

Abstract
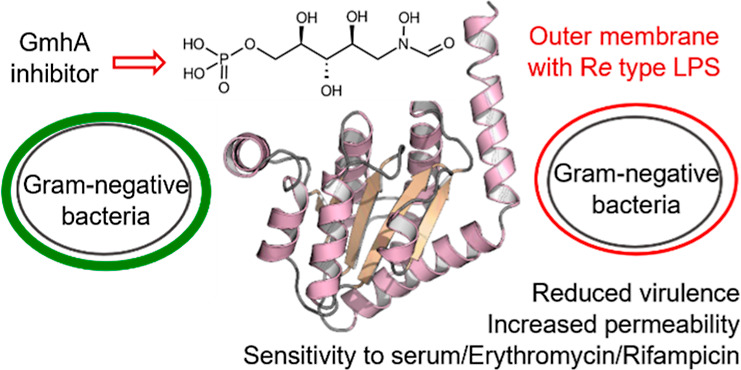
Inhibition of the biosynthesis of bacterial heptoses opens novel perspectives for antimicrobial therapies. The enzyme GmhA responsible for the first committed biosynthetic step catalyzes the conversion of sedoheptulose 7-phosphate into d-glycero-d-manno-heptose 7-phosphate and harbors a Zn2+ ion in the active site. A series of phosphoryl- and phosphonyl-substituted derivatives featuring a hydroxamate moiety were designed and prepared from suitably protected ribose or hexose derivatives. High-resolution crystal structures of GmhA complexed to two N-formyl hydroxamate inhibitors confirmed the binding interactions to a central Zn2+ ion coordination site. Some of these compounds were found to be nanomolar inhibitors of GmhA. While devoid of HepG2 cytotoxicity and antibacterial activity of their own, they demonstrated in vitro lipopolysaccharide heptosylation inhibition in Enterobacteriaceae as well as the potentiation of erythromycin and rifampicin in a wild-type Escherichia coli strain. These inhibitors pave the way for a novel treatment of Gram-negative infections.
Introduction
Infections caused by Gram-negative pathogens are presently of significant medical importance due to increasing bacterial resistance with life-threatening consequences for patients.1 Development of new antimicrobial drugs in a forthcoming post-antibiotic era is thus an urgent task to be accomplished by joint efforts of the pharmaceutical industry and academia. In particular, novel modes of action need to be elaborated to overcome the rapid adaptation of many bacteria to conventional antibiotics.
The bacterial lipopolysaccharide (LPS) is located in the outer leaflet of the outer membrane of Gram-negative bacteria and exerts a barrier function against many antibacterial agents. LPS is an amphipathic molecule consisting of the endotoxically active lipid A, a core oligosaccharide domain, and for some Gram-negative bacteria the surface exposed polysaccharide O-antigen.2 The inner core of LPS is made of 2 to 3 phosphorylated heptoses. These residues together with divalent cations are responsible for the stability of the outer leaflet and for its gatekeeper role against lipophilic compounds. As such, they constitute a key bottleneck for the penetration of antibacterials in the cytosol of Gram-negative bacteria. Bacterial knockout mutants being devoid of the heptose region of their LPS (termed deep-rough phenotype) show reduced virulence and biofilm formation, are sensitive to serum complement, and display a more permeable outer membrane allowing potentiation of antibiotics such as macrolides or rifamycins.3 Thus, inhibition of the biosynthetic pathways leading to the incorporation of heptoses in LPS and bacterial glycoproteins is regarded as a valuable option to develop antivirulence and potentiating therapies.4
The biosynthesis of these bacterial heptoses has been elucidated in the past decades and starts from the conversion of sedoheptulose 7-phosphate 1 into d-glycero-d-manno-heptose 7-phosphate 2 by the action of the ketose-aldose isomerase GmhA (Scheme 1).5
Scheme 1. Biosynthetic Pathways toward Nucleotide Activated Heptoses That Would Be Affected by Inhibiting the GmhA Reaction.
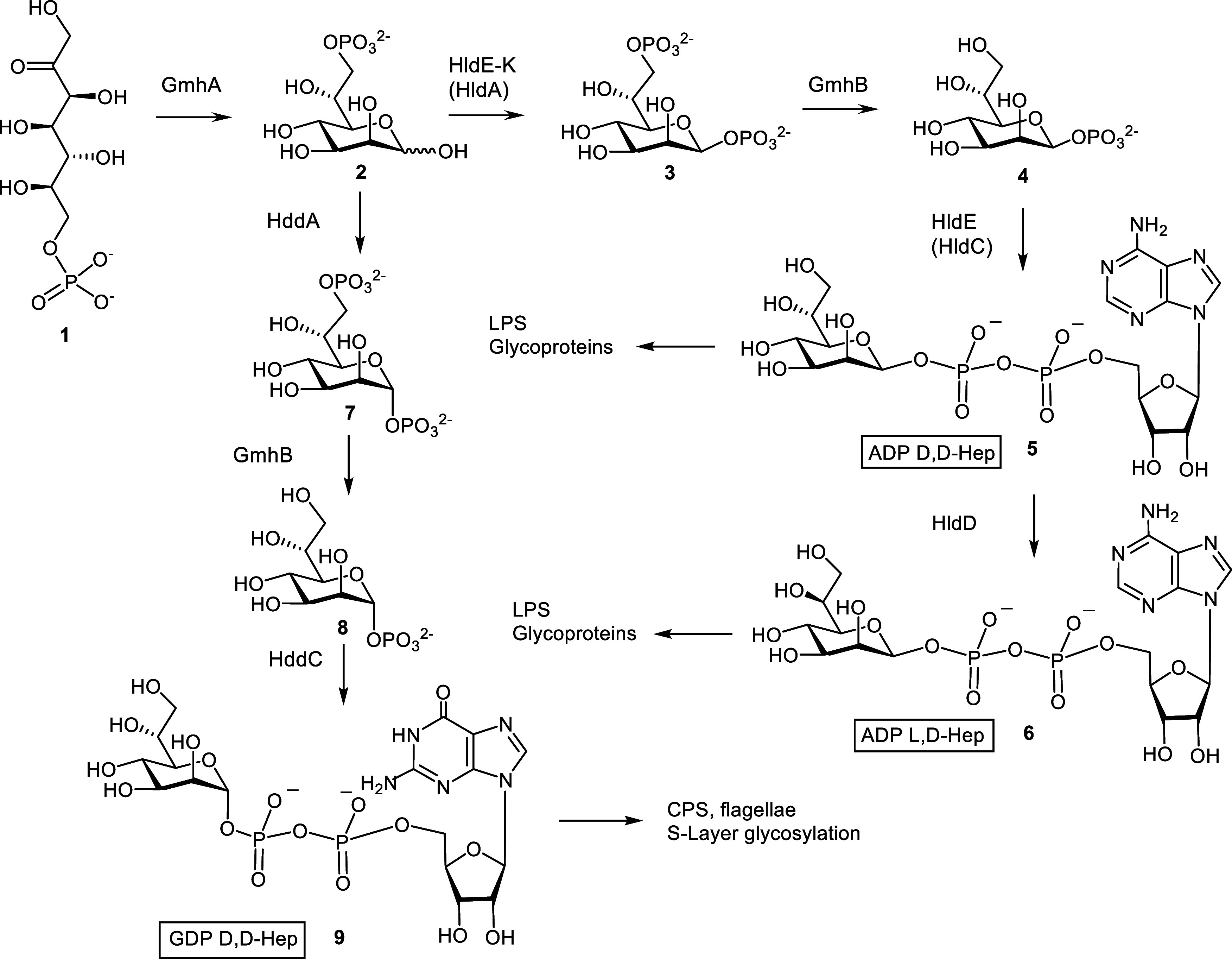
Next, the pathway diverges into two branches, eventually leading to the formation of ADP d-glycero- (5) and l-glycero-β-d-manno-heptose (6) involved in the assembly of bacterial LPS and alternatively produces GDP d-glycero-α-d-manno-heptose (9) needed for the biosynthesis of heptose constituents in capsular polysaccharides as well as for S-layer glycoproteins.6 As the first ketose to aldose isomerization step is common to the biosynthesis of all of these bacterial heptoses, inhibition of the sedoheptulose 7-phosphate isomerase reaction constitutes an attractive target to generate antimicrobial activities. This enzyme also called GmhA has been biochemically and structurally well characterized.7 Crystal structures of apo-forms have been reported for Escherichia coli,7Colwellia psychrerythraea, and Neisseria gonorrhoeae,8 as well as in liganded form with heptose 7-phosphate 2 for Burkholderia pseudomallei(9) and Pseudomonas aeruginosa.7 In addition, liganded structures containing sedoheptulose-7-phosphate 1 from E. coli, B. pseudomallei, and Mycobacterium tuberculosis have also been published.10 GmhA is well conserved among many Gram-negative bacteria but also some Gram-positive bacteria and shares little similarity with human enzymes, which reinforces its interest as a potential drug target.6a Previously, several small molecules have been generated as inhibitors of GmhA and the downstream kinases and transferases.11 The most potent GmhA inhibitor was identified as methyl 7-O-phosphoryl-d-glycero-d-gluco-heptopyranoside, a substrate analogue, with IC50 values in the low micromolar range.12 It was yet still too high to demonstrate heptosylation inhibition in a wild-type E. coli strain at 1 mM. In 2010, Harmer reported the presence of a zinc atom in B. pseudomallei GmhA as a key feature contributing to its catalytically active conformation.13 The presence of a metal at the heart of the active site was also postulated to be a general motif across a wide-spectrum of Gram-negative and Gram-positive bacterial GmhA as inferred from the excellent conservation of the zinc-binding side chains. The addition to substrate analogues of an hydroxamic acid moiety that is able to chelate zinc ions14 and therefore improve their potency would constitute a logical exploration strategy.
However, this strategy would make sense only if the designed GmhA inhibitors could address the well-known permeation and efflux challenge in Gram-negative bacteria and efficiently reach their target in the cytosol. Heptose analogues are small hydrophilic compounds that are expected to permeate easily into the periplasm through the outer membrane via the porins. Then, crossing the lipophilic inner membrane to reach the cytosol requires a priori an active transport mechanism. From the many transporter candidates for sugar import,15 the hexose-phosphate transporter UhpT would be considered the most plausible one as it can import a variety of phospho-sugars like pentose, hexose, and heptose phosphates but also phosphonate derivatives like fosfomycin and fosmidomycin. Importantly, the GmhA substrate sedoheptulose 7-phosphate 1 as well as the GmhA product d-glycero-d-manno-heptose 7-phosphate 2 are also transported by UhpT (Figure S3).
Based on our synthetic experience of heptose derivatives,16 we therefore decided to explore GmhA inhibition with phospho- or phosphono-sugars substituted with an hydroxamic acid moiety, postulated to reach their cytosolic target via porins and UhpT with the added advantage of excellent compound solubility. Herein, we describe the synthesis and structure–activity relationship of this novel series of GmhA inhibitors and two crystal structures of inhibitors liganded to GmhA from B. pseudomallei. We also report for the first time inhibition of LPS heptosylation on wild-type Enterobacteriaceae, leading to potentiation of macrolide and rifamycin antibiotics against Gram-negative pathogens.
Chemical Syntheses
For the synthesis of the N-hydroxyformate derivatives 17 and 20, the known 2,3,4-tri-O-benzyl ribose dithioacetal 10(17) was converted into the 7-O-triisopropylsilyl derivative 11 using triisopropylsilyl chloride in pyridine in the presence of N,N-dimethylaminopyridine. Next, the aldehyde was liberated by the action of N-bromosuccinimide in aqueous acetone to afford compound 12 (Scheme 2). Reductive amination of 12 was achieved first by treatment with benzyloxyamine in pyridine/MeOH, isolation of the crude oxime by column chromatography followed by reduction with sodium cyanoborohydride in acetic acid to give the benzyloxyamine derivative 13.18N-formylation was carried out by reaction with carbonyldiimidazole in formic acid/THF to produce compound 14. NMR-analysis of the tertiary amide was complicated by the occurrence of rotamers.19 In order to assign broadened 1H and 13C NMR signals, measurements had to be performed in deuterated toluene at 80 °C. In contrast, measurements performed at 40 °C resulted in two well separated spin systems. Removal of the silyl ether was smoothly accomplished in the presence of TBAF in THF to furnish primary alcohol 15. Subsequent phosphorylation of 15 was achieved by reaction with dibenzyl N,N-diisopropylphosphoramidite in the presence of 1H-tetrazole at room temperature, followed by oxidation with mCPBA at −78 °C.20 Phosphotriester 16 was isolated by HPLC purification, and catalytic hydrogenation of 16 in the presence of Pd-carbon using a solvent mixture of aqueous THF-HOAc followed by purification on a HILIC column afforded the target monophosphate 17. The modest yield was due to significant byproduct formation, in particular during longer reaction times, giving rise to deformylation as well as N–O reduction of the hydroxylamino group. Progress of the deprotection had thus to be checked by HILIC separation of aliquots using an analytical HILIC column and several cycles of HILIC runs had to be performed with concomitant purity assessment by NMR analysis prior to pooling of the individual fractions (see Figure S1, Supporting Information). Similar to the protected N-formyl derivatives 14–16, the deprotected derivative 17 was also present as a rotameric mixture, leading to two proton signals of the formyl unit at 8.35 and 7.90 ppm, respectively. To prove the dynamic exchange between the two rotamers, 1D gradient NOE-difference spectra according to Ley19 were measured. Irradiation of a solution of 17 at 7.90 ppm also led to a negative signal at the second formyl proton thus confirming that both protons are engaged in a chemical exchange equilibrium (Figure S2, Supporting Information).
