

Abstract
Sturge-Weber syndrome (SWS) is a rare, non-inherited neurovascular disorder characterized by abnormal vasculature in the brain, skin, and eye. Patients with SWS characteristically have facial capillary malformation, also known as port-wine birthmark, a leptomeningeal vascular malformation seen on contrast-enhanced MRI images, abnormal blood vessels in the eye, and glaucoma. Patients with SWS have impaired perfusion to the brain, and are at high risk of venous stroke and stroke-like episodes, seizures, and both motor and cognitive difficulties. While the activating R183Q GNAQ somatic mutation is the most common somatic mutation underlying SWS, recent research also implicates that GNA11 and GNB2 somatic mutations are related to SWS. Recent retrospective studies suggest the use of low dose aspirin and vitamin D in treatment for SWS, and prospective drug trials have supported the usefulness of cannabidiol and Sirolimus. Presymptomatic treatment with low-dose aspirin and anti-epileptic drugs shows promising results in delaying seizure onset in some patients. This review focuses on the latest progress in the field of research for Sturge-Weber syndrome and highlights directions for future research.
Graphical Abstract
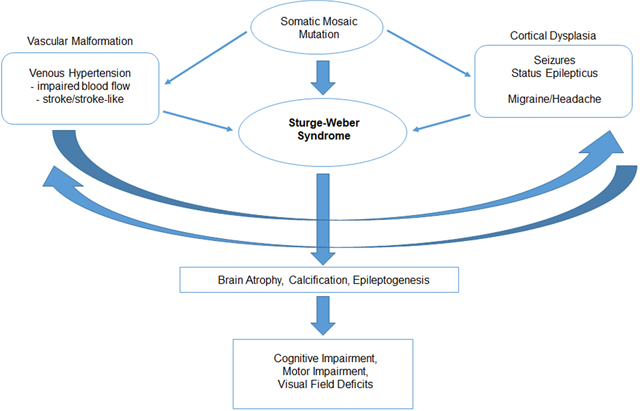
Sturge-Weber syndrome is known to be caused by somatic mosaic mutation during fetal development. An R183Q GNAQ mutation is most common, and result in a congenital leptomeningeal vascular malformation and cortial dysgenesis. These abnormalities result in venous hypertension, impaired blood flow, seizures and stroke. In time, these pathologic processes produce brain atrophy, calcification, and can epileptogenesis, and can lead to cognitive impairment, motor impairment, and visual deficits.
Introduction
Sturge-Weber syndrome (SWS) is a neurocutaneous disorder involving atypical formation and function of blood vessels in the skin, eye, and brain. SWS is usually associated with a facial capillary malformation (port-wine birthmark) on the upper face, leptomeningeal vascular malformation in the brain visible on contrast-enhanced MRI studies, and glaucoma (1). The neurological manifestations include strokes and stroke-like episodes (SLE), acquired hemiparesis, cerebral atrophy and calcifications, seizures, visual field deficits, and intellectual disorders (2). Patients with a facial port-wine birthmark also are at high risk for developing glaucoma (3, 4). As SWS is a multi-system disorder, with endocrine, psychiatric, ophthalmologic, rehabilitative, dermatologic and other medical issues often arising (2), patients with SWS are best followed by a multidisciplinary team of providers including neurologist, ophthalmologist, dermatologist, neuropsychologist, physical therapist, and occupational therapist.
Recent identification of the underlying somatic mosaic mutation and putative hyperactivity of the downstream pathways, as well as the development of biomarkers and outcome measures in SWS, has facilitated the recent prospective drug trials for these patients. While R183Q GNAQ mutation is commonly associated with SWS (5), recent discoveries suggest that mutations in GNA11 and GNB2 are also related to SWS (6, 7). Quantitative electroencephalography (EEG) (8) and transcranial doppler (9) have recently been suggested to be possible biomarkers in aiding diagnosis and follow up with SWS patients in clinical settings. More centers are implementing low dose aspirin for reduction of stroke-like episodes and seizures in SWS patients (10). Also in the last few years, oral drug trials for SWS have shown promising results in the treatment of stroke and stroke-like episodes, seizure management, and cognitive improvements (11, 12). The purpose of this review is to summarize recent updates in the field of SWS research important in the treatment of patients with SWS, and to lay the foundation for future multi-centered drug trials aimed at preventing stroke injury and preserving neurologic function in SWS.
Gene Mutation/ Molecular Aspect
Sturge-Weber syndrome is most commonly associated with a R183Q somatic nonsynonymous mosaic mutation in GNAQ (5). GNAQ encodes Gαq, a member of the q class of G-protein alpha subunits. Gαq mediates signals between G protein coupled receptors and downstream effector proteins. In SWS, the R183Q mutation of GNAQ is predicted to hyperactivate the downstream Ras/Raf/MEK/ERK pathway and other key pathways such as mTOR activity (Figure 1). The R183 GNAQ residue is located in the core of hydrophobic cleft between two inter-domain linkers that connect the GTPase and helical domains of Gαq. Computational analysis of the R183Q GNAQ mutation (13) suggests that the mutation impairs formation of the hydrogen bond between R183 residue and GDP molecule, which destabilizes the inactive GDP-bond conformation of the Gαq mutant. This alteration is predicted to decrease auto-hydrolysis of GTP to GDP, re-assembly of the trimeric G-protein complex, and inactivation of Gαq, resulting in a constitutive hyper-activation of downstream pathways. How this dysregulation of downstream pathways result in the vascular malformations associated with SWS is the subject of recent and ongoing studies (14, 15).
Figure 1:

Diagram of molecular pathways which are likely hyper-activated by SWS somatic mutation in the protein Gαq. This figure also indicates several classes of inhibitors, including mTOR inhibitor such as Sirolimus, which are being tested, or may be studied in future clinical drug trials for SWS.
The underlying somatic mutation most likely occurs at an early stage of embryonic development (16). Individuals with SWS have mosaic pattern of mutation, meaning that they have cells with normal copy of the gene interspersed with cells containing abnormal copies. In SWS, the somatic mutation is generally localized to the involved tissue of the head, and are therefore not inherited (5). Patients can have variability in severity of symptoms, in part depending on the extent of their involvement. Somatic mutations occurring later in fetal development more likely have lesser extent of involvement and involve fewer cell types; earlier timing of the mutation is more likely to involve brain, skin and eye involvement and a greater range of cell types (17).
Recent findings suggest that other somatic activating mutations may be less commonly underlying SWS. Thorpe et al. reported that mutations in the paralogue, GNA11 (which is similar to GNAQ), can also lead to Sturge-Weber syndrome (6). Furthermore, through deep DNA sequencing from skin biopsies, Fjaer et al reported that a somatic mutation in GNB2 (p.Lys78Glu) which encodes a beta chain of the same G-protein complex might be related to SWS. While previous research focused on regulation of downstream MAPK signaling (15) in the pathogenesis of SWS vascular abnormalities, results from GNB2 suggest that Yes-associated protein (YAP) in the Hippo signaling pathway might be involved in pathogenesis of SWS phenotypes (7). Further research is needed in the fields of genetics and pathogenesis of SWS. Table 1 presents gene mutations that are reported to be associated with SWS and other capillary malformation-related conditions. We do not routinely do genetic testing in patients with typical SWS. Genetic testing in this situation generally requires a skin biopsy; however, this is a rapidly evolving area.
Table 1.
