

Abstract
Motivation
Over the past decade, single-cell transcriptomic technologies have experienced remarkable advancements, enabling the simultaneous profiling of gene expressions across thousands of individual cells. Cell type identification plays an essential role in exploring tissue heterogeneity and characterizing cell state differences. With more and more well-annotated reference data becoming available, massive automatic identification methods have sprung up to simplify the annotation process on unlabeled target data by transferring the cell type knowledge. However, in practice, the target data often include some novel cell types that are not in the reference data. Most existing works usually classify these private cells as one generic ‘unassigned’ group and learn the features of known and novel cell types in a coupled way. They are susceptible to the potential batch effects and fail to explore the fine-grained semantic knowledge of novel cell types, thus hurting the model’s discrimination ability. Additionally, emerging spatial transcriptomic technologies, such as in situ hybridization, sequencing and multiplexed imaging, present a novel challenge to current cell type identification strategies that predominantly neglect spatial organization. Consequently, it is imperative to develop a versatile method that can proficiently annotate single-cell transcriptomics data, encompassing both spatial and non-spatial dimensions.
Results
To address these issues, we propose a new, challenging yet realistic task called universal cell type identification for single-cell and spatial transcriptomics data. In this task, we aim to give semantic labels to target cells from known cell types and cluster labels to those from novel ones. To tackle this problem, instead of designing a suboptimal two-stage approach, we propose an end-to-end algorithm called scBOL from the perspective of Bipartite prototype alignment. Firstly, we identify the mutual nearest clusters in reference and target data as their potential common cell types. On this basis, we mine the cycle-consistent semantic anchor cells to build the intrinsic structure association between two data. Secondly, we design a neighbor-aware prototypical learning paradigm to strengthen the inter-cluster separability and intra-cluster compactness within each data, thereby inspiring the discriminative feature representations. Thirdly, driven by the semantic-aware prototypical learning framework, we can align the known cell types and separate the private cell types from them among reference and target data. Such an algorithm can be seamlessly applied to various data types modeled by different foundation models that can generate the embedding features for cells. Specifically, for non-spatial single-cell transcriptomics data, we use the autoencoder neural network to learn latent low-dimensional cell representations, and for spatial single-cell transcriptomics data, we apply the graph convolution network to capture molecular and spatial similarities of cells jointly. Extensive results on our carefully designed evaluation benchmarks demonstrate the superiority of scBOL over various state-of-the-art cell type identification methods. To our knowledge, we are the pioneers in presenting this pragmatic annotation task, as well as in devising a comprehensive algorithmic framework aimed at resolving this challenge across varied types of single-cell data. Finally, scBOL is implemented in Python using the Pytorch machine-learning library, and it is freely available at https://github.com/aimeeyaoyao/scBOL.
Keywords: single-cell and spatial transcriptomics data, universal cell type identification, bipartite prototype alignment
INTRODUCTION
The capability to perform high-throughput assays for determining gene expression profiles at the resolution of individual neural cells has only been realized within the preceding decade. This advancement has emanated from the confluence of next-generation sequencing technologies, the refinement of molecular biology techniques for subnanomolar quantities of starting material and the expansion of computational analyses to manage the increased dataset sizes from numerous samples [1, 2]. Currently, the most developed technique for single-cell investigations, which has been integrated with genome-scale analysis, is single-cell RNA sequencing (scRNA-seq) [3]. Despite confronting challenges such as data sparsity and lower detection efficiency, scRNA-seq has proven invaluable, affording the quantification of several thousand transcripts across thousands of individual cells within a solitary experimental framework [4]. The swift advancement in scRNA-seq technologies has precipitated a plethora of discoveries within a remarkably brief period. These discoveries encompass the identification of elusive cell populations [5], the elucidation of intricate gene regulatory networks in action [6] and the detailed delineation of cellular transitions throughout organismal development [7]. Additionally, emergent spatial transcriptomics techniques, such as in situ hybridization-based [8, 9], sequencing-based [10, 11] and imaging-based [12, 13], are now layering these genetic data with a crucial spatial dimension. These technologies differ in terms of cell resolution, spatial dimension and sequencing throughput. Such spatially resolved transcriptomics data reveal the interplay between cellular spatial arrangement and functionality, an aspect pivotal for delving into the aberrant cellular networks located adjacent to pathological features [14, 15]. By amalgamating high-fidelity gene expression snapshots with spatial context, we stand on the verge of significantly enhancing our understanding of single-cell biology, potentially revolutionizing the field [16, 17].
Cell type identification is a fundamental procedure in analyzing single-cell and spatial transcriptomics data since many subsequent downstream explorations are based on the specific cell types, such as cell–cell communications and gene–gene interactions [18, 19]. The traditional cell type identification procedure first clusters the cell population and then finds the marker genes specific to each cluster, finally manually annotating the cells according to the ontological functions of their marker genes [20, 21]. For example, Seurat uses the community discovery algorithm Louvain to cluster the shared neighbor graph between cells and performs differential expression analysis based on the non-parametric Wilcoxon rank sum test [22]. STAGATE adopts an attention mechanism to adaptively learn the similarity of neighboring cells/spots, and an optional cell type-aware module to integrate the pre-clustering of gene expressions [23]. GraphST applies graph self-supervised contrastive learning to learn informative and discriminative cell representations by minimizing the embedding distance between spatially adjacent cells [24]. However, on the one hand, the use of marker genes differs greatly in various experiments, making it difficult for us to directly compare associated cell types. On the other hand, with the increasing size of the genetic data [25], the manual task of finding marker genes to annotate cells becomes increasingly burdensome and time-consuming. Moreover, for non-experts, it is not a trivial matter to understand the functional biology of marker genes, because ample knowledge of these genes usually requires a large amount of literature review and long-term accumulation [26, 27].
As the Human Cell Atlas and Mouse Organogenesis Cell Atlas projects move forward, more and more well-established large-scale annotated datasets have emerged in the single-cell community [28, 29]. For example, Tabula Muris [30] serves as an extensive repository of single-cell transcriptomic data derived from the model organism, Mus musculus. This collection encompasses approximately 100 000 cells sourced from 20 different organs and tissues. Using these well-labeled datasets as the reference data, researchers turn to develop automatic cell type identification algorithms based on cell classification techniques to annotate cell types in the unlabeled target data [31, 32]. They transfer the cell type label knowledge learned from the reference data to the target data. Specifically, when a laboratory needs to annotate a newly acquired dataset from pancreatic tissue sequenced by 10x Genomics, they could search for another existing labeled dataset from the same tissue yet different sequencing techniques like Smart-seq2 to facilitate the cell type annotation process. Moreover, in the common scenario where only a minority of cells in a dataset can be readily annotated in advance due to the availability of clear markers or prior characteristics, we could also use them as a reference to guide the remaining annotation process for the entire cell population. In general, this automatic annotation strategy through label transfer makes the original time-consuming process much more convenient and effective.
Assume that 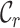 and
and 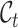 represent the label sets of reference and target data, respectively. Earlier developed methods are based on the close-set assumption, i.e.
represent the label sets of reference and target data, respectively. Earlier developed methods are based on the close-set assumption, i.e. 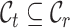 [33–35]. For example, scSemiCluster utilizes structure similarity regularization on the reference data to restrict the clustering solutions of the target data [36]. To be honest, this assumption is difficult to satisfy for data in the real scenario, because the target data usually contain extra cell types absent from the reference data in practical applications [32, 37, 38]. For ease of writing, the cell types shared by reference and target data are called common cell types, while the cell types only in target data are called novel or private cell types. To cover a more realistic situation, the partial overlap scenario is introduced, i.e.
[33–35]. For example, scSemiCluster utilizes structure similarity regularization on the reference data to restrict the clustering solutions of the target data [36]. To be honest, this assumption is difficult to satisfy for data in the real scenario, because the target data usually contain extra cell types absent from the reference data in practical applications [32, 37, 38]. For ease of writing, the cell types shared by reference and target data are called common cell types, while the cell types only in target data are called novel or private cell types. To cover a more realistic situation, the partial overlap scenario is introduced, i.e. 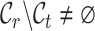 ,
, 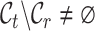 ,
, 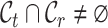 , and several methods are proposed to adapt this task [39–44]. For example, MARS introduces a meta-learning framework to obtain cell-type knowledge by identifying commonalities in the meta-dataset [40]. scArches uses transfer learning and parameter optimization to enable reference building and contextualization of target data [45]. scNym applies semi-supervised learning and adversarial learning techniques to integrate gene expression knowledge from different datasets [46]. SpaGCN aggregates gene expression of each spot from its neighboring spots to enable the identification of spatial domains with coherent expression and histology [47]. Spatial-ID combines the existing knowledge of reference scRNA-seq data and the spatial information of spatial transcriptomics data to achieve supervised cell typing [48–50]. However, one major drawback of these methods is that they annotate target cells from novel cell types using a generic ‘unassigned’ label, without further fine-grained procedure for them. It might be argued that we could use these methods to first find ‘unassigned’ cells and then apply clustering techniques to divide them into groups. Unfortunately, our experiments would show that such a two-stage approach does not work well. Recently, STELLAR, a geometric deep learning method for spatial transcriptomics datasets, was proposed to automatically assign cells to cell types present in the reference data and discover novel cell types and cell states in the target data [51]. Although it shows some promising results, STELLAR takes little effort to align the common cell types and separate the novel cell types from them in the feature space. Besides, the lack of label supervision for novel cell types will cause the model to be biased toward the known cell types, thus further generating an imbalanced prediction state. More importantly, few algorithms in the community are available for unified cell annotation and clustering on both single-cell and spatial transcriptomics data.
, and several methods are proposed to adapt this task [39–44]. For example, MARS introduces a meta-learning framework to obtain cell-type knowledge by identifying commonalities in the meta-dataset [40]. scArches uses transfer learning and parameter optimization to enable reference building and contextualization of target data [45]. scNym applies semi-supervised learning and adversarial learning techniques to integrate gene expression knowledge from different datasets [46]. SpaGCN aggregates gene expression of each spot from its neighboring spots to enable the identification of spatial domains with coherent expression and histology [47]. Spatial-ID combines the existing knowledge of reference scRNA-seq data and the spatial information of spatial transcriptomics data to achieve supervised cell typing [48–50]. However, one major drawback of these methods is that they annotate target cells from novel cell types using a generic ‘unassigned’ label, without further fine-grained procedure for them. It might be argued that we could use these methods to first find ‘unassigned’ cells and then apply clustering techniques to divide them into groups. Unfortunately, our experiments would show that such a two-stage approach does not work well. Recently, STELLAR, a geometric deep learning method for spatial transcriptomics datasets, was proposed to automatically assign cells to cell types present in the reference data and discover novel cell types and cell states in the target data [51]. Although it shows some promising results, STELLAR takes little effort to align the common cell types and separate the novel cell types from them in the feature space. Besides, the lack of label supervision for novel cell types will cause the model to be biased toward the known cell types, thus further generating an imbalanced prediction state. More importantly, few algorithms in the community are available for unified cell annotation and clustering on both single-cell and spatial transcriptomics data.
Given the analysis presented, the motivation for developing our universal annotation framework is 3-fold: Firstly, the challenges inherent in conventional manual annotation methodologies, which rely heavily on marker gene identification, coupled with the rich repository of thoroughly annotated databases, underscore the necessity for automated annotation solutions. Secondly, there is an evident demand not only for the assignment of known cell type labels in reference data but also for a more nuanced partition of unidentified cell types in the target data. This calls for a sophisticated, integrated algorithm capable of fulfilling both substantive requirements. Thirdly, the limitation of prevailing annotation strategies to single data types severely constrains their utility in broader practical applications. A model competent in concurrently analyzing diverse data forms would substantially economize resources and enhance user-friendliness. Therefore, here we propose an end-to-end algorithm called scBOL, a flexible deep-learning tool for universal cell type identification for both single-cell and spatial transcriptomics data. Using the well-labeled reference data, scBOL transfers its annotations to the part of the aligned target data and clusters the cells of novel cell types that only existed in the target data. The reference and target data can belong to different dissection regions, different donors or different tissue types. Specifically, scBOL offers a flexible annotation framework that is adaptable to both non-spatial scRNA-seq data and spatially resolved transcriptomics data through the construction of varied network architectures. For non-spatial scRNA-seq data, we apply the denoising autoencoder to extract the cell representation by compressing gene expression profiles, while for spatial transcriptomics data, we employ the graph convolutional neural network (GCN) to simultaneously leverage molecular information and additional spatial context of cells.
