

Abstract
Glioblastoma is the most common and aggressive malignant brain tumor and has limited treatment options. Hence, innovative approaches are urgently needed. Oncolytic virus therapy is emerging as a promising modality for cancer treatment due to its tumor-specific targeting and immune-stimulatory properties. In this study, we developed a new generation of oncolytic herpes simplex virus C5252 by deletion of a 15-kb internal repeat region and both copies of γ34.5 genes. Additionally, C5252 was armed with anti-programmed cell death protein 1 antibody and interleukin-12 to enhance its therapeutic efficacy for glioblastoma immune-virotherapy. In vitro and in vivo experiments demonstrate that C5252 has a remarkable safety profile and potent anti-tumor activity against glioblastoma. Mechanistic studies demonstrated that C5252 specifically induces cell apoptosis by caspase-3/7 activation via downregulating ciliary neurotrophic factor receptor α. Furthermore, the enhanced anti-tumor therapeutic efficacy of C5252 in a subcutaneous glioblastoma model and an orthotopic glioblastoma model was confirmed. Moreover, syngeneic mouse models showed that the murine surrogate of C5252 has superior anti-tumor activity compared to the unarmed backbone virus, with enhanced immune activation. Taken together, our findings support C5252 as a promising therapeutic option for glioblastoma treatment, positioning it as a highly promising candidate for clinical translation.
Keywords: glioblastoma, GBM, oncolytic herpes simplex virus, oHSV, immunotherapy, IL-12, checkpoint inhibitor
Graphical abstract
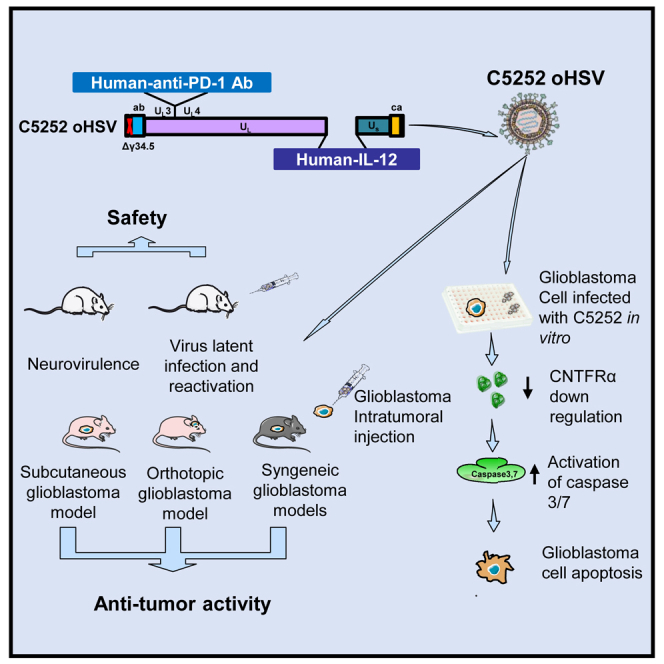
Wang and colleagues’ innovative oncolytic virus C5252, armed with anti-PD-1 antibody and IL-12, demonstrates safety and potent anti-glioblastoma activity in vitro and in vivo. Mechanistic studies reveal apoptosis induction via caspase-3/7 and CNTFRα downregulation. Superior efficacy in mouse models positions C5252 for clinical translation in glioblastoma treatment.
Introduction
Malignant gliomas, as the most aggressive primary brain tumors, account for 30% of all primary central nervous system (CNS) tumors in adults, exhibiting poor survival with a median of 12–18 months.1,2 In children, CNS tumors represent 25% of all childhood malignancies and remain the leading cause of cancer-related morbidity and mortality3; despite the implementation of various intricate treatment modalities, numerous children continue to contend with enduring and potentially incapacitating conditions.4 Nowadays, glioblastoma (GBM) remains one of the most challenging malignancies to treat. The main causes of these failures include GBM molecular heterogeneity,5,6 a “cold” immunosuppressive tumor microenvironment (TME),7,8 and the presence of the blood-brain barrier, which hinders drug penetration within brain tissue.9 Nevertheless, advances in multimodal treatment options and drug development come from innovative treatment strategies for GBM, such as multispecific “off-the-shelf” chimeric antigen receptor (CAR)-T therapy, oncolytic viral therapy, and autologous dendritic cell vaccination.
With regards to oncolytic viral therapy, a number of oncolytic vectors including engineered forms of retrovirus, adenovirus, adeno-associated virus, and herpes simplex virus type 1 (HSV-1)10 have been studied in the pursuit of novel therapies for GBM for the past several decades. Some of these oncolytic viruses have been investigated in several clinical trials of GBM and showed promising results. Among them, DELYTACT oncolytic virus G47Δ, the third generation oncolytic HSV-1 (oHSV), has received conditional and time-limited approval in Japan for the treatment with malignant gliomas based on the phase 2 study results. The backbone of G47Δ, G207 (derived from wild-type strain F), as the first oHSV developed to treat GBM, has deletions of two γ34.5 genes, which are neuropathogenic and overcome host protein shutoff and innate signaling.11 G207 has been studied in multiple clinical trials for GBM, including a recent pediatric trial with promising results.12 Despite some promising efficacy being shown,12,13 current evidence indicates that oHSV monotherapy needs to be enhanced, due to the failures of oHSV immunovirotherapy including an immunosuppressive TME and inadequate immune responses, as well as low viral replication and spread.14 Thus, combination therapy of oHSV with other immunotherapies or oncolytic viruses armed with therapeutic payload genes for local expression has emerged.15,16
Based on the G207 backbone, another oHSV, M032, was modified to express secreted interleukin-12 (IL-12), which promotes an immune response and produces an anti-angiogenic effect.17 M032 is currently being studied in phase 1 brain tumor clinical trials in children (ClinicalTrials.gov: NCT02457845) and adults (ClinicalTrials.gov: NCT02062827), respectively.18 Another ongoing evaluation of the combination of pembrolizumab with M032 in patients with recurrent/progressive GBM, anaplastic astrocytoma, or gliosarcoma is in phase 1/2 trial (NSC733972). In addition, Xin Xie et al. engineered two oncolytic viruses expressing a mouse anti-programmed cell death protein 1 (PD-1) antibody (Ab) or mouse IL-12, respectively, both of which demonstrated substantial anti-tumor effects. The combination of both viruses generated a vaccine-like response and significantly extended the overall survival of mice, which suggested that combining both engineered oncolytic viruses may offer a robust immunotherapeutic strategy for patients with cancer.19 To further enhance the oncolytic and immune efficacy of oHSVs, we genetically engineered a new generation of oHSV, C5252, which can deliver IL-12 and anti-PD-1 Ab into GBM while simultaneously eliminating both copies of the γ34.5 genes and a 15-kb internal repeat (IR) region. The safety and anti-tumor efficacy were confirmed in this study.
