

Abstract
BACKGROUND
Group 1 pulmonary arterial hypertension (PAH) is a progressive fatal condition characterized by right ventricular (RV) failure with worse outcomes in connective tissue disease (CTD). Obstructive sleep apnea and sleep-related hypoxia may contribute to RV dysfunction, though the relationship remains unclear.
OBJECTIVES
The aim of this study was to prospectively evaluate the association of the apnea-hypopnea index (AHI) and sleep-related hypoxia with RV function and survival.
METHODS
Pulmonary Vascular Disease Phenomics (National Heart, Lung, and Blood Institute) cohort participants (patients with group 1 PAH, comparators, and healthy control participants) with sleep studies were included. Multimodal RV functional measures were examined in association with AHI and percentage of recording time with oxygen saturation <90% (T90) per 10-unit increment. Linear models, adjusted for demographics, oxygen, diffusing capacity of the lungs for carbon monoxide, pulmonary hypertension medications, assessed AHI and T90, and RV measures. Log-rank test/Cox proportional hazards models adjusted for demographics, oxygen, and positive airway pressure were constructed for transplantation-free survival analyses.
RESULTS
Analysis included 186 participants with group 1 PAH with a mean age of 52.6 ± 14.1 years; 71.5% were women, 80.8% were Caucasian, and there were 43 events (transplantation or death). AHI and T90 were associated with decreased RV ejection fraction (on magnetic resonance imaging), by 2.18% (−2.18; 95% CI: −4.00 to −0.36; P = 0.019) and 0.93% (−0.93; 95% CI: −1.47 to −0.40; P < 0.001), respectively. T90 was associated with increased RV systolic pressure (on echocardiography), by 2.52 mm Hg (2.52; 95% CI: 1.61 to 3.43; P < 0.001); increased mean pulmonary artery pressure (on right heart catheterization), by 0.27 mm Hg (0.27; 95% CI: 0.05 to 0.49; P = 0.019); and RV hypertrophy (on electrocardiography), 1.24 mm (1.24; 95% CI: 1.10 to 1.40; P < 0.001). T90, but not AHI, was associated with a 17% increased 5-year risk for transplantation or death (HR: 1.17; 95% CI: 1.07 to 1.28). In non-CTD-associated PAH, T90 was associated with a 21% increased risk for transplantation or death (HR: 1.21; 95% CI: 1.08 to 1.34). In CTD-associated PAH, T90 was associated with RV dysfunction, but not death or transplantation.
CONCLUSIONS
Sleep-related hypoxia was more strongly associated than AHI with measures of RV dysfunction, death, or transplantation overall and in group 1 non-CTD-associated PAH but only with RV dysfunction in CTD-associated PAH. (Pulmonary Vascular Disease Phenomics Program [PVDOMICS]; NCT02980887)
Keywords: connective tissue disease–associated pulmonary arterial hypertension, obstructive sleep apnea, pulmonary arterial hypertension, pulmonary hypertension, right ventricular dysfunction, sleep-related hypoxia
World Symposium on Pulmonary Hypertension group 1 pulmonary arterial hypertension (PAH) is a progressive, ultimately fatal condition characterized by elevated pulmonary vascular resistance leading to right ventricular (RV) failure.1–6 Given high PAH-related morbidity and mortality, the identification of risk factors contributing to its pathophysiology and progression remains a high priority. Obstructive sleep apnea (OSA) and sleep-related hypoxia, via pathways of upregulation of systemic inflammation, vascular remodeling, and vasoconstriction,7,8 may represent key targets to mitigate PAH-associated morbidity and mortality.9,10
OSA results in cyclic increases in intrathoracic pressure due to repetitive forced inspiration against a closed airway, thereby leading to repeated increases in pulmonary arterial and right atrial pressure.11,12 OSA and sleep-related hypoxia are prevalent in pulmonary hypertension.13–21 Hypoxia promotes pulmonary arterial fibroblast production and proliferation and release of chemokines and cytokines that contribute to pulmonary vascular remodeling.8,22–27 Although a general association of OSA and sleep-related hypoxia relative to pulmonary hypertension has been reported,17–21,28,29 the contribution of OSA and intermittent hypoxia to PAH evolution and impact on mortality remains unclear. Moreover, although connective tissue disease (CTD)–associated PAH is the second most common etiology of group 1 PAH after idiopathic PAH and has a higher mortality rate,30 it is unknown if OSA-induced up-regulation of inflammation and sleep-related hypoxia contribute further to adverse outcomes in patients with CTD-associated PAH.
The association between OSA defined by the apnea-hypopnea index (AHI) and the severity of pulmonary hypertension is unclear. Reports favor an association between sleep-related hypoxia and severity of pulmonary hypertension more consistently than with AHI, but study limitations preclude definitive conclusions. For example, a small study involving different etiologies of precapillary pulmonary hypertension identified an association of AHI and percentage of time spent at oxygen saturation (SpO2) <90% (T90) relative to mean pulmonary artery pressure (mPAP) and right atrial pressure.28 However, other studies of overall precapillary pulmonary hypertension and group 1 PAH showed an association of nocturnal hypoxia (mean SpO2 <90% or T90), but not AHI, with pulmonary hypertension severity.17,19,29 These studies were mainly retrospective with small sample sizes, incompletely accounting for confounding variables. These reports were also not specific to group 1 PAH and did not assess group 1 PAH subgroup differences, particularly CTD-associated PAH.
We prospectively investigated the relationship of OSA and sleep-related hypoxia with RV dysfunction and transplantation-free survival in patients with group 1 PAH. We leveraged data from the PVDOMICS (Pulmonary Vascular Disease Phenomics Program), involving rigorous phenotyping of multimodal RV measures, prospective design, and data collection in comparator and control groups, thereby overcoming the limitations of existing studies and offering unique insights. We hypothesized that: 1) RV structural, hemodynamic, and electrophysiological measures are associated with OSA defined by AHI and sleep-related hypoxia defined by T90 as primary predictors independent of confounding influences; 2) AHI and T90 predict group 1 PAH transplantation-free survival; 3) the associations of AHI and T90 with survival are modulated by RV functional and structural alterations; and 4) the associations of AHI and T90 with transplantation-free survival are stronger for CTD-associated PAH than non-CTD-associated PAH.30
METHODS
COHORT DESCRIPTION.
The PVDOMICS clinical research network is a National Heart, Lung, and Blood Institute–funded, prospective, longitudinal cohort study (NCT02980887) that enrolled participants from November 30, 2016, to October 18, 2019. Protocol details have previously been published.10,31–34 The protocol was approved at each institution by the local Institutional Review Board, and informed consent was obtained from all participants.
Enrolling centers recruited persons ≥18 years of age in groups 1 to 5, referred for right heart catheterization (RHC) for clinical purposes, who were able to complete diagnostic testing (see the Supplemental Appendix for full eligibility criteria). The present study involved 3 groups: patients with group 1 PAH according to the World Symposium on Pulmonary Hypertension guidelines with mPAP ≥25 mm Hg, pulmonary artery wedge pressure ≤15 mm Hg, and pulmonary vascular resistance >3.0 WU35,36; a comparator group with mPAP <25 mm Hg and group 1 risk factors or exercise-induced pulmonary hypertension; and healthy control subjects with normal cardiopulmonary findings and without end-organ disease (Supplemental Appendix).
Participants underwent a comprehensive clinical phenotyping protocol including review of their medical history, demographics, and, when not contraindicated, transthoracic echocardiography, cardiac magnetic resonance imaging (CMR), RHC and 12-lead electrocardiography (ECG) (Supplemental Appendix). Healthy control subjects underwent the same evaluation except that they did not undergo RHC.
Overnight sleep monitoring was performed upon enrollment with the NOX-T3 (CareFusion) portable home sleep study system (type 3 sleep study). If patients were already using nocturnal supplemental oxygen and/or positive airway pressure, or were prescribed oxygen, this was documented and used during the sleep study. Similar sleep studies performed within 1 year of enrollment were accepted if there were no significant changes in weight, nocturnal oxygen, or sleep-disordered breathing therapy. Sleep studies, apnea, and hypopnea were scored according to the American Academy of Sleep Medicine guidelines, with hypopnea defined by a ≥30% reduction in peak signal excursion lasting ≥10 seconds and associated with ≥3% oxygen desaturation.37–39 Interscorer and intrascorer reliability was >90% for all sleep study measures (intraclass correlation coefficients, 0.92 [95% CI: 0.80–0.97] for AHI and 1.00 [95% CI: 0.99–1.00] for T90).
STATISTICAL ANALYSIS.
The primary predictors of right heart measures were sleep-related hypoxia and OSA defined by T90 and AHI, respectively. For comparisons across World Symposium on Pulmonary Hypertension group 1, comparator, and healthy control patients, analysis of variance or the Kruskal-Wallis test was performed for continuous variables and the Pearson chi-square test or Fisher exact test for categorical variables. Logistic and linear regression models adjusted for age, sex, race, body mass index, pulmonary hypertension medications, supplemental oxygen were used to assess the associations of OSA and sleep-related hypoxia cardiac indexes. Additional models were adjusted for diffusing capacity of the lungs for carbon monoxide (DlCO) and left ventricular diastolic dysfunction by lateral E/e′ ratio. DlCO was included to account for sleep-related hypoxia due to ventilation-perfusion mismatch and barriers to gas exchange specific to the pathobiology of PAH and chronic intermittent daytime hypoxemia not captured by a single SpO2 measurement.40 Adjustment for lateral E/e′ ratio was performed to account for left-sided diastolic dysfunction. Given existing literature and biologic plausibility, T90 and the AHI are presented per 10-unit increase.19,21,28,29 We considered alternate measures of sleep-related hypoxia (ie, mean and minimum SpO2). The primary cardiac measure outcomes in each structural and physiological domain were RV systolic pressure (on echocardiography), RV ejection fraction (on CMR), mPAP (on RHC), and RV hypertrophy (on ECG). We also examined secondary RV function measures yet to be reported in relation to OSA and sleep-related hypoxia but recognized to hold prognostic value in pulmonary hypertension (Supplemental Appendix).
