

Abstract
Parkinson’s disease is pathologically defined by the death of dopaminergic neurons within the pars compacta of the substantia nigra. To date, the etiology of this multifaceted disease remains largely unclear, which may contribute in part to a current lack of disease-modifying therapies. Recent advances in single-cell and spatial genomic profiling tools have provided powerful new ways to measure cellular state changes in brain diseases. Here, we describe how these tools have offered insight into these complex disorders and highlight a recently performed comprehensive study of dopaminergic neuron susceptibility in Parkinson’s. The data generated by this recent work provide evidence for the role of specific pathways and common genetic variants in giving rise to the loss of a critical dopaminergic subtype in Parkinson’s. We conclude by outlining a set of basic and translational opportunities that arise from those data and insights gathered from this work.
Background
Parkinson’s disease (PD) is a common movement disorder whose most consistent pathological correlate is the death of dopaminergic (DA) neurons residing within the substantia nigra pars compacta. The loss of these cells is responsible for the pathognomonic motor symptoms of PD1, and is observed in both the 10% of PD cases that are familial–which are largely caused by highly-penetrant mutations in a handful of genes2–and the 90% of sporadic PD cases of unknown etiology. Numerous cellular mechanisms have been identified that may contribute to PD-associated DA neuron death such as dysfunctional autophagy3, loss of calcium homeostasis, mitochondrial dysfunction, and protein misfolding4,5. Beyond the substantia nigra, other subcortical brain regions, contain neurons susceptible to PD-associated degeneration, including the dorsal motor nucleus of the vagus nerve (DMNV)6–10, the nucleus basalis of Meynert11–13, the medial/dorsal raphe14–17, the locus coeruleus12,18–20, and the pedunculopontine nucleus (PPN)14,15,21–24 (Figure 1), which could additionally explain some aspects of movement issues (especially in the case of the PPN25–27), as well as non-movement related symptomatology of PD, such as autonomic and olfactory dysfunction28. The pathogenic reasons for the vulnerability of these specific populations–compared with the hundreds of thousands of other neuronal cell types in the brain–remains a fundamental question in PD biology whose answer could unlock new potential treatment targets.
Figure 1. Single-cell and spatial profiling of Parkinson’s disease-susceptible brain regions.
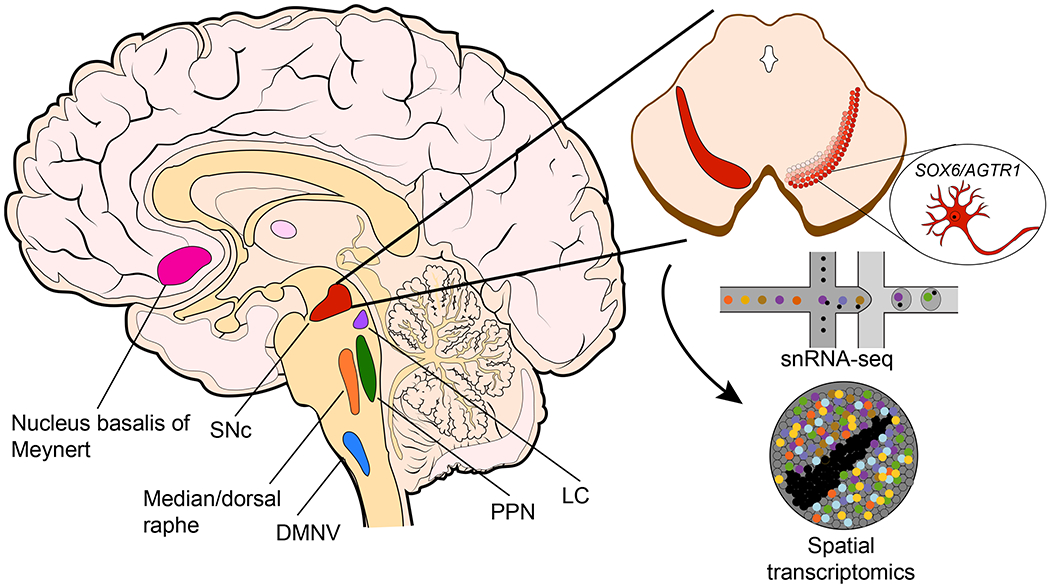
(Left) Schematic of sagittal cut of human brain with colored regions of brain with evidence of neurodegeneration in Parkinson’s disease. (Upper right) Axial cross-section of midbrain with substantia nigra pars compacta colored in shades of red. Darker shades indicate location of more severely-affected neurons. (Bottom right) Schematic for droplet-based single-nucleus RNA-sequencing (snRNA-seq) and Slide-seq/spatial transcriptomics. (Bubble inset) Cartoon of the most susceptible (SOX6/AGTR1) neuron subtype identified in Kamath et al. SNc = substantia nigra pars compacta, DMNV = dorsal motor nucleus of the vagus nerve, PPN = peduncolopontine nucleus, LC = locus coeruleus.
