

Abstract
Currently, over 500 rare genetic bone disorders are identified. These diseases are often accompanied by dental abnormalities, which are sometimes the first clue for an early diagnosis. However, not many dentists are sufficiently familiar with phenotypic abnormalities and treatment approaches when they encounter patients with rare diseases. Such patients often need dental treatment but have difficulties in finding a dentist who can treat them appropriately. Herein we focus on major dental phenotypes and summarize their potential causes and mechanisms, if known. We discuss representative diseases, dental treatments, and their effect on the oral health of patients and on oral health-related quality of life. This review can serve as a starting point for dentists to contribute to early diagnosis and further investigate the best treatment options for patients with rare disorders, with the goal of optimizing treatment outcomes.
Keywords: dental abnormality, dental phenotype, dental treatment, oral health-related quality of life, rare genetic bone diseases, rare genetic bone disorders, rare hereditary bone diseases, rare hereditary bone disorders
1 |. INTRODUCTION
A “rare disease” is defined as any disease or condition that affects fewer than 200,000 people in the United States, or less than one person in 1250, according to the Rare Diseases Act of 2002 (Schieppati et al., 2008). According to the NIH Office of Rare Disease Research, more than 500 “rare genetic bone diseases” have been identified. Most of these conditions become apparent early in life and are present throughout life. They are often accompanied by dental abnormalities, which are sometimes the first clue for an early diagnosis. Most dental phenotypes in rare genetic bone diseases include delayed tooth eruption, congenitally missing teeth, supernumerary teeth, or enamel hypoplasia (Table 1). This review focuses on rare diseases carrying one of those four dental phenotypes. In Table 2, we further summarize the mutations, craniofacial, and dental phenotypes of these diseases.
TABLE 1.
Summary of major dental phenotypes associated by rare genetic bone diseases.
| Dental phenotype | Representative disease | OMIM | Reference |
|---|---|---|---|
| Delayed tooth eruption | Cleidocranial dysplasia | 119600 | (Callea et al., 2012) |
| Cranio-metaphyseal Dysplasia | 123000 | (Dutra et al., 2013) | |
| Osteopetrosis | 259700 | (Athanasiadou et al., 2020) | |
| Orofaciodigital syndrome | 311200 | (Belengeanu et al., 2019) | |
| Pseudohypoparathyroidism | 103580 | (Hejlesen et al., 2019) | |
| Pycnodysostosis | 265800 | (Alves Pereira et al., 2008) | |
| Congenitally missing teeth | Cherubism | 118400 | (Papadaki et al., 2012) |
| Ectodermal dysplasia | 129810 | (Hanisch, Sielker, et al., 2019) | |
| Fraser syndrome | 219000, 617666, 617667 | (Kunz et al., 2020) | |
| Oculodentodigital dysplasia | 164200 | (Paznekas et al., 2009) | |
| Oligodontia | 106600 | (Aarts et al., 2023) | |
| Osteogenesis imperfecta type IB, II, III, IVB | 166200, 166210, 259420, 166220 | (Taqi et al., 2021) | |
| Osteopetrosis | 259700, 259730, 166600 | (Athanasiadou et al., 2020) | |
| Trichorhinophalangeal syndrome | 190350 | (Forys-Dworniczak et al., 2019) | |
| Supernumerary teeth | Cleidocranial dysplasia | 119600 | (Callea et al., 2012) |
| Opitz BBB/G syndrome | 300000 | (Cammarata-Scalisi et al., 2018) | |
| Orofaciodigital syndrome | 311200 | (Belengeanu et al., 2019) | |
| Robinow syndrome autosomal dominant | 180700 | (Jain et al., 2017) | |
| Trichorhinophalangeal syndrome | 190350 | (Forys-Dworniczak et al., 2019) | |
| Enamel hypoplasia | Ectodermal dysplasia | 129810 | (Itin & Fistarol, 2004) |
| Ellis-van Creveld syndrome | 225500 | (Baujat & Le Merrer, 2007) | |
| Kenny-Caffey syndrome | 244460 | (Moussaid et al., 2012) | |
| Oculodentodigital dysplasia | 164200 | (Harting et al., 2019) | |
| Orofaciodigital syndrome | 311200 | (Belengeanu et al., 2019) | |
| Pycnodysostosis | 265800 | (Alves Pereira et al., 2008) |
TABLE 2.
