

SUMMARY
Without new transcription, gene expression across the oocyte-to-embryo transition (OET) relies instead on regulation of mRNA poly(A) tails to control translation. However, how tail dynamics shape translation across the OET in mammals remains unclear. We perform long-read RNA sequencing to uncover poly(A) tail lengths across the mouse OET and, incorporating published ribosome profiling data, provide an integrated, transcriptome-wide analysis of poly(A) tails and translation across the entire transition. We uncover an extended wave of global deadenylation during fertilization in which short-tailed, oocyte-deposited mRNAs are translationally activated without polyadenylation through resistance to deadenylation. Subsequently, in the embryo, mRNAs are readenylated and translated in a surge of global polyadenylation. We further identify regulation of poly(A) tail length at the isoform level and stage-specific enrichment of mRNA sequence motifs among regulated transcripts. These data provide insight into the stage-specific mechanisms of poly(A) tail regulation that orchestrate gene expression from oocyte to embryo in mammals.
Graphical abstract
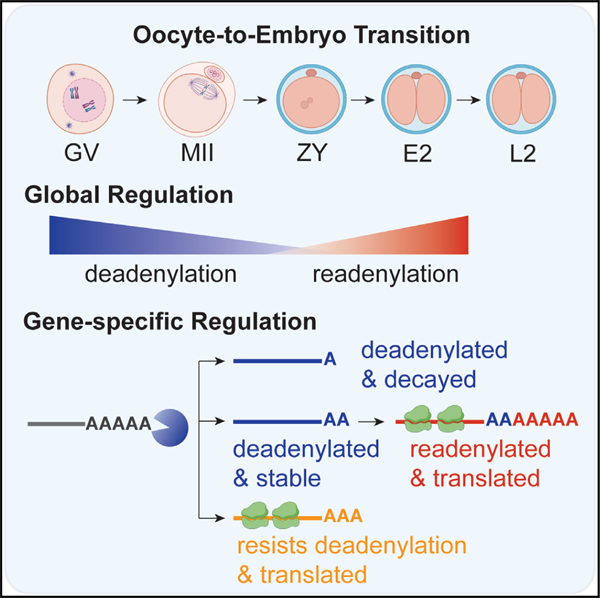
In brief
Lee et al. provide a transcriptome-wide analysis of poly(A) tail and translation dynamics in mice. These data uncover an extended wave of global deadenylation that sets up a switch in translation control, with mRNAs translationally activated by resistance to global deadenylation in the oocyte and by readenylation in the embryo.
INTRODUCTION
The transition from a fully differentiated oocyte to a totipotent embryo (oocyte-to-embryo transition [OET]) capable of driving development of an entirely new organism is one of the most dynamic transitions in biology and, remarkably, occurs in the absence of de novo transcription. The OET begins with global transcriptional silencing in the fully grown germinal vesicle oocyte and ends with full reactivation of transcription in the early embryo (major embryonic genome activation [EGA]). Critical events during this window include oocyte maturation (meiosis I), fertilization (meiosis II), reprogramming to totipotency, the first mitotic embryo cleavage, and EGA1,2 (Figure 1A). Although this period of transcriptional silence in development is highly conserved from worms to humans,1–3 the molecular mechanisms required to orchestrate gene expression across this transition remain poorly understood, particularly in mammals.
Figure 1. mRNA poly(A) tails are dynamically regulated across the OET at both the global and gene-specific levels.
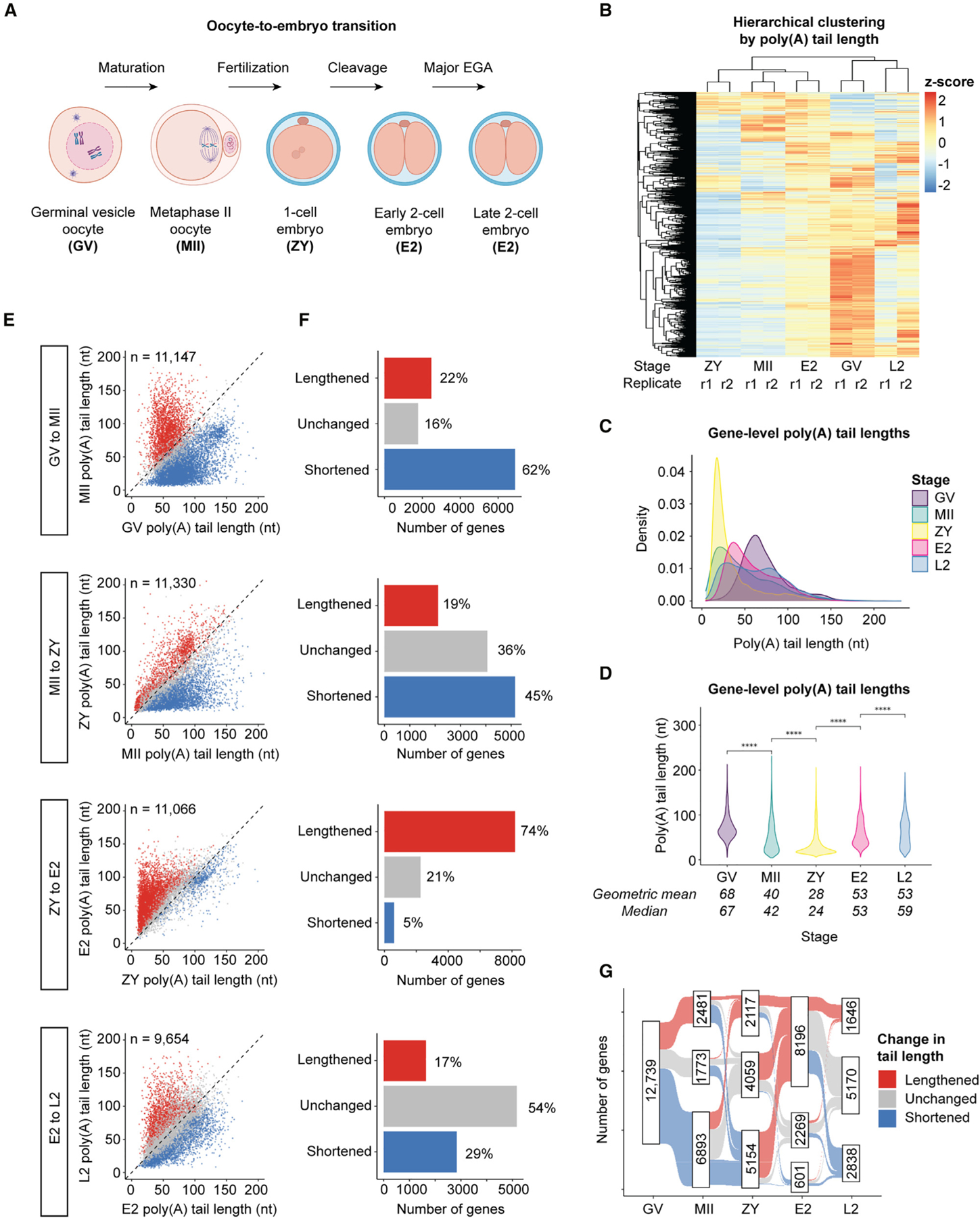
(A) Developmental stages and transitions profiled by Nanopore PCR-cDNA sequencing.
(B) Hierarchical clustering of developmental stages and 2 biological replicates for each stage by poly(A) tail length.
(C) Density plots showing global distributions of gene-level mean poly(A) tail lengths at each stage.
(D) Violin plots showing global distributions of gene-level mean poly(A) tail lengths at each stage. Pairwise two-sided Wilcoxon tests are shown for each stage transition (****p ≤ 0.0001). Means/medians of distributions are provided.
(C and D) Only genes with ≥10 polyadenylated reads in both replicates combined were included.
(E) Scatterplots showing mean poly(A) tail lengths for genes with significantly increased (lengthened, red), decreased (shortened, blue), or unchanged (gray) tail lengths at each stage transition (adjusted p < 0.05, one-sided Wilcoxon test).
(F) Number of genes in each category in (E).
(G) Sankey diagram showing changes in poly(A) tail length for maternal mRNAs across the OET. n, number of genes.
One post-transcriptional mechanism known to play a critical role during the OET involves regulation of mRNA polyadenosine (poly(A)) tail length in the cytoplasm to control translation.4 The poly(A) tail activates mRNA translation through binding by poly(A) binding protein (PABP), which, together with cap binding proteins, acts to recruit ribosomes and facilitate translation initiation.5 Discoveries in Xenopus more than 30 years ago demonstrated that “dormant” oocyte (maternal) mRNAs are deadenylated in the cytoplasm, stored in an unusually stable state with short poly(A) tails in the growing oocyte, and activated for translation later during the OET by cytoplasmic polyadenylation.6–10 Many other studies over the last 30 years have established a highly conserved role for cytoplasmic polyadenylation in the activation of gene expression during the OET. Cytoplasmic deadenylation plays a similarly critical regulatory role in gene repression.11,12 Deadenylation and short poly(A) tails can trigger rapid decay,13,14 and waves of deadenylation and decay in the maturing oocyte and early embryo eliminate the majority of maternal mRNA before major EGA.1,15–17 However, unlike in somatic cells, early studies posited that deadenylation can also occur uncoupled from decay during oocyte maturation18–20 but were unable to determine the specific mRNAs involved or explore the biological role of this uncoupling.
Gene-specific regulation of mRNA poly(A) tail length across the OET is orchestrated by RNA binding proteins (RBPs) and other RNA-regulatory factors. A growing number of sequence motifs within 3′ untranslated regions (UTRs) have been identified and are known to be bound by specific RBPs to regulate polyadenylation, deadenylation, and stability.6,21–24 The motif and RBP most well studied during the OET is the cytoplasmic polyadenylation element (CPE) and CPE binding protein 1 (CPEB1), which play critical roles in the deadenylation and storage of maternal mRNAs during oocyte growth and in mediating cytoplasmic polyadenylation during oocyte maturation.6,25,26 However, CPEB1 becomes undetectable after fertilization,24 suggesting that other regulatory pathways are important in the embryo. While elegant studies have examined 3′ UTR sequence motifs associated with mRNA stability across the OET in zebrafish27,28 or with mRNA translation specifically in the oocyte in Xenopus29 or mice,24,30,31 analyses of the transcriptome-wide landscape of 3′ UTR motifs associated with poly(A) tail regulation across the OET in mammals have not been possible.
While many developmental events across the OET are highly evolutionarily conserved, the specific factors and pathways that regulate these processes are much less conserved between organisms.32–42 Previous studies in mice have been limited to single genes or a limited number of developmental stages due to the relatively small number of oocytes and embryos available for analysis.20,24,26,43–49 Within the past decade, approaches to capture and accurately determine the length of the homopolymeric poly(A) tail transcriptome wide have been developed and used to examine maternal mRNA tail dynamics across the OET in frogs, zebrafish, and flies.29,50–52 More recently, long-read sequencing protocols, which can sequence even very long poly(A) tails with isoform-level resolution, have emerged,53–57 including sensitive approaches that allow tail length determination in the small number of oocytes and embryos that can be obtained in mammals, including humans.58–61 Parallel advances in ribosome profiling allow investigation of the coupling between poly(A) tail regulation and ribosome association.24,26,51,62–64 However, the transcriptome-wide landscape of mRNA poly(A) tail lengths across the entire OET in mice—and how changes in poly(A) tails affect translational efficiency across the OET in any mammal—has remained unclear.
Here, we leverage a recently developed Nanopore PCR-cDNA long-read sequencing approach to determine and compare poly(A) tail lengths and mRNA abundance transcriptome wide across five stages of the OET in mice. Incorporating these data with recently published ribosome profiling data,62 we provide an integrated, transcriptome-wide analysis of the relationship between mRNA poly(A) tail length and translational efficiency across the entire OET. Our findings uncover global, gene-specific, and isoform-specific patterns in tail length dynamics driving gene expression regulation and provide an unprecedented view of gene expression and the mechanisms by which it is regulated across the mammalian OET.
RESULTS
Determination of poly(A) tail lengths transcriptome wide across the mammalian OET
To determine and compare mRNA tail lengths transcriptome wide at each stage across the OET in a small number of cells, we leveraged a recently developed Nanopore long-read PCR-cDNA protocol together with a published algorithm able to determine the length of homopolymeric poly(A) sequences in Nanopore raw data.65 This protocol involves ligation of a 3′ adapter to mRNA before reverse transcription, allowing efficient capture of mRNAs with poly(A) tails as short as 10 nucleotides in a small number of cells (Figure S1A). Sequencing standards with poly(A) tails of known length demonstrated that this approach was accurate, reproducible, and able to distinguish differences of only 10 nucleotides (e.g., 40 vs. 50) (Figures S1B–S1D). We obtained high genome coverage with total RNA input as low as 200 ng from HeLa cells, with 13,727 genes represented by polyadenylated reads and a gene-level mean tail length of 94 nucleotides (Figure S1E), consistent with previous reports.45,51,53,55
We next profiled poly(A) tail lengths transcriptome wide at five developmental stages spanning the OET: the fully grown germinal vesicle oocyte (GV), MII oocyte (MII), zygote (ZY), early 2-cell embryo (E2; before major EGA), and late 2-cell embryo (L2; after major EGA) (Figure 1A). At each stage, two biological replicates were sequenced with ~400 oocytes or embryos each. We obtained an average of 12,204 genes represented by polyadenylated reads in both replicates at each stage and 8,490 genes represented by at least 10 polyadenylated reads in both replicates (Data S1). Principal-component analysis (PCA) showed that transcriptomes for biological replicates clustered together, whereas those for different stages were distinct, with the stages before (GV, MII, and ZY) and after (E2 and L2) EGA more closely clustered, as expected (Figure S1F). Read counts for spike-in control RNAs66,67 correlated well with input concentrations for each stage and replicate (Figure S2A). Comparing poly(A) tail length, replicates also clustered together by both PCA (Figure S1G) and hierarchical clustering (Figure 1B). Overall, replicates demonstrated high correlations in both measured abundance and poly(A) tail length (Figures S2B and S2C).
Comparison across consecutive stages allowed us to determine changes in tail length during four developmental transitions—oocyte maturation (GV–MII), fertilization (MII–ZY), the first mitotic cleavage and minor EGA (ZY–E2), and major EGA (E2–L2) (Figure 1A). To validate our findings, we examined poly(A) tail length changes for all mRNAs shown previously by orthogonal approaches to be polyadenylated or deadenylated during mouse oocyte maturation that we could identify.20,24,26,43,46–49,68–74 We found that 90% (9 of 10) of mRNAs shown previously to be polyadenylated and 93% (28 of 30) shown previously to be deadenylated also demonstrated significant increases and decreases in poly(A) tail length, respectively, in our data (Figures S3A and S3B). These data provide robust validation of this approach to determine and compare poly(A) tail lengths transcriptome-wide across the OET.
Poly(A) tail lengths are globally regulated in a stage-specific manner
We first compared global poly(A) tail length distributions at the gene level. Mean poly(A) tail lengths were longest at the GV stage, with a mean of 68 nt (Figures 1C and 1D), and then globally shortened from GV to MII, consistent with a known wave of mRNA deadenylation and decay during oocyte maturation.17,20 Unexpectedly, global tail lengths are significantly shortened even further in an extended wave of global deadenylation during fertilization. Mean poly(A) tail lengths at ZY were significantly shorter than at any other stage, with a mean of 28 nt and only ~5% of genes having their longest tails at ZY. Tails globally lengthened again from ZY to E2 and E2 to L2, with a mean length of 53 nt at both stages (Figures 1C and 1D), which may represent cytoplasmic polyadenylation of existing maternal transcripts, new transcription, or both. Tail lengthening at EGA onset is evolutionarily conserved,51,75 and newly transcribed mRNAs in somatic cells typically have longer tails relative to older cytoplasmic mRNAs.76–78 These data demonstrate that mRNA tail lengths across the OET are not random but are, instead, highly regulated at the global level in a stage-specific manner. A resource containing measured poly(A) tail lengths for all genes at each developmental stage is provided (Data S1).
