

Abstract
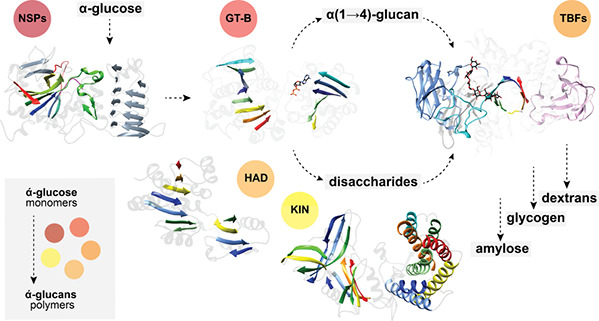
Bacteria have acquired sophisticated mechanisms for assembling and disassembling polysaccharides of different chemistry. α-d-Glucose homopolysaccharides, so-called α-glucans, are the most widespread polymers in nature being key components of microorganisms. Glycogen functions as an intracellular energy storage while some bacteria also produce extracellular assorted α-glucans. The classical bacterial glycogen metabolic pathway comprises the action of ADP-glucose pyrophosphorylase and glycogen synthase, whereas extracellular α-glucans are mostly related to peripheral enzymes dependent on sucrose. An alternative pathway of glycogen biosynthesis, operating via a maltose 1-phosphate polymerizing enzyme, displays an essential wiring with the trehalose metabolism to interconvert disaccharides into polysaccharides. Furthermore, some bacteria show a connection of intracellular glycogen metabolism with the genesis of extracellular capsular α-glucans, revealing a relationship between the storage and structural function of these compounds. Altogether, the current picture shows that bacteria have evolved an intricate α-glucan metabolism that ultimately relies on the evolution of a specific enzymatic machinery. The structural landscape of these enzymes exposes a limited number of core catalytic folds handling many different chemical reactions. In this Review, we present a rationale to explain how the chemical diversity of α-glucans emerged from these systems, highlighting the underlying structural evolution of the enzymes driving α-glucan bacterial metabolism.
1. Introduction to Glucans in Chemistry and Biology
1.1. Historical Perspective
Glucans are glucose homopolymers that cumulatively represent one of the largest deposits of biological carbon in nature. These ubiquitous polymers, whose primary examples are cellulose, starch, and glycogen, play essential roles in the carbon cycle and biosphere transformations. The omnipresent nature of these polysaccharides makes us overlook they are the main constituent of natural raw materials gathered by man since prehistoric times; glucans are the main energy component in food crops and the main constituent of wood for fuel and shelter. Thus, the transformation of glucan materials spans the whole of human existence. Early records on the extraction of cellulose fibers for flax linen, fermentation of starchy grains for beer brewing, and purification of starch used as glue or cosmetic powder can be traced to ancient civilizations. Arguably, uncountable observations of natural and man-made glucan transformations predate modern investigations.
The beginning of glucan chemistry can be stated with the discovery of glucose as a substance purified from raisins by Andreas Marggraf in 1747 and the subsequent discovery of the conversion of starch to glucose by acids by Gottlieb Kirchhof in 1811.1 In 1833, Anselme Payen and Jean-François Persoz isolated a substance that accelerated the transformation of starch into maltose, the diastase, which was the first enzyme produced in concentrated form. In 1860, Pierre Berthelot isolated the enzyme invertase that hydrolyzes sugar cane into glucose and fructose. In 1837, Payen also discovered cellulose composed of glucose residues and isomeric with starch.2 Jacob Berzelius, who coined the concepts of catalyst and polymers, aware of Kirchhof and Payen’s findings, stated, “...chemical processes in living nature, we regard them in a new light. For example, since nature has placed diastase around the eyes of potatoes...we find that the insoluble starch in the tuber is changed to gum and sugar by catalytic power...” and “...One can hardly assume that this catalytic process is the only one...”3 These early findings paved the way to advance the view of life based on enzyme-driven chemical transformations.
Most of the early observations on the biochemistry of glucans were extracted from fermentation studies. Louis Pasteur discovered the production of dextran during the fermentation of wine, which Philippe van Tieghem later assigned to a bacterial activity.4 Adrian Brown found that bacteria can also synthesize cellulose.5 In 1865, Claude Bernard discovered glycogen as an energy reserve substance in liver tissue.6 The occurrence of glycogen in bacterial cells was later confirmed by Arthur Meyer.7 Wilhelm Kühne advance the concept of the separation between a ferment (zyme) and the active component for these conversions, the enzymes. In 1878, Wilhelm Kühne used for the first time the word “enzyme” to describe the ability of yeast to produce alcohol from sugars. In 1897, Eduard Buchner discovered that yeast extract with no living cells can form alcohol from a sugar solution. The conclusion was that biochemical processes do not necessarily require living cells, but are driven by special substances, enzymes, formed in cells, ending the “vitalist” view of living processes.
The modern vision of glucan and sugar biochemistry was established early in the 20th century. Emil Fisher in his Nobel lecture already stated that polysaccharides were nothing other than the glucosides of the sugars.8 This idea was confirmed by Walter Haworth, Edmund Hirst, and co-workers with the first description of the chemical architecture of several glucans, among other polysaccharides.9,10 Later, Haworth received the Nobel Prize in Chemistry for his work on carbohydrates, the structure of complex sugars, and the structure of Vitamin C. Previously in 1894, Fisher proposed the “lock-and-key” model for enzyme function,11−13 and soon after, Leonor Michaelis and Maud Menten presented their seminal work on enzyme kinetics working on invertase.14 Jakub Parnas and Tadeusz Baranowski discovered phosphorolysis in glycogen metabolism,15 while Arthur Harden and Hans von Euler-Chelpin used fermentation of sugar and fermentative enzymes, identifying the role of phosphate in accelerating sugar fermentation, receiving the Nobel Prize in Chemistry 1929.16,17 Harden also reported that acellular fermentation was maintained due to the presence of glycogen, needing a factor “co-zymase”, the NADH.18 The studies of Gustav Embden, Otto Meyerhof, and Parnas on glucose and glycogen fermentation were critical in articulating the first reported pathway: glycolysis.19 Later in 1952, an alternative glycolytic pathway was described by Nathan Entner and Michael Doudoroff in Pseudomonas saccharophila.20 Charles Hanes observed the differential endo- and exohydrolytic action of α- and β-amylases.21 He also proposed the first tridimensional structure of a macromolecule, the helical amylose, and synthesized the first macromolecule in vitro: starch.22
Glycogen study was greatly propelled by Gerty Cori and Carl Cori, who reported the very first purification to homogeneity of a glycogen phosphorylase (GP).23 They discovered that GP can catalyze both the synthesis and degradation of glycogen, being activated by AMP.24 This activation was the first report of an allosteric effector, a concept developed by Jacques Monod, Jeffries Wyman, and Jean-Pierre Changeux.25 In 1956, Edwin Krebs and Edmond Fischer reported the discovery of the GP-kinase, revealing the importance of posttranslational modification in metabolic regulation.26 Meanwhile, Luis Leloir and co-workers revealed the origin of glycogen and starch metabolism with the discovery of the sugar-nucleotides, their synthetic enzymes, including the glycogen synthase.27,28 Earl Sutherland reported the mechanism of hormone-induced glycogen breakdown in the liver, linking the action of the endocrine system with glycogen metabolism.29
Altogether, glucans markedly impact and transverse our understanding of key chemical transformations in living matter. With this perspective, we review from the fundamentals of glucan chemistry to the biology of these polymers, providing a framework to conceptualize how the chemical diversity of α-glucans emerges from the structural evolution of the enzymatic machinery in bacteria.
1.2. The Origin of Glucan Unit: d-Glucose
The emergence of sugars under abiotic conditions from formaldehyde onto alumina and aluminosilicates into monosaccharides represents a model that could explain the primordial origin of these molecules.30 Sugars could also have been interconverted before living organisms arose, possibly giving rise to various types of monosaccharides.31,32 In present living organisms, 3 of the 16 possible aldohexoses are physiologically relevant, d-glucose, d-galactose, and d-mannose, all of which displaying d-isomery (Figure 1). d-Glucose was selected by nature as the central molecule in energy metabolism (Figure 1A). d-Glucose is the most abundant in all life kingdoms, pointing to a central role of this sugar at the origin of life (Figure 1B). There is no final understanding on how hexoses d-isomery prevailed in nature, and how d-glucose became the most common monosaccharide in life. d-Isomery asymmetry may have been selected and perpetuated with the emergence of proteo- or ribo-enzymes,33 as with other synthetic asymmetric catalysts.34 Nevertheless, symmetry-breaking events appear to be a distinct possibility within self-organizing chemical systems. This suggests that homochirality might have been a prevalent trait among the initial biopolymers, possibly evolving alongside their self-replication capabilities at the origin of protometabolism.33 The high intrinsic stability of d-glucose likely played a role in its selection. It is worth noting that the keto-hexose fructose can be converted to stable aldohexoses, i.e., glucose and mannose (2-epi-glucose), by Lobry de Bruyn-Van Ekenstein rearrangement35 (Figure 1D). Interestingly, d-glucose synthesis (gluconeogenesis), glycolysis, and the pentose pathways presumably existed in prebiotic metabolism as they have been assessed to be generated in abiotic conditions.36 In addition, genomic analysis of enzymes of the Embden-Meyerhof-Parnas pathway from archaea and hyperthermophilic bacteria support a gluconeogenic origin of metabolism.37 Arguably, the selection and centrality of d-glucose occurred in the transition between prebiotic and biotic world.
Figure 1.

Glucose, the central sugar in life. (A) Fischer projections of d-glucose (yellow shade), its non-biological occurring stereoisomer l-glucose (red), and the most common monosaccharides, hexoses and pentoses, found in nature. Differences between d-glucose with close related hexoses, d-mannose, d-galactose, and d-fructose, are highlighted in yellow. Note that the orientation of the last stereocenter is responsible for the d- and l-denominations of these sugars. (B) A schematic sequence of reactions of formaldehyde to form glycolaldehyde and subsequent aldoses and ketoses, the so-called formose reactions, that have been proposed as the abiotic origin of sugars. (C) Equilibrium of cyclization of linear d-glucose in solution. Glucose cyclizes, and the hydroxyl group on either position C4 or C5 undergoes an intramolecular reaction with the C1 carbonyl group of the aldehyde. As a result, the product formed is a hemiacetal resulting in either a 5- or 6-membered ring, in which the resulting hydroxyl group could present two orientations α- or β- concerning the ring plane. (D) Reactivity of d-glucose in solution leads to common sugars (i) d-fructose by an enediol rearrangement, and (ii) epimerization to d-mannose via an enolate intermediate. (E) Damaging of d-glucose with proteins, showing the reaction with lysine side chains via Schiff base formation and Amadori rearrangement, which ends in cyclic fructosamine. (F) Structure of common glucose disaccharides, showing maltose and cellobiose reducing disaccharides (grey shade) free anomeric hydroxyl group that can undergo a reducing reaction, while non-reducing disaccharides trehalose and sucrose (blue shade) compromise both anomeric carbons, therefore unavailable for reaction. (G) Overall metabolism of glucose. The glycolic and gluconeogenesis pathways (yellow), showing the classic Embden–Meyerhof–Parnas pathway (EMP) and its variant Entner–Doudoroff pathway (ED; dashed box). This pathway integrates main pathways such as the Pentose phosphate pathway (PP; red shade), the tricarboxylic Krebs cycle, the photosynthetic Calvin cycle (green shade), and the glyconeogenesis/glycogenolysis pathways (blue shade). Other metabolic pathways are shown linked to this central energy metabolism (black dashed). Other molecules can serve as energy deposits (red letters). It is worth noting that glycogen is the energy closest storage molecule, directly linked to the initial point of glycolysis.
1.3. d-Glucose Biochemistry
d-Glucose participates in several acid–base catalyzed reactions, including mutarotation, enolization, and β-elimination, and also has reducing power. d-Glucose generally presents a cyclic pyranose conformation in equilibrium with minor amounts of tautomeric linear and cyclic furanose forms. The cyclization results from an intramolecular hemiacetal formation whose hydroxyl group can take two anomeric positions leading to the α-d-glucose and β-d-glucose forms that exist in a mutarotation equilibrium (Figure 1C). The β-d-glucose predominates because the hydroxyl group of the anomeric carbon is in the more stable equatorial position. Notably, the hemiacetal confers a highly reactive character to the C1 that often participates in d-glucose enzymatic transformations. d-Glucose is the preferred energy source through glycolysis and the Krebs cycle, mediating the biosynthesis of several key compounds in the cell (Figure 1G). d-Glucose is converted into other major hexoses as d-fructose by isomerization, and d-galactose and d-mannose by epimerization. In addition, d-glucose is converted via the pentose pathway to d-ribose, a central constituent of nucleic acids, while fatty acids and most amino acids can be synthesized from d-glucose. Therefore, d-glucose is a molecule that plays a central role in producing all major components of the cell: proteins, lipids, nucleic acids, and polysaccharides. Supporting this notion, most organisms can synthesize d-glucose de novo via gluconeogenesis to maintain homeostasis.38 In addition, several microorganisms can grow using d-glucose as the sole energy and carbon source.39 Despite this importance, it is worth noting that free d-glucose accumulation in the cell can be harmful to the cell machinery due to its reactivity favors the formation of non-enzymatic covalent adducts with proteins and DNA via Schiff base and Amadori rearrangement40,41 (Figure 1E). In addition, high levels of intracellular glucose induce high osmotic pressure that is not compatible with cellular life. From an evolutionary perspective, the acquisition of mechanisms to safely store d-glucose may provide an advantage to a living organism when competing for environmental glucose.
1.4. Glucose Disaccharides
The condensation of monosaccharides to form disaccharides is the minimal event for the polymerization of sugars. The most common disaccharides comprise at least one d-glucose moiety, possibly due to the abundance of this monosaccharide in nature (Table 1). The biosynthesis of disaccharides involves the linkage between the anomeric carbon of a first sugar with any of the hydroxyl groups of a second monosaccharide forming a glycosidic bond. In principle, since the bonding can involve hydroxyl groups in different positions and in two anomeric configurations, the condensation between two monosaccharides can produce an extensive disaccharide repertoire. According to their redox capacity, disaccharides are classified in (i) reducing, where only one anomeric carbon is compromised in the linkage, and (ii) non-reducing, where both anomeric carbons are linked to each other (Figure 1F).
Table 1. Most Common Glucose Disaccharides.
| Name | Monosaccharide 1 (glycosyl) | Monosaccharide 2 | Glycosidic bond | |
|---|---|---|---|---|
| Non-reducing | Trehalose | Glucose | Glucose | α(1→1)α |
| Sucrose | Glucose | Fructose | α(1→2)β | |
| Reducing | Maltose | Glucose | Glucose | α(1→4) |
| Isomaltose | Glucose | Glucose | α(1→6) | |
| Cellobiose | Glucose | Glucose | β(1→4) | |
| Lactose | Galactose | Glucose | β(1→4) |
The metabolism and function of disaccharides containing d-glucose are diverse across organisms. The non-reducing disaccharides trehalose and sucrose play a central role in energy storage in several living organisms due their stability,42 also having properties that aid the cells in enduring environmental stress, such as protecting membranes and proteins from freezing and dehydration.43 Furthermore, due to their small size, disaccharides are used to transport carbohydrates; trehalose is present in high concentration in insects’ hemolymph, while sucrose is transported in plants’ phloem. Trehalose synthesis is widely distributed in nature and found in Bacteria, Archaea, and Eukaryota.44,45 Sucrose synthesis is restricted to plants and some photosynthetic bacteria,46,47 while most organisms can use it as a carbon and energy source. In contrast, disaccharides with a reducing end are not the best suited as storage compounds due to their reactive nature. This is the case for maltose and cellobiose, disaccharides that originate from the breakdown of glucans,48,49 and lactose, a special reducing disaccharide synthesized exclusively by mammals as the main energy component of milk, also fermented by microorganisms.50
1.5. Glucose Polysaccharides
Glucans are broadly classified according to the anomeric configuration of the d-glucose moieties in α-glucans, β-glucans, and mixed α/β-glucans (Figure 2).51 Glucans can also be classified as branched or unbranched polysaccharides according to the presence/absence of ramifications.51 Glucans can adopt a high degree of structural complexity, such as the ramified glycogen comprising α(1→4) backbones with α(1→6) branches while its topoisomer starch is composed of two different α-glucans, amylose, a linear unbranched α(1→4)-glucan, and amylopectin, a branched chain containing α(1→4) and α(1→6)-glucose linkages (Figure 2A). It is worth noting that, under a common denomination, a particular glucan can display structural variability, size, or branching level due to organism-specific biosynthetic machinery or the metabolic state of the organism.52,53 α-Glucans are generally viewed as polymers with an energy storage function in the cell, like glycogen and starch; however, they also play other important roles, including structural roles as exopolysaccharides, or as cell wall components. Similarly, β-glucans are usually associated with extracellular structural functions. This is well represented by cellulose, the most abundant biomolecule in the biosphere, and the main component of the plant cell wall (Figure 2A,B). β-Glucans can play other functions, including (i) cellulose as a fibrous structural component of bacterial biofilms, it forms a mechanically strong hydrogel with high water adsorption capabilities,54 (ii) cyclic β-glucans act as messengers in plant-microbe interactions,55 (iii) internal and external energy storages.56 The fungal cell wall is composed of a polysaccharide-based three-dimensional network that is continuously adapting to growth and environmental conditions and is essential for cell survival (1). The central core consists of a branched β(1→3)-glucan with 3% to 4% interchain linked (via β(1→4)-linkage) to chitin. Laminarin, a branched β(1→3)-glucan from brown algae, and paramylon, a β(1→3)-glucan synthesized by the flagellate Euglena, both are internal energy reserves with parallel function to glycogen. The curdlan-like β(1→3)-glucan exopolysaccharides are used as external energy storage molecules in Cellulomonas flavigena(57) (Figure 2A,B).
Figure 2.

Structure of glucans. (A) Chemical structure of common glucans produced by bacteria. Arrows indicate the main bonds found in amylose, amylopectin, dextran, altenan, and mutan, all representative α-glucans. Amylose and amylopectin are framed by a continuous line, indicating their structures can be found in glycogen, capsular α-glucans, and starch. Structures of cellulose and curdlan (framed by a dotted box) are also presented as examples of β-glucans. (B) Two perpendicular views of the three-dimensional structures obtained by X-ray crystal diffraction of (i) the double helical structure of the α-glucan amylose, (ii) the triple helical arrangement of curdlan, and (iii) the linear chains of cellulose, showing individual glucan chains in black, grey, and white carbon bonds. Structures have been obtained from PolySac3DB (https://polysac3db.cermav.cnrs.fr/home.html). (C–E) The biological organization of glucans. (C) Diagram showing the structural arrangement of macro and microfibrils formed by cellulose chains. (D) Diagrams showing the structural arrangement of starch, from left to right, (i) the starch granule, (ii) the lamellas, and (iii) the amylose and amylopectin complex. (E) Diagrams showing the structural arrangement of glycogen, from left to right, (i) the tiered model of glycogen, comprising the non-branched A-chains and branched B-chains (dashed red box), and (ii) the β-particle and its assembly product, the α particle. Proteins associated with particles are shown as red spots.
The chemistry and resulting architecture of glucans can certainly be correlated to the biological role they play. For instance, in (1→4) linkages, the α(1→4) anomeric configuration results in a helical structure of amylose, a component of glycogen and starch, while the β(1→4) anomeric configuration results in the classical straight chain polymer of cellulose (Figure 2B). This difference makes cellulose perfectly insoluble,58 forming crystalline matrices and fibers with high tensile strength and resistance to enzymatic digestion (Figure 2B,C), desired properties of a structural component. It is worth noting that the extra degrees of freedom provided by the rotation about the C5 and C6 bonds gives (1→6) linked homoglucans higher solution entropy values (Figure 2A).59 Altogether, glucan functions concern not solely the anomeric configuration (α or β) of the polymer but also the architecture and cellular localization. In the following sections, we will focus on α-glucans and related metabolism to discuss the biological machinery that generates their diversity.
2. Architecture of α-Glucans
2.1. Overview of Eukaryotic and Prokaryotic α-Glucans and Their Localization
Glycogen represents a form of soluble α(1→4)-glucan comprising α(1→6) branches of bacterial or heterotrophic eukaryotic origin60 (Figure 2A), whose main characteristic is to form non-crystalline particles with a wide range of size. Overall, glycogen synthesis is mainly based on the use of nucleotide-diphospho-sugar (NDP-sugar) donors, UDP-glucose in heterotrophic eukaryotes, and ADP-glucose in bacteria.61 In eukaryotic cells, glycogen particles present a buried protein, glycogenin (GN), that acts as the primer for glycogen synthesis, remaining covalently bound at the particle core.62 Different types of glycogen particles are observed in eukaryotic cells, classified as α-, β-, and γ-particles, representing different levels of α-glucan polymers organization63 (Figure 2E). The β-granules are individual glycogen particles comprising several protein-rich γ-particles that act as subunits, while α-granules are a structure of clustered β-granules glued together by the GN.64 The occurrence of “glycosomes”, dedicated dynamic organelles for glycogen metabolism, has been suggested,65 comprising a glycogen-protein complex, where the protein component provides the enzymatic machinery of the organelle, and glycogen is the product of its synthetic activity.65 Glycogen is also present as α- and β-granules in several bacterial species, including mycobacteria, streptomyces, and enterobacteria.66,67 However, since bacterial glycogen does not present any protein content to cluster the β-particles, the similar shape of eukaryotic and prokaryotic granules seems to appear as a result of convergent evolution.66 Starch can be considered a topoisomer of glycogen, primarily known for its ability to create insoluble crystalline structures. The starch granule is insoluble in water and densely packed, but still accessible to the plants’ metabolic enzymes. Plants and green algae form starch in plastid compartments such as the chloroplast of the leaf or amyloplasts.68 Plant starch consists of two types of molecules tightly clustered together, the linear α(1→4)-amylose and amylopectin containing α(1→6)-linked branches of α(1→4)-glucan69 (Figure 2D). One of the low energy conformations of the flexible amylose chain leads to single strands that readily form rigid double helices. These double helices associate in pairs, stabilized by hydrogen bonds and van der Waals forces. These pairs associate to give the A or B structures, depending on their chain length and water content.70 On the other hand, Floridian starch is present in the cytoplasm of glaucophytes and red algae, forming crystalline granules with radially oriented fibrils and concentric layers of amylopectin.71,72 The metabolism of these two types of starch relies on different nucleotide sugar donors, ADP-glucose for the plastidial plant starch and UDP-glucose for the cytosolic Floridian starch.73 Interestingly, in cyanobacteria, α-glucan was observed in two forms, glycogen or crystalline semi-amylopectin, also called cyanobacterial starch,74,75 indicating an evolutionary transition from soluble α-glucan states toward crystalline α-glucan storages in photosynthetic organisms. These pieces of evidence in the evolution of organisms show that starch metabolism is important to trace the cyanobacterial origin of plastid endosymbionts into photosynthetic eukaryotes.76−78
α-Glucans are major components of the cell wall, capsules, or slimes on the exterior of fungi and bacterial cells, displaying diverse α-linkages.79 Fungi present several types of α-glucan polysaccharides as extracellular or cell wall components, mostly forming linear backbones, some made only with homogeneous linkages, such as α(1→3), α(1→4), or α(1→6), and also mixed linkages of alternating α(1→3) and α(1→6),80 like in alternan (Figure 2A). Solid-state NMR studies showed that α-glucans associations with other polysaccharides allow the formation of a rigid and impermeable scaffold protecting fungal cells from external stresses.81 Bacteria present two main types of α-glucans outside the cell envelope, capsular α-glucans and dextrans.82,83 The capsular α-glucans are present in Actinobacteria and resemble glycogen but with shorter α(1→4) linear chains,84 while dextrans refer to a large and diverse group of α-glucans with different linkages, including dextrans, alternans, mutans, and reuteran, that form polymer matrix biofilms providing protection to colonizing bacteria85−87 (Figure 2A).
2.2. The Architecture of α-Glucans in Bacteria
2.2.1. The Cytosolic Bacterial Glycogen
Glycogen represents the most important intracellular carbon and energy storage polymer in bacteria. Glycogen follows the three principles for a compound to be considered an energy reserve: (i) the compound accumulates when there is a surplus in energy, (ii) the compound is used when the energy supply from exogenous sources is insufficient for optimal maintenance of the cell, and (iii) the compound provides an advantage compared with an organism lacking this storage mechanism.85,88 Glycogen particles accumulate in bacteria preponderantly during the stationary phase, in the presence of an excess carbon source, and under environmental conditions of slow growth or no growth,89−91 playing a major role in awakening from dormancy.92Escherichia coli mutants lacking functional enzymes associated with glycogen biosynthesis can grow as well as their wild type parent strains, indicating that glycogen is not required for bacterial growth.88 However, glycogen accumulation prolongs the survival rate, resistance to starvation, low temperatures, and desiccation of bacteria compared to mutants without glycogen.93−95 These observations suggest that under conditions of no available carbon source, glycogen is probably utilized to preserve cell integrity, providing the energy required by the bacteria for maintenance.96
The glycogen architecture and chemical structure were originally investigated and discussed based on animal glycogen and plant starch. Early investigations based on stepwise enzymatic degradation97,98 revealed that these α-glucan polysaccharides are composed of irregular tree-like structures as originally proposed by Meyer99 instead of comb-like (each side branch arises from a single main branch) or laminated structures (branch arises from a preceding branch). This tree-like structure is composed of linear chains of α(1→4)-linkages with α(1→6)-linkages at branching points with an apparent random arrangement,66 in contrast to the high-ordered structures required to form crystalline arrays as observed in starch. The α(1→4)-linked chains’ average span comprised 8–12 glucosyl residues.91,100 This short chain length enhances bacterial viability by altering glycogen degradation rate.101 Bacterial and eukaryotic glycogen branching account for 7–10% of the total linkages.78 As a consequence, the branched polymer results in a highly water-soluble 3D fractal-like structure,102 allowing the storage of large amounts of glucose, without causing osmotic stress. Specifically, the high number of terminal non-reducing glucose units are readily accessible to hydrolytic enzymes in case the bacterium needs energy because of starvation. Biophysical characterization and visualization of glycogen extracted from bacteria by electron microscopy revealed a size of ca. 20 nm for β-particles and ca. 40 nm for α-particles, the latter showing a rosette-like appearance (Figure 2).66 α-Particles can break into β-particles. α-Particles can show two structural states, (i) fragile (high-density) and (ii) stable (low-density).66 In animals, it has been observed that the small β-particles degrade more easily to glucose than α-particles.103 Recent studies in bacteria indicate that α-particles states are modulated by the association of enzymes, accounting for fragile and stable states due to changes in average chain length, allowing for the control of glycogen storage and degradation.104
The internal structure and architecture of the glycogen particle were first conceptualized by the “tiered model” based on the molecular arrangement of the α-glucan presenting two chain types: unbranched A-chains and branched B-chains105−107 (Figure 2E). Specifically, branches in B-chains are uniformly distributed, comprising two branches that generate further A- or B-chains. Mathematical calculations indicate that glycogen can arrange in up to 12 concentric tiers.108 This branching spherical growth of the particle leads to a progressively more packed structure toward the periphery allowing only A-chains in the most external tier while preventing the addition of further tiers due to the lack of space for the enzymes to process the polysaccharide, therefore self-limiting the size. Thus, the glycogen particle displays a molecular size between 107 and 108 Da comprising ca. 55,000 glucose units distributed in 12 tiers, and 20 to 50 nm in size.107–109 While the “tiered model” offers a fascinating perspective on understanding the organization of glucan chains within glycogen particles, it is worth noting that a combination of experimental and computational approaches has challenged the fractal-like organization of glycogen particle.102 Surprising findings from small angle X-ray scattering analysis (SAXS) of β-particles have revealed a high density at the center of particles, contradicting the notion of a fractal-like organization and instead suggesting a randomly branched polymer.110 Recent Monte Carlo simulations of the glycogen particle biosynthetic process support these experimental data, showing that enzymatic activities and hindrance may control the chain length, radial density distributions, and particle size.111
2.2.2. The Chemical Structure of α-Glucans as Extracellular Components
Glycogen was first recognized as an intracellular polymeric material thought to function as an inert storage deposit for carbon and energy. In recent years, however, it became evident that some bacteria can deposit polymers with a glycogen-like architecture outside the cells. Furthermore, the synthesis and deposition of extracellular α-glucans exhibiting a variety of structures differing from glycogen have been long known from bacteria producing an extracellular matrix and forming biofilms. These extracellular α-glucans are either synthesized outside the cells employing secreted polymerases or are first synthesized intracellularly and subsequently secreted (Figure 3). Their structures and physicochemical properties differ strongly from intracellular α-glucans as they fulfill other functions outside of the cells (Table 2).
Figure 3.

Diversity of metabolic arrangements for α-glucan and disaccharide pathways in the context of prokaryotes. (A) The classical glycogen metabolism (GlgC-GlgA-GlgB) and independent trehalose metabolism (OtsA-OtsB) pathways in E. coli. (B) The glycogen metabolism via maltose 1-phosphate (M1P; GlgM-GlgE) is connected with the production of extracellular capsular α-glucan in M. tuberculosis. Essential wiring of the trehalose metabolism (TreS-Pep2/Mak; TreZ-TreY) permits the redundancy for the production of trehalose or α-glucan. (C) The glycogen metabolism in P. aeruginosa highlights the use of an UGPase (lack of GlgC/AGPase) for the GlgA-dependent biosynthesis of α(1→4)-glucan. It is worth noting the presence of GlgE, concurrent in the synthesis of α(1→4)-glucan via maltose 1-phosphate and TreS-Pep2/Mak. P. aeruginosa lacks a direct/dedicated pathway for the biosynthesis of trehalose, relying on maltose interconversions via the MalQ and TreZ-TreY pathways. (D) The glycogen metabolism of S. venezuelae relies on the trehalose pathway via an OtsA GDP-glucose-dependent synthesis. (E) An overall view of the cyanobacteria classical glycogen metabolism (GlgC-GlgA-GlgB). The sucrose synthesis is via SPS/SPP or SuS, and the alternative production of α(1→4)-glucan via amylosucrase (AMS). (F) An overall view of the archaea classical glycogen metabolism (GlgC-GlgA-GlgB). Note the alternative production of trehalose via TreT. (G) Charts showing the different folds of enzymes geometric symbols used for NDP-sugar pyrophosphorylases (blue hexagons), GT-B GTs (yellow circles), TIM barrel folds (red boxes), HAD domain phosphorylases (green parallelograms), and the maltokinase (violet trapezium). Nucleotide sugars are indicated with small colored rhombuses according to the legend.
Table 2. Representative α-d-Glucans Chemical Diversity and Their Distribution in Nature.
| Eukaryotes | Name | Geometry | Backbone | Branching | Location/Function |
|---|---|---|---|---|---|
| Animals, fungi and protozoa | Glycogen | Branched | (1→4)-α-d-glucan | (1→6)-α-d-glucan | Intracellular. Cytosol. Storage |
| Higher plants and green algae | Starch amylopectin | Branched | (1→4)-α-d-glucan | (1→6)-α-d-glucan | Intracellular. Plastids. Storage |
| Starch amylose | Linear | (1→4)-α-d-glucan | Intracellular. Plastid. Storage | ||
| Red algae, glaucophytes | Floridean starch | Branched | (1→4)-α-d-glucan | (1→6)-α-d-glucan | Intracellular. Cytosol. Storage |
| Fungi | Nigeran | Linear | Alternating (1→3)α(1→4)-α-d-glucan | Wall component | |
| Fungi | Pseudonigeran | Linear | (1→3)-α-d-glucan | Wall component | |
| Fungi | Pullulan | Linear | (1→4)α(1→4)(1→6)-α-d-glucan | Extracellular | |
| Prokaryotes | |||||
| Bacteria | Glycogen | Branched | (1→4)-α-d-glucan | (1→6)-α-d-glucan | Intracellular. Cytosol. Storage |
| Archaea | Glycogen | Branched | (1→4)-α-d-glucan | (1→6)-α-d-glucan | Intracellular. Cytosol. Storage |
| Bacteria | Capsular α-glucan | Branched | (1→4)-α-d-glucan | (1→6)-α-d-glucan | Extracellular Capsular component |
| Bacteria | Dextran | Branched | (1→6)-α-d-glucan | α(1→2,3,4) | Exopolysaccharide |
| Bacteria | Alternan | Branched | Alternating α(1→3)α(1→6)- d-glucan | α(1→3) | Exopolysaccharide |
| Bacteria | Mutan | Branched | α(1→3)- d-glucan | α(1→6) | Exopolysaccharide |
| Bacteria | Reuteran | Branched | (1→4)-α-d-glucan, including α(1→6) | α(1→6) | Exopolysaccharide |
| Bacteria | Amylose | linear | (1→4)-α-d-glucan | Exopolysaccharide |
2.2.3. The Capsule of Mycobacterium tuberculosis
The ability to form a capsule surrounding the cells is a feature frequently found among pathogenic bacteria and, in most cases, an important virulence factor. In contrast to most other capsule-forming bacteria, the presence of an outermost capsular layer surrounding cells of the human pathogen Mycobacterium tuberculosis has been a matter of debate for many years. In contrast to typical bacterial capsules, the mycobacterial capsular layer is thin, not visible using light microscopy, and has a loosely-attached structure sensitive to agitation and the presence of detergents typically added to liquid culture media to minimize clumping of mycobacterial cells. More recently, ultrastructural studies employing advanced cryo-electron microscopy techniques could unambiguously prove the capsule’s existence in M. tuberculosis and visualize it in a close-to-native state.112 The mycobacterial capsule is mainly composed of neutral polysaccharides and additionally also contains proteins and lower amounts of lipids.113,114 The capsular polysaccharides identified in M. tuberculosis comprise three types of polymers: (i) a branched, high-molecular-weight α-d-glucan mainly comprising an α(1→4)-linked core with α(1→6)-branches every 5 or 6 residues by mono- and di- glucosides, with a molecular mass estimated to be ca. 100 kDa by gel permeation chromatography;113−116 (ii) a d-arabino-d-mannan heteropolysaccharide exhibiting an apparent molecular weight of 13 kDa;117 and (iii) a d-mannan homopolysaccharide with an apparent molecular weight of 4 kDa exhibiting α(1→6)-glycosidic linkages with α(1→2)-branches.117 Of the three mentioned polysaccharides, α-glucan is the major capsular polysaccharide constituent of M. tuberculosis, representing up to 80% of the extracellular polysaccharides.113,115,116 Using an α-glucan-specific monoclonal antibody, the production of extracellular α-glucan material has also been demonstrated to occur during infection for M. tuberculosis cells grown in mice.118 Structural analyses of the intracellular (i.e., glycogen) and extracellular α-glucans produced by slow-growing mycobacteria revealed a similar composition and architecture indicative of a common biosynthetic origin.116,117 However, depending on the analytical methods used, also differences were reported with capsular α-glucan possessing a higher molecular mass and a more compact spatial organization than the glycogen isolated from the same species, which has led to the speculation that specific enzymes might be responsible for the synthesis of each polymer.115 More recently, a combination of enzymatic characterizations and biochemical analyses of mutant strains resulted in the discovery of the metabolic network and configuration of pathways required for intra- and extracellular α-glucans in M. tuberculosis(119) (Figure 3B). This study unambiguously showed that both forms of α-glucan polymers are synthesized by the same enzymatic machinery. Synthesis occurs intracellularly, and a portion of the produced material is exported to yield the extracellular α-glucan that build up the capsule. As will be elaborated in more detail in section 3.2, α-glucan in M. tuberculosis is produced by iterative cooperation of the maltose 1-phosphate-dependent maltosyltransferase GlgE and the branching enzyme GlgB (Figure 3B).84 The polymer produced in this iterative process comprises C chains of DP ∼9, A and B chains of DP ∼7–8, and a mean number of branches per B chain of 1.2–1.6, which is considerably lower than the value of 1.8–1.9 reported for classical glycogens. Thus, the resulting molecule is a high-molecular weight glycogen-like polysaccharide of ∼5 × 106 Da but has a much less arboreal structure compared to glycogen described from other bacteria and from eukaryotic organisms and exhibits an A:BC chain ratio that is the smallest reported for α-glucans.84,120 Both the intracellular and extracellular polymers isolated from M. tuberculosis cells comprised β-particles that have diameters ranging from ∼30 to ∼60 nm and occasionally aggregate into larger α particles. The synthetic material produced in vitro using purified M. tuberculosis GlgE and GlgB proteins and maltose 1-phosphate as a substrate formed β-particles with similar diameter and morphology as the biological polymers isolated from M. tuberculosis cells.84 With an intracellular biosynthetic origin of all α-glucans in M. tuberculosis, the existence of a transport mechanism(s) for translocation of the polymer to the capsular space has to be postulated. However, such an α-glucan transporter has not been identified yet. It is tempting to speculate that the peculiar structure of M. tuberculosis α-glucan with a reduced degree of branching and a less arboreal architecture might be a feature facilitating export. However, no reports are available yet addressing the impact of α-glucan structure on extracellular deposition. M. tuberculosis has been shown to release extracellular vesicles that originate from the cytoplasmic membrane and contain cytosolic cargo. While it is theoretically conceivable that α-glucan is packed into the lumen of such vesicles for secretion, ELISA employing an α-glucan-specific monoclonal antibody could not detect this polysaccharide in purified vesicle preparations.121 Finally, theoretically it is possible that capsular α-glucan is produced extracellularly by secreted GlgE and GlgB proteins in a nucleoside-sugar-free biochemical reaction. However, this would necessitate secretion of substantial amounts of maltose 1-phosphate as substrate for the maltosyltransferase GlgE, but detection of this phosphosugar in cell-free culture supernatants has never been reported.
2.2.4. α-Glucans in Biofilm Forming Bacteria
Many bacteria are capable of forming biofilms, which are microbial communities characterized by their adhesion to solid surfaces of biotic and abiotic origin. An essential element in establishment and maintenance of a biofilm is the production of an extracellular matrix of exopolymeric substances (EPS) consisting of polysaccharides, proteins, DNA, and lipids. The EPS surrounds the microorganisms lending structural integrity, mechanical stability, and cohesiveness to the biofilm. The composition of the exopolymeric polysaccharides is diverse and can be complex comprising a mixture of several different types of molecules. Various types of α-glucans have been reported as a minor or major component of exopolymeric polysaccharides for some biofilm-producing bacteria (Table 2).
A group of bacteria notoriously forming α-glucan-containing biofilms are lactic acid bacteria. Synthesis of α-d-glucans by lactic acid bacteria occurs extracellularly using sucrose as a substrate and only requires secretion of single GH70 glucansucrase enzymes, which employ an α-retaining double displacement mechanism. The specificities of these glucansucrases differ, leading to production of various types of α-glucans with diverse linkages of the glucose units as well as the branching pattern. Thus, these extracellular α-glucans also exhibit very different physicochemical properties. Based on the linkage composition, these α-d-glucan polysaccharides are classified into dextran with mainly α(1→6) linkages, mutan with predominate α(1→3) linkages, alternan with alternating α(1→6) and α(1→3) linkages, and reuteran with mainly α(1→4) linkages.122−125 In addition, some lactic acid bacteria produce and secrete GH70 branching sucrases that can add single α(1→2) or α(1→4)-branched residue using dextran as an acceptor, resulting in highly branched polysaccharides with comb-like structure.126
Dextran is a homopolysaccharide which is composed of d-glucose monomers with mainly consecutive α(1→6) linkages in the backbone and branches connected via α(1→3) and occasionally α(1→2) and α(1→6) linkages (Figure 2A).127 The sucrose-dependent GH70 glucansucrase enzymes that synthesize dextran are termed dextransucrases.128Leuconostoc mesenteroides produces dextran consisting of 95% α(1→6) linkages and 5% α(1→3) branching linkages.123 Concerning the length of the branches, differing values have been reported depending on the studied strain of L. mesenteroides. 40% of the branching side chains of dextran produced by L. mesenteroides strain NRRL B-512 contain only one glucosyl unit, while 45% of the branching side chains possess two glucosyl units, and the remaining are longer than two glucosyl units.129 Similarly, branches from dextran obtained from strain L. mesenteroides NRRL B-1397 were reported to possess α(1→2) branches comprising just one glucosyl unit and longer α(1→3) branches comprising on average five glucosyl units,130 whereas dextran from L. mesenteroides strain NRRL B-512F was demonstrated to contain side chains up to 33 glucose residues.131 The molecular weight of dextran produced by dextransucrase enzymes from different bacteria generally varies in the range of 9–500 × 106 Da depending on the producing strains and enzymes. Dextran produced by L. mesenteroides exhibits a molecular weight of >2.0 × 106 Da, while that produced by Weissella cibaria was described to have a higher-molecular weight of 4 × 108 Da.128,132Oenococcus kitaharae DSM17330 synthesizes a dextran of over 109 Da, which is the largest dextran reported to date.133 Linear dextrans exhibiting exclusively α(1→6) linkages are very flexible polymers that are generally highly soluble in water. The water solubility of different dextrans is modulated by their branching linkage pattern and degree of branching.134 The high-molecular weight dextran from O. kitaharae DSM17330 was reported to exhibit a gel-like behavior.133 Cryo-TEM and dynamic light scattering analysis revealed that the dextran synthesized in vitro by the dextransucrase of L. mesenteroides strain D9909 displayed well-defined spheroidal particles in solution, with diameters ranging from 100 to 450 nm.135
Mutans are water-insoluble α-glucans, mainly consisting of consecutive α(1→3) linkages in the glucan chain backbone but may also contain a minority of consecutive α(1→6) linkages as well as α(1→3) and α(1→6) branches (Figure 2A).136 Mutans are generally produced from Streptococcus strains by specific sucrose-dependent GH70 glucansucrase enzymes termed mutansucrases and are associated with the development of dental caries.137 Structural analysis of a water-insoluble mutan produced by Streptococcus mutans strain 6715 revealed the presence of 67% of continuous α(1→3) linkages in the backbone and 33% α(1→6) linkages extending linearly from the branches.138Streptococcus salivarius strain HHT produced a water-insoluble mutan containing a high proportion of 80% of α(1→3) linkages and short side chains of α(1→4) and α(1→6) linkages.139 The molecular weight of mutans produced by S. mutans strains has been reported to be in the range of ca. 2.4 × 103 Da.140 Scanning electron microscopic examination of the mutan produced by S. mutans strain 20381 revealed a fibrillary structure consisting of granular formations.141
Reuterans are glucopolysaccharides found in Lactobacillus reuteri consisting of alternating consecutive α(1→4) linkages and single α(1→6) linkages in the glucan chain backbone as well as α(1→6) branches. The specific sucrose-dependent GH70 glucansucrase enzymes capable of forming alternating α(1→4) and α(1→6) linkages for synthesis of reuteran are termed reuteransucrases.142 Structural analysis of the reuteran produced by the reuteransucrase GtfA of L. reuteri strain 121 revealed a composition of 58% α(1→4) and 42% α(1→6) linkages, with a molecular weight of 34.6 × 106 Da.143,144 It is built up from maltose, maltotriose, and maltotetraose building blocks connected by single α(1→6) linkages, with some of the α(1→4) linked building blocks carrying α(1→6) branches.143 The reuteran polysaccharide produced by the reuteransucrase GtfO of L. reuteri strain ATCC 55730 is composed of 80% α(1→4) linkages and 20% α(1→6) linkages, suggesting the presence of longer stretches with consecutive α(1→4) linkages instead of alternating α(1→4) and α(1→6) linkages.128 Similarly, the exopolysaccharide produced by L. reuteri strain SK24.003 possesses predominantly α(1→4) linkages (80%) and a lower degree of α(1→6) linkages with a molecular weight of 43.1 × 106 Da and a radius of gyration of 43.6 nm.145 Structural modelling of reuteran from L. reuteri strain SK24.003 revealed that its alternating α(1→4)/α(1→6) backbone and branches are packed into a helical groove, generating a helical conformation in solution.146
Alternan is a high-molecular weight α-d-glucan homopolymer containing alternating α(1→6) and α(1→3) linkages (Figure 2A). The production of alternan has mainly been reported for strains of L. mesenteroides and Leuconostoc citreum. The specific sucrose-dependent GH70 glucansucrase enzymes capable of forming alternating α(1→6) and α(1→3) linkages for synthesis of alternan are termed alternansucrases.147 Alternan is a branched α-d-glucan with 7–11% 3,6-sustituted-d-glucosyl residues.148L. citreum strain ABK-1 encodes an alternansucrase that catalyzes the synthesis of an α-d-glucan with 60% α(1→6) linkages and 40% α(1→3) linkages.149 The molecular sizes of alternans produced by L. citreum strains SK24.002 and L3C1E7 were determined as 46.2 × 106 Da and 5.88 × 106 Da, respectively.150,151
In addition to typical glucansucrases, some lactic acid bacteria, particularly strains of L. citreum, have been reported to produce and secrete a distinct group of GH70 family enzymes, designated branching sucrases.126 Unlike typical glucansucrases, branching sucrases use sucrose as a substrate but do not catalyze α-d-glucan polysaccharide formation. They rather hydrolyze sucrose and transfer the glucosyl moiety to introduce branches into α-glucans produced by regular GH70 glucansucrases of the same bacterial strain.126,152 When dextran is provided as an acceptor substrate, branching sucrases catalyze the synthesis of α(1→2) or α(1→3) linked single glucosyl unit branches onto dextran, generating highly branched dextran with a comb-like structure, where up to 50% of the glucosyl monomers of the linear glucan backbone carry branches.126 The presence of α(1→2) or α(1→3) branches in branched dextran renders the polymer resistant to the hydrolysis by digestive enzymes of the gastrointestinal tract of mammals.153–155
In addition to capsular α-glucans present in M. tuberculosis as described above in section 2.2.3, extracellular α-d-glucan homopolymers with a glycogen-like structure comprising an α(1→4) linked core with α(1→6) branches have also been reported for some biofilm-forming bacteria. Species of the genus Neisseria express a GH13 amylosucrase, which is a sucrose-dependent α-amylase family enzyme that catalyzes the synthesis of a high-molecular weight linear α(1→4) linked core.156 First, this enzyme has been identified in the cytosol in some Neisseria species, leading to intracellular glycogen production.157 Later, this enzyme was also found to be secreted by Neisseria polysaccharea isolated from the throats of healthy children leading to extracellular polymer formation.158In vitro, in the presence of an activator α-glucan starter molecule (e.g., glycogen), purified amylosucrase protein catalyzes the synthesis of a linear amylose-like polysaccharide composed of only α(1→4) glucosidic linkages using sucrose as the substrate, exhibiting a DP of ∼55.159,160 In contrast, the α-glucan isolated from cells of N. polysaccharea, N. perflava, and others isolated from human dental plaque have been reported to comprise between 6 and 9% α(1→6)-linked branches,161−164 indicative of the presence of a branching enzyme acting on the linear α(1→4)-linked polysaccharide produced by GH13 amylosucrases.165 While initially thought to be restricted to species of the genus Neisseria, more recently GH13 amylosucrases have also been described for bacteria outside this genus such as various Deinococcus species, Arthrobacter chlorophenolicus, Alteromonas macleodii, Methylobacillus flagellatus, the cyanobacterium Synechococcus sp., and the halotolerant methanotrophic bacterium Methylomicrobium alcaliphilum, indicating that this mechanism of producing glycogen-like α-glucan is more widespread.126
For members of the genus Aeromonas, which are Gram-negative, water-borne bacteria that are ubiquitously found in aquatic environments, a surface α-glucan consisting of α(1→4)-linked glucosyl units with α(1→6)-branches has been described for A. piscicola AH-3 and Aeromonas hydrophila strains AH-1 and PPD134/91.166−168 The α-glucan is produced intracellularly by the UDP-glucose pyrophosphorylase (UGPase) GlgC and the glycogen synthase GlgA. In contrast to the classical GlgC-GlgA pathway for glycogen biosynthesis, Aeromonas GlgC synthesizes UDP-glucose instead of ADP-glucose, while Aeromonas GlgA can utilize both UDP-glucose and ADP-glucose as substrates.166−168 For A. hydrophila AH-3, it was demonstrated that the intracellularly produced glycogen-like α-glucan is exported to the cell surface involving WecP,168 which is the enzyme catalyzing the transfer of N-acetylgalactosamine to undecaprenyl phosphate to initiate O-antigen lipopolysaccharide (LPS) biosynthesis.169 The exported glycogen-like α-glucan is attached to the surface involving the O34-antigen polysaccharide ligase WaaL,168 which is the enzyme that ligates the O-antigen LPS to the LPS-core.170
Presence of extracellular α-glucan consisting of α(1→4)-linked glucosyl units with α(1→6) branches has further been described for the Gram-negative bacterium Pseudomonas fluorescens.171 In addition, an α(1→4)-linked α-glucan has been identified as part of the EPS produced by the biofilm-forming plant pathogen Pseudomonas syringae pv. actinidiae strain NZ V-13.172 While α-glucan biosynthesis has not been investigated in P. fluorescens and P. syringae pv. actinidiae yet, the configuration of pathways leading to α-glucan formation in the related bacterium Pseudomonas aeruginosa PAO1 has recently been elucidated, revealing a central role of the GlgE pathway in this organism.173 Thus, it is likely that α-glucan formation occurs on a similar pathway in all Pseudomonads (Figure 3C). For P. aeruginosa PAO1, however, it is unclear whether intracellularly produced α-glucan is secreted to become surface-exposed similar to P. fluorescens and P. syringae pv. actinidiae.
3. α-Glucan Metabolism
3.1. The Classical GlgC-GlgA Pathway in Bacteria
The classical bacterial glycogen metabolic pathway involves genes encoding the action of five enzymes. Three enzymes are involved in the anabolic route, (i) AGPase (glgC) activating the glucose moiety, (ii) glycogen synthase (glgA; GS) generating α(1→4)-linked linear glucose chains, and (iii) glycogen branching enzyme (glgB; GBE) introducing α(1→6)-linked glucan branches; while two enzymes are involved in the catabolic route, (i) glycogen debranching enzyme (glgX; GDE) cleaving α(1→6)-linked glucan branches, and (ii) glycogen phosphorylase (glgP; GP)61 (Figure 3A). Genes involved in the classical pathway of glycogen metabolism are often clustered in a single operon.174−177 In the case of the paradigmatic E. coli, these genes are located in a cluster of 15 kb organized in two neighboring operons.89,91,178 The most frequent order for the glgA/B/C triplet is BCA but the order CBA and BAC are also observed. The glgP and glgX genes are not always present near the glgA/B/C triplet, but if they are, their order is highly variable.179 The duplication of genes involved in glycogen metabolism was observed in several bacterial species, likely providing (i) functional redundancy, (ii) different kinetics, substrate specificities and/or expression profiles, and (iii) functional promiscuity/specialization of genes as a source of diversity and evolution of the metabolic pathways.180,181 Interestingly, certain bacterial species display additional functions clustered in the glg operon, as the amy (α-amylase) and pgm/phx (phosphoglucomutase) genes.182
3.1.1. Biosynthesis
Glucose 1-phosphate plays a central role in the metabolism of glycogen (Figure 3A).183 Glucose 1-phosphate is produced by the phosphoglucomutase (Pgm) from glucose 6-phosphate. In turn, glucose 6-phosphate is produced either by phosphorylation of extracellular glucose by the hexokinase, or from the final steps of gluconeogenesis by transforming fructose 6-phosphate by the phosphoglucoisomerase.38 The activation of glucose 1-phosphate is the first committed and rate-limiting step of the classic glycogen biosynthetic pathway, involving the formation of ADP-glucose mediated by AGPase. AGPase catalyzes a condensation reaction between ATP and glucose 1-phosphate releasing pyrophosphate (PPi) diphosphate and ADP-glucose, requiring Mg2+ for activity.61,184 AGPase displays a bi–bi mechanism with ATP binding first, followed by glucose 1-phosphate and by the ordered release of PPi and ADP-glucose.185 Importantly, AGPase displays positive and negative allosteric regulation.186−188 The second step is carried out by GS, which generates linear α(1→4)-linked glucose chains.61,189 It is well established that the initiation step of glycogen biosynthesis in yeast and mammals requires the action of the enzyme glycogenin (GN), which is considered the first acceptor of glucose units.190,191 GN catalyzes an autoglycosylation reaction, the transfer of a glucose residue from UDP-glucose to a tyrosine residue (Tyr195 in human GN1).192 Fully sequenced genomes of bacteria known to accumulate glycogen have failed to reveal the presence of GN homologs.193 It was found that GS from Agrobacterium tumefaciens can not only elongate α(1→4)-linked glucans but also generate the primer required for the elongation process by catalyzing its own glycosylation.194 The oligosaccharides formed by GS were composed of two to nine glucose residues and, in addition, this α-glucan was released from the enzyme.194 Thus, it was proposed that bacterial GSs use this de novo synthesis mechanism in the absence of available soluble α(1→4)-glucans to provide itself with an initial substrate. It is speculated that GS preferentially catalyzes an elongation reaction of (i) malto-oligosaccharide primers generated during the initiation step or (ii) glycogen, inducing an apparent inhibition of the initiation reaction.194 The third step catalyzed by the GBE enzyme produces α(1→6)-linked glucan branches in the polymer. Biochemical and structural data indicate that GBEs only act on long polymers to transfer chains no shorter than six units and preferring chains eight or more sugars in length.195
3.1.2. Degradation
The recovery of glucose 1-phosphate from the glycogen degradation pathway is carried out by GP.196 GP catalyzes the reversible phosphorolysis of α(1→4)-glucans to obtain glucose 1-phosphate,197 requiring pyridoxal phosphate (PLP) as a prosthetic group.198 GlgP acts directly on the glycogen polymer, while MalP most likely catabolize soluble malto-oligosaccharides. The majority of characterized polyglucan phosphorylases are unable to act on chains smaller than five glucose residues in length. GlgP is unable to bypass or hydrolyze the α(1→6) linkages and therefore stops two, three, or four residues from the first α(1→6) branch encountered to generate the so-called β-limit dextrin.199 Glucose 1-phosphate is subsequently converted to glucose 6-phosphate to enter into the glycolytic pathway providing energy to the cell. Due to the high degree of branching present in the glycogen molecule, a second type of enzyme is required to cleave the α(1→6)-glucosidic bonds that remain uncleaved by GP. Bacterial GDEs display only α(1→6)-glucosidase activity. GDE cleaves the α(1→6)-glucosidic linkage between these glucose residues and the linear α(1→4)-glucan chains of glycogen and relies on MalP and GP to process the remaining α(1→4)-bonds in this short-released chain.
3.1.3. Regulation
The main regulatory step in the bacterial glycogen biosynthetic pathway is carried out by AGPase.61 This markedly contrast with the metabolic regulation of glycogen biosynthesis and degradation mechanisms in eukaryotic cells.109 AGPase has the ability to sense the energy status of the cell controlling its enzymatic activity by the action of allosteric regulators.185,187 AGPase activators are metabolites that reflect signals of high carbon and energy content of a particular bacteria or tissue, whereas inhibitors of the enzyme indicate low metabolic energy levels.78,200 Based on the regulatory profiles to different allosteric effector AGPases were classified into nine different classes.200
Bacterial AGPases are encoded by a single gene, producing a native homotetrameric protein (α4) with a molecular mass of ca. 200 kDa.61,186,188 The paradigmatic bacterial AGPase from E. coli (EcAGPase), is positively regulated by glycolytic intermediates, including fructose 1,6-bisphosphate (FBP) as the main activator with pyruvate acting synergistically, and negatively regulated by AMP generated from the general metabolism.187,200,201 Covalent binding of pyridoxal resulted in permanent activation of EcAGPase, while the presence of FBP protected the enzyme from binding to the compound, allowing the identification of Lys39 as a key residue in the activation mechanism.202,203 The structural and mechanistic aspects of this exquisite allosteric regulation arise from the AGPase tetrameric architecture that will be discussed in Section 5.1.1.1. Interestingly, histidine phosphotransporter protein (HPr), a protein associated with the PTS system and subject to phosphorylation/phosphorolysis modification, interacts with E. coli GP (EcGP) regulating its oligomeric status and enzymatic activity.204,205 Therefore, this second point of regulation in the classical pathway prevents glycogen synthesis from acting like a futile cycle, tightly controlling the state of the pathway.
Finally, the glycogen metabolism regulation in E. coli also involves a complex assemblage of factors that are adjusted to the physiological and energetic status of the cell.91 At the level of gene expression, several factors have been described to control bacterial glycogen accumulation, including (i) the PhoP–PhoQ regulatory system,206 (ii) the carbohydrate phosphotransferase system (PTS),207 (iii) the carbon storage regulator CsrA,208,209 and (iv) and the cAMP-CRP responsive inner-membrane nucleoside transporters.210
3.2. The GlgE Pathway in Bacteria
The GlgC-GlgA pathway has been thought to be the only route for synthesizing intracellular glycogen-like α-glucan consisting of α(1→4)-linked glucosyl units with α(1→6)-branches in bacteria. However, in 2010, the presence of an alternative route that is not relying on a nucleoside diphosphate-activated donor substrate was discovered for glycogen-like α-glucan biosynthesis in mycobacteria, the GlgE pathway.211,212 Subsequently, it was found that the GlgE pathway is almost as frequently distributed among bacteria as the GlgC-GlgA pathway, being present in 14% of sequenced bacterial and archaeal genomes, whereas the GlgC-GlgA pathway is found in 20% of bacterial genomes.213 The GlgE pathway is very intimately connected and interrelated both in terms of biosynthesis and degradation with trehalose metabolism, as trehalose is a precursor for the formation of the activated donor substrate for GlgE, maltose 1-phosphate, and the glycogen-like α-glucan produced by the GlgE pathway can readily be mobilized to yield trehalose (Figure 3B).
3.2.1. Biosynthesis
The biosynthesis of α-glucans via the GlgE pathway has first and best been studied in actinomycetes and particularly in mycobacteria (Figure 3B). In the GlgE pathway, α(1→6)-branched α(1→4)-glucans are produced by iterative cooperation of two essential enzymes, the maltosyltransferase GlgE (systematic name (1→4)-α-d-glucan:phosphate α-d-maltosyltransferase) and the branching enzyme GlgB.84,212 Both enzymes are GH13 family members.226 The maltosyltransferase GlgE uses maltose 1-phosphate as the activated donor substrate to produce linear α(1→4)-linked maltooligosaccharides.212 As soon as GlgE has formed a linear chain of ∼16 glucosyl residues, GlgB introduces an α(1→6)-branch of ∼7–8 glucosyl residues in length employing a strictly intrachain transfer mechanism. GlgE then preferentially extends the newly formed branch until it is long enough to undergo branching again by GlgB. Only occasionally, GlgE also extends previous branches so that they might become long enough to allow a second branching by GlgB. Therefore, each branched chain mostly carries just one further branch. These specificities of GlgE and GlgB promote an iterative process that results in a glucan polymer that exhibits a significantly lower degree of branching, resulting in a less pronounced arboreal structure compared to glycogen from other bacteria and from mammals.84 The branching enzyme GlgB from bacteria employing the GlgE pathway does not substantially differ from that of bacteria using the classical GlgC-GlgA pathway for glycogen formation,214,215 indicating that the evolution of the maltosyltransferase GlgE was key for the establishment of the GlgE pathway. Consistent with the strictly iterative process of synthesis of a glycogen-like polysaccharide via the GlgE pathway, the genes encoding GlgE and GlgB are co-transcribed as part of an operon in mycobacteria and all other bacteria possessing the GlgE pathway.213 However, while protein structures of GlgE216−221 and GlgB215 have individually been resolved from different organisms possessing the GlgE pathway, it is unknown whether direct physical interaction between both proteins occurs to mediate the iterative synthesis of α(1→6)-branched α(1→4)-glucans. GlgE from actinomycetes has been shown to form a homodimer and to catalyze the α-retaining transfer of maltosyl units from α-maltose 1-phosphate to maltooligosaccharides using a double-displacement mechanism, i.e., a ping-pong mechanism involving the release of the glucan extended by a maltosyl unit prior to the next reaction.212,221In vitro experiments using only purified GlgE and GlgB proteins recombinantly expressed from M. tuberculosis as well as maltose 1-phosphate as the donor substrate resulted in the formation of high-molecular-weight α(1→6)-branched α(1→4)-glucan particles resembling in chemical structure and supramolecular architecture those natively isolated from cells of M. tuberculosis and Streptomyces venezuelae,84 indicating that GlgE can initiate de novo α-glucan synthesis without the need of a primer, similar to the priming function of GlgA activity in the GlgC-GlgA pathway.194 In absence of maltose 1-phosphate substrate and presence of maltooligosaccharides, GlgE can also mediate disproportionation by transfer of maltosyl units from the non-reducing end of a donor molecule to the non-reducing end of an acceptor maltooligosaccharide.212 In addition to GlgE from actinomycetes, the maltosyltransferase has also biochemically been studied in great detail in recombinantly expressed form from P. aeruginosa strain PAO1173 (Figure 3C) and Estrella lausannensis(222) revealing very similar enzymatic characteristics of the GlgE proteins of different origin.
3.2.2. Formation of the Maltose 1-Phosphate Donor Substrate
In contrast to glucosyltransferases such as the glycogen synthase GlgA that uses nucleoside diphosphate-coupled glucosyl units such as UDP-glucose or ADP-glucose as activated donor substrate, GlgE employs maltose 1-phosphate as activated donor substrate. Two alternative routes for biosynthesis of maltose 1-phosphate have been described, the GlgC-GlgM and the TreS-Pep2 (named TreS-Mak in some organisms) pathways. In actinomycetes such as M. tuberculosis, both pathways operate simultaneously, while other bacteria possessing the GlgE pathway only employ the TreS-Pep2 / TreS-Mak route (Figure 3B,C).
Both routes are closely connected to trehalose metabolism. Trehalose is an α,α(1→1)-linked glucose dimer that is abundant in many pro- and eukaryotic organisms. Trehalose is not representing an α-glucan sensu stricto and plays a broad variety of biological functions dependent on the producing organism, most of them being unrelated to α-glucan metabolism as has already extensively been reviewed.223 However, due to its important connection to the GlgE pathway, trehalose biosynthesis pathways will briefly be described here. Bacteria possessing the GlgE pathway for α-glucan production employ one or both of two alternative routes for de novo synthesis of trehalose. The most widespread route in prokaryotes is the OtsA-OtsB pathway. The trehalose 6-phosphate synthase OtsA (a Leloir-type glycosyltransferase) catalyzes the transfer of nucleoside diphosphate-activated glucose to glucose 6-phosphate to yield trehalose 6-phosphate with release of nucleoside diphosphate. In most cases, OtsA uses UDP-glucose. However, OtsA of M. tuberculosis (Rv3490) has been demonstrated to exhibit a 10-fold higher affinity for ADP-glucose than for UDP-glucose.224 The trehalose 6-phosphate phosphatase OtsB then catalyzes the dephosphorylation of trehalose 6-phosphate to release free trehalose and inorganic phosphate. The M. tuberculosis genome encodes two genes with homology to bacterial trehalose 6-phosphate phosphatases, otsB1 (Rv2006) and otsB2 (Rv3372). The encoded OtsB1 protein is much larger than OtsB2 (1327 aa vs. 391 aa, respectively). However, only the essential OtsB2 expresses trehalose 6-phosphate phosphatase activity and is relevant for trehalose biosynthesis225 while a ΔotsB1 mutant of M. tuberculosis showed no obvious phenotype so that the function of OtsB1 is yet unknown.226 In the alternative route, the TreY-TreZ pathway, the maltooligosyltrehalose synthase TreY converts the terminal α(1→4)-glycosidic linkage at the reducing end of a linear α(1→4)-glucan into an α-1,1-bond yielding maltooligosyltrehalose. Maltooligosyltrehalose trehalohydrolase TreZ then hydrolytically liberates trehalose. As this pathway requires linear glucans, branched α-glucans first need to be processed by glycogen phosphorylase GlgP, which reduces the branch length by releasing glucose 1-phosphate, followed by debranching enzyme TreX, which hydrolyzes the α(1→6)-glycosidic branch linkages. TreX, TreY, and TreZ all belong to the GH13 protein family227 (Figure 3).
In the most common route for maltose 1-phosphate biosynthesis, the TreS-Pep2/TreS-Mak pathway, the trehalose synthase TreS mediates the conversion of trehalose to maltose by converting the α(1→1)-bond into an α(1→4)-glycosidic bond. Subsequently, maltose is rapidly and quantitatively phosphorylated in an ATP-dependent reaction to maltose 1-phosphate by the maltose kinase (systematic name ATP:α-maltose 1-phosphotransferase), which is named Pep2 or Mak depending on the organism.228,229 Previously, TreS was thought to exclusively mediate trehalose formation from maltose. However, although the equilibrium of purified TreS favors the formation of trehalose from maltose in vitro, flux through TreS in vivo is in the opposite direction whenever a maltose kinase is expressed,230 driven by the rapid and irreversible ATP-dependent phosphorylation of the formed maltose to maltose 1-phosphate by the maltose kinase.216,229 The observed finding of the direction of flux through TreS for consumption of trehalose is supported by the fact that TreS and Pep2/Mak are expressed as a fusion protein in many organisms with the exception of actinobacteria (i.e., mycobacteria and Streptomycetes).213 Recombinant bifunctional TreS-Mak from Estrella lausannensis has been enzymatically characterized and was found to exhibit an apparent molecular weight of 256 kDa. The TreS-Mak fusion protein produced maltose 1-phosphate in the presence of nucleoside triphosphates and trehalose concentration resembling physiological intracellular conditions.222 Likewise, the TreS-Pep2 fusion protein from P. aeruginosa strain PAO1 has been reported to exhibit similar enzymatic properties and to synthesize maltose 1-phosphate from trehalose and ATP, although the supramolecular architecture has not been studied.173 In contrast, TreS and Pep2 are expressed as individual proteins in actinobacteria. Here, they form a hetero-octameric complex composed of four subunits of TreS and four subunits of Pep2 with an apparent molecular weight of ca. 490 kDa, in which a homotetramer of TreS forms a platform to recruit dimers of Pep2 via a specific interaction domain as observed in Mycolicibacterium smegmatis.231,232
More recently, a second route for formation of maltose 1-phosphate was discovered in M. tuberculosis, the GlgC-GlgM pathway119 (Figure 3B). While M. tuberculosis GlgM (Rv1212c) was previously believed to be a glycogen synthase and named GlgA accordingly, it was shown that GlgM is a maltose 1-phosphate-producing glucosyltransferase (formal name ADP-α-d-glucose:α-d-glucose-1-phosphate 4-α-d-glucosyltransferase) three orders of magnitude more efficient at transferring glucose from ADP-glucose to glucose 1-phosphate than to glycogen.119 Recently, GlgM from the related bacterium M. smegmatis has been crystallized, and enzymatically characterized revealing very similar properties to the enzyme from M. tuberculosis.233 GlgM from mycobacteria is a GT4 family enzyme employing an α-retaining mechanism, while bona fide bacterial glycogen synthases are GT5 family members. Bioinformatic analysis of bacterial genomes revealed that ∼32% of all annotated GlgA homologues exhibit GT4 membership. It is striking that in every case where GlgA and GlgE coexist in Gram-positive bacteria (typically actinomycetes including mycobacteria and streptomycetes), the GlgA belongs exclusively to the GT4 family and never to the GT5 family. This coexistence strongly suggests that all these GlgA homologues actually have no glycogen synthase activity but rather represent maltose 1-phosphate-producing ADP-α-d-glucose:α-d-glucose 1-phosphate 4-α-d-glucosyltransferases and should be named GlgM accordingly.119 This finding also implies that the proportion of microbes that possess the classical GlgC-GlgA glycogen pathway is only ∼20% and thus lower than previously estimated.213 GlgM prefers ADP-glucose as the donor substrate and can use UDP-glucose at ca. 10-fold lower efficiency. Thus, GlgM is essentially linked to GlgC for the production of ADP-glucose. Similarly, the affinity of trehalose 6-phopshate synthase OtsA from M. tuberculosis for ADP-glucose was found to be one order of magnitude higher than for UDP-glucose.224 Mutational studies in M. smegmatis revealed that GlgM and OtsA are the main consumers of ADP-glucose in this organism as revealed by substantial intracellular ADP-accumulation in the M. smegmatis ΔglgA(u) ΔotsA double mutant. Furthermore, it was found that there is a redirection of the flux of ADP-glucose since neither the M. smegmatis ΔglgA nor the ΔotsA single mutants accumulated detectable amounts of ADP-glucose. When GlgM is inactivated, ADP-glucose is redirected through OtsA promoting trehalose formation, whereas the increased trehalose level in turn stimulates maltose 1-phosphate synthesis via TreS-Pep2.119 Thus, the TreS-Pep2 and GlgC-GlgM pathways for maltose 1-phosphate synthesis are linked via the shared use of the intermediate ADP-glucose by GlgM and OtsA. The flux of ADP-glucose seems to be sufficiently redirected such that the net rate of maltose 1-phosphate generation and α-glucan accumulation can be balanced to some extent when one of the two routes is perturbed.119
3.2.3. Degradation
The structure of glycogen-like α-glucan produced by the GlgE pathway is virtually indistinguishable from those produced by the classical GlgC-GlgA pathway. Therefore, the GlgE pathway does not require a special set of enzymes for the degradation of the produced polysaccharide but relies on the same machinery as the GlgC-GlgA pathway. In fact, one set of degradative enzymes is also present in organisms such as P. aeruginosa PAO1 where both pathways have been reported to operate in parallel.173 The α(1→6)-branched α(1→4)-glucans produced by either pathway can be principally degraded to glucose 1-phosphate for energy production or to feed primary metabolism, or to trehalose (Figure 3). For this, glycogen phosphorylase GlgP reduces the branch length by releasing glucose 1-phosphate from the non-reducing ends of α-glucan chains until the branch length reaches DP4, where GlgP is unable to further degrade external chains due to steric constraints in the vicinity of branch points.199 The glycogen debranching enzyme TreX (named GlgX in other organisms) hydrolyzes the α(1→6) links in branched α-glucan. The enzyme has been shown to have a preference for short branch lengths of DP4 such as those produced by GlgP. Thus, TreX/GlgX is not active on glycogen with full-length branches, thereby preventing a futile cycle.234,235 Consequently, complete degradation of glycogen requires a close cooperation between GlgP and TreX/GlgX. Linear α(1→4) linked maltooligosaccharides produced by GlgP and TreX/GlgX can either be completely degraded to glucose 1-phosphate by GlgP or converted to trehalose by the TreY-TreZ pathway, where the maltooligosyltrehalose synthase TreY converts the terminal α(1→4)-glycosidic linkage at the reducing end into an α(1→1)-bond yielding maltooligosyltrehalose followed by release of free trehalose by the maltooligosyltrehalose trehalohydrolase TreZ. Although the OtsA-OtsB2 pathway dominates for trehalose synthesis in vitro and in vivo in a mouse infection model, the importance of glycogen-like α-glucan as a source for trehalose in vivo has been demonstrated for M. tuberculosis and M. smegmatis, for which trehalose is an essential metabolite.225,230 Not only does the combined inactivation of the otsA and treY-treZ genes result in an auxotrophic mutant requiring supplementation with exogenous trehalose but also blocking glycogen synthesis in combination with loss of otsA, i.e., in ΔotsA ΔglgC or ΔotsA ΔglgM double mutants.119,225 In P. aeruginosa strain PAO1, the α-glucanotransferase MalQ is a maltooligosaccharide disproportionating enzyme that is capable of producing maltose when maltotriose is provided as a donor molecule, which will be generated during disproportionation of maltohexaose as a substrate. Thus, when coupled to Pep2 activity and ATP as a substrate as a virtually irreversible sink, it was shown that MalQ is sufficient to produce maltose 1-phosphate from maltohexaose. Thereby, MalQ can bypass the TreY-TreZ-TreS reactions to feed the GlgE pathway with maltose from maltooligosaccharides in this organism.173 In summary, both the biosynthesis and degradation of glycogen-like α-glucan produced by the GlgE pathway are intimately interrelated with trehalose metabolism since trehalose can be readily converted to α-glucan via TreS-Pep2/Mak and GlgE-GlgB and recycled by GlgP-TreX/GlgX and TreY-TreZ.213
3.2.4. Regulation
The regulation of the GlgE pathway regarding both the biosynthesis and mobilization of the glycogen-like α-glucan has very sparsely been studied. As mentioned above, due to the fact that biosynthesis and degradation occur in the same cellular compartment and the close link with trehalose metabolism, tight regulation is required to avoid futile cycles of parallel synthesis and mobilization. However, how this is mediated is largely unknown.
A key enzyme of the GlgE pathway is the essential maltosyltransferase GlgE. It was reported that enzymatic activity of GlgE from M. tuberculosis and other actinomycetes is negatively regulated in vitro and in vivo through phosphorylation by the serine/threonine protein kinase PknB. PknB specifically phosphorylates one serine (Ser-85) and six threonine (Thr-10, Thr-59, Thr-148, Thr-191, Thr-193, and Thr-370) residues.236 Negative regulation of GlgE by protein phosphorylation is a mechanism distinct from the regulation of the classical GlgC-GlgA-dependent glycogen biosynthetic pathway that involves allosteric regulation by metabolic intermediates.200 However, it is still unclear how phosphorylation of GlgE by PknB is regulated and whether this negative regulation of enzymatic activity can be reverted by dephosphorylation. A temperature-sensitive GlgE mutant of M. smegmatis was found to be rescued by overexpression of the garA gene, which is a forkhead-associated (FHA) domain protein that is a target of PknB itself.237 Since FHA domain proteins have been described to be modules that bind to phospho-Thr residues in signaling cascades, it was conceivable that GarA might directly interact with phosphorylated GlgE. However, no direct interaction of the GarA protein with GlgE neither in phosphorylated nor non-phosphorylated form could be detected. Most likely, GarA in artificially high concentration can act as decoy for PknB-mediated GlgE phosphorylation and plays no role in regulation of the GlgE pathway in vivo.238 Genes of the GlgE pathway from M. tuberculosis such as glgE, glgB, and treS have been shown to be upregulated under conditions mimicking lysosomal stress conditions implying increased production of glycogen-like α-glucans.239 Furthermore, increased capsular α-glucan levels have been reported for M. tuberculosis under phosphate-limiting conditions.240 However, for both cases, the underlying signaling mechanism has not been elucidated, and it is also unknown if and how increased production of glycogen-like α-glucans might provide a specific advantage under these stress conditions. In yet another study, blocking GlgE enzymatic activity and maltose 1-phosphate accumulation in M. tuberculosis has been reported to elicit a pleiotropic stress response that included the upregulation of the genes encoding the TreX-TreY-TreZ branch for trehalose accumulation.212 Theoretically, this could mean the existence of a feedback-loop that can sense reduced cellular α-glucan levels and responds to it by promoting maltose 1-phopshate production via the TreS-Pep2/Mak branch from trehalose. However, since trehalose also functions a compatible solute and general small-molecule stress protectant in many bacteria,241 the upregulation of trehalose biosynthesis in maltose 1-phosphate-stressed M. tuberculosis cells might simply represent a misled general stress response rather than a specific regulatory mechanism involved in α-glucan metabolism.212
M. tuberculosis has been shown to engage two alternative routes for providing the maltose 1-phosphate building block of the GlgE pathway (Figure 3B). However, the TreS-Pep2 and GlgC-GlgM pathways do not equally contribute to maltose 1-phosphate production and are obvious subject to regulation. The TreS-Pep2 pathway dominates in culture while the GlgC-GlgM-dependent route is more important during infection. The molecular basis underlying these regulatory mechanisms under different physiological conditions remains to be fully elucidated.119 In mycobacteria, there is an apparent rechanneling of ADP-glucose between the TreS-Pep2/Mak and the GlgC-GlgM pathways such that the net production maltose 1-phosphate can largely be compensated for when one of these pathways is perturbed. The accumulation of ADP-glucose was detected only in a M. smegmatis ΔglgM ΔotsA double mutant, while none was detected in the ΔglgM and ΔotsA single mutants. These findings are consistent with the flux of ADP-glucose being redirected through OtsA when GlgM is inactive, leading to an enhanced generation of trehalose and subsequent conversion to maltose 1-phosphate via the TreS-Pep2/Mak pathway.119
Similar to regulation of glycogen biosynthesis in bacteria employing the classical GlgC-GlgA pathway, regulation of the GlgE pathway might also take place by allosteric regulation of enzymes controlling metabolic flux of intermediates. For the classical GlgC-GlgA pathway, regulation occurs at the level of the AGPase GlgC, which is typically activated by metabolites of glycolytic pathways such as fructose 6-phosphate, fructose 1,6-bisphosphate, or pyruvate, and inhibited by AMP, ADP, and/or Pi.200 A very similar allosteric regulation might occur at the stage of GlgC for those bacteria possessing the GlgC-GlgM route for maltose 1-phosphate synthesis. For example, it has been shown that purified AGPase GlgC from M. tuberculosis was allosterically activated primarily by phosphoenolpyruvate and glucose 6-phosphate, while the enzyme from Streptomyces coelicolor exhibited sensitivity to allosteric regulation by mannose 6-phosphate, phosphoenolpyruvate, fructose 6-phosphate, and glucose 6-phosphate, whereas NADPH was a main inhibitor.242 Furthermore, the other route for maltose 1-phosphate synthesis from trehalose via the TreS-Pep2/Mak pathway might be subject to indirect allosteric regulation since the activity of trehalose 6-phosphate synthase OtsA from M. tuberculosis involved in providing the trehalose substrate for TreS-Pep2/Mak has been described to be increased by fructose 6-phosphate.224
3.3. The Sucrose to α-Glucan Pathway
The sucrose-dependent glycogen pathway is unique among biosynthetic pathways discussed so far. Indeed, the glycogen is synthesized directly from sucrose through the combined actions of amylosucrase and branching enzymes without using an activated nucleotide-sugar or a phosphorylated sugar as building blocks.243 Initially found in the culture of Neisseria perflava,244 the majority of the amylosucrase studies have been conducted in N. polysaccharea. The genus Neisseria consists primarily of commensal Gram-negative cocci and two pathogenic species: Neisseria meningitidis and Neisseria gonorrhoeae. The term amylosucrase was coined by Herhe to describe a novel enzyme capable of synthesizing α-polysaccharides from sucrose.244 Early studies suggested, like other sucrose-utilizing enzymes such as dextransucrase, that amylosucrase can synthesize both α(1→4) and α(1→6) O-glucosidic bonds.243,245 Nonetheless, recombinant amylosucrases of N. polysaccharea and Deinococcus radiodurans formed linear α(1→4) glucan chains with no detectable α(1→6) linkage from sucrose.159 Two years later, Büttcher and coworkers characterized the branching enzyme of Neisseria denitrificans and demonstrated the stimulation of branching enzyme activity on amylosucrase activity.165 The branching enzyme, in fact, causes an exponential rise in the number of non-reducing ends. Each new extremity becomes a potential acceptor for the covalently attached glucosyl residue to amylosucrase. Interestingly, in a survey of the arrangement of glycogen genes in Neisseria suica (Figure 4), both glgB and malQ genes are fused to produce a large protein with a branching enzyme domain at the C-terminus and an α(1→4) glucanotransferase domain at the N-terminus. Currently, the amylosucrase (EC 2.4.1.4) is a member of the extensive GH13 α-amylase superfamily that establishes a transient covalent linkage with the glucosyl residue from sucrose and releases fructose.246
Figure 4.