Even within the nigra, molecularly- and spatially-defined DA neuronal populations show variable rates of loss, evidence of which dates back to stereotactic studies from the early 20th century29. Those neurons more resistant to PD-associated degeneration are found more frequently in the dorsal, as opposed to ventral, tier of the A9 catecholaminergic region of the substantia nigra30. Functionally, these neurons have been shown to project to the ventral striatum in rodent studies, suggesting an alternate function more consistent with modulation of reward behavior31 similar to their tegmental area counterparts32. In addition, early histological studies of PD brains suggested that the disease-resistant DA neurons exhibit molecular differences compared to their more susceptible counterparts, including a higher baseline expression of the gene CALB1, first described by Yamada and colleagues33–35,33,34 and further corroborated by more recent work36. The pattern of resistance of the calbindin-expressing DA neurons was recapitulated in toxin-based rodent and non-human primate models of PD which mirrored the movement phenomenon of PD35,37, undergirding the primacy of the loss of non-calbindin expressing neurons in PD pathogenesis.
To clarify and expand upon these molecular differences, later efforts capitalized on laser capture microdissection and RNA-sequencing based techniques to extensively profile midbrain DA neuron populations in healthy rodent and human brain38–41. With laser-capture microdissection, these profiles could be further separated spatially, thereby revealing key molecular differences between putatively susceptible neurons. Further efforts aimed to catalog the molecular pathways which were dysregulated in those dopaminergic neurons in PD brains42,43. These early studies laid evidence for a number of translational theories about the molecular basis of susceptibility and particularly pathways which were amenable to therapeutic interventions44,45. For example, the consistent identification of CALB1 as a marker for neurons resistant to PD has spawned a number of intriguing hypotheses about the role of calcium in disease4,46. One such theory maintains that calcium itself catalyzes the aggregation of alpha-synuclein which may in turn lead to oxidative stress and neuronal damage47,48. Yet another maintains that the slow oscillatory nature of L-type calcium channels, unique to DA neurons, may lead to an excess of mitochondrial oxidant stress and subsequent dysfunction. The dysfunction of mitochondria in DA neurons, a consistent molecular pathology49,50, ultimately leads to the demise of these cells when calcium-buffering elements are unavailable.
The importance of calcium regulation to PD pathogenesis was ultimately put to the test in a set of clinical trials measuring the safety profiles51,52 and efficacy53 of isradipine, an often-used dihydropyridine calcium-channel blocker (CCB) agent, in reducing PD symptoms. While these trials did not meet their primary endpoint, post-hoc analyses suggested that isradipine significantly delayed the need for levodopa therapy in those PD patients receiving adequate pharmacological dosing of calcium blockade54. Such results are congruent with observational work55,56 suggesting that CCBs are in fact most effective at reducing the incidence of PD and, as such, may continue to warrant investigation especially in prodromal PD patient cohorts.
On the flip side of the same coin, efforts have been made to understand the valence of the specific molecules that define susceptible DA neuronal populations. The gene ALDH1A1, for example, encodes an aldehyde dehydrogenase enzyme, and marks neurons that are preferentially lost in PD57. Interestingly, impaired metabolism of dopamine by aldehyde dehydrogenase has been shown to produce neurotoxic byproducts and the inhibition of this molecule has been shown to bring about dopaminergic cell loss58. This would suggest that certain intrinsic capacities of dopamine neurons to metabolize the molecule dopamine itself are related to the pathogenesis of PD via the buildup of toxic substrates over the course of years59,60. The enzyme has also been shown to participate in the production and metabolism of GABA in midbrain DA neurons, adding further complexity to its functional roles in vivo61. It remains to be understood what other specific molecules are responsible or protective of DA neurons in PD-associated degeneration.
Finally, one major pitfall of prior bulk sequencing technologies in understanding brain cell type function and dysfunction is a lack of precision. The human brain is composed of a remarkable diversity of cell types, even with anatomically- and functionally-defined regions such as the nigra. These bulk sequencing techniques required pre-defined aggregation of samples (e.g. by location in the form of laser capture microscopy), preventing a bottom-up ascertainment of cellular diversity, and thereby an unbiased assessment of specific neuron types that are lost in neurodegeneration.