Poly(A) tail lengths are selectively regulated at the gene-specific level
We next asked whether poly(A) tails for specific mRNAs are selectively polyadenylated or deadenylated against these global trends (Figures 1E and 1F). Indeed, during the extended wave of deadenylation from GV to ZY, approximately one-third and one-half of mRNAs escape the global deadenylation from GV to MII and MII to ZY, respectively, having tails that are significantly polyadenylated or unchanged. In contrast, from ZY to E2, while three-quarters of all mRNAs are significantly polyadenylated during this short, 6-h window, the remaining one-quarter have tails that are significantly decreased or unchanged. From E2 to L2, the global increase in tail length is modest. However, examination at the gene level reveals dynamic changes, with almost half of all mRNA tails undergoing significant increases or decreases, consistent with the opposing effects of maternal mRNA clearance and EGA at this stage. These data demonstrate that a substantial proportion of mRNAs at each developmental transition are protected from—and selectively regulated in a manner opposite to—the global waves of deadenylation and polyadenylation in the oocyte and embryo. Integrating these data to map the changes in poly(A) tail length across the OET for each mRNA within the maternal pool in the GV oocyte revealed dynamic, gene-specific patterns (Figure 1G), which are examined more closely below. A resource detailing stage-specific changes in poly(A) tail length for each gene across the OET is provided (Data S1).
Polyadenylated mRNAs are enriched for stage-specific roles in development
To determine whether mRNAs selectively polyadenylated and deadenylated at each stage play important roles in development, we conducted Gene Ontology (GO) overrepresentation analysis to identify gene sets enriched in mRNAs lengthened or shortened across each transition. During oocyte maturation from GV to MII, polyadenylated mRNAs were enriched for roles in meiosis (e.g., “sister chromatid segregation” and “cell cycle phase transition”), consistent with progression from prophase I to metaphase II across this transition. In contrast, during fertilization from MII to ZY, mRNAs with lengthened tails were highly enriched for roles related to epigenetic modification (e.g., “chromatin organization” and “histone modification”), replication (e.g., “DNA replication”), and translation (e.g., “regulation of translation” and “translational elongation”), consistent with reprogramming, preparation for mitosis, and increased protein synthesis at this stage64 (Figure S4A).
In the early embryo, mRNAs polyadenylated from ZY to E2 were enriched for roles in mRNA production (e.g., “RNA splicing”), translation (e.g., “ribosome biogenesis” and “cytoplasmic translation”), mitosis (e.g., “mitotic cell cycle phase transition”), and epigenetic modification (e.g., “chromatin remodeling” and “histone modification”), consistent with minor EGA, mitosis, and continued chromatin reprogramming. From E2 to L2, polyadenylated mRNAs were enriched for roles in transcription (e.g., “transcription initiation,” “transcription elongation,” and “RNA splicing”), translation (e.g., “cytoplasmic translation” and “translational initiation”), and epigenetic modification (e.g., “chromatin organization”), consistent with major EGA, increased translational activity, and continued chromatin remodeling (Figure S4A).
In contrast to polyadenylation, mRNAs significantly deadenylated across each stage transition were less consistently enriched for stage-specific developmental factors (Figure S4B). This is consistent with the possibility that deadenylation is a more global and less selective process. However, that genes polyadenylated at each transition were enriched for factors involved in important and temporally relevant pathways suggests that polyadenylation is a specific process and identifies these genes as candidate factors important for development during the OET. A resource detailing the gene sets enriched in mRNAs with and without significant changes in poly(A) tail length across each stage transition is provided (Data S2).
Poly(A) tail lengths are regulated at the transcript isoform-specific level
Taking advantage of our long-read sequencing approach, we asked whether tail lengths are also regulated in an isoform-specific manner. On average, almost half (49%) of the genes at each stage had more than one detectable isoform (Figure S5C). As observed at the gene level, examination of poly(A) tails at the isoform level showed that replicates also clustered together both by PCA (Figure S5A) and hierarchical clustering (Figure S5B), and similar stage-specific patterns of regulation were observed both at the global and isoform-specific levels (Figures S5D–S5F).
We next asked whether tail lengths for different gene isoforms are differentially regulated. Looking first within individual stages, a substantial number of genes had significantly different poly(A) tail length distributions between different isoforms (Figure 2A). The number and proportion of genes with differentially regulated isoforms were higher at GV, MII, and ZY relative to E2 and L2 and generally reflected differences in the prevalence of genes with multiple isoforms (Figure S5C). Genes with multiple isoforms with significantly different tail length profiles at specific developmental stages included genes with well-known roles in OET development, including Cpeb1 and Btg4 (Figure 2B; Data S1).
Figure 2. mRNA poly(A) tails are dynamically regulated across the OET at the transcript isoform-specific level.
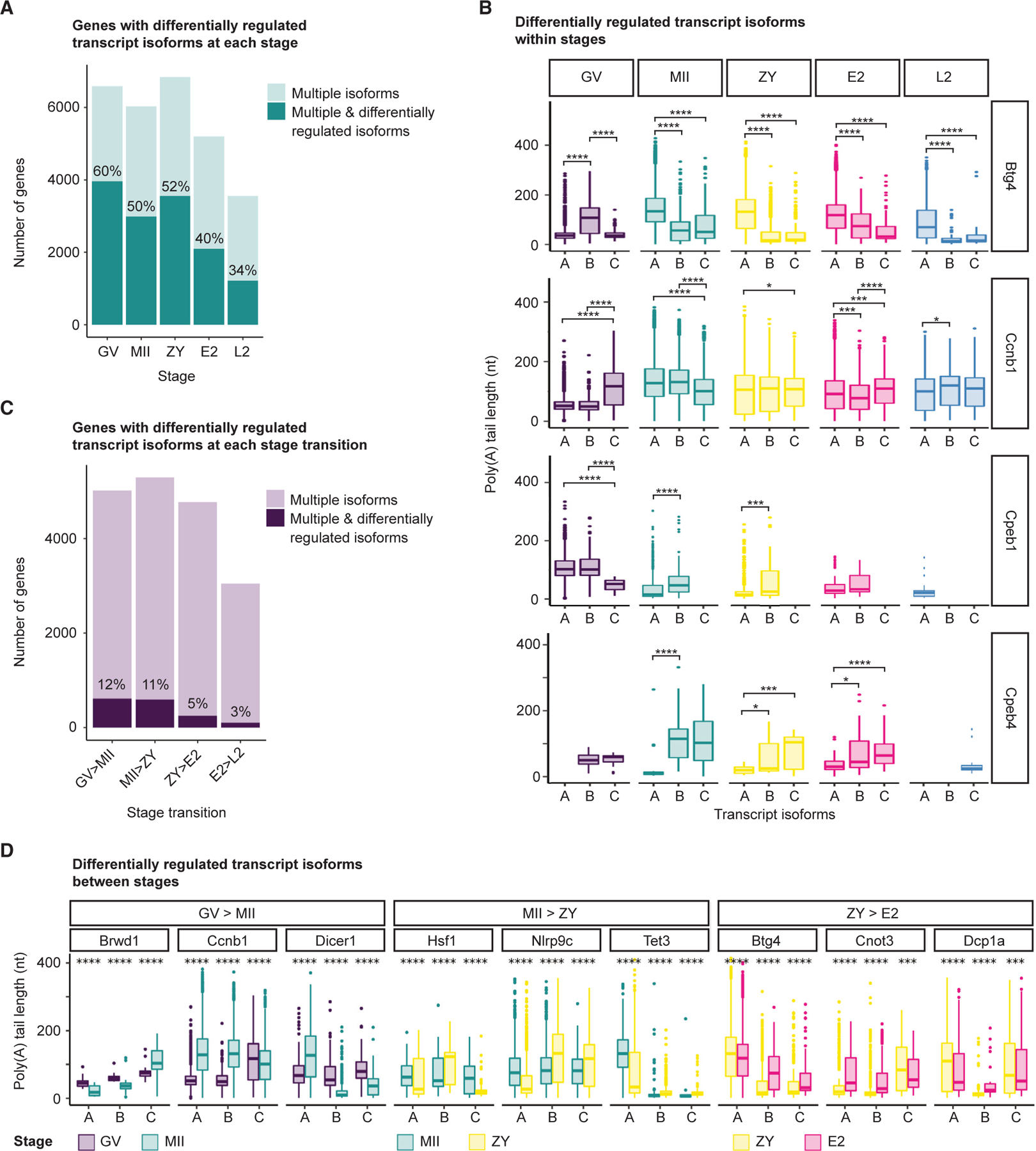
(A and B) Number of genes with multiple isoforms (light teal) and, of those, number of genes with significantly different poly(A) tail length distributions between different isoforms (dark teal) at each stage, with examples of the latter shown in (B).
(C and D) Number of genes with multiple isoforms (light purple) and, of those, number of genes with poly(A) tail lengths for different isoforms regulated in opposite directions (dark purple) at each stage transition, with examples of the latter shown in (D).
(B and D) A maximum of 3 isoforms are shown, only isoforms with R10 polyadenylated reads were included, and pairwise two-sided Wilcoxon tests are shown (*p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001). Ensembl transcript IDs are provided in Data S1.
Looking between stages, we conservatively defined differentially regulated genes as those with isoform tail lengths regulated in opposite directions between consecutive stages (polyadenylated vs. deadenylated; STAR Methods). Despite these strict criteria, between 105 and 610 genes (3%–12%) had differentially regulated isoforms at each stage transition (Figure 2C). Genes demonstrating differential isoform tail length regulation between stages also included maternal genes known to play critical roles in the oocyte or early embryo, including Brwd1, Dicer1, Hsf1, Nlrp3, and Tet3, among others (Figure 2D; Data S1). Complete lists of isoform-level mean poly(A) tail lengths and changes in tail length as well as genes with differentially regulated isoforms within and between stages are provided (Data S3).
Stage-specific 3′ UTR motif enrichment in polyadenylated and deadenylated mRNAs
A long-standing question is how specific subsets of maternal mRNAs are activated and repressed at specific stages to drive development across the OET. 3′ UTR sequence motifs and the RBPs that bind them play major roles in recruiting or blocking factors that regulate mRNA poly(A) tail length, stability, and translation.6,21–24 However, the pattern of 3′ UTR motifs enriched in mRNAs with regulated tails across the mammalian OET remains unclear. To address this gap, we integrated three publicly available databases of known RNA motifs79–81 and scanned the 3′ UTR sequences of mRNAs significantly polyadenylated or deadenylated across each stage transition for enrichment of these motifs (STAR Methods). The most highly enriched motifs at each stage for which a role in mRNA stability and/or translation regulation has been established previously are shown in Figures 3A and 3B. As validation, our analyses identified CPE as the most highly enriched 3′ UTR motif among mRNAs polyadenylated during oocyte maturation.6,25,26 Further, motifs bound by IGFBP2, for which knockout leads to defects in EGA and 2-cell arrest in mice,82 were specifically enriched among mRNAs polyadenylated between ZY and E2, just prior to major EGA. Among deadenylated mRNAs, AU-rich 3′ UTR motifs predicted to be bound by the RNA decay activator ZFP36L2, which is also required for development past the 2-cell stage,83 were also specifically enriched from ZY to E2 (Data S4).
Figure 3. Top 3′ UTR motifs enriched among mRNAs with regulated poly(A) tails.
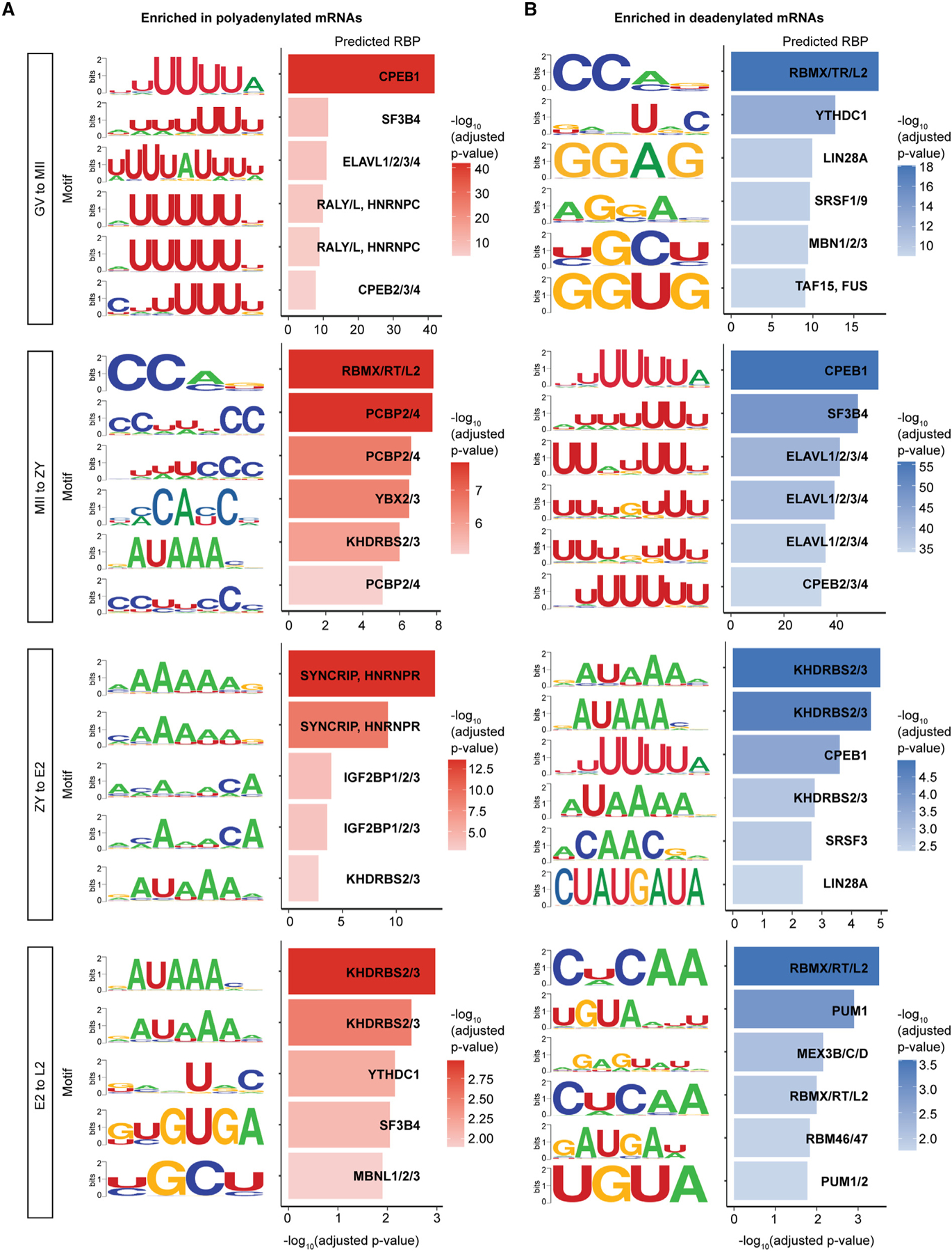
The most highly enriched 3′ UTRs motifs, for which a role in mRNA stability and/or translation regulation has been demonstrated previously, among mRNAs significantly polyadenylated (A) and deadenylated (B) at each stage transition. RBP, RNA binding protein.