Gene mutations of SWS and other capillary malformations
| Capillary Malformations | Mutated Genes |
|---|---|
| Sturge-Weber Syndrome | GNAQ (5), GNA11 (6), GNB2 (7) |
| Capillary Malformation-AVM Syndrome | RASA1 (18), EPHB4 (19) |
| Microcephaly-Capillary Malformation Syndrome (MIC-CAP) | STAMBP (20) |
| PIK3CA-related Overgrowth Spectrum (PROS) | PIK3CA (21) |
| Capillary Malformation with Undergrowth (CMU) | DDR2, GRHL2, PIK3CA (22), TEK, GNAQ, GNA11(23) |
| Isolated Port-Wine Birthmarks | GNAQ (5), GNA11 |
Clinical Presentation
Presentation of skin involvement:
Sturge-Weber syndrome presents with a facial capillary malformation at birth, which indicates increased risk of SWS brain and eye involvement. Facial port-wine birthmark/capillary malformation in the forehead area, eyelid, or temple region is associated with high risk of SWS (24, 25), with a 20–50% chance of being diagnosed with SWS brain involvement, depending on the size, location and extent of skin involvement. In exceptional cases, patients may have frontonasal PWB. PWB can present as red or pink, and the color will briefly lighten when the birthmark is compressed. While SWS skin involvement typically is located on the upper face, location, extent, and color of PWB can be highly variable. Study by Dymerska et al suggests that size of facial PWB correlates with neurological severity ratings for patients ages 6 and above with SWS brain involvement. The size and location of PWB in combination with brain MRI images can be helpful in predicting the severity of neurological dysfunction (24). Some patients (about 10%) have isolated SWS brain or eye involvement, without a facial port-wine birthmark (26).
Presentation of eye involvement:
Glaucoma is the most common ocular complication in patients with SWS, with a 30–70% prevalence rate in patients with both upper and lower eyelid birthmark involvement. Glaucoma (eye damage due to increased intraocular pressure) can develop at any time, with the most common periods being in infancy and in young adulthood (1). Patients develop glaucoma in early infancy in 60% of the cases, while 40% of the patients show later onset. In SWS patients, open-angle glaucoma is the most common form of glaucoma. Symptoms of open-angle glaucoma can include vision loss, development of dilated conjunctival vessels, eye pain, excessive tearing and, in infants, eye enlargement (buphthalmos) (27). If left untreated, glaucoma can threaten vision by causing ischemic injury to the optic nerve (2).
Presentation, Diagnosis, and Symptoms of brain involvement:
Patients with SWS brain involvement most commonly present with seizures in infancy. Most often, these patients will present with focal motor seizures, which can be subtle in infants, or complex partial seizures where consciousness is impaired (2). More rarely the seizures can present with infantile spasms or myoclonic epilepsy (28, 29). Status epilepticus is frequently associated with stroke-like episodes, particularly in young patients (30). Seizures occur in 75% of patients with unilateral brain involvement and in 95% of those with bilateral brain involvement (31). While not as common, it is possible for infants with SWS brain involvements to present with early handedness or visual gaze preference rather than acute seizures (2). Neurologic deficit can result from venous strokes, stroke-like episodes, seizures or migraines, often with progression of the brain injury on neuroimaging(2).
Sturge-Weber syndrome brain involvement is marked by a leptomeningeal vascular malformation on contrast enhanced MRI images (Figure 2, Panels A-B) (32). Dilated deep draining vessels underlying the affected cortical region and enhancement of the choroid plexus on the involved side are also often present in older children and adults. Cortical and subcortical calcifications can be diagnosed with non-contrast head CT (Figure 2, Panel C) (33) or sometimes on MRI. Because presymptomatic diagnosis of SWS brain involvement on brain MRI has low sensitivity in the newborn period, an early negative contrast enhanced MRI image does not exclude the possibility of SWS brain involvement for infants (2). However, a normal neurologic exam, no history of seizure onset, and a negative MRI image with contrast after a year of age generally excludes SWS brain involvement. MRI sequences should include SWI, T1 spin echo post-contrast, T2 post contrast flair, diffusion weighted imaging (DWI) and apparent diffusion coefficient (ADC) to aid diagnosis of brain involvement. Over time, patients frequently acquire brain atrophy, calcification, loss of white matter, and impaired perfusion of the affected region (34). With leptomeningeal venous malformations inducing destruction of effective superficial venous pathways, venous pressure results in an alternate drainage route with dilated deep draining vessels demonstrated on neuroimaging (35).
Figure 2:

Neuroimaging in 3 patients with Sturge-Weber syndrome. Stars indicate affected areas. A: Axial T1 post-contrast image showing leptomeningeal enhancement over the right hemisphere. B: Susceptibility weighted imaging (SWI) of another patient showing greatly dilated deep draining vessels draining to the deep venous system. C: Head CT, non-contrast, from another patient showing calcification in affected areas.
Stroke-like are characterized by transient episodes of hemiparesis, with or without speech impairments, and are considered to be the most enigmatic severe neurological symptoms for patients with SWS (36). Stroke-like episodes are called “stroke-like” as, in the acute phase, these episodes clinically resemble ischemic strokes, but have variable clinical course and often fully resolve in a period of days, weeks or a few months. One study reported median recovery time of 24 hours (37); this publication grouped all transient episodes of neurologic impairment in SWS, including episodes after seizures that resolve by 24 hours later, and included episodes associated with headaches (37). This classification acknowledges the role of the vascular malformation and perfusion impairments in these SWS-related episodes, as compared to transient weakness after focal seizure or with a migraine in other patients.
Over time and with multiple events, weakness may be permanently acquired, and further study is needed to determine factors that predict, or contribute to, prolonged stroke-like episodes and permanent neurologic impairment. In a retrospective cohort study, 54.5% of transient hemiplegic episodes had onset associated with seizures, 18.1% with blow to head or fall, and 6% with brain surgery (37). As seizures and head injuries are commonly associated with onset of stroke-like episodes, aggressive seizure management, as well as head injury prevention could prevent neurologic deterioration(36). SWS stroke-like episodes are different from strokes in that their brain diffusion weighted MRI rarely shows acute evidence of stroke. Perfusion imaging demonstrates impaired venous drainage in the affected brain regions, which is worsened by seizures, and results in impaired arterial perfusion (38). The decision to obtain rapid MRI during a SLE in a patient with SWS depends on patient age, the severity of the SLE symptoms, and how long the symptoms have been present for; imaging is frequently done in young patients, those with significant weakness or loss of language, or with more prolonged duration of symptoms. Further study of this area is needed and current research is underway through the Brain Vascular Malformation Consortium.
SWS related stroke-like episodes can be triggered by seizures, head trauma or illness but can also occur without an apparent trigger. Furthermore, the differential for stroke-like episodes includes complicated migraine and Todd’s Paralysis; episodes with prominent headaches are likely the former, while episodes lasting less than 24 hours in duration are often referred to as the later. EEG has been reported to be helpful in the differential diagnosis; however, in practice, many patients have features of seizures and migraines, at least initially, during these events. To avoid stroke-like episodes and worsening of neurologic deficits (39), patients with SWS are advised to avoid recreational activities with significant risk of head injuries, address any symptoms of illness, and obtain recommended vaccinations (2). Treatment includes aggressive use of fluids, treatment of fever, treatment of nausea and vomiting if present, and physical therapy and occupational therapy if recovery is slow (40).