Single-cell genomic profiling identifies a uniquely susceptible population to Parkinson’s disease
The advent of single-cell sequencing has allowed for the rapid development of high-dimensional datasets that enable systematic definitions of cell types within complex tissues62,63. Application of these tools to the human brain has focused on uncovering processes and cell types that underlie disease64. Recent efforts have begun to use these high-dimensional analyses to offer insights into the molecular basis of neurodegenerative disease. Single-cell sequencing studies, for example, have nominated specific neuron types that are vulnerable to degeneration in Alzheimer’s disease65 and multiple sclerosis66. In addition, these data have helped determine specific AD-associated angiogenic vascular cells67, identified common signatures associated with tau tangles68, a hallmark of AD, and determined specific molecular profiles of reactive glial cells in association with amyloid and tau aggregates69. These studies have continued to expand in size and scope, with additional modalities such as measurements of open chromatin70, offering additional insights into epigenetic and protein abundance changes in association with complex degenerative diseases. The vast majority of these efforts have focused on cortex, owing in part due to the intrinsically smaller length scale of variability (compared to less stereotyped areas such as the midbrain) and the existence of highly detailed cortical cell atlases in several mammalian species64,71,72.
Apart from traditional single-cell sequencing methods, the advent of spatial molecular profiling73–78 has been employed in the identification of patterns and cell types in complex tissue offering exciting opportunities for understanding disease biology. New ‘-omic’ spatial technologies broadly fall within two categories: capture-based methods that rely on a spatially-defined indexed surface to locally capture mRNA from tissue, and probe-based technologies in which molecules are tagged with fluorescent probes based on hybridization in situ78. To date, both approaches have revealed a census of cells in complex regions within the rodent and human brain, thereby revealing previously known and, as-of-yet, unexplored gradients and molecular patterns79–82. More recent work has aimed at defining spatially-variable changes that might result from a tissue-wide perturbation, such as in response to traumatic brain injury77 and aging83 or in the vicinity of local amyloid plaque buildup84. The additional advances made in computational tools to investigate and understand these data have allowed for the better assignment of cell-cell interactions, tissue organization, and differential expression within and between cell types across experimental conditions85. Finally, spatial profiling methods have recently begun to expand outside of ascertainment of RNA molecules, with the introduction of spatial DNA86 and antibody profiling87. Undoubtedly, these newer approaches will begin to reveal more hidden information about the molecular basis of brain diseases and fundamental biology about cell types in the brain.
The application of these spatial and single-cell methodologies to PD-relevant tissues has been especially revelatory with the single-cell characterization of mouse midbrain DA neurons88–90. These studies to date have identified numerous molecularly-defined populations that reside within specific areas of the rodent midbrain, corroborating prior sequencing and histological work that separate broad populations into calbindin-positive and aldehyde dehydrogenase-expressing subtypes88. Further comparisons between rodent and ESC-derived cell populations have identified early developmental differences in DA populations that might underlie the vast diversity of their mature successors88. In addition, profiling studies of the substantia nigra, both in health91,92 and disease93, have defined mature DA neuron subtypes and attempted to understand what specific populations are gained or lost in the process of disease. These studies have also pointed to specific processes that occur within cell types, such as the reactivity of glial populations93. One key limitation, however, to these studies is the under-representation of midbrain dopaminergic neurons in human tissue samples–they are surrounded by large white matter tracts and therefore make up a tiny fraction (<1%) of the surrounding cells–making it challenging to comprehensively define DA subpopulations in the human, or to make robust claims about affected molecular pathways in PD subjects.
The dataset provided by Kamath et al94–utilizing a novel sorting procedure to enrich DA neurons in human tissue– sampled a sufficient number of DA neurons across subjects to enable a comprehensive annotation classifying them into 10 molecularly-defined subpopulations. Computational integration of these data with those generated from similar regions in other mammalian species, such as those in the rodentia and scandentia orders, identified one population that was solely present in humans and non-human primates, suggesting intrinsic differences between the nigral cytoarchitecture of humans and that of model organisms. Further, spatial profiling of primate brains revealed that these 10 subpopulations fell along a dorsal-ventral gradient, consistent with prior studies suggesting calbindin-expressing DA neurons were more restricted to the dorsal tier of the A9 catecholaminergic region. These subpopulations broadly fell within a gradient of SOX6 and CALB1 expression, with more specific markers including TRHR, GEM, PART1, and DDT, to name a few. Interestingly, these other subpopulations also showed a gradation of susceptibility that matched well with increasing expression of CALB1 and a stronger dorsal tier preference for more resistant populations. Finally, while future work is needed to better characterize the precise surviving fraction in these populations in a large sample of PD cases, further studies have already corroborated the identification of these 10 subpopulations, in addition to identifying new types across the midbrain95.
Importantly, comparisons of the relative composition of these 10 subpopulations between neurotypical controls and individuals that died of PD or LBD identified one population that was significantly lost in the degenerative disease process, marked by the expression of an angiotensin type 1 receptor, AGTR1 (Figure 1). This same population was also marked by ALDH1A1, a previously-identified marker of susceptibility, but was non-specific to the specifically degenerating population in this study. In addition, other dopaminergic populations were relatively increased in abundance in association with disease, suggesting these types are preferentially preserved. Indeed, those preserved cells not only exist along the dorsal tier, but also express the marker, CALB1, as demarcating resistance33. Transcription factor-based pathway analysis identified a number of significantly induced transcription factor programs, driven by genes known to play roles in PD such as TP53 and NR2F2, among others. Importantly, these programs were only activated in the DA neuron population most susceptible to loss, providing an internal control on the valence of these pathways in the process of neurodegeneration. Finally, the same population with the most significant degree of loss additionally showed the strongest enrichment for common variant risk of genes expressed in this cell type, suggesting cell-intrinsic processes underlie PD-associated degeneration.