Results
Construction and identification of oHSV C5252 in vitro
In this study, oHSV C5252, a genetically modified replication-competent HSV-1, was designed to remove two copies of the γ34.5 genes in both the terminal repeat region and the IR region. Meanwhile, C5252 was modified by introducing a cassette encoding the heterodimer of IL-12 to replace the IR region, and an antigen-binding fragment (Fab) of an anti-human PD-1 Ab was inserted between UL3 and UL4 (Figure 1A). Here, another oHSV used, R361620 (Figure 1A), also deleted two copies of the γ34.5 genes. The DELYTACT oncolytic virus G47Δ has been modified from R3616 and approved in Japan for malignant glioma treatment. Thus, to confirm the safety and efficacy of C5252 for glioma treatment, we used oHSV R3616 as a first-generation control. As shown in Figure 1B, the growth kinetics analysis in Vero cells reveals that both C5252 and R3616 exhibit lower viral yields compared to wild-type HSV(F) infection. What’s more, C5252 showed relatively lower viral yields than R3616, confirming the genetic attenuation of C5252. Further confirmation of the genetic attenuation of C5252 and R3616 was obtained by evaluating the accumulation of viral proteins using specific Abs against viral α (ICP0, ICP4, ICP27), β (ICP8), and γ (US11, γ34.5) gene products. The results demonstrate significantly reduced protein expression levels in C5252 compared to wild-type HSV-1. Similarly, R3616 showed lower protein expression levels for all proteins, except for ICP4 and ICP8, when compared to wild-type HSV-1. Importantly, both C5252- and R3616-infected samples exhibited no detectable expression of the γ34.5 gene, confirming the successful deletion of the γ34.5 genes in these strains (Figure 1C). The expression of payload genes of C5252, including IL-12 p70 and anti-PD-1 Ab, were detected via ELISA, and the average amounts were 401.69 and 242.52 pg/mL, respectively (Figure 1D). The standard curves for IL-12 p70 and the anti-PD-1 Ab are shown in the Figure S2.
Figure 1.

Virus construction and identification
(A) Schematic representation of the virus genome. Wild-type HSV-1(F) is the prototype strain used in this laboratory and R3616 has a deletion of two copies of γ34.5. C5252 is a genetically modified replication-competent HSV-1 in which the internal repeat (IR) sequence (IR region) of 15 kb and the remaining copy of γ34.5 genes in the terminal repeat (TR) region were deleted, resulting in the removal of both copies of the γ34.5 genes as well as one copy each of diploid genes α0, α4, and LAT. The IR region is replaced with an expression cassette of heterodimer of IL-12. Another expression cassette of antigen-binding fragment (Fab) of an anti-PD-1 Ab is inserted into the genome between UL3 and UL4. (B) Growth curves. Vero cells were exposed to HSV-1(F), R3616, or C5252 (MOI = 0.1) over 48 h. Virus progeny were collected at 3, 6, 12, 24, and 48 h and titered via plaque assay using Vero cells (∗p < 0.05). (C) Accumulation of viral protein. Vero cells were infected with HSV-1(F), R3616, or C5252 (MOI = 1) for 6, 12, and 24 hpi. Cell samples were collected at indicated time points. Proteins were separated on 10% denaturing gels and analyzed via immunoblotting with antibodies targeting ICP0, ICP4, ICP27, ICP8, US11, γ34.5, and GAPDH. (D) The expression of human IL-12 p70 and anti-PD-1 Ab in HSV-1(F)-, R3616-, and C5252-infected Vero cells. Vero cells were mock infected (Mock) or infected with of HSV-1(F), R3616, or C5252. After 48 h, cell culture media were collected. Human IL-12 and anti-PD-1 Ab levels were quantified using ELISA, following a standard curve constructed with purified IL-12 protein and anti-PD-1 Ab as described in the materials and methods.
oHSV C5252 has a higher cytotoxic activity and lower replication efficiency in GBM cells
To evaluate the replication and cell-killing ability of C5252 in GBM cells, different GBM cell lines including A172, D54, U138, U87, and D548 were infected with C5252 for 48 h at a multiplicity of infection (MOI) of 0.1. The results showed that C5252 had a lower replication ability compared to oHSV R3616 in most of the GBM cells, with a significant decrease in progeny yield observed in A172, U138, U87 (p < 0.05), and D54 cells (p < 0.01) (Figure 2A); each result is presented as the mean ± standard deviation of three replicates. On the other hand, as shown in Figure 2C, C5252 infection for 48 h resulted in increased cytotoxicity in A172, D54, U87 (p < 0.05), and U138 (p < 0.001) glioma cells when compared to R3616 at an MOI = 0.1. Similarly, at an MOI = 1, C5252 showed lower replication efficiency but higher cell-killing ability compared to R3616 (Figures 2B and 2D). Specifically, C5252 replication was significantly reduced in A172 (p < 0.01), D54 and D458 (p < 0.05) cells, while the cytotoxicity was significantly enhanced in A172 and U87 (p < 0.05), U138 and D458 (p < 0.01) cells. Intriguingly, wild-type HSV-1(F) exhibited the highest replication ability in all tested cell lines, but demonstrated lower cytotoxicity compared to C5252. These results suggest that C5252 has a remarkable cytotoxic effect with relatively low efficiency of viral propagation in GBM cells, indicating its potential as a safe and effective treatment option for GBM. Moreover, at 24 h post-infection (hpi) (MOI = 1), lower virus replication and higher cytotoxic activity of C5252 are shown, which is consistent with that at 48 hpi (Figures S3A and S3B).
Figure 2.

Virus replication and cytotoxicity in glioblastoma cells
(A) Glioblastoma cells (A172, D54, U138, U87, and D458) were exposed to 0.1 PFU of HSV-1(F), R3616, or C5252 per cell. Virus progeny were collected at 48 hpi and titrated using Vero cells. (B) Similar to (A) but cells were exposed to 1.0 PFU per cell. (C) Glioblastoma cells were infected with HSV-1(F), R3616, or C5252 at 0.1 PFU per cell, and cytotoxicity was assessed via CCK8 assay at 48 hpi. (D) Similar to (C) but cells exposed to 1.0 PFU per cell. Data represent mean ± SD; N.S. p > 0.05, ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Student’s t test (two-tailed) was used for statistical analysis.
