

Abstract
Mitochondria have emerged as key participants in and regulators of myocardial injury during ischemia and reperfusion. This review examines the sites of damage to cardiac mitochondria during ischemia and focuses on the impact of these defects. The concept that mitochondrial damage during ischemia leads to cardiac injury during reperfusion is addressed. The mechanisms that translate ischemic mitochondrial injury into cellular damage, during both ischemia and early reperfusion, are examined. Next, we discuss strategies that modulate and counteract these mechanisms of mitochondrial-driven injury. The new concept that mitochondria are not merely stochastic sites of oxidative and calcium-mediated injury but that they activate cellular responses of mitochondrial remodeling and cellular reactions that modulate the balance between cell death and recovery is reviewed, and the therapeutic implications of this concept are discussed.
Keywords: oxidative phosphorylation, electron transport chain, reactive oxygen species, cardiolipin, fatty acid oxidation, ubiquinol:cytochrome c oxidoreductase
INTRODUCTION
Mitochondria are critical targets and the root of tissue injury, particularly cardiac injury (the focus of this review), during ischemia and subsequent reperfusion, sometimes termed I/R (1, 2). Occlusion of a coronary artery results in severe, abrupt limitation of blood flow to the myocardium. Sudden occlusion occurring in clinical settings is most often due to thrombosis of a coronary artery, underlain by an atherosclerotic plaque. A severe reduction in blood flow, usually to 10% or less of normal resting perfusion, sets in motion deleterious cellular processes, especially damage to mitochondria, which result in eventual cardiomyocyte death. The imbalance of energy supply and demand is greatest in the endocardium, leading to the onset of cell death in this innermost region of the ventricular wall, starting approximately 20 min after flow cessation. Cell death then proceeds in a transmural manner across the ventricular wall from the endocardium (next to the ventricular cavity) to the epicardium (outermost layer) over approximately 6 h (3, 4). At any given time following coronary occlusion, there are regions of dead myocardium, reversibly injured myocardium, and hypoperfused but viable myocardium. The aim of restoration of blood flow by the clinical use of emergency cardiac catheterization and coronary stent deployment or by administration of thrombolytic therapy is to restore arterial patency and flow—reperfusion—to limit the progression of ischemic necrosis. The goal is to apply this critical therapy within 90 min of clinical presentation, ideally within 6 h from the onset of artery thrombosis. Timely reperfusion results in improved left ventricular postinfarction contractile function and improved patient survival. Although resumption of flow limits damage from ischemia, salvage of jeopardized cardiac myocytes that have undergone cell injury during the ischemic period, but not yet sustained ischemic cell death, is frustratingly suboptimal (3). In fact, additional myocyte death occurs during the reperfusion period (2), not from ischemic necrosis, but from altered metabolism of restored oxygen and substrates by ischemia-damaged mitochondria. In experimental models, 25–40% of eventual cardiac cell death is related to reperfusion-induced death. Damage to cardiac mitochondria that leads to deranged mitochondrial metabolism during the early reperfusion period is a key contributing mechanism to this second occurrence in the progression of myocardial cell death. Nonetheless, despite timely and successful reperfusion, many patients with acute coronary thrombosis leading to acute myocardial infarction sustain substantial damage to cardiac tissue.
In the setting of severe myocardial ischemia, damage to the electron transport chain (ETC) occurs mainly during the ischemic period (1). Reperfusion of myocardium that contains mitochondria damaged by the preceding period of severe ischemia leads to early mitochondrial-driven injury with excessive production of reactive oxygen species (ROS) and dysregulation of calcium, leading to abnormal permeation, along with swelling and disruption of mitochondria by mitochondrial permeability transition pore (MPTP) (5, 6). The mechanisms of early reperfusion oxidative and calcium-mediated injury have been discussed extensively by others and are summarized only briefly in this review (2, 6). These mechanisms provided an initial concept of the early phase of reperfusion injury (2). Mitochondria participate in the activation of programmed cell death (7), either from MPTP or via selective signaling-pathway permeation of the outer mitochondrial membrane [so-called mitochondrial outer membrane permeabilization (MOMP)] (8). Either occurrence can lead to the release of mitochondrial proteins that activate cell death mechanisms, including apoptosis, necrosis, and programmed necrosis (necroptosis) (9). Although most prominent during reperfusion, evidence of activation of programmed cell death is apparent during ischemia (10). This review presents data showing that the ETC is an upstream activator of these processes and examines the sites of damage to cardiac mitochondria during ischemia, with special emphasis on the ETC. The ETC first drives damage to itself, leading to additional damage to other biochemical targets and compartments of mitochondria. In this review, we examine the mechanisms that translate ischemic mitochondrial injury into cellular damage during ischemia and early reperfusion. Mitochondria are not merely stochastic nodes of oxidative and calcium-mediated injury to the myocyte; dysfunctional mitochondria activate mitochondrial remodeling and cellular reactions that modulate the balance between cell death and recovery. These ideas have therapeutic implications, as discussed below.
NORMAL MITOCHONDRIAL PHYSIOLOGY
Mitochondria are the sites of oxidative metabolism in cardiomyocytes. Cardiac mitochondria exist in two functionally distinct populations: subsarcolemmal mitochondria (SSM) located immediately underneath the plasma membrane and interfibrillar mitochondria (IFM) situated among the myofibrils (11, 12) (Figure 1a). In addition to energy production, mitochondria contribute to the regulation of cell function via synthetic pathways and are involved in cell stress pathways. Mitochondrial structure provides compartmentation of these tasks (11) (Figure 1b). The outer mitochondrial membrane surrounds the organelle and has among its constituents a voltage-dependent anion channel (VDAC) that provides a route for metabolic substrates (e.g., pyruvate, glutamate, and malate) and nucleotides (e.g., ADP and ATP) to gain entrance to the intermembrane space. The inner membrane is a selectively impermeable barrier with specific transporters and translocases that facilitate the passage of the aforementioned substrates into the matrix, where they are metabolized by enzymes that include those in the tricarboxylic acid (TCA) cycle and fatty acid oxidation, as well as synthetic and antioxidant enzymes. The TCA cycle and fatty acid oxidation generate reduced nicotinamide adenine dinucleotide (NADH) and reduced flavin adenine dinucleotide (FADH2) for oxidation by the ETC (Figures 1c and 2a).
Figure 1.
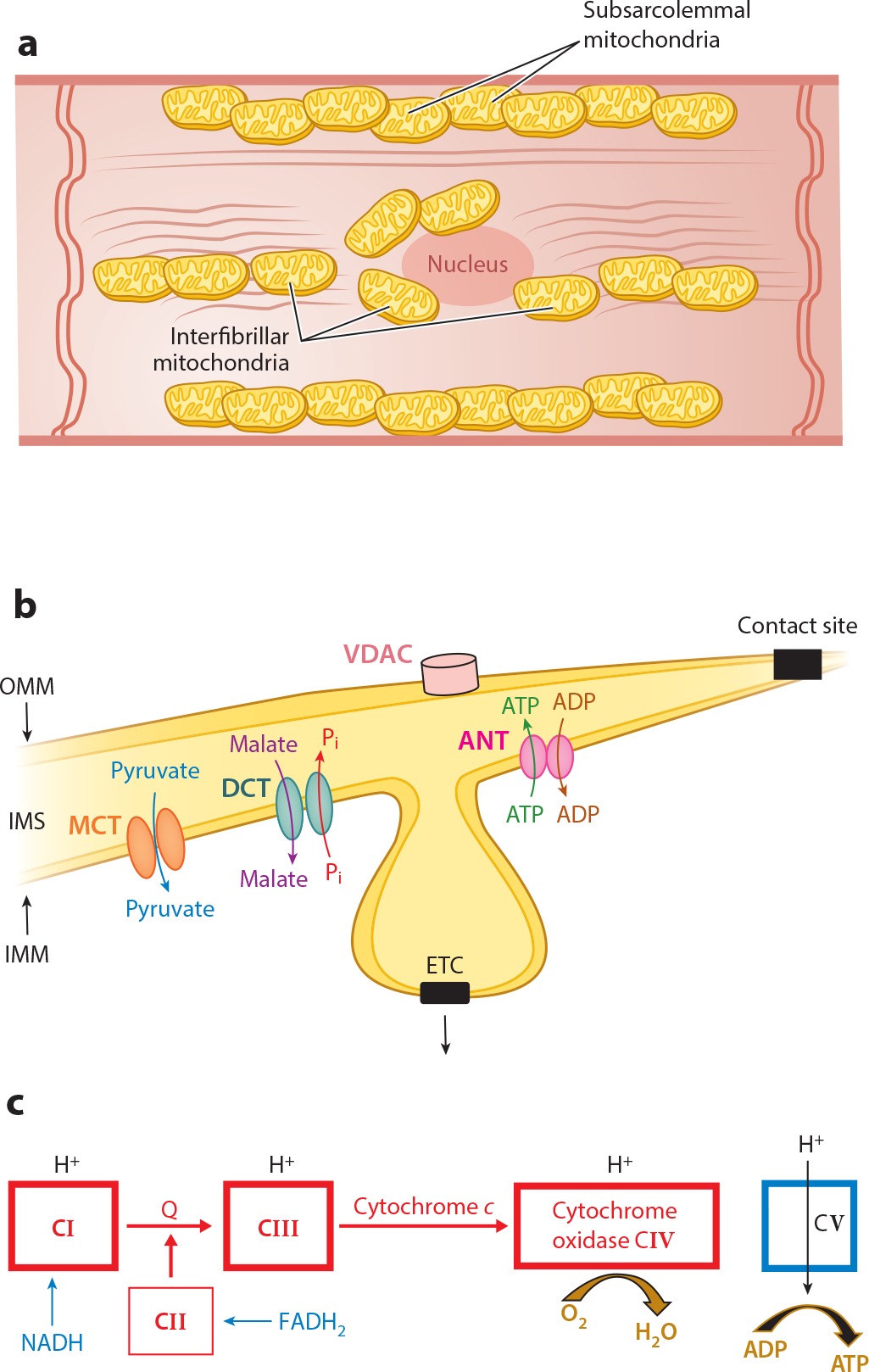
Structure and function of normal mitochondria. (a) The locations of subsarcolemmal and interfibrillar mitochondria in the cardiomyocyte are depicted schematically. The various cellular components are not drawn to scale. (b) Shown are the locations of the voltage-dependent anion channel (VDAC) in the outer mitochondrial membrane (OMM); the electron transport chain (ETC) in the cristae of the inner mitochondrial membrane (IMM) with the adenine nucleotide translocase (ANT), monocarboxylate transporter (MCT), and dicarboxylate translocase (DCT) in the IMM; the intermembrane space (IMS); and contact sites as fusion points of the outer and inner boundary membrane. (c) The ETC. Reduced nicotinamide adenine dinucleotide (NADH) donates an electron to complex I (CI) with flow through to complex IV (CIV) coupled with H+ pumping into the IMS at CI, CIII, and CIV. Reduced flavin adenine dinucleotide (FADH2) feeds electrons into CII with flow to CIV. However, no H+ is pumped into the IMS at complex II. H+ moves into the matrix coupled with the phosphorylation of ADP to ATP by CV. Other abbreviation: Q, coenzyme Q.
Figure 2.
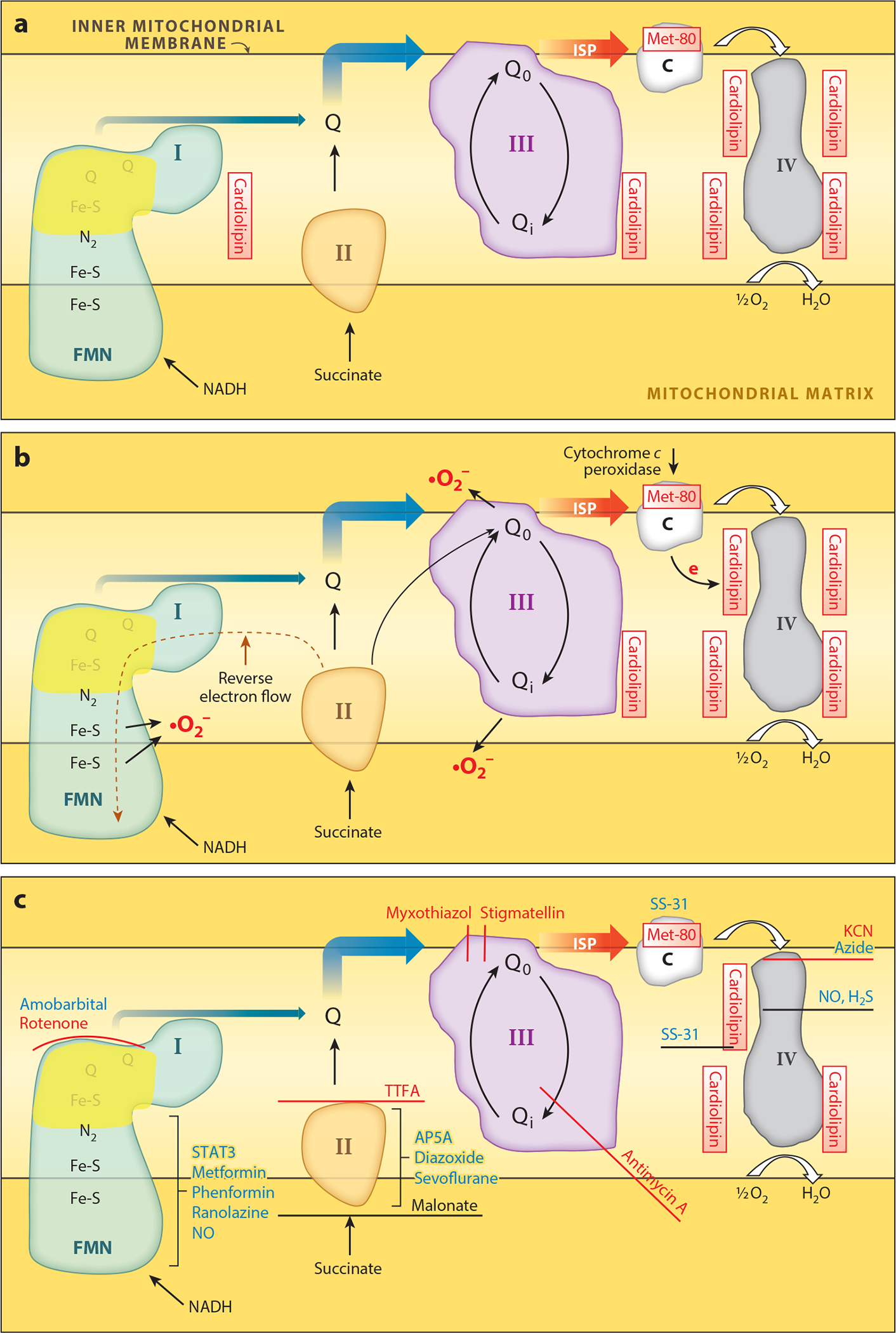
Sites of ischemia-mediated damage to electron transport. (a) Sites of ischemia damage to the electron transport chain (ETC) complexes. (b) Sites within these complexes where electrons leak to form superoxides and subsequently other reactive oxygen species, including hydrogen peroxide (H2O2). (c) Sites of action of classical chemical inhibitors (red) and potential therapeutic agents that modulate the ETC (blue). Potential therapeutic modulators of complex I include amobarbital acting at the quinol binding site (39), SNO donors (99), and nitric oxide (NO) (100) which likely interact with iron-sulfur (Fe-S) centers, and STAT3 (121). Other compounds that inhibit complex I partially and that have been reported to result in cardioprotection include metformin (31), phenformin (31), and ranolazine (97) (specific sites not depicted). Malonate competes for oxidation of succinate by complex II (29), whereas AP5A (60), diazoxide (200), and sevoflurane (201) alter other mechanisms. The peptide SS-31 binds to cardiolipin and also inhibits cytochrome c peroxidase (83, 86, 202–204). Other abbreviations: AP5A, diadenosine pentaphosphate; C, cytochrome c; FMN, flavin mononucleotide; I, II, III, and IV, complexes I, II, III, and IV; ISP, iron-sulfur protein; NADH, nicotinamide adenine dinucleotide; Q, coenzyme Q; STAT3, signal transducer and activator of transcription 3; TTFA, thenoyltrifluoroacetone.