Arrangement of glycogen metabolizing genes in Neisseria sicca. Colors indicate the family members according to the CAZy classification: glgP, glycogen phosphorylase (GT35 family: dark green background); glgX, glycogen debranching enzyme; glgB, glycogen branching enzyme and amylosucrase (GH13 family: blue background); malQ, α(1→4)-glucanotransferase (GH77 family: light green background). Arrows indicate transcriptional orientation. Parallel black lines represent a physical separation between the genes. The white arrows represent unrelated glycogen genes that encode a subunit of the HlyD family efflux transporter (WP_003761172.1) and a hypothetical protein (WP_003761174.1). glgP; glycogen phosphorylase; glgX, glycogen debranching enzyme; glgB, glycogen branching enzyme; malQ, α(1→4)-glucanotransferase.
Initially described in the Neisseria genus, the expanding number of complete bacterial genomes revealed a widespread distribution of putative amylosucrase sequences across prokaryotes. As depicted in Figure 5, the phylogenetic tree was inferred with putative-amylosucrases identified in various bacteria classes, including the large Terrabacteria group [D. radiodurans,247Arthrobacter,248Synechococcus PCC7002,249Cellulomonas,250Calidithermus timidus,251Truepera252]; γ-proteobacteria [Alteromonas macleodii253]; β-proteobacteria [Methylobacillus254]; and α-proteobacteria [Methylomicrobium alcaliphilum255].
Figure 5.
Maximum likelihood of putative-amylosucrases using substitution model LG4X implemented in IQTREE. The selected sequences were retrieved from the NCBI database and then aligned and then trimmed using MAFFT (v7.450) and TrimAl (V1.3) implemented in the webserver Phylemon 2 (http://phylemon2.bioinfo.cipf.es/), respectively. Black circles symbolize ultrafast bootstrap support values greater than or equal to 70. The scale bar indicates the number of substitutions per site. PVC stands for Planctomycetes, Verrumicrobia Chlamydiae. FCB stands for Fibrobacter Chlorobi Bacteroides. The amylosucrase in red indicate that they have been characterized enzymatically.
Few reports have been published on the physiological function of amylosucrase in prokaryotes. In N. polysaccharea, the amylosucrase contributes to intracellular glycogen synthesis to compensate for the lack of glycogen synthase activity and the synthesis of linear polysaccharides outside the cell. Amylosucrase has recently been hypothesized to be involved in the cyanobacterium’s sucrose metabolism pathway, Synechococcus PCC7002.249 This assumption is based upon the fact that amylosucrase and sucrose-related genes are arranged in an operon (spsA-sppA-frkA-amsA: sucrose synthase, sucrose phosphate phosphatase, fructokinase, and amylosucrase). Sucrose biosynthesis is an osmotic protectant tightly regulated in response to environmental changes. Therefore, sucrose homeostasis is maintained by a balance of synthesis and degradation, either with glycogen serving as a buffer or through the invertase activity (EC3.2.1.26) that hydrolyzes sucrose into glucose and fructose. In terms of energy consumption, hexokinase and fructokinase will consume one ATP each for recycling both glucides. Interestingly, transferring the glucosyl residue of sucrose onto the glycogen pool decreases ATP consumption by 50% as well as the osmotic pressure, avoiding the glucogenesis route. This alternate pathway provides a significant advantage to bacteria thriving in a poor carbon environment.
Over the past years, amylosucrases have attracted more attention due to their capability of isomerization and transglycosylation. To date, many efforts are pursued to engineer amylosucrase enzyme with better thermal stability properties,256,257 higher yield of sweeteners production through isomerization (turanose: glucose α(1→3)-fructose), trehalulose (glucose α(1→1)-fructose), and to improve their ability to glycosylate unnatural acceptors.258
3.4. Archaeal Glycogen: The Origins
In recent years, metagenome-assembled genomes have considerably increased our understanding of microbial diversity on Earth, particularly in the kingdom of Archaea, the third branch of the tree of life. According to collected samples, the population of Archaea and their phylogenetic topology are continuously expanding and changing. The Archaea kingdom is now constituted of the Euryarchaeota, the TACK group (Thaumarchaeota, Aigarchaeota, Crenarchaeota, and Korarchaeota), the DPANN (Daipherotrites, Parvarchaeota, Aenigmarchaeota, Nanoarchaeota Nanohaloarchaeota), and more recently the Asgard superphylum, which comprises Lokiarchaeota, Odinarchaeota, Thorarchaeota, and Heimdallarchaeota phyla.259 Remarkably, these archaea share unexpected similarities with eukaryotic cells.260 This discovery gives us new insights into the origin of eukaryotic cells. Despite intense debates, a growing body of phylogenomic evidence supports the idea that the Eukaryota phylum emerged from a proto-eukaryotic cell related to the archaeal Asgard superphylum, suggesting a two-domain rather than a three-domain tree of life (). In Archaea, the glycogen metabolism pathway and its regulation are mainly unexplored. The first evidence of glycogen in Archaea came from the pioneering work of König in 1982. König and colleagues observed and biochemically characterized the glycogen accumulation in numerous archaea, including the thermoacidophilic archaea Sulfolobus acidocaldarius, Thermoproteus tenax, Desulfurococcus mucosus, and Desulfurococcus mobilis (TACK group), and Thermoplasma acidophilum (Euryarchaeota).261
Exploration of the glycogen metabolism route in Archaea is currently facilitated by the use of annotated databases and blast search, notwithstanding the necessity of this laborious purifying and characterization effort for confirming glycogen synthesis. In this Review, putative glycogen-metabolizing enzymes were retrieved from the UniProt and CAZy databases for each representative Archaea phylum. Table 3 summarizes the glycogen gene content and the number of isoforms. Several members of the TACK, Asgard, Euryarchaeota, and DPANN superphyla lack all genes related to carbohydrate metabolism, as seen in this table. It is worth noting that other molecules, such as polyphosphate granules, polyhydroxyalkanoates, and triacylglycerol, have been found as potential energy storage alternatives.262 Moreover, several studies demonstrate the importance of syntrophic relationships among microbial communities. A recent analysis of the interaction between Candidatus Nanohalobium constans and Halomicrobium, two halophilic archaea, provides an outstanding example.263
Table 3. Archaea Glycogen-Metabolizing Enzymes Contenta.

Representatives of each archaeal phylum were chosen based on their availability in the CAZy or Uniprot databases. TACK group and DPANN are superphyla that include several phyla whose first letters form the acronyms: Thaumarchaeota, Aigarchaeota, Crenarchaeota, and Korarchaeota (TACK) and Daipherotrites, Parvarchaeota, Aenigmarchaeota, and Nanoarchaeota Nanohaloarchaeota (DPANN). The glycogen-metabolizing enzymes and their isoform numbers were classified. For the sake of clarity, background colors were employed. For instance, enzymes belonging to the GH13 family have a blue background. The number associated with GT or GH represents the family. GlgA-GT3 and GlgA-GT5: glycogen synthase activities; GT4: glycosyltrasferase; GlgB-GH13: branching enzyme; Pul./Amylopul./neopul.-GH13: pullulanase/amylopullulanase/neopullulanase; GlgX/TreX: debranching enzyme; TreY-GH13: maltooligosyl trehalose synthase; TreZ: maltooligosyl trehalose hydrolase; GT35-GlgP/MalP are acronyms for glycogen phosphosphorylase and maltodextrin phosphorylase; GBE-GH57: glycogen branching enzyme; Amp./amp-cd.-GH57 amylopullulanse/amylopullulanase cyclodextrinase; DBE-GH133: amylo-α(1→6) glucosidase. Question marks indicate that the function of GT4 in glycogen metabolism requires clarification. In the column GH13 total number, an asterisk denotes the presence of trehalose synthase (TreS). The letter “i” indicates the presence of one inactive isoform.
3.4.1. The Formation of α(1→4) Bonds in Archaea
Like prokaryotes and eukaryotes, the synthesis of α(1→4)-glucan chains relies mainly on the nucleotide-sugar-dependent glycogen pathway. In contrast, only a few archaea belonging to the superphylum Euryarchaeota (Picrophilus torridus, Acidiplasma cupricumulans, Cuniculiplasma divulgatum) utilize the alternate glycogen route, known as the GlgE pathway (Table 3). Two families, GT3 and GT5, of the 115 GT families reported in the CAZy Database catalyze the transfer of the glucose moiety of NDP-glucose onto the non-reducing ends of growing α(1→4)-glucan chains. GlgA-GT3 and GlgA-GT5 differ in their preference for the nucleotide-sugar donor and in their capacity to initiate de novo glucan chain synthesis. In contrast to ADP-glucose-dependent GlgA-GT5, heterotrophic eukaryotes GS-GT3 utilizes UDP-glucose and requires a primer, i.e., short glucan, to initiate glycogen synthesis. A GT belonging to family 8, termed glycogenin, specifically synthesizes the primer, composed of 12 to 15 glucose residues, by successive self-glycosylation reactions. Both GlgA-GT5 and GlgA-GT3 are represented among Archaea according to the CAZy database. GlgA-GT5 glycogen synthases are widely distributed in the TACK group and to a lesser extent in Euryarchaeota, whereas GlgA-GT3 glycogen synthases are confined to the DPANN group and Euryarchaeota.
Archaeal GlgA-GT5 have been characterized biochemically in Sulfolobus acidocaldarius, Thermococcus hydrothermalis, and Pyrococcus furiosus.261,264,265 These studies demonstrate that archaeal GlgA-GT5 utilizes either ADP-glucose or UDP-glucose as a nucleotide sugar, and other nucleotide diphosphate glucose derivatives such as GDP-glucose for Pyrococcus furiosus glycogen synthase.265 Intriguingly, Sueda and colleagues have recently proven that the NDP-sugar pyrophosphorylase gene of Haloarcula japonica encodes a GDP-glucose pyrophosphorylase (GGPase) activity and not an ADP- or UGPase, as was expected.266 Because the GlgA-GT5 has not been characterized enzymatically, the selectivity of glycogen synthase toward GDP-glucose is unknown.
The putative GlgA-GT3 activities of archaea have not yet been enzymatically characterized. However, the presence of glycogen in methanogenic M. thermophila TM1 indicates that glycogen synthase GT3 is almost certainly functioning.267 To better understand the evolution of GlgA-GT3, a phylogenetic tree was inferred with glycogen synthase-GT3 sequences retrieved from PVC (Planctomycetes, Verrumicrobia, Chlamydiae), Eukaryota, DPANN, Euryarchaeota, and FCB group. As depicted in Figure 6, all Euryarchaeota GlgA-GT3 sequences are monophyletic and phylogenetically distinct from DPANN and other GlgA-GT3 sequences. This highlights a unique evolution of GlgA-GT3 in this group, which may be associated with the connection between glycogen and methanogenesis pathways in Methanotrix sp., Methanococcoides sp., and Methanosarcina sp. (Methanosarcinales order).268 In contrast, putative GlgA-GT3 sequences of the DPANN group are more closely related to PVC and heterotrophic eukaryotic organisms. GlgA-GT3 sequences belonging to Bacteroidetes members (FCB group) are nested inside the DPANN (Figure 6). A plausible hypothesis is that Bacteroidetes gained the GlgA-GT3 gene from the DPANN through horizontal gene transfer. It is worth noting that Bacteroidetes have conserved a fragment of the GlgA-GT5 gene in their genome.
Figure 6.
Maximum likelihood phylogeny of glycogen synthase from the GT3 family based on the LG4X substitution model implemented in IQTREE. The chosen sequences were obtained from the NCBI database, aligned, and trimmed using MAFFT (v7.450) and TrimAl (V1.3) implemented in the website Phylemon 2 (http://phylemon2.bioinfo.cipf.es/). Similar taxa were collapsed for better readability. The numbers in parentheses represent the number of taxa in the clade that has been collapsed. The colors represent glycogen synthases GT3 from Euryarchaeota (blue), DPANN (yellow), Bacteroidetes (grey), PVC (red), and Eukaryota (green). The scale of the tree indicates a single substitution per site. Black circles symbolize ultrafast bootstrap support values greater than or equal to 80. The scale bar indicates the number of substitutions per site.
As stated previously, heterotrophic eukaryotic glycogen synthases-GT3 require glycogenin activity to initiate glucan synthesis. Thus, a question naturally arises on the presence of glycogenin-like proteins in archaea. Interestingly a glycogenin-like protein was annotated (LC1Nh 1199) in Candidatus Nanohalobium constans. However, a homology search using the Hidden Markov model did not reveal any hit with eukaryotic glycogenin. To date, the existence of glycogen-like in DPANN remains questionable, and the enzymatic properties of archaeal GlgA-GT3 remain unknown.
It should be stressed that P. torridus, C. divulgatum, A. cupricumulans, and Methanoculleus bourgensis (Euryarchaeota) lack glycogen synthase activity and instead utilize the GlgE glycogen pathway, which is based on a putative maltose 1-phosphate:maltosyltransferase (Table 3).
As mentioned in section 3.2, maltose 1-phosphate serves as a building block for the GlgE pathway. The latter can be derived either from trehalose followed by two enzymes: a trehalose synthase (TreS) directly isomerizes maltose into trehalose (TreS-GH13 family), and then a maltokinase phosphorylates maltose into maltose 1-phosphate, or directly by the transglycosylation between ADP-glucose and glucose 1-phosphate via GlgM activity.
Except for vertebrates, trehalose is a ubiquitous disaccharide present in all living organisms. In response to environmental stress, it is produced via many routes. In Archaea, numerous trehalose pathways have been identified, including the TreT pathway, discovered in archaea and thermophilic bacteria, which synthesizes trehalose from NDP-glucose and glucose,269−271 the TreS pathway, mentioned above, found in P. torridus,272 and the TreX-TreY-TreZ path that converts glycogen into trehalose using three successive enzymes: TreX (glycogen debranching enzyme), TreY (maltooligosyl trehalose synthase), and TreZ (maltooligosyl trehalose hydrolase).273
In the archaeal strains that harbor putative GlgE proteins, both putative TreS and Mak proteins were found in P. torridus (TreS, WP 011176870.1; Mak, WP 011176871.1), in A. cupricumulans (TreS WP 201779456.1; Mak, KQB33759.1), in C. divulgatum (TreS, SIM87424.1 A0A1N5WSF9), suggesting that GlgE path may operate. In P. torridus, a gene encoding a GT4 polypeptide (WP 011178040.1) forms a cluster with genes encoding putative amylo-α(1→6) glucosidase type debranching enzyme-GH133 and GH57-amylase (Figure 7). The GT4 family consists of several glycosyl transferase activities, including the recently identified GlgM enzyme of mycobacteria. However, the diversity of GT4 activities and the low level of similarity with mycobacterium GlgM (about 20%) make it difficult to conclude if GT4 gene encodes a GlgM-like activity.
Figure 7.

Arrangement of glycogen metabolizing genes in Archaea. The glycogen related genes were annotated according to the CAZy classification. Directional arrows denote transcriptional direction. Physical segregation between the genes is shown by parallel black lines. The white arrows represent unrelated glycogen genes. For clarity, the colored backgrounds of glycogen-related genes correspond to the enzymatic activities listed in Table 3. The number associated with GT or GH represents the family. GT3, GT3-glycogen synthase; GT4, NDP-glycosyl transferase; glgA, GT5-glycogen synthase; glgB, GH13-branching enzyme; glgX/treX, debranching enzyme; treS, trehalose synthase; treY, maltooligosyl trehalose synthase; treZ, maltooligosyl trehalose hydrolase; glgP, glycogen phosphosphorylase; GH57-BE, glycogen branching enzyme; GH57-GH, glycosyl hydrolase; GH57-amy, α-amylase-GH57; GH57-amp, amylopullulanase-GH57; GH15, glucoamylase; glgC, AGPase; GH133, amylo-α(1→6) glucosidase; glgE, maltose 1-phosphate transferase; malQ, α(1→4) glucanotransferase.
Another intriguing situation is observed for the methanogenic archaea Methanoculleus bourgensis DSM3045 (Table 3; Figure 7). A survey of the genomic organization of the glycogen genes highlights an annotated gene (ID: IJ7JAJ9) that encodes a putative glycosyltransferase family 4 (GT4) nested among glgB and putative-amylase (GH57) genes. As revealed by a Blast search, the GT4 gene (BN140 RS11275) is confined to a few Euryarchaeal species. Given the absence of glycogen synthase GT3 or GT5 in the M. methanoculleus genome, it is tempting to speculate that the GT4 gene encodes an uncharacterized glycogen synthase.
3.4.2. The Formation of α(1→6) Bonds in Archaea
The branching enzymes first cleave an α(1→4) glycosidic bond inside a glucan chain and subsequently transfer the glucosyl chain to the adjacent chain at the so-called branching point, or α(1→6) position. According to CAZy classification, branching enzymes fall into two glycosyl hydrolase families: GH13 and GH57. The GH57 was established due to an increasing number of enzymes lacking conserved regions characteristic of the GH13 family.274 Members of the GH57 family, which adopt the (β/α)7-barrel fold and five conserved domains,275 have been identified in archaea and a few eubacterial taxa, including cyanobacteria.276 Currently, the GH57 family encompasses various activity such as α(1→4)-glucanotransferase, amylopullulanase, β-galactosidase, maltogenic amylase, amylopullulanase-cyclodextrinase, α-amylase, and glycogen branching enzymes.277 3D structures have been elucidated for several branching enzymes: Thermococcus kodakarensis,278Thermotoga maritima,279Thermus thermophilus,280 and Pyrococcus horikoshii.281
As indicated in Table 3, the glycogen branching enzymes GH13 and GH57 are restricted to Asgard and Euryarchaeota (for review, see ref (282)). Unlike the branching enzyme GH13, the archaeal branching enzymes GH57 of P. horikoshii and Thermus thermophilus possess both amylase and branching enzyme activities.280,281 The balance between amylase and branching enzyme activities in P. horikoshii is associated with the occurrence of a flexible loop that modifies access to the catalytic site.281 Astonishingly, no branching enzyme GH57 or GH13 activity was observed in the TACK group despite the presence of glycogen,261 strongly suggesting the existence of undiscovered branching enzyme activity. A phylogenetic tree of archaeal GH57-proteins was inferred based on the archaea listed in Table 3 and supplemented with additional sequences (Figure 8). The enzymatic properties were established according to the annotation of the CAZy database and bioinformatics approach.283Table 3 provides an overview of Archaea’s GH57-enzyme content. A striking observation is that α-amylase/α-amylase-like GH57 is preserved in all archaea lacking a “classical” branching enzyme (Table 3; Figure 8). Could this amylase-GH57 be the missing branching enzyme? It is important to note that only a few enzymatic characterizations have been conducted on these GH57-amylases. Kim and coworkers reported the characterization of the GH57-amylase (AAB99631.1) of Methanocaldococcus janaschii.284 However, the enzymatic characterization mainly focused on the hydrolysis activity of the recombinant GH57-amylase toward various polysaccharides. Considering that archaeal branching enzymes exhibit dual functions of amylase and branching enzyme activities, further investigations are warrented to determine whether the GH57-amylase of M. janaschii harbors a branching enzyme activity.
Figure 8.

Maximum likelihood phylogeny of the archaeal GH57 family using the LG4X substitution model implemented in IQTREE. The chosen sequences were obtained from the NCBI database, aligned, and trimmed using MAFFT (v7.450) and TrimAl (V1.3) implemented in the website Phylemon 2 (http://phylemon2.bioinfo.cipf.es/). GH57 enzymes subjected to a detailed biochemical characterization are pointed by black stars. Black circles symbolize ultrafast bootstrap support values greater than or equal to 70. The scale of the tree indicates the number of substitution per site.
The characterization of a series of null mutants defective in the glycogen metabolism pathway of Sulfolobus acidocaldarius DSM 639 has provided some insight.285 Functional classification of enzymes can be achieved through glycogen content assays. Consequently, a mutation in either an anabolic or catabolic enzyme leads to a decrease or an excess of glycogen, respectively. All glycogen phenotypes were in line with the presumed function of the catabolic glucoamylase-GH15 and anabolic glycogen synthase mutants, except for the null GH57-amylase mutant, which displayed a dramatic decrease in glycogen level.285 This atypical phenotype could be explained by the lack of branches within the storage polysaccharide, rendering it extremely sensitive to intracellular hydrolase activities. Interestingly, a similar phenotype was documented in the Arabidopsis branching enzyme mutant, wherein the chloroplast harbors multiple β-amylase activities that prevent the accumulation of linear glucan chains.286
3.4.3. The Catabolism Pathway in Archaea
The glycogen catabolism pathway involves a series of enzymes that hydrolyze α(1→4) and α(1→6) bonds. To complete glycogen digestion in E. coli, glycogen phosphorylase (GlgP) collaborates with the debranching enzyme (GlgX). Glycogen phosphorylase (GT35) converts the reducing end of glucan and orthophosphate into glucose 1-phosphate. The α(1→6) linkages or branching points impede further phosphorylase action, leading to the formation of phosphorylase limit-dextrin, which contains short-branched glucans (3 to 4 residues glucose). The debranching enzyme specifically trims these short glucan chains (GlgX), thus preventing a futile cycle of synthesis and degradation, as the GlgX is synthesized during glycogen synthesis. Maltodextrin phosphorylase (MalP) and α(1→4) glucanotransferase (MalQ) further contribute to the catabolism of the short glucans released by GlgX. This enzyme plays a central role in the short glucan metabolism during glycogen breakdown and in the maltose catabolism pathway by converting short glucans into longer glucans which are more accessible to maltodextrin phosphorylase for hydrolysis. It should be stressed that the null malQ mutant of E. coli cannot grow in the presence of maltose. In Enterobacteriaceae, the catabolic enzymes result in the release of glucose 1-phosphate and glucose from glycogen. Given this information, it is interesting to explore the catabolic process in Archaea.
3.4.4. An Alternate Mechanism for Glycogen Breakdown in Archaea
Table 3 illustrates the variations in the number of glycogen phosphorylase genes in various archaeal strains. For instance, Acidianus hospitalis and Stygiolobus azoricus (TACK) lack glycogen phosphorylase activity, while Methanotrix soehngensis and Methanoculleus bourgensis (Euryarchaeota) possess two and three glycogen phosphorylases, respectively. These isoforms likely mirror their affinities for linear glucans and branched polysaccharides, as described for the maltodextrin and glycogen phosphorylase activities of E. coli. More significantly, the absence of glycogen phosphorylase activity in certain archaeal strains reveals a unique catabolic pathway where, at variance with enterobacteria, glycogen phosphorylase does not drive the glycogen degradation process but appears mostly devoted to the metabolism of imported short glucan.287 In line with this finding, a biochemical characterization of glgP null mutant of S. acidocaldarius displays a normal amount of glycogen in comparison to the wild-type strain, reinforcing the idea that GlgP is not a crucial enzyme in glycogen breakdown.285
The designations GlgX and TreX were assigned to the debranching enzymes based on their presence in the glycogen or trehalose operons. Both enzymes are considered to be identical in terms of enzymatic properties. However, the biochemical analysis of Sulfolobus sp. TreX reveals a bifunctional enzyme with α(1→4) glucanotransferase and amylo-α(1→6) glucosidase activity.288,289 Like in mammals, the α(1→4) glucanotransferase firstly cleaves the α(1→4) linkage prior to the last glucose residue hooked in α(1→6) linkage and transfers the glucan chain to the non-reducing end of an adjacent chain. Thanks to amylo-α(1→6) glucosidase, the residual branched glucose residue is eliminated. As described, the debranching step does not produce any short chains. A notable observation is the wide distribution of genes that encode amylo-α(1→6) glucosidase (EC3.2.1.33; GH133) activity among Archaea. The characterization of the null amylo-α(1→6) glucosidase mutant (GH133: AAY80544.1) of Sulfolobus, designated GlgX by the authors, results in an excess glycogen phenotype, suggesting its participation in the glycogen catabolism pathway.285
In contrast to E. coli, archaea possess additional hydrolytic enzymes, including glucoamylase-GH15 and different glycosyl hydrolases GH57, which are controlled by catabolic inhibition.290 These enzymes are secreted and have a function in starch utilization.291,292 The amylopullulanase mutant of Sulfolobus acidocaldarius DSM639, for instance, lost the capacity to thrive on a minimum medium containing starch as the only carbon source.293 Lee and colleagues have demonstrated that the starch medium stimulates the production of many extracellular catabolic enzymes in P. furiosus, including maltogenic amylase-GH57 and amylopullulanase-GH57.287 Two specialized transporters for trehalose/maltose and maltodextrin are then used to import maltose and maltodextrin inside P. furiosus.294 In the case of P. furiosus and other archaea lacking α(1→4) glucanotransferase activity GH77 or GH57, maltodextrins are likely further processed by glucoamylase-GH15/maltodextrin phosphorylase. Interestingly, in contrast to glycogen phosphorylase of Thermococcus litoralis, which liberates glucose 1-phosphate from maltodextrin containing at least four glucose residues,295 glycogen phosphorylases of P. furiosus can digest a glucan chain up to maltose, allowing complete hydrolysis into glucose by a glucoamylase-GH15.296
4. Chemical Reactions in α-Glucan Synthesis and Degradation
The evolution of enzymatic reactions and catalytic/molecular/structural mechanisms play key roles in the articulation of (new) metabolic pathways. α-Glucan metabolism offers a remarkable framework of catalysts using a defined building block, α-d-glucose, and a few ancillary substrates for its activation and polymerization. Altogether, each of these enzymes can be considered a piece of machinery dedicated to preserving the stereochemistry of α-d-glucose. Although certain underlying mechanisms are common among enzymes involved in bacterial α-glucan metabolism, as will be evident later, the overview presented here points to fundamental aspects of the building block processing. Therefore, the enzymatic reactions comprise three groups: (i) the synthesis of gluco-saccharide donors, (ii) the formation of α-glucosyl bonds between donors and sugar acceptors, and (iii) the disruption of glucosyl bonds. Here we present these chemical reactions and base mechanisms that present the enzymes-domain that perform such functions (Figure 9).
Figure 9.


