

Abstract
The calcium-sensing receptor (CaSR)-specific allosteric modulator cinacalcet has revolutionized the treatment of secondary hyperparathyroidism in patients with chronic kidney disease. However, its application is limited to patients with end-stage renal disease because of hypocalcemic side effects presumably caused by CaSR-mediated calcitonin secretion from thyroid parafollicular C-cells. These hypocalcemic side effects might be dampened by compounds that bias the signaling of CaSR, causing similar therapeutic effects as cinacalcet without stimulating calcitonin secretion. Because biased signaling of CaSR is poorly understood, the objective of the present study was to investigate biased signaling of CaSR by using rat medullary thyroid carcinoma 6-23 cells as a model of thyroid parafollicular C-cells. By doing concentration-response experiments we focused on the ability of two well known CaSR agonists, calcium and strontium, to activate six different signaling entities: Gq/11 signaling, Gi/o signaling, Gs signaling, extracellular signal-regulated kinases 1 and 2 (ERK1/2) signaling, intracellular calcium ([Ca2+]i) mobilization, and calcitonin secretion. The experiments showed that strontium biases CaSR signaling toward ERK1/2 signaling and possibly another pathway independent of Gq/11 signaling and [Ca2+]i mobilization. It is noteworthy that the potency of strontium-stimulated calcitonin secretion was elevated compared with calcium. Combining these results with experiments investigating signaling pathway components involved in calcitonin secretion, we found that the enhanced potency of strontium-mediated calcitonin secretion was caused by a different signaling pattern than that produced by calcium. Together, our results suggest that calcitonin secretion can be affected by CaSR-stimulated signaling bias, which may be used to develop novel drugs for the treatment of secondary hyperparathyroidism.
Introduction
The calcium-sensing receptor (CaSR) belongs to family C of the G protein-coupled receptor (GPCR) superfamily. Like other family C GPCRs, CaSR consists of a large bilobed extracellular amino-terminal domain structurally similar to the Venus flytrap domain motif of bacterial periplasmatic binding proteins, a cysteine-rich domain, a seven-transmembrane-spanning domain, and an intracellular carboxyl-terminal domain. CaSR is a pleiotropic receptor able to activate several G proteins, including Gq/11, Gi/o, G12/13, and, in rare cases, Gs (Kifor et al., 2001; Almadén et al., 2002; Huang et al., 2004; Mamillapalli et al., 2008). In addition, CaSR interacts with several intracellular adapter proteins, including β-arrestins and filamin A (Awata et al., 2001; Hjälm et al., 2001; Bouschet et al., 2007). The most important physiological function of CaSR is to maintain calcium homeostasis, which is achieved primarily through the negative regulation of parathyroid hormone (PTH) secretion from the parathyroid glands (Brown et al., 1993; Chang et al., 2008). However, CaSR is expressed and has physiological important functions in several other tissues, including thyroid gland, kidneys, and bones (Riccardi et al., 1995; Liu et al., 2003; Kantham et al., 2009).
Extracellular calcium ([Ca2+]e) is the endogenous ligand of CaSR, and disruption of CaSR function can severely affect calcium homeostasis (Pollak et al., 1993; Ho et al., 1995; Chang et al., 2008). However, CaSR is activated by various other cations, including metal ions (magnesium and strontium), polyamines (spermine and spermidine), and aminoglycoside (neomycin, gentamicin, and tobramycin) (Riccardi et al., 1995; Ruat et al., 1996; Quinn et al., 1997; Ward et al., 2002; Thomsen et al., 2012). In addition, CaSR activity is modulated allosteric by aromatic l-amino acids and several phenylalkylamine compounds (Conigrave et al., 2007; Davey et al., 2012).
Because of its function as a powerful regulator of PTH secretion, CaSR has proven to be a convenient drug target in PTH-related diseases. This has led to the discovery of the CaSR-specific allosteric modulator cinacalcet, which is used clinically to reduce PTH secretion related to primary and secondary hyperparathyroidism in patients suffering from chronic kidney disease (Nemeth et al., 2004; Torres, 2006). However, its application for the treatment of secondary hyperparathyroidism has been limited to patients with end-stage renal disease on hemodialysis because of the adverse hypocalcemic side effects associated with it (Chonchol et al., 2009). CaSR not only inhibits PTH secretion but also enhances the secretion of calcitonin from thyroid parafollicular C-cells (Nemeth et al., 2004). Calcitonin is a calcium-decreasing hormone; therefore, elevated calcitonin secretion is believed to be an important event involved in cinacalcet-associated hypocalcemia (Henley et al., 2011). Thus, to treat secondary hyperparathyroidism in lower-stage chronic kidney disease it is highly desirable to develop a positive modulator, which inhibits PTH secretion without causing hypocalcemic side effects mediated by increased calcitonin secretion.
One way this may be accomplished is to search for compounds that bias the signaling of CaSR toward signaling pathways involved in the inhibition of PTH secretion while having a minimal effect on signaling pathways involved in calcitonin secretion. Biased agonists or allosteric modulators are molecules that stabilize GPCRs in different active conformations compared with the active conformation stabilized by its endogenous ligand (Rajagopal et al., 2010; Kenakin, 2011). These alternative active GPCR conformations affect intracellular signaling pathways differently compared with the “native” active conformation, resulting in different cellular responses. So far, biased signaling has been intensively studied for several GPCRs. However, limited studies have investigated biased signaling of CaSR, and all have used a HEK293 cell model overexpressing exogenous CaSR (Chattopadhyay et al., 2007; Makita et al., 2007; Davey et al., 2012; Thomsen et al., 2012). Therefore, the importance of signaling bias of CaSR in tissues and cells expressing CaSR endogenously is not known.
In the present study, we investigated biased signaling of CaSR in rat medullary thyroid carcinoma 6-23 cells, which express CaSR endogenously. The 6-23 cell line is a well suited model of thyroid parafollicular C-cells because they secrete calcitonin in response to CaSR activation (Nemeth et al., 2004). To investigate biased signaling of CaSR we used extracellular strontium ([Sr2+]e), which previously has been indicated to be a biased agonist of CaSR in HEK293-CaSR cells (Chattopadhyay et al., 2007). Here, we found that [Sr2+]e biases the signaling of CaSR toward ERK1/2 signaling and possibly another pathway independently of Gq/11 signaling and intracellular calcium ([Ca2+]i) mobilization. The signaling bias resulted in an enhanced potency of [Sr2+]e-mediated calcitonin secretion compared with [Ca2+]e-mediated secretion. To our knowledge, this is the first time biased signaling of CaSR has been demonstrated in a cell model endogenously expressing CaSR. Furthermore, our results show that biased signaling of CaSR can have important physiological consequences that may be exploited for the development of new CaSR-targeting drugs.
Materials and Methods
Materials.
Rat medullary thyroid carcinoma 6-23 cells (clone 6) were purchased from the American Type Culture Collection (Manassas, VA). A rat calcitonin IRMA kit was purchased from Immutopics (San Clemente, CA). Dulbecco's modified Eagle's medium (DMEM) + GlutaMAX-I + 4.5g/l d-glucose + sodium pyruvate, DMEM without l-glutamine, sodium pyruvate, and CaCl2, horse serum, penicillin, streptomycin, l-glutamine, Dulbecco's phosphate-buffered saline (DPBS), Hanks' balanced salt solution (HBSS) without CaCl2 and MgCl2, 0,05% trypsin-EDTA, probenecid, and a Fluo-4 NW calcium assay kit were purchased from Invitrogen (Paisley, UK). CaCl2, SrCl2, nimodipine, O-tricyclo[5.2.1.02,6]dec-9-yl dithiocarbonate (D609), wortmannin, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002), 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD98059), 1-O-octadecyl-2-O-methyl-sn-glycero-3-phosphorylcholine (ET-18-OCH3), 2-aminoethoxydiphenyl borate (2-APB), 1-[6-[((17β)-3-methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-1H-pyrrole-2,5-dione (U73122), pertussis toxin, 1,2-bis(2-aminophenoxy)ethane-N,N, N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM), cell dissociation solution, HEPES, forskolin, 3-isobutyl-1-methylxanthine (IBMX), and poly-d-lysine were purchased from Sigma-Aldrich (St Louis, MO). A RNeasy kit and QuantiTect reverse transcription kit were purchased from QIAGEN (Dorking, Surrey, UK). The IP-One assay kit, cAMP dynamic 2 assay kit, and Cellul'ERK assay kit were purchased from Cisbio (Bagnols, France). 4-Chloro-N-[(1S,2S)-2-[[(1R)-1-(1-naphthalenyl)ethyl]amino]cyclohexyl]-benzamide hydrochloride (Calhex 231) and octreotide were purchased from Tocris Bioscience (Bristol, UK). Cinacalcet was provided by LEO Pharma A/S (Ballerup, Denmark).
Detection of CaSR mRNA by RT-PCR.
A total of 5 × 106 cells was harvested, and RNA was isolated by using the RNeasy kit (QIAGEN). Next, 1 μg of RNA was used to generate cDNA by using the QuantiTect reverse transcription kit (QIAGEN). The CaSR cDNA was amplified by using previously reported primers (Smajilovic et al., 2006): sense (bp 1668–1689), 5′-CTATCATCAACTGGCACCTCTC-3′ and antisense (bp 1929–1950), 5′-TTGTCACAGGCACTCGCATCTG-3′. Control PCR was performed with primers specific for the ubiquitously expressed, endogenous β-actin gene: sense, 5′-ACCCTCATAGATGGGCACAG-3′ and antisense, 5′-TGACCCAGATCATGTTTGAGA-3′. The size of the amplified cDNA was verified on a 1% agarose gel. The presence of a 283-bp amplified product was indicative of a positive PCR arising from the presence of a CaSR-related sequence within the cDNA.
Calcitonin Assay.