Basic and translational impact
The identification of a specific population of dopaminergic neurons and their markers provides a launching point for a range of basic and translational avenues. These areas are ripe for insight and offer therapeutic and biomarker potential. First, the substantial contrast in the heritable risk of PD versus that of Alzheimer’s disease (AD) suggests that there are markedly different processes that underlie these diseases. While AD common variant risk primarily points to an immune process, evidence suggests the PD genetic risk seems to converge on DA neurons91,94,96–99. It is important to note that the heritability enrichment does not mean that any individual allele acts in DA neurons; indeed, several elegant studies have described activities of particular risk loci in non-neuronal populations100–102. Rather, the result suggests that human genetic risk has a significant component that acts in DA neurons. Importantly, this convergence of risk on the most susceptible neuron populations suggests an ability to further test epistatic models of PD. While PD is pathognomically defined by the loss of these neurons, some histological studies suggest that other neurons die at similar rates over the course of this illness12,19. A brain-wide single-cell sequencing survey of PD brains will help uncover the particular cell populations that are similarly most vulnerable, and assess their underlying enrichment of genetic risk. Comparison of all of these vulnerable populations–including the AGTR1-expressing ventral tier dopaminergic neurons–should help to identify molecular similarities that are likely drivers of cell-intrinsic processes associated with PD-induced degeneration.
Second, the identification of particular neuron types that uniquely decline in PD versus other neurodegenerative conditions offers a springboard for determining diagnostic biomarkers more specific to the underlying neurobiology of PD. Indeed, antibodies against the protein AGTR1–the gene we find to be among the most enriched in the degenerating population–have been shown to be elevated in the serum of 117 individuals with PD as compared to a group of 106 age-matched controls 103. Interestingly, this same study also showed a strong correlation between autoantibodies against the angiotensin II receptor type 1 and serum inflammatory cytokines in PD patients. Such findings are consistent with the study authors’ proposed mechanism that the death of these AGTR1-expressing DA neurons causes a release of intracellular antigens and subsequent antibody formation by infiltrating B cells103. Interestingly, recent work would suggest a role for the adaptive immune system, via T cell brain parenchymal entry and recognition of alpha synuclein, in neuronal degeneration and the pathogenesis of PD and LBD104,105. Future approaches could be aimed at mining transcriptional data gathered from various brain regions to identify candidate biomolecules based on marker genes defining the most susceptible neuron types in PD and other degenerative conditions. Additional paired postmortem brain and CSF or serum samples would allow for matched data to validate these computational approaches to identifying disease biomarkers.
Third, comparisons of the most susceptible neuronal populations between PD-affected brains and age-matched controls allows for the identification of molecular pathways altered in the course of neuronal decline. Two specific transcription factors with prior evidence for relevance to PD pathogenesis, encoded by the genes TP53 and NR2F2, rose to significance in regards to their programmatic activity in disease. Specifically, knockout of the protein Tp53 in dopaminergic neurons prevented progression of neurodegeneration in a mitochondrial toxin-based model of PD106. Further, nuclear p53 has been shown to play a critical role in regulating autophagy, a key cellular pathway which mediates neuronal health, via the PD-associated protein PINK1107. Additionally, the transcription factor Nr2f2 has been shown to be induced in publicly available transcriptomic datasets of nigral DA neurons from PD patients108. Over-expression of this key transcription factor accelerated mitochondrial dysfunction and neurodegeneration in a mitochondrial pathology-based model of PD108.
Finally, the identification of specific markers of the dying population may offer insights into druggable pathways. Increasing evidence points to counter-regulatory functions of the dopamine production pathway and the renin-angiotensin-aldosterone system (RAAS)109. Within the nigra, activation of the RAA system has been implicated in production of oxidative stress and the knockdown of the angiotensin II type 1 receptor within a zebrafish model of PD ameliorated nigral degeneration110. From a clinical standpoint, observational studies of individuals with PD suggest a disease-modifying therapeutic effect of commonly-prescribed angiotensin receptor blockers (ARBs)111. Given that these medications are quite well tolerated in large swaths of the population, such drugs could be more easily evaluated for therapeutic efficacy in PD populations with randomized controlled trials. Additional mining of pathways specifically defining other dying neuron populations may offer similarly fruitful repurposed drug candidates.