Downregulation of CNTFRα expression enhances apoptotic cell death in GBM cells infected with C5252
To investigate why C5252 has lower replication ability but higher cytotoxic activity, additional studies were conducted. GBM cell lines A172, D54, U138, U87, and D458 were infected with C5252, R3616, or wild-type HSV-1(F) at an MOI of 5. Caspase-3/7 activity, which indicates apoptotic cell death, was measured 8 h after infection. The results in Figure 3A show that C5252 infection induced a significant increase in caspase-3/7 activity compared to R3616 and wild-type HSV-1(F). Specifically, in the case of A172 and D54 cell lines, the increase was highly significant (p < 0.01), while for U138 and D458, it was statistically significant (p < 0.05). These findings suggest that C5252 induces a higher level of cell death due to increased activation of caspase-3/7. To further explore why GBM cells were sensitive to C5252-induced apoptotic cell death, RNA deep sequencing was used to analyze the expression profiles between C5252- and R3616-infected A172 cells (data not shown). Notably, the expression of ciliary neurotrophic factor receptor α (CNTFRα) in A172 cells infected with C5252 showed a significant decrease compared to that of R3616 infection. The qPCR assay further confirmed that the CNTFRα mRNA level was significantly decreased in C5252-infected cells, particularly in A172, D54, U138, and U87 cells (Figure 3B). Thus, to explore whether CNTFRα can be considered as one of the factors responsible for increased apoptosis in C5252-infected cells, a rescue experiment was performed (Figures 3C and S4 A). As shown in Figure 3D, a significant difference was observed when transfected with a control plasmid pcDNA3.1 among C5252, R3616, and wild-type HSV-1(F)-infected cells, aligning with the findings presented in Figure 3A. However, when CNTFRα expression was rescued during infection through transfection (Pc-CNTFRα), no significant difference in caspase-3/7 activity was observed in the C5252, R3616, or HSV-1(F) infection groups. Furthermore, the knockdown of CNTFRα using small interfering RNA (siRNA), which mimics the C5252-induced downregulation of CNTFRα, resulted in a significant increase in caspase-3/7 activity (Figures 3E, 3F, and S4B). All these results provide further confirmation of the role of CNTFRα downregulation in enhancing caspase-3/7 activation during C5252 infection. CNTFRα is the receptor of CNTF, which promotes the survival of various neuronal cell populations, including glioma cells.21 Furthermore, the CNTF/CNTFR pathway exerts an anti-apoptotic effect by suppressing caspase-3 activation, and knockdown of CNTFRα significantly increased cell apoptosis in glioma cells.22,23 Thus, the downregulation of CNTFRα by C5252 infection, leading to increased caspase activation, contributes to its higher cytotoxic activity.
Figure 3.

Caspase-3/7 activity and CNTFRα expression during C5252 infection in glioblastoma cells
(A) Caspase-3/7 activity in glioblastoma cells. Caspase-3/7 activity was assessed at 8 h post-infection (hpi) with HSV-1(F), R3616, or C5252 in glioblastoma cells. Luminescence-based measurements are expressed as relative light units (RLUs). Data are from three independent trials and presented as mean ± SD. Statistically significant differences between C5252 and R3616 indicated (N.S. p > 0.05, ∗p < 0.05, and ∗∗p < 0.01). (B) CNTFRα mRNA levels in glioblastoma cells. Glioblastoma cells were infected with 0.1 PFU of HSV-1(F), R3616, or C5252 per cell. At 48 hpi, mRNA levels of CNTFRα were measured and normalized to HSV-1(F)-infected cells. Data are from three independent trials and presented as mean ± SD. Statistically significant differences between C5252 and R3616 are indicated (∗p < 0.05). (C) CNTFRα overexpression in glioblastoma cells. CNTFRα overexpression was achieved using the Pc-CNTFRα plasmid. Real-time PCR assessed the fold change in CNTFRα mRNA levels compared to control (Pc-DNA3.1)-transfected cells. Statistically significant differences between Pc-CNTFRα and Pc-DNA3.1 are indicated (∗p < 0.05 and ∗∗p < 0.01). (D) Caspase-3/7 activity in overexpressed CNTFRα glioblastoma cells. After plasmid transfection, cells were infected with 1.0 PFU of HSV-1(F), R3616, or C5252 per cell. Caspase-3/7 activity was assessed 24 h post-infection. Data are from three independent trials and presented as mean ± SD. Statistically significant differences between C5252 and R3616 are indicated (∗p < 0.05 and ∗∗p < 0.01). (E) CNTFRα knockdown in glioblastoma cells. Glioblastoma cells were transfected with siRNA targeting CNTFRα (si-CNTFRα) or non-target siRNA (si-NT). CNTFRα downregulation efficiency was assessed by real-time PCR. Statistically significant differences between si-CNTFRα and si-NT are indicated (∗p < 0.05 and ∗∗p < 0.01). (F) Caspase-3/7 activity in CNTFRα knockdown glioblastoma cells. Caspase-3/7 activity was measured using the Caspase-Glo 3/7 kit in CNTFRα knockdown glioblastoma cells. Results are presented as RLUs. Data are from three independent trials and presented as mean ± SD. Statistically significant differences between C5252 and R3616 are indicated (∗p < 0.05 and ∗∗p < 0.01).
The safety evaluation of C5252 in vivo
To assess the toxicity of oHSV C5252 in the intracranial cavity, BALB/c mice were grouped and received a single intracranial injection of oHSV C5252 with an initial dose level of 1.5 × 102 plaque-forming units (PFUs), which was then escalated by one-log increments until the dose of 1.5 × 105 PFUs. After the intracranial injection, the animals were observed daily for 14 days, and survival data were analyzed to calculate LD50. As previous reported,24 R3616 was used as a positive control. As shown in Figure 4A, the LD50 values of both oHSV C5252 and R3616 are over 1.5 × 105 PFUs, while that of wild-type HSV-1(F) is only 1.88 × 102 PFUs, indicating an over 800-fold attenuation when compared to the wild-type virus. Furthermore, there were no changes in physical appearance or behavioral abnormalities observed in the C5252- or R3616-treated animals, indicating that C5252 has a comparable safety profile to R3616.
Figure 4.

The safety evaluation of C5252 in vivo
(A) The results of LD50 after intracranial injection. Neurovirulence was observed after intracranial administration of HSV-1(F), R3616, and C5252. LD50 values were determined for each group. LD50 for C5252 exceeded the maximum administered dose (1.5 × 105 PFUs/animal), while HSV-1(F) had an LD50 of 1.88 × 102 PFUs/animal. (B) Quantification of viral DNA expression in mice TG during latency. BALB/c mice were infected with HSV-1(F) or C5252 via the corneal route. TGs were extracted at various time points, and viral genome DNA copy numbers were quantified and normalized to cellular DNA. (C) TGs excised 30 days after inoculation of HSV-1(F) were processed immediately or after 24 h of incubation with anti-NGF antibody. Fold changes in mRNA levels encoding ICP27, TK, UL41, VP16, and LAT compared to 0 h are shown (∗p < 0.05 and ∗∗p < 0.01). (D) TGs excised 30 days after inoculation of C5252 were processed immediately or after 24 h of incubation with anti-NGF antibody. Fold changes in mRNA levels encoding ICP27, TK, UL41, VP16, and LAT compared to 0 h are shown. Data are presented as mean ± SD.
It is important to investigate whether C5252, as an oHSV, can establish latency in neurons. Therefore, we employed the commonly used mouse trigeminal ganglion (TG) infection model by corneal inoculation25,26 to assess the latency and reactivation properties of C5252. BALB/c mice were grouped and inoculated with C5252 and HSV-1(F). TG was extracted at indicated time points, and DNA was quantified using qPCR to determine the viral DNA copy number, as illustrated in Figure 4B. The wild-type HSV-1(F) virus exhibited the classic latency pattern, with viral DNA levels reaching a peak within 14 days before declining. In contrast, C5252 displayed a distinct pattern compared to HSV-1(F); viral DNA levels remained below the limit of quantitation (44 copies/50 ng DNA) at all measured time points.
