

Abstract
Proper regulation of the immune system is required for protection against pathogens and preventing autoimmune disorders. Inborn errors of the immune system due to inherited or de novo germline mutations can lead to the loss of protective immunity, aberrant immune homeostasis, and the development of autoimmune disease, or combinations of these. Forward genetic screens involving clinical material from patients with primary immunodeficiencies (PIDs) can vary in severity from life-threatening disease affecting multiple cell types and organs to relatively mild disease with susceptibility to a limited range of pathogens or mild autoimmune conditions. As central mediators of innate and adaptive immune responses, T cells are critical orchestrators and effectors of the immune response. As such, several PIDs result from loss of or altered T cell function. PID-associated functional defects range from complete absence of T cell development to uncontrolled effector cell activation. Furthermore, the gene products of known PID causal genes are involved in diverse molecular pathways ranging from T cell receptor signaling to regulators of protein glycosylation. Identification of the molecular and biochemical cause of PIDs can not only guide the course of treatment for patients, but also inform our understanding of the basic biology behind T cell function. In this chapter, we review PIDs with known genetic causes that intrinsically affect T cell function with particular focus on perturbations of biochemical pathways.
1. INTRODUCTION
This theme has been at the centre of all my research…, both because of its intrinsic fascination and my conviction that a knowledge of sequences could contribute much to our understanding of living matter.
Frederick Sanger
T lymphocyte cells (T cells) play a central role in the adaptive immune response, coordinating immune functions comprising both humoral and cell-mediated responses to a myriad of immunogenic challenges including infection and cancer. Therefore, understanding how these cells work in humans requires a detailed molecular approach combining cellular immunology, clinical investigation, and human genetics. Normally, mechanisms of central and peripheral tolerance limit the response to self, thus preventing autoimmune disease. These varied and at times conflicting roles require many levels of control to appropriately modulate T cell-mediated immune responses. These include regulation of T cell development, maintenance of quiescence and self-tolerance, initiation and maintenance of T cell activation in response to cognate antigen, migration to effector sites, effector function, differentiation, and maintenance of a memory population. Multiple genes regulate these steps in ways critical to proper immune responses. One method of identifying important regulators of T cell function is a genetic approach involving the investigation of individuals with inborn errors of immunity. These primary immunodeficiencies (PIDs) result in diverse and overlapping clinical phenotypes including immunodeficiency (susceptibility to malignancy or infection), abnormal cellular homeostasis, autoimmunity, autoinflammation, and allergy. PIDs represent a forward genetic screen of nature, meaning that a phenotype is first identified—often provoked by infectious and noninfectious immunological challenges that the patients encounter—and then the pathophysiological attributes in immunity and ultimately gene variants that correlate with disease are interrogated. Within the last decade, the development of inexpensive technologies for sequencing the DNA of the human genome or exome (the coding portion of the genome) have allowed for the rapid identification of the genetic variants underlying various PIDs. This coupled with the vast reach of clinical medicine to identify, characterize, and follow longitudinally patient phenotypes has facilitated a powerful new approach to understand the effect of genotype variation on human immune function. In this chapter, we will discuss the biochemical and molecular basis of disease in PIDs caused in whole or in part by defects in T cell function.
2. PIDs
PIDs are most easily understood when they are caused by highly penetrant single-gene errors. Clinical manifestations of PIDs usually present in childhood and can be due to either de novo or hereditary mutations with various Mendelian modes of inheritance (MOIs). Affected genes can be specifically required for a certain immune response, formation of a single immunological cell type, or may more broadly affect a common cellular process necessary for proper immunological function. As such, PIDs can present with an extremely narrow clinical presentation, such as susceptibility to a single class of pathogen, or with wide-ranging clinical immune phenotypes with syndromic (nonimmune) features. The variants that cause PIDs are generally highly deleterious to the function of the encoded protein and, as such, are relatively rare (<0.1%) within a given population compared to less deleterious variants. This contrasts with the relatively common variants (≥0.1%) that contribute to the susceptibility to more common immunological diseases such as type 1 diabetes mellitus (T1DM) and inflammatory bowel disease (IBD). Though the single-gene defects giving rise to PIDs tend to be severe, there can be significant differences in penetrance (the proportion of individuals with a disease genotype having the corresponding clinical phenotype) and expressivity (the constellation and severity of clinical symptoms). The International Union of Immunological Societies Expert Committee for Primary Immunodeficiencies keeps a continuously updated list of known PID genes, which, as of writing this review, has a total of 354 immunological diseases with a known genetic cause, accounting for nearly 10% of all single gene disorders that have been defined (http://www.iuisonline.org).
Despite the relative rarity of PIDs compared to other immunological disorders, there are a multitude of benefits that can be gained by studying the underlying genetic and biochemical mechanism of PID pathogenesis. First, it provides an opportunity to gain novel insight into regulation of the human immune system. Second, it can offer great benefit to the patients who carry the burden of these disorders. Identification of the causal variant, MOI, and mechanism of action may provide clinicians with guidance in selecting a targeted therapy and allows for informed genetic counseling to the family with critical information relevant to life decisions. Third, we also gain scientific insights into the molecular pathways that mediate infectious and autoimmune disease at the population level. Fourth, by genetically “solving” PIDs, new immune genes have been discovered, new functions of known genes have been elucidated, and knowing how genes affect human immunology enables us to better annotate the human genome (Casanova, Abel, & Quintana-Murci, 2013). Finally, a key benefit of studying PIDs is understanding the variability of the causal genetic lesions generated by random mutation in the human population which cannot be derived from experimental animal models. These experiments of nature can lead to similar clinical disease caused by completely different genes and demonstrate that the same gene can cause various clinical phenotypes dependent on the type of mutation. This can allow for identification of gene functions in the biological context of both loss-of-function (LOF) and gain-of-function (GOF) mutations, thus taking the study of a gene well past where the usefulness of studying traditional knockout mice ends. Additionally, the constant exposure of humans to environmental influences and a multitude of infectious agents can help identify gene function that would not be immediately apparent in simple inbred mouse models that are housed under specific pathogen-free conditions.
Pathogenic DNA variants, excluding copy number variation or complete deletion of the genetic locus, can be broken down into three categories: LOF, hypomorphic mutations (partial LOF), and GOF. GOF mutations can be further divided into two types: increased activity of a traditional function and gain of a novel function. Fitting into one of these operational categories are multiple types of DNA variants that include coding variants such as nonsense, frameshift, canonical splice site mutations, loss of the initiation codon, larger insertions or deletions, and missense mutations, as well as noncoding variants in promoter or enhancer regions, that may alter the transcription of the gene, or intronic variants that may introduce a new splice site. Further complicating the genetic analysis of PID patients is that a given pathogenic genetic variant can cause either a dominant or recessive manifestation of disease, in which one or two alleles must be affected, respectively. Furthermore, different PIDs with distinct MOI may arise from mutations in the same gene. Examples of this will be discussed on a case-by-case basis in the following section. A recessive MOI is generally caused by LOF variants while a dominant MOI can be caused by LOF variants in which one allele is insufficient or actively interferes with the function of the other allele (haploinsufficiency or dominant interference, respectively) or by GOF variants. Undoubtedly, PIDs may be caused by the interaction of multiple genes or by alterations of the intergenic regions; however, the tools are still being developed to address those possibilities.
PID patients are typically characterized by their clinical and cellular phenotypic presentation prior to any knowledge of their genotype, and this information can often influence the prioritization of potential causal variants. For example, variants may affect known PID genes or lie in genes that have already been associated with a specific biochemical pathway, a particular cell type, or a given clinical presentation (Bousfiha et al., 2015; Picard et al., 2015). Currently, there are eight categories of PIDs based upon phenotypic classification: immunodeficiencies affecting cellular and humoral immunity, combined or severe combined immune deficiency (CID/SCID) with or without associated/syndromic features, antibody deficiencies, diseases of immune dysregulation, defects in phagocyte number/function, defects in innate immunity, autoinflammatory disorders, and complement deficiencies. Additionally, there are intriguing somatic mutation-based disorders or autoantibody-induced conditions that mimic PIDs that will not be discussed here. Because of the pivotal role of T cells in innate and adaptive immune responses, we will focus in this review on a large variety of PIDs that result from loss of, or altered T cell function and developmental T cell defects that range from complete absence of T cells to uncontrolled effector cell activation.
3. PIDs OF T CELL FUNCTION
In this section, we will discuss T cell-intrinsic PIDs, characterizing these mutations by the molecular mechanism that is disrupted in each PID rather than by the associated clinical or cellular phenotype. PIDs affecting T cell function that are caused by alterations in antigen-presentation cells (APCs) or other external determinants of T cell development/function will not be discussed and some have been reviewed elsewhere (Conley et al., 2009). Importantly, several proteins have roles in multiple pathways and may be classified in one or more of the functional categories we define. For simplicity, we will only mention a given protein under a single category, but will discuss noted areas of overlap with other categories when present and OMIM disease identifiers will be provided, where available.
3.1. Defects in T Cell Development
3.1.1. Thymic Development
Deleterious gene variants that alter thymus formation can affect T cell development. Of the known genes associated with thymic development, four cause a recognized PID. Haploinsufficiency of the T-box transcription factor 1 (TBX1), caused by a 1.5–3.0 Mb deletion at chromosome 22q11.2, causes DiGeorge syndrome (OMIM 188400), which involves thymic hypoplasia and T cell deficiencies ranging from relatively mild to a SCID-like phenotype in athymic individuals (Davies, 2013; Jerome & Papaioannou, 2001; Lischner & Huff, 1975; Yagi et al., 2003). Mutations in the sema domain, immunoglobulin domain (Ig), short basic domain, secreted (semaphorin) 3E protein (SEMA3E) are required for proper angiogenesis. Chromodomain-helicase-DNA-binding protein 7 (CHD7), the chromatin remodeling protein and likely transcription factor (TF), has an essential role in tissue patterning. Mutations in either SEMA3E or CHD7 cause autosomal dominant CHARGE syndrome and are associated with thymic hypoplasia or aplasia and a variety of associated T cell defects (OMIM 214800) (Bajpai et al., 2010; Gu et al., 2005; Jongmans et al., 2006; Lalani et al., 2004; Martin, Sheldon, & Gorski, 2001; Van Nostrand et al., 2014; Wong, Scholvinck, Lambeck, & van Ravenswaaij-Arts, 2015). Mutations in the master regulator of thymic epithelial cell formation, Forkhead Box N1 (FOXN1), cause an autosomal recessive SCID disorder mimicking that of Nude mice with a severe T cell immunodeficiency due to abrogated thymus formation (OMIM 601705) (Frank et al., 1999; Pignata et al., 1996; Romano et al., 2013; Rota & Dhalla, 2017). In these cases of athymia, thymic transplant can correct the immune defect, since there is no underlying intrinsic defect in T cell development (Markert et al., 2007, 2011).
3.1.2. VDJ Recombination
During development of T and B cells, the germline locus of the T cell antigen receptor (TCR) and B cell antigen receptor (BCR) undergo genetic recombination of the V, D, and J loci to form unique protein-coding genes for each of the receptor chains. Since only one or two of these alleles are completed in each cell, this generates a diverse repertoire of clonal lymphocytes harboring different antigen receptor specificities needed for broad immune protection. As such, severe defects in genes required for VDJ recombination lead to a T−/B−/natural killer (NK)+ SCID. Hypomorphic mutations in several of these genes lead to defects in selection, but allow some B and T cells to develop, but with reduced or autoreactive repertoires, forming the basis for Omenn syndrome and combined cellular and humoral immune defects with hranulomas (CCHIDG). The exact course of disease depends on the severity of the mutation, and, in the case of hypomorphic mutations that are incompletely penetrant, other factors such as genetic background and infectious history. Biallelic LOF and hypomorphic mutations in both recombination activating gene (RAG) 1 and 2, which together form the catalytic core of the RAG DNA cleavage complex, have been described to cause all three diseases: SCID (OMIM 601457), Omenn (OMIM 603554), and CCHIDG (OMIM 233650) (Schuetz et al., 2008; Schwarz et al., 1996; Villa et al., 1998). Following DNA cleavage, the Artemis/DNA-dependent protein kinase, catalytic subunit (DNA-PKcs) complex forms a key 5′−3′ exonuclease that is required for nonhomologous end-joining (NHEJ)-mediated DNA repair. This exonuclease activity is important for the removal of DNA hairpins created by the RAG complex (Ma, Pannicke, Schwarz, & Lieber, 2002; Ma, Schwarz, & Lieber, 2005). Mutations in Artemis and DNA-PKcs cause SCID with increased sensitivity to non-RAG-initiated double-strand breaks, such as those caused by radiation (OMIM 602450 and 615966, respectively) (Moshous et al., 2001; van der Burg et al., 2009). Artemis mutations can also cause Omenn syndrome (Ege et al., 2005). During recombination, after exonuclease activity, the double-strand break must be repaired to link VDJ segments to create open reading frames for immunoreceptor proteins. NHEJ factor 1 (NHEJ1), which was first identified via genetic screening of SCID patients, and DNA ligase IV forms a complex necessary for NHEJ following RAG-mediated double-strand breaks (Ahnesorg, Smith, & Jackson, 2006; Grawunder, Zimmer, Fugmann, Schwarz, & Lieber, 1998). Mutations in both proteins cause radiosensitive SCID with syndromic features (OMIM 611291 and 606593, respectively) (Buck et al., 2006; van der Burg et al., 2006). Thus, mutations in various proteins involved in the cutting, processing, and rejoining of double-strand breaks during T and B cell development give rise to an overlapping family of disorders. One interesting question that remains is why there is such a wide range of clinical phenotypes associated with RAG mutations; one might infer that these proteins have multiple roles in the metabolism of the genome or other cellular functions (Notarangelo, Kim, Walter, & Lee, 2016). Complete LOF mutations in RAG are simpler to understand, as these result in complete lack of T and B cell development, resulting in loss of adaptive immunity. Disease caused by hypomorphic mutations are more complex, because these variants result in decreased T and B cell selection as well as lead to the loss of regulatory cell populations and expansion of autoreactive cell clones in a lymphopenic environment. This likely accounts for the diverse autoimmune phenotypes associated with Omenn syndrome. Meanwhile, less severe hypomorphic mutations can lead to mild defects in T and B cell selection allowing a diverse repertoire, lymphadenopathy, and milder immunodeficiencies developed later in life, such as CCHIDG.
3.2. Mutations Affecting TCR Signaling
Several PIDs have been associated with deleterious variants leading to aberrations of proximal TCR-mediated signaling (Fig. 1) (Notarangelo, 2013). Importantly, many of these mutations lead to general loss of the T cell compartment and consequent deficits in immunity in affected individuals, but can also lead to autoimmune conditions through loss of Tregs, altered thymic selection leading to formation of autoreactive T cells, or altered/enhanced TCR signaling that cause aberrant T cell activation, effector function, and memory formation.
Fig. 1.
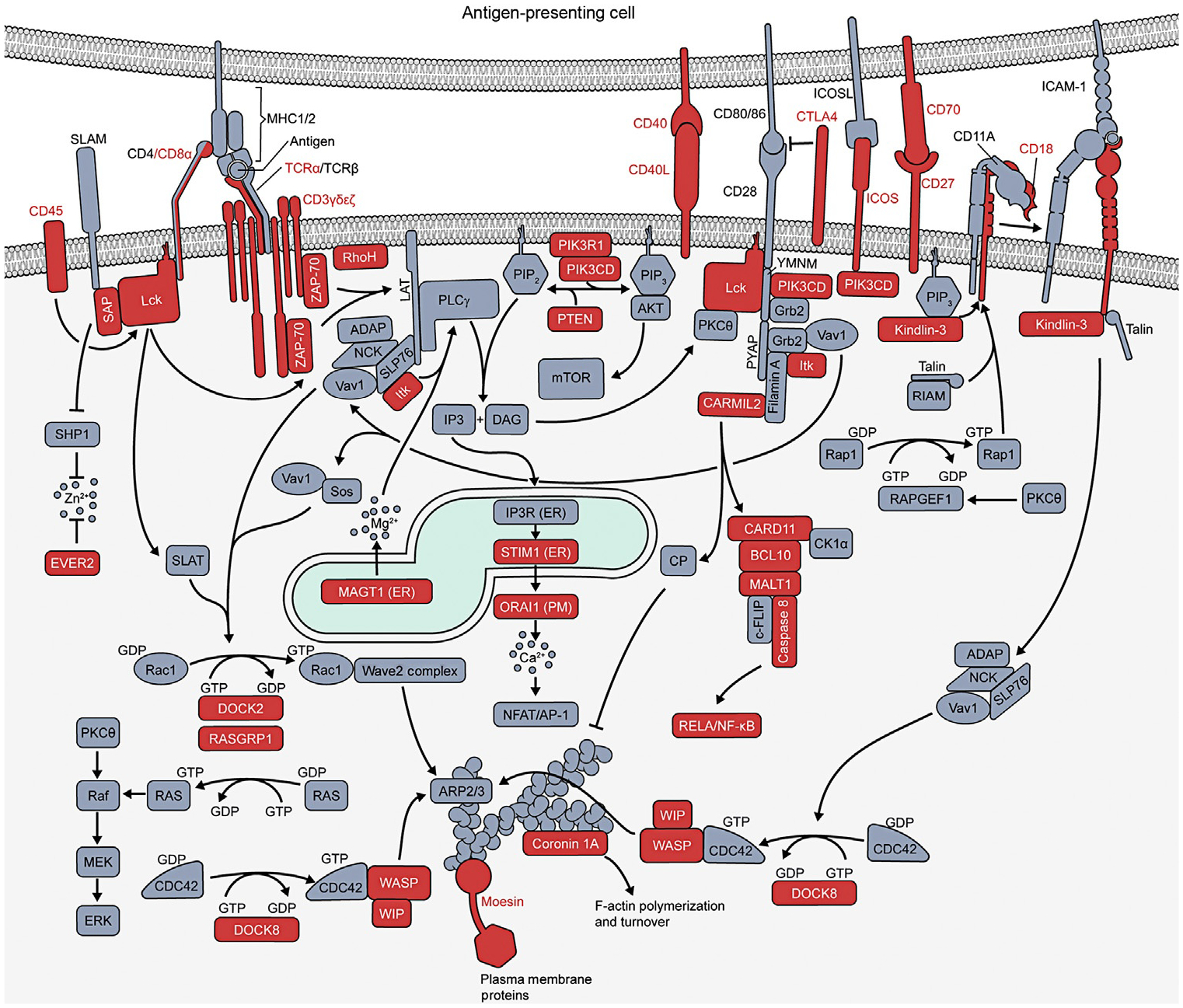
Primary immune deficiencies in T cell signaling and actin regulatory pathways. Schematic of signaling downstream of the T cell receptor, costimulatory, and adhesion molecules. Mutations in molecules shaded in red are known to cause a primary immune disease.
3.2.1. TCR Chains
T cells recognize antigen through binding of the TCR to antigenic peptides presented by major histocompatibility complex (MHC) molecules on APCs. The TCR is a multimeric complex consisting of the peptide–MHC binding chains TCRα and TCRβ associated with signaling chains comprising cluster of differentiation (CD)3ε, CD3δ, CD3γ, and CD3ζ in a 1:1:2:1:1:2 ratio. Deleterious variants affecting each of these proteins, except TCRβ, have been associated with autosomal recessive SCID and autoimmune disease.
Homozygous LOF TCRα mutations cause loss of TCRαβ-expressing T cells and features of both autoimmunity and immunodeficiency (OMIM 615387). Patients, mostly children, are susceptible to recurrent respiratory tract infections, varicella and Epstein–Barr virus (EBV) infections, otitis media, candidiasis, diarrhea, and failure to thrive and can variably develop hypereosinophila, autoantibodies, eczema, vitiligo, autoimmune hemolytic anemia, lymphadenopathy, and organomegally (Morgan et al., 2011). Loss of either CD3ε (OMIM 615615) or CD3δ (OMIM 615617) results in the selective loss of T cells, and patients present with diminished class-switched immunoglobulins, recurrent respiratory and ear infections, along with recurrent gasteroenteritis (Dadi, Simon, & Roifman, 2003; Dave et al., 1997; de Saint Basile et al., 2004; Gil et al., 2011; Soudais, de Villartay, Le Deist, Fischer, & Lisowska-Grospierre, 1993). Loss of CD3ζ (OMIM 610163) results in erythroderma, diarrhea, pulmonary infections with susceptibility to pseudomonas, herpes simplex virus (HSV), and candida infections of the mouth and skin (Rieux-Laucat et al., 2006). Finally, loss of CD3γ (OMIM 615607) can result in combined immunodeficiencies and autoimmunity including susceptibility to viral, bacterial, and fungal infections, low immunoglobulin G (IgG) levels, autoantibodies, autoimmune hemolytic anemia, and enterocolitis (Arnaiz-Villena et al., 1992; Recio et al., 2007). As noted earlier, there is considerable variation in cellular phenotypes depending on the affected CD3 chain as well as variable expressivity within a given genetic defect. For instance, some mutations in CD3δ result in complete loss of T cells while others leave γδ+ T cell development intact. This may be due to the degree that different mutations affect CD3δ association with either αβ or γδ chains. Additionally, in multiplex kindreds with identical CD3γ mutations, different mutation-bearing patients can develop either mild or severe disease, likely indicating the presence of modifying genetic variants or the influence of infectious history. This degree of intrakindred variable expressivity is also reminiscent of other disorders such as hypomorphic RAG mutations discussed earlier and could be influenced by the individual T cell population that is selected in the thymus. In general, CD3δ mutations are more deleterious than CD3γ mutations (Dadi et al., 2003; Dave et al., 1997; Recio et al., 2007). The differences possibly reflect varying signaling or complex formation requirements for individual chains and may be predisposed by the severity of the causal variant, with point mutants that disrupt recruitment of downstream signaling molecules potentially being less deleterious than complete protein deficiency.
3.2.2. CD8α
An autosomal recessive disease of recurrent bacterial and viral lung infections is caused by biallelic deleterious variants in CD8α, encoding the CD8α chain of the coreceptor for TCR interaction with class I MHC (OMIM 608957). The CD8 and CD4 coreceptors are important for TCR signaling by binding the constant portion of the MHCI and MHCII molecules, respectively, and recruiting the lymphocyte-specific protein tyrosine kinase (Lck) to the TCR complex where it initiates downstream signaling by phosphorylating CD3 chains and downstream kinases (Fig. 1). Interestingly, CD8α deficiency may have incomplete penetrance. For example, two healthy siblings of one patient have been identified as being homozygous for the same disease allele (de la Calle-Martin et al., 2001; Mancebo et al., 2008). The reason for this remains to be determined.
3.2.3. CD45
An autosomal recessive Tlow/NK+/B+ SCID phenotype (OMIM 608971) is caused by biallelic LOF mutations in the gene encoding the protein phosphatase CD45. Patients exhibit a combination of rash, fever, hepatosplenomegaly, diffuse lymphadenopathy, pneumonitis, pancytopenia, B cell lymphoma, and neonatal cytomegalovirus (CMV) infections. Patient immunophenotyping reveals very low αβ-positive T cells with normal or elevated NK cells and normal or elevated B cells with defective germinal center formation and low Ig levels. The diffuse lymphadenopathy was B cell dependent. Importantly, patient T cells failed to respond to mitogens (Kung et al., 2000; Roberts et al., 2012; Tchilian et al., 2001). Interestingly, cd45−/− mice also show arrested T cell development at the transition from double positive to single positive cells, phenocopying Lck-deficient mice. This appears to be a gene dosage effect, because heterozygous mice have a moderate decrease in single positive T cells compared to controls (Kishihara et al., 1993). Further analysis revealed that in vitro B cell activation in response to surface Ig-crosslinking to be defective in knockout cells as well as an accumulation of pro-B cells, suggesting a B cell-intrinsic role for CD45 (Fleming, Milne, & Paige, 2004). These data suggest a possible unappreciated B cell defect in CD45-deficient patients, though this has not been previously described. In addition to the autosomal recessive disease, heterozygous CD45 splice site mutations are associated with the development of familial multiple sclerosis, though this association needs further investigation, and heterozygous LOF mutations occur with less than expected frequency, possibly indicating an unrealized clinical condition associated with CD45 haploinsufficiency (ExAC) (Jacobsen et al., 2000).
CD45 is a protein phosphatase known to positively regulate TCR signaling by removing an inhibitory phosphate at Y505 on Lck (Burns, Sakaguchi, Appella, & Ashwell, 1994; Koretzky, Kohmetscher, & Ross, 1993). It should be noted that CD45 can also remove activating phosphates both within Lck and the TCR, and Lck isolated from cd45−/− thymocytes show increased kinase activity, indicating that the exact consequences of CD45 deficiency on signaling dynamics are complicated and not fully understood (D’Oro & Ashwell, 1999; Furlan, Minowa, Hanagata, Kataoka-Hamai, & Kaizuka, 2014). CD45 also promotes NK cell cytokine production and the inhibition of JAK/STAT signaling (Hesslein, Takaki, Hermiston, Weiss, & Lanier, 2006; Irie-Sasaki et al., 2001). Interestingly, decreased CD45 expression and CD45 mutations are associated with T and B acute lymphoblastic leukemia where its absence contributes to increased JAK/STAT signaling with the consequence of sensitivity of the transformed cells to JAK inhibitors (Nakamura et al., 2001; Porcu et al., 2012; Raponi et al., 2015). This is of particular interest as one of the described SCID cases developed splenomegaly and lymphadenopathy dependent on a B cell lymphoma. Thus, dysregulation of the JAK/STAT pathway may also be a feature of CD45 deficiency. Further analysis of patients with CD45 deficiency will likely yield interesting insights into the B and NK cell defects and the role of elevated JAK/STAT signaling in the patient phenotypes.
3.2.4. Lck
Lck is a tyrosine kinase that is recruited to the cytoplasmic tails of the CD4 and CD8 coreceptors and is thus recruited to the TCR complex upon coreceptor binding to MHC. Active Lck directly phosphorylates the immunoreceptor tyrosine-based activation motifs (ITAMs) on the CD3 chains of the TCR complex. This is the earliest known phosphorylation event upon TCR engagement and is essential for the antigen-mediated activation of T cells. Lck-dependent ITAM phosphorylation results in the recruitment of zeta-chain-associated protein kinase 70 (ZAP-70) through its Src homology 2 (SH2) domain and its subsequent phosphorylation and activation by Lck (Salmond, Filby, Qureshi, Caserta, & Zamoyska, 2009).
Biallelic LOF mutations in LCK lead to an autosomal recessive PID (OMIM 615758). In 2012, a single 15-month-old baby was discovered to harbor a homozygous Lck missense mutation (L341P), which results in production of an unstable and inactive Lck protein. TCR signaling was absent in the patient and in a model system of Lck-deficient Jurkat T cells reconstituted with the Lck variant (L341P) (Hauck et al., 2012). The patient suffered from diarrhea and failure to thrive, recurrent upper respiratory infections, multiple modular skin lesions characterized by infiltration of macrophages, and CD3+ T cell as well as neutrophil necrosis. Additionally, the patient had autoimmune cytopenias due to autoantibody formation. Immunophenotyping showed reduced CD4+ and CD8+ T cells with the remaining CD8 cells manifesting a central memory or exhausted memory (TEMRA) cellular phenotype. Three additional patients were identified with T cell-specific immunodeficiencies, two with loss of Lck exon 7 and reduced protein expression, and one with normal Lck protein levels but reduced kinase activity. In each of these patients, the decrease in Lck expression/activity correlated with reduced signaling and cellular activation following TCR stimulation, suggesting the presence of hypomorphic mutations, though the causal variant was not identified in any of these patients, leaving them without genetic confirmation of Lck deficiency (Goldman et al., 1998; Hubert et al., 2000; Sawabe et al., 2001). Of these three patients, the one with defective kinase activity was largely asymptomatic at 66 years old, but showed reduced CD4+ T cell numbers with an increased memory phenotype and unaffected CD8+ T cells. By contrast, the two patients with Lck lacking exon 7 showed selective CD4+ T cell loss accompanied by lymphadenopathy, low IgG, diarrhea, failure to thrive, candidiasis, and sepsis. Together, these data suggest that Lck deficiency may cause a selective T cell defect, preferentially affecting CD4+ T cells and resulting in decreased or absent TCR signaling. The cellular and clinical phenotype likely depends on the causal mutation. Those that permit a significant level of residual Lck kinase activity likely cause a less severe cellular and clinical phenotype. On the other hand, severely deleterious variants likely limit thymic output allowing for lymphopenia-induced proliferation and expansion of self-reactive T cells that may account for the increased proportion of TEMRA cells in some patients. Interestingly, the L341P mutation positive cells did not undergo restimulation-induced cell death (RICD) which, when combined with the requirement of Lck signaling for apoptosis mediated by the tumor necrosis factor receptor superfamily member 6 (FAS) receptor, may account for the observed lymphadenopathy, though establishing such a conclusion requires further investigation (Akimzhanov & Boehning, 2015).
Currently, only biallelic null or hypomorphic Lck mutations have been associated with a PID. Because Lck is closely regulated and maintained in an inactive state through phosphorylation of Y505, there remains the possibility of a dominant PID caused by either dominant-interfering variants, such as K273R, or GOF mutations, such as mutations of Y505. This is exemplified by the apparent oncogenicity of Lck activating mutations and the presence of such mutations in a T cell leukemia cell line (Laham, Mukhopadhyay, & Roberts, 2000; Wright, Sefton, & Kamps, 1994).
3.2.5. ZAP-70
ZAP-70 is a tyrosine kinase critical for TCR signaling. It is first recruited to phosphorylated ITAMs on CD3ζ and is subsequently phosphorylated and activated by Lck. Once activated, it phosphorylates linker for activation of T cells (LAT) and SH2 domain containing leukocyte protein of 76 kDa protein (SLP-76), both critical adaptor molecules in the TCR signaling pathway (Chan, Iwashima, Turck, & Weiss, 1992; Wang et al., 2010; Zhang, Sloan-Lancaster, Kitchen, Trible, & Samelson, 1998). Biallelic LOF mutations in ZAP-70 cause an autosomal recessive disease characterized as a selective deficiency of CD8+ T cells due to abnormal thymic selection (OMIM 269840) (Arpaia, Shahar, Dadi, Cohen, & Roifman, 1994; Elder et al., 1994). Although the number of CD4+ T cells is normal in ZAP70-deficient patients, their function is not. They exhibit abnormal calcium flux in response to TCR engagement, demonstrating defective TCR signaling (Chan et al., 1994). Clinically, ZAP70-deficient patients present early in life with a history of recurrent infections including viral lung disease, candidiasis, diarrhea, panhypogammaglobulinemia, and silent brain infarcts (Akar et al., 2015; Arpaia et al., 1994; Chan et al., 1994; Elder et al., 1994; Liu et al., 2017). Hypomorphic mutations with some residual kinase activity can result in a milder phenotype with late-onset immunodeficiency associated with skin and lung infections. These hypomorphic ZAP-70 patients have modestly reduced T cell counts and reduced TCR-dependent signaling (Picard et al., 2009a).
An additional autoimmune syndrome has recently been described due to the unique combination of coinherited activating and hypomorphic mutations in ZAP-70 (OMIM 617006) (Chan et al., 2016). The first of two siblings born to nonconsanguineous parents developed nephrotic syndrome, blistering skin disease, autoantibodies to clotting factor VIII resulting in bruising and hemarthrosis, and inflammatory colitis. The second sibling developed generalized bullous pemphigoid and failure to thrive due to inflammatory colitis. In both cases, there was no evidence of altered T cell numbers or immunodeficiency; however, successful hematopoietic stem cell transplantation (HSCT) proved curative. Both patients possessed compound heterozygous mutations in the ZAP-70 SH2 and kinase domains. The SH2 mutant did not associate with CD3ζ and resulted in diminished signaling while the kinase domain mutant resulted in loss of autoinhibition and enhanced ZAP-70, SLP76, and LAT phosphorylation after TCR stimulation. This example is particularly interesting because the R360P mutation in the catalytic domain enhanced kinase activity but was not capable of causing a dominant disease by itself (the single mutation positive sister and father were phenotypically normal). Apparently, the absence of WT protein was necessary to allow the activating variant to induce disease, possibly through preferential recruitment to CD3ζ in the presence of the SH2 variant protein. It is possible that a more strongly activating mutation may cause a similar disease in an autosomal dominant MOI.