Conclusion
Parkinson’s disease is a multifaceted and devastating degenerative disorder with no disease-modifying therapies available, in large part due to our incomplete understanding of the key molecular events that occur in vulnerable neuronal populations. Nigral dopaminergic neurons are one of the most clearly affected cell types in the PD brain and have manifest clinical relevance. Single-cell and spatial sequencing technologies provide an unprecedented opportunity to directly study, in detail, the molecular events that occur within human brain cells affected by disease. These methods have increasingly begun to be applied to PD with success in identifying vulnerable cell populations and intrinsic signaling processes that are likely to be pathogenically important. Additional profiling across other modalities, including a combination of spatial, epigenetic, and protein measurements, and in other affected brain regions will no doubt offer a clear picture of the key molecular events in PD. In turn, we see such data as integral to the development of therapeutic interventions and diagnostic metrics.
Funding Sources:
This work was supported by NIH NIGMS T32GM007753/T32GM144273 (to T.K.) and NIH NIA grant F30AG069446 (to T.K.).
Footnotes
Financial Disclosures: In the past 12 months, T.K. has received honoraria from Cajal Neurosciences. E.Z.M. is a consultant for Curio Bioscience.
References
- 1.Ma SY, Röyttä M, Rinne JO, Collan Y & Rinne UK Correlation between neuromorphometry in the substantia nigra and clinical features in Parkinson’s disease using disector counts. J. Neurol. Sci 151,83–87 (1997). [DOI] [PubMed] [Google Scholar]
- 2.Kouli A, Torsney KM & Kuan W-L Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. (Codon Publications, 2018). [PubMed] [Google Scholar]
- 3.Lynch-Day MA, Mao K, Wang K, Zhao M & Klionsky DJ The role of autophagy in Parkinson’s disease. Cold Spring Harb. Perspect. Med 2, a009357 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Surmeier DJ Calcium, ageing, and neuronal vulnerability in Parkinson’s disease. Lancet Neurol. 6, 933–938 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Surmeier DJ Determinants of dopaminergic neuron loss in Parkinson’s disease. FEBS J. 285, 3657–3668 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rajput AH & Rozdilsky B Dysautonomia in Parkinsonism: a clinicopathological study. J. Neurol. Neurosurg. Psychiatry 39, 1092–1100 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gaspar P & Gray F Dementia in idiopathic Parkinson’s disease. A neuropathological study of 32 cases. Acta Neuropathol. 64, 43–52 (1984). [DOI] [PubMed] [Google Scholar]
- 8.Eadie MJ THE PATHOLOGY OF CERTAIN MEDULLARY NUCLEI IN PARKINSONISM. Brain 86, 781–792 (1963). [DOI] [PubMed] [Google Scholar]
- 9.Saper CB, Sorrentino DM, German DC & de Lacalle S Medullary catecholaminergic neurons in the normal human brain and in Parkinson’s disease. Ann. Neurol 29, 577–584 (1991). [DOI] [PubMed] [Google Scholar]
- 10.Benarroch EE, Schmeichel AM, Sandroni P, Low PA & Parisi JE Involvement of vagal autonomic nuclei in multiple system atrophy and Lewy body disease. Neurology 66, 378–383 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Arendt T, Bigl V, Arendt A & Tennstedt A Loss of neurons in the nucleus basalis of Meynert in Alzheimer’s disease, paralysis agitans and Korsakoff’s Disease. Acta Neuropathol. 61, 101–108 (1983). [DOI] [PubMed] [Google Scholar]
- 12.Zarow C, Lyness SA, Mortimer JA & Chui HC Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch. Neurol 60, 337–341 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Nakano I & Hirano A Parkinson’s disease: neuron loss in the nucleus basalis without concomitant Alzheimer’s disease. Ann. Neurol 15, 415–418 (1984). [DOI] [PubMed] [Google Scholar]
- 14.Halliday GM et al. Midbrain neuropathology in idiopathic Parkinson’s disease and diffuse Lewy body disease. J. Clin. Neurosci 3, 52–60 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Gai WP, Halliday GM, Blumbergs PC, Geffen LB & Blessing WW Substance P-containing neurons in the mesopontine tegmentum are severely affected in Parkinson’s disease. Brain 114 (Pt 5), 2253–2267 (1991). [DOI] [PubMed] [Google Scholar]
- 16.Yamamoto T & Hirano A Nucleus raphe dorsalis in parkinsonism-dementia complex of Guam. Acta Neuropathol. 67, 296–299 (1985). [DOI] [PubMed] [Google Scholar]
- 17.Paulus W & Jellinger K The neuropathologic basis of different clinical subgroups of Parkinson’s disease. J. Neuropathol. Exp. Neurol 50, 743–755 (1991). [DOI] [PubMed] [Google Scholar]
- 18.Chan-Palay V & Asan E Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer type and in Parkinson’s disease with and without dementia and depression. J. Comp. Neurol 287, 373–392 (1989). [DOI] [PubMed] [Google Scholar]
- 19.Huynh B, Fu Y, Kirik D, Shine JM & Halliday GM Comparison of Locus Coeruleus Pathology with Nigral and Forebrain Pathology in Parkinson’s Disease. Mov. Disord 36, 2085–2093 (2021). [DOI] [PubMed] [Google Scholar]
- 20.Bertrand E, Lechowicz W, Szpak GM & Dymecki J Qualitative and quantitative analysis of locus coeruleus neurons in Parkinson’s disease. Folia Neuropathol. 35, 80–86 (1997). [PubMed] [Google Scholar]
- 21.Zweig RM, Jankel WR, Hedreen JC, Mayeux R & Price DL The pedunculopontine nucleus in Parkinson’s disease. Ann. Neurol 26, 41–46 (1989). [DOI] [PubMed] [Google Scholar]