Reactions and catalytic mechanism of enzymes in the α-glucan and disaccharide metabolism. Intervening chemical groups and molecule motifs from the substrates and products representations are colored in red. (A) Top view, the general reaction for nucleotide glucose biosynthesis mediated by nucleotide glucose pyrophosphorylases. The generic nucleotide triphosphate reacts with glucose 1-phosphate (glucose 1P) in the presence of Mg2+ to bring forth the corresponding nucleotide diphospho-glucose and pyrophosphate. Frequently used nucleotide bases are indicated on the right on a blue-shaded box. Bottom view, the catalytic mechanism of phosphotransfer mediated by the nucleotide glucose pyrophosphorylase, involves a couple of lysine residues that polarize the phosphoester bonds. (B) Top view, the general reaction for glucose transfer mediated by glycosyltransferases. The generic nucleotide diphosphate-glucose and an acceptor bearing a nucleophilic hydroxyl group react to produce a nucleotide diphosphate and the glucosylated product. Examples of acceptor substrates are displayed on the right on a yellow shaded box with the reactive hydroxyl group marked with a rectangular box. Bottom view, the proposed SNi-type catalytic mechanism for most of the retaining glycosyltransferases. (C) Top view, the general reaction for the phosphorolysis of a non-reducing end of an α-glucan to form glucose 1-phosphate. Bottom view, the 5'-phosphate group of the essential cofactor pyridoxal phosphate functions as an acid-base to promote attack by the substrate phosphate on the the α-glucan anomeric carbon. (D) Top view, the general reaction of disaccharide 6-phosphate phosphatases. Frequently used substrates are indicated on the right on a green shaded box. Bottom view, the catalytic mechanism showing a group of carboxilic groups from acid residues (Asp/Glu) that coordinates a Mg2+ atom, with one also acting in the nucleophilic attack of the phosphate group to formg a phospho-protein covalent intermediate. In a second step, an activated water molecule attacks the covalent intermediate, releasing phosphate. (E) Top view, the general reaction of maltokinase, showing the reaction between the ATP with maltose to bring forth maltose 1-phosphate. Bottom view, the proposed catalytic mechanism showing the nucleophilic attack of the anomeric OH group of maltose to the γ-phosphate group of ATP. (F) Top view, A general reaction for the catalysis mediated by TIM-barrel folds in the context of α-glucan metabolism. Depending on the nature of the reactions presented in the red shaded box, substrates (A and B) outcomes products (C and D). Central view, a general two-step double inversion SN2 mechanism, showing the participating acidic residues acting as a base and forming the covalent intermediate. Possible hydrogens or α-linked-chains substitutions in the α-glucose moiety are indicated as R groups in the pyranose ring (R2, R4, and R6) to represent the variable nature of the substrates under this mechanism. Bottom view, a brief list of substrates (A and B) and products (C and D), highlighting in different shade colors the other moieties exchanged in the reaction, permitting to read from top to bottom different reactions. For example, α(1→4)-glucan (substrate A) reacts with water (substrate B) bringing a remaining α(1→4)-glucan (product C) and maltose (product D), whereas maltose 1-phosphate (substrate A) and α(1→4)-glucan (substrate B) brings forth an elongated α(1→4)-glucan chain (product C) and phosphate (product D).
4.1. The Synthesis of Glucose Nucleotide Donors
The synthesis of NDP-glucose donors follows a mechanism parallel to other nucleotide-activated compounds.297,298 Two enzymes are dedicated to the synthesis of ADP-glucose and UDP-glucose, the two major glucose donors in bacterial α-glucan metabolism, AGPase (EC 2.7.7.27)184 and UGPase (EC 2.7.7.9),299−301 respectively. GGPase, which synthesizes GDP-glucose (EC 2.7.7.34), can also play a role in the synthesis of trehalose in a few bacteria.302,303
NDP-glucose donors are synthesized as a condensation reaction between NTP and glucose 1-phosphate following a bimolecular nucleophilic substitution SN2(P) that exchanges the phosphate-ester bond from the NTP to the glucose 1-phosphate.304 The transfer of the nucleoside monophosphate occurs via the nucleophilic attack of an oxygen atom from the PO4 group in glucose 1-phosphate to the phosphorus atom in the α-PO4 from NTP, producing the displacement of PPi via a pentavalent phosphate transition state, and leading to the inversion of the NTP α-PO4 upon nucleophilic attack. Notably, the reaction is held in the presence of a divalent metal cation with a strong preference for Mg2+.305,306 The divalent metal cation minimizes the repulsion between different PO4 groups and lowers the energetic barrier of the enzymatic triphosphate hydrolysis, weakening the leaving-group bond, stabilizing the transition state, and strengthening the interaction with the nucleophile.307 In addition, positively charged residues in the active site coordinate the substrate's PO4 groups in place.308 Finally, proton exchange is balanced by the solvent. Overall, the energy balance in breaking and forming the phosphodiester group is near zero; therefore, the reaction is reversible. Nevertheless, hydrolysis of pyrophosphate catalyzed by inorganic pyrophosphatases pulls this reaction to completion309 (Figure 9A).
4.2. Glucosyl Transfer Reactions
4.2.1. Glucosyl Transfer from an NDP-Glucose Donor
NDP-sugar-dependent glycosyltransferases (GTs) act on a broad range of acceptors to create new glycosidic linkages, generating a significant amount of structural diversity in biological systems.310−312 The transfer of a glycosyl group can proceed with retention or inversion of the anomeric configuration of the product with respect the donor substrate, and consequently, GTs are classified as ‘retaining’ or ‘inverting’.310,313−315 In reactions involving α-glucan biosynthesis from an NDP-α-d-glucose donor, a ‘retention’ mechanism is obliged. The reaction mechanism for ‘retaining’ GTs has been a matter of debate. By analogy with glycoside hydrolases, a double-displacement mechanism was first proposed.310,313−315 This mechanism requires a suitable positioned nucleophile, typically Glu or Asp, in the catalytic center that mediates the formation of a glycosyl-enzyme intermediate during the first part of the reaction, resulting in a first anomeric inversion. In the second part of the reaction, the activated acceptor substrate attacks the glycosyl-enzyme intermediate, resulting in the formation of a product with total retention of the anomeric configuration.314 Amino acid sequence alignments do not show consistently conserved amino acid residues for such an important role. Members of family 6 GT were first predicted to display such a putative nucleophile.315,316 Quantum mechanics/molecular mechanics (QM/MM) metadynamics analysis of the bovine α(1→3)-galactosyltransferase (α3GalT) GT6 mechanism supports that the donor saccharide forms a covalent Glu-Gal intermediate adduct prior to transfer to the acceptor.316,317 However, experiments with human GTA/GTB GT6s found that mutating Glu303 to Cys or Asp only slightly slows down the reaction.318,319 Native ternary complexes of the bovine α3GalT GT6 in the presence of UDP-Gal, lactose, and the divalent cation cofactor, revealed the substrates are organized very similarly to other ‘retaining’ GTs, with the putative nucleophile Glu317 participating in a hydrogen bond with the acceptor β-Gal O4 atom, supporting a role in acceptor binding.320,321 As an alternative possibility, an unusual single-displacement mechanism named as ‘front-face’ or SNi, substitution nucleophilic internal-like mechanism, was proposed, in which the nucleophilic attacks from the same face as the leaving group departure. Specifically, the acceptor hydroxyl nucleophile is deprotonated by the donor β-phosphate oxygen and attacks the anomeric carbon atom of the sugar donor from the same side as the leaving nucleotide, and involves a short-lived oxocarbenium ion intermediate.310,316 Several structural snapshots of enzyme-donor-acceptor Michaelis complexes for ‘retaining’ GTs strongly support the SNi-type mechanism, including the glucosyl-3-phosphoglycerate synthase (GpgS) from M. tuberculosis and the xyloside α-(1→3)-xylosyltransferase XXYLT from Mus musculus(310,322) (Figure 9B).
Very recently, mass spectrometry analyses of WbbB from family GT99, an enzyme that adds a terminal β-Kdo (3-deoxy-d-manno-oct-2-ulosonic acid) residue to the O-antigen saccharide, forms a covalent adduct between the catalytic nucleophile, Asp232, and Kdo.323 Similarly, the formation of covalent adducts with Kdo were reported in a member of the GT107 family, KpsC, a dual-module enzyme that is essential for biosynthesis of ‘group 2’ capsular polysaccharides in E. coli and other Gram-negative pathogens.324 Crystal structures of WbbB and KpsC variants revealed the presence of Kdo-adducts, supporting the occurrence of a double displacement mechanism in this two GT families, rather than the generally observed SNi mechanism.323,324 This suggests that despite the considerable divergence between WbbB (GT99) and KpsC (GT107), the presentation mode of Kdo in the active site is likely a key feature of the mechanism in both enzyme families. The evolutionary origins of these enzymes remain an intriguing area of research that encourages further study and exploration.323,324
The SNi mechanism is proposed to occur in several enzymes in the α-glucan metabolism, where the nucleophilic attack comes from different acceptors leading to other glucoside products. Examples of these enzymatic activities are (i) glycogen synthase GS (EC: 2.4.1.11 and EC: 2.4.1.21),325−327 where the attack of the OH4 group from the non-reducing end of α(1→4)-glucan elongates the glucan chain; (ii) the maltose 1-phosphate synthase GlgM (EC: 2.4.1.342;119 where the attack comes from the OH4 group from DG1P; (iii) the trehalose 6-phosphate synthase OtsA (EC: 2.4.1.15)328 and the trehalose synthase TreT (EC 2.4.1.245)271 where the attack comes from the OH1 group from either α-d-glucose or DG6P; and (iv) the sucrose synthase SUS (EC: 2.4.1.13),329 and sucrose-phosphate synthase SPS (EC 2.4.1.14),330 where the OH2 group attack comes from either d-fructose or F6P.
4.2.2. Maltose Transfer
A particular reaction for generating a new bond between two α-d-glucose moieties is catalyzed by the α(1→4)-glucan:maltose-1-phosphate maltosyltransferase GlgE (EC 2.4.99.16),211,331 a GH13 family enzyme246 which transfers maltose from maltose 1-phosphate to the non-reducing end of an α-glucan chain. This enzymatic reaction follows a SN2-type double displacement mechanism that retains the configuration of the α-glucose, and has experimentally been well supported at structural level.216,218,219,221 Specifically, in the first step, an aspartic acid residue acts as a nucleophile/base attacking the C1 position of maltose 1-phosphate while a second acidic residue donates a proton to the phosphate group to facilitate the release of the leaving group. This step ends in a transient covalent intermediate with the aspartate bound to C1, with the inversion of the anomeric configuration of the participating glucose moiety in maltose. In a second step, the OH group from the C4 non-reducing end of the glucan acceptor attacks the C1 generating the new glycosidic linkage. This second attack causes the scission of the covalent-intermediate bonding regenerating the nucleophile/base, leading to the second inversion of the glucose anomeric configuration, releasing an α(1→4)-glucan elongated with two glucose moieties (Figure 9F).
4.2.3. Glucose Transfer from Sucrose
Another mechanistic alternative for α-glucan elongation is presented by the so-called glucansucrases, a group of bacterial enzymes that use sucrose as a donor for α-glucan synthesis. Here the glycosyl transfer occurs through a concerted mechanism that involves sucrose hydrolysis generating a glucosyl-protein covalent intermediate, and the subsequent transfer of the glucose moiety to the acceptor.85,126,332,333 These enzymes belong to the GH13 and GH70 families, displaying a mechanism similar to hydrolase and transglycosylase reactions. A glutamic acid residue protonates the sucrose oxygen linkage assisting the sucrose glycosidic bond break, releasing a fructosyl group. An aspartic acid residue located on the opposite side of the sugar ring attacks the glucose C1 leading to the formation of a covalent glycosyl-enzyme intermediate (with transient change in the anomeric configuration of glucose). The intermediate is rapidly broken by the nucleophilic attack from an OH group that is activated by the glutamic acid residue, transferring the glucose while reversing the anomeric confirguration, an overall retaining mechanism.334 Typical activities include (i) amylosucrase (EC 2.4.1.4)243,335 that forms α(1→4)-glucan linkages; (ii) dextransucrases (EC 2.4.1.5;336,337 that produce α(1→6)-glucan linkages; and (iii) mixed mechanism enzymes as alternansucrases (E.C. 2.4.1.140)338 synthesizing α-glucans with alternating α(1→3) and α(1→6), α(1→4)-glycosidic bonds, and reuteransucrase) producing α(1→4) and α(1→6) linkages resulting in no heterogeneous structure with no repeating units339 (Figure 9F).
4.3. Glucosidic Bond Breakage
4.3.1. Glucosidic Bond Hydrolysis
Glycosidic bond hydrolysis can take place at the extremes or within the α-glucan chain, depending on the enzyme architecture and specificity. Interestingly, many of these enzymes rely on a common reaction involving a double displacement mechanism with the formation and hydrolysis of a covalent glucosyl-enzyme intermediate. Indeed, all the glycoside hydrolases discussed here are comprised in the GH13 family, displaying a retaining mechanism.340 In a first step, the glucose anomeric carbon undergoes a nucleophilic attack by a catalytic aspartate residue with the concomitant donation of a proton to the oxygen glycosyl linkage from a glutamic acid residue. The attack leads to the formation of the glucosyl-enzyme covalent intermediate displaying the inversion of the glucose anomeric center. In a second step, the intermediate undergoes the nucleophilic attack of the anomeric center by a water molecule activated by the glutamic acid residue resulting in an oxocarbenium state that transitions into the inversion of the anomeric center, releasing α-d-glucose.61,341 Enzymes displaying this mechanism include (i) α-amylases MalS, AmyA, AmyQ (EC 3.2.1.1),342−344 which hydrolyzes α(1→4)-glucan linkages; (ii) α(1→6)-glucosidases (EC 3.2.1.10, EC 3.2.1.20, EC 3.2.1.70);345,346 (iii) trehalose 6-phosphate hydrolase TreA/TreC (EC 3.2.1.93);347 (iv) TreY that releases trehalose from α(1→4)-glucan-α-d-glucanosyl-trehalose (EC 3.2.1.141);348 and (v) glycogen debranching enzyme GlgX that hydrolyzes α(1→6)-glucan linkages (EC 3.2.1.68)235 (Figure 9F).
4.3.2. Glucosidic-Phosphate Bond Hydrolysis
The removal of phosphoryl groups from trehalose 6-phosphate and sucrose 6-phosphate is the final step for synthesizing the corresponding disaccharides. These reactions are catalyzed by two enzymes belonging to the haloacid dehydrogenase (HAD) superfamily, (i) trehalose 6-phosphate phosphatase (EC: 3.1.3.12; OtsB or TPP)349−351 and (ii) sucrose 6-phosphate phosphatase (EC: 3.1.3.24, SPP).352,353 The scission of the phosphoryl group from the sugar relies on a divalent metal cation, Mg2+, for activity, which is coordinated to a pair of carboxylates group from aspartic residues. The catalytic mechanism was extensively studied using the phosphoserine phosphatase homologue as a model (PSP).354,355 PSP hydrolyses the phosphoryl group of phospho-l-serine to produce serine with the generation of a phosphor-enzyme intermediate, which is further hydrolized leading to the product release. An oxyanion hole comprising two aspartic acids and a lysine residue and the divalent metal cation Mg2+, stabilizes the negative charge of the deprotonated oxygen, while a third aspartic acid residue performs a nucleophilic attack into the 5′-phosphate group leading to a putative pentavalent transition state. Finally, in PSP, an aspartate residue assists in protonating the serine product, activating/deprotonating water that attacks the intermediate releasing phosphate. The dephosphorylation of the disaccharide phosphate mediated by TPP and SPP presumably follows the same mechanism, not only due to the strict conservation of the catalytic residues in the active site but also supported by single-point mutagenesis analysis349,352,356 (Figure 9D).
4.3.3. Phosphorolysis
A basic mechanism for breaking the glycosidic bonds of α(1→4)-glucan consists of phosphorolysis, exhibited by enzymes that present pyridoxal phosphate (PLP) as a covalently linked coenzyme in the active site, facilitating acid/base catalysis.198,357−359 This is the mechanism exhibited by bacterial glycogen phosphorylase (GP) and maltodextrin phosphorylase (MalP; EC 2.4.1.1), homologs of the classical eukaryote GPs.196 Specifically, an inorganic phosphate anion receives a proton from the PLP phosphate group. This exchange is stabilized by the coordination of PLP by protein basic groups. Concomitantly, the α(1→4)-glucan substrate interacts with the phosphate bringing the glycosidic bond close to the PLP, resulting in the transfer of a proton from the phosphate group to the oxygen linkage breaking the α(1→4)-glycosyl linkage generating a 4′-OH group nonreducing end of the leaving chain. As a result, a C1 carbocation intermediate is generated at the non-reducing end of the glucose moiety, which is attacked by the phosphate anion in a nucleophilic addition that produces the second product glucose 1-phosphate with retention of the α configuration. Importantly, this is a reversible reaction mechanism, that can perform an α(1→4)-glucan synthase activity acting as a glucose 1-phosphate-dependent glycosyltransferase in vitro(196,360) (Figure 9C).
In addition to the described mechanism, a group of enzymes of the GH13 family in bacteria display disaccharide phosphorylase activities.246 Specifically, some bacteria bear sucrose phosphorylases (EC 2.4.1.7),361,362 and the sucrose 6(F)-phosphate phosphorylase S6FP (EC 2.4.1.329).363 These enzymes also break down the glycosyl bond in a PLP-independent manner. Their phosphorolysis mechanism is similar to previously described hydrolytic enzymes, involving a double displacement mechanism but with Pi as an attacking group (Figure 9F).
4.4. Cut and Paste Enzymes
4.4.1. Trans-glycosylation
A group of enzymes of the GH13 and GH77 families have the ability to reprocess segments of the α-glucan chain.246,364 These enzymes display an α-glucan transferase activity based on the classic hydrolytic double inversion mechanism. After hydrolysis, the catalysis continues with the synthesis of a new glycosyl bond since the covalent intermediate is attacked by a OH group of an α-glucan acceptor, which is a better nucleophile than water. It is worth noting that hydrolysis can also compete. Therefore, these transglycosylation reactions present a kinetically controlled mechanism.365 Specifically, the first step of the reaction involves the nucleophilic attack of the anomeric carbon by an aspartic acid group, and the concomitant donation of a proton from a glutamic acid residue to the glycosidic oxygen leading to the formation of a glycosyl-enzyme intermediate. In the second step, the glutamate residue deprotonates an OH group of the α-glucan acceptor which attacks the anomeric carbon releasing the final product with retention of the anomeric configuration.341,366 Two enzymes display this mechanism: (i) the glycogen branching enzyme GlgB (EC 2.4.1.18)364 transfers a segment of an α-(1→4)-glucan chain to an internal OH6 group in the same or neighboring α-glucan chain, forming a new branch joined with an α(1→6)-glycosidic bond, and (ii) the amylomaltase 4-α-glucanotransferase MalQ (EC 2.4.1.25)341,366,367 that transfers an oligo α(1→4)-glucan to a new position in an acceptor, which may be glucose or a (1→4)-α-d-glucan (Figure 9F).
4.4.2. Isomerization
A special mechanism in saccharide remodeling involves the glycosyl-bond isomerization between glucose moieties. Two enzymes belonging to GH13 family246 display this capacity in the context of α-glucan metabolism: (i) maltooligosyl trehalose synthase TreY (EC 5.4.99.15)368 converts the glucosidic bond between the two last glucose residues of an α(1→4)-glucan chain into an α(1→1) bond, generating a non-reducing end, and (ii) trehalose synthase TreS (EC 5.4.99.16)232,369 catalyzes the conversion of the α(1→4) bond in maltose to an α(1→1) bond forming trehalose. Therefore, these enzymes are isomerases with intramolecular transferase activity. In the case of TreS, a reaction with 5-fluoroglycosyl fluorides leads to a covalent glycosyl-enzyme intermediate consistent with a two-step, double displacement mechanism.370 In the case of TreY, a pocket at the active site cavity recognizes the substrate enabling the rotation of the separated +1 glucose to form the α(1→1) bond368 (Figure 9F).
4.5. Maltoside Phosphorylation
Besides GlgM NDP-sugar-dependent synthesis of maltose 1-phosphate, some bacteria produce this donor by the action of a maltokinase (Mak/pep2, EC 2.7.1.175).229,371 Interestingly, in some organisms Mak form a complex with TreS, an oftentimes fused forming a unique protein Mak/TreS.213 Mak uses ATP as the main phosphate donor and the divalent metal cation Mg2+ for maximal enzyme activity. The OH1 group of maltose is oriented towards a catalytic aspartate residue, which act as a base to abstract the proton, attacking the γ-phosphate of ATP. A second aspartate residue positions the divalent metal cation, coordinating the ATP phosphate similarly to the aminoglycoside phosphotransferase (EC 2.7.1.95)372−376 (Figure 9E).
5. The Catalytic Folds Observed in Prokaryotic α-Glucan Processing Enzymes
The tertiary structure of the catalytic domains of enzymes involved in the α-glucan and disaccharide metabolism is substantially dominated by the α/β topology, mainly comprising Rossmann-like and TIM barrel folds. This aligns with the assumption that this structural class of globular proteins appeared early in evolution.377 A view on how metabolic pathways evolve conceptualizes survival of the fittest applies not only to genes, and to their encoded enzymes, but also their metabolites. This hypothesis proposes that the origin of new metabolites and new enzymes in a network arise from enzyme-metabolite coevolution, driving enzyme recruitment and pathway evolution.378 In this context, we present selected structural information from bacterial α-glucan and disaccharide processing enzymes. Structures are presented clustered by catalytic fold, enzymatic reaction, and metabolites to reflect underlying structural evolutive commonalities on the enzymatic pieces of the machinery handling the α-glucose moiety.
5.1. The Rossmann Fold
First described for the structure of lactate dehydrogenase (LDH),379 Rossmann folds (Figure 10) are widespread across the whole metabolism in all living organisms and are the most frequent protein fold in the Protein Data Bank.380 The Rossmann fold was identified by comparing the tertiary structure of LDH with other nucleotide-cofactor binding enzymes,379 revealing the arrangement between β-strands and α-helices with a common handedness for the conserved nucleotide-binding motif, suggesting a very distant divergent evolution or a convergence folding-to-function in these enzymes. Indeed, this fold was estimated present at a precellular life origin, according to measurements of evolutionary distance based on structural sequence aligned structures of nucleotide binding enzymes, including dehydrogenases, kinases, and flavodoxins.381 Currently, these views are still under debate as some structural analysis suggests these types of folds could have evolved independently multiple times into different Rossmann-like groups revealing convergent evolution.380 In contrast, other studies indicate the existence of an ancestral βαβ segment that comprises the core resulting to be the seed for the evolution of these enzymes.382
Figure 10.

Structure, catalysis, and regulation of NDP-glucose pyrophosphorylases. (A) Schematic representation of the secondary structure of AGPase showing bars representing α-helices and arrows as β-strands. The highlighted domains show the Rossmann fold catalytic domain (yellow background) and the C-terminal left-handed β-helix domain (orange). Arrows of the central β-sheet of the Rossmann fold domain are colored according to the key bar indicating the direction N-terminal to C-terminal. (B) Two views of the overall architecture of AGPase from E. coli as visualized by CryoEM (PDB 6R8U; grey).186 (C) Close views of the active sites of selected UGPases showing the superposition of complexes with ligands. Left panel, the structural superposition of the UGPase from Sphingomonas elodea in complex with glucose 1-phosphate (PDB 2UX8, grey)420 with that of Burkholderia ambifaria in complex with UTP (PDB 5VE7, yellow).668Right panel, the structural superposition of the UGPase from Corynebacteria glutamicum in complex with UDP-glucose and Mg2+ (PDB 2PA4, grey)422 with that of Acinetobacter baumanii in complex with UTP and pyrophosphate (PDB 6IKZ, yellow).669 (D) Structural superposition of two AGPase complexes from S. tuberosum with ATP (PDB 1YP3, grey)404 and ADP-glucose (PDB 1YP4, yellow).404 Equivalent residues shown in (C) and (D) are highlighted by colored arrows. (E) The allosteric regulatory mechanism of AGPases. Two views of a selected protomer of the tetrameric AGPase from E. coli in complex with the preferred allosteric positive regulator FBP, as observed by X-ray crystallography and CryoEM; N-terminal domain, yellow; C-terminal domain, orange). The ADP-glucose ligand was placed based on the AGPase complex from S. tuberosum (PDB 1YP4).404 The secondary structure elements involved in the regulatory mechanism, the SM (green) communicates the regulatory cleft providing an intra-protomer signal. The RL1 (pink) and RL2 loops (red) are displayed in open conformation. (F) Structural superposition of the preferred positive and negative allosteric regulators, FBP and AMP, respectively, as observed in the corresponding X-ray crystallography and CryoEM structures.186,188,408
The classical Rossmann fold topology comprises a three-layered α/β/α sandwich formed by consecutive βαβαβ super-secondary structures, in which β-strands assemble forming a central β-sheet, with strand order classical topology 321456.383 The βαβ segment is proposed as the minimal Rossmann-like motif where β-strands and α-helices are packed by lateral interactions connected by loops of variable size. The connecting loops between certain secondary structure elements comprised in the α/β/α sandwich topology and the central β-sheet contribute to build up a cleft that accommodates the active site. Some of these loops are key elements for the binding of phosphorylated nucleoside substrates in other enzymes including dehydrogenases, NTPases and kinases.384−386 Known as Phosphate-binding-loops (P-loops) and Glycine-rich-loops (G-loops),387 these structural elements contain conserved sequences so-called Walker-motifs, comprising the motif A, GxxGxGK[S/T] and motif B, DxxG where x is any residue and h correspond to a hydrophobic residue.384−386 Interestingly, the possibility of transferring the biochemical function by insertion of these loops in small designed proteins suggests these minimal elements emerged for nucleotide binding and catalysis, evolving later upon acquisition of higher sequence and structural complexity.388
5.1.1. Nucleotidyl-Transferases NDP-Sugar Pyrophosphorylases
NDP sugars in α-glucan metabolism are synthesized by a set of enzymes known as nucleotide-diphosphate-sugar pyrophosphorylases (NSPases), which are metal-dependent nucleotidyl-transferases comprising a catalytic Rossmann fold domain. Two NSPases are mainly implicated in the α-glucan and glucose disaccharide metabolism in bacteria, (i) AGPase (EC: 2.7.7.27) and UGPase (EC: 2.7.7.9), involved in the biosynthesis of ADP-glucose and UDP-glucose, respectively. Both nucleotide sugars differ in the spectrum of reactions they are involved in, with ADP-glucose being specific to α-glucan and some glucose disaccharides in bacteria, archaea, and plants;61 whereas UDP-glucose is the commonly used precursor of glycogen in heterotrophic eukaryotes, while also used in glycoprotein, glycolipid, cellulose, callose, sucrose, and trehalose synthesis.109,389 Interestingly, other NSPases have the ability of transferring cytidylyl- (EC: 2.7.7.33);390 guanidylyl-303 and thymidylyl groups (EC: 2.7.7.24),391 to glucose 1-phosphate revealing the centrality of nucleotide-activated glucose in the bacterial metabolism (Figures 10 and 11).
Figure 11.

Oligomeric states of NDP-glucose pyrophosphorylases. Selected structures highlight this family's common architecture, presenting from left to right a detail of the protomer and two orthogonal views of the oligomeric assembly. The reference protomer catalytic Rossmann-fold domain is shown in yellow, and when present, the C-terminal LβH domain is in orange. (A) The structure of the UGPase from Corynebacteria glutamicum in complex with magnesium and UDP-glucose highlighting the absence of a C-terminal domain and two views of the tetrameric architecture with other protomers in grey (PDB 2PA4).422 (B) The structure of E. coli AGPase in complex with AMP (PDB 6R8U)186 and ADP-glucose by superposition with AGPase from S. tuberosum (PDB 1YP4).404 On the right, the two views of the tetrameric architecture of EcAGPase show the other three protomers in grey, with the C-terminal domain in dark grey. (C) The archaeal structure of the Sulfurisphaera tokodaii glucose 1-phosphate thymidylyltransferase (PDB 5Z09)424 in complex with UTP and two views of the trimeric arrangement. Note the longer C-terminal LβH compared to the AGPase, and the difference in the participation in the oligomeric arrangement. (D) The structure of the human UGPase in complex with UDP-glucose (PDB 4R7P).418 Note the difference in the relative orientation of the C-terminal domain and its impact in the octameric oligomerization.
The first reported structure of a NSPase family member was that of the N-acetylglucosamine 1-phosphate uridyltransferase GlmU from E. coli, involved in peptidoglycan synthesis. GlmU protomer is a two-domain protein whose N-terminal domain corresponds to the signature NSPase fold while the C-terminal domain is a Left-Handed β-Helix (LβH).298 The NSPase central core domain consists of a Rossmann fold comprising seven strands mixed β-sheet (topology 3214657, underlined antiparallel) decorated by alternating α-helices. NSPase catalytic domains have been previously described as glycosyltransferase A-like (GT-A-like) domains, based on their structural similarity to other members of the GT-A superfamily,188 supporting a distant evolutionary relationship. Many NSPases display a C-terminal extension comprised by the LβH fold, belonging to the widespread β-Solenoid fold architecture, particularly the class flat triangular “T-type” cross-section associated with bacterial transferases.392 Specifically, the LβH is formed by the stacking of triangular coils comprising successive β-strand linear segments and β-arcs that change the main chain direction in ca. 60°.393 Stacked coils form a coordinated network of hydrogen bonds creating β-sheet walls resulting in a triangular prism shape. The internal amino acid side chains are packed to the central axis of the LβH, generating a compact hydrophobic core. Interestingly, the LβH domain of GlmU also comprises a glucosamine-1-phosphate acetyltransferase activity (EC 2.3.1.157) unique in the NSPase family.394 It is worth noting that the UDP-N-acetylglucosamine 3-O-acyltransferase LpxA from E. coli (EC: 2.3.1.129)395 also comprises an LβH domain involved in catalysis, highlighting the participation of such domains in nucleotide-sugar derivative synthesis. Importantly, in most NSPases the LβH domain presents a different orientation with respect to the GT-A like fold, enabling different forms of oligomerization. LβH oligomerization includes (i) top-to-top dimerization as observed in archeal GDP-mannose pyrophosphorylase,396 (ii) base-to-base dimerization allowing tetramerization as observed in the AGPase from E. coli,188 (iii) side-by-side parallel trimerization as in the case of GlmU from E. coli,298 and (iv) top-to-top dimerization allowing octamerization as in eukaryotic UGPases.397,398
5.1.1.1. ADP-Glucose Pyrophosphorylase
Most bacterial AGPases are characterized as homotetramers (α4), although some species have been reported to exhibit heterotetramers187,200,399−402 (Figure 11). In contrast, plant AGPases display heterotetrameric architectures built of small and large subunits (α2β2). However, in plant non-photosynthetic tissues, homotetrameric forms have also been reported.403 AGPase regulatory properties emerge from the communication across the tetramer;61,186,404,405 therefore, such variability in the AGPase architecture and subunits arrangement accounts for differential regulatory properties in different organisms, highlighting the prominent function as a control enzyme.
Several crystal structures of bacterial AGPases have been reported, including (i) AGPase from A. tumefaciens AGPase (AtAGPase) in complex with sulfate in the allosteric cleft or the allosteric activator pyruvate,405,406 (ii) AtAGPase mutant S72C,407 and (iii) EcAGPase in complex with the preferred physiological positive and negative allosteric regulators, FBP and AMP, respectively.188,408 More recently, the CryoEM structures of the EcAGPase in complex with FBP and AMP unveiled the allosteric regulatory mechanism of this paradigmatic model.186 It is worth noting that the crystal structure of a recombinant truncated homotetrameric form of the small subunit of the AGPase from Solanum tuberosum (StAGPase) was solved in its unliganded form, and in complex with either ATP (ATP in an off-catalytic position), or ADP-glucose and ADP, highlighting the binding of substrates and products into the active site.404 Interestingly, a sulfate ion was found in these structures in positions presumably related to the allosteric inhibitor, phosphate. The small subunit of StAGPase is evolutionarily related to the cyanobacterial enzyme, reflected by the high percentage of identity among the protomers.409
The N-terminal NSPase catalytic domain contains the active site where ATP and glucose 1-phosphate accommodate. The nucleotide moiety occupies the N-terminal region of the domain, being surrounded by three key loops involved in nucleotide binding, catalysis and allosteric regulation, the nucleotide-binding G-rich loop and the regulatory loops 1 and 2 (RL1 and RL2).188,404,410 The G-rich loop is part of an insertion composed of short secondary structure elements, the so-called sensory motif,188 that faces both the active and the regulatory sites, comprising key lysine residues involved in catalysis and allosteric regulation (Figure 10). The glucose 1-phosphate moiety binds to a deep pocket located in the C-terminal region of the NSPase domain.188,298,391,404,411,412 The C-terminal displays the triangular prism LβH domain arranged in close contact with the GT-A like catalytic domain.188
The AGPase protomers build into a physiological and functional homotetrameric structure that can be viewed as a dimer of dimers. The most important contribution to the dimer interface is the triangular base of the LβH prism of each protomer resulting in two antiparallel β sheets.188,186 The tetramer assembles mainly by interactions between the N-terminal GT-A-like domains from different dimers, showing a D2 symmetry. Four allosteric sites are located in the corresponding clefts between the GT-A-like and LβH domains of neighboring protomers from different dimers.186,188 Specifically, EcAGPase structures in complex with the preferred positive and negative physiological allosteric regulators, FBP and AMP, revealed that the corresponding binding sites partially overlap.186,188 The allosteric clefts display a conserved positively charged pocket where a phosphate group of FBP and AMP binds.404,405,407,408 As expected, certain residues located in the allosteric cleft, interact differently with the positive and negative regulators, such as Lys39 placed in the sensory motif and associated to the activation mechanism.203,400,410 In contrast, Arg130 located in a neighbor protomer enables a stacking interaction with the negative regulator AMP promoting the crosstalk with the RL2 loop in active site. Strikingly, CryoEM structures of EcAGPase in the presence of FBP and AMP, respectively, revealed the activation and inhibition signal transduction mechanisms. The allosteric regulators promote a conformational switch of the regulatory loop RL2, from an AMP-‘locked’ to an FBP-‘free’ state, modulating ATP binding and modulating the enzymatic activity of AGPase.186,188,408 Recently, the crystal structure of AtAGPase was solved in the presence of the positive regulator pyruvate.406 Although pyruvate is a weak activator by itself, it synergically enhances the FBP activation.201 The homotetrameric AtAGPase binds two molecules of pyruvate in a planar conformation. The pyruvate binding site is located in a fissure between two contiguous LβH domains of the same dimer.
5.1.1.2. UDP-Glucose Pyrophosphorylase
UDP-glucose was the first nucleotide sugar discovered and arguably the most extended glucose donor in all metabolic pathways.413,414 UGPases are present in all kingdoms of life.389 UGPase protomers comprise a canonical NSPase domain, with or without a C-terminal LβH domain, showing diverse forms of oligomerization, from monomers415,416 to octamers398,417,418 (Figures 10 and 11).
Bacterial UGPases, often referred to as GalU/GalF, have been extensively studied, with several structures reported including (i) UGPase from C. glutamicum in its unliganded form and in complex with UDP-glucose and Mg2+,419 (ii) UGPase from Sphingomonas elodea in complex with glucose 1-phosphate,420 (iii) UGPase from Helicobacter pylori in its unliganded form and in complex with UDP-glucose and Mg2+,421 (iv) UGPase from Burkholderia ambifaria in complex with UTP (PDB code 5VE7), and UGPase from E. coli in its unliganded form.422 Most bacterial UGPases display a NSPase domain and a long C-terminal α-helical hairpin instead of an LβH domain. Two protomers interact by the nucleotide-binding region of the catalytic domain, whereas the corresponding α-helical hairpins hooks each-other dimer, forming a D2 symmetry homotetramer. In contrast, the archeal glucose/glucosamine-1-phosphate uridylyltransferase from Sulfolobus tokodaii displays a C-terminal LβH by which the enzyme trimerizes similarly to GlmU.423,424 The catalytic site, including the G-rich loop and key catalytic residues, is essentially preserved among UGPases and AGPases, supporting a common mechanism. Major differences are observed in other loops, including the RL2 loop, whose equivalent in bacterial UGPases is shorter, reflecting its participation/specialization in the allosteric regulation mechanism of AGPases. The fact that certain NSPses comprise or not the LβH domain associated with the catalytic domain in different organisms might suggest that, during evolution, the common ancestor of these enzymes acquired the LβH domain before acquiring a high specificity for sugar residues and nucleotides.
5.1.2. The GT-B Fold Enzymes
GTs are classified in families based on the amino acid sequence similarities.314,425 GTs utilize a donor substrate containing a substituted phosphate leaving group. The most common sugar donor substrates are nucleotide-diphospho-sugars. There are marked contrasts between the diversity of three-dimensional fold observed for glycoside hydrolases with respect to the limited folds used by GTs. Specifically, only three major folds have been described for GTs, GT-A, GT-B, and GT-C, with some families adopt other unique folds.312,314,315,426,427 GT-A and GT-B comprise NDP-sugar-dependent GTs, the so-called Leloir GTs, consisting of two abutting or separated Rossmann fold domains, respectively.310,314 Leloir GTs have evolved and diversified outcoming enzymes each specifically selected to construct a particular glycosidic bond. This selection is driven by their specificity toward a defined nucleotide sugar donor scaffold and the acceptor. In the context of a particular sugar motif such as glucose, the pathway that the sugar follows is determined by the activating nucleotidic scaffold, which provides a means for its recognition by the GT, therefore partitioning the sugar motif into different areas of the metabolism. GT-B GTs are the only class present in the α-glucan and glucose disaccharide metabolism, following a retaining catalytic mechanism.313 Interestingly, phylogenetic analysis of the GT-B fold families highlights the retaining and inverting mechanisms, which agrees well with root separation in two phylogenetic branches, with few exceptions of inverting GT-B inside the retaining branch,428 thus supporting an ancestral relationship between mechanism and enzyme family.
The GT-B fold was first reported for the structure of the rabbit (Oryctolagus cuniculus) muscle glycogen phosphorylase (OcGP; EC: 2.4.1.1; GT-35).196 The structural arrangement of the two Rossmann fold domains generates an inter-domain cleft where the active site is located.429 The crystal and CryoEM structures of GT-B family members revealed that both domains display a high level of flexibility, essential for substrate recognition and catalysis61,189−191,430−432 (Figure 12). Although GPs reverse reactions exhibit a retaining mechanism, the main physiological function is phosphorolysis assisted by PLP as a covalently attached cofactor. Interestingly, the first sensu stricto reported NDP-sugar-dependent GT-B structure corresponds to the T4 bacteriophage β-glucosyltransferase BGT (EC: 2.4.1.27; GT-63) which transfers glucosidic residues to DNA through an inverting mechanism.433,434
Figure 12.

Structure and catalysis of GT-B fold. (A) Representation of the secondary structure of GT-B fold as a diagram showing bars representing α-helices and arrows as β-strands. The domains are highlighted, showing the N-terminal acceptor binding domain (yellow background) and the C-terminal nucleotide binding domain (orange background). For both Rossmann-fold domains, the strands of the central β-sheet are colored independently according to key bar indicating the direction N-terminal C-terminal. Note the C-terminal α-helix completes the N-terminal domain. (B) Two views of the architecture of A. tumefaciens glycogen synthase in complex with ADP (PDB 1RZU),189 displaying the classic GT-B architecture. (C) Open-to-close motion (indicated by a curve dashed arrow) as observed by the superposition of tructure of M. thermoresistibile OtsA apo and in complex with ADP and glucose 6-phosphate (PDB 5JIJ and 5JIO).440 Both structures central β-sheets are colored (yellow, N-terminal domain, orance, C-terminal domain), the C-terminal α-helix in red, and the rest in semitransparent. Both structures are superimposed on the C-terminal domain to highlight the relative movement of domains. (D) On the right side, the superposition of M. thermoresistibile OtsA as reference (PDB 5K44)440 with superimposed substrates ADP and glucose 6-phosphate (PDB 5JIO, ligands in black),440 ADP-glucose (PDB 5K41, ligand in green),440 and GDP-glucose (PDB 5K42, ligand semitransparent).440 Note the GDP-glucose nucleotide ring in a displaced position with respect the preferred substrate ADP-glucose. The left panel shows the reference structure MtOtsA in complex with trehalose 6-phosphate (PDB 5K44, ligand in pink)440 in the superposition with ADP (PDB 5JIO). (E) Open-to-close motion by the superposition of the structure of Halothermothrix orenii sucrose 6-phosphate synthase (SPS) in complex with sucrose 6-phosphate (PDB 2R68)436 and its close plant homolog from A. thaliana Sucrose Synthase (SuSy) in complex with the UDP-glucose breakdown products UDP and a d-glucosyl derived intermediate (PDB 3S28).446 The coloring is equivalent to C, but here both structures are superposed on the N-terminal domain. (F) In the right panel, the superposition of substrates using AtSuSy as reference in complex with UDP and fructose (PDB 3S27, ligands in pink),446 superposed ligands from breakdown product of the UDP-glucose, UDP and the glucosyl hydrolytic derivative (PDB 3S28),446 and the HoSPS in complex with fructose 6-phosphate (PDB 2R66, ligand in green).436 Note the similar positioning of the sugar substrate in both enzymes. The left panel shows the user the reference structure AtSuSy the superposition (PDB 3S28, ligands in black)446 and the superposition with the HoSPS in complex with the product sucrose 6-phosphate (PDB 2R68, ligands in white).436 Note the superposition of the fructose motif between both enzymes. (G) Open-to-close motion by the superposition of the structure of glycogen synthases from A. tumefaciens in complex with ADP (PDB 1RZU, ligand in black)189 and E. coli in complex with maltotriose and ADP (PDB 3CX4, ligands in white).455 The same coloring is used to depict the structures; here both structures are superposed on the C-terminal domain showing the sample placement of the nucleotide. Note the coincident position of the nucleotide. (H) In the right panel, the superposition of substrates using E. coli as reference GS in complex with ADP-glucose breakdown products ADP and a derived d-glucosyl intermediate (PDB 3GUH, ligands in pink),456 the complex with ADP and maltotriose (PDB 3CX4).455 The sugar product from ADP-glucose hydrolisis occupies the position suggesting the ready for transference to maltotriose. The left panel shows the use of the same reference structure (PDB 3GUH),456 the superposition of ligands from starch synthase from Cyanobacterium sp. CLg1 bound to ADP, and the glucan analog acarbose (PDB 6GNF, ligands in green),450 mimicking the product post glucosyl transfer. (I) Open-to-close motion by the superposition of the structure of E. coli MalP with cofactor PLP in complex with maltose and sulfate (PDB 1AHP, ligands black)431 and Corynebacterium callunae with PLP cofactor (PDB 2C4M, to be published, ligands black).670 Structures are superposed on the C-terminal domain showing the ovelapping placement of PLP. (J) In the right panel, the superposition of substrates using EcMalP as reference with cofactor PLP in complex with pentaoligosaccharide (PDB 1E4O, ligand color black)468 and in complex with pentaoligosaccharide (not shown) and phosphate (PDB 1L6I, ligands white).359 The phosphate is placed near the position for phosphorolysis to occur. The left panel shows the use of the same reference structure (PDB 1E4O),468 the superposition with EcMalP in complex with glucose 1-phosphate (PDB 1L5V, ligands in pink),359 and EcMalP in complex with maltotetraose (PDB 2AZD),469 showing the location of post reaction products. In panels D, F, H, and J, colored arrows indicate the conservation of conserved residues at the active site. In particular, residues implicated in catalysis are lysine (pink arrow), arginine (red arrow), and glutamic acid (grey arrow). Blue arrows point to conserved residues interacting with glucose residues.
GTs involved in the metabolism of α-glucans and glucose disaccharide are classified into five GT-B families, including the GT3, GT4, GT5, GT20, and GT35425 (Figure 12). A phylogenetic analysis of the GT-B fold landscape using Hidden Markov Models showed that these five families are clustered together, revealing a close ancestral relationship.435 More recently, a deep-learning based study revealed that GT families involved in the metabolism of α-glucan and glucose disaccharide comprise two clusters: (i) GT-B0, includes families GT4, GT5, and GT20, comprises disaccharide synthesis enzymes and the bacterial and plant GSs, and (ii) GT-B1, which consists of GT3 and GT35 enzymes, comprising heterotrophic eukaryote GSs and glucan phosphorylases.312
5.1.2.1. Maltose 1-Phosphate Synthase GlgM
The α-maltose 1-phosphate synthase GlgM belongs to the GT4 family (Figure 13A). As described earlier, GlgM uses ADP-glucose and glucose-1P as the main substrates (EC: 2.4.1.342).119 GlgM also uses UDP-glucose as a donor with glucose 1-phosphate. However, it is less efficient. Surprisingly, UDP-glucose is not used as a donor when glycogen is used as the acceptor, further restricting the ability of the enzyme to generate glycogen.119,224 The crystal structure of GlgM from M. smegmatis is determined in its unliganded form.233 GlgM comprises a homodimer in which three parallel α-helices from the N-terminal domain of one subunit interact in an antiparallel fashion with the equivalent helices in a noncrystallographic two-fold-related subunit to give a six-helix bundle. The structural comparison of GlgM with bacterial GSs (GT-5) revealed that the C-terminal domains architecture are essentially preserved, with several residues participating in ADP-glucose binding essentially conserved. In contrast, the N-terminal domains show more significant differences in secondary structure. The central parallel β-sheet is two strands shorter in GlgM relative to the bacterial GlgA GSs, a characteristic also observed in other GT4 family members including sucrose phosphate synthase.233,436 The fact that GlgM is mostly present in Actinobacteria,119 and not present in other GlgE containing bacteria such as Chlamydiales,222 raises the question about the evolutive origin of GlgM, either by speciation from other GT-B disaccharide synthase or a reduction of function from an ancestral GS.
Figure 13.

Oligomeric states of GT-B fold enzymes. A gallery of structures and oligomeric states of the GT-B. Selected structures highlight the typical architecture of GT-B enzymes presenting from left to right a detail of the protomer and a view of the oligomeric assembly. In the top of each panel, the reference protomer structure is presented colored in yellow for the N-terminal domain and orange for the C-terminal domain, except for the C-terminal α-helix shown in red. Accessory domains are depicted in different colors. On the bottom of the panel, the oligomeric arrangement with the rest of the protomers is shown in different grey shades. Symmetry axes are shown (oval shape two-fold symmetry, and triangle three-fold symmetry). (A) Structure of M. smegmatis α-maltose 1-phosphate synthase GlgM and it dimer highlighting the N-terminal domain oligomerization interaction (PDB 6TVP, GT-4).233 (B) Two views of the monomeric structure of H. orenii Sucrose Phosphate Synthase (SPS) in complex with sucrose 6-phosphate (PDB 2R68, GT-4).436 (C) N. europaea Sucrose Synthase SuSy tetramer (PDB 4RBN, GT-4).444 Note the oligomerization role of the two N-terminal domains (highlighted in light green and light blue) and the N-terminal domain. (D) Archaeal P. horikoshii trehalose synthase TreT and its dimer presenting a C-terminal interaction (PDB 2XA2, GT-4).269 (E) Structure of Burkholderia xenovorans OtsA tetramer (PDB 5VOT, to be published, GT-20).671 The tetrameric oligomerization involves both N- and C-terminal domains. (G) Structure of the A. tumefaciens GS (PDB 1RZU, GT-5).189 The dimeric form appears in the crystal structure, possibly representing a physiological assembly. F. Archeal P. abyssi GS and trimeric assembly mediated by N-terminal interactions (PDB 3FRO, GT-5).458 (H) Eukariotic S. cerevisiae GS in basal state in complex with UDP and tetramer (PDB 3O3C, GT-3).432 Note the long α-helix at the C-terminal domain providing interaction for the oligomerization. (I) EcMalP an its dimer (PDB 1AHP, GT-35),431 the oligomerization mediated by N-terminal with an α-helical insertion playing a prominent role. (J) Eukaryotic O. cuniculus GP (PDB 9GPB, GT-35).672 Note the high similitude between EcMalP protomer and dimerization with the OcGP tetramer when considered as dimers of dimers.
5.1.2.2. Trehalose 6-Phosphate Synthase OtsA and Trehalose Synthase TreT
Two groups of bacterial GT-B enzymes account for the synthesis of the α,α-trehalose scaffold using either glucose 6-phosphate or α-glucose as acceptors, the so-called trehalose 6-phosphate synthase (T6PS; OstA) or trehalose synthase (TS; TreT), respectively (Figure 12 and 13).
T6PSs belong to the GT20 family, comprising two activities, UDP-glucose-dependent (EC 2.4.1.15) and ADP-glucose-dependent enzymes (EC: 2.4.1.347). T6PSs have been extensively studied, with several structures reported including (i) OtsA from E. coli in its unliganded form and in complex with either UDP and glucose 6-phosphate, UDP-glucose, UDP-2-deoxy-2-fluoroglucose, or a glycomimetic inhibitor,437−439 (ii) OtsA from S. venezuelae in its unliganded form, a GDP-glucose dependent enzyme,303 and (iii) OtsA from Mycobacterium thermoresistibile in its unliganded form and in complex with either ADP and glucose 6-phosphate, ADP-glucose, GDP-glucose, glucose 6-phosphate, trehalose 6-phosphate, trehalose, or ADP and fructose 6-phosphate.440 Interestingly, thermophilic bacteria and archaea TreTs (EC: 2.4.1.245) belong to the GT4 family and use several NDP-glucose donors with similar efficiency, including ADP-glucose, UDP-glucose, and GDP-glucose.441,442 Two archeal TreT three dimensional structures have been experimentally determined by X-ray crystallography, including (i) TreT from P. horikoshii in its unliganded form and in complex with either UDP, or UDP-glucose,269 and (ii) TreT from Thermoproteus uzoniensis in complex with either UDP-glucose or trehalose.443
Although OtsA and TreT show a conserved GT-B fold, comparison of the individual protomers highlights important structural differences due mainly to the presence of additional β-strands flanking the central β-sheet and insertions in the N-terminal domain of the GT20 family, regions involved in OtsA tetramerization. In addition, OtsA from M. thermoresistibile complexes with ADP-glucose or GDP-glucose highlight subtle differences in the binding site that explains the preference for ADP-glucose.440 Interestingly, three single-point mutations allow the conversion of the donor substrate preference to UDP-glucose.
5.1.2.3. Sucrose 6-Phosphate Synthase and Sucrose Synthase
There are two highly related enzymes implicated in the synthesis of sucrose in bacteria, the sucrose synthase (SuSy; EC 2.4.1.13) and the sucrose-phosphate synthase (SPS; EC: 2.4.1.14), both belonging to the GT4 family (Figures 12 and 13). There are several three dimensional structures experimentally determined up to date, including (i) SuSy from the chemolithoautotroph Nitrosomonas europaea in its unliganded form,444 (ii) the halophilic anaerobic bacteria Halothermothrix orenii SPS in its unliganded form and in complex with fructose 6-phosphate,436 and (iii) the SPS from the cyanobacteria Thermosynechococcus elongatus in its unliganded form and in complex with hydrolyzed UDP-glucose.445 In addition, the homologous plant enzyme SuSy-1 from Arabidopsis thaliana was reported in complexes with UDP and fructose.446 It is worth noting that SuSy enzymes can use sucrose to reversibly produce the nucleotide-sugar de novo, linked to starch biosynthesis in heterotrophic tissues of plants,447 and other polysaccharides in cyanobacteria.329 Interestingly, cyanobacteria SPSs can use UDP-glucose and ADP-glucose as substrates.47,448
Most of the reported structures display a single canonical GT-B fold; nevertheless, SuSys form A. thaliana and N. europaea display two additional contiguous N-terminal domains, so-called sucrose synthase (SSN). Notably, the SSN-1 accessory domain comprises α-helices and four β-strands whereas SSN2 is formed by a five α-helices bundle, involved in enzyme tetramerization. Besides these differences, the structural superposition of all SuSy and SPS enzymes discloses a common binding site to accommodate the substrates sucrose and NDP-glucose.
5.1.2.4. Bacterial Glycogen Synthase
Compared to disaccharide synthases, GSs are more sophisticated enzymes capable of processive catalysis by associating with the nascent α-glucan and performing rounds of glycosyl-transferand α(1→4)-glucan polymerization.109,449 Prokaryotic GSs (GlgA) belong to the GT5 family and comprise three subgroups according to the nucleotide-sugar donor specificity, including (i) UDP-glucose (EC: 2.4.1.11), (ii) ADP-glucose (EC: 2.4.1.21), and (iii) UDP-glucose/ADP-glucose (EC: 2.4.1.11/EC: 2.4.1.21). In addition, plant GSs, so-called starch synthases (EC: 2.4.1.21), are also classified in the GT5 family, resembling bacterial GS, particularly close to cyanobacterial GSs.450 On the other hand, heterotrophic eukaryote GSs (EC: 2.4.1.11) are classified into the GT3 family, preserving the overall architecture of the catalytic core when compared with the bacterial and plant homologues, but displaying important insertions of secondary structural elements.61,192,432,451 This architecture enables the necessary conformational changes for allosteric regulation, such as the activation by glucose 6-phosphate and inhibition by covalent phosphorylation109,452 also facilitating the interaction with glycogenin190,191,453,454 (Figures 12 and 13).
Several bacterial three dimensional structures of full-length GSs have been experimentally determined by X-ray crystallography, revealing catalytic and substrate specificity aspects, including (i) GS from A. tumefaciens in its unliganded form and in complex with ADP,189 (ii) GS from E. coli in complex with ADP and glucose, ADP, and oligosaccharides.455,456 In addition, the granule bound starch synthase CLg1GBSS from Cyanobacterium sp. was reported in complex with ADP and acarbose.450 Furthermore, crystal structures of archeal GS from Pyrococcus abyssi were solved in its unliganded form and in complex with 1,5-anhydro-d-fructose, and bound to an α(1→4)-glucan in a non-catalytic glycogen-binding site.449,457,458 There is limited information about the oligomeric state of bacterial GSs, with early reports pointing to the existence of GS from E. coli as dimers, trimers, and tetramers.459 Nevertheless, reported structures on GS from E. coli revealed only monomeric forms, whereas the asymmetric crystal unit points to a possible dimer of GS from A. tumefaciens. Interestingly, crystal structures and electron microscopy studies on archeal GS from P. abyssi revealed functional trimerization mediated by the N-terminal domains.457 Heterotrophic eukaryote GSs of the GT3 family revealed additional α-helices in the GT-B fold core allowing functional tetramerization.61,109,432 All structural studies on GSs from the GT3 and GT5 families support the occurrence of an open-to-close movement critical during catalysis and regulatory mechanisms.61,189−192,432,451,454,456
5.1.2.5. Glycogen Phosphorylase and Maltodextrine Phosphorylase
From its elucidation,460,461 eukaryotic glycogen phosphorylase (GP) is one of the most studied enzymes with ca. 250 structures currently deposited in the Protein Data Bank (PDB; https://www.rcsb.org/). Nevertheless, these enzymes, belonging to the GT35 family, have been structurally studied to a lesser extent in the case of prokaryotes. There are two homologous α(1→4)-glucan phosphorylases in bacteria: (i) the maltose-inducible maltodextrin phosphorylase MalP, dedicated to the processing of environmental extracellular α-glucans as a source of carbon and energy and having a reduced activity against glycogen,462−464 and (ii) the glycogen phosphorylase GP (GlgP) specialized in the recovery of glucose from intracellular glycogen storages.462,465,466
The structural analysis of these bacterial enzymes relies mostly on the reported crystal structures of MalP from E. coli due to the lack of a bonafide experimentally determined bacterial GP structure. The crystal structures comprise (i) MalP from E. coli in complex with either maltose, acarbose, tetra- or penta-thio-oligosaccharides and phosphate, glucose 1-phosphate, glucose 1-phosphate, and maltopentaose;359,431,467-468,469 (ii) the starch phosphorylase from Corynebacterium callunae (PDB code, 2C4M, to be published); and (iii) the α(1→4)-glucan phosphorylase from S. mutans. The typical two Rossmann-fold domains GT-B architecture comprises a central catalytic cleft with the C-terminal domain containing the covalently attached PLP prosthetic group site198,359,431,467−469 (Figures 12 and 13).
As in other GT-B enzymes, the active site is located in a deep cleft between the N- and C-terminal domains.198,431 PLP lies covalently attached to C-terminal domain lysine residue facing the catalytic center (K646 in MalP from E. coli), whereas the α(1→4)-glucan substrate binds mainly to the N-terminal domain, which in the ‘close’ conformation forms a channel between both domains.359 This channel imposes a minimum length for the linear oligosaccharide to reach the catalytic center, correlating with the reduction of glycogen chains up to four glucose residues from the branch point.359 In MalP from E. coli, three highly conserved lysine residues, K534, K540, and K554, coordinate the PLP phosphate group and the arrangement of the phosphate moiety toward the reactive glycosidic bond. All the structures on MalP from E. coli support a model in which substrate binding and product release require the occurrence of open-to-close motion.198,359,431,467−469
5.1.3. Rossmann-like HAD Domain Disaccharide Phosphatases
Haloacid dehydrogenase superfamily (HAD) comprises a diverse group of enzymes that catalyze the cleavage of substrate C-Cl, P-C, and P-OP bonds, including different substrate specificities and activities, such hydrolases, ATPases, isomerases, transferases, and different types of phosphatases.470 All HAD enzymes comprise (i) a catalytic core Rossmann-like fold domain comprising a central parallel β-sheet (topology 32145) surrounded by α-helices354,355 and (ii) a small CAP domain responsible for the diversification of substrate specificity within the family.471 The two domains face each other and are separated by a cleft, with the enzyme adopting both open and closed forms that facilitate substrate recognition and product release472 (Figure 14).
Figure 14.

Structure and catalysis of HAD-fold phosphatases. (A) The secondary structure of HAD phosphatases is represented as a diagram showing bars representing α-helices and arrows as β-strands. The domains are highlighted to show the N- and C-terminal Rossmann-like folds, in yellow and orange background correspondingly. Arrows of the central β-sheet are colored according to the color key bar indicating the direction N-terminal-to-C-terminal. (B) Two views of the architecture of the Synechocystis sp. sucrose phosphatase (SPP) (PDB 1S2O)352 presenting the canonical HAD phosphatase two domain arrangement, where the β-sheets of both Rossmann-like folds are independently colored from the direction N-terminal C-terminal. (C) The structure of S. typhimurium trehalose 6-phosphate phosphatase (StT6PP) in complex with trehalose 6-sulfate (Tre6S) and Mg2+ showing the classical two domain architecture of the catalytic fold HAD phosphatases. (D) Structure of the Synechocystis sp. sucrose-phosphatase (SPP) in complex with sucrose 6-phosphate (PDB 1U2T).352 (E) Structure of the trehalose 6-phosphate phosphatase (OtsB2) from M. tuberculosis (PDB 5GVX),349 showing the N-terminal accessory domain in light blue. Note the strictly conserved architecture of the StT6PP and OtsB2 catalytic fold and the subtle differences with SPP. (F) The open-to-close motion of the HAD domain phosphatases (indicated by a curve dashed arrow) as observed by the superposition of the structure of SeT6PP in complex with Tre6S and Mg2+ (PDB 6UPC)350 and the T. acidophilum T6PPb in complex with Mg2+ (PDB 1U02).473 Both structures have been superposed by their N-terminal domain β-sheets (yellow). (G) In the left panel, detail of the active site of StT6PP in complex with the substrate analog Tre6S and Mg2+ (PDB 6UPC)350 representing the coordination of the substrate pre-reaction. In the right panel, StT6PP in complex with the product trehalose (PDB 6UPC).350 (H) Similar to F, the open-to-close motion by superposition of Synechocystis sp. SPP. F. in complex with Mg2+ (PDB 1S2O)352 and in complex with sucrose 6-phosphate (PDB 1U2T).352 (I) In the left panel, detail of the active site of Synechocystis sp. SPP in complex with the substrate sucrose 6-phosphate (PDB 1U2T), and in the right panel with the product sucrose (PDB 1TJ4).352 In F and H, conserved residues interacting with substrates and implicated in catalysis are indicated by corresponding colored arrows.
5.1.3.1. Trehalose 6-phosphate phosphatase OtsB2
Several crystal structures of bacterial trehalose 6-phosphate phosphatases (T6PP; OtsB2) provide insights into substrate specificity and catalytic mechanism of trehalose dephosphorylation, including (i) T6PP from Salmonella enterica in its unliganded form and in complex with either Mg2+, trehalose 6-sulfate and Mg2+, and trehalose and Mg2+,350 (ii) T6PP from Burkholderia pseudomallei in complex with Mg2+,351 (iii) T6PP from M. tuberculosis in complex with trehalose 6-sulfate and Mg2+,349 and (iv) an archeal T6PP from T. acidophilum in complex with glycerol and Mg2+ (Figure 14).473 The HAD-like domain displays the Rossmann-like domain with a central β-sheet which is extended with a β-hairpin motif forming a β-meander before the connection with the CAP-domain. The CAP domain shows a characteristic 2-layer sandwich α–β plaits. Interestingly, OtsB2 from M. tuberculosis comprises an extra N-terminal domain connected to the HAD-like domain by a long loop, remaining adjacent to the active site.349 The N-terminal domain truncation results in 70% loss of the enzymatic activity. T6PP complexes with trehalose 6-sulfate and trehalose show the disaccharide bound into the cleft in an extended conformation, oriented by charged residues from both the HAD-like and the CAP domains.349,350 As presented before in Section 4.3.3, the catalytic mechanism of TPP, as other HAD phosphatases, involves the formation of a covalent enzyme-substrate intermediate through the nucleophilic attack of a conserved aspartate, assisted by a lysine, on the phosphorus atom from the substrate. This nucleophilic residue belongs to long loop emerging from the HAD-domain first β-strand contains a DxDx(T/V) motif participating in the coordination of the divalent metal cation Mg2+. Moreover, the active site, resembling that of other HAD phosphatases, includes other two aspartic acids which coordinate Mg2+ for dephosphorylation.349
5.1.3.2. Sucrose 6-phosphate phosphatase
To date, the sucrose 6-phosphate phosphatase (S6PP) from cyanobacterial Synechocystis sp. is the only reported enzyme of this class.352,474 This enzyme has been extensively studied including experimental structural data of its unliganded form and complexes with sucrose, sucrose 6-phosphate, sucrose and phosphate, glucose, trehalose, cellobiose, and maltose.352,474 S6PPs share the structural fold observed for T6PPs, showing a high degree of similarity with the corresponding HAD-like and CAP domains. Nevertheless, subtle changes can be observed including a short α-helix in the transition between the HAD-like and CAP domains that replaces the last β-sheet in T6PPs. S6PPs present a shorter loop emerging from the HAD-domain first β-strand that account for a more open cleft, compared to T6PPs. The S6PP complexes with sucrose and sucrose 6-phosphate indicate that this loop is not required to position the disaccharide into the substrate binding site since the sucrose arranges in a different orientation when compared with trehalose 6-phosphate in T6PP.352,474 The glucose moiety of sucrose binds buried into the cleft plays a critical role in substrate recognition and specificity. Supporting this notion, the affinity for the glucose moiety accounts for inhibition by sucrose and glucose.352,474 The structural and biochemical data support a common catalytic mechanism for S6PPs and T6PPs mediated by the nucleophilic attack of the substrate to form a covalent phospho-enzyme intermediate.
5.3.4. Maltokinase Mak/Pep2
Currently, several crystal structures of the mycobacterial enzyme have been reported, including Mak/Pep2 from M. tuberculosis in its apo form and in complex with maltose,375 Mak/Pep2 from Mycobacterium vanbaalenii in its apo form, and in complex with ATP, and the non-hydrolizable ATP analog AppCp,228 and Mak/Pep2 from M. smegmatis, as a component of the Mak/Pep2-TreS complex.232 The structure of Mak/Pep2 is curious in the context of other enzyme folds participating in the α-glucan metabolism. Mak/Pep2 resembles the bacterial 5-methylthioribose kinase, despite low global amino acid sequence identity,228 displaying many conserved structural motifs associated with nucleotide binding and enzymatic activity.228,475
The mycobacterial Mak/Pep2 structures reveal a two-domain fold architecture comprising the N-terminal CAP subdomain, containing three antiparallel β-strands that form a curved β-sheet and enclosing the N-terminal α-helix and a β-hairpin. A second intermediate subdomain contains a central seven-stranded antiparallel β-sheet having two lateral α-helical segments. Finally, the C-terminal domain is composed of two central four α-helical bundles, a β-hairpin, and a two-stranded β-sheets375,228 (Figure 15). Both N- and C-terminal domains remain strongly bound by salt bridges and hydrogen bonds. The active site of Mak/Pep2 enzymes is located within a deep pocket between the intermediate subdomain and the C-terminal domain. Reported structural complexes in the presence of ATP and non-hydrolizable analogs clearly define the site where the adenine group locates. Importantly, a strictly conserved Lys149 interacts directly with the α-phosphate, and the γ-phosphate is hold by the so-called ‘P-loop’ residue Ser136. Mg2+ sites comprise side chains of Gln310 and Asp322, making bridges with ATP phosphates. On the other hand, maltose accommodates mainly into a pocket located in the C-terminal domain.375
Figure 15.