Motif enrichment in the 3′ UTRs of mRNAs with regulated tails was strikingly stage specific, particularly for polyadenylated mRNAs, with only motifs for CELF3/4/6, SYNCRIP/HNRNPR, SF3B4, and KHDRBS2/3 showing enrichment at more than one stage (Figure 3A; Data S4). Intriguingly, CPE was enriched among polyadenylated mRNAs during oocyte maturation, as expected, but not at any other subsequent stage, pointing to important roles of other motifs and RBPs in the embryo. Consistent with this idea, the most highly enriched motifs from GV to MII were U rich (e.g., the CPE), but those most enriched from MII to ZY were more C rich (Figure 3A). Stage-specific motif enrichment was also seen in deadenylated mRNAs (Figure 3B), and motifs enriched in polyadenylated mRNAs at one stage were commonly enriched in deadenylated mRNAs in the subsequent stage. A complete list of 3′ UTR motifs enriched among polyadenylated and deadenylated mRNAs at each stage transition is provided (Data S4).
Poly(A) tail length is positively coupled with translational efficiency across the OET
A key question is whether changes in poly(A) tail length drive changes in translational efficiency at each stage of the OET in mammals. Tail length and translational efficiency are coupled during the OET in Drosophila, Xenopus, and zebrafish50–52 but become uncoupled at variable points before gastrulation.51,52 In mammals, coupling has been demonstrated in mouse GV and MII oocytes;26,62 however, the relationship between tail length and translational efficiency during fertilization and in the early mouse embryo remains largely unexplored.
To determine how changes in poly(A) tail length affect translational efficiency in the mammalian oocyte and early embryo, we integrated our measured mRNA poly(A) tail lengths with published translational efficiencies measured at the same developmental stages.62 We found that poly(A) tail length and translational efficiency are positively correlated at each of the five developmental stages from GV to L2 (Figures 4A, S6A, and S6B). There was no clear decrease in coupling following EGA, suggesting that uncoupling of tail length and translational efficiency in mice occurs after L2. Changes in tail length and translational efficiency were also coupled across all four developmental transitions (Figures 4B, S6C, and S6D). Of note, translational efficiency increased for the vast majority of genes from ZY to E2 (Figure 4B, ZY–E2), a trend examined more closely below. These data demonstrate that poly(A) tail length is an important determinant of translational efficiency across the mammalian OET.
Figure 4. Poly(A) tail length positively correlates with translational efficiency during the OET.
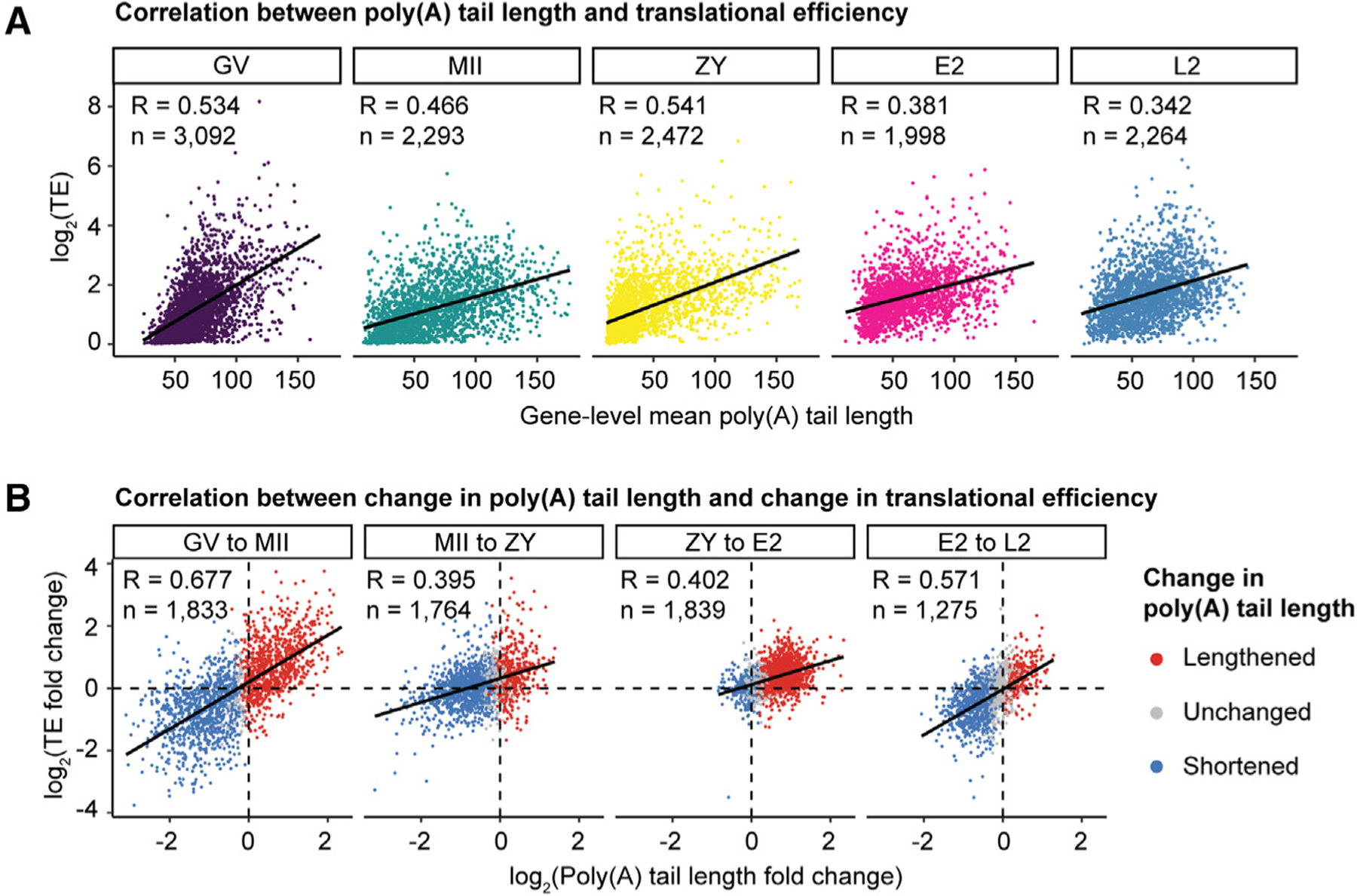
(A) Correlation between poly(A) tail length and translational efficiency at each developmental stage.
(B) Correlation between change in poly(A) tail length and change in translational efficiency at each stage transition, colored by change in tail length. n, number of genes; R, Pearson correlation coefficient; TE, translational efficiency.
Gene-specific uncoupling of deadenylation and decay during oocyte maturation
Consistent with early studies,18–20 our transcriptome-wide analyses demonstrated that a large subset of mRNAs deadenylated from GV to MII is not degraded during oocyte maturation but, instead, remains stable and undergoes further tail changes at subsequent stages (Figure 1G). To investigate the biological role of this uncoupling, we identified deadenylated mRNAs that are degraded versus those that escape decay by comparing changes in poly(A) tail length with changes in mRNA abundance. During oocyte maturation, we identified 6,709 mRNAs that were significantly deadenylated, of which 3,793 (57%) also showed a decrease in abundance. Because our approach can capture mRNAs with short tails, this decrease most likely represents mRNA decay (“deadenylated-decayed” mRNAs). In contrast, the remaining 2,916 deadenylated genes (43%) demonstrated no evident downregulation (Figure 5A), consistent with uncoupling of deadenylation and decay (“deadenylated-stable” mRNAs). As validation, we observed overall similar patterns of uncoupling both at the global and gene-specific levels with analysis of a published short-read RNA sequencing (RNA-seq) dataset despite differences expected with oligo(dT) priming84 (Figures S7A and S7B). Repeating these analyses for later stages revealed a similar pattern of deadenylated-decayed and deadenylated-stable mRNAs at each subsequent stage transition of the OET (Figures 5A and S7A). However, because fewer genes are deadenylated overall after ZY (Figures 1E and 1F) and analyses of RNA abundance at these stages are confounded by EGA, we focused further analyses on mRNAs deadenylated during oocyte maturation.
Figure 5. Deadenylation and readenylation of maternal mRNAs in the oocyte and embryo.
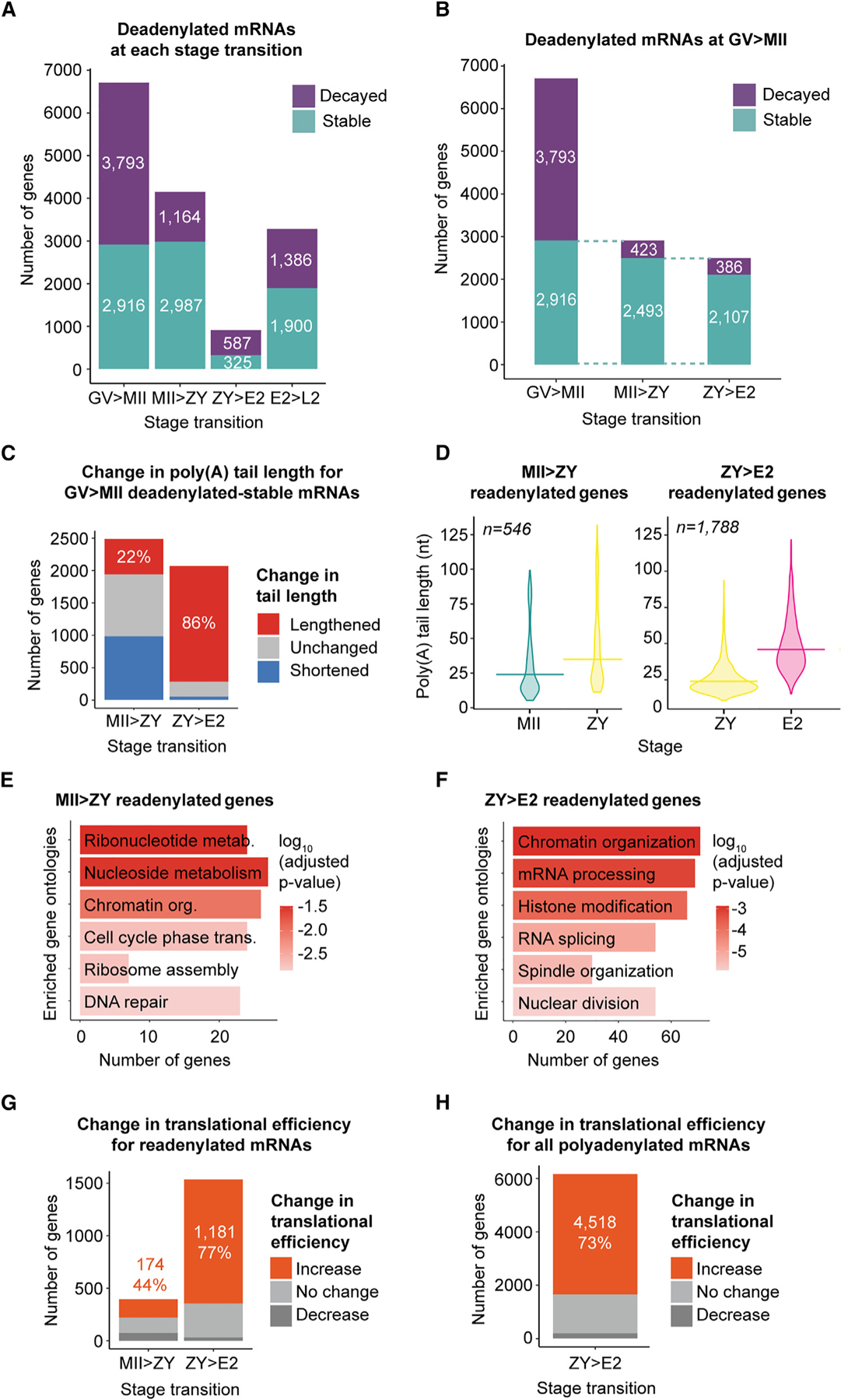
(A) Number of deadenylated-decayed (purple) or -stable (teal) genes at each stage transition.
(B) Number of deadenylated-stable genes from GV to MII that remain stable or are decayed from MII to ZY and from ZY to E2.
(C–F) For (C) and (D), MII>ZY, and (E), genes examined were deadenylated-stable from GV to MII and remained stable from MII to ZY. For (C) and (D), ZY>E2, and (F), genes examined were deadenylated-stable from GV to MII and remained stable from MII to ZY and from ZY to E2.
(C) Number of these genes with different changes in poly(A) tail length from MII to ZY (left) or ZY to E2 (right). The percentage of readenylated (lengthened) genes is indicated.
(D) Poly(A) tail lengths at MII, ZY, and/or E2 stages for genes that are readenylated from MII to ZY (left) or ZY to E2 (right). Horizontal lines indicate geometric means.
(E and F) Gene ontologies enriched in genes readenylated (E) from MII to ZY and (F) from ZY to E2.
(G) Number of readenylated genes with different concurrent changes in TE from MII to ZY (left) or ZY to E2 (right).
(H) Number of all polyadenylated genes with different concurrent changes in TE from ZY to E2.
(G and H) The number and percentage of genes with increased TE are indicated, and genes were filtered by ≥1 fragments per kilobase per million (FPKM) in the RNA-seq dataset used to calculate TE.62 n, number of genes.
To begin to explore the mechanism by which deadenylated-decayed and deadenylated-stable mRNAs are differentially regulated, we asked whether the stable subset retained longer poly(A) tails than the degraded subset. Indeed, at the MII stage, mean tail lengths for deadenylated-stable mRNAs were significantly longer (Figure S7C). However, tail lengths for both groups were relatively short, and the magnitude of the difference was small, with a mean of 30 nt in the stable subset compared to 22 nucleotides in the degraded subset. That the mean tail lengths hover just above and below the predicted binding foot-print for a single cytoplasmic poly(A) binding protein (PABP) (estimated to be 27 nucleotides in mammals85) for the stable and degraded subsets, respectively, suggests that deadenylation past the length required for a single PABP to bind might play a critical role in determining mRNA fate. This idea is consistent with findings in somatic cells, where mRNAs with tails of ~25 nucleotides or less are destabilized when PABPC levels are reduced but in seeming contrast to findings in Xenopus oocytes, where PABPC does not contribute significantly to mRNA stability.86 These findings suggest that small differences in poly(A) tail length might play an important role in determining mRNA stability in the mammalian oocyte.
We next investigated the biological processes in which the deadenylated-decayed versus deadenylated-stable gene subsets are involved to gain insight into the potential role for deadenylation-decay uncoupling. Genes for mRNAs deadenylated-stable during oocyte maturation were enriched for factors important for development in the zygote and later embryo stages, including those with roles in replication, transcription, and reprogramming (Figure S7D). In contrast, deadenylated-decayed mRNAs were involved in processes known to be downregulated during oocyte maturation87 (Figure S7E). This specificity suggests that stabilization of deadenylated mRNAs during oocyte maturation is a selective process and that deadenylated-stable mRNAs may be retained for translational activation in the early embryo. A resource delineating the subsets of mRNAs deadenylated with or without decay at each stage transition is provided (Data S1).
Deadenylation primes global polyadenylation and translational activation before EGA
We next determined the fate of these deadenylated-stable mRNAs after oocyte maturation at the next developmental stage—in the ZY. We found that 2,493 of 2,916 (85%) of these genes remained stable during fertilization from MII to ZY (Figure 5B, MII>ZY), and that 546 (22%) showed tail lengthening again during this transition (Figures 5C and 5D, MII>ZY). This tail lengthening almost certainly represents readenylation for the majority of these maternal mRNAs because new transcription in the ZY is low, and only 118 (22%) overlap with a list of >4,000 previously predicted minor EGA genes.88 In the context of global deadenylation (Figures 1E and 1F), these readenylated mRNAs represent one-quarter (26%) of all genes polyadenylated from MII to ZY. These readenylated genes were enriched for factors involved in important processes for early embryo development, including DNA replication, transcriptional reactivation, mitosis, reprogramming, and translation64,89 (Figure 5E). Integration with published ribosome profiling data62 revealed that the vast majority of these readenylated mRNAs (75%) were associated with ribosomes at ZY, and almost half (44%) of these genes demonstrated an increase in translational efficiency between MII and ZY (Figure 5G, MII>ZY).
