

Abstract

Lysine dioxygenase (KDO) is an important enzyme in human physiology involved in bioprocesses that trigger collagen cross-linking and blood pressure control. There are several KDOs in nature; however, little is known about the factors that govern the regio- and stereoselectivity of these enzymes. To understand how KDOs can selectively hydroxylate their substrate, we did a comprehensive computational study into the mechanisms and features of 4-lysine dioxygenase. In particular, we selected a snapshot from the MD simulation on KDO5 and created large QM cluster models (A, B, and C) containing 297, 312, and 407 atoms, respectively. The largest model predicts regioselectivity that matches experimental observation with rate-determining hydrogen atom abstraction from the C4–H position, followed by fast OH rebound to form 4-hydroxylysine products. The calculations show that in model C, the dipole moment is positioned along the C4–H bond of the substrate and, therefore, the electrostatic and electric field perturbations of the protein assist the enzyme in creating C4–H hydroxylation selectivity. Furthermore, an active site Tyr233 residue is identified that reacts through proton-coupled electron transfer akin to the axial Trp residue in cytochrome c peroxidase. Thus, upon formation of the iron(IV)-oxo species in the catalytic cycle, the Tyr233 phenol loses a proton to the nearby Asp179 residue, while at the same time, an electron is transferred to the iron to create an iron(III)-oxo active species. This charged tyrosyl residue directs the dipole moment along the C4–H bond of the substrate and guides the selectivity to the C4-hydroxylation of the substrate.
Introduction
Lysine hydroxylases are vital enzymes for human health that trigger collagen cross-linking processes of protein chains but also have been linked to blood pressure control and cardiac development in embryos.1−3 However, high expression of lysine hydroxylases may lead to cancer development.4,5 Several lysine hydroxylases have been characterized in various species, and generally, they are highly stereo- and regioselective and typically hydroxylate a lysine residue at the C3-, C4-, or C5-position.6−9 Thus, some lysine hydroxylases bind a collagen or protein chain on the interface of an enzymatic dimer and hydroxylate individual lysine residues of the chain, while others operate on isolated lysine amino acids.10 As a consequence, these lysine hydroxylases show major differences in substrate activation and product distributions.
One class of lysine hydroxylases is the nonheme iron/α-ketoglutarate (αKG, also called 2-oxoglutarate)-dependent lysine dioxygenases (KDOs). Nonheme iron and αKG-dependent dioxygenases are common enzymes in many biosystems involved in biosynthesis reactions of natural products, including hormones and antibiotics.11−21 They all have a central iron(II) atom that is bound to the protein through interactions with the side chains of two histidine and one carboxylate-based, e.g., Asp or Glu, residue. Indeed, the structure of KDO5 displayed in Figure 1 shows the facial 2-His/1-Glu binding orientation of iron(II), where the metal is covalently bound to His176, Glu178, and His312.22,23 The cosubstrate αKG binds as a bidentate ligand trans to the His176 and Glu178 groups. Substrate Lys is missing from the X-ray crystal structure, but the active site has a number of polar residues that are likely involved in substrate binding and positioning, namely, Asp179, Asp260, and Arg338.
Figure 1.

Active site of KDO5 (PDB ID: 6EUR) and the general reaction catalyzed by the enzyme.
There are various KDO isozymes labeled KDO1–KDO6 that all react with a free lysine substrate at a nonheme iron center. However, KDO1 gives S-3-hydroxylysine as a product, whereas the other isozymes react with a free lysine amino acid on the iron(II) center with αKG and dioxygen to form R-4-hydroxylysine regio- and stereoselectively. To understand this high regio- and stereoselectivity, we performed a detailed computational study of KDO5 enzymes. Similar to other nonheme iron and αKG-dependent dioxygenases, the reaction of dioxygen with αKG on a nonheme iron(II) center initially forms an iron(IV)-oxo species, succinate, and CO2.12−21,24−26 For various nonheme iron dioxygenases, the iron(IV)-oxo species has been experimentally trapped and characterized with spectroscopic methods. In all cases, the iron was found to be in a quintet spin ground state and reacts through hydrogen atom abstraction from the substrate.27−30 Despite the fact that the iron(IV)-oxo species of KDO5 has never been trapped and characterized, biochemical studies have been reported on its activity, substrate scope, and biochemistry.31,32 Moreover, engineered KDOs gave insight into substrate binding and orientation.33
To gain insight into the high selectivity of KDO5 enzymes, we decided to perform a computational study into lysine activation by KDO5 enzymes using the PDB 6EUR X-ray crystal structure as a starting point.22,23 The work shows that key hydrogen bonding and polar residues in the substrate binding pocket direct the local electric field and dipole moment and trigger a proton-coupled electron transfer from an active site Tyr residue to a neighboring Asp residue that guides the reaction to C4-hydroxylation even though it is not a thermodynamically favored pathway.
Methods
Model Setup
The model setup follows previous quantum cluster model calculations from our group and will be briefly summarized here.34,35 We started from the 6EUR PDB file,22,23 which is a substrate-devoid but iron(II)- and α-ketoglutarate (αKG)-bound enzymatic tetramer of KDO. We selected chain A of the protein for our work and removed glycerol molecules from the structure. The missing loop in the protein from residues 230–240 was inserted using a homology match found with the UniProt software package for patching missing loops from the AlphaFold model AF-J3BZS6-F1.36 Next, substrate lysine was docked into the substrate binding pocket using the Autodock Vina software package,37 and the most stable conformation was selected. Thereafter, hydrogen atoms were added in Chimera under pH 7 conditions,38 whereby all Lys and Arg residues of the protein chain were in their protonated states, while all Asp and Glu residues were in their deprotonated state. Histidine residues were manually inspected, and all internal histidine residues were taken in their singly protonated states. The structure was solvated with TIP3P-defined water molecules in a cubic box with a 10 Å radius from the outside of the protein.
Molecular Dynamics Simulations
Parameters of the metal with its first-coordination sphere were obtained from the MCPB program, while the Amber force field was used on protein atoms.39,40 The enzyme structure was equilibrated for 1 ns and heated to 298 K over a period of 1 ns using the ff14SB force field.41 Thereafter, molecular dynamics simulation was performed for 50 ns in Amber without constraints under NVT conditions; see the Supporting Information for details.40 The MD simulation converged the protein to a stable structure, and an analysis of geometries during the MD simulation shows that the system is highly rigid and, particularly, the substrate is bound tightly within the substrate-binding pocket. Geometrically, the last point of the MD simulation shows only minor differences with respect to the original crystal structure coordinates as shown from the overlay of the two structures in Figure S4, Supporting Information.
Cluster Model Setup
After the MD simulation, the last step of the equilibrated structure was used to create several cluster models. These cluster models capture all interactions of the substrate and oxidant in the protein and have been shown to accurately reproduce the experimental structure, spectroscopic parameters, and kinetics as well as product distributions.42−47 The cluster models were created based on the first- and second-coordination spheres of the iron and substrate, and they are shown in Figure 2. We initially did exploratory calculations using cluster model A of 297 atoms and an overall charge of −1. Model A included the iron(IV)-oxo(succinate) group, where we truncated succinate to acetate. The axial histidine residue (His312) was shortened to methylimidazole, while the other protein ligands of the metal were part of the peptide chain His176-Thr177-Glu178-Asp179-Ala180-Phe181-Leu182 with Thr177 truncated to Gly. In addition, part of the substrate binding pocket was included in the model, namely, the chains Asp230-Ala231-Asn232-Tyr233 and Arg338-Met339-Met340 with the residues Ala231 and Met339 truncated to a glycine residue. The Gln144, His152, and Asp260 side chains were also included in the model, as well as four water molecules. We further expanded the model to 312 atoms with the amino acid residue Arg145 as it has been implicated in substrate recognition and may be involved in the assistance of pulling the substrate into the protein structure.22 Model B therefore has an overall neutral charge. Models A and B were calculated in the lowest energy singlet, triplet, quintet, and septet spin states. None of the models use constraints during the geometry optimizations. A comparison of the optimized geometries with crystal structure coordinates showed minor deviations with respect to the starting structures.
Figure 2.
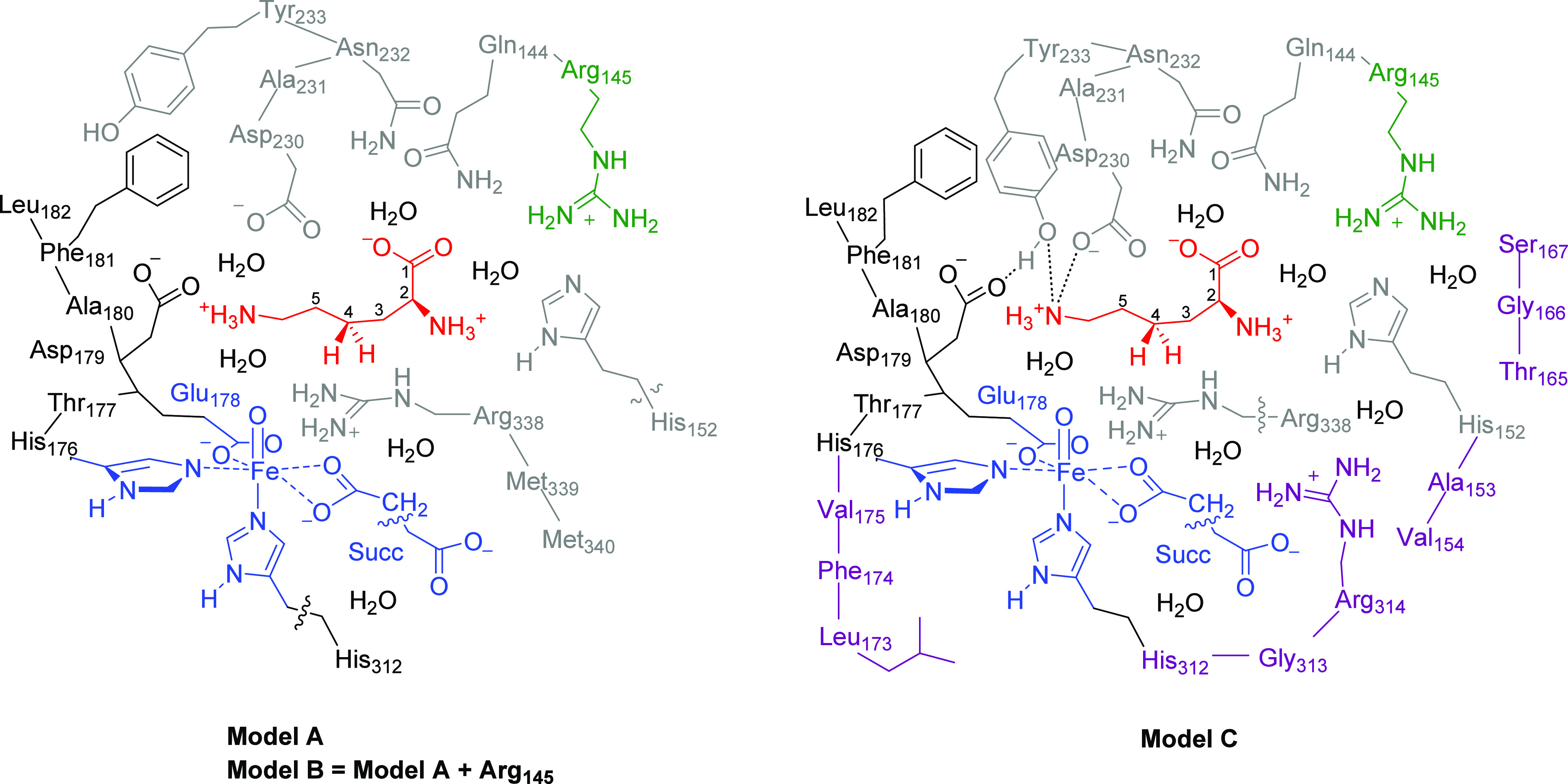
Cluster models A, B, and C of KDO5 as studied in this work. Model C is based on model B with the second-coordination sphere expanded, as highlighted in purple. Wiggly lines identify where bonds were cut and replaced by C–H.
Finally, a larger model was created, namely, model C with 407 atoms, which included the iron(IV)-oxo(succinate) active site with succinate truncated to acetate. The environment was incorporated through the peptide chain from Leu173 to Leu182 and the protein chains Gln144-Arg145, His152-Ala153-Val154, Thr165-Gly166-Ser167, Asp230-Ala231-Asn232-Tyr233, and His312-Gly313-Arg314. The residues Phe174, Val175, Thr177, and Ala231 were truncated to Gly residues. The model also included the side chain of Arg338 and seven water molecules. Model C had an overall charge of +2 and was calculated in the quintet spin state only (see below). A final model C2 was explored, which includes model C expanded with the full succinate group and the guanidinium group of Arg334.