Two days before the experiment, cells were washed once in DPBS and detached with trypsin solution. The detached cells were resuspended in DMEM without CaCl2 containing 10% horse serum and plated in collagen-coated 48-well plates (BD Biosciences Discovery Labware, Bedford, MA) at a concentration of 500,000 cells/well. The cells were incubated at 37°C for 24 h, then the media were changed with DMEM without CaCl2 containing 2% horse serum and incubated for another 24 h. At the day of the experiment media were aspirated from the cells, and 500 μl of new DMEM without CaCl2 containing 2% horse serum and the studied concentrations of CaCl2 or SrCl2 with/without cinacalcet were added. The cells were incubated at 37°C for 4 h, and the media were collected. When different signaling pathways were inhibited, the cells were preincubated with the inhibitors for 10 min before the addition of CaCl2 or SrCl2. Gi/o signaling was inhibited by 24-h preincubation of pertussis toxin before the experiments. The concentration of calcitonin secreted by the cells was determined by using the Rat Calcitonin IRMA Kit (Immutopics).
IP1 Assay.
On the day of the experiment subconfluent 6-23 cells were washed once with DPBS and detached from the cell culture plate by dissociation buffer (Sigma-Aldrich). The detached cells were resuspended in DMEM and centrifuged at 1000 rpm for 10 min. After centrifugation the supernatant was discharged, and the cells were resuspended in assay buffer (20 mM HEPES + 0.1% bovine serum albumin in HBSS buffer, pH 7.4) at a concentration of 3,000,000 cells/ml. In a 384-well OptiPlate (PerkinElmer Life and Analytical Sciences, Waltham, MA) 2 μl of ligand buffer containing ligand was mixed with 10 μl of cell suspension. The plate was sealed and incubated at 37°C for 1 h, followed by 15-min incubation at room temperature. Next, 10 μl of detection reagents (lysis buffer containing 2.5% Eu-anti-IP1 antibody and 2.5% IP1-d2) (Cisbio) was added, and the plate was incubated for 1 h at room temperature. The plate was read on an EnVision multilabel reader (PerkinElmer Life and Analytical Sciences) where the wells were excited with light at 340 nm and emission light was measured at 615 and 665 nm. The time resolved-fluorescence resonance energy transfer (TR-FRET) 665/615-nm ratio was used to calculate IP1 concentrations from a standard curve generated by IP1 standards provided by the manufacturer (Cisbio).
Fluo-4 NW Calcium Assay.
On the day before the experiment, cells were washed once in DPBS and detached with trypsin solution. The detached cells were resuspended in DMEM and plated in a 96-well Optilux microplate (BD Biosciences Discovery Labware) at a concentration of 150,000 cells/well. On the day of the assay the media were removed, and the cells were washed once in DPBS (Invitrogen). Next, 100 μl of dye loading solution (Fluo-4 NW dye + 2.5 mM probenecid + 20 mM HEPES in HBSS buffer, pH 7.4) (Invitrogen) was added to each well, and the plate was incubated at 37°C for 30 min. The plate was incubated at room temperature for 30 min and transferred to a NOVOstar microplate reader (BMG Labtech GmbH, Offenburg, Germany). Twenty five microliters of ligand buffer (20 mM HEPES in HBSS buffer, pH 7.4) containing the agonist of interest was added to each well, and responses were recorded by using excitation/emission wavelengths of 485 and 520 nm, respectively. Responses (Δfluorescence units) were calculated as peak fluorescence after agonist addition − fluorescence before agonist addition.
cAMP Dynamic 2 Assay.
On the day of the experiment subconfluent 6-23 cells were washed once with DPBS and detached from the cell culture plate by using dissociation buffer (Sigma-Aldrich). In experiments where Gi/o signaling was inhibited, the cells were preincubated for 24 h with pertussis toxin. Cells were centrifuged and resuspended in assay buffer (500 nM IBMX + 20 mM HEPES in HBSS buffer, pH 7.4) at a concentration of 800,000 cells/ml. In a white small-volume 384-well plate (Greiner Bio-One GmbH, Frickenhausen, Germany), 5 μl of ligand buffer with forskolin (500 nM IBMX + 20 μM forskolin + 20 mM HEPES in HBSS buffer, pH 7.4) or without forskolin (500 nM IBMX + 20 mM HEPES in HBSS buffer, pH 7.4, respectively) for measurements of Gi/o or Gs signaling, respectively, containing ligand with or without Calhex 231 was mixed with 5 μl of cell suspension. The plate was sealed and incubated at room temperature for 30 min. Next, 5 μl of lysis buffer containing 2.5% cAMP-d2 was added to each well followed by the addition of 5 μl of lysis buffer containing 2.5% Eu3+-anti-cAMP antibody (Cisbio). The plate was incubated for 1 h at room temperature. The plate was read on an EnVision multilabel reader (PerkinElmer Life and Analytical Sciences) where the wells were excited with light at 340 nm, and emission light was measured at 615 and 665 nm. The TR-FRET 665/615-nm ratio was applied to calculate cAMP concentrations from a standard curve generated by cAMP standards provided by the manufacturer (Cisbio).
Cellular ERK Assay.
On the day before the experiment 100,000 cells/well of 6-23 cells were plated on poly-d-lysine-coated 96-well plates (Becton Dickinson, Meylan Cedex, France) and incubated until the next day in culture medium. On the day of the assay each well was washed with DPBS and stimulated with 50 μl of stimulation buffer (20 mM HEPES in HBSS buffer, pH 7.4) containing ligand for 30 min at room temperature. Next, the ligand solution was aspirated, and 50 μl of lysis buffer (74% MilliQ water + 25% lysis solution + 1% blocking reagents) (Cisbio) was added to each well. The plate was incubated for 1 h at room temperature on a micro plate shaker at 450 rpm. In a white small-volume 384-well plate (Greiner Bio-One GmbH), 4 μl of detection solution (2.5% antiphospho-ERK1/2-d2 and 2.5% anti-ERK1/2-Eu3+ in ERK detection buffer) (Cisbio) was mixed with 16 μl of the lysis solution that had been transferred from the 96-well plate. The plate was then incubated in the dark for 2 h at room temperature. Finally, the plate was read on an EnVision multilabel reader (PerkinElmer Life and Analytical Sciences) where the wells were excited with light at 340 nm, and emitted light was measured at 615 and 665 nm. The TR-FRET 665/615-nm ratio, which is proportional to the ERK1/2 phosphorylation, was used to measure ERK1/2 phosphorylation. The ERK1/2 response was calculated as fold over basal ERK1/2 response (R/RBasal).
Data Analysis.
All pharmacological data were analyzed by using GraphPad Prism 5.0a for Mac OS X (GraphPad Software, Inc., San Diego, CA). Concentration-response curves generated in the different assays were fitted by nonlinear regression using the equation for sigmoidal concentration-response with variable slope:
where X is the logarithm of the ligand concentration, R is the response, Rmax is the maximal response, Rmin is the minimal response, EC50 is the agonist concentration that produces half of the maximal response, and nH describes steepness of the curve, the Hill number. To test for signaling bias, we used a previously reported method: calculation of bias factors (BF) (Drake et al., 2008; Thomsen et al., 2012). Using BFs, a biased agonist is defined as an agonist that has a statistically different BF compared with the BF of the endogenous agonist calcium. The potency BF (pBF) of an agonist is calculated as the ratio between EC50 of two different signaling pathways (pathway1 and pathway2). However, for analysis of the statistic difference between pBF of [Ca2+]e and pBF of [Sr2+]e we use the negative logarithm of the pBF or:
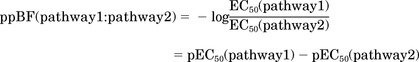 |
This is necessary because statistical information of S.E.M. is available for pEC50 but not EC50. Statistical analyses of the results were performed where appropriate. Student's t tests was conducted where appropriate. One-way ANOVA was conducted, followed by Dunnett's test where appropriate. Significance was set at p < 0.05. All p > 0.05 are considered not significant.
Results
Calcitonin Secretion, Gq/11 Signaling, and [Ca2+]i Mobilization in Response to CaSR Activation by [Ca2+]e and [Sr2+]e.
As a model for thyroid parafollicular C-cells, we used rat medullary thyroid carcinoma 6-23 cells, which previously have been demonstrated to express CaSR (Nemeth et al., 2004). We confirmed RNA expression of CaSR in 6-23 cells by RT-PCR (Fig. 1). These cells respond to [Ca2+]e by secreting calcitonin, and this response can be modulated by the CaSR-specific positive allosteric modulator cinacalcet (Nemeth et al., 2004). Therefore, this cell line is a well suited model for studying CaSR-mediated calcitonin secretion in vitro.
Fig. 1.

RT-PCR demonstrating CaSR expression in 6-23 cells. RT-PCR was performed on total RNA extracted from 6-23 cells (A), HEK293-rCaSR cells (B), HEK293 cells (C), rat kidney as a positive control (D), and water as a negative control (E). RT-PCR was performed as described under Materials and Methods by using primers specific for the rat CaSR gene (top) or the ubiquitously expressed, endogenous β-actin (bottom).
First, we confirmed the ability of the CaSR agonists, [Ca2+]e and [Sr2+]e, to cause calcitonin secretion in 6-23 cells. Both [Ca2+]e and [Sr2+]e were able to cause calcitonin secretion with similar efficacies in a concentration-dependent manner (Fig. 2A; Table 1). However, [Sr2+]e was slightly more potent than [Ca2+]e. Both [Ca2+]e- and [Sr2+]e-mediated responses could be modulated by cinacalcet, demonstrating that the responses were caused by CaSR activation (Fig. 2B).
TABLE 1.
Agonist potencies (EC50) and maximal responses (Rmax) for calcitonin secretion, IP1 accumulation and intracellular calcium response in 6-23 cells.