Headaches are another neurological symptom that can have moderate to severe impact on the quality of life for patients with SWS. A visual or sensory-motor aura can occur with these headaches, which if prolonged and prominent, can be referred to as a stroke-like episode. Vasomotor disturbance within and around the vascular malformation is hypothesized to lead to oligemia and to trigger cortical spreading depression (41). While no clear consensus on treatments for SWS-related headaches exists, ibuprofen and acetaminophen are widely used (42). The use of Flunarizine as preventative and abortive treatment for migraine has shown promising results as well (43). Triptans have also been safely used in patients with SWS and migraines with variable effectiveness (42).
Intellectual and language impairments are frequent in patients with SWS. Cognitive outcomes were associated with number of affected lobes, and bilateral brain involvement with more severe intellectual disability and language disorder (44). Earlier seizure onset was associated with intellectual and language disabilities; active epilepsy was associated with language disorder. Patients with PWB showed higher rates of intellectual disability and language disorder than patients without PWB, who had more favorable outcomes (45); this association may result from the mutation occurring later on in the fetal development in those without birthmark, which would result in fewer cell types affected. Absence of facial port-wine birthmark is associated with later age for seizure onset, which in turn is a predictor of cognitive and neurologic outcomes (44).
Recent studies suggest that SWS patients may be at higher risk of Autism Spectrum Disorder (ASD). Twenty-four percent of 92 children with SWS seen at a national referral center in England between 2002 and 2015 had diagnosis of autism spectrum disorder; 45% of the 92 patients had evidence of social communication difficulties (46). Patients with SWS reported other social-behavioral problems as well: 50% of the patients reported significant behavioral difficulties while 26% reported sleep difficulties. While difficulties with social communication, behavioral difficulties, and sleep difficulties were closely associated with one another, they were not significantly associated with epilepsy severity. Autism spectrum disorder was more likely to be present in patients with bilateral brain involvement.
The natural history of Sturge-Weber Syndrome is highly variable. While some patients continue to suffer significant neurologic deficits, others do well. In a study that analyzed physical and family history variables for 277 SWS patients (44), brain involvement and PWB were associated with SWS symptom severity. While bilateral brain involvement was associated with learning disorder and intellectual disability, extent of skin capillary malformation was associated with epilepsy and likelihood of glaucoma surgery. Earlier seizure onset was associated with learning disorder, intellectual disability, stroke-like episodes, symptomatic stroke, hemiparesis, visual field deficit, and brain surgery; this data reinforces the prior studies suggesting that age of seizure onset is important to natural history of the disease (47, 48). Patients with family history of seizure reported earlier seizure onset, while patients with family history of vascular malformations were more likely to report symptomatic strokes. The impact of family history of seizures and stroke on outcome in SWS patients requires further investigation; this association suggests the presence of inherited seizure and stroke susceptibility genes interacting with SWS brain involvement to result in more severe neurologic impairment.
A survey distributed to SWS patients and their families revealed that in 52 adults, patients with seizures had a greater chance of developmental delay, more emotional and behavioral problems, and were less likely to be employed (47). As adults, 39% of the 52 patients were financially self-sufficient and 55% were or could get married. Ten of the 52 patients had liveborn children as well. This survey was completed by SWS patients, unless they were physically or mentally incapable of completing the survey; relatives or caretakers completed the survey in those cases. As most research on natural history of SWS is cross-sectional, further prospective, longitudinal research is needed. Data from a multicenter effort to study the progression of SWS has closed and results are currently being analyzed (https://clinicaltrials.gov/ct2/show/NCT01425944).
Treatment
The standard neurologic treatment for Sturge-Weber syndrome focuses on managing specific symptoms such as seizure management, occupational therapy, and physical therapy.
Treatments for Neurologic Symptoms:
Treatment options for seizures in Sturge-Weber include using anticonvulsants and more invasive approaches including hemispherectomy, focal resection, and VNS. According to a multicenter cross-sectional questionnaire, levetiracetam, oxcarbazepine, and low-dose aspirin were the most frequently used medications (10). Forty-eight percent of patients reported being on levetiracetam, 45% of patients were on low-dose aspirin, and 40% reported being on oxcarbazepine. Subjects with bilateral brain involvement, early onset of seizures, and a family history of seizures were using a greater number of anticonvulsants. Patients with a history of neurosurgery were more likely to report no current anti-seizure medication use. Surgery should be considered for patients with medically refractory seizures and unilateral brain involvement (49). Bilaterally affected patients with SWS are generally not considered good surgical candidates, as they are at high risk of poor neurologic and cognitive outcomes. Hemispherectomy should only be considered for bilaterally involved patients with severe disabling seizures coming from one side of the brain; surgery in these patients is considered palliative (50). According to a questionnaire, 81% of 32 SWS patients who had hemispherectomies reported being seizure-free, with 53% being off of anticonvulsants and more recent studies confirm these findings (49, 51).
We recommend low-dose aspirin for almost all young patients under 3 years of age who have 3 or more lobes of brain involvement; it may be recommended to older or less severely involved patients who are having issues with uncontrolled seizures, stroke-like episodes, or focal neurologic deficits. Use of low dose aspirin is growing internationally in patients with SWS, particularly in centers who see larger numbers of these patient; a recent multi-centered study indicated that aspirin use is common (10). However, use of low-dose aspirin is not universal. Anecdotally bleeding can be a concern, patients with SWS rarely can developed a subdural hematoma, presumably due to hemispheric atrophy and tearing of bridging veins. The precise mechanism of low-dose aspirin in SWS remains uncertain and continued research is needed in this area.
Recent biomarker development for SWS:
Magnetic resonance imaging with contrast is used to diagnose patients with leptomeningeal vascular malformation, dilated deep draining vessels, and brain atrophy seen in SWS brain involvement (2). Extent and location of MRI brain involvement has been correlated with intelligence quotient (52). MRI, EEG, and neuropsychology evaluation of 33 young subjects suggested that frontal-lobe abnormality rather than side or hemispheric extent of brain involvement is associated with IQ. Recently, an effort has been made to diagnose infants pre-symptomatically using neuroimaging clinical biomarkers (53). Machine learning approaches have been proposed for biomarker development to predict the age of seizure onset (53). As early diagnosis and treatment may be crucial for delaying seizure onset (54) and earlier seizure onset is associated with lower cognitive quality of life (55), a consensus on how best to make early diagnosis on neuroimaging remains an important research goal.
Since MRI has low sensitivity for SWS brain involvement in young infants, EEG and quantitative EEG can screen for evidence of SWS brain involvement (8). EEG evaluates for excessive spike, sharps, slowing and ictal activity in these patients; qEEG can quantify power on both sides of the brain and screens for significant asymmetry of power (8, 56, 57). This approach to qEEG added about 40% diagnostic information in screening for risk of SWS brain involvement, compared to prediction based on the size and extent of the birthmark alone. qEEG can be helpful in terms of determining the optimal timing of diagnostic MRI and in risk prediction for infants born with a high-risk facial capillary malformation (8).
Research suggests that transcranial doppler (TCD) can also be utilized in evaluation and follow-up of patients with SWS by monitoring flow velocity (decrease) and pulsitility index (increase) (9, 58). As neurological decline in patients with SWS is associated with progressive venous hypertension and impaired perfusion, TCD has studied the correlation of asymmetry of flow velocity in the middle cerebral arteries and both clinical and radiological presentations in SWS patients. The results indicated that the percentage of MCA velocity asymmetry detected in TCD is positively correlated with seizure frequencies and clinical severity of SWS symptoms. In some cases, therapeutic and clinical changes were also associated with changes in TCD asymmetry as well. More research is necessary in the use of TCD in assessments of patients with SWS as TCD is a noninvasive method of screening that could be beneficial for younger patients.