- 22.Jellinger K. The pedunculopontine nucleus in Parkinson’s disease, progressive supranuclear palsy and Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 51, 540–543 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rinne JO, Ma SY, Lee MS, Collan Y & Röyttä M Loss of cholinergic neurons in the pedunculopontine nucleus in Parkinson’s disease is related to disability of the patients. Parkinsonism Relat. Disord 14, 553–557 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Schmeichel AM et al. Mesopontine cholinergic neuron involvement in Lewy body dementia and multiple system atrophy. Neurology 70, 368–373 (2008). [DOI] [PubMed] [Google Scholar]
- 25.French IT & Muthusamy KA A Review of the Pedunculopontine Nucleus in Parkinson’s Disease. Front. Aging Neurosci 10, 99 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia-Rill E, Houser CR, Skinner RD, Smith W & Woodward DJ Locomotion-inducing sites in the vicinity of the pedunculopontine nucleus. Brain Res. Bull 18, 731–738 (1987). [DOI] [PubMed] [Google Scholar]
- 27.Skinner RD, Kinjo N, Henderson V & Garcia-Rill E Locomotor projections from the pedunculopontine nucleus to the spinal cord. Neuroreport 1, 183–186 (1990). [DOI] [PubMed] [Google Scholar]
- 28.Giguère N, Burke Nanni S & Trudeau L-E On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front. Neurol 9, 455 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hassler R, Mundinger F & Riechert T Stereotaxis in Parkinson Syndrome. (Springer Berlin Heidelberg; ). [Google Scholar]
- 30.Fu H, Hardy J & Duff KE Selective vulnerability in neurodegenerative diseases. Nat. Neurosci 21, 1350–1358 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderegg A, Poulin J-F & Awatramani R Molecular heterogeneity of midbrain dopaminergic neurons--Moving toward single cell resolution. FEBS Lett. 589, 3714–3726 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haber SN & Knutson B The reward circuit: linking primate anatomy and human imaging. Neuropsychopharmacology 35, 4–26 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Damier P, Hirsch EC, Agid Y & Graybiel AM The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 122 ( Pt 8), 1437–1448 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Damier P, Hirsch EC, Agid Y & Graybiel AM The substantia nigra of the human brain. I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D(28K) immunohistochemistry. Brain 122 ( Pt 8), 1421–1436 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Yamada T, McGeer PL, Baimbridge KG & McGeer EG Relative sparing in Parkinson’s disease of substantia nigra dopamine neurons containing calbindin-D28K. Brain Res. 526, 303–307 (1990). [DOI] [PubMed] [Google Scholar]
- 36.Pereira Luppi M. et al. Sox6 expression distinguishes dorsally and ventrally biased dopamine neurons in the substantia nigra with distinctive properties and embryonic origins. Cell Rep. 37, 109975 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider JS, Yuwiler A & Markham CH Selective loss of subpopulations of ventral mesencephalic dopaminergic neurons in the monkey following exposure to MPTP. Brain Res. 411, 144–150 (1987). [DOI] [PubMed] [Google Scholar]
- 38.Simunovic F. et al. Gene expression profiling of substantia nigra dopamine neurons: further insights into Parkinson’s disease pathology. Brain 132, 1795–1809 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grimm J, Mueller A, Hefti F & Rosenthal A Molecular basis for catecholaminergic neuron diversity. Proc. Natl. Acad. Sci. U. S. A 101, 13891–13896 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dong X. et al. Enhancers active in dopamine neurons are a primary link between genetic variation and neuropsychiatric disease. Nat. Neurosci 21, 1482–1492 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monzón-Sandoval J. et al. Human-Specific Transcriptome of Ventral and Dorsal Midbrain Dopamine Neurons. Ann. Neurol 87, 853–868 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cantuti-Castelvetri I. et al. Effects of gender on nigral gene expression and parkinson disease. Neurobiol. Dis 26, 606–614 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elstner M. et al. Expression analysis of dopaminergic neurons in Parkinson’s disease and aging links transcriptional dysregulation of energy metabolism to cell death. Acta Neuropathol. 122, 75–86 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Chung CY, Koprich JB, Hallett PJ & Isacson O Functional enhancement and protection of dopaminergic terminals by RAB3B overexpression. Proc. Natl. Acad. Sci. U. S. A 106, 22474–22479 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chung CY et al. Cell type-specific gene expression of midbrain dopaminergic neurons reveals molecules involved in their vulnerability and protection. Hum. Mol. Genet 14, 1709–1725 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Surmeier DJ et al. Calcium and Parkinson’s disease. Biochem. Biophys. Res. Commun 483, 1013–1019 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lautenschläger J. et al. C-terminal calcium binding of α-synuclein modulates synaptic vesicle interaction. Nat. Commun 9, 712 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rcom-H’cheo-Gauthier AN, Osborne SL, Meedeniya ACB & Pountney DL Calcium: Alpha-Synuclein Interactions in Alpha-Synucleinopathies. Front. Neurosci 10, 570 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dryanovski DI et al. Calcium entry and α-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J. Neurosci 33, 10154–10164 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wallace DC A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet 39, 359–407 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simuni T. et al. Tolerability of isradipine in early Parkinson’s disease: a pilot dose escalation study. Mov. Disord 25, 2863–2866 (2010). [DOI] [PubMed] [Google Scholar]