Cristae, inward folds of the inner membrane, house the ETC and phosphorylation apparatus (Figure 1b). The ETC consists of four multiprotein complexes (complexes I–IV) composed of mitochondria- and nuclear-encoded subunits. Each complex contains catalytic and regulatory subunits for a specific reaction sequence for electron transfer (Figure 1c). The ETC also contains the mobile electron carriers coenzyme Q and cytochrome c (Figures 1c and 2). NADH is oxidized by complex I, leading to the reduction of coenzyme Q, which is then oxidized by complex III, resulting in the reduction of cytochrome c. The latter is oxidized by complex IV, transferring the electrons to oxygen to form water. These three complexes actively transport hydrogen ions from the matrix to the cytosolic side of the inner membrane, capturing and storing the energy as a proton gradient. Complex II oxidizes succinate produced in the TCA cycle and reduces coenzyme Q, which enters the ETC at complex III.
The phosphorylation apparatus, located in the inner membrane, consists of complex V (ATP-ase), a phosphate transporter, and adenine nucleotide translocase (ANT) (11). Complex V, situated in the cristae, harnesses the proton gradient to phosphorylate ADP to ATP. Inorganic phosphate and ADP enter the matrix via inner membrane transporters. These processes couple respiration to phosphorylation, that is, oxidative phosphorylation (OXPHOS). Myocyte energy demand determines the rate of OXPHOS.
DAMAGE TO MITOCHONDRIA DURING CARDIAC ISCHEMIA
Mitochondria sustain progressive injury during cardiac ischemia. Injury progresses both in a regional subpopulation–dependent manner as well as to specific sites in the ETC within both populations of mitochondria. The progression of ischemic injury is more rapid in SSM than in IFM (13). SSM have a decreased capacity for calcium accumulation compared to IFM and are more susceptible to calcium overload–mediated cytochrome c release and damage (14). In the canine heart subjected to regional ischemia, OXPHOS is decreased to a greater extent in SSM than IFM (15, 16).
Mitochondrial electron transport sustains progressive damage during ischemia (13, 17–19) (Figure 2a). OXPHOS is preserved in both SSM and IFM in periods of ischemia of <20 min that do not result in cell death. Oxidation of the complex I substrate glutamate decreases relatively early, by 15 min, in the course of ischemia, whereas the oxidation of the complex II donor succinate decreases as ischemic periods lengthen to 30–60 min (11, 18), suggesting that damage occurs distal to complex I (18). Damage also occurs to the phosphorylation apparatus, including complex V (18) and the adenine nucleotide transporter (20). As ischemia is extended, complex III (19) and cytochrome oxidase (complex IV) are also damaged (11, 13). Following 45 min of ischemia in the rabbit heart, OXPHOS is decreased in SSM, but not in IFM, for all substrates tested, thus localizing the defect to cytochrome c and cytochrome oxidase. Mitochondrial respiration remains well coupled with preserved ADP:O ratios and unchanged rates of State 4 respiration. Uncoupled respiration is also decreased, thereby localizing the defect to the ETC (13, 19).
MITOCHONDRIAL MORPHOLOGY WITH ISCHEMIA
Electron microscopy reveals that during ischemia, SSM and IFM remain intact and their appearance is similar to that in nonischemic hearts (Figure 3a,b) (13); similar findings occur with isolated SSM and IFM (Figure 3c,d). Isolated mitochondria generally appear intact, with slight dilatation of the cristae and swelling of some organelles. Mitochondria isolated following ischemia show only decreased matrix density and minor vacuolization of cristae compared to nonischemic controls. Even after 45 min of ischemia (13, 17), morphological evidence of irreversible injury to myocytes and isolated mitochondria is absent. Dimethyl sulfoxide and acrolein added to fixative speeds its penetration of solid tissues compared to conventional fixatives (19), thus averting prominent morphological changes in ischemic cardiomyocytes (21).
Figure 3.
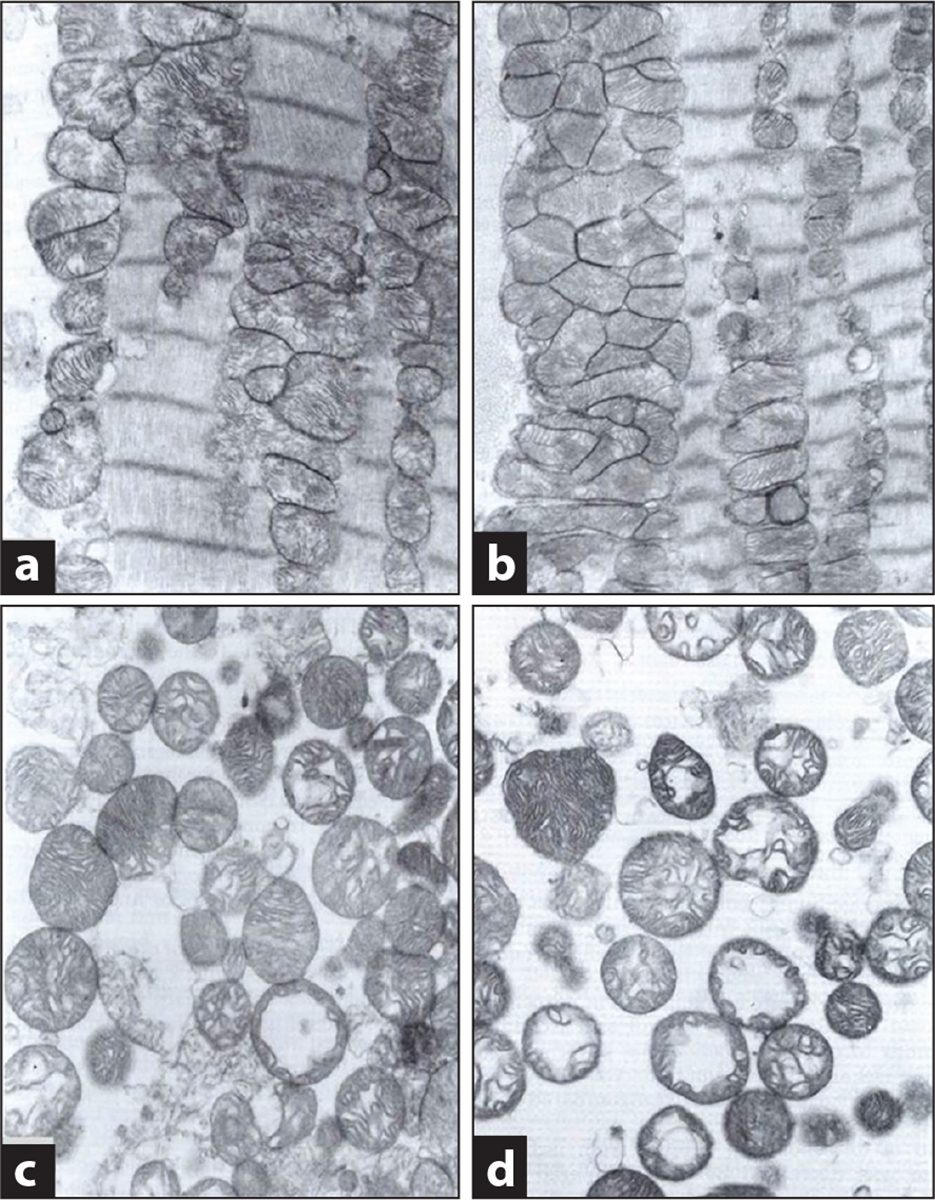
Mitochondrial morphology is preserved following ischemia alone. Electron micrographs of in situ subsarcolemmal mitochondria (SSM) and interfibrillar mitochondria (IFM) in nonischemic controls (a) and myocytes following 45 min of stop-flow ischemia (b) in the rabbit heart. Mitochondria are intact and, following ischemia, exhibit minimal change, confirming their morphological integrity at the end of the ischemic period. Micrographs of isolated SSM from nonischemic control (c) and ischemic hearts (d) confirm purity of the isolated mitochondrial population and reinforce the paucity of ischemia-induced damage to these organelles. Figure courtesy of American Physiological Society, copyright 1997 (13).
MOLECULAR PATHOLOGY OF ELECTRON TRANSPORT CHAIN DAMAGE
Complex I
Complex I activity decreases during ischemia (18, 19), in both SSM and IFM (22). Complex I activity is also altered by posttranslational modifications, including S-nitrosylation (23) and phosphorylation (24). Ischemic damage causes an increase in generation of ROS from complex I (25).
NADH ferricyanide oxidoreductase enzyme activity reflects the activity of NADH dehydrogenase, the initial component of complex I (18, 26). Ischemia does not decrease activity in either SSM or IFM, indicating that damage occurs at more distal sites in the complex (22, 25). In contrast, others have reported a decrease in the NADH dehydrogenase component of the complex (18) due to the loss of the flavin mononucleotide (FMN) coenzyme (Figure 2a) (18). An explanation for the different observations is not immediately apparent but may be due in part to differing models of ischemia.
Complex I undergoes conformational change in response to hypoxia and ischemia, shifting from the normal, active A form to the deactive, or dormant, D form when the iron-sulfur clusters and FMN are in the reduced state and the terminal acceptor ubiquinol pool is reduced (27). Conversion to the D form exposes oxidative-sensitive sulfhydryl groups (27), including cysteine 39 of mitochondria-encoded NDUF3 as well as cysteines on NDUF9 and NDUF1 (28). Reversible glutathionylation of these residues occurs as part of an antioxidant defense mechanism that protects complex I. Reaction with oxidants can trap complex I in the D form, leading to persistent decreased activity. In the absence of oxidation of the sulfhydryl groups, incubation of complex I with substrate leads to the regeneration of the A form and restores activity. Interactions of complex I with mitochondria-localized thioredoxin-2 or glutaredoxin-2 may enhance protection of complex I. The D form is favored, at least during stop-flow ischemia (27, 28). The D form blunts reverse electron transport that generates superoxide (29, 30) and inhibits ROS production via decreased forward flow (27). The role of complex I blockade with rotenone or amobarbital in favoring the A to D transition is the subject of current work. Phenformin, a biguanide, favors the transition of complex I to the D form (31). Metformin in high doses inhibits complex I partially in ischemic hearts and, if given for 5 min at the onset of reperfusion, decreases cardiac damage, as does amobarbital (32–34). Thus, modulation of the A to D transition of complex I may emerge as a key factor in cardioprotection.
Complex I produces ROS (26, 35, 36) (Figure 2b). Candidate sites for ROS production are FMN in NADH dehydrogenase (37), iron-sulfur cluster N2, and the two tightly bound ubiquinones located distal to the path of electron flow (26). In the baseline state, inhibition of NADH dehydrogenase decreases ROS generation, indicating that the site is distal to the FMN (26), although other investigators previously assigned ROS production to this site (38). Ischemia increases ROS generation from complex I despite preserved NADH dehydrogenase activity (25), localizing this site distal to FMN (26). Rotenone blocks complex I at the ubiquinone acceptor site (39). Inhibition by rotenone increased ROS generation from complex I after ischemia, indicating that the site is proximal to the ubiquinone binding site (26). Based on the use of forward and reverse electron flow (REF) (40, 41), ROS production after ischemia is likely from either the N2 iron-sulfur site or the tightly bound quinones within complex I (26).
Complex III
Complex III activity decreases during ischemia (18, 19). Mammalian complex III contains three catalytic subunits: cytochrome b, cytochrome c1, and the iron-sulfur protein (ISP). The ISP contains a 2Fe-2S cluster coordinated by two cysteines and two histidines (42). The contents of cytochromes b and c1 were unchanged by ischemia in both SSM and IFM from the rat heart (19). The content of ISP was measured by assessing the electron paramagnetic resonance (EPR) signal generated by the 2Fe-2S cluster in submitochondrial particles from nonischemic controls and from rat hearts following ischemia, which decreased the g 1.79 and g 1.89 EPR signals of the ISP dramatically in both SSM and IFM (19). Ischemia did not decrease the content of ISP peptide despite a marked decrease of the EPR signal of the ISP; the other 11 subunits of the complex, including the redox centers cytochrome b and c1, were unaltered (19) (Figure 2a). Complex III remains a key site for oxidant generation during and following ischemia. Most of the ROS generated at complex III are from the Qo (outer quinol binding site) oxidation center, although the Qi (quinol site internal) oxidation center also contributes (43, 44). Reduced coenzyme Q binds to the Qo center on cytochrome b and undergoes oxidation, providing the site of entry for electrons into complex III. This oxidation generates two electrons with the transfer of one electron to ISP and the other electron to the Qi center on the matrix side of cytochrome b (Figure 2a). Ischemic damage to complex III allows adequate residual electron flow into the complex for ROS production, probably because complex III exists as a dimer. Inactivation of one of the Qo sites in a dimer facilitates ROS generation from complex III Qi centers (45) (Figure 2b).
Transient, reversible blockade at the Qo site of complex III is an appealing therapeutic intervention. Myxothiazol and stigmatellin, irreversible inhibitors and mitochondrial poisons, attenuate ROS production from complex III (46). In contrast, a reversible inhibitor of the Qo site has the potential to inhibit ROS production and blunt reperfusion injury.
Complex IV–Cardiolipin
Activity of Complex IV, composed of 13 peptide subunits, requires the integrity of catalytic subunits (47) and regulatory and structural subunits (48). Complex IV is located in respirasomes with complexes I and III that optimize channeling of respiratory substrates (49). Ischemia does not deplete or inactivate subunit peptides (13). Cardiolipin is the only phospholipid that is decreased in content; this loss in SSM occurs over an interval that parallels the decrease in OXPHOS through cytochrome oxidase (17), providing a mechanism for the complex IV defect (Figure 2a). Mitochondrial cardiolipin content also decreases during ischemia in isolated rat heart (50, 51) and in vivo canine heart (52, 53). Fusion of isolated rat heart mitochondria with cardiolipin-containing liposomes restores cytochrome oxidase activity (51).
Loss of cardiolipin from SSM during ischemia plays an important role in mitochondrial damage. The high content of oxidatively sensitive linoleic acid (C18:2) acyl groups (54) renders cardiolipin susceptible to oxidative damage, but the unaltered composition of cardiolipin following ischemia excludes the presence of oxidatively altered acyl groups (17). However, oxidatively generated peroxy groups in cardiolipin are unstable, may decompose rapidly, and lead to cardiolipin destruction (55). Alternatively, oxidized acyl residues may react directly with proteins, leading to covalent phospholipid-protein complexes that render cardiolipin no longer extractable by organic solvents (56). Thus, oxidative processes cannot be excluded merely by the failure to detect oxidized cardiolipin.
Complex II
Complex II is relatively resistant to ischemic damage (13). Its activity decreases in rat heart mitochondria during prolonged global ischemia (Figure 2a) and decreases further after reperfusion (57). This decreased activity is related to a reduction of S-glutathionylation in the 70-kDa flavin protein (58) and increased oxidation and nitration of complex II, impairing its interaction with complex III (59). Modulation of complex II activity decreases cardiac injury during I/R and contributes to opening of the mitochondrial KATP channel (60) and inhibition of ROS generation (61). Defects in mammalian complex II increase ROS production, likely from auto-oxidation of FADH2 or FADH•− semiquinone (62).
In intact heart mitochondria, ROS can be generated through REF from complex II to complex I using succinate as substrate (41, 61) (Figure 2b). This generation is affected by the inner membrane potential (mΔΨ) (63) and by inhibitors of complex I (41) and complex II (61). With succinate as a substrate, inhibition by oligomycin increases REF-mediated ROS generation (61) by increasing mΔΨ. In contrast, depolarization of the inner membrane inhibits REF-mediated ROS generation completely (41, 63). Following prolonged ischemia, mΔΨ is depolarized in heart mitochondria (25, 41, 64). REF-mediated ROS generation can still occur in the heart following mild ischemia (29) that may not have led to depolarization of the inner membrane. Preventing REF-mediated ROS production decreases cardiac injury during I/R (29, 30). The role of REF-mediated ROS generation in cardiac injury might depend on the degree of ischemic damage sustained by the mitochondria.