Very few fMRI studies have been done in Sturge-Weber syndrome (59). This is because patients with SWS have very abnormal neural-vascular responses, suggesting that increased or decreased blood flow to a brain region in SWS does not necessarily accurately reflect increases or decreases in neuronal activity. Seizures are associated with decreased blood flow rather than increased blood flow, and neural-plasticity, coupled with abnormal neuro-vascular responses, may result in atypical findings on fMRI that are difficult to interpret (60).
Interictal 2-deoxy-2[18 F]fluoro-D-glucose (FDG)-PET scans, of 27 children with unilateral SWS and age-matched controls, indicated hemispheric differences in metabolic function (61). Verbal IQ was notably higher than non-verbal in the majority of the SWS group, while affected hemisphere FDG SPM(t) scores correlated with IQ in subjects with left-sided, but not right sided, SWS brain involvement. Normal interhemispheric metabolic connectivity was decreased in the SWS group, and unaffected right hemisphere regional FDG SPM(t) scores were negatively correlated with IQ. This study inferred that left-hemispheric lesions in SWS may reshape verbal functions, while negatively impacting non-verbal cognitive abilities.
Outcome measures for SWS treatment trials
Neuropsychological testing:
A study of intellectual and adaptive functioning in children and young adults with SWS found that patients with SWS are at risk of impaired intellectual and adaptive functioning. Patients displayed significantly lower functioning compared to normative data on neuropsychological evaluation: 32% of the sample presented impaired intellectual functioning while 58% displayed impaired adaptive functioning (62).
NIH Toolbox:
The National Institutes of Health toolbox has shown promising results in assessing neuropsychological outcomes in patients with SWS. The NIH Toolbox includes several domains such as cognitive, executive function, attention, episodic memory, language skills, and working memory to allow for assessment of neuropsychological functioning across several domains for patients with SWS. As the NIH Toolbox has a “low test floor,” children with developmental disorders and cognitive and functional impairments can be assessed (11). The NIH Toolbox was recently used in a prospective drug trial to assess the effect of Sirolimus in patients with SWS with cognitive impairments (11). The NIH Toolbox was also recently used in trial of Epidiolex for refractory seizures in SWS (12). Utilizing the National Institutes of Health’s Pediatric Quality of Life in Neurologic Disorders (NeuroQoL), pediatric patients with SWS were reported to have significantly lower t-scores for cognitive function quality of life (55). Earlier seizure onset was associated with lower cognitive quality of life.
SWS Neuroscore:
A composite measure of neurological status (SWS-NRS) was successfully used to measure hemiparesis and seizure frequency to predict functioning level. Hemiparesis status predicted overall adaptive functioning level, while seizure frequency predicted intellectual functioning (62). The SWS Neuroscore has been validated by EEG, quantitative EEG and multiple neuroimaging measures including MRI and transcranial doppler (9, 56, 63); it has also been used in drug trials for Epidiolex and Sirolimus to assess changes in seizure, hemiparesis, visual field cut, and cognitive function (11, 12).
Motor Assessments:
Motor impairments can be quantified and assessed in clinical trials in children with SWS using the Modified Ranking Scale, Pediatric Evaluation of Disability Index, Modified House Functional Classification, and a modified version of the Erhardt Developmental Prehension Assessment. Results indicated that inter-rater reliability was high across all measures in the testing battery, and the study measures demonstrated correlations with the SWS Neuroscore(64). These motor assessment measures were used in a drug trial of Epidiolex for cognitive impairments, which has closed, and the data is currently being analyzed (https://clinicaltrials.gov/ct2/show/NCT04447846).
Recent Studies of Other Neurologic Treatments:
Low-dose aspirin:
Low dose aspirin has shown promising results in reduction of strokes and stroke-like episodes, and in seizure control in patients with SWS. This dual positive effect may reflect prevention of thrombosis in the setting of venous stasis, thereby improving blood-flow to the brain, and reducing the risk of seizures (65). In one series of 6 well-described patients on chronic low-dose aspirin, only one patient had a stroke-like event while none had status epilepticus; all demonstrated improvements in their stroke-like episodes and in seizure control(66). A retrospective study of aspirin use in patients with SWS suggested that most patients taking low-dose aspirin had well controlled epilepsy and surgery was not pursued (65). A questionnaire that collected information on low-dose aspirin use reported a significant reduction in number of stroke-like episodes after aspirin use (67). The mean number of reported stroke-like episodes in 26 patients was reduced from 1.1 to 0.3 per month in the year after starting aspirin therapy; seven of the 26 patients reported cessation of stroke-like episodes, while 2 subjects reported onset of stroke-like episode after starting aspirin therapy. This study also indicated a significant reduction in median number of seizure episodes from 3 episodes to 1 episode a month (67). Further studies are needed, to determine the mechanism of low-dose aspirin effects upon seizures and stroke-like episodes in patients with SWS, as well as studies directly evaluating low-dose aspirin to prevent brain injury in SWS.
Presymptomatic Treatment:
As SWS has a progressive nature, early intervention may be important. Infants who received presymptomatic treatment, with low-dose aspirin and a seizure medication, reported lower seizure scores and older age of seizure onset (54) than case-matched controls matched for age and extent of brain involvement. Four out of eight infants treated presymptomatically with low-dose aspirin and low-dose anti-epileptic drugs had not develop seizures at ages 14 to 39 months. As seizures can lead to cognitive impairment and worse quality of life for patients with SWS, presymptomatic treatment may improve outcome. Patients with prophylactic phenobarbital exhibited a trend for less intellectual disability compared to controls matched for extent of involvement, but matching for age was not done (68). Use of Sirolimus in combination with aspirin to prevent seizures in an infant with SWS has been reported (69). Patients prophylactically treated with valproate or carbamazepine did not show significant difference in epilepsy onset compared to patients who were not treated prophylactically (70). Further prospective studies are needed to determine the optimal presymptomatic approach.
Vitamin D Deficiency Treatment:
Vitamin D deficiency and insufficiency are common in patients with Sturge-Weber patients (71). In fifty-eight patients with SWS, 66% had vitamin D levels below the normal range; this was more common in African American patients and in patients with more severe brain involvement. When these patients were treated, an increase in vitamin D level was associated with an improvement in hemiparesis; patients with early onset of seizures and bilateral brain involvement demonstrated improvements in hemiparesis. Vitamin D deficiency and insufficiency is common in pediatric epilepsy patients taking seizure medications (72, 73). However, as Vitamin D deficiency has also been associated with endothelial dysfunction (74), regulation of inflammatory processes, and nerve health (75), vitamin D has been suggested to be a useful therapeutic method for patients with SWS (71). Further in vivo and in vitro studies are needed to establish causal relationship.
