

Abstract
Genome-wide association studies (GWAS) have identified more than 200 common genetic variants independently associated with colorectal cancer (CRC) risk, but the causal variants and target genes are mostly unknown. We sought to fine-map all known CRC risk loci using GWAS data from 100,204 cases and 154,587 controls of East Asian and European ancestry. Our stepwise conditional analyses revealed 238 independent association signals of CRC risk, each with a set of credible causal variants (CCVs), of which 28 signals had a single CCV. Our cis-eQTL/mQTL and colocalization analyses using colorectal tissue-specific transcriptome and methylome data separately from 1299 and 321 individuals, along with functional genomic investigation, uncovered 136 putative CRC susceptibility genes, including 56 genes not previously reported. Analyses of single-cell RNA-seq data from colorectal tissues revealed 17 putative CRC susceptibility genes with distinct expression patterns in specific cell types. Analyses of whole exome sequencing data provided additional support for several target genes identified in this study as CRC susceptibility genes. Enrichment analyses of the 136 genes uncover pathways not previously linked to CRC risk. Our study substantially expanded association signals for CRC and provided additional insight into the biological mechanisms underlying CRC development.
Subject terms: Cancer epidemiology, Epidemiology
Here, the authors perform large trans-ancestry fine-mapping analyses identifying large numbers of association signals and putative target genes for colorectal cancer risk, advancing our understanding of the genetic and biological basis of this cancer.
Introduction
Colorectal cancer (CRC) is one of the most common malignancies worldwide1. Inherited genetic factors play an important role in the development of CRC2. Since 2007, genome-wide association studies (GWAS) have identified over 200 common genetic variants independently associated with CRC risk3–7. These GWAS, however, typically only reported the most significantly associated variant (the lead variant) at each risk locus. Statistical fine-mapping analyses of known risk loci can identify additional association signals independent of the lead variant.
Approximately 90% of GWAS-identified risk variants for CRC are located in noncoding or intergenic regions, and target genes for most of these risk variants remain unknown. Well-powered fine-mapping analyses, particularly those using data from multi-ancestry populations, can facilitate the identification of credible causal variants (CCVs) in each region. Previous genetic studies have provided strong evidence that regulatory variants in linkage disequilibrium (LD) with GWAS-identified risk variants drive the associations of genetic variants with cancer risk by modulating the expression of susceptibility genes8–11. Therefore, integrating functional genomic data to interrogate CCVs in each independent risk-associated signal could help to identify putative causal variants and target genes for CRC risk. Herein, we conducted large trans-ancestry fine-mapping analyses of all currently known CRC risk regions, using GWAS data from 100,204 CRC cases and 154,587 controls of East Asian and European ancestry, to identify independent association signals and their target genes for CRC risk.
Results
Identification of independent association signals with CRC risk
We conducted fine-mapping analyses using GWAS summary statistics from 100,204 CRC cases and 154,587 controls (73% European and 27% East Asian ancestry) (Fig. 1, Supplementary Data 1). In our recent trans-ancestry meta-analysis of GWAS, we identified 205 genetic variants independently associated with CRC risk7. We aggregated regions flagged by these variants into 143 risk regions, each containing at least a 1 Mb interval centered on the most significant association (Supplementary Data 2). Among them, 40 regions harbor at least two reported independent risk associations. All risk regions were autosomal, except the one at Xp22.2. For subsequent analyses, we focused on the 142 regions located on the autosomes.
Fig. 1. Schematic diagram of the study design.
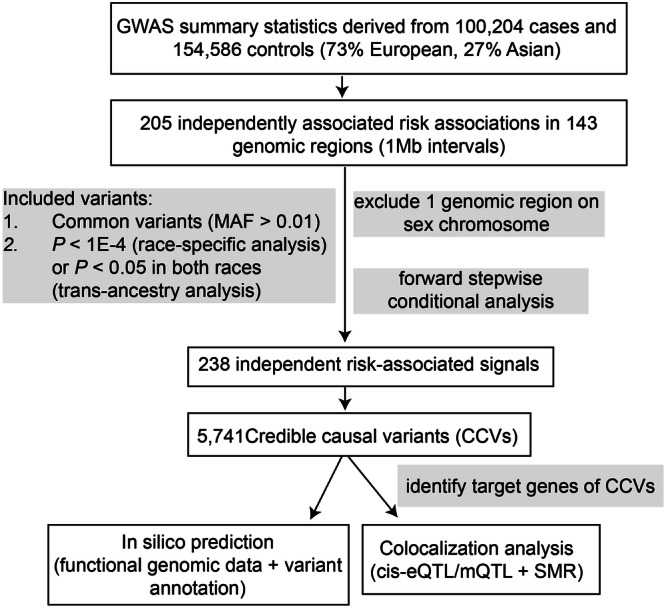
We conducted fine-mapping analyses using GWAS summary statistics from 100,204 cases and 154,587 controls. All 205 genetic variants were aggregated to 143 risk regions containing at least a 1 megabase (Mb) interval centered on the most significant association. This study focused on 142 risk regions located on the autosomes. In forward stepwise conditional analysis, we included common variants (minor allele frequency (MAF) > 0.01) with associations at P < 0.05 in both populations for the trans-ancestry analysis and with associations at P < 1 × 10−4 in each population for race-specific analysis. The threshold of conditional P < 1 × 10−6 was used to determine independent risk-associated signals. For credible causal variants (CCVs) for each independent signal, we conducted in-silico analyses with functional genomic data generated in CRC-related tissues/cells and colocalization of expression/methylation quantitative trait loci (e/mQTL) with GWAS signals to identify putative target genes for CCVs using the Summary-data-based Mendelian Randomization (SMR) approach.
We used forward stepwise conditional analyses to identify independent association signals in each region in each population, conditioning on the most significant association from the trans-ancestral summary statistics (Supplementary Fig. 1, Methods). We then meta-analyzed the conditioned data using the fixed-effects inverse variance weighted model. We considered the threshold of conditional P < 1 × 10−6 to determine independent significant associations to balance both Type 1 and 2 errors, as recommended by a previous fine-mapping study in breast cancer12. At this threshold, we identified 171 independent association signals in 122 regions (Fig. 2, Supplementary Data 3). To identify possible ancestry-specific association signals, we conducted similar analyses using only summary statistics from each population, conditioning on the ancestry-specific most significant association. Using the same threshold, we identified 198 and 45 independent association signals in European and East Asian descendants, respectively (Supplementary Data 4 and 5). Of them, 60 signals in European and 7 in East Asian were not detected in the trans-ancestry analysis above, suggesting them as potential ancestry-specific risk signals (Fig. 2).
Fig. 2. Independent association signals for colorectal cancer risk.

Numbers of fine-mapping regions and numbers of independent association signals identified through forward stepwise conditional analyses. The second bar for “Trans-ancestry”, “European” and “East Asian” also shows the number of regions with 1, 2, or 3+ signals per region. The green color indicates the number of independent association signals previously reported or not yet reported. The blue color indicates the number of independent associaiton signals in each risk region.
In total, we identified 238 independent association signals either from trans-ancestry or ancestry-specific analysis at these 142 regions (Fig. 2). A total of 94 regions (66.2%) contained only a single association signal, while the remaining 48 regions (33.8%) consisted of multiple independent association signals. Among the 238 independent association signals, 191 signals had lead variants that were correlated with previously GWAS-reported risk variants7 (LD r2 > 0.1 in either of East Asian or European-ancestry population). The remaining 47 independent signals (19.7%) have not been previously reported, including 18 from trans-ancestry, 28 from European-specific, and one from East Asian-specific analyses (Fig. 2, Table 1). Among these 47 signals, 31 demonstrated significant associations with conditional P < 1 × 10−7, including 28 signals reached genome-wide significance.
Table 1.
Independent association signals uncovered at known CRC risk loci in conditional analyses (conditional P < 1 × 10−6)
| Fine-mapping region | SNP | Chr | Position | Nearby gene | Alleles | AF | Single-SNP analysis | Joint analysis | Group | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P valuea | OR (95% CI) | P valueb | ||||||||