Structure and catalysis of Maltokinase. (A) Representation of the secondary structure of Maltokinase showing bars representing α-helices and arrows as β-strands. The background colors indicate the CAP domain (blue), the intermediate domain (yellow), and the C-terminal domain (orange). Arrows of the central β-sheet and the C-terminal α-helices are colored according to the color key bar indicating the direction N- to C-terminal. (B) Two views of the architecture of MaK/Pep2 from M. vanbaalenii. The secondary structure elements are colored according to the scheme shown in panel A (PDB 4U98).228 (C) Comparison of the maltokinase protomer by superposition of MvMak in complex with ATP analog AppCp (PDB 4U98, colors)228 and M. tuberculosis MaK (PDB 4O7P, grey)375 showing the alternative intermediate domain β-sheet difference (red arrow) between monomeric and dimeric maltokinases. (D) Structure of the dimer of MtMaK complexed with maltose presents the reference protomer in colors and the second protomer in grey (PDB 4O7P).375 The intermediate domain swapped β-sheet is indicated with the arrow. (E) Detail of the active site by superposition of MvMak in complex with ATP analog AppCp (PDB 4U98)228 and maltose from MtMaK complex (PDB 4O7P).375 (F) Two views of the octameric complex of M. smegmatis TreS with Mak showing the central TreS tetramer in grey. (G) Detail of the MsTreS-Mak complex highlighting the active site of both enzymes located on distant protomers reveals the impossibility of substrate channeling between sites.
The N-terminal CAP subdomain presents large B-factors, suggesting conformational flexibility.228 The possible structural/biological function of this subdomain is regulation as well as representing an anchoring point to TreS.231,232 It is worth noting that Mak/Pep2 and TreS form a hetero-octameric complex.231,232 Although the crystal structure of the Mak/Pep2:TreS complex appears to rule out substrate channeling between the corresponding active sites,231 the structural arrangement possibly enhances the enzymes recruitment facilitating the succession of isomerization (TreS) and phosphorylation (Mak/Pep2) reactions to transform trehalose into maltose 1-phosphate. In addition, formation of the complex may provide a mean for allosteric effects, as observed for similar complexes.232,476 Specifically, the Mak/Pep2:TreS complex has an overall diamond-shape, where the TreS tetramer forms the core. Each TreS protomer pairs with a Pep2/Mak protomers at the apices of the tetramer resulting in a 4 + 4 configuration,232 mainly mediated by interactions with the C-terminal domain of TreS and Mak/Pep2 N-terminal CAP subdomain. Interestingly, both enzymes are part of a single polypeptide in some members of the phylum Chlamydiae,222 although presenting a monomeric form might present a similar relative arrangement.
5.2. TIM Barrel Fold Enzymes (TBF) and α-Glucan Metabolism
The TIM-barrel is a versatile scaffold present in many enzymes involved in pathways of the central metabolism.477 TIM is an acronym for the glycolytic enzyme triose-phosphate isomerase, the first reported crystal structure for the scaffold.478 TIMs are α/β proteins displaying an (α/β)8 topology. Specifically, the central barrel core comprises eight parallel β-strands. Each β-strand interacts with a corresponding α-helix in antiparallel orientation surrounding the barrel core. Overall, α-helices and β-strands display an angle with the β-barrel central axis,479 whereas the overall fold shows a degree of elliptical shape.480
TIM barrel fold (TBF) structures revealed a toroidal shape with pseudo-C8-fold symmetry that defines two faces, where the catalytic face is oriented at the C-terminal end of the β-strands.479 As a result of the overall directionality of the β-barrel and α-helices, TIM barrel domains have a global dipole across the toroid, although it does not correlate with the location of the active site.481,482 In addition to the important amino acid sequence differences between TBF enzymes, the overall shape dictates the conservation of intrinsic domain dynamics.483 Moreover, TBF enzyme families, such as GHs, exhibit preserved long-range electrostatic interactions among charged residues directed toward the barrel center and the interaction energy in the structural residue network.483 The clustering of TBF enzymes according to geometrical characteristics such as the length of helices and sheets and the location of the elliptical axes indicates that GH α-amylases (GH13) have commonalities with distant enzymes including glycolytic enzymes such as enolase and pyruvate kinase.480
As a result of the relevance of α-glucans in biology and the evolutionary versatility of these enzymes, TBF α-glucan processing enzymes present a large number of functions in terms of substrate specificity and α-glycosidic bond reactions.484,485 Several of these functions are markedly improved by accessory domains such as Carbohydrate Binding Modules (CBM), which themselves comprise a large diversity.486 These modules involved in the polymer binding contribute to enzyme-polymer heterogeneous catalysis, as stated by the Sabatier principle, meaning enzymes interact with substrates with intermediary strength, permitting binding association-dissociation kinetics are optimal to permit effective catalysis.487,488 The double inversion mechanism is central to all activities, and the reaction can be either under thermodynamic or kinetic control.246,313,340 For example, the comparison of debranching enzymes architecture and accessory domains belonging to very different TBF GH-fold families reveals considerable differences,489 whereas debranching/branching enzymes can be clustered inside the same GH subfamily with the seemingly same structure.235,490,491 Furthermore, it has been proven that subtle differences as a single point mutation can account for changes in function.492 Since function may hold for structural extreme differences, but nuances can also alter the outcome function, it is challenging to extract a rationale using a reductionist approach. With this in mind, in the following sections, we present the overall makeup of GH families’ structural organization for selected functions. Later, we present a parallel comparison of TBF-substrate complexes to highlight commonalities in enzyme-metabolite coevolution for these enzymes.378
5.2.1. TIM-Barrel α-Glucan GH13 Family
TBF enzymes classified into the GH13 family comprise the most significant number of specificities related to α-glucan metabolism.246,340,493 The GH13 family is one of the most studied from a structural point of view, and it is further subdivided into several subfamilies. Structures corresponding to selected functions associated to glycogen and disaccharide metabolism include (i) 4-α-glucanotransferase (EC 2.4.1.25);289 (ii) GlgE α(1→4)-glucan phosphate α-maltosyltransferase (EC 2.4.99.16);216−219,221,494 (iii) GlgB glycogen branching enzyme (EC 2.4.1.18); (iv) amylosucrase (EC 2.4.1.4),156,332,333,495−497 sucrose phosphorylase (EC 2.4.1.7);362,498−500 (v) sucrose 6-phosphate phosphorylase (EC 2.4.1.329);501 (vi) AmyA α-amylase (EC 3.2.1.1);502−505 (vii) GlgX glycogen debranching enzyme (EC:3.2.1.196);235 and (viii) malto-oligosyltrehalose trehalohydrolase TreZ (EC 3.2.1.141),348 trehalose synthase TreS (EC 5.4.99.16),231,369 and malto-oligosyltrehalose synthase TreY (EC 5.4.99.15).506
The canonical GH13 enzyme comprises a characteristic TBF (β/α)8 barrel domain (A) including the catalytic center of the enzyme and an accessory domain (B) inserted into the loop between the β3-strand and α3-helix of the barrel, whereas a third C-terminal domain (C) completes the enzyme core architecture.507,508 The GH13 presents a TBF with a linear order of secondary structures, with a topology N-β1-α1-β2-α2-β3-α3-β4-α4-β5-α5-β6-α6-β7-α7-β8-α8-C. The B-domain presents a variable structure and length, whereas the C domain is formed by a β-sandwich domain. In addition to the core fold, often one or two additional C-terminal domains, D and E, are present.493 The E domain is characterized as a starch-binding domain. These domains correlate with GH13 function, specificity, and taxonomy493 (Figure 16).
Figure 16.

Structure and catalysis of TIM barrel fold enzymes. (A) The secondary structure of TBF enzyme families is represented as a diagram showing bars representing α-helices and arrows as strands. From left to right, the scheme offers the four GH families present in the α-glucan metabolism, GH13, GH70, GH77, and GH57. Strands in the (α/β)8-barrel are highlighted in colors according to N- and C-terminal color key. For the GH13, accessory domains are shown as colored circles, including the N-terminal domain (blue), inserted β3-strand α3-helix domain B (cyan), and the C-terminal β-sandwich domain C (maroon). The GH70 architecture presents three associated domains formed by N-terminal and C-terminal not consecutive segments shown as semicircles called domain V (orange and red), domain IV (dark and bright yellow) and domain B (dark and light green), and domain C inserted between α6 and β6 (maroon). Note the alternative arrangement of (α/β)8 barrel starting with an α-helix instead of a β-strand as GH13. GH77 scheme shows an N-terminal domain (blue circle) before the catalytic domain, whereas the (β/α)8 barrel presents subdomains inserted. Subdomain B2 is indicated by a yellow circle, whereas the subdomains B1 (cyan) and B3 (pink) are formed by two semicircles. Note for the GH77 the lack of the C-terminal domain C compared to the GH13 family. The GH57 scheme shows the incomplete TBF (β/α)7 displaying several secondary elements extra associated with the catalytic fold. Note the β-strands colored with the same color scheme, presenting seven in the barrel and an antiparallel β-strand not belonging to the barrel (yellow). (B) From left to right, two views of the structure of a GH13 branching enzyme from Cyanothece sp. in complex with maltohexaose (PDB 5GQV),673 and in the left panel, a close-up of the TBF active site. The view shows domains colored according to the scheme presented in A. Note an extra N-terminal domain colored in black. The close-up of the GH13 catalytic site indicates the position of strand β1 as a reference, with the rest of the barrel strand numbering increasing counter-clockwise. Catalytic triad residues are indicated in emerging loops extensions in strand β4, aspartate nucleophile (magenta arrow), in β5 glutamate acid/base (pink arrow), and in β7 aspartic stabilizer (blue arrow). (C) Same as in A, but the structure of GH70 L. citreum dextransucrase DSR-M inactive mutant N-terminal truncated mutant E715Q in complex with isomaltotetraose (PDB 6HTV).512 The catalytic domain permutation places the catalytic triad in different C-terminal loop strands in the barrel. Note a glutamine indicated by the pink arrow appears in the position of the acid/base due to the corresponding mutation E715Q. (D) Similar for the structure of the GH77 E. coli amylomaltase MalQ in complex with the pseudo-heptasaccharide (PDB 4S3R).366 (E) Structure of the GH57 Thermococcus litoralis 4-α-glucanotransferase complexed with acarbose (PDB 1K1Y).522 From B to E, note the close-up of the left panel; the catalytic domain is oriented to place the bound polysaccharide in the same overall orientation.
5.2.2. The Alternative TBF GH70 Family
The GH70 family comprises a number of enzymes that use sucrose as a sugar donor to synthesize α-glucans, so-called glucansucrases (GSuc), oftentimes catalyzing more than one type of glucosyl transfer. Selected activities include (i) α(1→6)-dextransucrases (EC 2.4.1.5),509−512 α(1→3)/ α(1→6)-alternansucrase (EC 2.4.1.140),338,513 and the α(1→4)/α(1→6)-reuteransucrase (EC 2.4.1.-).514
GSuc enzymes are usually large extracellular proteins that consist of four different regions, including (i) a signal peptide, (ii) an N-terminal variable region (VR) containing other repeat units, (iii) the TBF catalytic domain, and (iv) a C-terminal glucan-binding domain (GBD) also comprising repeating units. GH70s have a characteristic circularly permuted TBF compared to the GH13 topology.126 The GH70 (α/β)8 barrel starts with an α-helix that corresponds to α3 in the GH13 (instead of starting in β1), in a relative order N-α3-β4-α4-β5-α5-β6-α6-β7-α7-β8-α8-β1-α1-β2-α2-β3-C. Nevertheless, these enzymes still present the same catalytic amino acid motifs of the GH13 family enzymes, with TBF sequence position accounting for the circular permutation. In addition, crystal structures of truncated GH70 GSuc enzymes reveal five domains in a U-shaped form, with equivalent A, B, and C domains as observed in the GH13 members making the catalytic core, and domains IV and V are unique to GH70 enzymes.126 Interestingly, domain B is composed of two discontinuous inserts and is next to domain A, while domain C is a single domain formed by the N- and C-termini and is involved in substrate binding. Domain IV is involved in oligomerization, whereas domain V is located at the C-terminus and is believed to be involved in glucan binding126 (Figure 16).
5.2.3. The TBF GH13-like GH77 Family
Reported bacterial crystal structures of the GH77 family members are limited to the 4-α-glucanotransferase (EC 2.4.1.25).341,366,515−517 The GH77 TBF has a similar topology to the GH13 (N-β1-α1-β2-α2-β3-α3-β4-α4-β5-α5-β6-α6-β7-α7-β8-α8-C), reflecting a high evolutionary connection.518 Nevertheless, the separated classification family is supported by the divergence in several structural factors.485,519 The core of the GH77 enzymes comprises the TBF with three insertions, resulting in the formation of three subdomains so-called B1, B2, and B3. The B1 and B3 subdomains correspond to domains B and C in the GH13 family. The B2 domain is unique to the GH77 family, possibly playing a role in substrate recognition. In addition, the B3 subdomain does not present the classic β-sandwich as in domain C of GH13 enzymes. This structural arrangement of accessory domains generates a narrower and deeper catalytic cleft compared to GH13 enzymes, where domains clamp the α-glucan substrates. Moreover, although the catalytic triad is conserved, some GH77 enzymes present differences in the residue environment at the catalytic site.520 Finally, some enzymes in this family present an additional N-terminal immunoglobulin-like fold domain presumably implicated in α-glucan binding519 (Figure 16).
5.2.4. The Incomplete TBF GH57 Family
The GH57 family comprises an expanding number of activities since the discovery of α-amylases with unrelated sequences to the GH13 family.274,521 There are a limited number of bacterial and archeal GH57 enzymes with known three dimensional structures associated to the α-glucan metabolism: the 4-α-glucanotransferase (EC 2.4.1.25)522 and the glycogen branching enzyme (EC 2.4.1.18).281,364 The reported enzymes comprise an N-terminal incomplete TBF, containing a short helix (often called subdomain B) emerging from β2, an intermediate five-helix domain, and a C-terminal β-super sandwich domain.523 The incomplete TBF has a (β/α)7 topology including a β-barrel with a distorted toroidal shape. The β6 does not participate in the barrel, being located in an antiparallel orientation with respect to β7. Therefore, the lack of a strand in the β-barrel generates a more open conformation because there are two gaps between β3 and β4 and between β7 and β8.523 As in the other GHs, the TBF C-terminal β-strands define the active site, displaying a more significant loop variability, including the insertion of additional secondary elements (Figure 16).
6. Polysaccharides at the Dawn of Life
How polymers metabolism has emerged is a central question in biology. In the case α-glucans, their ubiquitous presence in present biology suggests that they have deep evolutionary roots, possibly dating back to the earliest life forms or before. Miller and Urey, whose famous experiment supported the prebiotic chemical origin of life, proposed that polymerization may be catalyzed by adsorption on clay and mineral surfaces.524 Orgel and colleagues proposed abiotic polymerization of amino acids and nucleotides on mineral surfaces, suggesting that these rocky interphases could provide a “library” for molecular evolution.525,526 Polysaccharides may arise first among other polymers in prebiotic times, suggesting that they were present in a glycoworld from which the RNA world emerged.527,528 Furthermore, it has been speculated that high-energy poly-phosphates may be served as a scaffold for the assembly of major polymers,527,529 thus facilitating the condensation of the first polysaccharides.
The putative occurrence of neutral polysaccharides, such as glucans, might help in original phase separation in the primordial soup, allowing the concentration of other charged proto-biopolymers and coalescence of dispersed particles.528 Indeed, carbohydrate polymers may appear on prebiotic earth as a prerequisite for life’s subsequent development.527 Long and colleagues showed that dextrans can contribute in creating compartments with properties of synthetic cells.530 Although lessons and the latest advances from synthetic chemistry pin-point first principles for synthesizing α-glucans,531,532 it appears that further exploration of α-glucan prebiotic origin is needed, which potentially could offer fascinating views. The presumed existence of prebiotic glucose, as well as the demonstrated abiotic phosphorylation of glucose forming α-d-glucose 1-phosphate,533,534 opens the door to think about the fate of these sugars phosphates in abiotic times.
6.1. Extracellular Polysaccharides and Biofilms at the Origin of Living Cells
The evolution of organisms selected confirmed metabolic processes taking advantage of available compounds and synthesizing others that benefit survival under environmental pressure in a defined niche. α-Glucan metabolism provides a framework for understanding how physiological functions associated with cellular structure and energy storage exhibited in different bacterial lifestyles. Biofilms seem to be the most frequent lifestyle, with ca. 80% of bacterial and archaeal cells living in this form, which may serve as diversity incubators; planktonic lifestyle might be a secondary form.535,536 Biofilms provide functions such as harvest and access to nutrients, shelter protection against environmental stresses, water retention to maintain extracellular enzyme activities, social cooperation, and horizontal gene transfer.537,538 The occurrence of biofilm structures appears early in the fossil record (ca. 3.2 giga annum; Ga), a characteristic shared by prokaryote ‘living fossils’ present in hot springs and sea hydrothermal vents;539 bacteria and archaea lineages emerged at a similar time frame (ca. 3.4 Ga).540 Diverse glucans have been shown to form biofilms. β-Glucans, such as cellulose541,542 and β-(1→3)-glucans,543 are common biofilm-forming exopolysaccharides. α-Glucans119,544−548 are components of the extracellular matrix, capsules and biofilms, often linked to glycogen metabolism. Interestingly, hot spring-living Geobacillus tepidamans forms a glucan-containing biofilm,549 whereas Geobacillus sp. expresses an extracellular thermostable (4→6)-α-glucanotransferase.550 Moreover, biofilm-forming uncultivated SM1 Euryarchaeon isolated from subsurface biotopes of sulfidic springs can synthesize glycogen.551 Based on these pieces of evidence, the presence of α-glucans in ancestral biofilms can be conjectured.
6.2. Considerations on the Origin of Glycogen Metabolism in Bacteria
The emergence of metabolic systems and their evolution represents a central question in biochemistry and biology. Efforts are dedicated to reconstruct the metabolic make-up of the Last Universal Common Ancestor (LUCA), the hypothetical prokaryotic common organism antecedent to all existing life. Inferring gene repertoires arising billions of years ago is extraordinarily challenging since orthologous genes/proteins are subject to continuous vertical inheritance, gene loss, and horizontal gene transfer.552 Nevertheless, it could be argued that primitive α-glucan pathways appeared early in the gene makeup of organisms due to the importance of survival to bacteria.94 Supporting this notion, glycogen metabolism provides an advantage in enduring famine, resisting different abiotic stresses and helping in colonizing niches.82,95,173,544,553 The structure of bacterial glycogen with a small average chain degrades slowly and serves as a resilience mechanism, prolonging survival.101 Finally, glycogen plays a role in awakening bacteria from dormancy, a near-dead state.554 Therefore, glycogen metabolism could have played a role in the early stages of life evolution.
From the point of view of synthetic biology, it has been proposed that the construction of small bacterial genomes containing a minimal set of proteins deduced from existing genomes can reveal the essential set of survival genes compatible with cellular life,555 thus strongly suggesting a requisite gene repertoire in the LUCA. Nevertheless, the metabolic routes are strongly context-dependent, and it seems difficult to find protein-coding genes that are entirely conserved in all prokaryotic genomes.556 The minimal genome synthetic approach appears unsuitable for evaluating early glycogen metabolism since it is not essential in some bacteria.557,558 Interestingly, the presence of GBEs and GDEs in all kingdoms of life indicates glycogen metabolism is an early process;559 while GP has also been predicted to be part of the LUCA.560 Furthermore, meta-consensus analysis of metabolic pathways predicted early enzyme functions and properties that may exist in the core metabolism of early life forms, with several enzymes related to starch metabolism.561
The LUCA has been predicted/proposed to be an anaerobe.562−564 Several strict anaerobes are known to produce glycogen,565,566 while glycogen metabolism enzymes were identified in anaerobic hyperthermophile bacteria from the genera Thermotogales and Aquifex.567,568 The reconstruction of the tree of life also suggests that the LUCA was a thermophile,569 which is consistent with the hypothesis of the origin of life at hydrothermal vents.570Thermotogales and Aquifex are at the base of the bacterial clade571 and therefore are thought to resemble ancient bacteria. In addition, the anaerobic thermophilic archaea Methanocaldococcus jannaschii shares a metabolism similar to bacteria, comprising genes of the glycogen metabolism,572 supporting the hypothesis that α-glucan metabolism predates separation between archaea and bacteria. Interestingly, symbiosis between nanohaloarchaeon and haloarchaeon is based on utilization of different polysaccharides including glycogen.263 The giant tube-worm hydrothermal vents-living Riftia pachyptila, utilizes glycogen as the main anaerobic substrate during extended anaerobiosis and a key molecule in chemoautotrophic endosymbionts,573 suggesting glycogen plays relevant roles in extreme niches associated with life origin. Altogether, these views support glycogen metabolism as an ancient process.
6.3. The α-Glucan Pathways in the Light of Metabolism Evolution Models
From the putative LUCA metabolism, enzymatic networks have diversified into apparently disparate systems. Several models for the evolution of pathways attempt to explain the emergence of metabolic networks on prebiotic and ancestral cellular metabolism.574 In this regard, the α-glucan and glucose disaccharide metabolism present a framework that could be evaluated a priori by these models while subjected to a simple chemical scaffold. Hereof, we discuss the emergence of the α-glucan biosynthetic machinery based on these models (Figure 17).
Figure 17.

Evolutionary basis of α-glucan chemistry and metabolic pathway diversity. Schematics depict an exercise based on different aspects and models for the evolution of metabolic pathways onto α-glucan metabolism. (A) Hypothetical scenario for the divergent evolution of one gene into three enzymes with different functions. Considering organisms with a coding gene (grey arrow) for an enzyme responsible for processing an α-glucoside A into B. Due to gene cumulative mutations and selection by environmental pressure (in the same or different organisms) the enzyme gene diverges into three distinct gene/enzymes (red arrows and boxes), with a different active site suited for different cellular functions involving α-glucosyl bond processing. Over time, these mutations became fixed in the population, creating three distinct enzyme lineages. This case may reflect the diversification of the GH13 family into different functions. (B) Hypothetical case of convergent evolution, involving three different species of bacteria that live in similar environments where the processing of an α-glucan provides an adaptive advantage to survive. Each species had evolved independently and contained genes (grey arrows with different red hatching) that do not share a recent common ancestor, encoding enzymes responsible for processing unrelated α-glucosides. The selection of mutations in these three genes converges on enzymes with the same function. This case may reflect the convergence of enzymes from GH13, GH77, and GH57 families into enzymes 4-α-glucanotransferase (EC 2.4.1.25). (C) A speculative scenario of an anterograde evolution pathway581 in which an organism utilizes a branched α-glucans using a gene/enzyme (red arrow and box) that processes the substrate in linear chains to be able to harvest it. An event of gene duplication and posterior divergence results in a second gene/enzyme (pink arrow and box) that processes the linear α-glucan into maltose, providing an advantage due, i.e., cluster to the same operon for utilization of nutrients. The same process repeats to acquire a gene/enzyme (purple) to transform maltose into trehalose, reaching a more efficient way to produce the functional disaccharide. (D) Speculative scenario of retrograde evolution575 where an extracellular α-glucan is critical for survival, which can be produced by an organism gene/enzyme (red arrow and box) from oligosaccharides obtained from the environment. An undergoing depletion of the disaccharide generates the selection, under growing pressure, of a new gene/enzyme (pink arrow and box) originated by gene duplication and divergence, that can synthesize oligosaccharides from disaccharides available in the environment. The same process repeats to acquire a gene/enzyme (purple) to generate disaccharides from monosaccharides. (E) A hypothetical case of an ancestral gene encoding promiscuous enzyme (blue arrow and hexagon),582,583 i.e., an NSPase produces different sugar donors NDP-glucose. The enzyme through gene duplication and subsequent divergence evolves into three differentiated enzymes (light, medium, and dark blue arrow and hexagons) with higher specificity to synthesize ADP-glucose, UDP-glucose, and GDP-glucose. (F) The differentiation of promiscuous enzymes recruited in an ancestral pathway (blue hexagon, and yellow circle) under gene duplication and subsequent divergence can lead to the arrangement of specific enzymes into different pathways “patchwork”. (G) Hypothetical enzyme-substrate co-evolution involving an enzyme (yellow circle) that synthesizes a linear α-glucan in a pathway comprising a second enzyme (red box) to generate branched α-glucans. Eventual errors or promiscuity lead the first enzyme to generate the byproduct maltose 1-phosphate. In a second moment, the new metabolite scaffolds the evolution of a specific enzyme (salmon circle). A higher level of maltose 1-phosphate is removed by unspecific enzymes by oligomerization. In a third moment, the enzyme (pink box) is recruited and evolves to catalyze such reaction specifically.
The pioneer retrograde model proposed by Horowitz, based on the Oparine-Haldane “primordial soup” hypothesis,570 proposed the metabolic evolution of a rudimentary heterotrophic organism under significant selective pressure due to the depletion of the prebiotic compound supply.575 Under such circumstances, the primordial cells acquired mechanisms to synthesize functional molecules by assembling the enzymatic machinery in a retrograde manner, originating the metabolism that frees them from exogenous sources of such compounds. In the retrograde view, α-glucans metabolism could arise by harvesting hypothetical proto-glucan compounds as an energy source or making part of functional compartments/biofilm in primordial cell communities. Once depleted, selective pressure enforced the selection of synthetic machinery to obtain in a retrograde manner the enzymes to synthesize first the α-glucan polymers or disaccharides, then nucleotide sugars, and so on. Phylogenetic studies comparing E. coli enzymes and pathways universally present in all domains of life indicate that the catabolism of α-glucans is among the most conserved pathways with enzymes present in Archaea, Bacteria, and Eucarya.576 Moreover, the presence of an analogous enzymatic machinery to the glycogen synthesis for the usage of external α-glucan, such as the maltose/maltodextrin utilization system present in distant bacteria,464,577−579 could indicate that the evolution of the α-glucan metabolism first associated to external sources. The presumed LUCA containing α(1→4)-glucan-phosphorylases is part of the GT35 family (MalP/GP).560 The reversibility of these enzymes360,580 insinuates an ancient pivot function between synthesis and degradation. During evolution, PLP could be incorporated into a proto-GT-35 NDP-sugar-dependent enzyme, surrogating a nucleoside monophosphate, and resulting in MalP/GP.314 This appears as a sensible explanation in the context of almost the whole GT-B family using nucleotide-activated sugars.
Contrary to the retrograde model, Granick proposed that biochemical pathways extended in an anterograde fashion, acquiring successive steps in the metabolic pathway to produce a functional metabolite which provides a survival advantage.581 Exopolysaccharides may be originated from early communities to form a matrix of biofilms as a survival strategy, allowing colonization of surfaces. From this point of view, the metabolism could have extended from gluconeogenesis toward α-glucans synthesis, possibly supported by activities from the nucleotide and RNA metabolism based on meta-consensus enzyme functions (EC: 2.4.1.- and 2.7.7.-).561 The putative LUCA presents NSPases and related GT-2 enzymes, including UTP-glucose 1-phosphate thymidylyltransferase.560,563 Moreover, phylogenetic analysis highlights GT folds go back in evolution to the LUCA.428 Therefore, this view could support nucleotide-sugars and disaccharides first, instead of a retrograde view of glucans first.
In the evolution of pathways, innovations are the driving force for enzyme function specialization that permits the synthesis of new specific metabolites. Ycas proposed earlier enzymes were less specific (more promiscuous) to catalyze several related reactions with a smaller number of enzymes.582 Independently, and along the same reasoning, Jensen postulated a “Recruitment Hypothesis” in which pathway assembly occurs by recruitment of such primitive enzymes with a wide range of chemically related substrates, also known as the “Patchwork Hypothesis”.583 The evolution of an enzyme into diverse enzymes with specific traits arising from gene duplication of the ancestral enzyme can be viewed as functional specialization, or division of labor, an evolutionary mechanism observed from molecules to organisms.584 Promiscuity outcome includes a ‘Flexible Metabolome’, a concept that suggests genetic and metabolic pathways are cross-wired in diverse unpredicted forms, therefore inherently ambiguous and stochastic.585 Importantly, enzyme promiscuity appears widespread in modern α-glucan metabolism. NSPases and GT-B enzymes often present some degree of usage of alternative nucleotide triphosphate or nucleotide sugars, respectively. For example, (i) OtsA M. thermoresistibile can use ADP-glucose and GDP-glucose,440 and (ii) GS from P. abyssi can use UDP-glucose and ADP-glucose.457 Moreover, NSPase promiscuity has been reported in several bacterial enzymes, including the AGPase from Rhodococcus jostii that can use ATP, GTP, and CTP, as well as diverse hexose 1-phosphates.586 On the other hand, TIM barrel folds bear a large diversity of functions for α-glucan processing. In the case of GBEs and GDEs (GH13), both are related to gene duplication and paralogous.559 Furthermore, functional and phylogenetic studies show the majority of these enzymes classified into the GH13 family comprising 35 subfamilies likely monofunctional, while others appear polyspecific,246 which correlates with the promiscuity to specificity evolution. Moreover, the GH13 family is a member of the clan GH-H, which also contains GH70 (glucansucrases) and GH77 (amylomaltases) families, supporting the evolutionary relatedness of these enzymes.518
6.4. Structural Relationships between the Enzyme Folds Involved in α-Glucan Biosynthetic Pathways to Other Metabolic Pathways
The catalytic folds involved in α-glucan and glucose disaccharide synthesis are widespread in the other areas of the metabolism, which could be thought to represent distant evolutionary relationships or convergent evolution. Rossmann-like folds are thought to have evolved prior to LUCA from a primordial generic nucleotide-binding domain, which participates in five out of eight enzymatic reactions of the oldest metabolic Wood-Ljungdahl pathway.380,563 TIM barrels have been hypothesized to allow early evolution of protein-mediated catalysis since they are flexible to incorporate different cofactors, providing a scaffold to transition from ribozymes and peptides catalysts to modern protein enzymes.587 Rossmann-like folds and TIM barrels can be considered the oldest folds based on the hypothesis that protein folds are more prevalent and more widely shared the more ancestral they are.377
In the case of the NSPase fold, the N-terminal region of the active site, participating in the nucleotide-binding, shows high degree of similarity to enzymes in distant regions of the metabolism, including (i) the molybdopterin guanine dinucleotide synthesis enzyme MobA, a key component in molybdenum cofactor synthesis,588,589 and (ii) the bifunctional methylerythritol 2,4-cyclodiphosphate synthase of Campylobacter jejuni, which transfers cytidil- groups to 2-C-methyl-d-erythritol 4-phosphate to form CDP-ME2P in the isoprenoid biosynthesis.590 Interestingly, the C-terminal pocket portion accounting for the sugar-binding in the NSPase folds presents the largest variability which correlates with differences in the sugar substrate, including (i) the N-acetylmuramic acid α-1-phosphate uridylyltransferase MurU (EC: 2.7.7.99) involved in peptidoglycan metabolism,591 and (ii) the nucleotide monophosphate CMP-Kdo synthetase (EC: 2.7.7.38), that activates the modified sugar Kdo for the synthesis of bacterial cell wall lipopolysaccharides.592 Altogether, the N-terminal region presents variability associated with nucleotide usage, whereas the C-terminal region of the cavity highlights a broad variability associated to several other areas of the metabolism. Finally, the NSPase domain shares similarities to enzymes with other functions. Interestingly, the N-terminal eukaryotic translation initiation factor eIF2B presents an extraordinary degree of architectural similitude.593
The GT-B fold, comprising the two articulated Rossmann domains, represents an interesting case since several bi-substrate reactions can profit from the open-to-close mechanism. Therefore, this architecture could have emerged by convergent evolution or very distant divergent evolution in several areas of the metabolism.380 Indeed, a resembling GT-B-like structural organization is observed in non-GT-B enzymes. Many examples can be extracted from glycolytic enzymes such as (i) the phosphofructokinase that catalyzes the ATP-dependent phosphorylation of fructose 6-phosphate to fructose 1,6-bisphosphate (EC: 2.7.1.11),594 (ii) the phosphoglycerate mutase that catalyzes the transfer of phosphate from 3-phosphoglycerate acid to form 2-phosphoglycerate (5.4.2.12),595 and (iii) the phosphoglycerate kinase (EC: 2.7.2.3) that catalyzes the reversible transfer of a phosphate group from 1,3-bisphosphoglycerate to ADP producing 3-phosphoglycerate and ATP.596 Other examples include (i) UDP-glucose 6-dehydrogenase that converts UDP-glucose into UDP-glucuronic acid, the precursor in the biosynthesis of bacterial exopolysaccharides (EC: 1.1.1.22),597 and (ii) 3-d-phosphoglycerate dehydrogenase that converts d-3-phosphoglycerate to phosphohydroxypyruvate in the serine synthesis pathway (EC: 1.1.1.95).598
The TIM barrel fold is the scaffold of at least 15 different enzymatic functions in all life kingdoms.477,599 Network interaction-conservation phylogenetic analysis allows classification of different groups of TIM barrel fold enzymes, including remote homologues, highlighting structural and functional evolutionary relationships.479 Due to the relationship of the α-glucan to the glycolysis pathways, it is worth mentioning three glycolytic enzymes comprising this fold, (i) the triosephosphate isomerase that interconverts dihydroxyacetone phosphate and d-glyceraldehyde 3-phosphate (EC 5.3.1.1),600 (ii) the pyruvate kinase that catalyzes the final step of glycolysis producing ATP and pyruvate from ADP and phosphoenolpyruvate (PK, EC: 2.7.1.40),601 and (iii) the 2-Keto-3-deoxy-6-phosphogluconate (KDPG) aldolase that transforms KDPG into pyruvate and d-glyceraldehyde-3-phosphate (EC 4.1.2.14) in the alternative glycolysis Entner–Doudoroff pathway.602 Other remarkable enzymes harboring this fold in other parts of the metabolism are (i) the α-subunit of the tryptophan synthase (EC: 4.2.1.20),603 and (ii) the iron-sulfur-flavoprotein trimethylamine dehydrogenase (EC: 1.5.8.2) part of an electron transfer complex.604
6.5. The Big Picture of Bacterial α-Glucan and Glucose Disaccharide Metabolisms
The joint analysis of the α-glucan and glucose disaccharide biosynthetic pathways reveals some interesting relationships. At first glance, the synthesis of these molecules follows a similar overall ‘vertical’ pathway comprising (i) the nucleotide-glucose synthesis mediated by NSPases, (ii) the synthesis of an α-glucosyl-bond mediated GT-B fold enzymes, that may be or not be followed by the removal of phosphate by a phosphatase, and (iii) the editing and interconversion of the α-glucose moieties by TIM barrel enzymes. In that sense, only Pep2/Mak appears foreign to this picture and may represent a novelty in this metabolism acquired after LUCA, as suggested by the lack of detection of this enzyme in Themotagae and Aquifex.228 Interestingly, this order suggests that the grouping of the early part of the metabolism is mediated by Rossmann fold enzymes (nucleotide-sugar dependent metabolism), while the latest part of the metabolism is driven by the TIM barrel enzymes (nucleotide-sugar independent metabolism; Figure 18).
Figure 18.