Summary of mutations and phenotypes for several rare genetic bone diseases.
| Disease (OMIM) | Gene name | Protein name | Protein’s function | Craniofacial phenotype | Dental phenotype | Dental treatment considerations | Reference |
|---|---|---|---|---|---|---|---|
| Cherubism (118400) | SH3BP2 | SH3 domain binding protein 2 | Positively regulation of transcriptional activity in T, NK, and basophilic cells | Fibro-osseous bilateral and symmetrical expansions of jaws Impaired vision and hearing |
Malocclusion by malformation of the jaw (major phenotype) Premature loss of deciduous teeth Congenitally missing teeth |
Orthodontic treatment is better after growth is completed and when disease regressed | (Faircloth et al., 1991) (Pontes et al., 2007) (Papadaki et al., 2012) |
| Cleidocranial dysplasia (119600) | RUNX2 | RUNX Family Transcription Factor 2 | Osteoblastic differentiation and skeletal morphogenesis | Increased head circumference Large fontanelles High palate or Cleft palate |
Delayed tooth eruption (major phenotype) Supernumerary teeth Malformed teeth |
Extraction of deciduous teeth does not necessarily induce the eruption of the permanent teeth | (Cooper et al., 2001) (Callea et al., 2012) (Thaweesapphithak et al., 2022) |
| Cranio-metaphyseal dysplasia (123000) | ANKH | ANKH Inorganic Pyrophosphate Transport Regulator | Control of pyrophosphate levels Transporter of small molecules such as citrate, ATP, and so forth |
Skull and jawbone hyperostosis Paranasal bossing Macrocephaly Hypertelorism Hearing loss, conductive deafness, and tinnitus |
Excess mineralization of teeth (major phenotype) Delayed tooth eruption (major phenotype) Unerupted teeth |
Special attention should be taken to potential root resorption caused by orthodontic force. | (Hayashibara et al., 2000) (Dutra et al., 2013) (Szeri et al., 2020) (Szeri et al., 2022) |
| Ectodermal dysplasia (129810) | EDA | Ectodysplasin A | Regulation of interactions between ectoderm and the mesoderm | Hypotrichosis Cleft formation |
Congenitally missing teeth (major phenotype) Enamel hypoplasia Microdontia |
High risk of dental caries secondary to enamel hypoplasia | (Itin & Fistarol, 2004) (Hanisch, Sielker, et al., 2019) |
| Ehlers–Danlos syndrome (130080) | COL3A1 matrix | Collagen Type III Alpha 1 Chain | Formation of Type III collagen | Severe generalized gingival edema and erythema | Pulp stones, pulpal calcifications, and abnormal root morphologies (major phenotype) Temporomandibular joint dysfunction Severe periodontitis |
Difficulty in endodontic therapy Needing frequent respites during dental treatment |
(Malfait et al., 2017) (Stock et al., 2021) (Baart & van Hagen, 2000) (Baujat & Le Merrer, 2007) |
| Ellis-van Creveld syndrome (225500) | EVC | EvC Ciliary Complex Subunit 1 | Regulation of Sonic Hedgehog signaling pathway | Fusion of the middle part of the upper lip to the labial sulcus | Malpositioned teeth (major phenotype) Conical teeth Enamel hypoplasia Malocclusion Prematurely erupted |