Time-to-event analysis was performed using Cox proportional hazards models to evaluate associations of OSA and sleep-related hypoxia with transplantation-free survival. The proportional hazards assumption was met for all models. Cox models were adjusted for age, sex, body mass index, supplemental oxygen, DlCO, unrepaired shunt, and positive airway pressure. Kaplan-Meier survival curves and log-rank tests were used to assess the associations of sleep indexes with death or transplantation (lung and/or heart). In sensitivity analyses, we excluded those without intracardiac shunt repair. In secondary analyses, we evaluated the associations of T90 and AHI with right heart measures in CTD-associated and non-CTD-associated PAH and transplantation or death. In exploratory analyses, we examined the statistical interaction of OSA indexes and RV function in relation to transplantation-free survival. Analyses were performed using SAS version 9.4 (SAS Institute).
RESULTS
OVERALL PARTICIPANT CHARACTERISTICS.
Figure 1 shows the distribution of study participants (patients with group 1 PAH, comparators, and healthy control subjects). Demographics of the final analytical sample compared with those who did not have sleep studies were similar except for higher body mass index (30.3 ± 7.6 kg/m2 vs 27.4 ± 7.1 kg/m2; P < 0.001) (Supplemental Table 1). Age at the time of enrollment was greatest among the comparators (approximately 60 years) and similar in the PAH group and healthy control subjects (approximately 50 years), and participants were predominantly women. Ethnicity, race, and functional class (healthy control subjects excluded) did not differ significantly. The proportion of patients with CTD was higher in the comparator group than the PAH group (78% vs 25%), as group 1 PAH comparators were identified by the presence of group 1-associated conditions, including CTD (Table 1, Supplemental Table 2).
FIGURE 1. Participant Enrollment.

Diagram depicting patient enrollment in PVDOMICS (Pulmonary Vascular Disease Phenomics Program), including all recruited participants with group 1 pulmonary arterial hypertension (PAH), healthy control subjects, and comparators who underwent sleep study testing. *Mean pulmonary artery pressure ≥25 mm Hg with pulmonary vascular resistance >3.0 WU. §Mean pulmonary artery pressure <25 mm Hg and group 1 risk factors or exercise-induced pulmonary hypertension. CTD = connective tissue disease.
TABLE 1.
Summary Characteristics of Patients With Group 1 PAH, Healthy Control Subjects, and Comparators
|
P Value |
||||||||
|---|---|---|---|---|---|---|---|---|
| PAH (n = 186) | Healthy Control (n = 78) | Comparator (n = 32) | PAH vs Comparator | All 3 Groups | ||||
|
| ||||||||
| Demographics | ||||||||
| Age at enrollment, y | 186 | 52.6 ± 14.1a | 78 | 48.2 ± 14.7a | 32 | 60.1 ± 12.2b,c | 0.005d | <0.001d |
| Male | 186 | 53 (28.5) | 78 | 24 (30.8) | 32 | 6 (18.8) | 0.25e | 0.43e |
| Hispanic ethnicity | 183 | 21 (11.5) | 78 | 9 (11.5) | 30 | 3 (10.0) | 0.99g | 0.97e |
| Race (Black, White, other) | 182 | 78 | 31 | 0.19g | 0.30g | |||
| Black or African American | 22 (12.1) | 9(11.5) | 6 (19.4) | |||||
| White | 147 (80.8) | 67 (85.9) | 25 (80.6) | |||||
| Other | 13 (7.1) | 2 (2.6) | 0 (0.00) | |||||
| Body mass index, kg/m2 | 186 | 30.3 ± 7.6 | 78 | 28.3 ± 5.9 | 32 | 28.9 ± 6.3 | 0.32d | 0.090d |
| PH medicationsf | 186 | 154 (82.8)a,b | 78 | 0 (0.00)c | 32 | 4 (12.5)c | <0.001e | <0.001e |
| Age at diagnosis of PH, y | 186 | 47.1 ± 15.8 | ||||||
| Years of PH at time of enrollment | 186 | 3.8 (0.83–9.0) | ||||||
| NYHA functional class | 185 | 0 | 31 | 0.19h | 0.19h | |||
| I | 23 (12.4) | 2 (6.5) | ||||||
| II | 76 (41.1) | 20 (64.5) | ||||||
| III | 77 (41.6) | 9 (29.0) | ||||||
| IV | 9 (4.9) | 0 (0.00) | ||||||
| 6MWD, m | 171 | 382.2 ± 128.8b | 78 | 524.7 ± 94.6a,c | 32 | 375.9 ± 113.0b | 0.80d | <0.001d |
| Etiology of PHi | ||||||||
| Idiopathic pulmonary arterial hypertension | 186 | 89 (47.8) | 32 | 0 (0.00) | <0.001e | |||
| Connective tissue disease | 186 | 44 (23.7) | 32 | 25 (78.1) | <0.001e | |||
| Systemic sclerosis | 44 | 21 (11.3) | 32 | 12 (37.5) | <0.001g | |||
| Systemic lupus erythematosus | 44 | 9 (4.8) | 32 | 4 (12.5) | 0.10g | |||
| Sjogren's syndrome | 44 | 6 (3.2) | 32 | 3 (9.4) | 0.13g | |||
| Rheumatoid arthritis | 44 | 4 (2.2) | 32 | 5 (15.6) | 0.004g | |||
| Mixed connective tissue disease | 44 | 4 (2.2) | 32 | 5 (15.6) | 0.004g | |||
| Antisynthetase syndrome | 44 | 0 (0.00) | 32 | 1 (3.1) | 0.15g | |||
| Congenital heart disease | 186 | 17(9.1) | 32 | 5 (15.6) | 0.34g | |||
| Familial pulmonary arterial hypertension | 186 | 16 (8.6) | 32 | 0 (0.00) | 0.14g | |||
| Portal hypertension | 186 | 10 (5.4) | 32 | 0 (0.00) | 0.36g | |||
| Shunt repaired | 186 | 7 (3.8) | 32 | 2 (6.3) | 0.62g | |||
| Shunt unrepaired | 186 | 14 (7.5) | 32 | 3 (9.4) | 0.72g | |||
| Drug-induced pulmonary arterial hypertension | 186 | 6 (3.2) | 32 | 1 (3.1) | 0.99g | |||
| Human immunodeficiency virus | 186 | 4 (2.2) | 32 | 0 (0.00) | 0.99g | |||
| Pulmonary veno-occlusive disease | 186 | 4 (2.2) | 32 | 0 (0.00) | 0.99g | |||
| Pulmonary capillary hemangiomatosis | 186 | 3(1.6) | 32 | 0 (0.00) | 0.99g | |||
| Schistosomiasis | 186 | 0 (0.00) | 32 | 0 (0.00) | ||||
| Other | 186 | 0 (0.00) | ||||||
Values are n, mean ± SD, n (column %), or median (Q1-Q3). Post hoc pairwise comparisons were performed using Bonferroni adjustment.
Significantly different from comparator group.
Significantly different from healthy control group.
Significantly different from PAH group.
Analysis of variance.
Pearson chi-square test.
Included PH medications of endothelin receptor antagonists, phosphodiesterase 5 inhibitors, soluble guanylate cyclase stimulators, and calcium-channel blockers for PH (see the Supplemental Appendix for medication specifics).
Fisher exact test.
Kruskal-Wallis test.
Participants may have had more than one noted etiology for PAH.
PAH = pulmonary arterial hypertension; PH = pulmonary hypertension; 6MWD = 6-minute walk distance.
SLEEP STUDY CHARACTERISTICS.
Ninety-two participants with PAH (49.7%), 31 control subjects (39.7%), and 17 comparators (54.8%) had OSA (AHI ≥5), whereas 41 participants with PAH (22.2%) had AHI ≥15, similar to comparators and more than healthy control subjects (Table 2). Central respiratory events were infrequent. Median resting awake SpO2 was lower in patients with PAH vs healthy control subjects vs comparators: 96% (Q1-Q3: 93.0% to 98.0%) vs 98% (Q1-Q3: 97.0% to 99.0%) vs 98% (Q1-Q3: 98.0% to 99.5%), respectively (P < 0.001). Percentage predicted DlCO was lower in patients with PAH vs healthy control subjects and comparators: 57.6% ± 21.1% vs 90.3% ± 16.5% and 75% ± 20.4%, respectively. Median T90 was 37.0% (Q1-Q3: 2.2% to 87.3%) of recording time in participants with PAH vs 0.12% (Q1-Q3: 0.00% to 2.8%) in healthy control subjects and 2.8% (Q1-Q3: 0.19% to 13.1%) in comparators (P < 0.001), despite the use of nocturnal oxygen or positive airway pressure in nearly 39.8% of participants with PAH vs 5.1% in healthy control subjects and 6.3% in comparators during the sleep study. In PAH, daytime awake SpO2 and sleep-related oxygenation (T90) were inversely correlated (Spearmen correlation coefficient = −0.34; 95% CI: −0.48 to −0.19; P < 0.001).
TABLE 2.
