

Abstract
Hypoxia is a hallmark of cancer development. However, the molecular mechanisms by which hypoxia promotes tumor metastasis are not fully understood. In this study, we demonstrate that hypoxia promotes breast cancer metastasis through suppression of ΔNp63α in a HIF1α-independent manner. We show that hypoxia-activated XBP1s forms a stable repressor protein complex with HDAC2 and EZH2 to suppress ΔNp63α transcription. Notably, H3K27ac is predominantly occupied on the ΔNp63 promoter under normoxia, while H3K27me3 on the promoter under hypoxia. We show that XBP1s binds to the ΔNp63 promoter to recruit HDAC2 and EZH2 in facilitating the switch of H3K27ac to H3K27me3. Pharmacological inhibition or the knockdown of either HDAC2 or EZH2 leads to increased H3K27ac, accompanied by the reduced H3K27me3 and restoration of ΔNp63α expression suppressed by hypoxia, resulting in inhibition of cell migration. Furthermore, the pharmacological inhibition of IRE1α, but not HIF1α, upregulates ΔNp63α expression in vitro and inhibits tumor metastasis in vivo. Clinical analyses reveal that reduced p63 expression is correlated with the elevated expression of XBP1, HDAC2, or EZH2, and is associated with poor overall survival in human breast cancer patients. Together, these results indicate that hypoxia-activated XBP1s modulates the epigenetic program in suppression of ΔNp63α to promote breast cancer metastasis independent of HIF1α and provides a molecular basis for targeting the XBP1s/HDAC2/EZH2-ΔNp63α axis as a putative strategy in the treatment of breast cancer metastasis.
Subject terms: Metastasis, Epigenetics
Introduction
Hypoxia, a hallmark of solid tumors, impacts various aspects of tumor biology, including tumor survival, growth, autophagy, metabolic reprogramming, tumor metastasis, and immune reactivity [1]. It is well-documented that hypoxia activates the hypoxia-inducible factor 1 α (HIF1α) to regulate the expression of a subset of genes involved in angiogenesis, tumor growth, and metastasis among others [2]. HIF1α targeted genes includes angiogenesis-associated VEGF, ANGPT1/2 and PDGF, EMT-associated Snail1, Twist1, ZEB1/2, CCL2 and LOX, or glucose-metabolism associated GLUT1/3 [3, 4].
Hypoxia can also activate the unfolded protein response (UPR) [5]. Hypoxia inhibits the function of ERO1α, an oxidoreductase important for proper disulfide bond formation and protein folding, and thus triggers endoplasmic reticulum (ER) stress and activation of UPR [6]. UPR comprises three independent sensors: PERK (PKR-like ER kinase), IRE1α (inositol-requiring protein 1), and ATF6α (activating transcription factor 6 α). Activation of IRE1α leads to a conformational change that activates its RNase activity to excise a 26-nucleotide intron of the mRNA of X-box binding protein 1 (XBP1) and produces a more stable and active protein known as XBP1s. XBP1s functions as a transcription factor to regulate a subset of genes involved in various biological processes including development, and immune response [7, 8]. Activated PERK phosphorylates eukaryotic translation initiator factor 2α (eIF2α), which leads to the inhibition of protein synthesis. ATF6α is a bZIP transcription factor that regulates the expression of genes of the ER‑associated degradation (ERAD) pathway [9].
The p53-related transcription factor p63 plays an essential role in diverse functions, including embryonic development, epidermal stem cell regeneration and differentiation, cell growth, and survival [10]. Deregulated p63 expression is associated with various human diseases, including developmental diseases, cancer, aging, and metabolic disorders [11, 12]. ΔNp63α is the prominent p63 isoform expressed in epithelial cells and has been documented as a master regulator of cell adhesion [13]. ΔNp63 transactivates a serial of cell adhesion genes, including E-cadherin, ITGA6, ITGB1, ITGB4, desmoplakin, Par3, fibronectin, LAMC2 involved in cell-cell and cell-matrix adhesion [13, 14]. Clinical analyses and various studies demonstrate that ΔNp63α can transactivate the expression of MKP3, CD82, or AMPK, and functions as a critical metastasis suppressor [15–17].
ΔNp63α expression is tightly controlled by various signaling, including oncogenic activation, Hippo, and TGF-β [18–22]. However, whether ΔNp63α is intimately involved in hypoxia-mediated tumor metastasis remains unknown. In this study, we show that hypoxia inhibits ΔNp63α to promote cell motility and tumor metastasis in a HIF1α-independent but XBP1s-dependent manner. We provide evidence that both the HIF1α and XBP1s-ΔNp63α pathways are pivotal in hypoxia-induced tumor metastasis.
Results
Hypoxia promotes cell motility through inhibition of ΔNp63α expression in a HIF1α-independent manner
It has been well-documented that hypoxia can promote tumor growth and metastasis in a HIF1α-dependent manner [3]. ΔNp63α is a critical tumor metastasis suppressor in response to various signaling including oncogenic activation, Hippo, and TGF-β [18–20]. However, whether ΔNp63α is intimately involved in hypoxia-mediated tumor metastasis remains unknown. We thus investigated the connection between hypoxia and ΔNp63 protein expression in breast cancer cell HCC1806-derived subcutaneous tumor. As shown in Fig. 1A, immunohistochemistry (IHC) analyses showed that the ΔNp63 protein expression was significantly reduced in the inner layers of the solid tumor, which often endure hypoxia, suggesting that hypoxia may be associated with the downregulation of ΔNp63. We then examined the effects of hypoxia on the expression of ΔNp63 in immortalized human breast epithelial MCF-10A or triple-negative breast cancer (TNBC) HCC1806 cells in vitro. Both MCF-10A and HCC1806 cells predominantly express ΔNp63α (Supplementary Fig. 1A), a major p63 protein isoform expressed in epithelial cells [13]. As shown in Fig. 1B, C, while hypoxia upregulated HIF1α protein expression, as expected, it also led to a significant reduction of ΔNp63α and ΔNp63β expression at both mRNA and protein levels, concomitant with downregulation of ΔNp63 transcriptional targets, as exemplified by E-cadherin and Par3 [18]. As expected, hypoxia significantly promoted cell motility, as shown by increased migration, invasion, and wound-healing in both MCF-10A and HCC1806 cells (Supplementary Fig. 1B–E).
Fig. 1. Hypoxia inhibits ΔNp63α expression via EZH2-mediated upregulation of H3K27me3 in promoting cell motility in a HIF1α-independent manner.

A HCC1806 cells were subcutaneously injected into female 6-week-old Nude mice. Tumor was subjected to ΔNp63 staining by using IHC (left panel); the expression of ΔNp63 in different regions was quantified by AOD (right panel). B, C MCF-10A or HCC1806 cells were cultured under normoxia (20% O2) or hypoxia (1% O2) for 24 h, then subjected to immunoblot (B) and qPCR analyses (C). D, E HCC1806 cells stably expressing empty vector (EV), ΔNp63α or ΔNp63β were cultured under hypoxia or normoxia for 24 h, and were then subjected to immunoblot (D) or transwell assays for cell migration (E). F HCC1806 cells stably expressing a control shRNA (shC) or a shRNA specific for HIF1α (shHIF1α-#1 and shHIF1α-#2) were cultured under hypoxia for 24 h, and were then subjected to immunoblot analysis. G, H HCC1806 cells stably expressing a control shRNA (shC) or a shRNA specific for HIF1α (shHIF1α-#1) with a shRNA specific for p63 (shp63) or expressing vector for ΔNp63α (WT and R304W) were cultured under hypoxia for 24 h, and were then subjected to immunoblot (G) and transwell analyses (H). I MCF-10A or HCC1806 cells were cultured under normoxia or hypoxia for 24 h, then subjected to immunoblot analysis. J–L HCC1806 cells were cultured under hypoxia or normoxia, and infected with the virous expressed a control shRNA (shC) or a shRNA specific for EZH2 (shEZH2-#1 and shEZH2-#2), and were then subjected to immunoblot (J), qPCR (K) or transwell assays (L). Results were presented as means ± SD from three independent experiments in triplicates. **P < 0.01, *P < 0.05, NS no significance.