Furthermore, TGs were excised at 30 days post-infection and immediately incubated in a medium containing an anti-nerve growth factor (NGF) Ab for 24 h to induce reactivation from latency. Total RNA was extracted, and mRNA levels for viral α (ICP27), β (TK), and γ (UL41, VP16) gene products, as well as latency-associated transcript (LAT), were quantified as described in the materials and methods. The LAT regulates the establishment of the latent state and is required for >65% of the latent infections established by HSV-1. Moreover, LAT can protect sensory neurons and enhance the establishment of latency in the peripheral nervous system.27 The results showed a dramatic increase in ICP27, TK, UL41, and VP16 mRNAs and a decrease in LAT in HSV-1(F)-infected samples after 24 h of incubation with anti-NGF compared to the excision time (0 h), indicating reactivation from latency (Figure 4C). However, there was only a slight increase observed in the levels of ICP27 and TK mRNA in C5252 samples, while UL41 and VP16 mRNA were not detectable (Figure 4D). All these results suggested C5252’s inability to establish latency and reactivate, consistent with a previous report in which selective deletion of the γ34.5 gene abolishes this capacity.28 Taken together, these findings suggest that C5252 has a lower toxicity and diminished ability to establish latency and reactivate in the brain, which makes it a safe candidate for the treatment of GBM.
The anti-tumor efficacy of C5252 in subcutaneous and orthotopic GBM models
In vitro studies have shown that C5252 has higher cytotoxic activity against GBM cells than R3616. To further evaluate the anti-tumor activity of C5252 in vivo, U87 and D54 subcutaneous xenografts were utilized. Intratumoral injections of C5252 and R3616 were administered once a week, and tumor volume was measured. As shown in Figures 5A and 5B, C5252 was significantly more effective than R3616 in reducing tumor volume in both models, consistent with the in vitro findings. In the next step, the luciferase (Luc) gene-expressing human U-87MG-Luc cells were intracranially injected into nude mice to create an orthotopic model of human GBM . Two weeks after tumor inoculation, C5252 or phosphate-buffered saline (PBS) as a vehicle control was intracerebrally injected. Tumor progression was monitored by Luc-based imaging (Figures 5C and S5A). C5252 treatment significantly inhibited the progression of GBM and prolonged the survival of the tumor-bearing mice (Figure 5D). The anti-tumor activity was further validated in an immunocompetent orthotopic mouse model treated with murinized C8282 virus. Murine GBM CT-2A cells expressing GFP and Luc were intracranially injected into C57BL/6 mice to establish an orthotopic model of murine GBM, as described in the materials and methods. C8282 or PBS as a vehicle control was intracerebrally injected once a week three times. Tumor progression and survival rates were monitored (Figures 5E and S5B). The results showed that C8282 treatment markedly inhibited the progression of GBM and prolonged the survival of the tumor-bearing mice (Figure 5F).
Figure 5.

The anti-tumor therapeutic efficacy of C5252 in a subcutaneous glioblastoma model and an orthotopic glioblastoma model
(A and B) Subcutaneous glioblastoma model. Subcutaneous U87 (A) and D54 (B) tumors were established in BALB/c-derived nude mice. Tumor-bearing mice were treated with intratumoral injections of control, R3616 (5 × 106 PFUs/animal), or C5252 (5 × 106 PFUs/animal) on specified days. Tumor volumes are represented as mean ± SD for 6 animals per group. Statistically significant differences between C5252 and R3616 group are indicated (∗p < 0.05). (C) C5252 in orthotopic tumor model. Female BALB/c nude mice received intracerebral inoculation with U-87MG-Luc tumor cells. Mice were treated with intratumoral injections of C5252 (3 × 105 PFUs/animal in 5 μL) at specific intervals. Tumor volume (ROI) is shown as mean ± SD. Statistically significant differences between C5252 and control group are indicated (∗p < 0.05). The Luc images are presented in Figure S5A. (D) Survival curves. Survival curves demonstrating the significant survival benefit of C5252 treatment are shown. Statistically significant differences between C5252 and control group are indicated (∗∗p < 0.01) using Kaplan-Meier analysis. (E) C8282 in orthotopic tumor model. Female C57BL/6 mice received intracerebral inoculation with CT-2A-GFP-Luc tumor cells (2 × 103 cells/animal). Mice were treated with intratumoral injections of C8282 (1 × 105 PFUs/animal in 5 μL) at specific intervals. Tumor volume (ROI) is shown as mean ± SD. The images are presented in Figure S5B. (F) Survival curves. Survival curves demonstrating the significant survival benefit of C8282 treatment are shown. Statistically significant differences between C8282 and control group are indicated (∗∗p < 0.01) using Kaplan-Meier analysis.
The anti-tumor activity of the murine version of C5252 in immunocompetent GBM models
To assess the efficacy of our oHSV in a mouse model with fully functioning immune systems, we developed C8282, a variant of C5252 specifically designed for use in mice. To create C8282, the human IL-12 and anti-PD-1 Ab were replaced with their murine equivalents. In addition, we generated C1212 as a control virus to evaluate the combined effect of the immune-activating payload genes, IL-12 and anti-PD-1 Ab. Figure S1 provides details on the construction and identification of both C1212 and C8282. We performed intratumoral injections of C8282 every 3 days for a total of six injections, which significantly reduced tumor volume in the mouse CT-2A GBM syngeneic model compared to the backbone C1212 (Figure 6A). This suggests that the inclusion of IL-12 and anti-PD-1 Ab payload genes further enhanced the anti-tumor efficacy. On the final day of the experiment, the tumors in the mice were dissected, and the images are shown in Figure S6. To investigate whether C8282 treatment increased immune activation against tumors and resulted in greater anti-tumor activity, we used a CT-2A syngeneic mouse model and injected C1212, C8282 oHSV, or a vehicle control once. Tumors were excised at specified time points after single injection, and the production of key indicators of immune activation, interferon (IFN)-γ and tumor necrosis factor α (TNF-α), was measured using ELISA. Our results showed that the expression levels of IFN-γ and TNF-α in tumors peaked 1 day after virus injection and gradually decreased thereafter. Moreover, C8282 induced higher levels of IFN-γ and TNF-α compared to the C1212 group at 1 day post-infection and with a longer induction of TNF-α. These findings confirm that oHSV carrying IL-12 and anti-PD-1 Ab genes induced stronger immunotherapy against GBM.
Figure 6.

In vivo anti-tumor efficacy with syngeneic mouse model
(A) C57BL/6 mice were used as the syngeneic host for the CT-2A cell line. In tumor treatment studies, C57BL/6 mice (n = 6 per group) were treated with C8282 or C1212 virus (1 × 107 PFUs/mouse) via intratumoral injection on days 1, 4, 7, 10, 13, and 16 six times in total. The sizes of tumors were measured on days 1, 4, 7, 10, 13, 16, and 19 before every administration using a caliper, and the volume was calculated as (length × width2) × 0.5. The results are shown as mean tumor volume (mm3) ± SD (n = 6). Statistically significant differences between C8282 and C1212 group were analyzed by Student’s t test (two-tailed) using GraphPad Prism software (∗p < 0.05). On the final day of the experiment, the tumors in the mice were dissected, and the images are shown in the Figure S6. Murine IFN-γ (B) and murine TNF-α (C) production in CT-2A syngeneic mice infected with control (DPBS+10% glycerin), C1212, or C8282 (1 × 107 PFUs/mouse) on days 0. CT-2A tumor samples from each mouse were collected on days 1, 3, 6, and 14 following infection (n = 3 mice per time point) for the determination of murine IFN-γ and murine TNF-α levels by ELISA. The results are reported as pg/g of tumor.