Scheme 2. Synthesis of the N-formyl-N-hydroxy Inhibitors 17 and 20.
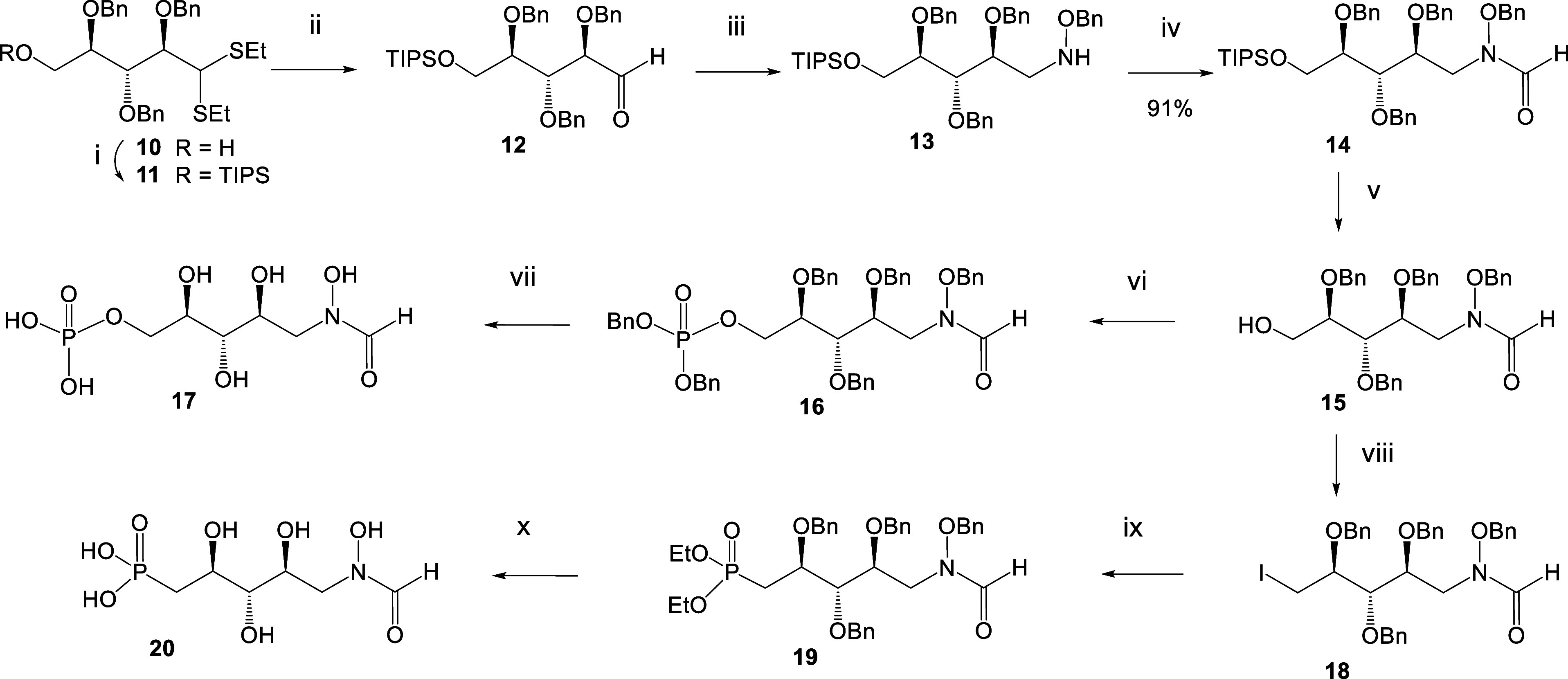
Reagents and Conditions: (i) TIPSCl, DMAP, pyr, 93%; (ii) NBS, aq. Me2CO, 97%; (iii) 1. H2NOBn, pyr., MeOH 2. NaCNBH3, AcOH, 75% over two steps; (iv) CDI, HCOOH, THF, 0 °C, 94%; (v) TBAF, THF, 87%; (vi) 1. 1H-tetrazole, phosphoramidite, rt. 2. mCPBA, TEA, −78 °C, 93%; (vii) Pd/C, H2, aq. THF-HOAc rt, 30%; (viii) I2, Imidazole, P(Ph)3, 66%; (ix) P(OEt)3, 44%; (x) 1. TMSBr, pyr. 2. Pd(OH)2/C, H2, aq. THF, 1% AcOH, 23%.
For the synthesis of the shortened C-phosphonate analogue 20, alcohol 15 was treated with triphenyl phosphine, imidazole, and iodine to afford the deoxy-iodo derivative 18. The structure of the iodo-derivative was confirmed by the high-field-shifted 13C NMR signal of C-5 at 9 ppm. Alternative approaches utilizing displacement of a 5-O-substituted triflate, mesylate, or tosylate group were less efficient due to concomitant formation of a cyclic C-glycosidic byproduct. Next, the Arbuzov reaction of 18 in triethylphosphite at 150 °C gave C-phosphonate 19. Global deprotection of 19 involved cleavage of the ethyl phosphonate using the McKenna reaction21 with trimethylsilyl bromide in pyridine followed by hydrogenation in the presence of Pd-hydroxide. Again, this step also generated byproducts due to the loss of the N-hydroxy as well as the formyl group and formation of reductive amination products. Isolation of the target compound 20 had thus to rely on HILIC-separation and careful NMR analysis of separated fractions prior to pooling and lyophilization. Structures of compounds 17 and 20 were eventually fully confirmed by NMR analysis, which revealed 31P NMR signals for 17 at 2.97/2.92 and 20.54 ppm for compound 20, respectively.
In order to study the impact of the distal fragment, additional analogues containing a hydrolytically stable C-phosphonate group with or without additional hydroxyl groups at the chain extension were synthesized (Scheme 3). Oxidation of the primary alcohol 15 under Dess-Martin conditions proceeded smoothly to give the aldehyde 21 which was directly used for the chain elongation steps. Wittig–Horner reaction of 21 with the lithium salt of tetrabenzylphosphonate 22(22) at −78 °C gave the E-configured alkene 23. Deprotection of 23 was performed with and without preserving the double bond. Hydrogenation of 23 in the presence of Pd(OH)2 again met with difficulties due to significant byproduct formation. Repeated HILIC chromatography enabled the isolation of a small amount of pure N-formyl derivative 24, which was fully characterized by NMR and HRMS analysis. Alternatively, benzyl groups were removed by the action of the Lewis acid SnCl4,23 followed by extensive HILIC purification of the reaction mixture. The presence of the double bond in 25 was confirmed by the characteristic 1H and 13C NMR data (see Supporting Information). Introduction of additional hydroxy groups was accomplished by the catalytic cis-dihydroxylation of 23 with NMO and potassium osmate in aqueous acetone.
Scheme 3. Synthesis of the N-formyl-N-hydroxy Inhibitors 24, 25, 27, and 31.
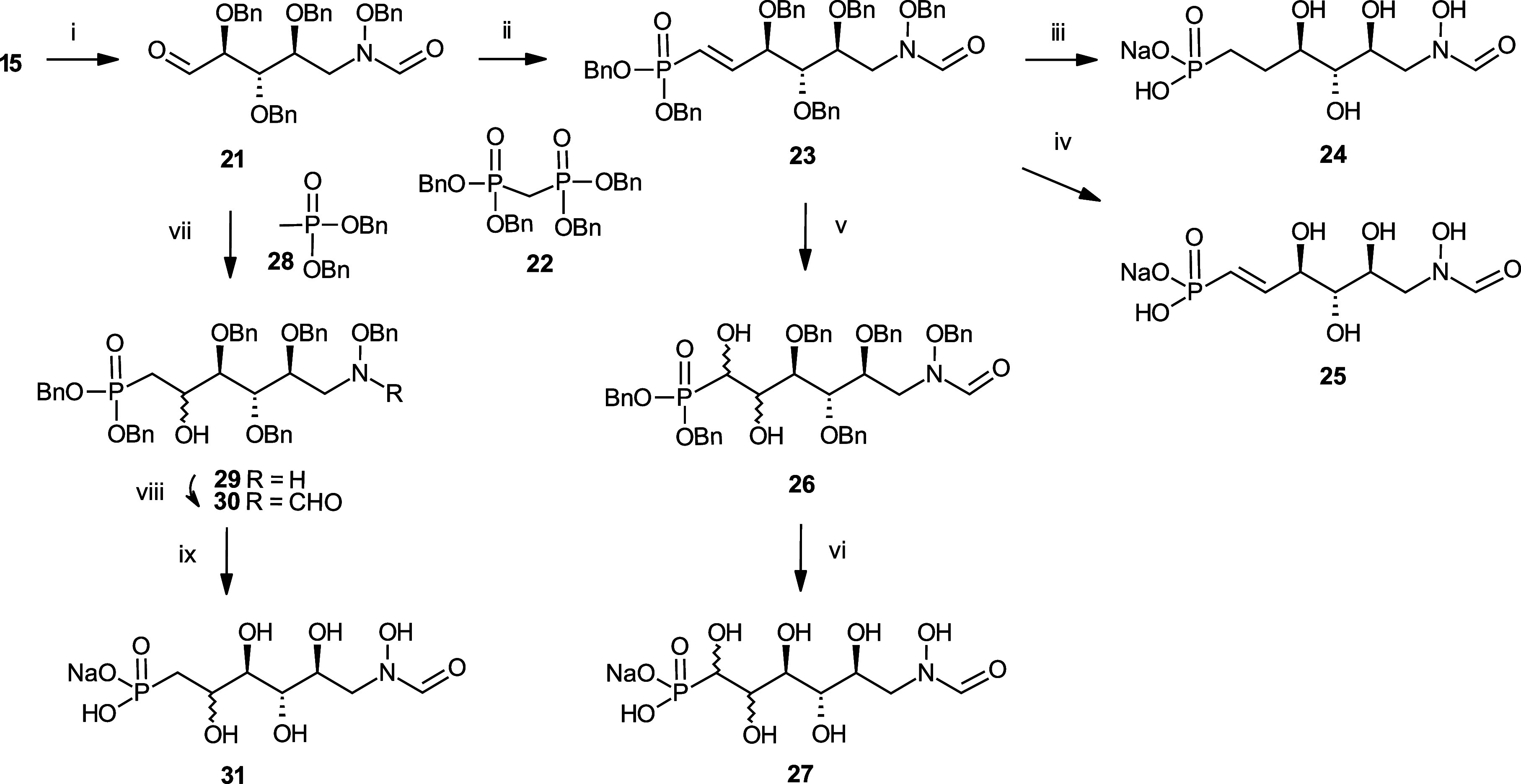
Reagents and Conditions: (i) Dess-Martin ox, 100%; (ii) 22, BuLi, THF, 76%; (iii) Pd(OH)2, aq. THF, MeOH, 16%; (iv) SnCl4, 54%; (v) NMO, K2OsO4, 79%; (vi) Pd(OH)2, aq. THF, 24%; (vii) 28, BuLi, THF, −78 °C, 22%; (viii) CDI, 73%; (ix) Pd(OH)2, aq. THF, 9%.
The diastereomeric mixture 26 was not separated and was directly subjected to deprotection as described above to furnish compound 27. Introduction of a single hydroxy group was carried out by reaction of the aldehyde 21 with the lithium salt of dibenzyl methanephosphonate 28 in THF, which resulted in a low yield of the N-formyl product 30. In addition, the deformylated derivative 29 was isolated in 22% yield and unreacted aldehyde 21 was recovered in 28% yield. In order to generate additional amounts of the formyl derivative 30, formylation of 29 was performed with formic acid and carbonyldiimidazole that gave 30. Deprotection of 30 by hydrogenolysis in the presence of Pd(OH)2 afforded 31 as a diastereomeric mixture.
Synthesis of distal hydroxyl analogue 41 started with conversion of 32 to triflate 33 with trifluoromethanesulfonic anhydride (Scheme 4). Nucleophilic substitution of 33 with [diethylphosphono(benzyloxy)methyl]lithium afforded 34, which was debenzylated under catalytic hydrogenation conditions. 35 was demethylated using Amberlyst 15, and the resulting ribose analogue 36 was converted into 37 using benzylhydroxylamine. Oxime 37 was then reduced using borane in THF, and the corresponding hydroxylamine 38 was carbonylated with trifluoroethyl formate to afford 39. Debenzylation under catalytic hydrogenation conditions followed by hydrolysis of the diethylphosphonate using TMSBr produced 41.
Scheme 4. Synthesis of the N-formyl-N-hydroxy Inhibitor 41.