| region_1 | rs11579545 | 1 | 22249333 | HSPG2 | T/C | 0.445 | 0.96 (0.95–0.98) | 4.34E−07 | 0.96 (0.95–0.98) | 5.63E−07 | Trans-ancestry |
| region_1 | rs112191583 | 1 | 22554378 | MIR4418 | T/C | 0.974 | 0.88 (0.83–0.92) | 1.19E−07 | 0.87 (0.83–0.92) | 5.29E−08 | Trans-ancestry |
| region_1 | rs12137525 | 1 | 22584118 | MIR4418 | T/C | 0.107 | 1.07 (1.04–1.09) | 2.90E−08 | 1.08 (1.06–1.11) | 1.14E−11 | European |
| region_9 | rs12122827 | 1 | 202172769 | LGR6 | T/G | 0.715 | 1.04 (1.02–1.06) | 9.44E−07 | 1.05 (1.03–1.06) | 7.94E−08 | European |
| region_22 | rs2554878 | 3 | 41200064 | RP11-372H2.1 | T/G | 0.036 | 1.12 (1.08–1.16) | 5.85E−09 | 1.12 (1.08–1.16) | 3.75E−09 | Trans-ancestry |
| region_27 | rs9283588 | 3 | 133874566 | RYK | A/G | 0.715 | 1.06 (1.04–1.07) | 3.43E−10 | 1.04 (1.03–1.06) | 7.32E−07 | Trans-ancestry |
| region_30 | rs902443 | 4 | 105888417 | RP11-556I14.1 | A/T | 0.536 | 1.04 (1.03–1.06) | 1.49E−11 | 1.04 (1.03–1.06) | 1.26E−11 | Trans-ancestry |
| region_36 | rs582489 | 5 | 39908712 | GCSHP1 | T/C | 0.570 | 0.97 (0.96–0.99) | 8.23E−05 | 0.96 (0.94–0.97) | 7.29E−09 | European |
| region_36 | rs77781678 | 5 | 40626064 | SNORA63 | T/C | 0.020 | 0.84 (0.79–0.89) | 2.09E−09 | 0.84 (0.79–0.89) | 1.75E−09 | European |
| region_43 | rs4714081 | 6 | 11977905 | RP11-456H18.1 | A/G | 0.451 | 0.96 (0.95–0.97) | 2.50E−09 | 0.96 (0.95–0.97) | 1.21E−09 | Trans-ancestry |
| region_43 | rs4714350 | 6 | 12270290 | EDN1 | A/T | 0.283 | 0.96 (0.94–0.97) | 8.60E−09 | 0.96 (0.95–0.98) | 4.43E−07 | Trans-ancestry |
| region_43 | rs17615624 | 6 | 12376025 | RN7SKP293 | C/G | 0.975 | 0.87 (0.83–0.91) | 7.29E−09 | 0.88 (0.84–0.92) | 2.28E−07 | European |
| region_44 | rs3094576 | 6 | 29516242 | OR2I1P | A/C | 0.131 | 0.94 (0.92–0.96) | 1.83E−07 | 0.94 (0.92–0.96) | 2.26E−08 | European |
| region_44 | rs2517671 | 6 | 29937977 | MICD | A/G | 0.591 | 0.96 (0.95–0.98) | 2.35E−08 | 0.96 (0.95–0.97) | 2.87E−09 | Trans-ancestry |
| region_45 | rs6920820 | 6 | 30969938 | MUC22 | C/G | 0.980 | 0.84 (0.79–0.9) | 6.87E−08 | 0.8 (0.75–0.85) | 1.89E−12 | European |
| region_45 | rs9264180 | 6 | 31219902 | HLA-C | A/C | 0.570 | 1.03 (1.02–1.05) | 1.71E−06 | 1.04 (1.02–1.05) | 5.62E−07 | Trans-ancestry |
| region_45 | rs9265501 | 6 | 31297568 | XXbac-BPG248L24.10 | A/G | 0.678 | 0.88 (0.85–0.92) | 3.05E−10 | 0.88 (0.84–0.91) | 5.21E−11 | European |
| region_45 | rs116000952 | 6 | 32541270 | HLA-DRB1 | T/G | 0.843 | 0.92 (0.89–0.96) | 5.74E−06 | 0.9 (0.87–0.94) | 1.50E−08 | European |
| region_45 | rs2858331 | 6 | 32681277 | XXbac-BPG254F23.7 | A/G | 0.601 | 1.03 (1.02–1.05) | 1.18E−05 | 1.05 (1.04–1.07) | 2.67E−13 | Trans-ancestry |
| region_50 | rs13204733 | 6 | 55566108 | RP11-228O6.2 | A/G | 0.858 | 0.94 (0.92–0.96) | 4.20E−08 | 0.93 (0.91–0.95) | 1.17E−12 | European |
| region_60 | rs10089517 | 8 | 60178721 | SNORA51 | A/C | 0.380 | 1.03 (1.02–1.05) | 7.44E−07 | 1.03 (1.02–1.05) | 2.81E−07 | Trans-ancestry |
| region_61 | rs117310502 | 8 | 117593052 | EIF3H | A/G | 0.048 | 0.92 (0.89–0.96) | 9.36E−05 | 0.88 (0.85–0.92) | 4.03E−10 | European |
| region_61 | rs72681666 | 8 | 117641754 | EIF3H | T/C | 0.043 | 1.09 (1.05–1.13) | 1.57E−05 | 1.12 (1.08–1.17) | 6.99E−10 | European |
| region_61 | rs1793717 | 8 | 118278575 | SNORA31 | A/C | 0.629 | 1.03 (1.02–1.05) | 6.90E−05 | 1.04 (1.03–1.06) | 1.55E−07 | European |
| region_62 | rs79122086 | 8 | 128397907 | CASC8 | T/G | 0.840 | 0.92 (0.9–0.93) | 5.46E−20 | 0.94 (0.93–0.96) | 9.34E−10 | Trans-ancestry |
| region_62 | rs77569096 | 8 | 128468955 | CASC8 | A/G | 0.763 | 0.92 (0.9–0.94) | 2.06E−15 | 0.93 (0.91–0.95) | 2.67E−12 | European |
| region_68 | rs4994332 | 9 | 137117194 | RP11-145E17.2 | T/C | 0.423 | 0.97 (0.96–0.98) | 4.05E−05 | 0.96 (0.95–0.97) | 9.08E−08 | European |
| region_74 | rs117746067 | 10 | 101222300 | RP11-441O15.3 | A/G | 0.101 | 1.06 (1.03–1.08) | 3.64E−06 | 1.08 (1.05–1.1) | 1.74E−09 | European |
| region_80 | rs9795065 | 11 | 74376844 | POLD3 | T/C | 0.981 | 1.19 (1.13–1.25) | 5.37E−13 | 1.17 (1.12–1.23) | 6.06E−11 | Trans-ancestry |
| region_85 | rs1003563 | 12 | 6424577 | PLEKHG6 | A/G | 0.433 | 0.95 (0.94–0.97) | 1.67E−12 | 0.95 (0.94–0.96) | 1.23E−14 | Trans-ancestry |
| region_106 | rs68097734 | 14 | 92717447 | RP11-472N19.3 | T/C | 0.496 | 1.06 (1.03–1.09) | 7.71E−06 | NA | − | Asian |
| region_108 | rs28630996 | 15 | 32993860 | SCG5 | A/T | 0.713 | 0.9 (0.89–0.92) | 1.25E−32 | 0.93 (0.91–0.94) | 3.02E−17 | Trans-ancestry |
| region_108 | rs144674978 | 15 | 33149751 | FMN1 | T/C | 0.013 | 1.34 (1.25–1.43) | 1.11E−18 | 1.23 (1.15–1.31) | 3.82E−10 | European |
| region_109 | rs3784710 | 15 | 68072458 | MAP2K5 | T/C | 0.763 | 1.05 (1.03–1.07) | 1.32E−07 | 1.05 (1.03–1.07) | 1.34E−08 | European |
| region_111 | rs12913420 | 15 | 90797010 | RP11-697E2.6 | C/G | 0.376 | 1.04 (1.03–1.06) | 2.29E−09 | NA | − | Trans-ancestry |
| region_114 | rs11117455 | 16 | 86179919 | RP11-805I24.4 | T/C | 0.181 | 1.04 (1.02–1.06) | 7.52E−06 | 1.05 (1.03–1.07) | 6.97E−07 | European |
| region_115 | rs73975588 | 17 | 816741 | NXN | A/C | 0.874 | 1.09 (1.07–1.12) | 6.62E−16 | 1.07 (1.04–1.09) | 6.08E−09 | European |
| region_117 | rs112592783 | 17 | 70633625 | LINC00511 | T/C | 0.175 | 1.05 (1.03–1.07) | 5.87E−09 | 1.05 (1.03–1.07) | 5.95E−09 | Trans-ancestry |
| region_120 | rs4939821 | 18 | 46371993 | CTIF | T/C | 0.304 | 0.91 (0.89–0.92) | 4.12E−32 | 0.96 (0.94–0.97) | 1.89E−07 | European |
| region_121 | rs72971616 | 19 | 987366 | WDR18 | T/G | 0.063 | 0.92 (0.89–0.95) | 4.89E−06 | NA | − | European |
| region_126 | rs12460535 | 19 | 49098750 | SULT2B1 | A/G | 0.349 | 0.96 (0.95–0.98) | 5.76E−07 | NA | − | European |
| region_127 | rs8099852 | 19 | 58895221 | RPS5 | T/C | 0.546 | 1.03 (1.02–1.05) | 5.15E−07 | NA | − | Trans-ancestry |
| region_133 | rs1971480 | 20 | 48897080 | RP11-290F20.3 | T/G | 0.672 | 0.96 (0.95–0.98) | 8.61E−07 | 0.96 (0.94–0.97) | 6.50E−09 | European |
| region_133 | rs149942633 | 20 | 48983073 | RP11-290F20.2 | T/C | 0.153 | 1.12 (1.08–1.16) | 1.93E−08 | 1.1 (1.05–1.14) | 3.94E−06 | European |
| region_133 | rs6126008 | 20 | 49075315 | COX6CP2 | A/T | 0.660 | 0.96 (0.94–0.97) | 1.07E−08 | 0.96 (0.94–0.97) | 3.74E−09 | European |
| region_134 | rs34161672 | 20 | 56020599 | RBM38 | A/G | 0.321 | 1.04 (1.02–1.05) | 2.40E−06 | 1.04 (1.03–1.06) | 1.64E−07 | European |
| region_142 | rs78106213 | 22 | 46121230 | ATXN10 | T/G | 0.693 | 1.04 (1.03–1.06) | 2.41E−07 | 1.05 (1.03–1.06) | 8.20E−09 | European |
All independent association signals presented in this table are those not previously reported.
Chr and Position GRCh37, Alleles risk allele/Reference allele, AF Allele frequency, OR odds ratio, CI confidence interval.
aP value derived from trans-ancestry or ancestry-specific meta-analysis under the fixed-effects inverse variance weighted model.
bP value derived from conditional analysis conditioning on all other independent association signals in each fine-mapping region. “NA”—Only a single association signal was detected in the fine-mapping region in the analysis group.
Identification of credible causal variants (CCVs) for independent association signals
To identify CCVs for each independent association signal, we conducted conditional analysis with adjustment of the lead variants for other signals in the same risk region. We conducted this analysis for trans-ancestral independent signals separately for each population to account for differences in the LD structure and then meta-analyzed conditioned results. Using a similar approach conducted in breast cancer12, we defined variants as CCVs if they satisfied conditional P values within two orders of magnitude of the most significant association, conditioning on all other independent association signals. We identified a total of 5741 CCVs for the 238 signals, with the number of CCVs per signal ranging from 1 to 249 (median: 11 CCVs per signal) (Supplementary Data 6). For 28 risk signals, only a single CCV was identified, suggesting that these CCVs are likely to be the causal variants for these signals (Table 2).
Table 2.