Our algorithm consists of three main parts. First, inspired by the inductive bias of class-wise closeness, we mine the mutual nearest clusters as the underlying common cell types across reference and target data. Then we detect the cycle-consistent anchor cells from the matched clusters to uncover the data intrinsic structure connection at both the semantic level and sample level. Indeed, we show that this strategy can align the cell types in the reference dataset with the same cell types in the target dataset accurately and can effectively solve the batch effect problem. Secondly, to improve the compact and discriminative ability of the learned feature space, we design a neighbor-aware prototypical learning paradigm by encouraging the cell type assignment consistency between samples and their nearest neighbors. At the same time, we transfer the cell type-specific knowledge through a semantic-aware anchor-prototype alignment regularizer to improve the model’s generalization ability on known cell types. Lastly, for the challenging target cluster number estimation problem, instead of artificially specifying or directly giving a relatively large value, we introduce a cross-data consensus score to tackle it from the perspective of anchor agreement degree.
To thoroughly assess scBOL’s performance, we selected a diverse range of comparison baselines and established both intra-data and inter-data benchmarks utilizing an extensive collection of highly imbalanced scRNA-seq and spatial transcriptomics datasets. Our comprehensive experimental results confirm scBOL’s utility relative to other leading cell type identification algorithms. Moreover, detailed ablation studies reveal the contributions of scBOL’s components to its overall effectiveness. From a practical perspective, the efficacy of scBOL is crucial in utilizing reference datasets produced under varying conditions, which might not encompass the complete spectrum of cell types present in the target condition.
METHOD
We first give some notations. In our cell type identification task, we are provided with some labeled reference data 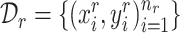 and unlabeled target data
and unlabeled target data 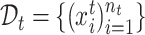 . For spatial transcriptomics data, the spatial coordinates of cells are
. For spatial transcriptomics data, the spatial coordinates of cells are 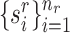 and
and 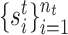 for reference and target data, respectively. Furthermore, the reference and target data can be drawn from the same or different datasets. So there may exist gene expression distribution differences between
for reference and target data, respectively. Furthermore, the reference and target data can be drawn from the same or different datasets. So there may exist gene expression distribution differences between 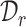 and
and 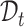 . We use
. We use 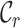 to denote the annotated cell type set which contains the known cell types of labeled data and employ
to denote the annotated cell type set which contains the known cell types of labeled data and employ 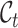 to represent the unannotated cell type set consisting of the cell types in unlabeled data. Note that in our setting, we do not know the exact relationship between
to represent the unannotated cell type set consisting of the cell types in unlabeled data. Note that in our setting, we do not know the exact relationship between 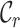 and
and 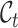 . Particularly, we use
. Particularly, we use 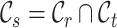 to denote the common cell types shared by
to denote the common cell types shared by 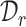 and
and 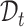 , and use
, and use 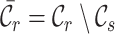 and
and 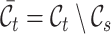 to denote the cell type sets private to the labeled and unlabeled data, respectively. Our goal is to annotate the target cells with either one of the known labels in
to denote the cell type sets private to the labeled and unlabeled data, respectively. Our goal is to annotate the target cells with either one of the known labels in 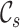 or the clustering labels in
or the clustering labels in 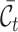 . We train the model on
. We train the model on 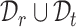 and evaluate on
and evaluate on 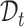 .
.
It is necessary to pre-process the transcriptome data profiling before further analysis. To ensure methodological rigor, the datasets employed in this study were subjected to stringent quality control measures and structured as count matrices. Uniformity across raw cell type annotations was achieved by leveraging the Cell Ontology framework [52], a meticulously curated and structured vocabulary for cell types. Subsequently, genes expressed in less than one cell were excluded, alongside cells exhibiting no gene expression, to refine the dataset. To address the challenges associated with the numerical optimization of neural networks, it is imperative to convert discrete datasets into a continuum of smooth data. This transformation process involves a two-step normalization procedure. Initially, the total expression level of each cell is normalized to its median value. Following this, a natural logarithm transformation is applied to these normalized expression values to stabilize variance across the dataset. Given that the majority of genes provide limited utility in distinguishing and characterizing cell types, a selection criterion was imposed to distill the dataset further. This was achieved by isolating the top genes exhibiting the most significant variability, determined by their rank in normalized dispersion values. After the log transformation, the data were standardized to z-scores, enabling each of the chosen genes to have a mean of zero and a unit variance. The entire preprocessing pipeline was executed utilizing the Scanpy software package [25]. For the subsequent analysis, the refined dataset, now suitably preprocessed, served as the input for neural network modeling. Moreover, the corresponding original count data were employed alongside the preprocessed data to enhance the robustness of the modeling approach.
For scRNA-seq data, considering their discrete and sparse traits, we assume that 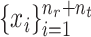 follows a zero-inflated negative binomial distribution and use an autoencoder model to denoise data [53]. Inspired by the self-supervised learning [54, 55], we use a mask-based data augmentation strategy to generate another view
follows a zero-inflated negative binomial distribution and use an autoencoder model to denoise data [53]. Inspired by the self-supervised learning [54, 55], we use a mask-based data augmentation strategy to generate another view 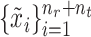 of gene expression, which can capture the correlations across genes better, which can be seen in the supplementary materials. For spatial transcriptomics data, given the spatial cell coordinates, we can construct a reference cell graph
of gene expression, which can capture the correlations across genes better, which can be seen in the supplementary materials. For spatial transcriptomics data, given the spatial cell coordinates, we can construct a reference cell graph 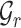 and a target cell graph
and a target cell graph 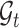 , where the nodes represent the cells and the edges connect the spatially close cells. Given these two graphs, we use a GCN to map the cells into a joint embedding space that captures spatial and molecular similarities between the cells [56] (see Figure 1). The specific graph construction procedure can be seen in the supplementary materials.
, where the nodes represent the cells and the edges connect the spatially close cells. Given these two graphs, we use a GCN to map the cells into a joint embedding space that captures spatial and molecular similarities between the cells [56] (see Figure 1). The specific graph construction procedure can be seen in the supplementary materials.
Figure 1.

An overview of scBOL. (A) scBOL performs annotation on the scRNA-seq data or spatial transcriptomics data. The input of scRNA-seq data is the gene expression matrix and the autoencoder is used on it to denoise data and capture biological information. In contrast, the input of spatial transcriptomics data includes gene expression profile and spatial cell coordinates and GCN is used on it to map the cells into a joint embedding space. (B) Our annotation task is the same for two kinds of data, that is, automatically assign cells to cell types present in the reference data, and assign cluster labels to the cells of novel cell types. Moreover, for the private cell types that only exist in the reference data, scBOL does not group cells in the target data with them. The reference and target classifiers are connected to the output of the embedding space and 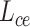 ,
, 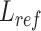 and
and 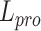 are the loss functions of scBOL. (C) Samples and prototypes in reference and target data are distinguished by different shapes and colors, respectively. The cell type relationship between reference and target data is partially overlapping, that is, there are overlapping cell types in them, as well as their own private cell types. (D) d1. The bipartite matching principle is proposed to link clusters from the same cell types by exploiting semantic-level cycle consistency. d2. Since prototype-level bipartite matching may bring too much noise and make the model unstable, the cycle consistency constraint is also given on the sample level to further consider the semantic relationship between the cells and prototypes. (E) We propose to impose the prototypical regularizer on the reference and target cells to drive features within the same cluster to become more aggregated and features in different clusters further apart. (F) The number of novel cell types is estimated by consensus score, calculated based on the common semantic anchor.
are the loss functions of scBOL. (C) Samples and prototypes in reference and target data are distinguished by different shapes and colors, respectively. The cell type relationship between reference and target data is partially overlapping, that is, there are overlapping cell types in them, as well as their own private cell types. (D) d1. The bipartite matching principle is proposed to link clusters from the same cell types by exploiting semantic-level cycle consistency. d2. Since prototype-level bipartite matching may bring too much noise and make the model unstable, the cycle consistency constraint is also given on the sample level to further consider the semantic relationship between the cells and prototypes. (E) We propose to impose the prototypical regularizer on the reference and target cells to drive features within the same cluster to become more aggregated and features in different clusters further apart. (F) The number of novel cell types is estimated by consensus score, calculated based on the common semantic anchor.
To assign an annotation label for each cell, existing works usually use both 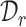 and
and 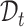 to learn a unified classifier on the latent embedding feature space
to learn a unified classifier on the latent embedding feature space 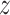 and then generate the predicted labels for
and then generate the predicted labels for 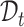 . However, such a transfer strategy may be susceptible to batch effects and has a potential risk of damaging the intrinsic structure discrimination on
. However, such a transfer strategy may be susceptible to batch effects and has a potential risk of damaging the intrinsic structure discrimination on 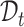 . Besides, since the novel cell types exist in
. Besides, since the novel cell types exist in 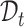 , this training manner makes it difficult to decouple the semantic-specific knowledge between the known and novel cell types. Based on this analysis, we are motivated to directly uncover the intrinsic discrimination via constrained generative clustering on the target data with structural regularization induced by reference data. Specifically, we learn two sets of parameterized cell type prototypes for the reference and target data, respectively (see Figure 1). For reference data, we take the average of cell embeddings belonging to the same cell types as the initialization of reference prototypes
, this training manner makes it difficult to decouple the semantic-specific knowledge between the known and novel cell types. Based on this analysis, we are motivated to directly uncover the intrinsic discrimination via constrained generative clustering on the target data with structural regularization induced by reference data. Specifically, we learn two sets of parameterized cell type prototypes for the reference and target data, respectively (see Figure 1). For reference data, we take the average of cell embeddings belonging to the same cell types as the initialization of reference prototypes 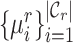 , where
, where 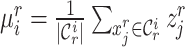 denotes the labeled prototype of
denotes the labeled prototype of 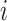 -th known cell type and
-th known cell type and 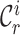 denotes the set of cells from
denotes the set of cells from 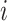 -th known cell type. For target data, we first perform k-means clustering to categorize them into
-th known cell type. For target data, we first perform k-means clustering to categorize them into 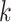 clusters
clusters 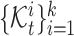 , where
, where 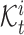 represents the set of cells from
represents the set of cells from 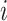 -th target cluster. And their clustering labels are denoted as
-th target cluster. And their clustering labels are denoted as 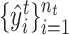 . The value of
. The value of 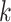 can be estimated and entered into the model as a prior. The specific estimation method will be introduced later. Then we take the average of target cell embeddings belonging to the same cluster as the initialization of target prototypes
can be estimated and entered into the model as a prior. The specific estimation method will be introduced later. Then we take the average of target cell embeddings belonging to the same cluster as the initialization of target prototypes 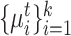 , where
, where 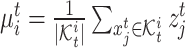 .
.
Semantic anchor selection for bipartite alignment
The main challenge of our task is how to effectively match the common cell types between 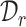 and
and 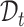 and separate the private cell types within them. Instead of introducing extra network parameters for common cell detection, we aim to mine both common cell types and individual private cell types simultaneously with discriminative clusters. So a question naturally arises: how to associate the common clusters that represent the same cell types from both
and separate the private cell types within them. Instead of introducing extra network parameters for common cell detection, we aim to mine both common cell types and individual private cell types simultaneously with discriminative clusters. So a question naturally arises: how to associate the common clusters that represent the same cell types from both 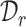 and
and 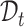 . To achieve this goal, we propose a bipartite matching principle to link clusters from the same cell types by exploiting the semantic-level cycle consistency.
. To achieve this goal, we propose a bipartite matching principle to link clusters from the same cell types by exploiting the semantic-level cycle consistency.
Specifically, for each target prototype 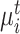 , we search for its nearest prototype
, we search for its nearest prototype 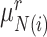 in the reference data by cosine distance. The same procedure is implemented for each reference prototype
in the reference data by cosine distance. The same procedure is implemented for each reference prototype 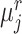 and we can obtain its nearest target prototype
and we can obtain its nearest target prototype 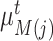 . If a target prototype
. If a target prototype 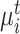 and a reference prototype
and a reference prototype 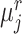 reach bipartite matching, i.e. both act as the other’s nearest prototype simultaneously,
reach bipartite matching, i.e. both act as the other’s nearest prototype simultaneously,
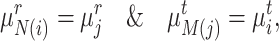 |
(1) |
then such a pair of clusters is recognized as common clusters. The intuition here is simple: cluster prototypes from the common cell type usually lie close enough to be associated compared with the other clusters representing private cell types [57]. Enabled by this motivation, we can further identify common cells based on the matched clusters from 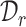 and
and 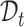 . However, simply unifying the cells belonging to the common clusters does not consider the semantic relationship between the cells and prototypes, leading to the high noise in cell division. To alleviate this issue, we further give the cycle consistency constraint as the sample level. For each target cell
. However, simply unifying the cells belonging to the common clusters does not consider the semantic relationship between the cells and prototypes, leading to the high noise in cell division. To alleviate this issue, we further give the cycle consistency constraint as the sample level. For each target cell 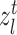 from paired clusters
from paired clusters 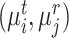 , we search for its nearest reference prototype
, we search for its nearest reference prototype 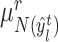 and then determine if it holds the corresponding cluster label in reference data. When the consensus is reached, i.e.
and then determine if it holds the corresponding cluster label in reference data. When the consensus is reached, i.e. 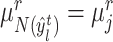 , then target cell
, then target cell 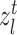 can be regarded as the common semantic anchor in target data. Similarly, we also collect the common semantic anchors in reference data. For convenience, we give some mathematical symbols to illustrate them. Assume that the one-to-one mapping function from the target label set to the reference label set is
can be regarded as the common semantic anchor in target data. Similarly, we also collect the common semantic anchors in reference data. For convenience, we give some mathematical symbols to illustrate them. Assume that the one-to-one mapping function from the target label set to the reference label set is 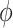 , then for target cell
, then for target cell 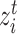 ,
, 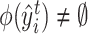 if and only if
if and only if 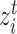 is a target semantic anchor. Similarly, for
is a target semantic anchor. Similarly, for 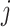 -th reference cell,
-th reference cell, 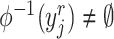 if and only if
if and only if 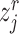 is a reference semantic anchor. It should be explained that
is a reference semantic anchor. It should be explained that 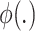 obtains a known cell type label in the reference data, and
obtains a known cell type label in the reference data, and 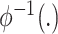 obtains a cluster label in the target data.
obtains a cluster label in the target data.