Therapies to decrease REF-mediated ROS can be approached in several ways. Blockade of complex II with malonate or diadenosine pentaphosphate (60) has clinical potential, but thenoyltrifluoroacetone, an irreversible inhibitor, has little utility. 3-Nitropropionic acid, an inhibitor of complex II, produces ROS and can result in cytoprotection (65). As noted above, relative uncoupling of mΔΨ will also attenuate REF. Safe approaches to uncouple respiration have been described (66, 67).
Cytochrome c
Ischemia results in the loss of cytochrome c from mitochondria (7, 10, 13) into the cytosol (7). Concomitant with cardiolipin loss, cytochrome c decreases in SSM at 30 min of ischemia (17). Unlike IFM, SSM in vitro exhibit a greater tendency to release cytochrome c (12) and to sustain rapid progression of ischemic damage (13), including the loss of cytochrome c (17). SSM are more sensitive to cytochrome c release by elevated external calcium (14) and thus more likely to activate cardiomyocyte death programs.
Cardiolipin provides binding sites on the inner membrane for cytochrome c (68), so a decrease in its content favors the loss of cytochrome c, a process that occurs during I/R (68). Thus, the decrease in mitochondrial content of cytochrome c is linked to the depletion of cardiolipin, followed by permeabilization of the outer membrane, either via proapoptotic peptides (69) or by MPT (10).
MITOCHONDRIAL INJURY DURING ISCHEMIA–ELECTRON TRANSPORT CHAIN–DRIVEN INJURY
Ischemic damage to mitochondria is mediated at least in part by mitochondria themselves (70). Blockade of the ETC with rotenone or amobarbital protects them against ischemic damage. When the ETC in the intact heart is inhibited with rotenone immediately before ischemia, the content of cardiolipin, cytochrome c, and OXPHOS through cytochrome oxidase is preserved (70) and ischemic damage is markedly attenuated (Figure 2c). It is not surprising that preservation by rotenone of cardiolipin during ischemia preserves cytochrome c content.
Amobarbital (Figure 2c), a short-acting barbiturate, inhibits complex I reversibly at the rotenone site (39). Amobarbital treatment of the isolated rat heart just before ischemia protected OXPHOS, including cytochrome oxidase; preserved cytochrome c content during subsequent ischemia; and protected respiration with glutamate, a complex I substrate (71). Amobarbital washes out of mitochondria rapidly during isolation; its reversible blockade of respiration at complex I protects complex I from ischemic damage (71). Preservation of OXPHOS with substrates for complex III and IV shows that the distal ETC is also protected. Thus, blockade of electron transport at complex I using two chemically dissimilar compounds immediately before ischemia protects against ischemic damage to the ETC.
SITES IN THE ELECTRON TRANSPORT CHAIN THAT GENERATE ISCHEMIC DAMAGE
Historically, the study of oxidative damage to the myocardium focused on the reperfusion phase. Oxidative damage also can occur during ischemia, as oxygen remains available early in the process (72) because cytochrome oxidase does not consume all of the residual oxygen rapidly. In line with this observation, ROS production increases during global ischemia (73). Reduced electron flow occurs owing to the decreasing concentration of oxygen (74) as well as the ETC damage (13, 18, 19) (Figure 2b). With complete anoxia, ROS production ceases, paradoxically attenuating injury in isolated cardiac myocytes (75).
ROS production and Ca2+ loading were lessened during ischemia in guinea pig hearts treated with amobarbital (73). Blocking electron transport during ischemia preserves respiratory function in isolated mitochondria (71), decreases ROS production, and reduces Ca2+ accumulation in intact hearts (73). The importance of ROS production by mitochondria is supported by cardiac protection resulting from treatment with mitochondria-targeted antioxidants (76, 77).
Mitochondria are the primary source of ROS production during ischemia (75, 78) (Figure 2b). Superoxide production during simulated ischemia in cardiac myocytes is decreased by blockade of electron transfer into complex III (75, 78), thereby lessening the amount of molecular oxygen that can form superoxide (46). Antimycin A, an inhibitor of complex III, blocks distal to the Qo site of the complex and augments the net production of ROS from complex III (46) (Figure 2c). Inhibition of cytochrome oxidase increases ROS generation from upstream redox sites (78).
We investigated the role of electron transport distal to the Qo site of complex III in the damage to cardiolipin during ischemia. Treatment with antimycin A before ischemia preserved the content of cardiolipin in SSM and decreased the loss of cytochrome c in a fashion similar to the protection with rotenone (70), suggesting that ischemic damage occurs distal to the Qo site of complex III.
Cytochrome oxidase might produce ROS (79) and toxic NO from nitrates during hypoxia (80). In isolated rabbit hearts that had undergone 30 min of global stop-flow ischemia, we blocked the ETC either at complex I using amobarbital or at cytochrome oxidase using azide before ischemia. Azide treatment (Figure 2c) did not protect against ischemia-induced decreases in respiration (81) or in cytochrome c in SSM. In contrast, blockade of complex III at the Qi site attenuated the depletion of cardiolipin and loss of cytochrome c during ischemia (70). These observations localize the site that depletes cardiolipin and cytochrome c to the ETC segment encompassing the complex III Qi site and cytochrome c.
Cardiolipin interacts with cytochrome c via nonionic (82) and electrostatic mechanisms to bind it to the inner mitochondrial membrane (68). In pathological settings (e.g., oxidative stress), cytochrome c undergoes a change in tertiary structure (82). Oxidation of a key methionine residue (Met-80), a ligand of the heme group of cytochrome c, occurs in concert with the change in its tertiary structure (83). The iron coordination in the heme of cytochrome c is disrupted (83, 84), allowing H2O2 to interact with the heme iron, in turn facilitating electron transfer to and peroxidation of acyl groups of cardiolipin (84) (Figure 2b).
Formation of the cytochrome c cardiolipin peroxidase results in a marked shift in redox potential, with cytochrome c being unable to be reduced by complex III (84) and with blockade of electron flow into complex III. Once ischemic damage to the ETC forms the peroxidase, the sites of the ETC for ROS production likely shift from complex III to complex I. An increase in intramitochondrial ROS generation, coupled with the electron flow into the cytochrome c segment, is required to oxidize Met-80 (85). Following I/R, methionine oxidation in cytochrome c is increased. Thus, oxidized cytochrome c is an intriguing therapeutic target.
Short peptides consisting of alternating aromatic and charged amino acids cross cell membranes and are incorporated into mitochondria. Such peptides, including SS-31, are inserted into mitochondrial membranes and protect cardiolipin from oxidation by inhibiting cytochrome c cardiolipin peroxidase (86). SS-31 reduces infarct size in I/R rodent models, supporting a role for cytochrome c cardiolipin peroxidase in injury during I/R (87). Clinical trials are in progress to test whether SS-31 can ameliorate heart failure and genetic mitochondrial disease and reduce infarct size (88).
The p66shc protein, located in the mitochondrial intermembrane space, generates H2O2 using electrons transferred from reduced cytochrome c (89) and activates apoptosis (90). Durations of ischemia that lead to cell death are associated with phosphorylation and migration of p66shc from the cytosol to mitochondria (91). p66shc is an alternative to complex III for ROS production during ischemia (85). Complex III or p66shc can potentially generate ROS concomitant with electron flow through cytochrome c to form the peroxidase to oxidize cardiolipin, leading to decreased content of the latter (17).
MITOCHONDRIAL DAMAGE OCCURS PREDOMINANTLY DURING ISCHEMIA
During ischemia of the isolated perfused rabbit heart (Table 1), SSM lose cardiolipin and cytochrome c, with a decrease in cytochrome oxidase OXPHOS. Reperfusion does not lead to additional damage in the distal ETC (92). OXPHOS through cytochrome oxidase and the content of cytochrome c does not decrease further during reperfusion. Thus, impairment of cardiolipin, cytochrome c, and cytochrome oxidase occurs during ischemia rather than during reperfusion. Decreased cardiolipin content leads to mitochondrial release of cytochrome c (68), decreasing respiration and activating programmed cell death (7). The decrease in electron flux through cytochrome oxidase enhances ROS production from proximal redox centers in the ETC during reperfusion (93). Complexes I and III are key sites that generate ROS that induce mitochondrial damage and myocyte injury during I/R (36, 93, 94).
Table 1.
Cardiac ischemia-mediated mitochondrial injury in different species
| Species | Time course of ischemia | Mitochondrial damage | Advantages | Disadvantage |
|---|---|---|---|---|
| Mouse (34, 116, 125) | Mixed population of mitochondria at 25–30 min ischemia | OXPHOS ↓, cytochrome c ↓, AIF ↓, MPTP ↑ | Genetic models; mechanism testing | Small heart size |
| Rat (19,22) | SSM ≈ IFM at 30 min ischemia | OXPHOS ↓, cytochrome c 4, AIF ↓, cardiolipin ↓, MPTP ↑ | Commonly used in vivo and in vitro | Fast heart rate |
| Guinea pig (91) | SSM injury at 30 min ischemia; IFM spared | p66shc phosphorylation ↑ | Larger heart than rat | Fast heart rate |
| Rabbit (17) | SSM injury at 30 min ischemia; IFM spared | OXPHOS ↓, cytochrome c ↓, cardiolipin ↓, MPTP ↑ | Slow heart rate; more similar to human | Slow heart rate |
| Canine (53) | SSM injury at 60 min ischemia; IFM spared | OXPHOS ↓, CPT-1 ↓ | First large animal model | Expensive, highly sentient animal model |
| Pig (124) | SSM injury at 90 min ischemia; IFM spared | OXPHOS ↓, MPTP ↑ | Mimic of human model | Expensive |
Abbreviations: AIF, apoptosis-inducing factor; CPT-1, carnitine palmitoyl transferase-1; IFM, interfibrillar mitochondria; MPTP, mitochondrial permeability transition pore; OXPHOS, oxidative phosphorylation; SSM, subsarcolemmal mitochondria.
In sum, ischemic damage to mitochondria leads to ROS production (78), favors onset of MPTP (7), and results in the release of cytochrome c (7), all of which are mechanisms of mitochondria-driven cell death during reperfusion (1, 93) (Figure 4). Mitochondrial self-injury during ischemia is thus a major mechanism in cardiomyocyte damage during I/R.
Figure 4.
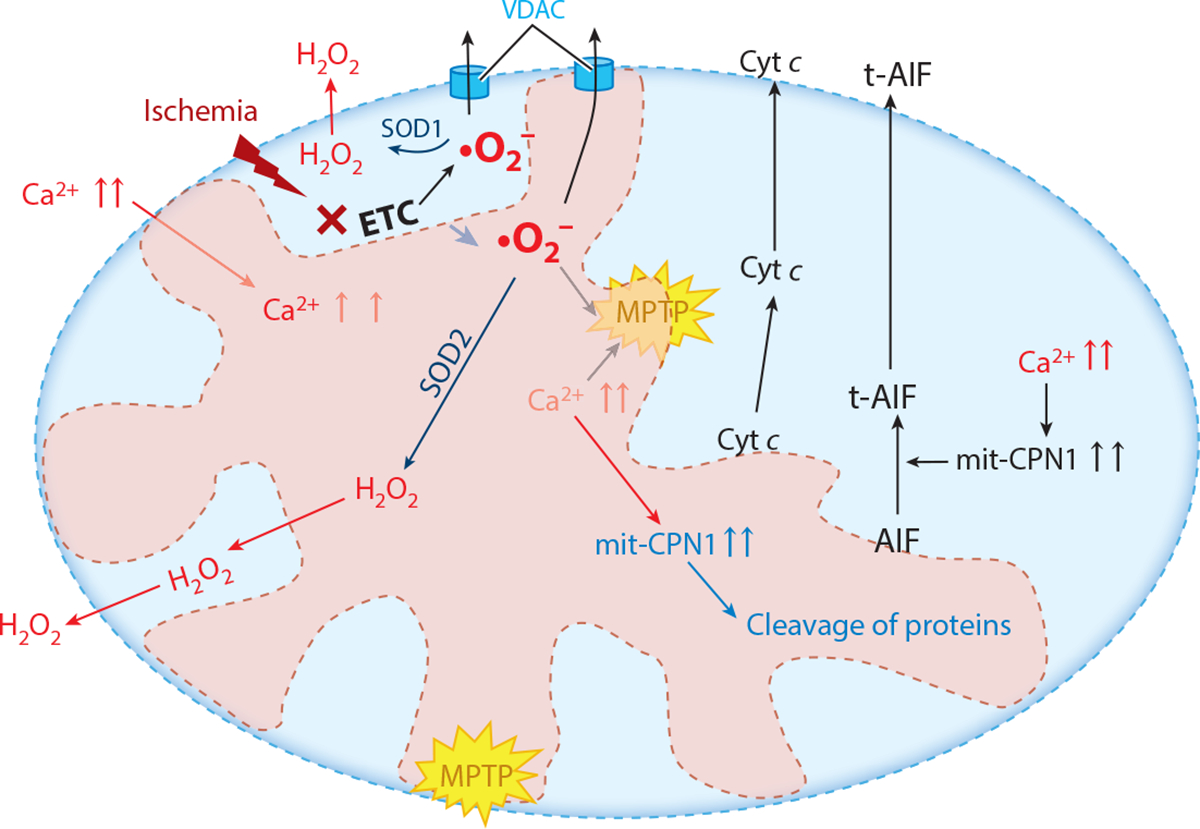
Mechanisms of mitochondrial-driven injury that result from ischemic damage to mitochondria, especially the mitochondrial electron transport chain (ETC). These include activation of the mitochondrial permeability transition pore (MPTP) and permeabilization of the outer mitochondrial membrane, leading to release of cytochrome c (Cyt c) and activation of mitochondrial calpains (mit-CPN1), which leads to the cleavage and release of truncated apoptosis-inducing factor (t-AIF) and the export of reactive oxygen species generated by the ETC by the voltage-dependent anion channel (VDAC), including mitochondrial contact sites. Antioxidant systems include superoxide dismutase 2 (SOD2) in the mitochondrial matrix and superoxide dismutase 1 (SOD1) in the intermembrane space. Superoxide (•O2−) in the mitochondrial matrix is converted to hydrogen peroxide (H2O2) by SOD2, whereas superoxide in the intermembrane space is converted by SOD1 to H2O2.
CARDIAC PROTECTION FROM ATTENUATION OF MITOCHONDRIAL-DRIVEN MITOCHONDRIAL INJURY
If the ETC remains intact and essentially undamaged at the onset of reperfusion, will myocyte injury occur? Blockade using amobarbital protects electron transport during ischemia (22, 70), supporting the idea that disruption of the ETC is the major source of mitochondrial damage (93). Other studies have documented that mitochondria, when free of ischemic damage at the onset of reperfusion (71), largely avoid cardiac injury (22). Amobarbital treatment (Figure 2c) preserves the rate and coupling of OXPHOS in both SSM and IFM following reperfusion (22). Thus, protection of the ETC by reversible blockade during ischemia is carried forward into reperfusion. Reversible blockade of electron transport during ischemia protected complex I (22) as well as the distal ETC, with preserved respiration through complex III and cytochrome oxidase and improved retention of cytochrome c (22).
Several mechanisms may explain why reperfusion of cardiomyocytes that contain mitochondria with essentially normal electron transport decreases cardiac injury (Figure 4). First, ROS production from both complex I and complex III is decreased (22). Preservation of respiratory function thus decreases mitochondrial production and release of ROS during reperfusion. Second, blockade of electron transport during ischemia enhances retention of cytochrome c during reperfusion (22), probably owing to maintenance of inner mitochondrial membrane integrity (17, 22, 68). Cytochrome c delocalized from a damaged inner membrane is released from mitochondria when outer membrane permeability increases (68). Electron transport blockade during ischemia preserves outer membrane integrity after reperfusion (22).
Early reperfusion after amobarbital treatment that led to retained mitochondrial function is associated with less mitochondrial ROS production and Ca2+ loading (73). A decrease in ischemic damage to the ETC preserves the antiapoptotic signaling from mitochondria (95) and decreases susceptibility to the onset of MPTP (64), retaining inner and outer membrane integrity. Blockade of electron transport before ischemia markedly reduces infarct size and improves contractile recovery during reperfusion (22, 73, 96). Protection of cardiac mitochondria during ischemia is carried forward into the early reperfusion period and supports the hypothesis that mitochondrial damage occurs predominantly during ischemia and mitochondria-mediated cardiomyocyte injury occurs largely during reperfusion.