Integrated summary of α-glucan metabolic pathways in bacteria. The scheme reflects the overlay of different metabolic routes and enzymes discussed, evoking the idea of an underlying common theme supported by the evolution of an ancestral metabolism. The prokaryotic cell is depicted as enclosed by its membrane (double dotted boundary line) where glucose and other sugars enter and are transformed along the gluconeogenesis pathway eventually leading to the synthesis of glucose 1-phosphate, which is subsequently activated by forming sugar donors NDP-glucose (ADP-glucose or UDP-glucose). Different pathways for intra- and extracellular α-glucan synthesis depart from top to bottom, traversing the synthesis of glucose disaccharides. The area of the metabolism comprises reactions requiring nucleotide-sugar donors (violet background shade) and independent nucleotide-sugar (light blue background shade). The parallel biosynthetic routes follow similar transformations catalyzed by enzymes with typical catalytic domain architecture. Rossmann folds enzymes, preeminently NDP-sugar transferases (blue hexagons), and GT-B fold (yellow circles) drive transformation involving nucleotide derivatives. In contrast, TIM barrel folds (red boxes) take control of all nucleotide-independent transformations. Other intermediate reactions are driven by HAD domain phosphorylases (green parallelograms), which catalyze disaccharide dephosphorylation, and the phosphorylation of maltose carried out by the Maltokinase (violet trapezium).
A horizontal view of the overall metabolism discloses the direction of evolutionary specialization of the different enzymes participating in the α-glucan and disaccharide pathways, suggesting the common ancestry of these enzymes. Interestingly, as we analyzed, in some species, the level of specialization can virtually separate these pathways; meanwhile, in others, it can be intertwined by the enzyme promiscuity, permitting a level of redundancy that can offer an evolutive advantage. This horizontal view also accounts for the specialization of GT-B disaccharide synthases. It can be speculated an ancestral enzyme to GlgM, a processive GS, and a processive GP that also acquired PLP to achieve the α-glucan depolymerization. In this general context, the synthesis of internal glycogen appears as a specialization that allows virtually enclosing this α-glucan energy storage broadly distributed in bacteria, implying an important evolutive advantage, whereas the GlgE pathway appears mostly centered on actinobacteria (Figure 18).605
Finally, TIM barrels appear associated to the intracellular and extracellular milieu, suggesting a primitive function of the corresponding polysaccharides associated with external energy sources or structural functions. Most of the interconversions mediated by the TIM barrel enzymes in this metabolism are phosphate-independent, and may be originated as part of the recent proposed ancient phosphate-free cryptic core metabolism.606 GlgE and S6FP are exceptions to this rule and their functions may have emerged in modern metabolism (Figure 18).
7. α-Glucans and Microbial Pathogenicity
Genes encoding enzymes involved in biosynthesis and mobilization of α-glucans are widespread among bacterial pathogens employing very diverse pathophysiological strategies. The specific contribution of α-glucan metabolism to virulence, pathogenesis, and potentially immune evasion has been investigated for several important pathogenic bacteria, and our knowledge on these aspects is summarized in the following sub-sections.
7.1. Mycobacterium Tuberculosis
The human pathogen M. tuberculosis produces α(1→4)-linked and α(1→6)-branched glycogen-like α-glucan exclusively via the GlgE pathway (Figure 3B). Two alternative routes, the TreS-Pep2/Mak and the GlgC-GlgM pathways, provide the substrate maltose 1-phosphate that is converted to the branched polymer by iterative cooperativity between the maltosyltransferase GlgE and the branching enzyme GlgB. The polymer is synthesized intracellularly, and then a portion of it is secreted by an unknown transport mechanism to form the major constituent of the capsular layer.119
Synthesis of glycogen-like α-glucan is relevant for pathogenesis of M. tuberculosis in two aspects. First, several genes involved in the α-glucan metabolic network are essential and, thus, represent potential drug target candidates. The maltosyltransferase GlgE was found to be essential because loss of enzymatic activity leads to intracellular build-up of its substrate maltose 1-phosphate, which is toxic to the bacterial cells for reasons that have not been fully elucidated yet.212 Similarly, the branching enzyme GlgB is essential because it is required for iterative production of the glycogen-like α-glucan polymer in cooperation with GlgE. Loss of GlgB enzymatic activity curtails GlgE activity since GlgE alone can only produce linear α(1→4)-linked maltooligosaccharides that become insoluble and provide less non-reducing ends than branched glucans to be extended by GlgE. Due to these reasons, inactivation of GlgB also causes accumulation of toxic levels of maltose 1-phosphate.212 In addition, a similar sugar phosphate-related toxicity supports essentiality of the trehalose 6-phosphate phosphatase OtsB2 in M. tuberculosis, which produces the trehalose substrate for the TreS-Pep2/Mak pathway, as inactivation causes accumulation of its substrate trehalose 6-phosphate.225 Furthermore, while the genes encoding trehalose 6-phosphate synthase OtsA and maltose 1-phosphate-producing glucosyltransferase GlgM are individually dispensable, their combined inactivation is synergistically lethal likely because of accumulation of their joined substrate ADP-glucose reaching toxic levels.119
Second, glycogen-like α-glucan was shown to be important for full virulence of M. tuberculosis in a mouse infection model. A mutant lacking the gene encoding maltose 1-phosphate-producing glycosyltransferase GlgM (described as glycogen synthase GlgA before knowing the configuration of α-glucan metabolic pathways in M. tuberculosis) that has reduced levels of capsular α-glucan showed impaired virulence in mice.82 With the recent elucidation of the complete metabolic network and pathways required for α-glucan production in M. tuberculosis, it was possible to generate defined mutants completely devoid of α-glucan in a rational way. This was mediated by blocking maltose 1-phosphate synthesis in a ΔglgC ΔtreS double mutant. This double mutant exhibited significant reduction in virulence in mice in contrast to the corresponding single mutants. Since these mutations affected both intracellular and capsular α-glucan likewise, which likely fulfill different biological functions, it is unknown to which degree the loss of each α-glucan type contributed to the observed attenuation phenotype. As the outermost layer of the mycobacterial cell wall and first line of contact with host cells, capsular α-glucans may be important for M. tuberculosis pathogenesis by interacting with mammalian host cells and influencing the immune response to M. tuberculosis. In fact, in vitro experiments using purified capsular α-glucan from mycobacteria demonstrated that it can interact with complement receptor 3, thus mediating binding of M. tuberculosis to phagocytic cells.607,608 Capsular α-glucan has also been reported to block dendritic cell functions609 and to interact with the C-type lectin receptor DC-SIGN on dendritic cells.610 While it is intriguing to speculate that M. tuberculosis uses capsular α-glucans to modulate and evade antimicrobial immune effector functions of phagocytic cells, there is a puzzling complexity and reciprocal promiscuity regarding interaction of mycobacterial capsule components with immune receptors, i.e., one molecule can be a ligand for several receptors and one receptor can be triggered by several ligands.611 Unless the still pending discovery of the mechanism of secretion will allow the generation of M. tuberculosis mutants specifically defective only in capsular glycogen-like α-glucans that still produce normal levels of the intracellular polymer, the precise function and importance of capsular glycogen-like α-glucans for pathogenesis of M. tuberculosis and other pathogenic mycobacteria remain elusive.
7.2. Chlamydiae
Chlamydiae are Gram-negative bacteria that employ an obligate intracellular lifestyle, which is linked to a massive genome reduction and a biphasic developmental cycle that includes two distinct stages: the elementary body and the reticulate body. While the elementary body is a non-dividing and infectious form adapted to extracellular survival, the reticulate body constitutes a replicating form that is surrounded by a membrane forming an inclusion.612−614 The order Chlamydiales includes the Chlamydiaceae family and several family-level lineages collectively called Chlamydia-related bacteria.615 The Chlamydiaceae family includes human pathogens such as Chlamydia trachomatis, which is the most common sexually transmitted infection leading to infertility, extrauterine pregnancy, and miscarriages in females; Chlamydia pneumoniae, which causes acute respiratory diseases of the upper and lower respiratory tract, such as pneumonia and bronchitis; and Chlamydia psittaci, which is a leading cause of zoonotic avian chlamydiosis and mainly causes respiratory infections followed by systemic dissemination to the heart, liver, and gastro-intestinal tract. Furthermore, different species of the Chlamydiaceae family can also infect different animals such as Chlamydia muridarum, which infects rodents.616
While for other intracellular bacteria the adaptation to a strict intracellular life-style led to the loss of glycogen metabolism pathway,557 nearly all sequenced chlamydial genomes with the exception of Criblamydiaceae and Waddliaceae families maintained the GlgC-GlgA-GlgB dependent glycogen pathway.222Chlamydia trachomatis and the closely related rodent pathogen Chlamydia muridarum are unique amongst Chlamydiae phylum as they accumulate α(1→4)-linked and α(1→6)-branched glycogen-like α-glucan not only intracellularly but also extracellularly in the inclusion lumen.617,618 Glycogen accumulates first in the inclusion lumen and only later in elementary bodies. While extracellular glycogen deposition was initially thought to result from bacterial cell lysis,617 it was recently shown to represent a specific process involving two mechanisms.619 First, the host glycogen synthase Gys1, which is known to tightly bind to glycogen,109 is translocated into the vacuole, resulting in concomitant bulk import of host glycogen. Translocation of cytoplasmic glycogen likely occurs through invagination of the inclusion membrane, but the underlying mechanisms remain elusive yet. Bulk uptake of host-derived glycogen is a minor pathway for glycogen accumulation in the inclusion lumen and contributes only to ca. 20% of total glycogen accumulation.619 In contrast, the main source of intraluminal glycogen accumulation was found to result from de novo synthesis of glycogen that is mediated by secretion of the glycogen synthase GlgA620 and the branching enzyme GlgB621 by the bacteria inside the inclusion. Furthermore, the host transporter SLC35D2 and possibly further transporters are recruited to the inclusion membrane to allow translocation of UDP-glucose from the host cell cytoplasm into the inclusion lumen.619 While glycogen synthase GlgA from other bacteria typically prefers ADP-glucose as the substrate, GlgA from C. trachomatis can also utilize host-derived UDP-glucose to mediate glycogen formation in the inclusion lumen.619 Consistently, in contrast to GlgA and GlgB, chlamydial GlgC was not found to be secreted and remains intracellularly in the bacterial cells, highlighting the fact that intraluminal glycogen synthesis relies on host-derived UDP-glucose as the substrate.619 While secretion of GlgA, GlgB, and other proteins into the inclusion lumen was reported to be mediated by the bacterial type 3 secretion system,619 another study suggested that C. trachomatis employs a plasmid-dependent and type 3 secretion system-independent mechanism to export GlgA.622 Early during infection, the chlamydial cell population largely consists of reticulate bodies, and intraluminal glycogen accumulation via de novo synthesis and bulk import sets in between 16 and 20 h post infection.619 Later during the infection cycle, reticulate bodies start converting into elementary bodies, which are associated with mobilization of intraluminal glycogen. For this, the chlamydial cells secrete the enzymes required for glycogen degradation, i.e., the debranching enzyme GlgX and the glycogen phosphorylase GlgP into the inclusion lumen, likely in a type 3 secretion system-dependent manner.619 The resulting depolymerization end product, glucose 1-phosphate, is converted to glucose 6-phosphate by the chlamydial phosphoglucomutase MrsA, which is secreted as well.619 Glucose 6-phosphate can be imported into C. trachomatis cells by the hexose phosphate transporter UhpC and can be used as source of energy and carbon. Later during conversion of reticulate into elementary bodies, the type 3 secretion system is turned off, allowing for intrabacterial activity of the glycogen metabolism enzymes, and glycogen accumulation in the bacteria.619 The temporal separation of glycogen synthesis and subsequent mobilization allows C. trachomatis to sequester a host metabolite and accumulate it extracellularly in an osmotic inert storage form and to rapidly depolymerize and convert it into a readily usable molecule for the bacterium. While the precise role and importance of glycogen synthesis and degradation for virulence of C. trachomatis has not been fully elucidated yet, it was demonstrated that loss of the glgA gene in C. muridarum resulted in a significant reduction of pathogenicity in the genital tract in a mouse infection model, demonstrating the role of GlgA in C. muridarum-mediated induction of hydrosalpinx in mice, which is similar to the pathophysiology of C. trachomatis infection in women. The reduced infectivity of the C. muridarum glgA mutant was associated with a decreased ability to trigger inflammation.623 Consistent with this finding, treatment of human monocytic cell line THP-1 with GlgA protein from C. trachomatis elicited the expression of proinflammatory cytokines interleukin-8 (IL-8), interleukin-1beta (IL-1β), and tumor necrosis factor alpha (TNF-α) in a TLR2- and TLR4-dependent manner.624
While nearly all sequenced chlamydial genomes comprise the genes for a fully functional GlgC-GlgA-GlgB dependent glycogen pathway,222 the Criblamydiaceae and Waddliaceae families form an exception since the GlgC-GlgA pathway is defective. This is due to genomic rearrangements causing deletion of both the glgC and glgP genes and a fusion of the glgA and glgB genes rendering the branching enzyme domain inactive.222 Instead, a complete and functional GlgE pathway was identified in five chlamydial species distributed in Criblamydiaceae, Waddliaceae, and Parachlamydiaceae families including the emerging human pathogen Waddlia chondrophila,625,626 comprising genes encoding the maltosyltransfrase GlgE, the branching enzyme GlgB2, and a bifunctional TreS-Mak fusion protein, mediating glycogen biosynthesis from trehalose.222 Furthermore, these organisms also encode the enzymatic machinery for degradation of glycogen comprising glycogen phosphorylase GlgP2, debranching enzyme GlgX, and α(1→4)-glucanotransferase MalQ. Thus, glycogen metabolism is remarkably preserved also in environmental Chlamydiae suggesting an important function for their intracellular lifestyle.222 However, no specific glycogen-defective mutants are available yet to address the role a glycogen metabolism for infectivity and pathogenesis.
Altogether, while not strictly essential for viability, the capability to synthesize and to mobilize glycogen-like α-glucan appears to be a conserved metabolic feature of Chlamydiae that contributes to the virulence potential of pathogenic members of this phylum. This might represent a storage function particularly important regarding fueling essential metabolic pathways in the extracellular forms, i.e., elementary bodies of Chlamydiales.
7.3. Pseudomonas aeruginosa
P. aeruginosa is an important human pathogen and one of the most common causes of nosocomial infections. It is an opportunistic pathogen that primarily affects immunocompromised individuals, notably patients with immunodeficiencies and traumatic burn wounds. It is also a major pneumonia agent causing both acute and chronic lung infections, the latter particularly associated with cystic fibrosis and the ability of the bacterium to form biofilms.627 Due to its high intrinsic antibiotic resistance, P. aeruginosa infections are difficult to eradicate.628P. aeruginosa is also ubiquitously found in the environment in soil and aquatic habitats, and strains of P. aeruginosa as well as of related species such as P. syringae can be pathogenic to plants.629,630
P. aeruginosa is peculiar in producing α(1→4)-linked and α(1→6)-branched glycogen-like α-glucan employing both the GlgE pathway and the classical glycogen synthase GlgA as has recently been reported for strain PAO1 (Figure 3C).173 Production of glycogen through GlgA is surprising since P. aeruginosa PAO1 lacks AGPase GlgC. However, GlgA was shown to be specific for UDP-glucose and thus contrast to typical bacterial glycogen synthases that prefer ADP-glucose. UDP-glucose is produced in this strain from UTP and glucose 1-phosphate by the UGPase GalU to be used as substrate for GlgA and other enzymes.173 More surprisingly, it was shown that GlgA can produce linear α(1→4)-linked α-glucan with a DP >40, but this polymer is readily degraded and metabolized to trehalose employing the TreY-TreZ pathway in a cellular context before glycogen particles are formed.173 In fact, this is the only route to trehalose formation in P. aeruginosa PAO1, which lacks the OtsA-OtsB pathway that is dominant in most other bacteria. Part of the trehalose synthesized from GlgA-produced α(1→4)-linked α-glucan is then converted to α(1→4)-linked and α(1→6)-branched glycogen-like α-glucan by the GlgE pathway comprising a bifunctional TreS-Pep2 fusion protein producing maltose 1-phosphate from trehalose and ATP, the maltosyltransferase GlgE and the branching enzyme GlgB in a process very similar to the one described for M. tuberculosis.173 There is a second route to maltose 1-phosphate in P. aeruginosa PAO1 that involves the α-glucanotransferase MalQ, which can produce maltose by transferring a glucose moiety from the non-reducing end of a maltooligosaccharide when glucose is used as an acceptor molecule or when maltotriose is used as the donor substrate. The maltokinase activity of the TreS-Pep2 fusion protein can then yield maltose 1-phosphate in presence of ATP. Thus, MalQ is able to bypass the TreY-TreZ-TreS reactions to provide maltose 1-phosphate for the GlgE pathway.173 α(1→4)-linked and α(1→6)-branched glycogen-like α-glucan that accumulates in P. aeruginosa PAO1 is almost exclusively produced by the GlgE pathway, whereas linear α(1→4)-glucan produced by GlgA is demonstrated to accumulate only when TreY-TreZ is inactivated. Consistent with its role in trehalose production, the glgA gene is localized in an operon together with the treY and treZ genes in addition to glgX and malQ, separated from the GlgE pathway operon comprising the genes treS-pep2, glgE, and glgB, while the glgP gene is an orphan.173 It remains elusive why in P. aeruginosa PAO1 only the α(1→4)-glucan produced by GlgA is virtually quantitatively degraded and converted to trehalose, while the GlgE pathway produces polymeric glycogen particles. It appears to be a futile cycle to build linear α-glucan only for synthesis of trehalose and subsequently to produce α-glucan from it again. But in the face of a lack of the OtsA-OtsB route for trehalose, coexpression of the glgA, treY, and treZ genes from the same operon and possibly direct interaction of the corresponding proteins might enable efficient trehalose formation. Trehalose is not quantitatively, but only partially, converted to glycogen-like α-glucan and accumulates in the cytoplasm of the bacterial cells itself in response to certain stress conditions.173 Theoretically, specificity of GlgA for UDP-glucose might allow P. aeruginosa PAO1 to sequester this metabolite from host cells during infection similar to what has been described above for C. trachomatis. However, this would require the ability of P. aeruginosa PAO1 to take up extracellular UDP-glucose, but no such activity has ever been reported for this bacterium.
Both trehalose synthesized by the GlgA-TreY-TreZ route and glycogen-like α-glucan synthesized by the GlgE pathway play important, yet distinct, roles in stress protection in P. aeruginosa PAO1. Trehalose is specifically required for tolerance to osmotic stress, whereas the GlgE-derived α-glucan mediates desiccation tolerance. In turn, GlgE-derived α-glucan has a minor effect on osmotic sensitivity, while trehalose does not contribute directly to the desiccation response.173 Both trehalose and glycogen-like α-glucan were shown to be equally important for survival on abiotic surfaces.173 However, it remains to be elucidated to which degree each molecule contributes to virulence and pathogenesis of P. aeruginosa PAO1 in various infection models. Previously, deletion of the treS and treY-treZ genes, respectively, in P. aeruginosa strain PA14 was shown to abolish trehalose production and cause attenuation of virulence in a plant infection model, implicating trehalose as a stress protectant and possible virulence factor.631 Similarly, treS gene disruption in P. syringae pv. tomato resulted in osmotic sensitivity and attenuation during plant infection.632 However, the conclusions back at the time were drawn without knowledge of the full network and close interaction of the metabolism of trehalose and glycogen-like α-glucan in Pseudomonas. Given that the mentioned Pseudomonas strains very likely exhibit the same metabolic configuration as P. aeruginosa strain PAO1, the mutations in both cases would also lead to abolition of α-glucan biosynthesis. Thus, the virulence phenotypes previously attributed to trehalose could in fact rather be mediated at least partially by α-glucan. In contrast to the reported role in phytopathogenesis, deletions of the treS and treY-treZ genes in P. aeruginosa strain PA14 did not attenuate growth in a range of animal infection models.631 However, the used models were unlikely to reflect the significant osmolarity and desiccation stresses present during cystic fibrosis in humans. Therefore, the importance of glycogen-like α-glucan for human pathogenesis of P. aeruginosa remains to be fully investigated.
7.4. Biofilm-Forming Bacteria (Streptococcus, Neisseria, Aeromonas)
The ability to form biofilms is an important virulence trait of several pathogenic bacteria. While most of them produce an extracellular matrix composed of polysaccharides other than α-glucan, α-glucan is an important component of the EPS of bacterial biofilms related to dental plaque formation and caries development. Within the complex oral microbiome, the facultatively anaerobic gram-positive coccoid bacterium S. mutans is a major producer of EPS, the composition of which is complex and changes dynamically depending on food uptake. Within the mixture of EPS, extracellular DNA, and lipoteichoic acid, α-glucan can comprise between 10 and 20% of the dry weight of dental plaque.633−637 Within the complex and dynamically changing polymicrobial oral microbiome, S. mutans is not always the most abundant species, and many other bacteria contribute to acidogenesis and cariogenicity.638,639 However, S. mutans is a major EPS producer and rapidly modulates the formation of cariogenic biofilms when appropriate dietary substrates (i.e., sucrose and starch) are available.640−643 The role of extracellular α-glucan production by S. mutans in caries development has extensively been reviewed recently.137 Briefly, S. mutans secretes three different glucosyltransferases, which have distinct but partially overlapping, redundant functions. GtfB (formerly known as Gtf-I) and GtfC (Gtf-SI) are mutansucrases that synthesize water-insoluble mutan, which is α-d-glucan mainly composed of α(1→3) linkages.644,645 The secreted mutansucrases are incorporated into pellicle (particularly GtfC) but also adsorb on the bacterial cell surface (mainly GtfB), and both utilize dietary sucrose to synthesize water-insoluble mutan in situ. Furthermore, the secreted mutansucrases can also adsorb onto the surface of other oral microorganisms that do not produce glucosyltransferases themselves, turning them into mutan producers. The water-insoluble mutans facilitate adherence of microorganisms to the smooth surface of the tooth enamel. Concomitantly, salivary α-amylase in the pellicle digests dietary starch, releasing maltose and maltooligosaccharides that can serve as acceptors particularly for surface-adsorbed GtfB and are incorporated into the mutan polymer. S. mutans produces and secretes a third glucansucrase enzyme (GtfD, formely known as Gtf-S) catalyzing the synthesis of water-soluble dextran-like α-d-glucan mainly containing α(1→6) linkages,646 which can serve as primers for GtfB.137 The parallel formation of both water-soluble dextran and water-insoluble mutan enhances the adherence of the oral microorganisms to the smooth enamel surface and promotes dental plaque formation. Accordingly, it was shown that the sucrose-dependent adherence of S. mutans to the tooth surface was related to all three glucansucrase enzymes at an optimum ratio.647 The insoluble mutan molecules provide ample binding sites for S. mutans and other oral bacteria mediating tight bacterial clustering and adherence to the tooth enamel. This is supported by the secretion of several nonglucosyltransferase glucan-binding proteins, inactivation of which has been shown to result in impaired biofilm formation and altered architecture.83 The α-glucan-containing EPS formed in situ enables the assembly of a spatially heterogeneous and cohesive multicellular structure and development of a cariogenic biofilm.648 The importance of α-glucan production for virulence and cariogenicity of S. mutants has been demonstrated by studies of mutants lacking glucansucrase activities. Inactivation of any of the glucansucrases GtfB, GtfC, or GtfD resulted in a reduction of smooth-surface carious lesions in a rat model system.649 Similarly, although the nature of mutations is not known, glucan synthesis-defective mutants of S. mutans were shown to cause reduced caries development in rats.650,651
In addition to extracellular mutan and dextran-like α-glucan, S. mutans also produces intracellular glycogen employing the classical GlgC-GlgA pathway, which may also be important for virulence. A mutant defective in intracellular glycogen accumulation exhibited reduced cariogenic potential in rats.652 Conversely, a transposon mutant overproducing glycogen showed increased cariogenicity.653 However, it remains unclear whether glycogen accumulation contributes to biofilm formation and how it can influence virulence of S. mutans during growth in an environment rich in carbohydrates.
Different Neisseria species such as N. polysaccharea, N. mucosa, and N. perflava can also be found in human dental plaque. Neisseria secrete a GH13 amylosucrase to produce an extracellular glycogen-like polymer mainly composed of α(1→4) linkages with few α(1→6) branches.161−164 While this suggests that extracellular glucan produced by these Neisseria contribute to biofilm and dental plaque formation, no glucan deficient mutants have been described to address their relevance for inhabiting the oral environment.
Members of the genus Aeromonas are gram-negative, water-borne bacteria that are ubiquitously found in aquatic environments. As many strains are able to grow and to produce exotoxins at low temperatures, they are mostly infective to poikilothermic animals, but some species are also emerging as human pathogens causing systemic as well as gastrointestinal infections.654Aeromonas species produce a surface α-glucan consisting of α(1→4)-linked glucosyl units with α(1→6)-branches that is intracellularly synthesized by the UGPase GlgC and the glycogen synthase GlgA before being exported in a WecP-dependent fashion.166−168 The surface-exposed glycogen-like α-glucan is important for biofilm formation as was revealed by a GlgA deficient mutant of A. hydrophila.168 While this suggests that glycogen-like α-glucan may contribute to virulence of this species, no studies in relevant infection models have been performed to address this hypothesis.
7.5. Enteric Bacteria (Escherichia coli, Salmonella enterica, Vibrio cholerae)
Despite providing a mucus layer rich in carbohydrates, lipids, and proteins, the mammalian gastrointestinal tract represents an environment characterized by fluctuating availability of nutrients. Thus, both commensal and pathogenic bacteria in the gut are regularly facing prolonged periods of restricted nutrient availability and even starvation.655,656 To cope with a nutrient-limiting environment and to compete with other microorganisms, the ability to accumulate intracellular carbon and energy storages during nutrient-rich periods and to remobilize them during hunger phases is believed to be essential for persistence of intestinal bacteria. Glycogen is thought to be the primary carbon and energy storage molecule for enteric bacteria that employ the classical GlgC-GlgA pathway for glycogen synthesis (Figure 3A).464 Furthermore, after shedding with feces, glycogen may be important for extracellular survival and possibly promotes dissemination of pathogenic enteric bacteria.93
To assess the importance of glycogen biosynthesis and degradation in E. coli, ΔglgA and ΔglgP mutants were assessed in a streptomycin-treated mouse infection model and revealed that both are necessary for efficient colonization of the intestine by E. coli.657 In contrast, conflicting data exist regarding the role of glycogen metabolism for virulence of the closely related gram-negative bacterium S. enterica. Supporting the hypothesis of a central role of glycogen for virulence and infectivity of enteric bacteria, one study showed a correlation between the amount of glycogen stored during the preincubation step and the 50% lethal dose in for S. enterica serovar Enteritidis in a chicken infection model.544 In contrast, in another study, no significant differences in intestinal colonization or virulence in chicken were observed for deletion mutants in the glgC gene encoding AGPase generated in three different S. enterica serovars (Gallinarum, Pullorum, Typhimurium).553 However, while the glgC mutant strains were thought to be deficient in glycogen accumulation because they cannot make the glycogen precursor ADP-glucose, more recent findings suggest that glgC mutants of E. coli and S. enterica serovar Typhimurium possess an alternative pathway for ADP-glucose formation and still can accumulate glycogen under certain conditions.658 While this alternative route has not been identified and characterized enzymatically yet, it is possible that the importance of glycogen accumulation for virulence and infectivity in E. coli and S. enterica is underestimated when the assessment is based on the phenotype of GlgC-deficient mutants. In favor of this, studies with mutants of the pathogenic enteric bacterium Vibrio cholerae defective in glycogen synthesis or degradation support the importance of glycogen metabolism for pathogenesis.95 Like other enteric bacteria, V. cholerae employs the classical GlgC-GlgA pathway for glycogen synthesis but is peculiar in possessing two genes coding for AGPase, glgC1 and glgC2. V. cholerae mutants defective in glycogen biosynthesis (ΔglgC1 ΔglgC2 double mutant) or degradation (ΔglgX) demonstrated that glycogen prolongs survival in nutrient-poor environments that are known ecological niches of V. cholerae, including pond water and rice-water stool. Additionally, defects in glycogen metabolism strongly attenuated pathogenesis of V. cholerae in an infant mouse transmission model of cholera. Lending further credence to the hypothesis of glycogen being an important contributor to virulence, glycogen granules can be found inside bacterial cells present in rice-water stool from cholera patients.95 Together, these findings implicate glycogen metabolism as a relevant survival strategy employed by enteric bacteria to survive the dramatic changes encountered in the host and the environment. This not only applies to pathogenic enteric bacteria, since glycogen has also been shown to support gut retention of probiotic bacteria in a germ-free mouse model, yielding a significant fitness disadvantage of a ΔglgA mutant strain of the gram-positive lactic acid bacterium Lactobacillus acidophilus.659
8. Concluding Remarks
More than a century of lessons about the biosynthesis of α-glucans has disclosed some functional roles and some patterns in the metabolism of these polysaccharides in bacteria. As we highlight in this Review, we show how bacterial metabolism evolved to handle the α-d-glucose scaffold to build assorted α-glucans with different functions, including energy storage or extracellular structural components as key elements in matrix biofilms formation, which provide protection to colonizing bacteria, and also as virulence factors. Taken together, the chemical landscape from α-d-glucose to disaccharides and α-glucans, the analysis of their metabolism and the structural/functional diversity of the intervenient enzymes, we provide an overall picture reflecting the underlying evolution of such enzymatic machinery (Figure 18). Our current view unveils a complexity in α-glucans diversity, although as we delve deeper into the fundamentals of heterogeneity, we encounter a nuanced landscape formed by few enzymatic folds linked by the evolution of the metabolism. Further examining the synergies of non-catalytic domains, including CBMs, for such α-glucans diversity, highlights the need for further research.
There is still an immense unexplored metabolic diversity landscape in the biosphere, likely influenced differently by such polymers in their niche and their metabolic blueprint. For example, the human gut microbiome, arguably the most studied ecosystem, is composed of ca. 3500 species,660 whose population composition is affected by the structure of dietary α-glucans.661 This is due to the different arsenal of enzymes of the diverse bacterial species for their processing;662 therefore, environmental α-glucans architecture acts as an environmental selective pressure, a fact that is already used to shift bacterial populations as medical treatment.663,664 In that sense, of special relevance is the so-called resistant starch, a type of starch that resists digestion in the small intestine, progressing to the large intestine where it acts as a prebiotic, feeding the beneficial bacteria in the gut microbiota.674,675 The use of resistant starch by gut bacteria involves several degradation steps, favoring the production of short-chain fatty acids such as butyrate that serves as a primary energy source for colon cells.676 Importantly, this process helps to maintain a healthy balance of gut bacteria, contributes to gut health by promoting bowel regularity, and can enhance the immune system. Therefore, foods high in resistant starch are indicated in a balanced diet for maintaining overall health. Extrapolating this to the biosphere, where diverse calculations predict the existence of ca. 9 million eukaryotic species and ca. 1 trillion microbial species,665,666 the dimension of the biosphere metabolic landscape suggests that we are only observing the tip of the iceberg. Moreover, most of our knowledge on α-glucans metabolism belongs to bacteria species cultured in the laboratory; nevertheless, the existence of the so-called “microbiological dark matter” represents one-quarter of the population of microbes belonging to phyla with no cultured relatives,667 indicating that future research will greatly expand on this metabolism architecture, possibly finding different rewirings of their metabolism, also offering an immense repertoire of new biotechnological opportunities.
Acknowledgments
This work was supported by the MICINN grant PID2022-138694OB-I00 (M.E.G.) and National Institutes of Health grant R01AI149297 (M.E.G.); the Jürgen Manchot Foundation (R.K.) and the CNRS, the Université de Lille CNRS, and the ANR grants “Animalga” (ANR-21-CE20-0051; C.C.). J.O.C. acknowledges financial support from the IKUR Strategy (Ikerbasque Foundation and Fundación Biofísica Bizkaia) and the Department of Education of the Basque Government.
Biographies
Javier O. Cifuente graduated in Biochemistry at the Universidad Nacional de La Plata, Argentina (2006). He earned a Ph.D. in Biological Sciences under the direction of Dr. Ricardo M. Gomez at the Institute of Biotechnology and Molecular Biology (2009). He conducted postdoctoral research under the direction of Prof. Susan Hafenstein, at Penn State University College of Medicine, USA, (2009–2012). He continued his postdoctoral training under the guidance of Prof. Marcelo E. Guerin at the Structural Glycobiology Laboratory, (Unit of Biophysics, UPV/EHU-CSIC, CIC bioGUNE and IIS-BioCruces Bizkaia), Spain (2014–2023). He is a postdoctoral researcher under the direction of Prof. Iban Ubarretxena-Belandia, in the Structural Biology of Disease Mechanisms Laboratory, at the Biofisika Institute (UPV/EHU-CSIC).
Christophe Colleoni obtained his PhD in microbial genetics under the supervision of Prof. Steven Ball. He completed a post-doctoral fellowship (1999–2003) on the structural and biochemical characterization of storage polysaccharides produced by various sweet-corn cultivars under Prof. Alan Myers in the Roy J. Carver Department of Biochemistry, Biophysics, and Molecular Biology (BBMB) at Iowa State University, USA. He is a full professor at the University of Lille, France, and leader of the Integrative Biology of Storage Polysaccharides (IBSP) team at the Institute for Structural and Functional Glycobiology (CNRS-UM8576). His research focuses on the evolution of storage polysaccharide metabolisms in prokaryotes and microalgae.
Rainer Kalscheuer graduated in Biology from the University of Münster, Germany (1998). He obtained his Ph.D. in Biology under the supervision of Prof. Alexander Steinbüchel at the Institute of Molecular Microbiology and Biotechnology, University of Münster, Germany (2003). Following a postdoctoral period at the same institute (2003–2005), he conducted postdoctoral studies under the direction of Prof. William R. Jacobs Jr. at the Howard Hughes Medical Institute at the Albert Einstein College of Medicine, Bronx, United States (2005–2009). He was leader of the Jürgen Manchot Research Group for Molecular Host-Pathogen Interaction at the Institute für Medical Microbiology and Hospital Hygiene, Heinrich Heine University Düsseldorf, Germany (2010–2015). Since 2015, he is professor at the Institue of Pharmaceutical Biology and Biotechnology, Heinrich Heine University Düsseldorf, Germany.
Marcelo E. Guerin graduated in Pharmacy from the University of Buenos Aires, Argentina (1997). He earned his Ph.D. in Chemistry under the direction of Prof. Armando J. Parodi, at the Glycobiology Laboratory, the Leloir Institute of Buenos Aires, Argentina (2002). He carried out postdoctoral studies in the Structural Biochemistry Unit, under the direction of Prof. Pedro M. Alzari, at the Institut Pasteur Paris, France (2003–2007). Additionally, he conducted postdoctoral studies in the Mycobacteria Research Laboratories, under the direction of Prof. Mary Jackson, at Colorado State University, United States (2008–2009). He is a full research professor at the Spanish National Research Council (CSIC) and leader of the Structural Glycobiology Laboratory (2009–present), currently at the Institute of Molecular Biology of Barcelona (IBMB), Spain.
Author Contributions
CRediT: Javier Orlando Cifuente conceptualization, data curation, investigation, writing-original draft, writing-review & editing; Christophe Colleoni conceptualization, data curation, funding acquisition, investigation, writing-original draft, writing-review & editing; Rainer Kalscheuer conceptualization, data curation, funding acquisition, investigation, writing-original draft, writing-review & editing; Marcelo E. Guerin conceptualization, data curation, funding acquisition, investigation, writing-original draft, writing-review & editing.
The authors declare no competing financial interest.
References
- Herstein B. The Centenary of Glucose and the Early History of Starch. J. Ind. Eng. Chem. 1911, 3, 158–168. 10.1021/ie50027a007. [DOI] [Google Scholar]
- Fisher C. H.Anselm Payen Pioneer in Natural Polymers and Industrial Chemistry. In Pioneers in Polymer Science; Seymour R. B., Mark H. F., Pauling L., Fisher C. H., Stahl G. A., Sperling L. H., Marvel C. S., Carraher C. E., Seymour R. B., Eds.; Springer Netherlands: Dordrecht, 1989; pp 47–61. [Google Scholar]
- Jorpes J. E.Jac. Berzelius. In His Life and Work; University of California Press, 1970; pp 21–25. [Google Scholar]
- Verma M. L.; Kumar S.; Jeslin J.; Dubey N. K.. Chapter 5. Microbial Production of Biopolymers with Potential Biotechnological Applications. In Biopolymer-Based Formulations; Pal K., Banerjee I., Sarkar P., Kim D., Deng W.-P., Dubey N. K., Majumder K., Eds.; Elsevier, 2020; pp 105–137. [Google Scholar]
- Brown A. J. XLIII.—On an Acetic Ferment which forms Cellulose. J. Chem. Soc. Trans. 1886, 49, 432–439. 10.1039/CT8864900432. [DOI] [Google Scholar]
- Young F. G. Claude Bernard and the Discovery of Glycogen: A Century of Retrospect. Br. Med. J. 1957, 1, 1431–1437. 10.1136/bmj.1.5033.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis I. M. The Cytology of Bacteria. Bacteriol. Rev. 1941, 5 (3), 181–230. 10.1128/br.5.3.181-230.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer E.Syntheses in the Purine and Sugar Group. Nobelstiftelsen. Nobelprize.org, 1902. [Google Scholar]
- Haworth W. N.; Machemer H. 323. Polysaccharides. Part X. Molecular Structure of Cellulose. J. Chem. Soc Resumed. 1932, 2270–2270. 10.1039/jr9320002270. [DOI] [Google Scholar]
- Haworth W. N.; Hirst E. L.; Webb J. I. CCCXXIX.—Polysaccharides. Part V. Glycogen. J Chem Soc 1929, 0, 2479–2485. 10.1039/JR9290002479. [DOI] [Google Scholar]
- Cramer F. Biochemical Correctness: Emil Fischer’s Lock and Key Hypothesis, a Hundred Years after—an Essay. Locky Key - Hundred Years After 1995, 69, 193–203. 10.1016/0031-6865(95)00012-X. [DOI] [Google Scholar]
- Fischer E. Einfluss Der Configuration Auf Die Wirkung Der Enzyme. Berichte Dtsch. Chem. Ges. 1894, 27, 2985–2993. 10.1002/cber.18940270364. [DOI] [Google Scholar]
- Koshland D. E. The Key–Lock Theory and the Induced Fit Theory. Angew. Chem. Int. Ed. Engl. 1995, 33, 2375–2378. 10.1002/anie.199423751. [DOI] [Google Scholar]
- Johnson K. A.; Goody R. S. The Original Michaelis Constant: Translation of the 1913 Michaelis-Menten Paper. Biochemistry 2011, 50, 8264–8264. 10.1021/bi201284u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colowick S. P.; Sutherland E. W. Polysaccharide Synthesis from Glucose by Means of Purified Enzymes. J. Biol. Chem. 1942, 144, 423–437. 10.1016/S0021-9258(18)72525-8. [DOI] [Google Scholar]
- Von Euler-Chelpin H.Fermentation of Sugars and Fermentative Enzymes; Nobelstiftelsen. Nobelprize.org, 1930. [Google Scholar]
- Harden A.The Function of Phosphate in Alcoholic Fermentation. Nobelstiftelsen. Nobelprize.org, 1929. [Google Scholar]
- Kohler R. E. The Background to Arthur Harden’s Discovery of Cozymase. Bull. Hist. Med. 1974, 48, 22–40. [PubMed] [Google Scholar]
- Chandel N. S. Glycolysis. Cold Spring Harb. Perspect. Biol. 2021, 13, a040535 10.1101/cshperspect.a040535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Entner N.; Doudoroff M. Glucose and Gluconic Acid Oxidation of Pseudomonas saccharophila. J. Biol. Chem. 1952, 196, 853–862. 10.1016/S0021-9258(19)52415-2. [DOI] [PubMed] [Google Scholar]
- Hanes C. S. The Breakdown and Synthesis of Starch by an Enzyme System from Pea Seeds. Proc. R. Soc. B Biol. Sci. 1940, 128, 421–450. [Google Scholar]
- Tze-Fei Wong J. Charles Samuel Hanes. Trends Biochem. Sci. 1979, 4, N130–N131. 10.1016/0968-0004(79)90434-1. [DOI] [Google Scholar]
- Green A. A.; Cori G. T. Crystalline Muscle Phosphorylase I. Preparation, Properties, and Molecular Weight. J. Biol. Chem. 1943, 151, 21–29. 10.1016/S0021-9258(18)72110-8. [DOI] [Google Scholar]
- Helmreich E.; Cori C. F. The Role of Adenylic Acid in the Activation of Phosphorylase*. Proc. Natl. Acad. Sci. U.S.A. 1964, 51, 131–138. 10.1073/pnas.51.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J.; Wyman J.; Changeux J. P. On the Nature of Allosteric Transitions: A Plausible Model. J. Mol. Biol. 1965, 12, 88–118. 10.1016/S0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- Krebs E. G.; Fischer E. H. The Phosphorylase b to a Converting Enzyme of Rabbit Skeletal Muscle. Biochim. Biophys. Acta 1956, 20, 150–157. 10.1016/0006-3002(56)90273-6. [DOI] [PubMed] [Google Scholar]
- Recondo E.; Leloir L. F. Adenosine Diphosphate Glucose and Starch Synthesis. Biochem. Biophys. Res. Commun. 1961, 6, 85–88. 10.1016/0006-291X(61)90389-8. [DOI] [PubMed] [Google Scholar]
- Leloir L. F.; Olavarría J. M.; Goldemberg S. H.; Carminatti H. Biosynthesis of Glycogen from Uridine Diphosphate Glucose. Arch. Biochem. Biophys. 1959, 81, 508–520. 10.1016/0003-9861(59)90232-2. [DOI] [PubMed] [Google Scholar]
- Kresge N.; Simoni R. D.; Hill R. L. Earl W. Sutherland’s Discovery of Cyclic Adenine Monophosphate and the Second Messenger System. J. Biol. Chem. 2005, 280, e39–e40. 10.1016/S0021-9258(19)48258-6. [DOI] [Google Scholar]
- Gabel N. W.; Ponnamperuma C. Model for Origin of Monosaccharides. Nature 1967, 216, 453–455. 10.1038/216453a0. [DOI] [PubMed] [Google Scholar]
- Saravanamurugan S.; Paniagua M.; Melero J. A.; Riisager A. Efficient Isomerization of Glucose to Fructose over Zeolites in Consecutive Reactions in Alcohol and Aqueous Media. J. Am. Chem. Soc. 2013, 135, 5246–5249. 10.1021/ja400097f. [DOI] [PubMed] [Google Scholar]
- Cleaves H. J. The Prebiotic Geochemistry of Formaldehyde. Precambrian Res. 2008, 164, 111–118. 10.1016/j.precamres.2008.04.002. [DOI] [Google Scholar]
- Ruiz-Mirazo K.; Briones C.; De La Escosura A. Prebiotic Systems Chemistry: New Perspectives for the Origins of Life. Chem. Rev. 2014, 114, 285–366. 10.1021/cr2004844. [DOI] [PubMed] [Google Scholar]
- Trost B. M. Asymmetric Catalysis: An Enabling Science. Proc. Natl. Acad. Sci. USA. 2004, 101, 5348–5355. 10.1073/pnas.0306715101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi J. Origin of Saccharides and Their Roles on Evolution of Life. Orig. Life Evol. Biosphere 1996, 26, 352–353. 10.1007/BF02459804. [DOI] [Google Scholar]
- Keller M. A.; Turchyn A. V.; Ralser M. Non-Enzymatic Glycolysis and Pentose Phosphate Pathway-like Reactions in a Plausible Archean Ocean. Mol. Syst. Biol. 2014, 10 (4), 725. 10.1002/msb.20145228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronimus R. S.; Morgan H. W. Distribution and Phylogenies of Enzymes of the Embden-Meyerhof-Parnas Pathway from Archaea and Hyperthermophilic Bacteria Support a Gluconeogenic Origin of Metabolism. Archaea 2003, 1, 162593 10.1155/2003/162593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson D.; Cox M.. Chapter 14. Glycolysis, Gluconeogenesis, and the Pentose Phosphate Pathway. In Lehninger Principles of Biochemistry .; Freeman W. H., 2017.
- Shimizu K.3 - Metabolic Regulation by Global Regulators in Response to Culture Environment. In Bacterial Cellular Metabolic Systems; Shimizu K., Ed.; Woodhead Publishing, 2013; pp 95–213. [Google Scholar]
- Rabbani N.; Thornalley P. J. Protein Glycation – Biomarkers of Metabolic Dysfunction and Early-Stage Decline in Health in the Era of Precision Medicine. Redox Biol. 2021, 42, 101920 10.1016/j.redox.2021.101920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironova R.; Niwa T.; Handzhiyski Y.; Sredovska A.; Ivanov I. Evidence for Non-Enzymatic Glycosylation of Escherichia coli Chromosomal DNA. Mol. Microbiol. 2005, 55, 1801–1811. 10.1111/j.1365-2958.2005.04504.x. [DOI] [PubMed] [Google Scholar]
- Wolfenden R.; Lu X.; Young G. Spontaneous Hydrolysis of Glycosides. J. Am. Chem. Soc. 1998, 120, 6814–6815. 10.1021/ja9813055. [DOI] [Google Scholar]
- Olsson C.; Swenson J. Structural Comparison between Sucrose and Trehalose in Aqueous Solution. J. Phys. Chem. B. 2020, 124, 3074–3082. 10.1021/acs.jpcb.9b09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T.; Amrein H. Gluconeogenesis: An Ancient Biochemical Pathway with a New Twist. Fly (Austin) 2017, 11, 218–223. 10.1080/19336934.2017.1283081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avonce N.; Mendoza-Vargas A.; Morett E.; Iturriaga G. Insights on the Evolution of Trehalose Biosynthesis. BMC Evol. Biol. 2006, 6, 109. 10.1186/1471-2148-6-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn J. E. Evolution of Sucrose Synthesis. Plant Physiol. 2002, 128, 1490–1500. 10.1104/pp.010898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porchia A. C.; Salerno G. L. Sucrose Biosynthesis in a Prokaryotic Organism: Presence of Two Sucrose-Phosphate Synthases in Anabaena with Remarkable Differences Compared with the Plant Enzymes. Proc. Natl. Acad. Sci. U. S. A. 1996, 93 (24), 13600–13604. 10.1073/pnas.93.24.13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynd L. R.; Weimer P. J.; van Zyl W. H.; Pretorius I. S. Microbial Cellulose Utilization: Fundamentals and Biotechnology. Microbiol. Mol. Biol. Rev. 2002, 66 (4), 506–577. 10.1128/MMBR.66.3.506-577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Sharkey T. D. The Importance of Maltose in Transitory Starch Breakdown. Plant Cell Environ. 2006, 29, 353–366. 10.1111/j.1365-3040.2005.01480.x. [DOI] [PubMed] [Google Scholar]
- Iskandar C. F.; Cailliez-Grimal C.; Borges F.; Revol-Junelles A. M. Review of Lactose and Galactose Metabolism in Lactic Acid Bacteria Dedicated to Expert Genomic Annotation. Trends Food Sci. Technol. 2019, 88, 121–132. 10.1016/j.tifs.2019.03.020. [DOI] [Google Scholar]
- Synytsya A.; Novak M. Structural Analysis of Glucans. Ann. Transl. Med. 2014, 2, 17. 10.3978/j.issn.2305-5839.2014.02.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T.; Sasaki S.; Utsumi Y.; Fujita N.; Umeda K.; Sawada T.; Kubo A.; Abe J. I.; Colleoni C.; Ball S.; et al. Comparison of Chain-Length Preferences and Glucan Specificities of Isoamylase-Type α-Glucan Debranching Enzymes from Rice, Cyanobacteria and Bacteria. PLoS ONE 2016, 11, e0157020 10.1371/journal.pone.0157020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Liu Q.; Hu J.; Asenso J.; Wise M. J.; Wu X.; Ma C.; Chen X.; Yang J.; Tang D. Structure and Evolution of Glycogen Branching Enzyme N-Termini from Bacteria. Front. Microbiol. 2019, 9, 3354. 10.3389/fmicb.2018.03354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Römling U.; Galperin M. Y. Bacterial Cellulose Biosynthesis: Diversity of Operons, Subunits, Products, and Functions. Trends Microbiol. 2015, 23, 545–557. 10.1016/j.tim.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanke A.; Malisic M.; Wawra S.; Zuccaro A. Unraveling the Sugar Code: The Role of Microbial Extracellular Glycans in Plant-Microbe Interactions. J. Exp. Bot. 2021, 72, 15–35. 10.1093/jxb/eraa414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinke N.; Vidal-Melgosa S.; Schultz-Johansen M.; Hehemann J. H. Biocatalytic Quantification of α-Glucan in Marine Particulate Organic Matter. MicrobiologyOpen 2022, 11, e1289 10.1002/mbo3.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siriwardana L. S.; Gall A. R.; Buller C. S.; Esch S. W.; Kenyon W. J. Factors Affecting Accumulation and Degradation of Curdlan, Trehalose and Glycogen in Cultures of Cellulomonas Flavigena Strain KU (ATCC 53703). Antonie Van Leeuwenhoek 2011, 99, 681–695. 10.1007/s10482-010-9544-z. [DOI] [PubMed] [Google Scholar]
- Strachan J. Solubility of Cellulose in Water. Nature 1938, 141, 332–333. 10.1038/141332b0. [DOI] [Google Scholar]
- Whistler R. L.Solubility of Polysaccharides and Their Behavior in Solution. Carbohydrates in Solution. 1973, Chapter 14. pp 242–255. https://doi.org/10.1021/ba-1971-0117.ch014.
- Kerly M. The Solubility of Glycogen. Biochem. J. 1930, 24, 67–76. 10.1042/bj0240067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuente J. O.; Comino N.; Trastoy B.; D’Angelo C.; Guerin M. E. Structural Basis of Glycogen Metabolism in Bacteria. Biochem. J. 2019, 476, 2059–2092. 10.1042/BCJ20170558. [DOI] [PubMed] [Google Scholar]
- Smythe C.; Cohen P. The Discovery of Glycogenin and the Priming Mechanism for Glycogen Biogenesis. Eur. J. Biochem. 1991, 200, 625–631. 10.1111/j.1432-1033.1991.tb16225.x. [DOI] [PubMed] [Google Scholar]
- Prats C.; Graham T. E.; Shearer J. The Dynamic Life of the Glycogen Granule. J. Biol. Chem. 2018, 293, 7089–7098. 10.1074/jbc.R117.802843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X.; Sullivan M. A.; Nada S. S.; Deng B.; Schulz B. L.; Gilbert R. G. Proteomic Investigation of the Binding Agent between Liver Glycogen β Particles. ACS Omega 2018, 3, 3640–3645. 10.1021/acsomega.8b00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybicka K. K. Glycosomes--the Organelles of Glycogen Metabolism. Tissue Cell 1996, 28, 253–265. 10.1016/S0040-8166(96)80013-9. [DOI] [PubMed] [Google Scholar]
- Wang L.; Liu Q.; Tan X.; Wang Z.; Wang M.; Wise M. J.; Li C.; Ma C.; Li E.; Deng B.; et al. Molecular Structure of Glycogen in Escherichia coli. Biomacromolecules 2019, 20, 2821–2829. 10.1021/acs.biomac.9b00586. [DOI] [PubMed] [Google Scholar]
- Rashid A. M.; Batey S. F. D.; Syson K.; Koliwer-Brandl H.; Miah F.; Barclay J. E.; Findlay K. C.; Nartowski K. P.; Khimyak Y. Z.; Kalscheuer R.; et al. Assembly of α-Glucan by GlgE and GlgB in Mycobacteria and Streptomycetes. Biochemistry 2016, 55, 3270–3284. 10.1021/acs.biochem.6b00209. [DOI] [PubMed] [Google Scholar]
