

Over 55 million people globally live with Alzheimer's disease (AD) or related dementias (ADRD) and the number is expected to double every twenty years. Until recently, only symptomatic treatments were available to patients with AD, including acetylcholine esterase inhibitors, of which the last one, galantamine, was approved by the US Food and Drug Administration (FDA) in 2001, and the noncompetitive N-methyl-D-aspartate receptor antagonist, memantine, approved in 2003. Thus, for nearly 20 years, despite numerous clinical trials, the landscape for developing new therapy looked barren and grim (Cummings et al., 2022). Then, in June 2021, FDA granted accelerated approval to aducanumab (AduhelmTM), an anti-amyloid-β (Aβ) monoclonal antibody (mAb) targeting “protofibrils”, which were described in the 1990s (Walsh et al., 1997) and shown later to be key neurotoxins. Aducanumab became the first approved drug addressing the underlying pathology of AD, albeit with substantial adverse effects. Due to the side effects and relatively weak efficacy data, the FDA approval was highly controversial (Kuller and Lopez, 2021): Ten of the eleven members of the scientific advisory committee voted against approval and the eleventh voted uncertain. Following the FDA's decision, three committee members quit in protest.
Yet better news was coming. In January 2023, FDA granted similar accelerated approval to lecanemab (LeqembiTM), another anti-Aβ protofibril mAb using a similar mechanism and causing similar side effects, however, this time with less controversy because the clinical trial data demonstrated a clear reduction in the progression of memory loss (van Dyck et al., 2023). FDA's Accelerated Approval Program supports drugs that both treat serious conditions and reach a marker predicting clinical benefit. This pathway brings a drug to market faster than traditional approval, but predicts, rather than demonstrates, clinical benefit. Therefore, companies must provide post-marketing studies confirming clinical benefits to obtain traditional approval. After additional review, in July 2023, the FDA granted full approval to lecanemab for the treatment of early-stage AD.
This landmark decision supports the “amyloid cascade hypothesis” in its revised form focusing on Aβ oligomers (Kirkitadze et al., 2002) and paves the way for approval of other disease-modifying therapeutics for AD. Interestingly, soon after the accelerated approval of lecanemab, the FDA denied accelerated approval for donanemab, a third mAb in this class, due to an inadequate size of the clinical trial. Donanemab will be considered for traditional approval at the end of 2023 when more data become available. In contrast, due to the controversy surrounding it, aducanumab likely will not receive full approval.
Lecanemab and aducanumab remove toxic Aβ protofibrils from the AD brain yet cause significant side effects called amyloid-related imaging abnormalities (ARIA) potentially triggering headaches, worsening confusion, dizziness, visual disturbance, nausea, and seizures. Moreover, a meta-analysis of clinical trials testing potential therapeutics for AD, including aducanumab, lecanemab, and donanemab found that ARIA-causing mAbs may cause accelerated brain atrophy (Alves et al., 2023). Thus, the battle against AD is not over, and the pros and cons of these treatments must be weighed carefully by patients and their caregivers.
The major global threat posed by ADRD has led to substantial investment by governments around the world supporting a highly active preclinical and clinical drug discovery and development pipeline. In August 2023, there were 3163 active ADRD clinical trials globally. Therapeutics are divided into two broad categories: small molecules and biologics. ‘Small molecules’ are organic compounds of molecular mass < 1 kDa whereas biologics are larger, e.g., antibodies and oligonucleotides. Small molecules currently make up > 90% of therapeutics on the market (Gurevich and Gurevich, 2014). Their small size facilitates permeating through barriers and reaching their targets and their relative simplicity lowers the cost of bulk production. Biologics are more difficult to produce in high quantity and quality but have superior target specificity compared to small molecules. In recent years, nearly 30% of all new, approved drugs have been biologics, though this trend may be driven by higher profit margins of biologics than by the underlying biology. Biologics currently comprise 2% of drugs on the market yet account for 37% of spending (Makurvet, 2021).
For ADRD, the recent mAb success is promising and supports the development of improved compounds targeting not only toxic Aβ oligomers, but also toxic oligomers and aggregates of tau and potentially other amyloidogenic proteins, e.g., α-synuclein and TAR DNA binding protein-43 (Bitan, 2019). The mechanism by which the mAb therapies work is not well understood. Their very low blood-brain barrier permeability limits their efficacy and to avoid catabolism in the digestive system, they are delivered intravenously, necessitating frequent patient visits to specialized clinics. In addition, though ARIA are mostly considered a low-to-moderate risk, in some cases they may be lethal. In the lecanemab phase-3 clinical trial, patients homozygous for apolipoprotein E ε4 had an increased incidence of ARIA leading Biogen/Eisai to warn against the use of the drug in these patients. Thus, now that biologics have provided proof-of-concept, small-molecule drugs using similar modes of intervention are welcome.