Independent association signals with a single CCV
| Fine-mapping region | SNP | Chr | Position | Alleles | AF | OR (95% CI) | P valuea | Putative target gene(s)b |
|---|---|---|---|---|---|---|---|---|
| European-specific analysis | ||||||||
| region_45 | rs116000952 | 6 | 32541270 | T/G | 0.843 | 0.92 (0.89–0.96) | 5.74E−06 | – |
| region_45 | rs6920820 | 6 | 30969938 | C/G | 0.980 | 0.84 (0.79–0.90) | 6.87E−08 | LINC00243 |
| region_61 | rs72681666 | 8 | 117641754 | T/C | 0.043 | 1.09 (1.05–1.13) | 1.57E−05 | – |
| region_62 | rs77569096 | 8 | 128468955 | A/G | 0.763 | 0.92 (0.90–0.94) | 2.06E−15 | – |
| region_84 | rs3217810 | 12 | 4388271 | T/C | 0.127 | 1.13 (1.11–1.16) | 1.96E−26 | – |
| region_108 | rs144674978 | 15 | 33149751 | T/C | 0.013 | 1.34 (1.25–1.43) | 1.11E−18 | – |
| region_133 | rs149942633 | 20 | 48983073 | T/C | 0.153 | 1.12 (1.08–1.16) | 1.93E−08 | – |
| Trans-ancestry analysis | ||||||||
| region_1 | rs112191583 | 1 | 22554378 | T/C | 0.974 | 0.88 (0.83–0.92) | 1.19E−07 | – |
| region_24 | rs704417 | 3 | 64252424 | T/C | 0.546 | 1.05 (1.03–1.06) | 4.35E−10 | – |
| region_27 | rs113569514 | 3 | 133748789 | T/C | 0.763 | 1.08 (1.07–1.10) | 1.92E−21 | SLCO2A1 |
| region_29 | rs2578155 | 4 | 94836291 | C/G | 0.503 | 1.04 (1.03–1.06) | 1.09E−09 | – |
| region_42 | rs9379084 | 6 | 7231843 | A/G | 0.144 | 0.93 (0.91–0.95) | 2.39E−12 | RREB1 |
| region_46 | rs16878812 | 6 | 35569562 | A/G | 0.892 | 1.09 (1.07–1.12) | 7.62E−15 | FKBP5 |
| region_48 | rs6933790 | 6 | 41672769 | T/C | 0.788 | 1.08 (1.06–1.10) | 2.66E−20 | – |
| region_61 | rs4129064 | 8 | 117735666 | T/G | 0.734 | 1.06 (1.04–1.07) | 1.01E−09 | – |
| region_62 | rs6983267 | 8 | 128413305 | T/G | 0.508 | 0.86 (0.85–0.87) | 1.65E−122 | MYC |
| region_72 | rs704017 | 10 | 80819132 | A/G | 0.473 | 0.92 (0.91–0.93) | 1.97E−38 | – |
| region_84 | rs12818766 | 12 | 4376091 | A/G | 0.215 | 1.10 (1.08–1.12) | 1.81E−29 | – |
| region_89 | rs7398375 | 12 | 57540848 | C/G | 0.651 | 1.07 (1.05–1.09) | 3.70E−19 | LRP1 |
| region_94 | rs11067228 | 12 | 115094260 | A/G | 0.560 | 0.95 (0.94–0.97) | 2.50E−13 | – |
| region_96 | rs116964464 | 13 | 27543193 | T/C | 0.035 | 1.11 (1.07–1.15) | 4.83E−09 | USP12 |
| region_99 | rs7325844 | 13 | 73625133 | A/G | 0.639 | 1.05 (1.04–1.07) | 1.28E−12 | – |
| region_104 | rs35107139 | 14 | 54419106 | A/C | 0.550 | 0.92 (0.91–0.93) | 4.22E−36 | – |
| region_105 | rs8020436 | 14 | 59208437 | A/G | 0.370 | 1.06 (1.05–1.08) | 1.27E−17 | – |
| region_108 | rs17816465 | 15 | 33156386 | A/G | 0.193 | 1.09 (1.07–1.10) | 5.73E−20 | – |
| region_116 | rs1078643 | 17 | 10707241 | A/G | 0.765 | 1.09 (1.07–1.11) | 2.31E−27 | – |
| region_132 | rs6066825 | 20 | 47340117 | A/G | 0.662 | 1.08 (1.07–1.10) | 2.13E−32 | – |
| region_136 | rs1741640 | 20 | 60932414 | T/C | 0.208 | 0.88 (0.86–0.89) | 8.15E−55 | LAMA5, CABLES2 |
Chr and Position GRCh37, Alleles risk allele/Reference allele, AF Allele frequency, OR odds ratio, CI confidence interval. aP value derived from trans-ancestry or European-ancestry meta-analysis under the fixed-effects inverse variance weighted model; b”-“ – No target genes were prioritized for the variant in this study.
For the 138 independent association signals identified in both trans-ancestry and European-ancestry specific analyses (Supplementary Data 7), trans-ancestry analyses identified a smaller-sized set of CCVs (mean = 23.2, median = 8.5), compared with European-ancestry specific analysis (mean = 31.08, median = 15) (paired Wilcoxon test, P = 4.9 × 10−7). Interestingly, a single CCV was identified for 10 signals in trans-ancestry analysis, while multiple CCV for them in European-ancestry specific analysis, highlighting the value of using multi-ancestry data to reduce the number of CCVs in fine-mapping analysis. For instance, signal 1 in region_42 included 16 CCVs in the European set (lead variant: rs41302867), but only one variant in the trans-ancestry set (rs9379084). The variant rs9379084 is a predicted-deleterious missense variant (p.Asp1171Asn) of the RREB1 gene which plays a regulatory role in Ras/Raf-mediated cell differentiation13, a pathway well known to be implicated in CRC development.
Identification of target genes for CCVs
Of the 5741 CCVs identified in this study, 3716 (64.7%) are located in regions with at least one of six genomic features (open chromatin, transcribed regions of active genes, promoter, enhancer, repressed gene regulatory elements, and transcription factor (TF) binding sites) (Supplementary Data 6 and 8). To identify putative target genes of these CCVs, we used functional genomic data generated in CRC-related tissues/cells to conduct in-silico analyses with a modified INQUISIT pipeline12 (Methods, Supplementary Data 9). We identified 72 putative target genes via CCVs located in distal enhancer elements (Supplementary Data 10), 48 genes via CCVs located in proximal promoter elements (Supplementary Data 11), and 19 genes that could be targeted by CCVs in coding regions (i.e., deleterious missense, stop_gained, and start_lost) (Supplementary Data 12). In total, we identified 128 genes associated with CCVs for 76 independent association signals, with a range from one to five putative target genes per signal. Of them, 52 independent association signals contain only a single putative target gene.
We also conducted cis-expression quantitative trait loci (cis-eQTL) analyses to identify target genes using four transcriptome datasets derived from either normal colon tissues or tumor-adjacent normal colon tissues from 1299 individuals from the Genotype-Tissue Expression (GTEx) project (n = 368 individuals predominantly of European ancestry), the BarcUVa-Seq project (n = 423 individuals of European ancestry), the Colonomics project (n = 144 individuals of European ancestry), and the Asia Colorectal Cancer Consortium (ACCC) (n = 364 individuals of East Asian ancestry) (Methods). At Bonferroni-corrected P < 0.05, we identified 153 genes associated with the lead variants, including 127 genes in 65 independent association signals and 30 in 15 signals identified from trans-ancestry and European-ancestry specific analyses, respectively. We also identified the PPP1R21 gene in a potential Asian-specific risk signal (lead variant rs77272589) (Supplementary Data 13). Out of the 153 genes, 37 had been previously identified by eQTL analysis5,10,11. For independent association signals identified in European and trans-ancestry analyses, we further performed cis-methylation quantitative trait loci (cis-mQTL) analyses using two methylation datasets generated from 321 individuals from the GTEx project (n = 189 individuals predominantly of European ancestry) and the Colonomics project (n = 132 individuals of European ancestry). We found that DNA methylation levels at CpG sites for 84 genes were associated with 71 independent association signals, including 14 genes identified in previous mQTL analysis11 (Supplementary Data 14).
We next conducted colocalization analyses for identified likely target genes in significant eQTL/mQTLs above using the Summary-data-based Mendelian Randomization (SMR) approach (Methods). Through the integration of eQTL/mQTL results and GWAS associations signals, we identified 205 genes at Bonferroni-corrected PSMR < 0.05 (Supplementary Data 15–19), including 150 genes from the eQTL analysis and 84 genes from the mQTL analysis. Of these, 45 (21.9%) genes were also identified as targets of CCVs by in-silico analyses based on functional genomic data as described above, and 29 genes were identified in both mQTL and eQTL analyses. That is in line with previous observations in the overlap fraction between mQTL and eQTL14. We considered genes with evidence of only mQTL colocalization, as the enrichment of mQTLs in gene regulatory elements, as well as their implications in other molecular phenotypes, such as chromatin accessibility14,15. Notably, of the 55 genes only identified in the mQTL analysis, seven genes were supported by the above in silico analyses with functional genomic data, and 22 genes showed association with CRC risk in previous TWAS and eQTL colocalization analysis7,11,16,17.
In total, we identified 288 putative target genes for 140 independent association signals based on functional genomics data and/or colocalization analysis. For 35 of these signals, multiple target gene candidates were detected per signal, suggesting that some may be false positives (Supplementary Data 20). To minimize false positive findings, we further prioritized target gene candidates by analyzing associations of genes with CRC risk based on previous transcriptome-wide association studies (TWAS) and colocalizations between eQTL and CRC GWAS signals7,11,16,17 (Methods). Finally, we obtained a credible set of 136 protein-coding genes for 124 independent association signals. Among them, 56 genes were not previously identified as potential targets for CRC risk associations, including nine genes in eight previously unreported association signals in this study (Table 3). The remaining 80 genes were previously reported as potential CRC susceptibility genes, and our study provided additional supporting evidence (Table 4)7,11,16,17.
Table 3.
The 56 CRC susceptibility gene candidates not previously reported
| Fine-mapping region | Gene | Lead variant | Distal | Proximal | Coding | Colocalization (eQTL) | Colocalization (mQTL) |
|---|---|---|---|---|---|---|---|
| region_1 | CELA3B | rs11579545 | + | ||||
| region_1 | HSPG2 | rs11579545 | + | + | + | ||
| region_5 | PTGER3 | rs2651244 | + | ||||
| region_7 | TNFSF18 | rs10489274 | + | ||||
| region_9 | LGR6 | rs12122827 | + | ||||
| region_10 | CNTN2 | rs12078075 | + | + | |||
| region_12 | FMN2 | rs2078095 | + | ||||
| region_14 | PPP1R21 | rs77272589 | + | + | |||
| region_16 | LCT | rs1446585 | + | ||||
| region_21 | GOLGA4 | rs1800734 | + | ||||
| region_21 | MLH1 | rs1800734 | + | + | |||
| region_24 | ADAMTS9 | rs6445418 | + | ||||
| region_24 | PRICKLE2 | rs704417 | + | ||||
| region_27 | SLCO2A1 | rs113569514 | + | ||||
| region_28 | LRRC34 | rs10936599 | + | ||||
| region_28 | ACTRT3 | rs10936599 | + | + | |||
| region_28 | MYNN | rs10936599 | + | + | |||
| region_34 | HPGD | rs1426947 | + | ||||
| region_42 | LY86 | rs1294438 | + | ||||
| region_44 | OR2I1P | rs73402748 | + | ||||
| region_46 | SRPK1 | rs16878812 | + | ||||
| region_49 | RUNX2 | rs57939401 | + | ||||
| region_55 | IGFBP3 | rs80077929 | + | ||||
| region_62 | MYC | rs4733655, rs6983267 | + | ||||
| region_63 | CDKN2B | rs7859362 | + | ||||
| region_63 | MTAP | rs7859362 | + | ||||
| region_68 | VAV2 | rs7038489 | + | ||||
| region_73 | KIF20B | rs140356782 | + | ||||
| region_73 | PANK1 | rs140356782 | + | + | |||
| region_74 | GOT1 | rs117746067 | + | ||||
| region_75 | BORCS7 | rs12268849 | + | ||||
| region_75 | AS3MT | rs12268849 | + | + | |||
| region_79 | ANO1 | rs10751097 | + | ||||
| region_92 | NTN4 | rs11108175 | + | ||||
| region_93 | CUX2 | rs3858704 | + | ||||
| region_94 | TBX3 | rs7300312, rs11067228 | + | + | |||
| region_96 | USP12 | rs116964464 | + | ||||
| region_101 | IRS2 | rs1078563 | + | ||||
| region_101 | COL4A2 | rs4773184 | + | ||||
| region_107 | BCL11B | rs80158569 | + | ||||
| region_108 | GOLGA8N | rs56338436 | + | ||||
| region_110 | SENP8 | rs8031386 | + | + | |||
| region_111 | CIB1 | rs12913420 | + | + | |||
| region_111 | ZNF774 | rs7179095 | + | ||||
| region_119 | MYL12A | rs1612128 | + | ||||
| region_119 | MYL12B | rs1612128 | + | ||||
| region_119 | TGIF1 | rs1612128 | + | ||||
| region_125 | B3GNT8 | rs1963413 | + | ||||
| region_133 | CEBPB | rs1971480 | + | ||||
| region_134 | RBM38 | rs34161672 | + | ||||
| region_134 | BMP7 | rs6014965 | + | + | |||
| region_138 | LSS | rs9983528 | + | + | |||
| region_138 | PCNT | rs9983528 | + | + | |||
| region_138 | SPATC1L | rs9983528 | + | + | |||
| region_142 | WNT7B | rs62228060 | + | ||||
| region_142 | ATXN10 | rs78106213 | + |
The lead variant for each gene is presented by independent association signals. Supporting evidence for the likely target gene is presented as follows: “Distal”—the CCV(s) located in distal enhancer elements of the gene; “Proxmial”—the CCV(s) located in proximal promoter element of the gene; “Coding”—the CCV is potential loss-of-function variants of the gene; “Colocalization (eQTL)”—target genes identified from eQTL colocalization analysis; “Colocalization (mQTL)”—target genes identified from mQTL colocalization analysis. “+” indicates the presense of supportive evidence.