Because a large proportion of genes that were deadenylated-stable during oocyte maturation remained stable through fertilization (2,493 genes; Figure 5B, MII>ZY), we also investigated the fate of these genes at the next developmental transition—from ZY to E2—and found that 2,107 (85%) remain stable during this transition (Figure 5B, ZY>E2). Strikingly, 1,788 (85%) of these mRNAs have tails that are significantly lengthened during this transition (Figures 5C and 5D, ZY>E2). Again, this tail lengthening likely represents readenylation of maternal mRNAs because only 20% overlap with predicted minor EGA genes.88 The vast majority of these readenylated mRNAs are also associated with ribosomes at the E2 stage (89%) and demonstrate increased translational efficiency between ZY and E2 (77%) (Figure 5G, ZY>E2). These genes are enriched for factors with important roles in the early embryo, including mRNA processing, histone modification, and cell division (Figure 5F).
That maternal mRNAs readenylated and translationally activated from ZY to E2 are enriched for factors with important roles in the early embryo suggests a selective process. However, readenylation of these mRNAs coincides with a global wave of polyadenylation we detected during this short, 6-h developmental window in which poly(A) tails for almost 75% of mRNAs (8,196 genes) are significantly lengthened (Figures 1E–1G), suggesting the presence of a more non-specific polyadenylation mechanism from ZY to E2. Only 22% of this large population of mRNAs overlap with predicted minor EGA genes,88 and only 10% overlap with predicted major EGA genes,64 suggesting that the majority represent readenylation of stored maternal mRNAs. Most notably, 81% of these readenylated mRNAs (6,623 genes) were detectable in ribosomes at the E2 stage, and, of those, 73% (4,518 of 6,178 genes with measurable translational efficiency) demonstrated increased translational efficiency between ZY and E2 (Figure 5H), consistent with the global increase in translational efficiency at this specific stage noted above (Figure 4B, ZY>E2). While global polyadenylation in the early human embryo has recently been reported,61 the scope, timing, and synchronicity of this global wave of polyadenylation—as well as the resulting surge in global translational activation captured just prior to major EGA—were unexpected. Together, these results support a model in which thousands of maternal mRNAs are deadenylated and selectively stabilized from GV to MII and readenylated and translationally activated from MII to ZY and, even more markedly, during the wave of global polyadenylation from ZY to E2 before the onset of major EGA.
Resistance to global deadenylation activates translation without polyadenylation
Closer examination of the relationship between tail length and translation at each stage revealed stage-specific differences. Most strikingly, for any given level of translational efficiency, mRNAs at ZY were able to achieve the same level of translational efficiency with significantly shorter poly(A) tails (Figure 6A) and become translationally activated with significantly smaller increases in poly(A) tail length relative to other stages (Figure S8A). These findings suggested that the tail length of an mRNA relative to the global pool of mRNAs at a given stage is a greater determinant of translational efficiency than its absolute poly(A) tail length. To further investigate this possibility, we examined change in tail length and change in translational efficiency at each stage more closely and found that most mRNAs demonstrated coordinated increases or decreases in both tail length and translational efficiency, as expected (Figure 4B). However, during fertilization, the global trend line was visibly shifted toward the upper left quadrant (Figure 4B, MII–ZY), pointing again to a unique relationship between tail length and translational efficiency at ZY and to a subset of mRNAs with increased translational efficiency despite decreased tail length.
Figure 6. Global deadenylation reorders relative mRNA poly(A) tail length distributions to activate translation without polyadenylation.
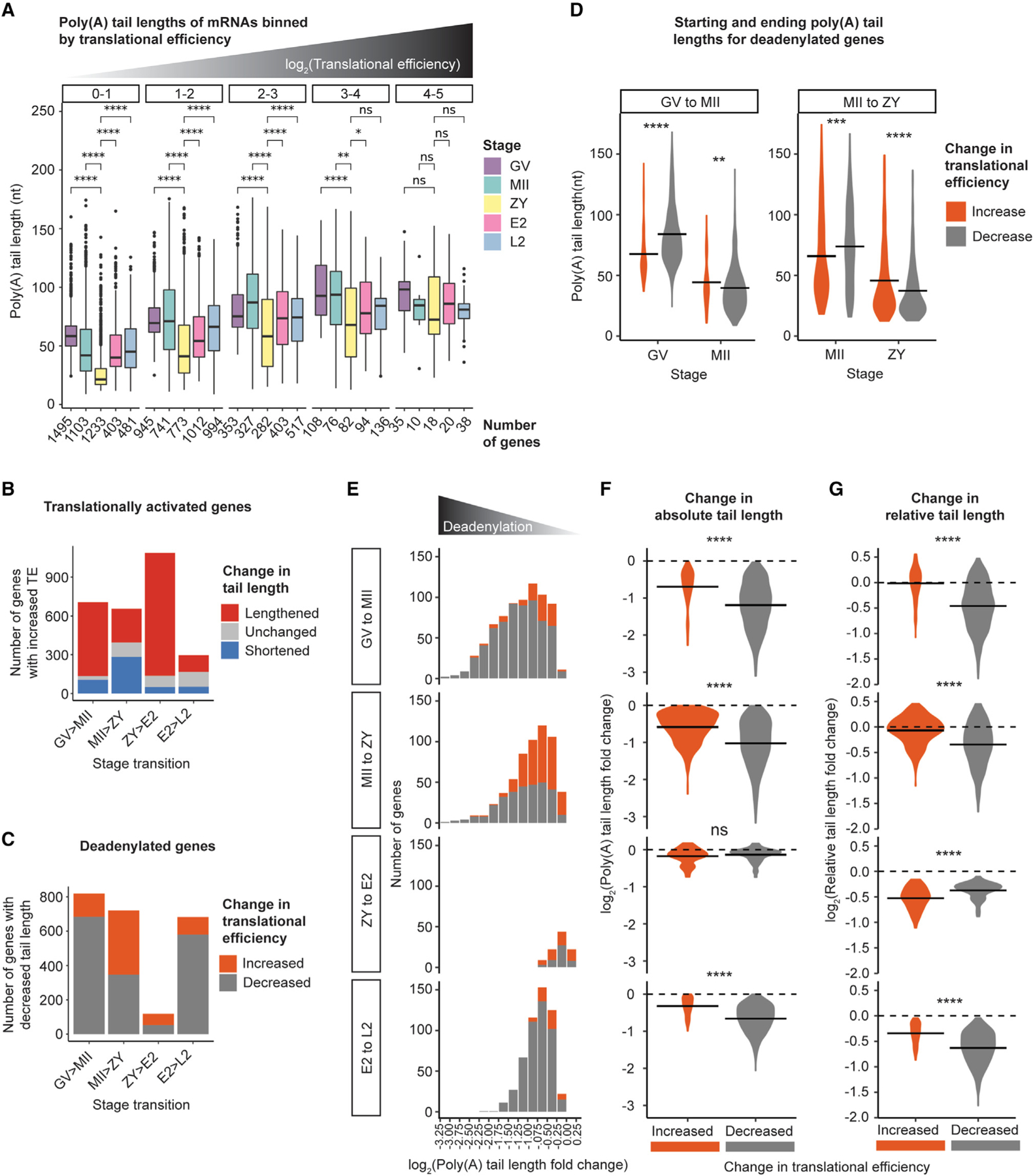
(A) Poly(A) tail lengths of genes binned by TE (plot facets) at each developmental stage. Pairwise two-sided Wilcoxon tests are shown for all stages compared with ZY (ns, p > 0.05; *p ≤ 0.05; **p ≤ 0.01; ****p ≤ 0.0001). The number of genes in each bin is indicated below the x axis. Only bins with R10 genes are plotted.
(B) Number of translationally activated genes with lengthened (red), shortened (blue), and not significantly changed (gray) tail lengths across each stage transition.
(C) Number of deadenylated-activated (orange) or -repressed (gray) genes across each stage transition.
(D) Mean poly(A) tail lengths for deadenylated-activated or -repressed genes at each stage from GV to MII and MII to ZY.
(E) Number of deadenylated-activated (orange) or -repressed (gray) genes, binned by magnitude of deadenylation across each stage transition.
(F and G) Log2 fold change in absolute (F) or relative (G) tail length for deadenylated-activated (orange) or -repressed (gray) genes across each stage transition. For (D, F, and G), horizontal lines indicate arithmetic means and two-sided Wilcoxon tests are shown (ns, p > 0.05; **p ≤ 0.01; ***p ≤ 0.001; ****p ≤ 0.0001). For (B)–(G), to include genes translationally activated or repressed despite no significant change in tail length, the adjusted p value cutoff for classifying genes as “deadenylated” was removed.
To quantify the scope and stage specificity of this population of mRNAs with an inverse tail length-translational efficiency relationship, we independently examined mRNAs translationally activated and mRNAs deadenylated across each developmental transition. Indeed, mRNAs that were translationally activated but deadenylated (“deadenylated-activated” mRNAs) were identified during both oocyte maturation and fertilization—the transitions marked by global deadenylation—but most pronounced from MII to ZY (Figure 6B). In fact, between MII and ZY, 60% of mRNAs with increased translational efficiency demonstrated no accompanying increase in poly(A) tail length. Similarly, examination of all mRNAs deadenylated during each transition revealed mRNAs with a paradoxical increase in translational efficiency from GV to MII and from MII to ZY (Figure 6C). This trend, again, was most prominent from MII to ZY, during which almost 50% of deadenylated mRNAs also showed increased translational efficiency. Deadenylated-activated mRNAs from GV to MII were enriched for factors that reflect the switch to mitosis after fertilization (e.g., “mitotic cell cycle phase transition” and “mitotic sister chromatid segregation”) and, from MII to ZY, for factors involved in transcription and DNA replication in the early embryo (e.g., “RNA splicing” and “regulation of DNA repair”) (Figure S8B). In contrast, deadenylated genes with decreased translational efficiency (“deadenylated-repressed” mRNAs) from GV to MII were enriched for genes involved in processes known to be downregulated during oocyte maturation (e.g., “cellular respiration” and “cytoplasmic translation”) and, from MII to ZY, for genes no longer needed after fertilization (e.g., “egg activation” and “regulation of fertilization”) (Figure S8C). This specificity suggests that translational activation of this subset of deadenylated mRNAs is a selective and regulated process.
To explore the mechanism underlying this inverse tail length-translation relationship, we next compared tail length dynamics for deadenylated-activated versus deadenylated-repressed mRNAs. Interestingly, deadenylated-activated mRNAs from GV to MII and MII to ZY began each transition with significantly shorter tails on average relative to deadenylated-repressed mRNAs but ended each transition with longer tails (Figure 6D). Although longer than tails for deadenylated-repressed genes, poly(A) tails for deadenylated-activated genes were still relatively short, with mean tail lengths of ~45 nt. These findings suggested that deadenylated-activated mRNAs—despite an overall decrease in tail length—retained longer tails relative to deadenylated-repressed mRNAs through relative protection from global deadenylation. Consistent with this mechanism, deadenylated-activated mRNAs during global deadenylation were highly enriched among those with the smallest decreases in tail length (Figure 6E) and, compared with deadenylated-repressed mRNAs, showed smaller decreases in absolute tail length (Figure 6F). We further quantified the poly(A) tail length rank order by calculating each gene’s mean tail length relative to the global mean at each stage (“relative tail length”; STAR Methods). This comparison showed that deadenylated-activated mRNAs also had smaller decreases in relative tail length compared with deadenylated-repressed mRNAs (Figure 6G). In fact, the average change in relative tail length for deadenylated-activated mRNAs was near zero, with a substantial subset of deadenylated-activated genes having an overall increase in relative tail length at both stage transitions. In contrast, deadenylation does not appear to be a significant mechanism for translational activation after the ZY stage, pointing to a unique and transient shift in the translational regulatory regime in the late oocyte and early embryo. Together, these data support a mechanism where maternal mRNAs can be translationally activated—not by cytoplasmic polyadenylation—but by relative protection from the extended wave of global deadenylation during the early mammalian OET.
Poly(A) tail and translational efficiency dynamics for maternal effect genes
In our analyses of all genes, we identified multiple mechanisms by which changes in tail length regulate mRNA translation and stability. To examine the potential importance of these mechanisms in development across the OET, we asked whether mRNAs encoding 51 known maternal effect genes (MEGs), maternal genes known to play critical roles in development during the OET90–92 demonstrate tail length dynamics consistent with regulation by these mechanisms. Overall, poly(A) tail lengths and change in tail length for this set of MEGs demonstrated the same global, gene-specific, and isoform-specific trends observed for all genes (Figures 2C–2F, 7A–7C, S9A–S9C, and S10D). Two notable differences were that MEG mRNAs were (1) more likely to be polyadenylated between GV and E2 relative to the rest of the transcriptome (Figures 7D and S10E) and (2) twice as likely to be deadenylated from E2 to L2 compared with all genes (Figures 7D and S10F).
Figure 7. TE of maternal effect genes (MEGs) critical for development is regulated by dynamic changes in poly(A) tail length.
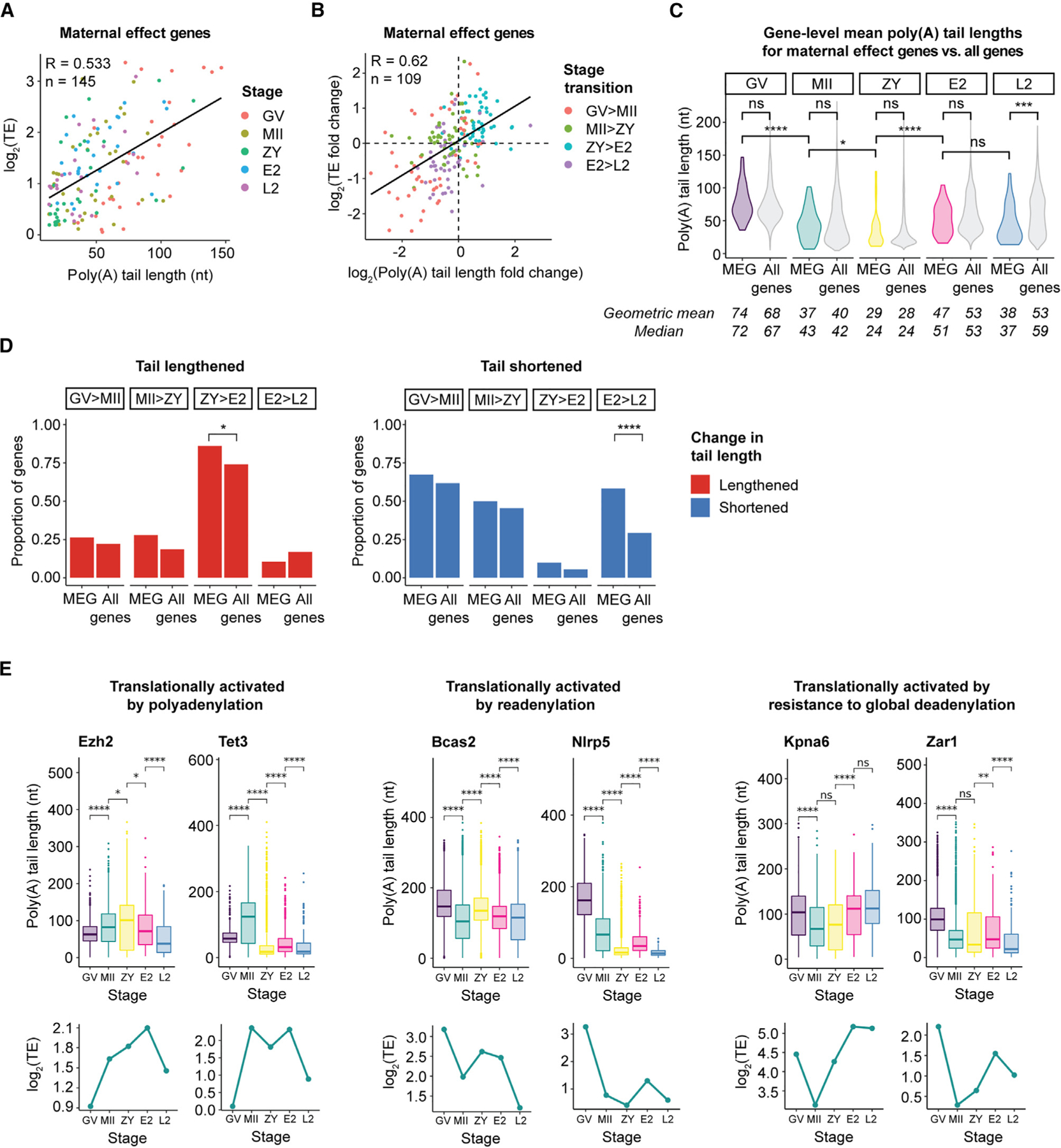
(A) Correlation between poly(A) tail length and TE for MEGs.