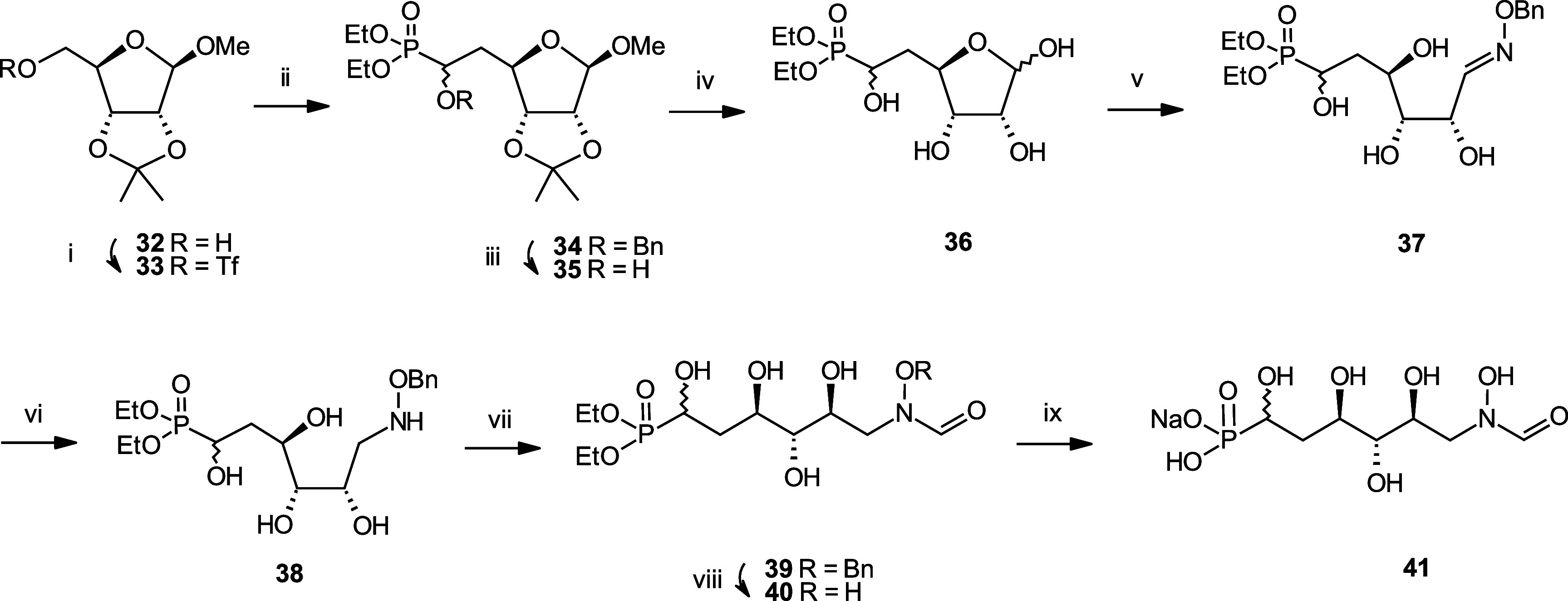
Reagents and Conditions: (i) Tf2O, DCM, −10 °C, 86%; (ii) Diethyl [(benzyloxy)methyl]Phosphonate, nBuLi, diisopropylamine, THF, −78 °C, 61%; (iii) H2, 10 bar, Pd/C, EtOH, 50 °C, 95%; (iv) Amberlyst 15, water, 60 °C; (v) Benzylhydroxylamine, NaHCO3, MeOH, water, 60 °C, 56% over two steps; (vi) 1. BH3.THF, THF 0 °C. 2. MeOH, 60 °C, 78%; (vii) Trifluoroethyl formate, THF, 60 °C, 65%; (viii) H2, 1 bar, Pd/C, MeOH, rt, 97%; (ix) 1. TMSBr, pyr, DCM, 5 °C. 2. NaHCO3, MeOH, water, 45%.
To study the impact of the absolute configuration of hydroxyl substituents, 54, the 2-epimeric form of 17 was made (Scheme 5). Synthesis started with the conversion of tetrahydroxypentanal 42 into dithiane 43 using ethanethiol. After protection of the primary alcohol using TIPSCl and benzylation of the remaining hydroxyl groups with benzyl bromide, 45 was converted into aldehyde 46 using N-bromosuccinimide. Reducing the level of amination with benzylhydroxylamine and sodium cyanoborohydride afforded 47. Carbonylation with carbonyldiimidazole followed by TBAF mediated silyl deprotection produced 49, which was oxidized into aldehyde 50 using Dess-Martin periodinane. Wittig reaction with tetramethyl methylenediphosphonate, phosphonic ester deprotection using TMSBr, and final debenzylation under catalytic hydrogenation conditions afforded 54.
Scheme 5. Synthesis of the N-formyl-N-hydroxy Inhibitor 54.
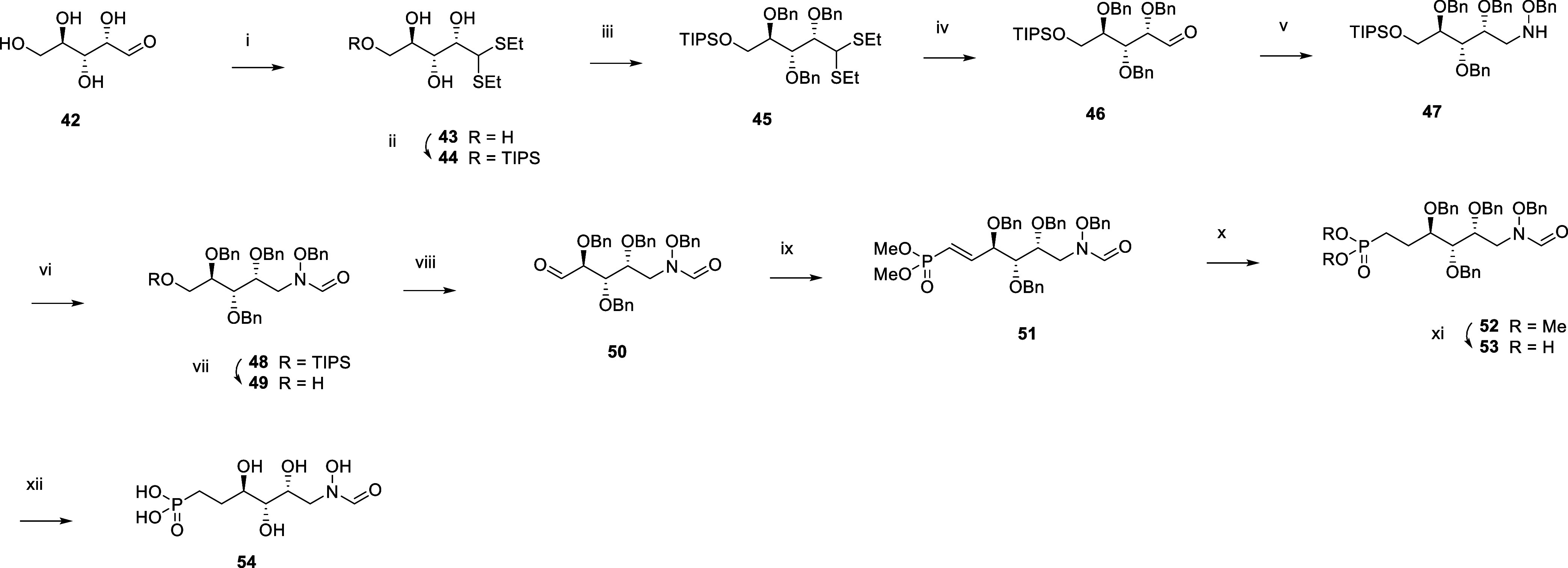
Reagents and Conditions: (i) EtSH, HCl, 0 °C, 83%; (ii) TIPSCl, DMAP, pyr, rt, 87%; (iii) Benzyl bromide, NaH, DMF, 0 °C to rt, 51%; (iv) NBS, acetone, water, 0 °C; (v) 1. H2NOBn.HCl, pyr, MeOH, 60 °C. 2. NaCNBH3, AcOH, rt, 60% over two steps; (vi) CDI, HCO2H, TEA, THF, 0 °C to rt, 77%; (vii) TBAF, THF, rt, 91%; (viii) DMP, DCM, 0 °C to rt, 48%; (ix) Tetramethyl methylenediphosphonate, NaH, Et2O, 0 °C, 55%; (x) H2, 1 bar, Pt/C, EtOAc, rt, 85%; (xi) TMSBr, pyr, DCM, 0 °C to rt, 78%; (xii) H2, 1 bar, Pd(OH)2, THF, AcOH, water, rt, 7%.
To study the impact of removing a hydroxyl group on the inhibitory activity, analogue 68 was made (Scheme 6). Deoxygenation of glucofuranose 56 was achieved with carbon disulfide, sodium hydride, and methyl iodide to afford dithiocarbonate 57, which was submitted to radical reduction with tributyltin hydride and AIBN. Selective acetonide deprotection using aqueous sulfuric acid in methanol was followed by oxidative cleavage with sodium periodate to afford 60. Wittig reaction with tetramethyl methylenediphosphonate, subsequent catalytic hydrogenation to reduce the carbon–carbon double bond, and acetonide deprotection using acetic acid yielded 63, which was further converted into 68 using the reaction suite analogous to the one described in Scheme 4.
Scheme 6. Synthesis of the N-formyl-N-hydroxy Inhibitor 68.
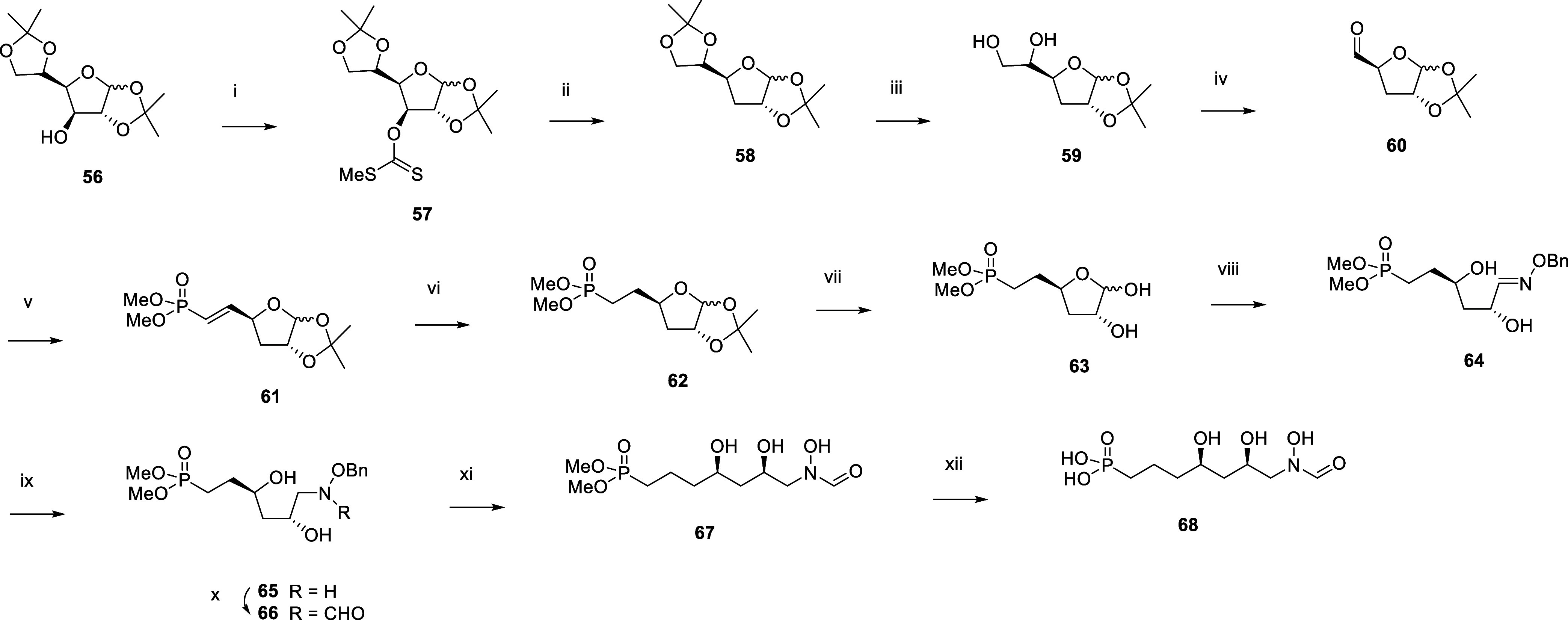
Reagents and Conditions: (i) NaH, CS2, MeI, imidazole, THF, 0 °C to rt; (ii) Bu3SnH, AIBN, toluene, reflux, 76% over two steps; (iii) aq. H2SO4, MeOH, 0 °C, 60%; (iv) NaIO4, MeOH, rt; (v) Tetramethyl methylenediphosphonate, NaH, Et2O, 0 °C to rt, 60% over two steps; (vi) H2, 1 bar, Pd/C, MeOH, rt; (vii) aq. AcOH, 80 °C, 40% over two steps; (viii) H2NOBn.HCl, pyr, MeOH, 60 °C, 67%; (ix) NaCNBH3, AcOH, 0 °C to rt, 88%; (x) Trifluoroethyl formate, THF, 65 °C, 51%; (xi) H2, 1 bar, Pd(OH)2, THF, AcOH, water, rt; (xii) TMSBr, pyr, DCM, 0 °C, 12% over two steps.