Table 4.
The 80 previously reported CRC susceptibility genes supported in this study
| Fine-mapping region | Gene | Lead variant | Distal | Proximal | Coding | Colocalization (eQTL) | Colocalization (mQTL) |
|---|---|---|---|---|---|---|---|
| region_1 | WNT4 | rs6426749 | + | ||||
| region_2 | FHL3 | rs61776719 | + | + | |||
| region_8 | LAMC1 | rs8179460 | + | + | |||
| region_9 | LMOD1 | rs12137232 | + | + | |||
| region_15 | ACTR1B | rs11692435 | + | + | |||
| region_18 | STK39 | rs4668039 | + | + | + | ||
| region_20 | TMBIM1 | rs3731861 | + | + | + | + | |
| region_23 | SFMBT1 | rs2001732, rs2581817 | + | + | |||
| region_26 | BOC | rs73235124 | + | ||||
| region_30 | TET2 | rs2047409, rs902443 | + | + | |||
| region_31 | UGT8 | rs3924508 | + | ||||
| region_35 | TERT | rs2735940 | + | ||||
| region_40 | CDX1 | rs2302275 | + | ||||
| region_41 | ERGIC1 | rs472959 | + | ||||
| region_42 | RREB1 | rs9379084 | + | ||||
| region_43 | EDN1 | rs2070699 | + | ||||
| region_43 | HIVEP1 | rs4714081 | + | + | |||
| region_47 | CDKN1A | rs9470361 | + | ||||
| region_48 | TFEB | rs6933790 | + | ||||
| region_52 | DCBLD1 | rs6911915 | + | ||||
| region_53 | TCF21 | rs151127921 | + | ||||
| region_54 | GNA12 | rs1182197 | + | + | + | ||
| region_55 | TBRG4 | rs67681615 | + | ||||
| region_55 | TNS3 | rs6948177 | + | ||||
| region_56 | ABHD11 | rs7806956 | + | + | |||
| region_57 | TRIM4 | rs2527927 | + | + | |||
| region_62 | POU5F1B | rs6983267 | + | ||||
| region_64 | DCAF12 | rs11557154 | + | ||||
| region_68 | BRD3 | rs11789898 | + | + | |||
| region_70 | BAMBI | rs1773860 | + | ||||
| region_71 | ASAH2B | rs10740013 | + | ||||
| region_72 | ZMIZ1 | rs704017 | + | ||||
| region_74 | ENTPD7 | rs35564340 | + | ||||
| region_76 | TCF7L2 | rs4554812 | + | ||||
| region_78 | TMEM258 | rs174570 | + | ||||
| region_81 | TRPC6 | rs2186607 | + | ||||
| region_82 | ARHGAP20 | rs3087967 | + | ||||
| region_82 | FDX1 | rs3087967 | + | ||||
| region_83 | BCL9L | rs497916 | + | ||||
| region_85 | PLEKHG6 | rs10849434, rs1003563 | + | + | + | ||
| region_88 | CERS5 | rs11169572 | + | ||||
| region_88 | ATF1 | rs11169572 | + | + | |||
| region_88 | DIP2B | rs11169572 | + | + | |||
| region_89 | LRP1 | rs7398375 | + | + | + | ||
| region_91 | TSPAN8 | rs11178634 | + | + | |||
| region_98 | SMAD9 | rs12427846 | + | + | + | ||
| region_99 | KLF5 | rs1304959, rs78341008 | + | ||||
| region_102 | NIN | rs1042266 | + | ||||
| region_102 | ABHD12B | rs1042266 | + | + | |||
| region_102 | PYGL | rs1042266 | + | + | |||
| region_103 | NID2 | rs1151580 | + | + | |||
| region_104 | BMP4 | rs1957628, rs35107139 | + | + | |||
| region_105 | DACT1 | rs8020436 | + | + | |||
| region_108 | GREM1 | rs16970016 | + | ||||
| region_109 | SMAD6 | rs3809570 | + | + | |||
| region_109 | SMAD3 | rs56324967 | + | ||||
| region_112 | ZFP90 | rs9924886 | + | ||||
| region_112 | CDH1 | rs9924886 | + | + | + | ||
| region_115 | NXN | rs11247566 | + | ||||
| region_117 | SOX9 | rs112592783 | + | ||||
| region_118 | METRNL | rs35204860 | + | + | |||
| region_120 | SMAD7 | rs4939821, rs2337113 | + | + | |||
| region_122 | FUT3 | rs10409772 | + | + | |||
| region_124 | RHPN2 | rs28840750 | + | + | |||
| region_126 | FUT2 | rs12460535 | + | + | |||
| region_127 | TRIM28 | rs11670192 | + | ||||
| region_127 | ZNF584 | rs8099852, rs11670192 | + | + | |||
| region_128 | BMP2 | rs990999 | + | ||||
| region_130 | MAP1LC3A | rs6059938 | + | ||||
| region_130 | MYH7B | rs6059938 | + | ||||
| region_131 | TOX2 | rs6073241 | + | + | |||
| region_132 | PREX1 | rs6066825 | + | ||||
| region_133 | PARD6B | rs6091213 | + | ||||
| region_133 | PTPN1 | rs6091213 | + | ||||
| region_135 | GNAS | rs8121252 | + | ||||
| region_136 | RBBP8NL | rs1741640 | + | ||||
| region_137 | STMN3 | rs6089763 | + | ||||
| region_139 | ZNRF3 | rs4616575 | + | + | |||
| region_140 | PDGFB | rs130651 | + | ||||
| region_142 | RIBC2 | rs6007600 | + | + |
The lead variant for each gene is presented by independent association signals. Supporting evidence for the likely target gene is presented as follows: “Distal”—the CCV(s) located in distal enhancer elements of the gene; “Proxmial”—the CCV(s) located in proximal promoter element of the gene; “Coding”—the CCV is potential loss-of-function variants of the gene; “Colocalization (eQTL)”—target genes identified from eQTL colocalization analysis; “Colocalization (mQTL)”—target genes identified from mQTL colocalization analysis. “+” indicates the presense of supportive evidence.
Using scRNA-seq data to evaluate gene expression pattern by cell types
To investigate potential underlying cell types of putative susceptibility genes that contribute to CRC development, we analyzed single-cell RNA-seq (scRNA-seq) datasets from normal colon tissues obtained from 31 participants included in the Colorectal Molecular Atlas Project18 (Methods). Of the 136 identified genes, 17 genes exhibited significantly differential expression in specific cell types compared to the other cell types at |log2 fold change (FC)| > 1 and a nominal P < 0.05 (Supplementary Data 21). Nine of these genes (DIP2B, CIB1, HPGD, CDKN2B, TMEM258, MYL12A, MYL12B, CDKN1A, and TMBIM1) showed a distinct expression pattern in specific absorptive cells (ABS) cell, underscoring the relevance of this cell type underlying CRC development.
Using whole exome sequencing data to evaluate pathogenic variants in target genes with CRC risk
We used whole exome sequencing data from 3362 CRC cases and 133,742 controls of European ancestry in the UK Biobank (UKBB) to evaluate the association of CRC risk with putative candidate genes identified our study using burden tests by aggregating either loss of function (pLOF) or pLOF and deleterious missense variants (Dmis) jointly in each gene (Methods). Of these 136 genes, MLH1 was significantly associated with CRC risk with P = 1.35 × 10−7 when considering only pLOF in tests (at Bonferroni-corrected threshold, 0.05/136 testing). Additional nine genes (TNFSF18, LRP1, SMAD9, PDGFB, CIB1, STK39, IGFBP3, FUT2, and FUT3) showed nominal P < 0.05 significance considering only pLoF or combination of pLoF and Dmis, whereas no significance was detected for the remaining genes.
Biological significance of the target genes for CCVs
We utilized Enrichr19–21 to analyze multiple pathway databases and identify enriched biological pathways among the 136 credible target genes (Methods). At a false-discovery rate (FDR) < 0.05, 126 pathways showed significant enrichment (Supplementary Data 22). Our findings were in line with our prior study18 and highlighted the enriched signaling pathways such as TGF-β, BMP, Wnt, Hippo, and TNF-α/NF-κB, which are known to play a crucial role in the development and progression of colorectal cancer19,20. Of the 56 genes not previously reported, nine genes (TGIF1, CDKN2B, MYC, BMP7, WNT7B, PRICKLE2, LGR6, CEBPB, and IRS2) were mapped to these pathways (Table 5). Additionally, we identified several significant pathways, including those related to cancer, pluripotency of stem cells, epithelial–mesenchymal transition, extracellular matrix organization, adipogenesis, senescence, and autophagy in cancer. Interestingly, we also identified the glycolysis pathway, which provides energy support for cancer cells, as a significant pathway not previously reported. Four previously unreported genes, GOT1, IGFBP3, IRS2, and LCT, were mapped to glycolysis, supporting their association with CRC risk.
Table 5.