Potencies are in μM for all assays. Rmax levels are in pg/ml for the calcitonin assay and nM for the IP1 assay and have been normalized to Δfluorescence units of 10 mM [Ca2+]e in the intracellular calcium assay.
| Assay | [Ca2+]e |
[Sr2+]e |
||
|---|---|---|---|---|
| Rmax ± S.E.M. | EC50 (pEC50± S.E.M.) | Rmax ± S.E.M. | EC50 (pEC50 ± S.E.M.) | |
| Calcitonin | 313 ± 20 | 2440 (2.61 ± 0.13) | 304 ± 24 | 794 (3.10 ± 0.13) |
| IP1 | 858 ± 150 | 4810 (2.32 ± 0.01) | 787 ± 46 | 8320 (2.08 ± 0.01) |
| Intracellular calcium | 100 ± 3.50 | 1470 (2.83 ± 0.09) | 44.3 ± 1.18 | 3050 (2.52 ± 0.05) |
Fig. 2.

Characterization of concentration-dependent [Ca2+]e- and [Sr2+]e-mediated calcitonin secretion, IP1 accumulation, and [Ca2+]I mobilization in 6-23 cells. A, concentration-response curves of [Ca2+]e- and [Sr2+]e-mediated calcitonin secretion. B, concentration-response curves of allosteric modulation of [Ca2+]e- and [Sr2+]e-stimulated calcitonin secretion with cinacalcet. Responses are normalized to the calcitonin secretory response at 10 mM cation. C, concentration-response curves of [Ca2+]e- and [Sr2+]e-mediated IP1 accumulation. D, concentration-response curves of [Ca2+]e- and [Sr2+]e-mediated [Ca2+]i mobilization. Graphs are representative of three independent experiments.
To investigate biased signaling of the CaSR we focused on the ability of [Ca2+]e and [Sr2+]e to activate two additional signaling entities: Gq/11 signaling and [Ca2+]i mobilization. Activity of Gq/11 signaling was quantified by the measurement of IP1 accumulation. IP1 is a breakdown product of inositol 1,4,5-triphosphate (IP3) whose degradation is inhibited by lithium ions present in the assay buffer. IP3 is generated by phosphatidylinositol-specific phospholipase C (PI-PLC), which is activated directly by Gq/11 protein. Thus, IP1 can be used as a measurement of specific activity of the Gq/11 protein. On the other hand, [Ca2+]i mobilization can be caused by the release of calcium from intracellular stores as a result of Gq/11 signaling and/or by entry of extracellular calcium through calcium channels located in the plasma membrane. Therefore, [Ca2+]i mobilization does not represent activity of a specific signaling pathway.
In 6-23 cells, both [Ca2+]e and [Sr2+]e caused a concentration-dependent accumulation of IP1 with equal efficacy (Fig. 2C; Table 1). In this case, the potency of [Ca2+]e was significantly higher than the potency of [Sr2+]e, which has been reported previously (Chattopadhyay et al., 2007; Thomsen et al., 2012). Furthermore, both cations stimulated [Ca2+]i mobilization in a concentration-dependent manner with [Ca2+]e being slightly more potent than [Sr2+]e. It is noteworthy that [Ca2+]e was more than 2-fold more efficacious in causing [Ca2+]i mobilization compared with [Sr2+]e (Fig. 2D; Table 1).
Biased Agonism of CaSR.
In the same way that [Ca2+]i mobilization does not represent activity of a specific signaling pathway, calcitonin secretion represents an endpoint of activity by multiple signaling pathways. The higher potency of [Sr2+]e-mediated calcitonin secretion compared with [Ca2+]e cannot be explained by either Gq/11 signaling or [Ca2+]i mobilization, because the potency rank orders of the two cations in these two experiments were reversed. These results show that [Sr2+]e-stimulated calcitonin secretion is caused, at least in part, through another signaling pathway independently of Gq/11 signaling and [Ca2+]i mobilization. This indicates that [Sr2+]e stabilizes a different active CaSR conformation than [Ca2+]e, which interacts and activates at least one different signaling pathway involved in calcitonin secretion. The biased signaling of CaSR stimulated by [Sr2+]e was further indicated by calculating the pBFs, pBF(IP1:calcitonin) and pBF([Ca2+]i:calcitonin) (Table 2). The negative logarithms of the pBF(IP1:calcitonin) [ppBF(IP1:calcitonin)] and pBF([Ca2+]i:calcitonin) [ppBF([Ca2+]i:calcitonin)] for each agonist were statistically different from each cation (p = 0.015 and 0.018, respectively), which implies that [Sr2+]e biases the signaling of CaSR toward calcitonin secretion (see Materials and Methods).
TABLE 2.
pBFs comparing potency for calcitonin secretion, Gq/11 signaling, and [Ca2+]i mobilization
The pBF of an agonist is calculated as the ratio between EC50 between two different signaling pathways (pathway1:pathway2). However, for analysis of statistic difference between pBF of calcium and pBFs for all other agonists the negative logarithm of pBF or ppBF(pathway1:pathway2) = pEC50(pathway1) − pEC50(pathway2) was used. Statistical comparison of ppBFs for [Sr2+]e to pBFs of [Ca2+]e was performed by using Student's t tests.
| Agonist | pBF(IP1:Calcitonin) (ppBF(IP1:Calcitonin) ± S.E.M.) | pBF([Ca2+]i:Calcitonin) (ppBF([Ca2+]i:Calcitonin) ± S.E.M.) | pBF([Ca2+]i:IP1) (ppBF([Ca2+]i:IP1)± S.E.M.) |
|---|---|---|---|
| [Ca2+]e | 2.0 (−0.295 ± 0.125) | 0.60 (0.22 ± 0.153) | 0.31 (−0.515 ± 0.089) |
| [Sr2+]e | 10 (−1.02 ± 0.128)* | 3.8 (−0.585 ± 0.138)* | 0.37 (−0.435 ± 0.053) |
, p < 0.05.
Another interesting observation of the concentration-response experiments was that [Ca2+]e was twice as efficacious in stimulating [Ca2+]i mobilization compared with [Sr2+]e, whereas both cations were equally efficacious in stimulating Gq/11 signaling and calcitonin secretion. However, this difference may not be caused by signaling bias. Gq/11/PI-PLC activity increases [Ca2+]i by producing IP3, which binds IP3 receptors on intracellular calcium stores, stimulating release of calcium into the cytosol. Because both cations were equally efficacious in causing IP1 accumulation, they were also equally efficacious in causing the release of calcium from intracellular stores. This implies that the residual ability of [Ca2+]e to stimulate [Ca2+]i mobilization comes from extracellular calcium entering the cell through calcium channels located in the plasma membrane. The endogenous agonist of CaSR is [Ca2+]e; thus, extracellular calcium will be present in the assay buffer of all experiments assessing the function of this cation. However, experiments assessing the function of [Sr2+]e had no extracellular calcium present in the assay buffer; therefore, we cannot exclude that [Sr2+]e also activates calcium channels in the plasma membrane.
Involvement of PI-PLC and [Ca2+]i Mobilization in [Ca2+]e- and [Sr2+]e-Mediated Calcitonin Secretion.
To further investigate the biased agonistic nature of [Sr2+]e and test our hypothesis that [Sr2+]e-mediated calcitonin secretion is caused by a signaling pathway independently of Gq/11 signaling and [Ca2+]i mobilization, we sought to identify the signaling pathways involved in [Ca2+]e- and [Sr2+]e-mediated calcitonin secretion by using pre-evaluated inhibitors of pathway components (Table 3).
TABLE 3.
Summary of inhibitors used to identify signaling pathways involved in [Ca2+]e- and [Sr2+]e-stimulated calcitonin secretion
| Inhibitor | Mode of Action | Relative Selectivity |
|---|---|---|
| U73122 | Noncompetitive inhibitor of PI-PLC (Smallridge et al., 1992) | Other effects have been reported, including inhibition of adenosine A1 receptor (Walker et al., 1998) and plasma membrane Ca2+ channels (Berven and Barritt, 1995; Pulcinelli et al., 1998) |
| ET-18-OCH3 | Inhibitor of PI-PLC (Powis et al., 1992) | Activity on platelet-activating factor receptor and Fas/CD95 has been well described (Mollinedo et al., 2004) |
| 2-APB | Modulator of IP3 receptor on intracellular calcium stores (Maruyama et al., 1997) | Other effects have been reported, including uncoupling of gap junction channels formed by certain connexins (Bai et al., 2006) |
| Nimodipine | L-type calcium channel blocker (McGehee et al., 1997) | Has been demonstrated to inhibit equilibrative nucleotide transporter-1 and amyloid β-stimulated IL-1β release (Li et al., 2012; Sanz et al., 2012) |
| BAPTA-AM | Chelator of intracellular divalent cations with high selectivity for [Ca2+]i (Tsien, 1980; Wie et al., 2001) | Other effects have been reported, including calcium-independent cytoskeleton disassembly (Saoudi et al., 2004) |
| D609 | Competitive inhibitor of PC-PLC (Farooqui and Horrocks, 2005) | Has been reported to inhibit sphingomyelin synthase and glucosylceramide synthase (Milhas et al., 2012) |
| Pertussis toxin | Inhibitor of ADP-ribosylation of Gi/o proteins (Mangmool and Kurose, 2011) | Other effects are known, including binding and activation of membrane receptors such as Toll-like receptor 4 (Mangmool and Kurose, 2011) |
| PD98059 | Noncompetitive inhibitor of MEK (Akella et al., 2008) | Controversy exists over whether PD98059 has nonspecific effects on voltage-sensitive calcium channels (Hu et al., 1998; Lagaud et al., 1999) |
| Wortmannin | Noncompetitive inhibitor of PI-3K (Stein, 2001) | Shows poor selectivity among different PI-3K isoforms (Stein, 2001) |
| LY294002 | Competitive inhibitor of PI-3K (Stein, 2001) | Has been reported to inhibit casein kinase 2 and mammalian target of rapamycin and displays poor selectivity among different PI-3K isoforms (Stein, 2001) |
Gq/11 protein and subsequently PI-PLC activation cause the production of IP3 and diacylglycerol, which modulates activity of downstream signaling molecules. CaSR is a well known activator of the Gq/11/PI-PLC signaling pathway, which is important in various physiological CaSR-regulated processes, including the inhibition of PTH secretion from parathyroid cells (Kifor et al., 2001). We have already established that both [Ca2+]e and [Sr2+]e activate Gq/11/PI-PLC signaling; therefore, we investigated whether this pathway is involved in calcitonin secretion. This was achieved by pretreating 6-23 cells for 10 min with three different inhibitors of the Gq/11/PI-PLC signaling pathway: two specific inhibitors of the PI-PLC enzyme (U73122 and Et-18-OCH3) and one specific inhibitor of the IP3 receptors on intracellular calcium stores (2-APB). U73122 and Et-18-OCH3 significantly reduced secretion by 62 and 57%, respectively, in the case of [Ca2+]e and 59 and 52%, respectively, in the case of [Sr2+]e (Fig. 3). 2-APB resulted in significant inhibition of [Ca2+]e- and [Sr2+]e-induced calcitonin secretion by 36 and 28%, respectively- This indicates that both [Ca2+]e- and [Sr2+]e-mediated calcitonin secretion in 6-23 cells is caused partly by Gq/11/PI-PLC signaling. In addition, this result further confirms that both cations are equally efficacious and stimulate Gq/11/PI-PLC signaling, because all inhibitors had similar effects on calcitonin secretion stimulated by both cations.