Possibility of having heart disease | |
| Fraser syndrome (219000) | FRAS1 | Fraser Extracellular Matrix Complex Subunit 1 | Regulation of epidermal-basement membrane adhesion | Cleft lip and palate | Congenitally missing teeth (major phenotype) | Periodontal treatment is important due to easy developing chronic lung disease. | (Keene & Day, 2011) (Gallottini et al., 2018) (Kunz et al., 2020) |
| (617667) | GRIP1 | Glutamate Receptor Interacting Protein 1 | Transmission across chemical synapses and glutamate binding | High palate | Short dental root | ||
| (617666) | FREM2 | FRAS1 Related Extracellular Matrix 2 | Maintenance of the integrity of the skin epithelium and renal epithelia | Ankyloglossia Middle and outer ear malformations Congenital deafness |
Malocclusion Microdontia Dental crowding |
||
| Kenny Caffey syndrome (244460) | TBCE | Tubulin Folding Cofactor E | Maintenance of the neuronal microtubule network | Micrognathia Delayed closure of the fontanel Macrocephaly |
Oligodontia (major phenotype) Abnormal root morphology Delayed eruption Enamel hypoplasia |
Implant placement is not a good choice due to poor Vitamin D levels | (Tahseen et al., 1997) (Demir et al., 2007) (Moussaid et al., 2012) |
| Oculodentodigital dysplasia (164200) | GJA1 | Gap Junction Protein Alpha 1 | Hemichannel and gap junction protein | Narrow nose Short palpebral fissures Flat face |
Enamel hypoplasia (major phenotype, 40%) Microdontia (21%) Congenitally missing teeth (7%) Pulp stones (2%) |
High risk of dental caries secondary to enamel hypoplasia Thin dentinal walls |
(Loddenkemper et al., 2002) (Paznekas et al., 2009) (Harting et al., 2019) (Jensen, 2021) |
| Opitz BBB/G syndrome (145410) | SPECC1L | Sperm Antigen With Calponin Homology And Coiled-Coil Domains 1 Like | Actin cytoskeleton organization and microtubule stabilization | Cleft lip and palate (major phenotype, 50%) Hypertelorism Hooded eyelids Micrognathia |
Supernumerary teeth Malformed teeth |
Possibility of having mental retardation (above 50%) | (da Silva Dalben et al., 2008) (Regan et al., 2017) (Cammarata-Scalisi et al., 2018) |
| Orofaciodigital syndrome (311200) | OFD1 | OFD1 Centriole and Centriolar Satellite Protein | Regulation of Wnt signaling pathway | Cleft lip and palate (major phenotype) Hypertelorism Defect of the middle ear |
Congenitally missing teeth Supernumerary teeth Malocclusion Delayed tooth eruption Abnormal dentition Enamel hypoplasia Unerupted teeth |
High risk of having cleft lip or palate | (Gunbay et al., 1996) (Driva et al., 2004) (Belengeanu et al., 2019) |
| Osteogenesis imperfecta (166200) | COL1A1 | Collagen Type I Alpha 1 Chain | Formation of Type I collagen | Underdeveloped nasomaxillary complex | Malocclusion by malformation of the jaw (major phenotype) | Care must be taken in the use of orthodontic appliances because of the brittleness of the teeth | (Wieczorek & Loster, 2013) (Ibrahim et al., 2019) (Taqi et al., 2021) |