3.2.6. RhoH
Ras homologue family member H (RhoH) is a hematopoietic-specific small guanosine triphosphate hydrolase (GTPase), which, due to loss of amino acid residues conserved in enzymatically active GTPases, can bind but not hydrolyze GTP. Hence, it is generally considered to be constitutively active (Li et al., 2002). A mutagenesis screen demonstrated that RhoH was an essential negative regulator of the integrin LFA-1 (Cherry, Li, Schwab, Lim, & Klickstein, 2004). Studies in rhoh−/− mice showed that RhoH is essential for T cell positive selection by directing the localization of Lck and ZAP-70 to the immunological synapse (IS) as well as promoting Lck’s interaction with and phosphorylation of CD3ζ-inducing downstream signaling events (Chae, Siefring, Hildeman, Gu, & Williams, 2010; Dorn et al., 2007; Gu et al., 2006). The interaction of ZAP-70 with RhoH is mediated through a pseudo-ITAM motif in RhoH and influenced by Lck activity. Interestingly, RhoH has opposite effects on Ras-related protein 1 (Rap1) and downstream integrin activation depending on the stimulus with RhoH upregulating TCR-mediated signaling and downregulating CXCR4-dependent signaling, possibly through the regulation of ZAP-70 localization (Baker et al., 2012).
Two siblings from a single consanguineous family have been described with homozygous nonsense mutations in RhoH (Crequer et al., 2012). T cells from the patients show reduced ZAP-70 phosphorylation, both at baseline and upon TCR engagement. This almost completely abrogates T cell proliferation in response to TCR stimulatory antibodies. In addition, while these patients were not T cell lymphopenic, in contrast to rhoh knockout mice, they did have reduced naïve T cells and enhanced T cell memory and TEMRA populations, possibly as a consequence of lymphopenia-induced proliferation. As in the mice, RhoH deficiency alters thymic selection as patients display abnormal Vαβ repertoires. Despite the profound signaling defect and loss of naïve T cells, the patients’ clinical phenotype was remarkably restricted, presenting with epidermodysplasia verruciformis, a rare disorder characterized by increased susceptibility to ß-papillomaviruses. However, one patient also presented with bronchopulmonary disease and Burkitt lymphoma. This may be B cell intrinsic because hypermutation of RhoH has been previously associated with B cell lymphomas in otherwise healthy individuals (Pasqualucci et al., 2001).
3.2.7. Itk
Interleukin-2-inducible T cell kinase (Itk) is a Tec family kinase that is recruited to sites of TCR activation by binding PtdIns(3,4,5)P3 (PIP3) through its pleckstrin homology (PH) domain. Once recruited, Itk interacts with the SLP76/LAT complex and is phosphorylated and activated by Lck. Following Lck-mediated phosphorylation, Itk then mediates its own autophosphorylation and that of its downstream target phospholipase C, gamma 1 (PLCγ) (Andreotti, Schwartzberg, Joseph, & Berg, 2010). Itk activity is required for T cell activation, effector function, and differentiation through its phosphorylation of PLCγ and downstream diacylglycerol (DAG) and calcium (Ca2+)-mediated signaling. Biallelic LOF mutations in Itk result in a fatal EBV-associated lymphoproliferative disease associated with increased risk of mononucleosis, Hodgkin’s lymphoma, lymphadenopathy, splenomegaly, hypogammaglobulinemia, progressive CD4+ T cell loss, recurrent infections, autoimmune disorders, and cytopenias (OMIM 613011) (Ghosh, Bienemann, Boztug, & Borkhardt, 2014; Huck et al., 2009; Linka et al., 2012; Stepensky et al., 2011). Patient cells and patient-associated Itk variants induce mild defects in Ca2+ flux downstream of TCR activation. Itk deficiency likely results in diminished CD8+ T cell ability to kill EBV-infected B cells leading to the narrow clinical phenotype. Interestingly, both Itk deficiency and MAGT1 deficiency, as discussed later, result in diminished PLCγ activity and a selective susceptibility to EBV infection. These two disorders also share a CD4+ T cell lymphopenia, suggesting Itk and PLCγ are important for CD4+ T cell selection, activation, and/or long-term survival.
3.2.8. Disorders of Phosphoinositide Signaling (PIK3CD, PIK3R1, and PTEN)
Phosphoinositide signaling is regulated by specific phosphoinositide kinases and phosphatases activated downstream of antigen receptors, coreceptors, and G protein-coupled receptors (GPCRs). These proteins are critical for the regulation of multiple immune functions, including T cell activation and migration, and have been associated with a family of related primary immune disorders (Lucas, Chandra, Nejentsev, Condliffe, & Okkenhaug, 2016).
PIK3CD encodes for phosphoinositide 3-kinase delta (PI3Kδ or P110δ), the leukocyte-specific class 1 PI3K capable of phosphorylating PtdIns(4,5)P2 (PIP2) to produce PIP3 in response to activating receptors (Vanhaesebroeck et al., 1997). PIP3, in turn, recruits protein kinase B (Akt) through its PH domain, leading to its activation and downstream signaling to multiple effector pathways, including the mechanistic target of rapamycin (mTOR) pathway (Vanhaesebroeck, Stephens, & Hawkins, 2012). In 2013, two groups described an autosomal dominant disease caused by heterozygous activating mutations in PI3Kδ (OMIM 615513) (Angulo et al., 2013; Lucas et al., 2014a). These mutations lead to a CID characterized by defects in both T cells and B cells leading to frequent chest infections and bronchiectasis, viral infections (herpes, EBV, and CMV), as well as mucosal lymphoid aggregates, lymphadenopathy, decreased naïve T cells, and increased terminally differentiated effector T cells. The elucidation of the molecular and genetic mechanism of this condition defined a new disease, called “p110δ Activating mutation causing Senescent T cells, Lymphadenopathy, And Immunodeficiency” (PASLI) disease or “Activated PI3Kδ Syndrome” (APDS). A large cohort study has since shown that there is considerable clinical variability with a mixture of autoimmunity, immunodeficiency, developmental delay, and susceptibility to lymphoma (Coulter et al., 2017). Patient mutations resulted in enhanced activation of Akt and mTOR signaling, both at baseline and upon antigen stimulation. This results in enhanced effector function, as measured by IFNγ production and glycolytic function. Interestingly, patient cells from peripheral blood mononuclear cells (PBMCs) did not proliferate as efficiently, and had increased RICD, presumably due to the presence of increased effector memory and senescent TEMRA cells. Rapamycin treatment was able to partially rescue naïve T cell percentages and decreases terminally differentiated effector cells, providing proof that pharmacological inhibition of the affected pathway may have great clinical benefit to these patients. Moreover, a specific enzymatic inhibitor of p110δ is showing great promise in a clinical treatment trial of PASLI patients (Rao et al., 2017).
A single patient has been described with biallelic PIK3CD LOF mutations leading to a disease characterized by hypogammaglobulinemia, sinopulmonary infections, and septic arthritis (Lucas et al., 2016). This may more closely phenocopy the knockout mouse that shows reduced activation of B and T cells following antigen receptor engagement than the activating mutations do (Okkenhaug et al., 2002).
The PIK3R1 gene encodes several different regulatory subunits (p85α/p55α/p50α) of class I PI3Ks by utilizing different transcriptional start sites. The regulatory subunit p85α, the main subunit known to bind PI3Kδ, controls PI3K activation in three ways: by inhibiting degradation of the catalytic subunit, inhibiting the baseline catalytic activity, and by aiding in the localization of the catalytic subunit to the plasma membrane upon TCR engagement (Lucas et al., 2016). Autosomal dominant mutations in PIK3R1 have been associated with two separate diseases. The first, short stature, hyper-extensibility, hernia, ocular depression, Rieger anomaly, and teething delay (SHORT) syndrome, is a developmental disorder with no known immunological component (OMIM 269880). Missense, nonsense, and frameshift mutations have been associated with SHORT syndrome, with several characterized mutations/patient samples showing enhanced baseline pAkt signaling and reduced insulin-induced Akt phosphorylation. Whether this represents a conditioned tachyphylaxis or a reduced ability of p85α to mediate the recruitment and activation of PI3K remains to be seen (Chudasama et al., 2013; Chung & Gibson, 2014; Dyment et al., 2013; Thauvin-Robinet et al., 2013). The second disease is characterized by varying degrees of autoimmunity, immunodeficiency, malignancy, growth retardation, and neurodevelopmental delay (OMIM 616005) (Elkaim et al., 2016). In all cases, heterozygous splice site mutations in the acceptor or donor sites surrounding exon 11 result in exon skipping and loss of amino acids 434–475 in the inter-SH2 domain. This domain is critical for negative regulation of PI3K catalytic activity. The resulting protein is still capable of stabilizing and recruiting the catalytic subunit, but no longer maintains it in an inactive state in the absence of a stimulatory event (Lucas et al., 2016). Patients exhibit sinopulmonary infections, likely due to B cell deficiency associated with hyper-IgM syndrome, conjunctivitis, autoimmunity (arthritis, IBD, thrombocytopenia, lymphadenopathy, and splenomegaly), and short stature (Elkaim et al., 2016; Lucas et al., 2014b; Petrovski et al., 2016). On a cellular level these patients, like PI3Kδ patients, exhibit increased pAkt and mTOR activation, both at baseline and following TCR stimulation (Deau et al., 2014; Lucas et al., 2014b; Petrovski et al., 2016). This is again accompanied by loss of naïve and expansion of terminally differentiated and senescent T cells. Interestingly, there is an overlap in the clinical features of both dominant PIK3R1 disorders as short stature and poor growth are characteristic of both diseases, but whether any single mutation can lead to a combined clinical presentation remains to be seen. Rapamycin (sirolimus) or a specific p110δ may be effective in ameliorating the clinical phenotype.
Interestingly, complete absence of p85α, due to a homozygous stop codon, results in decreased p110δ protein expression in all immunological cell types tested, but caused only a selective loss of the B cell lineage, suggesting that other PI3Ks may substitute in during T cell development (OMIM 615214) (Conley et al., 2012).
Specific kinase-activating mutations in PIK3CD and PIK3R1 result in overt activation of the PI3K pathway by upregulating PIP3 production. A similar increase in PIP3 is observed when the PIP3 phosphatase, phosphatase and tensin homologue (PTEN), is lost. Inactivating mutations in PTEN have long been associated with cancer cell growth, including growth of the Jurkat T leukemia cell line. Recently, a PID caused by PTEN mutations has been described (OMIM 601728) (Hollander, Blumenthal, & Dennis, 2011; Song, Salmena, & Pandolfi, 2012). In screening patients with similar clinical manifestations to PIK3CD GOF and PIK3R1 LOF, Tsujita et al. identified two patients with heterozygous de novo LOF mutations in PTEN. These patients had recurrent Staphylococcus infection, pancytopenia, pneumonia and respiratory tract infections, hepatosplenomegaly, lymphadenopathy, an inverted CD4+/CD8+ ratio, decreased naïve T cell numbers, and reduced numbers of memory and class-switched B cells (Tsujita et al., 2016). Importantly, patient cells showed reduced PTEN expression and enhanced pAkt and mTOR signaling, thus suggesting functional haploinsufficiency. Interestingly, autosomal dominant LOF mutations have already been associated with several genetic disorders, though only rarely with defined immunological phenotypes, ex. Cowden’s syndrome (CS, OMIM 158350) (Browning, Chandra, Carbonaro, Okkenhaug, & Barwell, 2015; Eng, 2003; Ruschak, Kauh, & Luscombe, 1981). Importantly, in the study by Tsujita et al., one patient had macrocephaly and mental retardation while the second had mild mental retardation, which are clinical symptoms of CS, possibly indicating that these are variable clinical phenotypes of the same genetic disorder. Fitting with this, two CS patients with no overt immunological phenotype showed enhanced PI3K pathway activation in T cells. Interestingly, there is little correlation between mutation location/type and clinical manifestation, suggesting highly variable clinical expressivity for PTEN haploinsufficiency whose cause is not currently understood. As in the case of PIK3R1 and PIK3CD disorders, sirolimus or p110δ inhibitors likely represent targeted therapeutic options for PTEN haploinsufficiency-associated diseases (Schmid et al., 2014; Squarize, Castilho, & Gutkind, 2008).
3.2.9. ORAI-1 and STIM1 (Disorders of Ca2+ Flux)
Following PLCγ activation and cleavage of PIP2 to DAG and inositol triphosphate (IP3), IP3 binds to IP3 receptors on the endoplasmic reticulum (ER) leading to release of Ca2+ from endoplasmic stores. This triggers the release of Ca2+ from the EF-hand of stromal interaction molecule 1 (STIM1), its conformational change, and oligomerization. Activated STIM1 then induces the influx of extracellular Ca2+ by activating the calcium release-activated calcium (CRAC) channel protein 1 (ORA-I), located on the plasma membrane (Vig et al., 2006). The influx of Ca2+ through activated CRAC channels acts as a key second messenger in TCR signaling (Feske, Skolnik, & Prakriya, 2012; Soboloff, Rothberg, Madesh, & Gill, 2012). Hence, the proper function of both STIM1 and ORAI-1 channels is essential for regulation of Ca2+ signaling. Interestingly, both autosomal recessive and autosomal dominant diseases can arise due to mutations in STIM1 and ORAI-1 with similar clinical features.
Biallelic LOF variants in ORAI-1 cause a SCID-like autosomal recessive disease with syndromic features including muscle weakness and dysplastic dental enamel (OMIM 612782) (Feske et al., 1996, 2006; McCarl et al., 2009). Two patients born to consanguineous parents presented within 2 weeks of birth with failure to thrive, rotavirus infection, stomatitis and aphthous ulcers, mycobacterial infections, sepsis, muscular hypotonia, and intermittent fever. While T cell and NK cell counts were normal, indicating lymphocyte ontogeny, patient T cells largely failed to respond to stimulation through the TCR or with phorbol myristate acetate and ionomycin (PMA/I), showing defective proliferation, cytokine production, and NF-AT activation (Feske et al., 1996). Patients carried a R91W substitution in ORAI-1 that caused a loss of extracellular Ca2+ influx following normal ER store release. This was rescued with reconstitution with wild-type (WT) ORAI1. Interestingly, heterozygous individuals showed a decreased calcium flux, suggesting a gene dosage effect (Feske et al., 2006). Identification of additional patients has expanded the clinical phenotype to include chronic diarrhea, candidiasis, pneumonia, pyelonephritis, toxoplasma encephalitis, and cytomegalovirus infection with one patient developing autoimmune neutropenia and thrombocytopenia (McCarl et al., 2009).
An autosomal dominant disease due to mutations in ORAI-1 primarily affecting muscle tissue with similarities to Stormorken syndrome has been described (OMIM 615883) (Endo et al., 2015; Garibaldi et al., 2017; Nesin et al., 2014). The patient phenotype is characterized by tubular aggregate myopathy, congenital miosis, and hypocalcemia. These mutations result in either prolonged Ca2+ entry following stimulation or the spontaneous entry of Ca2+ from the extracellular space to the cytoplasm. Interestingly no hematopoietic abnormalities have been associated with this disease, suggesting that the increased Ca2+ flux is either cell type-specific or not severe enough to cause enhanced immune cell activation. Nonetheless, the immunological features of these patients should be examined over time to see if there is a late-onset immunological phenotype associated with ORAI-1 activating mutations.
Homozygous deleterious variants in STIM1 give rise to an immunodeficiency with syndromic features including muscle hypotonia and defects in tooth enamel, similar to that seen in ORAI-1 patients (OMIM 612783) (Picard et al., 2009b; Wang et al., 2014). Clinical manifestations include recurrent urinary tract infections, otitis media, pneumonia, bacterial sepsis, and increased susceptibility to viral infection including chickenpox, CMV, EBV, and disseminated Kaposi sarcoma indicating severe immune dysfunction. Unexpectedly, the patients also developed autoimmune manifestations including thrombocytopenia, lymphadenopathy, and hepatosplenomegaly (Byun et al., 2010). Patient cells were found to have defective extracellular Ca2+ entry leading to poor activation of NK and T cells. Interestingly, patient T cells showed an increase in terminally differentiated and exhausted cells, suggesting a paradoxical altered stimulation or expansion in vivo due to a lymphopenic state (Fuchs et al., 2012; Parry et al., 2016; Picard et al., 2009b).
In addition to the autosomal recessive disease caused by LOF mutations in STIM1, an autosomal dominant disorder caused by GOF mutations in STIM1 has been described with similar clinical presentation to ORAI1 autosomal dominant disease (OMIM 160565) (Morin et al., 2014; Nesin et al., 2014; Noury et al., 2017). GOF mutations can lead to Stormorken syndrome with miosis (OMIM 185070) thrombocytopenia, hyperactive platelets, hypocalcemia, muscle fatigue, asplenia, and ichthyosis. Causal mutations include a missense mutation in STIM1 (R304W) that likely affects the CC1 autoinhibitory domain, thus leading to a constitutively active STIM1 molecule, as well as a D84E missense mutation in the EF-hand, presumed to reduce baseline Ca2+ binding thus facilitating STIM1 aggregation. There was a corresponding increase in baseline cytoplasmic Ca2+ levels in patient fibroblasts and enhanced STIM1 aggregation in unstimulated cells. Although immunological phenotyping was not performed, it would be interesting to see if T and NK cells show a hyperactive phenotype. Why STIM1 hyperactive mutations lead to thrombocyte defects and bleeding disorders while ORAI1 mutations do not remain an interesting question, and may have to do with the dynamics of extracellular Ca2+ entry caused by the causal variant.
3.2.10. EVER2
Like calcium, other divalent cations including zinc (Zn2+) and magnesium (Mg2+) are also important for T cell-mediated signaling (see MAGT1 deficiency) (Li et al., 2011; Yu et al., 2011). Primary epidermodysplasia verruciformis, characterized by eruptions of wart-like protrusions caused by human papilloma virus (HPV) infection, is caused by biallelic LOF in EVER1 or EVER2, ER resident proteins predicted to be transmembrane channels (OMIM 226400) (Horton & Stokes, 2014). T cells from EV patients show diminished responses to mitogenic stimuli, though the mechanism is incompletely understood (de Pereira, Carrasco, Neto, Rady, & Tyring, 2003; Prawer et al., 1977). EVER1 and EVER2 interact with the Zn2+ transporter ZnT-1 and modulate its function to block Zn2+-dependent TF activity (Lazarczyk et al., 2008). Interestingly, the HPV16 E5 protein inhibited this interaction and increased Zn2+-dependent transcription, suggesting this pathway is targeted by HPV to mediate its own survival or replication. In T cells, EVER1 and EVER2 proteins are expressed at baseline but are rapidly downregulated following activation, correlating with a rise in intracellular Zn2+ concentration (Lazarczyk et al., 2012). Consistent with this, B and T cells from EVER2-deficient patients had increased intracellular Zn2+. Altogether, this suggests that EVER proteins function to inhibit ZnT-1 and limit intracellular Zn2+ concentrations. Loss of protein expression, whether during T cell activation or in EVER-deficient patients, abrogates this inhibition causing Zn2+ accumulation. As Zn2+ modulates early TCR signaling through inhibition of src homology region 2 domain-containing phosphatase-1 (SHP1) recruitment to the TCR, prolonged/elevated Zn2+ concentrations at baseline may alter signaling or induce T cell anergy. Further work is needed to determine the role of EVER proteins in regulating Zn2+-dependent T cell activation and how elevated Zn2+ concentrations effect T cell function over the prolonged periods, though it is tempting to speculate that altered Zn2+ homeostasis accounts for the T cell proliferative deficiencies in EVER2-deficient patients.
3.2.11. SH2D1A
An excellent example of how PIDs can guide investigators to discover new genes and signaling pathways is hemizygous LOF variants in the gene SH2D1A and its product signaling lymphocytic activation molecule (SLAM)-associated protein (SAP). These variants were identified as the genetic cause of X-linked lymphoproliferative disease (XLP) (OMIM 308240). XLP is defined by uncontrolled EBV infection including fatal mononucleosis, vigorous polyclonal expansion of T and B cells, acquired hypogammaglobulinemia, reduced NK cell functionality, and malignant lymphoma (Coffey et al., 1998; Sayos et al., 1998). SAP binds phosphorylated tyrosine residues on the cytoplasmic tail of SLAM family members and recruits the tyrosine kinases Lck and Fyn to initiate downstream signaling. In addition, SAP binding blocks recruitment of the SHP1 and SHP2 phosphatases to SLAM receptors. SAP deficiency in XLP patients converts SLAM family members from stimulatory to inhibitory receptors through decreased Lck recruitment and increased phosphatase recruitment (Cannons, Tangye, & Schwartzberg, 2011; Katz, Krummey, Larsen, Stinson, & Snow, 2014; Latour et al., 2003; Nichols, Ma, Cannons, Schwartzberg, & Tangye, 2005). Interestingly, uncontrolled EBV infection in XLP is likely a byproduct of the B cell tropism exhibited by EBV, as SLAM receptor signaling is essential for CD8+ T cell-mediated killing of target B cells, but not of other cell types (Palendira et al., 2011). In addition to the inability of XLP CD8+ T cells to control EBV-infected B cells, RICD is defective in patient T cells, possibly accounting for a portion of the uncontrolled T cell response characteristic of XLP as reactivated cells do not undergo apoptosis in response to antigen, which is a normal control mechanism limiting the immune response (Snow et al., 2009). This was due to increased recruitment of SHP1 to the SLAM family receptor CD352 and SHP1-dependent down-modulation of TCR-mediated signaling. Interestingly, silencing of CD352 also reduced RICD, suggesting it normally initiates an activating signal that can contribute to TCR-mediated cellular activation, likely through Lck recruitment and activation (Katz et al., 2014). Pharmacological inhibition of SHP1/SHP2 or diacylglycerol kinase alpha (DGKα), which is also upregulated in T cells from XLP patients, may yield a targeted way of reversing the signaling abnormalities associated with SAP deficiency (Ruffo et al., 2016).
3.2.12. PKCdelta
Biallelic LOF variants in PRKCD, the gene encoding protein kinase C delta (PKCδ), cause an autosomal recessive disorder characterized primarily by B cell hyperactivation, with mild defects in T cell activation (OMIM 615559) (Belot et al., 2013; Kuehn et al., 2013; Salzer et al., 2013a). Patients suffer from recurrent infections, lupus-like autoimmunity with autoantibody production, chronic lymphadenopathy, and splenomegaly derived mainly from a hyperactivation/proliferation of patient B cells in the absence of PKCδ.
3.3. Defects in Costimulatory Pathways
While the main T cell activating signal is unquestionably delivered through the TCR, stimulation of other molecules, called costimulatory molecules, are absolutely required for full T cell activation, effector function, memory formation, and the prevention of T cell anergy (Chen & Flies, 2013). These costimulatory molecules, of which CD28 is the best recognized, are cell surface receptors that recognize ligands that are upregulated on APCs or target cells upon infection. This costimulatory signal can involve signaling intermediates that are either shared with the TCR or unique to costimulation. In addition to the ability to receive costimulatory signals, T cells can also deliver costimulatory signals during an immune response. In this section, we will discuss PIDs associated with costimulatory signals provided to and by T cells (Fig. 1).
3.3.1. CARMIL2/RLTPR Deficiency
Capping protein regulator and myosin 1 linker 2 (CARMIL2/RLTPR) (OMIM 610859) is best known as a negative-regulator of actin capping protein (CP), which binds to the barbed end of actin filaments and prevents monomer addition and is highly expressed in the immune system (Lanier, Kim, & Cooper, 2015; Liang, Niederstrasser, Edwards, Jackson, & Cooper, 2009). The essential role of CARMIL2 in CD28-mediated signaling and nTreg formation was originally identified in a rescue screen for mice expressing LAT (Y136F) (Liang et al., 2013). CARMIL2 translocates to CD28/CD80 microclusters at the IS and is essential for the recruitment of PKC-θ and CARD-containing MAGUK protein 1 (CARD11). Interestingly, the mutant form of CARMIL2 identified in this rescue screen was stable, recruited to CD28/80 complexes, and could still bind CP, suggesting additional roles of CARMIL2 other than the regulation of actin filaments. These points were later clarified as CARMIL2 was found to act as a scaffolding protein within the CD28 pathway, bridging CD28 to CARD11, thus inducing CARD11-mediated NF-κB activation (Roncagalli et al., 2016). Biallelic mutations in CARMIL2 have recently been identified as a cause of an autosomal recessive autoimmune disease (Schober et al., 2017; Sorte et al., 2016; Wang et al., 2016). In one study, the patients suffered from mucocutaneous candidiasis, multifocal tuberculosis, recurrent bacterial lung infections, subcutaneous Staphylococcus, asthma associated with severe allergic skin lesions comprising psoriasiform hyperplastic epidermis and spongiosis, with superficial perivascular CD8+ T cell infiltrates and EBV viremia in some patients (Wang et al., 2016). A second report described patients with a similar phenotype comprising failure to thrive, chronic diarrhea, recurrent skin (staphylococcus, skin warts, and eczema), and upper respiratory infections (Schober et al., 2017). Interestingly, this report identified disseminated EBV+ smooth muscle tumors in all patients, presenting variably in the gut, liver, brain, spleen, and kidney explaining the susceptibility to EBV as seen in the first study. All patients had biallelic mutations leading to the loss or reduction of CARMIL2/RLTPR protein with a recessive MOI. Immunophenotyping of patients revealed reduced Treg numbers and an increased percentage of naïve CD4+ and CD8+ T cells coupled with reduced memory populations. There was an apparent defect in CD28-mediated T cell costimulation. While stimulation through CD3 alone or with PMA/I proceeds normally, patient T cells did not upregulate CD25 or CD69, make TNF, induce p65 (RELA) phosphorylation or undergo proliferative expansion when CD28 activating antibodies were added to CD3 stimulation. Patients also displayed B and NK cell-intrinsic defects, though the addition of IL2 to the culture medium rescued both NKG2D expression and degranulation in NK cells and CD8+ T cells, possibly by compensating for defects in CD28-mediated costimulation. While no defects in cellular migration or synapse formation were described in either CARMIL2-deficient mice or humans, it may be interesting to determine if CARMIL2 is also involved in these actin-dependent processes, given the role of CAMRIL2 in the migration of other cell types.
3.3.2. CTLA4
Immune homeostasis is a balance between the ability to receive costimulatory signals and dampen the immune response by inhibiting CD28-dependent signaling as a form of checkpoint regulation. This is largely accomplished through the immune checkpoint protein CTLA4 expressed on Tregs and activated T cells. CTLA4 binds CD80 and CD86 with high affinity, effectively competing with CD28 ligand binding. CTLA4 binding removes CD80 and CD86 from the cell surface by transendocytosis to dampen the T cell immune response (Sansom, 2015; Tivol et al., 1995; Walker & Sansom, 2011; Waterhouse et al., 1995). Coding variants in CTLA4 have long been identified as potential risk alleles in common autoimmune disorders such as Graves’ disease, Celiac disease, T1DM, and systemic lupus erythematosus (Barreto et al., 2004; Nistico et al., 1996; Ueda et al., 2003). More recently, an autosomal dominant disease, termed “CTLA-4 haploinsufficiency with autoimmunity and infiltration” (CHAI) disease has been described due to heterozygous LOF variants in CTLA4 (OMIM 616100) (Kuehn et al., 2014; Schubert et al., 2014). Like CTLA4-deficient mice, patients develop a T cell lymphocytic infiltrative disease in several tissues including intestines, lungs, bone marrow, central nervous system, and kidneys with lymphadenopathy and splenomegaly associated with reduced CTLA4 expression levels. On a cellular level, patient Tregs express less FOXP3 and CD25 and are less inhibitory than control Tregs while conventional T cells are hyperproliferative in response to antigenic stimulation. Importantly, this disease can be treated with CTLA4-Ig, which replaces, pharmaceutically, the natural function of CTLA-4 in patients (Lee et al., 2016). CTLA4 deficiency, like other forms of autoimmune lymphoproliferative disease (see below), is incompletely penetrant, with only about 50% of mutation-bearing individuals showing clinically appreciable disease despite reduced CTLA4 expression. Whether these clinically asymptomatic individuals have protective variants in other genes or the disease is precipitated by susceptibility genes or an immunological/environmental event remains to be explored.
3.3.3. ICOS
Inducible T cell costimulatory (ICOS) is upregulated on the surface of activated T cells, particularly T follicular helper cells (Tfh), with structural and sequence homology to CD28 and CTLA-4 (Hutloff et al., 1999). Ligation of ICOS with an activating antibody provides a strong costimulatory signal to responding T cells leading to enhanced proliferation, primarily through PI3K recruitment to the YMFM motif in the cytoplasmic tail of ICOS (Harada et al., 2003; Yong, Salzer, & Grimbacher, 2009). ICOS is particularly important for the development of T cell-dependent antibody responses and the germinal center reaction through a combination of ICOS-dependent Tfh formation, migration into the germinal center, and costimulatory events necessary for the delivery of T cell help to GC B cells (Dong, Temann, & Flavell, 2001; Wikenheiser & Stumhofer, 2016; Xu et al., 2013; Yong et al., 2009). Biallelic LOF mutations in ICOS lead to autosomal recessive combined variable immunodeficiency (CVID) with recurrent bacterial infections, sinusitis, bronchiectasis, pneumonia, GI infections with diarrhea, hepatosplenomegaly, and in some cases malignancy caused by decreased class-switched and memory B cells, decreased serum immunoglobulins, and attenuated germinal center formation (OMIM 607594) (Grimbacher et al., 2003; Salzer et al., 2004; Warnatz et al., 2006). Although T cell populations were initially reported to be intact in ICOS-deficient patients, it was later discovered that patient T cells did not produce IL-10 or IL-17 upon stimulation with antibodies against CD3/28 or CD3/ICOS. Additionally, patients have a significant reduction in CXCR5+ Tfh cells, indicating an essential role of ICOS signaling in the differentiation of these cells in vivo (Bossaller et al., 2006; Warnatz et al., 2006). Thus, it seems likely that the B cell defects in ICOS patients arise from a selective defect in the formation, migration, or function of Tfh cells.
3.3.4. CD40 Ligand (CD40LG/CD154)
Hemizygous LOF mutations in CD40LG, encoding the T cell-expressed TNF-like ligand for CD40 (CD154), cause an X-linked hyper-IgM syndrome characterized by recurrent bacterial infections, ulcerative stomatitis, gingivitis, due to low levels of class-switched antibodies, loss of germinal center reactions, and low B cell isotype switching and memory formation but no appreciable defect in T cell populations (OMIM 308230) (Allen et al., 1993; DiSanto, Bonnefoy, Gauchat, Fischer, & de Saint Basile, 1993; Korthauer et al., 1993). Upon activation, T cells upregulate CD154 which is capable of binding the TNF receptor superfamily member CD40 (formerly known as TNFRSF5) on the surface of B cells and other APCs. During T-dependent B cell responses in the germinal center reaction, ligation of CD40 by CD154 at the IS induces multiple signaling pathways in the responding B cells, including TRAF-dependent NF-κB signaling, PI3K activation, JNK activation, and JAK/STAT signaling. These signaling events enhance B cell adhesion, survival, proliferation, class switching, and affinity maturation, while lack of CD40 engagement can lead to apoptosis of activated B cells (Elgueta et al., 2009). Thus, the presence of CD154 on CD4+ Tfh cells is absolutely critical for their effector function.