We next examined which isoform of ΔNp63 is attributed to hypoxia-induced cell motility. Restored expression of ΔNp63α, not ΔNp63β, effectively restored E-cadherin and Par3 expression, accompanied by complete rescued hypoxia-induced cell migration (Fig. 1D, E and Supplementary 1F, G). These results indicate that the downregulation of ΔNp63α is responsible for hypoxia-induced cell motility. Notably, ablation of HIF1α had little effect on the hypoxia-induced downregulation of ΔNp63α (Fig. 1F, G), suggesting that hypoxia downregulates ΔNp63α and promotes cell migration in a HIF1α-independent manner. To verify that hypoxia-mediated suppression of ΔNp63α and increased cell migration can be HIF1α-independent, we knocked down HIF1α under hypoxia. Our results showed that ablation of HIF1α significantly inhibited hypoxia-induced upregulation of VEGFA expression, as expected, it also dramatically inhibited hypoxia-induced cell migration (Fig. 1G, H). However, ablation of HIF1α had little effect on the protein levels of ΔNp63α as well as E-cadherin and Par3, which were repressed upon hypoxia. In addition, restored expression of the wild type of ΔNp63α, but not ΔNp63α-R304W defective in transactivation function [23], further enhanced inhibition of cell migration upon knockdown of HIF1α (Fig. 1H). Furthermore, treatment of DMOG, a potent HIF1α protein stabilizer [24], led to upregulated HIF1α protein levels, yet it failed to affect the hypoxia-mediated reduction of ΔNp63α (Supplementary Fig. 1H). Together, these results indicate that hypoxia promotes cell motility through both HIF1α-dependent and HIF1α-independent pathways, in later of which hypoxia suppresses ΔNp63α expression and consequently the elevation of cell motility.
Hypoxia upregulates H3K27me3 resulting in suppression of ΔNp63α expression and elevated cell migration independent of HIF1α
It has been reported that hypoxia can alter the epigenetic landscape [25]. Given that hypoxia leads to the suppression of ΔNp63α transcription, we wondered whether epigenetic regulation plays a role in hypoxia-mediated suppression of ΔNp63α. As shown in Fig. 1I, we first examined the expression of two well-documented repression marks [26], trimethylated H3K27 (H3K27me3) and H3K9 (H3K9me3). Notably, hypoxia led to an increase of H3K27me3, but not H3K9me3, concomitant with downregulation of ΔNp63α expression. To investigate whether H3K27me3 plays a causal role in hypoxia-induced downregulation of ΔNp63α, we examined the effect of inhibition of EZH2, a key enzyme in catalyzing H3K27me3 [27], on ΔNp63α expression. As shown in Fig. 1J, K and Supplementary Fig. 1I, J, either knockdown or pharmacological inhibition of EZH2 [28], completely blocked the hypoxia-induced upregulation of H3K27me3, effectively restored ΔNp63α expression and significantly suppressed hypoxia-induced cell invasion (Fig. 1L and Supplementary Fig. 1K). Interestingly, inhibition of EZH2 by GSK126 led to dramatically elevated ΔNp63α expression in triple-negative breast cancer MDA-MB-231 cells, which express little ΔNp63α protein (Supplementary Fig. 1L, M), suggesting that EZH2-mediated epigenetic regulation is pivotal in determining ΔNp63α expression. Importantly, while GSK126 could upregulate ΔNp63α expression and inhibit hypoxia-mediated cell migration (Supplementary Fig. 1N, O), the knockdown of HIF1α had little effect on the levels of H3K27me3 and ΔNp63α expression (Supplementary Fig. 1N), indicating that hypoxia-induced upregulation of H3K27me3 plays a key role in the suppression of ΔNp63α expression and the elevation of cell motility independent of HIF1α.
Hypoxia-activated IRE1α-XBP1s resulting in elevation of H3K27me3 and suppression of ΔNp63α expression
Hypoxia is well-documented to induce endoplasmic reticulum (ER) stress, which often activates three pathways including RNA-dependent protein kinase-like ER kinase (PERK), inositol-requiring enzyme 1α (IRE1α), and activating transcription factor 6 α (ATF6α) [29]. To investigate whether hypoxia-induced ER stress plays a role in this process, we knocked down individual ER stress sensors, including PERK, IRE1α, or ATF6α, respectively. As shown in Fig. 2A, depletion of IRE1α blocked hypoxia-induced elevation of H3K27me3, accompanied by the downregulation of ΔNp63α expression. By contrast, depletion of PERK had little effect on H3K27me3 or ΔNp63α expression. Interestingly, the knockdown of ATF6α could only block hypoxia-induced downregulation of ΔNp63α, but had little effect on hypoxia-induced elevation of H3K27me3 levels, suggesting that ATF6α regulates ΔNp63α expression independent of H3K27me3. Together, these data indicate that IRE1α plays an important role in the hypoxia-induced upregulation of H3K27me3 resulting in the suppression of ΔNp63α expression. Thus, we hypothesized that IRE1α is critical in connecting hypoxia to the epigenetic regulation of ΔNp63α.
Fig. 2. Hypoxia activates XBP1s to inhibit ΔNp63α expression in promoting cell invasion by EZH2.

A HCC1806 cells infected with lentivirus expressing either of two different shRNAs specific for IRE1α (shIRE1α-#1 and shIRE1α-#2), ATF6α (shATF6α-#1 and shATF6α-#2), PERK (shPERK-#1 and shPERK-#2) or a control shRNA (shC) were cultured under hypoxia for 24 h, and were subjected to immunoblot analysis. B–D HCC1806 cells stalely expressing shC or shXBP1 (#1 or #2) were cultured under hypoxia for 24 h, and were subjected to immunoblot (B), qPCR (C), or transwell assays (D). E–G HCC1806 cells stalely expressing empty vector (EV), XBP1s, or XBP1u were subjected to immunoblot (E), qPCR (F), or transwell assays (G). H–J MCF-10A or HCC1806 cells stably expressing empty vector (EV), XBP1s or XBP1s-ΔNp63α were subjected to immunoblot (H), transwell (I), and xCELLigence RTCA assays (J). K–M HCC1806 cells stalely expressing an empty vector (EV) or XBP1s were infected with lentivirus encoding a shRNA specific for EZH2 (shEZH2-#1 and shEZH2-#2), or a control shRNA (shC). 48 h post-infection, cells were subjected to immunoblot (K), qPCR (L), or transwell assays (M). Results were presented as means ± SD from three independent experiments in triplicates. **P < 0.01, NS no significance.
It has been well-established that activated IRE1α can exert its function through XBP1s [30]. We therefore examined whether XBP1s, derived from hypoxia-mediated IRE1α activation, plays a role in regulating ΔNp63α expression. As shown in Fig. 2B, C, hypoxia upregulated XBP1s, accompanied by the elevation of H3K27me3 and downregulation of ΔNp63α transcription, both of which were effectively rescued upon knockdown of XBP1. Notably, the knockdown of XBP1 completely inhibited hypoxia-induced cell invasion (Fig. 2D). Depletion of XBP1 in highly motile MDA-MB-231 cells could also lead to a significant reduction of H3K27me3 and upregulation of ΔNp63α expression, resulting in effective inhibition of cell invasion (Supplementary Fig. 2A, B). Further studies showed that ectopic expression of XBP1s, but not XBP1u, resulted in a significant elevation of H3K27me3, accompanied by reduced ΔNp63α expression and increased HCC1806 cell invasion and migration (Fig. 2E–G and Supplementary Fig. 2C). Immunofluorescent analyses showed that ectopic expression XBP1s in HCT1806 cells led to an accumulation of H3K27me3 in the nuclei (Supplementary Fig. 2D), concomitant with reduced nuclear ΔNp63α expression (Supplementary Fig. 2E). To substantiate the role of the XBP1s in the hypoxia-mediated suppression of ΔNp63α expression, we employed thapsigargin, an ER stress inducer [31], to induce the expression of XBP1s. As shown in Supplementary Fig. 2F–H, while thapsigargin led to activation of XBP1s resulting in upregulation of GRP78, as expected, it also markedly upregulated H3K27me3, concomitant with inhibited ΔNp63α expression and elevated cell migration. We next examined the causal role of ΔNp63α on the biological impacts of the hypoxia-XBP1s axis-induced cell motility. As shown in Fig. 2H–J, ectopic expression of XBP1s, again, led to a significant elevation of H3K27me3 and reduced expression of ΔNp63α (Fig. 2H), accompanied by increased cell migration (Fig. 2J) and invasion (Fig. 2I), which was effectively rescued by restored ΔNp63α expression, concomitant with the recovered expression of E-cadherin and Par3, the key downstream effectors of ΔNp63α (Fig. 2H).