It is important to highlight the distinct contrasts between the Mutual Nearest Neighbor (MNN) method [58] and our proposed bipartite matching method. Firstly, regarding semantic-level alignment, the MNN method prioritizes individual samples, whereas our bipartite matching approach focuses on prototypical representations. Secondly, in terms of sample-level alignment, the MNN method seeks the closest neighbor for each sample, as opposed to the bipartite matching method, which aims to identify the nearest prototype corresponding to each sample. These divergent approaches in alignment at varying levels suggest that our method of anchor selection is likely to demonstrate enhanced robustness in comparison with the traditional MNN approach.
Intra-data neighbor-aware prototypical learning
We learn a shared latent space 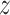 that extracts embedding features in both reference and target data. At the early training stage, the features are not so discriminative and the boundaries between clusters are not so clear. Some existing works use instance-wise discrimination techniques to learn a compact embedding space where all cells are well separated [59, 60]. Despite the promising results, these approaches have a fundamental weakness: the semantic structure of the whole data is not encoded by the learned representations. Therefore, we need to exploit the global and local semantic cell type structure and drive features within the same cluster to become more aggregated and features in different clusters further apart. Here we propose to impose the prototypical regularizer on the reference and target cells to uncover the intrinsic structure of the data. Specifically, for
that extracts embedding features in both reference and target data. At the early training stage, the features are not so discriminative and the boundaries between clusters are not so clear. Some existing works use instance-wise discrimination techniques to learn a compact embedding space where all cells are well separated [59, 60]. Despite the promising results, these approaches have a fundamental weakness: the semantic structure of the whole data is not encoded by the learned representations. Therefore, we need to exploit the global and local semantic cell type structure and drive features within the same cluster to become more aggregated and features in different clusters further apart. Here we propose to impose the prototypical regularizer on the reference and target cells to uncover the intrinsic structure of the data. Specifically, for 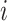 -th reference cell with the embedding feature
-th reference cell with the embedding feature 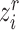 , we compute the similarity probability distribution between
, we compute the similarity probability distribution between 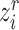 and
and 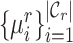 as
as 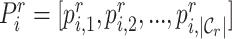 , with
, with
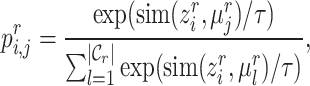 |
(2) |
where 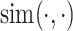 represents the cosine similarity and
represents the cosine similarity and 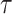 is a temperature factor. Similarly, for another augmented view
is a temperature factor. Similarly, for another augmented view 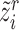 , we can also get its similarity vector
, we can also get its similarity vector 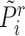 , where
, where 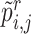 is calculated by replacing
is calculated by replacing 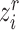 with
with 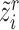 in Equation (2). Since the reference data are well-labeled, we give the prototypical learning loss on them via the cross-entropy function,
in Equation (2). Since the reference data are well-labeled, we give the prototypical learning loss on them via the cross-entropy function,
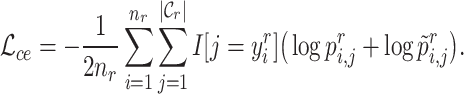 |
(3) |
For target data, the clustering label divides each cell into a prototype in a hard way and is highly noisy in the early stage of training. Using them exclusively for supervised learning on target data may lead to error accumulation and propagation as the model is trained. So instead we introduce a neighbor-aware prototypical learning paradigm that encourages the consistency of assignment distribution between nearest neighbors. To achieve this, we first divide the target data into confident set 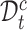 and fuzzy set
and fuzzy set 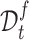 based on a reliable score. This score is obtained by calculating the ratio of the distance between the sample and its cluster center and the distance between the sample and the nearest non-self-cluster center. The smaller the score, the more reliable the cluster label of the sample. In each training epoch, we default to select the top
based on a reliable score. This score is obtained by calculating the ratio of the distance between the sample and its cluster center and the distance between the sample and the nearest non-self-cluster center. The smaller the score, the more reliable the cluster label of the sample. In each training epoch, we default to select the top 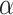 % samples with the lowest score in each cluster into
% samples with the lowest score in each cluster into 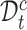 , otherwise into
, otherwise into 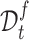 . Then, for samples in
. Then, for samples in 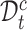 , we use their clustering labels to supervise learning their representations, while for samples in
, we use their clustering labels to supervise learning their representations, while for samples in 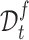 , we pursue that the similar cells should have the similar prototypical assignment distribution. Concretely, we also use the Gaussian kernel function to measure the similarity between each target cell and target prototypes,
, we pursue that the similar cells should have the similar prototypical assignment distribution. Concretely, we also use the Gaussian kernel function to measure the similarity between each target cell and target prototypes,
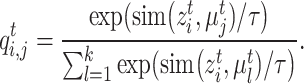 |
(4) |
Given the two branch target embeddings, we can search their nearest neighbor set in each branch by cosine distance. For ease of writing, we denote the closest cell as 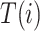 and
and 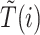 for
for 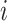 -th target cell. Then the neighbor-aware prototypical learning objective within
-th target cell. Then the neighbor-aware prototypical learning objective within 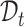 can be written as
can be written as
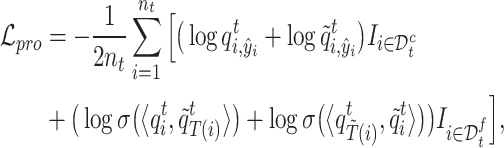 |
(5) |
where 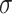 is sigmoid function and
is sigmoid function and 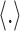 refers to the inner product operation. And
refers to the inner product operation. And 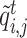 is obtained similar to
is obtained similar to 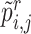 . By minimizing
. By minimizing 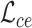 and
and 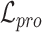 , we can improve the inter-cluster separability and intra-cluster compactness, thus improving the discriminability of feature space.
, we can improve the inter-cluster separability and intra-cluster compactness, thus improving the discriminability of feature space.
Cross-data semantic-aware prototypical learning
So far, we have only discussed the intra-data learning strategies and have not yet touched on cross-data alignment learning, especially on common cell types. When there are batch effects across reference and target data, the model does not necessarily project the same cell type from different data to the same area well. In this case, the prediction accuracy of the model on the common cell types would be greatly reduced. To alleviate this issue, here we aim to match the common cell types to transfer the semantic knowledge from reference data to target data, thereby improving the generalization ability of the model on these cell types. By utilizing the mined semantic anchors as matching bridges, we propose to perform cross-data instance-prototype representation learning to explicitly enforce learning cell type-aligned features. It is commonly hypothesized in transfer learning that the importance of samples varies for learning transferable models. A simple strategy to implement this hypothesis is to re-weight instances based on their similarities to the reference object [57]. Here we also employ this strategy in our model. Specifically, for any target cell 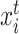 , we compute its similarity to the reference prototypes
, we compute its similarity to the reference prototypes 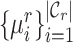 , based on the following transformed distance:
, based on the following transformed distance:
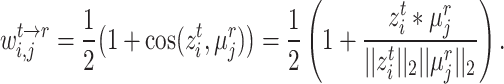 |
(6) |
Similarly, we can also calculate the similarity 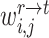 between reference cell
between reference cell 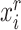 and target prototype
and target prototype 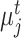 . Then we can further obtain the probability assignment distribution between target cells and reference prototypes by the Gaussian kernel function,
. Then we can further obtain the probability assignment distribution between target cells and reference prototypes by the Gaussian kernel function,
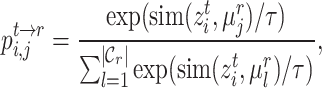 |
(7) |
The same procedure can be applied to reference cells and target prototypes to get their assignment distribution 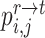 . Based on the cross-data semantic anchor cells, we design the following weighted cross-entropy objective to transfer the cell type-specific knowledge across reference and target data,
. Based on the cross-data semantic anchor cells, we design the following weighted cross-entropy objective to transfer the cell type-specific knowledge across reference and target data,
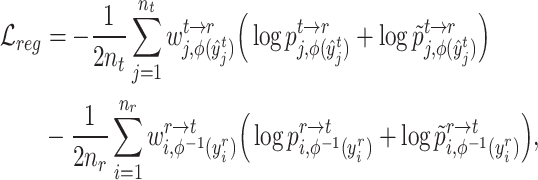 |
(8) |
where 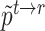 and
and 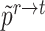 are obtained by substituting the augmented cell features into the corresponding equations. By minimizing
are obtained by substituting the augmented cell features into the corresponding equations. By minimizing 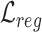 , the alignment of the common cell types between reference and target data can be achieved, allowing the model to better learn generalizable features.
, the alignment of the common cell types between reference and target data can be achieved, allowing the model to better learn generalizable features.
Overall loss. For non-spatial scRNA-seq data, together with the data denoising loss 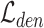 (see supplementary materials), we give the training objective as
(see supplementary materials), we give the training objective as
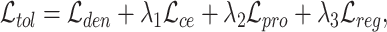 |
(9) |
where 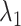 ,
, 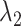 and
and 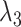 are three weight hyperparameters. For spatial transcriptomics data, we do not use the data denoising loss and just combine
are three weight hyperparameters. For spatial transcriptomics data, we do not use the data denoising loss and just combine 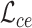 with
with 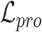 and
and 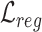 as the overall training objective.
as the overall training objective.
Estimating cell type number by consensus score
In the single-cell clustering and annotation field, cell type number estimation is always a challenging and under-investigated problem. This problem is also not solved yet in our task: how to determine the target cluster number without knowing the true value? The traditional approach is to apply the clustering evaluation criterion to estimate the cluster number [61]. However, they cannot directly consider the cross-data knowledge. So here we propose a consensus score that utilizes the ratio of semantic anchor to determine the target cluster number.
Concretely, given a pair of bipartite-matched prototypes 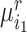 and
and 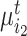 , their corresponding cluster sizes are
, their corresponding cluster sizes are 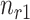 and
and 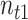 , respectively. Assume that there are
, respectively. Assume that there are 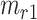 semantic anchor cells in cluster
semantic anchor cells in cluster 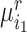 to be matched with
to be matched with 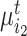 and
and 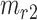 semantic anchor cells in cluster
semantic anchor cells in cluster 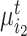 to be matched with
to be matched with 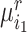 , then the consensus score of
, then the consensus score of 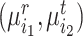 is defined as
is defined as 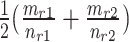 . Finally, we calculate the averaged consensus scores of all matched pairs of clusters as the evaluation metric. To specify the target cluster number
. Finally, we calculate the averaged consensus scores of all matched pairs of clusters as the evaluation metric. To specify the target cluster number 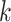 , we perform multiple clusterings with different
, we perform multiple clusterings with different 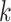 values and then determine the optimal one according to the consensus score.
values and then determine the optimal one according to the consensus score.
PERFORMANCE EVALUATION
Dataset composition
To enhance the comprehensiveness of cell annotation, our study delineates the experimental framework into two principal categories: intra-data annotation and inter-data annotation. The latter approach is specifically designed to mitigate batch effects that frequently arise between reference and target datasets. In the domain of intra-data annotation, our collection comprises five scRNA-seq datasets alongside one spatial transcriptomics dataset. These datasets exhibit a range of complexities, with total cell counts spanning from 6000 to 110 000. Additionally, they utilize diverse sequencing technologies and are derived from a variety of tissue types. To ensure broad applicability, we have categorized the cell types into three distinct classes. This classification scheme is elucidated in the supplementary tables provided. We operate under the assumption that for common cell types, both reference and target data comprise an equivalent proportion, accounting for 50% of the total cells in the pooled dataset. Moving to inter-data annotation, our selection encompasses five pairs of scRNA-seq datasets, in addition to one pair derived from spatial transcriptomics. Each pair is composed of a reference dataset paired with a corresponding target dataset. The cell counts within these pairs range from several thousand to tens of thousands. The number of cell types in the reference dataset is set to be approximately half that of the target dataset to demonstrate robustness in cases of unequal cell type distribution. Details pertinent to these datasets, including their specific attributes and configurations, are thoroughly documented in the supplementary materials accompanying this publication.