Significant enrichment in biological pathways
| Pathwaysa | Genesb |
|---|---|
| TGF-beta signaling | BAMBI, BMP2, BMP4, BMP7, CDH1, CDKN2B, GREM1, MYC, RUNX2, SMAD3, SMAD6, SMAD7, SMAD9, TGIF1 |
| Hippo signaling | BMP2, BMP4, BMP7, CDH1, GNAS, MYC, PARD6B, SMAD3, SMAD7, TCF7L2, WNT4, WNT7B |
| TNF-alpha Signaling via NF-kB | TGIF1, BMP2, CDKN1A, EDN1, CEBPB, SMAD3, MYC, IRS2 |
| BMP signaling | BMP2, SMAD6, RUNX2, SMAD9, SMAD7 |
| Pluripotency of stem cells | POU5F1B, BMP4, SMAD3, MYC, WNT7B, SMAD9, TBX3, WNT4, PDGFB, SMAD6, SMAD7, TCF7L2 |
| Epithelial–mesenchymal transition | SMAD3, CDH1, RUNX2, GREM1, COL4A2, LRP1, IGFBP3, LAMC1, NID2, WNT7B, WNT4 |
| Extracellular matrix organization | BMP4, BMP2, COL4A2, PDGFB, NTN4, LAMC1, HSPG2, NID2, BMP7, ADAMTS9 |
| Senescence and Autophagy | BMP2, CDKN1A, CEBPB, SMAD3, MAP1LC3A, IGFBP3, CDKN2B, MYC, KLF5 |
| DNA damage response | TCF7L2, CDKN1A, SMAD3, MYC, WNT7B, WNT4 |
| Cell cycle | CDKN1A, CDKN2B, SMAD3, MYC |
| Focal adhesion | COL4A2, PDGFB, LAMC1, MYL12A, MYL12B, VAV2 |
| Adherens junction | PTPN1, TCF7L2, SMAD3, CDH1 |
| Glycolysis | GOT1, IGFBP3, IRS2, SOX9, PYGL, LCT |
| Proteoglycans in cancer | CDKN1A, MYC, WNT7B, HSPG2, WNT4, VAV2 |
| Androgen Response | HPGD, ZMIZ1, STK39, MYL12A |
| Sphingolipid Metabolism | UGT8, CERS5 |
| Other cancer related pathwaysc | TCF7L2, CDKN1A, EDN1, CDKN2B, SMAD3, WNT7B, PTGER3, PDGFB, LAMC1, MLH1, BMP4, BMP2, COL4A2, TERT, CDH1, MYC, GNA12, GNAS, WNT4, ATF1, CEBPB, BCL11B, HPGD, IGFBP3, RUNX2, ZMIZ1, SMAD6, SMAD7, VAV2, TFEB |
aGenes from the same or similar pathway item in multiple databases were combined.
bGenes identified in this study for each pathway item are highlighted in bold.
cGenes from all general cancer-related pathways (i.e., pathway in cancer, colorectal cancer) identified in multiple databases were combined.
In addition, we performed functional annotation analysis on each credible target gene and assigned them to previously described cellular processes18 (Supplementary Fig. 2). Of the 56 genes not previously reported, 26 were found to be involved in these cellular processes. Specifically, five genes were related to stemness/differentiation, one gene was linked to adhesion/migration, and six genes were associated with proliferation. Interestingly, we also identified an additional cellular process, post-translation modifications (PTMs) of protein, which included three genes (DACF12, USP12, and SENP8). These findings suggest potential critical roles of PTMs in the development of CRC.
Discussion
Our study, including approximately 254,000 individuals of East Asian and European ancestry, represents the largest study conducted to fine-map CRC risk-associated genomic regions using GWAS data. We identified 238 independent association signals at conditional P value < 1 × 10−6, including 47 signals not reported previously. Furthermore, integrating functional genomic data and results from cis-eQTL/mQTL and colocalization analyses, we identified 136 putative CRC susceptibility genes, including 56 genes that had not been previously reported. Notably, these identified genes are significantly enriched in several major CRC signaling pathways and other cancer-related pathways. Our findings not only significantly expanded the number of associated signals for CRC, but also provide substantial data to advance our understanding of CRC biology.
The integration of comprehensive functional genomic data from relevant colon tissues and cell lines, as well as genetic associations data, can facilitate the identification of potential target genes for CRC risk. Our study significantly extends previous efforts7,11,16,17 by identifying 56 target gene candidates not previously reported for CRC risk, over half of which (29/56, 51.8%) are involved in the enriched biological pathways. For instance, eight target genes (TGIF1, CDKN2B, LGR6, MYC, PRICKLE2, WNT7B, BMP7, and TBX3) identified in this study may regulate normal intestinal homeostasis as they play roles in signaling pathways (i.e., Wnt and BMP) and pluripotency of stem cells. LGR6, for instance, is part of a G-protein-coupled receptor family and marks stem cells in the epidermis22. It activates a novel β-catenin/TCF7L2/LGR6-positive feedback loop in LGR6high cervical cancer stem cells (CSCs) to enhance the properties of cancer stem cells, including self-renewal, differentiation, and tumorigenicity23. Silencing of LGR6 resulted in the inhibition of stemness by repressing Wnt/β-catenin signaling in ovarian cancer24. TBX3, a transcriptional repressor, regulates stem cell maintenance by controlling stem cell self-renewal and differentiation, and reduced expression levels of TBX3 are associated with reduced pluripotency of stem cells25,26. MYC and WNT7B are implicated in the signaling related to the self-renewal and differentiation of cancer stem cells27. Here, we linked MYC and WNT7B with credible causal variants of CRC risk associations through functional genomic interaction. Our findings also indicated the relevance of glycolysis to CRC risk associations, a metabolic pathway critical in early CRC tumorigenesis by supporting the energetic and biosynthetic demands of CRC cells28,29. It should be noted that future studies are needed to validate chromatin interactions between identified CCVs and their target genes in this study by employing chromatin conformation capture technology such as in situ Hi-C, Capture Hi-C (CHi-C), and HiChIP.
Additional evidence supports some of the candidate target genes identified in our study as possible CRC susceptibility genes. In our differential gene expression analysis among normal colon mucosa, adenoma, and adenocarcinoma using gene expression data from 135 normal colon mucosas, 218 colon adenomas, and 2760 colon adenocarcinomas, we observed that 26 genes showed significant differential expression between adenoma and normal colon tissues, while 31 genes showed significant differential expression between carcinoma and adenoma tissues (adjusted P < 0.05) (Supplementary Data 20). Interestingly, three stemness/differentiation-related genes, including LRRC34, CEBPB, and TBX3, showed significant changes in their expression levels in adenoma compared to normal colon mucosa. Additionally, 34 (60.7%) of not previously identified genes have been implicated in cancer-related functions in in vitro or in vivo functional experimental studies in CRC or other cancer types (Supplementary Data 20). These results provide further evidence supporting the potential involvement of these genes in CRC progression. Despite the above supportive evidence, it remains necessary to evaluate the functions of identified putative CRC susceptibility genes through both in vitro and in vivo assays in future investigations.
The trans-ancestry and ancestry-specific fine-mapping analyses conducted in this study not only enabled the discovery of independent association signals that are shared across populations of European and East Asian ancestry, but also revealed ancestry-specific signals. The larger sample size of the European-ancestry study enabled us to identify a larger number of independent association signals than the study conducted on Asians. However, there are some ancestry-specific signals identified in this study, which is most likely due to differences in LD structures and allele frequency between these two populations. Indeed, we observed distinct differences in the allele frequency for most ancestry-specific signals, as shown in Supplementary Data 4 and 5. For instance, the lead variant of 24 European ancestry-specific signals (40%, 24/60) is not detected among East Asian-ancestry populations. On the other hand, fine-mapping analyses capitalizing on ancestry differences in LD structure can substantially reduce the credible set size compared to European-ancestry specific analysis. This highlights the value of multi-ancestry fine-mapping over single-ancestry analysis. Our analysis is limited to two ancestry groups. Further studies should increase the diversity of genetic data, including those from other racial groups.
In summary, our large trans-ancestry fine-mapping analysis has identified large numbers of not previously reported independent association signals for CRC risk and refined the majority of the previously reported association signals. By leveraging data from two ancestries, we further defined putative causal variants underlying CRC risk signals. Our study has also uncovered a credible set of target genes. These findings offer a significant advancement in our understanding of the genetic and biological processes underlying CRC and provide a roadmap for further investigation of variants and genes identified in our study.
Methods
GWAS data and meta-analysis
The GWAS data used in this study comprised 100,204 CRC cases and 154,587 controls (Supplementary Data 1), which were grouped into 31 GWAS analytical units based on the study or genotyping platform as consistent with the original reports. Of them, 17 datasets were derived from populations of European descent and 14 were from populations of Asian descent. These 31 GWAS datasets were meta-analyzed under the fixed-effects inverse variance weighted model implemented in METAL30. Further details regarding each analytical unit and meta-analysis were described in Supplementary Note.
Identifying independent association signals
A total of 205 independent genetic associations have been reported for CRC risk by GWAS7. To define fine-mapping regions for CRC, we aggregated these risk variants using bedtools. Specifically, we identified 1 megabase (Mb) intervals centered on the risk variants, and if there were regions of overlap, we combined them into a single interval over 1 Mb. In total, we determined 143 fine-mapping regions, including 142 on autosomes and one on chromosome X (Supplementary Data 2). Our fine-mapping analysis and downstream analyses focused on the 142 genomic risk regions on autosomes.
To identify distinct association signals within each risk region, we conducted a forward stepwise conditional analysis for summary statistics from the trans-ancestral meta-analysis, using GCTA-COJO31,32. We included common variants (MAF > 0.01) with associations at P < 0.05 in both populations. To account for differences in the LD structure, we conducted conditional analysis in each population for each fine-mapping region, conditioning on the most significant association from the trans-ancestral summary statistics. We then meta-analyzed the conditioned results using the fixed-effects inverse variance weighted model with METAL. To identify potential ancestry-specific independent signals, we also performed conditional analysis in each population, conditioning on the ancestry-specific most significant association. Common variants (MAF > 0.01) with association at P < 1 × 10−4 in each population were included. For LD estimation, we used genotyping data from 6684 unrelated samples of Asian descent33, and 503 European samples in the 1000 Genome project as the reference.
Following a previous study conducted for breast cancer12, we applied the conditional P value < 1 × 10−6 to define the independent signal. For each region, we first adjusted for the most significant association and then added any additional variant that remained an independent signal at the conditional P value < 1 × 10−6 to the conditional set. We then repeated the conditional analysis until no more variants met the significance threshold. In regions with multiple independent signals, we determined the index variant for each signal through a process of conditional analysis, adjusting for the index variants of the other signals. This process was repeated until the set of index variants were stabilized. The variant with the strongest residual association was defined as the index for the signal.
For independent association signals identified in ancestry-specific analyses, we compared them with those from trans-ancestry analyses by assessing correlations between their lead variants within each risk region. If a signal was consistently found in both ancestry-specific and trans-ancestry analyses (i.e., the same lead variant or correlated lead variants with LD r2 > 0.1 in each corresponding population), we considered it as a sharing signal between Asian and European-ancestry populations. Otherwise, they were defined as ancestry-specific signals.
Identifying a set of CCVs of each independent signal
To determine the CCVs of each independent signal, we used the approach described in a previous study for breast cancer12. Specifically, variants that have a conditional P value within two orders of magnitude of the most significant association, conditioning on all other independent association signals, were defined as CCVs.
RNA-seq data analysis
We conducted mRNA sequencing on tumor-adjacent normal colon tissues obtained from 364 East Asians patients with colorectal cancer who participated in the ACCC. Furthermore, we included RNA-seq data from normal colon tissues from 423 individuals of European ancestry who participated in the BarcUVa-Seq project. Included subjects, library preparation and sequencing of colon tissue samples in the ACCC and the BarcUVa-Seq project have been presented in Supplementary Note.