Recent Prospective Drug Trials for SWS brain involvement
Cannabidiol Treatment:
In a recently published study on Epidiolex for refractory seizures in SWS, subjects reported seizure reduction and improved quality of life (12). Subjects also reported subjective improvements in fine and gross motor skills, speech, and cognitive ability. Subjects also showed improvements in weight gain, level of alertness, communication, mood, strength or balance, and mobility. Adverse events included tiredness, behavioral issues, and temporary increase in seizures. CBD may be safe and effective as adjunctive medicine for patients with SWS and treatment resistant seizures. Cannabidiol (CBD), a cannabinoid without psychoactive properties, is a safe and effective treatment option for patients with treatment-resistant epilepsies (76, 77). Having neuroprotective properties, CBD is suggested to block the mammalian target of rapamycin pathway and have antioxidant and anti-inflammatory properties. CBD inhibits Gαq coupled receptors, GPR55 and TRPB1 to increase extracellular adenosine and GABA receptor function and, as a result, decreases seizures (78, 79) Another prospective drug trial of CBD (Epidiolex) for subjects with SWS and cognitive impairments, but controlled seizures, has closed and submitted for publication. Further study is needed.
Treatment with mTOR inhibitor:
Cutaneous vascular malformation samples from SWS patients revealed increased expression of phosphorylated S6, a downstream target of the mTOR pathway (80). In addition, leptomeningeal endothelial cells in the vascular malformation also express p-S6 (81). As the somatic mutation in GNAQ probably increases ERK activation, which in turn can activate mTOR pathways in affected vascular cells, patients with SWS may benefit from treatment with an mTOR inhibitor (5). Sirolimus is a direct inhibitor of mTOR pathway (82), and has been used to treat animal models and patients with complicated vascular malformation disorders (83, 84). It is hypothesized that Sirolimus can reduce the risk of stroke, by normalizing the function of affected vascular cells, including endothelial cells. Furthermore, Sirolimus may benefit seizure control (84).
In a small open-label prospective study targeting cognitive impairments, subjects taking oral Sirolimus showed significant improvement in neuroquality of life assessment subscales measuring anger, cognitive function, and depression (11). Subjects who experienced stroke-like episodes before and during the study showed shorter duration in their events. Subjects showed increased mean processing speed score after being on Sirolimus for 6 months and anecdotally reported improved school reports, reading and vocabulary. Overall, Sirolimus was considered safe and tolerable, and adverse events considered related to Sirolimus were mostly mild. Further research on the use of Sirolimus for SWS patients is needed, especially targeting strokes and stroke-like episodes.
Future prospects for SWS research:
As the previous drug trials on Sirolimus and Epidiolex are limited by small sample sizes, multi-centered drug trials with larger sample size are necessary to fully determine the safety and effectiveness of these and other proposed new treatment approaches. Adding a placebo group in future drug trial designs will also likely be important in establishing new treatment approaches. Continued efforts in understanding of the underlying genetic mutations and pathogenic mechanisms involved in SWS is essential, as the development of clinically-relevant in vitro and in vivo models will likely aid the development of new targeted treatments. Efforts to establish these networks and follow up multi-centered, placebo-controlled trials are underway.
Early diagnosis of SWS brain involvement and prediction of seizure onset and other neurologic outcomes is becoming increasingly urgent. As effective treatment approaches are developed, it is likely that new interventions, possibly able to delay or prevent seizure onset and thus decrease cognitive deterioration, will be considered. Small trials should continue, and when results are promising, multicenter efforts must be implemented to further assess these approaches.
Conclusions:
Sturge-Weber Syndrome is a multi-systems disorder characterized by a leptomeningeal vascular malformation in the brain, port-wine birthmark, and abnormal blood vessels in the eye. Because of the often progressive and potentially devastating nature of the disease, patients are encouraged to continuously follow with a team of specialists based on each patient’s symptoms. While R183Q GNAQ mutation is widely considered to be the SWS gene mutation, recent research suggests that GNA11 and GNB2 (p.Lys78Glu) somatic mutations are also associated with SWS. Further research is needed to improve the understanding the underlying genetic pathology behind SWS, the natural history from longitudinal studies, and both biomarker and outcome development. New advances are reported in care for patients with SWS. Low dose aspirin is utilized by several, but not all centers, and data suggests that it may reduce both stroke-like episodes and seizure symptoms. Drug trials for Sirolimus and cannabidiol have indicated improvements in cognition and seizure frequency respectively. Presymptomatic treatment with low-dose aspirin and low-dose anti-epileptic drugs may delay early seizure onset. Future research focusing on multi-centered drug trials and genetic models will bring significant advances in patient care and our understanding of Sturge-Weber syndrome.
Acknowledgements:
The authors would like to thank all patients and families for their support and involvement in clinical research to further our understanding of Sturge-Weber syndrome.
Funding:
We acknowledge past and current grant funding from the Brain Vascular Malformation Consortium which is funded by the National Institute of Neurological Disorders and Stroke and the National Center for Advancing Translational Sciences, the Celebrate Hope Foundation, and the Faneca 66 Foundation. We also acknowledge past grant funding and study drug received from GW/Jazz Pharmaceuticals, and from Pfizer.
Non-standard Abbreviations and Acronyms
- SWS
Sturge-Weber Syndrome
- PWB
Port-wine birthmark
- SLE
Stroke-like episode
- MRI
Magnetic resonance imaging
- EEG
Electroencephalography
- TCD
Transcranial doppler
- ADC
Apparent diffusion coefficient
- CBD
Cannabidiol
Footnotes
Disclosures:
Dr. Comi is an inventor on a patent for Epidiolex treatment in SWS (20200138738) and a patent for GNAQ mutation in SWS (20200370118). She previously gave compensated talks for GW/Jazz Pharmaceuticals from 2018-2019.