ReversibleinhibitionofcomplexIattherotenonesitehaspotentialtherapeuticutility.Blockade at this site attenuates REF (40, 41) and decreases ROS production from complex III (36). Amobarbital has limited utility. Rotenone, an irreversible inhibitor, is toxic during the reoxygenation of reperfusion. Other approaches to attenuate complex I activity reversibly include intracellular acidification at the onset of reperfusion (34), high-dose metformin (32), ranolazine (97), Mito-SNO (98), 3-mercaptopropionylglycine (99), and nitrite (100). Metformin and ranolazine are clinically used drugs.
MITOCHONDRIAL PERMEABILITY TRANSITION DURING REPERFUSION
MPTP are nonselective pores located in the inner mitochondrial membrane (2). Opening of MPTP depolarizes mΔΨ, impairing OXPHOS and ATP production. MPTP opening, a pathological event, results in an influx of water and solutes into the mitochondrial matrix, causing mitochondrial swelling and eventual rupture of the outer membrane, which in turn leads to the loss of intermembrane space proteins, including cytochrome c and apoptosis-inducing factor (AIF). The release of cytochrome c and AIF activates caspase-dependent and caspase-independent cell death programs, leading to apoptosis (101). The precise structure of MPTP is unclear. Regulation of MPTP opening is related to cyclophilin D within the matrix (102). Proteins of the inner and outer mitochondrial membrane include VDAC and ANT, which were considered to be MPTP components, but genetic studies do not support this idea (103). Complex V is reportedly a component of the MPTP (104), and I/R alters complex V subunits (104, 105).
The role of MPTP opening in I/R injury has been reviewed extensively (106). The opening of MPTP can occur during ischemia (7) and is prominent during reperfusion (107). Increased calcium availability during reperfusion enhances mitochondrial calcium loading (73) and, in concert with mitochondrial oxidative damage, leads to MPTP opening (107). As discussed above, ROS production and calcium loading during reperfusion are lessened when mitochondrial function remains normal during reperfusion (73); MPTP opening during early reperfusion is decreased when mitochondria are protected by blockade of electron transport during ischemia (64). Based on their greater sensitivity to calcium-mediated damage (14), MPTP during reperfusion may largely involve SSM. MPTP is not necessarily irreversible during reperfusion, as closure of the MPTP is enhanced by pyruvate oxidation (6). Inhibition of MPTP opening decreases cell death (6), supporting the idea that MPTP contributes to cardiac injury.
The importance of mitochondrial calcium loading during early reperfusion is supported by the finding that interventions to attenuate myocardial calcium loading result in cardiac protection. These pharmacological treatments include blockade of the mitochondrial calcium uptake uniporter using ruthenium red (108, 109), blockade of sodium-hydrogen exchange (110), and blockade of sodium-calcium exchange (111).
Cyclosporin A binds to cyclophilin D and decreases markedly the susceptibility of mitochondria to MPTP, thus leading to a decrease in cell death. Cyclosporin A protects myocardium when given either before ischemia or at the onset of reperfusion (112). Initial human trials showed a benefit of cyclosporin A given at the onset of reperfusion (113), supporting the clinical importance of mitochondria as generators of cardiac reperfusion injury. As measured in the intact heart (73), protection of mitochondrial function during ischemia by reversible inhibition of electron transport (71) reduces mitochondrial calcium loading during reperfusion, decreases oxidant production (22), and prevents MPTP opening (64). In sum, ischemia-damaged ETC is a key factor in inducing MPTP opening during reperfusion.
MODULATION OF ISCHEMICALLY DAMAGED MITOCHONDRIA DURING REPERFUSION PROTECTS AGAINST CONSEQUENCES OF ISCHEMIA-INDUCED MITOCHONDRIAL DAMAGE
At the onset of reperfusion, transient and reversible blockade of the proximal ETC decreases myocardial injury (33, 93, 114), although the extent of protection is less than if intervention occurs before ischemia (114). Thus, early reperfusion is a critical window of opportunity for interventions to attenuate electron transport–dependent cardiac injury even though ischemia-mediated mitochondrial damage has already occurred (64, 115). Ischemic damage to the distal ETC emerges as a link between ischemia and the mitochondrial-driven myocyte injury that occurs during reperfusion (Figure 4).
In addition to decreased OXPHOS, reperfusion leads to the loss of key signaling proteins located within mitochondria (7, 116) (Figure 4). Cytochrome c and AIF, located within the intermembrane space (117), contribute to the function of the ETC (101). The release of these proteins from mitochondria in response to MPTP or MOMP triggers programmed cell death networks. Release of cytochrome c promotes caspase-mediated cell death programs (7), whereas release of AIF enhances caspase-independent cell death programs (117). Interventions applied at the onset of reperfusion thus might be able to block the loss of these proteins from mitochondria and, consequently, programmed cell death (101).
RECRUITMENT OF TRANSCRIPTION FACTORS TO MODULATE ELECTRON TRANSPORT
Mitochondria-localized transcription factors can modulate ETC function and alter mitochondrial function by interacting with the apoptotic machinery or changing mitochondrial RNA expression (118). Signal transducer and activator of transcription 3 (STAT3), the best-studied transcription factor during ischemia, regulates the expression of genes that encode proteins involved in cellular responses to stress (119) and protection from injury (120). Mitochondrial STAT3 regulates electron transport by nontranscriptional mechanisms (121) and interacts with matrix-localized cyclophilin D, an MPTP component (122). Genetic deletion of STAT3 impairs complexes I and II (123) and increases sensitivity to calcium overload that leads to MPTP opening (122).
Cardiac overexpression of a mitochondrial-targeted, transcriptionally inactive form of STAT3 (MLS-STAT3E) leads to persistent, partial blockade of complex I, which protects it from ischemic damage (Figure 2c), decreases ROS production following ischemia, and enhances mitochondrial retention of cytochrome c (121). MLS-STAT3E response during ischemia is reminiscent of approaches using pharmacological blockade of complex I (1), which protects mitochondria and the myocardium (124). Overexpression of MLS-STAT3E can reduce cardiac injury after I/R (125). In line with these observations, cardioprotection in an in vivo pig model of regional myocardial I/R (considered similar to I/R in humans) leads to phosphorylation of mitochondrial STAT3, protection of complex I, and increased calcium tolerance (124). Enhancing mitochondrial STAT3 activation and content during ischemia or early reperfusion may be a way to harness intrinsic protective mechanisms. Activation of STAT3 to alter mitochondrial metabolism during ischemia or at the time of reperfusion using leptin (126), oncostatin M (127), or even tumor necrosis factor-α (128) is a future possibility.
PRECONDITIONING AND MITOCHONDRIAL FUNCTION
Ischemic preconditioning (IPC), wherein brief periods of ischemia followed by reperfusion are applied immediately before sustained ischemia, reduces infarct size by decreasing apoptosis and necrosis (129). The early phase of IPC protection results from activation of G protein–coupled receptors (129) that trigger cardioprotective signaling pathways (129, 130) and ultimately inhibit MPTP (131, 132). In addition, signaling ROS generated in mitochondria during IPC activate mitochondrial ATP-dependent potassium (m-KATP) channels, which prevent mitochondrial Ca+2 overloading (133). The early phase of preconditioning involves signaling that impinges on mitochondria to modulate function and cardioprotection, including preservation of OXPHOS during reperfusion. IPC and blockade of electron transport thus achieve cardioprotection by different mechanisms that modulate mitochondrial metabolism during ischemia. Therapeutic approaches based on IPC mechanisms are limited by the inability to predict the onset of coronary thrombosis. Anesthetic preconditioning, especially with sevoflurane, activates these mechanisms, exhibits efficacy, and is likely applicable to surgical settings when the onset of myocardial ischemia is known (111, 134, 135). The repurposing of agents that elicit IPC responses as chronic, preemptive therapeutics in high-risk patients merits consideration. Diazoxide (136) and nicorandil (137) are two clinically used agents that elicit IPC responses.
REMOTE CONDITIONING
Protection against I/R injury to the heart might be provided by applying cycles of I/R to seemingly unrelated vascular beds. Such remote conditioning produces changes in blood and neurons that are transmitted to the heart. The precise mechanisms by which this protection is conferred have not been identified in their entirety, although some are known (see 138–140). This protection appears to be independent of the time when cardiac ischemia will occur or has occurred (141–143). Proposed triggers include small-molecule receptor agonists (144–146). Researchers have proposed humeral, neural, exosomal, and cellular mechanisms to explain the communication between distant organs and the myocardium (141, 147, 148). Neural pathways are increasingly supported as an obligate mediator in the protective signaling (149, 150). Remote conditioning maintains the integrity of mitochondrial structure, attenuates loss of cytochrome c, and inhibits MOMP (151).
In the setting of coronary artery bypass grafting, the benefits of remote conditioning have been disappointing (152–158). However, there is still substantial enthusiasm for remote conditioning in myocardial protection in the treatment of coronary occlusion because of its ease of application, minimal adverse effects, and low cost. Notably, remote conditioning in ST segment myocardial infarction has had some encouraging results (157, 158). Because numerous factors that are released as the result of conditioning have been identified, the possibility exists that pharmacological mimetics could be developed; such agents would enhance the practicality and effectiveness of conditioning as therapy.
MITOCHONDRIA-LOCALIZED CALPAINS AND APOPTOSIS-INDUCING FACTOR: SENSING MITOCHONDRIAL CALCIUM OVERLOAD INJURY
Calpains are a family of Ca2+-dependent cysteine proteases generally considered to be localized in the cytoplasm; this includes calpain I (μ-calpain), which is activated in the presence of micromolar (μM) calcium (159, 160) and is regulated by the endogenous inhibitor calpastatin (160, 161). Calpain 10 and μ-calpain localize to mitochondria (162, 163), the former in the matrix (164). AIF must be detached from the inner mitochondrial membrane via cleavage to truncated AIF (t-AIF) by mitochondrial-localized μ-calpain (116, 162) with subsequent release when the outer membrane is breached by MPTP or MOMP (Figure 4). Mitochondrial calpains constitute a calcium-responsive signaling system that activates caspase-independent cell death programs. Activation of mitochondrial calpains appears to increase the susceptibility to MPTP. Mitochondrial calpain activity is inhibited by intracellular acidic pH, the endogenous inhibitor calpastatin (162), and pharmacological inhibitors (165), all potential means of reducing caspase-independent cell death (118). Cell-permeable calpain inhibitors (e.g., MDL-28170) have a favorable short-term toxicity profile, do not impair cardiac contractile function, and are chemical templates for the development of clinical agents (166).
MITOCHONDRIAL DYNAMICS
Although mitochondria can be modulated with ischemia-damaged ETC to reduce cardiomyocyte injury during early reperfusion (33, 93, 114), longer-term cardiomyocyte survival and recovery from ischemia require that the damaged mitochondria be repaired, removed, or replaced (Figure 5). Mitochondrial repair or removal involves fusion, fission, and autophagy. Mitochondrial fusion proteins include mitofusin 1 (Mfn1), Mfn2, and optic atrophy 1 (OPA1). Mitochondrial fission proteins include dynamin-related protein 1 (Drp1), mitochondrial fission protein 1 (Fis1), mitochondrial fission factor (MFF), and mitochondrial dynamics proteins of 49 and 51 kDa (MiD49/51) (167–169).
Figure 5.
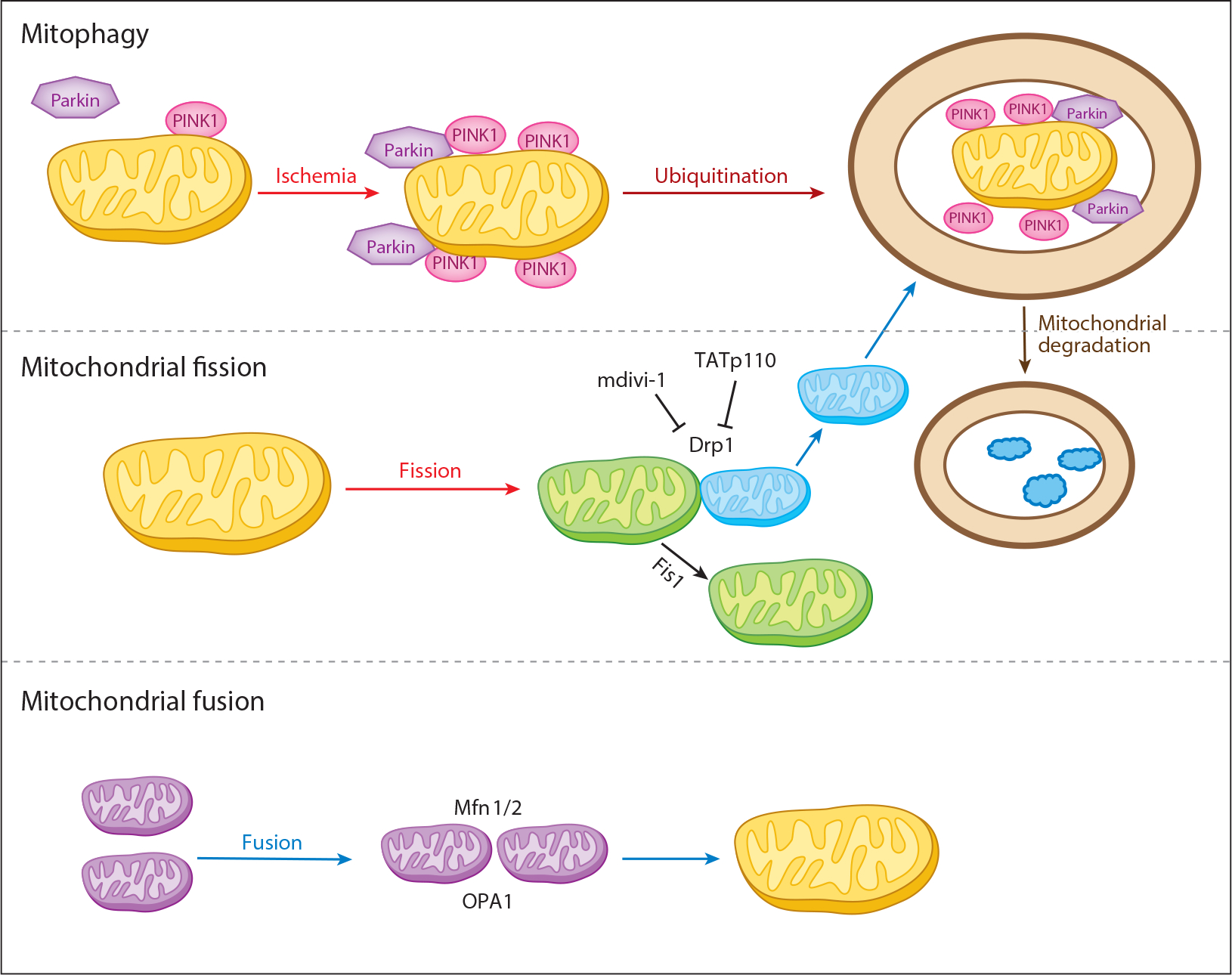
Mitochondrial dynamics. Mitophagy (mitochondrial autophagy) is mediated by Parkin and PINK1. Mitochondrial fission with Drp1 and Fis1 is a key component of the division of mitochondria. Drp1 can be inhibited by mdivi-1 and TATp110 peptides. Mitochondrial fusion utilizes the outer mitochondrial membrane Mfn1/2 and the inner membrane OPA1. Fission of damaged mitochondria can lead to destruction of the daughter organelles by mitophagy. Abbreviations: Drp1, dynamin-related protein 1; Fis1, mitochondrial fission protein 1; Mfn1/2, mitofusin 1 and 2; OPA1, optic atrophy 1; Parkin, component of a multiprotein E3 ubiquitin ligase complex; PINK1, PTEN-induced putative kinase 1.