Fig. 3.

Inhibition of Gq/11/PI-PLC signaling pathway of [Ca2+]e- or [Sr2+]e-mediated calcitonin secretion in 6-23 cells. The inhibition of the Gq/11/PI-PLC signaling pathway was achieved by 10-min preincubation of two PI-PLC inhibitors, U73122 and Et-18-OCH3, and one IP3 receptor inhibitor, 2-APB, before cation stimulation. Responses are normalized to the calcitonin secretory response at 10 mM cation. Statistical comparisons of the values were performed by using a one-way ANOVA of each condition, and Dunnett's post-tests were used to compare DMSO control response for each cation to the inhibitors' responses for each cation (n = 6). ***, p < 0.001; **, p < 0.01; *, p < 0.05.
Next, we focused on the role of [Ca2+]e- and [Sr2+]e-stimulated [Ca2+]i mobilization in calcitonin secretion. It is believed that increases in [Ca2+]i are a key event in [Ca2+]e-mediated calcitonin secretion, because [Ca2+]i triggers the exocytosis machinery of the secretory vesicles (Tucker et al., 2004). Both [Ca2+]e and [Sr2+]e cause [Ca2+]i mobilization; therefore, we hypothesized that [Ca2+]i mobilization induced by either cation would be involved in calcitonin secretion. This was confirmed by inhibiting rises of [Ca2+]i in general with the intracellular calcium chelator BAPTA-AM. The ability of [Ca2+]e to cause calcitonin secretion was almost completely inhibited by BAPTA-AM (82% reduction), confirming that [Ca2+]i mobilization caused by [Ca2+]e is the key event in calcitonin secretion in 6-23 cells (Fig. 4). On the other hand, BAPTA-AM inhibited only 48% of the [Sr2+]e-stimulated calcitonin secretion. The difference in dependence on [Ca2+]i mobilization in calcitonin secretion between the two cations fits well with our initial observation that [Ca2+]e was twice as efficacious in stimulating [Ca2+]i mobilization compared with [Sr2+]e. Again, the [Ca2+]i mobilization involved in [Sr2+]e-stimulated calcitonin secretion must be derived from intracellular calcium stores triggered by Gq/11/PI-PLC activity, because no extracellular calcium was present in the assay buffer. Because BAPTA-AM only partly inhibited [Sr2+]-mediated secretion it confirms that a signaling pathway independent of Gq/11/PI-PLC signaling and [Ca2+]i mobilization is involved in [Sr2+]e-stimulated calcitonin secretion.
Fig. 4.

Inhibition of [Ca2+]e- or [Sr2+]e-stimulated calcitonin secretion by molecules regulating [Ca2+]i and PC-PLC. The inhibition of [Ca2+]i was achieved by 10-min preincubation of BAPTA-AM before cation stimulation. The inhibition of L-type calcium channel was achieved by 10-min preincubation of nimodipine before cation stimulation. The inhibition of PC-PLC signaling pathway was achieved by 10-min preincubation of D609 before cation stimulation. Responses are normalized to the calcitonin secretory response at 10 mM cation. Statistical comparisons of the values were performed by using a one-way ANOVA of each condition, and Dunnett's post-tests were used to compare DMSO control response for each cation to the inhibitors' responses for each cation (n = 6). ***, p < 0.001.
We have shown that both cations stimulate Gq/11/PI-PLC activity to a similar extent, which indicates that a similar part of [Ca2+]i-dependent, [Ca2+]e-mediated calcitonin is a result of calcium release from intracellular stores compared with [Sr2+]e-stimulated secretion. Therefore, our results suggest that a residual [Ca2+]i-dependent calcitonin response to [Ca2+]e exists, which presumably is caused by entry of extracellular calcium through calcium channels in the plasma membrane. In primary parafollicular sheep cells it has been shown that CaSR activation leads to increased [Ca2+]i and plasma membrane L-type calcium channels are responsible for this increase (McGehee et al., 1997). Thus, we investigated the residual [Ca2+]i-dependent calcitonin response to [Ca2+]e further by focusing on the ability of the L-type calcium channel-specific inhibitor nimodipine to reduce the calcitonin secretion stimulated by [Ca2+]e and [Sr2+]e. Nimodipine significantly reduced the [Ca2+]e-induced calcitonin secretion by 49% while having no effect on [Sr2+]e-induced secretion (Fig. 4). This and the results on Gq/11/PI-PLC signaling clearly demonstrate that [Ca2+]e-mediated calcitonin secretion is caused by increased [Ca2+]i coming from both intracellular stores and calcium influx by L-type calcium channels. Furthermore, it has been shown that CaSR activates L-type calcium channels through a signaling pathway initiated by phosphatidylcholine-specific phospholipase C (PC-PLC) activity (McGehee et al., 1997). Therefore, we tested whether the PC-PLC-specific inhibitor D609 specifically could inhibit [Ca2+]e-stimulated calcitonin secretion. As reported previously, D609 inhibited [Ca2+]e-induced calcitonin secretion significantly but by only 20% (Fig. 4). D609 had no effect on [Sr2+]e-induced calcitonin secretion.
Gi/o Protein and Gs Protein Signaling in Response to CaSR Activation by [Ca2+]e and [Sr2+]e.
Thus far, we have shown that [Sr2+]e is a biased agonist of CaSR, which in addition to Gq/11 signaling activates another signaling pathway that is independent of [Ca2+]i mobilization and leads to enhanced potency of calcitonin secretion. Therefore, to further characterize the signaling bias of [Sr2+]e and identify [Sr2+]e-specific pathways involved in calcitonin secretion we focused on other pathways known to be activated by CaSR. It is well known that CaSR directly couples and activates Gi/o protein, causing inhibition of the adenylate cyclase (AC) and a subsequent reduction in cAMP concentration (Chakravarti et al., 2012). Therefore, to measure CaSR-mediated Gi/o signaling we tested the ability of [Ca2+]e and [Sr2+]e to inhibit cAMP production. In resting cells it is difficult to observe the inhibitory effect of Gi/o protein on the AC because the basal concentration of cAMP is very low. Therefore, the AC was preactivated with 10 μM forskolin, raising the basal intracellular cAMP concentration to 201 ± 19 nM. In these cells, [Ca2+]e had no effect on forskolin-stimulated cAMP production (Fig. 5A). It is noteworthy that [Sr2+]e inhibited forskolin-stimulated cAMP production in a concentration-dependent manner with a maximal inhibition of 65% and a EC50 of 340 μM (Fig. 5A; Table 4). To test whether the inhibitory effect of [Sr2+]e on forskolin-stimulated cAMP production was mediated by Gi/o signaling 6-23 cells were pretreated for 24 h with the Gi/o protein-specific inhibitor pertussis toxin. However, inhibition of Gi/o signaling had no effect on [Sr2+]e-mediated inhibition of forskolin-stimulated cAMP production in 6-23 cells (Fig. 5C). Furthermore, preincubation with pertussis toxin had no effect on either [Ca2+]e- or [Sr2+]e-mediated calcitonin secretion, further indicating that [Sr2+]e stimulation does not activate Gi/o signaling (Fig. 6). In addition, octreotide, an agonist of the Gi/o protein-coupled somatostatin receptor, which is expressed and functional in 6-23 cells, inhibited forskolin-stimulated cAMP production (Fig. 5C) (Zink et al., 1992). In this context, preincubation with pertussis toxin completely blocked the effect of octreotide, confirming the functionality of pertussis toxin in 6-23 cells (Fig. 5C).
TABLE 4.
Agonist potencies (EC50) and maximal inhibition and responses (Imax/Rmax) for forskolin-stimulated cAMP production and ERK1/2 phosphorylation
Potencies are in μM for all assays. Imax/Rmax levels are in nM for cAMP inhibition, and fold over basal ERK1/2 response for the ERK1/2 assay.
| Assay | [Ca2+]e |
[Sr2+]e |
||
|---|---|---|---|---|
| Imax or Rmax ± S.E.M. | EC50 (pEC50 ± S.E.M.) | Imax or Rmax ± S.E.M. | EC50 (pEC50 ± S.E.M.) | |
| cAMP | N.A. | N.A. | 65 ± 2 | 339 (3.47 ± 0.05) |
| ERK1/2 | 3.8 ± 0.4 | 3040 (2.52 ± 0.06) | 3.2 ± 0.2 | 405 (3.39 ± 0.14) |
N.A., no activity.
Fig. 5.