Computational Methods
The Gaussian-09 software package was used for all quantum chemical calculations discussed here.48 Following previous experience with cluster models of nonheme iron dioxygenases,34,49 we utilized the unrestricted B3LYP density functional method in combination with LANL2DZ (with electron core potential) on iron and 6-31G on the rest of the atoms, denoted basis set BS1.50−54 Dispersion corrections were included with the GD3 approach and Becke–Johnson damping factor.55 To correct the energetics, single point calculations with LACV3P+ (with electron core potential) on iron and 6-311+G* on the rest of the atoms (BS2) were performed. Frequency calculations were performed on all local minima and transition states, and it was confirmed that local minima had real frequencies only, while the transition states had a single imaginary mode for the expected vibration along the reaction coordinate. To test the effect of the basis set, we also did geometry optimizations with def2-SVP on all atoms followed by single point calculations with def2-TZVP. These calculations obtained results similar to those with basis sets BS1 and BS2. Test calculations using an implicit solvent model with a dielectric constant mimicking chlorobenzene were performed on key transition state structures for model C, which did not change the ordering of the transition states and predicted the same selectivity; see the Supporting Information.
Results
System Setup and MD Simulations
We started the work with a set of molecular mechanics and molecular dynamics (MD) simulations on the protein structure based on chain A of the X-ray crystal structure of KDO5 solved with bound αKG (PDB: 6EUR). Initially, we docked substrate lysine into the active site; see the Supporting Information for details. The docking pose with the highest score was then selected for further setup and MD simulations. The root-mean-square deviation (RMSD) of the structure converges within a few nanoseconds of MD simulation as shown in Figure 3 and reaches a stable plateau, which applies to the RMSD of the protein, substrate, and ligand components. In general, the structure is highly rigid and little deviation between the crystal structure and the final structure of the MD simulation is seen. The only motion seen during the MD simulation relates to the position of the termini of the protein chain; however, these ends are far from the active site. We then analyzed the substrate positions from each individual snapshot during the MD simulations and measured distances between the substrate and active site amino acids, as defined in Tables S2 and S3 (Supporting Information). Thus, during the full MD simulation, the carboxylic acid group of Asp179 is within 2 Å of the substrate ammonium terminus that holds the substrate in a tight orientation and position (Figure 3b). This is common in amino acid hydroxylases and seen before in, e.g., viomycin biosynthesis enzyme VioC.22,23,56 Another close amino acid residue from the substrate is the side chain of Arg338 that is held within 2–3 Å of the substrate and forms a strong salt bridge interaction with its carboxylate group. Although the Asn232 side chain points into the active site, its distance to the substrate is further than 3 Å, and therefore, it may not be an important residue for substrate positioning and catalysis.
Figure 3.

(a) RMSD plot of the MD simulation on a substrate-bound KDO5 structure. (b) Distance plots of key active site amino acid residues with respect to the substrate. (c) Substrate–oxidant C3–O, C4–O, and C5–O distances during the MD simulation.
As MD trajectories sometimes see major changes in the position of the substrate,47,57−60 we analyzed the substrate position with respect to the iron(IV)-oxo species during the MD simulation, and we display the C3–O, C4–O, and C5–O interactions in Figure 3c. After about 40 ns, the substrate–oxidant interaction stabilizes with the C4 atom closest to the iron(IV)-oxo species. In the MD run between 100 and 130 ns, however, the C4 and C5 atoms of the substrate are at similar distances. In the final stages of the MD simulation, i.e., after 130 ns, the substrate falls into a position that is closest to the iron(IV)-oxo species with the C4 atom of the substrate nearest to the oxidant. In those structures, the system is clearly set up for C4–H activation as the C3 and C5 atoms are much further away.
DFT Cluster Calculations on the Reactants
Based on the last snapshot of the MD simulation, we created three QM cluster models that describe the metal and substrate and their first- and second-coordination spheres, namely, model A (297 atoms, total charge of −1), model B (312 atoms, total charge of 0), and model C (407 atoms, total charge of +2). DFT-optimized geometries of the iron(IV)-oxo species of KDO for models A, B, and C are shown in Figure 4. The quintet spin state is the ground state for all models, whereby for models A and B, the system has four unpaired electrons in metal-based molecular orbitals and configuration π*xy1 π*xz1 π*yz1 σ*x2–y21. These orbitals represent interactions of the atomic 3d orbitals on the iron with first-coordination sphere atoms. In particular, the antibonding interactions of the atomic 3dxy, 3dxz, and 3dyz orbitals with 2p orbitals on the oxo group yield the molecular π*xy, π*xz, and π*yz orbitals. In addition, there is a singly occupied orbital (σ*x2–y2) for the σ-type antibonding interaction in the equatorial plane with the side chains of His176, Glu178, and succinate. The σ*z2 orbital is virtual and located along the Nax–Fe–O axis with Nax being the axial nitrogen atom from His312. The orbital occupation and spin ground state observed for KDO5 models A and B match experimental work on analogous nonheme iron dioxygenases27−30 but also reproduce previous computational studies on other nonheme iron systems that gave an iron(IV)-oxo species with the quintet spin ground state.61−83 The alternative singlet and triplet spin states were also calculated but found to be higher in energy for model C by 22.3 and 5.3 kcal mol–1, respectively. These large spin state energy gaps imply that KDO will react on a dominant quintet spin state surface through single-state reactivity.
Figure 4.
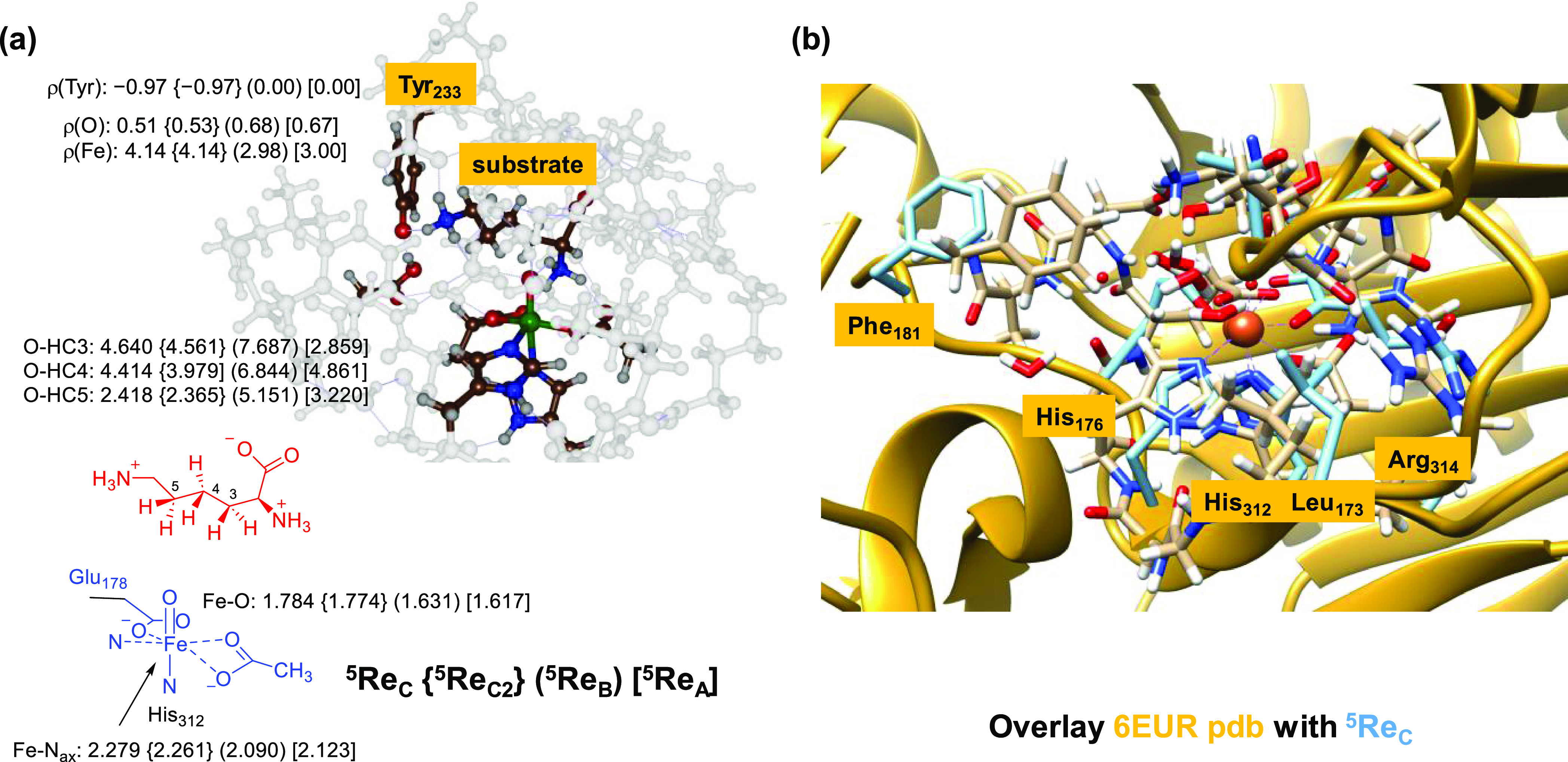
(a) UB3LYP-D3/BS1-optimized geometries of the iron(IV)-oxo species of KDO5 for models A, B, and C with bond lengths in Å. (b) Overlay of the 5ReC structure (blue) with the crystal structure coordinates of the 6EUR PDB (gold).
Interestingly, in model C, electron transfer has taken place from an active site Tyr residue (Tyr233) to the iron to create an iron(III)-oxo species and tyrosyl radical. Thus, Tyr233 has donated a proton to the Asp179 carboxylic acid group, and at the same time, an electron is transferred from the Tyr side chain to iron. As a consequence, the metal has five unpaired electrons, and there is considerable spin density found on Tyr233, where a down-spin electron is located with ρ = −0.97, while the Tyr residue shows no unpaired spin density in the iron(IV)-oxo complexes in models A and B. As such, in model C, the metal is in the oxidation state iron(III) with orbital occupation π*xy1 π*xz1 π*yz1 σ*x2–y21 σ*z21. The reactant complex 5ReC therefore can be described as [FeIII(O)---Tyr•]. We tried to swap molecular orbitals for 5ReC and create an iron(IV)-oxo with a neutral Tyr residue; however, during geometry optimization, the proton moved back to the carboxylate of Asp179 and the electron transferred to iron. The electronic configuration was also obtained with alternative computational approaches and protocols. Furthermore, expanding model C with the complete succinate and the Arg334 side chain (model C2) did not change the structure dramatically and resulted in a similar electronic configuration with a radical on Tyr233, a proton on Asp179, and an iron(III)-oxo group; see Figure 4. Therefore, in our large KDO5 model, the iron(IV)-oxo species is a fleeting intermediate that rapidly converged to an iron(III)-oxo species due to deprotonation of Tyr233 by the Asp179 residue and electron transfer. Interestingly, the Tyr-Asp couple does not seem conserved within the class of lysine dioxygenases;84 hence, these enzymes may operate through a different mechanism. Unfortunately, there are no reports of the iron-oxo species in KDO5 being trapped and characterized by electron paramagnetic resonance (EPR) and/or Mössbauer spectroscopy. However, a ground state for [FeIII(O)---Tyr•] should give it a distinct EPR and Mössbauer spectrum that is different from an [FeIV(O)---Tyr] configuration.
In the heme enzyme cytochrome c peroxidase (CcP), similar valence tautomerism occurs and the active species is an iron(IV)-oxo(heme) with a nearby Trp radical, i.e., [FeIV(O)(heme)---Trp•+].85 DFT calculations showed this state to be close in energy to an electronic state with an iron(IV)-oxo heme cation radical and closed-shell Trp group, i.e., [FeIV(O)(heme•+)---Trp], whereby external perturbations, e.g., a cation binding site, near the heme determined its ground state.86,87 In addition, work on biomimetic model complexes has shown that the second-coordination sphere can change the electronic configuration of a metal complex due to long-range electrostatic interactions through valence tautomerism.88−93 As such, valence tautomerism should also be possible in enzymatic systems despite the fact that there is no experimental evidence in the literature of iron(III)-oxo species. Several biomimetic iron(III)-oxo complexes have been characterized in recent years and show differences in reactivity to iron(IV)-oxo complexes.94−98
The four optimized iron(IV)-oxo species have a very similar first-coordination sphere of the metal with its direct ligands particularly in the equatorial plane. The axial nitrogen atom of His312 (Nax) is at a distance of 2.279 Å from the iron atom in model C, while it is at 2.090 and 2.123 Å for models B and A, respectively. This is not surprising as 5ReC has the σ*z2 orbital occupied with one electron that causes antibonding interactions along the O–Fe–Nax axis. As a result, also the Fe–O interaction is elongated to 1.784 Å for model C, while it is 1.631 Å for model B and 1.617 Å for model A. Therefore, the different models give a first-coordination sphere geometry of the active site with bond lengths that match the orbital occupation and configuration.