Evaluated baselines
We aim to establish a new practical cell type identification task for which few ready-to-use baselines exist. So we extend the recently published scRNA-seq clustering and annotation methods as the comparison baselines. For the clustering of scRNA-seq data, we select scCNC [62] and scDECL [63], since they use both 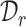 and
and 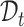 in training under the semi-supervised learning setting, while other methods only train on
in training under the semi-supervised learning setting, while other methods only train on 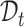 in the unsupervised scenario. We also compare with STAGATE [23], a customized clustering method for spatial transcriptomics data, which integrates the reference data and target data for representation learning. For the annotation of scRNA-seq data, we choose MARS [40], ItClust [34], scNym [46] and scArches [45] since they can detect the ‘unassigned’ cells. Specifically, we first use them to classify target cells into known cell types and identify the ‘unassigned’ group. Next, we apply k-means clustering on the ‘unassigned’ group to obtain novel clusters. For spatial transcriptomics data annotation, we choose STELLAR [51] as the compared baseline, because it can simultaneously identify the known cell types and discover novel cell types. The further running details of these methods can be seen in the supplementary materials.
in the unsupervised scenario. We also compare with STAGATE [23], a customized clustering method for spatial transcriptomics data, which integrates the reference data and target data for representation learning. For the annotation of scRNA-seq data, we choose MARS [40], ItClust [34], scNym [46] and scArches [45] since they can detect the ‘unassigned’ cells. Specifically, we first use them to classify target cells into known cell types and identify the ‘unassigned’ group. Next, we apply k-means clustering on the ‘unassigned’ group to obtain novel clusters. For spatial transcriptomics data annotation, we choose STELLAR [51] as the compared baseline, because it can simultaneously identify the known cell types and discover novel cell types. The further running details of these methods can be seen in the supplementary materials.
Evaluation metrics
In all experiments conducted, the results presented are the mean values computed over three independent trials. Concerning scBOL and other comparative annotation baselines, we assess classification performance for common cell types and evaluate clustering efficacy for novel cell types. But for clustering baselines, since they cannot recognize the common cell types, we report the clustering accuracy on both common and novel cell types. To calculate clustering accuracy, the Hungarian algorithm is utilized to address the optimal assignment problem [64]. When reporting accuracy on all cell types, we solve the optimal assignment problem on both common and novel cell types.
Implementation details
Our algorithm is mainly done in Python and is based on the PyTorch framework. We conducted the experiments with two Tesla A100 GPUs and the detailed version of the package used has been given on GitHub. For scRNA-seq data, the two layers of the encoder are sized 512 and 256, respectively, and the decoder has the reverse structure of the encoder. The bottleneck layer has a size of 128. The training mini-batch size is set to 256, and the optimizer is Adam with a learning rate of 1e-4. The temperature 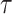 in prototypical learning is set to 1.0, and the sample selection ratio
in prototypical learning is set to 1.0, and the sample selection ratio 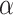 is set to 20. The loss weight
is set to 20. The loss weight 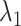 ,
, 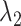 and
and 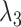 are all set to 1.0. We first train the whole model using
are all set to 1.0. We first train the whole model using 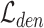 loss with 600 epochs. Then, we apply the k-means algorithm on target embeddings to obtain cluster centers as the initial values of target prototypes. The initialization of reference prototypes can be obtained by the mean values of reference embeddings based on ground-truth labels. Finally, we train the model with the overall loss
loss with 600 epochs. Then, we apply the k-means algorithm on target embeddings to obtain cluster centers as the initial values of target prototypes. The initialization of reference prototypes can be obtained by the mean values of reference embeddings based on ground-truth labels. Finally, we train the model with the overall loss 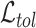 until the predictions no longer change. For spatial transcriptomics data, we use a graph convolutional layer with a hidden dimension of feature size in all layers. A cluster sampler first clusters the input graph into subgraphs and then assigns the subgraphs into mini-batches. The model is trained for 100 epochs by Adam optimizer with an initial learning rate of 1e-3 and weight decay of 0. We set the temperature
until the predictions no longer change. For spatial transcriptomics data, we use a graph convolutional layer with a hidden dimension of feature size in all layers. A cluster sampler first clusters the input graph into subgraphs and then assigns the subgraphs into mini-batches. The model is trained for 100 epochs by Adam optimizer with an initial learning rate of 1e-3 and weight decay of 0. We set the temperature 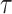 as 0.1 and the sample selection ratio as 0.05. The loss weight hyperparameters are set to the same values in scRNA-seq data.
as 0.1 and the sample selection ratio as 0.05. The loss weight hyperparameters are set to the same values in scRNA-seq data.
RESULTS COMPARISON
Intra-data experiment on scRNA-seq dataset
In summary, scBOL consistently outperforms competing algorithms on five authentic datasets (Figure 2A). Notably, scBOL’s coverage profile resembles a pentagon, suggesting its robust performance across these datasets. scBOL ranks within the top two for both known and novel accuracy metrics when compared with other methodologies. These findings corroborate our hypothesis that utilizing cycle-consistent anchor cells from aligned clusters enhances the model’s capacity to accurately map common cell types and identify new ones. It is significant that scNym matches scBOL’s high performance in known accuracy, and occasionally surpasses it in specific datasets. Nevertheless, scNym’s weaker novel accuracy performance, where scBOL remains a strong contender, suggests a limitation. Specifically, scNym’s tendency to classify novel cell types as common ones reduces its applicability. Similarly, scCNC and ItClust face challenges; they often rely on artificial thresholds and other means to differentiate between common and novel cell types, resulting in a skewed emphasis on annotating common types at the expense of discovering novel ones. ItClust, in particular, demonstrates the poorest performance in both known and novel accuracy, potentially due to suboptimal parameter initialization in its target network. scArches also yields less favorable results for both accuracy measures, handicapped by its assumption that a low-dimensional latent space conforms to a Gaussian mixture model, which significantly constrains its representational capabilities.
Figure 2.

Radar plots of scBOL and other six methods measured by three kinds of accuracy on five scRNA-seq datasets. A. Radar plots for intra-data experiments. B. Radar plots for inter-data experiments. M-SS2 to M-10X: Mammary Smart-seq2 as reference data and Mammary 10x as target data. Mu to Ba: Muraro as reference data and Baron as target data. V-SS2 to V-10X: Vento-Tormo Smart-seq2 as reference data and Vento-Tormo 10x as target data. P to M: Plasschaert as reference data and Montoro as target data. H-l to H-r: Haber largecell as reference data and Haber region as target data.
A comparative analysis with three additional clustering methods—MARS, scCNC and scDECL—reveals their inability to match scBOL’s effectiveness, largely because they do not leverage label information from the reference dataset. These methods merely assign cluster labels to samples without providing annotations, limiting their utility. scBOL, by contrast, navigates these challenges by executing bipartite alignment at both the cluster and sample levels between the target and reference datasets, employing neighbor-aware and semantic-aware prototypical learning.
Moreover, an intrinsic compromise exists between aligning common cell types and discovering novel ones. scBOL uniquely balances this compromise well, as evidenced by its superior overall accuracy results. scBOL’s preeminence in partitioning common and novel cell types is evident in intra-data experiments. The methodology’s explicit alignment of common cell types between datasets and facilitation of clustering for distinct private cell types in the embedding space further reinforce its superiority.
Inter-data experiment on scRNA-seq dataset
To rigorously evaluate the efficacy of scBOL in cross-data applications, where reference and target datasets may originate from differing tissues or donors, we conducted experiments across five dataset groups (Figure 2B). Despite all methods being susceptible to batch effects, scBOL consistently outperformed the others across three accuracy metrics, corroborating its robustness in mitigating batch variations. This superior performance likely stems from scBOL’s strategy of linking cluster identifiers through mutual nearest prototypes and the utilization of shared semantic anchors, which collectively contribute to effective batch effect amelioration.
While scNym exhibits commendable performance in the domain of known accuracy, rivaling that of scBOL, its limitations become apparent in the realm of novel cell type accuracy. The algorithm’s tendency to misclassify cells within ambiguous regions as common, rather than novel, cell types serves to underscore these shortcomings. Moreover, scNym and similar existing annotation tools often overlook the necessity of assigning specific cluster labels to novel cell types, instead choosing to default to broad, undefined categories for such cells. MARS, another noteworthy technique, displays distinct capabilities in novel cell type accuracy. However, its effectiveness diminishes when discerning known cell types, which could be attributed to the method’s strategy of independently training reference and target datasets. In addition, as a distinguishing clustering approach, MARS is restricted to offering generic cluster labels without providing precise cell type identification. The performance of the remaining four methodologies shows notable variability across different datasets, as evidenced by the substantial deviations from idealized pentagon shapes in their graphical representations. This variability suggests a strong dependency on dataset characteristics and highlights opportunities for enhancement in both their annotation and clustering capacities. In sum, scBOL emerges as the preeminent solution among the tested methods, eclipsing both annotation and clustering alternatives in terms of performance. Its efficacy in neutralizing batch effects, accurately conforming common cell types and adeptly clustering novel cell types firmly establishes scBOL as a veritable tool for integrated single-cell analysis.
To facilitate a more intuitive understanding of the association between predictive outcomes, we employed a Sankey diagram for comparative analysis of the performance of scBOL and three alternative well-regarded methodologies (Figure 3A). The diagram offers compelling visual evidence that substantiates our assessment: scBOL is capable of assigning pertinent cluster labels to target private cells and delivers precise annotations for cells with known cell types. In contrast, the other techniques exhibit varying levels of performance deterioration, which underscores the suboptimal efficacy of their two-stage approach that combines clustering with annotation. More specifically, ItClust and scNym partially succeed in correctly categorizing certain cell types; this limitation possibly arises from the arbitrary nature of their threshold determinations and misguided presumptions regarding the structure of the embedding space. Furthermore, ItClust’s methodology of independently training reference and target datasets renders it susceptible to batch effects. scArches, conversely, exhibits subpar performance, failing to consistently identify endothelial cells. This inadequacy is attributed to its flawed foundational hypotheses and illogical approaches to the alignment of shared cell types.
Figure 3.

Inter-data experiments scRNA-seq data. A. Mapping relationship among prediction results of scBOL and other three methods via Sankey plots for the experiment where Mammary Smart-seq2 is the reference data and Mammary 10x is the target data. B. Visualization plots via UMAP calculated using the latent representations of scBOL and the other seven methods colored by cell types for the experiment where Vento-Tormo Smart-seq2 is reference data and Vento-Tormo 10x is target data.
The visualization plots produced employing UMAP, grounded on the latent representations derived from each algorithm, demonstrate that scBOL proficiently discerns the heterogeneous cell populations (Figure 3B). This proficiency facilitates cell clustering predicated on intrinsic biological characteristics rather than confounding batch effects. In stark contrast, alternative approaches amalgamate cell types that are phenotypically similar yet distinct, such as decidual natural killer cells and trophoblast cells, as well as T cells and natural killer cells. This amalgamation betrays a shortfall in the specificity of their respective learning algorithms, which may stem from constraints in sample size precluding the accurate disambiguation of these subtle cell groups. Among them, MARS exhibits the poorest performance, displaying negligible discriminative capacity, likely a repercussion of its approach of bifurcating the training process between reference and target datasets, culminating in model overfitting. scNym and scArches struggle to capture the intricate biometrics of some phenotypically overlapping cell types, potentially attributable to their inadequate assimilation of the global data structure. Moreover, the graphical representation elucidates that the embedding outputs of MARS and scArches are derived from disparate batches and do not converge seamlessly within the embedding domain, signifying their ineptitude in overcoming the batch effect. Conversely, scBOL adeptly integrates samples from heterogeneous batches in the embedding realm, starkly juxtaposing its efficacy to that of its counterparts in resolving the batch effect. In summation, scBOL boasts a significant edge in the arena of generalized annotation. Globally, scBOL not only orchestrates cell type alignment and robustly ameliorates batch effects via cluster associations across reference and target datasets but also employs prototypical learning stratagems based on sample confidence to holistically delineate cell typologies. Locally, scBOL enhances the alignment of common cell types between the reference and the target sets and dispels batch influences by employing semantic anchors, attesting to its comprehensive capability in cell type annotation.
Robustness analysis in scRNA-seq dataset
Here we investigate the resilience of the scBOL model across different configurations by variably adjusting key parameters such as the count of novel cell types within the target dataset and the labeled ratio. These modifications are anticipated to exert considerable influence on the outcomes yielded by the model. It is pertinent to note that the nature of our task aligns with transductive learning as opposed to inductive learning. Accordingly, both the reference dataset and the target dataset are employed as training datasets, while the target dataset alone is designated for testing purposes. Consequently, the composition of both the training and testing datasets correlates directly with the authentic dataset utilized in our study.