| (166210) (259420) (166220) |
COL1A2 | Collagen Type I Alpha 2 Chain | Hearing loss and otosclerosis Craniofacial growth deficiency |
Unerupted teeth Congenitally missing teeth Ectopic eruption Dentinogenesis imperfecta Crown fracture |
Early exposure to bisphosphonate treatment may increase the risk of developing unerupted teeth | ||
| Osteopetrosis (259700) | CLCN7 | Chloride Voltage-Gated Channel 7 | Transportation of chloride ions and transmission of electrical signals | Diffuse osteosclerotic lesions (major phenotype) | Congenitally missing teeth | Osteomyelitis often occurs after odontogenic infections and tooth extraction | (Lam et al., 2007) (Vinay et al., 2011) (Athanasiadou et al., 2020) |
| (259730) | TC/RG1 | T Cell Immune Regulator 1, ATPase H+ Transporting V0 Subunit A3 | Transportation of protons across the membrane | Jas fractures or osteomyelitis | Enamel hypoplasia | High risk of dental caries secondary to enamel hypoplasia | |
| (166600) | IKBKG | Inhibitor Of Nuclear Factor Kappa B Kinase Regulatory Subunit Gamma | Activation of NF-kappaB | Visual impairment from optic nerve compression Facial paralysis Macrocephaly |
Abnormal root morphology Delayed tooth eruption Rampant caries |
Many cases are first identified by dentists | |
| Pseudohypoparathyroidism (103580) | GNAS1 | GNAS Complex Locus | Regulation of the activity of hormones | Round facies | Pulp calcification (major phenotype, 76%) Enamel hypoplasia Abnormal root morphology Delayed tooth eruption |
Possibility of having diabetes mellitus or hypertension Possibility of having neurocognitive impairment |
(Reis et al., 2016) (Hejlesen et al., 2019) |
| Pycnodysostosis (265800) | CTSK | Cathepsin K | Regulation of bone remodeling | Diffuse osteosclerotic lesions (major phenotype) Short maxilla and mandible Deep and narrow palate Facial dysmorphia Long wide-based nose |
Malocclusion by malformation of the jaw Obliterated pulp chambers Enamel hypoplasia Delayed tooth eruption Dental crowding |
Osteomyelitis often occurs after odontogenic infections | (O’Connell et al., 1998) (Landa et al., 2000) (Alves Pereira et al., 2008) |
| Robinow syndrome autosomal dominant (180700) | WNT5A | Wnt Family Member 5A | Regulation of developmental pathways during embryogenesis | Macrocephaly Hypertelorism Wide and depressed nasal bridge Large mouth Prominent eyes |
Oligodontia (major phenotype) Supernumerary teeth Alveolar ridge deformation Dental crowding Delayed tooth eruption |
Possibility of having heart disease Possibility of having mental retardation (20%) |
(Nualart Grollmus et al., 2007) (Jain et al., 2017) (Cammarata-Scalisi et al., 2018) |
