restricted CTL epitopes of HPV 16 E7 and influenza matrix protein, elicited multiple effector CTL responses (measured by ex vivo IFN-γ ELISpot) and memory CTL responses (measured by 51Cr-release cytotoxicity and tetramer staining following in vitro restimulation of splenocytes). The memory responses were enhanced by a ‘boost’ with recombinant vaccinia virus expressing the same epitopes (Fig. 4). While the responses were readily detectable in mice immunised i.d., neither effector nor memory CTL responses were detectable in mice immunised i.m. with polytope DNA alone, though a memory response was recalled by boosting with the recombinant vaccinia (Fig. 4). The responses we demonstrate to DNApoly1-encoded epitopes were long lived, being easily demonstrable 16 weeks after immunisation (Fig. 5). (Our data do not address whether these represent effector memory (TEM) or central memory (TCM) responses or both. Both memory subsets occur in the spleen, and are efficient at becoming effector killer cells after stimulation with antigen [32]).
The magnitude of responses to individual epitopes following DNApoly1 immunisation approximated in general to those seen by immunisation with corresponding peptides (Fig. 2a, c). Immunisation with a molar excess of peptide under the appropriate conditions (i.e. plus a Th epitope and Th1-predisposing adjuvant, as used in our experiments), provides a standard against which polytope and other immunisation modalities may be compared.
Responses to E7/Db epitope and flu/A2 epitope were ‘strong’, while much ‘weaker’ responses were seen to epitope E7/A2/2 in DNApoly1 immunised mice (Fig. 2). We hypothesised that a dominant E7-directed E7/Db CTL response may have impaired a subdominant response to E7/A2/2. Thus, we sought to enhance the E7/A2/2 response by immunising HLA-A-2–transgenic HHD mice which are incapable of making murine (H-2)–restricted responses, but to no avail (Fig. 3). The possibility remains that the E7/A2/2 response remained low in DNApoly1-immunised HHD mice because of an immunodomination effect [44] exerted by the ‘strong’ A2-restricted flu/A2 response. At least in some reports in the literature, removal of a dominant epitope from a polytope vaccine did not improve responses to a subdominant epitope within the same vaccine [20]. The observation that E7/A2/2 elicits low responses in a variety of immunisation scenarios (including when given as peptide) in both A2-transgenic mice and HLA-A*0201 humans [17], suggests that E7/A2/2 is constitutively weakly immunogenic, i.e. does not display immunodominance [44].
We demonstrate that the E7-directed response induced by immunisation with DNApoly1 was functional in vivo to effect a log-fold reduction in vaccinia infectious units in the ovary following a challenge with recombinant vaccinia expressing the polytope (Vacpoly1) (Fig. 7). We also demonstrate that intradermal immunisation with DNApoly1 at the tail base afforded complete and long-lived protection against challenge with an HPV 16 E7-expressing tumour, TC-1 (Fig. 8). Immunisation with DNApoly1 was also effective in a therapeutic setting. Although DNApoly1 immunisation did not always eradicate tumour completely, it did significantly reduce tumour growth (Fig. 9).
We have no data to indicate the relative contribution of CTL directed to E7/Db, E7/ A2/2 and flu/A2, to the protective responses we observe to proliferation of Vacpoly1 in immunised A2-transgenic mice. In the tumour model, protection was clearly mediated via the E7/Db response, since TC-1 tumour does not express the HLA-A2 class I molecule (or the flu/A2 epitope). Whether an HLA-A*0201–restricted response to E7 epitopes contributed to protection is clearly relevant to a polytope approach to human HPV-associated neoplastic disease. The capacity of polytope-encoded HLA-A*0201–restricted E7 epitopes (including E7/A2/2) to elicit protective and therapeutic responses to tumour has been clearly demonstrated in a model in which the immunodominant E7/Db epitope was excluded from a polytope immunogen, and from the challenge tumour which expressed a mutated E7 devoid of the E7/Db epitope [10]. Thus it is clear that HLA-A*0201–restricted anti-E7 responses can have an in vivo protective effect.