Summary Sleep Study and Oxygenation Characteristics of Patients With Group 1 PAH, Healthy Control Subjects, and Comparators
|
P Value |
||||||||
|---|---|---|---|---|---|---|---|---|
| PAH (n = 186) | Healthy Control (n = 78) | Comparator (n = 32) | PAH vs Comparator | All 3 Groups | ||||
|
| ||||||||
| Sleep study characteristics | ||||||||
| AHI | 185 | 4.4 (1.07–13.5) | 78 | 3.3 (1.3–8.8) | 31 | 8.1 (2.5–16.6) | 0.23a | 0.091a |
| ≥5 | 185 | 92 (49.7) | 78 | 31 (39.7) | 31 | 17 (54.8) | 0.60b | 0.23b |
| ≥15 | 185 | 41 (22.2)c | 78 | 7 (9.0)d | 31 | 8 (25.8) | 0.65b | 0.027b |
| Central apnea index | 183 | 0.00 (0.00–0.30) | 78 | 0.00 (0.00–0.28) | 31 | 0.16 (0.00–0.65) | 0.031a | 0.071a |
| Obstructive AHI | 182 | 4.6 (1.2–13.6) | 78 | 3.6 (1.4–9.6) | 31 | 8.1 (2.4–16.7) | 0.31a | 0.18a |
| Percentage of recording time at <90% SpO2 | 180 | 37.0 (2.2–87.3)c,e | 78 | 0.12 (0.00–2.8)d,e | 30 | 2.8 (0.19–13.1)c,d | <0.001a | <0.001a |
| Oxygen desaturation index 3%, events/h | 176 | 10.6 (5.1–19.3)c | 78 | 6.6 (4.3–12.8)d | 28 | 11.6 (6.0–21.4) | 0.91a | 0.016a |
| Mean SpO2, % | 185 | 90.0 (87.0–93.0)c,e | 78 | 94.0 (92.0–95.0)d | 31 | 94.0 (92.0–95.0)d | <0.001a | <0.001a |
| Lowest SpO2, % | 182 | 83.0 (78.0–87.0)c,e | 77 | 88.0 (84.0–90.0)d | 30 | 85.0 (82.0–88.0)d | 0.014a | <0.001a |
| Oxygen use | 186 | 78 | 32 | <0.001g | <0.001b | |||
| No oxygen or PAP use during the night of the sleep study | 92 (49.5)c,e | 74 (94.9)d | 29 (90.6)d | |||||
| Acute O2 or PAP the night of sleep study but no O2 prescription | 22 (11.8) | 4 (5.1) | 2 (6.3) | |||||
| Prescribed O2 long term | 20 (10.8) | 0 (0.00) | 1 (3.1) | |||||
| Prescribed O2 long term and using at sleep study | 52 (28.0) | 0 (0.00) | 0 (0.00) | |||||
| Oxygenation characteristics | ||||||||
| SpO2 at rest on room air, % | 171 | 96.0 (93.0–98.0)c,e | 78 | 98.0 (97.0–99.0)d | 32 | 98.0 (98.0–99.5)d | <0.001a | <0.001a |
| Diffusing capacity of the lungs for carbon monoxide, % predicted | 183 | 57.6 ± 21.1c,e | 77 | 90.3 ± 16.5d,e | 30 | 75. ± 20.4c,d | <0.001f | <0.001f |
Values are n, median (Q1-Q3), n (column %), or mean ± SD. Post hoc pairwise comparisons were performed using Bonferroni adjustment.
Kruskal-Wallis test.
Pearson chi-square test.
Significantly different from healthy control group.
Significantly different from PAH group.
Significantly different from comparator group.
Analysis of variance.
Fisher exact test. AHI = apnea-hypopnea index; PAH = pulmonary arterial hypertension; PAP = positive airway pressure; SpO2 = oxygen saturation.
RIGHT-SIDED CARDIAC STRUCTURAL, HEMODYNAMIC, AND ELECTROPHYSIOLOGICAL MEASURES.
RV imaging measures had expected differences between participants with PAH and comparators, including by echocardiography (higher RV systolic pressure, greater RV wall thickness, lower tricuspid annular plane systolic excursion, greater global RV free wall peak longitudinal strain [3 and 6 segments]) and by CMR (lower RV ejection fraction, increased RV end-systolic volume index, and increased RV mass) (Table 3) in those with PAH. RHC revealed expected differences between participants with PAH and comparators (mPAP 44.2 mm Hg vs 16.6 mm Hg, pulmonary vascular resistance 7.1 WU vs 1.7 WU) and lower cardiac output and cardiac index (Table 3). Comparators and healthy control subjects showed similar imaging results, but the latter did not undergo RHC. RV hypertrophy, right bundle branch block, and right-axis deviation on ECG were more evident among patients with PAH than other groups.
TABLE 3.
Summary Cardiac Characteristics of Patients With Group 1 PAH, Healthy Control Subjects, and Comparators
|
P Value |
||||||||
|---|---|---|---|---|---|---|---|---|
| PAH (n = 186) | Healthy Control (n = 78) | Comparator (n = 32) | PAH vs Comparato | All 3 Groups | ||||
|
| ||||||||
| Echocardiography | ||||||||
| Primary variable | ||||||||
| RV systolic pressure, mm Hg | 162 | 65.3 ± 22.8a,b | 60 | 24.3 ± 4.9b,c | 26 | 32.9 ± 9.6a,c | <0.001d | <0.001d |
| Secondary variables | ||||||||
| RV end-diastolic area, cm/m2 | 168 | 27.6 (22.5–36.1)a,b | 74 | 18.1 (15.0–20.1)c | 26 | 16.7 (14.3–20.8)c | <0.001e | <0.001e |
| RV end-diastolic basal dimension, cm | 177 | 4.6 ± 0.87a,b | 76 | 3.6 ± 0.49c | 28 | 3.8 ± 0.66c | <0.001d | <0.001d |
| RV diastolic wall thickness, cm | 169 | 0.61 (0.51–0.74)a,b | 71 | 0.46 (0.40–0.52)c | 28 | 0.47 (0.41–0.53)c | <0.001e | <0.001e |
| Global RV free wall peak longitudinal strain (3 segments), % | 152 | −18.4 ± 5.6a,b | 59 | −25.8 ± 6.2c | 21 | −25.1 ± 5.4c | <0.001d | <0.001d |
| Global RV free wall peak longitudinal strain (6 segments), % | 149 | −17.1 ± 4.7a,b | 59 | −23.9 ± 5.2c | 21 | −23.0 ± 4.4c | <0.001d | <0.001d |
| RV fractional shortening, % | 168 | 29.7 ± 10.0a,b | 73 | 43.4 ± 5.6c | 26 | 42.2 ± 6.8c | <0.001d | <0.001d |
| RV outflow tract velocity-time integral, cm | 174 | 14.3 (11.8–17.2)a | 74 | 15.7 (14.4–17.3)c | 29 | 16.1 (15.1–17.7) | 0.020e | 0.002e |
| Tricuspid annular plane systolic excursion, cm | 166 | 1.9 ± 0.48a,b | 74 | 2.3 ± 0.41c | 25 | 2.2 ± 0.38c | 0.001d | <0.001d |
| Tricuspid valve regurgitation degree | 181 | 76 | 29 | 0.55f | <0.001g | |||
| None or trace | 44 (24.3)a | 53 (69.7)b,c | 9 (31.0)a | |||||
| Mild | 85 (47.0) | 23 (30.3) | 14 (48.3) | |||||
| Moderate | 42 (23.2) | 0 (0.00) | 6 (20.7) | |||||
| Severe | 10 (5.5) | 0 (0.00) | 0 (0.00) | |||||
| Lateral E/e′ ratio | 173 | 7.1 (5.6–9.8)a,b | 78 | 6.3 (5.4–7.6)b,c | 29 | 9.6 (7.5–12.2)a,c | 0.007e | <0.001e |
| Left atrial volume index, mL/m2 | 159 | 23.1 (19.9–28.9)a | 77 | 20.8 (18.0–23.4)c | 29 | 23.1 (19.3–27.4) | 0.83e | 0.003e |
| Cardiac magnetic resonance imaging | ||||||||
| Primary variable | ||||||||
| RV ejection fraction, % | 139 | 37.6 ± 12.1a,b | 68 | 56.3 ± 6.1c | 23 | 54.3 ± 7.8c | <0.001d | <0.001d |
| Secondary variables | ||||||||
| RV end-diastolic volume index, mL/m2 | 139 | 100.5 (78.5–129.1)a,b | 68 | 72.6 (60.5–79.0)c | 23 | 69.3 (59.7–87.7)c | <0.001e | <0.001e |
| RV end-systolic volume index, mL/m2 | 139 | 59.4 (42.4–86.8)a,b | 68 | 31.0 (26.5–37.2)c | 23 | 33.3 (24.7–44.0)c | <0.001e | <0.001e |
| RV stroke volume index, mL/m2 | 139 | 37.6 ± 10.5 | 68 | 39.6 ± 7.9 | 23 | 40.0 ± 10.9 | 0.32d | 0.28d |
| RV mass, g | 139 | 43.4 ± 22.1a,b | 68 | 26.5 ± 10.0c | 23 | 24.4 ± 7.7c | <0.001d | <0.001d |
| RV peak global longitudinal systolic strain, % | 114 | −15.8 ± 5.1a,b | 60 | −20.4 ± 5.4c | 20 | −21.3 ± 4.9c | <0.001d | <0.001d |
| RA ejection fraction, % | 138 | 36.4 (27.9–45.9)a | 68 | 45.2 (37.0–52.1)c | 22 | 40.0 (30.8–47.8) | 0.40e | <0.001e |
| RA end-systolic volume index, mL/m2 | 138 | 39.7 (28.4–56.7)a | 68 | 26.8 (18.7–38.0)b,c | 22 | 35.1 (30.2–46.4)a | 0.22e | <0.001e |
| Left ventricular mass index, g/m2 | 140 | 41.9 (36.8–51.6) | 68 | 41.9 (37.7–49.2) | 23 | 39.4 (34.9–46.4) | 0.22e | 0.46e |
| Left ventricular end-diastolic volume index, mL/m2 | 140 | 62.4 (52.2–75.6)a | 68 | 72.3 (59.5–82.4)c | 23 | 64.7 (48.1–82.0) | 0.73e | 0.007e |
| Left ventricular end-systolic volume index, mL/m2 | 140 | 25.4 (20.7–32.1)a | 68 | 30.3 (23.5–35.2)c | 23 | 26.4 (18.9–31.4) | 0.98e | 0.013e |
| Right heart catheterization | ||||||||
| Primary variable | ||||||||
| Mean PA pressure (spontaneous breathing), mm Hg | 184 | 44.2 ± 15.3b | 0 | − | 31 | 16.6 ± 3.7c | <0.001d | |
| Secondary variables | ||||||||
| Mean RA pressure (spontaneous breathing), mm Hg | 183 | 7.9 ± 5.2b | 0 | − | 31 | 4.2 ± 2.4c | <0.001d | <0.001d |
| PCWP, mean (spontaneous breathing), mm Hg | 183 | 11.3 ± 5.5b | 0 | − | 31 | 7.8 ± 3.1c | <0.001d | <0.001d |
| Pulmonary vascular resistance (spontaneous breathing), WU | 180 | 7.1 ± 3.9b | 0 | − | 31 | 1.6 ± 0.75c | <0.001d | <0.001d |
| Cardiac output | 182 | 5.2 ± 1.6b | 0 | − | 31 | 6.1 ± 1.9c | 0.007d | 0.007d |
| Cardiac index, L/min/m2 | 182 | 2.7 ± 0.76b | 0 | − | 31 | 3.1 ± 0.91c | 0.004d | 0.004d |
| Electrocardiography | ||||||||
| Primary variable | ||||||||
| RV hypertrophy presence | 185 | 33 (17.8)a | 74 | 0 (0.00)c | 31 | 1 (3.2) | 0.035g | <0.001f |
| Secondary variables | ||||||||
| Right bundle branch block | 185 | 21 (11.4)a | 74 | 1 (1.4)c | 31 | 3 (9.7) | 0.99g | 0.034f |
| Right-axis deviation | 185 | 83 (44.9)a,b | 75 | 1 (1.3)c | 31 | 0 (0.00)c | <0.001f | <0.001f |
| RA enlargement | 185 | 21 (11.4)a | 74 | 0 (0.00)c | 31 | 0 (0.00) | 0.050f | 0.002f |
Values are n, mean ± SD, median (Q1-Q3), or n (column %). Post hoc pairwise comparisons were performed using Bonferroni adjustment.