The lysine substrate is bound in the substrate binding pocket and held by a salt bridge of its carboxylate group with the side chain of Arg145, and it forms hydrogen bonding interactions with the hydroxyl groups of Thr165 and Ser167. The terminal ammonium group of the substrate forms hydrogen bonding interactions with the carboxylic acid group of Asp230 and the phenol group of Tyr233 and two water molecules. A further hydrogen bonding network in the substrate and oxidant bonding pocket includes the side chains of Asp179, Asn232, and Arg338. As such, the substrate is tightly bound in the substrate binding pocket, which creates a highly rigid structure. These DFT calculations therefore support the results from the MD simulations that the substrate is tightly bound and highly rigid within the substrate binding pocket.
An overlay of the 5ReC-optimized geometry with the X-ray crystal structure coordinates of the 6EUR PDB file is shown on the right-hand side of Figure 4, while the overlay with the last snapshot from the MD simulation is shown in Figure S4 (Supporting Information). The match between the two structures is very good, and most protein backbone groups are in the same position in both systems. Several residues on the edge of the cluster model are highlighted in Figure 4 and are close to their starting position obtained from the crystal structure coordinates. The overlay of the two structures therefore confirms that the protein structure is tight and rigid and shows little flexibility as a function of time. Moreover, our models reflect the enzyme structure well and are expected to give an accurate reflection of the real system.
Hydrogen Atom Abstraction from Lys
Next, we calculated the hydrogen atom abstraction pathways of substrate Lys on the C3–H, C4–H, and C5–H positions as described in Scheme S1 (Supporting Information). In previous work, we explored pro-R and pro-S hydrogen atom abstraction of taurine in taurine/αKG dioxygenase.45 As those calculations gave close energy barriers for pro-R and pro-S pathways, we considered one stereoisomer only here. In particular, we investigated hydrogen atom abstraction via transition state TS1 to form radical intermediate IM1. Thereafter, an OH rebound barrier via the transition state TS2 leads to alcohol products (Pr). The labeling system gives the spin multiplicity in superscript before the label and the position of the hydrogen on the substrate and the cluster model in subscript after the label. In all cases, hydrogen atom abstraction is rate-determining and the OH rebound has a much lower barrier. Table 1 summarizes the calculated barrier heights for hydrogen atom abstraction from the C3–H, C4–H, and C5–H positions of the Lys substrate by the iron-oxo species of KDO for models A, B, and C.
Table 1. Hydrogen Atom Abstraction Transition States Calculated for C3–H, C4–H, and C5–H Abstraction from Lysine by KDO5 Iron(IV)-oxo Speciesa.
| model A |
model B |
model C |
||||
|---|---|---|---|---|---|---|
| structure | ΔE + ZPE | ΔG | ΔE + ZPE | ΔG | ΔE + ZPE | ΔG |
| 5TS1HA,C3 | 17.3 | 14.6 | 24.6 | 27.8 | 26.7 | 28.6 |
| 5TS1HA,C4 | 17.0 | 18.5 | 24.2 | 28.2 | 24.7 | 24.8 |
| 5TS1HA,C5 | 14.4 | 13.1 | 13.5 | 18.6 | 26.7 | 27.0 |
Energies are calculated at the BS2 level of theory, while ZPE, thermal, and entropic corrections are obtained at the BS1 level of theory.
In models A and B, the lowest free energy of activation for the hydrogen atom abstraction barrier is via 5TS1HA,C5 with magnitudes of ΔG‡ = 13.1 kcal mol–1 (model A) and ΔG‡ = 18.6 kcal mol–1 (model B). In model B, the other hydrogen atom abstraction barriers have a free energy that is >9 kcal mol–1 higher. Consequently, model B predicts selective hydrogen atom abstraction from the C5–H position, and little or no alternative products are predicted for this model. This is in contrast to experimental data on this KDO structure that measured dominant C4–H hydroxylation products.6,32
In model A, the margins between the hydrogen atom abstraction barriers are much smaller and the C3–H hydrogen atom abstraction is only ΔG = 1.5 kcal mol–1 above the one for C5–H, and therefore, some C3–H may be expected, although the C4–H barrier is still ΔG = 5.4 kcal mol–1 higher in free energy than the C5–H hydrogen atom abstraction barrier. Nevertheless, both models A and B predict little or no C4-hydroxylation products, which disagrees with experiment for this particular isozyme.6−9 These models give the dominant C5-hydroxylation of Lys rather than the expected C4-hydroxylation. Models A and B differ from each other through the additional Arg residue in model B. The large rise in barrier heights in model B therefore is the result of the inclusion of this residue, which fixes the substrate in a tighter orientation that makes hydrogen atom abstraction from some positions more difficult. In particular, Arg145 strongly destabilizes the pathway for C3–H hydrogen atom abstraction. As both models A and B predict the wrong products, these models do not accurately mimic KDO activity. Therefore, we explored further expanded model C, which is an even larger cluster model with an additional Arg residue in the second coordination sphere and several chains surrounding the substrate binding pocket.
In model C, we find the lowest energy transition state via 5TS1HA,C4 with a free energy of activation of ΔG‡ = 24.8 kcal mol–1, while the barriers for C3–H and C5–H activation are 3.8 and 2.2 kcal mol–1 higher lying, respectively. Consequently, model C predicts dominant C4–H hydrogen atom abstraction, in agreement with experimental product distributions. Therefore, we will focus the rest of the paper on the model C results. Model C includes one additional Arg residue, namely, Arg314, and its electrostatic perturbations appear to destabilize the C5–H hydrogen atom abstraction pathway. As such, the two Arg residues (Arg145 and Arg314) are positioned so that they disfavor the hydrogen atom abstraction pathways from the C3–H and C5–H positions and enable efficient hydrogen atom abstraction from the C4–H position. These perturbations, however, do raise the barrier heights for all processes significantly but will succeed in a regioselective reaction pathway.
The full energy landscape for Lys hydroxylation at the C4–H and C5–H positions as calculated for model C is given in Figure 5. After the hydrogen atom abstraction, the structures relax to a radical intermediate; however, there is a small barrier for OH rebound, leading to alcohol products with large exothermicity. Consequently, we expect the radical intermediate to have a very short lifetime. The reactant of model C has an electronic configuration of π*xy1 π*xz1 π*yz1 σ*x2–y21 σ*z21, i.e., [FeIII(O)---Tyr•]. During the hydrogen atom abstraction step of the Lys hydroxylation mechanism, the Tyr group is reprotonated again and accepts an electron from substrate Lys so that we form an iron(III)-hydroxo group with a Lys radical. In all radical intermediates (5IM1C3, 5IM1C4, and 5IM1C5) for model C, there is a down-spin electron on the substrate and five up-spin unpaired electrons in metal-type orbitals. After the radical intermediates, facile OH rebound (with a negligible barrier) leads to alcohol product complexes with large exergonicity. Therefore, the radical intermediates will be short-lived and quickly convert into alcohol product complexes in an irreversible step.
Figure 5.
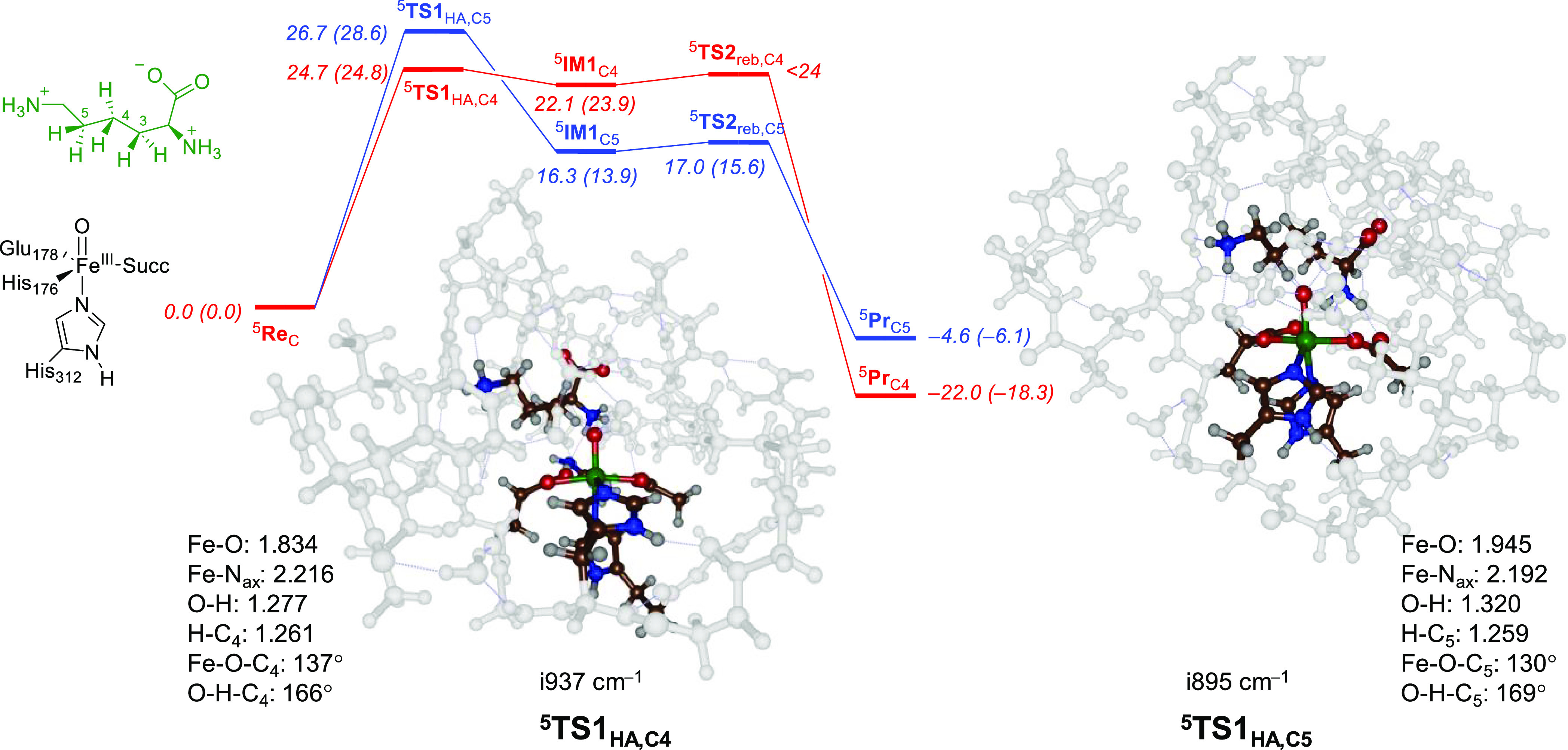
UB3LYP/BS1//UB3LYP/BS2-calculated free energy profile for Lys hydroxylation at the C4–H (red) and C5–H (blue) positions in KDO5. Energies are in kcal mol–1 with ΔE + ZPE (outside parentheses) and ΔG (in parentheses). Optimized transition state geometries give bond lengths in Å and angles in degrees.
Optimized geometries of 5TS1HA,C4 and 5TS1HA,C5 for model C are shown in Figure 5. In both structures, the Fe–O bond is long, probably due to occupation of the σ*z2 orbital with one electron. The 5TS1HA,C4 structure has the transferring hydrogen atom almost midway between the donor and the acceptor atom, i.e., the O–H distance is 1.277 Å and the C4–H distance is 1.261 Å. This is similar in 5TS1HA,C5, where the transferring hydrogen atom is at an O–H distance of 1.320 Å, while the C5–H distance is at 1.259 Å. Therefore, the C5–H hydrogen atom abstraction structure is reactant-like, whereas the C4–H structure is neither reactant- nor product-like but intermediate. Overall, the structural differences between the two transition states are small and similar angles and first-coordination sphere metal–ligand interactions are found. Moreover, optimized hydrogen atom abstraction transition state structures match previous calculations on nonheme iron enzymes and biomimetic models well.58−83,99−110 In both transition state structures, the angle between the substrate and the oxidant is similar. Thus, the Fe–O–C angle of the carbon atom that loses a hydrogen atom is 137° for 5TS1HA,C4, while it is 130° for 5TS1HA,C5. Also, the O–H–C angles are similar at 166° and 169°, respectively. Therefore, the mechanistic and energetic differences observed here are not the result of structural differences in the transition states.
Discussion
In this work, the regio- and chemoselective hydroxylation of a free Lys residue by KDO enzymes is explored. Three models were investigated, but only the largest model (model C) shows a preference for C4-hydroxylation products, while models A and B predict C5-hydroxylation. To understand the differences between the cluster models and, particularly, the hydrogen atom abstraction energetics, we analyzed the structure and electronic configuration of all species in detail.