The magnitude of the target private cell type count, denoted by 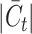 , critically influences the challenge associated with annotating established cell types and clustering novel cell types. It is therefore essential to investigate how fluctuations in
, critically influences the challenge associated with annotating established cell types and clustering novel cell types. It is therefore essential to investigate how fluctuations in 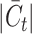 affect the accuracy of our methodology. We performed this analysis using the Quake 10x and Quake Smart-seq2 datasets, comprising 36 and 45 distinct cell types, respectively. For the Quake 10x dataset,
affect the accuracy of our methodology. We performed this analysis using the Quake 10x and Quake Smart-seq2 datasets, comprising 36 and 45 distinct cell types, respectively. For the Quake 10x dataset, 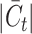 ranged across
ranged across 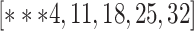 , while for Quake Smart-seq2, the range was
, while for Quake Smart-seq2, the range was 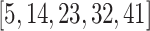 (Figure 4A, Figure 4B). Essentially, we altered the count of novel cell types in the test dataset (the target dataset) for this evaluation. Our results unequivocally demonstrate that scBOL consistently outperforms other comparative methodologies by significant margins, thereby highlighting its effectiveness in aligning common cell types and identifying new ones. The relative smoothness of scBOL’s performance curve further corroborates its resilience to variations in
(Figure 4A, Figure 4B). Essentially, we altered the count of novel cell types in the test dataset (the target dataset) for this evaluation. Our results unequivocally demonstrate that scBOL consistently outperforms other comparative methodologies by significant margins, thereby highlighting its effectiveness in aligning common cell types and identifying new ones. The relative smoothness of scBOL’s performance curve further corroborates its resilience to variations in 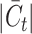 . On the contrary, the remaining six methodologies typically yield substandard outcomes. Specifically, the overall accuracy for scArches and scNym plummets as
. On the contrary, the remaining six methodologies typically yield substandard outcomes. Specifically, the overall accuracy for scArches and scNym plummets as 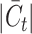 increases, attributed to their design prioritizing the annotation of common cell types and the potential increased interference from a higher
increases, attributed to their design prioritizing the annotation of common cell types and the potential increased interference from a higher 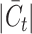 . While MARS, scCNC and scDECL exhibit relative stability as
. While MARS, scCNC and scDECL exhibit relative stability as 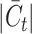 escalates, they tend to deliver suboptimal performance, often assigning generic cluster labels devoid of semantic content. Furthermore, the performance of ItClust is erratic, marked by a dramatic increase in Quake 10x and a notable decline in Quake Smart-seq2, which exposes its instability. From the evidence presented, we can deduce that scBOL yields a more consistent and robust performance compared with other benchmarked methods in response to variations in
escalates, they tend to deliver suboptimal performance, often assigning generic cluster labels devoid of semantic content. Furthermore, the performance of ItClust is erratic, marked by a dramatic increase in Quake 10x and a notable decline in Quake Smart-seq2, which exposes its instability. From the evidence presented, we can deduce that scBOL yields a more consistent and robust performance compared with other benchmarked methods in response to variations in 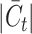 .
.
Figure 4.

Robustness analysis. (A, B) The trend of overall accuracy concerning the change of target private cell type numbers in Quake 10x and Quake Smart-seq2 datasets, respectively; (C, D) The trend of overall accuracy concerning the change of labeled ratio in Quake 10x and Quake Smart-seq2 datasets, respectively.
The ratio of labeled data is a critical factor that influences the extent to which knowledge from reference data can be applied to target data. To investigate this effect, we conducted a series of experiments on the Quake 10x and Quake Smart-seq2 datasets (Figure 4C, Figure 4D), varying the annotated data proportion across the spectrum of 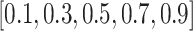 . Our analysis reveals that scBOL consistently outperforms the other baseline methods, sustaining its high performance irrespective of the annotated proportion. This highlights scBOL’s superiority and robustness. In stark contrast, the performance of the other six methodologies was impacted to varying degrees by the annotated proportion, underscoring their reliance on labeled information from reference data. Except for scCNC and scDECL, all methods exhibited a decline in overall accuracy as the annotated proportion increased. This suggests that an excessive volume of reference data may diminish the models’ capacity to identify new cell types. scCNC and scDECL, being unsupervised clustering approaches, operate independently of the labeled information within the reference dataset. This autonomy likely accounts for their performance fluctuations with varied annotated proportions and also suggests a relative loss of competitive edge due to the absence of labeling information. In summary, scBOL demonstrates that it can deliver reliable and impressive results without being susceptible to fluctuations in annotated data proportions. For reasons of brevity, additional experimental findings are presented in the supplementary materials.
. Our analysis reveals that scBOL consistently outperforms the other baseline methods, sustaining its high performance irrespective of the annotated proportion. This highlights scBOL’s superiority and robustness. In stark contrast, the performance of the other six methodologies was impacted to varying degrees by the annotated proportion, underscoring their reliance on labeled information from reference data. Except for scCNC and scDECL, all methods exhibited a decline in overall accuracy as the annotated proportion increased. This suggests that an excessive volume of reference data may diminish the models’ capacity to identify new cell types. scCNC and scDECL, being unsupervised clustering approaches, operate independently of the labeled information within the reference dataset. This autonomy likely accounts for their performance fluctuations with varied annotated proportions and also suggests a relative loss of competitive edge due to the absence of labeling information. In summary, scBOL demonstrates that it can deliver reliable and impressive results without being susceptible to fluctuations in annotated data proportions. For reasons of brevity, additional experimental findings are presented in the supplementary materials.
Intra-data experiment on the spatial transcriptomic dataset
In this study, we extended our evaluation of scBOL to single-cell transcriptome-imaging datasets, substituting the autoencoder with a graph convolutional network (GCN) (Figure 5). The integration of GCN allows scBOL to harness the spatial arrangement and molecular profiles of cells. We initiated our investigation with an intra-dataset analysis employing the Human BioMolecular Atlas Program (HuBMAP) dataset, which was generated using Cyclic-Immunofluorescence (CODEX) technology. Results demonstrate that scBOL outperforms competing methods, showcasing its robust capability to capture and integrate both spatial and gene expression information effectively. We subsequently compared scBOL with prominent scRNA-seq clustering and annotation algorithms, such as MARS, scNym and scArches, underlining the significance of incorporating spatial information. Relying solely on gene expression data proves insufficient to extract comprehensive biological insights; the concurrent utilization of spatial information significantly enhances clustering and annotation performance. Although MARS exhibits resilience to the absence of spatial data concerning known accuracy, it experiences pronounced degradation in both novel and overall accuracy, potentially attributable to the reference dataset predominantly covering common cell types. In stark contrast, scNym and scArches reveal subpar performances across all accuracy metrics, reinforcing the critical role that spatial context plays. STAGATE, a method for spatial clustering and integration, delivers markedly unsatisfactory results across all accuracy measures, with particular deficits in known accuracy. As an unsupervised method, it fails to leverage the annotated labels and spatial details contained within the reference dataset and may suffer from the stringent data quality requirements it imposes. STELLAR, a spatial annotation tool, matches scBOL’s performance concerning known accuracy yet falls short in novel accuracy. This shortfall may stem from its inaccurate estimations of the number of novel cell types. In summary, our comprehensive analysis positions scBOL as the superior analytical tool when confronted with spatial transcriptome datasets. Its remarkable performance is conclusively established against an array of benchmarks, including single-cell annotation, spatial clustering and spatial annotation methodologies, unequivocally highlighting scBOL’s excellence in this domain.
Figure 5.
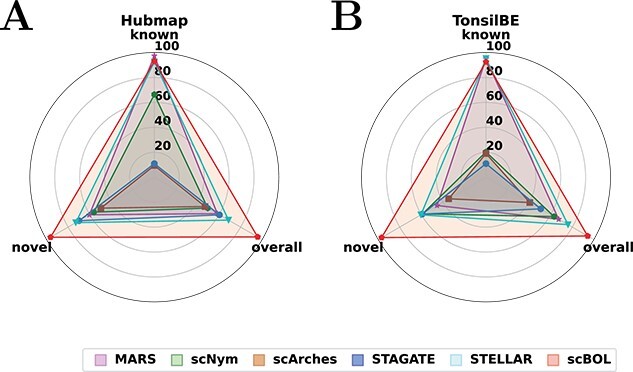
Radar plot of scBOL and other five competing methods measured by three kinds of accuracy. A. Radar plots on the Hubmap dataset for intra-data experiment. B. Radar plots for inter-data experiment, where Tonsil is the reference dataset and BE is the target dataset.
To provide a clearer visualization of annotation outcomes, we present Sankey diagrams that illustrate the correlation between actual and predicted cell types for each method (Figure 6A). These diagrams graphically depict the distribution of cell types and facilitate a comparative assessment of the efficacy of cell type annotations across various sample sizes. This approach is particularly valuable for identifying rare cell types as the visual representation can substantially enhance the interpretation of annotation accuracy. scBOL consistently achieves near-perfect identification, even of infrequent cell types such as Nerve and Endothelial, underscoring its proficiency in incorporating spatial information. Moreover, scBOL’s edge is evident in its capacity to detect and assign labels to novel cell types. Within the context of three newly identified cell types—Plasma, Smooth Muscle and Enterocyte—scBOL is the only method that can correctly classify all three simultaneously. This capability likely stems from scBOL’s algorithmic design, which is tailored to estimate the number of novel cell types and to ensure each cell type’s integrity while maintaining clear distinctions between different cell types on the global structural level. In contrast, both scNym and scArches fall short when analyzing spatial transcriptome data; these methods can identify only a limited subset of cell types. STELLAR, which is specifically designed for spatial transcriptomic analysis, also encounters challenges in this complex task. While STELLAR reliably identifies common cell types, it significantly errs with novel cell types, even in the presence of extensive samples, indicating a limitation in recognizing new cell categories. The information conveyed by the Sankey diagrams reaffirms that scBOL upholds its accuracy in annotating spatial transcriptome data regardless of cell category, size or uniqueness.
Figure 6.

Intra-data experiments on Hubmap spatial transcriptomics data. A. Mapping relationship among prediction results of scBOL and other three methods via Sankey plots. B. Visualization plots via UMAP for Hubmap experiments calculated using the latent representations of scBOL and other five methods colored by cell types.
The embedding space visualization via UMAP plots (Figure 6B) highlights the distribution of samples for scBOL and five alternative methods. The embedded representations derived by scBOL effectively retain critical cell-type-specific information, enabling distinct segregation of all cellular phenotypes. Remarkably, scBOL demonstrates its robust annotation capabilities even for underrepresented cell types, such as Nerve and Endothelial cells, by consistently achieving accurate identification, thereby showcasing its superior annotation prowess. Contrastingly, the alternative approaches, encompassing single-cell clustering, single-cell annotation, spatial data clustering and spatial data annotation methods, experience varying degrees of conflation, suggesting that prevailing methods predominantly struggle with the intricate challenges of cell type alignment and novel cell type clustering within the context of our study. Notably, MARS demonstrates a marked deficiency in spatial data analysis, evidenced by a complete amalgamation of cell types, which renders them indistinguishable—a clear indication of its inapplicability to spatial data. While scNym and scArches at times exhibit commendable performance with single-cell annotation, their efficacy diminishes notably when applied to spatial transcriptome data annotation, underscoring the imperative need to incorporate spatial context. STAGATE’s subpar performance, which is potentially attributable to both algorithmic design flaws and a total disregard for existing cell type labels, further emphasizes this point. Similarly, STELLAR’s limitations become conspicuous in the discovery and identification of novel cell types within the UMAP visualization. Novel cell types, such as SmoothMuscle and Endothelial cells, are often confounded in the embedding, challenging their recognition. In summary, benchmarking against these competitive methodologies showcases scBOL’s distinction in intra-data analyses. Such results affirm the effectiveness of scBOL’s dual-directional alignment and prototype learning strategies, consolidating its standing as a method of choice for intricate spatial transcriptomic data interrogation.
When cell-type annotations are projected back onto spatial coordinates, predictions made by scBOL demonstrate concordance with verified annotations. Furthermore, scBOL does not exhibit difficulties in identifying cell types within specific regions of the tissue sample (Figure 7). The evidence conclusively demonstrates the superiority of scBOL in accurately identifying common cell types and effectively clustering novel cell types within spatial data, thus highlighting its potential for widespread application in practical contexts. In contrast, the limitations of STELLAR are clearly illustrated in the provided images. Specifically, when analyzing plasma samples of substantial size, STELLAR predominantly erroneously assigns them to the category of macrophages, underscoring its deficiency in detecting novel cell types and its propensity to erroneously categorize them as common cell types. Additionally, this trend is reaffirmed by the consistent misclassification of the novel cell type ‘smooth muscle’ as ‘endothelial’ by STELLAR, which further corroborates our conclusions regarding its limitations in cell type discovery.