Significantly different from healthy control group.
Significantly different from comparator group.
Significantly different from PAH group.
Analysis of variance.
Kruskal-Wallis test.
Pearson chi-square test.
Fisher exact test.
PA = pulmonary artery; PAH = pulmonary arterial hypertension; PCWP = pulmonary capillary wedge pressure; RA = right atrial; RV = right ventricular.
SLEEP-DISORDERED BREATHING INDEXES AND RIGHT-SIDED CARDIAC FUNCTION MEASURES.
Of the primary RV structural and physiological measures assessed in patients with PAH, only RV ejection fraction (on CMR) was associated with increasing AHI (Table 4, Supplemental Table 3). Secondary RV measures associated with an increase in AHI included increased RV peak global longitudinal systolic strain (on echocardiography), decreased cardiac output (on RHC), and increased mean right atrial pressure and pulmonary vascular resistance (both on RHC).
TABLE 4.
Sleep-Disordered Breathing and Right-Sided Cardiac Measures in Patients With Group 1 Pulmonary Arterial Hypertension
| Apnea-Hypopnea Index, 10 Events per Hour Increment |
Percentage of Recording Time at <90% O2 Saturation, 10% Increment |
|||||
|---|---|---|---|---|---|---|
| n | Coefficient (95% CI) or OR (95% CI) | P Value | n | Coefficient (95% CI) or OR (95% CI) | P Value | |
|
| ||||||
| Structural/imaging measures | ||||||
| Primary variables | ||||||
| RV systolic pressure, mm Hga | 157 | 0.84 (−01.92 to 3.59) | 0.55 | 153 | 2.49 (1.58 to 3.40) | <0.001 |
| RV ejection fraction, %b | 135 | −2.09 (−3.93 to −0.24) | 0.027 | 133 | −0.94 (−1.47 to −0.40) | <0.001 |
| Secondary variables | ||||||
| Tricuspid annular plane systolic excursion, cma | 159 | −0.02 (−0.08 to 0.04) | 0.52 | 155 | −0.03 (−0.05 to −0.01) | 0.003 |
| Global RV free wall peak longitudinal strain (3 segments), %a | 146 | 0.47 (−0.25 to 1.20) | 0.20 | 141 | 0.39 (0.12 to 0.65) | 0.004 |
| Global RV free wall peak longitudinal strain (6 segments), %a | 144 | 0.40 (−0.18 to 0.97) | 0.18 | 139 | 0.26 (0.04 to 0.47) | 0.022 |
| RV end-diastolic area, cm/m2a | 161 | 1.07 (−0.06 to 2.20) | 0.063 | 156 | 0.67 (0.29 to 1.05) | <0.001 |
| RV fractional shortening, %a | 161 | −1.10 (−2.36 to 0.16) | 0.086 | 156 | −0.71 (−1.14 to −0.29) | 0.001 |
| RV end-diastolic basal dimension, cma | 170 | 0.08 (−0.01 to 0.18) | 0.076 | 165 | 0.05 (0.02 to 0.08) | 0.001 |
| RVD wall thickness, cma | 162 | 0.01 (−0.01 to 0.04) | 0.38 | 157 | 0.01 (0.00 to 0.02) | 0.018 |
| RVOT velocity-time integral, cma | 167 | −0.41 (−1.03 to 0.21) | 0.19 | 164 | −0.20 (−0.41 to 0.02) | 0.076 |
| RV end-diastolic volume index, mL/m2b | 135 | 2.94 (−4.14 to 10.03) | 0.41 | 133 | 3.86 (1.87 to 5.86) | <0.001 |
| RV end-systolic volume index, mL/m2b | 135 | 3.99 (−2.71 to 10.68) | 0.24 | 133 | 3.77 (1.88 to 5.65) | <0.001 |
| RV stroke volume index, mL/m2b | 135 | −1.04 (−2.68 to 0.60) | 0.21 | 133 | 0.10 (−0.40 to 0.59) | 0.70 |
| RV mass, gb | 135 | 1.34 (−1.98 to 4.66) | 0.43 | 133 | 2.10 (1.19 to 3.02) | <0.001 |
| RV peak global longitudinal systolic strain, %b | 110 | 0.94 (−0.01 to 1.89) | 0.052 | 108 | 0.24 (−0.03 to 0.52) | 0.076 |
| Hemodynamic measures | ||||||
| Primary variable | ||||||
| Mean PA pressure (spontaneous breathing), mm Hgc | 176 | 1.12 (−0.72 to 2.95) | 0.23 | 171 | 1.86 (1.29 to 2.42) | <0.001 |
| Secondary variables | ||||||
| Mean RA pressure (spontaneous breathing), mm Hgc | 175 | 0.61 (0.02 to 1.20) | 0.042 | 170 | 0.19 (−0.02 to 0.39) | 0.076 |
| Mean PCWP (a wave) (spontaneous breathing), mm Hgc | 175 | 0.23 (−0.43 to 0.90) | 0.49 | 170 | 0.28 (0.05 to 0.51) | 0.017 |
| Cardiac output, L/minc | 176 | −0.19 (−0.37 to −0.02) | 0.029 | 171 | 0.00 (−0.06 to 0.06) | 0.97 |
| Rest: cardiac index, L/min/m2c | 176 | −0.09 (−0.18 to −0.00) | 0.05 | 171 | 0.00 (−0.03 to 0.04) | 0.79 |
| PVR (spontaneous breathing), WUc | 174 | 0.47 (0.01 to 0.94) | 0.047 | 169 | 0.36 (0.20 to 0.51) | <0.001 |
| Electrophysiological measures | ||||||
| Primary variable | ||||||
| RV hypertrophyd | 177 | 1.08 (0.79 to 1.48) | 0.64 | 172 | 1.26 (1.11 to 1.44) | <0.001 |
| Secondary variables | ||||||
| Right bundle branch blockd | 177 | 1.05 (0.68 to 1.61) | 0.84 | 172 | 1.09 (0.96 to 1.24) | 0.20 |
| Right-axis deviationd | 177 | 1.14 (0.88 to 1.48) | 0.32 | 172 | 1.10 (1.01 to 1.21) | 0.035 |
Models were adjusted for age, sex, race, body mass index, pulmonary hypertension medications, supplemental oxygen use, and diffusing capacity of the lungs for carbon monoxide.
Obtained from echocardiogram.
Obtained from cardiac magnetic resonance imaging.
Obtained from right heart catheterization.
Obtained from electrocardiography.
PA = pulmonary artery; PCWP = pulmonary capillary wedge pressure; PVR = pulmonary vascular resistance; RA = right atrial; RV = right ventricular; RVD = right ventricular diastolic; RVOT = right ventricular outflow tract.