Figure 6 shows the electron transfer pathways calculated for the hydrogen atom abstraction mechanisms for model C of KDO. As reported above, instead of a reactant that is iron(IV)-oxo with a quintet spin ground state as is commonly observed for αKG-dependent nonheme iron dioxygenases,27,45,61−77,111,112 we find an iron(III)-oxo reactant species. This is the result of proton-coupled electron transfer from Tyr233 that has released a proton to the carboxylate group of Asp179 and an electron donated into the iron set of orbitals. Thus, the carboxylate group of Asp179 is within the hydrogen bonding distance (1.596 Å in 5ReC) from the phenol group of Tyr233, while on the other side of the Tyr residue, the terminal ammonia group of Lys is at a short distance in the other direction (1.95 Å). The terminal ammonia group of substrate Lys via a bridging water molecule connects to the oxo group of the iron(III)-oxo species through a hydrogen bonding network. We therefore anticipate that the mutation of Asp179 by, e.g., Asn179 will prevent the proton-coupled electron transfer from happening and presumably affects product distributions dramatically.
Figure 6.
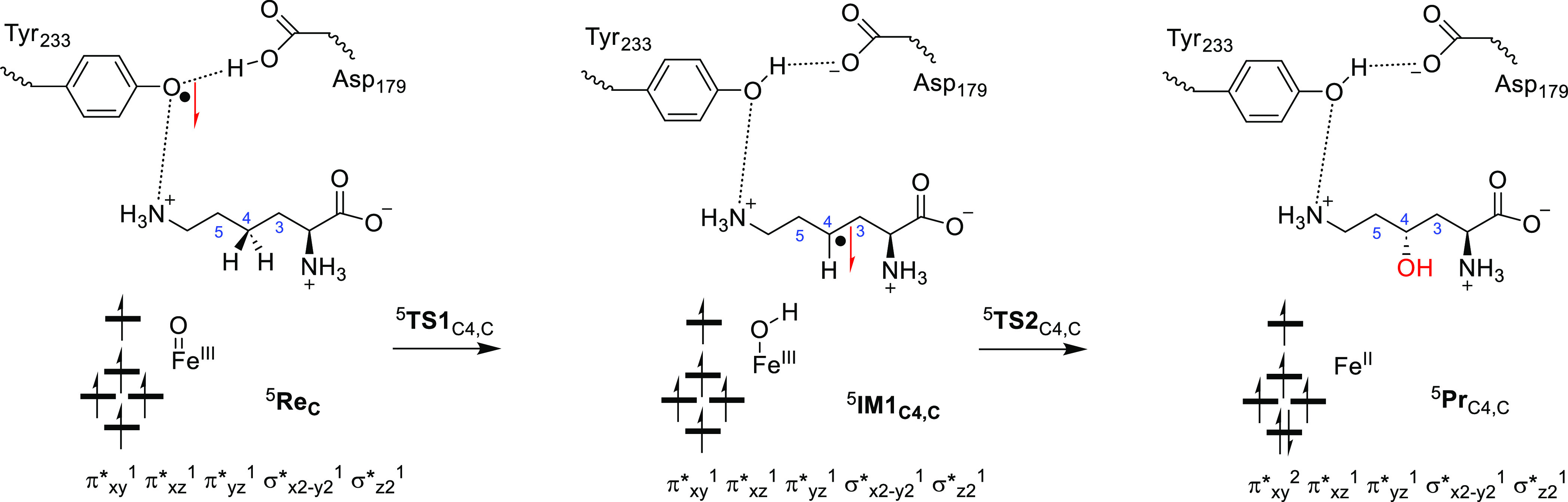
Electronic configuration of the intermediates along the hydroxylation mechanism for model C.
The reaction mechanism then continues with another proton-coupled electron transfer, whereby the iron(III)-oxo species abstracts the proton from the C4-position. At the same time, an electron is transferred from substrate Lys to the tyrosyl radical of Tyr233, which results in proton transfer from Asp179 to Tyr233. In the radical intermediate, therefore, the metal stays in the iron(III) oxidation state, but the radical disappears from the Tyr233 residue and accumulates on the substrate. Indeed, the group spin densities on iron are 4.1 in 5ReC and 4.2 in 5IM1C. On the other hand, a spin of −0.97 is found on the protein (mostly on Tyr233) in 5ReC, while no spin on the substrate is observed. In the radical intermediate, the spin on the substrate is −0.95, while the spin on Tyr has disappeared. Recent combined experimental and computational studies on a biomimetic iron(III)-oxo species, as compared to its iron(IV)-oxo analog, showed that both oxidation states react with toluene with similar hydrogen atom abstraction barriers.75 However, iron(III)-oxo by itself can only react as a one-electron oxidant and is not able to perform OH rebound reactions. In the KDO system discussed here, iron(III)-oxo has a nearby protein radical (on Tyr233) that can be used as the second oxidation equivalent during the reaction mechanism. Indeed, the OH rebound step transfers the final electron to the metal to form the iron(II)-alcohol complex to complete the catalytic cycle. It would be interesting to investigate what effect site-specific mutations of the Asp179 and Tyr233 groups will have on the general catalysis by KDO5 enzymes. We predict that mutation of the residues Asp179 and Phe233 will significantly affect the product distributions and regioselectivity of lysine dioxygenase as it will influence the dipole moment and local electric field effect inside the active site.
The active site structure and overall protein fold of KDO5 are not dramatically different from other nonheme iron dioxygenases and hence do not explain the PCET observation. In particular, a search for analogous enzyme structures shows that the closest enzyme structures of KDO5 are the structures of asparagine hydroxylase (AH, 25.9% homology) and taurine/α-ketoglutarate dioxygenase (TauD, 87.1% homology). Both AH and TauD are α-ketoglutarate-dependent nonheme iron dioxygenases that bind an iron(II) to a 2-His-1-carboxylate orientation of the protein. As such, all three enzymes utilize molecular oxygen and convert α-ketoglutarate to succinate concomitant to the C–H hydroxylation of a substrate. Figure 7 shows the comparison of the structures of KDO5 and asparagine dioxygenase as taken from the 6EUR and 2OG7 PDB files.22,23,113 The overlay of the two PDB files is shown in the top right corner of Figure 7 and shows the same structural features and protein fold for the two enzymes with loops, helices, and chains in similar position and orientation. The only difference appears to be an additional loop on the edge of the protein. When we zoom in into the active site region, the strong similarities between the two proteins become even more apparent (left-hand side of Figure 7). Two active site chains are highlighted in Figure 7, namely, part of the chain that includes the equatorial ligands and the part surrounding the axial ligand of the iron(II) ion. Thus, both proteins have the same 2-His/1-Glu coordination environment that holds the metal in position. They also both bind αKG in roughly the same position with the α-keto and carboxylate groups in the same plane as the equatorial ligands. The two protein structures have a protein loop surrounding the αKG cosubstrate that position it in the active site through interactions with the side chains of Leu and Arg residues: Leu152 and Arg289 in asparagine hydroxylase and Leu173 and Arg314 in KDO5. Therefore, the αKG binding loop is conserved among αKG-dependent nonheme iron dioxygenases as reported before.114 The only major difference between the two protein structures is the position of a carboxylate-containing residue (Asp and Glu) in the substrate binding pocket. Thus, in the position of Glu125 in the substrate binding pocket of asparagine dioxygenase, there is a Gln144 residue in KDO5, while in KDO5, there is a carboxylate in position 158 (Asp158), whereas asparagine dioxygenase has an Asn amino acid in that position. The difference in the position of an anionic amino acid in the substrate binding pocket will lead to differences in the charge and local electric field orientation in the protein. Previously, a similar difference between the VioC and NapI enzymes was identified as a cause to the dipole moment change and to explain the differences in reactivity of the two enzymes, whereby VioC acts as a C3-hydroxylase of free Arg substrate, while NapI reacts with l-Arg through desaturation of the C4–C5 bond.115
Figure 7.
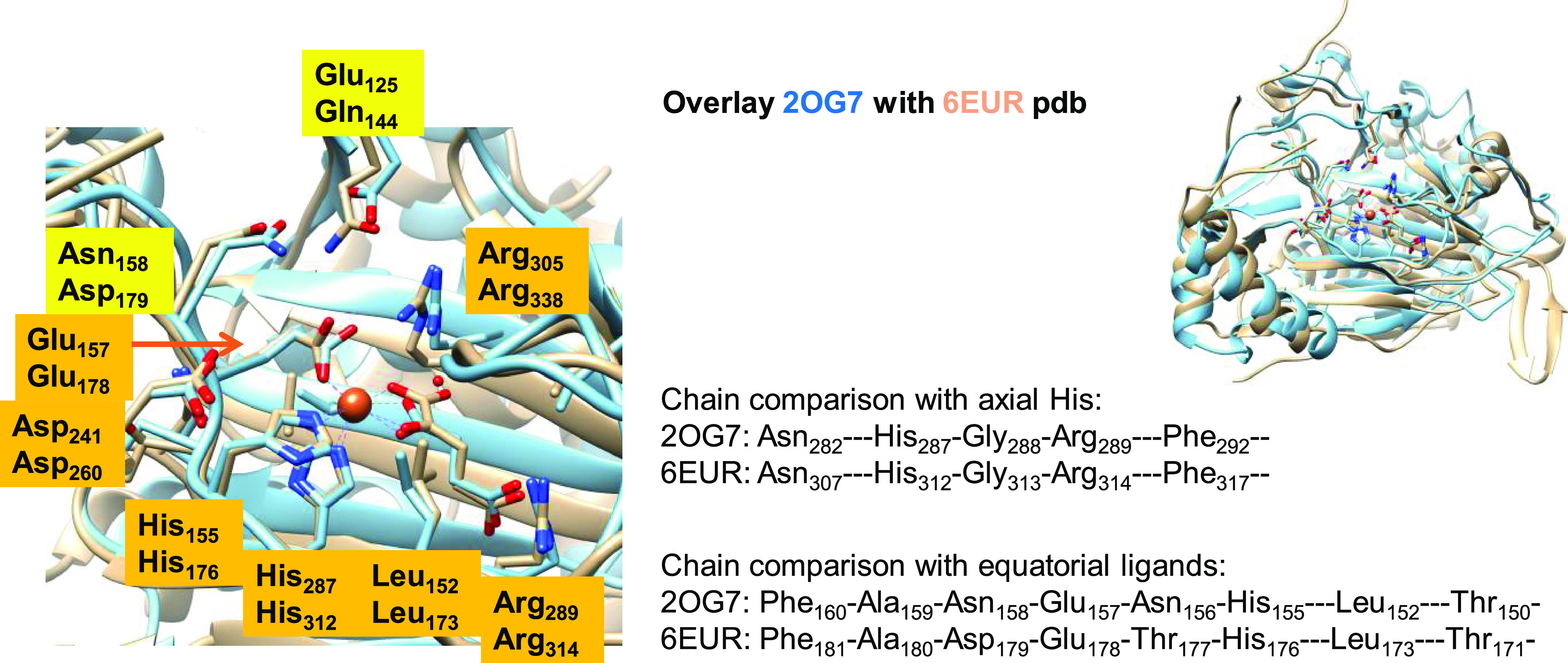
Structure comparison of KDO5 (6EUR PDB file) with asparagine hydroxylase (2OG7 PDB).
Next, we explored the electrostatics of the second coordination sphere of the model and the influence of the electric field effects. The overall dipole moment of structure 5ReC is shown in Figure 8, while those in models A and B are shown in Figure S8 (Supporting Information). The dipole moment of model C in the 5ReC state points perpendicular to the chain of atoms of the substrate along the C4–H bond. In recent work on the viomycin biosynthesis enzyme VioC, we showed that substrate arginine has its C3–H bond aligned with the dipole moment in the active site.116 This results in the weakening of the C3–H bond and strengthening of the C4–H bond of the substrate. In VioC, the dipole moment therefore guides the reaction selectivity and triggers a lesser thermodynamic favorable pathway. By contrast, in the naphthyridinomycin biosynthesis enzyme NapI, the dipole is along the substrate backbone and appear not to influence the C–H bond strengths in the substrate.117 As a consequence, in NapI, the weakest C–H bond in the gas phase is activated by the protein, whereas in VioC, one of the stronger bonds is activated instead. The dipole moment in model C is also perpendicular to the aromatic ring of Tyr233 and hence is likely to stabilize a radical center on this group and trigger an electron transfer from Tyr233 to iron(IV)-oxo and a simultaneous proton transfer from Tyr233 to Asp179. In model A, on the other hand, the dipole vector is perpendicular to the one in 5ReC and therefore keeps the nonheme center in an iron(IV)-oxo state that reacts differently with the substrate. In particular, in 5ReA, the C5–H bond is weakened, and abstraction of this hydrogen atom is preferential.
Figure 8.