Then, we focused our efforts on the synthesis of distal fluorinated analogues 76 and 84 (Schemes 7 and 8). The synthesis of monofluorinated 76 started with the conversion of intermediate 35 into 71 via its triflation with trifluoromethanesulfonic anhydride, followed by nucleophilic substitution with cesium fluoride. The reaction suite analogous to the one already described in Scheme 4 involving oxime formation, reduction, carbonylation, and phosphonate ester hydrolysis was used to produce 76. Difluorinated analogue 84 was made similarly from intermediate 77, which was obtained via nucleophilic substitution of triflate 33 with diethyl(difluoromethyl)phosphonate.
Scheme 7. Synthesis of the N-formyl-N-hydroxy Inhibitor 76.
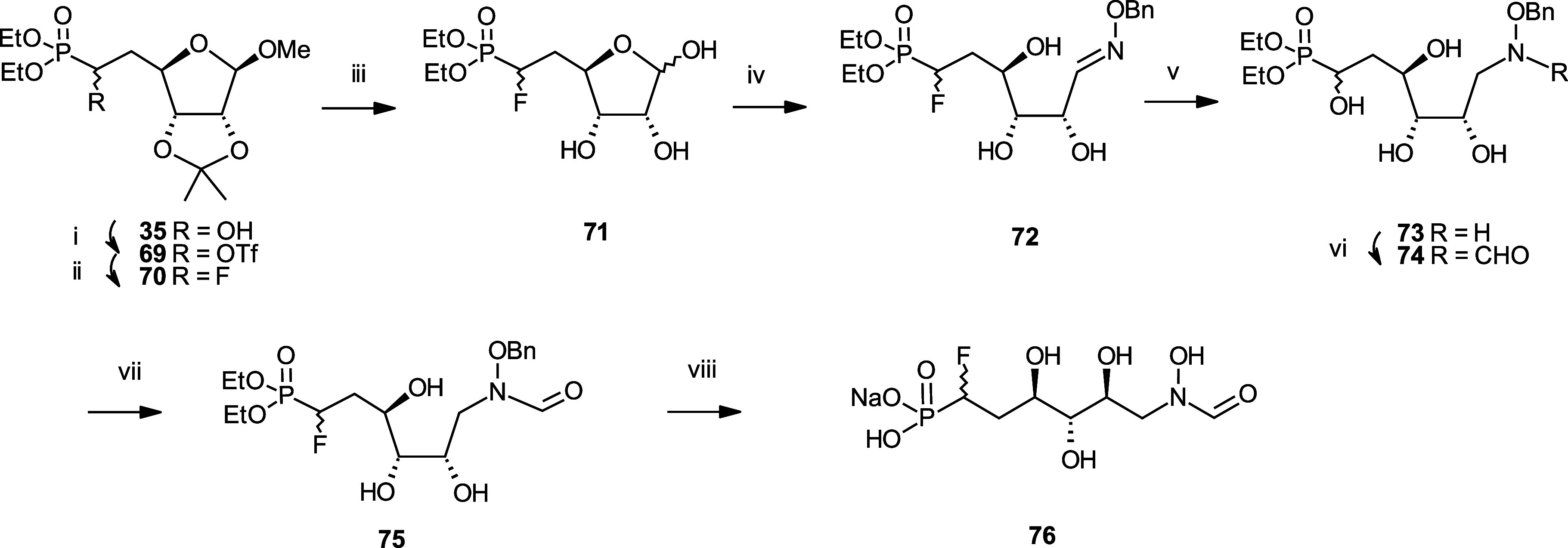
Reagents and Conditions: (i) Triflic anhydride, pyr, DCM, −10 °C; (ii) CsF, tBuOH, 40 °C, 58% over two steps; (iii) Amberlyst 15, water, 60 °C; (iv) H2NOBn.HCl, NaHCO3, EtOH, water, 60 °C, 81% over two steps; (v) 1. BH3.THF, THF, 0 °C. 2. MeOH, 60 °C, 71%; (vi) Trifluoroethyl formate, THF, 60 °C, 63%; (vii) H2, 1 bar, Pd/C, MeOH, rt, 90%; (viii) TMSBr, pyr, DCM, 0 °C, 40%.
Scheme 8. Synthesis of the N-formyl-N-hydroxy Inhibitor 84.
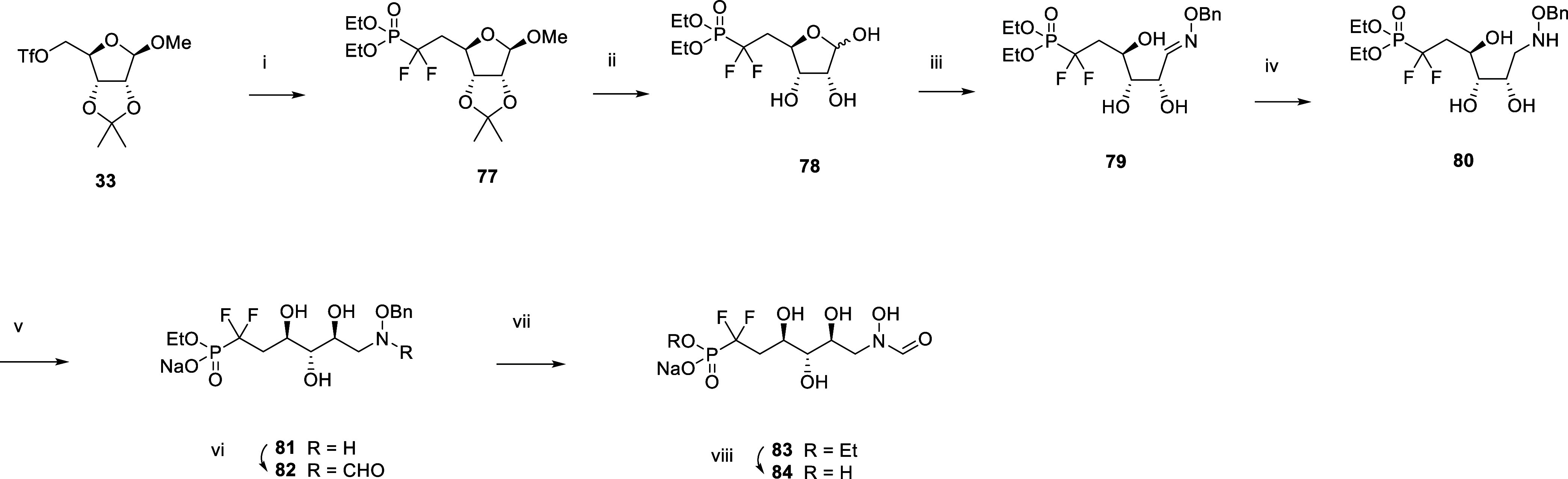
Reagents and conditions: (i) nBuLi, diisopropylamine, tris(N,N-tetramethylene)phosphoric acid triamine, diethyl(difluoromethyl)phosphonate, THF −78 °C, 18%; (ii) Amberlyst 15, water, 60 °C; (iii) H2NOBn.HCl, NaHCO3, water, EtOH, 60 °C, 73% over two steps; (iv) 1. BH3.THF, THF, 0 °C. 2. MeOH, 60 °C, 71%; (v) aq. NaOH, water, rt, 84%; (vi) Trifluoroethyl formate, THF, rt, 29%; (vii) H2, 1 bar, Pd/C, MeOH, rt; (viii) TMSBr, pyr, DCM, 0 °C, 66% over two steps.
The reverse form of hydroxamic acid 17 was also produced (analogue 96, Scheme 9). Synthesis started with the peracetylation of d-altrose 85 using acetic anhydride. Thioether protection of the anomeric position using thiophenol and boron trifluoride, followed by acetyl deprotection with triethylamine in water and methanol, afforded 88. The primary alcohol was silylated with TIPS chloride, and the remaining hydroxyl groups were benzylated with benzyl bromide. Deprotection of the primary alcohol with TBAF followed by phosphorylation with diphenyl phosphochloridate afforded 92. Conversion of thioether into hemiacetal 93 was achieved with N-bromosuccinimide. Oxidation with pyridinium chlorochromate afforded ester 94, which was debenzylated and dephenylated to 95 by catalytic hydrogenation. Final lactone transamidification with aqueous hydroxylamine afforded hydroxamate 96.
Scheme 9. Synthesis of the Hydroxamic Acid Inhibitor 96.
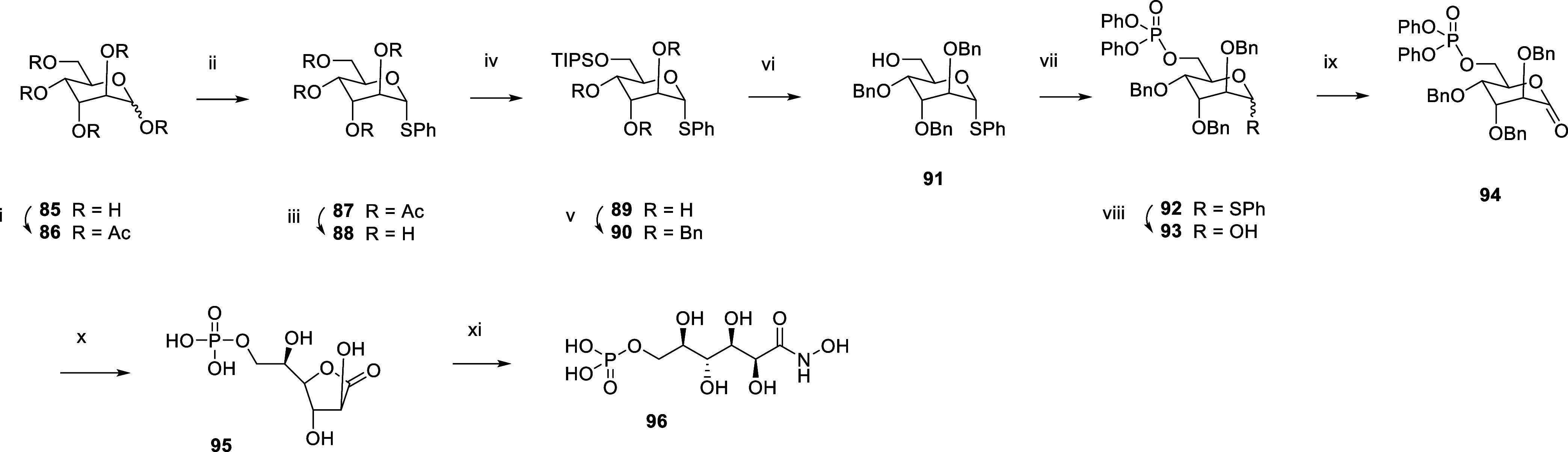
Reagents and Conditions: (i) Ac2O, pyr, DMAP, rt; (ii) PhSH, BF3. Et2O, DCM, 0 °C to rt, 82% over two steps; (iii) TEA, water, MeOH, rt, 91%; (iv) TIPSCl, DMAP, rt, 89%; (v) BnBr, NaH, DMF, rt, 96%; (vi) TBAF, THF, rt, 64%; (vii) Diphenyl phosphochloridate, TEA, DMAP, DCM, rt, 97%; (viii) NBS, acetone, water, −11 to 0 °C, 98%; (ix) PCC, DCM, rt, 65%; (x) 1. H2, 1 bar, Pd/C, THF, rt. 2. H2, 1 bar, PtO2, THF, rt, 90%; (xi) aq. NH2OH, rt, 88%.
Results And Discussion
On the basis of the critical presence of zinc in the catalytic site of GmhA13 and the hypothesis that this metal stabilizes the high energy enediolate intermediate as this is the case for other bacterial enzymes such as class II fructose bis-phosphate aldolases (Fbas)24 (Scheme 10), we have set out to exploit the strong zinc chelating ability of hydroxamic acids16 to design our inhibitors. Precedents such as phosphoglycolohydroxamic acid25 have been reported for the inhibition of Fbas, and N-formyl hydroxamate derivatives26 for the inhibition of peptide deformylase (PDF). Studies based on crystal structures, ab initio calculations, and IR-measurements of formylhydroxamic acid in gas phase indicated that the Z1-tautomeric form is the predominant one, thus being well suited for interaction with the catalytic Zn2+ ion.27
Scheme 10. Mechanism of Class II Fbas.