3.3.5. CD27/CD70
Stimulation of CD27, a member of the TNF family of receptors expressed on T, B, and NK cell populations, by its ligand CD70 (expressed on B cells and APCs) results in the activation of NF-κB and c-Jun N-terminal kinase (JNK) resulting in enhanced effector and memory differentiation (Denoeud & Moser, 2011; Hendriks et al., 2000). Biallelic LOF mutations in CD27 cause an EBV-associated lymphoproliferative disorder with symptoms varying from an asymptomatic memory B cell deficiency to EBV-driven hemophagocytosis, lymphoproliferation, hypogammaglobulinemia, and malignancy (OMIM 615122) (Salzer et al., 2013b; van Montfrans et al., 2012). Interestingly, while patient T cells had minor proliferative defects to ligands that can engage CD27 in vitro, EBV-specific T cells were detected in the memory compartment and showed normal effector function, so the reason for uncontrolled EBV-driven lymphoproliferation was not understood. This was clarified in two recent publications identifying and characterizing patients with a similar disease of immunodeficiency and EBV-driven malignancy in patients with biallelic LOF mutations in the CD27 ligand, CD70 (Abolhassani et al., 2017; Izawa et al., 2017). The authors show that EBV-specific T cells do not expand well when stimulated with EBV-infected B cells and CD8+ T cells had reduced cytolytic capacity against CD70-deficient target cells, likely due to reduced NKG2D and 2B4 expression. Importantly, this was phenocopied when CD27/CD70 interactions were blocked with inhibitory CD27 antibodies. It is likely that similar defects in EBV-specific T cell activation, proliferation, or effector function were simply unappreciated in CD27-deficient patients, possibly due to use of strong agonists to stimulate T cell responses.
3.4. Defects of Cytokine Signaling
Cytokines are key mediators of the innate and adaptive immune response. Chief among their T lymphocyte duties are T cell selection, maturation, survival, proliferation, effector function, and effector/memory differentiation. Several PIDs are due to cell-intrinsic defects in cytokine signaling. In the following section, we will discuss PIDs of cytokines, cytokine receptors, and their downstream Janus kinase (JAK) and signal transducer and activator of transcription (STAT) signaling mediators. Several additional PIDs are caused by variants in T cell-secreted cytokine signaling. However, we will not discuss these PIDs in detail because T cell cytokine production defects primarily affect other cell types leaving T cell function otherwise intact. Of the PIDs not described in detail below, several are notable including alterations in interferon gamma (IFNγ) pathway causing recurrent mycobacterial infections, the interleukin (IL)-10 pathway, associated with early onset IBD, and the IL-17 pathway (IL-17F and ACT1) which result in familial candidiasis.
3.4.1. CD132/Common Gamma Chain
LOF mutations in the common gamma chain (CD132) result in an X-linked SCID with frequent bacterial, viral, and fungal infections with defects in antibody responses, athymia, reduced NK cell numbers, and absent T lymphocytes (OMIM 300400) (Noguchi et al., 1993). CD132 is a cytokine receptor chain shared among multiple cytokine receptors including the those for IL-2, IL-4, IL-7, IL-9, IL-15, IL-21, and thymic stromal lymphopoietin (Rochman, Spolski, & Leonard, 2009). Several of these cytokines are critical for T cell development and survival, thus explaining the T cell SCID phenotype. As discussed below, defects in individual cytokine receptor chains that pair with CD132 cause more specific defects in T cell development or effector function.
3.4.2. IL7Rα
Biallelic LOF mutations in IL-7R alpha subunit result in an autosomal recessive T−/B+/NK+ SCID phenotype with absent peripheral T cells, opportunistic infections, splenomegaly and lymphadenopathy, candida, pneumonia, and diarrhea (OMIM 608971) (Puel, Ziegler, Buckley, & Leonard, 1998). This defect likely occurs during thymic development during which IL-7 is critical for survival and proliferation, though IL-7 is also essential for mature T cell survival and homeostasis in the periphery (Giliani et al., 2005).
3.4.3. CD25
Biallelic LOF mutations in the alpha subunit of IL2RA (CD25), which combines with IL2RB and CD132 to form a high-affinity IL-2 receptor, cause an autosomal recessive immunodeficiency with autoimmunity (OMIM 606367) (Caudy, Reddy, Chatila, Atkinson, & Verbsky, 2007; Goudy et al., 2013; Sharfe, Dadi, Shahar, & Roifman, 1997). Patients develop recurrent viral, bacterial, and fungal infections but also large-scale autoimmune disease including lung inflammation and infiltration, hepatosplenomegaly, enteropathy, eczema and dermatitis, and T1DM. Interestingly, these clinical features combine IPEX-like disease with a T cell-specific immunodeficiency. CD25 deficiency results in decreased RICD as well as reduced Treg function because IL-2 is required for both Treg survival and effector function (Barron et al., 2010; Pandiyan, Zheng, Ishihara, Reed, & Lenardo, 2007). Additionally, patient T cells fail to make the immunosuppressive cytokine IL-10 following TCR stimulation which may contribute to enterocolitis. Loss of T cell homeostasis, Treg function, and IL-10 production may account for the nonspecific T cell activation and expansion and IPEX-like autoimmunity associated with CD25 deficiency. CD25 is also required for TCR-induced proliferation, the absence of which leads to defective antigen-specific responses and the recurrent infections associated with CD25 deficiency. Thus CD25 is critical for IL-2 regulation of both effector and regulatory T cell function and overall control of the peripheral lymphoid compartment (Willerford et al., 1995).
3.4.4. IL21/IL21R
Biallelic LOF mutations in the IL-21 (OMIM 615767) and the IL-21 receptor (IL-21R) (OMIM 615207) cause a similar recessive immunodeficiency characterized by recurrent respiratory tract infections, hepatitis, liver fibrosis, recurrent diarrhea, septicemia, impaired B cell class switching, and impaired T cell responses to candida antigens (Kotlarz et al., 2013; Salzer et al., 2014). IL-21 signaling enhances T helper (Th)17 and Tfh differentiation while inhibiting Treg differentiation, which could account for the T cell abnormalities, and is possibly related to the development of colitis (Leonard & Wan, 2016; Tian & Zajac, 2016). IL-21 also enhances B cell class switching, which leads to a B cell autonomous defect in humoral immunity in these patients.
3.4.5. IL12RB1/IL-12B
IL-12 signaling is essential for the differentiation of naïve T cells into Th1 cells. Biallelic LOF mutations in the beta subunit of the IL-12 receptor (IL-12RB1) (OMIM 614891) or the p40 subunit of IL-12 (IL-12B) (OMIM 614890) result in a clinically limited PID with a selective susceptibility to mycobacteria, candida, and salmonella infections (Altare et al., 1998a, 1998b; de Jong et al., 1998; Picard et al., 2002). Patients with IL-12B deficiency do not produce IL-12 or the Th1 cytokine IFNγ which is necessary for the control of intracellular bacterial infections. IL-12RB-deficienct patients have reduced Th1 responses and IFNγ production upon stimulation. IL-21RB1 and IL-12B-deficient patients also had defects in IL-17 producing Th17 cell formation, thus accounting for the recurrent fungal infections (de Beaucoudrey et al., 2008). Importantly, the nature of the mutation in this pathway is critical for treatment, because only IL-12 deficient, and obviously not IL-12RB1 deficient, patients can be treated by the administration of exogenous IL-12.
3.4.6. TYK2
An autosomal recessive immunodeficiency with recurrent respiratory, viral, mycobacterial, and intracellular bacterial infections is caused by LOF mutations in tyrosine kinase 2 (TYK2), the JAK family member kinase associated with signaling downstream of IFNα, IL-12, IL-10, and IL-23, among others (OMIM 611521) (Kreins et al., 2015; Minegishi et al., 2006; Watford & O’Shea, 2006). As with mutations in other JAK/STAT proteins, the complex and varied patient phenotypes likely result from defects in signaling from multiple cytokine receptors that rely on TYK2. Thus, the viral and bacterial susceptibility of TYK2-deficient patients seem attributable to the loss of IFNα and IL-12 signaling, respectively. Indeed Tyk2-deficient cells have reduced IFNα, IL-10, and IL-12 receptor expression and diminished IFNα, IL-10, IL-23, and IL-12 signaling. Importantly, patients have diminished IL-12-dependent IFNγ production, but this was not as severe as in IL12R-deficient patients. Perhaps there is a degree of compensation by other JAK family members. Together, this indicates that TYK2 deficiency is indeed an amalgamation of incomplete signaling defects downstream of multiple cytokine receptors.
3.4.7. JAK1
JAK1 pairs with JAK3 to mediate signaling downstream of the common-gamma chain cytokine receptors (OMIM 147795) (Lin & Leonard, 2017). Three patients with JAK1 mutations from a single kindred presented with atopic dermatitis, allergies, hepatosplenomegaly, autoimmune thyroid disease, and failure to thrive caused by an autosomal dominant JAK1 mutation (Del Bel et al., 2017). The causal A634D missense mutation lies within the JAK1 inhibitory domain and enhances phosphorylation of STAT3 in T cells following IL-6 stimulation. Patients treated with ruxolitinib, a JAK1/2 inhibitor, showed marked clinical improvement after a month of treatment, again emphasizing the importance of understanding the molecular cause of disease in designing an individualized medical intervention (Del Bel et al., 2017).
3.4.8. JAK3
Biallelic LOF mutations in JAK3 cause an autosomal recessive SCID with recurrent upper respiratory tract infections, diarrhea, meningitis, absent peripheral lymph nodes, reduced T and NK cell numbers, reduced TCR-dependent T cell activation, and B cell activation deficiencies with hypogammaglobulinemia (OMIM 600802) (Macchi et al., 1995; Russell et al., 1995). As JAK3 is the sole JAK responsible for signaling through CD132, JAK3 deficiency largely phenocopied CD132 X-linked SCID (Lin & Leonard, 2017). In patient T cells, JAK3 deficiency results in abrogation of STAT protein phosphorylation after stimulation with all tested gamma chain cytokines, thus confirming the interrelated nature of these two PIDs.
3.4.9. STAT3
Deleterious variants in STAT3 have been linked to several separate PIDs. The first is caused by dominant negative (DN) mutations in STAT3 resulting in an autosomal dominant hyper-IgE syndrome (Job (pronounced with a long “o”) syndrome, OMIM 147060). Clinical features of Job syndrome include eczema, skin abscesses, Staphylococcus infection, recurrent fungal infections, increased IgE, and eosinophilia (Holland et al., 2007; Minegishi et al., 2007). Three concurrent reports demonstrated that DN STAT3 mutations specifically impaired the development and function of Th17 cells (de Beaucoudrey et al., 2008; Ma et al., 2008; Milner et al., 2008). Naïve T cells from Job syndrome patients were also defective in proliferation and differentiation into central memory T cells which leads to poor suppression of latently infected EBV and Zoster viruses (Siegel et al., 2011). The second PID caused by STAT3 variants is an autosomal dominant disease caused by activating STAT3 mutations which causes an infantile onset multisystem autoimmune disease (OMIM 615952) (Flanagan et al., 2014; Milner et al., 2015). Patients variably suffer from interstitial pneumonitis, autoimmune enteropathy, arthritis, eczema, T1DM, hypothyroidism, autoimmune cytopenias, hepatosplenomegaly, lymphadenopathy, and large granular lymphocytic T cell leukemia. Tregs from patients are reduced in number and functional markers, suggesting that decreased Treg function may contribute to the autoimmune disease. Interestingly, while STAT3 activity was increased, stimulation-dependent phosphorylation of STAT1 and STAT5 were diminished. The STAT1 and STAT5 defects are likely due to tachyphylaxis mediated by STAT3-driven expression of suppressor of cytokine signaling 3, thus dampening the activation of other STATs. Interestingly, pharmacological blockade of IL-6, which signals though STAT3, resulted in dramatic clinical improvement. Thus, the patient phenotype is due to an increase in STAT3 signaling and a decrease in sensitivity to other cytokines. A decrease in STAT5 activation downstream of IL-2 could explain the overlap in Treg phenotypes, which depend on IL-2 signaling, in patients with STAT5b deficiency and activating STAT3 variants (see below). Given the activating nature of the STAT3 variants in this disease, and the success of IL-6 blockade, ruxolitinib likely represents a therapeutic option for these patients.
3.4.10. STAT5b
Homozygous LOF mutations in STAT5b (activated downstream of IL-2, IL-4, and various growth hormones) are associated with an autosomal recessive disorder of growth hormone insensitivity, T cell lymphopenia, and Treg deficiency (OMIM 245590) (Cohen et al., 2006; Kofoed et al., 2003). Patient T cells do not upregulate CD25 upon stimulation to the same degree as control cells and express low levels of FOXP3 protein in freshly isolated CD25high cells, indicating loss of natural (n)Treg populations. Indeed, these Stat5b-deficient CD4+/CD25high nTregs are poorly suppressive in in vitro tests of Treg functionality. Interestingly, despite the lymphopenia and reduced nTreg activity, overt immunodeficiency or autoimmunity is so far not associated with STAT5b deficiency. It may be that STAT5a can largely compensate for STAT5b deficiency, as it does in mice (Moriggl et al., 1999). Interestingly, a recent study has identified a patient with an autoimmune lymphoproliferative syndrome (ALPS)-like disease but no growth hormone abnormality associated with a DN STAT5b variant (Majri et al., 2018).
3.5. Defects in Adhesion, Migration, or Cytoskeletal Organization
The immune system is unique in that it is not a static organ and must be able to respond to infection throughout the body. As such, cells of the immune system are highly motile and interact with multiple cell types to both migrate to their intended destination, receive activation signals, and exert effector function. These processes depend on coordinated function of the actin cytoskeleton and integrin cell adhesion molecules that are regulated downstream of both antigen receptors and GPCRs. These receptors coordinate actin responses through the activation of GTPase guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins that coordinate Rac1, CDC42, and RhoA GTP loading and thus activation of both actin nucleating complexes, such as the ARP2/3 complex, downstream of both CDC42 and Rac1, and myosin-mediated contractility, mediated by RhoA. In addition, other proteins, such as actin capping and uncapping proteins, are also regulated following receptor stimulation. These all lead to a highly dynamic actin network that is critical for T cell effector function (Comrie & Burkhardt, 2016). Here, we will review PIDs associated with defects in cytoskeletal regulators, chemokine signaling, and integrin-mediated cell adhesion (Fig. 1) (Moulding, Record, Malinova, & Thrasher, 2013).
3.5.1. RASGRP1
Homozygous LOF mutations in RAS guanyl releasing protein 1 (RASGRP1) cause recurrent bacterial and viral infections with low CD4+ T cells, B cells, and NK cells, and an inverted CD4+/CD8+ ratio with primarily effector or memory CD8+ T cells (Mao et al., 2017; Salzer et al., 2016). RASGRP1 is a Ras GEF with a DAG-binding domain that is highly expressed in hematopoietic lineages (OMIM 603962). Studies in knockout mice show reduced peripheral T cell populations due to defective Ras and extracellular signal-regulated kinase (ERK) activation leading to diminished proliferation and impaired thymocyte development (Dower et al., 2000). Conversely, overexpression of RASGRP1 enhanced ERK and Rac activation in Jurkat T cells, further solidifying the importance of RASGRP1 in this pathway (Ebinu et al., 2000). Fitting with this, CD4+ and CD8+ T cells from RASGRP1 patients had intact calcium flux responses but diminished ERK activation, CD69 upregulation, and proliferation upon TCR-induced activation (Mao et al., 2017; Salzer et al., 2016). Additionally, patient T cells had defective actin polymerization and reduced retrograde flow in migrating lymphocytes, which was associated with defects in RhoA activation and was reversible by lenalidomide treatment, which targets this pathway.
3.5.2. DOCK2
Dobbs et al. identified biallelic LOF mutations in dedicator of cytokinesis 2 (DOCK2), which encodes a hematopoietic-specific RAC1 GEF, as a cause of early onset and severe viral and bacterial infections (OMIM 616433) (Dobbs et al., 2015; Fukui et al., 2001). Rac1-GTP loading, chemotaxis, and actin polymerization downstream of chemokine receptor activation were defective in patient T cells. Perhaps more interestingly, patient NK cells had defective mitogen activation protein kinase kinase (MEK) and ERK signaling. These results are intriguing as DOCK2 and Rac1 are not known to directly affect the MEK/ERK pathway, which may suggest that DOCK2 acts as a GEF for Ras, like RASGRP1 (see above). Additionally, patient NK cells did not produce IFNγ after cytokine stimulation while patient fibroblasts did not produce interferon alpha (IFNα) following viral infection. These results may reflect altered intracellular trafficking of cytokine receptors and interferon molecules, since DOCK2 plays an essential role in molecular trafficking through regulation of microtubule dynamics (Tanaka et al., 2007). As DOCK2 activity is regulated by its association with ELMO1, and cannot assert its GEF activity in the absence of the Rac1/ELMO1/DOCK2 trimeric complex, it is possible that a similar PID could arise through LOF mutations in ELMO1 (Brugnera et al., 2002; Sanui et al., 2003; Stevenson et al., 2014).
3.5.3. DOCK8
Biallelic LOF mutations in another DOCK-180-related protein (DOCK8) cause a PID characterized by recurrent sinopulmonary and cutaneous bacterial and viral infections, and elevated IgE levels with severe atopy and anaphylaxis termed DOCK8 immunodeficiency syndrome (OMIM 243700) (Zhang et al., 2009). Patients have extremely low T and NK cell counts with poor ex vivo CD8+ T cell proliferation. This progressive loss of T and NK cells is due to both the aforementioned proliferative defects, and a failure in DOCK8-deficient T and NK cellular integrity when confined in a 3D environment, such as dermal tissue or an artificial collagen matrix, leading to cell elongation, nuclear deformation, and a form of cell death called cytothripsis (Zhang et al., 2014a). Interestingly, while DOCK8 and CDC42 knockdown resulted in a similar phenotype, knockdown of Wisckott–Aldrich syndrome protein (WASP), the main regulator of ARP2/3 downstream of CDC42, did not. This suggests that maintenance of cellular integrity in confined spaces is an actin-independent function of DOCK8/CDC42. Indeed, knockdown of PAK1/2, another downstream effector of CDC42, caused enhanced cytothripsis. Cellular elongation of DOCK8-deficient cells is accompanied by a deformation of the nucleus, suggesting that the target of PAK1/2 was critical for maintaining nuclear rigidity under mechanical stress. It is possible that PAK1/2, and by extension DOCK8, is regulating intermediate filaments such as lamins and vimentins to maintain nuclear integrity/rigidity. The lack of a chemotactic defect in DOCK8 T cells may also indicate that there is another GEF capable of regulating CDC42 and WASP activation as these are hallmarks of Wiskott–Aldrich syndrome (WAS), see below. DOCK8 may also regulate a specific subset of CDC42 effectors by acting as both a GEF and a molecular scaffold that bridges CDC42 to downstream effectors. DOCK8 and DOCK2 deficiencies also serve to highlight the great variety of immunodeficiencies that can arise from alterations to cytoskeletal dynamics, as defects in either a RAC or CDC42 GEF can cause such varied cellular and clinical phenotypes.
3.5.4. Coronin 1A
One aspect of the actin cytoskeleton in lymphocytes is its highly dynamic nature, both during migration and IS formation. In order to maintain this highly dynamic structure actin polymerization must be accompanied by turnover of old F-actin filaments and generation of G-actin monomers. The coronin family of proteins plays a role in both functions by promoting or substituting for ARP2/3 to initiate a highly flexible branched actin network while simultaneously protecting actin filaments from cofilin at the barbed end and enhancing cofilin activity and actin disassembly at the pointed end (Chan, Creed, & Bear, 2011). Coronin 1A is highly expressed in the immune system, and mice deficient in coronin 1A shows a selective loss of peripheral T cell numbers with defects in cellular polarization and migration in response to chemokine, but not TCR, stimulation (Foger, Rangell, Danilenko, & Chan, 2006). Interestingly, coronin 1A-deficient naïve T cells, but not effector or memory cells, underwent spontaneous caspase-mediated apoptosis. This increased cell death was due to high levels of polymerized actin at baseline in naïve T cells and could be reversed by actin-depolymerizing agents. Shiow et al. identified a coronin 1A E26K variant that causes coronin 1A mislocalization and peripheral T cell deficiency in mice (Shiow et al., 2008). The authors concurrently identified the first case of human coronin 1A deficiency resulting in a T−/B+/NK+ SCID phenotype with varicella infection (OMIM 615401). Identification of more patients, each with biallelic LOF mutations in coronin 1A indicated minor involvement of B and NK cells in some patients, and expanded the clinical phenotype to include EBV-associated B cell lymphoproliferation, HPV infection, extensive cutaneous warts, molluscum contagiosum, oral-cutaneous herpetic ulcers, disfiguring granulomatous tuberculoid leprosy, recurrent URT infections, and bronchiectasis (Moshous et al., 2013; Stray-Pedersen et al., 2014a; Yee et al., 2016). Like T cells from mouse models, patient T cells had increased baseline F-actin, decreased thymic output, and increased peripheral apoptosis. Interestingly, coronin 1A is known to play a role in TLR-2 signaling during Mycobacterium leprae infection, possible contributing to the patient phenotype in a T cell-independent manner (Tanigawa et al., 2009). While the role of coronin 1A in actin turnover, thymic output, and cell survival is consistently observed, there is a conflicting data concerning the importance of coronin 1A in TCR singling. Specifically, T cells from coronin 1A KO mice have a defect in TCR-mediated calcium flux this was not observed in patient’s cells and, moreover, patient cells are able to kill target cells efficiently (Shiow et al., 2008; Yee et al., 2016). How exactly increased F-actin accumulation may be linked to spontaneous apoptosis in coronin-1A-deficient T cells necessitates further study, as targeting this pathway may prove to be therapeutically beneficial to these patients. Interestingly, F-actin polymerization in coronin 1A KO cells causes high baseline JNK phosphorylation, suggesting a possible role of JNK in caspase-mediated apoptosis of these cells (Mugnier et al., 2008).
3.5.5. WASP
WASP is a hematopoietic-specific activator of the ARP2/3 complex which signals downstream of CDC42 (Thrasher, 2002). It is a good example of a previously unknown protein that was identified and functionally characterized through the study of PIDs. WASP was originally identified as the affected protein in WAS through a positional cloning strategy (Derry, Ochs, & Francke, 1994). Mutations in the WAS gene lead to three different X-linked recessive disorders: WAS, severe congenital neutropenia, and X-linked thrombocytopenia. We will discuss WAS in more detail. Depending on the causal variant, WAS can be associated with eczema, thrombocytopenia, recurrent infections, bloody diarrhea, autoimmunity, and hematopoietic malignancy. This disease usually follows a fatal course without HCST due to defects in multiple hematopoietic lineages including T, B, and NK cells (OMIM 301000). WASP is critical for T cell development, as well as multiple actin-mediated cellular functions, including microvilli formation (important for slow rolling and adhesion on endothelium), migration, actin polymerization, protein recruitment to the IS, and TCR-mediated cellular activation/proliferation (Bouma, Burns, & Thrasher, 2009; Thrasher & Burns, 2010). Of note, WASP is activated downstream of CDC42, for which DOCK8 is a critical GEF and activator. While the patients share some clinical overlap, DOCK8 cells are capable of normal chemotaxis suggesting intact cytoskeletal responses in these cells, either compensated by another CDC42 GEF or by RAC1-mediated activation of the WASP homologue, WAVE2. WASP-deficient T cells are not known to exhibit the stretched T cell morphology in 3D environments characteristic of DOCK8 T cells which supports the idea that DOCK8 deficiency involves nonactin-dependent pathogenic mechanisms.
3.5.6. WIP
WIP (WASP-interacting protein) was originally identified in a yeast two-hybrid screen for WASP-binding partners and induces actin polymerization in lymphoid cells when overexpressed (Ramesh, Anton, Hartwig, & Geha, 1997). WIP both regulates the localization of WASP and protects it from calpain-mediated cleavage and degradation (Chou et al., 2006). WIP, like WASP, regulates actin dynamics during T cell activation and is essential for both CD3/28-mediated CD25 upregulation and proliferation (Anton et al., 2002; Volkman, Prehoda, Scott, Peterson, & Lim, 2002). Given that a large percentage of WAS mutations occur within the WIP-interacting domain of WASP, it is perhaps unsurprising that homozygous WIP LOF variants cause a WAS-like immunodeficiency characterized by eczema, papulovascular lesions on the scalp, ulcerative lesions on the hard palate and tongue, thrombocytopenia, and respiratory distress (OMIM 614493) (Lanzi et al., 2012). WIP-deficient patients have reduced CD8+ T cell numbers and defective T and NK cell function. Importantly, both WIP and WASP proteins were undetectable in patient cells, and exogenous expression of WIP was able to rescue WASP expression—showing that they are obligate partners in a stable protein complex. WIP deficiency is thus an excellent example of how mutations in different proteins in the same pathway can present with identical clinical phenotypes.
3.5.7. Moesin
Hemizygous mutations in the X-linked gene encoding the Ezrin/Radixin/Moesin (ERM) family member moesin were identified in seven affected males from five kindreds suffering from profound T, B, and NK cell lymphopenia, hypogammaglobulinemia, monocytopenia, neutropenia, poor vaccine responses, bacterial infections, and zoster virus infections (OMIM 300988) (Delmonte et al., 2017; Lagresle-Peyrou et al., 2016). These variants lead to either unstable protein or truncated protein products in affected individuals. Moesin is involved in linking plasma membrane proteins to the underlying actin cytoskeleton through its FERM and actin-binding domain, respectively (Fehon, McClatchey, & Bretscher, 2010). This can control the mobility, localization, and function of key immunological regulators, such as the intercellular cell adhesion molecule (ICAM1) (Comrie, Li, Boyle, & Burkhardt, 2015). In addition, moesin regulates cortical actin networks and polarization of human T cells in response to CXCL12 (Brown et al., 2003). Moesin-deficient patient T cells are defective in antigen-induced proliferation. While conjugate formation and actin polymerization at the IS were generally intact, the authors did not look at the dynamic redistribution of ERM-binding partners during IS formation leaving open the possibility of more subtle defects in IS formation. Additionally, patient T cells had defective chemotaxis and enhanced integrin-mediated adhesion due to increased integrin expression and affinity maturation, though exactly how moesin regulates these integrin changes have not been worked out. Accompanying the patient lymphopenia was a relative loss of naïve T cells and an expansion of senescent T cells, possibly due to lymphopenia-induced expansion. It should be noted that senescent cells activate poorly, so whether the poor expansion of T cells from PBMCs represents a true signaling defect or a byproduct of increased T cell senescence is not currently known. The identification of moesin-deficient patients demonstrates the sometimes discordant phenotypes between mouse and man, as studies in moesin knockout mice have shown relatively mild immunological defects, likely due to redundant function of the related ezrin protein (Hirata et al., 2012). Nevertheless, moesin deficiency resulted in reduced thymic and bone marrow egress of T and B cells causing lymphopenia in mice. In humans, who may rely more on moesin, defects in migration from lymphoid organs may be compounded which could explain the severe lymphopenic status if hematopoietic progenitors cannot migrate to or from the thymus efficiently.
3.5.8. CXCR4
Activating mutations in the chemokine receptor CXCR4 cause an autosomal dominant disorder called WHIM syndrome (WHIMS) characterized by peripheral neutropenia due to retention in bone marrow, chronic HPV infection with skin warts, upper respiratory tract infections, and hypogammaglobulinemia (OMIM 193670). Patient mutations reduce CXCR4 internalization following ligand engagement, thus leading to enhanced stimulation by and migration toward CXCL12 (Balabanian et al., 2005; Hernandez et al., 2003; Lagane et al., 2008). As mentioned before, T cells are highly migratory and must also form a stable IS to become activated and exert effector functions. Thus, T cells must balance the two discordant functions of chemokine-driven “go” signals and TCR-mediated “stop” signals to function properly. While T cells from normal donors preferentially reduced their migratory behavior following stimulation by antigen-loaded APCs, WHIMS-T cells maintain their migratory behavior and will form shorter T/APC interactions (Kallikourdis et al., 2013). As T cells can receive chemokine-driven costimulatory signals and CXCL12 mediates aberrant costimulation of WHIM B cells, it will be interesting to see if WHIM T cells are hypersensitive to chemokine-driven costimulation (Molon et al., 2005; Roselli et al., 2017). The outcome of T cell antigen stimulation and the course of the T cell immune response in WHIMS are thus likely determined by the combined effects of enhanced chemokine-driven costimulatory signaling and reduced APC dwell time.
3.5.9. Leukocyte Adhesion Deficiencies I (CD18) and III (Kindlin-3)
Integral to cell migration and T cell effector function is the ability to adhere firmly to target cells during IS formation and endothelial cells during diapedesis (the crossing of endothelial barriers). These firm adhesions are mediated by cell adhesion molecules called integrins. There are defects in integrins in two known PIDS. In these leukocyte adhesion deficiencies, migration of both lymphocytes and neutrophils is affected leading to severe bacterial infections very early in life that are fatal if left untreated.
In leukocyte adhesion deficiency type I (LADI), biallelic LOF mutations in ITGB2, encoding the β2 integrin chain CD18 cause loss of the integrins LFA-1, Mac1, and αXβ2, all of which utilize CD18, from the surface of leukocytes (OMIM 116290). This results in defective adhesion to ICAM1 and fibronectin (Kishimoto, Hollander, Roberts, Anderson, & Springer, 1987). LADI patients develop recurrent bacterial infections, perirectal abscesses, gingivitis, periodontitis, and delayed separation of the umbilical cord primarily related to reduced neutrophil migration (Hayward et al., 1979). Currently, HSCT is required to correct this fatal PID, though gene therapy may also be an option (Bauer & Hickstein, 2000).
LADIII is caused by biallelic LOF mutations in the gene encoding kindlin-3 (OMIM 612840). LADIII is characterized by bleeding tendency due to platelet adhesion deficiency, neutropenia and lymphocytopenia, bacterial infections, pneumonia, and sepsis (Malinin et al., 2009; Pasvolsky et al., 2007; Svensson et al., 2009). Patient cells had defective integrin-mediated adhesion to and migration on ICAM1 surfaces. LADI was ruled out due to normal integrin receptor expression. LADIII cells, in contrast to LADI, show defects in adhesion mediated by multiple integrins that did not depend on CD18, including defects in adhesion mediated by LFA-1, MAC-1, VLA-4, and αVβ3. Functionally, kindlin-3 regulates both inside-out and outside-in integrin activation by binding the cytoplasmic tail of the β chain and enhancing the recruitment of talin, which links integrins to the cytoskeleton and stabilizes their activation (Hogg, Patzak, & Willenbrock, 2011). Kindlin-3 is highly expressed in the immune system, whereas kindlin-1 and -2 are expressed in nonimmune tissues, explaining why kindlin-3 mutations result primarily in an immunological phenotype. The essential role of integrins in development is reflected by the early lethality of talin and kindlin-1 knockout mice (Monkley et al., 2000; Ussar et al., 2008). LADIII was originally attributed to variants in CALDEG-GEF1 coinherited with Kindlin-3 mutations, though later studies demonstrated that overexpression of CALDEG-GEF1 could not rescue the associated phenotype, while reconstitution with Kindlin-3 rescued the adhesion and migratory defects (Pasvolsky et al., 2007; Svensson et al., 2009). This demonstrates the absolute requirement for functional validation of potentially causal variants, especially in the presence of multiple candidates that are coinherited with the disease phenotype (Abram & Lowell, 2009).