Mfn1 and Mfn2 are on the outer mitochondrial membrane and play a key role in regulating membrane fusion. Mfn2 is found on the endoplasmic reticulum (ER) and mitochondria-associated membranes (170). In addition to mitochondrial fusion, Mfn2 is involved in calcium signaling between the ER and mitochondria (171). Fusion of the inner mitochondrial membranes is regulated by OPA1, a GTPase present in these membranes. Mfn1 is required for OPA1 to induce mitochondrial fusion (172).
Drp1, a cytosolic protein, is essential for mitochondrial fission. Translocation of Drp1 from the cytosol to mitochondria is the initial step in fission. Drp1 is activated by phosphorylation (173). Activated Drp1 undergoes oligomerization and translocation to mitochondria (173). Fis1, MFF, MiD49, and MiD51 also participate in facilitating the translocation of Drp1 to mitochondria (167).
MITOCHONDRIAL FISSION AND FUSION DURING ISCHEMIA-REPERFUSION
The impairment of mitochondrial dynamic change contributes to cardiac injury during I/R (8, 167). Pharmacological inhibition of Drp1 using mdivi-1 increases the number of elongated IFM with decreased myocardial infarct size in the murine heart following I/R (174). Mfn2 content decreases in the rat heart following I/R, but the translocation of Drp1 to mitochondria increases, accompanied by decreased Mfn1 and Mfn2 and increased Fis1 and Drp1 (173). Thus, I/R favors fission. Drp1 inhibition, using adenoviral overexpression of a dominant-negative Drp1K38A, decreases cardiac injury (175). Nitrite treatment modulates mitochondrial morphology via protein kinase A–dependent phosphorylation of Drp1 that inhibits fission (175). In cardiomyocytes, H2O2 treatment increases fission and apoptosis and enhances MFF expression. Downregulation of MFF with siRNA inhibits mitochondrial fission and cell death. Increased MFF expression is also found in heart following I/R (176). These results support the idea that inhibition of mitochondrial fission protects myocytes during I/R. The role of fission and fusion in ischemia has not been studied, nor has protective intervention during early reperfusion with blockade of electron transport to enhance these processes.
REMOVAL BY MITOPHAGY
Mitophagy (mitochondrial autophagy) sequesters and eliminates damaged mitochondria (8, 177, 178) (Figure 5). Inhibition of mitophagy during I/R increases cardiac injury, implicating mitophagy as an important process that decreases cardiac injury. Paradoxically, excessive mitophagy in response to stress, including I/R, can increase cardiomyocyte death (179). During I/R, MPTP opening leads to depolarization of mΔΨ that is critical to triggering mitophagy by recruiting the E3 ubiquitin ligase Parkin from the cytosol to the mitochondrial surface via the PTEN-induced putative kinase 1 (PINK1) (180, 181). Upon mΔΨ depolarization, PINK1 import and degradation within mitochondria are inhibited; it accumulates on the outer mitochondrial membrane, resulting in recruitment of Parkin to the uncoupled mitochondria (182). Parkin attached to mitochondria ubiquitinates mitochondrial proteins, which are then degraded (8). Parkin can also be recruited to mitochondria through a PINK1-independent mechanism. Ubiquitinated mitochondrial proteins are recognized by the autophagy adaptor protein p62/sequestosome 1 (183); p62 bridges ubiquitinated proteins with LC3 on autophagosomes (184), which engulf the damaged mitochondria.
Genetic deletion of PINK1 and Parkin has shown that these proteins are required for mitophagy in response to I/R (185). Depolarized mitochondria, especially as an index of MPTP, render the cardiomyocyte at risk for necrotic cell death. PINK1 and Parkin thus provide a branch point for cell survival by enhancing mitophagy, an alternative to necrotic cell death (186). It is unclear if the modulation of mitochondrial metabolism that protects mitochondria at reperfusion is mediated in part by minimizing the need for remodeling of the mitochondria (autophagy, fusion, and fission) so that activation of excessive, damaged mitochondrial remodeling is averted. Attenuation of mitochondrial-driven injury at the onset of reperfusion should lead to mitochondrial fusion, which in turn will decrease the stimulus for activation of mitophagy, resulting in less tissue injury. Therapeutics directed at PINK1 and Parkin are being pursued; the impact of such agents on reperfusion pathophysiology and myocardial salvage will be of great interest. Alternatively, targeting m to depolarize and enhance mitophagy is a potential therapeutic avenue (187).
SUMMARY, FUTURE DIRECTIONS, AND THERAPEUTIC IMPLICATIONS
Ischemia leads to a mitochondrial phenotype that favors direct chemical- and signaling-mediated cardiomyocyte injury (Table 1). Ischemia results in the following:
Damage to the ETC that generates oxidant production, leading to impairment of mitochondrial components, including the chain itself (11).
Outer mitochondrial membrane changes that favor activation of programmed cell death (188), a phenotype that is protected partially by inhibition of ETC-driven mitochondrial injury.
Activation of calcium-mediated and oxidant-generated mitochondrial signaling that includes the pathological onset of MPTP and activation of mitochondria-localized calpains (189).
Formation and activation of mitochondria-based cell death signaling that includes formation of cytochrome c cardiolipin peroxidase and possible modification of mitochondrial peptides that promote MPTP activation (190).
These multiple mitochondrial responses signal to the cardiomyocyte that a critical portion of its mitochondria are injured. The cellular response involves activation of repair and compensatory systems at the transcriptional (191), posttranscriptional (192), and posttranslational levels (193, 194). Thus, ETC-dependent, ischemia-induced processes persist for prolonged periods of reperfusion. Blockade of electron transport induces protective changes in outer mitochondrial membrane proapoptotic peptides (95) and susceptibility to MPTP (64, 95), thus implying that ischemic alterations in nonoxidative mitochondrial functions result from damaged ETC. During early and later reperfusion, cardiomyocyte responses to ischemia-damaged mitochondria perpetuate mitochondrial damage well into the reperfusion period.
Recognition of ETC-driven responses that propagate mitochondrial-driven pathology in the myocyte suggests interventions that might attenuate the sequelae of ischemic injury during reperfusion. Cellular responses to the presence of ischemia-damaged mitochondria could be addressed during reperfusion to reach back into ischemia to avert the consequences of mitochondrial damage during reperfusion. Because mitochondria are major players in myocardial ischemic injury, there are several types of possible intervention, including the activation of mitophagy (8, 195) and of mitochondrial biogenesis (196, 197) by the use of a variety of currently available pharmacological agents. Also helpful might be interventions that modify function by posttranslational modification of mitochondrial proteins (198, 199) or modulation of transcription factors that regulate mitochondrial metabolism (118). Conceptual approaches to attenuate mitochondria-driven cardiomyocyte injury during ischemia as well as during reperfusion are summarized in Figure 6. Potential pharmacologic approaches that address these mechanisms of mitochondria-driven injury are listed in Table 2.
Figure 6.
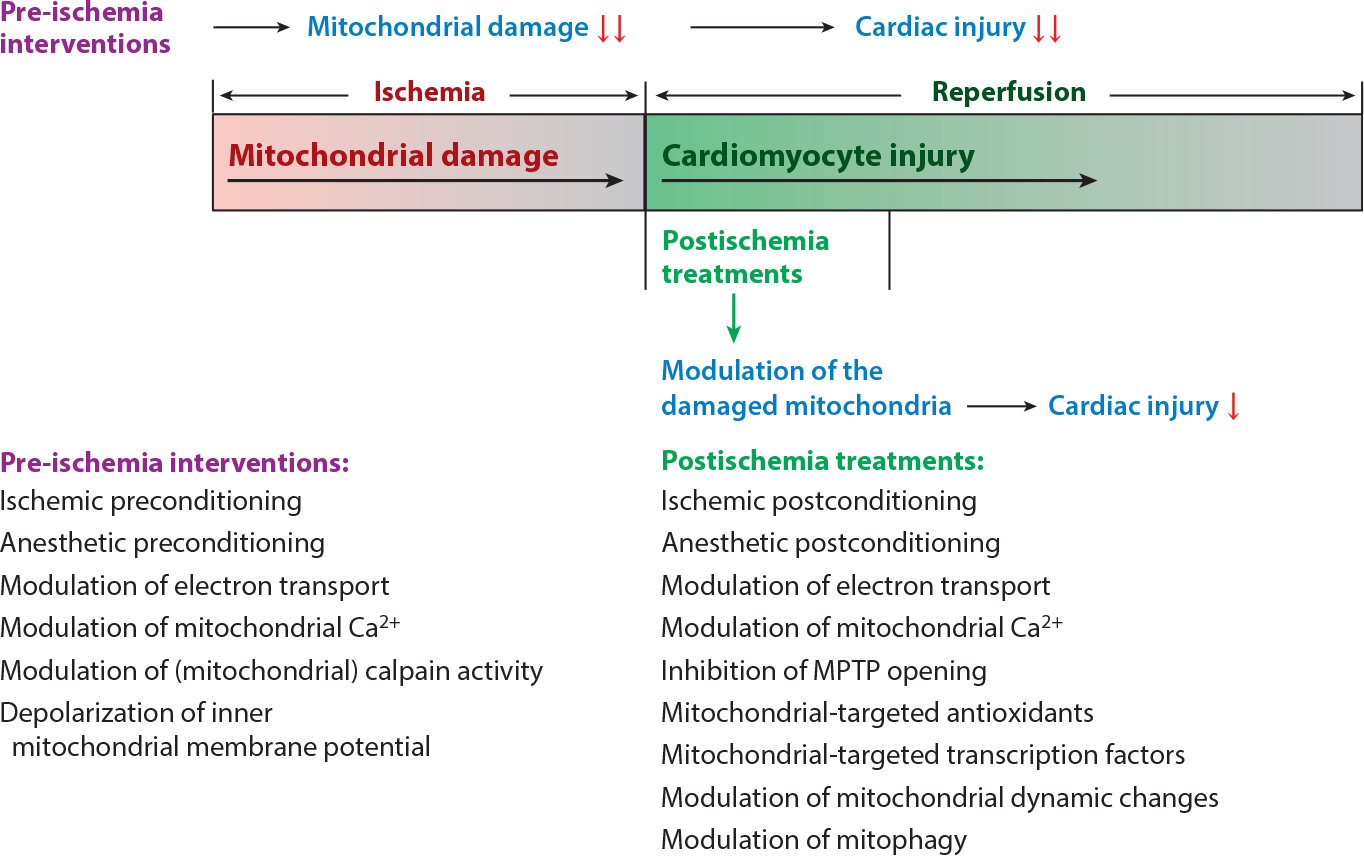
Strategies to decrease mitochondria and myocardial injury during ischemia-reperfusion. Mitochondria, which are damaged progressively during ischemia, set the stage for progressive myocardial injury during reperfusion. Therefore, pre-ischemic interventions, which include ischemic preconditioning (205), anesthetic preconditioning (111), modulation of electron transport (1), modulation of mitochondrial Ca2+ (108, 110, 111), modulation of mitochondrial calpain activity (206), and depolarization of inner mitochondrial membrane potential (66, 67), are more effective at reducing cardiac injury during reperfusion in that these interventions protect mitochondria from ischemic damage. Modulation of the ischemic-damaged mitochondria during early reperfusion, which include ischemic postconditioning (207–209), anesthetic postconditioning (134, 135), modulation of electron transport (93, 114), modulation of mitochondrial Ca2+ (110, 111), inhibition of mitochondrial permeability transition pore (MPTP) opening (112), mitochondrial-targeted antioxidants (86, 210), mitochondrial-targeted transcription factors (126, 127), modulation of mitochondrial dynamic changes (174), and modulation of mitophagy (187), can attenuate myocardial injury during reperfusion. Treatments during early reperfusion are less effective than interventions before ischemia but have more clinical relevance in that the imminent onset of a myocardial infarction is rarely known.
Table 2.
Pharmacological interventions to decrease cardiac injury during ischemia-reperfusion
| Drugs | Mechanisms of protection | Reference |
|---|---|---|
| Pre-ischemia interventions | ||
| Ischemic preconditioning | ||
| Dioxide | Katp channel opener, complex II inhibitor | 136 |
| Nicorandil | Katp channel opener | 137 |
| Anesthetic preconditioning | ||
| Sevoflurane | ROS ↓, intracellular Ca2+ ↓, mitochondrial Ca2+ ↓, complex II inhibitor | 111 |
| Modulation of electron transport | ||
| Amobarbital | Reversible complex I inhibitor, OXPHOS ↑, ROS ↓, MPTP ↓ | 22 |
| AP5A | Reversible complex II inhibitor, ROS ↓ | 60 |
| 3-Nitropropionic acid | Reversible complex II inhibitor | 65 |
| Ranolazine | Complex I inhibitor, ROS ↓, intracellular Ca2+ ↓, mitochondrial Ca2+ ↓ |
97 |
| Mito-SNO | Complex I inhibitor, ROS ↓ | 98 |
| 3-Mercaptopropionylglycine | Complex I inhibitor, ROS ↓ | 99 |
| Nitrite | Complex I inhibitor, ROS ↓, MPTP ↓ | 100 |
| Modulation of mitochondrial Ca2+ content | ||
| Ruthenium red | Mitochondrial Ca2+ uniporter inhibitor, mitochondrial Ca2+ ↓ | 108, 109 |
| Benzamide | Na+ -H+ exchange 1 inhibitor, intracellular Ca2+ ↓, mitochondrial Ca 2+ ↓ | 110 |
| KB-R7943 | Na+ -Ca2+ exchange inhibitor, intracellular Ca2+ ↓, mitochondrial Ca2+ ↓ | 111 |
| Modulation of cytosolic and mitochondrial calpain activity | ||
| MDL-28170 | Inhibition of mitochondrial calpain 1, OXPHOS ↑, MPTP ↓ | 162, 165 |
| Depolarization of inner mitochondrial membrane potential | ||
| Dinitrophenol | Prevention of reverse flow–induced ROS generation | 66,67 |
| Postischemia treatments | ||
| Ischemic postconditioning | ||
| Dioxide | Activation of RISK pathway, S-nitrosylation of mitochondrial protein | 208 |
| Hydrogen sulfide | Activation of the JAK2/STAT3 (SAFE) signaling pathway | 209 |
| Anesthetic preconditioning | ||
| Sevoflurane | Intracellular Ca2+ ↓, opening of mitochondrial KATP channel | 134, 135 |
| Modulation of electron transport | ||
| Amobarbital | Reversible complex I inhibitor, OXPHOS ↑, ROS ↓, MPTP ↓ | 33,93, 114 |
| Metformin | Complex I inhibitor, AMPK ↑, MPTP ↓ | 32 |
| Modulation of mitochondrial Ca2+ content | ||
| Benzamide | Na+ -H+ exchange 1 inhibitor, intracellular Ca2+ ↓, mitochondrial Ca2+ ↓ | 110 |
| Inhibition of MPTP opening | ||
| Cyclosporin A | Inhibition of MPTP opening | 112, 113 |
| Mitochondrial-targeted antioxidant | ||
| SS-31 | Cardiolipin content ↑, OXPHOS ↑, mitochondrial cytochrome c ↓ | 86 |
| MitoQ | ROS ↓, MPTP ↓ | 210 |
| Mitochondrial-targeted transcription factors | ||
| Oncostatin M | Activation of mitochondrial STAT3 | 127 |
| Leptin | Activation of mitochondrial STAT3 | 126 |
| Modulation of mitochondrial dynamic change | ||
| Mdivi-1 | Inhibition of mitochondrial fission | 176 |
| TATP110 | Inhibition of mitochondrial fission | 176 |
| Modulation of mitophagy | ||
| Chloroquine | Mitochondrial uncoupling, induction of autophagy | 187 |
Abbreviations: AMPK, AMP kinase; MPTP, mitochondrial permeability transition pore; OXPHOS, oxidative phosphorylation; ROS, reactive oxygen species.
ACKNOWLEDGMENTS
This work was supported by a R21AG049461 grant from the National Institute on Aging (C.L.H.); a Scientist Development Grant (11SDG5120011, Q.C.) and Grants-in-Aid (15GRNT24480123, Q.C.; 12GRNT20510024, C.L.H.) from the American Heart Association; a Virginia Common-wealth University Clinical and Translational Science Award (UL1TR000058 from the National Institutes of Health’s National Center for Advancing Translational Science, Q.C.); the Center for Clinical and Translational Research Endowment Fund of the Virginia Commonwealth University (Q.C.); the Office of Research and Development, Medical Research Service Merit Review Award (1IO1BX001355–01A1); Department of Veterans Affairs (E.J.L.); and the Virginia Commonwealth University Pauley Heart Center (Q.C., E.J.L.).