We sought to reproduce our findings using a second polytope, DNApoly2, in which the order of epitopes and epitope composition differed somewhat from DNApoly1 (Fig. 1). DNApoly2 also differed from DNApoly1in not having a PADRE Th epitope, or an ER-localising signal sequence. Results obtained using DNApoly2 for immunisation were disappointing. While DNApoly2 clearly elicited a response to an encoded ovalbumin epitope, responses to none of the E7 epitopes, or the flu/A2 epitope could be demonstrated (Fig. 6), even after boosting with Vacpoly1 (not shown). There are conflicting reports as to whether the precise position of epitopes in a given polytope construct, or the presence of interepitope-flanking sequences, or the inclusion of Th epitope(s), or the presence of ER localisation signal sequences are important to maximise CTL induction [6, 8, 19, 35, 39]. While it is clear that induction of some primary CTL responses may be Th-independent, or that ‘help’ may be provided by CpG motifs in the plasmid vector, inclusion of Th epitopes is emerging as a preferred option in polytope design [18, 19], as is inclusion of an ER signal sequence [19].
While the purpose of the present study was not to exhaustively investigate issues of polytope design, and our data allow no firm statements to be made regarding why the two polytopes we use differ in immunogenicity, nonetheless it is clear that DNApoly1 containing a different arrangement of epitopes, an ER signal sequence, and a Th epitope, was more effective at CTL induction than DNApoly2 which does not contain the latter two elements. A prevailing view is that until the rules (if any) for the construction of polytope vaccines are worked out, the design of particular polytope vaccines remains empirical [35], since testing of all possible formulations of any given polytope remains prohibitive.
In clinical trials, CTL induction in HPV16-associated cervical intraepithelial neoplasia stage 3 (CIN III) patients and cervical carcinoma patients who received peptides representing some of the epitopes included in our polytopes, has been encouraging [11, 25, 29, 40]. These observations indicate that the epitopes included in our polytope are clinically relevant. The data we and others [10, 41] present, suggest that a polytope approach to HPV-associated neoplastic disease may be appropriate. The lack of HLA-restricted E7 epitope immunodominance may not detract from a polytope approach, since effective protection against several diseases has been demonstrated by subdominant memory responses that are amplified after challenge [22, 24].
Acknowledgements
The work was funded by the National Health and Medical Research Council (Australia) and Queensland Cancer Fund. Dr Andreas Suhrbier provided pSTMPDV. We thank Ms Donna West and her staff for excellent animal husbandry. Publication number 207 of the Sir Alberts Sakzewski Virus Research Centre.
References
- 1.Ada Immunol Cell Biol. 1999;77:180. doi: 10.1046/j.1440-1711.1999.00803.x. [DOI] [PubMed] [Google Scholar]
- 2.Alexander Immunity. 1994;1:751. [Google Scholar]
- 3.Alexander J Immunol. 1997;159:4753. [Google Scholar]
- 4.Anderson J Exp Med. 1991;174:489. doi: 10.1084/jem.174.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borysiewicz Lancet. 1996;347:1523. doi: 10.1016/S0140-6736(96)90674-1. [DOI] [PubMed] [Google Scholar]