The hemorrhagic ARIA type, ARIA-H, is caused by ruptured capillaries, likely due to clearance of deposited vascular Aβ, a common comorbidity of AD called cerebral amyloid angiopathy (Greenberg et al., 2020). The high affinity of the mAbs for Aβ and their low blood-brain barrier permeability means they bind vascular Aβ deposits before and to a larger extent than Aβ in the brain parenchyma. Selectivity for Aβ oligomers improves the odds of targeting protofibrils in the parenchyma, yet the high concentration of the mAbs in the blood increases binding to the vascular deposits leading to ARIA. These concerns and the warning about potentially accelerating brain atrophy in a meta-analysis of multiple clinical trials (Alves et al., 2023) necessitate close monitoring of ARIA and brain atrophy in current and future mAb clinical trials.
Small-molecule drug candidates tested for AD to date have not been associated with ARIA. Most have addressed the production of Aβ and hyperphosphorylated tau, or the aggregation itself. Inhibitors/modulators of β- or γ-secretases, which produce Aβ by sequential cleavage of Aβ-protein precursor have been explored extensively, attempting to reduce the overall concentration of Aβ or the Aβ42/Aβ40 ratio (Vogt et al., 2023). Three β-secretase and three -secretase inhibitors demonstrated amyloid reduction in phase-3 clinical trials but ultimately failed. As the secretases are not specific to Aβ-protein precursor, their inhibition affected the processing of other substrates and in the case of γ-secretase inhibitors, even worsened patients' cognition.
The small molecule tramiprosate (homotaurine) inhibited Aβ aggregation in vitro and was safe in clinical trials but offered little symptom alleviation due to limited blood-brain barrier permeability and poorly understood mechanism in vivo. Its pro-drug, ALZ-801, is currently in a phase-3 clinical trial. Scyllo-inositol and epigallocatechin-3-gallate are small molecules that inhibited Aβ oligomerization and aggregation but failed in clinical trials due to unfavorable efficacy and pharmacokinetics (Yao et al., 2022).
Targeting hyperphosphorylated tau faces additional challenges because the oligomers and aggregates are intracellular and harder to target using antibodies. The complexity of tau, existing as six different isoforms undergoing numerous post-translational modifications further increases the challenge of targeting its self-assembly. Small-molecule tau phosphorylation, acetylation, and deglycosylation inhibitors have been tested for ADRD, including tideglusib and LiCl, inhibitors of glycogen synthase kinase 3β, a major kinase responsible for tau hyperphosphorylation, which did not produce significant cognitive improvement. Salsalate and MK-8719, which inhibit tau acetylation at Lys174 and deglycosylation by O-GlcNAcase, respectively, have progressed to phase-1 for AD or the rare tauopathy, progressive supranuclear palsy, respectively, following promising preclinical experiments (Wang et al., 2021). Methylene blue (RemberTM), an approved drug for other diseases, showed tau assembly inhibition and evidence of cognitive improvement in a phase-2 clinical trial yet failed in a phase-3 trial against behavioral-variant frontotemporal dementia, possibly because in some studies it was found to accelerate to tau aggregation.
Perhaps the failures of small-molecule drug candidates and the limited efficacy of antibodies targeting pathological forms of Aβ or tau are due to the very fact that they address either of these targets rather than both. The two proteins attack vulnerable neurons synergistically, suggesting that targeting one without the other would have a limited effect. A strategy that could target both proteins simultaneously is enhancing their clearance, not via specific agents, such as antibodies, but rather by facilitating their intracellular degradation by the autophagy-lysosomal pathway, which can degrade protein aggregates (Figure 1). A popular strategy for enhancing the autophagy-lysosomal pathway is mammalian target of rapamycin (mTOR) inhibition, which rapidly activates proteolysis. However, clinically, this could be problematic because increasing amounts of aggregated proteins delivered into already overwhelmed lysosomes could exacerbate, rather than ameliorate, the accumulation of the aggregates. Additionally, due to the central role of the mTOR pathway, its inhibition may lead to negative side effects concerning normal cell growth, metabolism, synaptic plasticity, and memory. Improving lysosomal function is promising for ADRD because the mechanism is well-understood, cell-autonomous, and does not cause the same side effects as immunotherapy-based approaches. In this context, small molecules called “molecular tweezers” developed by our group (Shahpasand-Kroner et al., 2023) may be a particularly attractive therapeutic option for AD because they target the self-assembly of both Aβ and hyperphosphorylated tau, and concentrate in the lysosomes, where they act as broad-spectrum facilitators of the degradation of amyloid proteins. They have also been shown to have a wide therapeutic margin in mice and are not expected to induce ARIA. Overall, we posit that the approval of lecanemab is the beginning of a road that likely will lead to the development of effective small-molecule agents targeting both amyloidogenic proteins instigating and propagating the neuropathology of AD, followed by similar strategies targeting additional amyloidogenic proteins in ADRD.
Figure 1.