3.6. Defects in TFs/TF Regulation
TFs control the rate of genetic transcription generally by binding-specific DNA sequences or to other DNA-binding proteins. By virtue of the specificity of binding to different promotor, enhancer, or suppressor DNA sequence motifs and the expression pattern of the various TFs and signals controlling their activation, TFs play an essential role in determination of tissue-specific transcriptional patterns. In the immune system, TFs control and enforce cell fate decisions, cell activation, differentiation, survival, and memory formation. In this section, we will discuss defects in immune-specific TFs, or their regulation, that cause various PIDs. Interestingly, several of these PIDs are caused by haploinsufficiency of the relevant TF, which is likely a function of tightly controlled TF expression.
3.6.1. FOXP3
A deadly X-linked autoimmune disorder characterized by early onset and severe IBD/colitis, candidiasis, eczema, atopy, T1DM, and hypothyroidism, autoimmune cytopenias, lymphadenopathy, and various other autoimmune conditions is caused by Forkhead box P3 (FOXP3) deficiency, the causative gene for a similar phenotype in scurfy mice (Bennett et al., 2001; Brunkow et al., 2001). This condition, termed IPEX or XLAAD disease (OMIM 304790), allowed for the first characterization of the FOXP3 gene, prior to understanding its role in immune function (Chatila et al., 2000). FOXP3 is now well understood to act as the master TF initiating the development and maintenance of regulatory T cells (Hori, Nomura, & Sakaguchi, 2003). Tregs, recruited to the sites of conventional T cell activation downmodulate the immune response through multiple effector pathways including IL-2 consumption and CTLA-4-mediated inhibition of CD28 ligands on APCs, among others (Josefowicz, Lu, & Rudensky, 2012). Tregs can also promote protective immune responses, especially to certain classes of fungi, which accounts of the candidiasis that occurs in IPEX disease (Pandiyan et al., 2011). Interestingly, IPEX disease shares several clinical similarities, particularly colitis, with PIDs associated with other Treg defects such as CTLA-4 deficiency, IL-2 signaling defects, and BACH2 deficiency (see below) thus highlighting the critical nature of this cell population to the control of human autoimmunity.
3.6.2. BACH2
Heterozygous mutations in transcriptional repressor BTB and CNC homology 2 (Bach2) cause a syndrome of early-onset colitis, splenomegaly, lymphadenopathy, immunoglobulin deficiency, and respiratory tract infections called BACH2-related immunodeficiency and autoimmunity (BRIDA) due to Bach2 haploinsufficiency (Afzali et al., 2017). Patients have specific and cell-intrinsic defects in CD27+ B cell populations, and the formation of FOXP3 positive Treg cells which is attributable to reduced Bach2 protein expression. These features are also observed in bach2 heterozygous mice. Given the defect in FOXP3 expression, we speculate that defective Treg cell number or function is the cause of patient colitis and lymphadenopathy in a partial phenocopy of IPEX disease, while the B cell and antibody defects may account for the patient’s respiratory infections. These effects are likely due to loss of Bach2-mediated transcriptional repression of Blimp1 or another Bach2 target, though the full transcriptional profile of Bach2-deficienct patients has yet to be determined.
3.6.3. BCL11B
An autosomal dominant disease caused by a heterozygous DN variant in B cell CLL/lymphoma 11B (BCL11B) causes a leaky SCID phenotype with syndromic features (OMIM 617237). Immunologically the patient had T cell lymphopenia, absent TREC formation and naïve CD4+ T cell populations, and impaired proliferative responses (Punwani et al., 2016). BCL11B encodes for a TF with multiple roles in T cell formation and function (Avram & Califano, 2014). BCL11B positively regulates T cell lineage commitment while inhibiting pluripotency during thymopoesis, a process that may determine the T cell-specific immunological phenotype. BCL11B also aids in the expansion of T cells in the periphery and induction of effector function, accounting for decreased patient T cell proliferation to mitogen.
3.6.4. NF-κB Family of TFs
The NF-κB family of TFs and their regulatory proteins are critical for the initiation and control of innate and adaptive immune responses, and over 20 PIDs have been associated with various defects in this pathway. As these PIDs have been reviewed recently and extensively, we will only discuss PIDs that were not included in the review by Zhang, Lenardo, and Baltimore (2017).
3.6.5. RELA
Badran et al. and Comrie et al. have described an autosomal dominant disorder caused by p65/RELA haploinsufficiency (Badran et al., 2017; Comrie et al., 2018). In the first report, the authors describe colitis and mucocutaneous ulcerations, both due to increased TNF-induced epithelial cell apoptosis. These patients benefited greatly pharmacological TNF inhibition. In contrast, Comrie et al. describe a patient with lymphadenopathy, autoimmune cytopenias, and increased T cell proliferation and effector function. While it may be too early to tell the full clinical spectrum caused by RELA haploinsufficiency, it should be noted that this variable clinical expressivity is similar to that described for p50 haploinsufficiecny (Badran et al., 2017). This can be contrasted with the relatively homogenous clinical phenotype of other patients with NF-κB-related PIDs, such as NEMO patients. This difference may arise because in p50 or RELA haploinsufficiency, the constellation of affected genes due to insufficient TF levels may largely depend on infectious history or genetic background, while patients with defects in NF-κB activators or effectors may more uniformly alter NF-κB-dependent transcriptional profile, independent of confounding determinants.
3.6.6. CARD11
CARD11 mutations cause both autosomal recessive (OMIM 615206) and autosomal dominant (OMIM 616452) disorders due to LOF and GOF mutations, respectively (Zhang et al., 2017). GOF mutations result in spontaneous CARD11 aggregation and NF-κB activation in B and T cells in B cell expansion with NF-κB and T cell anergy (BENTA) disease (Snow et al., 2012). While these GOF variants lead to spontaneous B cell activation, they result in diminished T cell responsiveness to TCR-mediated stimulation in the absence of appropriate costimulation, a form of stimulation that is known to cause T cell anergy. Recently an autosomal dominant PID associated with CARD11 hypomorphic mutations that cause dominant interference with the WT CARD11 allele-dependent activation of NF-κB has been described (OMIM 617638) (Ma et al., 2017). These patients presented with severe atopic dermatitis, allergies, elevated IgE, molluscum contagiosum infection, decreased B cell numbers, and defective T cell activation. Decreased NF-κB activation leads to reduced CD25 upregulation and proliferation following TCR-mediated T cell stimulation. Why this disease results in a different clinical phenotype than biallelic CARD11 LOF disease remains to be seen, though the DN mutations may allow for some residual CARD11 activity, possibly with altered kinetics, while LOF variants result in complete loss of CARD11-dependent NF-κB induction downstream of antigen receptors.
3.7. Defects in Apoptotic Pathways
Immune responses pose a particularly unique challenge in that T and B cells must expand exponentially to successfully combat infection; however, this expansion must also be reversed to achieve homeostasis following the resolution of infection. This contractile phase is partly due to cytokine withdrawal-induced apoptosis, in which T cell populations undergo apoptosis due to passive loss of proproliferative/prosurvival signals. Additionally, T cell populations are actively reduced through TCR signals, mediated in part by TNF-family receptors, which induce apoptosis (Zheng, Li, & Lenardo, 2017). Additionally, apoptosis must be inhibited in resting cells to prevent aberrant loss of cell populations. In this section, we will discuss PIDs associated with defects in the initiation and control of apoptosis and other forms of regulated cell death.
3.7.1. Somatic Defects in Cytokine Withdrawal-Mediated Apoptosis (NRAS/KRAS)
Following the clearance of pathogen effector cells no longer produce enough of the prosurvival/proliferative cytokine IL-2 to maintain the expanded populations and effector populations undergo growth factor/cytokine withdrawal-mediated apoptosis, shrinking the peripheral T cell pool to the size that can be maintained by homeostatic cytokines, such as IL-7 and IL-15. Somatic heterozygous activating variants in the Ras homologues NRAS and KRAS cause “RAS-associated leukoproliferative disorder,” or RALD (OMIM 614470) (Niemela et al., 2011; Oliveira et al., 2007; Takagi et al., 2011). These mutations lead to a clinical disease similar to ALPS (see below) with hepatosplenomegaly, autoimmune cytopenias, and lymphadenopathy caused by defective apoptosis following the acute removal of IL-2 from T cell in vitro cultures. This was due to NRAS/KRAS-mediated inhibition of BCL-2-interacting mediator of dell death (BIM) protein expression and could be rescued by inhibition of farnesylation, which is required for NRAS and KRAS localization and function, or PI3K/ERK inhibitors, two downstream RAS effectors. Importantly, germline activating mutations in NRAS and KRAS can cause Noonan syndrome, a developmental disorder with no appreciated immunological phenotype, though these mutations may be less activating than those causing RALD (Cirstea et al., 2010). It may be that the GOF mutations seen in RALD would otherwise be lethal if inherited in the germline configuration.
3.7.2. Death-Induced Signaling Complex Members (FAS/FASL/Caspase8/Caspase10/FADD)
One of the best described group of related human PIDs associated with apoptotic defects are caused by deleterious variants in the FAS signaling pathway that gives rise to ALPS. ALPS is characterized by nonmalignant lymphadenopathy and splenomegaly, expansion of a CD4−/8− αβ T cell population, development of autoimmune cytopenias, and defective FAS-induced apoptosis in vitro (Price et al., 2014). The FAS receptor (FAS) activation by FAS ligand (FASLG) results in trimerization of the receptor subunits and recruitment of Fas-associated protein with death domain (FADD) to the cytoplasmic tail. FADD bridges FAS to procaspases 8 and 10 to form the death-induced signaling complex (DISC) leading to the activation of these caspases and the induction of apoptosis (Peter & Krammer, 2003). FAS and FASLG are greatly upregulated after T cell activation and serve as an important checkpoint on T cell population size. As such, mutations in each component of the DISC complex have been associated with defective FAS-induced death and the development of ALPS or ALPS-like conditions (Rieux-Laucat, Le Deist, & Fischer, 2003).
In the most common form of ALPS, dominant-interfering mutations in FAS give rise to an autosomal dominant disease due to the incorporation of defective FAS chains into the FAS receptor trimers (OMIM 601859) (Fisher et al., 1995; Martin et al., 1999). The majority of these ALPS-causal variants result in the inability of FAS trimers to recruit FADD, though some inhibit the palmitoylation-dependent recruitment of FAS to lipid raft microdomains and an inability to fully process caspase-8 despite normal DISC formation (Cruz et al., 2016; Siegel et al., 2004). Importantly, FAS mutations can arise from both germline and somatic mutations, and thus represent Mendelian and non-Mendelian forms of the disease, respectively (Oliveira et al., 2010). Remarkably, for reasons that are obscure, patients with ALPS due to FAS mutations have very high levels of serum vitamin B12 which is almost pathognomonic for disease and can be used as a diagnostic tool when molecular confirmation is difficult to obtain, though direct confirmation of defects in FAS-mediated apoptosis remains an excellent clinical diagnostic approach (Lo et al., 2013). Dominant-interfering (OMIM 601859) and LOF mutations in FASLG cause autosomal dominant and autosomal recessive disease, respectively, though both are much less common than ALPS-associated FAS mutations (Magerus-Chatinet et al., 2013; Wu et al., 1996). Mutations in caspase 10 can generate a dominant-interfering protein that results in an autosomal dominant form of ALPS (OMIM 603909) (Wang et al., 1999). LOF mutations in caspase 8 also impair FAS-induced apoptosis as well as TCR- and BCR-induced activation of NF-κB (Chun et al., 2002; Su et al., 2005). As such, caspase 8-deficient patients suffer from lymphadenopathy, splenomegaly and other features reminiscent of ALPS combined with recurrent infections due to defects in NF-κB activation that are not seen in the caspase 10 patients. This unique clinical entity is called “caspase-8 deficiency syndrome” or CEDS (OMIM 607271). An autosomal recessive disease with a mild-ALPS phenotype and additional clinical features including severe bacterial and viral infections, recurrent hepatopathy and encephalopathy, as well as cardiac malformations results from biallelic LOF mutations in FADD (OMIM 613759) (Bolze et al., 2010). Despite the fact that cells from these patients are defective in FAS-mediated apoptosis only one patient has presented with ALPS-like features (elevated double negative T cells, high serum FASL, and high IL-10, with no lymphadenopathy), though the other three patients passed away prior to evaluation for these conditions. The absence of lymphadenopathy may be related to the importance of FADD in TCR-mediated T cell proliferation which likely serves to limit lymphoproliferation and T cell populations in FADD-deficient patients (Zhang, Cado, Chen, Kabra, & Winoto, 1998). Additionally, FADD-deficient cells were less susceptible to viral-mediated cell death and supported higher viral loads upon infection due to defective IFN signaling. Though not investigated, FADD does play a role in TNF signaling, which may account for the increased susceptibility to bacterial infections.
3.7.3. XIAP
In a disease whose molecular pathogenesis is related to that of ALPS, LOF mutations in X-linked inhibitor of apoptosis (XIAP) cause an X-linked recessive lymphoproliferative syndrome with recurrent infections, EBV viremia, fever, hemophagocytic lymphohistiocytosis, IBD, and loss of NKT cells (OMIM 300635) (Aguilar & Latour, 2015; Rigaud et al., 2006; Worthey et al., 2011). XIAP is thought to act by inhibiting caspase function downstream of death receptor engagement and can block FAS-mediated apoptosis (Bratton, Lewis, Butterworth, Duckett, & Cohen, 2002; Holcik & Korneluk, 2001). Functionally, XIAP-deficient patient cells show increased apoptosis mediated by FAS, TNSF10, and RICD. Thus, XIAP deficiency results in an inability to control infection through increased FAS-mediated death of effector cells, and enhanced RICD-mediated death of antigen-specific cells leading to functional immunodeficiency. XIAP also plays an important role in pattern recognition receptor signaling, which may contribute to the susceptibility to infection and early-onset IBD in these patients.
3.7.4. STK4
Serine/threonine-protein kinase 4 (STK4) is a proapoptotic and antiproliferative kinase best known for its involvement in the Hippo signaling pathway necessary for organ size control (Harvey, Zhang, & Thomas, 2013). This is accomplished by initiating a cellular signaling cascade that ultimately results in the phosphorylation and degradation of the transcriptional coactivators YAP/TAZ and loss of their proproliferative and antiapoptotic functions. Additionally, STK4 is required for the phosphorylation and activation of LC3 during autophagy, thus allowing for the fusion of autophagosomes with lysosomes (Wilkinson et al., 2015). Biallelic LOF mutations in STK4 cause an autosomal recessive PID characterized by atrial septal defects, progressive loss of T and B cells resulting in T/B lymphopenia, and intermittent neutropenia leading to the development of recurrent viral, bacterial, and fungal infections (OMIM 614868) (Abdollahpour et al., 2012). Fas- and staurosporine-induced apoptosis in T cells and neutrophils, respectively, was dramatically increased in patient cells compared to controls. This was associated with loss of plasma membrane potential and decreased Forkhead box 03 protein (Foxo3) expression. As STK4 phosphorylation, activation, and stabilization of Foxo3 are critical for the protection of naïve T cells from the damaging effects of reactive oxygen species (ROS) in mice, it is likely that STK4 regulation of Foxo3 is a critical regulator of the human disease (Choi et al., 2009). Additionally, STK4-deficient patient T cells do not divide well, primarily due to a massive increase in apoptosis associated with TCR stimulation, as well as decreased Bcl2 and Foxo1 expression (Nehme et al., 2012). Together, this suggests that STK4 deficiency results in increased T cell apoptosis through regulation of STK4 targets outside of the Hippo pathway, where STK4 normally promotes cell survival by activating Foxo3 and inducing antiapoptotic and anti-ROS target genes. Notably, activation of the Hippo pathway in T cells and induction of LC3 activation was not described in any of these studies, and altered Hippo signaling or decreased autophagy cannot be ruled out in this disease without further investigation. Interestingly, STK4 is most highly expressed in naïve T cells, which implies its loss may sensitize cells to RICD and Fas-mediated apoptosis in activated T cells as part of the normal immune response.
3.8. Defects in DNA Replication, Accessibility, Repair, and Telomere Maintenance
As a necessity of function, T and B cells, as well as their hematopoietic progenitor cells, undergo both constitutive division to maintain cell populations and rapid expansion during selection and immune responses. As such, they depend heavily on the ability to quickly and efficiently replicate DNA. During periods of such rapid proliferation and DNA synthesis, there is a high chance for DNA damage to occur. In these cases, repair of DNA damage is essential for cell cycle progression. Additionally, repeated cellular division of human immune cells often leads to shortened telomeres, an outcome of certain lymphoproliferative diseases and the natural aging of the immune system which characterizes a senescent cellular phenotype in aged T cells and in some PIDs. Because of this, maintenance of telomere length is critical to maintain hematopoietic lineages. Finally, in addition to DNA replication/repair/chromosomal stability, DNA accessibility must also be regulated to allow for specialized immune cell function. Defects in these processes can cause various PIDs and are discussed below.
3.8.1. POLEε
DNA polymerase epsilon (POLEε), a DNA polymerase composed of four subunits, synthesizes the forward strand during conventional DNA synthesis. Homozygous LOF mutations in POLE1, the catalytic subunit of POLEε, cause a syndromic immunodeficiency with facial dysmorphism, livedo, and short stature (FILS syndrome, OMIM 615139) (Pachlopnik Schmid et al., 2012). Immunologically, patients suffer from recurrent upper respiratory tract infections, pulmonary infections, bronchiectasis, and bacterial meningitis. Patients have decreased B cell and immunoglobulin levels, and a diverse T cell repertoire, but low naïve T cell counts and decreased T cell proliferation. Patient T cells proliferate poorly in response to TCR stimulation with fewer cells entering S phase, likely due to stalled/slow DNA replication. Unlike other cell types, T and B cells express more POLEε than POLEδ, a related DNA polymerase, suggesting that the T and B cell specificity of this immunodeficiency is a result of a cell-specific dependence on POLEε. Biallelic LOF mutations in the second subunit of POLEε, POLE2, have been described in a patient with facial dysmorphisms, combined immunodeficiency, and autoimmunity (Frugoni et al., 2016). The patient suffered from omphalitis, erythroderma, recurrent respiratory tract infections, T1DM, hepatomegaly, and hypothyroidism due to combined B and T cell defects including agammaglobulinemia, absence of circulating B cells, and T cell lymphopenia with reduced naïve and expanded effector memory cell populations. Undetectable T cell excision circles indicated decreased thymic output. The latter likely leads to lymphopenia with concomitant expansion and outgrowth of poorly selected autoreactive cells, thus accounting for the patients’ autoimmune phenotype. Also observed was a defect in cell cycle progression with reduced percentage of cells in S phase, similar to POLE1 patient cells.
3.8.2. DNA Ligase 1
While POLEε is required for forward-strand synthesis, DNA ligase 1 is required for joining of Okazaki fragments in the lagging strand. Biallelic LOF missense variants in DNA ligase 1 were found in a cell line derived from a single patient presenting with retarded growth, sun-sensitive facial erythema, and immunodeficiency characterized by decreased serum immunoglobulin levels, poor mitogen responses, and a deadly course of respiratory tract infections (Webster, Barnes, Arlett, Lehmann, & Lindahl, 1992). Though immunological samples were not available for further study, it is likely that DNA ligase 1 and POLEε patients share a related cellular phenotype, given the clinical similarities.
3.8.3. MYSM1
Myb like, SWIRM and MPN domain 1 (MYSM1) is an essential component of an H2A deubiquitinase complex that coordinates histone H1 disassociation and DNA accessibility, thus allowing for transcription initiation and elongation (Bahrami et al., 2017). Homozygous LOF mutations in MYSM1 cause a syndrome of progressive bone marrow failure, immunodeficiency, and developmental abnormalities. Immunologically, patients suffer from recurrent respiratory tract infections, with virtually absent mature and class-switched B cells, and reduced T and NK cells. Furthermore, T cells failed to proliferate robustly in response to mitogenic challenge (Alsultan, Shamseldin, Osman, Aljabri, & Alkuraya, 2013; Bahrami et al., 2017; Le Guen et al., 2015). MYSM1-deficient cells have increased p38 activation, cell cycle arrest, and increased apoptosis, presumably due to altered transcriptional activity. In one patient, a spontaneous genetic reversion caused recovery of all affected hematopoietic lineages, suggesting a strong survival advantage to WT cells over mutant cells. The exact genetic targets regulated by MYSM1 remain to be identified and may shed light onto the mechanism by which MYSM1 deficiency results in increased p38-induced apoptosis.
3.8.4. ATM
Biallelic LOF mutations in ataxia–telangiectasia mutated (ATM) serine/threonine kinase results in ataxia–telangiectasia (AT), characterized by cerebellar ataxia, telangiectasia, immune defects, and malignancy (OMIM 208900). ATM is recruited to, and activated by, sites of DNA double-strand breaks and is critical for the DNA damage response comprising inhibition of cell cycle progression, DNA repair, and apoptosis. The immune defects accompanying ATM deficiency include thymic hypoplasia, lymphopenia, reduced T cell numbers, and reduced B cell differentiation (Chopra et al., 2014; Staples et al., 2008). This is likely due to an inability to respond to double-strand breaks that occur during thymopoesis and T cell selection, resulting in apoptosis of T cell progenitors. Interestingly, AT patients present with a relative increase in γδ T cells compared to αβ T cells (Carbonari et al., 1990). This may reflect a less severe requirement of γδ T cells for ATM during development, though this remains to be seen.
3.8.5. Nibrin
Nibrin (NBN) deficiency results in the autosomal recessive disorder Nijmegen breakage syndrome, which is associated with recurrent pneumonia, GI infections, urinary tract infections, with reduced T cell numbers (OMIM 251260) (Varon et al., 1998). NBN, a component of the Mre11-Rad50-Nbs1 (MRN) complex, is critical for the response to DNA damage and maintenance of chromosomal integrity and telomere length (Lamarche, Orazio, & Weitzman, 2010). Interestingly, NBN activates both ATR and ATM by phosphorylation during DNA damage responses, placing it upstream of ATM in the DNA damage response pathway (Lee & Paull, 2004; Stiff et al., 2005). Consistent with this, Nijmegen breakage syndrome shares similar features to both AT and Seckel syndrome 1, an autosomal recessive disease characterized by multiple developmental abnormalities, but no known immunological features, due to hypomorphic ATR variants. Given the related nature of ATM and ATR kinases, and their respective roles in DNA damage responses, it is curious as it why ATM and NBN, but not ATR, deficiencies result in immunological deficiencies.
3.8.6. SMARCAL1
Biallelic LOF variants in SWI/SNF related, matrix-associated, actin-dependent regulator of chromatin, subfamily A-like 1 (SMARCAL1) cause an autosomal recessive disease characterized by several immunological defects including neutropenia, lymphopenia, and cytopenias with decreased T cell numbers and absent mitogenic responses, largely phenocopying ATM- and NBN-associated disease (OMIM 242900). SMARCAL1 is an ATP-driven annealing helicase involved in the reannealing of stably unwound DNA. It may also play an important role in DNA damage by maintaining genomic integrity at stalled replication forks and is activated by the ATM kinase, placing it downstream of NBN and ATM in the DNA damage response (Bansbach, Betous, Lovejoy, Glick, & Cortez, 2009; Boerkoel et al., 2002; Yusufzai & Kadonaga, 2008). Interestingly, ATR can also phosphorylate SMARCAL1 which is necessary for SMARCAL1-mediated prevention of replication fork collapse during DNA damage response deficiencies (Couch et al., 2013). This suggests that the ATR kinase may also play an important role in DNA damage response pathways in T cells, which may simply not be observed in Seckel syndrome I due to the incomplete loss of ATR function in this disease (OMIM 610600).
3.8.7. NSMCE3
Biallelic LOF variants in NSMCE3 cause an autosomal recessive syndrome immunologically characterized by thymic hypoplasia, recurrent pulmonary infections, and eczema (OMIM 617241) (van der Crabben et al., 2016). NSMCE3-deificient patients have severely reduced T cell numbers, with reduced T cell responses to multiple mitogenic and recall response stimuli. NSMCE3 is a critical component of the SMC5/6 DNA repair complex and supports mitotic proliferation. Defects in both functions likely contribute to the observed T cell defect, though the exact stages of T cell development and T cell activation that are affected remain to be elucidated.
It is interesting that not all genetic disorders caused by defects in DNA repair mechanisms are accompanied by a T or B cell-mediated immunodeficiency. Gene mutations affecting many members of the Fanconi anemia complex cause a bone marrow failure syndrome but are not associated with decreased T cell counts or function. Similarly, LOF variants in the gene ERCC6L2 which encodes a novel DNA repair factor, termed Hebo, cause a bone marrow failure syndrome without an appreciable T or B cell deficit (Tummala et al., 2014; Zhang et al., 2016). This would suggest that there is a special role for the ATM checkpoint kinase pathway in mediating T cell development or function, given the majority of defects in DNA damage response that cause T cell deficiency fall within this pathway.
3.8.8. Defects in DNA Methylation (DNMT3B, ZBTB24, CDCA7, and HELLS)
Mutations in four different proteins cause immunodeficiency-centromeric instability-facial anomalies syndrome (ICF1–4), which is characterized by a Tlow/NKlow/B+ CVID with reduced immunoglobulin levels and susceptibility to recurrent respiratory tract infections. Biallelic LOF mutations in the genes DNA methyltransferase 3 beta (DNMT3B), zinc finger and BTB domain containing 24 (ZBTB24), cell division cycle-associated protein 7 (CDCA7), and helicase, lymphoid specific (HELLS) are responsible for ICF1–4, respectively (OMIM 242860, 614069, 616910, and 616911) (de Greef et al., 2011; Hansen et al., 1999; Thijssen et al., 2015). While DNMT3B, ZBTB24, and HELLS play important roles in de novo DNA methylation and maintenance of DNA methylation, accounting for their shared phenotype, CDCA7’s best known role is as a Myc-induced positive regulator of Myc activity, whose activity can be downregulated by Akt-mediated phosphorylation (Gill, Gabor, Couzens, & Scheid, 2013; Xu et al., 1999). Interestingly, Myc can repress transcription by recruiting the DNA methyltransferase DNMT3A, a close homologue of DNMT3B (Brenner et al., 2005). This raises the possibility that all four proteins function within the same pathway, DNMT3B, ZBTB24, and HELLS directly regulating DNA methylation, while CDCA7 is required for Myc-dependent recruitment of one or more of these repressors. While the transcriptional repercussions of deficiencies in these proteins are likely to overlap with one-another, the mediators of the disease phenotype remain largely unknown.
3.8.9. Defects in Telomere Maintenance (DKC1 and RTEL1)
Telomeres are the hexapeptide repeats on the ends of chromosomes and associated protein complexes. Proper-telomere length is an actively maintained property and with multiple cell divisions resulting in telomere shortening opposed by the action of a family of proteins called telomerases that add the hexapeptide repeats to the 3′ end of telomeres. Proper length maintenance is critical, especially in quickly dividing cells, because telomeres that become too short result in the initiation of apoptosis or cellular senescence (Hodes, Hathcock, & Weng, 2002). In fact, short telomeres are a marker of multiple PIDs associated with hyperactivation and accumulation of senescent cells, such as PASLI. First identified in a screen of patients with X-linked recessive dyskeratosis congenital (XL-DC, OMIM 305000) and short telomeres, dyskerin pseudouridine synthase (DKC1) is an obligatory component of the telomerase complex (Heiss et al., 1998; Mochizuki, He, Kulkarni, Bessler, & Mason, 2004). In Hoyeraal–Hreidarsson syndrome, a severe early-onset form of XL-DC, a missense mutation in DKC1 was correlated with a complete absence of B and NK cells, with mild T cell defects (Cossu et al., 2002). Defects in another protein involved in telomere maintenance, RTEL1, causes decreased telomere length, bone marrow failure, and a severe B cell immunodeficiency, again with mild T cell defects (OMIM 615190) (Le Guen et al., 2013; Walne, Vulliamy, Kirwan, Plagnol, & Dokal, 2013). In a review of dyskeratosis congenital focused on immunological phenotypes, roughly 50% of patients had decreased T cell numbers (Solder, Weiss, Jager, & Belohradsky, 1998). Longitudinal studies in these patients may be particularly interesting, because telomere length in peripheral T cell populations is well known to shorten during the natural aging process. Thus, patients with DKC1 or RTEL1 mutations may have exacerbated T cell telomere loss and acquisition of senescent T cells as they age, even if this was not observed upon initial clinical presentation.
As with mediators of the DNA damage response, many more genetic defects in telomere maintenance result in bone marrow failure and cytopenias with no appreciated T or B cell defects. One possible reason for the presence of bone marrow failure, but not T or B cell defects, is that the progressive disease may largely take place after the development of a naïve T and B cell repertoire sufficient to fill the peripheral compartments.
3.9. Vesicular Trafficking and Protein Sorting
Mutations in genes that regulate the formation, translocation, fusion, or function of lytic granules can commonly impact the function of multiple immunological subsets, including the cytolytic activity of CD8+ cytotoxic T lymphocytes (CTLs), and ultimately cause PIDs with overlapping clinical features.
3.9.1. HPS2
An autosomal recessive form of Hermansky–Pudlak syndrome with immunodeficiency, including recurrent lung infections, recurrent bacterial infections, thrombocytopenia, with neutrophil, NK, and CTL functional defects (HPS2) is caused by biallelic LOF mutations in AP3B1 encoding the beta3A subunit of the adaptor protein (AP)-3 complex (OMIM 608233) (Dell’Angelica, Shotelersuk, Aguilar, Gahl, & Bonifacino, 1999; Huizing et al., 2002; Jung et al., 2006). Deficiency of this subunit results in diminished protein stability of the other complex subunits. Loss of the AP-3 complex causes enlargement of lytic granules in CD8+ T cells and their inability to move along microtubules to coalesce at the microtubule organizing center (MTOC) prior to MTOC polarization (Clark et al., 2003). This step is essential for granule delivery to the IS and failures to coalesce at the MTOC abolishes CTL cytolytic capacity. While CTL deficiency likely accounts for the immunodeficiency seen in HSP2 cases, trafficking of lysosomes in general is also perturbed, accounting for the wider HPS phenotype. While mutations in AP3B1 have been described in HPS2, it remains possible that mutations in other subunits of the AP-3 complex will cause a similar phenotype.
3.9.2. Lytic Granule Functional Deficiencies (Perforin-1, UNC13, MUNC13–4, Syntaxin-11, MUNC18–2, RAB27a, and LYST)
Familial hemophagocytic lymphohistiocytosis (FHL) is a hyperinflammatory state caused by the uncontrolled activation of lymphocytes and macrophages. In total, there are six known genetic causes of FHL, all encoding protein products critical for NK and CTL cytolytic function (OMIM PS267700). These proteins each provide a nonredundant function in the synthesis, trafficking, docking, priming, fusion, or effector function of cytotoxic granules, and loss of any single one results in defects in cytolytic activity. The inability to kill target cells results in prolonged APC/T cell contacts, hyperactivation and proliferation of CTLs, secretion of large quantities of IFNγ, macrophage activation, and cytokine storm, which together account for the patient phenotype (Sepulveda & de Saint Basile, 2017). FHL2 is an autosomal-recessive form of disease caused by biallelic mutations in Perforin-1, a pore-forming molecule packaged in cytolytic granules and excreted into the synaptic space where it acts on target cell membranes to initiate CTL-mediated killing (Podack et al., 1988; Stepp et al., 1999; Voskoboinik, Whisstock, & Trapani, 2015). Biallelic LOF variant in UNC13D, encoding Munc13–4, cause FHL3, where Munc13–4 plays an essential role in lytic granule fusion, but not granule polarizing or docking with the IS (Feldmann et al., 2003; Santoro et al., 2006). Munc13–4 is a Rab27a effector essential for a priming step prior to lytic granule fusion, likely by mediating Ca2+-dependent SNAP (soluble NSF attachment protein) receptor (SNARE) complex formation (Boswell et al., 2012; Menager et al., 2007). FHL4 is caused by LOF mutations in the gene encoding Syntaxin-11, an atypical member of the q-SNARE family required for SNARE complex formation and lytic granule fusion (Kogl et al., 2013; Muller et al., 2014; zur Stadt et al., 2005). Syntaxin-11 interacts with Munc18–2, whose loss causes FHL5 (Cote et al., 2009; zur Stadt et al., 2009). Munc18–2 is necessary for the localization of syntaxin-11 to the plasma membrane (Dieckmann, Hackmann, Arico, & Griffiths, 2015). Some causal mutations in both Syntaxin-11 and Munc18–2 abrogate the interaction between the two proteins, highlighting the requirement for both proteins in lytic granule function. Biallelic LOF variants in Rab27a and LYST also cause FHL in the context of Griscelli syndrome (OMIM 607624) and Chediak–Higashi syndrome (CHS, OMIM 214500), respectively (Menasche et al., 2000; Nagle et al., 1996). Rab27a, in addition to regulating Munc13–4, is required for the movement of lytic granules from the polarized MTOC to the plasma membrane, the last step prior to granule docking at the plasma membrane (Stinchcombe et al., 2001). The function of LYST, a large beach domain containing protein, remains poorly understood, though it may play important roles in regulating the general biogenesis, structure, and turnover of endosomal compartments, including lytic granules (Holland, Torgersen, Sandvig, & Simonsen, 2014; Sepulveda et al., 2015). Interestingly, LYST deficiency in CHS is also associated with defects in CTLA4 trafficking, where it accumulates in large endosomes, suggesting a possible role of CTLA4 deficiency in the lymphoproliferation seen in CHS (Barrat et al., 1999).