We next investigated whether EZH2 plays a direct role in XBP1s-mediated ΔNp63α inhibition. Depletion of EZH2 or pharmacological inhibition of EZH2 effectively restored the expression of ΔNp63α that was suppressed by XBP1s (Fig. 2K, L and Supplementary Fig. 2I), concurrently with significant inhibition of cell invasion (Fig. 2M and Supplementary Fig. 2J). Taken together, these results suggest that hypoxia activates XBP1s to elevate H3K27me3, resulting in the inhibition of ΔNp63α expression, which in turn promotes cell motility.
Hypoxia-activated XBP1s promotes epigenetic reprogramming by HDAC2-mediated downregulation of H3K27ac and upregulation of H3K27me3 resulting in suppression of ΔNp63α expression and elevation of cell migration
It is well known that, in addition to methylation, H3K27 can also be acetylated. Notably, H3K27ac and H3K27me3 are often reciprocally regulated and mutually exclusive [32]. Indeed, we observed a decrease of acetylated H3K27 (H3K27ac), concomitant with an increase of H3K27me3 in response to hypoxia (Fig. 3A). Since the acetylation modification on H3K27 is removed by histone deacetylases (HDACs) prior to the methylation of H3K27 [33], we rationalized that HDACs may play a role in hypoxia-induced upregulation of H3K27me3 and inhibition of ΔNp63α expression. Treatment with trichostatin A (TSA), a pan-inhibitor of class I, II, and IV HDACs, led to a marked increase in H3K27ac levels, as expected (Fig. 3A). Notably, TSA not only blocked hypoxia-mediated upregulation of H3K27me3 but also effectively restored expression of ΔNp63α, E-cadherin, and Par3, resulting in suppression of hypoxia-induced cell migration in HCC1806 cells (Fig. 3B, C). Interestingly, TSA significantly upregulated H3K27ac and downregulated H3K27me3, concomitant with a drastic upregulation of ΔNp63α expression and inhibition of cell migration in MDA-MB-231 cells under normoxia (Supplementary Fig. 3A–C), suggesting that the HDAC-H3K27ac axis plays a critical role in ΔNp63α-mediated cell motility. Furthermore, TSA largely reversed XBP1s-mediated suppression of ΔNp63α expression and reversal of XBP1s-mediated cell migration (Fig. 3D–F).
Fig. 3. Hypoxia downregulates H3K27ac and upregulates H3K27me3 via XBP1s-HDAC2 axis in promoting cell migration.

A–C HCC1806 cells treated with or without TSA (10 ng/mL) under hypoxia or normoxia for 48 h followed by immunoblot (A), qPCR (B), or transwell assays (C). D–F HCC1806 cells stably expressing empty vector (EV) or XBP1s were treated with or without TSA (10 ng/mL) and cultured under normoxia for 48 h. Cells were then subjected to immunoblot (D), qPCR (E), or transwell assays (F). G HCC1806 cells stably expressing XBP1s were infected with lentivirus expressing either of two different shRNAs specific for HDAC1 (shHDAC1-#1 and shHDAC1-#2), HDAC2 (shHDAC2-#1 and shHDAC2-#2), HDAC3 (shHDAC3-#1 and shHDAC3-#2), HDAC8 (shHDAC8-#1 and shHDAC8-#2), or a control shRNA (shC). Cells were then subjected to qPCR analysis. The cells stably expressing empty vector (EV) as a control. H, I HCC1806 stable cells expressing XBP1s were infected with lentivirus expressing either of two different shRNAs specific for HDAC2 (shHDAC2-#1 and shHDAC2-#2), or a control shRNA (shC), then subjected to immunoblot (H) and transwell assays (I), the cells stably expressing empty vector (EV) as a control. J, K HCC1806 cells treated with or without SCA (10 ng/mL) under hypoxia or normoxia for 48 h followed by immunoblot (J) or qPCR assays (K). Results are presented as means ± SD from three independent experiments in triplicates. **P < 0.01, *P < 0.05.
Since TSA selectively inhibits HDAC1, HDAC2, HDAC3, or HDAC8 [34], we next investigated which HDAC(s) plays a causal role in regulating H3K27ac and ΔNp63α expression. Depletion of HDAC2, but not HDAC1, HDAC3, or HDAC8, markedly reversed XBP1s-mediated suppression of ΔNp63α expression (Fig. 3G and Supplementary Fig. 3D). Importantly, the depletion of HDAC2 not only inhibited H3K27me3 but also restored ΔNp63α expression suppressed by XBP1s and completely blocked XBP1s-induced cell migration (Fig. 3H, I). Furthermore, inhibition of HDAC2 by a selective HDAC2 inhibitor, santacruzamate A (SCA) [35], led to an effective reverse of XBP1s-mediated or hypoxia-induced alteration of H3K27ac and H3K27me3, accompanied by the upregulation of ΔNp63α expression (Supplementary Fig. 3E, F and Fig. 3J, K). Together, these results suggest that hypoxia-induced cell migration is achieved through the elevation of XBP1s expression and epigenetic reprogramming, in which HDAC2-mediated downregulation of H3K27ac is critical in the inhibition of ΔNp63α gene transcription.
Hypoxia facilitates XBP1s interaction with HDCA2 and EZH2 to promote the switch of H3K27ac to H3K27me3 on the ΔNp63 promoter resulting in the suppression of ΔNp63 expression
We next investigated the molecular basis with which XBP1s promotes epigenetic reprogramming in the regulation of ΔNp63α gene transcription in response to hypoxia. As shown in Fig. 4A, hypoxia-activated XBP1s can interact with HDAC2 and EZH2. While HDAC2 interacts with EZH2 under normoxia, in keeping with previous reports [36, 37], HDAC2 also interacts with XBP1s under hypoxia (Fig. 4B). Notably, XBP1s was unable to complex with EZH2 in the absence of HDAC2 (Fig. 4C). On the contrary, depletion of EZH2 had little effect on the interaction between XBP1s and HDAC2 (Fig. 4D). Further studies showed that the C terminus of XBP1s was required in binding HDAC2 (Fig. 4E, F). To investigate whether hypoxia promotes the switch of H3K27ac to H3K27me3 on the ΔNp63 gene promoter, we performed ChIP-seq analyses in HCC1806 cells under normoxia or hypoxia. As shown in Fig. 4G, the H3K27ac mark was significantly enriched on the promoter region of the ΔNp63 gene under normoxia which was replaced by the H3K27me3 mark under hypoxia. These results suggest that hypoxia-activated XBP1s forms protein complexes with HDAC2-EZH2, resulting in the switch of H3K27ac to H3K27me3 on the promoter of the ΔNp63 gene.
Fig. 4. Hypoxia facilitates XBP1s interaction with the HDAC2-EZH2 complexes and loading on the ΔNp63 promoter resulting in switching H3K27ac to H3K27me3 to inhibit ΔNp63 transcription.

A HCC1806 cells were cultured under normoxia or hypoxia for 24 h, then subjected to anti-XBP1s (or anti-normal rabbit IgG) immunoprecipitation (IP), the co-precipitating endogenous HDAC2, EZH2 and XBP1 proteins were examined by immunoblot analysis (IB). B HCC1806 cells were cultured under normoxia or hypoxia for 24 h, then subjected to anti-HDAC2 (or anti-normal mouse IgG1) immunoprecipitation (IP); the co-precipitating endogenous XBP1, HDAC2, and EZH2 proteins were examined by immunoblot analysis (IB). C HCC1806 cells stably expressing shC or shHDAC2 (#1 and #2) were cultured under hypoxia for 24 h, then subjected to anti-XBP1s (or anti-normal rabbit IgG) immunoprecipitation (IP); the co-precipitating endogenous HDAC2, EZH2, and XBP1s proteins were examined by immunoblot analysis (IB). D HCC1806 cells stably expressing shC or shEZH2 (#1 and #2) were cultured under hypoxia for 24 h, then subjected to anti-HDAC2 (or anti-normal mouse IgG1) immunoprecipitation (IP); the co-precipitating endogenous EZH2 and XBP1s proteins were examined by immunoblot analysis (IB). E Schematic diagram of the full-length form of XBP1u and XBP1s, and truncated forms of XBP1s protein. The summary of XBP1 interaction with HDAC2 is based on (F). F HEK-293T cells, expressing Flag-tagged HDAC2 together with HA-tagged XBP1u, XBP1s, XBP1s-N or XBP1s-C, were subjected to anti-Flag immunoprecipitation (IP); the co-precipitating XBP1u, XBP1s, XBP1s-N and XBP1s-C proteins were examined by immunoblot analysis (IB). The asterisks indicate the nonspecific bands. G HCC1806 cells were grown under normoxia or hypoxia for 24 h, then subjected to chromatin immunoprecipitation (ChIP) of H3K27me3 or H3K27ac combined with sequencing analysis (ChIP-seq). H, I ChIP assay using indicated antibodies or a normal rabbit IgG were performed in HCC1806 cells which were cultured under normoxia or hypoxia for 24 h. Primers specific for P1, P2, Grp78, or NC (Negative control) were used. Data derived from PCR (H) or qPCR (I) analyses were shown.