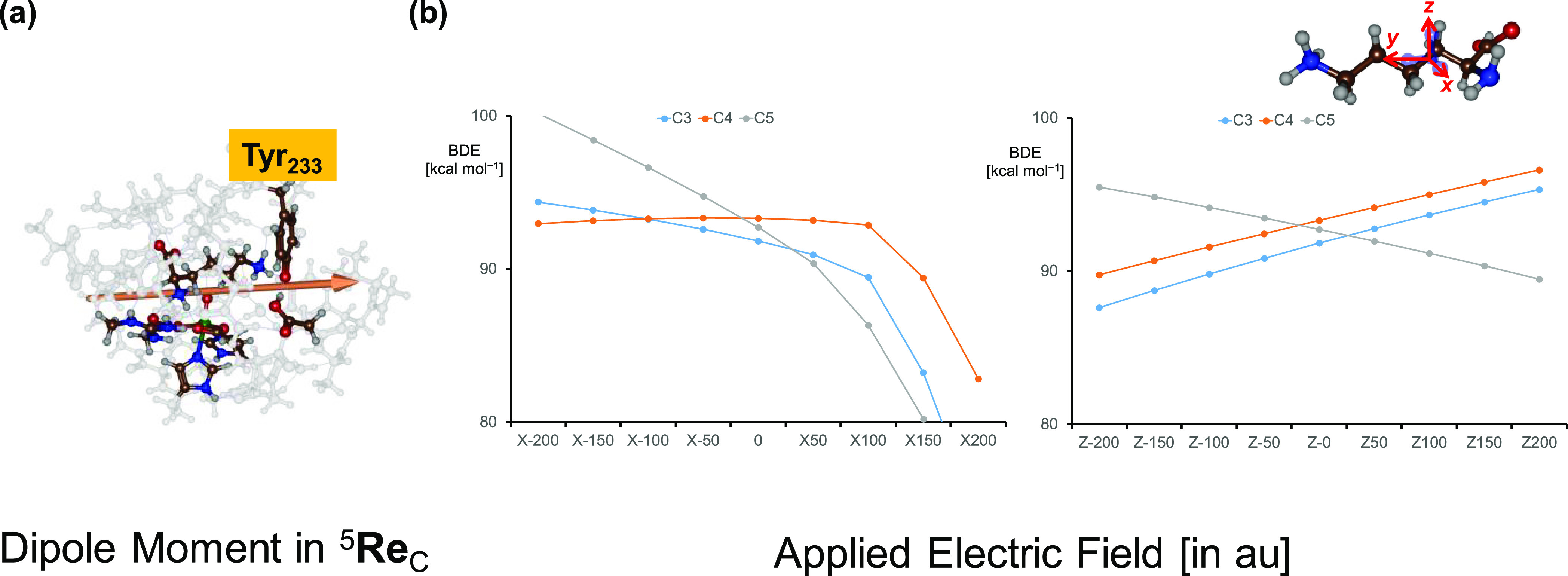
(a) Electric dipole moment in 5ReC. (b) Electric field effects along the x- or z-direction on the C–H BDEs of an isolated lysine substrate molecule with ΔG values (in kcal mol–1) as a function of the applied electric field.
To understand how the substrate C–H bond strengths are influenced by the environment and external perturbations, we calculated the C–H bond strengths of an isolated lysine molecule versus those of a hydrogen atom and lysine with one hydrogen atom removed. In the gas phase, the C3–H, C4–H, and C5–H bond dissociation free energies (BDEs) are ΔG = 91.8, 93.3, and 92.7 kcal mol–1, respectively. As such, in the gas-phase for an isolated Lys amino acid, the weakest C–H bond is the C3–H bond, and hence, hydrogen atom abstraction from the C3–H position should give the most exergonic reaction step. Based on the Bell–Evans–Polanyi principle, the most abundant product should therefore originate from C3–H hydrogen atom abstraction. Often, enzymes overcome the Bell–Evans–Polanyi rules by substrate and oxidant positioning and external perturbations in the substrate binding pocket and lead to products that are not expected from the thermochemistry of the reactions. This is sometimes termed negative catalysis.118 To overcome the large exergonicity of hydrogen atom abstraction from the C3–H position, external perturbations and substrate binding orientations may block this reaction channel.
To investigate the effect of external perturbations on lysine C–H bond strengths, we recalculated the C3–H, C4–H, and C5–H BDEs in the presence of an applied electric field along either the molecular x-, y-, or z-axis of the substrate. Previous calculations with electric field effects applied showed these perturbations to affect electronic properties and charge distributions and even lead to selectivity changes.88,119−127 The definitions of the x-, y-, and z-axes in the substrate are shown in Figure 8 with the y-axis along the carbon chain and the x- and z-axes along the C–H bonds. Figure 8b shows the BDE patterns as a function of the applied electric field along the x- and z-axes, which are both perpendicular to the carbon chain of the substrate. In the gas phase, i.e., with zero electric field, the C5–H position has the weakest bond followed by the C3–H and C4–H positions. A field along the positive x- or z-direction keeps these orderings, although the energy separations change. With a negative direction of the electric field (based on the definition in Gaussian) along the x-axis, however, C4–H becomes the weakest bond and C5–H the strongest bond. Under a local electric field effect in the protein, therefore, C4–H hydrogen atom abstraction can be favored over the C3–H and C5–H channels. On the other hand, a field along the z-axis in the negative electric field direction makes the C3–H bond of the substrate the weakest and the C5–H bond the strongest. These electric field calculations show that the charge distributions in the substrate binding pocket can be crucial in determining the weakest C–H bond of the substrate within the substrate binding pocket. The charge distributions ultimately determine the regio- and stereoselectivity of the reaction process.
To find out whether the BDEs of the substrate C–H bonds are indeed affected by the cluster models, we took the 5ReA (model A), 5ReB (model B), and 5ReC (model C) structures and ran a single point energy calculation in the sextet spin state with one hydrogen atom removed from the substrate from position C3–H, C4–H, or C5–H. Using 5ReA as a starting point, all substrate BDE values are calculated to be within 0.3 kcal mol–1 of each other. By contrast, for 5ReC, the weakest substrate bond is the C4–H interaction with a BDE of ΔE = 109.3 kcal mol–1. On the other hand, the C3–H and C5–H BDEs are ΔE = 112.1 and 112.9 kcal mol–1. Finally, for model B, the weakest C–H bond is the C3–H bond at 109.2 kcal mol–1, while the C4–H bond has a BDE of 110.1 kcal mol–1. Therefore, the dipole moment in model C and the local electric field strengths polarize the substrate and lead to preferential C4–H hydroxylation of substrate lysine, while that is not the case with the other cluster models.
We further applied electric field calculations on a minimal cluster model of iron(IV)-oxo with its first coordination sphere only. These calculations give a strong field effect on the reduction potential of the iron(IV)-oxo species with a field located along the molecular z-axis. This is not surprising as iron(IV)-oxo reduction leads to filling of the σ*z2 orbital with one electron, which is an orbital along the molecular z-axis. In addition, electric field effect calculations were done on a minimal model containing p-cresol and acetate. These calculations show that proton transfer can be triggered by an electric field effect parallel to the hydrogen bond axis. Our calculations therefore indicate that subtle perturbations from protein residues can trigger a proton transfer, electron transfer, or a combination of the two to obtain valence tautomerism in the active oxidant.
Conclusions
In this work, a computational study is presented on a 4-lysine dioxygenase. We set up three cluster models based on an MD simulation of the substrate-bound iron(IV)-oxo species. The MD shows tight substrate binding with strong interactions between the substrate and polar active site residues. For all three clusters, a full reaction mechanism for the hydroxylation of l-Lys on the C3-, C4-, and C5-positions was calculated. In general, the hydrogen atom abstraction transition state is rate-determining and the OH rebound step is much lower in energy. Only the largest cluster model predicts C4-hydroxylation, while the other two models predict C5-hydroxylation. As such, the polarity of the models and the second-coordination sphere of the substrate and oxidant influences reactivities and kinetics of the substrate activation processes. We then analyzed the structures and found that the dipole moment is different in the three cluster models and particularly points along the C4–H bond in model C, while it does not point along the C4–H bond in the other models. Further analysis of the structures shows that KDO reacts through negative catalysis where the electric dipole moment and charge distributions in the substrate binding pocket affect the various substrate C–H bond strengths and trigger the C4-hydroxylation mechanism. Our work shows that KDOs can be selectively engineered to give the preferred reaction pathways with substrate lysine or alternative substrates. Finally, in the largest cluster model, we observe unusual proton-coupled electron transfer where an active site Tyr residue is involved in the catalysis and reduces the iron from iron(IV) to iron(III) while donating its proton to a nearby Asp residue. This implies that in KDO5, substrate activation is performed by an iron(III)-oxo species rather than an iron(IV)-oxo species. Despite the lower valency of the metal center, the reaction still proceeds through substrate hydroxylation, which further shows that iron(III)-oxo species are capable of substrate oxidation events. This unusual proton-coupled electron transfer with assistance of an active site Tyr residue may be common in highly selective and energy-demanding (negative catalysis) reaction processes in nature, and we are searching for further applications of this interesting phenomenon.
Acknowledgments
The computational shared facilities at the University of Manchester are acknowledged for providing CPU time.
Glossary
Abbreviations
- αKG
α-ketoglutarate
- BDE
bond dissociation energy
- KDO
lysine dioxygenase
- PCET
proton-coupled electron transfer
- PDB
Protein Data Bank
- ZPE
zero-point energy
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c14574.