(B) Correlation between change in poly(A) tail length and change in TE for MEGs.
(A and B) Genes were filtered by ≥1 FPKM in the RNA-seq dataset used to calculate TE.62 n, number of reads; R, Pearson correlation coefficient; TE, translational efficiency.
(C) Violin plots showing global distributions of gene-level mean poly(A) tail lengths at each stage for MEGs (colored) compared with all genes (gray). Means/medians of distributions are provided. Only genes with ≥10 polyadenylated reads in both biological replicates combined were included. Pairwise two-sided Wilcoxon tests are shown (ns, p > 0.05; *p ≤ 0.05; ***p ≤ 0.001; ****p ≤ 0.0001).
(D) Proportion of MEGs or all genes with significantly (adjusted p < 0.05) lengthened (red, left) or shortened (blue, right) tail lengths at each stage transition. Only genes with R10 polyadenylated reads in both stages represented were included. Pairwise one-sided Fisher’s exact tests are shown (ns, p > 0.05; *p ≤ 0.05; ***p ≤ 0.001; ****p ≤ 0.0001).
(E) Poly(A) tail lengths (top) and translational efficiencies (bottom) of select MEGs across the OET. Each boxplot represents ≥20 polyadenylated reads. Pairwise two-sided Wilcoxon tests are shown (ns, p > 0.05; *p ≤ 0.05; **p ≤ 0.01; ****p ≤ 0.0001).
First, similar to our findings for all genes, a subset of MEGs demonstrated selective, stage-specific cytoplasmic polyadenylation amid the waves of global deadenylation from GV to MII and from MII to ZY (Figures S9B and S9C). Among MEGs with increases in both tail length and translational efficiency from GV to MII are the polycomb group protein Ezh2, critical for proper H3K9 and H3K27 methylation in the ZY,93 and Tet3, which is required for demethylation of the paternal genome following fertilization94 (Figure 7E). Additional examples are provided (Figure S10A).
Another subset of MEGs followed a second regulatory mechanism we observed in our analyses of all genes, with deadenylation without decay during oocyte maturation and readenylation from MII to ZY or, more prominently, from ZY to E2 (Figures 5C and 5D). In fact, MEGs are enriched among deadenylated-stable mRNAs during oocyte maturation compared with all genes (61% of MEGs vs. 43% of all genes) and among genes readenylated from MII to ZY (36% for MEGs vs. 23% for all genes), with no overlap with predicted minor EGA genes.88 Examples include Bcas2, which is important for DNA repair in zygotes and development past the 2- to 4-cell embryo stage,95 and Sebox, which is also required for development past the 2-cell stage96,97 (Figures 7E and S10B). Additional examples are provided (Figure S10B).
The vast majority of MEGs deadenylated-stable during oocyte maturation are significantly readenylated between ZY and E2 (19 of 21) (Figures S9B and S9C), consistent with an important role of the surge of polyadenylation and translational activation observed for all genes during this short stage transition (Figures 1E–1G and 4B). Only 2 of these MEGs (Ctcf and Gclm) overlap with a list of >4,000 predicted minor EGA genes88 and none overlap with >2,500 predicted major EGA genes,64 suggesting that these increases in tail length predominantly represent readenylation of maternal transcripts. Examples of MEGs that are deadenylated-stable during oocyte maturation and readenylated from ZY to E2 include Nlrp5 (also known as Mater), a critical component of the subcortical maternal complex (SCMC) required for development past the 2-cell stage,98,99 and Pms2, important in DNA mismatch repair in the early embryo100 (Figures 7E and S10B). Additional examples are provided (Figure S10B).
Finally, an additional subset of MEGs is translationally activated despite tail shortening during the global waves of deadenylation from GV to MII and MII to ZY (Figures 7B and 7E). As for all genes, this phenomenon was most pronounced from MII to ZY (with translational activation of 21% of deadenylated MEGs from GV to MII vs. 44% from MII to ZY). These deadenylated-activated MEGs demonstrate smaller decreases in both absolute and relative tail length compared with deadenylated-repressed MEGs during both oocyte maturation and fertilization, although the statistical power was limited given the small number of genes (Figures S9D and S9E). Examples of MEGs regulated by this noncanonical method include Rlim, a ubiquitin ligase required for imprinted X chromosome inactivation in mouse embryos, and Zar1, an RBP required for proper division from 1-cell to 2-cell embryo stages (Figures 7E and S10C). Additional examples are provided (Figures 7E and S10C; see also Rnf1, Spin1, and Trim28 in Figure S10A). A resource detailing poly(A) tail length and translational efficiency dynamics for known MEGs90–92 is provided (Data S1).
DISCUSSION
Although the link between maternal mRNA poly(A) tail length and translation during the OET was recognized more than 30 years ago,6 the transcriptome-wide landscape of poly(A) tail lengths across the OET in mice and how changes in tail length affect translation across the entire OET in any mammal have remained unclear. Here, we determine mRNA poly(A) tail lengths at five stages across the mouse OET and integrate these data with published ribosome profiling data62 to provide a transcriptome-wide analysis of poly(A) tail length and translational efficiency dynamics across the OET in a mammalian system. The Nanopore PCR-cDNA approach used in this study is a significant advance compared to earlier approaches because it allowed us to capture and sequence mRNAs with tails as short as 10 nucleotides and to do so with good genome coverage in a small number of cells. In addition, the long-read approach allows isoform-level resolution and sequencing of long tails not possible with short-read sequencing. These integrated data provide an unprecedented view of gene expression regulation across the OET, where changes in expression occur in large part without changes in RNA abundance.
One surprise finding in this study was the extended wave of global tail shortening from MII to ZY (Figures 1C and 1D). During oocyte maturation and fertilization, our analyses suggest a stage-specific role of global deadenylation in the translational activation of specific subsets of maternal mRNAs with short tails through a noncanonical mechanism without cytoplasmic polyadenylation. In fact, we found that more than half of mRNAs translationally activated during fertilization from MII to ZY have tails that are significantly deadenylated or unchanged (Figure 6B). With respect to mechanism, our findings suggest that these maternal mRNAs are translationally activated because they resist the more extensive deadenylation experienced by the rest of the maternal mRNA pool during global deadenylation. In this model, this extended wave of global deadenylation both (1) remodels the rank order of mRNA tail lengths and (2) lowers the minimum tail length required to achieve a certain level of translational efficiency so that short-tailed mRNAs can be efficiently translated in the MII oocyte and zygote.
This extended wave of global deadenylation from GV to ZY also broadly primes maternal mRNAs for readenylation and translational activation in the embryo. This wave of mRNA activation is most striking during the first embryo cleavage from ZY to E2, during which the vast majority of mRNAs deadenylated during oocyte maturation without decay are both readenylated and translationally activated during this short, 6-h developmental window (Figures 5C, 5D, and 5G, ZY>E2). The mechanism and developmental impact of this global wave of maternal gene activation immediately preceding major EGA are important unanswered questions.
Previous studies have been mixed as to the requirement of polyadenylation for translational activation during the OET. In Xenopus oocytes, early studies showed that the act of polyadenylation itself is important for translational activation even in the presence of a long poly(A) tail,101,102 while other studies suggested that the presence of a long tail is sufficient.103–105 During Drosophila egg activation, inhibition of a global polyadenylation wave by knockout of the cytoplasmic poly(A) polymerase Wispy did not decrease the correlation between poly(A) tail length and translational efficiency.52 These findings further demonstrated that polyadenylation itself is not required to establish differences in translational efficiency among mRNAs but that these differences can also be established via deadenylation of select transcripts. However, the precise role of deadenylation in defining the subset of mRNAs translationally activated in normal development was not clear because the knockout eggs were not competent to complete meiosis without cytoplasmic polyadenylation. In this study, we demonstrate—in wild-type cells—that the rank order of mRNA tail lengths and translational efficiency during the early mouse OET is largely defined by global deadenylation, with translational activation of select mRNAs demonstrating relative resistance to this deadenylation wave. Identifying the RNA-regulatory factors that function to activate translation during the OET—not by mediating polyadenylation but through relative protection from global deadenylation—will be an important future avenue of research.
Why might the translational efficiency of short-tailed mRNAs be higher in the zygote relative to other developmental stages? One potential reason is that, by significantly reducing the population of mRNAs with long tails during the extended wave of global deadenylation, mRNAs with short tails might better compete for a translation factor of limited abundance at these stages, such as PABP or ribosomes. In fact, it has been shown that PABP is present at limiting levels in Xenopus oocytes and that PABP abundance contributes to coupling and uncoupling of tail length and translational efficiency.86 Whether PABP levels are similarly limiting in mouse oocytes or embryos is unclear.
Identification of the specific mRNAs activated and repressed at each transition provides the opportunity to identify candidate factors driving early mammalian development and to uncover the post-transcriptional mechanisms that control their expression. With respect to factors in RNA regulation, we identify RBP motifs enriched among mRNAs selectively polyadenylated and deadenylated at each stage, uncovering candidate regulators of poly(A) tail length to drive future mechanistic studies. In addition, that mRNAs for many factors with known roles in OET development show differential, stage-specific isoform regulation raises questions as to how this regulation affects the expression and activity of these key developmental factors. With respect to development, we show that genes selectively activated at each stage are enriched for factors known to play key roles in important and temporally relevant developmental processes. As further validation, even within the small group of known MEGs, we find multiple examples of mRNAs with patterns of regulation consistent with each of the mechanisms observed in this study. A better understanding of the mechanisms that regulate expression of these genes is essential because these are candidates for factors important for female fertility.90–92
Together, our findings demonstrate the presence of an extended wave of global deadenylation that sets up a switch in translation control across the transition from oocyte to embryo. Maternal mRNA tail length and translational efficiency are dynamically ordered by global deadenylation and resistance to deadenylation in the oocyte and by broad readenylation of deadenylated-stable maternal mRNAs during a global wave of polyadenylation and translational activation in the embryo before major EGA. These divergent mechanisms in the oocyte and embryo support a model where the tail length of an mRNA relative to other mRNAs at a given stage is a greater determinant of translational efficiency than absolute poly(A) tail length or cytoplasmic polyadenylation itself. The datasets provided here offer a rich resource and insight into the complex post-transcriptional mechanisms by which cytoplasmic polyadenylation and deadenylation order and reorder the landscape of poly(A) tails to drive gene expression during the earliest stages of mammalian development.
Limitations of the study
Limitations of the approach used in this study include the inability to capture mRNAs with tails <10 nt. Therefore, it is possible that the true incidence of deadenylated and readenylated mRNAs is even higher than observed here and/or that the true mean tail length is shorter than the measurements in this study. Consistent with this possibility, it was shown during the completion of this study that maternal mRNAs can be partially degraded and readenylated after fertilization with exceptionally short tails.61,106 However, the biological role of these mRNAs is unclear, and the polyadenylated mRNAs evaluated in this study and in the ribosome profiling analyses with which it is integrated62 are expected to capture the poly(A) tail landscape that controls translation across the OET. We also observed a modest decline in measurement accuracy with increasing tail length (Figures S1B–S1D), which has been seen by others using similar approaches and is likely attributable to technical limitations in homopolymeric measurement53,107 and PCR amplification. However, while we report absolute tail lengths, our analyses focused on comparisons of tail length, where we were able to reliably detect differences of only 10 nucleotides (Figures S1B–S1D) and to validate >90% of changes previously observed using orthogonal approaches (Figure S3). During the completion of this study, another long-read approach, PAIso-seq2, was developed, which can capture mRNAs with no tails or tails with non-adenosine residues and can be used in single cells.57 For comparison, other short-read (including PAL-Seq,51 TAIL-Seq,45 and mTAIL-Seq50) and long-read (including FLAM-Seq,55 Nanopore direct RNA sequencing,54 Nano3P-Seq,53 PAIso-seq156) approaches either have insufficient genome coverage for use in a small number of cells45,50,51,53,54 or require longer tails for capture,55 although each has its own advantages to address different biological questions.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Heidi Cook-Andersen (hcookandersen@health.ucsd.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The Nanopore PCR-cDNA sequencing data associated with this study have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper also analyzes existing, publicly available data and these accession numbers for the datasets are listed in the key resources table. Additional data tables, including a summary of sequencing runs, gene-level poly(A) tail lengths at each stage, gene-level changes in poly(A) tail length at each stage transition, gene ontologies enriched in genes with poly(A) tail lengthening or shortening at each stage transition, isoform-level poly(A) tail lengths at each stage, isoform-level changes in poly(A) tail length at each stage transition, isoform-specific poly(A) tail length regulation both within and between stages, 3′ UTR sequence motifs enriched in mRNAs polyadenylated or deadenylated at each stage transition, lists of deadenylated-decayed vs. -stable genes at each stage transition, and poly(A) tail lengths and translational efficiencies for maternal effect genes at each stage are available in Data S1–S4.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| TRIzol | Thermo Fisher Scientific | 15-596-018 |
| Phenol: chloroform: iso-amyl alcohol (25:24:1) | VWR | VWRV0966-100ML |
| PMSG | Lee BioSolutions | 493-10 |
| Collagenase | Sigma-Aldrich | C9407 |
| M2 media | Sigma-Aldrich | M7167 |
| HCG | Sigma-Aldrich | C1063 |
| Hyaluronidase | Sigma-Aldrich | H4272 |
| Sodium acetate buffer solution | Sigma-Aldrich | S7899 |
| GenElute LPA | Sigma-Aldrich | 56575 |
| DMEM media | Thermo Fisher Scientific | 11995073 |
| FBS | Cytiva | SH30071.03HI |
| Penicillin/Streptomycin | Thermo Fisher Scientific | 15140122 |
| 0.25% Trypsin-EDTA | Thermo Fisher Scientific | 25200114 |
|
| ||
| Critical commercial assays | ||
|
| ||
| PCR-cDNA Sequencing Kit | Oxford Nanopore Technologies | SQK-PCS110 |
| PCR-cDNA Barcoding Kit | Oxford Nanopore Technologies | SQK-PCB109 |
| R9.4.1 flow cell | Oxford Nanopore Technologies | FLO-MIN106D |
| Minion sequencing device | Oxford Nanopore Technologies | MIN-101B |
| AMPure XP | Beckman Coulter | A63881 |
| RNA Clean & Concentrator-5 | Zymo Research | R1014 |
| DNAse I | Zymo Research | E1011-A |
| T4 DNA Ligase | NEB | M0202M |
| 5x quick ligation reaction buffer | NEB | E6058A |
| Lambda exonuclease | NEB | M0262S |
| USER Enzyme | NEB | M5505S |
| RNAClean XP | Beckman Coulter | A63987 |
| Maxima H Minus Reverse Transcriptase | Thermo Fisher Scientific | EP0751 |
| dNTP Mix | NEB | N0447S |
| LongAmp Hot Start Taq 2x Master Mix | NEB | M0533S |
| Exonuclease I | NEB | M0293S |
| RNaseOUT | Thermo Fisher Scientific | 10777019 |
| RNA 6000 Pico Kit | Agilent Technologies | 5067-1513 |
| High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 |
|
| ||
| Deposited data | ||
|
| ||
| Nanopore PCR-cDNA sequencing | This paper | GEO: GSE228001 |
| RNA-seq | Zhang et al.84 | GEO: GSE71434 |
| Ribosome profiling and RNA-seq | Xiong et al.62 | GEO: GSE165782 |
| RNA-seq | Zhang et al.64 | GEO: GSE169632 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Hela | ATCC | CCL-2 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57B/6J mice | Jackson Labs | 000664 |
| B6D2F1/J mice | Jackson Labs | 100006 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Polyadenylated standards (See method details for sequences) | This study | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Minknow (22.05.5) | Oxford Nanopore Technologies | https://community.nanoporetech.com/downloads |
| Guppy (6.0.6) | Oxford Nanopore Technologies | https://community.nanoporetech.com/downloads |
| Pychopper (2.5.0) | Oxford Nanopore Technologies | https://github.com/epi2me-labs/pychopper |
| minimap2 (2.17) | Li et al.108 | https://github.com/lh3/minimap2 |
| Salmon (0.14.1) | Patro et al.109 | https://github.com/COMBINE-lab/salmon |
| tailfindr (1.3) | Krause et al.65 | https://github.com/adnaniazi/tailfindr |
| DESeq2 (1.34.0) | Love et al.110 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| AME (5.0.2) | McLeay et al.111 | https://meme-suite.org/meme/tools/ame |
| clusterProfiler (4.2.2) | Yu et al.112 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
|
| ||
| Other | ||
|
| ||
| ERCC Spike-In RNAs | Thermo Fisher Scientific | 4456653 |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Isolation of mouse oocytes and embryos
Mouse oocytes were isolated from C57B/6J female mice. For GV oocytes, 4-week old females were stimulated by intraperitoneal injection of 5 IU pregnant mare serum gonadotropin (PMSG; Lee BioSolutions, 493-10), ovaries were harvested 48 h after PMSG injection, and denuded oocytes collected after brief digestion with trypsin (Thermo Fisher Scientific, 25200114), collagenase (Sigma-Aldrich, C9407), and DNAse in M2 media (Sigma-Aldrich, M7167). For MII oocytes, mice were injected with 5 IU HCG (Sigma-Aldrich, C1063) 48 h after PMSG induction and ovulated MII oocytes collected from the ampulla 15 h post-HCG and cumulus cells were removed by brief hyaluronidase (Sigma-Aldrich, H4272) digestion per standard protocol.