References
- 1.Comi AM. Sturge-Weber syndrome. Handb Clin Neurol. 2015;132:157–68. [DOI] [PubMed] [Google Scholar]
- 2.Comi AM. Presentation, diagnosis, pathophysiology, and treatment of the neurological features of Sturge-Weber syndrome. Neurologist. 2011;17(4):179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piram M, Lorette G, Sirinelli D, Herbreteau D, Giraudeau B, Maruani A. Sturge-Weber syndrome in patients with facial port-wine stain. Pediatr Dermatol. 2012;29(1):32–7. [DOI] [PubMed] [Google Scholar]
- 4.Marana Perez AI, Ruiz-Falco Rojas ML, Puertas Martin V, Dominguez Carral J, Carreras Saez I, Duat Rodriguez A, Sanchez Gonzalez V. Analysis of Sturge-Weber syndrome: A retrospective study of multiple associated variables. Neurologia. 2017;32(6):363–70. [DOI] [PubMed] [Google Scholar]
- 5.Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, North PE, Marchuk DA, Comi AM, Pevsner J. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368(21):1971–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorpe J, Frelin LP, McCann M, Pardo CA, Cohen BA, Comi AM, Pevsner J. Identification of a Mosaic Activating Mutation in GNA11 in Atypical Sturge-Weber Syndrome. J Invest Dermatol. 2021;141(3):685–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fjaer R, Marciniak K, Sundnes O, Hjorthaug H, Sheng Y, Hammarstrom C, Sitek JC, Vigeland MD, Backe PH, Oye AM, et al. A novel somatic mutation in GNB2 provides new insights to the pathogenesis of Sturge-Weber syndrome. Hum Mol Genet. 2021;30(21):1919–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gill RE, Tang B, Smegal L, Adamek JH, McAuliffe D, Lakshmanan BM, Srivastava S, Quain AM, Sebold AJ, Lin DDM, et al. Quantitative EEG improves prediction of Sturge-Weber syndrome in infants with port-wine birthmark. Clin Neurophysiol. 2021;132(10):2440–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Offermann EA, Sreenivasan A, DeJong MR, Lin DDM, McCulloch CE, Chung MG, Comi AM, National Institute of Health S, Rare Disease Clinical Research C, Brain, et al. Reliability and Clinical Correlation of Transcranial Doppler Ultrasound in Sturge-Weber Syndrome. Pediatr Neurol. 2017;74:15–23 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smegal LF, Sebold AJ, Hammill AM, Juhasz C, Lo WD, Miles DK, Wilfong AA, Levin AV, Fisher B, Ball KL, et al. Multicenter Research Data of Epilepsy Management in Patients With Sturge-Weber Syndrome. Pediatr Neurol. 2021;119:3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sebold AJ, Day AM, Ewen J, Adamek J, Byars A, Cohen B, Kossoff EH, Mizuno T, Ryan M, Sievers J, et al. Sirolimus Treatment in Sturge-Weber Syndrome. Pediatr Neurol. 2021;115:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaplan EH, Offermann EA, Sievers JW, Comi AM. Cannabidiol Treatment for Refractory Seizures in Sturge-Weber Syndrome. Pediatr Neurol. 2017;71:18–23 e2. [DOI] [PubMed] [Google Scholar]
- 13.Martins L, Giovani PA, Reboucas PD, Brasil DM, Haiter Neto F, Coletta RD, Machado RA, Puppin-Rontani RM, Nociti FH Jr., Kantovitz KR. Computational analysis for GNAQ mutations: New insights on the molecular etiology of Sturge-Weber syndrome. J Mol Graph Model. 2017;76:429–40. [DOI] [PubMed] [Google Scholar]
- 14.Huang L, Couto JA, Pinto A, Alexandrescu S, Madsen JR, Greene AK, Sahin M, Bischoff J. Somatic GNAQ Mutation is Enriched in Brain Endothelial Cells in Sturge-Weber Syndrome. Pediatr Neurol. 2017;67:59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wellman RJ, Cho SB, Singh P, Tune M, Pardo CA, Comi AM, Workgroup BS-WsP. Galphaq and hyper-phosphorylated ERK expression in Sturge-Weber syndrome leptomeningeal blood vessel endothelial cells. Vasc Med. 2019;24(1):72–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. 1987;16(4):899–906. [DOI] [PubMed] [Google Scholar]
- 17.Ye Z, McQuillan L, Poduri A, Green TE, Matsumoto N, Mefford HC, Scheffer IE, Berkovic SF, Hildebrand MS. Somatic mutation: The hidden genetics of brain malformations and focal epilepsies. Epilepsy Res. 2019;155:106161. [DOI] [PubMed] [Google Scholar]
- 18.Eerola I, Boon LM, Mulliken JB, Burrows PE, Dompmartin A, Watanabe S, Vanwijck R, Vikkula M. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73(6):1240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amyere M, Revencu N, Helaers R, Pairet E, Baselga E, Cordisco M, Chung W, Dubois J, Lacour JP, Martorell L, et al. Germline Loss-of-Function Mutations in EPHB4 Cause a Second Form of Capillary Malformation-Arteriovenous Malformation (CM-AVM2) Deregulating RAS-MAPK Signaling. Circulation. 2017;136(11):1037–48. [DOI] [PubMed] [Google Scholar]
- 20.McDonell LM, Mirzaa GM, Alcantara D, Schwartzentruber J, Carter MT, Lee LJ, Clericuzio CL, Graham JM Jr., Morris-Rosendahl DJ, Polster T, et al. Mutations in STAMBP, encoding a deubiquitinating enzyme, cause microcephaly-capillary malformation syndrome. Nat Genet. 2013;45(5):556–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Venot Q, Blanc T, Rabia SH, Berteloot L, Ladraa S, Duong JP, Blanc E, Johnson SC, Hoguin C, Boccara O, et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature. 2018;558(7711):540–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cubiro X, Rozas-Munoz E, Castel P, Roe Crespo E, Garcia-Melendo C, Puig L, Baselga E. Clinical and genetic evaluation of six children with diffuse capillary malformation and undergrowth. Pediatr Dermatol. 2020;37(5):833–8. [DOI] [PubMed] [Google Scholar]
- 23.Martinez-Glez V, Rodriguez-Laguna L, Viana-Huete V, Garcia Torrijos C, Hurtado B, Lapunzina P, Triana P, Lopez-Gutierrez JC. Segmental undergrowth is associated with pathogenic variants in vascular malformation genes: A retrospective case-series study. Clin Genet. 2022;101(3):296–306. [DOI] [PubMed] [Google Scholar]
- 24.Dymerska M, Kirkorian AY, Offermann EA, Lin DD, Comi AM, Cohen BA. Size of Facial Port-Wine Birthmark May Predict Neurologic Outcome in Sturge-Weber Syndrome. J Pediatr. 2017;188:205–9 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dutkiewicz AS, Ezzedine K, Mazereeuw-Hautier J, Lacour JP, Barbarot S, Vabres P, Miquel J, Balguerie X, Martin L, Boralevi F, et al. A prospective study of risk for Sturge-Weber syndrome in children with upper facial port-wine stain. J Am Acad Dermatol. 2015;72(3):473–80. [DOI] [PubMed] [Google Scholar]
- 26.Comi AM, Fischer R, Kossoff EH. Encephalofacial angiomatosis sparing the occipital lobe and without facial nevus: on the spectrum of Sturge-Weber syndrome variants? J Child Neurol. 2003;18(1):35–8. [DOI] [PubMed] [Google Scholar]
- 27.Sullivan TJ, Clarke MP, Morin JD. The ocular manifestations of the Sturge-Weber syndrome. J Pediatr Ophthalmol Strabismus. 1992;29(6):349–56. [DOI] [PubMed] [Google Scholar]