Evidence for the primary role of Zn2+ ion chelation in the active site by the N-formyl hydroxamate moiety was then obtained by crystallizing GmhA followed by soaking with inhibitors 17 and 84, respectively. The crystal structures of GmhA with compound 17 and compound 84 were both solved in the P42212 space group (Table S1, Supporting Information), revealing a single copy of GmhA, inhibitor, and zinc ion in the asymmetric unit (ASU) (Figure 1A,E,H). The unit cell contains 8 symmetry-related copies of the ASU, forming two identical GmhA tetramers. The GmhA tetramer contains 4 active sites that are each occupied by a zinc ion and an inhibitor (Figure 1B). Both inhibitors chelate the Zn2+ ion at two coordination sites via the hydroxamic acid moiety that is common to both molecules (Figure 1D,G). The Zn2+ metal center is further coordinated by other donor atoms from residues spanning two chains of GmhA, yielding a Zn complex with an octahedral geometry. An extensive hydrogen-bonding network involving GmhA residues and structured solvent molecules further stabilizes each inhibitor in the active site (Figure 1E,H). Compound 84 appears to have greater overall shape complementarity with the GmhA active site due to the additional “bulkiness” of the difluorophosphonate group compared to that of the phosphate group in 17. However, the phosphate group in 17 appears to accommodate additional H-bonds that are not possible with 84 (Figure 1C,E,F,H).
Figure 1.

Crystal structures of GmhA in complex with different orthosteric inhibitors. (A) ASU of a GmhA-inhibitor cocrystal structure. The ASU contains one copy of GmhA and one copy of inhibitor. (B) GmhA tetramer assembled from 4 symmetry related ASUs. Each of the 4 active sites contains residues from 3 separate chains of GmhA. (C–E) Co-crystal structure of GmhA with compound 17 deposited under PDB accession code 8V4J. (F–G) Co-crystal structure of GmhA with compound 84 deposited under PDB accession code 8V2T. (C,F) OMIT map verifying the presence of inhibitor in the crystal structure. Observed electron density is represented by a blue volume, while positive and negative features of the OMIT map are shown on green and red meshes, respectively. (D,G) Hydrogen-bonding and zinc-chelating interactions that stabilize the inhibitors in the GmhA active site. (E,H) 2D representation of the same interaction network. GmhA residues are colored according to the ASU, while the inhibitor molecule is labeled in magenta.
The GmhA inhibitory activity of our sedoheptulose-7-phosphate hydroxamic acid analogues was tested by using a luminescent assay (see the experimental section), which allowed the determination of IC50 values (Table 1). The ability of these analogues to inhibit LPS biosynthesis in wild-type E. coli was also visualized on silver-stained LPS SDS-PAGE electrophoretic images (Figure 2) using strain C7 (018:K1:H7), which is a newborn meningitis E. coli strain containing inner- and outer-core oligosaccharides as well as the O-antigen. Inhibition was monitored by formation of the heptose-deficient Re LPS, which only comprises lipid A and Kdo, and the EC50 data are reported in Table 1.
Table 1. E. coli GmhA and LPS Biosynthesis Inhibitory Activity of Sedoheptulose-7-Phosphate Hydroxamic Acid Analogues.

Inhibition of the enzymatic activity of E. coli GmhA by luminescence assay. Mean ± SD of at least 3 independent experiments.
Inhibition of E. coli LPS biosynthesis by SDS-PAGE electrophoresis. Mean ± SD of 3 independent experiments for 17 and 24, n = 1 for other compounds.
Figure 2.
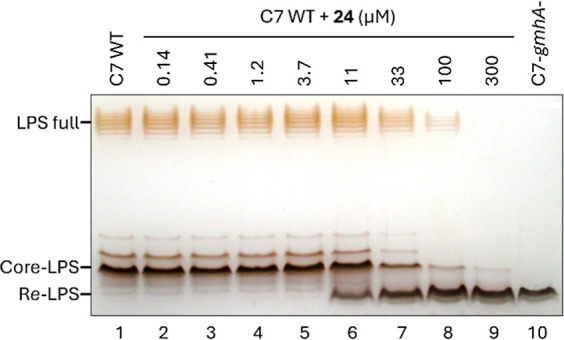
Silver-stained SDS-PAGE of the LPS of E. coli C7 grown with 0 to 300 μM compound 24 (columns 1 to 9) and control heptose-deficient Re-LPS of gmhA-deleted strain (column 10).
The hydroxamic acid analogue 96 of sedoheptulose-7-phosphate 1 proved to be a potent GmhA inhibitor with an IC50 in the low nanomolar range. This compound did not inhibit E. coli LPS biosynthesis up to 100 μM but started to affect bacterial growth above this concentration. The reverse N-formyl hydroxamate analogue 17 displayed an even better GmhA inhibitory activity at 2.4 ± 0.4 nM, with this time an observable effect on LPS biosynthesis at an EC50 of 20 ± 3 μM. This encouraging result prompted us to investigate its phosphonate analogue 24, which should be more stable to in vivo phosphatases than its phosphate counterpart. Gratifyingly, compound 24 also displayed low nanomolar GmhA inhibition at 3.0 ± 0.2 nM and LPS biosynthesis inhibition at 22 ± 4 μM. To further explore the structure–activity relationship around phosphonate analogue 24, we prepared and screened several closely related derivatives: the shorter analogue 20, the des-hydroxyl derivative 68, the diastereoisomer 54, the ene-analogue 25, and the hydroxylated derivatives 41, 31, and 27. Although inhibiting GmhA to various degrees, none of these analogues proved as potent as phosphonate 24. Of note, the des-hydroxyl derivative 68 displays a lower EC50/IC50 ratio than the other heptose analogues, which may reflect a better UhpT transport to the cytosol.
Since α-halogenated phosphonates are known to be potentially better bioisosteres of the phosphates than the nonhalogenated phosphonates,28 we turned our attention to the α-fluorinated analogues of phosphonate 24 in the hope of improving enzymatic inhibition and active transport. The parameters that may favor α-fluorinated phosphonates over their nonfluorinated congeners are a reduced pKa, an increased C–CHF–P or C–CF2–P dihedral angle, and the possibility of forming fluorine-hydrogen bonding. A pKa prediction analysis of phosphate 17 vs phosphonate 24 and its fluorinated analogues 76 and 84 indeed indicates that the monofluorinated phosphonate has closer pKa values to the phosphate than the nonhalogenated phosphonate (Figure 3). Although still displaying GmhA inhibition in the low nanomolar range, the monofluorinated diastereomeric 76 did not demonstrate any improvement over phosphonate 24, possibly due to stereoelectronic factors disfavoring fluorine vs hydrogen in this enzyme location, a trend confirmed by the even lower activity of its difluorinated congener 84.
Figure 3.

Predicted pKa of phosphate 17, phosphonate 24, monofluorophosphonate 76, and difluorophosphonate 84 according to ACD Laboratories V12.5.
Then, we investigated the cytotoxicity and the ability of such inhibitors to potentiate the antimicrobial activity of macrolide and rifamycin antibiotics. Phosphate 17 and phosphonate 24 were devoid of HepG2 cytotoxicity (CC50 > 1000 μM, Table 2), and, as expected by their mode of action, did not display any antibacterial activity (E. coli C7MIC > 1000 μM, Table 2). Yet at a concentration of 27 mg/L (100 μM), 17 displayed in the presence of the UhpT inducer glucose-6-phosphate (100 μM) a 32-fold potentiation of erythromycin (MIC from 32 to 1 mg/L) and a 16-fold potentiation of rifampicin (MIC from 4 to 0.25 mg/L) against E. coli C7. Against the same strain, the combination of 17 (300 μM) with rifampicin (1 mg/L) was rapidly bactericidal with more than 3 log reduction of the bacterial inoculum in less than 6 h and no detectable counts at 24 h, whereas the same compounds alone did not alter at all the bacterial growth (Figure 4). By inhibiting the first committed step in bacterial heptose biosynthesis in wild-type E. coli, this derivative is thus able to remove the hydrophilic outer core of Gram-negative bacteria and sensitize it back to a lipophilic macrolide and a rifamycin antibiotic.
Table 2. Minimum Inhibitory Concentration (MIC) on E. coli C7 and Cytotoxic Concentration CC50 on HepG2 Cells of Compounds 17 and 24 (n = 2).
| compound | MIC E. coli C7 (μM) | CC50 HepG2 (μM) |
|---|---|---|
| 17 | >1000 | >1000 |
| 24 | >1000 | >1000 |
Figure 4.

Combination of compound 17 with antibacterials. (A): E. coli C7 MIC of Erythromycin and Rifampicin alone or combined with 100 μM 17 (n = 1). (B): Time-kill kinetics of E. coli C7 with Rifampicin (RIF) alone or combined with compound 17. Bacterial enumeration replicates are represented in the plot. LOQ: limit of quantification.
Compound 17 was also tested against clinical isolates of E. coli,Escherichia cloacae, and K. pneumoniae for its ability to block LPS heptosylation (EC50). As detailed in Table 3, 17 in the presence of the UhpT inducer glucose-6-phosphate (100 μM) is a specific inhibitor of LPS heptosylation against various strains, including multiresistant clinical isolates expressing ESBL, AmpC, or NDM-1.
Table 3. EC50 of LPS Biosynthesis Inhibition of Clinical Isolates by Compound 17 in the Presence of 100 μM Glucose-6-Phosphate (n = 1).
| strain | resistance mechanism | EC50 of LPS heptosylation by 17 (μM) |
|---|---|---|
| E. coli S1 | ESBL | 21 |
| E. cloacae S2 | AmpC | 18 |
| K. pneumoniae S3 | NDM-1 | 21 |
Finally, E. coli C7 bacteria grown for 5 h in the presence of 0.3 and 1 mM compound 24 were as susceptible as the gmhA-deleted strain to 80% human serum, with a 3 log reduction of the bacterial load in 30 min (Figure 5). The treatment of E. coli with a GmhA inhibitor was therefore able to sensitize the strain to the antibacterial innate components present in the serum, such as the complement, thus validating the concept of bacterial antivirulence together with the permeabilizing effect to other antibacterial agents.
Figure 5.

Time-kill kinetics of E. coli C7 wild type or gmhA-deleted strain in the presence of 80% human serum untreated or heated (DC). Bacteria are pregrown in LB broth for 5 h in absence or presence of compound 24 at 0.3 and 1 mM. Bacterial enumeration replicates are represented on the plot. LOQ: limit of quantification.
Conclusions
We report here the synthesis and structure–activity relationship of various sedoheptulose 7-phosphate analogues containing a hydroxamic acid moiety designed to target the zinc-bearing catalytic site of GmhA as confirmed by two liganded high-resolution diffraction data. Some of these phosphoryl and phosphonyl derivatives demonstrate low nanomolar GmhA inhibition. They are also able to reach their cytosolic target and inhibit LPS biosynthesis in a wild-type E. coli strain at low micromolar concentrations. The most active hydroxamates are phosphates 17 and 96, phosphonate 24, and monofluorophosphonate 76, while distal hydroxylation or epimerization or chain shortening proved detrimental to inhibitory activity. In agreement with its mode of action, phosphoryl compound 17 does not display any antibacterial activity of its own but proves its ability to potentiate by 16 to 32-fold the activity of erythromycin and rifampicin on wild-type E. coli. These results pave the way toward novel possibilities of treating or preventing bloodstream infections caused by pathogenic Gram-negative bacteria without affecting their commensal flora, thus exerting potentially less selective pressure for antibiotic resistance than conventional antibacterial chemotherapy.
Experimental Section
Chemical Syntheses
The synthetic procedures for the preparation of compounds 41, 54, 68, 76, and 84 are fully described in the patent WO 2014/067904 A1.34 Experimental procedures and NMR spectra of compounds 10–27 and 85–96 have been compiled in the Supporting Information. If necessary, purity of compounds was checked by HPLC. All compounds are >95% purity.
Bacterial Strains Used in This Study
E. coli C7 (R. Debré hospital, Paris), E. coli S1, E. cloacae S2, and K. pneumoniae S3 (Necker hospital, Paris).
Purification of GmhA
E. coli BL21(DE3) (Novagen) cells were transformed with an expression plasmid, described in ref (13), containing full length GmhA from B. pseudomallei (strain K96423) with a TEV-cleaved, N-terminal hexa-histidine affinity tag. Cells were grown in Luria–Bertani (LB) broth supplemented with kanamycin (20 mg/L) to an optical density (OD600) of 0.5 prior to induction by the addition of 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). Following a 3 h induction at 37 °C, cells were harvested and resuspended in buffer (Buffer A) containing 20 mM Tris–HCl, pH 8.0, 750 mM KCl, 1 mM β-mercaptoethanol (BME), and 3 mM imidazole. Cells were lysed by three passes through a French pressure cell at 20,000 psi. Clarified lysate was loaded onto a HisTrap-HP nickel affinity column (GE healthcare, United States of America) equilibrated with Buffer A. The protein was eluted after step washes of Buffer A supplemented with 15, 30, and 60 mM imidazole. GmhA was eluted at a final concentration of 210 mM imidazole. Protein was then further purified by anion exchange chromatography. GmhA containing fractions were buffer exchanged into a buffer (Buffer B) containing 20 mM Tris–HCl, pH 8.0, 1 mM EDTA, 10 mM DTT, 100 mM KCl, and 10% v/v glycerol. This sample was then applied to a 10/100 Mono-Q hp column (GE healthcare, United States of America) and eluted with Buffer B via a linear salt gradient of 100–500 mM KCl. Fractions containing pure GmhA were pooled and buffer exchanged into a crystallization buffer containing 10 mM HEPES, pH 7.0, 500 mM KCl, and 10% w/v PEG 4000 for cryoprotection. Purified GmhA was then concentrated via centrifugation (Vivaspin) to a final concentration of 10 mg/mL and stored at −80 °C.