Figure 7.

CODEX image of Hubmap in spatial coordinates colored according to STELLAR predictions (left), scBOL predictions (middle) and ground-truth annotations (right).
Inter-data experiment on the spatial transcriptomic dataset
Buoyed by the promising outcomes from the intra-data transfer trials, we proceeded to execute inter-data experiments to evaluate the capacity of scBOL and comparative methodologies to mitigate batch effects (Figure 5). We employed expert-annotated samples from a singular donor for training and implemented STELLAR on unannotated samples originating from two additional donors. These datasets are characterized by variations arising from several parameters, including the timing of tissue collection, the individual conducting the staining and imaging processes and the distinct segmentation algorithms employed. These factors can potentially alter the distribution of markers, which in turn might impact the interpretability and uniformity of findings. Such discrepancies undeniably introduce heightened obstacles to the annotation task. Nonetheless, scBOL sustains superior performance across all three metrics of accuracy, reaffirming the robustness of the bidirectional alignment strategy employed at both the individual sample and cluster levels in addressing batch effects. Remarkably, scBOL stands out as the solitary methodology that does not exhibit a marked decline in performance in cross-data scenarios, whereas alternative approaches are substantially compromised by batch variations, particularly in terms of novel accuracy. Among these, scNym bears the brunt, with its known accuracy plummeting from approximately 80 to around 20. MARS and STELLAR continue to display a pronounced trade-off between aligning common cell types and identifying novel cell populations, with the presence of batch effects exacerbating their deficiencies in discovering new cell types. Hence, scBOL emerges as the preeminent performer in all evaluated aspects, unequivocally demonstrating its efficacy.
Sankey diagrams were utilized to analyze the proficiency of each method in establishing accurate cell type correspondences (Figure 8A). scBOL exhibited remarkable accuracy in mapping both common and novel cell types, demonstrating its adeptness at balancing the identification of these categories. Notably, scBOL’s performance remained robust even with cell types characterized by limited sample sizes, indicating its resistance to sample size imbalances. On the contrary, scNym failed to recognize cells labeled as pdpn, and scArches could not identify cells marked as pdpn or smoothmuscle. The inability of these methods to identify certain cell types could be attributed to insufficient sample numbers or inadequate batch effect correction, which hindered the integration of reference and target datasets. Additionally, some cell types may require spatial context for accurate identification. While STELLAR’s performance notably improved concerning the Hubmap dataset, it encountered significant challenges in learning data embeddings, such as incorrectly assigning disparate clustering labels to cells within the glandular_epi cluster. Complementary UMAP visualizations verified the predictive quality of each method (Figure 8B). Once again, scBOL distinguished itself by preserving cell-type-specific signatures and counteracting batch effects, unlike other methods, which struggled to delineate a coherent class structure. Notably, scBOL was the sole method capable of recognizing plasma cells. In stark contrast, scArches demonstrated a substantial deficiency in managing batch effects, leading to the erroneous classification of identical cell types from different batches. Similarly, scNym could not entirely segregate cell types within the embedding space, presenting a distorted view of cell type diversity. Moreover, batch effects caused scNym to bifurcate pdpn cells into two separate groups. STELLAR’s flawed identification of plasma cells further underscores the suboptimal nature of its performance.
Figure 8.

Inter-data experiments on Tonsil-BE spatial transcriptomics data, where using Tonsil as reference dataset and BE as target dataset. A. Mapping relationship among prediction results of scBOL and other three methods via Sankey plots. B. Visualization plots via UMAP for Tonsil-BE experiments calculated using the latent representations of scBOL and seven other methods colored by cell types.
Additionally, the CODEX images of Barrett’s esophagus (BE) were mapped using spatial coordinates, and the predictions for scBOL closely align with the ground-truth data, suggesting that scBOL’s accuracy is largely unimpacted by batch effects (Figure 9). Conversely, the results from STELLAR’s analysis are less than optimal. It incorrectly classified a substantial proportion of glandular epithelial cells as plasma cells (This is shown in the part of Figure 9 circled by an ellipse), which reveals its limitations in conducting detailed classification of novel cell types. In summarizing the above analysis, it becomes evident that scBOL excels in learning and accurately representing the embedding space of data while effectively mitigating batch effects. This underlines scBOL’s capacity for leveraging spatial information and underscores its superior performance in cross-dataset experimentation.
Figure 9.

CODEX image of BE in spatial coordinates colored according to STELLAR predictions (left), scBOL predictions (middle) and ground-truth annotations (right).
Robustness analysis in spatial transcriptomic dataset
The influence of the labeled data ratio on the annotation problem is pivotal, as it determines the extent of information transferred from reference to target data. We investigated the resilience of scBOL to variations in this parameter by modifying its value within the set 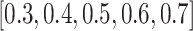 across two distinct datasets—Hubmap and Tonsil-BE (Figure 10A, Figure 10B). scBOL exhibited remarkable and consistent performance across both intra-dataset (Hubmap) and inter-dataset (Tonsil as the reference and BE as the target) evaluations, affirming its dominance and reliability. This impressive outcome is likely due to scBOL’s dual-feature extraction strategy that captures both global structure and individual sample characteristics, rendering it less prone to disruption. In contrast, other methods we examined, such as scArches and STELLAR, displayed pronounced variability across the experiments, underscoring their dependency on reference datasets. Although STELLAR, as an esteemed annotation approach, ranks just behind scBOL in overall accuracy, its inconsistency detracts from its competitive edge. This may stem from STELLAR’s inability to assimilate biological information from a macroscopic viewpoint. MARS, on the other hand, exhibited significant oscillations in performance on the Hubmap dataset, yet remained stable on the Tonsil-BE dataset. This discrepancy might be attributed to the abundant samples in the Tonsil reference dataset, which diminish the influence of varying labeled ratios. STAGATE, an unsupervised clustering method devoid of reference data reliance, also demonstrated performance fluctuations correlating with increased labeled ratio. The likely reason is that a higher labeled ratio decreases the number of samples requiring annotation, thus reducing its annotation burden. In summary, scBOL exemplifies superior stability when confronted with changes in the labeled data ratio in spatial transcriptomic data, enhancing its applicability in real-world multi-omics scenarios.
across two distinct datasets—Hubmap and Tonsil-BE (Figure 10A, Figure 10B). scBOL exhibited remarkable and consistent performance across both intra-dataset (Hubmap) and inter-dataset (Tonsil as the reference and BE as the target) evaluations, affirming its dominance and reliability. This impressive outcome is likely due to scBOL’s dual-feature extraction strategy that captures both global structure and individual sample characteristics, rendering it less prone to disruption. In contrast, other methods we examined, such as scArches and STELLAR, displayed pronounced variability across the experiments, underscoring their dependency on reference datasets. Although STELLAR, as an esteemed annotation approach, ranks just behind scBOL in overall accuracy, its inconsistency detracts from its competitive edge. This may stem from STELLAR’s inability to assimilate biological information from a macroscopic viewpoint. MARS, on the other hand, exhibited significant oscillations in performance on the Hubmap dataset, yet remained stable on the Tonsil-BE dataset. This discrepancy might be attributed to the abundant samples in the Tonsil reference dataset, which diminish the influence of varying labeled ratios. STAGATE, an unsupervised clustering method devoid of reference data reliance, also demonstrated performance fluctuations correlating with increased labeled ratio. The likely reason is that a higher labeled ratio decreases the number of samples requiring annotation, thus reducing its annotation burden. In summary, scBOL exemplifies superior stability when confronted with changes in the labeled data ratio in spatial transcriptomic data, enhancing its applicability in real-world multi-omics scenarios.
Figure 10.
(A, B) Robustness analysis: The trend of overall accuracy concerning the change of labeled ratio in the Hubmap dataset and Tonsil-BE dataset.
Validity of |𝒞t| estimation
The cardinality of the set 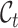 denotes the number of distinct cell types present within the target dataset. Accurately estimating this quantity is crucial for the identification of new cell types, thereby underscoring the necessity of validating the efficacy of the consensus score-based estimation methodology. In this regard, we employ two experimental datasets: Quake 10x and Quake Smart-seq2, which contain 36 and 45 cell types, respectively. Our investigation focuses on scenarios where the discrepancy, referred to as the ‘gap’ between the estimated and true values of
denotes the number of distinct cell types present within the target dataset. Accurately estimating this quantity is crucial for the identification of new cell types, thereby underscoring the necessity of validating the efficacy of the consensus score-based estimation methodology. In this regard, we employ two experimental datasets: Quake 10x and Quake Smart-seq2, which contain 36 and 45 cell types, respectively. Our investigation focuses on scenarios where the discrepancy, referred to as the ‘gap’ between the estimated and true values of 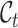 spans an array of values:
spans an array of values: 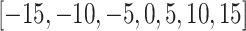 (Figure 11). Here, a ‘gap’ of 0 indicates a perfect match between the estimated count and the actual number of cell types. Examination of the results from both datasets reveals that the consensus score peaks when the gap equals 0; this phenomenon corroborates the reliability of our proposed estimation strategy.
(Figure 11). Here, a ‘gap’ of 0 indicates a perfect match between the estimated count and the actual number of cell types. Examination of the results from both datasets reveals that the consensus score peaks when the gap equals 0; this phenomenon corroborates the reliability of our proposed estimation strategy.
Figure 11.
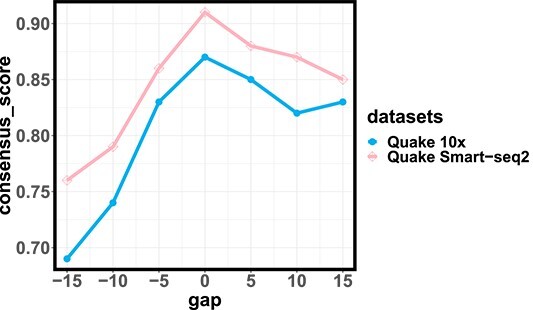
Variation in consensus score about gap measurement on two large-scale datasets: Quake 10x and Quake Smart-seq2.
Effect of 𝑳pro and 𝑳reg
In this section, we conduct ablation studies utilizing five pairs of scRNA-seq datasets to delve deeper into the impact of the intra-data neighbor-aware prototypical learning model, denoted as  , along with the cross-data semantic-aware prototypical learning model,
, along with the cross-data semantic-aware prototypical learning model, 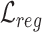 . The omission of
. The omission of 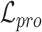 leads to a marked decline in scBOL’s overall accuracy, unequivocally affirming its integral role within the framework (Figure 12A). The benefits of
leads to a marked decline in scBOL’s overall accuracy, unequivocally affirming its integral role within the framework (Figure 12A). The benefits of 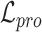 are particularly noteworthy when analyzing cross-dataset scenarios, such as when ‘Haber largecell’ serves as the reference dataset and ‘Haber region’ is the target dataset, or ‘Muraro’ and ‘Baron_human’ are employed as reference and target datasets, respectively. In a similar vein, excluding
are particularly noteworthy when analyzing cross-dataset scenarios, such as when ‘Haber largecell’ serves as the reference dataset and ‘Haber region’ is the target dataset, or ‘Muraro’ and ‘Baron_human’ are employed as reference and target datasets, respectively. In a similar vein, excluding 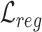 results in significant impairment in scBOL’s functionality, an effect that is starkly evident with cross-datasets pairings like ‘Muraro’ with ‘Baron_human’, or ‘Mammary Smart-seq2’ with ‘Mammary 10x’ as the reference and target datasets, respectively (refer to Figure 12B). The presence of
results in significant impairment in scBOL’s functionality, an effect that is starkly evident with cross-datasets pairings like ‘Muraro’ with ‘Baron_human’, or ‘Mammary Smart-seq2’ with ‘Mammary 10x’ as the reference and target datasets, respectively (refer to Figure 12B). The presence of 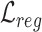 consistently bolsters annotation and clustering performance across all datasets, substantiating the meaningfulness of this enhancement. To draw things together, the data presented herein incontrovertibly affirm the critical nature of the two methodological advancements integrated into our training algorithm.
consistently bolsters annotation and clustering performance across all datasets, substantiating the meaningfulness of this enhancement. To draw things together, the data presented herein incontrovertibly affirm the critical nature of the two methodological advancements integrated into our training algorithm.
Figure 12.
(A, B) Ablation study: Comparing the accuracy on all cell types with or without 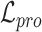 and
and  , respectively. M-SS2 to M-10X: Mammary Smart-seq2 as reference data and Mammary 10x as target data. Mu to Ba: Muraro as reference data and Baron as target data. V-SS2 to V-10X: Vento-Tormo Smart-seq2 as reference data and Vento-Tormo 10x as target data. P to M: Plasschaert as reference data and Montoro as target data. H-l to H-r: Haber largecell as reference data and Haber region as target data.