- 28.Fukuyama Y, Tsuchiya S. A study on Sturge-Weber syndrome. Report of a case associated with infantile spasms and electroencephalographic evolution in five cases. Eur Neurol. 1979;18(3):194–204. [DOI] [PubMed] [Google Scholar]
- 29.Ewen JB, Comi AM, Kossoff EH. Myoclonic-astatic epilepsy in a child with Sturge-Weber syndrome. Pediatr Neurol. 2007;36(2):115–7. [DOI] [PubMed] [Google Scholar]
- 30.Maria BL, Neufeld JA, Rosainz LC, Drane WE, Quisling RG, Ben-David K, Hamed LM. Central nervous system structure and function in Sturge-Weber syndrome: evidence of neurologic and radiologic progression. J Child Neurol. 1998;13(12):606–18. [DOI] [PubMed] [Google Scholar]
- 31.Bebin EM, Gomez MR. Prognosis in Sturge-Weber disease: comparison of unihemispheric and bihemispheric involvement. J Child Neurol. 1988;3(3):181–4. [DOI] [PubMed] [Google Scholar]
- 32.Jacoby CG, Yuh WT, Afifi AK, Bell WE, Schelper RL, Sato Y. Accelerated myelination in early Sturge-Weber syndrome demonstrated by MR imaging. J Comput Assist Tomogr. 1987;11(2):226–31. [DOI] [PubMed] [Google Scholar]
- 33.Marti-Bonmati L, Menor F, Poyatos C, Cortina H. Diagnosis of Sturge-Weber syndrome: comparison of the efficacy of CT and MR imaging in 14 cases. AJR Am J Roentgenol. 1992;158(4):867–71. [DOI] [PubMed] [Google Scholar]
- 34.Hu J, Yu Y, Juhasz C, Kou Z, Xuan Y, Latif Z, Kudo K, Chugani HT, Haacke EM. MR susceptibility weighted imaging (SWI) complements conventional contrast enhanced T1 weighted MRI in characterizing brain abnormalities of Sturge-Weber Syndrome. J Magn Reson Imaging. 2008;28(2):300–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bentson JR, Wilson GH, Newton TH. Cerebral venous drainage pattern of the Sturge-Weber syndrome. Radiology. 1971;101(1):111–8. [DOI] [PubMed] [Google Scholar]
- 36.Juhasz C. Toward a better understanding of stroke-like episodes in Sturge-Weber syndrome. Eur J Paediatr Neurol. 2020;25:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tillmann RP, Ray K, Aylett SE. Transient episodes of hemiparesis in Sturge Weber Syndrome - Causes, incidence and recovery. Eur J Paediatr Neurol. 2020;25:90–6. [DOI] [PubMed] [Google Scholar]
- 38.Aylett SE, Neville BG, Cross JH, Boyd S, Chong WK, Kirkham FJ. Sturge-Weber syndrome: cerebral haemodynamics during seizure activity. Dev Med Child Neurol. 1999;41(7):480–5. [PubMed] [Google Scholar]
- 39.Zolkipli Z, Aylett S, Rankin PM, Neville BG. Transient exacerbation of hemiplegia following minor head trauma in Sturge-Weber syndrome. Dev Med Child Neurol. 2007;49(9):697–9. [DOI] [PubMed] [Google Scholar]
- 40.Comi A. Current Therapeutic Options in Sturge-Weber Syndrome. Semin Pediatr Neurol. 2015;22(4):295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luat AF, Juhasz C, Loeb JA, Chugani HT, Falchek SJ, Jain B, Greene-Roethke C, Amlie-Lefond C, Ball KL, Davis A, et al. Neurological Complications of Sturge-Weber Syndrome: Current Status and Unmet Needs. Pediatr Neurol. 2019;98:31–8. [DOI] [PubMed] [Google Scholar]
- 42.Kossoff EH, Balasta M, Hatfield LM, Lehmann CU, Comi AM. Self-reported treatment patterns in patients with Sturge-Weber syndrome and migraines. J Child Neurol. 2007;22(6):720–6. [DOI] [PubMed] [Google Scholar]
- 43.Gallop F, Fosi T, Prabhakar P, Aylett SE. Flunarizine for Headache Prophylaxis in Children With Sturge-Weber Syndrome. Pediatr Neurol. 2019;93:27–33. [DOI] [PubMed] [Google Scholar]
- 44.Day AM, McCulloch CE, Hammill AM, Juhasz C, Lo WD, Pinto AL, Miles DK, Fisher BJ, Ball KL, Wilfong AA, et al. Physical and Family History Variables Associated With Neurological and Cognitive Development in Sturge-Weber Syndrome. Pediatr Neurol. 2019;96:30–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Powell S, Fosi T, Sloneem J, Hawkins C, Richardson H, Aylett S. Neurological presentations and cognitive outcome in Sturge-Weber syndrome. Eur J Paediatr Neurol. 2021;34:21–32. [DOI] [PubMed] [Google Scholar]
- 46.Gittins S, Steel D, Brunklaus A, Newsom-Davis I, Hawkins C, Aylett SE. Autism spectrum disorder, social communication difficulties, and developmental comorbidities in Sturge-Weber syndrome. Epilepsy Behav. 2018;88:1–4. [DOI] [PubMed] [Google Scholar]
- 47.Sujansky E, Conradi S. Outcome of Sturge-Weber syndrome in 52 adults. Am J Med Genet. 1995;57(1):35–45. [DOI] [PubMed] [Google Scholar]
- 48.Sujansky E, Conradi S. Sturge-Weber syndrome: age of onset of seizures and glaucoma and the prognosis for affected children. J Child Neurol. 1995;10(1):49–58. [DOI] [PubMed] [Google Scholar]
- 49.Arzimanoglou AA, Andermann F, Aicardi J, Sainte-Rose C, Beaulieu MA, Villemure JG, Olivier A, Rasmussen T. Sturge-Weber syndrome: indications and results of surgery in 20 patients. Neurology. 2000;55(10):1472–9. [DOI] [PubMed] [Google Scholar]
- 50.Tuxhorn IE, Pannek HW. Epilepsy surgery in bilateral Sturge-Weber syndrome. Pediatr Neurol. 2002;26(5):394–7. [DOI] [PubMed] [Google Scholar]
- 51.Kossoff EH, Buck C, Freeman JM. Outcomes of 32 hemispherectomies for Sturge-Weber syndrome worldwide. Neurology. 2002;59(11):1735–8. [DOI] [PubMed] [Google Scholar]
- 52.Bosnyak E, Behen ME, Guy WC, Asano E, Chugani HT, Juhasz C. Predictors of Cognitive Functions in Children With Sturge-Weber Syndrome: A Longitudinal Study. Pediatr Neurol. 2016;61:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vedmurthy P, Pinto ALR, Lin DDM, Comi AM, Ou Y, Group B-KSP-sBW. Study protocol: retrospectively mining multisite clinical data to presymptomatically predict seizure onset for individual patients with Sturge-Weber. BMJ Open. 2022;12(2):e053103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Day AM, Hammill AM, Juhasz C, Pinto AL, Roach ES, McCulloch CE, Comi AM, National Institutes of Health Sponsor: Rare Diseases Clinical Research Network B, Vascular Malformation Consortium SWSIG. Hypothesis: Presymptomatic treatment of Sturge-Weber Syndrome With Aspirin and Antiepileptic Drugs May Delay Seizure Onset. Pediatr Neurol. 2019;90:8–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harmon KA, Day AM, Hammill AM, Pinto AL, McCulloch CE, Comi AM, National Institutes of Health Rare Disease Clinical Research Consortium B, Vascular Malformation Consortium SWSIG. Quality of Life in Children With Sturge-Weber Syndrome. Pediatr Neurol. 2019;101:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hatfield LA, Crone NE, Kossoff EH, Ewen JB, Pyzik PL, Lin DD, Kelley TM, Comi AM. Quantitative EEG asymmetry correlates with clinical severity in unilateral Sturge-Weber syndrome. Epilepsia. 2007;48(1):191–5. [DOI] [PubMed] [Google Scholar]