Crystallization of GmhA
A solution of purified GmhA [10 mg/mL GmhA, 500 mM KCl, 10% (w/v) PEG4000, 10 mM HEPES, pH 7.0] was mixed in a 1:1 ratio with a crystallization solution (100 mM sodium acetate, pH 4.6) and sealed over a reservoir containing 800 μL of 1.5 M (NH4)2SO4. Crystals formed within 48 to 72 h of equilibration at 4 °C. Drops containing crystals were then soaked with 0.2 μL of inhibitor solution (50 mM 17 or 84, 50 mM HEPES, pH 7.5) for 90 min. Following soaking, crystals were harvested, flash frozen, and stored in Liquid N2 until data collection.
X-ray Diffraction Data Collection
Frozen crystals were mounted in a 100 K nitrogen cryostream at beamlines X-25 and X-29 of the National Synchrotron Light source at Brookhaven National Laboratories. X-ray diffraction data were collected over a total of 180° of rotation about the omega axis. Crystals containing 17 were diffracted at a wavelength of 1.075 Å at beamline X-29 with a frame width of 0.3°, while 84 was diffracted at 1.100 Å at beamline X-25 with a width of 0.5°. X-ray diffraction data were deposited to the Zenodo data repository.29,30
Structure Determination and Refinement
X-ray diffraction data from liganded 17 and 84 crystals were integrated and scaled using autoPROC (Global Phasing Ltd.) to a resolution limit of 1.31 and 1.40 Å, respectively.31 Phenix Phaser32 was used to calculate an initial electron density map by molecular replacement (MR) of an existing GmhA crystal structure (PDB: 2XBL).13 The structures of both liganded 17 and 84 crystals were solved in the P42212 space group, revealing a single copy of GmhA, inhibitor, and zinc ion in the ASU. The unit cell contained 8 symmetry-related copies of the ASU, forming two identical GmhA tetramers. The atomic coordinates of GmhA and inhibitor were then built and refined against the electron density using Phenix AutoBuild and Phenix Refine.32 The refined coordinates of GmhA in complex with 17 and 84 were deposited to the PDB under accession codes 8V4J and 8V2T, respectively.29,30 Intermolecular interactions between GmhA, inhibitor, and Zn were analyzed using the protein–ligand interaction profiler33 and visualized in PyMOL.
Inhibition of the Enzymatic Activity of GmhA (Luminescent Assay)
The assay buffer “AB” contained 50 mM HEPES, pH 7.5, 1 mM MnCl2, 25 mM KCl, 0.012% Triton-X100, 1 mM dithiothreitol (DTT), and 0.1 μM myelin basic protein (MBP, Sigma, ref M1891). The following components were added in a white polystyrene Costar plate up to a final volume of 30 μL: 10 μL of inhibitor dissolved in DMSO/water 50/50 and 20 μL of GmhA of E. coli in AB. After 30 min of preincubation at room temperature, 30 μL of the substrate mixture in AB were added in each well to give a final volume of 60 μL. This reaction mixture was then composed of 2 nM GmhA, 3 μM sedoheptulose-7-phosphate (Sigma), 3 μM ATP (Sigma), and 50 nM HldE of E. coli in assay buffer. After 30 min of incubation at room temperature, 100 μL of the revelation mix were added to a final volume of 160 μL, including the following constituents at the respective final concentrations: 10,000 light units/mL luciferase (Sigma), 20 μM D-luciferin (Sigma), and 100 μM N-acetylcysteamine (Aldrich). Luminescence intensity was immediately measured on a Luminoskan instrument (Thermofisher) and converted into inhibition percentages. For IC50 determinations, the inhibitor was tested at 6 to 10 different concentrations, and the related inhibitions were fitted using XLFit 5 (IDBS) to a classical Langmuir equilibrium model with Hill slope, according to the equation: %inhibition = 100 cn/(cn + IC50n), where c is the compound concentration and n is the Hill coefficient.
Inhibition of Bacterial LPS Biosynthesis: Bacterial Culture
The compounds to be tested were prepared in deionized water/DMSO (50/50) solutions and added (25 μL) in sterile culture microtubes. The test bacterial isolate was isolated on tryptic soy agar (TSA) overnight. Isolated colonies were cultured in 10 mL of LB medium at 37 °C up to an OD of typically 0.15. These exponentially growing bacteria were finally diluted to 5 × 105 cfu/mL and added in each well (225 μL) for incubation with the compounds at 37 °C for approximately 5 h, up to an OD of ≈0.2–0.4. Some test compounds, e.g., phospho-sugars are actively transported into the bacterial cytosol via the phospho-sugar transporter UhpT (Figure S3). 2.5 μL portion of a 10 mM water solution of glucose-6-phosphate (G6P, 100 μM final concentration, from Sigma) was added in the culture tube in order to induce UhpT expression. LPS extraction: Bacterial cultures were normalized via OD determination, pelleted, and washed with 1 mL of phosphate-buffered-saline (PBS). The pellets were then denatured for 10 min at 95–100 °C in 50 μL of 0.2% sodium dodecyl sulfate (SDS), β-mercaptoethanol 1%, glycerol 36%, Tris 30 mM, pH 7.4, and bromophenol blue 0.001%. Samples were cooled down to room temperature, supplemented with 1.5 μL of proteinase K at 20 mg/mL, incubated for 1 h at 55 °C, and centrifuged for 30 min at 13,000 rpm at 25 °C. The resulting supernatant, containing LPS, was finally analyzed by SDS-PAGE electrophoresis. LPS SDS-PAGE electrophoresis: Polyacrylamide gels (16%/4% acrylamide for separation and concentration, respectively) were prepared, loaded with 8 μL of LPS extracts, and migrated. Silver staining: Gels were incubated overnight in 5% acetic acid/40% ethanol/deionized water, treated by 1% periodic acid/5% acetic acid for 15 min, washed 4 times for 10 min in deionized water, and finally incubated for 18 min in the dark in a silver nitrate solution composed of 56 mL of 0.1 M NaOH (56 mL), 33% aq ammonia (4 mL), 5% AgNO3 (45 mL), and deionized water (195 mL). Gels were then washed extensively in deionized water for 30 min and incubated for 10–15 min (up to LPS band appearance) in the revelation mix composed of 300 mL of deionized water (300 mL), 36.5% formaldehyde (0.3 mL, Fluka), and 2.3 M citric acid (0.1 mL). The revelation was stopped by incubating the gels in 10% acetic acid for 5 min. Gels were finally washed in deionized water, numerized with a Samsung PL51 camera, and analyzed by ImageJ software. The percentage of inhibition of LPS heptosylation was defined as the relative area of the Re-LPS band compared to the cumulated areas of Re-LPS and Core-LPS bands.
MIC Determination of Antibiotics Combined with a GmhA Inhibitor Against E. coli C7 (018:K1:H7)
The compound to be tested was diluted in 50 mM HEPES buffer (pH 7.4) from 10 mM stock solution in DMSO. Five microliters of compound dilution or buffer were distributed in a sterile clear round-bottom 96-well polystyrene microplate (Corning). Five microliters of serial 2-fold dilutions of reference antibiotics [Erythromycin (Fluka), Rifampicin (Fluka)] in DMSO were added. An exponentially growing preculture of E. coli C7 (O18:K1:H7) in LB was diluted to 104 cfu/mL and supplemented with 100 μM G6P, and 90 μL of this suspension were added to the microplates. After overnight incubation at 37 °C, the MIC of the antibiotics were determined for each test compound concentration as the lowest antibiotic concentration for which no bacterial pellet is visible without magnification.
Time-Kill Kinetics of Rifampicin in Combination with Compound 17
Rifampicin (Fluka) was diluted in DMSO, and the compound to be tested was diluted in water from 50 mM stock solution in 50 mM HEPES buffer, pH 7.4. Ten microliter of DMSO was distributed in sterile 1.5 mL polypropylene microtubes (Eppendorf). 2.5 μL of compound dilution or water, 2.5 μL of Rifampicin or DMSO, 2.5 μL of 10 mM G6P in water (100 μM final concentration), and 230 μL of LB were added to the microtubes. E. coli C7 (O18:K1:H7) was grown in 5 mL of LB broth at 37 °C until mid-log phase. The suspension was then diluted in LB to obtain a 100× inoculum of 5 × 107 cfu/mL, 2.5 μL of which were added to each microtube (5 × 105 cfu/mL final inoculum). The microtubes were incubated at 37 °C, and aliquots of 10 μL were sampled at 0, 2, 4, 6, 8, and 24 h for bacterial enumeration in duplicate.
Engineering of gmhA-Deleted E. coli C7
The mutant was obtained by homologous recombination using the lambda Red recombinase system described by Datsenko and Wanner35 A chloramphenicol resistance cassette (CamR) was PCR-amplified from a pKD3 plasmid by Pfu DNA polymerase using primers with extensions homologous to the genetic environment upstream and downstream of the gmhA gene (forward primer: 5′-TGTTTACAATATAATTACAACAAAGCT CACATTGTTGC TGGTGTAGGCTGGAGCTGCTTC; reverse primer: 5′-TCAATTAACTGGATCAGGATATGGATCACTTTA ATGTGGACATATGAATATCCTCCTTAG), according to the supplier’s instructions, and purified using QIAquick PCR Purification Kit (Qiagen). E. coli C7 containing the curable thermosensitive helper plasmid pKD46 was grown at 30 °C with shaking in 70 mL of LB supplemented with 100 mg/L ampicillin and 1 mM l-arabinose to an OD of 0.5, washed thrice in 70, 35, and 17 mL of ice-cold 10% glycerol, and then resuspended in 180 μL of ice-cold 10% glycerol. Fifty five microliters of cells were electroporated with 5 μL of purified PCR product. The cells were resuspended in SOC medium, incubated for 1 h at 35 °C under shaking, then plated on LB agar containing 10 mg/L chloramphenicol. After overnight growth at 37 °C, isolated colonies were picked and checked for the insertion of the CamR cassette in the gmhA locus by colony PCR (forward primer: 5′-GTGATGATAT GGTTGTAGTGG; reverse primer: 5′-CATCCCGAGCAATTCG CACA). Clones with a PCR product of the expected 1.5 kb size corresponding to gmhA::CamR (vs 1.0 kb for gmhA) were grown three times overnight at 37 °C on nonselective agar, then checked again for chloramphenicol resistance and ampicillin susceptibility for insert stability and loss of the pKD46 thermosensitive plasmid, respectively. Selected clones were suspended in LB 20% glycerol and stored at −80 °C.
Sensitization to Killing by the Complement of Human Serum
The E. coli C7 wild type and gmhA-deleted strains were isolated from TSA plates and grown overnight in 10 mL of LB broth at 35 °C. The suspension was then diluted in LB to obtain an inoculum of 5 × 105 cfu/mL in 2 mL, complemented with compound 24 at a final concentration of 0 or 1 mM in 5% DMSO and glucose-6-phosphate at 100 μM. After 5 h of incubation under shaking at 37 °C, the bacterial inoculum was adjusted to 105 cfu/mL in 20% LB broth mixed with 80% human serum (Biopredic) and filtered on 0.45 μm pore size membranes and used as such, or further heated for 30 min at 56 °C for complement deactivation. A volume of 250 μL of each condition was finally incubated at 37 °C in microtubes, and aliquots of 10 μL were sampled at 0, 15, 30, 60, 120, and 180 min for bacterial enumeration in duplicate.
HepG2 Cytotoxicity
HepG2 cells (LGC Promochem, ref HB-8065) in EMEM medium supplemented with fetal bovine serum (10%), Ampicillin (100 mg/L), and Streptomycin (100 mg/L) were plated on cell culture plates (Costar 3596, 2 × 104 cells/well) and incubated overnight at 37 °C in 5% CO2. The culture medium was then aspirated and replaced by supplemented EMEM (210 μL including 2% DMSO) containing dilutions of compounds. Plates were incubated overnight at 37 °C in 5% CO2. Cell viability was determined by the Celltiter Aqueous One Cell Proliferation Assay (G3581–80, Promega) according to the manufacturer’s recommendations, using a Multiskan absorption reader (490 nm).
Acknowledgments
The authors thank Dr. Andreas Hofinger for recording NMR spectra. They are also grateful to Prof. Pierre Vogel’s helpful discussions on carbohydrate syntheses and Prof. Xavier Nassif for stimulating biological discussions.
Glossary
Abbreviations
- ADP
Adenosine diphosphate
- ASU
Asymmetric unit
- CDI
Carbonyldiimidazole
- CPS
Capsular Polysaccharide
- DMP
Dess-Martin periodinane
- Fbas
Fructose bis-phosphate aldolase
- Gmh
Glycero-manno-heptose
- HEPES
2-[4-(2-Hydroxyethyl)-1-piperazinyl]ethanesulfonic acid
- HILIC
Hydrophilic interaction liquid chromatography
- MR
Molecular replacement
Peptide deformylase
- S-Layer
Surface-Layer
- TEA
Triethylamine
Data Availability Statement
Authors will release the atomic coordinates and experimental data upon article publication.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c00037.