| Schwachman Diamond syndrome (260400) | SBDS | SBDS Ribosome Maturation Factor | Assembly of mature ribosomes and ribosome biogenesis | Ulceration of the oral mucosa | Congenitally missing teeth (major phenotype) Enamel hypoplasia |
Possibility of having blood disease (e.g., acute myelogenous leukemia, 15%−30%) | (Ho et al., 2007) (Dror et al., 2011) |
| Trichorhinophalangeal syndrome (190350) | TRPS1 | Transcriptional Repressor GATA Binding 1 | Regulation of chondrocyte proliferation and differentiation | Large and laterally protruding ears Bulbous nose Elongated upper lip Sparse scalp hair |
Supernumerary teeth (major phenotype) Congenitally missing teeth Enamel hypoplasia Taurodontism |
— | (Bauermeister & Letts, 1992) (Machuca et al., 1997) (Forys-Dworniczak et al., 2019) |
| X-linked hypophosphatemia (307800) | PHEX | Phosphate Regulating Endopeptidase Homolog X-Linked | Promotion of dentin mineralization and renal phosphate reabsorption | Not in particular | Enlarged dental pulp (major phenotype) Abnormal root morphology Enamel hypoplasia Delayed tooth eruption |
Risk of pulp necrosis without dental caries and trauma | (Zambrano et al., 2003) (Baroncelli et al., 2006) (Lee et al., 2017) |
2 |. MAJOR DENTAL PHENOTYPES IN RARE GENETIC BONE DISEASES
2.1 |. Delayed tooth eruption
Delayed tooth eruption is defined as the emergence of a tooth into the oral cavity significantly slower than the norm (Suri et al., 2004). Delayed eruption can cause an array of oral problems such as malocclusion, temporo-mandibular joint dysfunction, and masticatory dysfunction, which can affect psychological health and quality of life (QoL) into adulthood. Wise et al. elegantly outline how tooth eruption is critically dependent upon the presence of osteoclasts (Wise et al., 2002). Osteoclast precursors must be recruited into the dental follicle prior to the onset of tooth eruption. After fusion and differentiation into mature osteoclasts, they resorb alveolar bone to form an eruption pathway for the tooth to exit its bony crypt. Lack of functional osteoclasts can severely disrupt tooth eruption (Helfrich, 2005). Osteoclast formation is a complex process guided by multiple key factors. The chemokines tumor necrosis factor-alpha (TNF-α), interleukin-one alpha (IL-1α), and monocyte chemotactic protein-1 (MCP-1) recruit mononuclear cells to the dental follicle (Wise et al., 1999; Wise et al., 2002). After those osteoclast precursors have been recruited, the environment within the dental follicle must be suitable for promoting their fusion to form osteoclasts. This process requires macrophage colony-stimulating factor (M-CSF), receptor activator of nuclear factor kappa B (RANK), and receptor activator of nuclear factor kappa B ligand (RANKL) (Helfrich, 2003; Wise et al., 1999). Moreover, abnormal expression of or mutations in the genes coding for cellular Src kinase (c-SRC), TNF receptor-associated factor 6 (TRAF6), tartrate resistant acid phosphatase (TRAP), and cathepsin K (CTSK) can