, respectively. M-SS2 to M-10X: Mammary Smart-seq2 as reference data and Mammary 10x as target data. Mu to Ba: Muraro as reference data and Baron as target data. V-SS2 to V-10X: Vento-Tormo Smart-seq2 as reference data and Vento-Tormo 10x as target data. P to M: Plasschaert as reference data and Montoro as target data. H-l to H-r: Haber largecell as reference data and Haber region as target data.
These insights dovetail perfectly with the hypothetical outcomes initially proposed in our study. Specifically, the intra-data neighbor-aware prototypical learning hones in on both the global and local semantic structures of cell types, promoting within-cluster feature cohesion while distancing features across distinct clusters. On the other hand, cross-data semantic-aware prototypical learning compels the model to consciously seek alignment with cell type features by leveraging semantically linked anchors as connective conduits. Furthermore, our exploration extends to hyperparameter sensitivity, particularly the contrastive temperature, 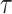 , and the sample selection ratio,
, and the sample selection ratio, 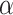 , detailed in the supplementary material. The robustness of our proposed method is underscored by consistently high overall accuracy across all cell types, notwithstanding fluctuations in these parameters.
, detailed in the supplementary material. The robustness of our proposed method is underscored by consistently high overall accuracy across all cell types, notwithstanding fluctuations in these parameters.
DISCUSSION
Identification methods for cell types within single-cell datasets have progressed significantly, transitioning from labor-intensive manual annotation techniques based on unsupervised clustering and the detection of marker genes to automated annotation methods that employ supervised classification algorithms to facilitate label transfer using well-characterized reference datasets. However, when the target dataset contains previously unidentified cell types absent in the reference dataset, a common approach is to first isolate these novel cell types as a distinct population and subsequently apply cluster analysis to categorize them. Although this two-stage approach is practical, it is not necessarily the most effective. Our integrative one-stage end-to-end methodology demonstrates superior accuracy and efficiency when compared with the modified two-stage techniques. Empirical evidence from prior experiments indicates that the precision of two-stage algorithms significantly lags behind that of our scBOL framework, particularly concerning the identification of novel cell types. The rational explanation for this is that the tasks of classifying established cell types and uncovering novel ones are intrinsically complementary, with both relying heavily on the robustness of the cell feature representation. A more nuanced understanding of the target data’s category structure is critical to achieving a more distinctive representation of cell features. Additionally, our one-stage end-to-end annotation strategy outpaces the two-stage method in terms of speed, by eliminating the need for a separate unsupervised cell clustering phase.
Despite the numerous benefits inherent in our methodological approach, we have yet to finalize cell annotations across the entirety of the target dataset. In particular, we need to identify marker genes in newly discovered cell clusters, which is a critical step in the biological analysis process. While this aspect is beyond the primary scope of this study, it remains an essential and intellectually stimulating topic for discussion. There may be skepticism among some members of the scientific community regarding the biological relevance of these novel cell clusters. To address such concerns, we have included a series of validation experiments in the supplementary materials. These experiments utilize differential gene expression analysis to evaluate the congruence between marker genes in our predicted cell clusters and those in well-established reference clusters. Empirical evidence demonstrates that the cell cluster predictions made by our scBOL algorithm show a high degree of marker gene similarity to the known clusters—a level of accuracy not replicated by competing algorithms. This high correlation between our predictions and the ground truth underscores the significance of the problem we are tackling and attests to the robustness of our proposed solution. Looking forward, there is an enticing opportunity to integrate genetic-level a priori knowledge into our framework, to reduce the need for extensive differential gene expression analysis when determining the marker genes of new cell clusters. This prospect poses a fascinating and worthwhile challenge, meriting deliberate reflection and potentially paving the way for additional scholarly inquiry.
The supplementary material includes several critical additional experiments. For instance, we adjusted our analytical framework, originally calibrated on known cell types, to encompass a range of novel cell types, to evaluate the potential influence of rare cellular subsets on our methodology. The findings demonstrate that scBOL is adept at precisely discerning cell types across the spectrum of prevalence, from high-density populations to those occurring in low abundance. Corresponding to our assessment of the labeled ratios in known cell types, we further investigated the influence that varying proportions of unique, target-specific cell types exert on scBOL’s performance. It was observed that an increased presence of novel cells might slightly compromise the identification accuracy for familiar cell types. Nevertheless, scBOL consistently maintains a robust and favorable balance in its performance for distinguishing both known and novel cell types. Moreover, we present scalability tests for scBOL and competing algorithms in the context of extensive datasets. While not the most expedient, our approach is far from the slowest, efficiently processing tens of thousands of cells within several hours. Crucially, we conducted comparative analyses using different algorithms on two distinct spatial transcriptomic datasets. Diverging from the Hubmap and TonsilBE datasets, which were generated via multiplexed imaging technology, we included a representative subset of seqFISH data—derived from in situ hybridization techniques—and Stereo-seq data—sourced from sequencing technologies. Impressively, scBOL sustained robust performance metrics on both datasets. This suggests that the intrinsic variance in spatial transcriptomics data types and their origins exerts minimal impact on the applicability of scBOL. This resilience of performance across diverse datasets aligns with our previously drawn conclusions from scRNA-seq data analysis.
Last but not least, we turn our attention to the development of algorithms within the single-cell community, particularly those harnessing deep learning technology. Notably, the versatility of the scBOL framework in addressing both scRNA-seq data and spatial transcriptomic data can be attributed to its innovative bipartite prototype alignment technique. This approach operates on the dimension of cell feature representation, a shared element across various foundational network modules. The inherent flexibility of scBOL suggests a potential for broader application across an array of single-cell omics data; a simple substitution of its core backbone with one tailored to a specific omics type would facilitate the generation of cell representations. We urge our fellow researchers in algorithm development to channel their expertise toward crafting algorithms of greater generality, capable of embracing the widest spectrum of data types. Concurrently, we must also ponder whether a singular network structure could proficiently accommodate the diversity inherent in single-cell data, in a manner reminiscent of how the transformer model has achieved paradigmatic status in the realms of natural language processing, computer vision and speech recognition [65–67]. We remain optimistic that an analogous architectural framework could simplify the challenge of adapting to various data forms and analytical tasks within our domain [41, 68, 69]. Furthermore, there is a clear necessity for additional work in creating algorithmic evaluation benchmarks. While this study has endeavored to establish a benchmark as inclusive as possible, we acknowledge its preliminary nature—it is but an initial foray into a domain rife with opportunities for refinement by the research community. Advancing the quality and comprehensiveness of these benchmarks necessitates collective effort, a goal we enthusiastically endorse.
CONCLUSION
The accelerated advancement of scRNA-seq has revolutionized our capacity to investigate gene expression heterogeneity at an individual cellular level. Simultaneously, the advent of spatial proteomic and RNA imaging technologies has catalyzed a paradigm shift in our comprehension of cells and molecules in their native context, underscoring the significance of their spatial attributes. In light of these developments, we have conceptualized scBOL, an innovative and adaptive deep-learning instrument engineered to universally identify cell types across both single-cell and spatial transcriptomics datasets. Distinguished from prevailing annotation tools—which are generally constrained to assigning a generic ‘unassigned’ label to unrecognized cell types—scBOL empowers researchers with versatile applicability to diverse datasets and supports more nuanced analytical approaches. The scBOL framework is predicated on four foundational strategies:
(1) Universality in Annotation: scBOL is adept at annotating both scRNA-seq and spatial transcriptomics data, due to its diverse network architectures. It employs a community-endorsed autoencoder to refine scRNA-seq data, filtering noise to reveal the underlying biological signals and capture the data’s intrinsic low-dimensional structure while deploying a GCN to discern spatial and molecular patterns within spatial transcriptomics data.
(2) Global-Local Semantic Structure Exploration: Unlike methods solely focusing on a singular level of abstraction, scBOL introduces an intra-data neighbor-aware prototypical learning approach. This method harnesses the entirety of the dataset’s semantic structure across both macroscopic and microscopic perspectives, thereby enhancing the granularity of analysis.
(3) Cross-Data Semantic Anchor Utilization: The framework features a novel, cross-data semantic-aware prototypical learning strategy, leveraging semantic anchors to align cell-type features. This alignment is pivotal for maintaining consistency across common cell types and also addresses the batch effect by performing a dual-orientation alignment at the prototype and sample levels.
(4) Refined Estimation of Novel Cell Populations: scBOL also offers an advanced method for estimating the prevalence of novel cell populations, which surpasses mere heuristic determination, facilitating more rational and precise outcomes.
Functioning as a transductive learning technique, scBOL integrates both reference and target datasets within the training phase, enhancing its predictive power and robustness. Overall, the scBOL annotation framework stands out for its exceptional flexibility and broad applicability across the realm of transcriptomics research.
scBOL has demonstrated its versatility across a diverse array of scenarios. Initially, our experimental outcomes utilizing scRNA-seq data and spatial transcriptomics evidence scBOL’s proficiency in identifying common cell types and delineating novel ones. What enhances its utility is the model’s capacity to interface with reference and target datasets from either identical or disparate tissues and donors, showcasing scBOL’s exceptional ability to mitigate batch effects. Furthermore, the robustness of scBOL has been rigorously confirmed through an analysis of the model’s performance sensitivity to variations in key parameters, reinforcing its suitability for practical applications. Visual aids, such as Sankey diagrams, UMAP visualizations and CODEX plots, corroborate scBOL’s competence in capturing both the intrinsic biological signals in gene expression profiles and the spatial information encoded by spatial coordinates. In addition, the progression of consensus scores about the variable 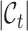 substantiates our approach for inferring the count of novel cell types.
substantiates our approach for inferring the count of novel cell types.
LIMITATIONS AND FUTURE IMPLICATIONS
Given these accomplishments, we aim to identify areas ripe for enhancement and propose several plausible avenues for future research endeavors.
Firstly, the current framework has demonstrated its efficacy with canonical datasets; nevertheless, the burgeoning interest in scalable neural networks to accommodate voluminous datasets has become increasingly prominent. Tackling the challenge of concurrently assimilating vast quantities of data across a diverse array of tissues and donors is not only worthwhile but also has the potential to broaden the applicability of the model by enhancing its capacity for generalization. Given the significance of this expansion, it is imperative to refine the generalized annotation problem and evolve the scBOL framework to encompass atlas-scale datasets. Such refinement would empower the model to achieve a more nuanced embedding representation of cell types, thereby elevating its utility across a wider range of applications and catalyzing the identification of hitherto undiscovered cell types within complex biological landscapes.
Secondly, scBOL operates as a transductive learning method, necessitating access to both reference and target datasets during its training phase. However, this requirement is often impractical in real-world scenarios. For one, reference data may not always be readily accessible. Moreover, the addition of new data for learning purposes demands their incorporation alongside reference data within the model, necessitating re-training—a resource-intensive process. Consequently, there is a crucial need to investigate an inductive learning approach. Such a method would require only the reference data for initial training, after which the resulting model would be capable of generalizing to target data without the need for further modification or re-training. This could significantly enhance the applicability and efficiency of the learning process in practical settings.
Thirdly, this study primarily addresses the challenge of annotating scRNA-seq and spatial transcriptomics data using the scBOL framework. Nevertheless, the advent of multi-omics technologies facilitates the simultaneous acquisition of diverse modalities of biological data, thereby enabling a more comprehensive characterization of cellular diversity. A critical trajectory for subsequent research endeavors involves the expansion of scBOL’s capabilities to encompass multi-omics datasets. This augmentation would enhance scBOL’s capacity to unravel cellular heterogeneity with greater efficiency by integrating and interpreting critical biological insights gleaned from various data dimensions.
Key Points
To our knowledge, we present the novel scBOL algorithm, which represents the inaugural unified approach to address the practical annotation problem in both scRNA-seq and spatial transcriptomics data.
scBOL deftly assigns distinct cluster labels to novel cell types, diverging from the simplistic assignment of a generic ‘unassigned’ label. This added capability poses a significantly higher level of difficulty, distinguishing our algorithm as a sophisticated advancement in the field.
scBOL introduces two innovative strategies: the intra-data neighbor-aware prototypical learning strategy and the cross-data semantic-aware prototypical learning strategy. They collectively enhance scBOL’s capability of comprehending semantic structures at both macroscopic and microscopic levels.
scBOL presents a straightforward and efficacious approach to the complex task of quantifying the total number of cell types within a given dataset.
Extensive experiments on scRNA-seq and spatial transcriptomics data demonstrate that scBOL effectively resolves the annotation conundrum, consistently delivering outstanding performance. Furthermore, scBOL exhibits superior capabilities in mitigating batch effects and demonstrates robustness across diverse data conditions.
Supplementary Material
ACKNOWLEDGEMENT
This work was supported by the National Key Research and Development Program of China (2021YFF1200902) and the National Natural Science Foundation of China (32270689, 12126305).
Yuyao Zhai is a doctoral candidate at the School of Mathematical Sciences, Peking University.