- 52.Parkinson Study Group. Phase II safety, tolerability, and dose selection study of isradipine as a potential disease-modifying intervention in early Parkinson’s disease (STEADY-PD). Mov. Disord 28, 1823–1831 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Parkinson Study Group STEADY-PD III Investigators. Isradipine Versus Placebo in Early Parkinson Disease: A Randomized Trial. Ann. Intern. Med 172, 591–598 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Venuto CS et al. Isradipine plasma pharmacokinetics and exposure-response in early Parkinson’s disease. Ann Clin Transl Neurol 8, 603–612 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ritz B. et al. L-type calcium channel blockers and Parkinson disease in Denmark. Ann. Neurol 67, 600–606 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pasternak B. et al. Use of calcium channel blockers and Parkinson’s disease. Am. J. Epidemiol 175, 627–635 (2012). [DOI] [PubMed] [Google Scholar]
- 57.Galter D, Buervenich S, Carmine A, Anvret M & Olson L ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol. Dis 14, 637–647 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Marchitti SA, Deitrich RA & Vasiliou V Neurotoxicity and metabolism of the catecholamine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol. Rev 59, 125–150 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Anderson DW, Schray RC, Duester G & Schneider JS Functional significance of aldehyde dehydrogenase ALDH1A1 to the nigrostriatal dopamine system. Brain Res. 1408, 81–87 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fitzmaurice AG et al. Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proc. Natl. Acad. Sci. U. S. A 110, 636–641 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim J-I et al. Aldehyde dehydrogenase 1a1 mediates a GABA synthesis pathway in midbrain dopaminergic neurons. Science 350, 102–106 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Regev A. et al. The Human Cell Atlas. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bakken T. et al. Evolution of cellular diversity in primary motor cortex of human, marmoset monkey, and mouse. BioRxiv (2020) doi: 10.1101/2020.03.31.016972. [DOI] [Google Scholar]
- 64.Hodge RD et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leng K. et al. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat. Neurosci 24, 276–287 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schirmer L. et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 573, 75–82 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lau S-F, Cao H, Fu AKY & Ip NY Single-nucleus transcriptome analysis reveals dysregulation of angiogenic endothelial cells and neuroprotective glia in Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A 117, 25800–25809 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Otero-Garcia M. et al. Molecular signatures underlying neurofibrillary tangle susceptibility in Alzheimer’s disease. Neuron 110, 2929–2948.e8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gerrits E. et al. Distinct amyloid-β and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol. 141, 681–696 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morabito S. et al. Single-nucleus chromatin accessibility and transcriptomic characterization of Alzheimer’s disease. Nat. Genet 53, 1143–1155 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bakken TE et al. Comparative cellular analysis of motor cortex in human, marmoset and mouse. Nature 598, 111–119 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lake BB et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol 36, 70–80 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eng C-HL et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 568, 235–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M & Cai L Single-cell in situ RNA profiling by sequential hybridization. Nature methods vol. 11 360–361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ståhl PL et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353, 78–82 (2016). [DOI] [PubMed] [Google Scholar]
- 76.Chen KH, Boettiger AN, Moffitt JR, Wang S & Zhuang X RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rodriques SG et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tian L, Chen F & Macosko EZ The expanding vistas of spatial transcriptomics. Nat. Biotechnol (2022) doi: 10.1038/s41587-022-01448-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shi H. et al. Spatial Atlas of the Mouse Central Nervous System at Molecular Resolution. bioRxiv 2022.06.20.496914 (2022) doi: 10.1101/2022.06.20.496914. [DOI] [Google Scholar]
- 80.Chen A. et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 185, 1777–1792.e21 (2022). [DOI] [PubMed] [Google Scholar]
- 81.Chen A. et al. Global Spatial Transcriptome of Macaque Brain at Single-Cell Resolution. bioRxiv 2022.03.23.485448 (2022) doi: 10.1101/2022.03.23.485448. [DOI] [Google Scholar]