Discussion
Pediatric and adult GBMs are highly aggressive brain tumors that continue to pose a major treatment challenge due to their high morbidity and mortality rates. However, recent developments in the field of engineered oHSV therapy have shown promise in treating these tumors. In this study, we constructed a novel oHSV C5252 by deleting the 15-kb IR region and both copies of the γ34.5 genes while also expressing human IL-12 and anti-PD-1 Ab. We then conducted preclinical safety and anti-tumor efficacy assessments of C5252, comparing it to the first-generation oHSV R3616, which also has two γ34.5 gene defects, and the backbone of G207, which is currently in clinical trials for GBM treatment in both children and adults. Our in vitro studies of C5252 revealed that though its replication ability was decreased, its cell-killing capability was significantly increased in GBM cells. Furthermore, C5252 infection specifically induced caspase-dependent apoptosis in GBM cells via downregulation of CNTFRα, although the underlying mechanisms need to be further explored. These findings suggest that C5252 has the potential to be a more effective treatment option for GBMs compared to other oHSV therapies currently in use. Future studies are needed to further explore the clinical efficacy and safety of C5252 in treating this devastating disease.
For the safety assessment of C5252, in vivo study showed that C5252 is highly attenuated with a LD50 over 1.5 × 105 PFUs, while that of HSV-1(F) is only 1.88 × 102 PFUs, which is consistent with another murine study in which 50% of mice died within 3–14 days after intracerebral inoculation of HSV-1(F) at a dose of 103 PFUs.29 What’s more, the viral DNA was detected to be lower than the limited detection during the latency phase, which further confirmed that the γ34.5-deleted oHSV was less capable of establishing latency or reactivating from latency.30 For the anti-tumor activity evaluation of C5252 in vivo, the data showed that oHSV C5252 has increased anti-tumor activity compared to control oHSV R3616 in both human orthotopic and subcutaneous GBM models mentioned. In the syngeneic mouse model, C8282, expressing murine IL-12 and anti-PD-1 Ab, also showed an enhanced reduction of tumor volume when compared to backbone virus C1212. The murine version of C8282 was constructed, as human IL-12 and anti-PD-1 Ab used in C5252 have no cross-reaction with their corresponding murine receptor or binding protein.31 The payload IL-12 is a cytokine with potent anti-tumor efficacy by induction of IFN-γ production and anti-angiogenic activity.32,33,34 In addition, IL-12 can reshape the TME, driving increased infiltration of proinflammatory CD4+ T cells and decreasing the numbers of regulatory T cells and the activation of the myeloid compartment.35,36 Giulia Agliardi et al. has shown that the local delivery of IL-12 may be an effective adjuvant for CAR-T cell therapy for GBM.37 The other payload of C5252, the immune checkpoint inhibitor (PD-1 Ab nivolumab), has been used in a phase 3 trial for GBM, which did not meet the primary endpoint.38 Thus, combination of PD-1 Ab with other therapies, including oHSV, e.g., M032 (NSC733972), is ongoing.
Taken together, our findings indicate that intracranial inoculation of C5252 and C8282 is safe and effective in GBM models. This preclinical safety data, along with the promising anti-tumor efficacy results, strongly support the translation of C5252 and the development of a phase 1 clinical trial in patients with recurrent or progressive GBM. C5252 expressing IL-12 and anti-PD-1 Ab represents a new generation of combination therapy for GBM with the potential to provide significant benefits to patients.
Materials and methods
Cell lines
Vero cells were purchased from the American Type Culture Collection. A172, D54, U138, U87, and D458 cells are commonly studied GBM cell lines that were obtained from JOINN Biologics. U-87MG-Luc tumor cells were purchased from Caliper Life Sciences. CT-2A cells were purchased from Shanghai Cell Bank. CT-2A-GFP-Luc tumor cells were purchased from Shenzhen SAFE Pharmaceutical. Vero cells were maintained in 5% (v/v) newborn bovine serum (NBCS, Gibco, Grand Island, NY, USA). GBM cell lines (A172, D54, U138, U87, D458, CT-2A, and CT-2A-GFP-Luc tumor cells) were maintained in DMEM (Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS; Gibco). U-87MG-Luc tumor cells were maintained in Eagle’s minimal essential medium (ATCC, Manassas, VA, USA) supplemented with 10% (v/v) FBS.
Virus
HSV-1(F), the prototype HSV-1 strain used in this laboratory. C5252 was constructed by deleting both copies of the γ34.5 gene and inserting an anti-human PD-1 Ab (PD-1 Fab) expression cassette between UL3 and UL4, and a modified IR region was replaced by an IL-12 expression cassette with the aid of a bacterial artificial chromosome system. C8282 is the murine version of C5252, insertion genes encoding murine IL-12 and anti-PD-1 Ab. C1212 is the backbone of C5252 oncolytic viruses. R3616 was reported elsewhere.24 The propagation of the virus and determination of its titer were performed using Vero cells.
Abs
Abs against ICP8, ICP0, and ICP4 were purchased from Abcam (Cambridge, UK). Abs against US11 have been described elsewhere.39 Abs against γ34.5 and ICP27 were kind gifts from Bernard Roizman (University of Chicago, Chicago, IL, USA). Additional Abs used in this study were anti-GAPDH (Sungene Biotech, Tianjin, China), anti-CNTFR (Abcam), and FLAG Tag Mouse monoclonal Ab (Beyotime, Shanghai, China).
Growth curve determination
Vero cells were seeded in a 6-well plate at densities of 1 × 106 cells per well. The cells were subsequently exposed to 0.1 PFU of HSV-1(F), R3616, or C5252 virus. The cells were harvested at 3, 6, 12, 24, and 48 hpi. Viral progeny was titrated by plaque assay following three freeze-thaw cycles.
Virus titer determination
A172, D54, U138, U87, or D458 cells were seeded in a 6-well plate at densities of 1 × 106 cells per well. The cells were subsequently exposed to 0.1 or 1.0 PFUs of HSV-1(F), R3616, or C5252 virus. The cells were harvested at 48 hpi. Viral progeny was titrated by plaque assay after three freeze-thaw cycles.
Plaque assay
The virus titer was determined on Vero cells. The growth curve sample and GBM cell lines samples were maintained in DMEM supplemented with 1% FBS plus 0.05% (w/v) human pooled immunoglobulin (WEIGUANG, Shenzhen, China) for 72 h. The cells were fixed with 4% (w/v) paraformaldehyde for 30 min, rinsed three times with PBS, and stained with Giemsa (DINGGUO, Beijing, China). The viral plaque was counted and visually confirmed.
Immunoblotting assays
The Vero cells were harvested at the indicated time points after infection by being scraped into the medium, rinsed with PBS, and solubilized in RIPA buffer (Beyotime) supplemented with PMSF (Beyotime) as specified by the manufacturer. Samples containing protein were denatured in 5×SDS loading buffer, boiled for 10 min, electrophoretically separated on a 10% denaturing polyacrylamide gel, electrically transferred to a nitrocellulose sheet, blocked, and then reacted overnight at 4°C with the appropriate primary Abs diluted in PBS with 1% non-fat milk. The protein bands were scanned with an Image Lab scanner (BioRad).