Liang Chen is a senior researcher at Huawei.
Minghua Deng is a professor at the School of Mathematical Sciences, Peking University.
Contributor Information
Yuyao Zhai, School of Mathematical Sciences, Peking University, Beijing, China.
Liang Chen, Huawei Technologies Co., Ltd., Beijing, China.
Minghua Deng, School of Mathematical Sciences, Peking University, Beijing, China; Center for Statistical Science, Peking University, Beijing, China; Center for Quantitative Biology, Peking University, Beijing, China.
References
- 1. Lin L, Li Y, Li S, et al. Comparison of next-generation sequencing systems. J Biomed Biotechnol 2012;2012:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Slatko Barton E, Gardner Andrew F, and Ausubel Frederick M. Overview of next-generation sequencing technologies. Curr Protoc Mol Biol, 122(1):e59, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kolodziejczyk AA, Kim JK, Svensson V, et al. The technology and biology of single-cell rna sequencing. Mol Cell 2015;58(4):610–20. [DOI] [PubMed] [Google Scholar]
- 4. Ding J, Adiconis X, Simmons SK, et al. Systematic comparison of single-cell and single-nucleus rna-sequencing methods. Nat Biotechnol 2020;38(6):737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tirosh I, Venteicher AS, Hebert C, et al. Single-cell rna-seq supports a developmental hierarchy in human oligodendroglioma. Nature 2016;539(7628):309–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Van de Sande B, Flerin C, Davie K, et al. A scalable scenic workflow for single-cell gene regulatory network analysis. Nat Protoc 2020;15(7):2247–76. [DOI] [PubMed] [Google Scholar]
- 7. Marques S, van Bruggen D, Vanichkina DP, et al. Transcriptional convergence of oligodendrocyte lineage progenitors during development. Dev Cell 2018;46(4):504–517.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moffitt JR, Bambah-Mukku D, Eichhorn SW, et al. Molecular, spatial, and functional single-cell profiling of the hypothalamic preoptic region. Science 2018;362(6416):eaau5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lohoff T, Ghazanfar S, Missarova A, et al. Integration of spatial and single-cell transcriptomic data elucidates mouse organogenesis. Nat Biotechnol 2022;40(1):74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen A, Liao S, Cheng M, et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using dna nanoball-patterned arrays. Cell 2022;185(10):1777–92. [DOI] [PubMed] [Google Scholar]
- 11. Maynard KR, Collado-Torres L, Weber LM, et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat Neurosci 2021;24(3):425–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lin J-R, Fallahi-Sichani M, Sorger PK. Highly multiplexed imaging of single cells using a high-throughput cyclic immunofluorescence method. Nat Commun 2015;6(1):8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Goltsev Y, Samusik N, Kennedy-Darling J, et al. Deep profiling of mouse splenic architecture with codex multiplexed imaging. Cell 2018;174(4):968–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen KH, Boettiger AN, Moffitt JR, et al. Spatially resolved, highly multiplexed rna profiling in single cells. Science 2015;348(6233):aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lewis SM, Asselin-Labat M-L, Nguyen Q, et al. Spatial omics and multiplexed imaging to explore cancer biology. Nat Methods 2021;18(9):997–1012. [DOI] [PubMed] [Google Scholar]
- 16. Rozenblatt-Rosen O, Regev A, Oberdoerffer P, et al. The human tumor atlas network: charting tumor transitions across space and time at single-cell resolution. Cell 2020;181(2):236–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang M, Eichhorn SW, Zingg B, et al. Spatially resolved cell atlas of the mouse primary motor cortex by merfish. Nature 2021;598(7879):137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Malte D. Current best practices in single-cell rna-seq analysis: a tutorial. Mol Syst Biol 2019;15(6):e8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen J, Liu W, Luo T, et al. A comprehensive comparison on cell-type composition inference for spatial transcriptomics data. Brief Bioinform 2022;23(4):bbac245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lähnemann D, Köster J, Szczurek E, et al. Eleven grand challenges in single-cell data science. Genome Biol 2020;21(1):1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhai Y, Chen L, Deng M. Generalized cell type annotation and discovery for single-cell RNA-seq data. In Proceedings of the AAAI Conference on Artificial Intelligence 2023 Jun 26 (Vol. 37, No. 4, pp. 5402–5410). [Google Scholar]
- 22. Satija R, Farrell JA, Gennert D, et al. Jeffrey a Farrell, David Gennert, Alexander F Schier, and Aviv Regev. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 2015;33(5):495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dong K, Zhang S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat Commun 2022;13(1):1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Long Y, Ang KS, Li M, et al. Spatially informed clustering, integration, and deconvolution of spatial transcriptomics with graphst. Nat Commun 2023;14(1):1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wolf FA, Angerer P, Theis FJ. Scanpy: large-scale single-cell gene expression data analysis. Genome Biol 2018;19(1):1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pliner HA, Shendure J, Trapnell C. Supervised classification enables rapid annotation of cell atlases. Nat Methods 2019;16(10):983–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhai Y, Chen L, Deng M. Realistic cell type annotation and discovery for single-cell RNA-seq data. In Proceedings of the Thirty-Second International Joint Conference on Artificial Intelligence 2023 Aug 19 (pp. 4967–74).
- 28. Regev A, Teichmann SA, Lander ES, et al. The human cell atlas. Elife 2017;6:e27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cao J, O’Day DR, Pliner HA, et al. A human cell atlas of fetal gene expression. Science 2020;370(6518):eaba7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Consortium TM, Overall coordination, Logistical coordination, et al. Single-cell transcriptomics of 20 mouse organs creates a tabula muris. Nature 2018;562(7727):367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao ZJ, Wei L, Lu S. et al. Searching large-scale scRNA-seq databases via unbiased cell embedding with Cell BLAST. Nat Commun 2020;11:3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xu C, Lopez R, Mehlman E, et al. Probabilistic harmonization and annotation of single–cell transcriptomics data with deep generative models. Mol Syst Biol 2021;(1):e9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Flores M, Liu Z, Zhang T, et al. Paniagua. Deep learning tackles single-cell analysis–a survey of deep learning for scrna-seq analysis. Brief Bioinform 2022;23(1):bbab531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hu J, Li X, Hu G, et al. Iterative transfer learning with neural network for clustering and cell type classification in single-cell rna-seq analysis. Nat Mach Intell 2020;2(10):607–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xu J, Zhang A, Liu F, et al. Ciform as a transformer-based model for cell-type annotation of large-scale single-cell rna-seq data. Brief Bioinform 2023;24(4):bbad195. [DOI] [PubMed] [Google Scholar]
- 36. Chen L, Wang W, Zhai Y, Deng M. Deep soft k-means clustering with self-training for single-cell rna sequence data. NAR Genomics Bioinf 2020;2(2):lqaa039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xiong Y-X, Zhang X-F. Scdot: enhancing single-cell rna-seq data annotation and uncovering novel cell types through multi-reference integration. Brief Bioinform 2024;25(2):bbae072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hu Y, Xiao K, Yang H, et al. Spatially contrastive variational autoencoder for deciphering tissue heterogeneity from spatially resolved transcriptomics. Brief Bioinform 2024;25(2):bbae016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liang C, Zhai Y, He Q, et al. Integrating deep supervised, self-supervised and unsupervised learning for single-cell rna-seq clustering and annotation. Genes 2020;11(7):792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brbić M, Zitnik M, Wang S, et al. Angela O Pisco, Russ B Altman, Spyros Darmanis, and jure Leskovec. Mars: discovering novel cell types across heterogeneous single-cell experiments. Nat Methods 2020;17(12):1200–6. [DOI] [PubMed] [Google Scholar]
- 41. Fischer F, Fischer DS, Biederstedt E, et al. Scaling cross-tissue single-cell annotation models bioRxiv. 2023.
- 42. Zhi-Hua D, Hu W-L, Li J-Q, et al. Scpml: pathway-based multi-view learning for cell type annotation from single-cell rna-seq data. Commun Biol 2023;6(1):1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhai Y, Liang C, Deng M. Scgad: a new task and end-to-end framework for generalized cell type annotation and discovery. Brief Bioinform 2023;24(2):bbad045. [DOI] [PubMed] [Google Scholar]
- 44. Zhai Y, Liang C, Deng M. Scevolve: cell-type incremental annotation without forgetting for single-cell rna-seq data. Brief Bioinform 2024;25(2):bbae039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lotfollahi M, Naghipourfar M, Luecken MD, et al. Mapping single-cell data to reference atlases by transfer learning. Nat Biotechnol 2022;40(1):121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kimmel JC, Kelley DR. Semisupervised adversarial neural networks for single-cell classification. Genome Research. 2021;31(10):1781–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hu J, Li X, Coleman K, et al. David J Irwin, Edward B lee, Russell T Shinohara, and Mingyao Li. Spagcn: integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat Methods 2021;18(11):1342–51. [DOI] [PubMed] [Google Scholar]
- 48. Shen R, Lin L, Wu Z, et al. Spatial-id: a cell typing method for spatially resolved transcriptomics via transfer learning and spatial embedding. Nat Commun 2022;13(1):7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fan Z, Luo Y, Lu H, et al. Spascer: spatial transcriptomics annotation at single-cell resolution. Nucleic Acids Res 2023;51(D1):D1138–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhong Z, Hou J, Yao Z, et al. Domain generalization enables general cancer cell annotation in single-cell and spatial transcriptomics. Nat Commun 2024;15(1):1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brbić M, Cao K, Hickey JW, et al. Annotation of spatially resolved single-cell data with stellar. Nat Methods 2022;19(11):1411–8. [DOI] [PubMed] [Google Scholar]
- 52. Cao Z-J, Lin W, Lu S, et al. Searching large-scale scrna-seq databases via unbiased cell embedding with cell blast. Nat Commun 2020;11(1):3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Eraslan G, Simon LM, Mircea M, et al. Single-cell rna-seq denoising using a deep count autoencoder. Nat Commun 2019;(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He K, Fan H, Wu Y, Xie S, Girshick R. Momentum contrast for unsupervised visual representation learning. In Proceedings of the IEEE/CVF conference on computer vision and pattern recognition 2020 (pp. 9729–9738).
- 55. Yoon J, Zhang Y, Jordon J, van der Schaar M. Vime: extending the success of self-and semi-supervised learning to tabular domain. Adv Neural Inf Process Syst 2020;33:11033–43. [Google Scholar]
- 56. Hamilton W, Ying Z, Leskovec J. Inductive representation learning on large graphs. Adv Neural Inf Process Syst 2017;30. [Google Scholar]
- 57.Tang H, Chen K, Jia K. Unsupervised domain adaptation via structurally regularized deep clustering. In Proceedings of the IEEE/CVF conference on computer vision and pattern recognition 2020 (pp. 8725–8735).
- 58. Haghverdi L, Lun ATL, Morgan MD, Marioni JC. Batch effects in single-cell rna-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol 2018;36(5):421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Han W, Cheng Y, Chen J, et al. Self-supervised contrastive learning for integrative single cell rna-seq data analysis. Brief Bioinform 2022;23(5):bbac377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang M, Yang Y, Xie C, et al. Contrastive learning enables rapid mapping to multimodal single-cell atlas of multimillion scale. Nat Mach Intell 2022;4(8):696–709. [Google Scholar]
- 61. Vieth B, Parekh S, Ziegenhain C, et al. A systematic evaluation of single cell rna-seq analysis pipelines. Nat Commun 2019;10(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang H-Y, Zhao J-P, Zheng C-H, Yan-Sen S. Sccnc: a method based on capsule network for clustering scrna-seq data. Bioinformatics 2022. [DOI] [PubMed] [Google Scholar]
- 63. Gan Y, Chen Y, Xu G, et al. Deep enhanced constraint clustering based on contrastive learning for scrna-seq data. Brief Bioinform 2023;24(4):bbad222. [DOI] [PubMed] [Google Scholar]
- 64. Kuhn HW. The hungarian method for the assignment problem. Naval Res Logist Q 1955;2(1–2):83–97. [Google Scholar]
- 65. Touvron H, Martin L, Stone K, et al. Llama 2: open foundation and fine-tuned chat models arXiv preprint arXiv:2307.09288. 2023.
- 66. Liu H, Li C, Wu Q, Lee YJ. Visual instruction tuning. Adv Neural Inf Process Syst 2024;36. [Google Scholar]
- 67. Zhu D, Chen J, Shen X, et al. Minigpt-4: enhancing vision-language understanding with advanced large language models arXiv preprint arXiv:2304.10592. 2023.
- 68. Theodoris CV, Xiao L, Chopra A, et al. Transfer learning enables predictions in network biology. Nature 2023;618(7965):616–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cui H, Wang C, Maan H, et al. Scgpt: toward building a foundation model for single-cell multi-omics using generative ai. Nat Methods 2024;1–11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.