3.9.3. LRBA
An autosomal recessive disorder caused by LOF mutations in the LPS-responsive beige-like anchor protein (LRBA) leading to complete loss of protein expression was first identified in a large consanguineous family with early onset IBD (EO-IBD) and combined immunodeficiency with autoimmunity (OMIM 614700). Patients with LATAIE (LRBA deficiency with autoantibodies, Treg defects, autoimmune infiltration, and enteropathy) disease had various combinations of early onset diarrhea, autoimmune pancytopenia, EBV-associated lymphoproliferation, and vitamin B12 deficiency. While some patients had low B cell and IgG/IgA levels, this was not consistent across all patients (Alangari et al., 2012). Further characterization of additional LATAIE patients expanded the clinical phenotype to include chronic lung infections and bronchiectasis and highlighted the importance of LRBA for B cell activation and class-switched antibody secretion (Lopez-Herrera et al., 2012). LRBA deficiency also caused decreased numbers of FOXP3+ Tregs that had lower expression of the Treg functional markers CTLA4/CD25 and impaired inhibitory function. This was associated with increased memory T cell accumulation (Charbonnier et al., 2015). Extended analysis of a large cohort of LATAIE patients demonstrated that most patients develop some combination of immune dysregulation, lymphadenopathy, infections, and hypogammaglobulinemia with defects in Tregs, B cell class switching, and plasmablast formation (Gamez-Diaz et al., 2016). Lo et al. discovered that LRBA associates with CTLA4 via the YVKM motif in the CTLA4 cytoplasmic tail and prevents its proteolytic turnover. Specifically, LRBA prevents the association of CTLA4 with AP-1, thus restricting its localization to recycling endosomes (Lo et al., 2015). In the absence of LRBA, CTLA4 is shuttled preferentially to the lysosomes where it is degraded. Critically, pulmonary lymphocytic infiltrates responded favorably to abatacept therapy, suggesting that the decrease in CTLA4 expression was responsible for the autoimmune symptoms. Interestingly, defects in autophagy and mTOR activation were also demonstrated in LRBA-deficient cells, suggesting additional roles of LRBA in regulation of the immune response (Charbonnier et al., 2015; Lopez-Herrera et al., 2012). Further analysis of LRBA deficiency will be required to understand how LRBA mediates its B cell-intrinsic functions and to what degree this is influenced by the loss of CTLA4 and consequent dysregulation of T cell immunity vis-à-vis the defects in autophagy and mTOR activation.
3.9.4. UNC119
Recently a 32-year-old female was described with an autosomal dominant disease consisting of shingles, oral hepatic lesions, otitis media, persistent fungal infections, and pulmonary infections with an idiopathic CD4+ T cell lymphopenia (OMIM 615518). The patient’s disease was associated with a heterozygous dominant-interfering mutation (G22V) in the unc-119 homologue A protein (UNC119) (Gorska & Alam, 2012). UNC119 acts as a trafficking chaperone for multiple proteins and is involved in endosome recycling through the activation of Rab11 (Jaiswal et al., 2016; Kobayashi, Kubota, Mori, McLaren, & Inana, 2003; Zhang et al., 2011). In T cells, UNC119 is recruited to the IS and interacts specifically with Lck via a proline-rich motif in UNC119 and the SH3 domain of Lck in an activation-dependent manner. This interaction is critical for TCR stimulation as the UNC119/Lck interaction directly contributes to the activation of this keystone src kinase during TCR signaling (Gorska, Stafford, Cen, Sur, & Alam, 2004). UNC119 is also critical for the proper trafficking of Lck to the IS since Lck will accumulate in Rab11+ endosomes in the absence of UNC119. This defect is likely due to loss of UNC119-mediated Rab11 activation and it can be overcome through the expression of constitutively active Rab11 or UNC119 reconstitution (Gorska, Liang, Karim, & Alam, 2009). The G22V mutation is unable to bind Lck, presumably because the mutation is directly N-terminal to the polyproline motif required for Lck association and activation. In overexpression systems, and in patient cells, G22V UNC119 caused decreased membrane targeting of Lck and increased Lck localization to Rab11+ endosomes, leading to reduced Lck activation and poor proliferative responses of patient T cells (Gorska & Alam, 2012). The two functions (UNC119-dependent Lck recruitment and activation of Rab11) may be linked as UNC119-mediated Lck recruitment to Rab11 endosomes may initiate Src kinase-dependent activation of Rab11 (Lee et al., 2013). Interestingly, heterozygous LOF UNC119 mutations cause cone-rod dystrophy and retinal degeneration without any immunological phenotype, suggesting multiple disease phenotypes depending on the nature of the UNC119 mutation (Kobayashi et al., 2000). Given how important proper trafficking and activation of Lck is to T cell function, it will be interesting to see if mutations in other proteins involved in the regulation of Rab11+ vesicular trafficking (such as family of Rab11-interacting protein (FIP)3) are associated with undiscovered PIDs (Bouchet et al., 2016, 2017).
3.10. Defects of Ubiquitination
3.10.1. ITCH
Ubiquitination of proteins can target them for proteasomal degradation, mediate protein–protein interactions, or alter activity and cellular localization. As such, proper regulation of ubiquitination is essential for cellular function and dysregulation of this pathway has been implicated in several human diseases (Popovic, Vucic, & Dikic, 2014). Several patients have been identified from an Amish cohort with homozygous truncating mutations in the E3 ubiquitin ligase, ITCH (OMIM 613385) (Lohr et al., 2010). These patients present with hepatosplenomegaly, chronic lung disease, and recurrent diarrhea with severe lymphocytic infiltrates. While more extensive immunophenotyping of T cells and other immune cell types is required to confirm T cell defects in humans, ITCH-deficient mice develop skewed T cell responses and T cell-dependent autoimmunity (Fang et al., 2002; Parravicini, Field, Tomlinson, Basson, & Zamoyska, 2008). Thus it is likely that these patients will have some level of T cell dysfunction.
3.11. Defects of Glycosylation
Proper glycosylation, the enzymatic process of attaching a carbohydrate to a molecule, is generally essential to the efficient functioning of the recipient. In the case of proteins, glycosylation can serve to ensure proper protein folding, stability, and function. As such, various immunodeficiencies have been identified that affect the glycosylation pathway (Monticelli, Ferro, Jaeken, Dos Reis Ferreira, & Videira, 2016).
3.11.1. MAGT1
In 2011, our lab identified hemizygous LOF mutations in the X-linked gene encoding magnesium transporter 1 (MAGT1) in patients with chronic EBV infection and lymphoma with reduced naïve T cells, low CD4+ T cell counts, and defective CTL cytotoxicity accompanied by deficits in NKG2D glycosylation and expression (X-linked magnesium deficiency with EBV and neoplasia, XMEN disease, OMIM 300853) (Chaigne-Delalande et al., 2013; Li et al., 2011). MAGT1-deficient cells have reduced basal intracellular Mg2+ levels and are defective in a TCR-mediated Mg2+ flux. This is consistent with MAGT1’s described role as a magnesium transporter (Goytain & Quamme, 2005). The MAGT1 protein regulates T cell activation at an early stage, specifically at the level of PLCγ phosphorylation and further downstream processes including p65 nuclear translocation, calcium flux, and activation marker upregulation. As previously mentioned, defects in Itk/PLCγ may preferentially lead to susceptibility to EBV infections, which further implicate this molecular pathway in the pathogenesis of XMEN disease. Interestingly, Mg2+ supplementation in vitro and in vivo rescues NKG2D expression, CTL function, and resulted in reduced EBV viral load, but does not affect early TCR-mediated signaling defects (Chaigne-Delalande et al., 2013). These results are consistent with MAGT1 regulating both a stable basal and transient TCR-induced pool of free Mg2+, with the former being important for NKG2D glycosylation/stability and rescued by Mg2+ supplementation and the latter important for TCR proximal signaling events and not rescued by Mg2+ supplementation. Interestingly, however, MAGT1 is a homologue of yeast OST3 and, along with a third homologous protein (TUSC3), performs an accessory function to STT3B-mediated glycosylation (Cherepanova & Gilmore, 2016; Kelleher & Gilmore, 2006). Thus, the MAGT1 protein may directly modulate glycosyltransferase activity independently, or in addition to, its role in Mg2+ regulation. This raises the additional question of whether MAGT1 is itself a magnesium channel or indirectly regulates Mg2+ levels by assisting in the glycosylation of an as-of-yet unidentified channel. Why exactly MAGT1 deficiency results in such a narrow clinical phenotype is incompletely understood, though it may be possible that TUSC3, a highly homologous protein encoded on chromosome 8, can compensate in other tissues in XMEN patients, thus confining the disease mainly to specific cells of the immune system.
3.11.2. PGM3
The conversion of GlcNAc-6-P into GlcNAc-1-P is catalyzed by phosphoacetylglucosamine mutase (PGM3) and is required for the synthesis of UDP-GlcNAc, a critical nucleotide sugar donor for multiple glycosylation pathways. Several studies have identified biallelic LOF mutations in PGM3 in an autosomal recessive PID characterized by recurrent bacterial, fungal, and viral infections, allergies, and T cell lymphopenia (OMIM 615816) (Lundin et al., 2015; Sassi et al., 2014; Stray-Pedersen et al., 2014b; Zhang et al., 2014b). Patients show reduced UDP-GlcNAc and decreased formation of complex N-glycans. Patient T cells skew toward Th2-type responses possibly accounting for the increased prevalence of allergies in patients with PGM3 deficiency. Why PGM3 deficiency causes specific T cell lymphopenia is incompletely understood, however, it is known that T cells upregulate UDP-GlcNAc and O-GlcNAcylation by O-GlcNAc glycosyltransferase (OGT) upon activation (Swamy et al., 2016). Additionally, the deletion of OGT in the thymus results in a strong block in thymic development while its deletion in ex vivo proliferating T cells resulted in diminished proliferation due to reduced c-Myc expression. Therefore, PGM3 deficiency may reduce O-GlcNAcylation due to decreased UDP-GlcNAc which could inhibit c-Myc in PGM3-deficient patients. Supplementation with exogenous GlcNAc increases intracellular UDP-GlcNAc levels in PGM3-deficient cells thereby bypassing PGM3 in the catalytic pathway and rescuing the deficient function in ex vivo cell cultures. Hence, this may represent a useful therapeutic strategy in PGM3 deficiency (Zhang et al., 2014b).
3.11.3. EXTL3
Biallelic LOF mutations in exostosin-like glycosyltransferase 3 (EXTL3) lead to an autosomal recessive disease of skeletal dysplasia and immunodeficiency characterized by T cell lymphopenia, expansion of a senescent CD8+ T cell population, reduced TCR-mediated T cell activation, Omenn syndrome, eosinophilia, and hyper-IgE (OMIM 617425) (Guo et al., 2017; Notarangelo, 2017; Oud et al., 2017; Volpi et al., 2017). These defects were linked to a decreased capacity for HSC differentiation and decreased thymopoesis, as well as reduced IL-2-dependent signaling. EXTL3 is an α1,4-N-acetylglucosaminyltransferase that is involved in heparan sulfate biosynthesis through glycan chain initiation and elongation. How exactly altered heparin sulfate composition observed in patient cells may be regulating these various processes remains to be seen, though it should be noted the HSCT was curative, marking the affected pathway as a T cell-intrinsic defect. It is possible that modification of certain signaling proteins by heparin sulfate can modulate their signaling capacity. Precedence for this idea is demonstrated by the requirement of CD47 modification by heparan sulfate for thrombospondin-1 to inhibit T cell activation (Kaur et al., 2011).
3.12. Defects in Autophagy and Protein Turnover
Autophagy, and more generally the turnover of cellular components, is dynamically regulated during T cell responses and is critical for T cell survival during multiple phases of the immune response including the contraction phase, effector to memory transition, and memory maintenance. Ultimately, defects in autophagy lead to the inability to control of chronic infections (Xu et al., 2014). Currently, there are two described PIDs with defects in autophagy and protein turnover arising from either EPG5 or TPP2 deficiency, respectively.
3.12.1. EPG5
Biallelic LOF mutations in the gene encoding Ectopic P-granules autophagy protein 5 homologue (EPG5) leads to a variable PID characterized by recurrent viral, bacterial, and fungal infections, and profound CD4+ T cell lymphopenia, coupled with multiple syndromic features (slow growth, microcephaly, cleft palate, and heart defects with corpus callosum agenesis, cataracts, and cardiomyopathy), termed VICI syndrome (OMIM 242840) (Byrne et al., 2016; Cullup et al., 2013; Ehmke et al., 2014). EPG5 deficiency causes the accumulation of impaired autolysosomes due to defects in the final maturation step of autophagosomes to autolysosomes necessary for cargo degradation (Tian et al., 2010). While variable immunodeficiency has been associated with VICI syndrome, it is not known if EPG5 regulates T cell memory formation or maintenance and if this is an important aspect of the immunodeficiency. Interestingly, patient fibroblasts show altered Akt/mTOR signaling, but this has not been evaluated in patient T cells where the mTOR pathway is critical for T cell effector responses. Further investigation into the PID associated with VICI syndrome would be necessary to understand EPG5’s role in T cell development, effector responses, and memory formation.
3.12.2. TPPII
During turnover in both the proteasome and the lysosome/autophagosome, proteins are first cleaved into long oligopeptides with subsequent processing by N-terminal tripeptide cleavage by tripeptidyl peptidases (Tomkinson, 1999). Biallelic LOF mutations in tripeptidyl peptidase II (TPPII) results in a recessive disease of immunodeficiency, autoimmunity, and developmental delay termed “TPPII-related immunodeficiency, autoimmunity, and neurodevelopmental delay with impaired glycolysis and lysosomal expansion” (TRIANGLE disease) (OMIM 190470). Patients develop recurrent viral and bacterial infections, cytopenias, lupus-like disease in the central nervous system, early-onset Evan’s syndrome, and autoimmune hepatitis (Lu et al., 2014). Patient CD8+ T cells largely expressed a senescent phenotype with poor proliferative responses (Stepensky et al., 2015). TPPII was observed to be essential for protein turnover and regulation of free amino acid levels as well as maintenance of baseline mTOR signaling. Interestingly, TPPII deficiency/inhibition resulted in increased lysosome biogenesis and modestly increased autophagosome formation in an apparent compensation for the deficiency in free amino acids. In turn, this leads to a secondary proteolytic loss of hexokinase 2 and decreased glycolysis/effector function in TPPII-deficient/inhibited cells. Why TPPII deficiency results in autoimmunity and a population of activated senescent CD8+ T cells, especially given reduced mTOR activation in TPPII-deficient cells, requires further investigation. Whether or not TPPII is important for memory formation or viral control in chronic infection remains to be elucidated, as does the exact role of autophagy and protein turnover in these processes.
3.13. Defects in Metabolic Pathways
Throughout ontogeny and during the mature lifecycle in the immune system, the metabolic properties of T cells are meticulously controlled to best support the cellular demands for function (Buck, O’Sullivan, & Pearce, 2015). Additionally, some metabolites that are produced at sites of rapid proliferation and apoptosis, such as in bone marrow and thymic environments, are toxic to lymphocytes and must be properly disposed of in order to ensure optimal lymphocyte survival and function. In the next section, we will discuss PIDs related to defects in metabolic pathways.
3.13.1. CTPS1
Cytidine 5′ triphosphate synthase 1 (CTPS1) is critical for the formation of CTP, a precursor of DNA, RNA, and phospholipids (Kursula et al., 2006). While this protein’s function is obvious, it did not have a selective function in the immune system until WES sequencing identified biallelic LOF mutations in CTPS1 as the cause of an autosomal recessive immunological disorder (OMIM 615897) (Martin et al., 2014). Patients presented with recurrent viral and bacterial infections, including severe EBV infection. Infectious susceptibility was traced to a T cell-specific lymphopenia, with specific loss of naïve T cells, increased memory T cells, and complete absence of invariant T cell populations. Interestingly, CTPS1 expression is low in all cell types, but is specifically and massively upregulated in antigen-stimulated T cells within days after activation. This implies that it is a special prerequisite during this period of rapid cell growth and division. T and B cell proliferation following in vitro antigen stimulation was markedly decreased in the absence of CTPS1. Critically, supplementation of T cells with CTP or cytidine rescued the T cell proliferation defect, suggesting this disorder could be circumvented by providing exogenous end products of CTPS1 function (Martin et al., 2014). As the plasma membrane is generally impermeable to nucleoside triphosphates, the use of concentrative or equilibrative nucleoside transporters is required for the uptake of these biomolecules. Interestingly, several of these transporters are regulated during immune cell activation and may ultimately prove to be the source of as-of-yet undefined PIDs (Pastor-Anglada et al., 2001).
3.13.2. AK2
Adenylate kinase 2 (AK2) catalyzes the conversion of ADP to ATP and AMP, and is thus crucial for the de novo biogenesis of AMP (Noma, 2005). Biallelic LOF mutations in AK2 causes reticular dysgenesis, a severe SCID phenotype with absence of granulocytes, severe lymphocyte deficiency, hypoplasia of the thymus and lymph nodes caused by an early arrest in myeloid lineage development and lymphoid maturation with loss of almost all major immune cellular subsets (OMIM 267500) (Lagresle-Peyrou et al., 2009; Pannicke et al., 2009). This was traced back to an increase in spontaneous apoptosis related to defects in mitochondrial function, oxidative phosphorylation, and ROS production due to altered homeostasis of ADP, ATP, and AMP (Six et al., 2015).
3.13.3. ADA
Adenosine deaminase (ADA) catalyzes the irreversible deamination of adenosine and deoxyadenosine in the purine catabolic pathway. ADA deficiency, which causes an accumulation of dATP, was first recognized to cause a SCID phenotype in the early 1970s (OMIM 102700) (Blackburn & Kellems, 2005). Since then, it has been appreciated that ADA deficiency can cause a range of clinical severity that largely depends on the quantity of residual ADA activity (Hershfield, 2003). In the absence of ADA, adenosine and deoxyadenosine build up in the bone marrow and thymus where these metabolites cause increased apoptosis along with inhibition of the activation and expansion of T lymphocytes (Bradford, Moretti, Carbonaro-Sarracino, Gaspar, & Kohn, 2017). Treatment for ADA–SCID has traditionally included HSCT, but has now been expanded to include a gene replacement therapy called Strimvelis, underscoring the growing importance of individualized genetic medicine for monogenic disorders.
3.13.4. PNP
Biallelic LOF mutations in purine nucleoside phosphorylase (PNP) cause an autosomal recessive SCID phenotype with frequent bacterial and viral infections, lymphopenia, a small or absent thymus, and decreased T cell proliferation and function, often accompanied by autoimmune disease (OMIM 613179) (Edwards, Hopkinson, & Harris, 1971; Markert, 1991). PNP catalyzes inosine to hypoxanthine and guanosine to guanine, which ultimately get degraded to uric acid. In the absence of PNP, there is a build-up of deoxy-GTP which proves highly toxic to T cell populations, thus leading to the T cell-specific deficiency.
3.13.5. MTHFD1
Biallelic LOF variants in methylenetetrahydrofolate dehydrogenase, cyclohydrolase, and formyltetrahydrofolate synthetase 1 (MTHFD1) result in an autosomal recessive PID characterized by recurrent lung infections, moniliasis, liver fibrosis, septic arthritis, multiple cytopenias, and low T, B, and NK cells, which are sometimes accompanied by autoimmunity (OMIM 617780) (Burda et al., 2015; Keller et al., 2013; Watkins et al., 2011). MTHFD1 is an enzyme that catalyzes three separate steps in folate metabolism by processing single carbon folate derivatives utilizing three separate protein domains (Hum, Bell, Rozen, & MacKenzie, 1988). Vitamin B12 and folate replacement in MTHFD1-deficient patients resulted in reconstitution of T and B cell counts, and rescued response to mitogenic stimuli. Thus, in metabolic disorders, such as MTHFD1 deficiency, supplementation of patients with end products of the pathway can overcome the endogenous deficiency in metabolite synthesis, though this likely depends on metabolite bioavailability.
3.13.6. TFRC
Biallelic LOF mutations in the transferrin receptor (TFRC), which is critical for iron uptake, results in an autosomal recessive PID characterized by mild anemia, intermittent neutropenia, and defective B and T cell activation and memory formation, despite normal lymphocyte counts (OMIM 616740) (Jabara et al., 2016; Lo, 2016). The identified TFRC mutation results in overall increased TFRC protein production and cell surface expression and, concomitantly, reduces receptor internalization following stimulation. The addition of iron citrate, which saturates the transferrin receptor and allows iron internalization independent of TFRC rescued T and B cell proliferation and class switching defects. This study illustrates how the molecular investigation of PIDs reveals previously unknown roles of known proteins in immune function, as this was the first appreciated role of TFRC in host defense. While TFRC deficiency is the first known PID associated with cell-intrinsic iron uptake, it has long been known that iron deficiency results in poor T and B cell responses and decreased cellularity (Bowlus, 2003).
3.14. Altered T Cell Effector Function in Complement Deficiencies
3.14.1. C3
While the complement pathway is well recognized to play an essential role in host defense, we have only recently begun to understand the role of complement proteins in the regulation of adaptive immune responses by modulating T cell effector function (Hess & Kemper, 2016; Liszewski et al., 2013). C3a, C3b, and C5a, along with their respective receptors C3aR, CD46, and C5aR1 influence the survival, metabolic program, and effector differentiation of T cells during stimulation. Interestingly, T cells from C3-deficient patients (OMIM 613779) show reduced IFNγ and IL-2 production, which is likely dependent on T cell-intrinsic C3 production.
3.14.2. CD55
The significance of complement signaling in the function of human T cells was demonstrated by Ozen et al. who recently described CD55 deficiency with hyperactivation of complement, angiopathic thrombosis, and protein-losing enteropathy (the CHAPLE syndrome) (Ozen et al., 2017). The CD55-deficient patients also exhibited T cell-intrinsic defects in IL-10 production and enhanced TNF production (OMIM 226300) (Ozen et al., 2017). CD55 is a cell surface complement inhibitory protein that is expressed on a wide range of cells including lymphocytes. It controls the level of opsonin and anaphylatoxin split products produced from the C3 and C5 proteins. Interestingly, while the enhanced TNF production was dependent on C5aR1, the IL-10 defect was not rescued by complement inhibition, and may reveal a CD55-dependent costimulatory pathway that is absent in patient cells. Importantly, administration of Eculizumab, a monoclonal antibody that prevents complement protein C5 cleavage, dramatically alleviates many symptoms related to CD55 deficiency, suggesting that the patient disease is, in-fact, mediated by overt complement activation (Kurolap et al., 2017).
3.15. Unknown Mechanism
In some cases, the causal gene of an immunodeficiency may be known while its biological purpose is not well understood. Such cases are excellent working examples of forward genetic screens of nature identifying genes of unknown function that are critical to human health and disease. In the next section, we will briefly describe two PIDs associated with mutations in genes whose function remains poorly understood.
3.15.1. SP110
Biallelic LOF mutations in SP110 nuclear body protein (SP110) cause an autosomal recessive disorder with veno-occlusive disease and immunodeficiency characterized by infection susceptibility, low T cell numbers, hypogammaglobulinemia, and decreased germinal center and plasma cell formation (OMIM 235550) (Roscioli et al., 2006; Wang, Ong, Roscioli, Cliffe, & Church, 2012). In its best known function, SP110 associates with the nuclear body and may either act as a nuclear hormone receptor or as an activator of NF-κB under certain circumstances, though neither role has been well validated (Bloch et al., 2000; Leu et al., 2017).
3.15.2. TTC7A
Biallelic mutations in tetratricopeptide repeat domain 7A (TTC7A) cause an autosomal recessive combined syndrome of immunodeficiency, autoimmunity, and intrinsic GI defects (OMIM 243150) (Chen et al., 2013). These patients have reduced B and T cell populations with poor antibody production and T cell proliferation to mitogen. Patient intestinal organoids showed defects in polarity formation with increased activation of RhoA targets—pERM and phosphorylated myosin light chain. Rho-associated protein kinase (ROCK) inhibition resulted in reversion of polarity, suggesting increased RhoA signaling may be the cause of patient disease (Bigorgne et al., 2014). Patient PBMCs also have increased spontaneous phosphorylation of ERM proteins and MLC, with increased TCR- or integrin-mediated T cell spreading. Additionally, patient cells express decreased chemokine receptors and migrated poorly compared to control cells. Finally, patient T cells were hyperproliferative, which could be corrected by ROCK inhibition (Lemoine et al., 2014). In a known function, TTC7A regulates PI4KA localization, with TTC7A knockdown resulting in poor PIP production and mutant TTC7A associating poorly with PI4KA (Avitzur et al., 2014). Despite these strong hints, the precise effect of TTC7A on RhoA and PI4KA function, and if these represent separate or linked biochemical functions of TTC7A, are undetermined.
4. CURRENT CHALLENGES/APPROACHES AND FUTURE CONSIDERATIONS
One of the greatest challenges facing clinicians and scientists in the identification and study of causal variants in PIDs, and genetic disease in general, is the need to supersede correlation to establish causality of potentially pathogenic variants. This is complicated by the extensive nucleotide variation in the human genome, much of which is mysterious or, at best benign polymorphism. Several things make this challenge more tractable. First, it is very useful to be able to evaluate genomic sequences of patients in the context of sequences of members, the nuclear family, especially parents or other affected individuals. This allows for the elimination of variants that do not track with disease, though one must be careful in eliminating potentially causal variants in the case of incompletely penetrant disease. Second, it helps to have sequences for unrelated individuals that have very closely matching disease manifestations, especially those involving biochemical or molecular similarities. Third, to possess an extended sequence database of individuals from the proband’s race/ethnic background is beneficial, as the frequency and identity of benign polymorphisms differ between ethnic populations. The use of next-generation sequencing techniques and the increasing deployment of whole-exome and whole-genome sequencing in the clinic has greatly increased the amount of genetic information to compare against and facilitated the identification of disease-associated variants. As investigators and clinicians look in to the future it will become increasingly necessary to adopt whole-genome sequencing which is essential to identify noncoding variants and provides better detection of exonic variants than whole-exome sequencing (Belkadi et al., 2015). The tradeoff for the better sequence data is the time and cost associated with whole-genome sequencing and the difficulty inherent in analyzing noncoding variants—both problems that will diminish with time.
Once a list of rare genetic variants that track with disease has been developed, there are usually multiple candidate variants remaining, especially when only a single affected individual is available for analysis. Prioritization of these candidate variants then occurs based on algorithms that predict a deleterious effect on protein function (PolyPhen/CADD, etc.), the observed vs expected mutation rates in the gene encoding the protein in large-scale genetic databases such as ExAC, the tissue expression pattern of the protein, the known or hypothetical roles of the protein, and the availability of genetic models in animals including mice, zebrafish, flies, etc. All of this information is subjectively analyzed and variants are ranked for follow-up analysis. Importantly, while coding variants (missense, nonsense, and splice site) are perhaps the easiest to identify and study, noncoding variants (promotor/enhancer/silencer mutations, copy number variation, intronic, and synonymous) must also be considered as these can affect transcription, translation, mRNA stability, or splicing of the affected gene, all with impacts on protein function. Another interesting way to prioritize candidate genes is the identification of genes likely to cause human disease based on their relationship with previously identified human PID genes. This approach has shown good predictive capacity in identifying genes associated with yet-undiscovered PIDs (Itan & Casanova, 2015).
Perhaps the easiest way to strengthen the argument that mutations in a given gene are the cause of disease is the identification of multiple individuals with a similar clinical phenotype with highly deleterious variants in the same gene. Still, this does not move past correlation to implicate causation. To move in this direction, additional laboratory testing must be done to establish that the given variant is deleterious to protein function and affects the biological pathway that is presumed to be associated with disease. Usually this first entails the identification of an in vitro cellular phenotype associated with disease followed by add back of WT protein to patient cells or introduction of patient protein/knockout of the gene in WT cells to determine if these correct or phenocopy patient cellular defects, respectively. This can be supplemented with pharmacological inhibition of the affected biochemical pathway and the generation/use of genetically engineered animal models.
Casanova et al. have established three general guidelines that should be viewed as prerequisites to identification and publication of causal genes in cases involving a single affected individual (Itan & Casanova, 2015). First, family and population genetics should indicate that the candidate genotype is monogenic and the potential causal variant does not occur in healthy individuals. Second, experimental and mechanistic studies must prove that the candidate variant is deleterious to the expression or function of the protein product. Finally, the connection of the candidate gene and patient phenotype must be established by known molecular interactions, expression patterns, and a relevant cellular or animal phenotype. Stated differently, the variant must be proven deleterious and absent from healthy individuals and the gene the variant resides in must be plausibly important for the disease mechanism. In cases where a single patient has already been published, follow-up reports are useful confirmations of gene–disease relationships and often provide insight into the clinical spectrum of disease. Hence, although specific gene implication will always involve a measure of correlation, if that can be established at the molecular, cellular, and organismal levels, the hypothesis of causality is substantially strengthened.
As mentioned throughout this review, identification of the causal variant can lead to targeted therapeutic interventions. This has proven to be one of the most exciting and gratifying aspects of genomic research. Those working in the field of PID research and immunology is especially fortunate since their “organ” of interest can be replaced through bone marrow transplantation. This enables a measure of manipulability that is unparalleled in other organs in the body. The development of new treatments or the deployment of known, but perhaps unobvious, treatments are facilitated by strong mechanistic evidence identifying the affected biochemical pathway in patient cells and the availability of interventions that can target this pathway. In many cases where disease is confined to the immune system and there is no known targeted therapeutic option, HSCT is indicated as the principle course of action. However, disease must be severe enough to warrant the inherent risk associated with HSCT. More recently, the treatment of several PIDs by genetic therapy based upon viral restitution of the gene/protein encoded by the affected genetic loci, has been successful (Mukherjee & Thrasher, 2013). In the future, gene editing by CRISPR/Cas9 and similar technologies will likely play an important role in correcting these monogenic disorders. Both gene therapy and genetic editing are made more attractive in PIDs than other primary genetic disorders because it is possible to virtually replace the all or part of the immune system with corrected material through radioablation and replacement of patient bone marrow, or simple transfer of corrected HSCs allowing for the outgrowth of corrected cells. The likelihood of success of the latter approach is augured by the presence of somatic reversion mutations in causative genes that correct the course of disease (Jing et al., 2014).