Human IL-12 p70 and anti-PD-1 Ab analysis
The human IL-12 p70 Quantikine ELISA Kit was purchased from R&D Systems (Minneapolis, MN, USA). The samples were quantified by ELISA according to the manufacturer’s instructions. Anti-PD-1 Ab analysis methods were reported elsewhere.40
CCK8 assay
The tumor cell-killing activity was assessed using a Cell Counting Kit 8 (CCK8) assay. Briefly, A172, D54, U138, U87, or D458 cells were plated in 96-well plates at 5 × 103 cells/well. The cells were infected with 0.1 or 1.0 PFUs/cell of HSV-1(F), R3616, or C5252 for 48 h. Cell viability was evaluated using CCK8 (Beyotime) according to the manufacturer’s instructions. The cell inhibition rate was calculated according to the following formula: cell inhibition rate (100%) = [(a value of target cell wells) − (a value of experimental wells)/(a value of target cell wells) − (a value of the blank wells)] × 100%. The assay was performed in triplicate.
Overexpression of CNTRFα
Overexpression of CNTFRα was achieved by utilizing the Pc-CNTFRα plasmid with a FLAG tag (YouBio, Hunan, China), while Pc-DNA3.1 served as the control plasmid. GBM cells were seeded in 12-well plates 1 day before transfection in DMEM containing 10% FBS. The following day, the aforementioned plasmids were transfected into the GBM cells using Lipofectamine 8000 (Beyotime), following the manufacturer’s instructions. After 48 h post-transfection, the cells were infected with 1 PFU of either R3616 or C5252 per cell.
Knockdown of CNTFRα in GBM cells by siRNA
The knockdown of CNTFRα was achieved using siRNAs (GenePharma, Shanghai, China). The sequences of the siRNA were as follows: CNTFRα siRNA (si-CNTFRα): 5′-CCUACAUUCCCAACACCUUTT-3′; and negative control (si-NT): 5′-UUCUCCGAACGUGUCACGUTT-3’.
The siRNA transfections were carried out using Lipofectamine 8000 (Beyotime) according to the manufacturer’s instructions.
Caspase-3/7 activity assays
A172, D54, U138, U87, or D458 cells were seeded at a density of 2 × 103 cells per well on 96-well plates and allowed to adhere overnight. Cells in triplicate wells were infected with 5 PFUs/cell of HSV-1(F), R3616, or C5252 virus, and the plates were incubated at 37°C with 5% CO2 for 8 h. After treatment, the Caspase-Glo 3/7 (Promega, Madison, WI, USA) was prepared according to the manufacturer’s guidelines, and 100 μL of the reagent was added per well and incubated for 1 h at room temperature in the dark. The luminescent signal was recorded by a multimode microplate reader (Synergy Multi-Mode Reader, BioTek).
RNA extraction and real-time PCR
A172, D54, U138, U87, or D458 cells were plated and infected with 0.1 PFU/cell of HSV-1(F), R3616, or C5252 for 48 h. The total RNAs from cells were isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). A total amount of 0.5 μg RNA was reverse transcribed to cDNA using the PCR RT kit (TOYOBO, Tokyo, Japan) according to the manufacturer’s instructions. Real-time PCR was performed using the StepOnePlus Real-Time PCR System (Applied Biosystems), and the amplifications were performed using the SYBR Premix (TaKaRa, Kusatsu, Japan). The mRNA expression levels were normalized by18S rRNA and analyzed using the 2−ΔΔCT method described elsewhere.41
The primers used for amplifications were as follows: CNTFRα-F: 5′-CTTACCCCAAGGGCTTCTACT-3′; CNTFRα-R: 5′-ACAGACCATAATTTTGGAGCCAT-3′; 18S-F: 5′-CTCAACACGGGAAACCTCAC-3′; and 18S-R: 5′-CGCTCCACCAACTAAGAACG-3′.
Animal models
All animal experiments were performed under protocols approved by the Institutional Animal Care and Use Committee of SAFE (Shenzhen, China) New Drug Research Technology Company. All mice used in this study were obtained from Vital River Laboratory Animal Technologies (Beijing, China).
Murine model assessment of neurovirulence following intracranial administration
Healthy female BALB/c mice with a body weight of about 18 g were selected randomly and divided into 3 groups (n = 8 per group): R3616 and C5252 groups (at the dosages of 1.5 × 105, 1.5 × 104, 1.5×103, and 1.5 × 102 PFUs/animal) and the HSV-1(F) group (at the dosages of 1.5 × 103, 1.5 × 102, 15, and 1.5 PFUs/animal). With an inserted syringe needle 3–4 mm below the point bregma, the test article was slowly injected into the brain. After intracranial injection, animals were observed daily for 14 days, and survival data were processed to calculate LD50.
Murine model of virus latent infection and reactivation
Five-week-old inbred female BALB/c mice were infected by 1 × 105 PFUs of HSV-1(F) virus and 1 × 106 PFUs of C5252 recombinant virus through the corneal route. On the indicated days after infection (1, 3, 7 14, and 30 days), TGs were extracted, and total DNA was isolated. Subsequently, a qPCR assay was performed using primers designed to target viral gD gene sequences to quantify the copy number of viral genome DNA. Bilateral trigeminal nerves were aseptic separated 30 days after infection, one side was cultured in 199V culture medium (Gibco,) containing 100 ng/mL anti-NGF Ab (Abcam) for 24 h to induce virus reactivation, and the other side of TG was treated as a control group (n = 4 per group). The TG RNA samples, either treated (24 h) or untreated (0 h) with an anti-NGF Ab, were extracted. Specific primers targeting HSV lytic genes (ICP27, VP16, TK, UL41) and LAT were used. The fold changes in the expression levels of these genes were calculated by comparing them to the baseline level at 0 h.
Murine model of subcutaneous GBM
The U87 and D54 tumor models were generated by subcutaneous implantation of 1 × 107 U87 cells and D54 cells, respectively, into BALB/c nude mouse flanks. When the mean tumor volumes reached about 80 mm3, the mice were randomly grouped and treated as indicated. In tumor treatment studies, BALB/c nude mice (n = 6 per group) were treated with R3616 or C5252 virus (5 × 106 PFUs/mouse) via intratumoral injection on days 1, 8, 15, and 22. The sizes of tumors were measured twice a week using a caliper, and the volume was calculated as (length × width2) × 0.5. The results are presented as the mean ± SD.
C5252 in murine model of orthotopic tumor
Female BALB/c nude mice were intracerebrally inoculated with 5 μL U-87MG-Luc tumor cells (2 × 105 cells/animal) in the left striatum. The mice were randomized into 2 groups of 8 mice in each group 2 weeks after inoculation according to IVIS (In Vivo Imaging Systems) results. Mice were treated with C5252 (3 × 105 PFUs/animal in 5 μL) every 3 days, 6 times in total. IVIS was performed once weekly to monitor tumor growth. Tumor volume (region of interest [ROI]) was presented as mean ± SD.