General methods, synthetic procedures for compounds 17, 20, 24, 25, 27, 31, and 96, HILIC reaction control of the final deprotection and stacked 1H NMR plot of individual fractions of compound 17, 1H, 13C, and 31P NMR spectra of compounds 17, 20, 24, 25, 27, 31 and 96, 1D NOE-difference spectrum of compound 17 with selective saturation of one formyl proton, synthetic procedure reference for compounds 41, 54, 68, 76 and 84, X-ray data collection and refinement statistics, role of UhpT phospho-sugar transporter in the transport of d-glycero-d-manno-heptose 7-phosphate (H7P), IC50 fits of E. coli GmhA in biochemical inhibition assays, and EC50 fits of LPS biosynthesis inhibition assays (PDF)
Molecular formula strings (CSV)
Accession Codes
Author Present Address
○ AB-Science, 3 Avenue George V, 75,008 Paris, France
Author Present Address
⧫ DRT 30 Rue Gambetta 40,100 Dax, France.
Author Present Address
†† Novartis 20 Rue Rudolf Diesel, 01630 Saint-Genis-Pouilly, France.
Author Present Address
‡‡ Debiopharm, Rue du Levant 146, CP368, 1920 Martigny, Switzerland.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Funding for work completed by R.S., C.B., and M.J. came from the Canadian Institutes of Health Research (MOP-89903).
The authors declare the following competing financial interest(s): When this study was performed F.M., D.A., S.F., D.B., and V.G. were Mutabilis full-time employees; M.Bl., M.Ba., M.H., and N.M.X. were full-time employees of the University of Natural Resources and Life Sciences, Vienna, Austria (funded by MUTABILIS, project no. 8389); J.M., E.A., and V.H. were Activation full-time employees, and F.L. was a full-time employee of Carbosynth.
Due to a production error, the version of this paper that was published ASAP April 10, 2024, contained the wrong versions of Schemes 3, 4, and 7. The corrected version was reposted April 11, 2024.
Supplementary Material
References
- 1a Fischbach M. A.; Walsh C. T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]; 1b Chellat M. F.; Raguž L.; Riedl R. Targeting antibiotic resistance. Angew. Chem., Int. Ed. 2016, 55, 6600–6626. 10.1002/anie.201506818. [DOI] [PMC free article] [PubMed] [Google Scholar]; 1c Culyba M. J.; Mo C. Y.; Kohli R. M. Targets for combating the evolution of acquired antibiotic resistance. Biochemistry 2015, 54, 3573–3582. 10.1021/acs.biochem.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]; 1d Brown E. D.; Wright G. D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. 10.1038/nature17042. [DOI] [PubMed] [Google Scholar]; 1e Taylor P. L.; Wright G. D. Novel approaches to discovery of antibacterial agents. Anim. Health Res. Rev. 2008, 9, 237–246. 10.1017/S1466252308001527. [DOI] [PubMed] [Google Scholar]; 1f Beyer P.; Paulin S. The antibacterial research and development pipeline needs urgent solutions. ACS Infect. Dis. 2020, 6, 1289–1291. 10.1021/acsinfecdis.0c00044. [DOI] [Google Scholar]
- 2a Raetz C. R.; Whitfield C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]; 2b Holst O.Structure of the lipopolysaccharide core region. In Bacterial lipopolysaccharides; Knirel Y. A., Valvano M., Eds.; Springer: Wien, NY, 2011, pp 21–39. [Google Scholar]
- 3a Brooke J. S.; Valvano M. A. Molecular cloning of the Haemophilus influenzae gmhA (lpcA) gene encoding a phosphoheptose isomerase required for lipopolysaccharide biosynthesis. J. Bacteriol. 1996, 178, 3339–3341. 10.1128/jb.178.11.3339-3341.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]; 3b Loutet S. A.; Flannagan R. S.; Kooi C.; Sokol P. A.; Valvano M. A. A complete lipopolysaccharide inner core oligosaccharide is required for resistance of Burkholderia cenocepacia to antimicrobial peptides and bacterial survival in vivo. J. Bacteriol. 2006, 188, 2073–2080. 10.1128/jb.188.6.2073-2080.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; 3c Vaara M. Antibiotic-supersusceptible mutants of Escherichia coli and Salmonella typhimurium. Antimicrob. Agents Chemother. 1993, 37, 2255–2260. 10.1128/AAC.37.11.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4a Escaich S. Antivirulence as a new antibacterial approach for chemotherapy. Curr. Opin. Chem. Biol. 2008, 12, 400–408. 10.1016/j.cbpa.2008.06.022. [DOI] [PubMed] [Google Scholar]; 4b Kuo C. J.; Chen J. W.; Chiu H. C.; Teng C. H.; Hsu T. I.; Lu P. J.; Syu W. Jr.; Wang S. T.; Chou T. C.; Chen C. S. Mutation of the enterohemorrhagic Escherichia coli core LPS biosynthesis enzyme rfaD confers hypersusceptibility to host intestinal innate immunity in vivo. Frontiers Cell. Infect. Microbiol. 2016, 6, 82. 10.3389/fcimb.2016.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]; 4c Clatworthy A. E.; Pierson E.; Hung D. T. Targeting virulence: a new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. 10.1038/nchembio.2007.24. [DOI] [PubMed] [Google Scholar]
- 5a Eidels L.; Osborn M. J. Lipopolysaccharide and aldoheptose biosynthesis in transketolase mutants of Salmonella typhimurium. Proc. Natl. Acad. Sci. U.S.A. 1971, 68, 1673–1677. 10.1073/pnas.68.8.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]; 5b Chiu S.-F.; Teng K.-W.; Wang P.-C.; Chung H.-Y.; Wang C.-J.; Cheng H.-C.; Kao M.-C. Helicobacter pylori GmhB enzyme involved in ADP-heptose biosynthesis pathway is essential for lipopolysaccharide biosynthesis and bacterial virulence. Virulence 2021, 12, 1610–1628. 10.1080/21505594.2021.1938449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6a Valvano M.; Messner P.; Kosma P. Novel pathways for biosynthesis of nucleotide-activated glycero-manno-heptose precursors of bacterial glycoproteins and cell surface polysaccharides. Microbiology 2002, 148, 1979–1989. 10.1099/00221287-148-7-1979. [DOI] [PubMed] [Google Scholar]; 6b Kneidinger B.; Graninger M.; Puchberger M.; Kosma P.; Messner P. Biosynthesis of Nucleotide-activated-glycero-d-manno-Heptose. J. Biol. Chem. 2001, 276, 20935–20944. 10.1074/jbc.M100378200. [DOI] [PubMed] [Google Scholar]; 6c Huddleston J. P.; Anderson T. K.; Girardi N. M.; Thoden J. B.; Taylor Z.; Holden H. M.; Raushel F. M. Biosynthesis of d-glycero-L-gluco-heptose in the capsular polysaccharides of Campylobacter jejuni. Biochemistry 2021, 60, 1552–1563. 10.1021/acs.biochem.1c00183. [DOI] [PMC free article] [PubMed] [Google Scholar]; 6d Benz I.; Schmidt M. A. Glycosylation with heptose residues mediated by the aah gene product is essential for adherence of the AIDA-I adhesin. Mol. Microbiol. 2001, 40, 1403–1413. 10.1046/j.1365-2958.2001.02487.x. [DOI] [PubMed] [Google Scholar]
- Taylor P. L.; Blakely K. M.; de Leon G. P.; Walker J. R.; McArthur F.; Evdokimova E.; Zhang K.; Valvano M. A.; Wright G. D.; Junop M. S. Structure and function of sedoheptulose-7-phosphate isomerase, a critical enzyme for lipopolysaccharide biosynthesis and a target for antibiotic adjuvants. J. Biol. Chem. 2008, 283, 2835–2845. 10.1074/jbc.m706163200. [DOI] [PubMed] [Google Scholar]
- Wierzbicki I. H.; Zielke R. A.; Korotkov K. V.; Sikora A. E. Functional and structural studies on the Neisseria gonorrhoeae GmhA, the first enzyme in the glycero-manno-heptose biosynthetic pathways, demonstrate a critical role in lipooligosaccharide synthesis and gonococcal viability. Microbiology Open 2017, 6, e432 10.1002/mbo3.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivoli M.; Pang J.; Harmer N. J. A half-site multimeric enzyme achieves its cooperativity without conformational changes. Sci. Rep. 2017, 7, e16529 10.1038/s41598-017-16421-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10a Yu C. K.; Wang C. J.; Chew Y.; Wang P. C.; Yin H. S.; Kao M. C. Functional characterization of Helicobacter pylori 26695 sedoheptulose 7-phosphate isomerase encoded by hp0857 and its association with lipopolysaccharide biosynthesis and adhesion. Biochem. Biophys. Res. Commun. 2016, 477, 794–800. 10.1016/j.bbrc.2016.06.137. [DOI] [PubMed] [Google Scholar]; 10b Do H.; Yun J. S.; Lee C. W.; Choi Y. J.; Kim H. Y.; Kim Y. J.; Park H.; Chang J. H.; Lee J. H. Crystal structure and comparative sequence analysis of GmhA from Colwellia psychrerythaea strain 34H provides insight into functional similarity with DiaA. Mol. Cells 2015, 38, 1086–1095. 10.14348/molcells.2015.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]; 10c Karan S.; Pratap B.; Yadav S. P.; Ashish F. N. U.; Saxena A. K. Structural and functional characterization of M. tuberculosis sedoheptulose-7-phosphate isomerase, a critical enzyme involved in lipopolysaccharide biosynthetic pathway. Sci. Rep. 2020, 10, 20813. 10.1038/s41598-020-77230-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11a Desroy N.; Denis A.; Oliveira C.; Atamanyuk D.; Briet S.; Faivre F.; Le Fralliec G.; Bonvin Y.; Oxoby M.; Escaich S.; Floquet S.; Drocourt E.; Vongsouthi V.; Durant L.; Moreau F.; Verhey T. B.; Lee T.-W.; Junop M. S.; Gerusz V. Novel HldE-K inhibitors leading to attenuated Gram negative bacterial virulence. J. Med. Chem. 2013, 56, 1418–1430. 10.1021/jm301499r. [DOI] [PubMed] [Google Scholar]; 11b Lee T.; Verhey T. B.; Antiperovitch P. A.; Atamanyuk D.; Desroy N.; Oliveira C.; Denis A.; Gerusz V.; Drocourt E.; Loutet S. A.; Hamad M. A.; Stanetty C.; Andres S.; Sugiman-Marangos S.; Kosma P.; Valvano M.; Moreau F.; Junop M. S. Structural–functional studies of Burkholderia cenocepaciad-glycero-β-d-manno-heptose 7-phosphate kinase (HldA) and characterization of inhibitors with antibiotic adjuvant and antivirulence properties. J. Med. Chem. 2013, 56, 1405–1417. 10.1021/jm301483h. [DOI] [PMC free article] [PubMed] [Google Scholar]; 11c De Leon G. P.; Elowe N. H.; Koteva K. P.; Valvano M. A.; Wright G. D. An in vitro screen of bacterial lipopolysaccharide biosynthetic enzymes identifies an inhibitor of ADP-heptose biosynthesis. Chem. Biol. 2006, 13, 437–441. 10.1016/j.chembiol.2006.02.010. [DOI] [PubMed] [Google Scholar]; 11d Johnsson R. E. Synthesis and evaluation of mannitol-based inhibitors for lipopolysaccharide biosynthesis. Int. J. Med. Chem. 2016, 2016, e3475235 10.1155/2016/3475235. [DOI] [PMC free article] [PubMed] [Google Scholar]; 11e Kosma P.; Blaukopf M.; Atamanyuk D.; Xavier N.; Gerusz V. Synthesis of 1,5-Anhydro-d-glycero-d-gluco-heptitol derivatives as potential inhibitors of bacterial heptose biosynthetic pathways. Synthesis 2017, 49, 5320–5334. 10.1055/s-0036-1591518. [DOI] [Google Scholar]; 11f Kim S.; Jo S.; Kim M. S.; Shin D. H. A study of inhibitors of d-glycero-β-d-manno-heptose-1-phosphate adenylyltransferase from Burkholderia pseudomallei as a potential antibiotic target. J. Enzyme Inhib. Med. Chem. 2021, 36, 776–784. 10.1080/14756366.2021.1900166. [DOI] [PMC free article] [PubMed] [Google Scholar]; 11g Milicaj J.; Hassan B. A.; Cote J. M.; Ramirez-Mondragon C. A.; Jaunbocus N.; Rafalowski A.; Patel K. R.; Castro C. D.; Muthyala R.; Sham Y. Y.; Taylor E. A. Discovery of first-in-class nanomolar inhibitors of heptosyltransferase I reveals a new aminoglycoside target and potential alternative mechanism of action. Sci. Rep. 2022, 12, 7302. 10.1038/s41598-022-10776-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durka M.; Tikad A.; Périon R.; Bosco M.; Andaloussi M.; Floquet S.; Malacain E.; Moreau F.; Oxoby M.; Gerusz V.; Vincent S. P. Systematic synthesis of inhibitors of the two first enzymes of the bacterial heptose biosynthetic pathway: towards antivirulence molecules targeting lipopolysaccharide biosynthesis. Chem.—Eur. J. 2011, 17, 11305–11313. 10.1002/chem.201100396. [DOI] [PubMed] [Google Scholar]