Glossary
- ETC
electron transport chain
- ROS
reactive oxygen species
- MPTP
mitochondrial permeability transition pore
- MOMP
mitochondrial outer membrane permeabilization
- SSM
subsarcolemmal mitochondria
- IFM
interfibrillar mitochondria
- OXPHOS
mitochondrial oxidative phosphorylation
- ISP
iron-sulfur protein
- STAT3
signal transducer and activator of transcription 3
- Drp1
dynamin-related protein 1
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Chen Q, Camara AK, Stowe DF, Hoppel CL, Lesnefsky EJ. 2007. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am. J. Physiol. Cell Physiol. 292:C137–47 [DOI] [PubMed] [Google Scholar]
- 2.Murphy E, Steenbergen C. 2008. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 88:581–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yellon DM, Hausenloy DJ. 2007. Myocardial reperfusion injury. N. Engl. J. Med. 357:1121–35 [DOI] [PubMed] [Google Scholar]
- 4.Reimer KA, Lowe JE, Rasmussen MM, Jennings RB. 1977. The wavefront phenomenon of ischemic cell death. 1. Myocardial infarct size versus duration of coronary occlusion in dogs. Circulation 56:786–94 [DOI] [PubMed] [Google Scholar]
- 5.Lesnefsky EJ, Chen Q, Hoppel CL. 2016. Mitochondrial metabolism in aging heart. Circ. Res. 118:1593–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halestrap AP, Clarke SJ, Javadov SA. 2004. Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc. Res. 61:372–85 [DOI] [PubMed] [Google Scholar]
- 7.Borutaite V, Jekabsone A, Morkuniene R, Brown GC. 2003. Inhibition of mitochondrial permeability transition prevents mitochondrial dysfunction, cytochrome c release and apoptosis induced by heart ischemia. J. Mol. Cell. Cardiol. 35:357–66 [DOI] [PubMed] [Google Scholar]
- 8.Kubli DA, Gustafsson AB. 2012. Mitochondria and mitophagy: the yin and yang of cell death control. Circ. Res. 111:1208–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kung G, Konstantinidis K, Kitsis RN. 2011. Programmed necrosis, not apoptosis, in the heart. Circ. Res. 108:1017–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borutaite V, Budriunaite A, Morkuniene R, Brown GC. 2001. Release of mitochondrial cytochrome c and activation of cytosolic caspases induced by myocardial ischaemia. Biochim. Biophys. Acta 1537:101–9 [DOI] [PubMed] [Google Scholar]
- 11.Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. 2001. Mitochondrial dysfunction in cardiac disease: ischemia-reperfusion, aging, and heart failure. J. Mol. Cell. Cardiol. 33:1065–89 [DOI] [PubMed] [Google Scholar]
- 12.Palmer JW, Tandler B, Hoppel CL. 1977. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J. Biol. Chem. 252:8731–39 [PubMed] [Google Scholar]
- 13.Lesnefsky EJ, Tandler B, Ye J, Slabe TJ, Turkaly J, Hoppel CL. 1997. Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. Am. J. Physiol. Heart Circ. Physiol. 273:H1544–54 [DOI] [PubMed] [Google Scholar]
- 14.Palmer JW, Tandler B, Hoppel CL. 1986. Heterogeneous response of subsarcolemmal heart mitochondria to calcium. Am. J. Physiol. Heart Circ. Physiol. 250:H741–48 [DOI] [PubMed] [Google Scholar]
- 15.Asemu G, O’Connell KA, Cox JW, Dabkowski ER, Xu W, et al. 2013. Enhanced resistance to permeability transition in interfibrillar cardiac mitochondria in dogs: effects of aging and long-term aldosterone infusion. Am. J. Physiol. Heart Circ. Physiol. 304:H514–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pauly DF, Kirk KA, McMillin JB. 1991. Carnitine palmitoyltransferase in cardiac ischemia: a potential site for altered fatty acid metabolism. Circ. Res. 68:1085–94 [DOI] [PubMed] [Google Scholar]
- 17.Lesnefsky EJ, Slabe TJ, Stoll MS, Minkler PE, Hoppel CL. 2001. Myocardial ischemia selectively depletes cardiolipin in rabbit heart subsarcolemmal mitochondria. Am. J. Physiol. Heart Circ. Physiol. 280:H2770–78 [DOI] [PubMed] [Google Scholar]
- 18.Rouslin W 1983. Mitochondrial complexes I, II, III, IV, and V in myocardial ischemia and autolysis. Am. J. Physiol. Heart Circ. Physiol. 244:H743–48 [DOI] [PubMed] [Google Scholar]
- 19.Lesnefsky EJ, Gudz TI, Migita CT, Ikeda-Saito M, Hassan MO, et al. 2001. Ischemic injury to mitochondrial electron transport in the aging heart: damage to the iron–sulfur protein subunit of electron transport complex III. Arch. Biochem. Biophys. 385:117–28 [DOI] [PubMed] [Google Scholar]
- 20.Duan J, Karmazyn M. 1989. Relationship between oxidative phosphorylation and adenine nucleotide translocase activity of two populations of cardiac mitochondria and mechanical recovery of ischemic hearts following reperfusion. Can. J. Physiol. Pharmacol. 67:704–9 [DOI] [PubMed] [Google Scholar]
- 21.Kalt MR, Tandler B. 1971. A study of fixation of early amphibian embryos for electron microscopy. J. Ultrastruct. Res. 36:633–45 [DOI] [PubMed] [Google Scholar]
- 22.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. 2006. Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion. J. Pharmacol. Exp. Ther. 319:1405–12 [DOI] [PubMed] [Google Scholar]
- 23.Burwell LS, Nadtochiy SM, Tompkins AJ, Young S, Brookes PS. 2006. Direct evidence for S-nitrosation of mitochondrial complex I. Biochem. J. 394:627–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen R, Fearnley IM, Peak-Chew SY, Walker JE. 2004. The phosphorylation of subunits of complex I from bovine heart mitochondria. J. Biol. Chem. 279:26036–45 [DOI] [PubMed] [Google Scholar]
- 25.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. 2008. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am. J. Physiol. Cell Physiol. 294:C460–66 [DOI] [PubMed] [Google Scholar]
- 26.Ohnishi ST, Ohnishi T, Muranaka S, Fujita H, Kimura H, et al. 2005. A possible site of superoxide generation in the complex I segment of rat heart mitochondria. J. Bioenerg. Biomembr. 37:1–15 [DOI] [PubMed] [Google Scholar]
- 27.Galkin A, Abramov AY, Frakich N, Duchen MR, Moncada S. 2009. Lack of oxygen deactivates mitochondrial complex I: implications for ischemic injury? J. Biol. Chem. 284:36055–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Babot M, Labarbuta P, Birch A, Kee S, Fuszard M, et al. 2014. ND3, ND1 and 39 kDa subunits are more exposed in the de-active form of bovine mitochondrial complex I. Biochim. Biophys. Acta. 1837:929–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, et al. 2014. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515:431–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pell VR, Chouchani ET, Murphy MP, Brookes PS, Krieg T. 2016. Moving forwards by blocking backflow: the yin and yang of MI therapy. Circ. Res. 118:898–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuzaki S, Humphries KM. 2015. Selective inhibition of deactivated mitochondrial complex I by biguanides. Biochemistry 54:2011–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lesnefsky EJ, Thompson J, Hu Y, Chen Q. 2016. Administration of metformin during early reperfusion decreases cardiac injury through inhibition of MPTP opening. FASEB J. 30:725.4 [Google Scholar]
- 33.Chen Q, Ross T, Hu Y, Lesnefsky EJ. 2012. Blockade of electron transport at the onset of reperfusion decreases cardiac injury in aged hearts by protecting the inner mitochondrial membrane. J. Aging Res. 2012:753949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu A, Szczepanek K, Maceyka MW, Ross T, Bowler E, et al. 2014. Transient complex I inhibition at the onset of reperfusion by extracellular acidification decreases cardiac injury. Am. J. Physiol. Cell Physiol. 306:C1142–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Q, Lesnefsky EJ. 2006. Depletion of cardiolipin and cytochrome c during ischemia increases hydrogen peroxide production from the electron transport chain. Free Radic. Biol. Med. 40:976–82 [DOI] [PubMed] [Google Scholar]
- 36.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. 2003. Production of reactive oxygen species by mitochondria: central role of complex III. J. Biol. Chem. 278:36027–31 [DOI] [PubMed] [Google Scholar]
- 37.Kudin AP, Bimpong-Buta NY, Vielhaber S, Elger CE, Kunz WS. 2004. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 279:4127–35 [DOI] [PubMed] [Google Scholar]
- 38.Okun JG, Lummen P, Brandt U. 1999. Three classes of inhibitors share a common binding domain in mitochondrial complex I (NADH:ubiquinone oxidoreductase). J. Biol. Chem. 274:2625–30 [DOI] [PubMed] [Google Scholar]
- 39.Chance B, Williams GR, Hollunger G. 1963. Inhibition of electron and energy transfer in mitochondria. I. Effects of Amytal, thiopental, rotenone, progesterone, and methylene glycol. J. Biol. Chem. 238:418–31 [PubMed] [Google Scholar]
- 40.Chouchani ET, Methner C, Nadtochiy SM, Logan A, Pell VR, et al. 2013. Cardioprotection by S-nitrosation of a cysteine switch on mitochondrial complex I. Nat. Med. 19:753–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ross T, Szczepanek K, Bowler E, Hu Y, Larner A, et al. 2013. Reverse electron flow-mediated ROS generation in ischemia-damaged mitochondria: role of complex I inhibition versus depolarization of inner mitochondrial membrane. Biochim. Biophys. Acta. 1830:4537–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Snyder CH, Gutierrez-Cirlos EB, Trumpower BL. 2000. Evidence for a concerted mechanism of ubiquinol oxidation by the cytochrome bc1 complex. J. Biol. Chem. 275:13535–41 [DOI] [PubMed] [Google Scholar]
- 43.Orr AL, Ashok D, Sarantos MR, Shi T, Hughes RE, Brand MD. 2013. Inhibitors of ROS production by the ubiquinone-binding site of mitochondrial complex I identified by chemical screening. Free Radic. Biol. Med. 65:1047–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Remmen H, Richardson A. 2001. Oxidative damage to mitochondria and aging. Exp. Gerontol. 36:957–68 [DOI] [PubMed] [Google Scholar]
- 45.Covian R, Trumpower BL. 2006. Regulatory interactions between ubiquinol oxidation and ubiquinone reduction sites in the dimeric cytochrome bc1 complex. J. Biol. Chem. 281:30925–32 [DOI] [PubMed] [Google Scholar]
- 46.Moghaddas S, Hoppel CL, Lesnefsky EJ. 2003. Aging defect at the QO site of complex III augments oxyradical production in rat heart interfibrillar mitochondria. Arch. Biochem. Biophys. 414:59–66 [DOI] [PubMed] [Google Scholar]
- 47.Babcock GT, Varotsis C. 1993. Discrete steps in dioxygen activation—the cytochrome oxidase/O2 reaction. J. Bioenerg. Biomembr. 25:71–80 [DOI] [PubMed] [Google Scholar]
- 48.Anthony G, Reimann A, Kadenbach B. 1993. Tissue-specific regulation of bovine heart cytochrome-c oxidase activity by ADP via interaction with subunit VIa. PNAS 90:1652–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosca MG, Vazquez EJ, Kerner J, Parland W, Chandler MP, et al. 2008. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc. Res. 80:30–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Edoute Y, van der Merwe E, Sanan D, Kotzé JCN, Steinmann C, Lochner A. 1983. Normothermic ischemic cardiac arrest of the isolated working rat heart: effects of time and reperfusion on myocardial ultrastructure, mitochondrial oxidative function, and mechanical recovery. Circ. Res. 53:663–78 [DOI] [PubMed] [Google Scholar]
- 51.Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Serena D, Ruggiero FM. 1999. Lipid peroxidation and alterations to oxidative metabolism in mitochondria isolated from rat heart subjected to ischemia and reperfusion. Free Radic. Biol. Med. 27:42–50 [DOI] [PubMed] [Google Scholar]
- 52.Vasdev SC, Biro GP, Narbaitz R, Kako KJ. 1980. Membrane changes induced by early myocardial ischemia in the dog. Can. J. Biochem. 58:1112–19 [DOI] [PubMed] [Google Scholar]
- 53.Kajiyama K, Pauly DF, Hughes H, Yoon SB, Entman ML, McMillin-Wood JB. 1987. Protection by verapamil of mitochondrial glutathione equilibrium and phospholipid changes during reperfusion of ischemic canine myocardium. Circ. Res. 61:301–10 [DOI] [PubMed] [Google Scholar]
- 54.Abramovitch DA, Marsh D, Powell GL. 1990. Activation of beef-heart cytochrome c oxidase by cardiolipin and analogues of cardiolipin. Biochim. Biophys. Acta 1020:34–42 [DOI] [PubMed] [Google Scholar]
- 55.O’Brien PJ. 1969. Intracellular mechanisms for the decomposition of a lipid peroxide. I. Decomposition of a lipid peroxide by metal ions, heme compounds, and nucleophiles. Can. J. Biochem. 47:485–92 [DOI] [PubMed] [Google Scholar]
- 56.Parinandi NL, Zwizinski CW, Schmid HH. 1991. Free radical-induced alterations of myocardial membrane proteins. Arch. Biochem. Biophys. 289:118–23 [DOI] [PubMed] [Google Scholar]
- 57.Pasdois P, Beauvoit B, Tariosse L, Vinassa B, Bonoron-Adèle S, Santos PD. 2006. MitoKATP-dependent changes in mitochondrial volume and in complex II activity during ischemic and pharmacological preconditioning of Langendorff-perfused rat heart. J. Bioenerg. Biomembr. 38:101–12 [DOI] [PubMed] [Google Scholar]
- 58.Chen YR, Chen CL, Pfeiffer DR, Zweier JL. 2007. Mitochondrial complex II in the post-ischemic heart: oxidative injury and the role of protein S-glutathionylation. J. Biol. Chem. 282:32640–54 [DOI] [PubMed] [Google Scholar]
- 59.Chen CL, Chen J, Rawale S, Varadharaj S, Kaumaya PP, et al. 2008. Protein tyrosine nitration of the flavin subunit is associated with oxidative modification of mitochondrial complex II in the post-ischemic myocardium. J. Biol. Chem. 283:27991–8003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wojtovich AP, Brookes PS. 2009. The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res. Cardiol. 104:121–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Drose S, Bleier L, Brandt U. 2011. A common mechanism links differently acting complex II inhibitors to cardioprotection: modulation of mitochondrial reactive oxygen species production. Mol. Pharmacol. 79:814–22 [DOI] [PubMed] [Google Scholar]
- 62.Chen YR, Zweier JL. 2014. Cardiac mitochondria and reactive oxygen species generation. Circ. Res. 114:524–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brand MD, Esteves TC. 2005. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab. 2:85–93 [DOI] [PubMed] [Google Scholar]
- 64.Chen Q, Paillard M, Gomez L, Li H, Hu Y, Lesnefsky EJ. 2012. Postconditioning modulates ischemia-damaged mitochondria during reperfusion. J. Cardiovasc. Pharmacol. 59:101–8 [DOI] [PubMed] [Google Scholar]
- 65.Ockaili RA, Bhargava P, Kukreja RC. 2001. Chemical preconditioning with 3-nitropropionic acid in hearts: role of mitochondrial KATP channel. Am. J. Physiol. Heart Circ. Physiol. 280:H2406–11 [DOI] [PubMed] [Google Scholar]
- 66.Perry RJ, Zhang D, Zhang XM, Boyer JL, Shulman GI. 2015. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347:1253–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Minners J, van den Bos EJ, Yellon DM, Schwalb H, Opie LH, Sack MN. 2000. Dinitrophenol, cyclosporin A, and trimetazidine modulate preconditioning in the isolated rat heart: support for a mitochondrial role in cardioprotection. Cardiovasc. Res. 47:68–73 [DOI] [PubMed] [Google Scholar]
- 68.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. 2002. Cytochrome c release from mitochondria proceeds by a two-step process. PNAS 99:1259–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Champattanachai V, Marchase RB, Chatham JC. 2008. Glucosamine protects neonatal cardiomyocytes from ischemia-reperfusion injury via increased protein O-GlcNAc and increased mitochondrial Bcl-2. Am. J. Physiol. Cell Physiol. 292:C178–87 [DOI] [PubMed] [Google Scholar]