Tables with absolute and relative energies, group spin densities and charges, and Cartesian coordinates of optimized geometries discussed in this work (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Devkota A. K.; Veloria J. R.; Guo H.-F.; Kurie J. M.; Cho E. J.; Dalby K. N. Development of a High-Throughput Lysyl Hydroxylase (LH) Assay and Identification of Small-Molecule Inhibitors against LH2. SLAS Discovery 2019, 24, 484–491. 10.1177/2472555218817057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasamatsu A.; Uzawa K.; Hayashi F.; Kita A.; Okubo Y.; Saito T.; Kimura Y.; Miyamoto I.; Oka N.; Shiiba M.; Ito C.; Toshimori K.; Miki T.; Yamauchi M.; Tanzawa H. Deficiency of Lysyl Hydroxylase 2 in Mice Causes Systemic Endoplasmic Reticulum Stress Leading to Early Embryonic Lethality. Biochem. Biophys. Res. Commun. 2019, 512, 486–491. 10.1016/j.bbrc.2019.03.091. [DOI] [PubMed] [Google Scholar]
- Li H.; Xu H.; Wen H.; Wang H.; Zhao R.; Sun Y.; Bai C.; Ping J.; Song L.; Luo M.; Chen J. Lysyl Hydroxylase 1 (LH1) Deficiency Promotes Angiotensin II (Ang II)–Induced Dissecting Abdominal Aortic Aneurysm. Theranostics 2021, 11, 9587–9604. 10.7150/thno.65277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Zhang T.; Guo L.; Wang Y.; Yang Y. Lysyl Hydroxylases Are Transcription Targets for GATA3 Driving Lung Cancer Cell Metastasis. Sci. Rep. 2018, 8, 11905. 10.1038/s41598-018-30309-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S.; Wu C.; Köhler F.; Meixensberger J.; Schopow N.; Kallendrusch S. Procollagen-Lysine, 2-Oxoglutarate 5-Dioxygenase Family: Novel Prognostic Biomarkers and Tumor Microenvironment Regulators for Lower-Grade Glioma. Front. Cell. Neurosci. 2022, 16, 838548 10.3389/fncel.2022.838548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud D.; Saaidi P.-L.; Monfleur A.; Harari M.; Cuccaro J.; Fossey A.; Besnard M.; Debard A.; Mariage A.; Pellouin V.; Petit J.-L.; Salanoubat M.; Weissenbach J.; de Berardinis V.; Zaparucha A. Synthesis of Mono- and Dihydroxylated Amino Acids with New a-Ketoglutarate-Dependent Dioxygenases: Biocatalytic Oxidation of C-H Bonds. ChemCatChem. 2014, 6, 3012–3017. 10.1002/cctc.201402498. [DOI] [Google Scholar]
- Hara R.; Yamagata K.; Miyake R.; Kawabata H.; Uehara H.; Kino K.; Liu S. J. Discovery of Lysine Hydroxylases in the Clavaminic Acid Synthase-Like Superfamily for Efficient Hydroxylysine Bioproduction. Appl. Environm. Microbiol. 2017, 83, e00693–17. 10.1128/AEM.00693-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud D.; Peruch O.; Saaidi P.-L.; Fossey A.; Mariage A.; Petit J.-L.; Salanoubat M.; Vergne-Vaxelaire C.; de Berardinis V.; Zaparucha A. Biocatalytic Approaches towards the Synthesis of Chiral Amino Alcohols from Lysine: Cascade Reactions Combining alpha-Keto Acid Oxygenase Hydroxylation with Pyridoxal Phosphate-Dependent Decarboxylation. Adv. Synth. Catal. 2017, 359, 1563–1569. 10.1002/adsc.201600934. [DOI] [Google Scholar]
- Rolf J.; Nerke P.; Britner A.; Krick S.; Lütz S.; Rosenthal K. From Cell-Free Protein Synthesis to Whole-Cell Biotransformation: Screening and Identification of Novel α-Ketoglutarate-Dependent Dioxygenases for Preparative-Scale Synthesis of Hydroxy-L-Lysine. Catalysts 2021, 11, 1038. 10.3390/catal11091038. [DOI] [Google Scholar]
- Guo H. F.; Tsai C. L.; Terajima M.; Tan X.; Banerjee P.; Miller M. D.; Liu X.; Yu J.; Byemerwa J.; Alvarado S.; Kaoud T. S.; Dalby K. N.; Bota-Rabassedas N.; Chen Y.; Yamauchi M.; Tainer J. A.; Phillips G. N.; Kurie J. M. Pro-Metastatic Collagen Lysyl Hydroxylase Dimer Assemblies Stabilized by Fe2+-Binding. Nat. Commun. 2018, 9, 512. 10.1038/s41467-018-02859-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield C. J.; Zhang Z. Structural and Mechanistic Studies on 2-Oxoglutarate-Dependent Oxygenases and Related Enzymes. Curr. Opin. Struc. Biol. 1999, 9, 722–731. 10.1016/S0959-440X(99)00036-6. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Brunold T. C.; Davis M. I.; Kemsley J. N.; Lee S. K.; Lehnert N.; Neese F.; Skulan A. J.; Yang Y. S.; Zhou J. Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes. Chem. Rev. 2000, 100, 235–349. 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- Costas M.; Mehn M. P.; Jensen M. P.; Que L. Jr. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104, 939–986. 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- Abu-Omar M. M.; Loaiza A.; Hontzeas N. Reaction Mechanisms of Mononuclear Non-Heme Iron Oxygenases. Chem. Rev. 2005, 105, 2227–2252. 10.1021/cr040653o. [DOI] [PubMed] [Google Scholar]
- Kovaleva E. G.; Lipscomb J. D. Versatility of Biological Non-Heme Fe(II) Centers in Oxygen Activation Reactions. Nat. Chem. Biol. 2008, 4, 186–193. 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser S. P.; Kumar D. (Eds.) Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reactions in Nature; Royal Society of Chemistry Publishing: Cambridge (UK), 2011. [Google Scholar]
- McDonald A. R.; Que L. Jr High-Valent Nonheme Iron-Oxo Complexes: Synthesis, Structure, and Spectroscopy. Coord. Chem. Rev. 2013, 257, 414–428. 10.1016/j.ccr.2012.08.002. [DOI] [Google Scholar]
- Nam W.; Lee Y.-M.; Fukuzumi S. Tuning Reactivity and Mechanism in Oxidation Reactions by Mononuclear Nonheme Iron(IV)-Oxo Complexes. Acc. Chem. Res. 2014, 47, 1146–1154. 10.1021/ar400258p. [DOI] [PubMed] [Google Scholar]
- Salminen A.; Kauppinen A.; Kaarniranta K. 2-Oxoglutarate-Dependent Dioxygenases Are Sensors of Energy Metabolism, Oxygen Availability, and Iron Homeostasis: Potential Role in the Regulation of Aging Process. Cell. Mol. Life Sci. 2015, 72, 3897–3914. 10.1007/s00018-015-1978-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White M. D.; Flashman E. Catalytic Strategies of the Non-Heme Iron Dependent Oxygenases and Their Roles in Plant Biology. Curr. Opin. Chem. Biol. 2016, 31, 126–135. 10.1016/j.cbpa.2016.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr C. Q.; Hausinger R. P. Amazing Diversity in BioChemical Roles of Fe(II)/2-Oxoglutarate Oxygenases. Trends Biochem. Sci. 2018, 43, 517–532. 10.1016/j.tibs.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastard K.; Isabet T.; Stura E. A.; Legrand P.; Zaparucha A. Structural Studies based on two Lysine Dioxygenases with Distinct Regioselectivity Brings Insights Into Enzyme Specificity within the Clavaminate Synthase-Like Family. Sci. Rep. 2018, 8, 16587. 10.1038/s41598-018-34795-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijnincx P. C. A.; van Koten G.; Klein Gebbink R. J. M. Mononuclear Non-Heme Iron Enzymes with the 2-His-1-Carboxylate Facial Triad: Recent Developments in Enzymology and Modeling Studies. Chem. Soc. Rev. 2008, 37, 2716–2744. 10.1039/b707179p. [DOI] [PubMed] [Google Scholar]
- Kal S.; Que L. Jr Dioxygen Activation by Nonheme Iron Enzymes with the 2-His-1-Carboxylate Facial Triad That Generate High-Valent Oxoiron Oxidants. J. Biol. Inorg. Chem. 2017, 22, 339–365. 10.1007/s00775-016-1431-2. [DOI] [PubMed] [Google Scholar]
- de Visser S. P. Mechanistic Insight on the Activity and Substrate Selectivity of Nonheme Iron Dioxygenases. Chem. Record 2018, 18, 1501–1516. 10.1002/tcr.201800033. [DOI] [PubMed] [Google Scholar]
- Price J. C.; Barr E. W.; Tirupati B.; Bollinger J. M.; Krebs C. The First Direct Characterization of a High-Valent Iron Intermediate in the Reaction of an α-Ketoglutarate-Dependent Dioxygenase: a High-Spin Fe(IV) Complex in Taurine/α-Ketoglutarate Dioxygenase (TauD) from Escherichia coli. Biochemistry 2003, 42, 7497–7508. 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- Proshlyakov D. A.; Henshaw T. F.; Monterosso G. R.; Ryle M. J.; Hausinger R. P. Direct Detection of Oxygen Intermediates in the Non-Heme Fe Enzyme Taurine/α-Ketoglutarate Dioxygenase. J. Am. Chem. Soc. 2004, 126, 1022–1024. 10.1021/ja039113j. [DOI] [PubMed] [Google Scholar]
- Riggs-Gelasco P. J.; Price J. C.; Guyer R. B.; Brehm J. H.; Barr E. W.; Bollinger J. M. Jr; Krebs C. EXAFS Spectroscopic Evidence for an Fe = O Unit in the Fe(IV) Intermediate Observed During Oxygen Activation by Taurine:α-Ketoglutarate Dioxygenase. J. Am. Chem. Soc. 2004, 126, 8108–8109. 10.1021/ja048255q. [DOI] [PubMed] [Google Scholar]
- Neidig M. L.; Brown C. D.; Light K. M.; Fujimori D. G.; Nolan E. M.; Price J. C.; Barr E. W.; Bollinger J. M.; Krebs C.; Walsh C. T.; Solomon E. I. CD and MCD of CytC3 and Taurine Dioxygenase: Role of the Facial Triad in α-KG-Dependent Oxygenases. J. Am. Chem. Soc. 2007, 129, 14224–14231. 10.1021/ja074557r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatuni A.; Renata H. Identification of a Lysine 4-Hydroxylase from the Glidobactin Biosynthesis and Evaluation of Its Biocatalytic Potential. Org. Biomol. Chem. 2019, 17, 1736–1739. 10.1039/C8OB02054J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seide S.; Arnold L.; Wetzels S.; Bregu M.; Gätgens J.; Pohl M. From Enzyme to Preparative Cascade Reactions with Immobilized Enzymes: Tuning Fe(II)/α-Ketoglutarate-Dependent Lysine Hydroxylases for Application in Biotransformations. Catalysts 2022, 12, 354. 10.3390/catal12040354. [DOI] [Google Scholar]
- Wang F.; Zhu M.; Song Z.; Li C.; Wang Y.; Zhu Z.; Sun D.; Lu F.; Qin H.-M. Reshaping the Binding Pocket of Lysine Hydroxylase for Enhanced Activity. ACS Catal. 2020, 10, 13946–13956. 10.1021/acscatal.0c03841. [DOI] [Google Scholar]
- Ghafoor S.; Mansha A.; de Visser S. P. Selective Hydrogen Atom Abstraction from Dihydroflavonol by a Non-Heme Iron Center Is the Key Step in the Enzymatic Flavonol Synthesis and Avoids Byproducts. J. Am. Chem. Soc. 2019, 141, 20278–20292. 10.1021/jacs.9b10526. [DOI] [PubMed] [Google Scholar]
- Louka S.; Barry S. M.; Heyes D. J.; Mubarak M. Q. E.; Ali H. S.; Alkhalaf L. M.; Munro A. W.; Scrutton N. S.; Challis G. L.; de Visser S. P. The Catalytic Mechanism of Aromatic Nitration by Cytochrome P450 TxtE: Involvement of a Ferric-Peroxynitrite Intermediate. J. Am. Chem. Soc. 2020, 142, 15764–15779. 10.1021/jacs.0c05070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt: the Universal Protein Knowledgebase. Nucleic Acids Res. 2023, 51, D523–D531. 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt J.; Santos-Martins D.; Tillack A. F.; Forli S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. 10.1021/acs.jcim.1c00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera--a Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Li P.; Merz K. M. MCPB.py: A Python Based Metal Center Parameter Builder. J. Chem. Inf. Model. 2016, 56, 599–604. 10.1021/acs.jcim.5b00674. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Ben-Shalom I. Y.; Brozell S. R.; Cerutti D. S.; Cheatham T. E. III; Cruzeiro V. W. D.; Darden T. A.; Duke R. E.; Ghoreishi D.; Gilson M. K.; Gohlke H.; Goetz A. W.; Greene D.; Harris R.; Homeyer N.; Huang Y.; Izadi S.; Kovalenko A.; Kurtzman T.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Liu J.; Luchko T.; Luo R.; Mermelstein D. J.; Merz K. M.; Miao Y.; Monard G.; Nguyen C.; Nguyen H.; Omelyan I.; Onufriev A.; Pan F.; Qi R.; Roe D. R.; Roitberg A.; Sagui C.; Schott-Verdugo S.; Shen J.; Simmerling C. L.; Smith J.; Salomon-Ferrer R.; Swails J.; Walker R. C.; Wang J.; Wei H.; Wolf R. M.; Wu X.; Xiao L.; York D. M.; Kollman P. A.. AMBER-2018; University of California: San Francisco, 2018. [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng X.; Himo F. Computational Study of Pictet-Spenglerase Strictosidine Synthase: Reaction Mechanism and Origins of Enantioselectivity of Natural and Non-Natural Substrates. ACS Catal. 2020, 10, 13630–13640. 10.1021/acscatal.0c03758. [DOI] [Google Scholar]