In contrast to AHI, T90 in the PAH group was significantly associated with all primary structural, hemodynamic, and electrophysiological right heart measures. Specifically, for every 10% increment in T90, RV systolic pressure assessed by echocardiography increased by 2.49 mm Hg (95% CI: 1.58 to 3.40; P < 0.001), RV ejection fraction assessed by CMR decreased by 0.94% (95% CI: −1.47 to −0.40; P < 0.001), mPAP on RHC increased by 1.86 mm Hg (95% CI: 1.29 to 2.42; P < 0.001), and the odds of RV hypertrophy assessed using ECG were 26% higher (OR: 1.26; 95% CI: 1.11 to 1.44; P < 0.001). Secondary RV measures were associated with T90, including tricuspid annular plane systolic excursion (on echocardiography), global RV free wall peak longitudinal strain (3 and 6 segments, on echocardiography), RV fractional shortening (on echocardiography), and pulmonary vascular resistance (on RHC) (Table 4, Figure 2). Similar findings were observed when: 1) adjusted for supplemental oxygen or positive airway pressure use during the sleep study; 2) left heart diastolic dysfunction was characterized by lateral E/e′ ratio; and 3) participants with unrepaired intracardiac shunts (n = 14) were excluded (Supplemental Tables 4 to 6).
FIGURE 2. Association of Primary RV Measures and Sleep Indices.

Scatterplot depiction of linear regression analysis of the association of primary right ventricular (RV) measures and apnea-hypopnea index (AHI) and the percentage of recording time at oxygen saturation <90% (T90): (A) echocardiographic RV systolic pressure (RVSP) and AHI, (B) RVSP and T90, (C) echocardiographic RV ejection fraction (RVEF) and AHI, (D) echocardiographic RVEF and T90, (E) right heart catheterization (RHC) mean pulmonary artery pressure (mPAP) and AHI, (F) RHC mPAP and T90, (G) electrocardiographic (ECG) RV hypertrophy and AHI, and (H) ECG RV hypertrophy and T90. The gray area indicates the 95% CI. PA = pulmonary artery.
SLEEP-DISORDERED BREATHING AND TRANSPLANTATION-FREE SURVIVAL.
There were 34 deaths and 9 transplantations, with a median follow-up duration of 48.3 months (Q1-Q3: 36.9–60.0 months). Sleep-related hypoxia (T90) was associated with transplantation-free survival. For each 10% increment in T90, the risk for transplantation or death increased by 12% (HR: 1.12; 95% CI: 1.04–1.22; P = 0.005) in unadjusted models and 17% (HR: 1.17; 95% CI: 1.07–1.28; P < 0.001) when adjusted for age, sex, body mass index, DlCO, and supplemental oxygen use, with similar findings when adjusted for overnight oxygen, unrepaired shunt, positive airway pressure, and lateral E/e′ ratio (Table 5, Supplemental Tables 7 to 9). Neither AHI nor oxygen saturation nadir was associated with transplantation-free survival, but mean SpO2 of >90% vs ≤90% was associated with a lower occurrence of transplantation or death (unadjusted HR: 0.43 [95% CI: 0.22–0.82; P = 0.010]; adjusted HR: 0.41 [95% CI: 0.20–0.83; P = 0.13]) (Table 5). A median T90 threshold >37% (P = 0.010) and mean SpO2 < 90% (P = 0.008) were associated with decreased transplantation-free survival (Figure 3). The linear trend of risk for transplantation or death by T90 quartile was significant with adjusted (P = 0.04) and unadjusted (P = 0.012) models, with T90 > 87.3% (Figure 4) associated with increased risk for transplantation or death (Table 5, Supplemental Tables 7 to 9). There was no statistically significant interaction of transplantation-free survival and sleep-related hypoxia or AHI and RV measures.
TABLE 5.
Cox Proportional Hazards Model of Transplantation or Death in Patients With Group 1 Pulmonary Arterial Hypertension
| Unadjusted | Adjusted for Age, Sex, BMI, and O2 Use | Adjusted for Age, Sex, BMI, Prescribed O2, and DlCO | |||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|||||||
| n | HR (95% CI) | P Value | n | HR (95% CI) | P Value | n | HR (95% CI) | P Value | |
|
| |||||||||
| AHI, 10 events/h increment | 185 | 0.94 (0.74–1.20) | 0.62 | 185 | 0.93 (0.70–1.23) | 0.60 | 182 | 1.01 (0.78–1.30) | 0.97 |
| Percentage of recording time at <90% SpO2, 10% increment | 180 | 1.12 (1.04–1.22) | 0.005 | 180 | 1.16 (1.06–1.27) | <0.001 | 177 | 1.17 (1.07–1.28) | <0.001 |
| Oxygen desaturation index (3%) by 10 events/h | 176 | 0.95 (0.79–1.16) | 0.63 | 176 | 1.04 (0.84–1.30) | 0.71 | 173 | 1.08 (0.88–1.33) | 0.47 |
| T90 >37% (median) vs ≤37% | 180 | 2.31 (1.19–4.46) | 0.013 | 180 | 2.31 (1.14–4.68) | 0.020 | 177 | 2.55 (1.25–5.21) | 0.010 |
| T90 by quartiles | 180 | 180 | 177 | ||||||
| >2.24%−37% vs 0%−2.24% | 1.14 (0.38–3.38) | 0.82 | 1.19 (0.40–3.59) | 0.75 | 1.52 (0.48–4.85) | 0.47 | |||
| >37%−87.3% vs 0%−2.24% | 2.06 (0.77–5.49) | 0.15 | 1.81 (0.65–5.06) | 0.26 | 2.32 (0.78–6.88) | 0.13 | |||
| >87.3%−100% vs 0%−2.24% | 2.90 (1.13–7.41) | 0.026 | 3.59 (1.33–9.70) | 0.012 | 4.38 (1.54–12.47) | 0.006 | |||
| Mean SpO2 > 90% (median) vs ≤90% | 185 | 0.43 (0.22–0.82) | 0.010 | 185 | 0.42 (0.21–0.84) | 0.014 | 182 | 0.41 (0.20–0.83) | 0.013 |
AHI = apnea-hypopnea index; BMI = body mass index; DlCO = diffusing capacity of the lungs for carbon monoxide; SpO2 = oxygen saturation; T90 = percentage of recording time with oxygen saturation < 90%.
FIGURE 3. Sleep Indexes and Transplant-Free Survival in Group 1 PAH.

Kaplan-Meier survival curves for transplantation or death in group 1 PAH depicted with months to heart or lung transplantation or death as associated with (A) T90, dichotomized at oxygen saturation (SaO2) of 37%, and (B) mean SaO2, dichotomized at 90%. Abbreviations as in Figures 1 and 2.
FIGURE 4. T90 and Transplantation-Free Survival in Group 1 PAH.
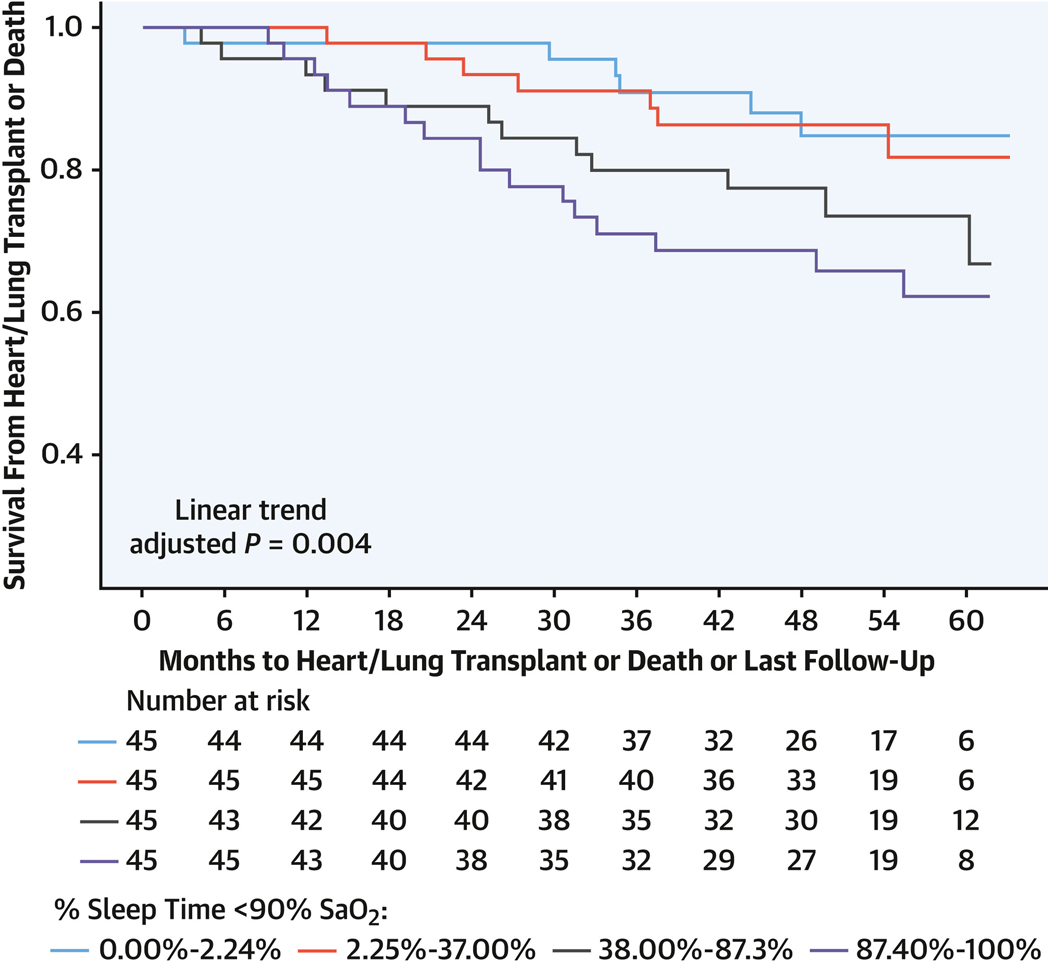
Kaplan-Meier survival curves for transplantation or death in group 1 PAH depicted with months to heart or lung transplantation or death as associated with T90 in quartiles with a significant linear adjusted trend (P = 0.0004). Abbreviations as in Figures 1 to 3.