Characterization of concentration-dependent [Ca2+]e- and [Sr2+]e-mediated effects on cAMP formation in 6-23 cells. A, concentration-response curves of [Ca2+]e- and [Sr2+]e-mediated inhibition of forskolin-stimulated cAMP production. Responses are normalized to the cAMP production in response to 10 μM forskolin. B, concentration-response experiments of [Ca2+]e- and [Sr2+]e-stimulated cAMP production. The cAMP production in response to 10 μM forskolin is plotted as well as a positive control of AC activity. Concentration-response curves are representative of three independent experiments. C, modulation of 10 mM [Sr2+]e-mediated inhibition of forskolin-stimulated cAMP production (empty bars) by pertussis toxin and Calhex 231 and modulation of 100 nM octreotide-mediated inhibition of forskolin-stimulated cAMP production (filled bars) by pertussis toxin. Statistical comparisons of the values were performed by using a one-way ANOVA of each condition, and Dunnett's post-tests were used to compare 10 mM [Sr2+]e control response to the inhibition by pertussis toxin and Calhex 231. Statistical comparison of the responses to 100 nM octreotide and 100 nM octreotide + 100 ng/ml pertussis toxin was done by using Student's t test (n = 8). ***, p < 0.001.
Fig. 6.

Inhibition of signaling pathway regulated by CaSR of [Ca2+]e- or [Sr2+]e-mediated calcitonin secretion in 6-23 cells. The inhibition of Gi/o protein signaling pathway was achieved by 24-h preincubation of the Gi/o protein inhibitor pertussis toxin. The inhibition of the PI3-K signaling pathway was achieved by 10-min preincubation of the PI3-K inhibitors wortmannin and LY294002. The inhibition of ERK1/2 phosphorylation was achieved by 10-min preincubation of the MEK1 inhibitor PD98059. Responses were normalized to the calcitonin secretory response at 10 mM cation. Statistical comparisons of the values were performed by using a one-way ANOVA of each condition, and Dunnett's post-tests were used to compare DMSO control response for each cation to the inhibitors responses for each cation (n = 6).
To investigate whether the inhibitory effect of [Sr2+]e on forskolin-stimulated cAMP production was mediated by CaSR 6-23 cells were treated with the CaSR-specific antagonist Calhex 231. Calhex 231 had no significant effect on [Sr2+]e-mediated inhibition of forskolin-induced cAMP production (Fig. 5C). This indicates that the inhibitory effect of [Sr2+]e on forskolin-induced cAMP production may be caused by a nonspecific mechanism independent of the CaSR, although a role for the CaSR cannot be excluded (see Discussion).
In malignant breast cancer cells, it has been shown that CaSR switches the usage of G protein and activates Gs protein, causing activation of the AC and production of cAMP (Mamillapalli et al., 2008). To test whether CaSR activates Gs signaling in 6-23 cells the ability of [Ca2+]e and [Sr2+]e to enhance cAMP production was measured in resting cells. Both [Ca2+]e and [Sr2+]e had no effect on cAMP production in resting cells, indicating no coupling of CaSR to the Gs protein in these cells (Fig. 5B).
Phosphorylation of Extracellular Signal-Regulated Kinase 1/2 in Response to CaSR Activation by [Ca2+]e and [Sr2+]e.
Activation of CaSR leads to the phosphorylation and activation of ERK1/2 through multiple signaling pathways, including Gq/11 signaling, Gi/o signaling, and β-arrestin (Kifor et al., 2001; Thomsen et al., 2012). Therefore, to further investigate biased signaling of [Sr2+]e we characterized the ability of [Ca2+]e and [Sr2+]e to activate ERK1/2. Both [Ca2+]e and [Sr2+]e activated ERK1/2 in a concentration-dependent manner with [Ca2+]e being slightly more efficacious than [Sr2+]e although not significantly different (Fig. 7; Table 4). As observed for calcitonin secretion, [Sr2+]e was more potent compared with [Ca2+]e indicating [Sr2+]e-stimulated bias toward ERK1/2 signaling (Fig. 7). This was confirmed by comparing ppBF(ERK1/2:[Ca2+]i) and ppBF (ERK1/2:IP1) values between the two cations, which showed significant differences (p = 0.0029 and 0.0019, respectively) (Table 5). There was no significant difference in ppBF(ERK1/2:calcitonin) values between the two agonists (Table 5).
TABLE 5.
pBFs comparing potency for ERK1/2 phosphorylation with calcitonin secretion, Gq/11 signaling, and [Ca2+]i mobilization
The pBF of an agonist is calculated as the ratio between EC50 between two different signaling pathways (pathway1:pathway2). However, for analysis of statistic difference between pBF of calcium and pBFs for all other agonists the negative logarithms of pBF or ppBF(pathway1:pathway2) = pEC50(pathway1) − pEC50(pathway2) were used. Statistical comparison of ppBFs for [Sr2+]e to ppBFs of [Ca2+]e was performed by using Student's t tests.
| Agonist | pBF(ERK1/2:Calcitonin) (ppBF(ERK1/2:calcitonin) ± S.E.M.) | pBF(ERK1/2:[Ca2+]i) (ppBF(ERK1/2:[Ca2+]i) ± S.E.M.) | pBF(ERK1/2:IP1) (ppBF(ERK1/2:IP1) ± S.E.M.) |
|---|---|---|---|
| [Ca2+]e | 1.2 (−0.095 ± 0.137) | 2.1 (−0.315 ± 0.105) | 0.63 (0.200 ± 0.056) |
| [Sr2+]e | 0.51 (−0.292 ± 0.190) | 0.13 (0.877 ± 0.150)** | 0.049 (1.31 ± 0.14)** |
, p < 0.01.
Fig. 7.

Characterization of concentration-dependent [Ca2+]e- and [Sr2+]e-mediated phosphorylation of ERK1/2 in 6-23 cells. Responses are plotted as fold over basal ERK1/2 response (R/RBasal). Graphs is representative of three independent experiments.
To test whether ERK1/2 is involved in [Sr2+]e-mediated calcitonin secretion 6-23 cells were pretreated for 10 min with the MEK1 inhibitor PD98059. However, inhibition of ERK1/2 phosphorylation did not reduce the [Ca2+]e- and [Sr2+]e-mediated calcitonin secretion in 6-23 cells (Fig. 6).
Involvement of Phosphatidylinositol 3-Kinase in [Ca2+]e- and [Sr2+]e-Induced Calcitonin Secretion.
Previously, it has been demonstrated that the PI3-K pathway is highly involved in [Ca2+]e-mediated calcitonin secretion in primary sheep parafollicular cells (Liu et al., 2000, 2003). Thus, to investigate whether this pathway is involved in either [Ca2+]e- or [Sr2+]e-mediated calcitonin secretion in 6-23 cells the cells were preincubated for 10 min with the PI3-K-specific inhibitors wortmannin and LY294002. However, the inhibitors did not reduce calcitonin secretion stimulated by either cation; therefore, PI3-K does not seems to be involved in [Ca2+]e- and [Sr2+]e-mediated calcitonin secretion in 6-23 cells (Fig. 6).
Discussion
Biased signaling is a well established concept within GPCR pharmacology, which proposes that GPCRs can adopt multiple active conformations rather than just one unitary active conformation (Liu et al., 2012). The various active conformations are stabilized by different ligands and affect GPCR signaling distinctly (Rajagopal et al., 2010; Kenakin, 2011). In this way, biased ligands promote different signaling patterns compared with endogenous ligands, which also may be associated with unique therapeutic properties. So far only limited studies investigating biased signaling of CaSR exist, and all of them were conducted in a HEK293 cell system stably overexpressing exogenous CaSR. Chattopadhyay et al. (2007) showed that [Sr2+]e behaved as a partial agonist of CaSR and was less potent in activating Gq/11 signaling compared with [Ca2+]e but behaved as a full agonist and with similar potency in causing ERK1/2 phosphorylation and activation of nonselective cation channels in HEK293-CaSR cells. This suggested that [Sr2+]e is a biased agonist of CaSR in HEK293-CaSR cells. Although other studies have not found these exact properties of [Sr2+]e using Chinese hamster ovary-CaSR cells, AtT-20 cells, and HEK293-CaSR cells, we used [Sr2+]e in the present study to investigate its biased agonistic properties on CaSR in rat medullary thyroid carcinoma 6-23 cells (Coulombe et al., 2004; Thomsen et al., 2012). Previously, 6-23 cells have proven to be a well suited model of thyroid parafollicular C-cells, because they express functional CaSR endogenously and respond to CaSR activation by secreting calcitonin (Nemeth et al., 2004). In the present study, endogenous CaSR expression in 6-23 cells was confirmed by RT-PCR, and we demonstrated that both [Ca2+]e and [Sr2+]e stimulated calcitonin secretion. Furthermore, both [Ca2+]e- and [Sr2+]e-mediated calcitonin secretion could be modulated by the CaSR-specific allosteric modulator cinacalcet, indicating that calcitonin secretion was mediated by CaSR activation by each cation.
To study biased signaling of CaSR, we first compared concentration-response experiments of [Ca2+]e and [Sr2+]e from three signaling entities: Gq/11 signaling, [Ca2+]i mobilization, and calcitonin secretion. It is noteworthy that in mediating Gq/11 signaling and [Ca2+]i mobilization [Ca2+]e was more potent than [Sr2+]e as reported previously (Chattopadhyay et al., 2007), whereas [Sr2+]e was more potent than [Ca2+]e in causing calcitonin secretion. Calcitonin secretion represents an endpoint of activity by several signaling pathways where both Gq/11 signaling and [Ca2+]i mobilization previously have been shown to be involved (Tamir et al., 1996; McGehee et al., 1997; Liu et al., 2000, 2003). However, neither Gq/11 signaling nor [Ca2+]i mobilization could explain the increased potency in calcitonin secretion of [Sr2+]e compared with [Ca2+]e; thus, our results imply that [Sr2+]e stabilizes an active CaSR conformation, which interacts with another signaling pathway other than the CaSR conformation stabilized by [Ca2+]e. This was further confirmed by pBFs that were statistically different between the two cations.