- Maddelein M. L.; Libessart N.; Bellanger F.; Delrue B.; D’Hulst C.; Van den Koornhuyse N.; Fontaine T.; Wieruszeski J. M.; Decq A.; Ball S. Toward an Understanding of the Biogenesis of the Starch Granule. Determination of Granule-Bound and Soluble Starch Synthase Functions in Amylopectin Synthesis. J. Biol. Chem. 1994, 269, 25150–25157. 10.1016/S0021-9258(17)31510-7. [DOI] [PubMed] [Google Scholar]
- Seung D. Amylose in Starch: Towards an Understanding of Biosynthesis, Structure and Function. New Phytol. 2020, 228, 1490–1504. 10.1111/nph.16858. [DOI] [PubMed] [Google Scholar]
- Imberty A.; Buléon A.; Tran V.; Pérez S. Recent Advances in Knowledge of Starch Structure. Starch - Stärke 1991, 43, 375–384. 10.1002/star.19910431002. [DOI] [Google Scholar]
- Sheath R. G.; Hellebust J. A.; Sawa T. Ultrastructure of the Floridean Starch Granule. Phycologia 1981, 20, 292–297. 10.2216/i0031-8884-20-3-292.1. [DOI] [Google Scholar]
- Patron N. J.; Keeling P. J. Common Evolutionary Origin of Starch Biosynthetic Enzymes in Green and Red Algae. J. Phycol. 2005, 41, 1131–1141. 10.1111/j.1529-8817.2005.00135.x. [DOI] [Google Scholar]
- Viola R.; Nyvall P.; Pedersén M. The Unique Features of Starch Metabolism in Red Algae. Proc. Biol. Sci. 2001, 268, 1417–1422. 10.1098/rspb.2001.1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki E.; Onoda M.; Colleoni C.; Ball S.; Fujita N.; Nakamura Y. Physicochemical Variation of Cyanobacterial Starch, the Insoluble α-Glucans in Cyanobacteria. Plant Cell Physiol. 2013, 54, 465–473. 10.1093/pcp/pcs190. [DOI] [PubMed] [Google Scholar]
- Nakamura Y.; Takahashi J. I.; Sakurai A.; Inaba Y.; Suzuki E.; Nihei S.; Fujiwara S.; Tsuzuki M.; Miyashita H.; Ikemoto H.; et al. Some Cyanobacteria Synthesize Semi-Amylopectin Type Alpha-Polyglucans Instead of Glycogen. Plant Cell Physiol. 2005, 46, 539–545. 10.1093/pcp/pci045. [DOI] [PubMed] [Google Scholar]
- Suzuki E.; Suzuki R. Variation of Storage Polysaccharides in Phototrophic Microorganisms. J. Appl. Glycosci. 2013, 60, 21–27. 10.5458/jag.jag.JAG-2012_016. [DOI] [Google Scholar]
- Sagan L. On the Origin of Mitosing Cells. J. Theor. Biol. 1967, 14, 255–274. 10.1016/0022-5193(67)90079-3. [DOI] [PubMed] [Google Scholar]
- Ball S.; Colleoni C.; Cenci U.; Raj J. N.; Tirtiaux C. The Evolution of Glycogen and Starch Metabolism in Eukaryotes Gives Molecular Clues to Understand the Establishment of Plastid Endosymbiosis. J. Exp. Bot. 2011, 62, 1775–1801. 10.1093/jxb/erq411. [DOI] [PubMed] [Google Scholar]
- Latgé J. P.; Beauvais A.; Chamilos G. The Cell Wall of the Human Fungal Pathogen Aspergillus fumigatus: Biosynthesis, Organization, Immune Response, and Virulence. Annu. Rev. Microbiol. 2017, 71, 99–116. 10.1146/annurev-micro-030117-020406. [DOI] [PubMed] [Google Scholar]
- Synytsya A.; Novák M. Structural Diversity of Fungal Glucans. Carbohydr. Polym. 2013, 92, 792–809. 10.1016/j.carbpol.2012.09.077. [DOI] [PubMed] [Google Scholar]
- Ghassemi N.; Poulhazan A.; Deligey F.; Mentink-Vigier F.; Marcotte I.; Wang T. Solid-State NMR Investigations of Extracellular Matrixes and Cell Walls of Algae, Bacteria, Fungi, and Plants. Chem. Rev. 2022, 122, 10036–10086. 10.1021/acs.chemrev.1c00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambou T.; Dinadayala P.; Stadthagen G.; Barilone N.; Bordat Y.; Constant P.; Levillain F.; Neyrolles O.; Gicquel B.; Lemassu A.; et al. Capsular Glucan and Intracellular Glycogen of Mycobacterium Tuberculosis: Biosynthesis and Impact on the Persistence in Mice. Mol. Microbiol. 2008, 70, 762–774. 10.1111/j.1365-2958.2008.06445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch D. J.; Fountain T. L.; Mazurkiewicz J. E.; Banas J. A. Glucan-Binding Proteins are Essential for Shaping Streptococcus mutans Biofilm Architecture. FEMS Microbiol. Lett. 2007, 268, 158–165. 10.1111/j.1574-6968.2006.00576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid A. M.; Batey S. F. D.; Syson K.; Koliwer-Brandl H.; Miah F.; Barclay J. E.; Findlay K. C.; Nartowski K. P.; Khimyak Y. Z.; Kalscheuer R.; et al. Assembly of α-Glucan by GlgE and GlgB in Mycobacteria and Streptomycetes. Biochemistry 2016, 55, 3270–3284. 10.1021/acs.biochem.6b00209. [DOI] [PubMed] [Google Scholar]
- Monsan P.; Remaud-Siméon M.; André I. Transglucosidases as Efficient Tools for Oligosaccharide and Glucoconjugate Synthesis. Curr. Opin. Microbiol. 2010, 13, 293–300. 10.1016/j.mib.2010.03.002. [DOI] [PubMed] [Google Scholar]
- Leathers T. D.; Côté G. L. Biofilm Formation by Exopolysaccharide Mutants of Leuconostoc mesenteroides Strain NRRL B-1355. Appl. Microbiol. Biotechnol. 2008, 78, 1025–1031. 10.1007/s00253-008-1384-7. [DOI] [PubMed] [Google Scholar]
- Doppalapudi S.; Katiyar S.; Domb A. J.; Khan W.. Biodegradable Natural Polymers. In Advanced Polymers in Medicine; Springer International Publishing, 2015; pp 33–66. https://doi.org/10.1007/978-3-319-12478-0_2. [Google Scholar]
- Eydallin G.; Viale A. M.; Morán-Zorzano M. T.; Muñoz F. J.; Montero M.; Baroja-Fernández E.; Pozueta-Romero J. Genome-Wide Screening of Genes Affecting Glycogen Metabolism in Escherichia coli K-12. FEBS Lett. 2007, 581, 2947–2953. 10.1016/j.febslet.2007.05.044. [DOI] [PubMed] [Google Scholar]
- Preiss J.Glycogen: Biosynthesis and Regulation. EcoSal Plus 2014, 6. https://doi.org/10.1128/ecosalplus.esp-0015-2014. 10.1128/ecosalplus.esp-0015-2014 [DOI] [PubMed] [Google Scholar]
- Preiss J.; Romeo T. Molecular Biology and Regulatory Aspects of Glycogen Biosynthesis in Bacteria. Prog. Nucleic Acid Res. Mol. Biol. 1994, 47, 299–329. 10.1016/S0079-6603(08)60255-X. [DOI] [PubMed] [Google Scholar]
- Wilson W. A.; Roach P. J.; Montero M.; Baroja-Fernández E.; Muñoz F. J.; Eydallin G.; Viale A. M.; Pozueta-Romero J. Regulation of Glycogen Metabolism in Yeast and Bacteria. FEMS Microbiol. Rev. 2010, 34, 952–985. 10.1111/j.1574-6976.2010.00220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz A.; Forchhammer K. Glycogen, a Major Player for Bacterial Survival and Awakening from Dormancy. Future Microbiol. 2017, 12, 101–104. 10.2217/fmb-2016-0218. [DOI] [PubMed] [Google Scholar]
- Wang M.; Liu Q.; Kang X.; Zhu Z.; Yang H.; Xi X.; Zhang X.; Du Y.; Guo M.; Tang D.; et al. Glycogen Metabolism Impairment via Single Gene Mutation in the glgBXCAP Operon Alters the Survival Rate of Escherichia coli Under Various Environmental Stresses. Front. Microbiol. 2020, 11, 588099 10.3389/fmicb.2020.588099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange R. E. Bacterial “Glycogen” and Survival. Nature 1968, 220, 606–607. 10.1038/220606a0. [DOI] [PubMed] [Google Scholar]
- Bourassa L.; Camilli A. Glycogen Contributes to the Environmental Persistence and Transmission of Vibrio cholerae. Mol. Microbiol. 2009, 72, 124–138. 10.1111/j.1365-2958.2009.06629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson J. F. Carbon and Energy Storage in Bacteria. J. Gen. Microbiol. 1963, 32, 171–176. 10.1099/00221287-32-2-171. [DOI] [PubMed] [Google Scholar]
- Illingworth B.; Larner J.; Cori G. T. Structure of Glycogens and Amylopectins: I. Enzymatic Determination of Chain Length. J. Biol. Chem. 1952, 199, 631–640. 10.1016/S0021-9258(18)38501-6. [DOI] [PubMed] [Google Scholar]
- Larner J.; Illingworth B.; Cori G. T.; Cori C. F. Structure of Glycogens and Amylopectins: II. Analysis by Stepwise Enzymatic Degradation. J. Biol. Chem. 1952, 199, 641–651. 10.1016/S0021-9258(18)38502-8. [DOI] [PubMed] [Google Scholar]
- Meyer K. H.; Bernfeld P.; Boissonnas R. A.; Gurtler P.; Noelting G. Starch Solutions and Pastes and their Molecular Interpretation. J. Phys. Colloid Chem. 1949, 53, 319–334. 10.1021/j150468a001. [DOI] [PubMed] [Google Scholar]
- Abdel-Akher M.; Smith F. The Repeating Unit of Glycogen. J. Am. Chem. Soc. 1951, 73, 994–996. 10.1021/ja01147a031. [DOI] [Google Scholar]
- Wang L.; Wise M. J. Glycogen with Short Average Chain Length Enhances Bacterial Durability. Naturwissenschaften 2011, 98, 719–729. 10.1007/s00114-011-0832-x. [DOI] [PubMed] [Google Scholar]
- Meléndez R.; Meléndez-Hevia E.; Canela E. I. The Fractal Structure of Glycogen: A Clever Solution to Optimize Cell Metabolism. Biophys. J. 1999, 77, 1327–1332. 10.1016/S0006-3495(99)76982-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.; Zhang P.; Li S.; Tan X.; Hu Z.; Deng B.; Wang K.; Li C.; Sullivan M. A.; Li E.; et al. Molecular-Size Dependence of Glycogen Enzymatic Degradation and Its Importance for Diabetes. Eur. Polym. J. 2016, 82, 175–180. 10.1016/j.eurpolymj.2016.07.017. [DOI] [Google Scholar]
- Li F.; Wang M. M.; Liu Q. H.; Ma Z. W.; Wang J. J.; Wang Z. Y.; Tang J. W.; Lyu J. W.; Zhu Z. B.; Wang L. Molecular Mechanisms of Glycogen Particle Assembly in Escherichia Coli. Carbohydr. Polym. 2023, 299, 120200 10.1016/j.carbpol.2022.120200. [DOI] [PubMed] [Google Scholar]
- Gunja-Smith Z.; Marshall J. J.; Mercier C.; Smith E. E.; Whelan W. J. A Revision of the Meyer-Bernfeld Model of Glycogen and Amylopectin. FEBS Lett. 1970, 12, 101–104. 10.1016/0014-5793(70)80573-7. [DOI] [PubMed] [Google Scholar]
- Goldsmith E.; Sprang S.; Fletterick R. Structure of Maltoheptaose by Difference Fourier Methods and a Model for Glycogen. J. Mol. Biol. 1982, 156, 411–427. 10.1016/0022-2836(82)90336-9. [DOI] [PubMed] [Google Scholar]
- Meléndez R.; Meléndez-Hevia E.; Cascante M. How Did Glycogen Structure Evolve to Satisfy the Requirement for Rapid Mobilization of Glucose? A Problem of Physical Constraints in Structure Building. J. Mol. Evol. 1997, 45, 446–455. 10.1007/PL00006249. [DOI] [PubMed] [Google Scholar]
- Melendez-Hevia E.; Waddell T. G.; Shelton E. D. Optimization of Molecular Design in the Evolution of Metabolism: The Glycogen Molecule. Biochem. J. 1993, 295, 477–483. 10.1042/bj2950477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach P. J.; Depaoli-Roach A. A.; Hurley T. D.; Tagliabracci V. S. Glycogen and Its Metabolism: Some New Developments and Old Themes. Biochem. J. 2012, 441, 763–787. 10.1042/BJ20111416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besford Q. A.; Zeng X.-Y.; Ye J.-M.; Gray-Weale A. Liver Glycogen in Type 2 Diabetic Mice Is Randomly Branched as Enlarged Aggregates with Blunted Glucose Release. Glycoconj. J. 2016, 33, 41–51. 10.1007/s10719-015-9631-5. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Nada S. S.; Tan X.; Deng B.; Sullivan M. A.; Gilbert R. G. Exploring Glycogen Biosynthesis through Monte Carlo Simulation. Int. J. Biol. Macromol. 2018, 116, 264–271. 10.1016/j.ijbiomac.2018.05.027. [DOI] [PubMed] [Google Scholar]
- Sani M.; Houben E. N.; Geurtsen J.; Pierson J.; de Punder K.; van Zon M.; Wever B.; Piersma S. R.; Jimenez C. R.; Daffe M.; et al. Direct Visualization by Cryo-EM of the Mycobacterial Capsular Layer: A Labile Structure Containing ESX-1-Secreted Proteins. PLoS Pathog 2010, 6, e1000794 10.1371/journal.ppat.1000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortalo-Magne A.; Dupont M.-A.; Lemassu A.; Andersen A. B.; Gounon P.; Mamadou D. Molecular Composition of the Outermost Capsular Material of the Tubercle Bacillus. Microbiol. Read. 1995, 141, 1609–1620. 10.1099/13500872-141-7-1609. [DOI] [PubMed] [Google Scholar]
- Lemassu A.; Ortalo-Magne A.; Bardou F.; Silve G.; Laneelle M. A.; Daffe M. Extracellular and Surface-Exposed Polysaccharides of Non-Tuberculous Mycobacteria. Microbiol. Read. 1996, 142, 1513–1520. 10.1099/13500872-142-6-1513. [DOI] [PubMed] [Google Scholar]
- Dinadayala P.; Sambou T.; Daffé M.; Lemassu A. Comparative Structural Analyses of the Alpha-Glucan and Glycogen from Mycobacterium bovis. Glycobiology 2008, 18, 502–508. 10.1093/glycob/cwn031. [DOI] [PubMed] [Google Scholar]
- Dinadayala P.; Lemassu A.; Granovski P.; Cerantola S.; Winter N.; Daffe M. Revisiting the Structure of the Anti-Neoplastic Glucans of Mycobacterium bovis Bacille Calmette-Guerin. Structural Analysis of the Extracellular and Boiling Water Extract-Derived Glucans of the Vaccine Substrains. J. Biol. Chem. 2004, 279, 12369–12378. 10.1074/jbc.M308908200. [DOI] [PubMed] [Google Scholar]
- Ortalo-Magne A.; Andersen A. B.; Daffe M. The Outermost Capsular Arabinomannans and Other Mannoconjugates of Virulent and Avirulent Tubercle Bacilli. Microbiol. Read. 1996, 142, 927–935. 10.1099/00221287-142-4-927. [DOI] [PubMed] [Google Scholar]
- Schwebach J. R.; Glatman-Freedman A.; Gunther-Cummins L.; Dai Z.; Robbins J. B.; Schneerson R.; Casadevall A. Glucan Is a Component of the Mycobacterium tuberculosis Surface That Is Expressed in Vitro and in Vivo. Infect. Immun. 2002, 70, 2566–2575. 10.1128/IAI.70.5.2566-2575.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koliwer-Brandl H.; Syson K.; van de Weerd R.; Chandra G.; Appelmelk B.; Alber M.; Ioerger T. R.; Jacobs W. R.; Geurtsen J.; Bornemann S.; Kalscheuer R. Metabolic Network for the Biosynthesis of Intra- and Extracellular α-Glucans Required for Virulence of Mycobacterium tuberculosis. PLoS Pathog. 2016, 12, 1–26. 10.1371/journal.ppat.1005768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manners D. J. Recent Developments in Our Understanding of Glycogen Structure. Carbohydr. Polym. 1991, 16, 37–82. 10.1016/0144-8617(91)90071-J. [DOI] [Google Scholar]
- Prados-Rosales R.; Baena A.; Martinez L. R.; Luque-Garcia J.; Kalscheuer R.; Veeraraghavan U.; Camara C.; Nosanchuk J. D.; Besra G. S.; Chen B.; et al. Mycobacteria Release Active Membrane Vesicles That Modulate Immune Responses in a TLR2-Dependent Manner in Mice. J. Clin. Invest. 2011, 121, 1471–1483. 10.1172/JCI44261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leemhuis H.; Pijning T.; Dobruchowska J. M.; van Leeuwen S. S.; Kralj S.; Dijkstra B. W.; Dijkhuizen L. Glucansucrases: Three-Dimensional Structures, Reactions, Mechanism, α-Glucan Analysis and Their Implications in Biotechnology and Food Applications. J. Biotechnol. 2013, 163, 250–272. 10.1016/j.jbiotec.2012.06.037. [DOI] [PubMed] [Google Scholar]
- Monchois V.; Remaud-Simeon M.; Russell R. R.; Monsan P.; Willemot R. M. Characterization of Leuconostoc Mesenteroides NRRL B-512F Dextransucrase (DSRS) and Identification of Amino-Acid Residues Playing a Key Role in Enzyme Activity. Appl. Microbiol. Biotechnol. 1997, 48, 465–472. 10.1007/s002530051081. [DOI] [PubMed] [Google Scholar]
- Korakli M.; Vogel R. F. Structure/Function Relationship of Homopolysaccharide Producing Glycansucrases and Therapeutic Potential of Their Synthesised Glycans. Appl. Microbiol. Biotechnol. 2006, 71, 790–803. 10.1007/s00253-006-0469-4. [DOI] [PubMed] [Google Scholar]
- Monchois V.; Willemot R. M.; Monsan P. Glucansucrases: Mechanism of Action and Structure-Function Relationships. FEMS Microbiol. Rev. 1999, 23, 131–151. 10.1111/j.1574-6976.1999.tb00394.x. [DOI] [PubMed] [Google Scholar]
- Moulis C.; André I.; Remaud-Simeon M. GH13 Amylosucrases and GH70 Branching Sucrases, Atypical Enzymes in Their Respective Families. Cell. Mol. Life Sci. CMLS 2016, 73, 2661–2679. 10.1007/s00018-016-2244-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue N.; Svensson B.; Bai Y. Structure, Function and Enzymatic Synthesis of Glucosaccharides Assembled Mainly by Alpha1 –> 6 Linkages - A Review. Carbohydr. Polym. 2022, 275, 118705 10.1016/j.carbpol.2021.118705. [DOI] [PubMed] [Google Scholar]
- Kralj S.; van Geel-Schutten G. H.; Dondorff M. M. G.; Kirsanovs S.; van der Maarel M.; Dijkhuizen L. Glucan Synthesis in the Genus Lactobacillus: Isolation and Characterization of Glucansucrase Genes, Enzymes and Glucan Products from Six Different Strains. Microbiol. Read. 2004, 150, 3681–3690. 10.1099/mic.0.27321-0. [DOI] [PubMed] [Google Scholar]
- Larm O.; Lindberg B.; Svensson S. Studies on the Length of the Side Chains of the Dextran Elaborated by Leuconostoc Mesenteroides NRRL B-512. Carbohydr. Res. 1971, 20, 39–48. 10.1016/S0008-6215(00)84947-2. [DOI] [PubMed] [Google Scholar]
- Miyaji H.; Misaki A.; et al. The Structure of a Dextran Produced by Leuconostoc mesenteroides NRRL B-1397: The Linkages and Length of the Branches. Carbohydr. Res. 1973, 31, 277–287. 10.1016/S0008-6215(00)86192-3. [DOI] [PubMed] [Google Scholar]
- Walker G. J.; Pulkownik A. Degradation of Dextrans by an Alpha-1,6-Glucan Glucohydrolase from Streptococcus mitis. Carbohydr. Res. 1973, 29, 1–14. 10.1016/S0008-6215(00)82066-2. [DOI] [PubMed] [Google Scholar]
- Li X.; Wang X.; Meng X.; Dijkhuizen L.; Liu W. Structures, Physico-Chemical Properties, Production and (Potential) Applications of Sucrose-Derived Alpha-d-Glucans Synthesized by Glucansucrases. Carbohydr. Polym. 2020, 249, 116818 10.1016/j.carbpol.2020.116818. [DOI] [PubMed] [Google Scholar]
- Vuillemin M.; Grimaud F.; Claverie M.; Rolland-Sabate A.; Garnier C.; Lucas P.; Monsan P.; Dols-Lafargue M.; Remaud-Simeon M.; Moulis C. A Dextran with Unique Rheological Properties Produced by the Dextransucrase from Oenococcus kitaharae DSM 17330. Carbohydr. Polym. 2018, 179, 10–18. 10.1016/j.carbpol.2017.09.056. [DOI] [PubMed] [Google Scholar]
- Xu L.; Zhang J. Bacterial Glucans: Production, Properties, and Applications. Appl. Microbiol. Biotechnol. 2016, 100 (21), 9023–9036. 10.1007/s00253-016-7836-6. [DOI] [PubMed] [Google Scholar]
- Semyonov D.; Ramon O.; Shoham Y.; Shimoni E. Enzymatically Synthesized Dextran Nanoparticles and their Use as Carriers for Nutraceuticals. Food Funct. 2014, 5 (10), 2463–2474. 10.1039/C4FO00103F. [DOI] [PubMed] [Google Scholar]
- Nisizawa T.; Imai S.; Araya S. Methylation Analysis of Extracellular Glucans Produced by Streptococcus mutans Strain JC 2. Arch. Oral. Biol. 1977, 22 (4), 281–285. 10.1016/0003-9969(77)90114-5. [DOI] [PubMed] [Google Scholar]
- Bowen W. H.; Koo H. Biology of Streptococcus mutans-Derived Glucosyltransferases: Role in Extracellular Matrix Formation of Cariogenic Biofilms. Caries Res. 2011, 45 (1), 69–86. 10.1159/000324598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis H. M.; Hines H. B.; Edwards J. R. Structural Elucidation of a Water-Insoluble Glucan Produced by a Cariogenic Oral Streptococcus. Carbohydr. Res. 1986, 156, 69–77. 10.1016/S0008-6215(00)90100-9. [DOI] [PubMed] [Google Scholar]
- Tsumuraya Y.; Misaki A. Structure of the Water-Insoluble Alpha-D-Glucan of Streptococcus salivarius HHT. Carbohydr. Res. 1979, 74, 217–225. 10.1016/S0008-6215(00)84778-3. [DOI] [PubMed] [Google Scholar]
- Lei L.; Yang Y.; Mao M.; Li H.; Li M.; Yang Y.; Yin J.; Hu T. Modulation of Biofilm Exopolysaccharides by the Streptococcus mutans vicX Gene. Front. Microbiol. 2015, 6, 1432–1432. 10.3389/fmicb.2015.01432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiater A.; Choma A.; Szczodrak J. Insoluble Glucans Synthesized by Cariogenic Streptococci: A Structural Study. J. Basic Microbiol. 1999, 39, 265–273. 10.1002/(SICI)1521-4028(199909)39:4<265::AID-JOBM265>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Meng X.; Pijning T.; Dobruchowska J. M.; Yin H.; Gerwig G. J.; Dijkhuizen L. Structural Determinants of Alternating (Alpha1 –> 4) and (Alpha1 –> 6) Linkage Specificity in Reuteransucrase of Lactobacillus reuteri. Sci. Rep. 2016, 6, 35261–35261. 10.1038/srep35261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen S. S.; Kralj S.; van Geel-Schutten I. H.; Gerwig G. J.; Dijkhuizen L.; Kamerling J. P. Structural Analysis of the Alpha-D-Glucan (EPS180) Produced by the Lactobacillus reuteri Strain 180 Glucansucrase GTF180 Enzyme. Carbohydr. Res. 2008, 343, 1237–1250. 10.1016/j.carres.2008.01.042. [DOI] [PubMed] [Google Scholar]
- Bai Y.; Dobruchowska J. M.; van der Kaaij R. M.; Gerwig G. J.; Dijkhuizen L. Structural Basis for the Roles of Starch and Sucrose in Homo-Exopolysaccharide Formation by Lactobacillus reuteri 35-5. Carbohydr. Polym. 2016, 151, 29–39. 10.1016/j.carbpol.2016.05.048. [DOI] [PubMed] [Google Scholar]
- Miao M.; Ma Y.; Jiang B.; Huang C.; Li X.; Cui S. W.; Zhang T. Structural Investigation of a Neutral Extracellular Glucan from Lactobacillus reuteri SK24.003. Carbohydr. Polym. 2014, 106, 384–392. 10.1016/j.carbpol.2014.01.047. [DOI] [PubMed] [Google Scholar]
- Miao M.; Ma Y.; Huang C.; Jiang B.; Cui S. W.; Zhang T. Physicochemical Properties of a Water Soluble Extracellular Homopolysaccharide from Lactobacillus reuteri SK24.003. Carbohydr. Polym. 2015, 131, 377–383. 10.1016/j.carbpol.2015.05.066. [DOI] [PubMed] [Google Scholar]
- Cote G. L.; Robyt J. F. Isolation and Partial Characterization of an Extracellular Glucansucrase from Leuconostoc mesenteroides NRRL B-1355 That Synthesizes an Alternating (1 Goes to 6), (1 Goes to 3)-Alpha-D-Glucan. Carbohydr. Res 1982, 101, 57–74. 10.1016/S0008-6215(00)80795-8. [DOI] [PubMed] [Google Scholar]
- Seymour F. R.; Slodki M. E.; Plattner R. D.; Jeanes A. Six Unusual Dextrans: Methylation Structural Analysis by Combined g.l.c.—m.s. of per-O-Acetyl-Aldononitriles. Carbohydr. Res. 1977, 53, 153–166. 10.1016/S0008-6215(00)88083-0. [DOI] [Google Scholar]
- Wangpaiboon K.; Padungros P.; Nakapong S.; Charoenwongpaiboon T.; Rejzek M.; Field R. A.; Pichyangkura R. An α-1,6-and α-1,3-Linked Glucan Produced by Leuconostoc Citreum ABK-1 Alternansucrase with Nanoparticle and Film-Forming Properties. Sci. Rep. 2018, 8, 1–10. 10.1038/s41598-018-26721-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingos-Lopes M. F. P.; Lamosa P.; Stanton C.; Ross R. P.; Silva C. C. G. Isolation and Characterization of an Exopolysaccharide-Producing Leuconostoc citreum Strain from Artisanal Cheese. Lett. Appl. Microbiol. 2018, 67, 570–578. 10.1111/lam.13073. [DOI] [PubMed] [Google Scholar]
- Miao M.; Jia X.; Jiang B.; Wu S.; Cui S. W.; Li X. Elucidating Molecular Structure and Prebiotics Properties of Bioengineered α-D-Glucan from Leuconostoc citreum SK24.002. Food Hydrocoll. 2016, 54, 227–233. 10.1016/j.foodhyd.2015.10.013. [DOI] [Google Scholar]
- Vuillemin M.; Claverie M.; Brison Y.; Severac E.; Bondy P.; Morel S.; Monsan P.; Moulis C.; Remaud-Simeon M. Characterization of the First Alpha-(1–>3) Branching Sucrases of the GH70 Family. J. Biol. Chem. 2016, 291, 7687–7702. 10.1074/jbc.M115.688044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valette P.; Pelenc V.; Djouzi Z.; Andrieux C.; Paul F.; Monsan P.; Szylit O. Bioavailability of New Synthesised Glucooligosaccharides in the Intestinal Tract of Gnotobiotic Rats. J. Sci. Food Agric. 1993, 62, 121–127. 10.1002/jsfa.2740620204. [DOI] [Google Scholar]
- Brison Y.; Laguerre S.; Lefoulon F.; Morel S.; Monties N.; Potocki-Veronese G.; Monsan P.; Remaud-Simeon M. Branching Pattern of Gluco-Oligosaccharides and 1.5kDa Dextran Grafted by the Alpha-1,2 Branching Sucrase GBD-CD2. Carbohydr. Polym. 2013, 94, 567–576. 10.1016/j.carbpol.2013.01.064. [DOI] [PubMed] [Google Scholar]
- Sarbini S. R.; Kolida S.; Naeye T.; Einerhand A.; Brison Y.; Remaud-Simeon M.; Monsan P.; Gibson G. R.; Rastall R. A. In Vitro Fermentation of Linear and Alpha-1,2-Branched Dextrans by the Human Fecal Microbiota. Appl. Env. Microbiol. 2011, 77, 5307–5315. 10.1128/AEM.02568-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skov L. K.; Mirza O.; Henriksen A.; De Montalk G. P.; Remaud-Simeon M.; Sarcabal P.; Willemot R. M.; Monsan P.; Gajhede M. Amylosucrase, a Glucan-Synthesizing Enzyme from the Alpha-Amylase Family. J. Biol. Chem. 2001, 276, 25273–25278. 10.1074/jbc.M010998200. [DOI] [PubMed] [Google Scholar]
- MacKenzie C. R.; McDonald I. J.; Johnson K. G. Glycogen Metabolism in the Genus Neisseria: Synthesis from Sucrose by Amylosucrase. Can. J. Microbiol. 1978, 24 (4), 357–362. 10.1139/m78-060. [DOI] [PubMed] [Google Scholar]
- Riou J. Y.; Guibourdenche M.; Popoff M. Y. A New Taxon in the Genus Neisseria. Ann. Microbiol. Paris 1983, 134, 257–267. 10.1016/S0769-2609(83)80038-6. [DOI] [PubMed] [Google Scholar]
- Buttcher V.; Welsh T.; Willmitzer L.; Kossmann J. Cloning and Characterization of the Gene for Amylosucrase from Neisseria polysaccharea: Production of a Linear Alpha-1,4-Glucan. J. Bacteriol. 1997, 179, 3324–3330. 10.1128/jb.179.10.3324-3330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potocki de Montalk G.; Remaud-Simeon M.; Willemot R.-M.; Sarcabal P.; Planchot V.; Monsan P. Amylosucrase from Neisseria polysaccharea: Novel Catalytic Properties. FEBS Lett. 2000, 471, 219–223. 10.1016/S0014-5793(00)01406-X. [DOI] [PubMed] [Google Scholar]
- MacKenzie C. R.; Perry M. B.; McDonald I. J.; Johnson K. G. Structure of the D-Glucans Produced by Neisseria perflava. Can. J. Microbiol. 1978, 24, 1419–1422. 10.1139/m78-227. [DOI] [PubMed] [Google Scholar]
- Riou J. Y.; Guibourdenche M.; Perry M. B.; MacLean L. L.; Griffith D. W. Structure of the Exocellular D-Glucan Produced by Neisseria polysaccharea. Can. J. Microbiol. 1986, 32, 909–911. 10.1139/m86-167. [DOI] [PubMed] [Google Scholar]
- Ruby J. D.; Shirey R. E.; Gerencser V. F.; Stelzig D. A. Extracellular Iodophilic Polysaccharide Synthesized by Neisseria in Human Dental Plaque. J. Dent. Res. 1982, 61, 627–631. 10.1177/00220345820610050101. [DOI] [PubMed] [Google Scholar]
- Birkhed D.; Rosell K. G.; Bowden G. H. Structure of Extracellular Polysaccharides Synthesized from Sucrose by Neisseria Isolated from Human Dental Plaque. Arch. Oral Biol. 1979, 24, 63–66. 10.1016/0003-9969(79)90176-6. [DOI] [PubMed] [Google Scholar]
- Büttcher V.; Quanz M.; Willmitzer L. Molecular Cloning, Functional Expression and Purification of a Glucan Branching Enzyme from Neisseria denitrificans 1.. Biochim. Biophys. Acta BBA - Protein Struct. Mol. Enzymol. 1999, 1432, 406–412. 10.1016/S0167-4838(99)00101-6. [DOI] [PubMed] [Google Scholar]
- Mendoza-Barberá E.; Merino S.; Tomás J. Surface Glucan Structures in Aeromonas Spp. Mar. Drugs 2021, 19, 649. 10.3390/md19110649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomas J. M. The Main Aeromonas Pathogenic Factors. ISRN Microbiol. 2012, 2012, 256261 10.5402/2012/256261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino S.; Bouamama L.; Knirel Y. A.; Senchenkova S. N.; Regué M.; Tomás J. M. Aeromonas Surface Glucan Attached through the O-Antigen Ligase Represents a New Way to Obtain UDP-Glucose. PLoS ONE 2012, 7, e35707 10.1371/journal.pone.0035707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino S.; Jimenez N.; Molero R.; Bouamama L.; Regue M.; Tomas J. M. A UDP-HexNAc:Polyprenol-P GalNAc-1-P Transferase (WecP) Representing a New Subgroup of the Enzyme Family. J. Bacteriol. 2011, 193, 1943–1952. 10.1128/JB.01441-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez N.; Canals R.; Lacasta A.; Kondakova A. N.; Lindner B.; Knirel Y. A.; Merino S.; Regue M.; Tomas J. M. Molecular Analysis of Three Aeromonas Hydrophila AH-3 (Serotype O34) Lipopolysaccharide Core Biosynthesis Gene Clusters. J. Bacteriol. 2008, 190, 3176–3184. 10.1128/JB.01874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiles F.; Polyakov P.; Humbert F.; Francius G. Production of Extracellular Glycogen by Pseudomonas fluorescens: Spectroscopic Evidence and Conformational Analysis by Biomolecular Recognition. Biomacromolecules 2012, 13, 2118–2127. 10.1021/bm300497c. [DOI] [PubMed] [Google Scholar]
- Ghods S.; Sims I. M.; Moradali M. F.; Rehm B. H. Bactericidal Compounds Controlling Growth of the Plant Pathogen Pseudomonas syringae Pv. Actinidiae, Which Forms Biofilms Composed of a Novel Exopolysaccharide. Appl. Env. Microbiol. 2015, 81, 4026–4036. 10.1128/AEM.00194-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock S. D.; Syson K.; Little R. H.; Ward D.; Sifouna D.; Brown J. K. M.; Bornemann S.; Malone J. G. Trehalose and α-Glucan Mediate Distinct Abiotic Stress Responses in Pseudomonas aeruginosa. PLoS Genet. 2021, 17, e1009524 10.1371/journal.pgen.1009524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel J. A.; Boels J. M.; Beldman G.; Venema G. Glycogen in Bacillus subtilis: Molecular Characterization of an Operon Encoding Enzymes Involved in Glycogen Biosynthesis and Degradation. Mol. Microbiol. 1994, 11, 203–218. 10.1111/j.1365-2958.1994.tb00301.x. [DOI] [PubMed] [Google Scholar]
- Lepek V. C.; D’Antuono A. L.; Tomatis P. E.; Ugalde J. E.; Giambiagi S.; Ugalde R. A. Analysis of Mesorhizobium loti Glycogen Operon: Effect of Phosphoglucomutase (Pgm) and Glycogen Synthase (g/gA) Null Mutants on Nodulation of Lotus tenuis. Mol. Plant-Microbe Interact. 2002, 15, 368–375. 10.1094/MPMI.2002.15.4.368. [DOI] [PubMed] [Google Scholar]
- Ugalde J. E.; Lepek V.; Uttaro A.; Estrella J.; Iglesias A.; Ugalde R. A. Gene Organization and Transcription Analysis of the Agrobacterium tumefaciens Glycogen (Glg) Operon: Two Transcripts for the Single Phosphoglucomutase Gene. J. Bacteriol. 1998, 180, 6557–6564. 10.1128/JB.180.24.6557-6564.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marroquí S.; Zorreguieta A.; Santamaría C.; Temprano F.; Soberón M.; Megías M.; Downie J. A. Enhanced Symbiotic Performance by Rhizobium tropici Glycogen Synthase Mutants. J. Bacteriol. 2001, 183, 854–864. 10.1128/JB.183.3.854-864.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romeo T.; Preiss J. Genetic Regulation of Glycogen Biosynthesis in Escherichia coli: In Vitro Effects of Cyclic AMP and Guanosine 5’-Diphosphate 3’-Diphosphate and Analysis of in Vivo Transcripts. J. Bacteriol. 1989, 171, 2773–2782. 10.1128/jb.171.5.2773-2782.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniélou Y. P.; Sagot M. F.; Boyer F.; Viari A. Bacterial Syntenies: An Exact Approach with Gene Quorum. BMC Bioinformatics 2011, 12, 193. 10.1186/1471-2105-12-193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg J.; Commichau F. M. Harnessing Underground Metabolism for Pathway Development. Trends Biotechnol. 2019, 37, 29–37. 10.1016/j.tibtech.2018.08.001. [DOI] [PubMed] [Google Scholar]
- Peracchi A. The Limits of Enzyme Specificity and the Evolution of Metabolism. Trends Biochem. Sci. 2018, 43, 984–996. 10.1016/j.tibs.2018.09.015. [DOI] [PubMed] [Google Scholar]
- Goh Y. J.; Klaenhammer T. R. A Functional Glycogen Biosynthesis Pathway in Lactobacillus acidophilus: Expression and Analysis of the Glg Operon. Mol. Microbiol. 2013, 89, 1187–1200. 10.1111/mmi.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfrom M. L.; Pletcher D. E. The Structure of the Cori Ester. J. Am. Chem. Soc. 1941, 63, 1050–1053. 10.1021/ja01849a044. [DOI] [Google Scholar]
- Espada J. Enzymic Synthesis of Adenosine Diphosphate Glucose from Glucose 1-Phosphate and Adenosine Triphosphate. J. Biol. Chem. 1962, 237, 3577–3581. 10.1016/S0021-9258(19)84491-5. [DOI] [Google Scholar]
- Paule M. R.; Preiss J. Biosynthesis of Bacterial Glycogen: X. The Kinetic Mechanism of Adenosine Diphosphoglucose Pyrophosphorylase from Rhodospirillum rubrum. J. Biol. Chem. 1971, 246, 4602–4609. 10.1016/S0021-9258(18)62053-8. [DOI] [Google Scholar]
- Cifuente J. O.; Comino N.; D’Angelo C.; Marina A.; Gil-Carton D.; Albesa-Jové D.; Guerin M. E. The Allosteric Control Mechanism of Bacterial Glycogen Biosynthesis Disclosed by cryoEM. Curr. Res. Struct. Biol. 2020, 2, 89–103. 10.1016/j.crstbi.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preiss J. Regulation of Adenosine Diphosphate Glucose Pyrophosphorylase. Adv. Enzymol. Relat. Areas Mol. Biol. 1978, 46, 317–381. 10.1002/9780470122914.ch5. [DOI] [PubMed] [Google Scholar]
- Cifuente J. O.; Comino N.; Madariaga-Marcos J.; López-Fernández S.; García-Alija M.; Agirre J.; Albesa-Jové D.; Guerin M. E. Structural Basis of Glycogen Biosynthesis Regulation in Bacteria. Structure 2016, 24, 1613–1622. 10.1016/j.str.2016.06.023. [DOI] [PubMed] [Google Scholar]
- Buschiazzo A.; Ugalde J. E.; Guerin M. E.; Shepard W.; Ugalde R. A.; Alzari P. M. Crystal Structure of Glycogen Synthase: Homologous Enzymes Catalyze Glycogen Synthesis and Degradation. EMBO J. 2004, 23, 3196–3205. 10.1038/sj.emboj.7600324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorvie T. J.; Loria P. M.; Tu M.; Han S.; Shrestha L.; Froese D. S.; Ferreira I. M.; Berg A. P.; Yue W. W. Molecular Basis for the Regulation of Human Glycogen Synthase by Phosphorylation and Glucose-6-Phosphate. Nat. Struct. Mol. Biol. 2022, 29, 628–638. 10.1038/s41594-022-00799-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr L.; Biswas D.; Daly L. A.; Browning C.; Vial S. C. M.; Maskell D. P.; Hudson C.; Bertrand J. A.; Pollard J.; Ranson N. A.; et al. Mechanism of Glycogen Synthase Inactivation and Interaction with Glycogenin. Nat. Commun. 2022, 13 (1), 3372. 10.1038/s41467-022-31109-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikwana V. M.; Khanna M.; Baskaran S.; Tagliabracci V. S.; Contreras C. J.; DePaoli-Roach A.; Roach P. J.; Hurley T. D. Structural Basis for 2’-Phosphate Incorporation into Glycogen by Glycogen Synthase. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 20976–20981. 10.1073/pnas.1310106111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox J.; Kennedy L. D.; Hawker J. S.; Ozbun J. L.; Greenberg E.; Lammel C.; Preiss J. De Novo Synthesis of Bacterial Glycogen and Plant Starch by ADPG: α-Glucan 4-Glucosyl Transferase. Ann. N. Y. Acad. Sci. 1973, 210, 90–103. 10.1111/j.1749-6632.1973.tb47564.x. [DOI] [PubMed] [Google Scholar]
- Ugalde J. E.; Parodi A. J.; Ugalde R. A. De Novo Synthesis of Bacterial Glycogen: Agrobacterium tumefaciens Glycogen Synthase Is Involved in Glucan Initiation and Elongation. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 10659–10663. 10.1073/pnas.1534787100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L.; Fawaz R.; Hovde S.; Gilbert L.; Chiou J.; Geiger J. H. Crystal Structures of Escherichia coli Branching Enzyme in Complex with Linear Oligosaccharides. Biochemistry 2015, 54, 6207–6218. 10.1021/acs.biochem.5b00228. [DOI] [PubMed] [Google Scholar]
- Buchbinder J. L.; Rath V. L.; Fletterick R. J. Structural Relationships Among Regulated and Unregulated Phosphorylases. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 191–209. 10.1146/annurev.biophys.30.1.191. [DOI] [PubMed] [Google Scholar]
- Kitaoka M.; Hayashi K. Carbohydrate-Processing Phosphorolytic Enzymes. Trends Glycosci. Glycotechnol. 2002, 14, 35–50. 10.4052/tigg.14.35. [DOI] [Google Scholar]
- Baranowski T.; Illingworth B.; Brown D. H.; Cori C. F. The Isolation of Pyridoxal-5-Phosphate from Crystalline Muscle Phosphorylase. Biochim. Biophys. Acta 1957, 25, 16–21. 10.1016/0006-3002(57)90410-9. [DOI] [PubMed] [Google Scholar]
- Alonso-Casajús N.; Dauvillée D.; Viale A. M.; Muñoz F. J.; Baroja-Fernández E.; Morán-Zorzano M. T.; Eydallin G.; Ball S.; Pozueta-Romero J. Glycogen Phosphorylase, the Product of the glgP Gene, Catalyzes Glycogen Breakdown by Removing Glucose Units from the Nonreducing Ends in Escherichia coli. J. Bacteriol. 2006, 188, 5266–5272. 10.1128/JB.01566-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballicora M. A.; Iglesias A. A.; Preiss J. ADP-Glucose Pyrophosphorylase, a Regulatory Enzyme for Bacterial Glycogen Synthesis. Microbiol. Mol. Biol. Rev. 2003, 67, 213–225. 10.1128/MMBR.67.2.213-225.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asención Diez M. D.; Aleanzi M. C.; Iglesias A. a.; Ballicora M. A. A Novel Dual Allosteric Activation Mechanism of Escherichia coli ADP-Glucose Pyrophosphorylase: The Role of Pyruvate. PLoS ONE 2014, 9, e103888 10.1371/journal.pone.0103888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons T. F.; Preiss J. Biosynthesis of Bacterial Glycogen. Isolation and Characterization of the Pyridoxal-P Allosteric Activator Site and the ADP-Glucose-Protected Pyridoxal-P Binding Site of Escherichia coli B ADP-Glucose Synthase. J. Biol. Chem. 1978, 253, 7638–7645. 10.1016/S0021-9258(17)34418-6. [DOI] [PubMed] [Google Scholar]
- Gardiol A.; Preiss J. Escherichia coli E-39 ADP Glucose Synthetase has Different Activation Kinetics from the Wild-Type Allosteric Enzyme. Arch. Biochem. Biophys. 1990, 280, 175–180. 10.1016/0003-9861(90)90533-5. [DOI] [PubMed] [Google Scholar]
- Seok Y. J.; Koo B. M.; Sondej M.; Peterkofsky A. Regulation of Escherichia coli Glycogen Phosphorylase Activity by HPr. J. Mol. Microbiol. Biotechnol. 2001, 3, 385–393. [PubMed] [Google Scholar]
- Seok Y.-J.; Sondej M.; Badawi P.; Lewis M. S.; Briggs M. C.; Jaffe H.; Peterkofsky A. High Affinity Binding and Allosteric Regulation of Escherichia coli Glycogen Phosphorylase by the Histidine Phosphocarrier Protein. HPr. J. Biol. Chem. 1997, 272, 26511–26521. 10.1074/jbc.272.42.26511. [DOI] [PubMed] [Google Scholar]
- Montero M.; Eydallin G.; Viale A. M.; Almagro G.; Muñoz F. J.; Rahimpour M.; Sesma M. T.; Baroja-Fernández E.; Pozueta-Romero J. Escherichia coli Glycogen Metabolism Is Controlled by the PhoP-PhoQ Regulatory System at Submillimolar Environmental Mg2+ Concentrations, and Is Highly Interconnected with a Wide Variety of Cellular Processes. Biochem. J. 2009, 424, 129–141. 10.1042/BJ20090980. [DOI] [PubMed] [Google Scholar]
- Deutscher J.; Francke C.; Postma P. W. How Phosphotransferase System-Related Protein Phosphorylation Regulates Carbohydrate Metabolism in Bacteria. Microbiol. Mol. Biol. Rev. 2006, 70, 939–1031. 10.1128/MMBR.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker C. S.; Morozov I.; Suzuki K.; Romeo T.; Babitzke P. CsrA Regulates Glycogen Biosynthesis by Preventing Translation of glgC in Escherichia coli. Mol. Microbiol. 2002, 44, 1599–1610. 10.1046/j.1365-2958.2002.02982.x. [DOI] [PubMed] [Google Scholar]
- Cashel M.; Gallant J. Two Compounds Implicated in the Function of the RC Gene of Escherichia Coli. Nature 1969, 221, 838–841. 10.1038/221838a0. [DOI] [PubMed] [Google Scholar]
- Almagro G.; Viale A. M.; Montero M.; Muñoz F. J.; Baroja-Fernández E.; Mori H.; Pozueta-Romero J. A cAMP/CRP-Controlled Mechanism for the Incorporation of Extracellular ADP-Glucose in Escherichia coli Involving NupC and NupG Nucleoside Transporters. Sci. Rep. 2018, 8, 15509. 10.1038/s41598-018-33647-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbein A. D.; Pastuszak I.; Tackett A. J.; Wilson T.; Pan Y. T. Last Step in the Conversion of Trehalose to Glycogen: A Mycobacterial Enzyme That Transfers Maltose from Maltose 1-Phosphate to Glycogen. J. Biol. Chem. 2010, 285, 9803–9812. 10.1074/jbc.M109.033944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalscheuer R.; Syson K.; Veeraraghavan U.; Weinrick B.; Biermann K. E.; Liu Z.; Sacchettini J. C.; Besra G.; Bornemann S.; Jacobs W. R. Jr. Self-Poisoning of Mycobacterium tuberculosis by Targeting GlgE in an Alpha-Glucan Pathway. Nat. Chem. Biol. 2010, 6, 376–384. 10.1038/nchembio.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra G.; Chater K. F.; Bornemann S. Unexpected and Widespread Connections between Bacterial Glycogen and Trehalose Metabolism. Microbiol. Read. Engl. 2011, 157, 1565–1572. 10.1099/mic.0.044263-0. [DOI] [PubMed] [Google Scholar]
- Garg S. K.; Alam M. S.; Kishan K. V.; Agrawal P. Expression and Characterization of Alpha-(1,4)-Glucan Branching Enzyme Rv1326c of Mycobacterium tuberculosis H37Rv. Protein Expr. Purif. 2007, 51, 198–208. 10.1016/j.pep.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Pal K.; Kumar S.; Sharma S.; Garg S. K.; Alam M. S.; Xu H. E.; Agrawal P.; Swaminathan K. Crystal Structure of Full-Length Mycobacterium tuberculosis H37Rv Glycogen Branching Enzyme. J. Biol. Chem. 2010, 285, 20897–20903. 10.1074/jbc.M110.121707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syson K.; Stevenson C. E. M.; Rejzek M.; Fairhurst S. A.; Nair A.; Bruton C. J.; Field R. A.; Chater K. F.; Lawson D. M.; Bornemann S. Structure of Streptomyces Maltosyltransferase GlgE, a Homologue of a Genetically Validated Anti-Tuberculosis Target. J. Biol. Chem. 2011, 286, 38298–38310. 10.1074/jbc.M111.279315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syson K.; Stevenson C. E.; Miah F.; Barclay J. E.; Tang M.; Gorelik A.; Rashid A. M.; Lawson D. M.; Bornemann S. Ligand-Bound Structures and Site-Directed Mutagenesis Identify the Acceptor and Secondary Binding Sites of Streptomyces Coelicolor Maltosyltransferase GlgE. J. Biol. Chem. 2016, 291, 21531–21540. 10.1074/jbc.M116.748160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenberger J. J.; Kumar Veleti S.; Wilson B. N.; Sucheck S. J.; Ronning D. R. Crystal Structures of Mycobacterium tuberculosis GlgE and Complexes with Non-Covalent Inhibitors. Sci. Rep. 2015, 5, 12830 10.1038/srep12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes V.; Blaszczyk M.; Maranha A.; Empadinhas N.; Blundell T. L. Structure of Mycobacterium thermoresistibile GlgE Defines Novel Conformational States That Contribute to the Catalytic Mechanism. Sci. Rep. 2015, 5, 17144 10.1038/srep17144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanna S.; Lindenberger J. J.; Gaitonde V. V.; Ronning D. R.; Sucheck S. J. Synthesis of 2-Deoxy-2,2-Difluoro-Alpha-Maltosyl Fluoride and Its X-Ray Structure in Complex with Streptomyces coelicolor GlgEI-V279S. Org Biomol Chem 2015, 13, 7542–7550. 10.1039/C5OB00867K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syson K.; Stevenson C. E. M.; Rashid A. M.; Saalbach G.; Tang M.; Tuukkanen A.; Svergun D. I.; Withers S. G.; Lawson D. M.; Bornemann S. Structural Insight into How Streptomyces coelicolor Maltosyl Transferase GlgE Binds α-Maltose 1-Phosphate and Forms a Maltosyl-Enzyme Intermediate. Biochemistry 2014, 53, 2494–2504. 10.1021/bi500183c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colpaert M.; Kadouche D.; Ducatez M.; Pillonel T.; Kebbi-Beghdadi C.; Cenci U.; Huang B.; Chabi M.; Maes E.; Coddeville B.; et al. Conservation of the Glycogen Metabolism Pathway Underlines a Pivotal Function of Storage Polysaccharides in Chlamydiae. Commun. Biol. 2021, 4, 296. 10.1038/s42003-021-01794-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalscheuer R.; Koliwer-Brandl H. Genetics of Mycobacterial Trehalose Metabolism. Microbiol Spectr. 2014, 2, MGM2-0002-2013. 10.1128/microbiolspec.MGM2-0002-2013. [DOI] [PubMed] [Google Scholar]
- Asención Diez M. D.; Demonte A. M.; Syson K.; Arias D. G.; Gorelik A.; Guerrero S. A.; Bornemann S.; Iglesias A. A. Allosteric Regulation of the Partitioning of Glucose-1-Phosphate between Glycogen and Trehalose Biosynthesis in Mycobacterium tuberculosis. Biochim. Biophys. Acta 2015, 1850, 13–21. 10.1016/j.bbagen.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte J.; Alber M.; Trujillo C. M.; Syson K.; Koliwer-Brandl H.; Deenen R.; Kohrer K.; DeJesus M. A.; Hartman T.; Jacobs W. R. Jr.; et al. Trehalose-6-Phosphate-Mediated Toxicity Determines Essentiality of OtsB2 in Mycobacterium tuberculosis In Vitro and in Mice. PLoS Pathog 2016, 12, e1006043 10.1371/journal.ppat.1006043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy H. N.; Stewart G. R.; Mischenko V. V.; Apt A. S.; Harris R.; McAlister M. S. B.; Driscoll P. C.; Young D. B.; Robertson B. D. The OtsAB Pathway Is Essential for Trehalose Biosynthesis in Mycobacterium tuberculosis. J. Biol. Chem. 2005, 280, 14524–14529. 10.1074/jbc.M414232200. [DOI] [PubMed] [Google Scholar]
- van Wyk N.; Drancourt M.; Henrissat B.; Kremer L. Current Perspectives on the Families of Glycoside Hydrolases of Mycobacterium tuberculosis: Their Importance and Prospects for Assigning Function to Unknowns. Glycobiology 2017, 27, 112–122. 10.1093/glycob/cww099. [DOI] [PubMed] [Google Scholar]
- Fraga J.; Maranha A.; Mendes V.; Pereira P. J. B.; Empadinhas N.; Macedo-Ribeiro S. Structure of Mycobacterial Maltokinase, the Missing Link in the Essential GlgE-Pathway. Sci. Rep. 2015, 5, 8026. 10.1038/srep08026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes V.; Maranha A.; Lamosa P.; da Costa M. S.; Empadinhas N. Biochemical Characterization of the Maltokinase from Mycobacterium bovis BCG. BMC Biochem. 2010, 11, 21. 10.1186/1471-2091-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miah F.; Koliwer-Brandl H.; Rejzek M.; Field R. A.; Kalscheuer R.; Bornemann S. Flux through Trehalose Synthase Flows from Trehalose to the Alpha Anomer of Maltose in Mycobacteria. Chem. Biol. 2013, 20, 487–493. 10.1016/j.chembiol.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy R.; Usha V.; Kermani A.; Scott D. J.; Hyde E. I.; Besra G. S.; Alderwick L. J.; Futterer K. Synthesis of Alpha-Glucan in Mycobacteria Involves a Hetero-Octameric Complex of Trehalose Synthase TreS and Maltokinase Pep2. ACS Chem. Biol. 2013, 8, 2245–2255. 10.1021/cb400508k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermani A. A.; Roy R.; Gopalasingam C.; Kocurek K. I.; Patel T. R.; Alderwick L. J.; Besra G. S.; Fütterer K. Crystal Structure of the TreS:Pep2 Complex, Initiating α-Glucan Synthesis in the GlgE Pathway of Mycobacteria. J. Biol. Chem. 2019, 294, 7348–7359. 10.1074/jbc.RA118.004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syson K.; Stevenson C. E. M.; Lawson D. M.; Bornemann S. Structure of the Mycobacterium smegmatis α-Maltose-1-Phosphate Synthase GlgM. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2020, 76 (4), 175–175. 10.1107/S2053230X20004343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauvillée D.; Kinderf I. S.; Li Z.; Kosar-Hashemi B.; Samuel M. S.; Rampling L.; Ball S.; Morell M. K. Role of the Escherichia coli glgX Gene in Glycogen Metabolism. J. Bacteriol. 2005, 187, 1465–1473. 10.1128/JB.187.4.1465-1473.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.-N.; Jung T.-Y.; Park J.-T.; Park B.-C.; Myung P. K.; Boos W.; Woo E.-J.; Park K.-H. Structural Rationale for the Short Branched Substrate Specificity of the Glycogen Debranching Enzyme GlgX. Proteins Struct. Funct. Bioinforma. 2010, 78, 1847–1855. 10.1002/prot.22697. [DOI] [PubMed] [Google Scholar]
- Leiba J.; Syson K.; Baronian G.; Zanella-Cleon I.; Kalscheuer R.; Kremer L.; Bornemann S.; Molle V. Mycobacterium tuberculosis Maltosyltransferase GlgE, a Genetically Validated Antituberculosis Target, Is Negatively Regulated by Ser/Thr Phosphorylation. J. Biol. Chem. 2013, 288, 16546–16556. 10.1074/jbc.M112.398503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger A. E.; Hatfull G. F. Exponential-Phase Glycogen Recycling Is Essential for Growth of Mycobacterium smegmatis. J. Bacteriol. 1999, 181, 6670–6678. 10.1128/JB.181.21.6670-6678.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syson K.; Batey S. F. D.; Schindler S.; Kalscheuer R.; Bornemann S. A Temperature-Sensitive Mycobacterium smegmatis glgE Mutation Leads to a Loss of GlgE Enzyme Activity and Thermostability and the Accumulation of Alpha-Maltose-1-Phosphate. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129783 10.1016/j.bbagen.2020.129783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W.; de Sessions P. F.; Teoh G. H.; Mohamed A. N.; Zhu Y. O.; Koh V. H.; Ang M. L.; Dedon P. C.; Hibberd M. L.; Alonso S. Transcriptional Profiling of Mycobacterium tuberculosis Exposed to In Vitro Lysosomal Stress. Infect. Immun. 2016, 84 (9), 2505–2523. 10.1128/IAI.00072-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Weerd R.; Boot M.; Maaskant J.; Sparrius M.; Verboom T.; van Leeuwen L. M.; Burggraaf M. J.; Paauw N. J.; Dainese E.; Manganelli R.; et al. Inorganic Phosphate Limitation Modulates Capsular Polysaccharide Composition in Mycobacteria. J. Biol. Chem. 2016, 291 (22), 11787–11799. 10.1074/jbc.M116.722454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbein A. D.; Pan Y. T.; Pastuszak I.; Carroll D. New Insights on Trehalose: A Multifunctional Molecule. Glycobiology 2003, 13, 17R–27R. 10.1093/glycob/cwg047. [DOI] [PubMed] [Google Scholar]
- Asencion Diez M. D.; Peiru S.; Demonte A. M.; Gramajo H.; Iglesias A. A. Characterization of Recombinant UDP- and ADP-Glucose Pyrophosphorylases and Glycogen Synthase to Elucidate Glucose-1-Phosphate Partitioning into Oligo- and Polysaccharides in Streptomyces coelicolor. J. Bacteriol. 2012, 194, 1485–1493. 10.1128/JB.06377-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada G.; Hehre E. J. New Studies on Amylosucrase, a Bacterial α-d-Glucosylase That Directly Converts Sucrose to a Glycogen-like α-Glucan. J. Biol. Chem. 1974, 249, 126–135. 10.1016/S0021-9258(19)43100-1. [DOI] [PubMed] [Google Scholar]
- Hehre E. J.; Hamilton D. M. Bacterial Synthesis of an Amylopectin-like Polysaccharide from Sucrose. J. Biol. Chem. 1946, 166, 777–778. 10.1016/S0021-9258(17)35217-1. [DOI] [PubMed] [Google Scholar]
- Tao B. Y.; Reilly P. J.; Robyt J. F. Neisseria perflava Amylosucrase: Characterization of Its Product Polysaccharide and a Study of Its Inhibition by Sucrose Derivatives. Carbohydr. Res. 1988, 181, 163–174. 10.1016/0008-6215(88)84032-1. [DOI] [PubMed] [Google Scholar]
- Stam M. R.; Danchin E. G. J.; Rancurel C.; Coutinho P. M.; Henrissat B. Dividing the Large Glycoside Hydrolase Family 13 into Subfamilies: Towards Improved Functional Annotations of Amylase-Related Proteins. Protein Eng. Des. Sel. 2006, 19, 555–562. 10.1093/protein/gzl044. [DOI] [PubMed] [Google Scholar]
- Pizzut-Serin S.; Potocki-Véronèse G.; Van Der Veen B. A.; Albenne C.; Monsan P.; Remaud-Simeon M. Characterisation of a Novel Amylosucrase from Deinococcus radiodurans. FEBS Lett. 2005, 579, 1405–1410. 10.1016/j.febslet.2004.12.097. [DOI] [PubMed] [Google Scholar]
- Seo D.-H.; Jung J.-H.; Choi H.-C.; Cho H.-K.; Kim H.-H.; Ha S.-J.; Yoo S.-H.; Cha J. Functional Expression of Amylosucrase, a Glucan-Synthesizing Enzyme, from Arthrobacter chlorophenolicus A6. J. Microbiol. Biotechnol. 2012, 22, 1253–1257. 10.4014/jmb.1201.01056. [DOI] [PubMed] [Google Scholar]
- Perez-Cenci M.; Salerno G. L. Functional Characterization of Synechococcus Amylosucrase and Fructokinase Encoding Genes Discovers Two Novel Actors on the Stage of Cyanobacterial Sucrose Metabolism. Plant Sci. 2014, 224, 95–102. 10.1016/j.plantsci.2014.04.003. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Xu W.; Bai Y.; Zhang T.; Jiang B.; Mu W. Identification of an α-(1,4)-Glucan-Synthesizing Amylosucrase from Cellulomonas carboniz T26. J. Agric. Food Chem. 2017, 65, 2110–2119. 10.1021/acs.jafc.6b05667. [DOI] [PubMed] [Google Scholar]
- Tian Y.; Xu W.; Guang C.; Zhang W.; Mu W. Thermostable Amylosucrase from Calidithermus timidus DSM 17022: Insight into Its Characteristics and Tetrameric Conformation. J. Agric. Food Chem. 2019, 67, 9868–9876. 10.1021/acs.jafc.9b04023. [DOI] [PubMed] [Google Scholar]
- Zhu X.; Tian Y.; Xu W.; Bai Y.; Zhang T.; Mu W. Biochemical Characterization of a Highly Thermostable Amylosucrase from Truepera radiovictrix DSM 17093. Int. J. Biol. Macromol. 2018, 116, 744–752. 10.1016/j.ijbiomac.2018.05.096. [DOI] [PubMed] [Google Scholar]
- Ha S.-J.; Seo D.-H.; Jung J.-H.; Cha J.; Kim T.-J.; Kim Y.-W.; Park C.-S. Molecular Cloning and Functional Expression of a New Amylosucrase from Alteromonas macleodii. Biosci. Biotechnol. Biochem. 2009, 73, 1505–1512. 10.1271/bbb.80891. [DOI] [PubMed] [Google Scholar]
- Jeong J.-W.; Seo D.-H.; Jung J.-H.; Park J.-H.; Baek N.-I.; Kim M.-J.; Park C.-S. Biosynthesis of Glucosyl Glycerol, a Compatible Solute, Using Intermolecular Transglycosylation Activity of Amylosucrase from Methylobacillus flagellatus KT. Appl. Biochem. Biotechnol. 2014, 173, 904–917. 10.1007/s12010-014-0889-z. [DOI] [PubMed] [Google Scholar]
- But S. Y.; Khmelenina V. N.; Reshetnikov A. S.; Mustakhimov I. I.; Kalyuzhnaya M. G.; Trotsenko Y. A. Sucrose Metabolism in Halotolerant Methanotroph Methylomicrobium alcaliphilum 20Z. Arch. Microbiol. 2015, 197, 471–480. 10.1007/s00203-015-1080-9. [DOI] [PubMed] [Google Scholar]
- Guérin F.; Barbe S.; Pizzut-Serin S.; Potocki-Véronèse G.; Guieysse D.; Guillet V.; Monsan P.; Mourey L.; Remaud-Siméon M.; André I.; et al. Structural Investigation of the Thermostability and Product Specificity of Amylosucrase from the Bacterium Deinococcus geothermalis. J. Biol. Chem. 2012, 287, 6642–6654. 10.1074/jbc.M111.322917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y.; Hou X.; Ni D.; Xu W.; Guang C.; Zhang W.; Chen Q.; Rao Y.; Mu W. Structure-Based Interface Engineering Methodology in Designing a Thermostable Amylose-Forming Transglucosylase. J. Biol. Chem. 2022, 298, 102074 10.1016/j.jbc.2022.102074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.; Xu M.; Lu C.; Chen L.; Xu A.; Fang J.; Chen H.; Lu Y.; Fan Y.; Chen X. Optimization of Whole-Cell Biotransformation for Scale-up Production of α-Arbutin from Hydroquinone by the Use of Recombinant Escherichia coli. AMB Express 2019, 9, 94. 10.1186/s13568-019-0820-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneron A.; Cruaud P.; Lovejoy C.; Vincent W. F. Genomic Evidence of Functional Diversity in DPANN Archaea, from Oxic Species to Anoxic Vampiristic Consortia. ISME Commun. 2022, 2, 4. 10.1038/s43705-022-00088-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eme L.; Spang A.; Lombard J.; Stairs C. W.; Ettema T. J. G. Archaea and the Origin of Eukaryotes. Nat. Rev. Microbiol. 2017, 15, 711–723. 10.1038/nrmicro.2017.133. [DOI] [PubMed] [Google Scholar]
- König H.; Skorko R.; Zillig W.; Reiter W. D. Glycogen in Thermoacidophilic Archaebacteria of the Genera Sulfolobus, Thermoproteus. Desulfurococcus and Thermococcus. Arch. Microbiol. 1982, 132, 297–303. 10.1007/BF00413378. [DOI] [Google Scholar]
- Wang L.; Liu Q.; Wu X.; Huang Y.; Wise M. J.; Liu Z.; Wang W.; Hu J.; Wang C. Bioinformatics Analysis of Metabolism Pathways of Archaeal Energy Reserves. Sci. Rep. 2019, 9, 1034. 10.1038/s41598-018-37768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Cono V.; Messina E.; Rohde M.; Arcadi E.; Ciordia S.; Crisafi F.; Denaro R.; Ferrer M.; Giuliano L.; Golyshin P. N.; et al. Symbiosis between Nanohaloarchaeon and Haloarchaeon Is Based on Utilization of Different Polysaccharides. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 20223–20234. 10.1073/pnas.2007232117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruyer S.; Legin E.; Bliard C.; Ball S.; Duchiron F. The Endopolysaccharide Metabolism of the Hyperthermophilic Archeon Thermococcus hydrothermalis: Polymer Structure and Biosynthesis. Curr. Microbiol. 2002, 44, 206–211. 10.1007/s00284-001-0029-1. [DOI] [PubMed] [Google Scholar]
- Zea C. J.; Pohl N. L. Unusual Sugar Nucleotide Recognition Elements of Mesophilic vs. Thermophilic Glycogen Synthases. Biopolymers 2005, 79, 106–113. 10.1002/bip.20338. [DOI] [PubMed] [Google Scholar]
- Sueda R.; Yoshida K.; Onodera M.; Fukui T.; Yatsunami R.; Nakamura S. Characterization of a GlgC Homolog from Extremely Halophilic Archaeon Haloarcula japonica. Biosci. Biotechnol. Biochem. 2021, 85, 1441–1447. 10.1093/bbb/zbab050. [DOI] [PubMed] [Google Scholar]
- Murray P. A.; Zinder St. H. Polysaccharide Reserve Material in the Acetotrophic Methanogen, Methanosarcina thermophila Strain TM-1: Accumulation and Mobilization. Arch. Microbiol. 1987, 147, 109–116. 10.1007/BF00415270. [DOI] [Google Scholar]