- 6.Chen J Virol. 1998;72:8301. doi: 10.1128/jvi.72.10.8301-8308.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chu Clin Exp Immunol. 2000;121:216. doi: 10.1046/j.1365-2249.2000.01293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Del Cell. 1991;66:1145. doi: 10.1016/0092-8674(91)90037-Y. [DOI] [PubMed] [Google Scholar]
- 9.Doan Virology. 1998;244:352. doi: 10.1006/viro.1998.9128. [DOI] [PubMed] [Google Scholar]
- 10.Eiben Cancer Res. 2002;62:5792. [PubMed] [Google Scholar]
- 11.Evans Cancer Res. 1997;57:2943. [PubMed] [Google Scholar]
- 12.Feltkamp Eur J Immunol. 1993;23:2242. doi: 10.1002/eji.1830230929. [DOI] [PubMed] [Google Scholar]
- 13.Feltkamp Eur J Immunol. 1995;25:2638. doi: 10.1002/eji.1830250935. [DOI] [PubMed] [Google Scholar]
- 14.Firat Eur J Immunol. 1999;29:3112. doi: 10.1002/(SICI)1521-4141(199910)29:10<3112::AID-IMMU3112>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 15.Fossum Cell Immunol. 1990;129:414. doi: 10.1016/0008-8749(90)90217-f. [DOI] [PubMed] [Google Scholar]
- 16.Gotch Nature. 1987;326:881. doi: 10.1038/326881a0. [DOI] [PubMed] [Google Scholar]
- 17.Greenstone Proc Natl Acad Sci U S A. 1998;95:1800. doi: 10.1073/pnas.95.4.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanke Nat Med. 2000;6:951. doi: 10.1038/79626. [DOI] [PubMed] [Google Scholar]
- 19.Ishioka J Immunol. 1999;162:3915. [PubMed] [Google Scholar]
- 20.Ji Hum Gene Ther. 1999;10:2727. doi: 10.1089/10430349950016474. [DOI] [PubMed] [Google Scholar]
- 21.Jungbluth Int J Cancer. 2001;92:856. doi: 10.1002/ijc.1282. [DOI] [PubMed] [Google Scholar]
- 22.Le Vaccine. 2001;19:4669. doi: 10.1016/S0264-410X(01)00243-2. [DOI] [PubMed] [Google Scholar]
- 23.Lin Cancer Res. 1996;56:21. [Google Scholar]
- 24.Moss Adv Cancer Res. 1996;69:213. doi: 10.1016/s0065-230x(08)60864-7. [DOI] [PubMed] [Google Scholar]
- 25.Muderspach Clin Cancer Res. 2000;6:3406. [PubMed] [Google Scholar]
- 26.Pascolo J Exp Med. 1997;185:2043. doi: 10.1084/jem.185.12.2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramsay Immunol Rev. 1999;171:27. doi: 10.1111/j.1600-065x.1999.tb01341.x. [DOI] [PubMed] [Google Scholar]
- 28.Ressing J Immunol. 1995;154:5934. [PubMed] [Google Scholar]
- 29.Ressing Cancer Res. 1996;56:582. [PubMed] [Google Scholar]
- 30.Rosenberg Nature. 2001;411:380. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 31.Scheffner Cell. 1993;75:495. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 32.Seder Nat Immunol. 2003;4:835. doi: 10.1038/ni969. [DOI] [PubMed] [Google Scholar]
- 33.Slebos Proc Natl Acad Sci U S A. 1994;91:5320. [Google Scholar]
- 34.Smith Clin Cancer Res. 2001;7:4253. [PubMed] [Google Scholar]
- 35.Suhrbier Expert Rev Vaccines. 2002;1:207. doi: 10.1586/14760584.1.2.207. [DOI] [PubMed] [Google Scholar]
- 36.Thomson Proc Natl Acad Sci U S A. 1995;92:5845. [Google Scholar]
- 37.Thomson J Immunol. 1996;157:822. [PubMed] [Google Scholar]
- 38.Thomson J Immunol. 1998;160:1717. [PubMed] [Google Scholar]
- 39.Uger J Immunol. 1997;158:685. [PubMed] [Google Scholar]
- 40.van Eur J Cancer. 1999;35:946. doi: 10.1016/s0959-8049(99)00048-9. [DOI] [PubMed] [Google Scholar]
- 41.Velders J Immunol. 2001;166:5366. doi: 10.4049/jimmunol.166.9.5366. [DOI] [PubMed] [Google Scholar]
- 42.Vitiello J Immunol. 1983;131:1635. [PubMed] [Google Scholar]
- 43.Von J Virol. 1994;68:2811. doi: 10.1128/jvi.68.5.2811-2821.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yewdell Annu Rev Immunol. 1999;17:51. doi: 10.1146/annurev.immunol.17.1.51. [DOI] [PubMed] [Google Scholar]