- Nandy A.; Adamji H.; Kastner D. W.; Vennelakanti V.; Nazemi A.; Liu M.; Kulik H. J. Using Computational Chemistry To Reveal Nature’s Blueprints for Single-Site Catalysis of C-H Activation. ACS Catal. 2022, 12, 9281–9306. 10.1021/acscatal.2c02096. [DOI] [Google Scholar]
- Gérard E. F.; Yadav V.; Goldberg D. P.; de Visser S. P. What Drives Radical Halogenation Versus Hydroxylation in Mono-nuclear Nonheme Iron Complexes? A Combined Experimental and Computational Study. J. Am. Chem. Soc. 2022, 144, 10752–10767. 10.1021/jacs.2c01375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali H. S.; de Visser S. P. Electrostatic Perturbations in the Substrate-Binding Pocket of Taurine/α-Ketoglutarate Dioxygenase Determine Its Selectivity. Chem. - Eur. J. 2022, 28, e202104167 10.1002/chem.202104167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himo F.; de Visser S. P. Status Report on the Quantum Chemical Cluster Approach for Modeling Enzyme Reactions. Commun. Chem. 2022, 5, 29. 10.1038/s42004-022-00642-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokkawes T.; de Visser S. P. Caffeine Biodegradation by Cytochrome P450 1A2. What Determines the Product Distributions?. Chem. - Eur. J. 2023, 29, e202203875 10.1002/chem.202203875. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Keith T.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.;. Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian-09, Revision D.01; Gaussian, Inc.: Wallingford CT, 2013. [Google Scholar]
- de Visser S. P.; Mukherjee G.; Ali H. S.; Sastri C. V. Local Charge Distributions, Electric Dipole Moments and Local Electric Fields Influence Reactivity Patterns and Guide Regioselectivities in α-Ketoglutarate-Dependent Nonheme Iron Dioxygenases. Acc. Chem. Res. 2022, 55, 65–74. 10.1021/acs.accounts.1c00538. [DOI] [PubMed] [Google Scholar]
- Becke A. D. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti Correlation-Energy Formula Into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Hay P. J.; Wadt W. R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. 10.1063/1.448799. [DOI] [Google Scholar]
- Ditchfield R.; Hehre W. J.; Pople J. A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. 10.1063/1.1674902. [DOI] [Google Scholar]
- Francl M. M.; Pietro W. J.; Hehre W. J.; Binkley J. S.; Gordon M. S.; DeFrees D. J.; Pople J. A. Self-Consistent Molecular Orbital Methods. XXIII. A Polarization-Type Basis Set for Second-Row Elements. J. Chem. Phys. 1982, 77, 3654–3658. 10.1063/1.444267. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Mitchell A. J.; Dunham N. P.; Martinie R. J.; Bergman J. A.; Pollock C. J.; Hu K.; Allen B. D.; Chang W.-C.; Silakov A.; Bollinger J. M. Jr.; Krebs C.; Boal A. K. Visualizing the Reaction Cycle in an Iron(II)- and 2-(Oxo)-glutarate-Dependent Hydroxylase. J. Am. Chem. Soc. 2017, 139, 13830–13836. 10.1021/jacs.7b07374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi S. S.; Jaber Sathik Rifayee S. B.; Waheed S. O.; Wildey J.; Warner C.; Schofield C. J.; Karabencheva-Christova T. G.; Christov C. Z. Can Second Coordination Sphere and Long-Range Interactions Modulate Hydrogen Atom Transfer in a Non-Heme Fe(II)-Dependent Histone Demethylase?. JACS Au 2022, 2, 2169–2186. 10.1021/jacsau.2c00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese A.; Waheed S. O.; Chaturvedi S. S.; DiCastri I.; LaRouche C.; Kaski B.; Lehnert N.; Li D.; Christov C. Z.; Karabencheva-Christova T. G. Revealing the catalytic strategy of FTO. Chem. Catal. 2023, 3, 100732 10.1016/j.checat.2023.100732. [DOI] [Google Scholar]
- Mokkawes T.; Lim Z. Q.; de Visser S. P. Mechanism of melatonin metabolism by CYP1A1. What determines the bifurcation pathways of hydroxylation versus de-formylation?. J. Phys. Chem. B 2022, 126, 9591–9606. 10.1021/acs.jpcb.2c07200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gérard E. F.; Mokkawes T.; Johannissen L. O.; Warwicker J.; Spiess R. R.; Blanford C. F.; Hay S.; Heyes D. J.; de Visser S. P. How Is Substrate Halogenation Triggered by the Vanadium Haloperoxidase from Curvularia inaequalis?. ACS Catal. 2023, 13, 8247–8261. 10.1021/acscatal.3c00761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borowski T.; Bassan A.; Siegbahn P. E. M. Mechanism of Dioxygen Activation in 2-Oxoglutarate-Dependent Enzymes: A Hybrid DFT Study. Chem. - Eur. J. 2004, 10, 1031–1041. 10.1002/chem.200305306. [DOI] [PubMed] [Google Scholar]
- Nemukhin A. V.; Topol I. A.; Cachau R. E.; Burt S. K. On the Nature of Oxoiron (IV) Intermediate in Dioxygen Activation by Non-Heme Enzymes. Theor. Chem. Acc. 2006, 115, 348–353. 10.1007/s00214-005-0047-3. [DOI] [Google Scholar]
- Sinnecker S.; Svensen N.; Barr E. W.; Ye S.; Bollinger J. M. Jr; Neese F.; Krebs C. Spectroscopic and Computational Evaluation of the Structure of the High-Spin Fe(IV)-Oxo Intermediates in Taurine: α-Ketoglutarate Dioxygenase from Escherichia coli and Its His99Ala Ligand Variant. J. Am. Chem. Soc. 2007, 129, 6168–6179. 10.1021/ja067899q. [DOI] [PubMed] [Google Scholar]
- Godfrey E.; Porro C. S.; de Visser S. P. Comparative Quantum Mechanics/Molecular Mechanics (QM/MM) and Density Functional Theory Calculations on the Oxo-Iron Species of Taurine/α-Ketoglutarate Dioxygenase. J. Phys. Chem. A 2008, 112, 2464–2468. 10.1021/jp710999v. [DOI] [PubMed] [Google Scholar]
- Chen H.; Lai W.; Yao J.; Shaik S. Perferryl FeV–Oxo Non-Heme Complexes: Do They Have High-Spin or Low-Spin Ground States?. J. Chem. Theory Comput. 2011, 7, 3049–3053. 10.1021/ct200614g. [DOI] [PubMed] [Google Scholar]
- Bushnell E. A. C.; Fortowsky G. B.; Gauld J. W. Model Iron–Oxo Species and the Oxidation of Imidazole: Insights Into the Mechanism of OvoA and EgtB?. Inorg. Chem. 2012, 51, 13351–13356. 10.1021/ic3021172. [DOI] [PubMed] [Google Scholar]
- Kulik H. J.; Drennan C. L. Substrate Placement Influences Reactivity in Non-Heme Fe(II) Halogenases and Hydroxylases. J. Biol. Chem. 2013, 288, 11233–11241. 10.1074/jbc.M112.415570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratter S. M.; Konstantinovics C.; DiGiuro C. L. M.; Leitner E.; Kumar D.; de Visser S. P.; Grogan G.; Straganz G. D. Inversion of Enantio-Selectivity of a Mononuclear Non-Heme Iron(II)-Dependent Hydroxylase by Tuning the Interplay of Metal-Center Geometry and Protein Structure. Angew. Chem., Int. Ed. 2013, 52, 9677–9681. 10.1002/anie.201304633. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9859–9863
- Latifi R.; Sainna M. A.; Rybak-Akimova E. V.; de Visser S. P. Does Hydrogen Bonding-Donation to Manganese(IV)-Oxo and Iron(IV)-Oxo Oxidants Affect the Oxygen Atom Transfer Ability? A Computational Study. Chem. - Eur. J. 2013, 19, 4058–4068. 10.1002/chem.201202811. [DOI] [PubMed] [Google Scholar]
- Wang B.; Usharani D.; Li C.; Shaik S. Theory Uncovers an Unusual Mechanism of DNA Repair of a Lesioned Adenine by AlkB Enzymes. J. Am. Chem. Soc. 2014, 136, 13895–13901. 10.1021/ja507934g. [DOI] [PubMed] [Google Scholar]
- Wójcik A.; Radoń M.; Borowski T. Mechanism of O2 Activation by α-Ketoglutarate Dependent Oxygenases Revisited. A Quantum Chemical Study. J. Phys. Chem. A 2016, 120, 1261–1274. 10.1021/acs.jpca.5b12311. [DOI] [PubMed] [Google Scholar]
- Álvarez-Barcia S.; Kästner J. Atom Tunneling in the Hydroxylation Process of Taurine/α-Ketoglutarate Dioxygenase Identified by Quantum Mechanics/Molecular Mechanics Simulations. J. Phys. Chem. B 2017, 121, 5347–5354. 10.1021/acs.jpcb.7b03477. [DOI] [PubMed] [Google Scholar]
- Timmins A.; Saint-André M.; de Visser S. P. Understanding How Prolyl-4-Hydroxylase Structure Steers a Ferryl Oxidant Toward Scission of a Strong C–H Bond. J. Am. Chem. Soc. 2017, 139, 9855–9866. 10.1021/jacs.7b02839. [DOI] [PubMed] [Google Scholar]
- Ali H. S.; Henchman R. H.; de Visser S. P. Mechanism of Oxidative Ring-Closure As Part of the Hygromycin Biosynthesis Step by a Nonheme Iron Dioxygenase. ChemCatChem. 2021, 13, 3054–3066. 10.1002/cctc.202100393. [DOI] [Google Scholar]
- Lu J.; Wang B.; Shaik S.; Lai W. QM/MM Calculations Reveal the Important Role of α-Heteroatom Substituents in Controlling Selectivity of Mononuclear Nonheme HppE-Catalyzed Reactions. ACS Catal. 2020, 10, 9521–9532. 10.1021/acscatal.0c01803. [DOI] [Google Scholar]
- Waheed S. O.; Varghese A.; Chaturvedi S. S.; Karabencheva-Christova T. G.; Christov C. Z. How Human TET2 Enzyme Catalyzes the Oxidation of Unnatural Cytosine Modifications in Double-Stranded DNA. ACS Catal. 2022, 12, 5327–5344. 10.1021/acscatal.2c00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojdyla Z.; Borowski T. Properties of the Reactants and Their Interactions Within and With the Enzyme Binding Cavity Determine Reaction Selectivities. The Case of Fe(II)/2-Oxoglutarate Dependent Enzymes. Chem. - Eur. J. 2022, 28, e202104106 10.1002/chem.202104106. [DOI] [PubMed] [Google Scholar]
- Yeh C.-C. G.; Ghafoor S.; Satpathy J. K.; Mokkawes T.; Sastri C. V.; de Visser S. P. A Cluster Model Study Into the Catalytic Mechanism of α-Ketoglutarate Biodegradation by the Ethylene-Forming-Enzyme Reveals Structural Differences with Nonheme Iron Hydroxylases. ACS Catal. 2022, 12, 3923–3937. 10.1021/acscatal.1c04029. [DOI] [Google Scholar]
- Vennelakanti V.; Li G. L.; Kulik H. J. Why Nonheme Iron Halogenases Do Not Fluorinate C-H Bonds: A Computational Investigation. Inorg. Chem. 2023, 62, 19768–197790. 10.1021/acs.inorgchem.3c03215. [DOI] [PubMed] [Google Scholar]
- Cao Y.; Valdez-Moreira J. A.; Hay S.; Smith J. M.; de Visser S. P. Reactivity Differences of Trigonal Pyramidal Nonheme Iron(IV)-Oxo and Iron(III)-Oxo Complexes: Experiment and Theory. Chem. - Eur. J. 2023, 29, e202300271 10.1002/chem.202300271. [DOI] [PubMed] [Google Scholar]
- Hirao H.; Kumar D.; Que L. Jr; Shaik S. Two-State Reactivity in Alkane Hydroxylation By Non-Heme Iron-Oxo Complexes. J. Am. Chem. Soc. 2006, 128, 8590–8606. 10.1021/ja061609o. [DOI] [PubMed] [Google Scholar]
- de Visser S. P. Differences In and Comparison of the Catalytic Properties of Heme and Non-Heme Enzymes With a Central Oxo-Iron Group. Angew. Chem., Int. Ed. 2006, 45, 1790–1793. 10.1002/anie.200503841. [DOI] [PubMed] [Google Scholar]
- Company A.; Feng Y.; Güell M.; Ribas X.; Luis J. M.; Que L.; Costas M. Olefin-Dependent Discrimination Between Two Nonheme HO-FeV=O Tautomeric Species In Catalytic H2O2 Epoxidations. Chem. - Eur. J. 2009, 15, 3359–3362. 10.1002/chem.200802597. [DOI] [PubMed] [Google Scholar]
- Neugebauer M. E.; Kissman E. N.; Marchand J. A.; Pelton J. G.; Sambold N. A.; Millar D. C.; Chang M. C. Y. Reaction Pathway Engineering Converts a Radical Hydroxylase Into a Halogenase. Nat. Chem. Biol. 2022, 18, 171–175. 10.1038/s41589-021-00944-x. [DOI] [PubMed] [Google Scholar]
- Sivaraja M.; Goodin D. B.; Smith M.; Hoffman B. M. Identification by ENDOR of Trp191 as the Free-Radical Site in Cytochrome c Peroxidase Compound ES. Science 1989, 245, 738–740. 10.1126/science.2549632. [DOI] [PubMed] [Google Scholar]
- Wirstam M.; Blomberg M. R. A.; Siegbahn P. E. M. Reaction Mechanism of Compound I Formation in Heme Peroxidases: A Density Functional Theory Study. J. Am. Chem. Soc. 1999, 121, 10178–10185. 10.1021/ja991997c. [DOI] [Google Scholar]