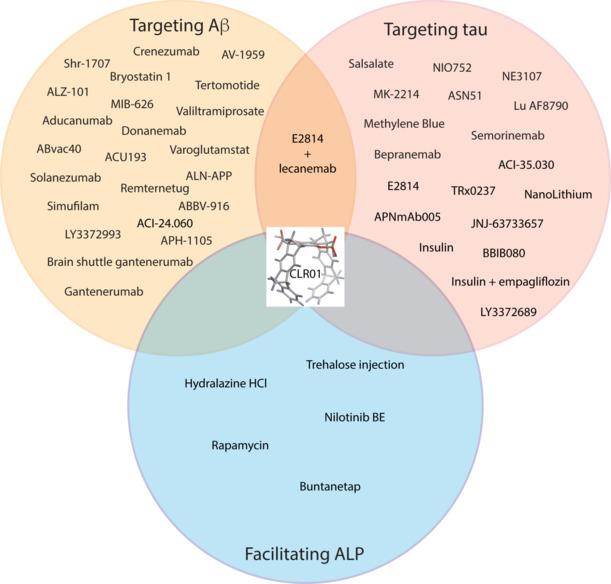
Drug candidates targeting Aβ and/or tau or facilitating degradation via the ALP.
The drugs listed in the three circles are currently in clinical trials. The molecular tweezer CLR01 (Shahpasand-Kroner et al., 2023) in the middle has been shown preclinically to target all three mechanisms. Created using Adobe Illustrator. Aβ: Amyloid-β; ALP: autophagy-lysosomal pathway.
This work was supported by NIH grants RF1NS126406 and R21NS130326 and by a generous gift from the Binder Foundation (to GB).
GB is an author and an inventor of International Patent PCT/EP2010/000437, US Patent No. US 8481484 B2, European Patent No. EP 2493859 A1, US Patent No. 10,918,657, International Patent Application No. PCT/US2019/039943, US Patent Application No. 17/255,963. EU Application No. 19824953.4, International Patent Application No. PCT/US2019/029221, EU application No. 19850534.9, International Patent Application No. PCT/US2019/029222, and EU application No. 19841367.6.
Footnotes
C-Editors: Zhao M, Sun Y, Qiu Y; T-Editor: Jia Y
References
- Alves F, Kalinowski P, Ayton S. Accelerated brain volume loss caused by anti-β-amyloid drugs: a systematic review and meta-analysis. Neurology. 2023;100:e2114–2124. doi: 10.1212/WNL.0000000000207156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitan G. Disease-modifying therapy for proteinopathies: can the exception become the rule? Prog Mol Biol Transl Sci. 2019;168:277–287. doi: 10.1016/bs.pmbts.2019.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings J, Lee G, Nahed P, Kambar M, Zhong K, Fonseca J, Taghva K. Alzheimer's disease drug development pipeline: 2022. Alzheimers Dement (N Y) 2022;8:e12295. doi: 10.1002/trc2.12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat Rev Neurol. 2020;16:30–42. doi: 10.1038/s41582-019-0281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich EV, Gurevich VV. Therapeutic potential of small molecules and engineered proteins. Handb Exp Pharmacol. 2014;219:1–12. doi: 10.1007/978-3-642-41199-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkitadze MD, Bitan G, Teplow DB. Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J Neurosci Res. 2002;69:567–577. doi: 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- Kuller LH, Lopez OL. ENGAGE and EMERGE: Truth and consequences? Alzheimers Dement. 2021;17:692–695. doi: 10.1002/alz.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makurvet FD. Biologics vs. small molecules: drug costs and patient access. Med Drug Discov. 2021 doi: 10.1016/j.medidd.2020.100075. [Google Scholar]
- Shahpasand-Kroner H, Siddique I, Malik R, Linares GR, Ivanova MI, Ichida J, Weil T, Munch J, Sanchez-Garcia E, Klarner FG, Schrader T, Bitan G. Molecular tweezers: supramolecular hosts with broad-spectrum biological applications. Pharmacol Rev. 2023;75:263–308. doi: 10.1124/pharmrev.122.000654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, Kanekiyo M, Li D, Reyderman L, Cohen S, Froelich L, Katayama S, Sabbagh M, Vellas B, Watson D, Dhadda S, Irizarry M, Kramer LD, Iwatsubo T. Lecanemab in early Alzheimer's disease. N Engl J Med. 2023;388:9–21. doi: 10.1056/NEJMoa2212948. [DOI] [PubMed] [Google Scholar]
- Vogt ACS, Jennings GT, Mohsen MO, Vogel M, Bachmann MF. Alzheimer's disease: a brief history of immunotherapies targeting amyloid β. Int J Mol Sci. 2023;24:3895. doi: 10.3390/ijms24043895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid β-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- Wang L, Bharti, Kumar R, Pavlov PF, Winblad B. Small molecule therapeutics for tauopathy in Alzheimer's disease: walking on the path of most resistance. Eur J Med Chem. 2021;209:112915. doi: 10.1016/j.ejmech.2020.112915. [DOI] [PubMed] [Google Scholar]
- Yao W, Yang H, Yang J. Small-molecule drugs development for Alzheimer's disease. Front Aging Neurosci. 2022;14:1019412. doi: 10.3389/fnagi.2022.1019412. [DOI] [PMC free article] [PubMed] [Google Scholar]