To substantiate the role of XBP1s in the regulation of the ΔNp63 gene transcription, we employed computer-aided analyses which revealed two putative XBP1s binding elements (CACGT), termed P1 and P2, on the ΔNp63 gene promoter (Supplementary Fig. 4A, B). The luciferase reporter activity assays showed that ectopic expression of XBP1s was able to inhibit the reporter activities of either ΔNp63-Gluc-WT or ΔNp63-Gluc-P2 Mut, but not ΔNp63-Gluc-P1 Mut (Supplementary Fig. 4C, D), suggesting that XBP1s binds to P1 site of the ΔNp63 gene promoter. Further ChIP assays showed that hypoxia promoted XBP1s bound to the Grp78 promoter, as expected. By contrast, hypoxia promoted XBP1s, HDAC2, and EZH2 bound to the P1 site of the ΔNp63 promoter (Fig. 4H, I). Together, these results indicate that hypoxia-activated XBP1s directly binds to the ΔNp63 promoter, recruiting HDAC2 and EZH2 to remove H3K27ac mark followed by adding H3K27me3 mark and consequently inhibiting ΔNp63α transcription.
The XBP1s-ΔNp63α axis critically regulates cancer cell motility and tumor metastasis associated with the clinical prognosis of human breast cancer patients
We further investigated the role of the XBP1s-ΔNp63α axis in tumor metastasis using tail vein-injection mouse metastasis models. As shown in Fig. 5A–C, mice injected with HCC1806 cells overexpressing XBP1s exhibited multiple metastatic nodules on the lung surfaces, which was effectively inhibited by simultaneous overexpression of ΔNp63α, indicating that XBP1s-mediated suppression of ΔNp63α is critically important in tumor metastasis. To investigate whether IRE1α-XBP1s-induced tumor metastasis is dependent on HIF1α, we examined the effects of PX478, a HIF1α inhibitor [38], or/and MKC8866, an IRE1α inhibitor [39], on the tumor metastasis in vivo. As shown in Fig. 5D, while PX478 could significantly inhibit HIF1α and its downstream target VEGFA, it had little effect on ΔNp63α expression. On the other hand, MKC8866, which specifically inhibits the activity of IRE1α leading to the blockage of XBP1s production [39], could dramatically upregulate the expression of ΔNp63α with little effect on the expression of HIF1α and VEGFA (Fig. 5D). Importantly, while the administration of PX478 or MKC8866 led to significant but incomplete inhibition of tumor metastasis, the combined inhibition of HIF1α and IRE1α by PX478/MKC8866 completely suppressed tumor metastasis in mouse models (Fig. 5E, F). These results strongly support the notion that both the HIF1α pathway and the IRE1α-XBP1s-ΔNp63α axis critically contribute to hypoxia-induced tumor metastasis.
Fig. 5. XBP1s inhibits ΔNp63a expression to promote cell migration and tumor metastasis.

A–C HCC1806 stable cells were injected in the tail vein of nude mice (6 mice per group). Lungs were dissected after 50 days, and inspected for metastatic nodules on the surface (A, B), then subjected to H&E staining for histological analysis (C). **P < 0.01. D MDA-MB-231 cells were treated with PX478 (10 μM), MKC8866 (5 μM) or/and DMSO as indicated for 48 h, and then subjected to immunoblot analysis. E, F MDA-MB-231 cells were injected in the tail vein of nude mice (5 mice per group), then 7 days after cell inoculation, these mice were treated with either PX478 (100 mg/kg, i.p.) or/and MKC8866 (300 mg/kg, oral) daily for another 14 days. Lungs were dissected on the day 30 after inoculation, then subjected to H&E staining for histological analysis (E), and the lung lesions were counted (F).
Clinical analyses showed that the elevated expression of XBP1, HDAC2, or EZH2 in human invasive lobular breast carcinoma was significantly associated with reduced TP63 expression (Fig. 6A). Further analyses revealed a remarkable negative relationship between the mRNA expression of TP63 and XBP1, EZH2, or HDAC2 (Fig. 6B). Consistently, immunohistochemical (IHC) analyses of human breast cancer biopsy samples (n = 60) showed an inverse correlation between ΔNp63 protein expression and XBP1s (Fig. 6C). Moreover, low expression of TP63 (P = 0.00057) or high expression of XBP1 (P = 1.4E-6) was associated with poor prognosis in breast cancer patients (Fig. 6D).
Fig. 6. Expression of TP63 is negatively correlated with XBP1/HDAC2/EZH2 in human breast cancer.

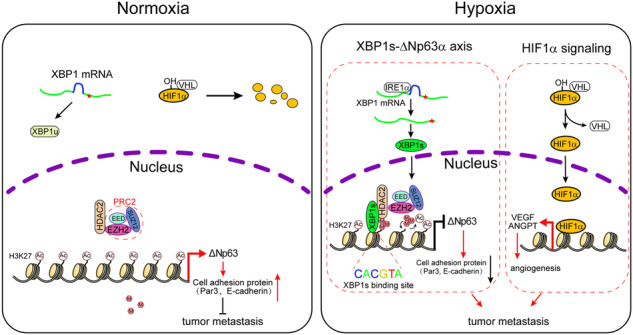
A, B The Oncomine Curtis breast cancer dataset was used to analyze TP63, XBP1, HDAC2, and EZH2 mRNA levels in normal breast samples and invasive lobular breast cancer samples (A). The same datasets were used for analyses of the Pearson correlation coefficient (R value) and a two-tail probability test (P value) (B). C Tissue microarray slides containing consecutive sections derived from human breast carcinoma were subjected to IHC staining (left) and to quantitative analyses (AOD) for protein expression of XBP1s and ΔNp63 (right). D The correlation between TP63, XBP1, HDAC2, or EZH2 mRNA levels and overall survival in breast cancer patients was analyzed using the Kaplan–Meier Plotter database, the log-rank test P values are shown. E A model depicts hypoxia-mediated epigenetic regulation of ΔNp63 through XBP1s and the role of ΔNp63 in cancer metastasis. Under normoxia, HIF1α is hydroxylated which promotes its binding to the von Hippel–Lindau (VHL) E3 ligase, resulting in its subsequent ubiquitylation and proteasomal degradation. XBP1 mRNA is translated into XBP1u (Asterisks represent the stop codon). HDAC2 interacts with the PRC2 complex (EED, EZH2 and SUZ12), but cannot bind to the ΔNp63 promoter. Under hypoxia, HIF1α cannot be hydroxylated leading to its dissociation with the VHL and stabilization. Stabilized HIF1α transactivates a subset of genes expression to promote tumor metastasis. Besides, hypoxia also activates IRE1α-XBP1s signaling. XBP1s interacts with HDAC2-PRC2 complex to switch H3K27ac to H3K27me3 on ΔNp63 promoter, leading to ΔNp63 expression reduced and consequent promoting tumor metastasis.