- 82.Maynard KR et al. Transcriptome-scale spatial gene expression in the human dorsolateral prefrontal cortex. Nat. Neurosci 24, 425–436 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Allen WE, Blosser TR, Sullivan ZA, Dulac C & Zhuang X Molecular and spatial signatures of mouse brain aging at single-cell resolution. Cell 186, 194–208.e18 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen W-T et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell 182, 976–991.e19 (2020). [DOI] [PubMed] [Google Scholar]
- 85.Walker BL, Cang Z, Ren H, Bourgain-Chang E & Nie Q Deciphering tissue structure and function using spatial transcriptomics. Commun Biol 5, 220 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhao T. et al. Spatial genomics enables multi-modal study of clonal heterogeneity in tissues. Nature 601,85–91 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu Y. et al. High-Spatial-Resolution Multi-Omics Sequencing via Deterministic Barcoding in Tissue. Cell 183, 1665–1681.e18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.La Manno G. et al. Molecular Diversity of Midbrain Development in Mouse, Human, and Stem Cells. Cell 167, 566–580.e19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tiklová K. et al. Single-cell RNA sequencing reveals midbrain dopamine neuron diversity emerging during mouse brain development. Nat. Commun 10, 581 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hook PW et al. Single-Cell RNA-Seq of Mouse Dopaminergic Neurons Informs Candidate Gene Selection for Sporadic Parkinson Disease. Am. J. Hum. Genet 102, 427–446 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Agarwal D. et al. A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat. Commun 11, 4183 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Welch JD et al. Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell 177, 1873–1887.e17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Smajić S. et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain 145, 964–978 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kamath T. et al. Single-cell genomic profiling of human dopamine neurons identifies a population that selectively degenerates in Parkinson’s disease. Nat. Neurosci 25, 588–595 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Siletti K. et al. Transcriptomic diversity of cell types across the adult human brain. bioRxiv 2022.10.12.511898 (2022) doi: 10.1101/2022.10.12.511898. [DOI] [PubMed] [Google Scholar]
- 96.Bryois J et al. Genetic identification of cell types underlying brain complex traits yields insights into the etiology of Parkinson’s disease. Nat. Genet 52, 482–493 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Blauwendraat C, Nalls MA & Singleton AB The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, 170–178 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nalls MA et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schilder BM, Navarro E & Raj T Multi-omic insights into Parkinson’s Disease: From genetic associations to functional mechanisms. Neurobiol. Dis 163, 105580 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Langston RG et al. Association of a common genetic variant with Parkinson’s disease is mediated by microglia. Sci. Transl. Med 14, eabp8869 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Diaz-Ortiz ME et al. GPNMB confers risk for Parkinson’s disease through interaction with α-synuclein. Science 377, eabk0637 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Andersen MS et al. Heritability Enrichment Implicates Microglia in Parkinson’s Disease Pathogenesis. Ann. Neurol 89, 942–951 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Labandeira CM et al. Angiotensin type-1 receptor and ACE2 autoantibodies in Parkinson’s disease. NPJ Parkinsons Dis 8, 76 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gate D. et al. CD4+ T cells contribute to neurodegeneration in Lewy body dementia. Science 374, 868–874 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sulzer D. et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 546, 656–661 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Qi X. et al. Dopaminergic neuron-specific deletion of p53 gene is neuroprotective in an experimental Parkinson’s disease model. J. Neurochem 138, 746–757 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Goiran T. et al. Nuclear p53-mediated repression of autophagy involves PINK1 transcriptional down-regulation. Cell Death Differ. 25, 873–884 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kao C-Y et al. Elevated COUP-TFII expression in dopaminergic neurons accelerates the progression of Parkinson’s disease through mitochondrial dysfunction. PLoS Genet. 16, e1008868 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Labandeira-Garcia JL et al. Aging, Angiotensin system and dopaminergic degeneration in the substantia nigra. Aging Dis. 2, 257–274 (2011). [PMC free article] [PubMed] [Google Scholar]
- 110.Kim G-HJ et al. A zebrafish screen reveals Renin-angiotensin system inhibitors as neuroprotective via mitochondrial restoration in dopamine neurons. Elife 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lin H-C et al. Association of Angiotensin Receptor Blockers with Incident Parkinson Disease in Patients with Hypertension: A Retrospective Cohort Study. Am. J. Med 135, 1001–1007 (2022). [DOI] [PubMed] [Google Scholar]