- 70.Lesnefsky EJ, Chen Q, Moghaddas S, Hassan MO, Tandler B, Hoppel CL. 2004. Blockade of electron transport during ischemia protects cardiac mitochondria. J. Biol. Chem. 279:47961–67 [DOI] [PubMed] [Google Scholar]
- 71.Chen Q, Hoppel CL, Lesnefsky EJ. 2006. Blockade of electron transport before cardiac ischemia with the reversible inhibitor amobarbital protects rat heart mitochondria. J. Pharmacol. Exp. Ther. 316:200–7 [DOI] [PubMed] [Google Scholar]
- 72.Ilangovan G, Liebgott T, Kutala VK, Petryakov S, Zweier JL, Kuppusamy P. 2004. EPR oximetry in the beating heart: myocardial oxygen consumption rate as an index of postischemic recovery. Magn. Reson. Med. 51:835–42 [DOI] [PubMed] [Google Scholar]
- 73.Aldakkak M, Stowe DF, Chen Q, Lesnefsky EJ, Camara AK. 2008. Inhibited mitochondrial respiration by amobarbital during cardiac ischaemia improves redox state and reduces matrix Ca2+ overload and ROS release. Cardiovasc. Res. 77:406–15 [DOI] [PubMed] [Google Scholar]
- 74.Chandel NS, Budinger GR, Schumacker PT. 1996. Molecular oxygen modulates cytochrome c oxidase function. J. Biol. Chem. 271:18672–77 [DOI] [PubMed] [Google Scholar]
- 75.Vanden Hoek TL, Li C, Shao Z, Schumacker PT, Becker LB. 1997. Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. J. Mol. Cell. Cardiol. 29:2571–83 [DOI] [PubMed] [Google Scholar]
- 76.Kelso GF, Porteous CM, Hughes G, Ledgerwood EC, Gane AM, et al. 2002. Prevention of mitochondrial oxidative damage using targeted antioxidants. Ann. N. Y. Acad. Sci. 959:263–74 [DOI] [PubMed] [Google Scholar]
- 77.Dongworth RK, Hall AR, Burke N, Hausenloy DJ. 2014. Targeting mitochondria for cardioprotection: examining the benefit for patients. Future Cardiol. 10:255–72 [DOI] [PubMed] [Google Scholar]
- 78.Becker LB, Vanden Hoek TL, Shao ZH, Li CQ, Schumacker PT. 1999. Generation of superoxide in cardiomyocytes during ischemia before reperfusion. Am. J. Physiol. Heart Circ. Physiol. 277:H2240–46 [DOI] [PubMed] [Google Scholar]
- 79.Pinakoulaki E, Pfitzner U, Ludwig B, Varotsis C. 2003. Direct detection of Fe(IV)=O intermediates in the cytochrome aa3 oxidase from Paracoccus denitrificans/H2O2 reaction. J. Biol. Chem. 278:18761–66 [DOI] [PubMed] [Google Scholar]
- 80.Castello PR, David PS, McClure T, Crook Z, Poyton RO. 2006. Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: implications for oxygen sensing and hypoxic signaling in eukaryotes. Cell Metab. 3:277–87 [DOI] [PubMed] [Google Scholar]
- 81.Chen Q, Yin G, Stewart S, Hu Y, Lesnefsky EJ. 2010. Isolating the segment of the mitochondrial electron transport chain responsible for mitochondrial damage during cardiac ischemia. Biochem. Biophys. Res. Commun. 397:656–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sparagna GC, Lesnefsky EJ. 2009. Cardiolipin remodeling in the heart. J. Cardiovasc. Pharmacol. 53:290–301 [DOI] [PubMed] [Google Scholar]
- 83.Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, et al. 2005. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 1:223–32 [DOI] [PubMed] [Google Scholar]
- 84.Vladimirov YA, Proskurnina EV, Izmailov DY, Novikov AA, Brusnichkin AV, et al. 2006. Cardiolipin activates cytochrome c peroxidase activity since it facilitates H2O2 access to heme. Biochem. (Mosc.) 71:998–1005 [DOI] [PubMed] [Google Scholar]
- 85.Aluri HS, Simpson DC, Allegood JC, Hu Y, Szczepanek K, et al. 2014. Electron flow into cytochrome c coupled with reactive oxygen species from the electron transport chain converts cytochrome c to a cardiolipin peroxidase: role during ischemia-reperfusion. Biochim. Biophys. Acta 1840:3199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Birk AV, Liu S, Soong Y, Mills W, Singh P, et al. 2013. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 24:1250–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kloner RA, Hale SL, Dai W, Gorman RC, Shuto T, et al. 2012. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria-targeting cytoprotective peptide. J. Am. Heart Assoc. 1:e001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gibson CM, Giugliano RP, Kloner RA, Bode C, Tendera M, et al. 2016. EMBRACE STEMI study: a Phase 2a trial to evaluate the safety, tolerability, and efficacy of intravenous MTP-131 on reperfusion injury in patients undergoing primary percutaneous coronary intervention. Eur. Heart J. 37:1296–303 [DOI] [PubMed] [Google Scholar]
- 89.Orsini F, Moroni M, Contursi C, Yano M, Pelicci P, et al. 2006. Regulatory effects of the mitochondrial energetic status on mitochondrial p66Shc. Biol. Chem. 387:1405–10 [DOI] [PubMed] [Google Scholar]
- 90.Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, et al. 2005. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122:221–33 [DOI] [PubMed] [Google Scholar]
- 91.Yang M, Stowe DF, Udoh KB, Heisner JS, Camara AK. 2014. Reversible blockade of complex I or inhibition of PKCβ reduces activation and mitochondria translocation of p66Shc to preserve cardiac function after ischemia. PLOS ONE 9:e113534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lesnefsky EJ, Chen Q, Slabe TJ, Stoll MS, Minkler PE, et al. 2004. Ischemia, rather than reperfusion, inhibits respiration through cytochrome oxidase in the isolated, perfused rabbit heart: role of cardiolipin. Am. J. Physiol. Heart Circ. Physiol. 287:H258–67 [DOI] [PubMed] [Google Scholar]
- 93.Ambrosio G, Zweier JL, Duilio C, Kuppusamy P, Santoro G, et al. 1993. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J. Biol. Chem. 268:18532–41 [PubMed] [Google Scholar]
- 94.Han D, Antunes F, Canali R, Rettori D, Cadenas E. 2003. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 278:5557–63 [DOI] [PubMed] [Google Scholar]
- 95.Chen Q, Lesnefsky EJ. 2011. Blockade of electron transport during ischemia preserves bcl-2 and inhibits opening of the mitochondrial permeability transition pore. FEBS Lett. 585:921–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tanaka-Esposito C, Chen Q, Lesnefsky EJ. 2012. Blockade of electron transport before ischemia protects mitochondria and decreases myocardial injury during reperfusion in aged rat hearts. Transl. Res.160:207–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aldakkak M, Camara AK, Heisner JS, Yang M, Stowe DF. 2011. Ranolazine reduces Ca2+ overload and oxidative stress and improves mitochondrial integrity to protect against ischemia reperfusion injury in isolated hearts. Pharmacol. Res. 64:381–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brown GC, Borutaite V. 2007. Nitric oxide and mitochondrial respiration in the heart. Cardiovasc. Res. 75:283–90 [DOI] [PubMed] [Google Scholar]
- 99.Nadtochiy SM, Burwell LS, Brookes PS. 2007. Cardioprotection and mitochondrial S-nitrosation: effects of S-nitroso-2-mercaptopropionyl glycine (SNO-MPG) in cardiac ischemia-reperfusion injury. J. Mol. Cell. Cardiol. 42:812–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shiva S, Sack MN, Greer JJ, Duranski M, Ringwood LA, et al. 2007. Nitrite augments tolerance to ischemia/reperfusion injury via the modulation of mitochondrial electron transfer. J. Exp. Med. 204:2089–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xu A, Szczepanek K, Hu Y, Lesnefsky EJ, Chen Q. 2013. Cardioprotection by modulation of mitochondrial respiration during ischemia-reperfusion: role of apoptosis-inducing factor. Biochem. Biophys. Res. Commun. 435:627–33 [DOI] [PubMed] [Google Scholar]
- 102.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, et al. 2005. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434:658–62 [DOI] [PubMed] [Google Scholar]
- 103.Halestrap AP. 2009. What is the mitochondrial permeability transition pore? J. Mol. Cell. Cardiol. 46:821–31 [DOI] [PubMed] [Google Scholar]
- 104.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, et al. 2013. Dimers of mitochondrial ATP synthase form the permeability transition pore. PNAS 110:5887–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sammut IA, Burton K, Balogun E, Sarathchandra P, Brooks KJ, et al. 2000.Time-dependent impairment of mitochondrial function after storage and transplantation of rabbit kidneys. Transplantation 69:1265–75 [DOI] [PubMed] [Google Scholar]
- 106.Gateau-Roesch O, Argaud L, Ovize M. 2006. Mitochondrial permeability transition pore and postconditioning. Cardiovasc. Res. 70:264–73 [DOI] [PubMed] [Google Scholar]
- 107.Crompton M 1999. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 341:233–49 [PMC free article] [PubMed] [Google Scholar]
- 108.Cao CM, Yan WY, Liu J, Kam KW, Zhan SZ, et al. 2006. Attenuation of mitochondrial, but not cytosolic, Ca2+ overload reduces myocardial injury induced by ischemia and reperfusion. Acta Pharmacol. Sin. 27:911–18 [DOI] [PubMed] [Google Scholar]
- 109.Amberger A, Weiss H, Haller T, Kock G, Hermann M, et al. 2001. A subpopulation of mitochondria prevents cytosolic calcium overload in endothelial cells after cold ischemia/reperfusion. Transplantation 71:1821–27 [DOI] [PubMed] [Google Scholar]
- 110.An J, Varadarajan SG, Camara A, Chen Q, Novalija E, et al. 2001. Blocking Na+/H+ exchange reduces [Na+]i and [Ca2+]i load after ischemia and improves function in intact hearts. Am. J. Physiol. Heart Circ. Physiol. 281:H2398–409 [DOI] [PubMed] [Google Scholar]
- 111.An J, Rhodes SS, Jiang MT, Bosnjak ZJ, Tian M, Stowe DF. 2006. Anesthetic preconditioning enhances Ca2+ handling and mechanical and metabolic function elicited by Na+-Ca2+ exchange inhibition in isolated hearts. Anesthesiology 105:541–49 [DOI] [PubMed] [Google Scholar]
- 112.Halestrap AP, Connern CP, Griffiths EJ, Kerr PM. 1997. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol. Cell. Biochem. 174:167–72 [PubMed] [Google Scholar]
- 113.Piot C, Croisille P, Staat P, Thibault H, Rioufol G, et al. 2008. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 359:473–81 [DOI] [PubMed] [Google Scholar]
- 114.Stewart S, Lesnefsky EJ, Chen Q. 2009. Reversible blockade of electron transport with amobarbital at the onset of reperfusion attenuates cardiac injury. Transl. Res. 153:224–31 [DOI] [PubMed] [Google Scholar]
- 115.Paillard M, Gomez L, Augeul L, Loufouat J, Lesnefsky EJ, Ovize M. 2009. Postconditioning inhibits mPTP opening independent of oxidative phosphorylation and membrane potential. J. Mol. Cell. Cardiol. 46:902–9 [DOI] [PubMed] [Google Scholar]
- 116.Chen Q, Paillard M, Gomez L, Ross T, Hu Y, et al. 2011. Activation of mitochondrial μ-calpain increases AIF cleavage in cardiac mitochondria during ischemia–reperfusion. Biochem. Biophys. Res. Commun. 415:533–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, et al. 2002. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297:259–63 [DOI] [PubMed] [Google Scholar]
- 118.Szczepanek K, Lesnefsky EJ, Larner AC. 2012. Multi-tasking: nuclear transcription factors with novel roles in the mitochondria. Trends Cell Biol. 22:429–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Negoro S, Kunisada K, Fujio Y, Funamoto M, Darville MI, et al. 2001. Activation of signal transducer and activator of transcription 3 protects cardiomyocytes from hypoxia/reoxygenation-induced oxidative stress through the upregulation of manganese superoxide dismutase. Circulation 104:979–81 [DOI] [PubMed] [Google Scholar]
- 120.Boengler K, Hilfiker-Kleiner D, Drexler H, Heusch G, Schulz R. 2008. The myocardial JAK/STAT pathway: from protection to failure. Pharmacol. Ther. 120:172–85 [DOI] [PubMed] [Google Scholar]
- 121.Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, et al. 2011. Mitochondrial-targeted signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J. Biol. Chem. 286:29610–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R. 2010. Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res. Cardiol. 105:771–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wegrzyn J, Potla R, Chwae YJ, Sepuri NBV, Zhang Q, et al. 2009. Function of mitochondrial Stat3 in cellular respiration. Science 323:793–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Heusch G, Musiolik J, Gedik N, Skyschally A. 2011. Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ. Res. 109:1302–8 [DOI] [PubMed] [Google Scholar]
- 125.Szczepanek K, Xu A, Hu Y, Thompson J, He J, et al. 2015. Cardioprotective function of mitochondrial-targeted and transcriptionally inactive STAT3 against ischemia and reperfusion injury. Basic Res. Cardiol. 110:53. [DOI] [PubMed] [Google Scholar]
- 126.Smith CC, Dixon RA, Wynne AM, Theodorou L, Ong SG, et al. 2010. Leptin-induced cardioprotection involves JAK/STAT signaling that may be linked to the mitochondrial permeability transition pore. Am. J. Physiol. Heart Circ. Physiol. 299:H1265–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Beigel F, Friedrich M, Probst C, Sotlar K, Göke B, et al. 2014. Oncostatin M mediates STAT3-dependent intestinal epithelial restitution via increased cell proliferation, decreased apoptosis and upregulation of SERPIN family members. PLOS ONE 9:e93498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lecour S, Suleman N, Deuchar GA, Somers S, Lacerda L, et al. 2005. Pharmacological preconditioning with tumor necrosis factor-α activates signal transducer and activator of transcription-3 at reperfusion without involving classic prosurvival kinases (Akt and extracellular signal–regulated kinase). Circulation 112:3911–18 [DOI] [PubMed] [Google Scholar]
- 129.Bolli R 2007. Preconditioning: a paradigm shift in the biology of myocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 292:H19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Minamino T 2012. Cardioprotection from ischemia/reperfusion injury: basic and translational research. Circ. J. 76:1074–82 [DOI] [PubMed] [Google Scholar]
- 131.Miura T, Tanno M. 2012. The mPTP and its regulatory proteins: final common targets of signalling pathways for protection against necrosis. Cardiovasc. Res. 94:181–89 [DOI] [PubMed] [Google Scholar]
- 132.Peart JN, Headrick JP. 2009. Clinical cardioprotection and the value of conditioning responses. Am. J. Physiol. Heart Circ. Physiol. 296:H1705–20 [DOI] [PubMed] [Google Scholar]
- 133.Yang X, Cohen MV, Downey JM. 2010. Mechanism of cardioprotection by early ischemic preconditioning. Cardiovasc. Drugs Ther. 24:225–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Varadarajan SG, An J, Novalija E, Stowe DF. 2002. Sevoflurane before or after ischemia improves contractile and metabolic function while reducing myoplasmic Ca2+ loading in intact hearts. Anesthesiology 96:125–33 [DOI] [PubMed] [Google Scholar]
- 135.Obal D, Dettwiler S, Favoccia C, Scharbatke H, Preckel B, Schlack W. 2005.The influence of mitochondrial KATP-channels in the cardioprotection of preconditioning and postconditioning by sevoflurane in the rat in vivo. Anesth. Analg. 101:1252–60 [DOI] [PubMed] [Google Scholar]
- 136.Coetzee WA. 2013. Multiplicity of effectors of the cardioprotective agent, diazoxide. Pharmacol. Ther. 140:167–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lesnefsky EJ. 2002. The IONA study: preparing the myocardium for ischaemia? Lancet 359:1262–63 [DOI] [PubMed] [Google Scholar]