- Harmer N. J. The structure of sedoheptulose-7-phosphate isomerase from Burkholderia pseudomallei reveals a zinc binding site at the heart of the active site. Mol. Biol. 2010, 400, 379–392. 10.1016/j.jmb.2010.04.058. [DOI] [PubMed] [Google Scholar]
- 14a Medina S. I.; Wu J.; Bode J. W. Nitrone protecting groups for enantiopure N-hydroxyamino acids: synthesis of N-terminal peptide hydroxylamines for chemoselective ligations. Org. Biomol. Chem. 2010, 8, 3405–3417. 10.1039/c004490c. [DOI] [PubMed] [Google Scholar]; 14b Robl J. A.; Simpkins L. M.; Asaad M. M. N -Formyl hydroxylamine containing dipeptides: generation of a new class of vasopeptidase inhibitors. Bioorg. Med. Chem. Lett. 2000, 10, 257–260. 10.1016/s0960-894x(99)00671-x. [DOI] [PubMed] [Google Scholar]; 14c Wu T. Y. H.; Hassig C.; Wu Y.; Ding S.; Schultz P. G. Design, synthesis, and activity of HDAC inhibitors with a N-formyl hydroxylamine head group. Bioorg. Med. Chem. Lett. 2004, 14, 449–453. 10.1016/j.bmcl.2003.10.055. [DOI] [PubMed] [Google Scholar]; 14d Yu S.-W.; Lee H.-Y.; Cho B.-H.; An K.-M.; Ryu J.-S.; Lee Y.-H.; Kang J.-H. Synthesis and biological evaluation of N-formyl hydroxylamine derivatives as potent peptide deformylase inhibitors. Bull. Korean Chem. Soc. 2006, 27, 1075–1078. 10.5012/bkcs.2006.27.7.1075. [DOI] [Google Scholar]; 14e Walz A. J.; Miller M. J. Synthesis and biological activity of hydroxamic acid-derived vasopeptidase inhibitor analogues. Org. Lett. 2002, 4, 2047–2050. 10.1021/ol025896m. [DOI] [PubMed] [Google Scholar]; 14f Wada C. K.; Holms J. H.; Curtin M. L.; Dai Y.; Florjancic A. S.; Garland R. B.; Guo Y.; Heyman H. R.; Stacey J. R.; Steinman D. H.; Albert D. H.; Bouska J. J.; Elmore I. N.; Goodfellow C. L.; Marcotte P. A.; Tapang P.; Morgan D. W.; Michaelides M. R.; Davidsen S. K. Phenoxyphenyl sulfone N-formylhydroxylamines (retrohydroxamates) as potent, selective, orally bioavailable matrix metalloproteinase inhibitors. J. Med. Chem. 2002, 45, 219–232. 10.1021/jm0103920. [DOI] [PubMed] [Google Scholar]; 14g Woo Y.-H.; Fernandes R. P. M.; Proteau P.-J. Evaluation of fosmidomycin analogs as inhibitors of the Synechocystis sp. PCC6803 1-deoxy-d-xylulose 5-phosphate reductoisomerase. Bioorg. Med. Chem. 2006, 14, 2375–2385. 10.1016/j.bmc.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 15a Marger M. D.; Saier M. H. A major superfamily of transmembrane facilitators that catalyse uniport, symport and antiport. Trends Biol. Sci. 1993, 18, 13–20. 10.1016/0968-0004(93)90081-W. [DOI] [PubMed] [Google Scholar]; 15b Kadner R. J.; Island M. D.; Dahl J. L.; Webber C. A. A transmembrane signalling complex controls transcription of the Uhp sugar phosphate transport system. Res. Microbiol. 1994, 145, 381–387. 10.1016/0923-2508(94)90085-X. [DOI] [PubMed] [Google Scholar]; 15c Lloyd A. D.; Kadner R. J. Topology of the Escherichia coli UhpT sugar-phosphate transporter analyzed by using TnphoA fusions. J. Bacteriol. 1990, 172, 1688–1693. 10.1128/jb.172.4.1688-1693.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]; 15d Fann M.-C.; Maloney P. C. Functional symmetry of UhpT, the sugar phosphate transporter of Escherichia coli. J. Biol. Chem. 1998, 273, 33735–33740. 10.1074/jbc.273.50.33735. [DOI] [PubMed] [Google Scholar]; 15e Eidels L.; Rick P. D.; Stimler N. P.; Osborn M. J. Transport of d-arabinose-5-phosphate and d-sedoheptulose-7-phosphate by the hexose phosphate transport system of Salmonella typhimurium. J. Bacteriol. 1974, 119, 138–143. 10.1128/jb.119.1.138-143.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16a Gerusz V.; Vincent S. P.; Oxoby M.; Atamanyuk D.; Moreau F.; Tikad A.; Andaloussi M.. New heptose derivatives and biological applications thereof. WO 2012073214, 2010.; 16b Atamanyuk D.; Gerusz V.. New monosaccharide derivatives and biological applications thereof. WO 2013178622, 2012.
- Schiesser C. H.; Zheng S.-L. Samarium(II) iodide mediated intramolecular homolytic substitution at selenium: Preparation of 5-seleno-d-pentopyranose sugars from common pentose starting materials. Tetrahedron Lett. 1999, 40, 5095–5098. 10.1016/S0040-4039(99)00990-9. [DOI] [Google Scholar]
- Jung S. H.; Ahn J. H.; Park S. K.; Choi J.-K. A practical and convenient procedure for the N-formylation of amines using formic acid. Bull. Korean Chem. Soc. 2002, 23, 149–150. 10.5012/bkcs.2002.23.1.149. [DOI] [Google Scholar]
- Hu D. X.; Grice P.; Ley S. V. Rotamers or diastereomers? An overlooked NMR solution. J. Org. Chem. 2012, 77, 5198–5202. 10.1021/jo300734r. [DOI] [PubMed] [Google Scholar]
- Bannwarth W.; Trzeciak A. A simple and effective chemical phosphorylation procedure for biomolecules. Helv. Chim. Acta 1987, 70, 175–186. 10.1002/hlca.19870700122. [DOI] [Google Scholar]
- 21a McKenna C. E.; Higa M. T.; Cheung N. H.; McKenna M.-C. The facile dealkylation of phosphonic acid dialkyl esters by bromotrimethylsilane. Tetrahedron Lett. 1977, 18, 155–158. 10.1016/S0040-4039(01)92575-4. [DOI] [Google Scholar]; 21b Rabinowitz R. The reactions of phosphonic acid esters with acid chlorides. A very mild hydrolytic route. J. Org. Chem. 1963, 28, 2975–2978. 10.1021/jo01046a008. [DOI] [Google Scholar]
- Liu W.; Hajibeigi A.; Lin M.; Rostollan C. L.; Kovacs Z.; Öz O. K.; Sun X. An osteoclast-targeting agent for imaging and therapy of bone metastasis. Bioorg. Med. Chem. Lett. 2008, 18, 4789–4793. 10.1016/j.bmcl.2008.07.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori H.; Nishida Y.; Ohrui H.; Meguro H. Regioselective de-O-benzylation with Lewis acids. J. Org. Chem. 1989, 54, 1346–1353. 10.1021/jo00267a022. [DOI] [Google Scholar]
- 24a Hill H. A.; Lobb R. R.; Sharp S. L.; Stokes A. M.; Harris J. I.; Jack R. S. Metal-replacement studies in Bacillus stearothermophilus aldolase and a comparison of the mechanisms of class I and class II aldolases. Biochem. J. 1976, 153, 551–560. 10.1042/bj1530551. [DOI] [PMC free article] [PubMed] [Google Scholar]; 24b Hall D. R.; Leonard G. A.; Reed C. D.; Watt C. I.; Berry A.; Hunter W. N. The crystal structure of Escherichiacoli class II fructose-1,6-bisphosphate aldolase in complex with phosphoglycolohydroxamate reveals details of mechanism and specificity. J. Mol. Biol. 1999, 287, 383–394. 10.1006/jmbi.1999.2609. [DOI] [PubMed] [Google Scholar]
- Collins K. D. An Activated Intermediate Analogue. J. Biol. Chem. 1974, 249, 136–142. 10.1016/S0021-9258(19)43101-3. [DOI] [PubMed] [Google Scholar]
- 26a Davies S. J.; Ayscough A. P.; Beckett R. P.; Clements J. M.; Doel S.; Pratt L. M.; Spavold Z. M.; Thomas S. W.; Whittaker M. Structure-activity relationships of the peptide deformylase inhibitor BB-3497: modification of the P2′ and P3′ side chains. Bioorg. Med. Chem. Lett. 2003, 13, 2715–2718. 10.1016/s0960-894x(03)00533-x. [DOI] [PubMed] [Google Scholar]; 26b Boularot A.; Giglione C.; Petit S.; Duroc Y.; Alves de Sousa R.; Larue V.; Cresteil T.; Dardel F.; Artaud I.; Meinnel T. Discovery and refinement of a new structural class of potent peptide deformylase inhibitors. J. Med. Chem. 2007, 50, 10–20. 10.1021/jm060910c. [DOI] [PubMed] [Google Scholar]
- 27a Sałdyka M.; Mielke Z. Cis-trans isomerism of the keto tautomer of formohydroxamic acid. Chem. Phys. Lett. 2003, 371, 713–718. 10.1016/S0009-2614(03)00333-6. [DOI] [Google Scholar]; 27b Kaur D.; Kohli R. Intra and intermolecular hydrogen bonding in formohydroxamic acid. Int. J. Quantum Phys. 2008, 108, 119–134. 10.1002/qua.21424. [DOI] [Google Scholar]; 27c Ventura O. N.; Rama J. B.; Turi L.; Dannenberg J. J. Gas-phase structure and acidity of formohydroxamic acid and formamide: a comparative ab initio study. J. Phys. Chem. 1995, 99, 131–136. 10.1021/j100001a023. [DOI] [Google Scholar]
- 28a Blackburn G. M.; Kent D. E.; Kolkmann F. The synthesis and metal binding characteristics of novel, isopolar phosphonate analogues of nucleotides. J. Chem. Soc., Perkin Trans. 1 1984, 1119–1125. 10.1039/p19840001119. [DOI] [Google Scholar]; 28b O’Hagan D.; Rzepa H. S. Some influences of fluorine in bioorganic chemistry. Chem. Commun. 1997, 645–652. 10.1039/a604140j. [DOI] [Google Scholar]; 28c Thatcher G. R. J.; Campbell A. S. Phosphonates as mimics of phosphate biomolecules: ab initio calculations on tetrahedral ground states and pentacoordinate intermediates for phosphoryl transfer. J. Org. Chem. 1993, 58, 2272–2281. 10.1021/jo00060a050. [DOI] [Google Scholar]; 28d Howard J. A. K.; Hoy V. J.; O’Hagan D.; Smith G. T. How good is fluorine as a hydrogen bond acceptor?. Tetrahedron 1996, 52, 12613–12622. 10.1016/0040-4020(96)00749-1. [DOI] [Google Scholar]
- Brown C.; Junop M.; Szabla R.. X-Ray Diffraction Images from a Crystal of GmhA with Inhibitor Mut148233 - PDB 8V4J; Zenodo, 2023. 10.5281/zenodo.10222806. [DOI] [Google Scholar]
- Brown C.; Junop M.; Szabla R.. X-Ray Diffraction Images from a Crystal of GmhA with Inhibitor Mut148591 - PDB 8V2T; Zenodo, 2023. 10.5281/zenodo.10201898. [DOI] [Google Scholar]
- Vonrhein C.; Flensburg C.; Keller P.; Sharff A.; Smart O.; Paciorek W.; Womack T.; Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. 2011, 67, 293–302. 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D.; Afonine P. V.; Baker M. L.; Bunkóczi G.; Chen V. B.; Croll T. I.; Hintze B.; Hung L.-W.; Jain S.; McCoy A. J.; Moriarty N. W.; Oeffner R. D.; Poon B. K. M.; Prisant G.; Read R. J.; Richardson J. S.; Richardson D. C.; Sammito M. D.; Sobolev O. V.; Stockwell D. H.; Terwilliger T. C.; Urzhumtsev A. G.; Videau L. L.; Williams C. J.; Adams P. D. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. 2019, 75, 861–877. 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salentin S.; Schreiber S.; Haupt V. J.; Adasme M. F.; Schroeder M. PLIP: fully automated protein-ligand interaction profiler. Nucleic Acid Res. 2015, 43, W443–W447. 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atamyanuk D.; Gerusz V.; Moreau F.; Henryon V.; Monbrun J.; Airiau E.. New antibacterial compounds and applications thereof. WO 2014/067904 A1, 2014.
- Datsenko K. A.; Wanner B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 6640–6645. 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Authors will release the atomic coordinates and experimental data upon article publication.