Future challenges facing researches studying PIDs include the need to develop methods for predicting the impact of variants on the function of intronic and other nonprotein-coding segments of the genome. Additionally, we will begin to understand the clinical variability seen in PIDs (variable expressivity and reduced penetrance) which likely occurs due to a confluence of Mendelian variant inheritance, infectious history, and alternate genetic variants that may modify the clinical phenotype. Understanding this matrix of factors that link genotype and phenotype will expedite the understanding and treatment of these diseases. Finally, as the field moves forward, we will likely encounter more case reports where a given individual is afflicted by more than one PID, with the clinical phenotype developing in an additive fashion where the patient presents with the full spectrum of symptoms from both PIDs, or a unique clinical spectrum arising from two or more PIDs affecting overlapping pathways. In such cases, it will be important to analyze patient mutations separately and in combination. Finally, research will gradually divulge how the combination of two genes, three genes, and, ultimately, polygenic constellations of variants will contribute to the pathogenesis of a disease and its response to treatment. It seems clear that the application of machine learning computational approaches will be essential to derive the most knowledge from the expanding global corpus of genomic DNA sequence data. Thus, although medicine and biomedical research have reached a watershed moment in which human investigation through the prism of genomics has greatly accelerated discovery, there is still a vast terra incognita ahead for future generations of immunological and genetic investigators.
REFERENCES
- Abdollahpour H, et al. (2012). The phenotype of human STK4 deficiency. Blood, 119, 3450–3457. 10.1182/blood-2011-09-378158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abolhassani H, et al. (2017). Combined immunodeficiency and Epstein–Barr virus-induced B cell malignancy in humans with inherited CD70 deficiency. The Journal of Experimental Medicine, 214, 91–106. 10.1084/jem.20160849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abram CL, & Lowell CA (2009). Leukocyte adhesion deficiency syndrome: A controversy solved. Immunology and Cell Biology, 87, 440–442. 10.1038/icb.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afzali B, et al. (2017). BACH2 immunodeficiency illustrates an association between super-enhancers and haploinsufficiency. Natural Immunity, 18, 813–823. 10.1038/ni.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar C, & Latour S (2015). X-linked inhibitor of apoptosis protein deficiency: More than an X-linked lymphoproliferative syndrome. Journal of Clinical Immunology, 35, 331–338. 10.1007/s10875-015-0141-9. [DOI] [PubMed] [Google Scholar]
- Ahnesorg P, Smith P, & Jackson SP (2006). XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell, 124, 301–313. 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- Akar HH, et al. (2015). Silent brain infarcts in two patients with zeta chain-associated protein 70 kDa (ZAP70) deficiency. Clinical Immunology, 158, 88–91. 10.1016/j.clim.2015.03.014. [DOI] [PubMed] [Google Scholar]
- Akimzhanov AM, & Boehning D (2015). Rapid and transient palmitoylation of the tyrosine kinase Lck mediates Fas signaling. Proceedings of the National Academy of Sciences of the United States of America, 112, 11876–11880. 10.1073/pnas.1509929112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alangari A, et al. (2012). LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. The Journal of Allergy and Clinical Immunology, 130, 481–488 e482. 10.1016/j.jaci.2012.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RC, et al. (1993). CD40 ligand gene defects responsible for X-linked hyper-IgM syndrome. Science, 259, 990–993. [DOI] [PubMed] [Google Scholar]
- Alsultan A, Shamseldin HE, Osman ME, Aljabri M, & Alkuraya FS (2013). MYSM1 is mutated in a family with transient transfusion-dependent anemia, mild thrombocytopenia, and low NK- and B-cell counts. Blood, 122, 3844–3845. 10.1182/blood-2013-09-527127. [DOI] [PubMed] [Google Scholar]
- Altare F, et al. (1998a). Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science, 280, 1432–1435. [DOI] [PubMed] [Google Scholar]
- Altare F, et al. (1998b). Inherited interleukin 12 deficiency in a child with bacille Calmette–Guerin and Salmonella enteritidis disseminated infection. The Journal of Clinical Investigation, 102, 2035–2040. 10.1172/JCI4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreotti AH, Schwartzberg PL, Joseph RE, & Berg LJ (2010). T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harbor Perspectives in Biology, 2, a002287. 10.1101/cshperspect.a002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo I, et al. (2013). Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science, 342, 866–871. 10.1126/science.1243292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton IM, et al. (2002). WIP deficiency reveals a differential role for WIP and the actin cytoskeleton in T and B cell activation. Immunity, 16, 193–204. [DOI] [PubMed] [Google Scholar]
- Arnaiz-Villena A, et al. (1992). Brief report: Primary immunodeficiency caused by mutations in the gene encoding the CD3-gamma subunit of the T-lymphocyte receptor. The New England Journal of Medicine, 327, 529–533. 10.1056/NEJM199208203270805. [DOI] [PubMed] [Google Scholar]
- Arpaia E, Shahar M, Dadi H, Cohen A, & Roifman CM (1994). Defective T cell receptor signaling and CD8+ thymic selection in humans lacking zap-70 kinase. Cell, 76, 947–958. [DOI] [PubMed] [Google Scholar]
- Avitzur Y, et al. (2014). Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology, 146, 1028–1039. 10.1053/j.gastro.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avram D, & Califano D (2014). The multifaceted roles of Bcl11b in thymic and peripheral T cells: Impact on immune diseases. Journal of Immunology, 193, 2059–2065. 10.4049/jimmunol.1400930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badran YR, et al. (2017). Human RELA haploinsufficiency results in autosomal-dominant chronic mucocutaneous ulceration. The Journal of Experimental Medicine, 214, 1937–1947. 10.1084/jem.20160724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahrami E, et al. (2017). Myb-like, SWIRM, and MPN domains 1 (MYSM1) deficiency: Genotoxic stress-associated bone marrow failure and developmental aberrations. The Journal of Allergy and Clinical Immunology, 140, 1112–1119. 10.1016/j.jaci.2016.10.053. [DOI] [PubMed] [Google Scholar]
- Bajpai R, et al. (2010). CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature, 463, 958–962. 10.1038/nature08733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CM, et al. (2012). Opposing roles for RhoH GTPase during T-cell migration and activation. Proceedings of the National Academy of Sciences of the United States of America, 109, 10474–10479. 10.1073/pnas.1114214109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balabanian K, et al. (2005). WHIM syndromes with different genetic anomalies are accounted for by impaired CXCR4 desensitization to CXCL12. Blood, 105, 2449–2457. 10.1182/blood-2004-06-2289. [DOI] [PubMed] [Google Scholar]
- Bansbach CE, Betous R, Lovejoy CA, Glick GG, & Cortez D (2009). The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes & Development, 23, 2405–2414. 10.1101/gad.1839909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrat FJ, et al. (1999). Defective CTLA-4 cycling pathway in Chediak–Higashi syndrome: A possible mechanism for deregulation of T lymphocyte activation. Proceedings of the National Academy of Sciences of the United States of America, 96, 8645–8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreto M, et al. (2004). Evidence for CTLA4 as a susceptibility gene for systemic lupus erythematosus. European Journal of Human Genetics, 12, 620–626. 10.1038/sj.ejhg.5201214. [DOI] [PubMed] [Google Scholar]
- Barron L, et al. (2010). Cutting edge: Mechanisms of IL-2-dependent maintenance of functional regulatory T cells. Journal of Immunology, 185, 6426–6430. 10.4049/jimmunol.0903940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer TR Jr., & Hickstein DD (2000). Gene therapy for leukocyte adhesion deficiency. Current Opinion in Molecular Therapeutics, 2, 383–388. [PubMed] [Google Scholar]
- Belkadi A, et al. (2015). Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proceedings of the National Academy of Sciences of the United States of America, 112, 5473–5478. 10.1073/pnas.1418631112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belot A, et al. (2013). Protein kinase cdelta deficiency causes Mendelian systemic lupus erythematosus with B cell-defective apoptosis and hyperproliferation. Arthritis and Rheumatism, 65, 2161–2171. 10.1002/art.38008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CL, et al. (2001). The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nature Genetics, 27, 20–21. 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- Bigorgne AE, et al. (2014). TTC7A mutations disrupt intestinal epithelial apicobasal ‘polarity. The Journal of Clinical Investigation, 124, 328–337. 10.1172/JCI71471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn MR, & Kellems RE (2005). Adenosine deaminase deficiency: Metabolic basis of immune deficiency and pulmonary inflammation. Advances in Immunology, 86, 1–41. 10.1016/S0065-2776(04)86001-2. [DOI] [PubMed] [Google Scholar]
- Bloch DB, et al. (2000). Sp110 localizes to the PML-Sp100 nuclear body and may function as a nuclear hormone receptor transcriptional coactivator. Molecular and Cellular Biology, 20, 6138–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerkoel CF, et al. (2002). Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nature Genetics, 30, 215–220. 10.1038/ng821. [DOI] [PubMed] [Google Scholar]
- Bolze A, et al. (2010). Whole-exome-sequencing-based discovery of human FADD deficiency. American Journal of Human Geneticse, 87, 873–881. 10.1016/j.ajhg.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossaller L, et al. (2006). ICOS deficiency is associated with a severe reduction of CXCR5 + CD4 germinal center Th cells. Journal of Immunology, 177, 4927–4932. [DOI] [PubMed] [Google Scholar]
- Boswell KL, et al. (2012). Munc13–4 reconstitutes calcium-dependent SNARE-mediated membrane fusion. The Journal of Cell Biology, 197, 301–312. 10.1083/jcb.201109132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchet J, et al. (2016). Rac1-Rab11-FIP3 regulatory hub coordinates vesicle traffic with actin remodeling and T-cell activation. The EMBO Journal, 35, 1160–1174. 10.15252/embj.201593274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchet J, et al. (2017). Rab11-FIP3 regulation of Lck endosomal traffic controls TCR signal transduction. Journal of Immunology, 198, 2967–2978. 10.4049/jimmunol.1600671. [DOI] [PubMed] [Google Scholar]
- Bouma G, Burns SO, & Thrasher AJ (2009). Wiskott–Aldrich syndrome: Immunodeficiency resulting from defective cell migration and impaired immunostimulatory activation. Immunobiology, 214, 778–790. 10.1016/j.imbio.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousfiha A, et al. (2015). The 2015 IUIS phenotypic classification for primary immunodeficiencies. Journal of Clinical Immunology, 35, 727–738. 10.1007/s10875-015-0198-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowlus CL (2003). The role of iron in T cell development and autoimmunity. Autoimmunity Reviews, 2, 73–78. [DOI] [PubMed] [Google Scholar]
- Bradford KL, Moretti FA, Carbonaro-Sarracino DA, Gaspar HB, & Kohn DB (2017). Adenosine deaminase (ADA)-deficient severe combined immune deficiency (SCID): Molecular pathogenesis and clinical manifestations. Journal of Clinical Immunology, 37, 626–637. 10.1007/s10875-017-0433-3. [DOI] [PubMed] [Google Scholar]
- Bratton SB, Lewis J, Butterworth M, Duckett CS, & Cohen GM (2002). XIAP inhibition of caspase-3 preserves its association with the Apaf-1 apoptosome and prevents CD95- and Bax-induced apoptosis. Cell Death and Differentiation, 9, 881–892. 10.1038/sj.cdd.4401069. [DOI] [PubMed] [Google Scholar]
- Brenner C, et al. (2005). Myc represses transcription through recruitment of DNA methyltransferase corepressor. The EMBO Journal, 24, 336–346. 10.1038/sj.emboj.7600509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MJ, et al. (2003). Chemokine stimulation of human peripheral blood T lymphocytes induces rapid dephosphorylation of ERM proteins, which facilitates loss of microvilli and polarization. Blood, 102, 3890–3899. 10.1182/blood-2002-12-3807. [DOI] [PubMed] [Google Scholar]
- Browning MJ, Chandra A, Carbonaro V, Okkenhaug K, & Barwell J (2015). Cowden’s syndrome with immunodeficiency. Journal of Medical Genetics, 52, 856–859. 10.1136/jmedgenet-2015-103266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugnera E, et al. (2002). Unconventional Rac-GEF activity is mediated through the Dock180–ELMO complex. Nature Cell Biology, 4, 574–582. 10.1038/ncb824. [DOI] [PubMed] [Google Scholar]
- Brunkow ME, et al. (2001). Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nature Genetics, 27, 68–73. 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- Buck MD, O’Sullivan D, & Pearce EL (2015). T cell metabolism drives immunity. The Journal of Experimental Medicine, 212, 1345–1360. 10.1084/jem.20151159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck D, et al. (2006). Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell, 124, 287–299. 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- Burda P, et al. (2015). Characterization and review of MTHFD1 deficiency: Four new patients, cellular delineation and response to folic and folinic acid treatment. Journal of Inherited Metabolic Disease, 38, 863–872. 10.1007/s10545-015-9810-3. [DOI] [PubMed] [Google Scholar]
- Burns CM, Sakaguchi K, Appella E, & Ashwell JD (1994). CD45 regulation of tyrosine phosphorylation and enzyme activity of src family kinases. The Journal of Biological Chemistry, 269, 13594–13600. [PubMed] [Google Scholar]
- Byrne S, et al. (2016). EPG5-related Vici syndrome: A paradigm of neurodevelopmental disorders with defective autophagy. Brain, 139, 765–781. 10.1093/brain/awv393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun M, et al. (2010). Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. The Journal of Experimental Medicine, 207, 2307–2312. 10.1084/jem.20101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannons JL, Tangye SG, & Schwartzberg PL (2011). SLAM family receptors and SAP adaptors in immunity. Annual Review of Immunology, 29, 665–705. 10.1146/annurev-immunol-030409-101302. [DOI] [PubMed] [Google Scholar]
- Carbonari M, et al. (1990). Relative increase of T cells expressing the gamma/delta rather than the alpha/beta receptor in ataxia-telangiectasia. The New England Journal of Medicine, 322, 73–76. 10.1056/NEJM199001113220201. [DOI] [PubMed] [Google Scholar]
- Casanova JL, Abel L, & Quintana-Murci L (2013). Immunology taught by human genetics. Cold Spring Harbor Symposia on Quantitative Biology, 78, 157–172. 10.1101/sqb.2013.78.019968. [DOI] [PubMed] [Google Scholar]
- Caudy AA, Reddy ST, Chatila T, Atkinson JP, & Verbsky JW (2007). CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. The Journal of Allergy and Clinical Immunology, 119, 482–487. 10.1016/j.jaci.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Chae HD, Siefring JE, Hildeman DA, Gu Y, & Williams DA (2010). RhoH regulates subcellular localization of ZAP-70 and Lck in T cell receptor signaling. PLoS One, 5, e13970, 10.1371/journal.pone.0013970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaigne-Delalande B, et al. (2013). Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science, 341, 186–191. 10.1126/science.1240094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KT, Creed SJ, & Bear JE (2011). Unraveling the enigma: Progress towards understanding the coronin family of actin regulators. Trends in Cell Biology, 21, 481–488. 10.1016/j.tcb.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AC, Iwashima M, Turck CW, & Weiss A (1992). ZAP-70: A 70 kd protein-tyrosine kinase that associates with the TCR zeta chain. Cell, 71, 649–662. [DOI] [PubMed] [Google Scholar]
- Chan AC, et al. (1994). ZAP-70 deficiency in an autosomal recessive form of severe combined immunodeficiency. Science, 264, 1599–1601. [DOI] [PubMed] [Google Scholar]
- Chan AY, et al. (2016). A novel human autoimmune syndrome caused by combined hypomorphic and activating mutations in ZAP-70. The Journal of Experimental Medicine, 213, 155–165. 10.1084/jem.20150888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbonnier LM, et al. (2015). Regulatory T-cell deficiency and immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like disorder caused by loss-of-function mutations in LRBA. The Journal of Allergy and Clinical Immunology, 135, 217–227. 10.1016/j.jaci.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatila TA, et al. (2000). JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. The Journal of Clinical Investigation, 106, R75–81. 10.1172/JCI11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, & Flies DB (2013). Molecular mechanisms of T cell co-stimulation and co-inhibition. Nature Reviews. Immunology, 13, 227–242. 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, et al. (2013). Whole-exome sequencing identifies tetratricopeptide repeat domain 7A (TTC7A) mutations for combined immunodeficiency with intestinal atresias. The Journal of Allergy and Clinical Immunology, 132, 656–664.e617. 10.1016/j.jaci.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanova NA, & Gilmore R (2016). Mammalian cells lacking either the cotranslational or posttranslocational oligosaccharyltransferase complex display substrate-dependent defects in asparagine linked glycosylation. Scientific Reports, 6, 20946. 10.1038/srep20946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry LK, Li X, Schwab P, Lim B, & Klickstein LB (2004). RhoH is required to maintain the integrin LFA-1 in a nonadhesive state on lymphocytes. Nature Immunology, 5, 961–967. 10.1038/ni1103. [DOI] [PubMed] [Google Scholar]
- Choi J, et al. (2009). Mst1-FoxO signaling protects Naive T lymphocytes from cellular oxidative stress in mice. PLoS One, 4, e8011. 10.1371/journal.pone.0008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra C, et al. (2014). Immune deficiency in Ataxia-Telangiectasia: A longitudinal study of 44 patients. Clinical and Experimental Immunology, 176, 275–282. 10.1111/cei.12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou HC, et al. (2006). WIP regulates the stability and localization of WASP to podosomes in migrating dendritic cells. Current Biology, 16, 2337–2344. 10.1016/j.cub.2006.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudasama KK, et al. (2013). SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. American Journal of Human Genetics, 93, 150–157. 10.1016/j.ajhg.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun HJ, et al. (2002). Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature, 419, 395–399. 10.1038/nature01063. [DOI] [PubMed] [Google Scholar]
- Chung BK, & Gibson WT (2014). Autosomal dominant PIK3R1 mutations cause SHORT syndrome. Clinical Genetics, 85, 228–229. 10.1111/cge.12262. [DOI] [PubMed] [Google Scholar]
- Cirstea IC, et al. (2010). A restricted spectrum of NRAS mutations causes Noonan syndrome. Nature Genetics, 42, 27–29. 10.1038/ng.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RH, et al. (2003). Adaptor protein 3-dependent microtubule-mediated movement of lytic granules to the immunological synapse. Nature Immunology, 4, 1111–1120. 10.1038/ni1000. [DOI] [PubMed] [Google Scholar]
- Coffey AJ, et al. (1998). Host response to EBV infection in X-linked lymphoproliferative disease results from mutations in an SH2-domain encoding gene. Nature Genetics, 20, 129–135. 10.1038/2424. [DOI] [PubMed] [Google Scholar]
- Cohen AC, et al. (2006). Cutting edge: Decreased accumulation and regulatory function of CD4+CD25 high T cells in human STAT5b deficiency. The Journal of Immunology, 177, 2770–2774. 10.4049/jimmunol.177.5.2770. [DOI] [PubMed] [Google Scholar]
- Comrie WA, & Burkhardt JK (2016). Action and traction: Cytoskeletal control of receptor triggering at the immunological synapse. Frontiers in Immunology, 7, 68. 10.3389/fimmu.2016.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comrie WA, Li S, Boyle S, & Burkhardt JK (2015). The dendritic cell cytoskeleton promotes T cell adhesion and activation by constraining ICAM-1 mobility. The Journal of Cell Biology, 208, 457–473. 10.1083/jcb.201406120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comrie WA, et al. (2018). RELA haploinsufficiency in CD4 lymphoproliferative disease with autoimmune cytopenias. The Journal of Allergy and Clinical Immunology, Epub ahead of print. 10.1016/j.jaci.2017.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley ME, et al. (2009). Primary B cell immunodeficiencies: Comparisons and contrasts. Annual Review of Immunology, 27, 199–227. 10.1146/annurev.immunol.021908.132649. [DOI] [PubMed] [Google Scholar]
- Conley ME, et al. (2012). Agammaglobulinemia and absent B lineage cells in a patient lacking the p85alpha subunit of PI3K. The Journal of Experimental Medicine, 209, 463–470. 10.1084/jem.20112533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossu F, et al. (2002). A novel DKC1 mutation, severe combined immunodeficiency (T+B-NK-SCID) and bone marrow transplantation in an infant with Hoyeraal–Hreidarsson syndrome. British Journal of Haematology, 119, 765–768. [DOI] [PubMed] [Google Scholar]
- Cote M, et al. (2009). Munc18–2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. The Journal of Clinical Investigation, 119, 3765–3773. 10.1172/JCI40732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch FB, et al. (2013). ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes & Development, 27, 1610–1623. 10.1101/gad.214080.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter TI, et al. (2017). Clinical spectrum and features of activated phosphoinositide 3-kinase delta syndrome: A large patient cohort study. The Journal of Allergy and Clinical Immunology, 139, 597–606 e594. 10.1016/j.jaci.2016.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crequer A, et al. (2012). Human RHOH deficiency causes T cell defects and susceptibility to EV-HPV infections. The Journal of Clinical Investigation, 122, 3239–3247. 10.1172/JCI62949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz AC, et al. (2016). Fas/CD95 prevents autoimmunity independently of lipid raft localization and efficient apoptosis induction. Nature Communications, 7, 13895. 10.1038/ncomms13895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullup T, et al. (2013). Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nature Genetics, 45, 83–87. 10.1038/ng.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadi HK, Simon AJ, & Roifman CM (2003). Effect of CD3delta deficiency on maturation of alpha/beta and gamma/delta T-cell lineages in severe combined immunodeficiency. The New England Journal of Medicine, 349, 1821–1828. 10.1056/NEJMoa031178. [DOI] [PubMed] [Google Scholar]
- Dave VP, et al. (1997). CD3 delta deficiency arrests development of the alpha beta but not the gamma delta T cell lineage. The EMBO Journal, 16, 1360–1370. 10.1093/emboj/16.6.1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies EG (2013). Immunodeficiency in DiGeorge syndrome and options for treating cases with complete athymia. Frontiers in Immunology, 4, 322. 10.3389/fimmu.2013.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Beaucoudrey L, et al. (2008). Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. The Journal of Experimental Medicine, 205, 1543–1550. 10.1084/jem.20080321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Greef JC, et al. (2011). Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. American Journal of Human Genetics, 88, 796–804. 10.1016/j.ajhg.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong R, et al. (1998). Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science, 280, 1435–1438. [DOI] [PubMed] [Google Scholar]
- de la Calle-Martin O, et al. (2001). Familial CD8 deficiency due to a mutation in the CD8 alpha gene. The Journal of Clinical Investigation, 108, 117–123. 10.1172/JCI10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira de Oliveira WR, Carrasco S, Neto CF, Rady P, & Tyring SK (2003). Non-specific cell-mediated immunity in patients with epidermodysplasia verruciformis. The Journal of Dermatology, 30, 203–209. [DOI] [PubMed] [Google Scholar]
- de Saint Basile G, et al. (2004). Severe combined immunodeficiency caused by deficiency in either the delta or the epsilon subunit of CD3. The Journal of Clinical Investigation, 114, 1512–1517. 10.1172/JCI22588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deau MC, et al. (2014). A human immunodeficiency caused by mutations in the PIK3R1 gene. The Journal of Clinical Investigation, 124, 3923–3928. 10.1172/JCI75746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bel KL, et al. (2017). JAK1 gain-of-function causes an autosomal dominant immune dysregulatory and hypereosinophilic syndrome. The Journal of Allergy and Clinical Immunology, 139, 2016–2020.e5. 10.1016/j.jaci.2016.12.957. [DOI] [PubMed] [Google Scholar]
- Dell’Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, & Bonifacino JS (1999). Altered trafficking of lysosomal proteins in Hermansky–Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Molecular Cell, 3, 11–21. [DOI] [PubMed] [Google Scholar]
- Delmonte OM, et al. (2017). First case of X-linked moesin deficiency identified after new-born screening for SCID. Journal of Clinical Immunology, 37, 336–338. 10.1007/s10875-017-0391-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denoeud J, & Moser M (2011). Role of CD27/CD70 pathway of activation in immunity and tolerance. Journal of Leukocyte Biology, 89, 195–203. 10.1189/jlb.0610351. [DOI] [PubMed] [Google Scholar]
- Derry JM, Ochs HD, & Francke U (1994). Isolation of a novel gene mutated in Wiskott–Aldrich syndrome. Cell, 79. following 922. [PubMed] [Google Scholar]
- Dieckmann NM, Hackmann Y, Arico M, & Griffiths GM (2015). Munc18–2 is required for Syntaxin 11 localization on the plasma membrane in cytotoxic T-lymphocytes. Traffic, 16, 1330–1341. 10.1111/tra.12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiSanto JP, Bonnefoy JY, Gauchat JF, Fischer A, & de Saint Basile G (1993). CD40 ligand mutations in x-linked immunodeficiency with hyper-IgM. Nature, 361, 541–543. 10.1038/361541a0. [DOI] [PubMed] [Google Scholar]
- Dobbs K, et al. (2015). Inherited DOCK2 deficiency in patients with early-onset invasive infections. The New England Journal of Medicine, 372, 2409–2422. 10.1056/NEJMoa1413462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Temann UA, & Flavell RA (2001). Cutting edge: Critical role of inducible costimulator in germinal center reactions. Journal of Immunology, 166, 3659–3662. [DOI] [PubMed] [Google Scholar]
- Dorn T, et al. (2007). RhoH is important for positive thymocyte selection and T-cell receptor signaling. Blood, 109, 2346–2355. 10.1182/blood-2006-04-019034. [DOI] [PubMed] [Google Scholar]
- D’Oro U, & Ashwell JD (1999). Cutting edge: The CD45 tyrosine phosphatase is an inhibitor of Lck activity in thymocytes. Journal of Immunology, 162, 1879–1883. [PubMed] [Google Scholar]
- Dower NA, et al. (2000). RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nature Immunology, 1, 317–321. 10.1038/79766. [DOI] [PubMed] [Google Scholar]
- Dyment DA, et al. (2013). Mutations in PIK3R1 cause SHORT syndrome. American Journal of Human Genetics, 93, 158–166. 10.1016/j.ajhg.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebinu JO, et al. (2000). RasGRP links T-cell receptor signaling to Ras. Blood, 95, 3199–3203. [PubMed] [Google Scholar]
- Edwards YH, Hopkinson DA, & Harris H (1971). Inherited variants of human nucleoside phosphorylase. Annals of Human Genetics, 34, 395–408. [DOI] [PubMed] [Google Scholar]
- Ege M, et al. (2005). Omenn syndrome due to ARTEMIS mutations. Blood, 105, 4179–4186. 10.1182/blood-2004-12-4861. [DOI] [PubMed] [Google Scholar]
- Ehmke N, et al. (2014). First description of a patient with Vici syndrome due to a mutation affecting the penultimate exon of EPG5 and review of the literature. American Journal of Medical Genetics. Part A, 164A, 3170–3175. 10.1002/ajmg.a.36772. [DOI] [PubMed] [Google Scholar]
- Elder ME, et al. (1994). Human severe combined immunodeficiency due to a defect in ZAP-70, a T cell tyrosine kinase. Science, 264, 1596–1599. [DOI] [PubMed] [Google Scholar]
- Elgueta R, et al. (2009). Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunological Reviews, 229, 152–172. 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkaim E, et al. (2016). Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase delta syndrome 2: A cohort study. The Journal of Allergy and Clinical Immunology, 138 210–218 e219. 10.1016/j.jaci.2016.03.022. [DOI] [PubMed] [Google Scholar]
- Endo Y, et al. (2015). Dominant mutations in ORAI1 cause tubular aggregate myopathy with hypocalcemia via constitutive activation of store-operated Ca(2)(+) channels. Human Molecular Genetics, 24, 637–648. 10.1093/hmg/ddu477. [DOI] [PubMed] [Google Scholar]
- Eng C (2003). PTEN: One gene, many syndromes. Human Mutation, 22, 183–198. 10.1002/humu.10257. [DOI] [PubMed] [Google Scholar]
- Fang D, et al. (2002). Dysregulation of T lymphocyte function in itchy mice: A role for Itch in TH2 differentiation. Nature Immunology, 3, 281–287. 10.1038/ni763. [DOI] [PubMed] [Google Scholar]
- Fehon RG, McClatchey AI, & Bretscher A (2010). Organizing the cell cortex: The role of ERM proteins. Nature Reviews. Molecular Cell Biology, 11, 276–287. 10.1038/nrm2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann J, et al. (2003). Munc13–4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell, 115, 461–473. [DOI] [PubMed] [Google Scholar]
- Feske S, Skolnik EY, & Prakriya M (2012). Ion channels and transporters in lymphocyte function and immunity. Nature Reviews. Immunology, 12, 532–547. 10.1038/nri3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, et al. (1996). Severe combined immunodeficiency due to defective binding of the nuclear factor of activated T cells in T lymphocytes of two male siblings. European Journal of Immunology, 26, 2119–2126. 10.1002/eji.1830260924. [DOI] [PubMed] [Google Scholar]
- Feske S, et al. (2006). A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature, 441, 179–185. 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- Fisher GH, et al. (1995). Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell, 81, 935–946. [DOI] [PubMed] [Google Scholar]
- Flanagan SE, et al. (2014). Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nature Genetics, 46, 812–814. 10.1038/ng.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming HE, Milne CD, & Paige CJ (2004). CD45-deficient mice accumulate pro-B cells both in vivo and in vitro. Journal of Immunology, 173, 2542–2551. [DOI] [PubMed] [Google Scholar]
- Foger N, Rangell L, Danilenko DM, & Chan AC (2006). Requirement for coronin 1 in T lymphocyte trafficking and cellular homeostasis. Science, 313, 839–842. 10.1126/science.1130563. [DOI] [PubMed] [Google Scholar]
- Frank J, et al. (1999). Exposing the human nude phenotype. Nature, 398, 473–474. 10.1038/18997. [DOI] [PubMed] [Google Scholar]
- Frugoni F, et al. (2016). A novel mutation in the POLE2 gene causing combined immunodeficiency. The Journal of Allergy and Clinical Immunology, 137 635–638.e1. 10.1016/j.jaci.2015.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs S, et al. (2012). Antiviral and regulatory T cell immunity in a patient with stromal interaction molecule 1 deficiency. Journal of Immunology, 188, 1523–1533. 10.4049/jimmunol.1102507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui Y, et al. (2001). Haematopoietic cell-specific CDM family protein DOCK2 is essential for lymphocyte migration. Nature, 412, 826–831. 10.1038/35090591. [DOI] [PubMed] [Google Scholar]
- Furlan G, Minowa T, Hanagata N, Kataoka-Hamai C, & Kaizuka Y (2014). Phosphatase CD45 both positively and negatively regulates T cell receptor phosphorylation in reconstituted membrane protein clusters. The Journal of Biological Chemistry, 289, 28514–28525. 10.1074/jbc.M114.574319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamez-Diaz L, et al. (2016). The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. The Journal of Allergy and Clinical Immunology, 137, 223–230. 10.1016/j.jaci.2015.09.025. [DOI] [PubMed] [Google Scholar]
- Garibaldi M, et al. (2017). A novel gain-of-function mutation in ORAI1 causes late-onset tubular aggregate myopathy and congenital miosis. Clinical Genetics, 91, 780–786. 10.1111/cge.12888. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Bienemann K, Boztug K, & Borkhardt A (2014). Interleukin-2-inducible T-cell kinase (ITK) deficiency—Clinical and molecular aspects. Journal of Clinical Immunology, 34, 892–899. 10.1007/s10875-014-0110-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil J, et al. (2011). A leaky mutation in CD3D differentially affects alphabeta and gammadelta T cells and leads to a Talphabeta-Tgammadelta + B + NK + human SCID. The Journal of Clinical Investigation, 121, 3872–3876. 10.1172/JCI44254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giliani S, et al. (2005). Interleukin-7 receptor alpha (IL-7Ralpha) deficiency: Cellular and molecular bases. Analysis of clinical, immunological, and molecular features in 16 novel patients. Immunological Reviews, 203, 110–126. 10.1111/j.0105-2896.2005.00234.x. [DOI] [PubMed] [Google Scholar]