- 138.Heusch G 2013. Remote conditioning: the future of cardioprotection? J. Cardiovasc. Med. 14:176–79 [DOI] [PubMed] [Google Scholar]
- 139.Heusch G 2015. Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 116:674–99 [DOI] [PubMed] [Google Scholar]
- 140.Li J, Rohailla S, Gelber N, Rutka J, Sabah N, et al. 2014. MicroRNA-144 is a circulating effector of remote ischemic preconditioning. Basic Res. Cardiol. 109:423. [DOI] [PubMed] [Google Scholar]
- 141.Basalay M, Barsukevich V, Mastitskaya S, Mrochek A, Pernow J, et al. 2012. Remote ischaemic pre- and delayed postconditioning – similar degree of cardioprotection but distinct mechanisms. Exp. Physiol. 97:908–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhu SB, Liu Y, Zhu Y, Yin GL, Wang RP, et al. 2013. Remote preconditioning, perconditioning, and postconditioning: a comparative study of their cardio-protective properties in rat models. Clinics 68:263–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Andreka G, Vertesaljai M, Szantho G, Font G, Piroth Z, et al. 2007. Remote ischaemic postconditioning protects the heart during acute myocardial infarction in pigs. Heart 93:749–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tapuria N, Kumar Y, Habib MM, Abu Amara M, Seifalian AM, Davidson BR. 2008. Remote ischemic preconditioning: a novel protective method from ischemia reperfusion injury—a review. J. Surg. Res. 150:304–30 [DOI] [PubMed] [Google Scholar]
- 145.Vinten-Johansen J, Shi W. 2013. The science and clinical translation of remote postconditioning. J. Cardiovasc. Med. 14:206–13 [DOI] [PubMed] [Google Scholar]
- 146.Przyklenk K, Whittaker P. 2013. Genesis of remote conditioning: action at a distance – ‘hypotheses non fingo’? J. Cardiovasc. Med. 14:180–86 [DOI] [PubMed] [Google Scholar]
- 147.Mastitskaya S, Marina N, Gourine A, Gilbey MP, Spyer KM, et al. 2012. Cardioprotection evoked by remote ischaemic preconditioning is critically dependent on the activity of vagalpre-ganglionic neurones. Cardiovasc. Res. 95:487–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lim SY, Yellon DM, Hausenloy DJ. 2010. The neural and humoral pathways in remote limb ischemic preconditioning. Basic Res. Cardiol. 105:651–55 [DOI] [PubMed] [Google Scholar]
- 149.Pickard JM, Davidson SM, Hausenloy DJ, Yellon DM. 2016. Co-dependence of the neural and humoral pathways in the mechanism of remote ischemic conditioning. Basic Res. Cardiol. 111:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mastitskaya S, Basalay M, Hosford PS, Ramage AG, Gourine A, Gourine AV. 2016. Identifying the source of a humoral factor of remote (pre)conditioning cardioprotection. PLOS ONE 11:e0150108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wang L, Oka N, Tropak M, Callahan J, Lee J, et al. 2008. Remote ischemic preconditioning elaborates a transferable blood-borne effector that protects mitochondrial structure and function and preserves myocardial performance after neonatal cardioplegic arrest. J. Thorac. Cardiovasc. Surg. 136:335–42 [DOI] [PubMed] [Google Scholar]
- 152.Rahman IA, Mascaro JG, Steeds RP, Frenneaux MP, Nightingale P, et al. 2010. Remote ischemic preconditioning in human coronary artery bypass surgery: from promise to disappointment? Circulation 122:S53–59 [DOI] [PubMed] [Google Scholar]
- 153.Karuppasamy P, Chaubey S, Dew T, Musto R, Sherwood R, et al. 2011. Remote intermittent ischemia before coronary artery bypass graft surgery: a strategy to reduce injury and inflammation? Basic Res. Cardiol. 106:511–19 [DOI] [PubMed] [Google Scholar]
- 154.Young PJ, Dalley P, Garden A, Horrocks C, La Flamme A, et al. 2012. A pilot study investigating the effects of remote ischemic preconditioning in high-risk cardiac surgery using a randomised controlled double-blind protocol. Basic Res. Cardiol. 107:256. [DOI] [PubMed] [Google Scholar]
- 155.Hausenloy DJ, Candilio L, Laing C, Kunst G, Pepper J, et al. 2011. Effect of remote ischemic preconditioning on clinical outcomes in patients undergoing coronary artery bypass graft surgery (ERICCA): rationale and study design of a multi-centre randomized double-blinded controlled clinical trial. Clin. Res. Cardiol. 101:339–48 [DOI] [PubMed] [Google Scholar]
- 156.Meybohm P, Zacharowski K, Cremer J, Roesner J, Kletzin F, et al. 2012. Remote ischaemic preconditioning for heart surgery. The study design for a multi-center randomized double-blinded controlled clinical trial—the RIPHeart-Study. Eur. Heart J. 33:1423–26 [PubMed] [Google Scholar]
- 157.Botker HE, Kharbanda R, Schmidt MR, Bottcher M, Kaltoft AK, et al. 2010. Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: a randomised trial. Lancet 375:727–34 [DOI] [PubMed] [Google Scholar]
- 158.Rentoukas I, Giannopoulos G, Kaoukis A, Kossyvakis C, Raisakis K, et al. 2010. Cardioprotective role of remote ischemic periconditioning in primary percutaneous coronary intervention: enhancement by opioid action. JACC Cardiovasc. Interv. 3:49–55 [DOI] [PubMed] [Google Scholar]
- 159.Goll DE, Thompson VF, Li H, Wei W, Cong J. 2003. The calpain system. Physiol. Rev. 83:731–801 [DOI] [PubMed] [Google Scholar]
- 160.Thompson VF, Lawson K, Goll DE. 2000. Effect of μ-calpain on m-calpain. Biochem. Biophys. Res. Commun. 267:495–99 [DOI] [PubMed] [Google Scholar]
- 161.Vosler PS, Brennan CS, Chen J. 2008. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol. Neurobiol. 38:78–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ozaki T, Tomita H, Tamai M, Ishiguro S. 2007. Characteristics of mitochondrial calpains. J. Biochem. 142:365–76 [DOI] [PubMed] [Google Scholar]
- 163.Joshi A, Bondada V, Geddes JW. 2009. Mitochondrial μ-calpain is not involved in the processing of apoptosis-inducing factor. Exp. Neurol. 218:221–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Arrington DD, Van Vleet TR, Schnellmann RG. 2006. Calpain 10: a mitochondrial calpain and its role in calcium-induced mitochondrial dysfunction. Am. J. Physiol. Cell Physiol. 291:C1159–71 [DOI] [PubMed] [Google Scholar]
- 165.Chen M, He H, Zhan S, Krajewski S, Reed JC, Gottlieb RA. 2001. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J. Biol. Chem. 276:30724–28 [DOI] [PubMed] [Google Scholar]
- 166.Randriamboavonjy V, Pistrosch F, Bolck B, Schwinger RH, Dixit M, et al. 2008. Platelet sarcoplasmic endoplasmic reticulum Ca2+-ATPase and μ-calpain activity are altered in type 2 diabetes mellitus and restored by rosiglitazone. Circulation 117:52–60 [DOI] [PubMed] [Google Scholar]
- 167.Ong SB, Hall AR, Hausenloy DJ. 2013. Mitochondrial dynamics in cardiovascular health and disease. Antioxid. Redox Signal. 19:400–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Marin-Garcia J, Akhmedov AT, Moe GW. 2013. Mitochondria in heart failure: the emerging role of mitochondrial dynamics. Heart Fail. Rev. 18:439–56 [DOI] [PubMed] [Google Scholar]
- 169.Vásquez-Trincado C, García-Carvajal I, Pennanen C, Parra V, Hill JA, et al. 2016. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 594:509–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.de Brito OM, Scorrano L. 2009. Mitofusin-2 regulates mitochondrial and endoplasmic reticulum morphology and tethering: the role of Ras. Mitochondrion 9:222–26 [DOI] [PubMed] [Google Scholar]
- 171.Piquereau J, Caffin F, Novotova M, Lemaire C, Veksler V, et al. 2013. Mitochondrial dynamics in the adult cardiomyocytes: which roles for a highly specialized cell? Front. Physiol. 4:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hwang SJ, Kim W. 2014. Mitochondrial dynamics in the heart as a novel therapeutic target for cardioprotection. Chonnam Med. J. 49:101–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zepeda R, Kuzmicic J, Parra V, Troncoso R, Pennanen C, et al. 2014. Drp1 loss-of-function reduces cardiomyocyte oxygen dependence protecting the heart from ischemia-reperfusion injury. J. Cardiovasc. Pharmacol. 63:477–87 [DOI] [PubMed] [Google Scholar]
- 174.Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. 2012. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 121:2012–22 [DOI] [PubMed] [Google Scholar]
- 175.Pride CK, Mo L, Quesnelle K, Dagda RK, Murillo D, et al. 2014. Nitrite activates protein kinase A in normoxia to mediate mitochondrial fusion and tolerance to ischaemia/reperfusion. Cardiovasc. Res. 101:57–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Long B, Wang K, Li N, Murtaza I, Xiao JY, et al. 2013. miR-761 regulates the mitochondrial network by targeting mitochondrial fission factor. Free Radic. Biol. Med. 65:371–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hill JA. 2011. Autophagy in cardiac plasticity and disease. Pediatr. Cardiol. 32:282–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Youle RJ, van der Bliek AM. 2012. Mitochondrial fission, fusion, and stress. Science 337:1062–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, et al. 2007. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 100:914–22 [DOI] [PubMed] [Google Scholar]
- 180.Narendra DP, Youle RJ. 2011. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid. Redox Signal. 14:1929–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, et al. 2010. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell. Biol. 189:211–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, et al. 2012. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 13:378–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. 2004. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell. Biol. 24:8055–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, et al. 2007. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282:24131–45 [DOI] [PubMed] [Google Scholar]
- 185.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, et al. 2013. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J. Biol. Chem. 288:915–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gottlieb RA, Mentzer RM Jr., Linton PJ. 2011. Impaired mitophagy at the heart of injury. Autophagy 7:1573–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zhang J, Nadtochiy SM, Urciuoli WR, Brookes PS. 2016. The cardioprotective compound cloxyquin uncouples mitochondria and induces autophagy. Am. J. Physiol. Heart Circ. Physiol. 310:H29–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Chipuk JE, Bouchier-Hayes L, Green DR. 2006. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 13:1396–402 [DOI] [PubMed] [Google Scholar]
- 189.Hausenloy DJ, Yellon DM. 2007. Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart Fail. Rev. 12:217–34 [DOI] [PubMed] [Google Scholar]
- 190.Hausenloy DJ, Yellon DM. 2003. The mitochondrial permeability transition pore: its fundamental role in mediating cell death during ischaemia and reperfusion. J. Mol. Cell. Cardiol. 35:339–41 [DOI] [PubMed] [Google Scholar]
- 191.Chiong M, Wang ZV, Pedrozo Z, Cao DJ, Troncoso R, et al. 2011. Cardiomyocyte death: mechanisms and translational implications. Cell Death Dis. 2:e244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ong SG, Hausenloy DJ. 2012. Hypoxia-inducible factor as a therapeutic target for cardioprotection. Pharmacol. Ther. 136:69–81 [DOI] [PubMed] [Google Scholar]
- 193.Wong R, Aponte AM, Steenbergen C, Murphy E. 2010. Cardioprotection leads to novel changes in the mitochondrial proteome. Am. J. Physiol. Heart Circ. Physiol. 298:H75–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Murphy E, Steenbergen C. 2011. What makes the mitochondria a killer? Can we condition them to be less destructive? Biochim. Biophys. Acta 1813:1302–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Gustafsson AB, Gottlieb RA. 2009. Autophagy in ischemic heart disease. Circ. Res. 104:150–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Dorn GWII Vega RB, Kelly DP. 2015. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. 29:1981–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Whitaker RM, Corum D, Beeson CC, Schnellmann RG. 2016. Mitochondrial biogenesis as a pharmacological target: a new approach to acute and chronic diseases. Annu. Rev. Pharmacol. Toxicol. 56:229–49 [DOI] [PubMed] [Google Scholar]
- 198.Lesnefsky EJ, He D, Moghaddas S, Hoppel CL. 2006. Reversal of mitochondrial defects before ischemia protects the aged heart. FASEB J. 20:1543–45 [DOI] [PubMed] [Google Scholar]
- 199.Gallogly MM, Shelton MD, Qanungo S, Pai HV, Starke DW, et al. 2010. Glutaredoxin regulates apoptosis in cardiomyocytes via NFκB targets Bcl-2 and Bcl-xL: implications for cardiac aging. Antioxid. Redox Signal. 12:1339–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Rousou AJ, Ericsson M, Federman M, Levitsky S, McCully JD. 2004. Opening of mitochondrial KATP channels enhances cardioprotection through the modulation of mitochondrial matrix volume, calcium accumulation, and respiration. Am. J. Physiol. Heart Circ. Physiol. 287:H1967–76 [DOI] [PubMed] [Google Scholar]
- 201.Riess ML, Eells JT, Kevin LG, Camara AK, Henry MM, Stowe DF. 2004. Attenuation of mitochondrial respiration by sevoflurane in isolated cardiac mitochondria is mediated in part by reactive oxygen species. Anesthesiology 100:498–505 [DOI] [PubMed] [Google Scholar]
- 202.Kagan VE, Bayir HA, Belikova NA, Kapralov O, Tyurina YY, et al. 2009. Cytochrome c/cardiolipin relations in mitochondria: a kiss of death. Free Radic. Biol. Med. 46:1439–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kagan VE, Borisenko GG, Tyurina YY, Tyurin VA, Jiang J, et al. 2004. Oxidative lipidomics of apoptosis: redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic. Biol. Med. 37:1963–85 [DOI] [PubMed] [Google Scholar]
- 204.Kagan VE, Tyurina YY, Bayir H, Chu CT, Kapralov AA, et al. 2006. The “pro-apoptotic genies” get out of mitochondria: oxidative lipidomics and redox activity of cytochrome c/cardiolipin complexes. Chem. Biol. Interact. 163:15–28 [DOI] [PubMed] [Google Scholar]
- 205.Murry CE, Jennings RB, Reimer KA. 1986. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 74:1124–36 [DOI] [PubMed] [Google Scholar]
- 206.Thompson J, Hu Y, Lesnefsky EJ, Chen Q. 2015. Activation of mitochondrial calpain and increased cardiac injury: beyond AIF release. Am. J. Physiol. Heart Circ. Physiol. 310:H376–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, et al. 2003. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 285:H579–88 [DOI] [PubMed] [Google Scholar]
- 208.Penna C, Perrelli MG, Tullio F, Angotti C, Camporeale A, et al. 2013. Diazoxide postconditioning induces mitochondrial protein S-nitrosylation and a redox-sensitive mitochondrial phosphorylation/translocation of RISK elements: no role for SAFE. Basic Res. Cardiol. 108:371. [DOI] [PubMed] [Google Scholar]
- 209.Luan HF, Zhao ZB, Zhao QH, Zhu P, Xiu MY, Ji Y. 2012. Hydrogen sulfide postconditioning protects isolated rat hearts against ischemia and reperfusion injury mediated by the JAK2/STAT3 survival pathway. Braz. J. Med. Biol. Res. 45:898–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kelso GF, Porteous CM, Hughes G, Ledgerwood EC, Gane AM, et al. 2002. Prevention of mitochondrial oxidative damage using targeted antioxidants. Ann. N. Y. Acad. Sci. 959:263–74 [DOI] [PubMed] [Google Scholar]