Another interesting result from the concentration-response experiments was that [Ca2+]e was twice as efficacious in causing [Ca2+]i mobilization but equally efficacious in causing Gq/11 signaling (measured as IP1 generation) and calcitonin secretion compared with [Sr2+]e. Although this might indicate signaling bias, it was presumably caused by extracellular calcium flowing into the cells through L-type calcium channels activated by CaSR. In all experiments investigating [Sr2+]e no extracellular calcium was present in the assay buffer; thus, it is possible that [Sr2+]e-activated CaSR also activates L-type calcium channels. Therefore, we cannot conclude that the enhanced efficacy of [Ca2+]e on [Ca2+]i mobilization was caused by signaling bias.
To further elucidate how [Sr2+]e biases the signaling of CaSR in 6-23 cells we continued our investigation by focusing on the molecular mechanism of [Ca2+]e- and [Sr2+]e-mediated calcitonin secretion. Multiple studies have been conducted to dissect the mechanism of [Ca2+]e-mediated calcitonin secretion in primary parafollicular sheep cells (Tamir et al., 1990, 1994, 1996; McGehee et al., 1997; Liu et al., 2000, 2003). It is believed that [Ca2+]e-stimulated CaSR couples to a G protein that activates the PC-PLC enzyme, initiating a signaling pathway, which ultimately leads to opening L-type calcium channels. This causes calcium influx and thereby increases [Ca2+]i mobilization, which triggers the secretion of the vesicular content (McGehee et al., 1997; Tucker et al., 2004).
It is not clear whether the activation of Gq/11 or Gi/o proteins is involved in calcitonin secretion of primary sheep parafollicular cells. Experiments have demonstrated that the inhibition of Gi/o protein and PI-PLC decreases the ability of high [Ca2+]e to cause acidification of vesicles containing calcitonin, a key step in hormone loading of secretory vesicles (Cidon et al., 1991; Tamir et al., 1996). However, another study showed that the inhibition of PI-PLC had no effect on vesicular secretion, indicating that the Gq/11/PI-PLC signaling pathway is not involved (McGehee et al., 1997). It is noteworthy that we demonstrated that PI-PLC partly causes calcitonin secretion in 6-23 cells because three different inhibitors of PI-PLC signaling significantly reduced both [Ca2+]e- and [Sr2+]e-stimulated calcitonin secretion.
Activation of PI-PLC and subsequently IP3 production leads to the release of calcium from intracellular stores into the cytoplasm. In our case, [Ca2+]e was twice as efficacious in causing [Ca2+]i mobilization compared with [Sr2+]e. This implied that [Ca2+]e-mediated calcitonin secretion to a higher extent depends on rises in [Ca2+]i than [Sr2+]e-mediated calcitonin secretion, which was confirmed by blocking all rises in [Ca2+]i by BAPTA-AM. It showed that [Ca2+]e-mediated calcitonin secretion almost entirely depends on rises in [Ca2+]i, whereas only approximately 50% of the [Sr2+]e-mediated calcitonin secretion is [Ca2+]i-dependent. By using inhibitors of both PC-PLC and L-type calcium channels, we showed that the superior dependence on [Ca2+]i in [Ca2+]e-stimulated calcitonin secretion was caused by the activity of PC-PLC signaling and opening of L-type calcium channels, resulting in an influx of extracellular calcium in combination with PI-PLC-mediated [Ca2+]i mobilization. Furthermore, because Gq/11/PI-PLC-stimulated [Ca2+]i mobilization accounts for only 50% of [Sr2+]e-stimulated calcitonin secretion, the results confirmed our hypothesis that [Sr2+]-mediated calcitonin secretion, at least in part, is caused by a signaling pathway independent of Gq/11 signaling and [Ca2+]i mobilization.
To search for the non-[Ca2+]i-dependent pathway of [Sr2+]e-mediated calcitonin secretion and further characterize [Sr2+]e-induced signaling bias we focused on pathways that have been associated with CaSR activation in thyroid parafollicular C-cells and other cellular systems. It is well known that CaSR couples and activates Gi/o protein signaling in various cell types, resulting in the inhibition of the AC and a subsequent reduction in cAMP formation (Chakravarti et al., 2012). In the present study we found that [Sr2+]e selectively inhibits forskolin-stimulated cAMP production, whereas [Ca2+]e had no effect on cAMP concentration. However, this cAMP inhibition by [Sr2+]e was not mediated through a Gi/o protein-dependent mechanism, because pretreatment with pertussis toxin had no effect. It has been reported that CaSR-mediated inhibition of forskolin-stimulated cAMP production, in fact, may be caused by increases in [Ca2+]i in some cells (Ortiz-Capisano et al., 2007a,b). In our case, this did not seem likely because [Ca2+]e is more than twice as efficacious in generating increases in [Ca2+]i compared with [Sr2+]e. A third option could be [Sr2+]-mediated activity of phosphodiesterases, which degrade cAMP through unidentified signaling. To investigate the involvement of CaSR in the [Sr2+]e-mediated cAMP response, 6-23 cells were treated with the CaSR-specific antagonist Calhex 231. Again, no significant effect was observed. Although this indicates a CaSR-independent response of [Sr2+]e, we cannot rule out that this effect is mediated by CaSR as well. Recently, it was found that both positive [cinacalcet and N-(2-chlorophenylpropyl)-1-(3-methoxyphenyl)ethylamine (NPS-R568)] and negative [N-(2-hydroxy-3-(2-cyano-3-chlorophenoxy)propyl)-1,1-dimethyl-2-(2-nephthyl)ethylamine (NPS-2143)] CaSR-specific modulators generated signaling bias of the CaSR when their allosteric modulation was characterized on three signaling entities: [Ca2+]i mobilization, ERK1/2 signaling, and the ability to generate plasma membrane ruffles (Davey et al., 2012). Therefore, signaling bias seems to be a general trait of CaSR-specific modulators; thus, we cannot assume that the antagonizing potential of Calhex 231 is equal over all CaSR-activated pathways, especially in the case of [Sr2+]e-activated CaSR, which is already stabilized in an alternative active conformation.
Another signaling component stimulated by CaSR activity is ERK1/2 (Kifor et al., 2001; Thomsen et al., 2012). We found that both [Ca2+]e and [Sr2+]e activated ERK1/2 in a concentration-dependent manner, but as for calcitonin secretion [Sr2+]e was more potent than [Ca2+]e. ERK1/2 activity was not involved in the [Sr2+]e-mediated bias observed on calcitonin secretion, because its inhibition by the MEK1 inhibitor PD59098 did not affect calcitonin secretion. However, by calculating pBFs we found that the [Sr2+]e-stimulated bias on ERK1/2 activity was similar to the [Sr2+]e-stimulated bias on calcitonin secretion. Therefore, it is possible that [Sr2+]e-stimulated bias toward calcitonin secretion is mediated by a signaling pathway upstream of ERK1/2 such as β-arrestins. Unfortunately, the activity and effects of β-arrestins are challenging to determine in nontransfected cell lines such as 6-23 cells because of a lack of selective pharmacological inhibitors. Thus they were not investigated.
Finally, it has been reported that CaSR coupling to Gβγ subunits activates PI3-K, which causes secretion of vesicular content in primary parafollicular sheep cells through a [Ca2+]i-independent pathway (Liu et al., 2000, 2003). In our studies, inhibition of PI3-K with wortmannin or LY294002 had no effect on either [Ca2+]e- or [Sr2+]e-mediated calcitonin secretion; thus, PI3-K is not involved in the [Sr2+]e-stimulated bias.
Our results on the biased agonism of [Sr2+]e in calcitonin-secreting 6-23 cells are interesting for various reasons. First, our results shows that biased signaling of CaSR is physiologically relevant, which may be exploited in the development of new CaSR-affecting drugs that selectively modulate the desired physiological effects. Second, strontium is the active ingredient of strontium ranelate, which is clinically useful for treating postmenopausal osteoporosis (Meunier et al., 2004). The Sr2+ ion targets bones where it is retained, increases the activity of osteoblasts, and inhibits the activity of osteoclasts through CaSR-dependent and -independent mechanisms (Chattopadhyay et al., 2007; Brennan et al., 2009; Fromigué et al., 2009; Caudrillier et al., 2010). Our results indicate that [Sr2+]e might stabilize CaSR in a different active conformation than [Ca2+]e. Therefore, it is possible that [Sr2+]e-mediated CaSR activation causes additional physiological responses in cells involved in bone metabolism compared with [Ca2+]e that account for [Sr2+]e's superior value as a therapeutic agent.
Acknowledgments
We thank Lenea Nørskov-Lauritsen for fruitful discussions.
This work has been financially supported by LEO Pharma A/S and grants from the Drug Research Academy, Aase og Ejner Danielsens Fond, and Beckett-Fonden.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
- CaSR
- calcium-sensing receptor
- GPCR
- G protein-coupled receptor
- PTH
- parathyroid hormone
- [Ca2+]e
- extracellular calcium
- [Sr2+]e
- extracellular strontium
- [Ca2+]i
- intracellular calcium
- DMEM
- Dulbecco's modified Eagle's medium
- DPBS
- Dulbecco's phosphate-buffered saline
- HBSS
- Hanks' balanced salt solution
- D609
- O-tricyclo[5.2.1.02,6]dec-9-yl dithiocarbonate
- LY294002
- 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one
- PD98059
- 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one
- ET-18-OCH3
- 1-O-octadecyl-2-O-methyl-sn-glycero-3-phosphorylcholine
- 2-APB
- 2-aminoethoxydiphenyl borate
- U73122
- 1-[6-[((17β)-3-methoxyestra-1,3,5[10]-trien-17-yl)amino]hexyl]-1H-pyrrole-2,5-dione
- BAPTA-AM
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester)
- IBMX
- 3-isobutyl-1-methylxanthine
- Calhex 231
- 4-chloro-N-[(1S,2S)-2-[[(1R)-1-(1-naphthalenyl)ethyl]amino]cyclohexyl]-benzamide hydrochloride
- TR-FRET
- time-resolved-fluorescence resonance energy transfer
- IP1
- inositol monophosphate
- BF
- bias factor
- pBF
- potency BF
- IP3
- inositol 1,4,5-triphosphate
- PI-PLC
- phosphatidylinositol-specific phospholipase C
- PC-PLC
- phosphatidylcholine-specific phospholipase C
- AC
- adenylate cyclase
- ERK1/2
- extracellular signal-regulated kinases 1/2
- HEK
- human embryonic kidney
- PI3-K
- phosphatidylinositol 3-kinase
- ANOVA
- analysis of variance
- DMSO
- dimethyl sulfoxide
- RT
- reverse transcription
- PCR
- polymerase chain reaction
- bp
- base pairs
- MEK1
- mitogen-activated protein kinase kinase 1
- NPS-R568
- N-(2-chlorophenylpropyl)-1-(3-methoxyphenyl)ethylamine
- NPS-2143
- N-(2-hydroxy-3-(2-cyano-3-chlorophenoxy)propyl)-1,1-dimethyl-2-(2-nephthyl)ethylamine.