The raw RNA-seq data were processed according to the pipeline of the GTEx Consortium. Sequencing reads were aligned to the reference genome GRCh37 (RNA-seq data from East Asians) or GRCh38 (RNA-seq data from the BarcUVa-Seq project) with STAR (v2.5.4)34. Quality control of aligned samples was performed using RNA-SeQC (v2.3.5)35. Samples that met any of the following criteria were removed: (1) <10 million mapped reads; (2) read mapping rate < 0.2; (3) intergenic mapping rate >0.4; (4) base mismatch rate >0.01 for read mate 1 or >0.02 for read mate 2; and (5) rRNA mapping rate >0.3. If the sample had replicated RNA-seq data, the one with the highest mapped reads was retained.
Gene-level expression quantification was performed using RNA-SeQC based on the GENCODE release 19 annotation (for RNA-seq data from East Asians) and the GENCODE release 26 annotation (for RNA-seq data from the BarcUVa-Seq project)36. The read counts and TPM values of genes were calculated using aligned reads with the following criteria: (1) reads were uniquely mapped; (2) aligned reads were properly paired; (3) the read alignment distance was <6. The genes with expression thresholds of ≥0.1 TPM in ≥20% of samples and ≥6 reads (unnormalized) in ≥20% of samples were selected. Quantile normalization of the gene expression was performed. We further performed rank-based inverse normal transformation for the expressions of genes across samples.
Cis-expression/methylation quantitative loci (cis-eQTL/mQTL) analysis
To identify target genes, we performed cis-eQTL analysis based on a linear regression framework10,11. Gene expression data from four expression datasets comprising a total of 1,299 individuals were used: 1) GTEx project of transverse colon tissues from 368 individuals predominantly of European ancestry, 2) Colonomics project of normal colon tissues or tumor-adjacent normal colon tissues from 144 individuals of European ancestry, 3) BarcUVa-Seq project of normal colon tissues from 423 individuals of European ancestry, and 4) ACCC of tumor-adjacent normal colon tissue from 364 CRC patients of East Asian ancestry. We obtained available cis-eQTL results for CCVs and their nearby genes (within 1 Mb to CCV) from the GTEx database (version 8) and the Colonomics project. Details for gene expression data and eQTL analysis in the Colonomics project are explained elsewhere37. For the analyses using the remaining two datasets, we conducted a linear regression analysis to assess the associations between CCV and the normalized expression levels of nearby genes (within 1 Mb to CCV), adjusting for age, gender, and five top principal components.
We conducted cis-mQTL analysis for CCVs identified in European and trans-ancestry analyses. To do this, we included methylation data obtained from a total of 321 individuals. These datasets consisted of 189 transverse colon tissues predominantly of European ancestry from GTEx, as well as normal colon tissues or tumor-adjacent normal colon tissues of 132 individuals of European ancestry from the Colonomics project. We extracted cis-mQTL results for CCVs and their nearby CpG sites (within 1 Mb to CCV) from the GTEx database (version 8)14. In the Colonomics project, a linear regression analysis was used to evaluate the associations between CCV and the normalized methylation levels of CpG sites (within 1 Mb to CCV), with adjustments of age, gender, and colon sites (right/left). Further details about the cis-mQTL in the Colonomics project can be found in previous studies37,38.
Meta-analysis of cis-eQTL/mQTL results
We performed a meta-analysis to integrate the summary cis-eQTL/mQTL results based on beta and p values from different datasets10,11. In brief, we calculated the z score from function qnorm(p/2)*sign(beta) and further converted the standard z score derived from sum(z*sqrt(N))/sqrt(sum(N)) with a normalized weighted sampled size. Here, beta and p value were derived from eQTL/mQTL results and N referred to the sample size for each dataset. The meta p value was derived from the standard z score. For independent signals detected in both European and Asian populations, the eQTL results from both populations were combined.
We adjusted the combined p-values of eQTL/mQTL results with the Bonferroni procedure. The procedure was conducted for index variants of independent association signals. The Bonferroni-adjusted P < 0.05 was applied to identify potential target genes for each signal.
Colocalization analyses between GWAS association signals and eQTL/mQTL signals
To identify putative target genes, we employed the SMR method to conduct a colocalization analysis39. We integrated GWAS summary statistics of CCVs and their associations with genes from eQTL/mQTL analysis described above. The results of meta-analyses on cis-eQTLs/mQTLs were used. Specifically, we have a statistic:
| 1 |
Here, Zzx and Zzy are the Z statistics for the GWAS summary statistics and the cis-eQTL/mQTL results, respectively. TSMR is the χ2 statistic, which tests the significance of bxy. The significant colocalized signals were determined based on the threshold of the Bonferroni-corrected PSMR < 0.05 within each independent signal.
Functional annotation of CCVs
We investigated whether each potential causal variant was mapped to gene regulatory regions (i.e., promoter or enhancer) (Supplementary Data 8). We obtained 351 chromatin immunoprecipitation sequencing (ChIP-seq) peak files for histone modification marks and transcription factors, and 25 DNase I hypersensitive sites sequencing peak files for chromatin accessibility, generated in normal colorectal epithelium and CRC cell lines from the Cistrome database40,41. Only peaks that met all six quality controls set recommended by Cistrome were analyzed. Additionally, we obtained available ChIP-seq data of histone modification marks from colon tissues, tumor tissues of CRC, and CRC cell lines from Gene Expression Omnibus (GEO), which included 16 from GSE13392842, 215 from GSE13688943 and 233 from GSE15661344. To generate coverage tracks Bigwig (bw) files for ChIP-seq data, we converted them to bedGraph files and then identified peaks with the subcommand bdgpeakcall from macs245. For each variant, we examined whether it was mapped to a peak region of histone modification marks, DNase I hypersensitive, or transcription factors binding sites using an in-house script.
In silico prediction of regulatory element-to-gene
Since the majority of the CCVs are located outside protein-coding regions, genes can potentially be regulated by CCVs located in distal enhancer elements and proximal promoter elements. Hence, we identified an extensive set of functional genomic data from normal colon tissues or tumor tissues of colorectal cancer or colorectal cancer cell lines (Supplementary Data 9). Subsequently, we conducted an in-silico analysis for each CCV-gene pair.
We used a variety of experimental and computational functional genomic data to identify target genes of CCVs in regulatory elements. Specifically, for distal regulatory elements, we utilized chromatin-chromatin interaction data from experiments or computational predictions. To do this, we downloaded 13 experimental chromatin-chromatin interaction datasets under accessions GSE13392842 and GSE13662943 from GEO, as well as two promoter capture Hi-C datasets from the previous study46. We combined this data with ChIP-seq data of the histone modification H3K27ac (an active enhancer mark) to identify enhancer-promoter loops. We defined these loops as interactions where one fragment overlapped an H3K27ac peak (enhancer-like) and the other fragment overlapped the promoter of a gene (the region from downstream 1 kb to upstream 100 bp around the transcription start site).
In addition to this, we downloaded experimentally confirmed enhancer-gene pairs from the ENdb database. We also obtained computational enhancer-promoter interactions from IM-PET47, FANTOM548,49, EnhancerAtlas50, and super-enhancer51,52. To further refine our analysis, we included topologically associating domain (TAD) boundaries in three colorectal cancer cell lines (HT29, LoVo, and DLD1)46,53. Finally, we examined the overlap between CCVs and enhancer elements. For proximal promoter elements, we analyzed CCVs located within gene promoter regions that intersected with ChIP-seq peaks of H3K4me3 (an activity promoter mark).
To identify potential loss-of-function variants and their corresponding targeted genes, we conducted variant annotation of CCVs using the Variant Effect Predictor (VEP) tool54. To predict the consequence of missense coding variants, we utilized PolyPhen-2 and SIFT. Furthermore, to evaluate splicing effects, MaxEntScan was used.
We scored CCV target genes using different criteria (Supplementary Data 9). For the potential target gene of CCV in distal enhancer elements, the gene was awarded two points or one point if there was evidence from experimental chromatin-chromatin interaction or computed interaction. The score was unweighted to three if both experimental and computational interaction were detected for the gene-CCV pair. If CCV interacted with genomic features (open chromatin, activity enhancer, and TF binding sites), the corresponding gene was further unweighted by one point. An additional point was awarded if there are at least two interactions for the CCV. If the gene were colorectal cancer or pan-cancer drivers55, they were up-weighted by an additional point. The score was down-weighted for the gene if the CCV-gene pair was separated by TAD or a lack of expression in colon tissues. Distal scores eventually ranged from 0 to 6. For the potential target gene of CCV in proximal promoter elements, the gene was awarded one point if CCV overlapped with binding sites of transcription factors. If genes were colorectal cancer or pan-cancer drivers, they were up-weighted with an additional point. A lack of its expression resulted in down-weighting to 10% as target genes. Proximal scores eventually ranged from 0 to 2. Genes predicted to be regulated targets of coding CCVs were awarded points based on the annotation as either of missense, nonsense, and predicted splicing alterations. The consequences of missense variants which probably are damaging or deleterious resulted in the addition of one point to the target gene. Further points were awarded to such a gene if it was colorectal cancer or pan-cancer drivers. A lack of expression reduced the score (the score was down to 10%). Coding scores ranged from 0 to 2. For the set of confident target genes, we defined such genes if it has a distal score >4 or a proximal score >1, or a coding score >1.
Credible set of susceptibility genes
To determine a set of credible genes for CRC susceptibility, we combined information on gene-CRC risk associations through TWAS and colocalization of eQTL signal with GWAS risk signals for genes that were present in both our study and previous investigations. We used three sets of previously identified genes below: (A) 155 effector genes identified through GWAS, TWAS, TIsWAS, and MWAS7; (B) 136, 26, and 48 genes identified through TWASs7,16,17; (C) 73 genes identified through colocalization analysis between eQTL and GWAS signals11 or genes associated with CRC risk at nominal P < 0.05 in the previous TWAS17. We considered the prioritization order as A > B > C for these three gene sets and focused on protein-coding genes outside the MHC region. For the independent association signals with multiple target gene candidates, we kept either genes with higher prioritization or all genes if there was no evidence from these three gene sets. For the independent association signals with a single gene, we kept it regardless of evidence from the gene sets.
Single-cell RNA-sequencing data analysis
We included single-cell RNA-sequencing datasets from colon tissues of 31 individuals who participated in the Colorectal Molecular Atlas Project (COLON MAP)18. We analyzed gene expression dataset for each individual’s cell and combined these datasets into a count matrix. We normalized the number of unique molecular identifiers (UMIs) per cell and converted it to transcripts per 10,000 transcripts (TP10K). Next, we applied a logarithmic transformation to the normalized values and got the log2(TP10K + 1) expression matrix for the downstream analyses. Further, we determined the 2000 most highly variable genes within the entire dataset and performed a principal component analysis (PCA). The top 30 and 40 principal components (PCs) were identified. Subsequently, we performed batch correction removal and utilized the top 40 batch-corrected components to construct a k-nearest neighbors graph of cell profiles with k = 9. We visualized the individual single-cell profiles using the Uniform Manifold Approximation and Projection (UMAP) and constructed the neighborhood graph using the Leiden graph-clustering method. Nine cell types were defined, including well-known major cell types such as absorptive cells (ABS), crypt top colonocytes (CT), enteroendocrine cells (EE), goblet cells (GOB), stem cells (STM), and others. We identified differentially expressed genes (DEGs) by comparing each cell type with all other cell types and calculated a P-value for each gene using Wilcoxon’s rank-sum test. The criteria |log2 fold change (FC)| > 1 and P < 0.05 were applied to determine genes with significantly differential expression between cell types.