Mouse embryos were isolated from B6D2F1/J female mice mated with C57B/6J males of known fertility at the time of hCG injection. 1-cell, early 2-cell, and late 2-cell embryos were collected at 27–28, 30–32, and 46–48 h post-HCG. All mice were maintained in agreement with protocols approved by the Institutional Animal Care and Use Committee at the University of California, San Diego.
Maintenance of HeLa cells
HeLa cells (ATCC) were grown in DMEM (Thermo Fisher Scientific) supplemented with 10% FBS (Cytiva) and 1% Penicillin/Streptomycin (Thermo Fisher Scientific), and were split using 0.25% Trypsin-EDTA (Thermo Fisher Scientific) approximately every two days, when cells reached 80% confluence. RNA was isolated from cells once they reached approximately 80% confluence, as described in method details below.
METHOD DETAILS
RNA isolation
Total RNA was isolated from HeLa cells or ~400 oocytes or embryos for each stage and replicate using Trizol reagent (Thermo Fisher Scientific, 15-596-018), followed by ethanol precipitation and RNA Clean & Concentrator-5 kit (Zymo Research, R1014) as per manufacturer’s protocol. RNA was eluted in 7 µl nuclease-free water and used immediately for Nanopore PCR-cDNA sequencing library prep.
Design of polyadenylated standards
DNA standards with the following sequences were obtained from IDT:
pA_standard_10:
CTTGCCTGTCGCTCTATCTTCTTTTTTTTTTACCAACGGCGACGAATAGTAGTTTACTTCCTCCCTGCGGGCCCCTCCTGAAGTGCCACCTATACGGCTTGTTGAAGCCGATTAGTACAATAGATTTATTCAACCCCAAAGGTCTACACTCCCGGCTTACTCTTAGCTGATATGTCGCGCAATATCAGCACCAACAGAAA.
pA_standard_40:
CTTGCCTGTCGCTCTATCTTCTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTCGGCAAAGAGACAATTATAGCGGCTAGGAACGCAACTAGTTATAACGAACGGCCTCGAATAGTAGAAAATATCCCTCCTCCGGGCACCTCCTGAAATGCCACATATTCGGGTTATTGGCAATATCAGCACCAACAGAAA.
pA_standard_50:
CTTGCCTGTCGCTCTATCTTCTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTAGAGAGCCAGCAACAATTGCAAATGTCAGATCAAAGTAATATTAGCAAACAATAAGTCCCTAACTAGTTGTGACCTTTTGTAAAGTGAATTTCATTATATATGCTGTGCAATATCAGCACCAACAGAAA.
pA_standard_70:
CTTGCCTGTCGCTCTATCTTCTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTGCAACGGGGAGCCGAGATTATTGAGTGATCACCAGTAGCTGTACTATTATATAAGCTATTAAAGATTGATCAAAGTAAACATACCGCGCAATATCAGCACCAACAGAAA.
pA_standard_100:
CTTGCCTGTCGCTCTATCTTCTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTCCCAAGCGAAAACGGGTGCGTGGACTAGCGAGGAGCAAACGAAAATTCTTGGCCTGCGCAATATCAGCACCAACAGAAA.
pA_standard_130:
CTTGCCTGTCGCTCTATCTTCTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTAGGAGGCAATTCTATAAGAATGCATACGCAATATCAGCACCAACAGAAA.
pA_standard_150:
CTTGCCTGTCGCTCTATCTTCTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTCGAGCACGCAATATCAGCACCAACAGAAA.
Nanopore PCR-cDNA sequencing library prep
For Nanopore PCR-cDNA sequencing of mouse oocytes and embryos, approximately 400 oocytes or embryos per biological replicate per stage were used per sequencing run. An identical amount of ERCC spike-in standards67 (6.5 pg) per cell was added to samples at the beginning of RNA extraction. For sequencing of HeLa, 200 ng of total RNA input was used. Nanopore PCR-cDNA sequencing (PCS) was performed with an early access PCR-cDNA Sequencing Kit (SQK-PCS110) as per manufacturer’s protocol (Figure S1A). This kit is most similar to the currently available cDNA-PCR Sequencing Kit (SQK-PCS114). Briefly, ligation of Nanopore’s proprietary cDNA-RT adapter (CRTA) to the 3′ end of polyadenylated mRNAs enriched for transcripts with a minimum poly(A) tail length of 10 nt. Following enzymatic digestion of the adapter, RT with strand switching was performed, followed by selection of full-length transcripts by PCR amplification for 18 cycles. Sequencing adapters were then attached, followed by loading of the library onto an R9.4.1 flow cell (FLO-MIN106D) and sequenced in a Minion sequencing device with Minknow software (version 22.05.5). Libraries comprised of polyadenylated DNA standards were prepared by PCR amplification of the above DNA standards using the following barcoded PCR primers from the Nanopore PCR-cDNA Barcoding Kit (SQK-PCB109), followed by attachment of sequencing adapters from the SQK-PCS110 kit and sequencing on the Minion as per the SQK-PCS110 protocol: pA_standard_10: BC01, pA_standard_40: BC02, pA_standard_50: BC03, pA_standard_70: BC04, pA_standard_100: BC05, pA_standard_130: BC06, and pA_standard_150: BC07.
Data processing
Raw fast5 files were basecalled with Guppy (version 6.0.6) using following parameters: –config dna_r9.4.1_450bps_hac.cfg –fast5_out –trim_strategy none –records_per_fastq 0 –device cuda:all:100% –recursive. For barcoded libraries comprised of polyadenylated DNA standards, the following additional Guppy parameter was used for demultiplexing: –barcode_kits SQK-PCB109. Full length reads with all adapter sequences (from the CRTA, PCR primers, and sequencing adapter) were filtered by Pychopper (version 2.5.0) with following parameters: -k PCS110. Only full-length reads with a minimum basecalling Q-value score of 9 and all adapter sequences were retained for further analysis. Reads were mapped to the GRCm38 mouse transcriptome with minimap2108 (version 2.17) using the following parameters: -a -x map-ont. Reads per transcript were quantified with Salmon (version 0.14.1) using the following parameters: salmon quant -l A –noErrorModel.
Tail length measurements and comparisons
Poly(A) tail lengths were measured using the find_tails and annotate_tails functions from tailfindr65 (version 1.3) with default parameters. Since tail length measurements correlated highly between biological replicates of the same stage (Figure S2C), polyadenylated reads from the same stage were combined for further analyses. Stage-specific gene/isoform-level poly(A) tail lengths were estimated as the geometric mean of all polyadenylated reads detected for each gene/isoform at each individual stage. For consecutive stage comparisons at the gene/isoform-level, genes/isoforms were filtered by a minimum number of 10 polyadenylated reads in each of the stages compared and change in poly(A) tail length was tested by one-sided Wilcoxon tests. Significant changes in tail length were defined by a maximum Benjamini-Hochberg adjusted p value cutoff of 0.05.
PCA and hierarchical clustering
Unsupervised hierarchical clustering of stages and biological replicates by poly(A) tail length was performed in R (version 4.1.2) with the pheatmap function from the pheatmap package (version 1.0.12) and the following parameters: scale = “row”, show_rownames = F. Principal component analysis (PCA) by poly(A) tail length was performed using the prcomp function in the stats package (version 4.1.2) and the following parameters: scale = T. PCA by gene expression was performed using the plotPCA function in DESeq2110 (version 1.34.0) and default parameters.
Gene ontology overrepresentation analyses
Gene ontology overrepresentation analysis was performed with the enrichGO function from the Bioconductor package clusterProfiler112 (version 4.2.2) using gene ontology biological process annotations and the following parameters: OrgDb = org.Mm.eg. db::org.Mm.eg.db, ont = “BP”, keyType = “ENSEMBL”. Significantly enriched gene ontologies were determined by a Benjamini-Hochberg adjusted p value cutoff of <0.05.
Isoform-specific analyses
For analyses of isoform-specific poly(A) tail length regulation within individual developmental stages, isoforms were filtered by a minimum number of 10 polyadenylated reads within each stage, poly(A) length distributions of different isoforms of the same gene were compared by Kruskal-Wallis tests, and genes with differentially regulated isoforms were defined as those with significantly different tail length distributions between different detected isoforms (Benjamini-Hochberg adjusted p value <0.05). For analyses of isoform-specific poly(A) tail length regulation between consecutive developmental stages, to exclude isoforms that did not pass the significance cutoff for change in tail length (Benjamini-Hochberg adjusted p value <0.05), genes with differential isoform regulation of poly(A) tail length were defined as genes with isoforms regulated in opposite directions (≥1 isoforms with significantly lengthened tails and ≥1 isoforms with significantly shortened tails).
Motif enrichment analyses
We integrated three publicly available databases (ATtRACT,80 CISBP-RNA,79 and oRNAment81) of known RNA sequence motifs and the RNA binding proteins predicted to bind them. We then used AME111 (version 5.0.2) to search for motifs significantly enriched in the 3′UTR sequences of transcripts with significantly lengthened/shortened poly(A) tails, compared to that of transcripts with unchanged tail lengths, using the following parameters: –method fisher. Significantly enriched motifs were determined by a Bonferroni adjusted p value cutoff of <0.05.
Differential expression analysis
Read counts were normalized with ERCC spike-in standards by dividing read counts for each gene by the mean read count amongst ERCCs. Differential expression analysis was performed using the DESeq function from DESeq2110 (version 1.34.0) with default parameters and differentially expressed genes (DEGs) were defined by a minimum fold change of 2x and those that did not meet this criteria were classified as stable. For validation of changes in RNA abundance using a previously published RNA-seq dataset from Zhang et al., 2016,84 DEGs in this dataset were defined by an adjusted p value <0.05 and those that did not meet these criteria were classified as stable.
Translational efficiency analyses
Translational efficiencies (TEs) were obtained from a previously published ribosome profiling and RNA-seq dataset from Xiong et al., 202262 and TE was calculated as the ratio of ribosome profiling to RNA-seq (FPKM +1/FPKM +1) as per the authors. Unless otherwise indicated, for comparisons of gene-level poly(A) tail length and translational efficiency, genes were filtered by a minimum of 25 FPKM in the RNA-seq dataset and 10 polyadenylated reads in our PCS dataset. For consecutive stage comparisons, genes were defined as translationally activated if they had a log2(TE fold change) ≥ 0.25 and translationally repressed if they had a log2(TE fold change) ≤ −0.25.
Relative tail length analyses
Tail lengths relative to the stage-specific global mean (aka “relative tail lengths”) were calculated for each gene as follows: (TLrel)gene X = (TLabs)gene X - (mean TL)stage Y + 70 nt, where (TLrel)gene X is the relative tail length for a given gene X; (TLabs)gene X is its absolute mean tail length; and (mean TL)stage Y is the mean of mean tail lengths amongst all genes at stage Y. Note that all means were geometric means and that the scalar of 70 nt was added to avoid taking the log of negative numbers.
Definition of maternal effect and newly transcribed genes
A list of 51 maternal effect genes were obtained from literature review of genes with known biological roles in mouse oocyte/embryo development.90–92 A previously published list of 4,039 predicted minor EGA genes was obtained from Abe et al., 2015.88 A previously published list of 2,518 predicted major EGA genes was obtained by reprocessing raw RNA-seq data from Zhang et al., 202264 and defining major EGA genes as genes upregulated in the 2-cell embryo stage relative to the zygote stage (FC ≥ 5, FDR <0.01, and 2-cell mean RPKM ≥1), as per the authors.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis and software used have already been detailed in the methods sections above. The statistical details of all experiments, including the statistical tests used, the number of data points (n), statistical measures (e.g., mean, median), and inclusion criteria for genes/isoforms are denoted in these methods as well as in relevant figures and figure legends. P-values were adjusted for multiple comparisons as described in the methods details above and an adjusted p value of <0.05 was considered statistically significant.
Supplementary Material
Highlights.