Discussion
Hypoxia has been documented as a critical driving force for tumor metastasis [1]. Hypoxia leads to the stabilization of transcription factor HIF1α, which in turn transactivates VEGF and ANGPT1/2 to promote angiogenesis, a key biological process for tumor growth and metastasis. In this study, we provide evidence that, in parallel with the HIF1α pathway, hypoxia can trigger epigenetic reprogramming involved in the XBP1s-HDAC2-EZH2 axis, and consequently, suppress ΔNp63α expression to facilitate breast cancer metastasis.
p63 protein contained two classes of isoforms, TAp63 and ΔNp63, which play distinct roles in various biological processes, including development, metabolism, epidermal mesenchymal transition, stemness, senescence, cell death, cell cycle arrest, and tumorigenesis [40]. Specifically conditional knockout mouse model demonstrated that depletion of TAp63 promotes sarcoma development and mammary tumorigenesis in vivo [41, 42]. Whereas ΔNp63 is highly expressed in various squamous cell carcinoma [43]. ΔNp63α is a pivotal metastasis suppressor [12] and is tightly regulated at transcriptional, post-transcriptional, translational, and post-translational levels [11]. Our recent work demonstrated that ΔNp63α is transcriptionally inhibited by several oncogenic signaling (Ras, PIK3CA, or HER2)-mediated tumor metastasis [18]. In addition, ΔNp63α can be transcriptionally regulated by Snail, ZEB2, E47, STAT3, SOX2 [44–48]. Notably, ΔNp63α can be regulated at the post-transcription levels, including mRNA stability and translational efficiency [49, 50]. ΔNp63α protein stability is tightly regulated by the post-translational modification. It has been shown that the E3 ubiquitin ligase ITCH, WWP1, FBXW7, Pirh2, or CHIP can bind to and destabilize ΔNp63α protein [51–55]. Deubiquitylase USP28, on the other hand, can remove the polyubiquitination chain to stabilize ΔNp63α protein [56]. In this study, we revealed another layer of regulation of ΔNp63α expression. We show that hypoxia-activated XBP1s can recruit HDAC2 and EZH2 to form a repressor complex on the promoter of ΔNp63 resulting in the suppression of the ΔNp63α transcription and consequently promoting breast cancer metastasis (Fig. 6E). Interestingly, our results indicate that under normoxia, H3K27ac is primarily found on the ΔNp63 promoter while hypoxia promotes a switch of H3K27ac to H3K27me3 for the execution of transcription suppression. Notably, hypoxia has been shown to trigger epigenetic change in a HIF1α-dependent or HIF1α-independent manner [57, 58]. For instance, hypoxia can inhibit the activity of an oxygen-dependent demethylase KDM6A (also termed UTX), leading to the elevation of H3K27me3 [25]. However, our results indicate that KDM6A is unlikely to be involved since hypoxia can still elevate H3K27me3 in KDM6A-deficient HCC1806 cells [59].
UPR (Unfolded Protein Response) signaling has been shown to play an important role in tumor metastasis [5]. UPR can be executed through three distinctive branches including PERK-eIF2α, ATF6α, or IRE1α-XBP1s. It has been reported that hypoxic can activate the PERK/eIF2α arm of the UPR signaling in promoting tumor metastasis through induction of the expression of LAMP3 [60]. However, depletion of PERK is unable to reverse the hypoxia-mediated suppression of the ΔNp63α expression (Fig. 2A), indicating that the UPR-PERK axis is not involved in the hypoxia-ΔNp63α axis-mediated tumor metastasis. Notably, the knockdown of ATF6α can effectively restore ΔNp63α expression even though the H3K27me3 levels remain comparable to the hypoxia induction, raising an interesting possibility that the UPR-ATF6α axis may regulate the ΔNp63α to impact cell migration and tumor metastasis independent of H3K27me3, which deserves further investigation.
XBP1s is a multitasking transcription factor that is a key component of the ER stress response and regulates a variety of different biological processes. XBP1s plays an important role in differentiation during liver, mammary gland development and B lymphocytes to plasma cells [61–63]. XBP1s can directly regulate the expression of lipogenic genes to facilitate lipogenesis and antigen presentation [62, 64]. XBP1s is also involved in the regulation of the glutamine influx that is necessary to sustain mitochondrial respiration in T cells [65]. XBP1s can also activate the transcription of c-Myc to promote cell proliferation of NK [66].
XBP1s has been shown as an essential survival factor and is required for tumor growth upon hypoxia [67]. Notably, Hypoxia promotes HIF1α-XBP1s interaction in facilitating the expression of HIF1α targeted genes and promoting metastasis in triple-negative breast cancer [68]. In line with this notion, we found that the knockdown of IRE1α results in the downregulation of VEGFA (Fig. 2A). In this study, our results indicate that another function of hypoxia-activated XBP1 is to engage epigenetic regulation of ΔNp63α expression to impact cell migration and tumor metastasis. Notably, the C-terminal of XBP1s physically interacts with HDAC2 and recruits EZH2 to form a repressor complex. Interestingly, pharmacological inhibition of IRE1α by MKC8866 markedly reduces the yield of XBP1s as expected, it dramatically elevates ΔNp63α expression while it has only mild effects on the HIF1α and VEGF protein levels (Fig. 5G). Pharmacological inhibition of HIF1α by PX478 effectively inhibits the expression of HIF1α-targeted genes and significantly suppresses tumor metastasis (Fig. 5G, H). Together, these results indicate that both HIF1α and the XBP1s-ΔNp63α pathways critically contribute to hypoxia-induced tumor metastasis. Notably, ER stress-induced activation of the IRE1α-XBP1s pathway can enhance tumor cell EMT and invasion by inducing the expression of Snail [69]. Thus, it is possible that activation of the IRE1α-XBP1s-Snail pathway may impact tumor metastasis upon hypoxia. However, our results indicate that the ectopic expression of ΔNp63α, but not ΔNp63β, completely rescues the expression of cell adhesion proteins E-cadherin and Par3, accompanied by complete inhibition of hypoxia-induced cell migration (Fig. 1D, E), supporting the notion that ΔNp63α is the key downstream effector of hypoxia in cell migration.
One important finding in this study is both the HIF1α signaling and the IRE1α-XBP1s-ΔNp63α pathway are pivotal in hypoxia-mediated tumor metastasis. Therefore, this study provides a rationale for exploring the strategy of combined inhibition of HIF1α and IRE1α/XBP1s in the effective suppression of hypoxia-induced metastasis.
Materials and methods
Cell culture
The MCF-10A, HCC1806, MDA-MB-231, and HEK-293T cell lines were obtained from the American Type Culture Collection (ATCC). MCF-10A cells were maintained in 1:1 mixture of DMEM and F12 medium (Invitrogen, Carlsbad, CA, USA), 5% horse serum (Invitrogen), 100 ng/mL cholera toxin (Sigma, St Louis, MO, USA), 10 μg/mL insulin (Sigma), 20 ng/mL epidermal growth factor (Invitrogen), 500 ng/mL hydrocortisone (Sigma). MDA-MB-231 and HEK293FT cells were cultured in DMEM (Gibco, Rockville, MD, USA) containing 10% FBS. HCC1806 cells were grown in RPMI-1640 (Gibco) containing 10% FBS. All cell lines were free from mycoplasma contamination and cultured supplemented with penicillin (100 U/mL)/streptomycin (100 μg/mL) at 37 °C in a humidified incubator under 5% CO2. For hypoxia treatment, cells were delivered into an anaerobic chamber maintained in a humidified atmosphere of 5% CO2, 1% O2, and 94% N2 at 37 °C.
Plasmid construction
The short hairpin RNAs (shRNAs) targeting human HIF1α, PERK, IRE1α, ATF6α, XBP1, HDAC1, HDAC2, HDAC3, HDAC8, or EZH2 were generated by insertion of specific oligoes into a pLKO.1-puromycin lentiviral vector (10878, Addgene), and pLKO.1-scramble (1864, Addgene, Cambridge, MA, USA) was a negative control vector containing scrambled shRNA. The oligoes used in this study were listed in Supplementary Table S1.
The constructs encoding human ΔNp63α, TAp63α, ΔNp63β or ΔNp63γ were described in previous study [70]. The open reading frame (ORF) of human XBP1s, XBP1u, or HDAC2 was purchased from Miaoling Plasmid Sharing Platform (miaolingbio.com, Wuhan, Hubei, China) and cloned into the lentiviral vector pLVX-puro (632164, Clontech, Mountain View, CA, USA). The truncation mutants (XBP1s-N and XBP1s-C) of XBP1s were subcloned into the pLVX-HA-puro vector. For promoter assay, a fragment of human ΔNp63 promoter containing XBP1s putative binding sites (P1 and P2) was inserted into the Gluc-On promoter reporter vector (pEZX-PG04, GeneCopoeia, Guangzhou, China) and designated as ΔNp63-Gluc-WT; the putative binding site P1 or P2 was mutated and designated as ΔNp63-Gluc-WT-P1 Mut or ΔNp63-Gluc-WT-P2 Mut, respectively. All the constructs including mutants were generated by KOD-Plus-Mutagenesis kit (SMK-101, Toyobo, Osaka, Japan) and confirmed by DNA sequencing. Specific primers for gene cloning were listed in Supplementary Table S2.
Lentiviral packaging and generation of stable cell lines
Recombinant lentiviruses particles were generated by transfecting HEK-293T cells with pMD2.G and psPAX2 packaging plasmids and the corresponding backbone plasmid using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Viruses were collected at 60 h post-transfection, filtered, and infected cells at 50% cell confluence in the presence of 10 μg/mL polybrene. Cells were screened with 2 μg/mL puromycin (A1113803, Gibco) or 10 μg/mL blasticidin (A1113903, Gibco) at 48 h post-infection for stable cells.