Burden test for credible susceptibility genes
We annotated all variants in the UKBB WES 200 K cohort with functional annotations from ANNOVAR56 based on the reference genome GRCh38. We only included rare loss-of-function (LoF) and deleterious missense (Dmis) variants with MAF < 0.01 in our gene-based test. LoF variants were those predicted as frameshift insertion/deletion, splice-site alteration, stop gain, and stop loss by ANNOVAR, and deleterious missense (Dmis) variants were those predicted as deleterious by MetaSVM57. We considered both LoF sets and damaging sets (LoF+ Dmis) within a gene for testing. For a given set, we collapsed rare variants within a gene as a single combined ‘mask’ and tested the association between the ‘mask’ genotype and the CRC phenotype using logistic regression after adjusting for sex, age, the interaction of sex and age, and the top four principal components.
Pathway analysis of credible susceptibility genes
To explore the potential biological roles of the identified CRC susceptibility genes, we analyzed their functional enrichment using the enrichR19–21 and various pathway databases, including WikiPathway, KEGG, MSigDB, and Reactome. The biological pathways (adjusted P < 0.05) were considered and presented.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
This research was supported primarily by US National Institutes of Health (NIH) grant R01CA188214 (to W.Z.), Anne Potter Wilson Chair endowment from the Vanderbilt University School of Medicine (to W.Z.), and NIH grant R37CA227130 and R01CA269589 (to X.G.). Sample preparation and genotyping assays at Vanderbilt University were conducted at the Survey and Biospecimen Shared Resources and Vanderbilt Microarray Shared Resource, supported in part by the Vanderbilt-Ingram Cancer Center (grant P30CA068485). Data analyses were performed on servers maintained by the Advanced Computing Center for Research and Education (ACCRE) at Vanderbilt University.
Author contributions
Wei Zheng and Xingyi Guo conceived and supervised the study, and acquired funding. Wei Zheng, Xingyi Guo, and Zhishan Chen designed the study with significant contribution from Ran Tao. Zhishan Chen carried out the main analysis. Chao Li, Quanhu Shen, and Ken S Lau contributed to single-cell RNA-seq analysis. Yuhan Xie and Hongyu Zhao contributed to whole exome sequencing analysis. Zhishan Chen, Xingyi Guo, and Wei Zheng interpreted results with help from other authors. Zhishan Chen, Xingyi Guo, Jeroen R Huyghe, Philip J Law, Ceres Fernandez-Rozadilla, Jie Ping, Guochong Jia, Maria N Timofeeva, Minta Thomas, Stephanie L Schmit, Virginia Díez-Obrero, Matthew Devall, Ferran Moratalla-Navarro, Juan Fernandez-Tajes, Sarah E W Briggs, Victoria Svinti, Kevin Donnelly, Yingchang Lu, Fredrick R Schumacher, Stephanie J Weinstein, Kala Visvanathan, Kostas K Tsilidis, Yu-Ru Su, Robert Steinfelder, Sonja I Berndt, Sushma S Thomas, Kimberly F Doheny, Tameka Shelford, Amit D Joshi, Anshul Kundaje, Christopher K Edlund, Andre Kim, Lori C Sakoda, Stephanie A Bien, Yi Lin, Conghui Qu, Chenxu Qu, Stuart Reid, and Li Hsu analyzed the data. Xingyi Guo, Ceres Fernandez-Rozadilla, Jirong Long, Matthew Devall, Claire Palles, Kitty Sherwood, Susan M Farrington, James Blackmur, Peter G. Vaughan-Shaw, Xiao-Ou Shu, Peter Broderick, James Studd, Tabitha A Harrison, David V Conti, Marilena Melas, Gad Rennert, Mireia Obón-Santacana, Vicente Martín-Sánchez, Jae Hwan Oh, Jeongseon Kim, Sun Ha Jee, Keum Ji Jung, Sun-Seog Kweon, Min-Ho Shin, Aesun Shin, Yoon-Ok Ahn, Dong-Hyun Kim, Isao Oze, Wanqing Wen, Keitaro Matsuo, Koichi Matsuda, Chizu Tanikawa, Zefang Ren, Yu-Tang Gao, Wei-Hua Jia, John L Hopper, Mark A Jenkins, Aung Ko Win, Rish K Pai, Jane C Figueiredo, Robert W Haile, Steven Gallinger, Michael O Woods, Polly A Newcomb, David Duggan, Jeremy P. Cheadle, Richard Kaplan, Rachel Kerr, David Kerr, Iva Kirac, Jan Böhm, Jukka-Pekka Mecklin, Pekka Jousilahti, Paul Knekt, Lauri A. Aaltonen, Harri Rissanen, Eero Pukkala, Johan G Eriksson, Tatiana Cajuso, Ulrika Hänninen, Johanna Kondelin, Kimmo Palin, Tomas Tanskanen, Laura Renkonen-Sinisalo, Satu Männistö, Demetrius Albanes, Edward Ruiz-Narvaez, Julie R Palmer, Daniel D Buchanan, Elizabeth A Platz, Cornelia M Ulrich, Erin Siegel, Stefanie Brezina, Andrea Gsur, Peter T Campbell, Jenny Chang-Claude, Michael Hoffmeister, Hermann Brenner, Martha L Slattery, John D Potter, Matthias B Schulze, Marc J Gunter, Neil Murphy, Antoni Castells, Sergi Castellví-Bel, Leticia Moreira, Volker Arndt, Anna Shcherbina, D. Timothy Bishop, Graham G Giles, Melissa C. Southey, Gregory E Idos, Kevin J McDonnell, Zomoroda Abu-Ful, Joel K Greenson, Katerina Shulman, Flavio Lejbkowicz, Kenneth Offit, Temitope O Keku, Bethany van Guelpen, Thomas J Hudson, Heather Hampel, Rachel Pearlman, Richard B Hayes, Marie Elena Martinez, Paul D. P. Pharoah, Susanna C Larsson, Yun Yen, Heinz-Josef Lenz, Emily White, Li Li, Elizabeth Pugh, Andrew T Chan, Marcia Cruz-Correa, Annika Lindblom, David J Hunter, Clemens Schafmayer, Peter C Scacheri, Robert E Schoen, Jochen Hampe, Zsofia K Stadler, Pavel Vodicka, Ludmila Vodickova, Veronika Vymetalkova, W. James Gauderman, David Shibata, Amanda Toland, Sanford Markowitz, Stephen J Chanock, Franzel van Duijnhoven, Edith JM Feskens, Manuela Gago-Dominguez, Alicja Wolk, Barbara Pardini, Liesel M FitzGerald, Soo Chin Lee, Shuji Ogino, Charles Kooperberg, Christopher I Li, Ross Prentice, Stéphane Bézieau, Taiki Yamaji, Norie Sawada, Motoki Iwasaki, Loic Le Marchand, Anna H Wu, Caroline E McNeil, Gerhard Coetzee, Caroline Hayward, Ian J Deary, Sarah E Harris, Evropi Theodoratou, Marion Walker, Li Yin Ooi, Qiuyin Cai, Malcolm G Dunlop, Stephen B Gruber, Richard S Houlston, Victor Moreno, Graham Casey, Ulrike Peters, Ian Tomlinson, and Wei Zheng recruited patients and collected samples. Zhishan Chen, Xingyi Guo, and Wei Zheng wrote the manuscript with substantial contributions from Ceres Fernandez-Rozadilla, Jie Ping, Guochong Jia, Jirong Long, Xiao-Ou Shu, Richard S Houlston, and Ian Tomlinson. All authors have reviewed and approved the final manuscript.
Peer review
Peer review information
Nature Communications thanks Jyotsna Batra and Juliet French for their contribution to the peer review of this work. A peer review file is available.
Data availability
The GWAS summary statistics are available at the GWAS catalog under accession number GCST90129505. The RNA-seq data and genotype data of subjects of East Asian ancestry from the ACCC is being deposited to NCBI database of Genotypes and Phenotypes (dbGaP, accession number phs002813.v1.p1). All requests to access these data could also be made by contacting Drs. Wei Zheng (wei.zheng@vanderbilt.edu) and Xingyi Guo (xingyi.guo@vumc.org). The data from the Genotype-Tissue Expression (GTEx, version 8) project used in this study are publicly available at the dbGaP under accession number phs000424.v8.p2 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000424.v8.p2). The transcriptome and genotype data as well as the sample covariates from the BarcUVa-Seq project can be accessed at the dbGaP under accession number phs003338.v1.p1 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs003338.v1.p1). The access to data from the Colonomics project could be requested by submission of an inquiry to Dr. Victor Moreno (v.moreno@iconcologia.net). The CRC-relevant epigenome and functional genomic data were obtained from the NCBI’s Gene Expression Omnibus database (GEO) under accession numbers: GSE133928, GSE136889, and GSE156613. Enhancer-promoter interaction data were obtained from the ENdb database (https://bio.liclab.net/ENdb/), 4Dgenome (https://4dgenome.research.chop.edu/), FANTOM5 (https://fantom.gsc.riken.jp/5/), EnhancerAtlas 2.0 (http://www.enhanceratlas.org/) and Super-enhancers (https://bio.liclab.net/sedb/ and https://www.cell.com/fulltext/S0092-8674(13)01227-0#supplementaryMaterial). Single-cell RNA-sequencing datasets from colon tissues of 31 individuals were obtained from the Colorectal Molecular Atlas Project (COLON MAP). Whole exome sequencing data from 137,104 individuals of European ancestry were obtained from the UK Biobank (https://www.ukbiobank.ac.uk/).
Code availability
The code used in this study is available at the GitHub repository https://github.com/zhishanchen/CRC_Finemapping58.
Competing interests
Antoni Castells is a consultant to Bayer Pharma AG, Boehringer Ingelheim and Pfizer Inc. for work unrelated to this manuscript. Anna Shcherbina is an employee at Insitro, including consulting fees from BMS. Heather Hampel is SAB for Invitae Genetics, Promega and Genome Medical, Stock/Stock options for Genome Medical and GI OnDemand. Rish K Pai collaborates with Eli Lilly, AbbVie, Allergan, Verily and Alimentiv, which includes consulting fees (outside the submitted work). Stephanie A Bien has a financial interest in Adaptive Biotechnologies. Stephen B Gruber is co-founder, Brogent International LLC. One of Zsofia K Stadler’s immediate family members serves as a consultant in ophthalmology for Alcon, Adverum, Gyroscope Therapeutics Limited, Neurogene and RegenexBio (outside the submitted work). Victor Moreno has research projects and owns stocks of Aniling. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-47399-x.