SLEEP-DISORDERED BREATHING AND CONNECTIVE TISSUE DISEASE SUBGROUP OF WORLD SYMPOSIUM ON PULMONARY HYPERTENSION GROUP 1.
Forty-four participants with group 1 PAH had CTD (Table 1), with 14 of 34 deaths overall and 1 transplantation. Of the primary RV measures, only electrocardiographic RV hypertrophy was different between patients with CTD-associated PAH and those with non-CTD-associated PAH (4.5% vs 22%, respectively; P = 0.008) (Supplemental Table 10). None of the adjusted associations of AHI or sleep-related hypoxia with primary RV measures showed differences between the CTD and non-CTD groups. However, positive associations of AHI and T90 with cardiac output in CTD-associated PAH and an inverse association with cardiac output in non-CTD-associated PAH (AHI: 0.21 [95% CI: −0.19 to 0.61] vs −0.23 [95% CI: −0.41 to −0.05] [P = 0.041]; T90: 0.12 [95% CI: −0.01 to 0.24] vs −0.04 [95% CI: −0.11 to 0.03] [P = 0.030]) was observed. A positive association with T90 and mean right atrial pressure in non-CTD-associated PAH but an inverse relationship in CTD-associated PAH was observed (0.29 [95% CI: 0.07 to 0.51] vs −0.20 [95% CI: −0.60 to 0.21]; P = 0.036) (Supplemental Table 11). In CTD-associated PAH, a trend toward an association of T90 in 10% increments (P = 0.058), dichotomized at 37% (P = 0.051), with transplantation-free survival was observed (Supplemental Table 12). The number of events (transplantation or death) was too few to perform adjusted models.
DISCUSSION
We leveraged a richly phenotyped cohort, PVDOMICS, which enabled for the first time a prospective examination of the associations between OSA and sleep-related hypoxia in group 1 PAH and in-depth structural, hemodynamic, and electrophysiological assessments of RV dysfunction as well as survival. We observed that: 1) increasing severity of sleep-related hypoxia was consistently associated with a range of RV dysfunction measures; 2) the frequency of respiratory events (AHI) was associated only with reduced RV ejection fraction and some secondary RV dysfunction measures; 3) increasing sleep-related hypoxia severity (10% increment in T90) was associated with a 17% increase in adjusted risk for transplantation or death over a median 4-year follow-up period; 4) OSA and sleep-related hypoxia were differentially associated with cardiac output and mean right atrial pressure in CTD and non-CTD groups; and 5) in the non-CTD group, sleep-related hypoxia portended worse transplantation-free sur vival (Central Illustration).
CENTRAL ILLUSTRATION. Sleep-Related Hypoxia, Right Ventricular Dysfunction, and Survival in Group 1 Pulmonary Arterial Hypertension.

Among participants with group 1 pulmonary arterial hypertension (PAH), sleep-related hypoxia defined by the percentage of recording time at oxygen saturation <90% (T90) was associated with worsening markers of right heart dysfunction and increased risk for death or transplantation. This association was not seen in sleep-disordered breathing defined by the apnea-hypopnea index (AHI). CTD = connective tissue disease; Echo = echocardiography; mPAP = mean pulmonary arterial pressure; MRI = magnetic resonance imaging; RHC = right heart catheterization; RV = right ventricular; TAPSE = tricuspid annular plane systolic excursion.
Our key finding that sleep-related hypoxia is associated with a range of measures of RV dysfunction as well as mortality in group 1 PAH is consistent with the concept that hypoxia contributes to pulmonary hypertension severity, increasing RV afterload and impairing RV function, hence predisposing to worse outcomes.19,28 Sleep-related hypoxia, a repetitive overnight stressor, can cause sustained pulmonary hypertension via pulmonary vasoconstriction and remodeling.8,41–47 Hypoxia promptly increases pulmonary arterial pressure, which is fully reversible with reoxygenation,28,29,47,48 as oxygen-sensing mechanisms and mediation of hypoxic pulmonary vasoconstriction reside in the pulmonary arterial smooth muscle cells.41–46 Moreover, hypoxic pulmonary vasoconstriction limits exercise capacity via increases in RV afterload.8,43,45,46 Profound hypoxia also results in dysregulation of metabolic pathways in the RV, leading to greater RV hypertrophy or fibrosis and pulmonary vascular remodeling.49,50
Persistence of the association of sleep-related hypoxia and measures of RV dysfunction even after accounting for DlCO suggests a hypoxic etiology independent of ventilation-perfusion mismatch, anemia, and circulatory or diffusion barriers.40 Furthermore, in our cohort, mean baseline awake SpO2 was normal in group 1 PAH (96%), in contrast to the high degree of sleep-related hypoxia. Only 11% of participants with group 1 PAH had been prescribed long-term supplemental oxygen for daytime oxygen needs. By controlling for nocturnal oxygen and positive airway pressure use during the sleep study as well as DlCO, a reflection of reduced gas exchange, we demonstrated a persistent association with nocturnal hypoxia and compromised RV function and worse survival. Results also persisted after accounting for left heart diastolic dysfunction, thereby substantiating a unique association with sleep-related hypoxia and RV dysfunction. Our results support the hypothesis that nocturnal hypoxia during sleep exerts clinically important pathophysiologic effects on the pulmonary arteries and RV without evidence of day-time awake hypoxia.
Although the associations of sleep-related hypoxia and an array of measures of RV dysfunction (echocardiography, CMR, ECG, and RHC) were more consistent than with AHI, AHI was significantly associated with greater reductions in RV ejection fraction and RV peak global longitudinal strain by CMR. These associations may reflect the negative influence of repetitive apneas and hypopneas on hemodynamic measures (cardiac output and pulmonary vascular resistance) (eg, from wide swings in intrathoracic pressure or autonomic nervous system fluctuations during respiratory events). These specific pathophysiologic consequences may operate independent of or synergistic to hypoxia leading to specific subtypes of RV cardiac structural alterations.
Our prospective findings provide needed confirmation and enhancement of prior retrospective work19 showing a stronger association of sleep-related hypoxia than OSA with survival, but in a larger cohort, better taking into consideration confounding factors not previously addressed and, for the first time, providing analysis of right-sided cardiac measures reported to influence survival, such as tricuspid annular plane systolic excursion and echocardiographic RV free wall peak longitudinal strain in relation to OSA and sleep-related hypoxia. Tricuspid annular plane systolic excursion serves as a readily attainable marker of RV systolic function by echocardiography51,52 and is associated with worse survival in patients with group 1 PAH when <1.8 cm.53 Additionally, echocardiographic RV free wall peak longitudinal strain (3 segments) ≤−19% was associated with a >3-fold risk for all-cause mortality across an array of pulmonary hypertension etiologies.54 Our finding of an association with sleep-related hypoxia and both tricuspid annular plane systolic excursion and RV strain is novel and underscores the potential pathophysiological significance and clinical prognostic value of sleep-related hypoxia in group 1 PAH.
Current guidelines recommend nocturnal oximetry or a sleep study in patients with pulmonary hypertension if there is a suspicion of OSA.55 Although many patients undergo polysomnography as part of pulmonary hypertension evaluation, management guidelines do not provide recommendations regarding systematic screening of sleep-related hypoxia, as there are no long-term data suggesting that long-term supplemental nocturnal oxygen therapy has sustained benefits on mitigating disease progression. Our findings, particularly that greater duration of hypoxia portended worse transplantation-free survival, suggest potential utility of routine screening of sleep-related hypoxia to inform risk stratification.
Finally, contrary to our a priori hypothesis, OSA and sleep-related hypoxia were associated with worse cardiac output and survival in non-CTD-associated PAH but not CTD-associated PAH. The smaller sample size and number of transplantations and deaths in the CTD group may have limited the ability to detect an association with sleep-related hypoxia and transplantation-free survival. However, our findings are consistent with reports of improved survival in systemic sclerosis group 1 PAH diagnosed after 2010 compared with prior years, presumably because of earlier recognition and improved treatments.56
STUDY STRENGTHS AND LIMITATIONS.
This study has several strengths that overcame prior studies’ limitations. The proximate timing of the collection of sleep-disordered breathing indexes along with a range of rigorously collected, multimodal RV indexes (overcoming challenges with measurement variability of imaging and physiological measures in prior clinical cohorts) provides a unique opportunity to elucidate influences of sleep-related hypoxic stresses on RV function, structure, and physiology. These findings are clinically significant, as RV failure is the primary contributor to pulmonary hypertension mortality. This work uniquely allowed the prospective investigation of the influence of OSA and sleep-related hypoxia on survival in group 1 PAH over an approximately 4-year period. We also had the advantage of examining OSA and hypoxic associations with RV indexes across comparator and healthy control groups with data collection concurrent with participants with group 1 PAH. We accounted for confounding factors including demographics, pulmonary hypertension medications, positive airway pressure, and supplemental oxygen. Finally, we examined PAH subgroups to elucidate sleep-disordered breathing relationships across PAH subtypes.
Although a relatively large number of participants with group 1 PAH were recruited, approximately 50% were missing sleep study data, with results more generalizable to obese individuals. Although a subset of participants used supplemental oxygen and/or positive airway pressure therapy, results were robust to statistical adjustment of this use. Residual confounding by this therapy remains possible, but this would be expected to bias findings toward the null. To account for confounding by obesity, intracardiac shunt, left cardiac diastolic dysfunction, and oxygen gas exchange barriers, models were adjusted for body mass index, lateral E/e′ ratio, positive airway pressure, shunt repair status, and DlCO, respectively, without substantive changes in findings. Although median daytime SpO2 was normal in our PAH cohort, we cannot exclude the possibility that continuous daytime oximetry could have detected hypoxic episodes that may contribute to the overall hypoxic burden.