- Santiago-Martínez M. G.; Encalada R.; Lira-Silva E.; Pineda E.; Gallardo-Pérez J. C.; Reyes-García M. A.; Saavedra E.; Moreno-Sánchez R.; Marín-Hernández A.; Jasso-Chávez R. The Nutritional Status of Methanosarcina acetivorans Regulates Glycogen Metabolism and Gluconeogenesis and Glycolysis Fluxes. FEBS J. 2016, 283, 1979–1999. 10.1111/febs.13717. [DOI] [PubMed] [Google Scholar]
- Woo E. J.; Ryu S. I.; Song H. N.; Jung T. Y.; Yeon S. M.; Lee H. A.; Park B. C.; Park K. H.; Lee S. B. Structural Insights on the New Mechanism of Trehalose Synthesis by Trehalose Synthase TreT from Pyrococcus horikoshii. J. Mol. Biol. 2010, 404, 247–259. 10.1016/j.jmb.2010.09.056. [DOI] [PubMed] [Google Scholar]
- Kouril T.; Zaparty M.; Marrero J.; Brinkmann H.; Siebers B. A Novel Trehalose Synthesizing Pathway in the Hyperthermophilic Crenarchaeon Thermoproteus tenax: The Unidirectional TreT Pathway. Arch. Microbiol. 2008, 190, 355–369. 10.1007/s00203-008-0377-3. [DOI] [PubMed] [Google Scholar]
- Qu Q.; Lee S. J.; Boos W. TreT, a Novel Trehalose Glycosyltransferring Synthase of the Hyperthermophilic Archaeon Thermococcus litoralis. J. Biol. Chem. 2004, 279, 47890–47897. 10.1074/jbc.M404955200. [DOI] [PubMed] [Google Scholar]
- Li N.; Wang H.; Li L.; Cheng H.; Liu D.; Cheng H.; Deng Z. Integrated Approach To Producing High-Purity Trehalose from Maltose by the Yeast Yarrowia Lipolytica Displaying Trehalose Synthase (TreS) on the Cell Surface. J. Agric. Food Chem. 2016, 64, 6179–6187. 10.1021/acs.jafc.6b02175. [DOI] [PubMed] [Google Scholar]
- Maruta K.; Mitsuzumi H.; Nakada T.; Kubota M.; Chaen H.; Fukuda S.; Sugimoto T.; Kurimoto M. Cloning and Sequencing of a Cluster of Genes Encoding Novel Enzymes of Trehalose Biosynthesis from Thermophilic Archaebacterium Sulfolobus acidocaldarius. Biochim. Biophys. Acta BBA - Gen. Subj. 1996, 1291, 177–181. 10.1016/S0304-4165(96)00082-7. [DOI] [PubMed] [Google Scholar]
- Henrissat B.; Bairoch A. Updating the Sequence-Based Classification of Glycosyl Hydrolases. Biochem. J. 1996, 316, 695–695. 10.1042/bj3160695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zona R.; Chang-Pi-Hin F.; O’Donohue M. J.; Janeček Š. Bioinformatics of the glycoside hydrolase family 57 and identification of catalytic residues in amylopullulanase from Thermococcus hydrothermalis. Eur. J. Biochem. 2004, 271, 2863–2872. 10.1111/j.1432-1033.2004.04144.x. [DOI] [PubMed] [Google Scholar]
- Colleoni C.; Suzuki E.. Storage Polysaccharide Metabolism in Cyanobacteria. In Essential reviews in experimental biology: Starch: Origins, Structure and metabolism; Society Experimental Biology, 2012. [Google Scholar]
- Janeček Š.; Martinovičová M. New Groups of Protein Homologues in the α-Amylase Family GH57 Closely Related to α-Glucan Branching Enzymes and 4-α-Glucanotransferases. Genetica 2020, 148, 77–86. 10.1007/s10709-020-00089-0. [DOI] [PubMed] [Google Scholar]
- Murakami T.; Kanai T.; Takata H.; Kuriki T.; Imanaka T. A Novel Branching Enzyme of the GH-57 Family in the Hyperthermophilic Archaeon Thermococcus kodakaraensis KOD1. J. Bacteriol. 2006, 188, 5915–5924. 10.1128/JB.00390-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickmanns A.; Ballschmiter M.; Liebl W.; Ficner R. Structure of the Novel α-Amylase AmyC from It Thermotoga maritima. Acta Crystallogr. Sect. D 2006, 62, 262–270. 10.1107/S0907444905041363. [DOI] [PubMed] [Google Scholar]
- Palomo M.; Pijning T.; Booiman T.; Dobruchowska J. M.; Van Der Vlist J.; Kralj S.; Planas A.; Loos K.; Kamerling J. P.; Dijkstra B. W.; et al. Thermus thermophilus Glycoside Hydrolase Family 57 Branching Enzyme. J. Biol. Chem. 2011, 286, 3520–3530. 10.1074/jbc.M110.179515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na S.; Park M.; Jo I.; Cha J.; Ha N.-C. Structural Basis for the Transglycosylase Activity of a GH57-Type Glycogen Branching Enzyme from Pyrococcus horikoshii. Biochem. Biophys. Res. Commun. 2017, 484, 850–856. 10.1016/j.bbrc.2017.02.002. [DOI] [PubMed] [Google Scholar]
- Suzuki E.; Suzuki R. Distribution of Glucan-Branching Enzymes among Prokaryotes. Cell. Mol. Life Sci. 2016, 73, 2643–2660. 10.1007/s00018-016-2243-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinovičová M.; Janeček Š. In Silico Analysis of the α-Amylase Family GH57: Eventual Subfamilies Reflecting Enzyme Specificities.. 3 Biotech 2018, 8, 307. 10.1007/s13205-018-1325-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. W.; Flowers L. O.; Whiteley M.; Peeples T. L. Biochemical Confirmation and Characterization of the Family-57-like α-Amylase of Methanococcus Jannaschii. Folia Microbiol. (Praha) 2001, 46, 467–473. 10.1007/BF02817988. [DOI] [PubMed] [Google Scholar]
- Lee A.; Bae E.; Park J.; Choi K.-H.; Cha J. Identification of the Genes Related to the Glycogen Metabolism in Hyperthermophilic Archaeon, Sulfolobus acidocaldarius. Front. Microbiol. 2021, 12, 661053 10.3389/fmicb.2021.661053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumez S.; Wattebled F.; Dauvillee D.; Delvalle D.; Planchot V.; Ball S. G.; D’Hulst C. Mutants of Arabidopsis Lacking Starch Branching Enzyme II Substitute Plastidial Starch Synthesis by Cytoplasmic Maltose Accumulation. Plant Cell 2006, 18 (10), 2694–2709. 10.1105/tpc.105.037671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.-S.; Shockley K. R.; Schut G. J.; Conners S. B.; Montero C. I.; Johnson M. R.; Chou C.-J.; Bridger S. L.; Wigner N.; Brehm S. D.; et al. Transcriptional and Biochemical Analysis of Starch Metabolism in the Hyperthermophilic Archaeon Pyrococcus furiosus. J. Bacteriol. 2006, 188, 2115–2125. 10.1128/JB.188.6.2115-2125.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H.-S.; Park J.-T.; Kang H.-K.; Cha H.; Kim D.-S.; Kim J.-W.; Park K.-H. TreX from Sulfolobus solfataricus ATCC 35092 Displays Isoamylase and 4-Alpha-Glucanotransferase Activities. Biosci. Biotechnol. Biochem. 2007, 71 (5), 1348–1352. 10.1271/bbb.70016. [DOI] [PubMed] [Google Scholar]
- Woo E.-J.; Lee S.; Cha H.; Park J.-T.; Yoon S.-M.; Song H.-N.; Park K.-H. Structural Insight into the Bifunctional Mechanism of the Glycogen-Debranching Enzyme TreX from the Archaeon Sulfolobus solfataricus. J. Biol. Chem. 2008, 283, 28641–28648. 10.1074/jbc.M802560200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bini E.; Blum P. Archaeal Catabolite Repression: A Gene Regulatory Paradigm. Adv. Appl. Microbiol. 2001, 50, 339–366. 10.1016/S0065-2164(01)50009-X. [DOI] [PubMed] [Google Scholar]
- Worthington P.; Hoang V.; Perez-Pomares F.; Blum P. Targeted Disruption of the Alpha-Amylase Gene in the Hyperthermophilic Archaeon Sulfolobus solfataricus. J. Bacteriol. 2003, 185, 482–488. 10.1128/JB.185.2.482-488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanFossen A. L.; Lewis D. L.; Nichols J. D.; Kelly R. M. Polysaccharide Degradation and Synthesis by Extremely Thermophilic anaerobes. Ann. N. Y. Acad. Sci. 2008, 1125, 322–337. 10.1196/annals.1419.017. [DOI] [PubMed] [Google Scholar]
- Choi K.-H.; Cha J. Membrane-Bound Amylopullulanase Is Essential for Starch Metabolism of Sulfolobus acidocaldarius DSM639. Extremophiles 2015, 19, 909–920. 10.1007/s00792-015-0766-x. [DOI] [PubMed] [Google Scholar]
- Jung J.-H.; Seo D.-H.; Holden J. F.; Park C.-S. Maltose-Forming α-Amylase from the Hyperthermophilic Archaeon Pyrococcus Sp. ST04. Appl. Microbiol. Biotechnol. 2014, 98, 2121–2131. 10.1007/s00253-013-5068-6. [DOI] [PubMed] [Google Scholar]
- Xavier K. B.; Peist R.; Kossmann M.; Boos W.; Santos H. Maltose Metabolism in the Hyperthermophilic Archaeon Thermococcus litoralis: Purification and Characterization of Key Enzymes. J. Bacteriol. 1999, 181, 3358–3367. 10.1128/JB.181.11.3358-3367.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizanur R. M.; Griffin A. K. K.; Pohl N. L. Recombinant Production and Biochemical Characterization of a Hyperthermostable α-Glucan/Maltodextrin Phosphorylase from Pyrococcus furiosus. Archaea 2008, 2, 169–176. 10.1155/2008/549759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattridge K. A.; Weber C. H.; Friesen J. A.; Sanker S.; Kent C.; Ludwig M. L. Glycerol-3-Phosphate Cytidylyltransferase. Structural Changes Induced by Binding of CDP-Glycerol and the Role of Lysine Residues in Catalysis. J. Biol. Chem. 2003, 278, 51863–51871. 10.1074/jbc.M306174200. [DOI] [PubMed] [Google Scholar]
- Brown K.; Pompeo F.; Dixon S.; Mengin-Lecreulx D.; Cambillau C.; Bourne Y. Crystal Structure of the Bifunctional N-Acetylglucosamine 1-Phosphate Uridyltransferase from Escherichia coli: A Paradigm for the Related Pyrophosphorylase Superfamily. EMBO J. 1999, 18, 4096–4107. 10.1093/emboj/18.15.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leloir L. F. Two Decades. Science. 1971, 172, 1299–1303. 10.1126/science.172.3990.1299. [DOI] [PubMed] [Google Scholar]
- Weissborn A. C.; Liu Q.; Rumley M. K.; Kennedy E. P. UTP: Alpha-D-Glucose-1-Phosphate Uridylyltransferase of Escherichia coli: Isolation and DNA Sequence of the galU Gene and Purification of the Enzyme. J. Bacteriol. 1994, 176, 2611–2618. 10.1128/jb.176.9.2611-2618.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K.; Narikawa R.; Ikeuchi M. CugP Is a Novel Ubiquitous Non-GalU-Type Bacterial UDP-Glucose Pyrophosphorylase Found in Cyanobacteria. J. Bacteriol. 2014, 196, 2348–2354. 10.1128/JB.01591-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbein A. D. Trehalose Phosphate Synthesis in Streptomyces hygroscopicus: Purification of Guanosine Diphosphate D-Glucose: D-Glucose-6-Phosphate 1-Glucosyl-Transferase. J. Bacteriol. 1968, 96, 1623–1631. 10.1128/jb.96.5.1623-1631.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asencion Diez M. D.; Miah F.; Stevenson C. E.M.; Lawson D. M.; Iglesias A. A.; Bornemann S. The Production and Utilization of GDP-Glucose in the Biosynthesis of Trehalose 6-Phosphate by Streptomyces venezuelae. J. Biol. Chem. 2017, 292, 945–954. 10.1074/jbc.M116.758664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bochove M. A.; Swart M.; Bickelhaupt F. M. Nucleophilic Substitution at Phosphorus (SN2@P): Disappearance and Reappearance of Reaction Barriers. J. Am. Chem. Soc. 2006, 128, 10738–10744. 10.1021/ja0606529. [DOI] [PubMed] [Google Scholar]
- Preiss J.; Shen L.; Greenberg E.; Gentner N. Biosynthesis of Bacterial Glycogen. IV. Activation and Inhibition of the Adenosine Diphosphate Glucose Pyrophosphorylase of Escherichia coli B.. Biochemistry 1966, 5, 1833–1845. 10.1021/bi00870a008. [DOI] [PubMed] [Google Scholar]
- Turnquist R. L.; Gillett T. A.; Hansen R. G. Uridine Diphosphate Glucose Pyrophosphorylase: Crystallization and Properties of the Enzyme from Rabbit Liver and Species Comparisons. J. Biol. Chem. 1974, 249, 7695–7700. 10.1016/S0021-9258(19)81292-9. [DOI] [PubMed] [Google Scholar]
- Van Bochove M. A.; Roos G.; Fonseca Guerra C.; Hamlin T. A.; Bickelhaupt F. M. How Mg2+ Ions Lower the SN2@P Barrier in Enzymatic Triphosphate Hydrolysis. Chem. Commun. 2018, 54, 3448–3451. 10.1039/C8CC00700D. [DOI] [PubMed] [Google Scholar]
- Führing J.; Cramer J. T.; Routier F. H.; Lamerz A. C.; Baruch P.; Gerardy-Schahn R.; Fedorov R. Catalytic Mechanism and Allosteric Regulation of UDP-Glucose Pyrophosphorylase from Leishmania major. ACS Catal. 2013, 3, 2976–2985. 10.1021/cs4007777. [DOI] [Google Scholar]
- Lahti R. Microbial Inorganic Pyrophosphatases. Microbiol. Rev. 1983, 47, 169–178. 10.1128/mr.47.2.169-178.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moremen K. W.; Haltiwanger R. S. Emerging Structural Insights into Glycosyltransferase-Mediated Synthesis of Glycans. Nat. Chem. Biol. 2019, 15, 853–864. 10.1038/s41589-019-0350-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rini J. M.; Moremen K. W.; Davis B. G.; Esko J. D.. Glycosyltransferases and Glycan-Processing Enzymes. In Essentials of Glycobiology; Varki A., Cummings R. D., Esko J. D., Stanley P., Hart G. W., Aebi M., Mohnen D., Kinoshita T., Packer N. H., Prestegard J. H., Schnaar R. L., Seeberger P. H., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor (NY), 2022. [PubMed] [Google Scholar]
- Taujale R.; Zhou Z.; Yeung W.; Moremen K. W.; Li S.; Kannan N. Mapping the Glycosyltransferase Fold Landscape Using Interpretable Deep Learning. Nat. Commun. 2021 121 2021, 12, 1–12. 10.1038/s41467-021-25975-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnott M. L. Catalytic Mechanisms of Enzymic Glycosyl Transfer. Chem. Rev. 1990, 90, 1171–1202. 10.1021/cr00105a006. [DOI] [Google Scholar]
- Lairson L. L.; Henrissat B.; Davies G. J.; Withers S. G. Glycosyltransferases: Structures, Functions, and Mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- Albesa-Jové D.; Guerin M. E. The Conformational Plasticity of Glycosyltransferases. Curr. Opin. Struct. Biol. 2016, 40, 23–32. 10.1016/j.sbi.2016.07.007. [DOI] [PubMed] [Google Scholar]
- Ardèvol A.; Iglesias-Fernández J.; Rojas-Cervellera V.; Rovira C. The Reaction Mechanism of Retaining Glycosyltransferases. Biochem. Soc. Trans. 2016, 44, 51–60. 10.1042/BST20150177. [DOI] [PubMed] [Google Scholar]
- Rojas-Cervellera V.; Ardèvol A.; Boero M.; Planas A.; Rovira C. Formation of a Covalent Glycosyl-Enzyme Species in a Retaining Glycosyltransferase. Chem. Weinh. Bergstr. Ger. 2013, 19, 14018–14023. 10.1002/chem.201302898. [DOI] [PubMed] [Google Scholar]
- Soya N.; Fang Y.; Palcic M. M.; Klassen J. S. Trapping and Characterization of Covalent Intermediates of Mutant Retaining Glycosyltransferases. Glycobiology 2011, 21, 547–552. 10.1093/glycob/cwq190. [DOI] [PubMed] [Google Scholar]
- Blackler R. J.; Gagnon S. M. L.; Polakowski R.; Rose N. L.; Zheng R. B.; Letts J. A.; Johal A. R.; Schuman B.; Borisova S. N.; Palcic M. M.; et al. Glycosyltransfer in Mutants of Putative Catalytic Residue Glu303 of the Human ABO(H) A and B Blood Group Glycosyltransferases GTA and GTB Proceeds through a Labile Active Site. Glycobiology 2016, 27 (4), 370–380. 10.1093/glycob/cww117. [DOI] [PubMed] [Google Scholar]
- Albesa-Jové D.; Sainz-Polo M. Á.; Marina A.; Guerin M. E. Structural Snapshots of α-1,3-Galactosyltransferase with Native Substrates: Insight into the Catalytic Mechanism of Retaining Glycosyltransferases. Angew. Chem. - Int. Ed. 2017, 56, 14853–14857. 10.1002/anie.201707922. [DOI] [PubMed] [Google Scholar]
- Gómez H.; Mendoza F.; Lluch J. M.; Masgrau L. QM/MM Studies Reveal How Substrate-Substrate and Enzyme-Substrate Interactions Modulate Retaining Glycosyltransferases Catalysis and Mechanism. Adv. Protein Chem. Struct. Biol. 2015, 100, 225–254. 10.1016/bs.apcsb.2015.06.004. [DOI] [PubMed] [Google Scholar]
- Albesa-Jové D.; Mendoza F.; Rodrigo-Unzueta A.; Gomollón-Bel F.; Cifuente J. O.; Urresti S.; Comino N.; Gómez H.; Romero-García J.; Lluch J. M.; et al. A Native Ternary Complex Trapped in a Crystal Reveals the Catalytic Mechanism of a Retaining Glycosyltransferase. Angew. Chem. Int. Ed Engl. 2015, 54, 9898–9902. 10.1002/anie.201504617. [DOI] [PubMed] [Google Scholar]
- Forrester T. J. B.; Ovchinnikova O. G.; Li Z.; Kitova E. N.; Nothof J. T.; Koizumi A.; Klassen J. S.; Lowary T. L.; Whitfield C.; Kimber M. S. The Retaining β-Kdo Glycosyltransferase WbbB Uses a Double-Displacement Mechanism with an Intermediate Adduct Rearrangement Step. Nat. Commun. 2022, 13, 6277. 10.1038/s41467-022-35491-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle L.; Ovchinnikova O. G.; Huang B.-S.; Forrester T. J. B.; Lowary T. L.; Kimber M. S.; Whitfield C. Mechanism and Linkage Specificities of the Dual Retaining β-Kdo Glycosyltransferase Modules of KpsC from Bacterial Capsule Biosynthesis. J. Biol. Chem. 2023, 299, 104609 10.1016/j.jbc.2023.104609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leloir L. F.; Goldemberg S. H. Synthesis of Glycogen from Uridine Diphosphate Glucose in Liver. J. Biol. Chem. 1960, 235, 919–923. 10.1016/S0021-9258(18)69451-7. [DOI] [PubMed] [Google Scholar]
- Leloir L. F.; Cardini C. E. Biosynthesis of Glycogen from Uridine Diphosphate Glucose. J. Am. Chem. Soc. 1957, 79, 6340–6341. 10.1021/ja01580a061. [DOI] [Google Scholar]
- Shen L.; Ghosh H. P.; Greenberg E.; Preiss J. Adenosine Diphosphate Glucose-Glycogen Transglucosylase in Arthrobacter Sp. NRRL B 1973. Biochim. Biophys. Acta - Enzymol. Subj. 1964, 89, 370–372. 10.1016/0926-6569(64)90232-9. [DOI] [PubMed] [Google Scholar]
- Cabib E.; Leloir L. F. The Biosynthesis of Trehalose Phosphate. J. Biol. Chem. 1958, 231, 259–275. 10.1016/S0021-9258(19)77303-7. [DOI] [PubMed] [Google Scholar]
- Curatti L.; Giarrocco L. E.; Cumino A. C.; Salerno G. L. Sucrose Synthase Is Involved in the Conversion of Sucrose to Polysaccharides in Filamentous Nitrogen-Fixing Cyanobacteria. Planta 2008, 228, 617–625. 10.1007/s00425-008-0764-7. [DOI] [PubMed] [Google Scholar]
- Mendicino J. Sucrose Phosphate Synthesis in Wheat Germ and Green Leaves. J. Biol. Chem. 1960, 235, 3347–3352. 10.1016/S0021-9258(18)64469-2. [DOI] [PubMed] [Google Scholar]
- Roujeinikova A.; Raasch C.; Burke J.; Baker P. J.; Liebl W.; Rice D. W. The Crystal Structure of Thermotoga maritima Maltosyltransferase and Its Implications for the Molecular Basis of the Novel Transfer Specificity. J. Mol. Biol. 2001, 312, 119–131. 10.1006/jmbi.2001.4944. [DOI] [PubMed] [Google Scholar]
- Jensen M. H.; Mirza O.; Albenne C.; Remaud-Simeon M.; Monsan P.; Gajhede M.; Skov L. K. Crystal Structure of the Covalent Intermediate of Amylosucrase from Neisseria polysaccharea. Biochemistry 2004, 43, 3104–3110. 10.1021/bi0357762. [DOI] [PubMed] [Google Scholar]
- Skov L. K.; Mirza O.; Sprogo̷e D.; Dar I.; Remaud-Simeon M.; Albenne C.; Monsan P.; Gajhede M. Oligosaccharide and Sucrose Complexes of Amylosucrase. Structural Implications for the Polymerase Activity. J. Biol. Chem. 2002, 277, 47741–47747. 10.1074/jbc.M207860200. [DOI] [PubMed] [Google Scholar]
- Jaña G. A.; Mendoza F.; Osorio M. I.; Alderete J. B.; Fernandes P. A.; Ramos M. J.; Jiménez V. A. A QM/MM Approach on the Structural and Stereoelectronic Factors Governing Glycosylation by GTF-SI from Streptococcus mutans. Org. Biomol. Chem. 2018, 16, 2438–2447. 10.1039/C8OB00284C. [DOI] [PubMed] [Google Scholar]
- Albenne C.; Skov L. K.; Mirza O.; Gajhede M.; Feller G.; D’Amico S.; André G.; Potocki-Véronèse G.; Van Der Veen B. A.; Monsan P.; et al. Molecular Basis of the Amylose-like Polymer Formation Catalyzed by Neisseria polysaccharea Amylosucrase. J. Biol. Chem. 2004, 279, 726–734. 10.1074/jbc.M309891200. [DOI] [PubMed] [Google Scholar]
- Chludzinski A. M.; Germaine G. R.; Schachtele C. F. Purification and Properties of Dextransucrase from Streptococcus mutans. J. Bacteriol. 1974, 118, 1–7. 10.1128/jb.118.1.1-7.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre E.; Bozonnet S.; Arcache A.; Willemot R. M.; Vignon M.; Monsan P.; Remaud-Simeon M. Role of the Two Catalytic Domains of Dsr-E Dextransucrase and Their Involvement in the Formation of Highly α-1,2 Branched Dextran. J. Bacteriol. 2005, 187, 296–303. 10.1128/JB.187.1.296-303.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina M.; Moulis C.; Monties N.; Pizzut-Serin S.; Guieysse D.; Morel S.; Cioci G.; Remaud-Siméon M. Deciphering an Undecided Enzyme: Investigations of the Structural Determinants Involved in the Linkage Specificity of Alternansucrase. ACS Catal. 2019, 9, 2222–2237. 10.1021/acscatal.8b04510. [DOI] [Google Scholar]
- Van Leeuwen S. S.; Kralj S.; Van Geel-Schutten I. H.; Gerwig G. J.; Dijkhuizen L.; Kamerling J. P. Structural Analysis of the A-D D-Glucan (EPS35-5) Produced by the Lactobacillus reuteri Strain 35-5 Glucansucrase GTFA Enzyme. Carbohydr. Res. 2008, 343, 1251–1265. 10.1016/j.carres.2008.01.044. [DOI] [PubMed] [Google Scholar]
- Henrissat B.; Davies G. Structural and Sequence-Based Classification of Glycoside Hydrolases. Curr. Opin. Struct. Biol. 1997, 7, 637–644. 10.1016/S0959-440X(97)80072-3. [DOI] [PubMed] [Google Scholar]
- Barends T. R. M.; Bultema J. B.; Kaper T.; Van Der Maarel M. J. E. C.; Dijkhuizen L.; Dijkstra B. W. Three-Way Stabilization of the Covalent Intermediate in Amylomaltase, an Alpha-Amylase-like Transglycosylase. J. Biol. Chem. 2007, 282, 17242–17249. 10.1074/jbc.M701444200. [DOI] [PubMed] [Google Scholar]
- Welker N. E.; Campbell L. L. Crystallization and Properties of α-Amylase from Five Strains of Bacillus amyloliquefaciens. Biochemistry 1967, 6, 3681–3689. 10.1021/bi00864a010. [DOI] [PubMed] [Google Scholar]
- Raha M.; Kawagishi I.; Muller V.; Kihara M.; Macnab R. M. Escherichia coli Produces a Cytoplasmic Alpha-Amylase. AmyA. J. Bacteriol. 1992, 174, 6644–6644. 10.1128/jb.174.20.6644-6652.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiess C.; Happersberger H. P.; Glocker M. O.; Spiess E.; Rippe K.; Ehrmann M. Biochemical Characterization and Mass Spectrometric Disulfide Bond Mapping of Periplasmic α-Amylase MalS of Escherichia coli. J. Biol. Chem. 1997, 272, 22125–22133. 10.1074/jbc.272.35.22125. [DOI] [PubMed] [Google Scholar]
- Plant A. R.; Parratt S.; Daniel R. M.; Morgan H. W. A Cell-Associated Oligo-1,6-α-Glucosidase from an Extremely Thermophilic Anaerobic Bacterium, Thermoanaerobium Tok6-B1. Biochem. J. 1988, 255, 865–868. 10.1042/bj2550865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai T.; Vo S. H.; Morinaka K.; Kobayashi T.; Ito S. Crystal Structure of GH13 Alpha-Glucosidase GSJ from One of the Deepest Sea Bacteria. Proteins 2008, 73, 126–133. 10.1002/prot.22044. [DOI] [PubMed] [Google Scholar]
- Lin M. G.; Chi M. C.; Naveen V.; Li Y. C.; Lin L. L.; Hsiao C. D. Bacillus licheniformis Trehalose-6-Phosphate Hydrolase Structures Suggest Keys to Substrate Specificity. Acta Crystallogr. Sect. Struct. Biol. 2016, 72, 59–70. 10.1107/S2059798315020756. [DOI] [PubMed] [Google Scholar]
- Okazaki N.; Tamada T.; Feese M. D.; Kato M.; Miura Y.; Komeda T.; Kobayashi K.; Kondo K.; Blaber M.; Kuroki R. Substrate Recognition Mechanism of a Glycosyltrehalose Trehalohydrolase from Sulfolobus solfataricus KM1. Protein Sci. 2012, 21, 539–552. 10.1002/pro.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan S.; Min H.; Liu T.; Jiang D.; Rao Z. Structural Insight into Dephosphorylation by Trehalose 6-Phosphate Phosphatase (OtsB2) from Mycobacterium tuberculosis. FASEB J. 2016, 30, 3989–3996. 10.1096/fj.201600463R. [DOI] [PubMed] [Google Scholar]
- Harvey C. M.; O’Toole K. H.; Liu C.; Mariano P.; Dunaway-Mariano D.; Allen K. N. Structural Analysis of Binding Determinants of Salmonella typhimurium Trehalose-6-Phosphate Phosphatase Using Ground-State Complexes. Biochemistry 2020, 59, 3247–3257. 10.1021/acs.biochem.0c00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthisawat S.; Gourlay L. J.; Bolognesi M.; Boonyuen U.; Vanaporn M. Functional and Structural Analysis of Trehalose-6-Phosphate Phosphatase from Burkholderia pseudomallei: Insights into the Catalytic Mechanism. Biochem. Biophys. Res. Commun. 2020, 523, 979–984. 10.1016/j.bbrc.2019.12.088. [DOI] [PubMed] [Google Scholar]
- Fieulaine S.; Lunn J. E.; Borel F.; Ferrer J. L. The Structure of a Cyanobacterial Sucrose-Phosphatase Reveals the Sugar Tongs That Release Free Sucrose in the Cell. Plant Cell 2005, 17, 2049–2049. 10.1105/tpc.105.031229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D. H.; Roberts A.; Jancarik J.; Yokota H.; Kim R.; Wemmer D. E.; Kim S.-H. Crystal Structure of a Phosphatase with a Unique Substrate Binding Domain from Thermotoga maritima. Protein Sci. 2003, 12, 1464–1472. 10.1110/ps.0302703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Kim R.; Jancarik J.; Yokota H.; Kim S. H. Crystal Structure of Phosphoserine Phosphatase from Methanococcus jannaschii, a Hyperthermophile, at 1.8 Å Resolution. Structure 2001, 9, 65–71. 10.1016/S0969-2126(00)00558-X. [DOI] [PubMed] [Google Scholar]
- Wang W.; Cho H. S.; Kim R.; Jancarik J.; Yokota H.; Nguyen H. H.; Grigoriev I. V.; Wemmer D. E.; Kim S. H. Structural Characterization of the Reaction Pathway in Phosphoserine Phosphatase: Crystallographic “Snapshots” of Intermediate States. J. Mol. Biol. 2002, 319, 421–431. 10.1016/S0022-2836(02)00324-8. [DOI] [PubMed] [Google Scholar]
- Miao Y.; Tenor J. L.; Toffaletti D. L.; Washington E. J.; Liu J.; Shadrick W. R.; Schumacher M. A.; Lee R. E.; Perfect J. R.; Brennan R. G. Structures of Trehalose-6-Phosphate Phosphatase from Pathogenic Fungi Reveal the Mechanisms of Substrate Recognition and Catalysis. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 7148–7153. 10.1073/pnas.1601774113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagaya M.; Fukui T. Catalytic Reaction of Glycogen Phosphorylase Reconstituted with a Coenzyme-Substrate Conjugate. J. Biol. Chem. 1984, 259, 4860–4865. 10.1016/S0021-9258(17)42925-5. [DOI] [PubMed] [Google Scholar]
- Schwarz A.; Pierfederici F. M.; Nidetzky B. Catalytic Mechanism of Alpha-Retaining Glucosyl Transfer by Corynebacterium callunae Starch Phosphorylase: The Role of Histidine-334 Examined through Kinetic Characterization of Site-Directed Mutants. Biochem. J. 2005, 387, 437–445. 10.1042/BJ20041593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geremia S.; Campagnolo M.; Schinzel R.; Johnson L. N. Enzymatic Catalysis in Crystals of Escherichia coli Maltodextrin Phosphorylase. J. Mol. Biol. 2002, 322, 413–423. 10.1016/S0022-2836(02)00771-4. [DOI] [PubMed] [Google Scholar]
- Szabó K.; Kandra L.; Gyémánt G. Studies on the Reversible Enzyme Reaction of Rabbit Muscle Glycogen Phosphorylase b Using Isothermal Titration Calorimetry. Carbohydr. Res. 2019, 477, 58–65. 10.1016/j.carres.2019.03.014. [DOI] [PubMed] [Google Scholar]
- Mueller M.; Nidetzky B. Dissecting Differential Binding of Fructose and Phosphate as Leaving Group/Nucleophile of Glucosyl Transfer Catalyzed by Sucrose Phosphorylase. FEBS Lett. 2007, 581, 3814–3818. 10.1016/j.febslet.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Sprogo̷e D.; Van Den Broek L. A. M.; Mirza O.; Kastrup J. S.; Voragen A. G. J.; Gajhede M.; Skov L. K. Crystal Structure of Sucrose Phosphorylase from Bifidobacterium adolescentis. Biochemistry 2004, 43, 1156–1162. 10.1021/bi0356395. [DOI] [PubMed] [Google Scholar]
- Verhaeghe T.; Aerts D.; Diricks M.; Soetaert W.; Desmet T. The Quest for a Thermostable Sucrose Phosphorylase Reveals Sucrose 6’-Phosphate Phosphorylase as a Novel Specificity. Appl. Microbiol. Biotechnol. 2014, 98, 7027–7037. 10.1007/s00253-014-5621-y. [DOI] [PubMed] [Google Scholar]
- Santos C. R.; Tonoli C. C. C.; Trindade D. M.; Betzel C.; Takata H.; Kuriki T.; Kanai T.; Imanaka T.; Arni R. K.; Murakami M. T. Structural Basis for Branching-Enzyme Activity of Glycoside Hydrolase Family 57: Structure and Stability Studies of a Novel Branching Enzyme from the Hyperthermophilic Archaeon Thermococcus kodakaraensis KOD1. Proteins 2011, 79, 547–557. 10.1002/prot.22902. [DOI] [PubMed] [Google Scholar]
- Bojarová-Fialová P.; Křen V.. Enzymatic Approaches to O-Glycoside Introduction: Glycosidases. In Comprehensive Glycoscience; Elsevier, 2007; pp 453–487. [Google Scholar]
- Weiss S. C.; Skerra A.; Schiefner A. Structural Basis for the Interconversion of Maltodextrins by MalQ, the Amylomaltase of Escherichia coli. J. Biol. Chem. 2015, 290, 21352–21364. 10.1074/jbc.M115.667337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresolin N. S.; Li Z.; Kosar-Hashemi B.; Tetlow I. J.; Chatterjee M.; Rahman S.; Morell M. K.; Howitt C. A. Characterisation of Disproportionating Enzyme from Wheat Endosperm. Planta 2006, 224, 20–31. 10.1007/s00425-005-0187-7. [DOI] [PubMed] [Google Scholar]
- Okazaki N.; Blaber M.; Kuroki R.; Tamada T. Crystal Structure of Glycosyltrehalose Synthase from Sulfolobus shibatae DSM5389. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2018, 74, 741–746. 10.1107/S2053230X1801453X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caner S.; Nguyen N.; Aguda A.; Zhang R.; Pan Y. T.; Withers S. G.; Brayer G. D. The Structure of the Mycobacterium smegmatis Trehalose Synthase Reveals an Unusual Active Site Configuration and Acarbose-Binding Mode. Glycobiology 2013, 23, 1075–1083. 10.1093/glycob/cwt044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Pan Y. T.; He S.; Lam M.; Brayer G. D.; Elbein A. D.; Withers S. G. Mechanistic Analysis of Trehalose Synthase from Mycobacterium smegmatis. J. Biol. Chem. 2011, 286, 35601–35609. 10.1074/jbc.M111.280362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehues B.; Jossek R.; Kramer U.; Koch A.; Jarling M.; Schröder W.; Pape H. Isolation and Characterization of Maltokinase (ATP:Maltose 1-Phosphotransferase) from Actinoplanes Missouriensis. Arch. Microbiol. 2003, 180, 233–239. 10.1007/s00203-003-0575-y. [DOI] [PubMed] [Google Scholar]
- Hon W. C.; McKay G. A.; Thompson P. R.; Sweet R. M.; Yang D. S.; Wright G. D.; Berghuis A. M. Structure of an Enzyme Required for Aminoglycoside Antibiotic Resistance Reveals Homology to Eukaryotic Protein Kinases. Cell 1997, 89, 887–895. 10.1016/S0092-8674(00)80274-3. [DOI] [PubMed] [Google Scholar]
- Burk D. L.; Hon W. C.; Leung A. K. W.; Berghuis A. M. Structural Analyses of Nucleotide Binding to an Aminoglycoside Phosphotransferase. Biochemistry 2001, 40, 8756–8764. 10.1021/bi010504p. [DOI] [PubMed] [Google Scholar]
- Fong D. H.; Berghuis A. M. Substrate Promiscuity of an Aminoglycoside Antibiotic Resistance Enzyme via Target Mimicry. EMBO J. 2002, 21, 2323–2331. 10.1093/emboj/21.10.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Guan X.; Shaw N.; Chen W.; Dong Y.; Xu X.; Li X.; Rao Z. Homotypic Dimerization of a Maltose Kinase for Molecular Scaffolding. Sci. Rep. 2014, 4, 6418. 10.1038/srep06418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K.-C.; Lyu S.-Y.; Liu Y.-C.; Chang C.-Y.; Wu C.-J.; Li T.-L. Insights into the Binding Specificity and Catalytic Mechanism of N-Acetylhexosamine 1-Phosphate Kinases through Multiple Reaction Complexes. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1401–1410. 10.1107/S1399004714004209. [DOI] [PubMed] [Google Scholar]
- Caetano-Anollés G.; Caetano-Anollés D. An Evolutionarily Structured Universe of Protein Architecture. Genome Res. 2003, 13, 1563–1571. 10.1101/gr.1161903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda-Garcia L.; Liebermeister W.; Tawfik D. S. Metabolite-Enzyme Coevolution: From Single Enzymes to Metabolic Pathways and Networks. Annu. Rev. Biochem. 2018, 87, 187–216. 10.1146/annurev-biochem-062917-012023. [DOI] [PubMed] [Google Scholar]
- Rao S. T.; Rossmann M. G. Comparison of Super-Secondary Structures in Proteins. J. Mol. Biol. 1973, 76, 241–256. 10.1016/0022-2836(73)90388-4. [DOI] [PubMed] [Google Scholar]
- Medvedev K. E.; Kinch L. N.; Schaeffer R. D.; Grishin N. V. Functional Analysis of Rossmann-like Domains Reveals Convergent Evolution of Topology and Reaction Pathways. PLoS Comput. Biol. 2019, 15, e1007569 10.1371/journal.pcbi.1007569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossmann M. G.; Moras D.; Olsen K. W. Chemical and Biological Evolution of Nucleotide-Binding Protein. Nature 1974, 250, 194–199. 10.1038/250194a0. [DOI] [PubMed] [Google Scholar]
- Longo L. M.; Jabłońska J.; Vyas P.; Kanade M.; Kolodny R.; Ben-Tal N.; Tawfik D. S. On the Emergence of P-Loop NTPase and Rossmann Enzymes from a Beta-Alpha-Beta Ancestral Fragment. eLife 2020, 9, e64415 10.7554/eLife.64415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev K. E.; Kinch L. N.; Dustin Schaeffer R.; Pei J.; Grishin N. V. A Fifth of the Protein World: Rossmann-like Proteins as an Evolutionarily Successful Structural Unit. J. Mol. Biol. 2021, 433, 166788 10.1016/j.jmb.2020.166788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J. E.; Saraste M.; Runswick M. J.; Gay N. J. Distantly Related Sequences in the Alpha- and Beta-Subunits of ATP Synthase, Myosin, Kinases and Other ATP-Requiring Enzymes and a Common Nucleotide Binding Fold. EMBO J. 1982, 1, 945–951. 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E. V. A Common Set of Conserved Motifs in a Vast Variety of Putative Nucleic Acid-Dependent ATPases Including MCM Proteins Involved in the Initiation of Eukaryotic DNA Replication. Nucleic Acids Res. 1993, 21, 2541–2547. 10.1093/nar/21.11.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leipe D. D.; Wolf Y. I.; Koonin E. V.; Aravind L. Classification and Evolution of P-Loop GTPases and Related ATPases. J. Mol. Biol. 2002, 317, 41–72. 10.1006/jmbi.2001.5378. [DOI] [PubMed] [Google Scholar]
- Kornev A. P.; Taylor S. S. Dynamics-Driven Allostery in Protein Kinases. Trends Biochem. Sci. 2015, 40, 628–647. 10.1016/j.tibs.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero Romero M. L.; Yang F.; Lin Y.-R.; Toth-Petroczy A.; Berezovsky I. N.; Goncearenco A.; Yang W.; Wellner A.; Kumar-Deshmukh F.; Sharon M.; et al. Simple yet Functional Phosphate-Loop Proteins. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E11943–E11950. 10.1073/pnas.1812400115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker D.; Kleczkowski L. A. UDP-Sugar Producing Pyrophosphorylases: Distinct and Essential Enzymes with Overlapping Substrate Specificities, Providing de Novo Precursors for Glycosylation Reactions. Front. Plant Sci. 2019, 9, 1822–1822. 10.3389/fpls.2018.01822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koropatkin N. M.; Holden H. M. Molecular Structure of α-d-Glucose-1-Phosphate Cytidylyltransferase from Salmonella typhi. J. Biol. Chem. 2004, 279, 44023–44029. 10.1074/jbc.M407755200. [DOI] [PubMed] [Google Scholar]
- Blankenfeldt W.; Asuncion M.; Lam J. S.; Naismith J. H. The Structural Basis of the Catalytic Mechanism and Regulation of Glucose-1-Phosphate Thymidylyltransferase (RmlA). EMBO J. 2000, 19, 6652–6663. 10.1093/emboj/19.24.6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajava A. V.; Steven A. C. Beta-Rolls, Beta-Helices, and Other Beta-Solenoid Proteins. Adv. Protein Chem. 2006, 73, 55–96. 10.1016/S0065-3233(06)73003-0. [DOI] [PubMed] [Google Scholar]
- Hennetin J.; Jullian B.; Steven A. C.; Kajava A. V. Standard Conformations of Beta-Arches in Beta-Solenoid Proteins. J. Mol. Biol. 2006, 358, 1094–1105. 10.1016/j.jmb.2006.02.039. [DOI] [PubMed] [Google Scholar]
- Olsen L. R.; Roderick S. L. Structure of the Escherichia coli GlmU Pyrophosphorylase and Acetyltransferase Active Sites. Biochemistry 2001, 40, 1913–1921. 10.1021/bi002503n. [DOI] [PubMed] [Google Scholar]
- Raetz C. R.; Roderick S. L. A Left-Handed Parallel Beta Helix in the Structure of UDP-N-Acetylglucosamine Acyltransferase. Science 1995, 270, 997–1000. 10.1126/science.270.5238.997. [DOI] [PubMed] [Google Scholar]
- Pelissier M. C.; Lesley S. a.; Kuhn P.; Bourne Y. Structural Insights into the Catalytic Mechanism of Bacterial Guanosine-Diphospho-D-Mannose Pyrophosphorylase and Its Regulation by Divalent Ions. J. Biol. Chem. 2010, 285, 27468–27476. 10.1074/jbc.M109.095182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeben A.; Plitzko J. M.; Körner R.; Böttcher U. M. K.; Siegers K.; Hayer-Hartl M.; Bracher A. Structural Basis for Subunit Assembly in UDP-Glucose Pyrophosphorylase from Saccharomyces cerevisiae. J. Mol. Biol. 2006, 364, 551–560. 10.1016/j.jmb.2006.08.079. [DOI] [PubMed] [Google Scholar]
- Han X.; D’Angelo C.; Otamendi A.; Cifuente J. O.; de Astigarraga E.; Ochoa-Lizarralde B.; Grininger M.; Routier F. H.; Guerin M. E.; Fuehring J.; et al. CryoEM Analysis of the Essential Native UDP-Glucose Pyrophosphorylase from Aspergillus nidulans Reveals Key Conformations for Activity Regulation and Function. mBio 2023, 14, e0041423 10.1128/mbio.00414-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugen T. H.; Ishaque A.; Preiss J. Biosynthesis of bacterial glycogen. Characterization of the Subunit Structure of Escherichia coli B Glucose-1-Phosphate Adenylyltransferase (EC 2.7.7.27). J. Biol. Chem. 1976, 251, 7880–7885. 10.1016/S0021-9258(19)57016-8. [DOI] [PubMed] [Google Scholar]
- Haugen T. H.; Preiss J. Biosynthesis of Bacterial Glycogen. The Nature of the Binding of Substrates and Effectors to ADP-Glucose Synthase. J. Biol. Chem. 1979, 254, 127–136. 10.1016/S0021-9258(17)30281-8. [DOI] [PubMed] [Google Scholar]
- Takata H.; Takaha T.; Okada S.; Takagi M.; Imanaka T. Characterization of a Gene Cluster for Glycogen Biosynthesis and a Heterotetrameric ADP-Glucose Pyrophosphorylase from Bacillus stearothermophilus. J. Bacteriol. 1997, 179, 4689–4698. 10.1128/jb.179.15.4689-4698.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejar C. M.; Ballicora M. a.; Gómez-Casati D. F.; Iglesias A. A.; Preiss J. The ADP-Glucose Pyrophosphorylase from Escherichia coli Comprises Two Tightly Bound Distinct Domains. FEBS Lett. 2004, 573, 99–104. 10.1016/j.febslet.2004.07.060. [DOI] [PubMed] [Google Scholar]
- Morell M. K.; Bloom M.; Knowles V.; Preiss J. Subunit Structure of Spinach Leaf ADP-glucose Pyrophosphorylase. Plant Physiol. 1987, 85, 182–187. 10.1104/pp.85.1.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X.; Ballicora M. a; Preiss J.; Geiger J. H. Crystal Structure of Potato Tuber ADP-Glucose Pyrophosphorylase. EMBO J. 2005, 24, 694–704. 10.1038/sj.emboj.7600551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupp-Vickery J. R.; Igarashi R. Y.; Perez M.; Poland M.; Meyer C. R. Structural Analysis of ADP-Glucose Pyrophosphorylase from the Bacterium Agrobacterium tumefaciens. Biochemistry 2008, 47, 4439–4451. 10.1021/bi701933q. [DOI] [PubMed] [Google Scholar]
- Hill B. L.; Mascarenhas R.; Patel H. P.; Asencion Diez M. D.; Wu R.; Iglesias A. A.; Liu D.; Ballicora M. A. Structural Analysis Reveals a Pyruvate-Binding Activator Site in the Agrobacterium tumefaciens ADP-Glucose Pyrophosphorylase. J. Biol. Chem. 2019, 294, 1338–1348. 10.1074/jbc.RA118.004246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alghamdi M. A.; Hussien R. A.; Zheng Y.; Patel H. P.; Asencion Diez M. D.; A Iglesias A.; Liu D.; Ballicora M. A. Site-Directed Mutagenesis of Serine-72 Reveals the Location of the Fructose 6-Phosphate Regulatory Site of the Agrobacterium tumefaciens ADP-Glucose Pyrophosphorylase. Protein Sci. Publ. Protein Soc. 2022, 31, e4376 10.1002/pro.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comino N.; Cifuente J. O.; Marina A.; Orrantia A.; Eguskiza A.; Guerin M. E. Mechanistic Insights into the Allosteric Regulation of Bacterial ADP-Glucose Pyrophosphorylases. J. Biol. Chem. 2017, 292, 6255–6268. 10.1074/jbc.M116.773408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgelis N.; Braun E. L.; Shaw J. R.; Hannah L. C. The Two AGPase Subunits Evolve at Different Rates in Angiosperms, yet They Are Equally Sensitive to Activity-Altering Amino Acid Changes When Expressed in Bacteria. Plant Cell 2007, 19 (5), 1458–1472. 10.1105/tpc.106.049676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa C. M.; Esper M. C.; Bertolo A.; Demonte A. M.; Aleanzi M.; Iglesias A. a.; Ballicora M. a. Understanding the Allosteric Trigger for the Fructose-1,6-Bisphosphate Regulation of the ADP-Glucose Pyrophosphorylase from Escherichia coli. Biochimie 2011, 93, 1816–1823. 10.1016/j.biochi.2011.06.029. [DOI] [PubMed] [Google Scholar]
- Brito J. a.; Borges N.; Vonrhein C.; Santos H.; Archer M. Crystal Structure of Archaeoglobus fulgidus CTP:Inositol-1-Phosphate Cytidylyltransferase, a Key Enzyme for Di-Myo-Inositol-Phosphate Synthesis in (Hyper)Thermophiles. J. Bacteriol. 2011, 193, 2177–2185. 10.1128/JB.01543-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejar C. M.; Jin X.; Ballicora M. A.; Preiss J. Molecular Architecture of the Glucose 1-Phosphate Site in ADP-Glucose Pyrophosphorylases. J. Biol. Chem. 2006, 281, 40473–40484. 10.1074/jbc.M607088200. [DOI] [PubMed] [Google Scholar]
- Caputto R.; Leloir L. F.; Cardini C. E.; Paladini A. C. Isolation of the Coenzyme of the Galactose Phosphate-Glucose Phosphate Transformation. J. Biol. Chem. 1950, 184, 333–350. 10.1016/S0021-9258(19)51153-X. [DOI] [PubMed] [Google Scholar]
- Leloir L. F. Far Away and Long Ago. Annu. Rev. Biochem. 1983, 52, 1–16. 10.1146/annurev.bi.52.070183.000245. [DOI] [PubMed] [Google Scholar]
- Steiner T.; Lamerz A.-C.; Hess P.; Breithaupt C.; Krapp S.; Bourenkov G.; Huber R.; Gerardy-Schahn R.; Jacob U. Open and Closed Structures of the UDP-Glucose Pyrophosphorylase from Leishmania major. J. Biol. Chem. 2007, 282, 13003–13010. 10.1074/jbc.M609984200. [DOI] [PubMed] [Google Scholar]
- Führing J.; Damerow S.; Fedorov R.; Schneider J.; Münster-Kühnel A.-K.; Gerardy-Schahn R. Octamerization Is Essential for Enzymatic Function of Human UDP-Glucose Pyrophosphorylase. Glycobiology 2013, 23, 426–437. 10.1093/glycob/cws217. [DOI] [PubMed] [Google Scholar]
- Kleczkowski L. A.; Geisler M.; Fitzek E.; Wilczynska M. A Common Structural Blueprint for Plant UDP-Sugar-Producing Pyrophosphorylases. Biochem. J. 2011, 439, 375–379. 10.1042/BJ20110730. [DOI] [PubMed] [Google Scholar]
- Führing J. I.; Cramer J. T.; Schneider J.; Baruch P.; Gerardy-Schahn R.; Fedorov R. A Quaternary Mechanism Enables the Complex Biological Functions of Octameric Human UDP-Glucose Pyrophosphorylase, a Key Enzyme in Cell Metabolism. Sci. Rep. 2015, 5, 9618–9618. 10.1038/srep09618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoden J. B.; Holden H. M. The Molecular Architecture of Glucose-1-Phosphate Uridylyltransferase. Protein Sci. 2007, 16, 432–440. 10.1110/ps.062626007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragão D.; Fialho A. M.; Marques A. R.; Mitchell E. P.; Sá-Correia I.; Frazão C. The Complex of Sphingomonas Elodea ATCC 31461 Glucose-1-Phosphate Uridylyltransferase with Glucose-1-Phosphate Reveals a Novel Quaternary Structure, Unique among Nucleoside Diphosphate-Sugar Pyrophosphorylase Members. J. Bacteriol. 2007, 189, 4520–4528. 10.1128/JB.00277-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.; Choi J.; Kim T.; Lokanath N. K.; Ha S. C.; Suh S. W.; Hwang H. Y.; Kim K. K. Structural Basis for the Reaction Mechanism of UDP-Glucose Pyrophosphorylase. Mol. Cells 2010, 29, 397–405. 10.1007/s10059-010-0047-6. [DOI] [PubMed] [Google Scholar]
- Thoden J. B.; Holden H. M. Active Site Geometry of Glucose-1-Phosphate Uridylyltransferase. Protein Sci. 2007, 16, 1379–1388. 10.1110/ps.072864707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Tsujimura M.; Akutsu J.; Sasaki M.; Tajima H.; Kawarabayasi Y. Identification of an Extremely Thermostable Enzyme with Dual Sugar-1-Phosphate Nucleotidylyltransferase Activities from an Acidothermophilic Archaeon, Sulfolobus tokodaii Strain 7. J. Biol. Chem. 2005, 280, 9698–9705. 10.1074/jbc.M411211200. [DOI] [PubMed] [Google Scholar]
- Honda Y.; Nakano S.; Ito S.; Dadashipour M.; Zhang Z.; Kawarabayasi Y. Improvement of ST0452 N-Acetylglucosamine-1-Phosphate Uridyltransferase Activity by the Cooperative Effect of Two Single Mutations Identified through Structure-Based Protein Engineering. Appl. Environ. Microbiol. 2018, 84, e02213-18 10.1128/AEM.02213-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drula E.; Garron M.-L.; Dogan S.; Lombard V.; Henrissat B.; Terrapon N. The Carbohydrate-Active Enzyme Database: Functions and Literature. Nucleic Acids Res. 2022, 50, D571–D577. 10.1093/nar/gkab1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly S. D.; Williams D. M.; Nothof J. T.; Kim T.; Lowary T. L.; Kimber M. S.; Whitfield C. The Biosynthetic Origin of Ribofuranose in Bacterial Polysaccharides. Nat. Chem. Biol. 2022, 18, 530–537. 10.1038/s41589-022-01006-6. [DOI] [PubMed] [Google Scholar]
- Cifuente J. O.; Schulze J.; Bethe A.; Di Domenico V.; Litschko C.; Budde I.; Eidenberger L.; Thiesler H.; Ramón Roth I.; Berger M.; et al. A Multi-Enzyme Machine Polymerizes the Haemophilus influenzae Type b Capsule. Nat. Chem. Biol. 2023, 19, 865–877. 10.1038/s41589-023-01324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K.; Madej T.; Bryant S. H.; Panchenko A. R. Functional States of Homooligomers: Insights from the Evolution of Glycosyltransferases. J. Mol. Biol. 2010, 399, 196–196. 10.1016/j.jmb.2010.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albesa-Jové D.; Giganti D.; Jackson M.; Alzari P. M.; Guerin M. E. Structure-Function Relationships of Membrane-Associated GT-B Glycosyltransferases. Glycobiology 2014, 24, 108–124. 10.1093/glycob/cwt101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson K. A. The Crystal Structure of Escherichia coli Maltodextrin Phosphorylase Provides an Explanation for the Activity without Control in This Basic Archetype of a Phosphorylase. EMBO J. 1997, 16, 1–14. 10.1093/emboj/16.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly M.; Watson K. A.; Schinzel R.; Palm D.; Johnson L. N. Oligosaccharide Substrate Binding in Escherichia coli Maltodextrin Phosphorylase. Nat. Struct. Biol. 1997, 4, 405–412. 10.1038/nsb0597-405. [DOI] [PubMed] [Google Scholar]
- Baskaran S.; Roach P. J.; DePaoli-Roach A. A.; Hurley T. D. Structural Basis for Glucose-6-Phosphate Activation of Glycogen Synthase. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 17563–17568. 10.1073/pnas.1006340107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrielink A.; Rüger W.; Driessen H. P.; Freemont P. S. Crystal Structure of the DNA Modifying Enzyme Beta-Glucosyltransferase in the Presence and Absence of the Substrate Uridine Diphosphoglucose. EMBO J. 1994, 13, 3413–3422. 10.1002/j.1460-2075.1994.tb06646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artymiuk P. J.; Rice D. W.; Poirrette A. R.; Willett P. β-Glucosyltransferase and Phosphorylase Reveal Their Common Theme. Nat. Struct. Biol. 1995 22 1995, 2, 117–120. 10.1038/nsb0295-117. [DOI] [PubMed] [Google Scholar]
- Huo L.; Zhang H.; Huo X.; Yang Y.; Li X.; Yin Y. pHMM-Tree: Phylogeny of Profile Hidden Markov Models. Bioinforma. Oxf. Engl. 2017, 33, 1093–1095. 10.1093/bioinformatics/btw779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua T. K.; Bujnicki J. M.; Tan T.-C.; Huynh F.; Patel B. K.; Sivaraman J. The Structure of Sucrose Phosphate Synthase from Halothermothrix orenii Reveals Its Mechanism of Action and Binding Mode. Plant Cell Online 2008, 20, 1059–1072. 10.1105/tpc.107.051193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson R. P.; Turkenburg J. P.; Charnock S. J.; Lloyd R.; Davies G. J. Insights into Trehalose Synthesis Provided by the Structure of the Retaining Glucosyltransferase OtsA. Chem. Biol. 2002, 9, 1337–1346. 10.1016/S1074-5521(02)00292-2. [DOI] [PubMed] [Google Scholar]
- Gibson R. P.; Tarling C. A.; Roberts S.; Withers S. G.; Davies G. J. The Donor Subsite of Trehalose-6-Phosphate Synthase: Binary Complexes with UDP-Glucose and UDP-2-Deoxy-2-Fluoro-Glucose at 2 A Resolution. J. Biol. Chem. 2004, 279, 1950–1955. 10.1074/jbc.M307643200. [DOI] [PubMed] [Google Scholar]
- Errey J. C.; Lee S. S.; Gibson R. R.; Fleites C. M.; Barry C. S.; Jung P. M. J.; O’Sullivan A. C.; Davis B. G.; Davies G. J. Mechanistic Insight into Enzymatic Glycosyl Transfer with Retention of Configuration through Analysis of Glycomimetic Inhibitors. Angew. Chem. - Int. Ed. 2010, 49, 1234–1237. 10.1002/anie.200905096. [DOI] [PubMed] [Google Scholar]
- Mendes V.; Acebrón-García-de-eulate M.; Verma N.; Blaszczyk M.; Dias M. V. B.; Blundell T. L.. Mycobacterial OtsA Structures Unveil Substrate Preference Mechanism and Allosteric Regulation by 2-Oxoglutarate and 2-Phosphoglycerate. mBio 2019, 10. 10.1128/mBio.02272-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu S. I.; Park C. S.; Cha J.; Woo E. J.; Lee S. B. A Novel Trehalose-Synthesizing Glycosyltransferase from Pyrococcus horikoshii: Molecular Cloning and Characterization. Biochem. Biophys. Res. Commun. 2005, 329, 429–436. 10.1016/j.bbrc.2005.01.149. [DOI] [PubMed] [Google Scholar]
- Nobre A.; Alarico S.; Fernandes C.; Empadinhas N.; da Costa M. S. A Unique Combination of Genetic Systems for the Synthesis of Trehalose in Rubrobacter xylanophilus: Properties of a Rare Actinobacterial TreT. J. Bacteriol. 2008, 190, 7939–7946. 10.1128/JB.01055-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestrom L.; Marsden S. R.; van der Eijk H.; Laustsen J. U.; Jeffries C. M.; Svergun D. I.; Hagedoorn P.-L.; Bento I.; Hanefeld U. Anomeric Selectivity of Trehalose Transferase with Rare L-Sugars. ACS Catal. 2020, 10, 8835–8839. 10.1021/acscatal.0c02117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R.; Asención Diez M. D.; Figueroa C. M.; Machtey M.; Iglesias A. A.; Ballicora M. A.; Liu D. The Crystal Structure of Nitrosomonas europaea Sucrose Synthase Reveals Critical Conformational Changes and Insights into Sucrose Metabolism in Prokaryotes. J. Bacteriol. 2015, 197, 2734–2746. 10.1128/JB.00110-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Yao Y.; Yang G.; Tang J.; Ayala G. J.; Li X.; Zhang W.; Han Q.; Yang T.; Wang H.; et al. Co-Crystal Structure of Thermosynechococcus elongatus Sucrose Phosphate Synthase With UDP and Sucrose-6-Phosphate Provides Insight Into Its Mechanism of Action Involving an Oxocarbenium Ion and the Glycosidic Bond. Front. Microbiol. 2020, 11, 1050. 10.3389/fmicb.2020.01050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y.; Anderson S.; Zhang Y.; Garavito R. M. The Structure of Sucrose Synthase-1 from Arabidopsis thaliana and Its Functional Implications. J. Biol. Chem. 2011, 286, 36108–36118. 10.1074/jbc.M111.275974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baroja-Fernández E.; Muñoz F. J.; Saikusa T.; Rodríguez-López M.; Akazawa T.; Pozueta-Romero J. Sucrose Synthase Catalyzes the de Novo Production of ADPglucose Linked to Starch Biosynthesis in Heterotrophic Tissues of Plants. Plant Cell Physiol. 2003, 44, 500–509. 10.1093/pcp/pcg062. [DOI] [PubMed] [Google Scholar]
- Curatti L.; Folco E.; Desplats P.; Abratti G.; Limones V.; Herrera-Estrella L.; Salerno G. Sucrose-Phosphate Synthase from Synechocystis Sp. Strain PCC 6803: Identification of the spsA Gene and Characterization of the Enzyme Expressed in Escherichia coli. J. Bacteriol. 1998, 180, 6776–6779. 10.1128/JB.180.24.6776-6779.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz A.; Martínez-Pons C.; Fita I.; Ferrer J. C.; Guinovart J. J. Processivity and Subcellular Localization of Glycogen Synthase Depend on a Non-Catalytic High Affinity Glycogen-Binding Site. J. Biol. Chem. 2011, 286, 18505–18514. 10.1074/jbc.M111.236109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M. M.; Ruzanski C.; Krucewicz K.; Striebeck A.; Cenci U.; Ball S. G.; Palcic M. M.; Cuesta-Seijo J. A. Crystal Structures of the Catalytic Domain of Arabidopsis Thaliana Starch Synthase IV, of Granule Bound Starch Synthase From CLg1 and of Granule Bound Starch Synthase I of Cyanophora paradoxa Illustrate Substrate Recognition in Starch Synthases. Front. Plant Sci. 2018, 9, 1138. 10.3389/fpls.2018.01138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskaran S.; Chikwana V. M.; Contreras C. J.; Davis K. D.; Wilson W. A.; DePaoli-Roach A. A.; Roach P. J.; Hurley T. D. Multiple Glycogen-Binding Sites in Eukaryotic Glycogen Synthase Are Required for High Catalytic Efficiency toward Glycogen. J. Biol. Chem. 2011, 286, 33999–34006. 10.1074/jbc.M111.264531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach P. J. Glycogen and Its Metabolism. Curr. Mol. Med. 2002, 2, 101–120. 10.2174/1566524024605761. [DOI] [PubMed] [Google Scholar]
- Zeqiraj E.; Tang X.; Hunter R. W.; Garcia-Rocha M.; Judd A.; Deak M.; von Wilamowitz-Moellendorff A.; Kurinov I.; Guinovart J. J.; Tyers M.; et al. Structural Basis for the Recruitment of Glycogen Synthase by Glycogenin. Proc. Natl. Acad. Sci. 2014, 111, E2831–E2840. 10.1073/pnas.1402926111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fastman N. M.; Liu Y.; Ramanan V.; Merritt H.; Ambing E.; DePaoli-Roach A. A.; Roach P. J.; Hurley T. D.; Mellem K. T.; Ullman J. C.; et al. The Structural Mechanism of Human Glycogen Synthesis by the GYS1-GYG1 Complex. Cell Rep. 2022, 40, 111041 10.1016/j.celrep.2022.111041. [DOI] [PubMed] [Google Scholar]
- Sheng F.; Yep A.; Feng L.; Preiss J.; Geiger J. H. Oligosaccharide Binding in Escherichia coli Glycogen Synthase. Biochemistry 2009, 48 (42), 10089–10097. 10.1021/bi900916t. [DOI] [PubMed] [Google Scholar]
- Sheng F.; Jia X.; Yep A.; Preiss J.; Geiger J. H. The Crystal Structures of the Open and Catalytically Competent Closed Conformation of Escherichia coli Glycogen Synthase. J. Biol. Chem. 2009, 284, 17796–17807. 10.1074/jbc.M809804200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horcajada C.; Guinovart J. J.; Fita I.; Ferrer J. C. Crystal Structure of an Archaeal Glycogen Synthase: Insights into Oligomerization and Substrate Binding of Eukaryotic Glycogen Synthases. J. Biol. Chem. 2006, 281, 2923–2931. 10.1074/jbc.M507394200. [DOI] [PubMed] [Google Scholar]
- Díaz A.; Díaz-Lobo M.; Grados E.; Guinovart J. J.; Fita I.; Ferrer J. C. Lyase Activity of Glycogen Synthase: Is an Elimination/Addition Mechanism a Possible Reaction Pathway for Retaining Glycosyltransferases?. IUBMB Life 2012, 64, 649–658. 10.1002/iub.1048. [DOI] [PubMed] [Google Scholar]
- Fox J.; Kawaguchi K.; Greenberg E.; Preiss J. Biosynthesis of Bacterial Glycogen. Purification and Properties of the Escherichia coli B ADPglucose:1,4-Alpha-D-Glucan 4-Alpha-Glucosyltransferase. Biochemistry 1976, 15, 849–857. 10.1021/bi00649a019. [DOI] [PubMed] [Google Scholar]
- Fletterick R. J.; Sygusch J.; Murray N.; Madsen N. B. Low-Resolution Structure of the Glycogen Phosphorylase Alpha Monomer and Comparison with Phosphorylase Beta. J. Mol. Biol. 1976, 103, 1–13. 10.1016/0022-2836(76)90048-6. [DOI] [PubMed] [Google Scholar]
- Fletterick R. J.; Sygusch J.; Semple H.; Madsen N. B. Structure of Glycogen Phosphorylase a at 3.0 A Resolution and Its Ligand Binding Sites at 6 A. J. Biol. Chem. 1976, 251, 6142–6146. 10.1016/S0021-9258(17)33070-3. [DOI] [PubMed] [Google Scholar]
- Schwartz M.; Hofnung M. Maltodextrin phosphorylase of Escherichia coli. Eur. J. Biochem. 1967, 2, 132–145. 10.1111/j.1432-1033.1967.tb00117.x. [DOI] [PubMed] [Google Scholar]
- Bukau B.; Ehrmann M.; Boos W. Osmoregulation of the Maltose Regulon in Escherichia coli. J. Bacteriol. 1986, 166, 884–891. 10.1128/jb.166.3.884-891.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boos W.; Shuman H. Maltose/Maltodextrin System of Escherichia coli: Transport, Metabolism, and Regulation. Microbiol. Mol. Biol. Rev. 1998, 62, 204–229. 10.1128/MMBR.62.1.204-229.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G. S.; Segel I. H. Purification and Properties of Glycogen Phosphorylase from Escherichia coli. Arch. Biochem. Biophys. 1968, 127, 175–186. 10.1016/0003-9861(68)90214-2. [DOI] [PubMed] [Google Scholar]
- Pulkownik A.; Walker G. J. Metabolism of the Reserve Polysaccharide of Streptococcus mitior (Mitis): Is There a Second Alpha-1,4-Glucan Phosphorylase?. J. Bacteriol. 1976, 127, 281–290. 10.1128/jb.127.1.281-290.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly M.; Watson K. A.; Johnson L. N. The Crystal Structure of the Escherichia coli Maltodextrin Phosphorylase-Acarbose Complex. Biochemistry 1999, 38, 5337–5345. 10.1021/bi9828573. [DOI] [PubMed] [Google Scholar]