C8282 in murine model of orthotopic tumor
Female C57BL/6 mice were intracerebrally inoculated with 10 μL CT-2A-GFP-Luc tumor cells (2 × 103 cells/animal) 2 mm to the right of the anterior fontanelle and 0.5 mm anterior to the coronal suture. The mice were randomized into 2 groups of 8 mice in each group after 2 weeks with inoculation according to IVIS (In Vivo Imaging Systems) results. Mice were treated with C8282 (1 × 105 PFUs/animal in 5 μL) 3 times total at a frequency of once a week. IVIS was performed once weekly to monitor tumor growth. Tumor volume (ROI) was presented as mean ± SD.
Murine model of syngeneic mice
The syngeneic mice models of CT-2A were generated by subcutaneous implantation of 5 × 105 CT-2A cells into C57BL/6 mice (n = 6 per group). Once the mice bearing tumors reached an average volume of 80 mm3, they were treated as indicated. In tumor treatment studies, C57BL/6 mice were treated with C8282 or C1212 virus (1 × 107 PFUs/mouse) via intratumoral injection on days 1, 4, 7, 10, 13, and 16. The sizes of tumors were measured on days 1, 4, 7, 10, 13, 16, and 19 using a caliper, and the volume was calculated as (length × width2) × 0.5. The results are presented as the mean ± SD.
IFN-γ and TNF-α analysis
CT-2A tumor samples from each mouse were collected on days 1, 3, 6, and 14 (each time point n = 3) following a single injection for the determination of mouse IFN-γ (Beyotime) and mouse TNF-α (Beyotime) levels by ELISA, according to the manufacturer’s instructions.
Data and code availability
The data and biological materials that support the findings of this study are available from the corresponding author upon request.
Acknowledgments
This research were supported by the Science and Technology Innovation Commission of Shenzhen, grant number JCYJ20220818102618040, and Longhua District Science and Innovation Commission Project Grants of Shenzhen, grant number 10162A20221027B1FA526.
Author contributions
Conceptualization, X.C., J.Z., and G.G.Z.; methodology, L.W. and X.Z.; validation, L.W. and X.Z., formal analysis, L.W.; investigation, L.W., X.Z., Y.L., and Y.H.; writing – original draft preparation, L.W., J.Z., and X.C.; writing – review & editing, L.W., J.Z., X.C., X.Z., G.G.Z., P.R., and Y.C.; supervision, J.Z., X.C., and G.G.Z.; project administration, J.Z., X.C., and G.G.Z. All authors read and agreed to the published version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omton.2024.200799.
Contributor Information
Peigen Ren, Email: pg.ren@siat.ac.cn.
Jing Zhao, Email: jingzhao@siitm.org.cn.
Grace Guoying Zhou, Email: zhoug@siitm.org.cn.
Supplemental information
References
- 1.Andreansky S.S., He B., Gillespie G.Y., Soroceanu L., Markert J., Chou J., Roizman B., Whitley R.J. The application of genetically engineered herpes simplex viruses to the treatment of experimental brain tumors. Proc. Natl. Acad. Sci. USA. 1996;93:11313–11318. doi: 10.1073/pnas.93.21.11313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gatto L., Franceschi E., Tosoni A., Di Nunno V., Bartolini S., Brandes A.A. Glioblastoma treatment slowly moves toward change: novel druggable targets and translational horizons in 2022. Expert Opin. Drug Discov. 2023;18:269–286. doi: 10.1080/17460441.2023.2174097. [DOI] [PubMed] [Google Scholar]
- 3.Friedman G.K., Pressey J.G., Reddy A.T., Markert J.M., Gillespie G.Y. Herpes simplex virus oncolytic therapy for pediatric malignancies. Mol. Ther. 2009;17:1125–1135. doi: 10.1038/mt.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Metzger S., Weiser A., Gerber N.U., Otth M., Scheinemann K., Krayenbühl N., Grotzer M.A., Guerreiro Stucklin A.S. Central nervous system tumors in children under 5 years of age: a report on treatment burden, survival and long-term outcomes. J. Neuro Oncol. 2022;157:307–317. doi: 10.1007/s11060-022-03963-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan G., Wang Y., Chen J., Zheng W., Liu C., Chen S., Wang L., Luo J., Li Z. Advances in drug development for targeted therapies for glioblastoma. Med. Res. Rev. 2020;40:1950–1972. doi: 10.1002/med.21676. [DOI] [PubMed] [Google Scholar]
- 6.Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., Cahill D.P., Nahed B.V., Curry W.T., Martuza R.L., et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McGranahan N., Furness A.J.S., Rosenthal R., Ramskov S., Lyngaa R., Saini S.K., Jamal-Hanjani M., Wilson G.A., Birkbak N.J., Hiley C.T., et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Touat M., Li Y.Y., Boynton A.N., Spurr L.F., Iorgulescu J.B., Bohrson C.L., Cortes-Ciriano I., Birzu C., Geduldig J.E., Pelton K., et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature. 2020;580:517–523. doi: 10.1038/s41586-020-2209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu W., Klockow J.L., Zhang M., Lafortune F., Chang E., Jin L., Wu Y., Daldrup-Link H.E. Glioblastoma multiforme (GBM): An overview of current therapies and mechanisms of resistance. Pharmacol. Res. 2021;171:105780. doi: 10.1016/j.phrs.2021.105780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chambers M.R., Bentley R.T., Crossman D.K., Foote J.B., Koehler J.W., Markert J.M., Omar N.B., Platt S.R., Self D.M., Shores A., et al. The One Health Consortium: Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Combination With a Checkpoint Inhibitor in Canine Patients With Sporadic High Grade Gliomas. Front. Surg. 2020;7:59. doi: 10.3389/fsurg.2020.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jahan N., Ghouse S.M., Martuza R.L., Rabkin S.D. In Situ Cancer Vaccination and Immunovirotherapy Using Oncolytic HSV. Viruses. 2021;13:1740. doi: 10.3390/v13091740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Powles T., Rosenberg J.E., Sonpavde G.P., Loriot Y., Durán I., Lee J.L., Matsubara N., Vulsteke C., Castellano D., Wu C., et al. Enfortumab Vedotin in Previously Treated Advanced Urothelial Carcinoma. N. Engl. J. Med. 2021;384:1125–1135. doi: 10.1056/NEJMoa2035807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Todo T., Ito H., Ino Y., Ohtsu H., Ota Y., Shibahara J., Tanaka M. Intratumoral oncolytic herpes virus G47Δ for residual or recurrent glioblastoma: a phase 2 trial. Nat. Med. 2022;28:1630–1639. doi: 10.1038/s41591-022-01897-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Totsch S.K., Schlappi C., Kang K.D., Ishizuka A.S., Lynn G.M., Fox B., Beierle E.A., Whitley R.J., Markert J.M., Gillespie G.Y., et al. Oncolytic herpes simplex virus immunotherapy for brain tumors: current pitfalls and emerging strategies to overcome therapeutic resistance. Oncogene. 2019;38:6159–6171. doi: 10.1038/s41388-019-0870-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao J., Yan Q. Cancer Epigenetics, Tumor Immunity, and Immunotherapy. Trends Cancer. 2020;6:580–592. doi: 10.1016/j.trecan.2020.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakashima H., Nguyen T., Chiocca E.A. Combining HDAC inhibitors with oncolytic virotherapy for cancer therapy. Oncolytic Virother. 