Transcriptome-wide poly(A) tail and translation dynamics from oocyte to embryo in mice
In the oocyte, translation is activated by resistance to global deadenylation
In the embryo, translation is driven by readenylation of stable deadenylated mRNAs
Specific 3′ UTR motifs are associated with poly(A) tail regulation at each stage
ACKNOWLEDGMENTS
This work was supported by the Burroughs Wellcome Fund Career Award for Medical Scientists, NIH R01 GM124519, NIH K12 HD007203, NIH F30 HD089579, and NIH R01 HD093846. Computational analyses were performed on the NSF’s Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) Expanse cluster at SDSC (allocation ID: BIO2100150), supported by NSF OAC-1928224. Figures 1A and S1A and the graphical abstract were created with BioRender. We thank Jens Lykke-Andersen, Amy Pasquinelli, and Olcay Soyalan for critical review of the manuscript and Kathleen Fisch, Gene Yeo, Jens Lykke-Andersen, Amy Pasquinelli, and members of the Cook-Andersen laboratory for helpful feedback during the project.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.113710.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Schultz RM, Stein P, and Svoboda P (2018). The oocyte-to-embryo transition in mouse: past, present, and future. Biol. Reprod 99, 160–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li L, Lu X, and Dean J (2013). The maternal to zygotic transition in mammals. Mol. Aspects Med 34, 919–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tadros W, and Lipshitz HD (2009). The maternal-to-zygotic transition: a play in two acts. Development 136, 3033–3042. [DOI] [PubMed] [Google Scholar]
- 4.Richter JD (1999). Cytoplasmic Polyadenylation in Development and beyond. Microbiol. Mol. Biol. Rev 63. 446–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Passmore LA, and Coller J (2022). Roles of mRNA poly(A) tails in regulation of eukaryotic gene expression. Nat. Rev. Mol. Cell Biol 23, 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mcgrew LL, Dworkin-Rastl E, Dworkin MB, and Richter JD (1989). Poly(A) elongation during Xenopus oocyte maturation is required for translational recruitment and is mediated by a short sequence element. Genes Dev 3, 803–815. [DOI] [PubMed] [Google Scholar]
- 7.Gross PR, Malkin LI, and Moyer WA (1964). Templates for the first proteins of embryonic development. Proc. Natl. Acad. Sci. USA 51, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spirin AS (1966). On “Masked” Forms of Messenger RNA in Early Embryogenesis and in Other Differentiating Systems. Curr. Top. Dev. Biol 1, 1–38. [PubMed] [Google Scholar]
- 9.Huarte J, Belin D, and Vassalli JD (1985). Plasminogen activator in mouse and rat oocytes: Induction during meiotic maturation. Cell 43, 551–558. [DOI] [PubMed] [Google Scholar]
- 10.Mendez R, and Richter JD (2001). Translational control by CPEB: a means to the end. Nat. Rev. Mol. Cell Biol 2, 521–529. [DOI] [PubMed] [Google Scholar]
- 11.Charlesworth A, Meijer HA, and de Moor CH (2013). Specificity Factors in Cytoplasmic Polyadenylation, 4 (Wiley Interdiscip Rev RNA), pp. 437–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Norbury CJ (2013). Cytoplasmic RNA: a case of the tail wagging the dog. Nat. Rev. Mol. Cell Biol 14, 643–653. [DOI] [PubMed] [Google Scholar]
- 13.Decker CJ, and Parker R (1993). A turnover pathway for both stable and unstable mRNAs in yeast: Evidence for a requirement for deadenylation. Genes Dev 7, 1632–1643. [DOI] [PubMed] [Google Scholar]
- 14.Couttet P, Fromont-Racine M, Steel D, Pictet R, and Grange T (1997). Messenger RNA deadenylylation precedes decapping in mammalian cells. Proc. Natl. Acad. Sci. USA 94, 5628–5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bachvarova R, and De Leon V (1980). Polyadenylated RNA of mouse ova and loss of maternal RNA in early development. Dev. Biol 74, 1–8. [DOI] [PubMed] [Google Scholar]
- 16.Pikó L, and Clegg KB (1982). Quantitative changes in total RNA, total poly(A), and ribosomes in early mouse embryos. Dev. Biol 89, 362–378. [DOI] [PubMed] [Google Scholar]
- 17.Bachvarova R, De Leon V, Johnson A, Kaplan G, and Paynton BV (1985). Changes in total RNA, polyadenylated RNA, and actin mRNA during meiotic maturation of mouse oocytes. Dev. Biol 108, 325–331. [DOI] [PubMed] [Google Scholar]
- 18.Gillian-Daniel DL, Gray NK, Aström J, Barkoff A, and Wickens M (1998). Modifications of the 5 Cap of mRNAs during Xenopus Oocyte Maturation: Independence from Changes in Poly(A) Length and Impact on Translation. Mol. Cell Biol 18, 6152–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voeltz GK, and Steitz JA (1998). AUUUA Sequences Direct mRNA Deadenylation Uncoupled from Decay during Xenopus Early Development. Mol. Cell Biol 18, 7537–7545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paynton BV, Rempel R, and Bachvarova R (1988). Changes in state of adenylation and time course of degradation of maternal mRNAs during oocyte maturation and early embryonic development in the mouse. Dev. Biol 129, 304–314. [DOI] [PubMed] [Google Scholar]
- 21.Sheets MD, Fox CA, Hunt T, Vande Woude G, and Wickens M (1994). The 3’-untranslated regions of c-mos and cyclin mRNAs stimulate translation by regulating cytoplasmic polyadenylation. Genes Dev 8, 926–938. [DOI] [PubMed] [Google Scholar]
- 22.Fox CA, and Wickens M (1990). Poly(A) removal during oocyte maturation: A default reaction selectively prevented by specific sequences in the 3’ UTR of certain maternal mRNAs. Genes Dev 4, 2287–2298. [DOI] [PubMed] [Google Scholar]
- 23.Reyes JM, and Ross PJ (2016). Cytoplasmic polyadenylation in mammalian oocyte maturation. Wiley. Interdiscip. Rev. RNA 7, 71–89. [DOI] [PubMed] [Google Scholar]
- 24.Chen J, Melton C, Suh N, Oh JS, Horner K, Xie F, Sette C, Blelloch R, and Conti M (2011). Genome-wide analysis of translation reveals a critical role for deleted in azoospermia-like (Dazl) at the -to-zygote transition. Genes Dev 25, 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richter JD (2007). CPEB: a life in translation. Trends Biochem. Sci 32, 279–285. [DOI] [PubMed] [Google Scholar]
- 26.Luong XG, Daldello EM, Rajkovic G, Yang CR, and Conti M (2020). Genome-wide analysis reveals a switch in the translational program upon oocyte meiotic resumption. Nucleic Acids Res 48, 3257–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vejnar CE, Abdel Messih M, Takacs CM, Yartseva V, Oikonomou P, Christiano R, Stoeckius M, Lau S, Lee MT, Beaudoin JD, et al. (2019). Genome wide analysis of 3′ UTR sequence elements and proteins regulating mRNA stability during maternal-to-zygotic transition in zebrafish. Genome Res 29, 1100–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi B, Ma C, Liu G, Guo Y, Zhang T, Li P, Sun B-F, Yang Y, Zhang N, Zhao Y-L, et al. (2019). RNA Structural Dynamics Regulates Early Embryogenesis Through Controlling Transcriptome Fate and Function. Cell. Mol. Biol. Lett 24, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang F, Wang W, Cetinbas M, Sadreyev R, and Blower M (2020). Genome-wide Analysis Identifies Cis-Acting Elements Regulating mRNA Polyadenylation and Translation during Vertebrate Oocyte Maturation. RNA 26, 324–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang C-R, Rajkovic G, Daldello EM, Luong XG, Chen J, and Conti M (2020). The RNA-binding protein DAZL functions as repressor and activator of mRNA translation during oocyte maturation. Nat. Commun 11, 1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dai XX, Jiang JC, Sha QQ, Jiang Y, Ou XH, and Fan HY (2019). A combinatorial code for mRNA 3′-UTR-mediated translational control in the mouse oocyte. Nucleic Acids Res 47, 328–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Christou-Kent M, Dhellemmes M, Lambert E, Ray PF, and Arnoult C (2020). Diversity of RNA-Binding Proteins Modulating Post-Transcriptional Regulation of Protein Expression in the Maturing Mammalian Oocyte. Cells 9, 662–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schall PZ, and Latham KE (2021). Essential shared and species-specific features of mammalian oocyte maturation-associated transcriptome changes impacting oocyte physiology. Am. J. Physiol. Cell Physiol 321, C3–C16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suh N, Baehner L, Moltzahn F, Melton C, Shenoy A, Chen J, and Blelloch R (2010). MicroRNA Function Is Globally Suppressed in Mouse Oocytes and Early Embryos. Curr. Biol 20, 271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giraldez AJ, Mishima Y, Rihel J, Grocock RJ, Van Dongen S, Inoue K, Enright AJ, and Schier AF (2006). Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science (1979) 312, 75–79. [DOI] [PubMed] [Google Scholar]
- 36.Walser CB, and Lipshitz HD (2011). Transcript clearance during the maternal-to-zygotic transition. Curr. Opin. Genet. Dev 21, 431–443. [DOI] [PubMed] [Google Scholar]
- 37.Pepling ME (2010). A novel maternal mRNA storage compartment in mouse oocytes. Biol. Reprod 82, 807–808. [DOI] [PubMed] [Google Scholar]
- 38.Benoit P, Papin C, Kwak JE, Wickens M, and Simonelig M (2008). PAP- and GLD-2-type poly(A) polymerases are required sequentially in cytoplasmic polyadenylation and oogenesis in Drosophila. Development 135, 1969–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barnard DC, Ryan K, Manley JL, and Richter JD (2004). Symplekin and xGLD-2 are required for CPEB-mediated cytoplasmic polyadenylation. Cell 119, 641–651. [DOI] [PubMed] [Google Scholar]
- 40.Latham KE (2023). Preimplantation embryo gene expression: 56 years of discovery, and counting. Mol. Reprod. Dev 90, 169–200. [DOI] [PubMed] [Google Scholar]
- 41.Nakanishi T, Kumagai S, Kimura M, Watanabe H, Sakurai T, Kimura M, Kashiwabara S.i., and Baba T (2007). Disruption of mouse poly(A) polymerase mGLD-2 does not alter polyadenylation status in oocytes and somatic cells. Biochem. Biophys. Res. Commun 364, 14–19. [DOI] [PubMed] [Google Scholar]
- 42.Yartseva V, and Giraldez AJ (2015). The Maternal-to-Zygotic Transition During Vertebrate Development: A Model for Reprogramming. Curr. Top. Dev. Biol 113, 191–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martins JPS, Liu X, Oke A, Arora R, Franciosi F, Viville S, Laird DJ, Fung JC, and Conti M (2016). DAZL and CPEB1 regulate mRNA translation synergistically during oocyte maturation. J. Cell Sci 129, 179218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldman DS, Kiessling AA, and Cooper GM (1988). Post-transcriptional processing suggests that c-mos functions as a maternal message in mouse eggs. Oncogene 3, 159–162. [PubMed] [Google Scholar]
- 45.Chang H, Lim J, Ha M, and Kim VN (2014). TAIL-seq: Genome-wide Determination of Poly(A) Tail Length and 3′ End Modifications. Mol. Cell 53, 1044–1052. [DOI] [PubMed] [Google Scholar]
- 46.Huarte J, Belin D, Vassalli A, Strickland S, and Vassalli JD (1987). Meiotic maturation of mouse oocytes triggers the translation and polyadenylation of dormant tissue-type plasminogen activator mRNA. Genes Dev 1, 1201–1211. [DOI] [PubMed] [Google Scholar]
- 47.Ma J, Fukuda Y, and Schultz RM (2015). Mobilization of Dormant Cnot7 mRNA Promotes Deadenylation of Maternal Transcripts During Mouse Oocyte Maturation. Biol. Reprod 93, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tay J, Hodgman R, and Richter JD (2000). The Control of Cyclin B1 mRNA Translation during Mouse Oocyte Maturation. Dev. Biol 221, 1–9. [DOI] [PubMed] [Google Scholar]
- 49.Yu C, Ji SY, Sha QQ, Dang Y, Zhou JJ, Zhang YL, Liu Y, Wang ZW, Hu B, Sun QY, et al. (2016). BTG4 is a meiotic cell cycle-coupled maternal-zygotic-transition licensing factor in oocytes. Nat. Struct. Mol. Biol 23, 387–394. [DOI] [PubMed] [Google Scholar]
- 50.Lim J, Lee M, Son A, Chang H, and Kim VN (2016). mTAIL-seq reveals dynamic poly(A) tail regulation in oocyte-to-embryo development. Genes Dev 30, 1671–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Subtelny AO, Eichhorn SW, Chen GR, Sive H, and Bartel DP (2014). Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature 508, 66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eichhorn SW, Subtelny AO, Kronja I, Kwasnieski JC, Orr-Weaver TL, and Bartel DP (2016). mRNA poly(A)-tail changes specified by deadenylation broadly reshape translation in Drosophila oocytes and early embryos. Elife 5, e16955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Begik O, Diensthuber G, Liu H, Delgado-Tejedor A, Kontur C, Niazi AM, Valen E, Giraldez AJ, Beaudoin JD, Mattick JS, and Novoa EM (2023). Nano3P-seq: transcriptome-wide analysis of gene expression and tail dynamics using end-capture nanopore cDNA sequencing. Nat. Methods 20, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Workman RE, Tang AD, Tang PS, Jain M, Tyson JR, Razaghi R, Zuzarte PC, Gilpatrick T, Payne A, Quick J, et al. (2019). Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods 16, 1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Legnini I, Alles J, Karaiskos N, Ayoub S, and Rajewsky N (2019). FLAM-seq: full-length mRNA sequencing reveals principles of poly(A) tail length control. Nat. Methods 16, 879–886. [DOI] [PubMed] [Google Scholar]
- 56.Liu Y, Zhang Y, Wang J, and Lu F (2022). Transcriptome-wide measurement of poly(A) tail length and composition at subnanogram total RNA sensitivity by PAIso-seq. Nat. Protoc 17, 1980–2007. [DOI] [PubMed] [Google Scholar]
- 57.Liu Y, Nie H, Zhang Y, Lu F, and Wang J (2023). Comprehensive analysis of mRNA poly(A) tails by PAIso-seq2. Sci. China Life Sci 66, 187–190. [DOI] [PubMed] [Google Scholar]
- 58.Liu Y, Nie H, Liu H, and Lu F (2019). Poly(A) inclusive RNA isoform sequencing (PAIso—seq) reveals wide-spread non-adenosine residues within RNA poly(A) tails. Nat. Commun 10, 5292–5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y, Nie H, Zhang C, Hou Z, Wang J, and Lu F (2021). Poly(A) Tail Length Is a Major Regulator of Maternal Gene Expression during the Mammalian Oocyte-To-Embryo Transition. Preprint at bioRxiv 10.1101/2021.08.29.458052. [DOI] [Google Scholar]
- 60.Liu Y, Jin J, Zhang Y, Wang L-Y, Zhang C, Hou Z, Li W, Liu Z, Lu F, and Wang J (2021). Conservation and divergence of poly(A) tail regulation during the mammalian oocyte-to-embryo transition. Preprint at bioRxiv 10.1101/2021.08.29.458065. [DOI] [Google Scholar]
- 61.Liu Y, Zhao H, Shao F, Zhang Y, Nie H, Zhang J, Li C, Hou Z, Chen ZJ, Wang J, et al. (2023). Remodeling of maternal mRNA through poly(A) tail orchestrates human oocyte-to-embryo transition. Nat. Struct. Mol. Biol 30, 200–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xiong Z, Xu K, Lin Z, Kong F, Wang Q, Quan Y, Fang J, Yu G, Liu B, Wang L, et al. (2022). Ultrasensitive Ribo-Seq Reveals Translational Landscapes during Mammalian Oocyte-To-Embryo Transition and Pre-implantation Development. Nat. Cell. Biol 24, 968–980. [DOI] [PubMed] [Google Scholar]
- 63.Zou Z, Zhang C, Wang Q, Hou Z, Xiong Z, Kong F, Wang Q, Song J, Liu B, Liu B, et al. (2022). Translatome and transcriptome co-profiling reveals a role of TPRXs in human zygotic genome activation. Science (1979) 378, 7923. [DOI] [PubMed] [Google Scholar]