- Gill RM, Gabor TV, Couzens AL, & Scheid MP (2013). The MYC-associated protein CDCA7 is phosphorylated by AKT to regulate MYC-dependent apoptosis and transformation. Molecular and Cellular Biology, 33, 498–513. 10.1128/MCB.00276-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman FD, et al. (1998). Defective expression of p56lck in an infant with severe combined immunodeficiency. The Journal of Clinical Investigation, 102, 421–429. 10.1172/JCI3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorska MM, & Alam R (2012). A mutation in the human uncoordinated 119 gene impairs TCR signaling and is associated with CD4 lymphopenia. Blood, 119, 1399–1406. 10.1182/blood-2011-04-350686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorska MM, Liang Q, Karim Z, & Alam R (2009). Uncoordinated 119 protein controls trafficking of Lck via the Rab11 endosome and is critical for immunological synapse formation. Journal of Immunology, 183, 1675–1684. 10.4049/jimmunol.0900792. [DOI] [PubMed] [Google Scholar]
- Gorska MM, Stafford SJ, Cen O, Sur S, & Alam R (2004). Unc119, a novel activator of Lck/Fyn, is essential for T cell activation. The Journal of Experimental Medicine, 199, 369–379. 10.1084/jem.20030589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudy K, et al. (2013). Human IL2RA null mutation mediates immunodeficiency with lymphoproliferation and autoimmunity. Clinical Immunology, 146, 248–261. 10.1016/j.clim.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goytain A, & Quamme GA (2005). Identification and characterization of a novel mammalian Mg2+ transporter with channel-like properties. BMC Genomics, 6, 48. 10.1186/1471-2164-6-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grawunder U, Zimmer D, Fugmann S, Schwarz K, & Lieber MR (1998). DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Molecular Cell, 2, 477–484. [DOI] [PubMed] [Google Scholar]
- Grimbacher B, et al. (2003). Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nature Immunology, 4, 261–268. 10.1038/ni902. [DOI] [PubMed] [Google Scholar]
- Gu C, et al. (2005). Semaphorin 3E and plexin-D1 control vascular pattern independently of neuropilins. Science, 307, 265–268. 10.1126/science.1105416. [DOI] [PubMed] [Google Scholar]
- Gu Y, et al. (2006). RhoH GTPase recruits and activates Zap70 required for T cell receptor signaling and thymocyte development. Nature Immunology, 7, 1182–1190. 10.1038/ni1396. [DOI] [PubMed] [Google Scholar]
- Guo L, et al. (2017). Identification of biallelic EXTL3 mutations in a novel type of spondylo-epi-metaphyseal dysplasia. Journal of Human Genetics, 62, 797–801. 10.1038/jhg.2017.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RS, et al. (1999). The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proceedings of the National Academy of Sciences of the United States of America, 96, 14412–14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada Y, et al. (2003). A single amino acid alteration in cytoplasmic domain determines IL-2 promoter activation by ligation of CD28 but not inducible costimulator (ICOS). The Journal of Experimental Medicine, 197, 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey KF, Zhang X, & Thomas DM (2013). The hippo pathway and human cancer. Nature Reviews. Cancer, 13, 246–257. 10.1038/nrc3458. [DOI] [PubMed] [Google Scholar]
- Hauck F, et al. (2012). Primary T-cell immunodeficiency with immunodysregulation caused by autosomal recessive LCK deficiency. The Journal of Allergy and Clinical Immunology, 130 1144–1152 e1111. 10.1016/j.jaci.2012.07.029. [DOI] [PubMed] [Google Scholar]
- Hayward AR, et al. (1979). Delayed separation of the umbilical cord, widespread infections, and defective neutrophil mobility. Lancet, 1, 1099–1101. [DOI] [PubMed] [Google Scholar]
- Heiss NS, et al. (1998). X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nature Genetics, 19, 32–38. 10.1038/ng0598-32. [DOI] [PubMed] [Google Scholar]
- Hendriks J, et al. (2000). CD27 is required for generation and long-term maintenance of T cell immunity. Nature Immunology, 1, 433–440. 10.1038/80877. [DOI] [PubMed] [Google Scholar]
- Hernandez PA, et al. (2003). Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nature Genetics, 34, 70–74. 10.1038/ng1149. [DOI] [PubMed] [Google Scholar]
- Hershfield MS (2003). Genotype is an important determinant of phenotype in adenosine deaminase deficiency. Current Opinion in Immunology, 15, 571–577. [DOI] [PubMed] [Google Scholar]
- Hess C, & Kemper C (2016). Complement-mediated regulation of metabolism and basic cellular processes. Immunity, 45, 240–254. 10.1016/j.immuni.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesslein DG, Takaki R, Hermiston ML, Weiss A, & Lanier LL (2006). Dysregulation of signaling pathways in CD45-deficient NK cells leads to differentially regulated cytotoxicity and cytokine production. Proceedings of the National Academy of Sciences of the United States of America, 103, 7012–7017. 10.1073/pnas.0601851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata T, et al. (2012). Moesin-deficient mice reveal a non-redundant role for moesin in lymphocyte homeostasis. International Immunology, 24, 705–717. 10.1093/intimm/dxs077. [DOI] [PubMed] [Google Scholar]
- Hodes RJ, Hathcock KS, & Weng NP (2002). Telomeres in T and B cells. Nature Reviews. Immunology, 2, 699–706. 10.1038/nri890. [DOI] [PubMed] [Google Scholar]
- Hogg N, Patzak I, & Willenbrock F (2011). The insider’s guide to leukocyte integrin signalling and function. Nature Reviews. Immunology, 11, 416–426. 10.1038/nri2986. [DOI] [PubMed] [Google Scholar]
- Holcik M, & Korneluk RG (2001). XIAP, the guardian angel. Nature Reviews. Molecular Cell Biology, 2, 550–556. 10.1038/35080103. [DOI] [PubMed] [Google Scholar]
- Holland P, Torgersen ML, Sandvig K, & Simonsen A (2014). LYST affects lysosome size and quantity, but not trafficking or degradation through autophagy or endocytosis. Traffic, 15, 1390–1405. 10.1111/tra.12227. [DOI] [PubMed] [Google Scholar]
- Holland SM, et al. (2007). STAT3 mutations in the hyper-IgE syndrome. The New England Journal of Medicine, 357, 1608–1619. 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- Hollander MC, Blumenthal GM, & Dennis PA (2011). PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nature Reviews. Cancer, 11, 289–301. 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori S, Nomura T, & Sakaguchi S (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science, 299, 1057–1061. 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- Horton JS, & Stokes AJ (2014). The transmembrane channel-like protein family and human papillomaviruses: Insights into epidermodysplasia verruciformis and progression to squamous cell carcinoma. Oncoimmunology, 3, e28288, 10.4161/onci.28288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert P, et al. (2000). Defective p56Lck activity in T cells from an adult patient with idiopathic CD4+ lymphocytopenia. International Immunology, 12, 449–457. [DOI] [PubMed] [Google Scholar]
- Huck K, et al. (2009). Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. The Journal of Clinical Investigation, 119, 1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizing M, et al. (2002). Nonsense mutations in ADTB3A cause complete deficiency of the beta3A subunit of adaptor complex-3 and severe Hermansky–Pudlak syndrome type 2. Pediatric Research, 51, 150–158. 10.1203/00006450-200202000-00006. [DOI] [PubMed] [Google Scholar]
- Hum DW, Bell AW, Rozen R, & MacKenzie RE (1988). Primary structure of a human trifunctional enzyme. Isolation of a cDNA encoding methylenetetrahydrofolate dehydrogenase-methenyltetrahydrofolate cyclohydrolase-formyltetrahydrofolate synthetase. The Journal of Biological Chemistry, 263, 15946–15950. [PubMed] [Google Scholar]
- Hutloff A, et al. (1999). ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature, 397, 263–266. 10.1038/16717. [DOI] [PubMed] [Google Scholar]
- Irie-Sasaki J, et al. (2001). CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature, 409, 349–354. 10.1038/35053086. [DOI] [PubMed] [Google Scholar]
- Itan Y, & Casanova JL (2015). Novel primary immunodeficiency candidate genes predicted by the human gene connectome. Frontiers in Immunology, 6, 142. 10.3389/fimmu.2015.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa K, et al. (2017). Inherited CD70 deficiency in humans reveals a critical role for the CD70–CD27 pathway in immunity to Epstein–Barr virus infection. The Journal of Experimental Medicine, 214, 73–89. 10.1084/jem.20160784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabara HH, et al. (2016). A missense mutation in TFRC, encoding transferrin receptor 1, causes combined immunodeficiency. Nature Genetics, 48, 74–78. 10.1038/ng.3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen M, et al. (2000). A point mutation in PTPRC is associated with the development of multiple sclerosis. Nature Genetics, 26, 495–499. 10.1038/82659. [DOI] [PubMed] [Google Scholar]
- Jaiswal M, et al. (2016). Novel biochemical and structural insights into the interaction of myristoylated cargo with Unc119 protein and their release by Arl2/3. The Journal of Biological Chemistry, 291, 20766–20778. 10.1074/jbc.M116.741827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerome LA, & Papaioannou VE (2001). DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nature Genetics, 27, 286–291. 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- Jing H, et al. (2014). Somatic reversion in dedicator of cytokinesis 8 immunodeficiency modulates disease phenotype. The Journal of Allergy and Clinical Immunology, 133, 1667–1675. 10.1016/j.jaci.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongmans MC, et al. (2006). CHARGE syndrome: The phenotypic spectrum of mutations in the CHD7 gene. Journal of Medical Genetics, 43, 306–314. 10.1136/jmg.2005.036061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu LF, & Rudensky AY (2012). Regulatory T cells: Mechanisms of differentiation and function. Annual Review of Immunology, 30, 531–564. 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, et al. (2006). Identification of a homozygous deletion in the AP3B1 gene causing Hermansky-Pudlak syndrome, type 2. Blood, 108, 362–369. 10.1182/blood-2005-11-4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallikourdis M, et al. (2013). The CXCR4 mutations in WHIM syndrome impair the stability of the T-cell immunologic synapse. Blood, 122, 666–673. 10.1182/blood-2012-10-461830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz G, Krummey SM, Larsen SE, Stinson JR, & Snow AL (2014). SAP facilitates recruitment and activation of LCK at NTB-A receptors during restimulation-induced cell death. Journal of Immunology, 192, 4202–4209. 10.4049/jimmunol.1303070. [DOI] [PubMed] [Google Scholar]
- Kaur S, et al. (2011). Heparan sulfate modification of the transmembrane receptor CD47 is necessary for inhibition of T cell receptor signaling by thrombospondin-1. The Journal of Biological Chemistry, 286, 14991–15002. 10.1074/jbc.M110.179663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelleher DJ, & Gilmore R (2006). An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology, 16, 47R–62R. 10.1093/glycob/cwj066. [DOI] [PubMed] [Google Scholar]
- Keller MD, et al. (2013). Severe combined immunodeficiency resulting from mutations in MTHFD1. Pediatrics, 131, e629–634. 10.1542/peds.2012-0899. [DOI] [PubMed] [Google Scholar]
- Kishihara K, et al. (1993). Normal B lymphocyte development but impaired T cell maturation in CD45-exon6 protein tyrosine phosphatase-deficient mice. Cell, 74, 143–156. [DOI] [PubMed] [Google Scholar]
- Kishimoto TK, Hollander N, Roberts TM, Anderson DC, & Springer TA (1987). Heterogeneous mutations in the beta subunit common to the LFA-1, mac-1, and p150,95 glycoproteins cause leukocyte adhesion deficiency. Cell, 50, 193–202. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kubota S, Mori N, McLaren MJ, & Inana G (2003). Photoreceptor synaptic protein HRG4 (UNC119) interacts with ARL2 via a putative conserved domain. FEBS Letters, 534, 26–32. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, et al. (2000). HRG4 (UNC119) mutation found in cone-rod dystrophy causes retinal degeneration in a transgenic model. Investigative Ophthalmology & Visual Science, 41, 3268–3277. [PubMed] [Google Scholar]
- Kofoed EM, et al. (2003). Growth hormone insensitivity associated with a STAT5b mutation. The New England Journal of Medicine, 349, 1139–1147. 10.1056/NEJMoa022926. [DOI] [PubMed] [Google Scholar]
- Kogl T, et al. (2013). Hemophagocytic lymphohistiocytosis in syntaxin-11-deficient mice: T-cell exhaustion limits fatal disease. Blood, 121, 604–613. 10.1182/blood-2012-07-441139. [DOI] [PubMed] [Google Scholar]
- Koretzky GA, Kohmetscher M, & Ross S (1993). CD45-associated kinase activity requires lck but not T cell receptor expression in the Jurkat T cell line. The Journal of Biological Chemistry, 268, 8958–8964. [PubMed] [Google Scholar]
- Korthauer U, et al. (1993). Defective expression of T-cell CD40 ligand causes X-linked immunodeficiency with hyper-IgM. Nature, 361, 539–541. 10.1038/361539a0. [DOI] [PubMed] [Google Scholar]
- Kotlarz D, et al. (2013). Loss-of-function mutations in the IL-21 receptor gene cause a primary immunodeficiency syndrome. The Journal of Experimental Medicine, 210, 433–443. 10.1084/jem.20111229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreins AY, et al. (2015). Human TYK2 deficiency: Mycobacterial and viral infections without hyper-IgE syndrome. The Journal of Experimental Medicine, 212, 1641–1662. 10.1084/jem.20140280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn HS, et al. (2013). Loss-of-function of the protein kinase C delta (PKCdelta) causes a B-cell lymphoproliferative syndrome in humans. Blood, 121, 3117–3125. 10.1182/blood-2012-12-469544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehn HS, et al. (2014). Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science, 345, 1623–1627. 10.1126/science.1255904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung C, et al. (2000). Mutations in the tyrosine phosphatase CD45 gene in a child with severe combined immunodeficiency disease. Nature Medicine, 6, 343–345. 10.1038/73208. [DOI] [PubMed] [Google Scholar]
- Kurolap A, et al. (2017). Loss of CD55 in eculizumab-responsive protein-losing enteropathy. The New England Journal of Medicine, 377, 87–89. 10.1056/NEJMc1707173. [DOI] [PubMed] [Google Scholar]
- Kursula P, et al. (2006). Structure of the synthetase domain of human CTP synthetase, a target for anticancer therapy. Acta Crystallographica. Section F, Structural Biology and Crystallization Communications, 62, 613–617. 10.1107/S1744309106018136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagane B, et al. (2008). CXCR4 dimerization and beta-arrestin-mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood, 112, 34–44. 10.1182/blood-2007-07-102103. [DOI] [PubMed] [Google Scholar]
- Lagresle-Peyrou C, et al. (2009). Human adenylate kinase 2 deficiency causes a profound hematopoietic defect associated with sensorineural deafness. Nature Genetics, 41, 106–111. 10.1038/ng.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagresle-Peyrou C, et al. (2016). X-linked primary immunodeficiency associated with hemizygous mutations in the moesin (MSN) gene. The Journal of Allergy and Clinical Immunology, 138, 1681–1689.e8. 10.1016/j.jaci.2016.04.032. [DOI] [PubMed] [Google Scholar]
- Laham LE, Mukhopadhyay N, & Roberts TM (2000). The activation loop in Lck regulates oncogenic potential by inhibiting basal kinase activity and restricting substrate specificity. Oncogene, 19, 3961–3970. 10.1038/sj.onc.1203738. [DOI] [PubMed] [Google Scholar]
- Lalani SR, et al. (2004). SEMA3E mutation in a patient with CHARGE syndrome. Journal of Medical Genetics, 41, e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamarche BJ, Orazio NI, & Weitzman MD (2010). The MRN complex in double-strand break repair and telomere maintenance. FEBS Letters, 584, 3682–3695. 10.1016/j.febslet.2010.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier MH, Kim T, & Cooper JA (2015). CARMIL2 is a novel molecular connection between vimentin and actin essential for cell migration and invadopodia formation. Molecular Biology of the Cell, 26, 4577–4588. 10.1091/mbc.E15-08-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzi G, et al. (2012). A novel primary human immunodeficiency due to deficiency in the WASP-interacting protein WIP. The Journal of Experimental Medicine, 209, 29–34. 10.1084/jem.20110896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latour S, et al. (2003). Binding of SAP SH2 domain to FynT SH3 domain reveals a novel mechanism of receptor signalling in immune regulation. Nature Cell Biology, 5, 149–154. 10.1038/ncb919. [DOI] [PubMed] [Google Scholar]
- Lazarczyk M, et al. (2008). Regulation of cellular zinc balance as a potential mechanism of EVER-mediated protection against pathogenesis by cutaneous oncogenic human papillomaviruses. The Journal of Experimental Medicine, 205, 35–42. 10.1084/jem.20071311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarczyk M, et al. (2012). EVER proteins, key elements of the natural anti-human papillomavirus barrier, are regulated upon T-cell activation. PLoS One, 7, e39995. 10.1371/journal.pone.0039995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guen T, et al. (2013). Human RTEL1 deficiency causes Hoyeraal–Hreidarsson syndrome with short telomeres and genome instability. Human Molecular Genetics, 22, 3239–3249. 10.1093/hmg/ddt178. [DOI] [PubMed] [Google Scholar]
- Le Guen T, et al. (2015). An in vivo genetic reversion highlights the crucial role of Myb-Like, SWIRM, and MPN domains 1 (MYSM1) in human hematopoiesis and lymphocyte differentiation. The Journal of Allergy and Clinical Immunology, 136 1619–1626.e5. 10.1016/j.jaci.2015.06.008. [DOI] [PubMed] [Google Scholar]
- Lee JH, & Paull TT (2004). Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science, 304, 93–96. 10.1126/science.1091496. [DOI] [PubMed] [Google Scholar]
- Lee Y, et al. (2013). UNC119a bridges the transmission of Fyn signals to Rab11, leading to the completion of cytokinesis. Cell Cycle, 12, 1303–1315. 10.4161/cc.24404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, et al. (2016). Abatacept alleviates severe autoimmune symptoms in a patient carrying a de novo variant in CTLA-4. The Journal of Allergy and Clinical Immunology, 137, 327–330. 10.1016/j.jaci.2015.08.036. [DOI] [PubMed] [Google Scholar]
- Lemoine R, et al. (2014). Immune deficiency-related enteropathy-lymphocytopenia-alopecia syndrome results from tetratricopeptide repeat domain 7A deficiency. The Journal of Allergy and Clinical Immunology, 134, 1354–1364.e6. 10.1016/j.jaci.2014.07.019. [DOI] [PubMed] [Google Scholar]
- Leonard WJ, & Wan CK (2016). IL-21 signaling in immunity [version 1; referees: 3 approved]. F1000Research, 5(F1000 Faculty Rev), 224. 10.12688/f1000research.7634.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JS, et al. (2017). SP110b controls host immunity and susceptibility to tuberculosis. American Journal of Respiratory and Critical Care Medicine, 195, 369–382. 10.1164/rccm.201601-0103OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, et al. (2002). The hematopoiesis-specific GTP-binding protein RhoH is GTPase deficient and modulates activities of other rho GTPases by an inhibitory function. Molecular and Cellular Biology, 22, 1158–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FY, et al. (2011). Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature, 475, 471–476. 10.1038/nature10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Niederstrasser H, Edwards M, Jackson CE, & Cooper JA (2009). Distinct roles for CARMIL isoforms in cell migration. Molecular Biology of the Cell, 20, 5290–5305. 10.1091/mbc.E08-10-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, et al. (2013). The lymphoid lineage-specific actin-uncapping protein Rltpr is essential for costimulation via CD28 and the development of regulatory T cells. Nature Immunology, 14, 858–866. 10.1038/ni.2634. [DOI] [PubMed] [Google Scholar]
- Lin JX, & Leonard WJ (2017). The common cytokine receptor gamma chain family of cytokines. Cold Spring Harbor Perspectives in Biology. 10.1101/cshperspect.a028449 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linka RM, et al. (2012). Loss-of-function mutations within the IL-2 inducible kinase ITK in patients with EBV-associated lymphoproliferative diseases. Leukemia, 26, 963–971. 10.1038/leu.2011.371. [DOI] [PubMed] [Google Scholar]
- Lischner HW, & Huff DS (1975). T-cell deficiency in DiGeorge syndrome. Birth Defects Original Article Series, 11, 16–21. [PubMed] [Google Scholar]
- Liszewski MK, et al. (2013). Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity, 39, 1143–1157. 10.1016/j.immuni.2013.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, et al. (2017). Novel compound heterozygous mutations in ZAP70 in a Chinese patient with leaky severe combined immunodeficiency disorder. Immunogenetics, 69, 199–209. 10.1007/s00251-017-0971-0. [DOI] [PubMed] [Google Scholar]
- Lo B (2016). The requirement of iron transport for lymphocyte function. Nature Genetics, 48, 10–11. 10.1038/ng.3478. [DOI] [PubMed] [Google Scholar]
- Lo B, et al. (2013). A rapid ex vivo clinical diagnostic assay for fas receptor-induced T lymphocyte apoptosis. Journal of Clinical Immunology, 33, 479–488. 10.1007/s10875-012-9811-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo B, et al. (2015). Autoimmune Disease. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science, 349, 436–440. 10.1126/science.aaa1663. [DOI] [PubMed] [Google Scholar]
- Lohr NJ, et al. (2010). Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. American Journal of Human Genetics, 86, 447–453. 10.1016/j.ajhg.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Herrera G, et al. (2012). Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. American Journal of Human Genetics, 90, 986–1001. 10.1016/j.ajhg.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, et al. (2014). Dual proteolytic pathways govern glycolysis and immune competence. Cell, 159, 1578–1590. 10.1016/j.cell.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas CL, Chandra A, Nejentsev S, Condliffe AM, & Okkenhaug K (2016). PI3Kdelta and primary immunodeficiencies. Nature Reviews. Immunology, 16, 702–714. 10.1038/nri.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas CL, et al. (2014a). Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nature Immunology, 15, 88–97. 10.1038/ni.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas CL, et al. (2014b). Heterozygous splice mutation in PIK3R1 causes human immunodeficiency with lymphoproliferation due to dominant activation of PI3K. The Journal of Experimental Medicine, 211, 2537–2547. 10.1084/jem.20141759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin KE, et al. (2015). Susceptibility to infections, without concomitant hyper-IgE, reported in 1976, is caused by hypomorphic mutation in the phosphoglucomutase 3 (PGM3) gene. Clinical Immunology, 161, 366–372. 10.1016/j.clim.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Pannicke U, Schwarz K, & Lieber MR (2002). Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell, 108, 781–794. [DOI] [PubMed] [Google Scholar]
- Ma Y, Schwarz K, & Lieber MR (2005). The Artemis:DNA-PKcs endonuclease cleaves DNA loops, flaps, and gaps. DNA Repair (Amst), 4, 845–851. 10.1016/j.dnarep.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Ma CS, et al. (2008). Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. The Journal of Experimental Medicine, 205, 1551–1557. 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CA, et al. (2017). Germline hypomorphic CARD11 mutations in severe atopic disease. Nature Genetics, 49, 1192–1201. 10.1038/ng.3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macchi P, et al. (1995). Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature, 377, 65–68. 10.1038/377065a0. [DOI] [PubMed] [Google Scholar]
- Magerus-Chatinet A, et al. (2013). Autoimmune lymphoproliferative syndrome caused by a homozygous null FAS ligand (FASLG) mutation. The Journal of Allergy and Clinical Immunology, 131, 486–490. 10.1016/j.jaci.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majri SS, et al. (2018). STAT5B: A differential regulator of the life and death of CD4(+) effector memory T cells. Journal of Immunology, 200, 110–118. 10.4049/jimmunol.1701133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinin NL, et al. (2009). A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nature Medicine, 15, 313–318. 10.1038/nm.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancebo E, et al. (2008). Gly111Ser mutation in CD8A gene causing CD8 immunodeficiency is found in Spanish gypsies. Molecular Immunology, 45, 479–484. 10.1016/j.molimm.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Mao H, et al. (2017). RASGRP1 mutation in autoimmune lymphoproliferative syndrome-like disease. The Journal of Allergy and Clinical Immunology. 10.1016/j.jaci.2017.10.026. [DOI] [PubMed] [Google Scholar]
- Markert ML (1991). Purine nucleoside phosphorylase deficiency. Immunodeficiency Reviews, 3, 45–81. [PubMed] [Google Scholar]
- Markert ML, et al. (2007). Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation: Outcome of 44 consecutive transplants. Blood, 109, 4539–4547. 10.1182/blood-2006-10-048652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markert ML, et al. (2011). First use of thymus transplantation therapy for FOXN1 deficiency (nude/SCID): A report of 2 cases. Blood, 117, 688–696. 10.1182/blood-2010-06-292490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DM, Sheldon S, & Gorski JL (2001). CHARGE association with choanal atresia and inner ear hypoplasia in a child with a de novo chromosome translocation t(2,7) (p14;q21.11). American Journal of Medical Genetics, 99, 115–119. [DOI] [PubMed] [Google Scholar]
- Martin DA, et al. (1999). Defective CD95/APO-1/Fas signal complex formation in the human autoimmune lymphoproliferative syndrome, type Ia. Proceedings of the National Academy of Sciences of the United States of America, 96, 4552–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, et al. (2014). CTP synthase 1 deficiency in humans reveals its central role in lymphocyte proliferation. Nature, 510, 288–292. 10.1038/nature13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarl CA, et al. (2009). ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. The Journal of Allergy and Clinical Immunology, 124 1311–1318.e7. 10.1016/j.jaci.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menager MM, et al. (2007). Secretory cytotoxic granule maturation and exocytosis require the effector protein hMunc13–4. Nature Immunology, 8, 257–267. 10.1038/ni1431. [DOI] [PubMed] [Google Scholar]
- Menasche G, et al. (2000). Mutations in RAB27A cause Griscelli syndrome associated with haemophagocytic syndrome. Nature Genetics, 25, 173–176. 10.1038/76024. [DOI] [PubMed] [Google Scholar]
- Milner JD, et al. (2008). Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature, 452, 773–776. 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner JD, et al. (2015). Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood, 125, 591–599. 10.1182/blood-2014-09-602763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minegishi Y, et al. (2006). Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity, 25, 745–755. 10.1016/j.immuni.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Minegishi Y, et al. (2007). Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature, 448, 1058–1062. 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- Mochizuki Y, He J, Kulkarni S, Bessler M, & Mason PJ (2004). Mouse dyskerin mutations affect accumulation of telomerase RNA and small nucleolar RNA, telomerase activity, and ribosomal RNA processing. Proceedings of the National Academy of Sciences of the United States of America, 101, 10756–10761. 10.1073/pnas.0402560101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molon B, et al. (2005). T cell costimulation by chemokine receptors. Nature Immunology, 6, 465–471. 10.1038/ni1191. [DOI] [PubMed] [Google Scholar]
- Monkley SJ, et al. (2000). Disruption of the talin gene arrests mouse development at the gastrulation stage. Developmental Dynamics, 219, 560–574. doi:. [DOI] [PubMed] [Google Scholar]
- Monticelli M, Ferro T, Jaeken J, Dos Reis Ferreira V, & Videira PA (2016). Immunological aspects of congenital disorders of glycosylation (CDG): A review. Journal of Inherited Metabolic Disease, 39, 765–780. 10.1007/s10545-016-9954-9. [DOI] [PubMed] [Google Scholar]
- Morgan NV, et al. (2011). Mutation in the TCRalpha subunit constant gene (TRAC) leads to a human immunodeficiency disorder characterized by a lack of TCRalphabeta+ T cells. The Journal of Clinical Investigation, 121, 695–702. 10.1172/JCI41931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriggl R, et al. (1999). Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity, 10, 249–259. [DOI] [PubMed] [Google Scholar]
- Morin G, et al. (2014). Gain-of-function mutation in STIM1 (P.R304W) is associated with Stormorken syndrome. Human Mutation, 35, 1221–1232. 10.1002/humu.22621. [DOI] [PubMed] [Google Scholar]
- Moshous D, et al. (2001). Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell, 105, 177–186. [DOI] [PubMed] [Google Scholar]
- Moshous D, et al. (2013). Whole-exome sequencing identifies coronin-1A deficiency in 3 siblings with immunodeficiency and EBV-associated B-cell lymphoproliferation. The Journal of Allergy and Clinical Immunology, 131, 1594–1603. 10.1016/j.jaci.2013.01.042. [DOI] [PubMed] [Google Scholar]
- Moulding DA, Record J, Malinova D, & Thrasher AJ (2013). Actin cytoskeletal defects in immunodeficiency. Immunological Reviews, 256, 282–299. 10.1111/imr.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugnier B, et al. (2008). Coronin-1A links cytoskeleton dynamics to TCR alpha beta-induced cell signaling. PLoS One, 3, e3467. 10.1371/journal.pone.0003467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, & Thrasher AJ (2013). Gene therapy for PIDs: Progress, pitfalls and prospects. Gene, 525, 174–181. 10.1016/j.gene.2013.03.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller ML, et al. (2014). An N-terminal missense mutation in STX11 causative of FHL4 abrogates syntaxin-11 binding to Munc18–2. Frontiers in Immunology, 4, 515. 10.3389/fimmu.2013.00515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagle DL, et al. (1996). Identification and mutation analysis of the complete gene for Chediak–Higashi syndrome. Nature Genetics, 14, 307–311. 10.1038/ng1196-307. [DOI] [PubMed] [Google Scholar]
- Nakamura A, et al. (2001). Prognostic impact of CD45 antigen expression in high-risk, childhood B-cell precursor acute lymphoblastic leukemia. Leukemia & Lymphoma, 42, 393–398. 10.3109/10428190109064596. [DOI] [PubMed] [Google Scholar]
- Nehme NT, et al. (2012). MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood, 119, 3458–3468. 10.1182/blood-2011-09-378364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesin V, et al. (2014). Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proceedings of the National Academy of Sciences of the United States of America, 111, 4197–4202. 10.1073/pnas.1312520111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols KE, Ma CS, Cannons JL, Schwartzberg PL, & Tangye SG (2005). Molecular and cellular pathogenesis of X-linked lymphoproliferative disease. Immunological Reviews, 203, 180–199. 10.1111/j.0105-2896.2005.00230.x. [DOI] [PubMed] [Google Scholar]
- Niemela JE, et al. (2011). Somatic KRAS mutations associated with a human nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis. Blood, 117, 2883–2886. 10.1182/blood-2010-07-295501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nistico L, et al. (1996). The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Belgian Diabetes Registry. Human Molecular Genetics, 5, 1075–1080. [DOI] [PubMed] [Google Scholar]
- Noguchi M, et al. (1993). Interleukin-2 receptor gamma chain mutation results in X-linked severe combined immunodeficiency in humans. Cell, 73, 147–157. [DOI] [PubMed] [Google Scholar]
- Noma T (2005). Dynamics of nucleotide metabolism as a supporter of life phenomena. The Journal of Medical Investigation, 52, 127–136. [DOI] [PubMed] [Google Scholar]
- Notarangelo LD (2013). Partial defects of T-cell development associated with poor T-cell function. The Journal of Allergy and Clinical Immunology, 131, 1297–1305. 10.1016/j.jaci.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notarangelo LD (2017). Expanding the spectrum of skeletal dysplasia with immunodeficiency: A commentary on identification of biallelic EXTL3 mutations in a novel type of spondylo-epi-metaphyseal dysplasia. Journal of Human Genetics, 62, 737–738. 10.1038/jhg.2017.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notarangelo LD, Kim MS, Walter JE, & Lee YN (2016). Human RAG mutations: Biochemistry and clinical implications. Nature Reviews. Immunology, 16, 234–246. 10.1038/nri.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noury JB, et al. (2017). Tubular aggregate myopathy with features of Stormorken disease due to a new STIM1 mutation. Neuromuscular Disorders, 27, 78–82. 10.1016/j.nmd.2016.10.006. [DOI] [PubMed] [Google Scholar]