Using type 3 home testing rather than attended polysomnography may have resulted in an underestimation of the true severity of OSA. This, combined with the small number of death and transplantation events, may have limited our ability to detect a statistically significant interaction of OSA with RV measures and survival. Additionally, event numbers in the analysis of transplantation-free survival in CTD-associated PAH may have been too few to fully characterize an association between sleep-related hypoxia and transplantation-free survival, which was observed in non-CTD-associated PAH. Finally, use of pulmonary hypertension medications may have increased the overall survival within the entire cohort, and although use of medications was controlled for in our analyses, the true association with sleep-related hypoxia may be underestimated.
CONCLUSIONS
In this multicenter, prospective cohort study, we show that increased sleep-related hypoxia was associated with worse right-sided cardiac structural, hemodynamic, and electrophysiological measures in group 1 PAH, even after adjustments for confounding factors, suggesting that hypoxia specific to sleep contributes to the pathogenesis of group 1 PAH. AHI was also associated with some specific measures of RV dysfunction, such as RV ejection fraction, recognized to hold prognostic significance in pulmonary hypertension, but overall findings were less consistent than with hypoxia. Sleep-related hypoxia was also independently associated with death or transplantation in non-CTD-associated group 1 PAH but not in CTD-associated PAH, possibly related to the smaller sample size and number of outcome events in the latter. These results are hypothesis generating and prompt further investigation to better elucidate underlying mechanisms of the association between sleep-related hypoxia and RV dysfunction, expound physiological contributors to sleep-related hypoxia (eg, ventilation-perfusion mismatch, sleep apnea–specific hypoxic burden), and clarify whether routine screening for sleep-related hypoxia and whether nocturnal supplemental oxygen can improve PAH outcomes. As continuous monitoring of daytime awake oxygenation was not performed, future studies should also examine the differential contributions of nocturnal and daytime hypoxia.
Supplementary Material
PERSPECTIVES.
COMPETENCY IN MEDICAL KNOWLEDGE:
Sleep-related hypoxia may contribute to RV dysfunction, increasing mortality in patients with Group 1 PAH.
TRANSLATIONAL OUTLOOK:
Future studies should focus on the pathophysiological links between sleep-related hypoxia and RV dysfunction and whether supplemental nocturnal oxygen improves outcomes in patients with Group 1 PAH.
ACKNOWLEDGMENTS
The authors thank the participants of the PVDOMICS cohort for contributing their valuable time to provide the data used for this work, which will allow us to advance insights in PH biology. The authors also acknowledge the work of research coordinator Joan Aylor and research polysomnologist Samantha Wells.
FUNDING SUPPORT AND AUTHOR DISCLOSURES
This study was supported by grants U01 HL125218 (principal investigator, Dr Rosenzweig), U01 HL125205 (principal investigator, Dr Frantz), U01 HL125212 (principal investigator, Dr Hemnes), U01 HL125208 (principal investigator, Dr Rischard), U01 HL125175 (principal investigator, Dr Hassoun), U01 HL125215 (principal investigator, Dr Leopold), and U01 HL125177 (principal investigator, Dr Beck) and the Pulmonary Hypertension Association. Dr Hill is a scientific advisory board member for Aerovate and Insmed; is a consultant for Bellerophon; and is a data and safety monitoring board member for Merck. Dr Finet has served as a clinical practice advisor for Wolters Kluwer Health-Lexicomp (forfeited compensation); and is an Item-Writing Task Force member for the American Board of Internal Medicine. Dr Kwon has received funding from the National Heart, Lung, and Blood Institute (grant 1R01HL170090–01); and has a research agreement with Circle Cardiovascular Imaging. Dr Beck has received support from the Pulmonary Hypertension Association. Dr Frantz has consulting, steering committee, and advisory board relationships with Altavant Sciences, Bayer, Gossamer Bio, Janssen, Shouti, the France Foundation, IQVIA, Tenax, UpToDate, and United Therapeutics. Dr Hassoun serves on a scientific steering committee for Merck Sharpe & Dohme; and is a scientific adviser for ARIA-CV (unrelated to the present work). Dr Hemnes serves as a consultant for Bayer, United Therapeutics, Janssen, GossamerBio, and Tenax Therapeutics; holds stock in Tenax Therapeutics; and has received grants from the National Institutes of Health, the Cardiovascular Medical Research and Education Fund, and Imara. Dr Horn has conducted research studies with Acceleron/Merck, Cereno, and Insmed. Dr Leopold is a consultant for Abbott Vascular; is a speaker for United Therapeutics; has received research funding from Astellas to her institution; and has received support from the American Heart Association (grant AIM 19AIML34980000; National Heart, Lung, and Blood Institute grant U01 HL125215). Dr Rischard has consulting relationships with Acceleron and United Therapeutics; is a steering committee member for Acceleron; and receives research support from Ismed, United Therapeutics, Bayer, Acceleron, Janssen, and Aadi Bioscience. Dr Mehra has received an honorarium from the American Academy of Sleep Medicine; has received funds for service on the American Board of Internal Medicine and as associate editor of the American Journal of Respiratory and Critical Care Medicine; has received National Institutes of Health funding; has received investigator-initiated research funds to her institution from Resmed, Inspire, and Sommetrics; and has received royalties from UpToDate. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
ABBREVIATIONS AND ACRONYMS
- AHI
apnea-hypopnea index
- CMR
cardiac magnetic resonance imaging
- CTD
connective tissue disease
- DlCO
diffusing capacity of the lungs for carbon monoxide
- ECG
electrocardiography
- mPAP
mean pulmonary artery pressure
- OSA
obstructive sleep apnea
- PAH
pulmonary arterial hypertension
- RHC
right heart catheterization
- RV
right ventricle/ventricular
- SpO2
oxygen saturation
- T90
percentage of time spent at oxygen saturation <90%
Footnotes
The authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the Author Center.
APPENDIX For a list of PVDOMICS Study Group members, supplemental methods, a list of pulmonary hypertension medications, additional baseline sleep study and cardiac results, supplemental tables, and supplemental references, please see the online version of this paper.
REFERENCES
- 1.Humbert M, Sitbon O, Yaïci A, et al. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010;36(3):549–555. [DOI] [PubMed] [Google Scholar]
- 2.Rådegran G, Kjellström B, Ekmehag B, et al. Characteristics and survival of adult Swedish PAH and CTEPH patients 2000–2014. Scand Cardiovasc J. 2016;50(4):243–250. [DOI] [PubMed] [Google Scholar]
- 3.Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL registry. Chest. 2012;142(2):448–456. [DOI] [PubMed] [Google Scholar]
- 4.Brewis MJ, Bellofiore A, Vanderpool RR, et al. Imaging right ventricular function to predict outcome in pulmonary arterial hypertension. Int J Cardiol. 2016;218:206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Humbert M, Farber HW, Ghofrani HA, et al. Risk assessment in pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Eur Respir J. 2019;53(6):1802004. 10.1183/13993003.02004-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boucly A, Weatherald J, Savale L, et al. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur Respir J. 2017;50(2):1700889. 10.1183/13993003.00889-2017 [DOI] [PubMed] [Google Scholar]
- 7.Ismail K, Roberts K, Manning P, Manley C, Hill NS. OSA and pulmonary hypertension: time for a new look. Chest. 2015;147(3):847–861. [DOI] [PubMed] [Google Scholar]
- 8.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006;99(7): 675–691. [DOI] [PubMed] [Google Scholar]
- 9.Badesch DB, Raskob GE, Elliott CG, et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL registry. Chest. 2010;137(2):376–387. [DOI] [PubMed] [Google Scholar]
- 10.Hemnes AR, Leopold JA, Radeva MK, et al. Clinical characteristics and transplant-free survival across the spectrum of pulmonary vascular disease. J Am Coll Cardiol. 2022;80(7):697–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vonk-Noordegraaf A, Haddad F, Chin KM, et al. Right heart adaptation to pulmonary arterial hypertension: physiology and pathobiology. J Am Coll Cardiol. 2013;62(25 suppl):D22–D33. [DOI] [PubMed] [Google Scholar]
- 12.Vonk Noordegraaf A, Chin KM, Haddad F, et al. Pathophysiology of the right ventricle and of the pulmonary circulation in pulmonary hypertension: an update. Eur Respir J. 2019;53(1):1801900. 10.1183/13993003.01900-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ulrich S, Fischler M, Speich R, Bloch KE. Sleep-related breathing disorders in patients with pulmonary hypertension. Chest. 2008;133(6):1375–1380. [DOI] [PubMed] [Google Scholar]
- 14.Minic M, Granton JT, Ryan CM. Sleep disordered breathing in group 1 pulmonary arterial hypertension. J Clin Sleep Med. 2014;10(3):277–283. 10.5664/jcsm.3528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rafanan AL, Golish JA, Dinner DS, Hague LK, Arroliga AC. Nocturnal hypoxemia is common in primary pulmonary hypertension. Chest. 2001;120(3):894–899. [DOI] [PubMed] [Google Scholar]