Quantitative RT-PCR
Total RNA was extracted from cells using TRIzol reagent (A33250, Invitrogen, Shanghai, China) and reverse-transcribed by using Rever Tra Acc qPCR RT Master Mix with gDNA Remover (Cat# FSQ-301, TOYOBO, Shanghai, China). qPCR was carried out for ΔNp63, Grp78, HDAC1, HDAC2, HDAC3, HDAC8, and GAPDH (The primer sequences used in the reactions were listed in Supplementary Table S1). The qPCR reactions were performed in CFX-960 Real-time PCR System (Bio-Rad, Hercules, CA, USA) and using SYBR Green Supermix (1725851, Bio-Rad) according to the manufacturer’s instructions. Relative quantitation values were normalized to GAPDH and calculated using the ΔΔCt method. qPCR primers used in this study were listed in Supplementary Table S3.
Immunoblot analysis and co-immunoprecipitation
For immunoblot analysis, cells were lysed in EBC250 lysis buffer (50 mM Tris pH 7.4, with 250 mM NaCl, 0.5% Nonidet P-40, 50 mM NaF, 0.5 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride (PMSF), 2 μg/mL aprotinin, and 2 μg/mL leupeptin) containing a protease inhibitor cocktail (Roche). Equal amounts of lysates were loaded, separated by SDS-PAGE, and transferred to PVDF membrane (Millipore, Darmstadt, Germany). Membranes were blocked in 5% nonfat milk in TBST, probed with the indicated primary antibodies and HRP-conjugated secondary antibody for subsequent detection by enhanced chemiluminescence. Antibodies for Histone H3 (4499, 1:1000), H3K9me3 (13969, 1:1000), H3K27me3 (9733, 1:1000), EZH2 (5246, 1:1000), HDAC2 (5113, 1:1000), HA-tag (3724, 1:1000), IRE1α (3294, 1:1000), PERK (5683, 1:1000), GRP78 (3183, 1:1000) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies for E-cadherin (ab40772, 1:1000), H3K27ac (ab4729, 1:1000), XBP1 (ab37152, 1:500), ATF6 (ab122897, 1:500) were purchased from Abcam (Cambridge, MA, USA). Antibody for Flag-tag (F1804, 1:1000) was purchased from Sigma. Antibody for XBP1s (619502, Biolegend, 1:500) was purchased from BioLegend. Antibody for p63 (381215, 1:1000) was purchased from ZEN-Bioscience (Chengdu, China). Antibody for Par3 (07-330, 1:1000) was purchased from Millipore. Antibodies for HIF1α (CY5197, 1:1000), HIF2α (CY5098, 1:1000), and GAPDH (AB0036, 1:5000) were purchased from Abways Technology (Shanghai, China).
For Co-immunoprecipitation experiments, cells were lysed in lysis buffer (50 mM Tris HCl, pH 7.4, with 150 mM NaCl, 1 mM EDTA, and 1% Nonidet P-40). Then the lysate was centrifuged at 15,000 × g at 4 °C to remove the precipitation. The supernatants were incubated with mouse-anti-XBP1s (647501, Biolegend, 1:50), mouse-anti-HDAC2 (5113, CST, 1:50), mouse-anti-Flag (F1804, Sigma, 1:50) or normal mouse IgG (sc-2025, Santa Cruz, 1:10) on a rotator overnight at 4 °C, followed by addition of protein A (sc-2001, Santa Cruz) or protein G (sc-2002, Santa Cruz) agarose beads and incubation for a further 2 h at 4 °C. After four washes in PBST, samples were eluted in 2× sample buffer (125 mM Tris HCl, pH 6.8, with 4% SDS, 20% (v/v) glycerol and 0.004% bromphenol blue) to each sample for 10 min at 100 °C, separated by SDS-PAGE and immunoblotted. Membrane was incubated with Rabbit-anti-XBP1s (ab37152, Abcam, 1:500) and other primary antibodies as described in immunoblot analysis.
Immunofluorescent analyses
For Immunofluorescent analyses, cells grown on coverslips were fixed with 4% polyformaldehyde in PBS, permeabilized with 0.1% Triton X-100 in PBS, blocked with 4% bovine serum albumin in PBS, hybridized with rabbit-anti-p63 (381215, ZEN-Bioscience, 1:200), and Rhodamine (TRITC)-conjugated donkey-anti-rabbit IgG (711-025-152, Jackson ImmunoRsearch, PA, USA) and counter-staining with DAPI (Beyotime) for subsequent detection. Coverslips were mounted with ProLong Gold antifade reagent (Invitrogen). Images were acquired using the Leica TCS SP5 II system.
Immunohistochemistry (IHC) staining
Human breast tumor tissue microarrays (HBreDuc060CD01) consisting of breast cancer specimens from different patients were purchased from Shanghai Outdo Biotech (Shanghai, China). IHC analyses were performed as previously described [71]. Tissue microarrays containing 60 breast cancer tissues were used to test the expression of ΔNp63 (619002, Biolegend, 1:100) and XBP1s (619502, Biolegend, 1:100). For quantitative analyses, slides were scanned through NanoZoomer (Hamamatsu, Japan). Scanned images were then subjected to integrated optical density (IOD) measurements using Image-Pro Plus 6.0 to calculate average optical density (AOD) using the formula: AOD = IOD/Area [72].
Wound-healing and Transwell assays
For wound-healing assay, cells were grown to 90% confluence, then scraped with a pipette tip, washed twice with PBS, and incubated in growth media containing 1% serum. At indicated time intervals, cells were photographed using a light microscope (Nikon Eclipse Ti-S/L 100).
Transwell assays for migration were performed in transwell inserts with a 6.5-mm, 8.0-μm-pore polycarbonate membrane, or Matrigel coated inserts for invasion assays (BD Biosciences). Briefly, cells were suspended in serum-free media and seeded into the inner chamber (MCF-10A, 5 × 104 cells per chamber; HCC1806, 1 × 105 cells per chamber). The outer chamber contained complete growth media. 24 h post-incubation, cells were fixed and stained with 0.5% crystal violet in 70% methanol. Cells on the inside of the membrane were removed with a cotton swab; the migrating/invading cells on the outside of the membrane were photographed and counted at least five random fields. Alternatively, the stained cells were also lysed with 2% SDS in PBS and subjected to spectrophotometric analysis at 570 nm [15].
xCELLigence real-time cell analysis (RTCA) for cell migration
To monitor cell migration in real time, xCELLigence RTCA instruments (ACEA Biosciences, CA, USA) were used according to the instructions of the supplier. MCF-10A (5 × 104) or HCC1806 (1 × 105) cells were seeded in CIM-Plate and monitored the cell index signals every 1 h for 48 h.
In vivo metastasis assays
Female 6-week-old nude mice (GemPharmatech Co., Ltd) were used for tumor metastasis in vivo. To validate the role of XBP1s-ΔNp63α axis in tumor metastasis, 18 mice were randomly divided into three groups and injected HCC1806 cells, which stably expressed empty vector (EV), XBP1s with or without ΔNp63α, through the lateral tail veins. To confirm activation of IRE1α-induced tumor metastasis is HIF1α-independent, 20 mice were injected with MDA-MB-231 cells through the lateral tail veins and randomly divided into four groups. Seven days after cell inoculation, these mice were treated with either PX478 (100 mg/kg, i.p.) or/and MKC8866 (300 mg/kg, oral) daily for another 14 days. Mice were monitored daily and euthanized as indicated days. The lungs were dissected, fixed, embedded in paraffin, and sectioned onto microscope slides for hematoxylin and eosin (H&E) staining prior to histological analysis. The numbers of metastatic nodules in the lungs per mouse were counted.
Luciferase reporter assays
Luciferase reporter assays were performed with Secrete-PairTM Dual Luminescence Assay Kit (GeneCopoeia, USA) according to the manufacturer’s instructions. Briefly, cells were co-transfected with 500 ng of ΔNp63-Gluc reporters (ΔNp63-Gluc-WT, ΔNp63-Gluc-P1 Mut or ΔNp63-Gluc-P2 Mut) and 750 ng of XBP1 expression plasmids (XBP1u, XPP1s or XBP1s-R326P) or empty vector (EV). Forty-eight hours post-transfection, cell culture media were collected and ΔNp63-Gluc and SEAP activities were measured. The ΔNp63-Gluc activity was normalized to SEAP activity.