Authorship Contributions
Participated in research design: Thomsen, Worm, Jacobsen, Stahlhut, Latta, and Bräuner-Osborne.
Conducted experiments: Thomsen, Worm, and Jacobsen.
Contributed new reagents or analytic tools: Thomsen, Worm, Stahlhut, Latta, and Bräuner-Osborne.
Performed data analysis: Thomsen, Worm, Jacobsen, Stahlhut, Latta, and Bräuner-Osborne.
Wrote or contributed to the writing of the manuscript: Thomsen, Worm, Jacobsen, Stahlhut, Latta, and Bräuner-Osborne.
References
- Akella R, Moon TM, Goldsmith EJ. (2008) Unique MAP kinase binding sites. Biochim Biophys Acta 1784:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almadén Y, Canalejo A, Ballesteros E, Añón G, Cañadillas S, Rodríguez M. (2002) Regulation of arachidonic acid production by intracellular calcium in parathyroid cells: effect of extracellular phosphate. J Am Soc Nephrol 13:693–698. [DOI] [PubMed] [Google Scholar]
- Awata H, Huang C, Handlogten ME, Miller RT. (2001) Interaction of the calcium-sensing receptor and filamin, a potential scaffolding protein. J Biol Chem 276:34871–34879. [DOI] [PubMed] [Google Scholar]
- Bai D, del Corsso C, Srinivas M, Spray DC. (2006) Block of specific gap junction channel subtypes by 2-aminoethoxydiphenyl borate (2-APB). J Pharmacol Exp Ther 319:1452–1458. [DOI] [PubMed] [Google Scholar]
- Berven LA, Barritt GJ. (1995) Evidence obtained using single hepatocytes for inhibition by the phospholipase C inhibitor U73122 of store-operated Ca2+ inflow. Biochem Pharmacol 49:1373–1379. [DOI] [PubMed] [Google Scholar]
- Bouschet T, Martin S, Kanamarlapudi V, Mundell S, Henley JM. (2007) The calcium-sensing receptor changes cell shape via a β-arrestin-1 ARNO ARF6 ELMO protein network. J Cell Sci 120:2489–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan TC, Rybchyn MS, Green W, Atwa S, Conigrave AD, Mason RS. (2009) Osteoblasts play key roles in the mechanisms of action of strontium ranelate. Br J Pharmacol 157:1291–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC. (1993) Cloning and characterization of an extracellular Ca2+-sensing receptor from bovine parathyroid. Nature 366:575–580. [DOI] [PubMed] [Google Scholar]
- Caudrillier A, Hurtel-Lemaire AS, Wattel A, Cournarie F, Godin C, Petit L, Petit JP, Terwilliger E, Kamel S, Brown EM, et al. (2010) Strontium ranelate decreases receptor activator of nuclear factor-κB ligand-induced osteoclastic differentiation in vitro: involvement of the calcium-sensing receptor. Mol Pharmacol 78:569–576. [DOI] [PubMed] [Google Scholar]
- Chakravarti B, Chattopadhyay N, Brown EM. (2012) Signaling through the extracellular calcium-sensing receptor (CaSR). Adv Exp Med Biol 740:103–142. [DOI] [PubMed] [Google Scholar]
- Chang W, Tu C, Chen TH, Bikle D, Shoback D. (2008) The extracellular calcium-sensing receptor (CaSR) is a critical modulator of skeletal development. Sci Signal 1:ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay N, Quinn SJ, Kifor O, Ye C, Brown EM. (2007) The calcium-sensing receptor (CaR) is involved in strontium ranelate-induced osteoblast proliferation. Biochem Pharmacol 74:438–447. [DOI] [PubMed] [Google Scholar]
- Chonchol M, Locatelli F, Abboud HE, Charytan C, de Francisco AL, Jolly S, Kaplan M, Roger SD, Sarkar S, Albizem MB, et al. (2009) A randomized, double-blind, placebo-controlled study to assess the efficacy and safety of cinacalcet HCl in participants with CKD not receiving dialysis. Am J Kidney Dis 53:197–207. [DOI] [PubMed] [Google Scholar]
- Cidon S, Tamir H, Nunez EA, Gershon MD. (1991) ATP-dependent uptake of 5-hydroxytryptamine by secretory granules isolated from thyroid parafollicular cells. J Biol Chem 266:4392–4400. [PubMed] [Google Scholar]
- Conigrave AD, Mun HC, Lok HC. (2007) Aromatic l-amino acids activate the calcium-sensing receptor. J Nutr 137:1524S–1527S; discussion 1548S. [DOI] [PubMed] [Google Scholar]
- Coulombe J, Faure H, Robin B, Ruat M. (2004) In vitro effects ofstrontium ranelate on the extracellular calcium-sensing receptor. Biochem Biophys Res Commun 323:1184–1190. [DOI] [PubMed] [Google Scholar]
- Davey AE, Leach K, Valant C, Conigrave AD, Sexton PM, Christopoulos A. (2012) Positive and negative allosteric modulators promote biased signaling at the calcium-sensing receptor. Endocrinology 153:1232–1241. [DOI] [PubMed] [Google Scholar]
- Drake MT, Violin JD, Whalen EJ, Wisler JW, Shenoy SK, Lefkowitz RJ. (2008) β-arrestin-biased agonism at the β2-adrenergic receptor. J Biol Chem 283:5669–5676. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Horrocks LA. (2005) Signaling and interplay mediated by phospholipases A2, C, and D in LA-N-1 cell nuclei. Reprod Nutr Dev 45:613–631. [DOI] [PubMed] [Google Scholar]
- Fromigué O, Haÿ E, Barbara A, Petrel C, Traiffort E, Ruat M, Marie PJ. (2009) Calcium sensing receptor-dependent and receptor-independent activation of osteoblast replication and survival by strontium ranelate. J Cell Mol Med 13:2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley C, 3rd, Yang Y, Davis J, Lu JY, Morony S, Fan W, Florio M, Sun B, Shatzen E, Pretorius JK, et al. (2011) Discovery of a calcimimetic with differential effects on parathyroid hormone and calcitonin secretion. J Pharmacol Exp Ther 337:681–691. [DOI] [PubMed] [Google Scholar]
- Hjälm G, MacLeod RJ, Kifor O, Chattopadhyay N, Brown EM. (2001) Filamin-A binds to the carboxyl-terminal tail of the calcium-sensing receptor, an interaction that participates in CaR-mediated activation of mitogen-activated protein kinase. J Biol Chem 276:34880–34887. [DOI] [PubMed] [Google Scholar]
- Ho C, Conner DA, Pollak MR, Ladd DJ, Kifor O, Warren HB, Brown EM, Seidman JG, Seidman CE. (1995) A mouse model of human familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Nat Genet 11:389–394. [DOI] [PubMed] [Google Scholar]
- Hu XQ, Singh N, Mukhopadhyay D, Akbarali HI. (1998) Modulation of voltage-dependent Ca2+ channels in rabbit colonic smooth muscle cells by c-Src and focal adhesion kinase. J Biol Chem 273:5337–5342. [DOI] [PubMed] [Google Scholar]
- Huang C, Hujer KM, Wu Z, Miller RT. (2004) The Ca2+-sensing receptor couples to Gα12/13 to activate phospholipase D in Madin-Darby canine kidney cells. Am J Physiol Cell Physiol 286:C22–C230. [DOI] [PubMed] [Google Scholar]
- Kantham L, Quinn SJ, Egbuna OI, Baxi K, Butters R, Pang JL, Pollak MR, Goltzman D, Brown EM. (2009) The calcium-sensing receptor (CaSR) defends against hypercalcemia independently of its regulation of parathyroid hormone secretion. Am J Physiol Endocrinol Metab 297:E915–E923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. (2011) Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther 336:296–302. [DOI] [PubMed] [Google Scholar]
- Kifor O, MacLeod RJ, Diaz R, Bai M, Yamaguchi T, Yao T, Kifor I, Brown EM. (2001) Regulation of MAP kinase by calcium-sensing receptor in bovine parathyroid and CaR-transfected HEK293 cells. Am J Physiol Renal Physiol 280:F291–F302. [DOI] [PubMed] [Google Scholar]
- Lagaud GJ, Lam E, Lui A, van Breemen C, Laher I. (1999) Nonspecific inhibition of myogenic tone by PD98059, a MEK1 inhibitor, in rat middle cerebral arteries. Biochem Biophys Res Commun 257:523–527. [DOI] [PubMed] [Google Scholar]