2015;4:183–191. doi: 10.2147/OV.S66081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patel D.M., Foreman P.M., Nabors L.B., Riley K.O., Gillespie G.Y., Markert J.M. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients with Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Hum. Gene Ther. Clin. Dev. 2016;27:69–78. doi: 10.1089/humc.2016.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friedman G.K., Bernstock J.D., Chen D., Nan L., Moore B.P., Kelly V.M., Youngblood S.L., Langford C.P., Han X., Ring E.K., et al. Enhanced Sensitivity of Patient-Derived Pediatric High-Grade Brain Tumor Xenografts to Oncolytic HSV-1 Virotherapy Correlates with Nectin-1 Expression. Sci. Rep. 2018;8:13930. doi: 10.1038/s41598-018-32353-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie X., Lv J., Zhu W., Tian C., Li J., Liu J., Zhou H., Sun C., Hu Z., Li X. The combination therapy of oncolytic HSV-1 armed with anti-PD-1 antibody and IL-12 enhances anti-tumor efficacy. Transl. Oncol. 2022;15:101287. doi: 10.1016/j.tranon.2021.101287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dambach M.J., Trecki J., Martin N., Markovitz N.S. Oncolytic viruses derived from the gamma34.5-deleted herpes simplex virus recombinant R3616 encode a truncated UL3 protein. Mol. Ther. 2006;13:891–898. doi: 10.1016/j.ymthe.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Weis J., Schönrock L.M., Züchner S.L., Lie D.C., Sure U., Schul C., Stögbauer F., Ringelstein E.B., Halfter H. CNTF and its receptor subunits in human gliomas. J. Neuro Oncol. 1999;44:243–253. doi: 10.1023/a:1006303221064. [DOI] [PubMed] [Google Scholar]
- 22.Adamus G., Sugden B., Shiraga S., Timmers A.M., Hauswirth W.W. Anti-apoptotic effects of CNTF gene transfer on photoreceptor degeneration in experimental antibody-induced retinopathy. J. Autoimmun. 2003;21:121–129. doi: 10.1016/s0896-8411(03)00092-1. [DOI] [PubMed] [Google Scholar]
- 23.Fan K., Wang X., Zhang J., Ramos R.I., Zhang H., Li C., Ye D., Kang J., Marzese D.M., Hoon D.S.B., Hua W. Hypomethylation of CNTFRalpha is associated with proliferation and poor prognosis in lower grade gliomas. Sci. Rep. 2017;7:7079. doi: 10.1038/s41598-017-07124-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chou J., Kern E.R., Whitley R.J., Roizman B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science. 1990;250:1262–1266. doi: 10.1126/science.2173860. [DOI] [PubMed] [Google Scholar]
- 25.Du T., Zhou G., Roizman B. Induction of apoptosis accelerates reactivation of latent HSV-1 in ganglionic organ cultures and replication in cell cultures. Proc. Natl. Acad. Sci. USA. 2012;109:14616–14621. doi: 10.1073/pnas.1212661109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du T., Zhou G., Roizman B. HSV-1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency-associated transcript and miRNAs. Proc. Natl. Acad. Sci. USA. 2011;108:18820–18824. doi: 10.1073/pnas.1117203108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thompson R.L., Sawtell N.M. The herpes simplex virus type 1 latency-associated transcript gene regulates the establishment of latency. J. Virol. 1997;71:5432–5440. doi: 10.1128/jvi.71.7.5432-5440.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitley R.J., Kern E.R., Chatterjee S., Chou J., Roizman B. Replication, establishment of latency, and induced reactivation of herpes simplex virus gamma 1 34.5 deletion mutants in rodent models. J. Clin. Invest. 1993;91:2837–2843. doi: 10.1172/JCI116527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sundaresan P., Hunter W.D., Martuza R.L., Rabkin S.D. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation in mice. J. Virol. 2000;74:3832–3841. doi: 10.1128/jvi.74.8.3832-3841.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He B., Gross M., Roizman B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA. 1997;94:843–848. doi: 10.1073/pnas.94.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hellums E.K., Markert J.M., Parker J.N., He B., Perbal B., Roizman B., Whitley R.J., Langford C.P., Bharara S., Gillespie G.Y. Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro Oncol. 2005;7:213–224. doi: 10.1215/S1152851705000074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bramson J.L., Hitt M., Addison C.L., Muller W.J., Gauldie J., Graham F.L. Direct intratumoral injection of an adenovirus expressing interleukin-12 induces regression and long-lasting immunity that is associated with highly localized expression of interleukin-12. Hum. Gene Ther. 1996;7:1995–2002. doi: 10.1089/hum.1996.7.16-1995. [DOI] [PubMed] [Google Scholar]
- 33.Manetti R., Parronchi P., Giudizi M.G., Piccinni M.P., Maggi E., Trinchieri G., Romagnani S. Natural killer cell stimulatory factor (interleukin 12 [IL-12]) induces T helper type 1 (Th1)-specific immune responses and inhibits the development of IL-4-producing Th cells. J. Exp. Med. 1993;177:1199–1204. doi: 10.1084/jem.177.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Voest E.E., Kenyon B.M., O'Reilly M.S., Truitt G., D'Amato R.J., Folkman J. Inhibition of angiogenesis in vivo by interleukin 12. J. Natl. Cancer Inst. 1995;87:581–586. doi: 10.1093/jnci/87.8.581. [DOI] [PubMed] [Google Scholar]
- 35.Tugues S., Burkhard S.H., Ohs I., Vrohlings M., Nussbaum K., Vom Berg J., Kulig P., Becher B. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015;22:237–246. doi: 10.1038/cdd.2014.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kueberuwa G., Kalaitsidou M., Cheadle E., Hawkins R.E., Gilham D.E. CD19 CAR T Cells Expressing IL-12 Eradicate Lymphoma in Fully Lymphoreplete Mice through Induction of Host Immunity. Mol. Ther. Oncolytics. 2018;8:41–51. doi: 10.1016/j.omto.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agliardi G., Liuzzi A.R., Hotblack A., De Feo D., Núñez N., Stowe C.L., Friebel E., Nannini F., Rindlisbacher L., Roberts T.A., et al. Intratumoral IL-12 delivery empowers CAR-T cell immunotherapy in a pre-clinical model of glioblastoma. Nat. Commun. 2021;12:444. doi: 10.1038/s41467-020-20599-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reardon D.A., Brandes A.A., Omuro A., Mulholland P., Lim M., Wick A., Baehring J., Ahluwalia M.S., Roth P., Bähr O., et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020;6:1003–1010. doi: 10.1001/jamaoncol.2020.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roller R.J., Roizman B. The herpes simplex virus 1 RNA binding protein US11 is a virion component and associates with ribosomal 60S subunits. J. Virol. 1992;66:3624–3632. doi: 10.1128/jvi.66.6.3624-3632.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan R., Zhou X., Chen X., Liu X., Tang Y., Ma J., Wang L., Liu Z., Zhan B., Chen H., et al. Enhancement of oncolytic activity of oHSV expressing IL-12 and anti PD-1 antibody by concurrent administration of exosomes carrying CTLA-4 miRNA. Immunotherapy. 2019;5:154. [Google Scholar]
- 41.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data and biological materials that support the findings of this study are available from the corresponding author upon request.