Chromatin immunoprecipitation (ChIP) and ChIP-Seq
ChIP assays were performed in HCC1806 cells, which were cultured under normoxic or hypoxic conditions, with ChIP-IT Kit (53009, Active Motif, USA) using antibodies specific for XBP1s (619502, Biolegend, 1:50), HDAC2 (57156, CST, 1:50), EZH2 (5246, CST, 1:100), H3K27me3 (9733, CST, 1:50), H3K27ac (ab4729, abcam, 1: 50) or normal rabbit IgG (2927, CST, 1:50), as described previously [18]. Immunoprecipitated DNA by antibodies for H3K27me3, H3K27ac, and normal rabbit IgG were used to construct sequencing libraries according to the protocol provided by the TruePrep DNA library Prep kit (Vazyme #TD501) and sequenced on Illumina Nova Seq6000 (Gene Denovo CO., Ltd., Guangzhou, China). The low-quality reads were filtered out by Trimmomatic (version 0.38). All of the ChIP-seq peaks were identified by MACS2 software (version 2.1.1.20160309) with default parameters (bandwidth, 300 bp; model fold, 5, 50; q value, 0.05). ChIP-seq data were deposited in the GEO database under the accession number GSE253833.
In addition, immunoprecipitated DNA was subjected to PCR experiments to amplify fragments of the ΔNp63 promoter elements using indicated primers as listed in Supplementary Table S3. The value of each ChIP sample was normalized to its corresponding input.
Human data from publicly available datasets
The microarray dataset of Curtis Breast was acquired from the Oncomine database (https://www.Oncomine.org/resource/login.html) by selecting the filter of breast cancer. Pearson’s correlation was used as an analysis of the correlation between TP63, XBP1, HDAC2, or EZH2. Kaplan–Meier survival graphs were generated from data available from the breast cancer (n = 1089) of Pan-cancer mRNA RNA-seq datasets in KM Plotter (www.kmplotter.com) [73].
Statistical analyses
Data from cell culture were performed in three independent experiments and presented as means ± SD. Except specific indication, the differences between two groups were tested using the two-tailed unpaired Student’s t test. Prior to differences analyses, the clinical data were performed homogeneity tests by using Levene’s test in SPSS 16 software. Statistical significance was determined as P < 0.05.
Supplementary information
Acknowledgements
We are grateful for members of Xiao lab for stimulating discussion during this study.
Author contributions
HC and SHY performed all the biochemical experiments. SHY, RDM, and LYD analyzed and processed the IHC data from cell lines. YY and MMN analyzed and processed the public database. HC, CX and ZXX analyzed all experiments, managed the overall study, and supervised manuscript preparation and submission. All authors have contributed to the final version of the manuscript and rebuttal.
Funding
This work was supported in part by National Natural Science Foundation of China (NSFC) (81903018, 92259102), National Key R&D Program of China (2022YFA1103700), Natural Science Foundation of Chengdu Medical College (CYZZD21-01), Introduction Foundation of High-level Talents of The First Affiliated Hospital of Chengdu Medical College (CYFY-GQ37) and Disciplinary Construction Innovation Team Foundation of Chengdu Medical College (CMCXK-2101).
Data availability
The data analyzed during this study are included in this published article and the supplemental data files.
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All human tissue research in this study had the approval of ethics committees of the Affiliated Taizhou People’s Hospital of Nanjing Medical University (Taizhou, China) and Shanghai Outdo Biotech (Shanghai, China). All participants provided informed consent. Animal care and experiments in this study were approved by the Institutional Animal Care and Use Committee (IACUC) of Chengdu Medical College, and the procedures were carried out according to the guidelines established by the China Council on Animal Care.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
6/20/2025
In this article the Supplementary fig 2 file has been updated.
Contributor Information
Hu Chen, Email: chenhu126@126.com.
Chuan Xu, Email: xuchuan100@163.com.
Zhi-Xiong Jim Xiao, Email: jimzx@scu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41418-024-01271-z.
References
- 1.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393–410. [DOI] [PubMed] [Google Scholar]
- 2.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8:967–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. 2016;352:175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Heer EC, Jalving M, Harris AL. HIFs, angiogenesis, and metabolism: elusive enemies in breast cancer. J Clin Investig. 2020;130:5074–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8:851–64. [DOI] [PubMed] [Google Scholar]
- 6.Koritzinsky M, Levitin F, van den Beucken T, Rumantir RA, Harding NJ, Chu KC, et al. Two phases of disulfide bond formation have differing requirements for oxygen. J Cell Biol. 2013;203:615–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. [DOI] [PubMed] [Google Scholar]
- 8.Glimcher LH. XBP1: the last two decades. Annal Rheumatic Dis. 2010;69:i67–71. [DOI] [PubMed] [Google Scholar]
- 9.Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013;12:703–19. [DOI] [PubMed] [Google Scholar]
- 10.Su X, Chakravarti D, Flores ER. p63 steps into the limelight: crucial roles in the suppression of tumorigenesis and metastasis. Nat Rev Cancer. 2013;13:136–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yi M, Tan Y, Wang L, Cai J, Li X, Zeng Z, et al. TP63 links chromatin remodeling and enhancer reprogramming to epidermal differentiation and squamous cell carcinoma development. Cell Mol Life Sci. 2020;77:4325–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergholz J, Xiao ZX. Role of p63 in development, tumorigenesis and cancer progression. Cancer Microenviron. 2012;5:311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carroll DK, Carroll JS, Leong CO, Cheng F, Brown M, Mills AA, et al. p63 regulates an adhesion programme and cell survival in epithelial cells. Nat Cell Biol. 2006;8:551–61. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y, Peng Y, Fan S, Li Y, Xiao ZX, Li C. A double dealing tale of p63: an oncogene or a tumor suppressor. Cell Mol Life Sci. 2018;75:965–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergholz J, Zhang Y, Wu J, Meng L, Walsh EM, Rai A, et al. ΔNp63a regulates Erk signaling via MKP3 to inhibit cancer metastasis. Oncogene. 2014;33:212–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu J, Liang S, Bergholz J, He H, Walsh EM, Zhang Y, et al. ΔNp63α activates CD82 metastasis suppressor to inhibit cancer cell invasion. Cell Death Dis. 2014;5:e1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yi Y, Chen D, Ao J, Zhang W, Yi J, Ren X, et al. Transcriptional suppression of AMPKα1 promotes breast cancer metastasis upon oncogene activation. Proc Natl Acad Sci USA. 2020;117:8013–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu L, Liang S, Chen H, Lv T, Wu J, Chen D, et al. ΔNp63α is a common inhibitory target in oncogenic PI3K/Ras/Her2-induced cell motility and tumor metastasis. Proc Natl Acad Sci USA. 2017;114:E3964–E3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Y, Li J, Gao Y, Luo Y, Luo H, Wang L, et al. Hippo kinases regulate cell junctions to inhibit tumor metastasis in response to oxidative stress. Redox Biol. 2019;26:101233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137:87–98. [DOI] [PubMed] [Google Scholar]
- 21.Fisher ML, Balinth S, Mills AA. p63-related signaling at a glance. J Cell Sci. 2020;133:jcs228015. [DOI] [PMC free article] [PubMed]
- 22.Yoh KE, Regunath K, Guzman A, Lee SM, Pfister NT, Akanni O, et al. Repression of p63 and induction of EMT by mutant Ras in mammary epithelial cells. Proc Natl Acad Sci USA. 2016;113:E6107–E6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramsey MR, Wilson C, Ory B, Rothenberg SM, Faquin W, Mills AA, et al. FGFR2 signaling underlies p63 oncogenic function in squamous cell carcinoma. J Clin Investig. 2013;123:3525–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Wang C, Chen X, Takada M, Fan C, Zheng X, et al. EglN2 associates with the NRF1-PGC1α complex and controls mitochondrial function in breast cancer. EMBO J. 2015;34:2953–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chakraborty AA, Laukka T, Myllykoski M, Ringel AE, Booker MA, Tolstorukov MY, et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science. 2019;363:1217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Z, Shilatifard A. Epigenetic modifications of histones in cancer. Genome Biol. 2019;20:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swigut T, Wysocka J. H3K27 demethylases, at long last. Cell. 2007;131:29–32. [DOI] [PubMed] [Google Scholar]
- 28.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–12. [DOI] [PubMed] [Google Scholar]
- 29.Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell. 2017;69:169–81. [DOI] [PubMed] [Google Scholar]