- de Visser S. P. What Affects the Quartet-Doublet Energy Splitting in Peroxidase Enzymes?. J. Phys. Chem. B 2005, 109, 11050–11057. 10.1021/jp053873u. [DOI] [PubMed] [Google Scholar]
- Shaik S.; de Visser S. P.; Kumar D. External Electric Field Will Control the Selectivity of Enzymatic-Like Bond Activations. J. Am. Chem. Soc. 2004, 126, 11746–11749. 10.1021/ja047432k. [DOI] [PubMed] [Google Scholar]
- Leeladee P.; Baglia R. A.; Prokop K. A.; Latifi R.; de Visser S. P.; Goldberg D. P. Valence Tautomerism in a High-Valent Manganese-Oxo Porphyrinoid Complex Induced by a Lewis Acid. J. Am. Chem. Soc. 2012, 134, 10397–10400. 10.1021/ja304609n. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Faponle A. S.; Barman P.; Vardhaman A. K.; Sastri C. V.; Kumar D.; de Visser S. P. Long-Range Electron Transfer Triggers Mechanistic Differences Between Iron(IV)-Oxo and Iron(IV)-Imido Oxidants. J. Am. Chem. Soc. 2014, 136, 17102–17115. 10.1021/ja508403w. [DOI] [PubMed] [Google Scholar]
- Yeh C. G.; Mokkawes T.; Bradley J. M.; Le Brun N. E.; de Visser S. P. Second Coordination Sphere Effects on the Mechanistic Pathways For Dioxygen Activation By a Ferritin: Involvement of a Tyr Radical and the Identification of a Cation Binding Site. ChemBioChem. 2022, 2022, e202200257 10.1002/cbic.202200257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon H.; Lee Y.-M.; Wu X.; Cho K.-B.; Sarangi R.; Nam W.; Fukuzumi S. Enhanced Electron-Transfer Reactivity of Nonheme Manganese(IV)-Oxo Complexes by Binding Scandium Ions. J, Am. Chem. Soc. 2013, 135, 9186–9194. 10.1021/ja403965h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess K. M.; Leach I. F.; Wijtenhorst L.; Lee H.; Klein J. E. M. N. Valence Tautomerism Induced Proton Coupled Electron Transfer: X-H Bond Oxidation with a Dinuclear Au(II) Hydroxide Complex. Angew. Chem., Int. Ed. 2024, 63, e202318916. 10.1002/anie.202318916. [DOI] [PubMed] [Google Scholar]
- MacBeth C. E.; Golombek A. P.; Young V. G. Jr; Yang C.; Kuczera K.; Hendrich M. P.; Borovik A. S. O2 Activation by Nonheme Iron Complexes: A Monomeric Fe(III)–Oxo Complex Derived From O2. Science 2000, 289, 938–941. 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]
- Matson E. M.; Park Y. J.; Fout A. R. Facile Nitrite Reduction in a Non-heme Iron System: Formation of an Iron(III)-Oxo. J. Am. Chem. Soc. 2014, 136, 17398–17401. 10.1021/ja510615p. [DOI] [PubMed] [Google Scholar]
- Andris E.; Navrátil R.; Jašík J.; Puri M.; Costas M.; Que L. Jr; Roithová J. Trapping Iron(III)-Oxo Species at the Boundary of the “Oxo Wall”: Insights into the Nature of the Fe(III)-O Bond. J. Am. Chem. Soc. 2018, 140, 14391–14400. 10.1021/jacs.8b08950. [DOI] [PubMed] [Google Scholar]
- Reed C. J.; Agapie T. A Terminal FeIII-Oxo in a Tetranuclear Cluster: Effects of Distal Metal Centers on Structure and Reactivity. J. Am. Chem. Soc. 2019, 141, 9479–9484. 10.1021/jacs.9b03157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez-Moreira J. A.; Beagan D. M.; Yang H.; Telser J.; Hoffman B. M.; Pink M.; Carta V.; Smith J. M. Hydrocarbon Oxidation by an Exposed, Multiply Bonded Iron(III) Oxo Complex. ACS Cent. Sci. 2021, 7, 1751–1755. 10.1021/acscentsci.1c00890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat I.; Company A.; Postils V.; Ribas X.; Que L.; Luis J. M.; Costas M. The Mechanism of Stereospecific C-H Oxidation by Fe(Pytacn) Complexes: Bioinspired Non-Heme Iron Catalysts Containing Cis-Labile Exchangeable Sites. Chem. - Eur. J. 2013, 19, 6724–6738. 10.1002/chem.201300110. [DOI] [PubMed] [Google Scholar]
- Prat I.; Mathieson J. S.; Güell M.; Ribas X.; Luis J. M.; Cronin L.; Costas M. Observation of Fe(V)=O Using Variable-Temperature Mass Spectrometry and Its Enzyme-Like C-H and C = C Oxidation Reactions. Nat. Chem. 2011, 3, 788–793. 10.1038/nchem.1132. [DOI] [PubMed] [Google Scholar]
- Hirao H.; Li F.; Que L. Jr.; Morokuma K. Theoretical Study of the Mechanism of Oxoiron(IV) Formation From H2O2 and a Nonheme Iron(II) Complex: O-O Cleavage Involving Proton-Coupled Electron Transfer. Inorg. Chem. 2011, 50, 6637–6648. 10.1021/ic200522r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H.; Guan J.; Zhang L.; Liu H.; Huang X. The Effect of the Axial Ligand on Distinct Reaction Tunneling For Methane Hydroxylation by Nonheme Iron(IV)-Oxo Complexes. Phys. Chem. Chem. Phys. 2012, 14, 12863–12874. 10.1039/c2cp42423a. [DOI] [PubMed] [Google Scholar]
- Ansari A.; Kaushik A.; Rajaraman G. Mechanistic Insights on the Ortho-Hydroxylation of Aromatic Compounds by Non-Heme Iron Complex: a Computational Case Study on the Comparative Oxidative Ability of Ferric-Hydroperoxo and High-Valent FeIV=O and FeV=O Intermediates. J. Am. Chem. Soc. 2013, 135, 4235–4249. 10.1021/ja307077f. [DOI] [PubMed] [Google Scholar]
- Gani T. Z. H.; Kulik H. J. Understanding and Breaking Scaling Relations in Single-Site Catalysis: Methane to Methanol Conversion by FeIV=O. ACS Catal. 2018, 8, 975–986. 10.1021/acscatal.7b03597. [DOI] [Google Scholar]
- Klein J. E. M. N.; Knizia G. cPCET versus HAT: A Direct Theoretical Method for Distinguishing X–H Bond-Activation Mechanisms. Angew. Chem., Int. Ed. 2018, 57, 11913–11917. 10.1002/anie.201805511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheaib K.; Mubarak M. Q. E.; Sénéchal-David K.; Herrero C.; Guillot R.; Clémancey M.; Latour J.-M.; de Visser S. P.; Mahy J.-P.; Banse F.; Avenier F. Selective Formation of an FeIVO or an FeIIIOOH Intermediate from FeII-H2O2: Controlled Heterolytic vs Homolytic O–O Bond Cleavage by the Second Coordination Sphere. Angew. Chem., Int. Ed. 2019, 58, 854–858. 10.1002/anie.201812724. [DOI] [PubMed] [Google Scholar]
- Andris E.; Segers K.; Mehara J.; Rulíšek L.; Roithová J. Closed Shell Iron(IV) Oxo Complex with an Fe-O Triple Bond: Computational Design, Synthesis, and Reactivity. Angew. Chem., Int. Ed. 2020, 59, 23137–23144. 10.1002/anie.202009347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Liu Y. Mechanistic Investigation of Isonitrile Formation Catalyzed by the Nonheme Iron/α-KG-Dependent Decarboxylase (ScoE). ACS Catal. 2020, 10, 2942–2957. 10.1021/acscatal.9b05411. [DOI] [Google Scholar]
- Bleher K.; Comba P.; Faltermeier D.; Gupta A.; Kerscher M.; Krieg S.; Martin B.; Velmurugan G.; Yang S. Non-Heme-Iron-Mediated Selective Halogenation of Unactivated Carbon-Hydrogen Bonds. Chem. - Eur. J. 2022, 28, e202103452 10.1002/chem.202103452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Song W.; Yao J.; Wu Z.; Lee Y.-M.; Wang Y.; Nam W.; Wang B. Hydrogen Bonding-Assisted and Nonheme Manganese-Catalyzed Remote Hydroxylation of C-H Bonds in Nitrogen-Containing Molecules. J. Am. Chem. Soc. 2023, 145, 5456–5466. 10.1021/jacs.2c13832. [DOI] [PubMed] [Google Scholar]
- de Visser S. P. Trends in Substrate Hydroxylation Reactions by Heme and Nonheme Iron(IV)-Oxo Oxidants Give Correlations Between Intrinsic Properties of the Oxidant With Barrier Height. J. Am. Chem. Soc. 2010, 132, 1087–1097. 10.1021/ja908340j. [DOI] [PubMed] [Google Scholar]
- Latifi R.; Bagherzadeh M.; de Visser S. P. Origin of the Correlation of the Rate Constant of Substrate Hydroxylation by Nonheme Iron(IV)-Oxo Complexes With the Bond-Dissociation Energy of the C–H Bond of the Substrate. Chem. - Eur. J. 2009, 15, 6651–6662. 10.1002/chem.200900211. [DOI] [PubMed] [Google Scholar]
- Strieker M.; Kopp F.; Mahlert C.; Essen L. O.; Marahiel M. A. Mechanistic and Structural Basis of Stereospecific Cβ-Hydroxylation in Calcium-Dependent Antibiotic, a Daptomycin-Type Lipopeptide. ACS Chem. Biol. 2007, 2, 187–196. 10.1021/cb700012y. [DOI] [PubMed] [Google Scholar]
- Ali H. S.; Henchman R. H.; de Visser S. P. What determines the selectivity of arginine dihydroxylation by the nonheme iron enzyme OrfP?. Chem. - Eur. J. 2021, 27, 1795–1809. 10.1002/chem.202004019. [DOI] [PubMed] [Google Scholar]
- Ali H. S.; de Visser S. P. Catalytic Divergencies in the Mechanism of L-Arginine Hydroxylating Nonheme Iron Enzymes. Front. Chem. 2024, 12, 1365494. 10.3389/fchem.2024.1365494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali H. S.; Henchman R. H.; Warwicker J.; de Visser S. P. How Do Electrostatic Perturbations of the Protein Affect the Bifurcation Pathways of Substrate Hydroxylation Versus Desaturation in the Nonheme Iron-Dependent Viomycin Biosynthesis Enzyme?. J. Phys. Chem. A 2021, 125, 1720–1737. 10.1021/acs.jpca.1c00141. [DOI] [PubMed] [Google Scholar]
- Ali H. S.; Warwicker J.; de Visser S. P. How Does the Nonheme Iron Enzyme NapI React Through L-Arginine Desaturation Rather Than Hydroxylation? A QM/MM Study. ACS Catal. 2023, 13, 10705–10721. 10.1021/acscatal.3c02262. [DOI] [Google Scholar]
- de Visser S. P.; Lin Y.-T.; Ali H. S.; Bagha U. K.; Mukherjee G.; Sastri C. V. Negative Catalysis or Non-Bell-Evans-Polanyi Reactivity by Metalloenzymes: Examples from Mononuclear Heme and Non-Heme Iron Oxygenases. Coord. Chem. Rev. 2021, 439, 213914 10.1016/j.ccr.2021.213914. [DOI] [Google Scholar]
- Hill N. S.; Coote M. L. Internal Oriented Electric Fields As a Strategy for Selectively Modifying Photochemical Reactivity. J. Am. Chem. Soc. 2018, 140, 17800–17804. 10.1021/jacs.8b12009. [DOI] [PubMed] [Google Scholar]
- Shaik S.; Danovich D.; Joy J.; Wang Z.; Stuyver T. Electric-Field Mediated Chemistry: Uncovering and Exploiting the Potential of (Oriented) Electric Fields to Exert Chemical Catalysis and Reaction Control. J. Am. Chem. Soc. 2020, 142, 12551–12562. 10.1021/jacs.0c05128. [DOI] [PubMed] [Google Scholar]
- Ma Z.; Nakatani N.; Fujii H.; Hada M. Effect of External Electric Fields on the Oxidation Reaction of Olefins by Fe(IV)OCl–Porphyrin Complexes. Bull. Chem. Soc. Jpn. 2020, 93, 187–193. 10.1246/bcsj.20190293. [DOI] [Google Scholar]
- Dixit V. A.; Warwicker J.; de Visser S. P. How Do Metal Ions Modulate the Rate-Determining Electron Transfer Step in Cytochrome P450 Reactions?. Chem. - Eur. J. 2020, 26, 15270–15281. 10.1002/chem.202003024. [DOI] [PubMed] [Google Scholar]
- Kirshenboim O.; Frenklah A.; Kozuch S. Switch Chemistry at Cryogenic Conditions: Quantum Tunnelling Under Electric Fields. Chem. Sci. 2021, 12, 3179–3187. 10.1039/D0SC06295B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S.; Vermeeren P.; Hamlin T. A.; Bickelhaupt F. M. How Oriented External Electric Fields Modulate Reactivity. Chem. - Eur. J. 2021, 27, 5683–5693. 10.1002/chem.202004906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.; Ali H. S.; de Visser S. P. Electrostatic Perturbations from the Protein Affect C-H Bond Strengths of the Substrate and Enable Negative Catalysis in the TmpA Biosynthesis Enzyme. Chem. - Eur. J. 2021, 27, 8851–8864. 10.1002/chem.202100791. [DOI] [PubMed] [Google Scholar]
- Peng W.; Yan S.; Zhang X.; Liao L.; Zhang J.; Shaik S.; Wang B. How Do Preorganized Electric Fields Function in Catalytic Cycles? The Case of the Enzyme Tyrosine Hydroxylase. J. Am. Chem. Soc. 2022, 144, 20484–20494. 10.1021/jacs.2c09263. [DOI] [PubMed] [Google Scholar]
- Chaturvedi S. S.; Jaber Sathik Rifayee S. B.; Ramanan R.; Rankin J. A.; Hu J.; Hausinger R. P.; Christov C. Z. Can an External Electric Field Switch Between Ethylene Formation and L-Arginine Hydroxylation in the Ethylene Forming Enzyme?. Phys. Chem. Chem. Phys. 2023, 25, 13772–13783. 10.1039/D3CP01899G. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.