- 57.Ewen JB, Kossoff EH, Crone NE, Lin DD, Lakshmanan BM, Ferenc LM, Comi AM. Use of quantitative EEG in infants with port-wine birthmark to assess for Sturge-Weber brain involvement. Clin Neurophysiol. 2009;120(8):1433–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jimenez-Legido M, Martinez-de-Azagra-Garde A, Bernardino-Cuesta B, Solis-Muniz I, Soto-Insuga V, Cantarin-Extremera V, Garcia-Salido A, Duat-Rodriguez A, Garcia-Penas JJ, Ruiz-Falco-Rojas ML. Utility of the transcranial doppler in the evaluation and follow-up of children with Sturge-Weber Syndrome. Eur J Paediatr Neurol. 2020;27:60–6. [DOI] [PubMed] [Google Scholar]
- 59.Jeong JW, Chugani HT, Behen ME, Guy W, Juhasz C. Quantitative Assessment of Brain Networks in Children With Sturge-Weber Syndrome Using Resting State Functional Magnetic Resonance Imaging (MRI). J Child Neurol. 2013;28(11):1448–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oguz KK, Senturk S, Ozturk A, Anlar B, Topcu M, Cila A. Impact of recent seizures on cerebral blood flow in patients with sturge-weber syndrome: study of 2 cases. J Child Neurol. 2007;22(5):617–20. [DOI] [PubMed] [Google Scholar]
- 61.Kim JA, Jeong JW, Behen ME, Pilli VK, Luat A, Chugani HT, Juhasz C. Metabolic correlates of cognitive function in children with unilateral Sturge-Weber syndrome: Evidence for regional functional reorganization and crowding. Hum Brain Mapp. 2018;39(4):1596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kavanaugh B, Sreenivasan A, Bachur C, Papazoglou A, Comi A, Zabel TA. [Formula: see text]Intellectual and adaptive functioning in Sturge-Weber Syndrome. Child Neuropsychol. 2016;22(6):635–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kelley TM, Hatfield LA, Lin DD, Comi AM. Quantitative analysis of cerebral cortical atrophy and correlation with clinical severity in unilateral Sturge-Weber syndrome. J Child Neurol. 2005;20(11):867–70. [DOI] [PubMed] [Google Scholar]
- 64.Reidy TG, Suskauer SJ, Bachur CD, McCulloch CE, Comi AM. Preliminary reliability and validity of a battery for assessing functional skills in children with Sturge-Weber syndrome. Childs Nerv Syst. 2014;30(12):2027–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lance EI, Sreenivasan AK, Zabel TA, Kossoff EH, Comi AM. Aspirin use in Sturge-Weber syndrome: side effects and clinical outcomes. J Child Neurol. 2013;28(2):213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Udani V, Pujar S, Munot P, Maheshwari S, Mehta N. Natural history and magnetic resonance imaging follow-up in 9 Sturge-Weber Syndrome patients and clinical correlation. J Child Neurol. 2007;22(4):479–83. [DOI] [PubMed] [Google Scholar]
- 67.Bay MJ, Kossoff EH, Lehmann CU, Zabel TA, Comi AM. Survey of aspirin use in Sturge-Weber syndrome. J Child Neurol. 2011;26(6):692–702. [DOI] [PubMed] [Google Scholar]
- 68.Ville D, Enjolras O, Chiron C, Dulac O. Prophylactic antiepileptic treatment in Sturge-Weber disease. Seizure. 2002;11(3):145–50. [DOI] [PubMed] [Google Scholar]
- 69.Triana Junco PE, Sanchez-Carpintero I, Lopez-Gutierrez JC. Preventive treatment with oral sirolimus and aspirin in a newborn with severe Sturge-Weber syndrome. Pediatr Dermatol. 2019;36(4):524–7. [DOI] [PubMed] [Google Scholar]
- 70.Bar C, Pedespan JM, Boccara O, Garcelon N, Levy R, Grevent D, Boddaert N, Nabbout R. Early magnetic resonance imaging to detect presymptomatic leptomeningeal angioma in children with suspected Sturge-Weber syndrome. Dev Med Child Neurol. 2020;62(2):227–33. [DOI] [PubMed] [Google Scholar]
- 71.Smegal L, Lin DD, Cho A, Cho S, Kalb L, Cohen B, Germain-Lee E, Comi A. Vitamin D and Neurological Status in Sturge-Weber Syndrome. Journal of Vascular Anomalies. 2021;2(4):e025. [Google Scholar]
- 72.Dura-Trave T, Gallinas-Victoriano F, Malumbres-Chacon M, Moreno-Gonzalez P, Aguilera-Albesa S, Yoldi-Petri ME. Vitamin D deficiency in children with epilepsy taking valproate and levetiracetam as monotherapy. Epilepsy Res. 2018;139:80–4. [DOI] [PubMed] [Google Scholar]
- 73.Likasitthananon N, Nabangchang C, Simasathien T, Vichutavate S, Phatarakijnirund V, Suwanpakdee P. Hypovitaminosis D and risk factors in pediatric epilepsy children. BMC Pediatr. 2021;21(1):432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tarcin O, Yavuz DG, Ozben B, Telli A, Ogunc AV, Yuksel M, Toprak A, Yazici D, Sancak S, Deyneli O, et al. Effect of vitamin D deficiency and replacement on endothelial function in asymptomatic subjects. J Clin Endocrinol Metab. 2009;94(10):4023–30. [DOI] [PubMed] [Google Scholar]
- 75.Bouillon R, Marcocci C, Carmeliet G, Bikle D, White JH, Dawson-Hughes B, Lips P, Munns CF, Lazaretti-Castro M, Giustina A, et al. Skeletal and Extraskeletal Actions of Vitamin D: Current Evidence and Outstanding Questions. Endocr Rev. 2019;40(4):1109–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Szaflarski JP, Bebin EM, Comi AM, Patel AD, Joshi C, Checketts D, Beal JC, Laux LC, De Boer LM, Wong MH, et al. Long-term safety and treatment effects of cannabidiol in children and adults with treatment-resistant epilepsies: Expanded access program results. Epilepsia. 2018;59(8):1540–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lattanzi S, Brigo F, Trinka E, Zaccara G, Cagnetti C, Del Giovane C, Silvestrini M. Efficacy and Safety of Cannabidiol in Epilepsy: A Systematic Review and Meta-Analysis. Drugs. 2018;78(17):1791–804. [DOI] [PubMed] [Google Scholar]
- 78.Castillo A, Tolon MR, Fernandez-Ruiz J, Romero J, Martinez-Orgado J. The neuroprotective effect of cannabidiol in an in vitro model of newborn hypoxic-ischemic brain damage in mice is mediated by CB(2) and adenosine receptors. Neurobiol Dis. 2010;37(2):434–40. [DOI] [PubMed] [Google Scholar]
- 79.Whyte LS, Ryberg E, Sims NA, Ridge SA, Mackie K, Greasley PJ, Ross RA, Rogers MJ. The putative cannabinoid receptor GPR55 affects osteoclast function in vitro and bone mass in vivo. Proc Natl Acad Sci U S A. 2009;106(38):16511–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shirazi F, Cohen C, Fried L, Arbiser JL. Mammalian target of rapamycin (mTOR) is activated in cutaneous vascular malformations in vivo. Lymphat Res Biol. 2007;5(4):233–6. [DOI] [PubMed] [Google Scholar]
- 81.McCann M, Cho A, Pardo CA, Phung T, Hammill A, Comi A. Phosphorylated-S6 Expression in Sturge-Weber Syndrome Brain Tissue. Journal of Vascular Anomalies. in press. [Google Scholar]
- 82.Van Damme A, Seront E, Dekeuleneer V, Boon LM, Vikkula M. New and Emerging Targeted Therapies for Vascular Malformations. Am J Clin Dermatol. 2020;21(5):657–68. [DOI] [PubMed] [Google Scholar]
- 83.Yin L, Ye S, Chen Z, Zeng Y. Rapamycin preconditioning attenuates transient focal cerebral ischemia/reperfusion injury in mice. Int J Neurosci. 2012;122(12):748–56. [DOI] [PubMed] [Google Scholar]
- 84.Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29(21):6964–72. [DOI] [PMC free article] [PubMed] [Google Scholar]