References
- 1.Sung H, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Jiao S, et al. Estimating the heritability of colorectal cancer. Hum. Mol. Genet. 2014;23:3898–3905. doi: 10.1093/hmg/ddu087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu Y, et al. Large-scale genome-wide association study of east asians identifies loci associated with risk for colorectal cancer. Gastroenterology. 2019;156:1455–1466. doi: 10.1053/j.gastro.2018.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huyghe JR, et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nat. Genet. 2019;51:76–87. doi: 10.1038/s41588-018-0286-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Law PJ, et al. Association analyses identify 31 new risk loci for colorectal cancer susceptibility. Nat. Commun. 2019;10:2154. doi: 10.1038/s41467-019-09775-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu Y, et al. Identification of Novel Loci and New Risk Variant in Known Loci for Colorectal Cancer Risk in East Asians. Cancer Epidemiol. Biomark. Prev. 2020;29:477–486. doi: 10.1158/1055-9965.EPI-19-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandez-Rozadilla C, et al. Deciphering colorectal cancer genetics through multi-omic analysis of 100,204 cases and 154,587 controls of European and east Asian ancestries. Nat. Genet. 2023;55:89–99. doi: 10.1038/s41588-022-01222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeng, C. et al. Identification of independent association signals and putative functional variants for breast cancer risk through fine-scale mapping of the 12p11 locus. Breast Cancer Res. 18 (2016). [DOI] [PMC free article] [PubMed]
- 9.Guo X, et al. A comprehensive cis-eQTL analysis revealed target genes in breast cancer susceptibility loci identified in genome-wide association studies. Am. J. Hum. Genet. 2018;102:890–903. doi: 10.1016/j.ajhg.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Z, et al. Identifying putative susceptibility genes and evaluating their associations with somatic mutations in human cancers. Am. J. Hum. Genet. 2019;105:477–492. doi: 10.1016/j.ajhg.2019.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan Y, et al. Multi-omics analysis to identify susceptibility genes for colorectal cancer. Hum. Mol. Genet. 2021;30:321–330. doi: 10.1093/hmg/ddab021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fachal L, et al. Fine-mapping of 150 breast cancer risk regions identifies 191 likely target genes. Nat. Genet. 2020;52:56–73. doi: 10.1038/s41588-019-0537-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thiagalingam A, et al. RREB-1, a novel zinc finger protein, is involved in the differentiation response to Ras in human medullary thyroid carcinomas. Mol. Cell. Biol. 1996;16:5335–5345. doi: 10.1128/MCB.16.10.5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oliva M, et al. DNA methylation QTL mapping across diverse human tissues provides molecular links between genetic variation and complex traits. Nat. Genet. 2023;55:112–122. doi: 10.1038/s41588-022-01248-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Y, et al. Joint analysis of GWAS and multi-omics QTL summary statistics reveals a large fraction of GWAS signals shared with molecular phenotypes. Cell Genom. 2023;3:100344. doi: 10.1016/j.xgen.2023.100344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo X, et al. Identifying novel susceptibility genes for colorectal cancer risk from a transcriptome-wide association study of 125,478 subjects. Gastroenterology. 2021;160:1164–1178.e6. doi: 10.1053/j.gastro.2020.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Z, et al. Novel insights into genetic susceptibility for colorectal cancer from transcriptome-wide association and functional investigation. J. Natl Cancer Inst. 2023 doi: 10.1093/jnci/djad178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen B, et al. Differential pre-malignant programs and microenvironment chart distinct paths to malignancy in human colorectal polyps. Cell. 2021;184:6262–6280.e26. doi: 10.1016/j.cell.2021.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen EY, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuleshov MV, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie Z, et al. Gene set knowledge discovery with Enrichr. Curr. Protoc. 2021;1:e90. doi: 10.1002/cpz1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung B, Staudacher JJ, Beauchamp D. Transforming growth factor β superfamily signaling in development of colorectal cancer. Gastroenterology. 2017;152:36–52. doi: 10.1053/j.gastro.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng Q, Li S, Ma H-M, Yang W-T, Zheng P-S. LGR6 activates the Wnt/β-catenin signaling pathway and forms a β-catenin/TCF7L2/LGR6 feedback loop in LGR6high cervical cancer stem cells. Oncogene. 2021;40:6103–6114. doi: 10.1038/s41388-021-02002-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruan X, et al. Silencing LGR6 attenuates stemness and chemoresistance via inhibiting Wnt/β-catenin signaling in ovarian cancer. Mol. Ther. Oncolytics. 2019;14:94–106. doi: 10.1016/j.omto.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong L, Lyu X, Faleti OD, He M-L. The special stemness functions of Tbx3 in stem cells and cancer development. Semin. Cancer Biol. 2019;57:105–110. doi: 10.1016/j.semcancer.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 26.Russell R, et al. A dynamic role of TBX3 in the pluripotency circuitry. Stem Cell Rep. 2015;5:1155–1170. doi: 10.1016/j.stemcr.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elbadawy M, Usui T, Yamawaki H, Sasaki K. Emerging roles of C-Myc in cancer stem cell-related signaling and resistance to cancer chemotherapy: A potential therapeutic target against colorectal cancer. Int. J. Mol. Sci. 2019;20:2340. doi: 10.3390/ijms20092340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Satoh K, et al. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc. Natl Acad. Sci. USA. 2017;114:E7697–E7706. doi: 10.1073/pnas.1710366114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ong ES, et al. Metabolic profiling in colorectal cancer reveals signature metabolic shifts during tumorigenesis. Mol. Cell. Proteom. 2010 doi: 10.1074/mcp.M900551-MCP200. [DOI] [PubMed] [Google Scholar]
- 30.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang J, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 2012;44:369–375. doi: 10.1038/ng.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang B, et al. Large-scale genetic study in East Asians identifies six new loci associated with colorectal cancer risk. Nat. Genet. 2014;46:533–542. doi: 10.1038/ng.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Graubert A, Aguet F, Ravi A, Ardlie KG, Getz G. RNA-SeQC 2: Efficient RNA-seq quality control and quantification for large cohorts. Bioinformatics. 2021;37:3048–3050. doi: 10.1093/bioinformatics/btab135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frankish A, et al. GENCODE: reference annotation for the human and mouse genomes in 2023. Nucleic Acids Res. 2023;51:D942–D949. doi: 10.1093/nar/gkac1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Díez-Villanueva A, et al. Identifying causal models between genetically regulated methylation patterns and gene expression in healthy colon tissue. Clin. Epigenetics. 2021;13:162. doi: 10.1186/s13148-021-01148-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Díez-Villanueva A, et al. DNA methylation events in transcription factors and gene expression changes in colon cancer. Epigenomics. 2020;12:1593–1610. doi: 10.2217/epi-2020-0029. [DOI] [PubMed] [Google Scholar]
- 39.Zhu Z, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016;48:481–487. doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
- 40.Zheng R, et al. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019;47:D729–D735. doi: 10.1093/nar/gky1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mei S, et al. Cistrome Data Browser: a data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res. 2017;45:D658–D662. doi: 10.1093/nar/gkw983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnstone SE, et al. Large-scale topological changes restrain malignant progression in colorectal cancer. Cell. 2020;182:1474–1489.e23. doi: 10.1016/j.cell.2020.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orouji E, et al. Chromatin state dynamics confers specific therapeutic strategies in enhancer subtypes of colorectal cancer. Gut. 2022;71:938–949. doi: 10.1136/gutjnl-2020-322835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Q-L, et al. Genome-wide profiling in colorectal cancer identifies PHF19 and TBC1D16 as oncogenic super enhancers. Nat. Commun. 2021;12:6407. doi: 10.1038/s41467-021-26600-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Orlando G, et al. Promoter capture Hi-C-based identification of recurrent noncoding mutations in colorectal cancer. Nat. Genet. 2018;50:1375–1380. doi: 10.1038/s41588-018-0211-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He B, Chen C, Teng L, Tan K. Global view of enhancer-promoter interactome in human cells. Proc. Natl Acad. Sci. USA. 2014;111:E2191–E2199. doi: 10.1073/pnas.1320308111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lizio M, et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol. 2015;16:22. doi: 10.1186/s13059-014-0560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andersson R, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gao T, Qian J. EnhancerAtlas 2.0: an updated resource with enhancer annotation in 586 tissue/cell types across nine species. Nucleic Acids Res. 2020;48:D58–D64. doi: 10.1093/nar/gkz980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hnisz D, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang Y, et al. SEdb: a comprehensive human super-enhancer database. Nucleic Acids Res. 2019;47:D235–D243. doi: 10.1093/nar/gky1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lajoie BR, Dekker J, Kaplan N. The Hitchhiker’s guide to Hi-C analysis: practical guidelines. Methods. 2015;72:65–75. doi: 10.1016/j.ymeth.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McLaren, W. et al. The ensembl variant effect predictor. Genome Biol. 17 (2016). [DOI] [PMC free article] [PubMed]
- 55.Bailey MH, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173:371–385.e18. doi: 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015;10:1556–1566. doi: 10.1038/nprot.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim S, Jhong J-H, Lee J, Koo J-Y. Meta-analytic support vector machine for integrating multiple omics data. BioData Min. 2017;10:2. doi: 10.1186/s13040-017-0126-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.zhishanchen. zhishanchen/CRC_Finemapping: crc_finemapping. (Zenodo, 2024). 10.5281/ZENODO.10645372
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The GWAS summary statistics are available at the GWAS catalog under accession number GCST90129505. The RNA-seq data and genotype data of subjects of East Asian ancestry from the ACCC is being deposited to NCBI database of Genotypes and Phenotypes (dbGaP, accession number phs002813.v1.p1). All requests to access these data could also be made by contacting Drs. Wei Zheng (wei.zheng@vanderbilt.edu) and Xingyi Guo (xingyi.guo@vumc.org). The data from the Genotype-Tissue Expression (GTEx, version 8) project used in this study are publicly available at the dbGaP under accession number phs000424.v8.p2 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000424.v8.p2). The transcriptome and genotype data as well as the sample covariates from the BarcUVa-Seq project can be accessed at the dbGaP under accession number phs003338.v1.p1 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs003338.v1.p1). The access to data from the Colonomics project could be requested by submission of an inquiry to Dr. Victor Moreno (v.moreno@iconcologia.net). The CRC-relevant epigenome and functional genomic data were obtained from the NCBI’s Gene Expression Omnibus database (GEO) under accession numbers: GSE133928, GSE136889, and GSE156613. Enhancer-promoter interaction data were obtained from the ENdb database (https://bio.liclab.net/ENdb/), 4Dgenome (https://4dgenome.research.chop.edu/), FANTOM5 (https://fantom.gsc.riken.jp/5/), EnhancerAtlas 2.0 (http://www.enhanceratlas.org/) and Super-enhancers (https://bio.liclab.net/sedb/ and https://www.cell.com/fulltext/S0092-8674(13)01227-0#supplementaryMaterial). Single-cell RNA-sequencing datasets from colon tissues of 31 individuals were obtained from the Colorectal Molecular Atlas Project (COLON MAP). Whole exome sequencing data from 137,104 individuals of European ancestry were obtained from the UK Biobank (https://www.ukbiobank.ac.uk/).
The code used in this study is available at the GitHub repository https://github.com/zhishanchen/CRC_Finemapping58.