- Watson K. A.; McCleverty C.; Geremia S.; Cottaz S.; Driguez H.; Johnson L. N. Phosphorylase Recognition and Phosphorolysis of Its Oligosaccharide Substrate: Answers to a Long Outstanding Question. EMBO J. 1999, 18, 4619–4632. 10.1093/emboj/18.17.4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagnolo M.; Campa C.; Zorzi R. D.; Wuerges J.; Geremia S. X-Ray Studies on Ternary Complexes of Maltodextrin Phosphorylase. Arch. Biochem. Biophys. 2008, 471, 11–19. 10.1016/j.abb.2007.11.023. [DOI] [PubMed] [Google Scholar]
- Koonin E. V.; Tatusov R. L. Computer Analysis of Bacterial Haloacid Dehalogenases Defines a Large Superfamily of Hydrolases with Diverse Specificity. Application of an Iterative Approach to Database Search. J. Mol. Biol. 1994, 244, 125–132. 10.1006/jmbi.1994.1711. [DOI] [PubMed] [Google Scholar]
- Lahiri S. D.; Zhang G.; Dai J.; Dunaway-Mariano D.; Allen K. N. Analysis of the Substrate Specificity Loop of the HAD Superfamily Cap Domain. Biochemistry 2004, 43, 2812–2820. 10.1021/bi0356810. [DOI] [PubMed] [Google Scholar]
- Park J.; Guggisberg A. M.; Odom A. R.; Tolia N. H. Cap-Domain Closure Enables Diverse Substrate Recognition by the C2-Type Haloacid Dehalogenase-like Sugar Phosphatase Plasmodium falciparum HAD1. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 1824–1834. 10.1107/S1399004715012067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao K. N.; Kumaran D.; Seetharaman J.; Bonanno J. B.; Burley S. K.; Swaminathan S. Crystal Structure of Trehalose-6-Phosphate Phosphatase-Related Protein: Biochemical and Biological Implications. Protein Sci. 2006, 15, 1735–1744. 10.1110/ps.062096606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieulaine S.; Lunn J. E.; Ferrer J.-L. Crystal Structure of a Cyanobacterial Sucrose-Phosphatase in Complex with Glucose-Containing Disaccharides. Proteins 2007, 68, 796–801. 10.1002/prot.21481. [DOI] [PubMed] [Google Scholar]
- Ku S.-Y.; Yip P.; Cornell K. A.; Riscoe M. K.; Behr J.-B.; Guillerm G.; Howell P. L. Structures of 5-Methylthioribose Kinase Reveal Substrate Specificity and Unusual Mode of Nucleotide Binding. J. Biol. Chem. 2007, 282, 22195–22206. 10.1074/jbc.M611045200. [DOI] [PubMed] [Google Scholar]
- Blackmore N. J.; Nazmi A. R.; Hutton R. D.; Webby M. N.; Baker E. N.; Jameson G. B.; Parker E. J. Complex Formation between Two Biosynthetic Enzymes Modifies the Allosteric Regulatory Properties of Both: An Example of Molecular Symbiosis. J. Biol. Chem. 2015, 290, 18187–18198. 10.1074/jbc.M115.638700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga R. K. The TIM-Barrel Fold: A Versatile Framework for Efficient Enzymes. FEBS Lett. 2001, 492, 193–198. 10.1016/S0014-5793(01)02236-0. [DOI] [PubMed] [Google Scholar]
- Banner D. W.; Bloomer A. C.; Petsko G. A.; Phillips D. C.; Pogson C. I.; Wilson I. A.; Corran P. H.; Furth A. J.; Milman J. D.; Offord R. E.; et al. Structure of Chicken Muscle Triose Phosphate Isomerase Determined Crystallographically at 2.5 Angstrom Resolution Using Amino Acid Sequence Data. Nature 1975, 255, 609–614. 10.1038/255609a0. [DOI] [PubMed] [Google Scholar]
- Vijayabaskar M. S.; Vishveshwara S. Insights into the Fold Organization of TIM Barrel from Interaction Energy Based Structure Networks. PLoS Comput. Biol. 2012, 8, e1002505 10.1371/journal.pcbi.1002505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber G. K.; Petsko G. A. The Evolution of Alpha/Beta Barrel Enzymes. Trends Biochem. Sci. 1990, 15, 228–234. 10.1016/0968-0004(90)90035-A. [DOI] [PubMed] [Google Scholar]
- Hol W. G.; Halie L. M.; Sander C. Dipoles of the Alpha-Helix and Beta-Sheet: Their Role in Protein Folding. Nature 1981, 294, 532–536. 10.1038/294532a0. [DOI] [PubMed] [Google Scholar]
- Vasanthi G.; Krishnaswamy S. Dipole Moment in TIM Alpha/Beta Fold Proteins. Indian J. Biochem. Biophys. 2003, 40, 194–202. [PubMed] [Google Scholar]
- Tiwari S. P.; Reuter N. Similarity in Shape Dictates Signature Intrinsic Dynamics Despite No Functional Conservation in TIM Barrel Enzymes. PLoS Comput. Biol. 2016, 12, e1004834 10.1371/journal.pcbi.1004834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel B. L.; Coutinho P. M.; Rancurel C.; Bernard T.; Lombard V.; Henrissat B. The Carbohydrate-Active EnZymes Database (CAZy): An Expert Resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233–238. 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGregor E. A.; Janecek S.; Svensson B. Relationship of Sequence and Structure to Specificity in the Alpha-Amylase Family of Enzymes. Biochim. Biophys. Acta 2001, 1546, 1–20. 10.1016/S0167-4838(00)00302-2. [DOI] [PubMed] [Google Scholar]
- Janeček Š.; Mareček F.; MacGregor E. A.; Svensson B. Starch-Binding Domains as CBM Families-History, Occurrence, Structure, Function and Evolution. Biotechnol. Adv. 2019, 37, 107451 10.1016/j.biotechadv.2019.107451. [DOI] [PubMed] [Google Scholar]
- Kari J.; Olsen J. P.; Jensen K.; Badino S. F.; Krogh K. B. R. M.; Borch K.; Westh P. Sabatier Principle for Interfacial (Heterogeneous) Enzyme Catalysis. ACS Catal. 2018, 8, 11966–11972. 10.1021/acscatal.8b03547. [DOI] [Google Scholar]
- Kari J.; Schaller K.; Molina G. A.; Borch K.; Westh P. The Sabatier Principle as a Tool for Discovery and Engineering of Industrial Enzymes. Curr. Opin. Biotechnol. 2022, 78, 102843 10.1016/j.copbio.2022.102843. [DOI] [PubMed] [Google Scholar]
- Mo̷ller M. S.; Henriksen A.; Svensson B. Structure and Function of α-Glucan Debranching Enzymes. Cell. Mol. Life Sci. 2016, 73, 2619–2641. 10.1007/s00018-016-2241-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abad M. C.; Binderup K.; Rios-Steiner J.; Arni R. K.; Preiss J.; Geiger J. H. The X-Ray Crystallographic Structure of Escherichia coli Branching Enzyme. J. Biol. Chem. 2002, 277, 42164–42170. 10.1074/jbc.M205746200. [DOI] [PubMed] [Google Scholar]
- Schumacher M. A.; Wörmann M. E.; Henderson M.; Salinas R.; Latoscha A.; Al-Bassam M. M.; Singh K. S.; Barclay E.; Gunka K.; Tschowri N. Allosteric Regulation of Glycogen Breakdown by the Second Messenger Cyclic Di-GMP. Nat. Commun. 2022, 13, 5834. 10.1038/s41467-022-33537-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leemhuis H.; Dijkstra B. W.; Dijkhuizen L. Mutations Converting Cyclodextrin Glycosyltransferase from a Transglycosylase into a Starch Hydrolase. FEBS Lett. 2002, 514, 189–192. 10.1016/S0014-5793(02)02362-1. [DOI] [PubMed] [Google Scholar]
- Janecek S.; Svensson B.; MacGregor E. A. Relation between Domain Evolution, Specificity, and Taxonomy of the Alpha-Amylase Family Members Containing a C-Terminal Starch-Binding Domain. Eur. J. Biochem. 2003, 270, 635–645. 10.1046/j.1432-1033.2003.03404.x. [DOI] [PubMed] [Google Scholar]
- Thanvi R.; Jayasinghe T. D.; Kapil S.; Obadawo B. S.; Ronning D. R.; Sucheck S. J. Synthesis of C7/C8-Cyclitols and C7N-Aminocyclitols from Maltose and X-Ray Crystal Structure of Streptomyces Coelicolor GlgEI V279S in a Complex with an Amylostatin GXG-like Derivative. Front. Chem. 2022, 10, 950433 10.3389/fchem.2022.950433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza O.; Skov L. K.; Remaud-Simeon M.; Potocki de Montalk G.; Albenne C.; Monsan P.; Gajhede M. Crystal Structures of Amylosucrase from Neisseria polysaccharea in Complex with D-Glucose and the Active Site Mutant Glu328Gln in Complex with the Natural Substrate Sucrose. Biochemistry 2001, 40, 9032–9039. 10.1021/bi010706l. [DOI] [PubMed] [Google Scholar]
- Champion E.; Guérin F.; Moulis C.; Barbe S.; Tran T. H.; Morel S.; Descroix K.; Monsan P.; Mourey L.; Mulard L. A.; et al. Applying Pairwise Combinations of Amino Acid Mutations for Sorting out Highly Efficient Glucosylation Tools for Chemo-Enzymatic Synthesis of Bacterial Oligosaccharides. J. Am. Chem. Soc. 2012, 134, 18677–18688. 10.1021/ja306845b. [DOI] [PubMed] [Google Scholar]
- Skov L. K.; Pizzut-Serin S.; Remaud-Simeon M.; Ernst H. A.; Gajhede M.; Mirza O. The Structure of Amylosucrase from Deinococcus radiodurans Has an Unusual Open Active-Site Topology. Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun. 2013, 69, 973–978. 10.1107/S1744309113021714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza O.; Skov L. K.; Sprogo̷e D.; van den Broek L. A. M.; Beldman G.; Kastrup J. S.; Gajhede M. Structural Rearrangements of Sucrose Phosphorylase from Bifidobacterium adolescentis during Sucrose Conversion. J. Biol. Chem. 2006, 281, 35576–35584. 10.1074/jbc.M605611200. [DOI] [PubMed] [Google Scholar]
- Kraus M.; Grimm C.; Seibel J. Redesign of the Active Site of Sucrose Phosphorylase through a Clash-Induced Cascade of Loop Shifts. Chembiochem Eur. J. Chem. Biol. 2016, 17, 33–36. 10.1002/cbic.201500514. [DOI] [PubMed] [Google Scholar]
- Kraus M.; Grimm C.; Seibel J. Switching Enzyme Specificity from Phosphate to Resveratrol Glucosylation. Chem. Commun. Camb. Engl. 2017, 53, 12181–12184. 10.1039/C7CC05993K. [DOI] [PubMed] [Google Scholar]
- Franceus J.; Capra N.; Desmet T.; Thunnissen A.-M. W. H. Structural Comparison of a Promiscuous and a Highly Specific Sucrose 6F-Phosphate Phosphorylase. Int. J. Mol. Sci. 2019, 20, 3906. 10.3390/ijms20163906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machius M.; Declerck N.; Huber R.; Wiegand G. Activation of Bacillus Licheniformis Alpha-Amylase through a Disorder-->order Transition of the Substrate-Binding Site Mediated by a Calcium-Sodium-Calcium Metal Triad. Struct. Lond. Engl. 1993 1998, 6, 281–292. 10.1016/S0969-2126(98)00032-X. [DOI] [PubMed] [Google Scholar]
- Machius M.; Wiegand G.; Huber R. Crystal Structure of Calcium-Depleted Bacillus licheniformis Alpha-Amylase at 2.2 A Resolution. J. Mol. Biol. 1995, 246, 545–559. 10.1006/jmbi.1994.0106. [DOI] [PubMed] [Google Scholar]
- Machius M.; Declerck N.; Huber R.; Wiegand G. Kinetic Stabilization of Bacillus Licheniformis Alpha-Amylase through Introduction of Hydrophobic Residues at the Surface. J. Biol. Chem. 2003, 278, 11546–11553. 10.1074/jbc.M212618200. [DOI] [PubMed] [Google Scholar]
- Linden A.; Mayans O.; Meyer-Klaucke W.; Antranikian G.; Wilmanns M. Differential Regulation of a Hyperthermophilic Alpha-Amylase with a Novel (Ca,Zn) Two-Metal Center by Zinc. J. Biol. Chem. 2003, 278, 9875–9884. 10.1074/jbc.M211339200. [DOI] [PubMed] [Google Scholar]
- Kobayashi M.; Kubota M.; Matsuura Y. Refined Structure and Functional Implications of Trehalose Synthase from Sulfolobus acidocaldarius. J. Appl. Glycosci. 2003, 50, 1–8. 10.5458/jag.50.1. [DOI] [Google Scholar]
- Matsuura Y.; Kusunoki M.; Harada W.; Kakudo M. Structure and Possible Catalytic Residues of Taka-Amylase A. J. Biochem. (Tokyo) 1984, 95, 697–702. 10.1093/oxfordjournals.jbchem.a134659. [DOI] [PubMed] [Google Scholar]
- Janecek S.; Svensson B.; Henrissat B. Domain Evolution in the Alpha-Amylase Family. J. Mol. Evol. 1997, 45, 322–331. 10.1007/PL00006236. [DOI] [PubMed] [Google Scholar]
- Vujicic-Zagar A.; Pijning T.; Kralj S.; López C. A.; Eeuwema W.; Dijkhuizen L.; Dijkstra B. W. Crystal Structure of a 117 kDa Glucansucrase Fragment Provides Insight into Evolution and Product Specificity of GH70 Enzymes. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 21406–21411. 10.1073/pnas.1007531107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K.; Ito S.; Shimamura T.; Weyand S.; Kawarasaki Y.; Misaka T.; Abe K.; Kobayashi T.; Cameron A. D.; Iwata S. Crystal Structure of Glucansucrase from the Dental Caries Pathogen Streptococcus mutans. J. Mol. Biol. 2011, 408, 177–186. 10.1016/j.jmb.2011.02.028. [DOI] [PubMed] [Google Scholar]
- Claverie M.; Cioci G.; Vuillemin M.; Monties N.; Roblin P.; Lippens G.; Remaud-Simeon M.; Moulis C. Investigations on the Determinants Responsible for Low Molar Mass Dextran Formation by DSR-M Dextransucrase. ACS Catal. 2017, 7, 7106–7119. 10.1021/acscatal.7b02182. [DOI] [Google Scholar]
- Claverie M.; Cioci G.; Guionnet M.; Schörghuber J.; Lichtenecker R.; Moulis C.; Remaud-Simeon M.; Lippens G. Futile Encounter Engineering of the DSR-M Dextransucrase Modifies the Resulting Polymer Length. Biochemistry 2019, 58, 2853–2859. 10.1021/acs.biochem.9b00373. [DOI] [PubMed] [Google Scholar]
- Molina M.; Moulis C.; Monties N.; Guieysse D.; Morel S.; Cioci G.; Remaud-Siméon M. A Specific Oligosaccharide-Binding Site in the Alternansucrase Catalytic Domain Mediates Alternan Elongation. J. Biol. Chem. 2020, 295, 9474–9489. 10.1074/jbc.RA120.013028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijning T.; Vujičić-Žagar A.; Kralj S.; Dijkhuizen L.; Dijkstra B. W. Structure of the α-1,6/α-1,4-Specific Glucansucrase GTFA from Lactobacillus reuteri 121. Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 1448–1454. 10.1107/S1744309112044168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przylas I.; Tomoo K.; Terada Y.; Takaha T.; Fujii K.; Saenger W.; Sträter N. Crystal Structure of Amylomaltase from Thermus aquaticus, a Glycosyltransferase Catalysing the Production of Large Cyclic Glucans. J. Mol. Biol. 2000, 296, 873–886. 10.1006/jmbi.1999.3503. [DOI] [PubMed] [Google Scholar]
- Roth C.; Weizenmann N.; Bexten N.; Saenger W.; Zimmermann W.; Maier T.; Sträter N. Amylose Recognition and Ring-Size Determination of Amylomaltase. Sci. Adv. 2017, 3, e1601386 10.1126/sciadv.1601386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumhom S.; Nimpiboon P.; Wangkanont K.; Pongsawasdi P. Streptococcus Agalactiae Amylomaltase Offers Insight into the Transglycosylation Mechanism and the Molecular Basis of Thermostability among Amylomaltases. Sci. Rep. 2021, 11, 6740. 10.1038/s41598-021-85769-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeček Š.; Gabriško M. Remarkable Evolutionary Relatedness among the Enzymes and Proteins from the α-Amylase Family. Cell. Mol. Life Sci. 2016, 73, 2707–2725. 10.1007/s00018-016-2246-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mareček F.; Mo̷ller M. S.; Svensson B.; Janeček Š. A Putative Novel Starch-Binding Domain Revealed by in Silico Analysis of the N-Terminal Domain in Bacterial Amylomaltases from the Family GH77. 3 Biotech 2021, 11, 229. 10.1007/s13205-021-02787-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godány A.; Vidová B.; Janecek S. The Unique Glycoside Hydrolase Family 77 Amylomaltase from Borrelia Burgdorferi with Only Catalytic Triad Conserved. FEMS Microbiol. Lett. 2008, 284, 84–91. 10.1111/j.1574-6968.2008.01191.x. [DOI] [PubMed] [Google Scholar]
- Janeček Š.; Svensson B. Amylolytic Glycoside Hydrolases. Cell. Mol. Life Sci. CMLS 2016, 73, 2601–2602. 10.1007/s00018-016-2240-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura H.; Fushinobu S.; Yamamoto M.; Kumasaka T.; Jeon B.-S.; Wakagi T.; Matsuzawa H. Crystal Structures of 4-Alpha-Glucanotransferase from Thermococcus litoralis and Its Complex with an Inhibitor. J. Biol. Chem. 2003, 278, 19378–19386. 10.1074/jbc.M213134200. [DOI] [PubMed] [Google Scholar]
- Miao M.; Jiang B.; Jin Z.; BeMiller J. N. Microbial Starch-Converting Enzymes: Recent Insights and Perspectives. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1238–1260. 10.1111/1541-4337.12381. [DOI] [PubMed] [Google Scholar]
- Miller S. L.; Urey H. C. Organic Compound Synthesis on the Primitive Earth. Science 1959, 130, 245–251. 10.1126/science.130.3370.245. [DOI] [PubMed] [Google Scholar]
- Ferris J. P.; Hill A. R.; Liu R.; Orgel L. E. Synthesis of Long Prebiotic Oligomers on Mineral Surfaces. Nature 1996, 381, 59–61. 10.1038/381059a0. [DOI] [PubMed] [Google Scholar]
- Orgel L. E. Polymerization on the Rocks: Theoretical Introduction. Orig. Life Evol. Biosphere J. Int. Soc. Study Orig. Life 1998, 28, 227–234. 10.1023/A:1006595411403. [DOI] [PubMed] [Google Scholar]
- Stern R.; Jedrzejas M. J. Carbohydrate Polymers at the Center of Life’s Origins: The Importance of Molecular Processivity. Chem. Rev. 2008, 108, 5061–5085. 10.1021/cr078240l. [DOI] [PubMed] [Google Scholar]
- Tolstoguzov V. Why Were Polysaccharides Necessary?. Orig. Life Evol. Biosphere 2004 346 2004, 34 (6), 571–597. 10.1023/B:ORIG.0000043127.18942.c4. [DOI] [PubMed] [Google Scholar]
- Brown M. R. W.; Kornberg A. Inorganic Polyphosphate in the Origin and Survival of Species. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 16085–16087. 10.1073/pnas.0406909101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M. S.; Jones C. D.; Helfrich M. R.; Mangeney-Slavin L. K.; Keating C. D. Dynamic Microcompartmentation in Synthetic Cells. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 5920–5925. 10.1073/pnas.0409333102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damager I.; Engelsen S. B.; Blennow A.; Lindberg Mo̷ller B.; Motawia M. S. First Principles Insight into the α-Glucan Structures of Starch: Their Synthesis, Conformation, and Hydration. Chem. Rev. 2010, 110, 2049–2080. 10.1021/cr900227t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Delbianco M.; Seeberger P. H. Automated Assembly of Starch and Glycogen Polysaccharides. J. Am. Chem. Soc. 2021, 143, 9758–9768. 10.1021/jacs.1c02188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degani C.; Halmann M. D-Glucose 1-Phosphate Formation by Cyanogen-Induced Phosphorylation of D-Glucose. Synthesis, Mechanism, and Application to Other Reducing Sugars. J. Chem. Soc. C Org. 1971, 1459–1465. 10.1039/j39710001459. [DOI] [Google Scholar]
- Nam I.; Lee J. K.; Nam H. G.; Zare R. N. Abiotic Production of Sugar Phosphates and Uridine Ribonucleoside in Aqueous Microdroplets. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 12396–12400. 10.1073/pnas.1714896114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Römling U. Is Biofilm Formation Intrinsic to the Origin of Life?. Environ. Microbiol. 2023, 25, 26. 10.1111/1462-2920.16179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penesyan A.; Paulsen I. T.; Kjelleberg S.; Gillings M. R. Three Faces of Biofilms: A Microbial Lifestyle, a Nascent Multicellular Organism, and an Incubator for Diversity. NPJ Biofilms Microbiomes 2021, 7, 80. 10.1038/s41522-021-00251-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoodley P.; Sauer K.; Davies D. G.; Costerton J. W. Biofilms as Complex Differentiated Communities. Annu. Rev. Microbiol. 2002, 56, 187–209. 10.1146/annurev.micro.56.012302.160705. [DOI] [PubMed] [Google Scholar]
- Flemming H.-C.; Wingender J.; Szewzyk U.; Steinberg P.; Rice S. A.; Kjelleberg S. Biofilms: An Emergent Form of Bacterial Life. Nat. Rev. Microbiol. 2016, 14, 563–575. 10.1038/nrmicro.2016.94. [DOI] [PubMed] [Google Scholar]
- Hall-Stoodley L.; Costerton J. W.; Stoodley P. Bacterial Biofilms: From the Natural Environment to Infectious Diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- Betts H. C.; Puttick M. N.; Clark J. W.; Williams T. A.; Donoghue P. C. J.; Pisani D. Integrated Genomic and Fossil Evidence Illuminates Life’s Early Evolution and Eukaryote Origin. Nat. Ecol. Evol. 2018, 2, 1556–1562. 10.1038/s41559-018-0644-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown R. M.; Willison J. H.; Richardson C. L. Cellulose Biosynthesis in Acetobacter Xylinum: Visualization of the Site of Synthesis and Direct Measurement of the in Vivo Process. Proc. Natl. Acad. Sci. U. S. A. 1976, 73, 4565–4569. 10.1073/pnas.73.12.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra D. O.; Richter A. M.; Hengge R. Cellulose as an Architectural Element in Spatially Structured Escherichia coli Biofilms. J. Bacteriol. 2013, 195, 5540–5554. 10.1128/JB.00946-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh M.; Stone B. A.; Stanisich V. A. Curdlan and Other Bacterial (1-->3)-Beta-D-Glucans. Appl. Microbiol. Biotechnol. 2005, 68, 163–173. 10.1007/s00253-005-1959-5. [DOI] [PubMed] [Google Scholar]
- Bonafonte M. A.; Solano C.; Sesma B.; Alvarez M.; Montuenga L.; Garcia-Ros D.; Gamazo C. The Relationship between Glycogen Synthesis, Biofilm Formation and Virulence in Salmonella enteritidis. FEMS Microbiol Lett 2000, 191, 31–36. 10.1111/j.1574-6968.2000.tb09315.x. [DOI] [PubMed] [Google Scholar]
- Jackson D. W.; Suzuki K.; Oakford L.; Simecka J. W.; Hart M. E.; Romeo T. Biofilm Formation and Dispersal under the Influence of the Global Regulator CsrA of Escherichia coli. J. Bacteriol. 2002, 184, 290–301. 10.1128/JB.184.1.290-301.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y.; Gangoiti J.; Dijkstra B. W.; Dijkhuizen L.; Pijning T. Crystal Structure of 4,6-α-Glucanotransferase Supports Diet-Driven Evolution of GH70 Enzymes from α-Amylases in Oral Bacteria. Struct. Lond. Engl. 1993 2017, 25, 231–242. 10.1016/j.str.2016.11.023. [DOI] [PubMed] [Google Scholar]
- Petruzzi B.; Briggs R. E.; Swords W. E.; de Castro C.; Molinaro A.; Inzana T. J. Capsular Polysaccharide Interferes with Biofilm Formation by Pasteurella multocida Serogroup A. mBio 2017, 8, e01843-17 10.1128/mBio.01843-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champion A. E.; Catanzaro K. C. F.; Bandara A. B.; Inzana T. J. Formation of the Francisella Tularensis Biofilm Is Affected by Cell Surface Glycosylation, Growth Medium, and a Glucan Exopolysaccharide. Sci. Rep. 2019, 9, 12252. 10.1038/s41598-019-48697-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambourova M.; Mandeva R.; Dimova D.; Poli A.; Nicolaus B.; Tommonaro G. Production and Characterization of a Microbial Glucan, Synthesized by Geobacillus Tepidamans V264 Isolated from Bulgarian Hot Spring. Carbohydr. Polym. 2009, 77, 338–343. 10.1016/j.carbpol.2009.01.004. [DOI] [Google Scholar]
- Te Poele E. M.; Van Der Hoek S. E.; Chatziioannou A. C.; Gerwig G. J.; Duisterwinkel W. J.; Oudhuis L. A. A. C. M.; Gangoiti J.; Dijkhuizen L.; Leemhuis H. GtfC Enzyme of Geobacillus Sp. 12AMOR1 Represents a Novel Thermostable Type of GH70 4,6-α-Glucanotransferase That Synthesizes a Linear Alternating (Α1 → 6)/(Α1 → 4) α-Glucan and Delays Bread Staling. J. Agric. Food Chem. 2021, 69, 9859–9868. 10.1021/acs.jafc.1c03475. [DOI] [PubMed] [Google Scholar]
- Probst A. J.; Weinmaier T.; Raymann K.; Perras A.; Emerson J. B.; Rattei T.; Wanner G.; Klingl A.; Berg I. A.; Yoshinaga M.; et al. Biology of a Widespread Uncultivated Archaeon That Contributes to Carbon Fixation in the Subsurface. Nat. Commun. 2014, 5, 5497. 10.1038/ncomms6497. [DOI] [PubMed] [Google Scholar]
- Koonin E. V. Comparative Genomics, Minimal Gene-Sets and the Last Universal Common Ancestor. Nat. Rev. Microbiol. 2003, 1, 127–136. 10.1038/nrmicro751. [DOI] [PubMed] [Google Scholar]
- McMeechan A.; Lovell M. A.; Cogan T. A.; Marston K. L.; Humphrey T. J.; Barrow P. A. Glycogen Production by Different Salmonella enterica Serotypes: Contribution of Functional glgC to Virulence, Intestinal Colonization and Environmental Survival. Microbiol. Read. Engl. 2005, 151, 3969–3977. 10.1099/mic.0.28292-0. [DOI] [PubMed] [Google Scholar]
- Klotz A.; Georg J.; Bučinská L.; Watanabe S.; Reimann V.; Januszewski W.; Sobotka R.; Jendrossek D.; Hess W. R.; Forchhammer K. Awakening of a Dormant Cyanobacterium from Nitrogen Chlorosis Reveals a Genetically Determined Program. Curr. Biol. 2016, 26, 2862–2872. 10.1016/j.cub.2016.08.054. [DOI] [PubMed] [Google Scholar]
- Mushegian A. The Minimal Genome Concept. Curr. Opin. Genet. Dev. 1999, 9, 709–714. 10.1016/S0959-437X(99)00023-4. [DOI] [PubMed] [Google Scholar]
- Acevedo-Rocha C. G.; Fang G.; Schmidt M.; Ussery D. W.; Danchin A. From Essential to Persistent Genes: A Functional Approach to Constructing Synthetic Life. Trends Genet. 2013, 29, 273–279. 10.1016/j.tig.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrissat B.; Deleury E.; Coutinho P. M. Glycogen Metabolism Loss: A Common Marker of Parasitic Behaviour in Bacteria?. Trends Genet. 2002, 18, 437–440. 10.1016/S0168-9525(02)02734-8. [DOI] [PubMed] [Google Scholar]
- Seibold G.; Dempf S.; Schreiner J.; Eikmanns B. J. Glycogen Formation in Corynebacterium glutamicum and Role of ADP-Glucose Pyrophosphorylase. Microbiol. Read. Engl. 2007, 153, 1275–1285. 10.1099/mic.0.2006/003368-0. [DOI] [PubMed] [Google Scholar]
- Zmasek C. M.; Godzik A. Phylogenomic Analysis of Glycogen Branching and Debranching Enzymatic Duo. BMC Evol. Biol. 2014, 14, 183–183. 10.1186/s12862-014-0183-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouzounis C. A.; Kunin V.; Darzentas N.; Goldovsky L. A Minimal Estimate for the Gene Content of the Last Universal Common Ancestor—Exobiology from a Terrestrial Perspective. Res. Microbiol. 2006, 157, 57–68. 10.1016/j.resmic.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Goldman A. D.; Baross J. A.; Samudrala R. The Enzymatic and Metabolic Capabilities of Early Life. PloS ONE 2012, 7, e39912 10.1371/journal.pone.0039912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa F. L.; Thiergart T.; Landan G.; Nelson-Sathi S.; Pereira I. A. C.; Allen J. F.; Lane N.; Martin W. F. Early Bioenergetic Evolution. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2013, 368, 20130088. 10.1098/rstb.2013.0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss M. C.; Sousa F. L.; Mrnjavac N.; Neukirchen S.; Roettger M.; Nelson-Sathi S.; Martin W. F. The Physiology and Habitat of the Last Universal Common Ancestor. Nat. Microbiol. 2016, 1, 1–8. 10.1038/nmicrobiol.2016.116. [DOI] [PubMed] [Google Scholar]
- Weiss M. C.; Preiner M.; Xavier J. C.; Zimorski V.; Martin W. F. The Last Universal Common Ancestor between Ancient Earth Chemistry and the Onset of Genetics. PLoS Genet. 2018, 14, e1007518 10.1371/journal.pgen.1007518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton J. D.; Cripps R. E. The Occurrence and Identification of Intracellular Polyglucose Storage Granules in Methylococcus NCIB 11083 Grown in Chemostat Culture on Methane. Arch. Microbiol. 1978 1171 1978, 117, 41–48. 10.1007/BF00689349. [DOI] [PubMed] [Google Scholar]
- Rozova O. N.; Ekimova G. A.; Molochkov N. V.; Reshetnikov A. S.; Khmelenina V. N.; Mustakhimov I. I. Enzymes of an Alternative Pathway of Glucose Metabolism in Obligate Methanotrophs. Sci. Rep. 2021, 11, 8795. 10.1038/s41598-021-88202-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuiyan S. H.; Rus’d A. A.; Kitaoka M.; Hayashi K. Characterization of a Hyperthermostable Glycogen Phosphorylase from Aquifex Aeolicus Expressed in Escherichia coli. J. Mol. Catal. B Enzym. 2003, 22, 173–180. 10.1016/S1381-1177(03)00029-8. [DOI] [Google Scholar]
- Zhang X.; Leemhuis H.; Janeček Š.; Martinovičová M.; Pijning T.; van der Maarel M. J. E. C. Identification of Thermotoga maritima MSB8 GH57 α-Amylase AmyC as a Glycogen-Branching Enzyme with High Hydrolytic Activity. Appl. Microbiol. Biotechnol. 2019, 103, 6141–6141. 10.1007/s00253-019-09938-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli F. D.; Doerks T.; Von Mering C.; Creevey C. J.; Snel B.; Bork P. Toward Automatic Reconstruction of a Highly Resolved Tree of Life. Science 2006, 311, 1283–1287. 10.1126/science.1123061. [DOI] [PubMed] [Google Scholar]
- Martin W.; Baross J.; Kelley D.; Russell M. J. Hydrothermal Vents and the Origin of Life. Nat. Rev. Microbiol. 2008, 6, 805–814. 10.1038/nrmicro1991. [DOI] [PubMed] [Google Scholar]
- Bocchetta M.; Gribaldo S.; Sanangelantoni A.; Cammarano P. Phylogenetic Depth of the Bacterial Genera Aquifex and Thermotoga Inferred from Analysis of Ribosomal Protein, Elongation Factor, and RNA Polymerase Subunit Sequences. J. Mol. Evol. 2000, 50, 366–380. 10.1007/s002399910040. [DOI] [PubMed] [Google Scholar]
- Bult C. J.; White O.; Olsen G. J.; Zhou L.; Fleischmann R. D.; Sutton G. G.; Blake J. A.; FitzGerald L. M.; Clayton R. A.; Gocayne J. D.; et al. Complete Genome Sequence of the Methanogenic Archaeon, Methanococcus jannaschii. Science 1996, 273, 1058–1073. 10.1126/science.273.5278.1058. [DOI] [PubMed] [Google Scholar]
- Sorgo A.; Gaill F.; Lechaire J. P.; Arndt C.; Bright M. Glycogen Storage in the Riftia Pachyptila Trophosome: Contribution of Host and Symbionts. Mar. Ecol. Prog. Ser. 2002, 231, 115–120. 10.3354/meps231115. [DOI] [Google Scholar]
- Muchowska K. B.; Varma S. J.; Moran J. Nonenzymatic Metabolic Reactions and Life’s Origins. Chem. Rev. 2020, 120, 7708–7744. 10.1021/acs.chemrev.0c00191. [DOI] [PubMed] [Google Scholar]
- Horowitz N. H. On the Evolution of Biochemical Syntheses. Proc. Natl. Acad. Sci. U.S.A. 1945, 31, 153–157. 10.1073/pnas.31.6.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peregrin-Alvarez J. M.; Tsoka S.; Ouzounis C. A. The Phylogenetic Extent of Metabolic Enzymes and Pathways. Genome Res. 2003, 13, 422–427. 10.1101/gr.246903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsmann A.; Lurz R.; Scheffel F.; Schneider E. Maltose and Maltodextrin Transport in the Thermoacidophilic Gram-Positive Bacterium Alicyclobacillus Acidocaldarius Is Mediated by a High-Affinity Transport System That Includes a Maltose Binding Protein Tolerant to Low pH. J. Bacteriol. 2000, 182, 6292–6292. 10.1128/JB.182.22.6292-6301.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai H.; Baumann M. J.; Petersen B. O.; Westphal Y.; Schols H.; Dilokpimol A.; Hachem M. A.; Lahtinen S. J.; Duus J. O.; Svensson B. The Maltodextrin Transport System and Metabolism in Lactobacillus acidophilus NCFM and Production of Novel α-Glucosides through Reverse Phosphorolysis by Maltose Phosphorylase. FEBS J. 2009, 276, 7353–7365. 10.1111/j.1742-4658.2009.07445.x. [DOI] [PubMed] [Google Scholar]
- Joyet P.; Mokhtari A.; Riboulet-Bisson E.; Blancato V. S.; Espariz M.; Magni C.; Hartke A.; Deutscher J.; Sauvageot N. Enzymes Required for Maltodextrin Catabolism in Enterococcus faecalis Exhibit Novel Activities. Appl. Environ. Microbiol. 2017, 83, 38–55. 10.1128/AEM.00038-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cori G. T.; Cori C. F. The Kinetics of the Enzymatic Synthesis of Glycogen from Glucose-1-Phosphate. J. Biol. Chem. 1940, 135, 733–756. 10.1016/S0021-9258(18)73136-0. [DOI] [Google Scholar]
- Granick S. Speculations on the Origins and Evolution of Photosynthesis. Ann. N. Y. Acad. Sci. 1957, 69, 292–308. 10.1111/j.1749-6632.1957.tb49665.x. [DOI] [PubMed] [Google Scholar]
- Yčas M. On Earlier States of the Biochemical System. J. Theor. Biol. 1974, 44, 145–160. 10.1016/S0022-5193(74)80035-4. [DOI] [PubMed] [Google Scholar]
- Jensen R. A. Enzyme Recruitment in Evolution of New Function. Annu. Rev. Microbiol. 1976, 30, 409–425. 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- Rueffler C.; Hermisson J.; Wagner G. P. Evolution of Functional Specialization and Division of Labor. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, E326–E335. 10.1073/pnas.1110521109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khersonsky O.; Tawfik D. S. Enzyme Promiscuity: A Mechanistic and Evolutionary Perspective. Annu. Rev. Biochem. 2010, 79, 471–505. 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- Cereijo A. E.; Asencion Diez M. D.; Dávila Costa J. S.; Alvarez H. M.; Iglesias A. A. On the Kinetic and Allosteric Regulatory Properties of the ADP-Glucose Pyrophosphorylase from Rhodococcus jostii: An Approach to Evaluate Glycogen Metabolism in Oleaginous Bacteria. Front. Microbiol. 2016, 7, 830–830. 10.3389/fmicb.2016.00830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman A. D.; Beatty J. T.; Landweber L. F. The TIM Barrel Architecture Facilitated the Early Evolution of Protein-Mediated Metabolism. J. Mol. Evol. 2016, 82 (1), 17–26. 10.1007/s00239-015-9722-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. L.; Bastian N. R.; Rajagopalan K. V. Molybdopterin Guanine Dinucleotide: A Modified Form of Molybdopterin Identified in the Molybdenum Cofactor of Dimethyl Sulfoxide Reductase from Rhodobacter Sphaeroides Forma Specialis Denitrificans. Proc. Natl. Acad. Sci. U. S. A. 1990, 87, 3190–3194. 10.1073/pnas.87.8.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson C. E. M.; Sargent F.; Buchanan G.; Palmer T.; Lawson D. M. Crystal Structure of the Molybdenum Cofactor Biosynthesis Protein MobA from Escherichia coli at Near-Atomic Resolution. Struct. Lond. Engl. 1993 2000, 8, 1115–1125. 10.1016/S0969-2126(00)00518-9. [DOI] [PubMed] [Google Scholar]
- Gabrielsen M.; Bond C. S.; Hallyburton I.; Hecht S.; Bacher A.; Eisenreich W.; Rohdich F.; Hunter W. N. Hexameric Assembly of the Bifunctional Methylerythritol 2,4-Cyclodiphosphate Synthase and Protein-Protein Associations in the Deoxy-Xylulose-Dependent Pathway of Isoprenoid Precursor Biosynthesis. J. Biol. Chem. 2004, 279, 52753–52761. 10.1074/jbc.M408895200. [DOI] [PubMed] [Google Scholar]
- Renner-Schneck M.; Hinderberger I.; Gisin J.; Exner T.; Mayer C.; Stehle T. Crystal Structure of the N-Acetylmuramic Acid α-1-Phosphate (MurNAc-Α1-P) Uridylyltransferase MurU, a Minimal Sugar Nucleotidyltransferase and Potential Drug Target Enzyme in Gram-Negative Pathogens. J. Biol. Chem. 2015, 290, 10804–10813. 10.1074/jbc.M114.620989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelakovic S.; Schulz G. E. The Structure of CMP:2-Keto-3-Deoxy-Manno-Octonic Acid Synthetase and of Its Complexes with Substrates and Substrate Analogs. J. Mol. Biol. 2001, 312, 143–155. 10.1006/jmbi.2001.4948. [DOI] [PubMed] [Google Scholar]
- Zyryanova A. F.; Weis F.; Faille A.; Alard A. A.; Crespillo-Casado A.; Sekine Y.; Harding H. P.; Allen F.; Parts L.; Fromont C.; et al. Binding of ISRIB Reveals a Regulatory Site in the Nucleotide Exchange Factor eIF2B. Science 2018, 359, 1533–1536. 10.1126/science.aar5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakihara Y.; Evans P. R. Crystal Structure of the Complex of Phosphofructokinase from Escherichia Coli with Its Reaction Products. J. Mol. Biol. 1988, 204, 973–994. 10.1016/0022-2836(88)90056-3. [DOI] [PubMed] [Google Scholar]
- Jedrzejas M. J.; Chander M.; Setlow P.; Krishnasamy G. Structure and Mechanism of Action of a Novel Phosphoglycerate Mutase from Bacillus stearothermophilus. EMBO J. 2000, 19, 1419–1431. 10.1093/emboj/19.7.1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach G.; Huber R.; Grättinger M.; Zaiss K.; Schurig H.; Jaenicke R.; Jacob U. Closed Structure of Phosphoglycerate Kinase from Thermotoga maritima Reveals the Catalytic Mechanism and Determinants of Thermal Stability. Structure 1997, 5, 1475–1483. 10.1016/S0969-2126(97)00297-9. [DOI] [PubMed] [Google Scholar]
- Chen Y.-Y.; Ko T.-P.; Lin C.-H.; Chen W.-H.; Wang A. H.-J. Conformational Change upon Product Binding to Klebsiella pneumoniae UDP-Glucose Dehydrogenase: A Possible Inhibition Mechanism for the Key Enzyme in Polymyxin Resistance. J. Struct. Biol. 2011, 175, 300–310. 10.1016/j.jsb.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Schuller D. J.; Grant G. A.; Banaszak L. J. The Allosteric Ligand Site in the Vmax-Type Cooperative Enzyme Phosphoglycerate Dehydrogenase. Nat. Struct. Biol. 1995, 2, 69–76. 10.1038/nsb0195-69. [DOI] [PubMed] [Google Scholar]
- Nagano N.; Orengo C. A.; Thornton J. M. One Fold with Many Functions: The Evolutionary Relationships between TIM Barrel Families Based on Their Sequences, Structures and Functions. J. Mol. Biol. 2002, 321, 741–765. 10.1016/S0022-2836(02)00649-6. [DOI] [PubMed] [Google Scholar]
- Alvarez M.; Zeelen J. P.; Mainfroid V.; Rentier-Delrue F.; Martial J. A.; Wyns L.; Wierenga R. K.; Maes D. Triose-Phosphate Isomerase (TIM) of the Psychrophilic Bacterium Vibrio Marinus. Kinetic and Structural Properties. J. Biol. Chem. 1998, 273, 2199–2206. 10.1074/jbc.273.4.2199. [DOI] [PubMed] [Google Scholar]
- Mattevi A.; Valentini G.; Rizzi M.; Speranza M. L.; Bolognesi M.; Coda A. Crystal Structure of Escherichia Coli Pyruvate Kinase Type I: Molecular Basis of the Allosteric Transition. Structure 1995, 3, 729–741. 10.1016/S0969-2126(01)00207-6. [DOI] [PubMed] [Google Scholar]
- Allard J.; Grochulski P.; Sygusch J. Covalent Intermediate Trapped in 2-Keto-3-Deoxy-6- Phosphogluconate (KDPG) Aldolase Structure at 1.95-A Resolution. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 3679–3684. 10.1073/pnas.071380898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee S.; Parris K. D.; Ahmed S. A.; Miles E. W.; Davies D. R. Exchange of K+ or Cs+ for Na+ Induces Local and Long-Range Changes in the Three-Dimensional Structure of the Tryptophan Synthase Alpha2beta2 Complex. Biochemistry 1996, 35, 4211–4221. 10.1021/bi952506d. [DOI] [PubMed] [Google Scholar]
- Barber M. J.; Neame P. J.; Lim L. W.; White S.; Matthews F. S. Correlation of X-Ray Deduced and Experimental Amino Acid Sequences of Trimethylamine Dehydrogenase. J. Biol. Chem. 1992, 267, 6611–6619. 10.1016/S0021-9258(19)50471-9. [DOI] [PubMed] [Google Scholar]
- Wang L.; Yang J.; Huang Y.; Liu Q.; Xu Y.; Piao X.; Wise M. J. Systematic Analysis of Metabolic Pathway Distributions of Bacterial Energy Reserves. G3 Bethesda Md 2019, 9, 2489–2496. 10.1534/g3.119.400123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldford J. E.; Hartman H.; Smith T. F.; Segrè D. Remnants of an Ancient Metabolism without Phosphate. Cell 2017, 168, 1126–1134. 10.1016/j.cell.2017.02.001. [DOI] [PubMed] [Google Scholar]
- Daffe M.; Etienne G. The Capsule of Mycobacterium tuberculosis and Its Implications for Pathogenicity. Tuber. Lung Dis. 1999, 79, 153–169. 10.1054/tuld.1998.0200. [DOI] [PubMed] [Google Scholar]
- Cywes C.; Hoppe H. C.; Daffe M.; Ehlers M. R. Nonopsonic Binding of Mycobacterium tuberculosis to Complement Receptor Type 3 Is Mediated by Capsular Polysaccharides and Is Strain Dependent. Infect. Immun. 1997, 65, 4258–4266. 10.1128/iai.65.10.4258-4266.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliardi M. C.; Lemassu A.; Teloni R.; Mariotti S.; Sargentini V.; Pardini M.; Daffe M.; Nisini R. Cell Wall-Associated Alpha-Glucan Is Instrumental for Mycobacterium tuberculosis to Block CD1 Molecule Expression and Disable the Function of Dendritic Cell Derived from Infected Monocyte. Cell. Microbiol. 2007, 9, 2081–2092. 10.1111/j.1462-5822.2007.00940.x. [DOI] [PubMed] [Google Scholar]
- Geurtsen J.; Chedammi S.; Mesters J.; Cot M.; Driessen N. N.; Sambou T.; Kakutani R.; Ummels R.; Maaskant J.; Takata H.; et al. Identification of Mycobacterial Alpha-Glucan as a Novel Ligand for DC-SIGN: Involvement of Mycobacterial Capsular Polysaccharides in Host Immune Modulation. J. Immunol. 2009, 183, 5221–5231. 10.4049/jimmunol.0900768. [DOI] [PubMed] [Google Scholar]
- Kalscheuer R.; Palacios A.; Anso I.; Cifuente J.; Anguita J.; Jacobs W. R. Jr.; Guerin M. E.; Prados-Rosales R. The Mycobacterium tuberculosis Capsule: A Cell Structure with Key Implications in Pathogenesis. Biochem. J. 2019, 476, 1995–2016. 10.1042/BCJ20190324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayramova F.; Jacquier N.; Greub G. Insight in the Biology of Chlamydia-Related Bacteria. Microbes Infect. 2018, 20, 432–440. 10.1016/j.micinf.2017.11.008. [DOI] [PubMed] [Google Scholar]
- Cosse M. M.; Hayward R. D.; Subtil A. One Face of Chlamydia trachomatis: The Infectious Elementary Body. Curr. Top. Microbiol. Immunol. 2016, 412, 35–58. 10.1007/82_2016_12. [DOI] [PubMed] [Google Scholar]
- Abdelrahman Y. M.; Belland R. J. The Chlamydial Developmental Cycle. FEMS Microbiol. Rev. 2005, 29, 949–959. 10.1016/j.femsre.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Everett K. D.; Bush R. M.; Andersen A. A. Emended Description of the Order Chlamydiales, Proposal of Parachlamydiaceae Fam. Nov. and Simkaniaceae Fam. Nov., Each Containing One Monotypic Genus, Revised Taxonomy of the Family Chlamydiaceae, Including a New Genus and Five New Species, and Standards for the Identification of Organisms. Int. J. Syst. Bacteriol. 1999, 49, 415–440. 10.1099/00207713-49-2-415. [DOI] [PubMed] [Google Scholar]
- Elwell C.; Mirrashidi K.; Engel J. Chlamydia Cell Biology and Pathogenesis. Nat. Rev. Microbiol. 2016, 14, 385–400. 10.1038/nrmicro.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappino M. L.; Dawson C.; Schachter J.; Nichols B. A. Cytochemical Localization of Glycogen in Chlamydia trachomatis Inclusions. J Bacteriol 1995, 177, 5358–5363. 10.1128/jb.177.18.5358-5363.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon F. B.; Quan A. L. Occurence of Glycogen in Inclusions of the Psittacosis-Lymphogranuloma Venereum-Trachoma Agents. J. Infect. Dis. 1965, 115, 186–196. 10.1093/infdis/115.2.186. [DOI] [PubMed] [Google Scholar]
- Gehre L.; Gorgette O.; Perrinet S.; Prevost M.-C.; Ducatez M.; Giebel A. M.; Nelson D. E.; Ball S. G.; Subtil A. Sequestration of Host Metabolism by an Intracellular Pathogen. eLife 2016, 5, e12552 10.7554/eLife.12552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C.; Lei L.; Peng B.; Tang L.; Ding H.; Gong S.; Li Z.; Wu Y.; Zhong G. Chlamydia trachomatis GlgA Is Secreted into Host Cell Cytoplasm. PLoS ONE 2013, 8 (7), e68764 10.1371/journal.pone.0068764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen B. D.; Valdivia R. H. Virulence Determinants in the Obligate Intracellular Pathogen Chlamydia Trachomatis Revealed by Forward Genetic Approaches. Proc Natl Acad Sci U. S. A. 2012, 109, 1263–1268. 10.1073/pnas.1117884109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L.; Yang C.; Patton M. J.; Smelkinson M.; Dorward D.; Ma L.; Karanovic U.; Firdous S.; McClarty G.; Caldwell H. D. A Chlamydial Plasmid-Dependent Secretion System for the Delivery of Virulence Factors to the Host Cytosol. mBio 2021, 12, e0117921 10.1128/mBio.01179-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C.; Wu H.; Sun Y.; Kong J.; Shao L.; Chen X.; Liu Q.; Liu Y. GlgA Plays an Important Role in the Induction of Hydrosalpinx by Chlamydia muridarum. Pathog. Dis. 2020, 78, ftaa027 10.1093/femspd/ftaa027. [DOI] [PubMed] [Google Scholar]
- Sun Z.; Li Y.; Chen H.; Xie L.; Xiao J.; Luan X.; Peng B.; Li Z.; Chen L.; Wang C.; Lu C. Chlamydia trachomatis Glycogen Synthase Promotes MAPK-Mediated Proinflammatory Cytokine Production via TLR2/TLR4 in THP-1 Cells. Life Sci. 2021, 271, 119181 10.1016/j.lfs.2021.119181. [DOI] [PubMed] [Google Scholar]
- Lamoth F.; Pillonel T.; Greub G. Waddlia: An Emerging Pathogen and a Model Organism to Study the Biology of Chlamydiae. Microbes Infect. 2015, 17, 732–737. 10.1016/j.micinf.2015.09.021. [DOI] [PubMed] [Google Scholar]
- Baud D.; Regan L.; Greub G. Emerging Role of Chlamydia and Chlamydia-like Organisms in Adverse Pregnancy Outcomes. Curr. Opin. Infect. Dis. 2008, 21, 70–76. 10.1097/QCO.0b013e3282f3e6a5. [DOI] [PubMed] [Google Scholar]
- Tuon F. F.; Dantas L. R.; Suss P. H.; Ribeiro V. S. T. Pathogenesis of the Pseudomonas aeruginosa Biofilm: A Review. Pathogens 2022, 11, 300. 10.3390/pathogens11030300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne A. J. K.; Ghali A. E.; Holger D.; Rebold N.; Rybak M. J. Therapeutic Strategies for Emerging Multidrug-Resistant Pseudomonas aeruginosa. Infect. Ther. 2022, 11, 661–682. 10.1007/s40121-022-00591-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. S.; Yoon S. J.; Park Y. J.; Kim S. Y.; Ryu C. M. Crossing the Kingdom Border: Human Diseases Caused by Plant Pathogens. Env. Microbiol. 2020, 22, 2485–2495. 10.1111/1462-2920.15028. [DOI] [PubMed] [Google Scholar]
- Cao H.; Baldini R. L.; Rahme L. G. Common Mechanisms for Pathogens of Plants and Animals. Annu. Rev. Phytopathol. 2001, 39, 259–284. 10.1146/annurev.phyto.39.1.259. [DOI] [PubMed] [Google Scholar]
- Djonovic S.; Urbach J. M.; Drenkard E.; Bush J.; Feinbaum R.; Ausubel J. L.; Traficante D.; Risech M.; Kocks C.; Fischbach M. A.; et al. Trehalose Biosynthesis Promotes Pseudomonas aeruginosa Pathogenicity in Plants. PLoS Pathog 2013, 9, e1003217 10.1371/journal.ppat.1003217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman B. C.; Chen C.; Beattie G. A. Identification of the Trehalose Biosynthetic Loci of Pseudomonas Syringae and Their Contribution to Fitness in the Phyllosphere. Env. Microbiol. 2010, 12, 1486–1497. 10.1111/j.1462-2920.2010.02171.x. [DOI] [PubMed] [Google Scholar]
- Emilson C. G.; Nilsson B.; Bowen W. H. Carbohydrate Composition of Dental Plaque from Primates with Irradiation Caries. J. Oral Pathol. 1984, 13, 213–220. 10.1111/j.1600-0714.1984.tb01419.x. [DOI] [PubMed] [Google Scholar]
- Mayhall C. W.; Butler W. T. The Carbohydrate Composition of Experimental Salivary Pellicles. J. Oral Pathol. 1976, 5, 358–370. 10.1111/j.1600-0714.1976.tb01787.x. [DOI] [PubMed] [Google Scholar]
- Critchley P. The Breakdown of the Carbohydrate and Protein Matrix of Dental Plaque. Caries Res. 1969, 3, 249–265. 10.1159/000259599. [DOI] [PubMed] [Google Scholar]
- Wood J. M. The State of Hexose Sugar in Human Dental Plaque and Its Metabolism by the Plaque Bacteria. Arch. Oral Biol. 1969, 14, 161–168. 10.1016/0003-9969(69)90060-0. [DOI] [PubMed] [Google Scholar]
- Gibbons R. J. Formation and Significance of Bacterial Polysaccharides in Caries Etiology. Caries Res. 1968, 2, 164–171. 10.1159/000259554. [DOI] [PubMed] [Google Scholar]
- Takahashi N.; Nyvad B. The Role of Bacteria in the Caries Process: Ecological Perspectives. J. Dent. Res. 2011, 90, 294–303. 10.1177/0022034510379602. [DOI] [PubMed] [Google Scholar]
- Valm A. M.; Welch J. L. M.; Rieken C. W.; Hasegawa Y.; Sogin M. L.; Oldenbourg R.; Dewhirst F. E.; Borisy G. G. Systems-Level Analysis of Microbial Community Organization through Combinatorial Labeling and Spectral Imaging. Proc Natl Acad Sci U. S. A. 2011, 108, 4152–4157. 10.1073/pnas.1101134108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh P. D.; Moter A.; Devine D. A. Dental Plaque Biofilms: Communities, Conflict and Control. Periodontol 2000 2011, 55, 16–35. 10.1111/j.1600-0757.2009.00339.x. [DOI] [PubMed] [Google Scholar]
- Leme A. F. P.; Koo H.; Bellato C. M.; Bedi G.; Cury J. A. The Role of Sucrose in Cariogenic Dental Biofilm Formation–New Insight. J. Dent. Res. 2006, 85, 878–887. 10.1177/154405910608501002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro C. C.; Tabchoury C. P.; Cury A. A. D. B.; Tenuta L. M.; Rosalen P. L.; Cury J. A. Effect of Starch on the Cariogenic Potential of Sucrose. Br. J. Nutr. 2005, 94, 44–50. 10.1079/BJN20051452. [DOI] [PubMed] [Google Scholar]
- Firestone A. R.; Schmid R.; Muhlemann H. R. Cariogenic Effects of Cooked Wheat Starch Alone or with Sucrose and Frequency-Controlled Feedings in Rats. Arch. Oral Biol. 1982, 27, 759–763. 10.1016/0003-9969(82)90026-7. [DOI] [PubMed] [Google Scholar]
- Ueda S.; Shiroza T.; Kuramitsu H. K. Sequence Analysis of the gtfC Gene from Streptococcus mutans GS-5. Gene 1988, 69, 101–109. 10.1016/0378-1119(88)90382-4. [DOI] [PubMed] [Google Scholar]
- Shiroza T.; Ueda S.; Kuramitsu H. K. Sequence Analysis of the gtfB Gene from Streptococcus mutans. J Bacteriol 1987, 169, 4263–4270. 10.1128/jb.169.9.4263-4270.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada N.; Kuramitsu H. K. Isolation and Characterization of the Streptococcus mutans gtfD Gene, Coding for Primer-Dependent Soluble Glucan Synthesis. Infect. Immun. 1989, 57 (7), 2079–2085. 10.1128/iai.57.7.2079-2085.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooshima T.; Matsumura M.; Hoshino T.; Kawabata S.; Sobue S.; Fujiwara T. Contributions of Three Glycosyltransferases to Sucrose-Dependent Adherence of Streptococcus mutans. J. Dent. Res. 2001, 80, 1672–1677. 10.1177/00220345010800071401. [DOI] [PubMed] [Google Scholar]
- Klein M. I.; Hwang G.; Santos P. H.; Campanella O. H.; Koo H. Streptococcus mutans-Derived Extracellular Matrix in Cariogenic Oral Biofilms. Front. Cell. Infect. Microbiol. 2015, 5, 10. 10.3389/fcimb.2015.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita Y.; Bowen W. H.; Burne R. A.; Kuramitsu H. K. Role of the Streptococcus mutans Gtf Genes in Caries Induction in the Specific-Pathogen-Free Rat Model. Infect. Immun. 1993, 61, 3811–3817. 10.1128/iai.61.9.3811-3817.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M. C.; Bozzola J. J.; Shechmeister I. L.; Shklair I. L. Biochemical Study of the Relationship of Extracellular Glucan to Adherence and Cariogenicity in Streptococcus mutans and an Extracellular Polysaccharide Mutant. J. Bacteriol. 1977, 129, 351–357. 10.1128/jb.129.1.351-357.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanzer J. M.; Freedman M. L.; Fitzgerald R. J.; Larson R. H. Diminished Virulence of Glucan Synthesis-Defective Mutants of Streptococcus mutans. Infect. Immun. 1974, 10, 197–203. 10.1128/iai.10.1.197-203.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris G. S.; Michalek S. M.; Curtiss R. Cloning of a Locus Involved in Streptococcus mutans Intracellular Polysaccharide Accumulation and Virulence Testing of an Intracellular Polysaccharide-Deficient Mutant. Infect. Immun. 1992, 60, 3175–3185. 10.1128/iai.60.8.3175-3185.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatafora G.; Rohrer K.; Barnard D.; Michalek S. A Streptococcus mutans Mutant That Synthesizes Elevated Levels of Intracellular Polysaccharide Is Hypercariogenic in Vivo. Infect. Immun. 1995, 63, 2556–2563. 10.1128/iai.63.7.2556-2563.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda J. M.; Abbott S. L. The Genus Aeromonas: Taxonomy, Pathogenicity, and Infection. Clin. Microbiol. Rev. 2010, 23, 35–73. 10.1128/CMR.00039-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabich A. J.; Jones S. A.; Chowdhury F. Z.; Cernosek A.; Anderson A.; Smalley D.; McHargue J. W.; Hightower G. A.; Smith J. T.; Autieri S. M.; et al. Comparison of Carbon Nutrition for Pathogenic and Commensal Escherichia coli Strains in the Mouse Intestine. Infect. Immun. 2008, 76, 1143–1152. 10.1128/IAI.01386-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D.-E.; Smalley D. J.; Tucker D. L.; Leatham M. P.; Norris W. E.; Stevenson S. J.; Anderson A. B.; Grissom J. E.; Laux D. C.; Cohen P. S.; Conway T. Carbon Nutrition of Escherichia coli in the Mouse Intestine. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 7427–7432. 10.1073/pnas.0307888101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S. A.; Jorgensen M.; Chowdhury F. Z.; Rodgers R.; Hartline J.; Leatham M. P.; Struve C.; Krogfelt K. A.; Cohen P. S.; Conway T. Glycogen and Maltose Utilization by Escherichia coli O157:H7 in the Mouse Intestine. Infect. Immun. 2008, 76, 2531–2540. 10.1128/IAI.00096-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morán-Zorzano M. T.; Alonso-Casajús N.; Muñoz F. J.; Viale A. M.; Baroja-Fernández E.; Eydallin G.; Pozueta-Romero J. Occurrence of More than One Important Source of ADPglucose Linked to Glycogen Biosynthesis in Escherichia coli and Salmonella. FEBS Lett. 2007, 581, 4423–4429. 10.1016/j.febslet.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Goh Y. J.; Klaenhammer T. R. Insights into Glycogen Metabolism in Lactobacillus Acidophilus: Impact on Carbohydrate Metabolism, Stress Tolerance and Gut Retention. Microb. Cell Factories 2014, 13, 94. 10.1186/s12934-014-0094-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leviatan S.; Shoer S.; Rothschild D.; Gorodetski M.; Segal E. An Expanded Reference Map of the Human Gut Microbiome Reveals Hundreds of Previously Unknown Species. Nat. Commun. 2022, 13, 1–14. 10.1038/s41467-022-31502-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren F. J.; Fukuma N. M.; Mikkelsen D.; Flanagan B. M.; Williams B. A.; Lisle A. T.; Cuív P. Ó.; Morrison M.; Gidley M. J.. Food Starch Structure Impacts Gut Microbiome Composition. mSphere 2018, 3. 10.1128/mSphere.00086-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaoutari A. E.; Armougom F.; Gordon J. I.; Raoult D.; Henrissat B. The Abundance and Variety of Carbohydrate-Active Enzymes in the Human Gut Microbiota. Nat. Rev. Microbiol. 2013, 11, 497–504. 10.1038/nrmicro3050. [DOI] [PubMed] [Google Scholar]
- Hooda S.; Vester Boler B. M.; Rossoni Serao M. C.; Brulc J. M.; Staeger M. A.; Boileau T. W.; Dowd S. E.; Fahey G. C.; Swanson K. S. 454 Pyrosequencing Reveals a Shift in Fecal Microbiota of Healthy Adult Men Consuming Polydextrose or Soluble Corn Fiber. J. Nutr. 2012, 142, 1259–1265. 10.3945/jn.112.158766. [DOI] [PubMed] [Google Scholar]
- Zmora N.; Zeevi D.; Korem T.; Segal E.; Elinav E. Taking It Personally: Personalized Utilization of the Human Microbiome in Health and Disease. Cell Host Microbe 2016, 19, 12–20. 10.1016/j.chom.2015.12.016. [DOI] [PubMed] [Google Scholar]
- Mora C.; Tittensor D. P.; Adl S.; Simpson A. G. B.; Worm B. How Many Species Are There on Earth and in the Ocean?. PLoS Biol. 2011, 9, e1001127 10.1371/journal.pbio.1001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locey K. J.; Lennon J. T. Scaling Laws Predict Global Microbial Diversity. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (21), 5970–5975. 10.1073/pnas.1521291113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd K. G.; Steen A. D.; Ladau J.; Yin J.; Crosby L.. Phylogenetically Novel Uncultured Microbial Cells Dominate Earth Microbiomes. mSystems 2018, 3. 10.1128/mSystems.00055-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abendroth J.; Higgins T. W.; Dranow D. M.; Lorimer D.; Edwards T. E.. Crystal structure of UTP-glucose-1-phosphate uridylyltransferase from Burkholderia ambifaria in complex with UTP. https://www.rcsb.org/structure/5ve7.
- Kang L. W.UDP-glucose pyrophosphorylase from Acinetobacter baumanii. https://doi.org/10.2210/pdb6IKZ/pdb.
- Purvis A.; Nidetzky B.; Watson K.. Starch Phosphorylase: Structural Studies Explain Oxyanion-Dependent Kinetic Stability and Regulatory Control. https://doi.org/10.2210/pdb2C4M/pdb.
- Mayclin S.J.; Dranow D.M.; Lorimer D.; Edwards T.E.. Crystal structure of an alpha,alpha-trehalose-phosphate synthase (UDP-forming) from Burkholderia xenovorans in complex with glucose-6-phosphate. https://doi.org/10.2210/pdb5V0T/pdb.
- Barford D.; Johnson L. N. The Allosteric Transition of Glycogen Phosphorylase. Nature 1989, 340, 609–616. 10.1038/340609a0. [DOI] [PubMed] [Google Scholar]
- Hayashi M.; Suzuki R.; Colleoni C.; Ball S. G.; Fujita N.; Suzuki E. Bound Substrate in the Structure of Cyanobacterial Branching Enzyme Supports a New Mechanistic Model. J. Biol. Chem. 2017, 292, 5465–5475. 10.1074/jbc.M116.755629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint H. J.; Scott K. P.; Duncan S. H.; Louis P.; Forano E. Microbial Degradation of Complex Carbohydrates in the Gut. Gut Microbes. 2012, 3, 289–306. 10.4161/gmic.19897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung D. H.; Park C. S. Resistant Starch Utilization by Bifidobacterium, the Beneficial Human Gut Bacteria. Food Sci. Biotechnol. 2023, 32, 441–452. 10.1007/s10068-023-01253-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerqueira F. M.; Photenhauer A. L.; Pollet R. M.; Brown H. A.; Koropatkin N. M. Starch Digestion by Gut Bacteria: Crowdsourcing for Carbs. Trends Microbiol. 2020, 28, 95–108. 10.1016/j.tim.2019.09.004. [DOI] [PubMed] [Google Scholar]