- Li RW, Yang C, Sit AS, Lin SY, Ho EY, Leung GP. (2012) Physiological and pharmacological roles of vascular nucleoside transporters. J Cardiovasc Pharmacol 59:10–15. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Horst R, Katritch V, Stevens RC, Wüthrich K. (2012) Biased signaling pathways in β2-adrenergic receptor characterized by 19F-NMR. Science 335:1106–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Hsiung S, Adlersberg M, Sacktor T, Gershon MD, Tamir H. (2000) Ca2+-evoked serotonin secretion by parafollicular cells: roles in signal transduction of phosphatidylinositol 3′-kinase, and the γ and ζ isoforms of protein kinase C. J Neurosci 20:1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu KP, Russo AF, Hsiung SC, Adlersberg M, Franke TF, Gershon MD, Tamir H. (2003) Calcium receptor-induced serotonin secretion by parafollicular cells: role of phosphatidylinositol 3-kinase-dependent signal transduction pathways. J Neurosci 23:2049–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makita N, Sato J, Manaka K, Shoji Y, Oishi A, Hashimoto M, Fujita T, Iiri T. (2007) An acquired hypocalciuric hypercalcemia autoantibody induces allosteric transition among active human Ca-sensing receptor conformations. Proc Natl Acad Sci U S A 104:5443–5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamillapalli R, VanHouten J, Zawalich W, Wysolmerski J. (2008) Switching of G-protein usage by the calcium-sensing receptor reverses its effect on parathyroid hormone-related protein secretion in normal versus malignant breast cells. J Biol Chem 283:24435–24447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangmool S, Kurose H. (2011) Gi/o protein-dependent and -independent actions of pertussis toxin (PTX). Toxins (Basel) 3:884–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K. (1997) 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J Biochem 122:498–505. [DOI] [PubMed] [Google Scholar]
- McGehee DS, Aldersberg M, Liu KP, Hsuing S, Heath MJ, Tamir H. (1997) Mechanism of extracellular Ca2+ receptor-stimulated hormone release from sheep thyroid parafollicular cells. J Physiol 502:31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier PJ, Roux C, Seeman E, Ortolani S, Badurski JE, Spector TD, Cannata J, Balogh A, Lemmel EM, Pors-Nielsen S, et al. (2004) The effects of strontium ranelate on the risk of vertebral fracture in women with postmenopausal osteoporosis. N Engl J Med 350:459–468. [DOI] [PubMed] [Google Scholar]
- Milhas D, Andrieu-Abadie N, Levade T, Benoist H, Ségui B. (2012) The tricyclodecan-9-yl-xanthogenate D609 triggers ceramide increase and enhances FasL-induced caspase-dependent and -independent cell death in T lymphocytes. Int J Mol Sci 13:8834–8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollinedo F, Gajate C, Martín-Santamaría S, Gago F. (2004) ET-18-OCH3 (edelfosine): a selective antitumour lipid targeting apoptosis through intracellular activation of Fas/CD95 death receptor. Curr Med Chem 11:3163–3184. [DOI] [PubMed] [Google Scholar]
- Nemeth EF, Heaton WH, Miller M, Fox J, Balandrin MF, Van Wagenen BC, Colloton M, Karbon W, Scherrer J, Shatzen E, et al. (2004) Pharmacodynamics of the type II calcimimetic compound cinacalcet HCl. J Pharmacol Exp Ther 308:627–635. [DOI] [PubMed] [Google Scholar]
- Ortiz-Capisano MC, Ortiz PA, Garvin JL, Harding P, Beierwaltes WH. (2007a) Expression and function of the calcium-sensing receptor in juxtaglomerular cells. Hypertension 50:737–743. [DOI] [PubMed] [Google Scholar]
- Ortiz-Capisano MC, Ortiz PA, Harding P, Garvin JL, Beierwaltes WH. (2007b) Adenylyl cyclase isoform V mediates renin release from juxtaglomerular cells. Hypertension 49:618–624. [DOI] [PubMed] [Google Scholar]
- Pollak MR, Brown EM, Chou YH, Hebert SC, Marx SJ, Steinmann B, Levi T, Seidman CE, Seidman JG. (1993) Mutations in the human Ca2+-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell 75:1297–1303. [DOI] [PubMed] [Google Scholar]
- Powis G, Seewald MJ, Gratas C, Melder D, Riebow J, Modest EJ. (1992) Selective inhibition of phosphatidylinositol phospholipase C by cytotoxic ether lipid analogues. Cancer Res 52:2835–2840. [PubMed] [Google Scholar]
- Pulcinelli FM, Gresele P, Bonuglia M, Gazzaniga PP. (1998) Evidence for separate effects of U73122 on phospholipase C and calcium channels in human platelets. Biochem Pharmacol 56:1481–1484. [DOI] [PubMed] [Google Scholar]
- Quinn SJ, Ye CP, Diaz R, Kifor O, Bai M, Vassilev P, Brown E. (1997) The Ca2+-sensing receptor: a target for polyamines. Am J Physiol Cell Physiol 273:C1315–C1323. [DOI] [PubMed] [Google Scholar]
- Rajagopal S, Rajagopal K, Lefkowitz RJ. (2010) Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat Rev Drug Discov 9:373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi D, Park J, Lee WS, Gamba G, Brown EM, Hebert SC. (1995) Cloning and functional expression of a rat kidney extracellular calcium/polyvalent cation-sensing receptor. Proc Natl Acad Sci U S A 92:131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruat M, Snowman AM, Hester LD, Snyder SH. (1996) Cloned and expressed rat Ca2+-sensing receptor. J Biol Chem 271:5972–5975. [DOI] [PubMed] [Google Scholar]
- Sanz JM, Chiozzi P, Colaianna M, Zotti M, Ferrari D, Trabace L, Zuliani G, Di Virgilio F. (2012) Nimodipine inhibits amyloid β-stimulated IL-1β release from microglia. Br J Pharmacol 10.1111/j.1476-5381.2012.02112.x. [DOI] [PMC free article] [PubMed]
- Saoudi Y, Rousseau B, Doussière J, Charrasse S, Gauthier-Rouvière C, Morin N, Sautet-Laugier C, Denarier E, Scaïfe R, Mioskowski C, et al. (2004) Calcium-independent cytoskeleton disassembly induced by BAPTA. Eur J Biochem 271:3255–3264. [DOI] [PubMed] [Google Scholar]
- Smajilovic S, Hansen JL, Christoffersen TE, Lewin E, Sheikh SP, Terwilliger EF, Brown EM, Haunso S, Tfelt-Hansen J. (2006) Extracellular calcium sensing in rat aortic vascular smooth muscle cells. Biochem Biophys Res Commun 348:1215–1223. [DOI] [PubMed] [Google Scholar]
- Smallridge RC, Kiang JG, Gist ID, Fein HG, Galloway RJ. (1992) U-73122, an aminosteroid phospholipase C antagonist, noncompetitively inhibits thyrotropin-releasing hormone effects in GH3 rat pituitary cells. Endocrinology 131:1883–1888. [DOI] [PubMed] [Google Scholar]
- Stein RC. (2001) Prospects for phosphoinositide 3-kinase inhibition as a cancer treatment. Endocr Relat Cancer 8:237–248. [DOI] [PubMed] [Google Scholar]
- Tamir H, Liu KP, Adlersberg M, Hsiung SC, Gershon MD. (1996) Acidification of serotonin-containing secretory vesicles induced by a plasma membrane calcium receptor. J Biol Chem 271:6441–6450. [DOI] [PubMed] [Google Scholar]
- Tamir H, Liu KP, Hsiung SC, Adlersberg M, Nunez EA, Gershon MD. (1990) Multiple signal transduction mechanisms leading to the secretion of 5-hydroxytryptamine by MTC cells, a neurectodermally derived cell line. J Neurosci 10:3743–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamir H, Piscopo I, Liu KP, Hsiung SC, Adlersberg M, Nicolaides M, al-Awqati Q, Nunez EA, Gershon MD. (1994) Secretogogue-induced gating of chloride channels in the secretory vesicles of parafollicular cells. Endocrinology 135:2045–2057. [DOI] [PubMed] [Google Scholar]
- Thomsen AR, Hvidtfeldt M, Bräuner-Osborne H. (2012) Biased agonism of the calcium-sensing receptor. Cell Calcium 51:107–116. [DOI] [PubMed] [Google Scholar]
- Torres PU. (2006) Cinacalcet HCl: a novel treatment for secondary hyperparathyroidism caused by chronic kidney disease. J Ren Nutr 16:253–258. [DOI] [PubMed] [Google Scholar]
- Tsien RY. (1980) New calcium indicators and buffers with high selectivity against magnesium and protons: design, synthesis, and properties of prototype structures. Biochemistry 19:2396–2404. [DOI] [PubMed] [Google Scholar]
- Tucker WC, Weber T, Chapman ER. (2004) Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science 304:435–438. [DOI] [PubMed] [Google Scholar]
- Walker EM, Bispham JR, Hill SJ. (1998) Nonselective effects of the putative phospholipase C inhibitor, U73122, on adenosine A1 receptor-mediated signal transduction events in Chinese hamster ovary cells. Biochem Pharmacol 56:1455–1462. [DOI] [PubMed] [Google Scholar]
- Ward DT, McLarnon SJ, Riccardi D. (2002) Aminoglycosides increase intracellular calcium levels and ERK activity in proximal tubular OK cells expressing the extracellular calcium-sensing receptor. J Am Soc Nephrol 13:1481–1489. [DOI] [PubMed] [Google Scholar]
- Wie MB, Koh JY, Won MH, Lee JC, Shin TK, Moon CJ, Ha HJ, Park SM, Kim HC. (2001) BAPTA/AM, an intracellular calcium chelator, induces delayed necrosis by lipoxygenase-mediated free radicals in mouse cortical cultures. Prog Neuropsychopharmacol Biol Psychiatry 25:1641–1659. [DOI] [PubMed] [Google Scholar]
- Zink A, Scherübl H, Kliemann D, Höflich M, Ziegler R, Raue F. (1992) Inhibitory effect of somatostatin on cAMP accumulation and calcitonin secretion in C-cells: involvement of pertussis toxin-sensitive G-proteins. Mol Cell Endocrinol 86:213–219. [DOI] [PubMed] [Google Scholar]