- 30.Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and immunosuppressive effects of endoplasmic reticulum stress in cancer. Cell. 2017;168:692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Denmeade SR, Mhaka AM, Rosen DM, Brennen WN, Dalrymple S, Dach I, et al. Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci Transl Med. 2012;4:140ra186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tie F, Banerjee R, Stratton CA, Prasad-Sinha J, Stepanik V, Zlobin A, et al. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development. 2009;136:3131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang X, Yan J, Zhang M, Wang Y, Chen Y, Fu X, et al. Targeting epigenetic crosstalk as a therapeutic strategy for EZH2-aberrant solid tumors. Cell. 2018;175:186–99.e119. [DOI] [PubMed] [Google Scholar]
- 34.Qian Y, Jung YS, Chen X. ΔNp63, a target of DEC1 and histone deacetylase 2, modulates the efficacy of histone deacetylase inhibitors in growth suppression and keratinocyte differentiation. J Biol Chem. 2011;286:12033–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao Y, Nihira NT, Bu X, Chu C, Zhang J, Kolodziejczyk A, et al. Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat Cell Biol. 2020;22:1064–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fukumoto T, Park PH, Wu S, Fatkhutdinov N, Karakashev S, Nacarelli T, et al. Repurposing Pan-HDAC inhibitors for ARID1A-mutated ovarian cancer. Cell Rep. 2018;22:3393–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tong ZT, Cai MY, Wang XG, Kong LL, Mai SJ, Liu YH, et al. EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and Snail to inhibit E-cadherin. Oncogene. 2012;31:583–94. [DOI] [PubMed] [Google Scholar]
- 38.Welsh S, Williams R, Kirkpatrick L, Paine-Murrieta G, Powis G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1alpha. Mol Cancer Therapeutics. 2004;3:233–44. [PubMed] [Google Scholar]
- 39.Zhao N, Cao J, Xu L, Tang Q, Dobrolecki LE, Lv X, et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J Clin Investig. 2018;128:1283–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Melino G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ. 2011;18:1487–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo X, Keyes WM, Papazoglu C, Zuber J, Li W, Lowe SW, et al. TAp63 induces senescence and suppresses tumorigenesis in vivo. Nat Cell Biol. 2009;11:1451–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Su X, Napoli M, Abbas HA, Venkatanarayan A, Bui NHB, Coarfa C, et al. TAp63 suppresses mammary tumorigenesis through regulation of the Hippo pathway. Oncogene. 2017;36:2377–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dotto GP, Rustgi AK. Squamous cell cancers: a unified perspective on biology and genetics. Cancer Cell. 2016;29:622–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Higashikawa K, Yoneda S, Tobiume K, Taki M, Shigeishi H, Kamata N. Snail-induced down-regulation of ΔNp63a acquires invasive phenotype of human squamous cell carcinoma. Cancer Res. 2007;67:9207–13. [DOI] [PubMed] [Google Scholar]
- 45.Gao Y, Zhang W, Han X, Li F, Wang X, Wang R, et al. YAP inhibits squamous transdifferentiation of Lkb1-deficient lung adenocarcinoma through ZEB2-dependent DNp63 repression. Nat Commun. 2014;5:4629. [DOI] [PubMed] [Google Scholar]
- 46.Xu J, Li F, Gao Y, Guo R, Ding L, Fu M, et al. E47 upregulates ΔNp63α to promote growth of squamous cell carcinoma. Cell Death Dis. 2021;12:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wing-Keung C, Pei-Min D, Hsin-Lun L, Jan-Kan C. Transcriptional activity of the ΔNp63 promoter is regulated by STAT3. J Biol Chem. 2008;283:7328–37. [DOI] [PubMed] [Google Scholar]
- 48.Bhattacharya S, Serror L, Nir E, Dhiraj D, Altshuler A, Khreish M, et al. SOX2 regulates P63 and stem/progenitor cell state in the corneal epithelium. Stem Cells. 2019;37:417–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang J, Jun Cho S, Chen X. RNPC1, an RNA-binding protein and a target of the p53 family, regulates p63 expression through mRNA stability. Proc Natl Acad Sci USA. 2010;107:9614–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mohibi S, Zhang J, Chen X. PABPN1, a target of p63, modulates keratinocyte differentiation through regulation of p63α mRNA translation. J Investig Dermatol. 2020;140:2166–77.e2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rossi M, Aqeilan RI, Neale M, Candi E, Salomoni P, Knight RA, et al. The E3 ubiquitin ligase Itch controls the protein stability of p63. Proc Natl Acad Sci USA. 2006;103:12753–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Y, Zhou Z, Chen C. WW domain-containing E3 ubiquitin protein ligase 1 targets p63 transcription factor for ubiquitin-mediated proteasomal degradation and regulates apoptosis. Cell Death Differ. 2008;15:1941–51. [DOI] [PubMed] [Google Scholar]
- 53.Galli F, Rossi M, D’Alessandra Y, De Simone M, Lopardo T, Haupt Y, et al. MDM2 and Fbw7 cooperate to induce p63 protein degradation following DNA damage and cell differentiation. J Cell Sci. 2010;123:2423–33. [DOI] [PubMed] [Google Scholar]
- 54.Jung YS, Qian Y, Yan W, Chen X. Pirh2 E3 ubiquitin ligase modulates keratinocyte differentiation through p63. J Investig Dermatol. 2013;133:1178–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu HH, Wang B, Armstrong SR, Abuetabh Y, Leng S, Roa WHY, et al. Hsp70 acts as a fine-switch that controls E3 ligase CHIP-mediated TAp63 and ΔNp63 ubiquitination and degradation. Nucleic Acids Res. 2021;49:2740–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prieto-Garcia C, Hartmann O, Reissland M, Braun F, Fischer T, Walz S, et al. Maintaining protein stability of ∆Np63 via USP28 is required by squamous cancer cells. EMBO Mol Med. 2020;12:e11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mahara S, Lee PL, Feng M, Tergaonkar V, Chng WJ, Yu Q. HIFI-α activation underlies a functional switch in the paradoxical role of Ezh2/PRC2 in breast cancer. Proc Natl Acad Sci USA. 2016;113:E3735–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shmakova A, Batie M, Druker J, Rocha S. Chromatin and oxygen sensing in the context of JmjC histone demethylases. Biochem J. 2014;462:385–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Investig. 2011;121:2750–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mujcic H, Nagelkerke A, Rouschop KM, Chung S, Chaudary N, Span PN, et al. Hypoxic activation of the PERK/eIF2α arm of the unfolded protein response promotes metastasis through induction of LAMP3. Clin Cancer Res. 2013;19:6126–37. [DOI] [PubMed] [Google Scholar]
- 61.Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J, et al. An essential role in liver development for transcription factor XBP-1. Genes Dev. 2000;14:152–7. [PMC free article] [PubMed] [Google Scholar]
- 62.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reimold AM, Ponath PD, Li YS, Hardy RR, David CS, Strominger JL, et al. Transcription factor B cell lineage-specific activator protein regulates the gene for human X-box binding protein 1. J Exp Med. 1996;183:393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, et al. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161:1527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Song M, Sandoval TA, Chae CS, Chopra S, Tan C, Rutkowski MR, et al. IRE1α-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature. 2018;562:423–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dong H, Adams NM, Xu Y, Cao J, Allan DSJ, Carlyle JR, et al. The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-Myc. Nat Immunol. 2019;20:865–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004;64:5943–7. [DOI] [PubMed] [Google Scholar]
- 68.Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature. 2014;508:103–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H, Chen X, Gao Y, Wu J, Zeng F, Song F. XBP1 induces snail expression to promote epithelial- to-mesenchymal transition and invasion of breast cancer cells. Cell Signalling. 2015;27:82–89. [DOI] [PubMed] [Google Scholar]
- 70.Wang L, Xia W, Chen H, Xiao Z-X. ΔNp63α modulates phosphorylation of p38 MAP kinase in regulation of cell cycle progression and cell growth. Biochem Biophys Res Commun. 2019;509:784–9. [DOI] [PubMed] [Google Scholar]
- 71.Sun S, Yi Y, Xiao Z-XJ, Chen H. ER stress activates TAp73α to promote colon cancer cell apoptosis via the PERK-ATF4 pathway. J Cancer. 2023;14:1946–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tilley WD, Lim-Tio SS, Horsfall DJ, Aspinall JO, Marshall VR, Skinner JM. Detection of discrete androgen receptor epitopes in prostate cancer by immunostaining: measurement by color video image analysis. Cancer Res. 1994;54:4096–102. [PubMed] [Google Scholar]
- 73.Nagy Á, Lánczky A, Menyhárt O, Győrffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep. 2018;8:9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data analyzed during this study are included in this published article and the supplemental data files.