- Okkenhaug K, et al. (2002). Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science, 297, 1031–1034. 10.1126/science.1073560. [DOI] [PubMed] [Google Scholar]
- Oliveira JB, et al. (2007). NRAS mutation causes a human autoimmune lymphoproliferative syndrome. Proceedings of the National Academy of Sciences of the United States of America, 104, 8953–8958. 10.1073/pnas.0702975104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira JB, et al. (2010). Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): Report from the 2009 NIH International Workshop. Blood, 116, e35–40. 10.1182/blood-2010-04-280347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oud MM, et al. (2017). Mutations in EXTL3 cause neuro-immuno-skeletal dysplasia syndrome. American Journal of Human Genetics, 100, 281–296. 10.1016/j.ajhg.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozen A, et al. (2017). CD55 deficiency, early-onset protein-losing enteropathy, and thrombosis. The New England Journal of Medicine, 377, 52–61. 10.1056/NEJMoa1615887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachlopnik Schmid J, et al. (2012). Polymerase epsilon1 mutation in a human syndrome with facial dysmorphism, immunodeficiency, livedo, and short stature (“FILS syndrome”). The Journal of Experimental Medicine, 209, 2323–2330. 10.1084/jem.20121303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palendira U, et al. (2011). Molecular pathogenesis of EBV susceptibility in XLP as revealed by analysis of female carriers with heterozygous expression of SAP. PLoS Biology, 9, e1001187. 10.1371/journal.pbio.1001187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandiyan P, Zheng L, Ishihara S, Reed J, & Lenardo MJ (2007). CD4+ CD25+ Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nature Immunology, 8, 1353–1362. 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- Pandiyan P, et al. (2011). CD4(+)CD25(+)Foxp3(+) regulatory T cells promote Th17 cells in vitro and enhance host resistance in mouse Candida albicans Th17 cell infection model. Immunity, 34, 422–434. 10.1016/j.immuni.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannicke U, et al. (2009). Reticular dysgenesis (aleukocytosis) is caused by mutations in the gene encoding mitochondrial adenylate kinase 2. Nature Genetics, 41, 101–105. 10.1038/ng.265. [DOI] [PubMed] [Google Scholar]
- Parravicini V, Field AC, Tomlinson PD, Basson MA, & Zamoyska R (2008). Itch−/− alphabeta and gammadelta T cells independently contribute to autoimmunity in Itchy mice. Blood, 111, 4273–7282. 10.1182/blood-2007-10-115667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry DA, et al. (2016). A homozygous STIM1 mutation impairs store-operated calcium entry and natural killer cell effector function without clinical immunodeficiency. The Journal of Allergy and Clinical Immunology, 137 955–957.e8. 10.1016/j.jaci.2015.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, et al. (2001). Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature, 412, 341–346. 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- Pastor-Anglada M, et al. (2001). Complex regulation of nucleoside transporter expression in epithelial and immune system cells. Molecular Membrane Biology, 18, 81–85. [DOI] [PubMed] [Google Scholar]
- Pasvolsky R, et al. (2007). A LAD-III syndrome is associated with defective expression of the Rap-1 activator CalDAG-GEFI in lymphocytes, neutrophils, and platelets. The Journal of Experimental Medicine, 204, 1571–1582. 10.1084/jem.20070058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter ME, & Krammer PH (2003). The CD95(APO-1/Fas) DISC and beyond. Cell Death and Differentiation, 10, 26–35. 10.1038/sj.cdd.4401186. [DOI] [PubMed] [Google Scholar]
- Petrovski S, et al. (2016). Dominant splice site mutations in PIK3R1 cause hyper IgM syndrome, lymphadenopathy and short stature. Journal of Clinical Immunology, 36, 462–471. 10.1007/s10875-016-0281-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard C, et al. (2002). Inherited interleukin-12 deficiency: IL12B genotype and clinical phenotype of 13 patients from six kindreds. American Journal of Human Genetics, 70, 336–348. 10.1086/338625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard C, et al. (2009a). Hypomorphic mutation of ZAP70 in human results in a late onset immunodeficiency and no autoimmunity. European Journal of Immunology, 39, 1966–1976. 10.1002/eji.200939385. [DOI] [PubMed] [Google Scholar]
- Picard C, et al. (2009b). STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. The New England Journal of Medicine, 360, 1971–1980. 10.1056/NEJMoa0900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard C, et al. (2015). Primary immunodeficiency diseases: An update on the classification from the International Union of Immunological Societies Expert Committee for primary immunodeficiency 2015. Journal of Clinical Immunology, 35, 696–726. 10.1007/s10875-015-0201-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignata C, et al. (1996). Congenital alopecia and nail dystrophy associated with severe functional T-cell immunodeficiency in two sibs. American Journal of Medical Genetics, 65, 167–170. . [DOI] [PubMed] [Google Scholar]
- Podack ER, et al. (1988). Structure, function and expression of murine and human perforin 1 (P1). Immunological Reviews, 103, 203–211. [DOI] [PubMed] [Google Scholar]
- Popovic D, Vucic D, & Dikic I (2014). Ubiquitination in disease pathogenesis and treatment. Nature Medicine, 20, 1242–1253. 10.1038/nm.3739. [DOI] [PubMed] [Google Scholar]
- Porcu M, et al. (2012). Mutation of the receptor tyrosine phosphatase PTPRC (CD45) in T-cell acute lymphoblastic leukemia. Blood, 119, 4476–4479. 10.1182/blood-2011-09-379958. [DOI] [PubMed] [Google Scholar]
- Prawer SE, et al. (1977). Depressed immune function in epidermodysplasia verruciformis. Archives of Dermatology, 113, 495–499. [PubMed] [Google Scholar]
- Price S, et al. (2014). Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood, 123, 1989–1999. 10.1182/blood-2013-10-535393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Ziegler SF, Buckley RH, & Leonard WJ (1998). Defective IL7R expression in T(−)B(+)NK(+) severe combined immunodeficiency. Nature Genetics, 20, 394–397. 10.1038/3877. [DOI] [PubMed] [Google Scholar]
- Punwani D, et al. (2016). Multisystem anomalies in severe combined immunodeficiency with mutant BCL11B. The New England Journal of Medicine, 375, 2165–2176. 10.1056/NEJMoa1509164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh N, Anton IM, Hartwig JH, & Geha RS (1997). WIP, a protein associated with Wiskott–Aldrich syndrome protein, induces actin polymerization and redistribution in lymphoid cells. Proceedings of the National Academy of Sciences of the United States of America, 94, 14671–14676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao VK, et al. (2017). Effective “activated PI3Kdelta syndrome”-targeted therapy with the PI3Kdelta inhibitor leniolisib. Blood, 130, 2307–2316. 10.1182/blood-2017-08-801191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raponi S, et al. (2015). CD45 antigen negativity in T-lineage ALL correlates with PTPRC mutation and sensitivity to a selective JAK inhibitor. British Journal of Haematology, 171, 884–887. 10.1111/bjh.13448. [DOI] [PubMed] [Google Scholar]
- Recio MJ, et al. (2007). Differential biological role of CD3 chains revealed by human immunodeficiencies. Journal of Immunology, 178, 2556–2564. [DOI] [PubMed] [Google Scholar]
- Rieux-Laucat F, Le Deist F, & Fischer A (2003). Autoimmune lymphoproliferative syndromes: Genetic defects of apoptosis pathways. Cell Death and Differentiation, 10, 124–133. 10.1038/sj.cdd.4401190. [DOI] [PubMed] [Google Scholar]
- Rieux-Laucat F, et al. (2006). Inherited and somatic CD3zeta mutations in a patient with T-cell deficiency. The New England Journal of Medicine, 354, 1913–1921. 10.1056/NEJMoa053750. [DOI] [PubMed] [Google Scholar]
- Rigaud S, et al. (2006). XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature, 444, 110–114. 10.1038/nature05257. [DOI] [PubMed] [Google Scholar]
- Roberts JL, et al. (2012). CD45-deficient severe combined immunodeficiency caused by uniparental disomy. Proceedings of the National Academy of Sciences of the United States of America, 109, 10456–10461. 10.1073/pnas.1202249109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochman Y, Spolski R, & Leonard WJ (2009). New insights into the regulation of T cells by gamma(c) family cytokines. Nature Reviews. Immunology, 9, 480–490. 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano R, et al. (2013). FOXN1: A master regulator gene of thymic epithelial development program. Frontiers in Immunology, 4, 187. 10.3389/fimmu.2013.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roncagalli R, et al. (2016). The scaffolding function of the RLTPR protein explains its essential role for CD28 co-stimulation in mouse and human T cells. The Journal of Experimental Medicine, 213, 2437–2457. 10.1084/jem.20160579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscioli T, et al. (2006). Mutations in the gene encoding the PML nuclear body protein Sp110 are associated with immunodeficiency and hepatic veno-occlusive disease. Nature Genetics, 38, 620–622. 10.1038/ng1780. [DOI] [PubMed] [Google Scholar]
- Roselli G, et al. (2017). CXCL12 mediates aberrant costimulation of B lymphocytes in warts, hypogammaglobulinemia, infections, myelokathexis immunodeficiency. Frontiers in Immunology, 8, 1068. 10.3389/fimmu.2017.01068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota IA, & Dhalla F (2017). FOXN1 deficient nude severe combined immunodeficiency. Orphanet Journal of Rare Diseases, 12, 6. 10.1186/s13023-016-0557-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffo E, et al. (2016). Inhibition of diacylglycerol kinase alpha restores restimulation-induced cell death and reduces immunopathology in XLP-1. Science Translational Medicine, 8, 321ra327. 10.1126/scitranslmed.aad1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruschak PJ, Kauh YC, & Luscombe HA (1981). Cowden’s disease associated with immunodeficiency. Archives of Dermatology, 117, 573–575. [PubMed] [Google Scholar]
- Russell SM, et al. (1995). Mutation of Jak3 in a patient with SCID: Essential role of Jak3 in lymphoid development. Science, 270, 797–800. [DOI] [PubMed] [Google Scholar]
- Salmond RJ, Filby A, Qureshi I, Caserta S, & Zamoyska R (2009). T-cell receptor proximal signaling via the Src-family kinases, Lck and Fyn, influences T-cell activation, differentiation, and tolerance. Immunological Reviews, 228, 9–22. 10.1111/j.1600-065X.2008.00745.x. [DOI] [PubMed] [Google Scholar]
- Salzer U, et al. (2004). ICOS deficiency in patients with common variable immunodeficiency. Clinical Immunology, 113, 234–240. 10.1016/j.clim.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Salzer E, et al. (2013a). B-cell deficiency and severe autoimmunity caused by deficiency of protein kinase C delta. Blood, 121, 3112–3116. 10.1182/blood-2012-10-460741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzer E, et al. (2013b). Combined immunodeficiency with life-threatening EBV-associated lymphoproliferative disorder in patients lacking functional CD27. Haematologica, 98, 473–478. 10.3324/haematol.2012.068791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzer E, et al. (2014). Early-onset inflammatory bowel disease and common variable immunodeficiency-like disease caused by IL-21 deficiency. The Journal of Allergy and Clinical Immunology, 133, 1651–1659.e12. 10.1016/j.jaci.2014.02.034. [DOI] [PubMed] [Google Scholar]
- Salzer E, et al. (2016). RASGRP1 deficiency causes immunodeficiency with impaired cytoskeletal dynamics. Nature Immunology, 17, 1352–1360. 10.1038/ni.3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom DM (2015). Immunology. Moving CTLA-4 from the trash to recycling. Science, 349, 377–378. 10.1126/science.aac7888. [DOI] [PubMed] [Google Scholar]
- Santoro A, et al. (2006). Novel Munc13–4 mutations in children and young adult patients with haemophagocytic lymphohistiocytosis. Journal of Medical Genetics, 43, 953–960. 10.1136/jmg.2006.041863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanui T, et al. (2003). DOCK2 regulates Rac activation and cytoskeletal reorganization through interaction with ELMO1. Blood, 102, 2948–2950. 10.1182/blood-2003-01-0173. [DOI] [PubMed] [Google Scholar]
- Sassi A, et al. (2014). Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. The Journal of Allergy and Clinical Immunology, 133, 1410–1419. 1419.e1–13. 10.1016/j.jaci.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawabe T, et al. (2001). Defect of lck in a patient with common variable immunodeficiency. International Journal of Molecular Medicine, 7, 609–614. [DOI] [PubMed] [Google Scholar]
- Sayos J, et al. (1998). The X-linked lymphoproliferative-disease gene product SAP regulates signals induced through the co-receptor SLAM. Nature, 395, 462–469. 10.1038/26683. [DOI] [PubMed] [Google Scholar]
- Schmid GL, et al. (2014). Sirolimus treatment of severe PTEN hamartoma tumor syndrome: Case report and in vitro studies. Pediatric Research, 75, 527–534. 10.1038/pr.2013.246. [DOI] [PubMed] [Google Scholar]
- Schober T, et al. (2017). A human immunodeficiency syndrome caused by mutations in CARMIL2. Nature Communications, 8, 14209. 10.1038/ncomms14209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, et al. (2014). Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nature Medicine, 20, 1410–1416. 10.1038/nm.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz C, et al. (2008). An immunodeficiency disease with RAG mutations and granulomas. The New England Journal of Medicine, 358, 2030–2038. 10.1056/NEJMoa073966. [DOI] [PubMed] [Google Scholar]
- Schwarz K, et al. (1996). RAG mutations in human B cell-negative SCID. Science, 274, 97–99. [DOI] [PubMed] [Google Scholar]
- Sepulveda FE, & de Saint Basile G (2017). Hemophagocytic syndrome: Primary forms and predisposing conditions. Current Opinion in Immunology, 49, 20–26. 10.1016/j.coi.2017.08.004. [DOI] [PubMed] [Google Scholar]
- Sepulveda FE, et al. (2015). LYST controls the biogenesis of the endosomal compartment required for secretory lysosome function. Traffic, 16, 191–203. 10.1111/tra.12244. [DOI] [PubMed] [Google Scholar]
- Sharfe N, Dadi HK, Shahar M, & Roifman CM (1997). Human immune disorder arising from mutation of the alpha chain of the interleukin-2 receptor. Proceedings of the National Academy of Sciences of the United States of America, 94, 3168–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiow LR, et al. (2008). The actin regulator coronin 1A is mutant in a thymic egress-deficient mouse strain and in a patient with severe combined immunodeficiency. Nature Immunology, 9, 1307–1315. 10.1038/ni.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RM, et al. (2004). SPOTS: Signaling protein oligomeric transduction structures are early mediators of death receptor-induced apoptosis at the plasma membrane. The Journal of Cell Biology, 167, 735–744. 10.1083/jcb.200406101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel AM, et al. (2011). A critical role for STAT3 transcription factor signaling in the development and maintenance of human T cell memory. Immunity, 35, 806–818. 10.1016/j.immuni.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Six E, et al. (2015). AK2 deficiency compromises the mitochondrial energy metabolism required for differentiation of human neutrophil and lymphoid lineages. Cell Death & Disease, 6, e1856. 10.1038/cddis.2015.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow AL, et al. (2009). Restimulation-induced apoptosis of T cells is impaired in patients with X-linked lymphoproliferative disease caused by SAP deficiency. The Journal of Clinical Investigation, 119, 2976–2989. 10.1172/JCI39518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow AL, et al. (2012). Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. The Journal of Experimental Medicine, 209, 2247–2261. 10.1084/jem.20120831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J, Rothberg BS, Madesh M, & Gill DL (2012). STIM proteins: Dynamic calcium signal transducers. Nature Reviews. Molecular Cell Biology, 13, 549–565. 10.1038/nrm3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solder B, Weiss M, Jager A, & Belohradsky BH (1998). Dyskeratosis congenita: Multisystemic disorder with special consideration of immunologic aspects. A review of the literature. Clinical Pediatrics, 37, 521–530. 10.1177/000992289803700901. [DOI] [PubMed] [Google Scholar]
- Song MS, Salmena L, & Pandolfi PP (2012). The functions and regulation of the PTEN tumour suppressor. Nature Reviews. Molecular Cell Biology, 13, 283–296. 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- Sorte HS, et al. (2016). A potential founder variant in CARMIL2/RLTPR in three Norwegian families with warts, molluscum contagiosum, and T-cell dysfunction. Molecular Genetics & Genomic Medicine, 4, 604–616. 10.1002/mgg3.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudais C, de Villartay JP, Le Deist F, Fischer A, & Lisowska-Grospierre B (1993). Independent mutations of the human CD3-epsilon gene resulting in a T cell receptor/CD3 complex immunodeficiency. Nature Genetics, 3, 77–81. 10.1038/ng0193-77. [DOI] [PubMed] [Google Scholar]
- Squarize CH, Castilho RM, & Gutkind JS (2008). Chemoprevention and treatment of experimental Cowden’s disease by mTOR inhibition with rapamycin. Cancer Research, 68, 7066–7072. 10.1158/0008-5472.CAN-08-0922. [DOI] [PubMed] [Google Scholar]
- Staples ER, et al. (2008). Immunodeficiency in ataxia telangiectasia is correlated strongly with the presence of two null mutations in the ataxia telangiectasia mutated gene. Clinical and Experimental Immunology, 153, 214–220. 10.1111/j.1365-2249.2008.03684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepensky P, et al. (2011). IL-2-inducible T-cell kinase deficiency: Clinical presentation and therapeutic approach. Haematologica, 96, 472–476. 10.3324/haematol.2010.033910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepensky P, et al. (2015). Early-onset Evans syndrome, immunodeficiency, and premature immunosenescence associated with tripeptidyl-peptidase II deficiency. Blood, 125, 753–761. 10.1182/blood-2014-08-593202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepp SE, et al. (1999). Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science, 286, 1957–1959. [DOI] [PubMed] [Google Scholar]
- Stevenson C, et al. (2014). Essential role of Elmo1 in Dock2-dependent lymphocyte migration. Journal of Immunology, 192, 6062–6070. 10.4049/jimmunol.1303348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiff T, et al. (2005). Nbs1 is required for ATR-dependent phosphorylation events. The EMBO Journal, 24, 199–208. 10.1038/sj.emboj.7600504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinchcombe JC, et al. (2001). Rab27a is required for regulated secretion in cytotoxic T lymphocytes. The Journal of Cell Biology, 152, 825–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stray-Pedersen A, et al. (2014a). Compound heterozygous CORO1A mutations in siblings with a mucocutaneous-immunodeficiency syndrome of epidermodysplasia verruciformis-HPV, molluscum contagiosum and granulomatous tuberculoid leprosy. Journal of Clinical Immunology, 34, 871–890. 10.1007/s10875-014-0074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stray-Pedersen A, et al. (2014b). PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. American Journal of Human Genetics, 95, 96–107. 10.1016/j.ajhg.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, et al. (2005). Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science, 307, 1465–1468. 10.1126/science.1104765. [DOI] [PubMed] [Google Scholar]
- Svensson L, et al. (2009). Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nature Medicine, 15, 306–312. 10.1038/nm.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swamy M, et al. (2016). Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nature Immunology, 17, 712–720. 10.1038/ni.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi M, et al. (2011). Autoimmune lymphoproliferative syndrome-like disease with somatic KRAS mutation. Blood, 117, 2887–2890. 10.1182/blood-2010-08-301515. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, et al. (2007). T helper type 2 differentiation and intracellular trafficking of the interleukin 4 receptor-alpha subunit controlled by the Rac activator Dock2. Nature Immunology, 8, 1067–1075. 10.1038/ni1506. [DOI] [PubMed] [Google Scholar]
- Tanigawa K, et al. (2009). Tryptophan aspartate-containing coat protein (CORO1A) suppresses toll-like receptor signalling in Mycobacterium leprae infection. Clinical and Experimental Immunology, 156, 495–501. 10.1111/j.1365-2249.2009.03930.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchilian EZ, et al. (2001). A deletion in the gene encoding the CD45 antigen in a patient with SCID. Journal of Immunology, 166, 1308–1313. [DOI] [PubMed] [Google Scholar]
- Thauvin-Robinet C, et al. (2013). PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. American Journal of Human Genetics, 93, 141–149. 10.1016/j.ajhg.2013.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thijssen PE, et al. (2015). Mutations in CDCA7 and HELLS cause immunodeficiency-centromeric instability-facial anomalies syndrome. Nature Communications, 6, 7870. 10.1038/ncomms8870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrasher AJ (2002). WASp in immune-system organization and function. Nature Reviews. Immunology, 2, 635–646. 10.1038/nri884. [DOI] [PubMed] [Google Scholar]
- Thrasher AJ, & Burns SO (2010). WASP: A key immunological multitasker. Nature Reviews. Immunology, 10, 182–192. 10.1038/nri2724. [DOI] [PubMed] [Google Scholar]
- Tian Y, & Zajac AJ (2016). IL-21 and T cell differentiation: Consider the context. Trends in Immunology, 37, 557–568. 10.1016/j.it.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, et al. (2010). C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell, 141, 1042–1055. 10.1016/j.cell.2010.04.034. [DOI] [PubMed] [Google Scholar]
- Tivol EA, et al. (1995). Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity, 3, 541–547. [DOI] [PubMed] [Google Scholar]
- Tomkinson B (1999). Tripeptidyl peptidases: Enzymes that count. Trends in Biochemical Sciences, 24, 355–359. [DOI] [PubMed] [Google Scholar]
- Tsujita Y, et al. (2016). Phosphatase and tensin homolog (PTEN) mutation can cause activated phosphatidylinositol 3-kinase delta syndrome-like immunodeficiency. The Journal of Allergy and Clinical Immunology, 138 1672–1680.e10. 10.1016/j.jaci.2016.03.055. [DOI] [PubMed] [Google Scholar]
- Tummala H, et al. (2014). ERCC6L2 mutations link a distinct bone-marrow-failure syndrome to DNA repair and mitochondrial function. American Journal of Human Genetics, 94, 246–256. 10.1016/j.ajhg.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda H, et al. (2003). Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature, 423, 506–511. 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- Ussar S, et al. (2008). Loss of Kindlin-1 causes skin atrophy and lethal neonatal intestinal epithelial dysfunction. PLoS Genetics, 4, e1000289. 10.1371/journal.pgen.1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burg M, et al. (2006). A new type of radiosensitive T-B-NK+ severe combined immunodeficiency caused by a LIG4 mutation. The Journal of Clinical Investigation, 116, 137–145. 10.1172/JCI26121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burg M, et al. (2009). A DNA-PKcs mutation in a radiosensitive T-B-SCID patient inhibits artemis activation and nonhomologous end-joining. The Journal of Clinical Investigation, 119, 91–98. 10.1172/JCI37141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Crabben SN, et al. (2016). Destabilized SMC5/6 complex leads to chromosome breakage syndrome with severe lung disease. The Journal of Clinical Investigation, 126, 2881–2892. 10.1172/JCI82890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Montfrans JM, et al. (2012). CD27 deficiency is associated with combined immunodeficiency and persistent symptomatic EBV viremia. The Journal of Allergy and Clinical Immunology, 129 787–793.e6. 10.1016/j.jaci.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand JL, et al. (2014). Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature, 514, 228–232. 10.1038/nature13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Stephens L, & Hawkins P (2012). PI3K signalling: The path to discovery and understanding. Nature Reviews. Molecular Cell Biology, 13, 195–203. 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, et al. (1997). P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proceedings of the National Academy of Sciences of the United States of America, 94, 4330–4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varon R, et al. (1998). Nibrin, a novel DNA double-strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell, 93, 467–476. [DOI] [PubMed] [Google Scholar]
- Vig M, et al. (2006). CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science, 312, 1220–1223. 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa A, et al. (1998). Partial V(D)J recombination activity leads to Omenn syndrome. Cell, 93, 885–896. [DOI] [PubMed] [Google Scholar]
- Volkman BF, Prehoda KE, Scott JA, Peterson FC, & Lim WA (2002). Structure of the N-WASP EVH1 domain–WIP complex: Insight into the molecular basis of Wiskott–Aldrich syndrome. Cell, 111, 565–576. [DOI] [PubMed] [Google Scholar]
- Volpi S, et al. (2017). EXTL3 mutations cause skeletal dysplasia, immune deficiency, and developmental delay. The Journal of Experimental Medicine, 214, 623–637. 10.1084/jem.20161525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskoboinik I, Whisstock JC, & Trapani JA (2015). Perforin and granzymes: Function, dysfunction and human pathology. Nature Reviews. Immunology, 15, 388–400. 10.1038/nri3839. [DOI] [PubMed] [Google Scholar]
- Walker LS, & Sansom DM (2011). The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nature Reviews. Immunology, 11, 852–863. 10.1038/nri3108. [DOI] [PubMed] [Google Scholar]
- Walne AJ, Vulliamy T, Kirwan M, Plagnol V, & Dokal I (2013). Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. American Journal of Human Genetics, 92, 448–453. 10.1016/j.ajhg.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Ong P, Roscioli T, Cliffe ST, & Church JA (2012). Hepatic veno-occlusive disease with immunodeficiency (VODI): First reported case in the U.S. and identification of a unique mutation in Sp110. Clinical Immunology, 145, 102–107. 10.1016/j.clim.2012.07.016. [DOI] [PubMed] [Google Scholar]
- Wang J, et al. (1999). Inherited human Caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell, 98, 47–58. 10.1016/S0092-8674(00)80605-4. [DOI] [PubMed] [Google Scholar]
- Wang H, et al. (2010). ZAP-70: An essential kinase in T-cell signaling. Cold Spring Harbor Perspectives in Biology, 2, a002279. 10.1101/cshperspect.a002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, et al. (2014). STIM1 and SLC24A4 are critical for enamel maturation. Journal of Dental Research, 93, 94S–100S. 10.1177/0022034514527971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, et al. (2016). Dual T cell- and B cell-intrinsic deficiency in humans with biallelic RLTPR mutations. The Journal of Experimental Medicine, 213, 2413–2435. 10.1084/jem.20160576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnatz K, et al. (2006). Human ICOS deficiency abrogates the germinal center reaction and provides a monogenic model for common variable immunodeficiency. Blood, 107, 3045–3052. 10.1182/blood-2005-07-2955. [DOI] [PubMed] [Google Scholar]
- Waterhouse P, et al. (1995). Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science, 270, 985–988. [DOI] [PubMed] [Google Scholar]
- Watford WT, & O’Shea JJ (2006). Human tyk2 kinase deficiency: Another primary immunodeficiency syndrome. Immunity, 25, 695–697. 10.1016/j.immuni.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Watkins D, et al. (2011). Novel inborn error of folate metabolism: Identification by exome capture and sequencing of mutations in the MTHFD1 gene in a single proband. Journal of Medical Genetics, 48, 590–592. 10.1136/jmedgenet-2011-100286. [DOI] [PubMed] [Google Scholar]
- Webster AD, Barnes DE, Arlett CF, Lehmann AR, & Lindahl T (1992). Growth retardation and immunodeficiency in a patient with mutations in the DNA ligase I gene. Lancet, 339, 1508–1509. [DOI] [PubMed] [Google Scholar]
- Wikenheiser DJ, & Stumhofer JS (2016). ICOS co-stimulation: Friend or foe? Frontiers in Immunology, 7, 304. 10.3389/fimmu.2016.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson DS, et al. (2015). Phosphorylation of LC3 by the Hippo kinases STK3/STK4 is essential for autophagy. Molecular Cell, 57, 55–68. 10.1016/j.molcel.2014.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willerford DM, et al. (1995). Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity, 3, 521–530. [DOI] [PubMed] [Google Scholar]
- Wong MT, Scholvinck EH, Lambeck AJ, & van Ravenswaaij-Arts CM (2015). CHARGE syndrome: A review of the immunological aspects. European Journal of Human Genetics, 23, 1451–1459. 10.1038/ejhg.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthey EA, et al. (2011). Making a definitive diagnosis: Successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genetics in Medicine, 13, 255–262. 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- Wright DD, Sefton BM, & Kamps MP (1994). Oncogenic activation of the Lck protein accompanies translocation of the LCK gene in the human HSB2 T-cell leukemia. Molecular and Cellular Biology, 14, 2429–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, et al. (1996). Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. The Journal of Clinical Investigation, 98, 1107–1113. 10.1172/JCI118892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu GL, et al. (1999). Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature, 402, 187–191. 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- Xu H, et al. (2013). Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature, 496, 523–527. 10.1038/nature12058. [DOI] [PubMed] [Google Scholar]
- Xu X, et al. (2014). Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nature Immunology, 15, 1152–1161. 10.1038/ni.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi H, et al. (2003). Role of TBX1 in human del22q11.2 syndrome. Lancet, 362, 1366–1373. [DOI] [PubMed] [Google Scholar]
- Yee CS, et al. (2016). Recurrent viral infections associated with a homozygous CORO1A mutation that disrupts oligomerization and cytoskeletal association. The Journal of Allergy and Clinical Immunology, 137, 879–888.e2. 10.1016/j.jaci.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong PF, Salzer U, & Grimbacher B (2009). The role of costimulation in antibody deficiencies: ICOS and common variable immunodeficiency. Immunological Reviews, 229, 101–113. 10.1111/j.1600-065X.2009.00764.x. [DOI] [PubMed] [Google Scholar]
- Yu M, et al. (2011). Regulation of T cell receptor signaling by activation-induced zinc influx. The Journal of Experimental Medicine, 208, 775–785. 10.1084/jem.20100031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusufzai T, & Kadonaga JT (2008). HARP is an ATP-driven annealing helicase. Science, 322, 748–750. 10.1126/science.1161233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Cado D, Chen A, Kabra NH, & Winoto A (1998). Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature, 392, 296–300. 10.1038/32681. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Lenardo MJ, & Baltimore D (2017). 30 years of NF-kappaB: A blossoming of relevance to human pathobiology. Cell, 168, 37–57. 10.1016/j.cell.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Sloan-Lancaster J, Kitchen J, Trible RP, & Samelson LE (1998). LAT: The ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell, 92, 83–92. [DOI] [PubMed] [Google Scholar]
- Zhang Q, et al. (2009). Combined immunodeficiency associated with DOCK8 mutations. The New England Journal of Medicine, 361, 2046–2055. 10.1056/NEJMoa0905506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, et al. (2011). UNC119 is required for G protein trafficking in sensory neurons. Nature Neuroscience, 14, 874–880. 10.1038/nn.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, et al. (2014a). DOCK8 regulates lymphocyte shape integrity for skin antiviral immunity. The Journal of Experimental Medicine, 211, 2549–2566. 10.1084/jem.20141307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. (2014b). Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. The Journal of Allergy and Clinical Immunology, 133, 1400–1409. 1409.e1–5. 10.1016/j.jaci.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, et al. (2016). A nonsense mutation in the DNA repair factor Hebo causes mild bone marrow failure and microcephaly. The Journal of Experimental Medicine, 213, 1011–1028. 10.1084/jem.20151183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Li J, & Lenardo M (2017). Restimulation-induced cell death: New medical and research perspectives. Immunological Reviews, 277, 44–60. 10.1111/imr.12535. [DOI] [PubMed] [Google Scholar]
- zur Stadt U, et al. (2009). Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18–2 and impaired binding to syntaxin 11. American Journal of Human Genetics, 85, 482–492. 10.1016/j.ajhg.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Stadt U, et al. (2005). Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Human Molecular Genetics, 14, 827–834. 10.1093/hmg/ddi076. [DOI] [PubMed] [Google Scholar]