- 16.Yan L, Luo Q, Zhao Z, et al. Nocturnal hypoxia in patients with idiopathic pulmonary arterial hypertension. Pulm Circ. 2020;10(3): 2045894019885364. 10.1177/2045894019885364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spiesshoefer J, Herkenrath S, Harre K, et al. Sleep-disordered breathing and nocturnal hypoxemia in precapillary pulmonary hypertension: prevalence, pathophysiological determinants, and clinical consequences. Respiration. 2021;100(9): 865–876. [DOI] [PubMed] [Google Scholar]
- 18.Murta MS, Duarte RLM, Waetge D, Gozal D, Cardoso AP, Mello FCQ. Sleep-disordered breathing in adults with precapillary pulmonary hypertension: prevalence and predictors of nocturnal hypoxemia. Lung. 2022;200(4):523–530. [DOI] [PubMed] [Google Scholar]
- 19.Nagaoka M, Goda A, Takeuchi K, et al. Nocturnal hypoxemia, but not sleep apnea, is associated with a poor prognosis in patients with pulmonary arterial hypertension. Circ J. 2018;82(12):3076–3081. [DOI] [PubMed] [Google Scholar]
- 20.Dumitrascu R, Tiede H, Eckermann J, et al. Sleep apnea in precapillary pulmonary hypertension. Sleep Med. 2013;14(3):247–251. [DOI] [PubMed] [Google Scholar]
- 21.Jilwan FN, Escourrou P, Garcia G, Jaïs X, Humbert M, Roisman G. High occurrence of hypoxemic sleep respiratory disorders in precapillary pulmonary hypertension and mechanisms. Chest. 2013;143(1):47–55. [DOI] [PubMed] [Google Scholar]
- 22.Stenmark KR, Mecham RP. Cellular and molecular mechanisms of pulmonary vascular remodeling. Annu Rev Physiol. 1997;59:89–144. [DOI] [PubMed] [Google Scholar]
- 23.Stenmark KR, Davie N, Frid M, Gerasimovskaya E, Das M. Role of the adventitia in pulmonary vascular remodeling. Physiology (Bethesda). 2006;21:134–145. [DOI] [PubMed] [Google Scholar]
- 24.Das M, Dempsey EC, Reeves JT, Stenmark KR. Selective expansion of fibroblast subpopulations from pulmonary artery adventitia in response to hypoxia. Am J Physiol Lung Cell Mol Physiol. 2002;282(5):L976–L986. [DOI] [PubMed] [Google Scholar]
- 25.Chen T, Yang C, Li M, Tan X. Alveolar hypoxia-induced pulmonary inflammation: from local initiation to secondary promotion by activated systemic inflammation. J Vasc Res. 2016;53(5–6): 317–329. [DOI] [PubMed] [Google Scholar]
- 26.Madjdpour C, Jewell UR, Kneller S, et al. Decreased alveolar oxygen induces lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2003;284(2):L360–L367. 10.1152/ajplung.00158.2002 [DOI] [PubMed] [Google Scholar]
- 27.Ali MH, Schlidt SA, Chandel NS, Hynes KL, Schumacker PT, Gewertz BL. Endothelial permeability and IL-6 production during hypoxia: role of ROS in signal transduction. Am J Physiol. 1999;277(5):L1057–L1065. [DOI] [PubMed] [Google Scholar]
- 28.Prisco DL, Sica AL, Talwar A, et al. Correlation of pulmonary hypertension severity with metrics of comorbid sleep-disordered breathing. Sleep Breath. 2011;15(4):633–639. [DOI] [PubMed] [Google Scholar]
- 29.Samhouri B, Venkatasaburamini M, Paz Y, et al. Pulmonary artery hemodynamics are associated with duration of nocturnal desaturation but not apnea-hypopnea index. J Clin Sleep Med. 2020;16(8):1231–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khanna D, Zhao C, Saggar R, et al. Long-term outcomes in patients with connective tissue disease-associated pulmonary arterial hypertension in the modern treatment era: meta-analyses of randomized, controlled trials and observational registries. Arthritis Rheumatol. 2021;73(5): 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hemnes AR, Beck GJ, Newman JH, et al. PVDOMICS: a multi-center study to improve understanding of pulmonary vascular disease through phenomics. Circ Res. 2017;121(10):1136–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jellis CL, Park MM, Abidov A, et al. Comprehensive echocardiographic evaluation of the right heart in patients with pulmonary vascular diseases: the PVDOMICS experience. Eur Heart J Cardiovasc Imaging. 2022;23(7):958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang WHW, Wilcox JD, Jacob MS, et al. Comprehensive diagnostic evaluation of cardiovascular physiology in patients with pulmonary vascular disease: insights from the PVDOMICS program. Circ Heart Fail. 2020;13(3):e006363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pulmonary Vascular Disease Phenomics Program (PVDOMICS). Accessed September 14, 2022. https://clinicaltrials.gov/ct2/show/NCT02980887
- 35.Galiè N, Simonneau G. The Fifth World Symposium on Pulmonary Hypertension. J Am Coll Cardiol. 2013;62(25 suppl):D1–D3. [DOI] [PubMed] [Google Scholar]
- 36.Galiè N, McLaughlin VV, Rubin LJ, Simonneau G. An overview of the 6th World Symposium on Pulmonary Hypertension. Eur Respir J. 2019;53(1):1802148. 10.1183/13993003.02148-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collop NA, Anderson WM, Boehlecke B, et al. Clinical guidelines for the use of unattended portable monitors in the diagnosis of obstructive sleep apnea in adult patients. Portable Monitoring Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med. 2007;3(7):737–747. [PMC free article] [PubMed] [Google Scholar]
- 38.Berry RB, Brooks R, Gamaldo C, et al. AASM scoring manual updates for 2017 (version 2.4). J Clin Sleep Med. 2017;13(5):665–666. 10.5664/jcsm.6576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kapur VK, Auckley DH, Chowdhuri S, et al. Clinical practice guideline for diagnostic testing for adult obstructive sleep apnea: an American Academy of Sleep Medicine clinical practice guideline. J Clin Sleep Med. 2017;13(3):479–504. 10.5664/jcsm.6506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graham BL, Brusasco V, Burgos F, et al. 2017 ERS/ATS standards for single-breath carbon monoxide uptake in the lung. Eur Respir J. 2017;49(1):1600016. 10.1183/13993003.00016-2016 [DOI] [PubMed] [Google Scholar]
- 41.Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev. 2012;92(1):367–520. Published correction appears in Physiol Rev. 2014;94(3):989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fishman AP. Vasomotor regulation of the pulmonary circulation. Annu Rev Physiol. 1980;42: 211–220. [DOI] [PubMed] [Google Scholar]
- 43.Fagan KA. Selected contribution: pulmonary hypertension in mice following intermittent hypoxia. J Appl Physiol (1985). 2001;90(6):2502–2507. [DOI] [PubMed] [Google Scholar]
- 44.Fagan KA, Fouty BW, Tyler RC, et al. The pulmonary circulation of homozygous or heterozygous eNOS-null mice is hyperresponsive to mild hypoxia. J Clin Invest. 1999;103(2):291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steudel W, Scherrer-Crosbie M, Bloch KD, et al. Sustained pulmonary hypertension and right ventricular hypertrophy after chronic hypoxia in mice with congenital deficiency of nitric oxide synthase 3. J Clin Invest. 1998;101(11):2468–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hales CA, Kradin RL, Brandstetter RD, Zhu YJ. Impairment of hypoxic pulmonary artery remodeling by heparin in mice. Am Rev Respir Dis. 1983;128(4):747–751. [DOI] [PubMed] [Google Scholar]
- 47.Dunham-Snary KJ, Wu D, Sykes EA, et al. Hypoxic pulmonary vasoconstriction: from molecular mechanisms to medicine. Chest. 2017;151(1):181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maggiorini M. High altitude-induced pulmonary oedema. Cardiovasc Res. 2006;72(1):41–50. [DOI] [PubMed] [Google Scholar]
- 49.Li Y, Ren W, Wang X, et al. MicroRNA-150 relieves vascular remodeling and fibrosis in hypoxia-induced pulmonary hypertension. Biomed Pharmacother. 2019;109:1740–1749. [DOI] [PubMed] [Google Scholar]
- 50.Choudhary G, Troncales F, Martin D, Harrington EO, Klinger JR. Bosentan attenuates right ventricular hypertrophy and fibrosis in normobaric hypoxia model of pulmonary hypertension. J Heart Lung Transplant. 2011;30(7):827–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rudski LG, Lai WW, Afilalo J, et al. Guidelines for the echocardiographic assessment of the right heart in adults: a report from the American Society of Echocardiography endorsed by the European Association of Echocardiography, a registered branch of the European Society of Cardiology, and the Canadian Society of Echocardiography. J Am Soc Echocardiogr. 2010;23(7): 685–788. [DOI] [PubMed] [Google Scholar]
- 52.Kaul S, Tei C, Hopkins JM, Shah PM. Assessment of right ventricular function using two-dimensional echocardiography. Am Heart J. 1984;107(3):526–531. [DOI] [PubMed] [Google Scholar]
- 53.Forfia PR, Fisher MR, Mathai SC, et al. Tricuspid annular displacement predicts survival in pulmonary hypertension. Am J Respir Crit Care Med. 2006;174(9):1034–1041. [DOI] [PubMed] [Google Scholar]
- 54.Haeck ML, Scherptong RW, Marsan NA, et al. Prognostic value of right ventricular longitudinal peak systolic strain in patients with pulmonary hypertension. Circ Cardiovasc Imaging. 2012;5(5): 628–636. [DOI] [PubMed] [Google Scholar]
- 55.Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022;43(38):3618–3731. [DOI] [PubMed] [Google Scholar]
- 56.Hassan HJ, Naranjo M, Ayoub N, et al. Improved survival for patients with systemic sclerosis-associated pulmonararterial hypertension: the Johns Hopkins Registry. Am J Respir Crit Care Med. 2023;207(3):312–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.