- 64.Zhang C, Wang M, Li Y, and Zhang Y (2022). Profiling and functional characterization of maternal mRNA translation during mouse maternal-to-zygotic transition. Sci. Adv 8, eabj3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krause M, Niazi AM, Labun K, Torres Cleuren YN, Muller FS, and Valen E (2019). tailfindr: alignment-free poly(A) length measurement for Oxford Nanopore RNA and DNA sequencing. RNA 25, 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baker SC, Bauer SR, Beyer RP, Brenton JD, Bromley B, Burrill J, Causton H, Conley MP, Elespuru R, Fero M, et al. (2005). The External RNA Controls Consortium: a progress report. Nat. Methods 2, 731–734. [DOI] [PubMed] [Google Scholar]
- 67.External RNA Controls Consortium (2005). Proposed methods for testing and selecting the ERCC external RNA controls. BMC Genom 6, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jiang X, Cheng Y, Zhu Y, Xu C, Li Q, Xing X, Li W, Zou J, Meng L, Azhar M, et al. (2023). Maternal NAT10 orchestrates oocyte meiotic cell-cycle progression and maturation in mice. Nat. Commun 14, 3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu Y, Lu X, Shi J, Yu X, Zhang X, Zhu K, Yi Z, Duan E, and Li L (2016). BTG4 is a key regulator for maternal mRNA clearance during mouse early embryogenesis. J. Mol. Cell Biol 8, 366–368. [DOI] [PubMed] [Google Scholar]
- 70.Sha QQ, Zhu YZ, Li S, Jiang Y, Chen L, Sun XH, Shen L, Ou XH, and Fan HY (2020). Characterization of zygotic genome activation-dependent maternal mRNA clearance in mouse. Nucleic Acids Res 48, 879–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhu Y, Wu W, Chen S, Zhang Z, Zhang G, Li J, and Jiang M (2022). Mettl3 downregulation in germinal vesicle oocytes inhibits mRNA decay and the first polar body extrusion during maturation. Biol. Reprod 107, 765–778. [DOI] [PubMed] [Google Scholar]
- 72.Zhao LW, Zhu YZ, Chen H, Wu YW, Pi SB, Chen L, Shen L, and Fan HY (2020). PABPN1L mediates cytoplasmic mRNA decay as a placeholder during the maternal-to-zygotic transition. EMBO Rep 21, 499566–e50015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takei N, Sato K, Takada Y, Iyyappan R, Susor A, Yamamoto T, and Kotani T (2021). Tdrd3 regulates the progression of meiosis II through translational control of Emi2 mRNA in mouse oocytes. Curr. Res. Cell Biol 2, 100009. [Google Scholar]
- 74.Rong Y, Ji SY, Zhu YZ, Wu YW, Shen L, and Fan HY (2019). ZAR1 and ZAR2 are required for oocyte meiotic maturation by regulating the maternal transcriptome and mRNA translational activation. Nucleic Acids Res 47, 11387–11402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chang H, Yeo J, Kim JG, Kim H, Lim J, Lee M, Kim HH, Ohk J, Jeon HY, Lee H, et al. (2018). Terminal Uridylyltransferases Execute Programmed Clearance of Maternal Transcriptome in Vertebrate Embryos. Mol. Cell 70, 72–82.e7. [DOI] [PubMed] [Google Scholar]
- 76.Eisen TJ, Eichhorn SW, Subtelny AO, Lin KS, Mcgeary SE, Gupta S, and Bartel DP (2020). The Dynamics of Cytoplasmic mRNA Metabolism. Mol. Cell 77, 786–799.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alles J, Legnini I, Pacelli M, and Rajewsky N (2023). Rapid nuclear deadenylation of mammalian messenger RNA. iScience 26, 105878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nicholson-Shaw AL, Kofman ER, Yeo GW, and Pasquinelli AE (2022). Nuclear and cytoplasmic poly(A) binding proteins (PABPs) favor distinct transcripts and isoforms. Nucleic Acids Res 50, 4685–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, et al. (2013). A compendium of RNA-binding motifs for decoding gene regulation. Nature 499, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Giudice G, Sánchez-Cabo F, Torroja C, and Lara-Pezzi E (2016). ATtRACT-a database of RNA-binding proteins and associated motifs. Database 2016. baw035–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Benoit Bouvrette LP, Bovaird S, Blanchette M, and Lécuyer E (2020). ORNAment: A database of putative RNA binding protein target sites in the transcriptomes of model species. Nucleic Acids Res 48, D166–D173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu HB, Muhammad T, Guo Y, Li MJ, Sha QQ, Zhang CX, Liu H, Zhao SG, Zhao H, Zhang H, et al. (2019). RNA-Binding Protein IGF2BP2/IMP2 is a Critical Maternal Activator in Early Zygotic Genome Activation. Adv. Sci 6, 1900295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ramos SBV, Stumpo DJ, Kennington EA, Phillips RS, Bock CB, Ribeiro-Neto F, and Blackshear PJ (2004). The CCCH tandem zinc-finger protein Zfp36l2 is crucial for female fertility and early embryonic development. Development 131, 4883–4893. [DOI] [PubMed] [Google Scholar]
- 84.Zhang B, Zheng H, Huang B, Li W, Xiang Y, Peng X, Ming J, Wu X, Zhang Y, Xu Q, et al. (2016). Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature Publishing Group 537, 553–557. [DOI] [PubMed] [Google Scholar]
- 85.Baer BW, and Kornberg RD (1983). The protein responsible for the repeating structure of cytoplasmic poly(A)-ribonucleoprotein. J. Cell Biol 96, 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xiang K, and Bartel DP (2021). The molecular basis of coupling between poly(A)-tail length and translational efficiency. Elife 10, 1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Su Y-Q, Sugiura K, Woo Y, Wigglesworth K, Kamdar S, Affourtit J, and Eppig JJ (2007). Selective degradation of transcripts during meiotic maturation of mouse oocytes. Dev. Biol 302, 104–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Abe KI, Yamamoto R, Franke V, Cao M, Suzuki Y, Suzuki MG, Vlahovicek K, Svoboda P, Schultz RM, and Aoki F (2015). The first murine zygotic transcription is promiscuous and uncoupled from splicing and 3′ processing. EMBO J 34, 1523–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Israel S, Ernst M, Psathaki OE, Drexler HCA, Casser E, Suzuki Y, Makalowski W, Boiani M, Fuellen G, and Taher L (2019). An integrated genome-wide multi-omics analysis of gene expression dynamics in the preimplantation mouse embryo. Sci. Rep 9, 13356–13415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Innocenti F, Fiorentino G, Cimadomo D, Soscia D, Garagna S, Rienzi L, Ubaldi FM, Zuccotti M, and SIERR. (2022). Maternal effect factors that contribute to oocytes developmental competence: an update. J. Assist. Reprod. Genet 39, 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mitchell LE (2022). Maternal effect genes: Update and review of evidence for a link with birth defects. HGG Adv 3, 100067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Condic ML (2016). The Role of Maternal-Effect Genes in Mammalian Development: Are Mammalian Embryos Really an Exception? Stem Cell Rev. Rep 12, 276–284. [DOI] [PubMed] [Google Scholar]
- 93.Erhardt S, Su IH, Schneider R, Barton S, Bannister AJ, Perez-Burgos L, Jenuwein T, Kouzarides T, Tarakhovsky A, and Surani MA (2003). Consequences of the depletion of zygotic and embryonic enhancer of zeste 2 during preimplantation mouse development. Development 130, 4235–4248. [DOI] [PubMed] [Google Scholar]
- 94.Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, Xie ZG, Shi L, He X, Jin SG, et al. (2011). The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477, 606–610. [DOI] [PubMed] [Google Scholar]
- 95.Xu Q, Wang F, Xiang Y, Zhang X, Zhao ZA, Gao Z, Liu W, Lu X, Liu Y, Yu XJ, et al. (2015). Maternal BCAS2 protects genomic integrity in mouse early embryonic development. Development 142, 3943–3953. [DOI] [PubMed] [Google Scholar]
- 96.Kim K-H, Kim E-Y, and Lee K-A (2008). SEBOX Is Essential for Early Embryogenesis at the Two-Cell Stage in the Mouse. Biol. Reprod 79, 1192–1201. [DOI] [PubMed] [Google Scholar]
- 97.Park M-W, Kim K-H, Kim E-Y, Lee S-Y, Ko J-J, and Lee K-A (2015). Associations Among Sebox and Other MEGs and its Effects on Early Embryogenesis. PLoS One 13, e0115050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bebbere D, Albertini DF, Coticchio G, Borini A, and Ledda S (2021). The subcortical maternal complex: emerging roles and novel perspectives. Mol. Hum. Reprod 27, gaab043. [DOI] [PubMed] [Google Scholar]
- 99.Tong Z-B, Gold L, Pfeifer KE, Dorward H, Lee E, Bondy CA, Dean J, and Nelson LM (2000). Mater, a maternal effect gene required for early embryonic development in mice. Nat. Genet 26, 267–268. [DOI] [PubMed] [Google Scholar]
- 100.Gurtu VE, Verma S, Grossmann AH, Liskay RM, Skarnes WC, and Baker SM (2002). Maternal Effect for DNA Mismatch Repair in the Mouse. Genetics 160. 271–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McGrew LL, and Richter JD (1990). Translational control by cytoplasmic polyadenylation during Xenopus oocyte maturation: characterization of cis and trans elements and regulation by cyclin/MPF. EMBO J 9, 3743–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Simon R, Tassan JP, and Richter JD (1992). Translational control by poly(A) elongation during Xenopus development: Differential repression and enhancement by a novel cytoplasmic polyadenylation element. Genes Dev 6, 2580–2591. [DOI] [PubMed] [Google Scholar]
- 103.Vassalli JD, Huarte J, Belin D, Gubler P, Vassalli A, O’Connell ML, Parton LA, Rickles RJ, and Strickland S (1989). Regulated polyadenylation controls mRNA translation during meiotic maturation of mouse oocytes. Genes Dev 3, 2163–2171. [DOI] [PubMed] [Google Scholar]
- 104.Paris J, and Richter JD (1990). Maturation-Specific Polyadenylation and Translational Control: Diversity of Cytoplasmic Polyadenylation Elements, Influence of Poly(A) Tail Size, and Formation of Stable Polyadenylation Complexes. Mol. Cell Biol 10, 5634–5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Barkoff A, Ballantyne S, and Wickens M (1998). Meiotic maturation in Xenopus requires polyadenylation of multiple mRNAs. EMBO J 17, 3168–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu Y, Zhang Y, Nie H, Liu Z, Wang J, and Lu F (2021). Re-polyadenylation occurs predominantly on maternal mRNA degradation inter-mediates during mammalian oocyte-to-embryo transition. Preprint at bioRxiv 10.1101/2021.08.29.458080. [DOI] [Google Scholar]
- 107.Niazi AM, Krause M, and Valen E (2021). Transcript Isoform-Specific Estimation of Poly(A) Tail Length by Nanopore Sequencing of Native RNA. In Methods in Molecular Biology (Humana Press Inc.), pp. 543–567. [DOI] [PubMed] [Google Scholar]
- 108.Li H (2018). Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 34, 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McLeay RC, and Bailey TL (2010). Motif Enrichment Analysis: a unified framework and an evaluation on ChIP data. BMC Bioinf 11, 165–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yu G, Wang LG, Han Y, and He QY (2012). ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Nanopore PCR-cDNA sequencing data associated with this study have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. This paper also analyzes existing, publicly available data and these accession numbers for the datasets are listed in the key resources table. Additional data tables, including a summary of sequencing runs, gene-level poly(A) tail lengths at each stage, gene-level changes in poly(A) tail length at each stage transition, gene ontologies enriched in genes with poly(A) tail lengthening or shortening at each stage transition, isoform-level poly(A) tail lengths at each stage, isoform-level changes in poly(A) tail length at each stage transition, isoform-specific poly(A) tail length regulation both within and between stages, 3′ UTR sequence motifs enriched in mRNAs polyadenylated or deadenylated at each stage transition, lists of deadenylated-decayed vs. -stable genes at each stage transition, and poly(A) tail lengths and translational efficiencies for maternal effect genes at each stage are available in Data S1–S4.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| TRIzol | Thermo Fisher Scientific | 15-596-018 |
| Phenol: chloroform: iso-amyl alcohol (25:24:1) | VWR | VWRV0966-100ML |
| PMSG | Lee BioSolutions | 493-10 |
| Collagenase | Sigma-Aldrich | C9407 |
| M2 media | Sigma-Aldrich | M7167 |
| HCG | Sigma-Aldrich | C1063 |
| Hyaluronidase | Sigma-Aldrich | H4272 |
| Sodium acetate buffer solution | Sigma-Aldrich | S7899 |
| GenElute LPA | Sigma-Aldrich | 56575 |
| DMEM media | Thermo Fisher Scientific | 11995073 |
| FBS | Cytiva | SH30071.03HI |
| Penicillin/Streptomycin | Thermo Fisher Scientific | 15140122 |
| 0.25% Trypsin-EDTA | Thermo Fisher Scientific | 25200114 |
|
| ||
| Critical commercial assays | ||
|
| ||
| PCR-cDNA Sequencing Kit | Oxford Nanopore Technologies | SQK-PCS110 |
| PCR-cDNA Barcoding Kit | Oxford Nanopore Technologies | SQK-PCB109 |
| R9.4.1 flow cell | Oxford Nanopore Technologies | FLO-MIN106D |
| Minion sequencing device | Oxford Nanopore Technologies | MIN-101B |
| AMPure XP | Beckman Coulter | A63881 |
| RNA Clean & Concentrator-5 | Zymo Research | R1014 |
| DNAse I | Zymo Research | E1011-A |
| T4 DNA Ligase | NEB | M0202M |
| 5x quick ligation reaction buffer | NEB | E6058A |
| Lambda exonuclease | NEB | M0262S |
| USER Enzyme | NEB | M5505S |
| RNAClean XP | Beckman Coulter | A63987 |
| Maxima H Minus Reverse Transcriptase | Thermo Fisher Scientific | EP0751 |
| dNTP Mix | NEB | N0447S |
| LongAmp Hot Start Taq 2x Master Mix | NEB | M0533S |
| Exonuclease I | NEB | M0293S |
| RNaseOUT | Thermo Fisher Scientific | 10777019 |
| RNA 6000 Pico Kit | Agilent Technologies | 5067-1513 |
| High Sensitivity DNA Kit | Agilent Technologies | 5067-4626 |
|
| ||
| Deposited data | ||
|
| ||
| Nanopore PCR-cDNA sequencing | This paper | GEO: GSE228001 |
| RNA-seq | Zhang et al.84 | GEO: GSE71434 |
| Ribosome profiling and RNA-seq | Xiong et al.62 | GEO: GSE165782 |
| RNA-seq | Zhang et al.64 | GEO: GSE169632 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Hela | ATCC | CCL-2 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57B/6J mice | Jackson Labs | 000664 |
| B6D2F1/J mice | Jackson Labs | 100006 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Polyadenylated standards (See method details for sequences) | This study | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Minknow (22.05.5) | Oxford Nanopore Technologies | https://community.nanoporetech.com/downloads |
| Guppy (6.0.6) | Oxford Nanopore Technologies | https://community.nanoporetech.com/downloads |
| Pychopper (2.5.0) | Oxford Nanopore Technologies | https://github.com/epi2me-labs/pychopper |
| minimap2 (2.17) | Li et al.108 | https://github.com/lh3/minimap2 |
| Salmon (0.14.1) | Patro et al.109 | https://github.com/COMBINE-lab/salmon |
| tailfindr (1.3) | Krause et al.65 | https://github.com/adnaniazi/tailfindr |
| DESeq2 (1.34.0) | Love et al.110 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| AME (5.0.2) | McLeay et al.111 | https://meme-suite.org/meme/tools/ame |
| clusterProfiler (4.2.2) | Yu et al.112 | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html |
|
| ||
| Other | ||
|
| ||
| ERCC Spike-In RNAs | Thermo Fisher Scientific | 4456653 |