block the development or function of mature osteoclasts and therefore their ability to resorb alveolar bone, resulting in delayed tooth eruption (Helfrich, 2003, 2005). Disorders associated with delayed tooth eruption include cleidocranial dysplasia (CCD), cranio-metaphyseal dysplasia (CMD), osteopetrosis, orofaciodigital syndrome, pseudohypoparathyroidism, and pycnodysostosis (PYCD) (Table 1). We will discuss two of those disorders in more detail.
CCD (OMIM 119600) is a rare autosomal dominant bone disease characterized by craniofacial and dental phenotypes, which include increased head circumference, large fontanelles, delayed tooth eruption, supernumerary teeth, or malformed teeth (Callea et al., 2012; Cooper et al., 2001; Thaweesapphithak et al., 2022) (Figure 1). Long bone phenotypes include hypoplastic or absent clavicles and malformations of the hand. The prevalence is one in a million live births (Dhiman et al., 2014; orpha.net). It affects both sexes with equal frequency; there is no sex or ethnicity predilection (Paul et al., 2015). Dental abnormalities are a major feature of CCD and occur in 93.5% of affected patients (Dhiman et al., 2014). CCD is caused by heterozygous loss of function in the Runt-related transcription factor 2 (RUNX2) gene. RUNX2 mutations disturb bone remodeling during tooth eruption through the RANK/RANKL/osteoprotegerin (OPG) signaling pathway and thus lead to delayed and abnormal tooth eruption in CCD patients (Liu et al., 2019).
FIGURE 1.
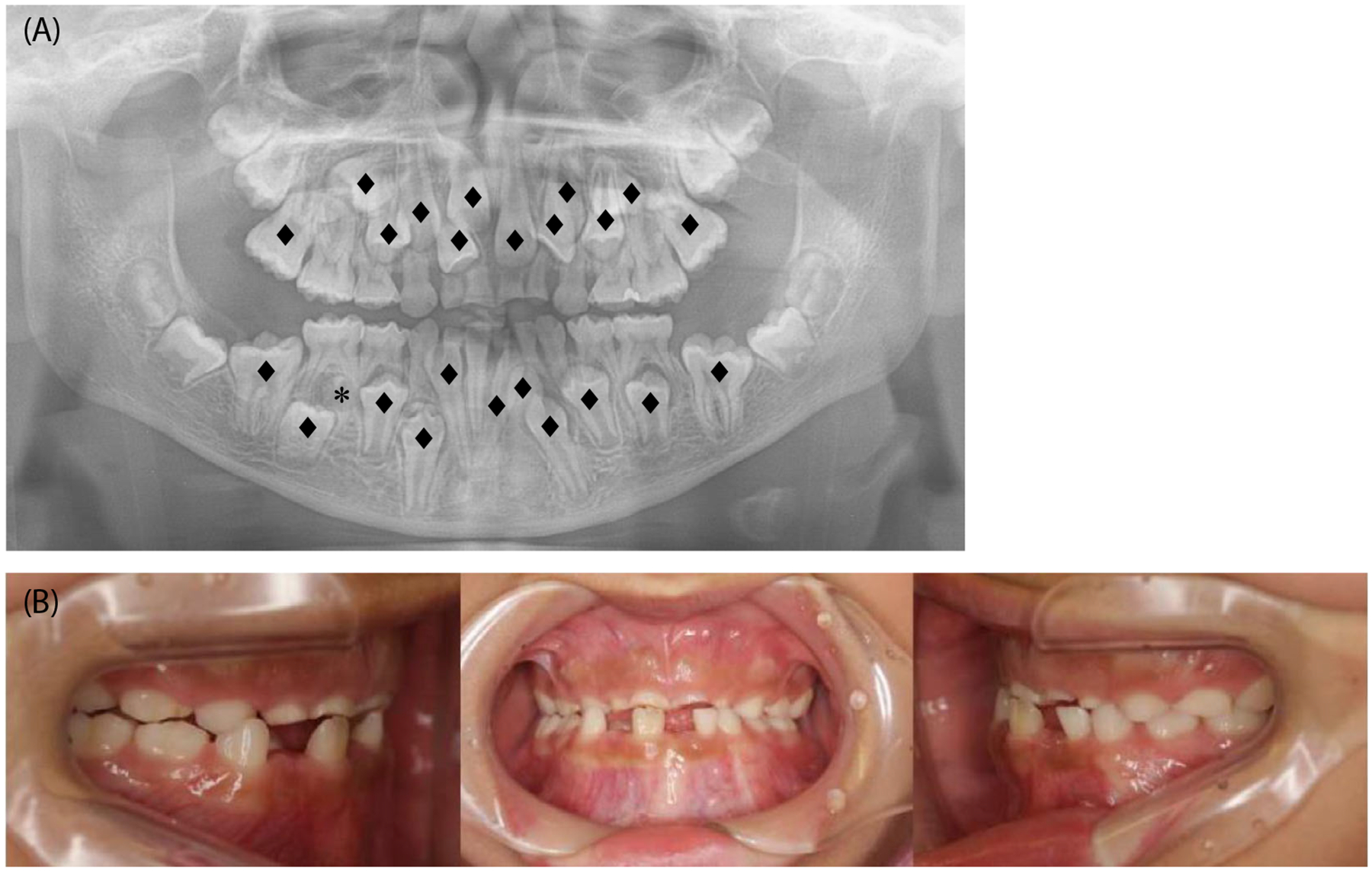
Intraoral images of a patient with cleidocranial dysplasia. (A) Orthopantomogram showing delayed tooth eruption of multiple teeth (♦) and a supernumerary tooth (*) of a 12-year-old patient. (B) Intraoral photographs showing retained deciduous teeth and class III malocclusion.
Osteopetrosis is a general term for a group of genetic diseases characterized by increased bone mass and bone density due to defective bone resorption (Stark & Savarirayan, 2009). Patients often suffer from progressive deafness, macrocephaly, visual impairment caused by optic nerve compression, delayed tooth eruption, congenitally missing teeth, and abnormal root morphology (Athanasiadou et al., 2020; Lam et al., 2007; Vinay et al., 2011). Osteopetrosis can be classified into three types on the basis of severity of symptoms and secondary clinical features, age of onset and mode of inheritance: autosomal recessive osteopetrosis (ARO; OMIM 259700), which is the most severe form; intermediate autosomal recessive osteopetrosis (IARO; OMIM 259730); and autosomal dominant osteopetrosis (ADO; OMIM 166600) (Palagano et al., 2018; Sobacchi et al., 2013). ADO affects approximately one in 20,000 people, while ARO affects approximately one in 250,000. Other types of osteopetrosis are less frequent. However, mutations in the Chloride Voltage-Gated Channel 7 (CLCN7) gene, a member of the voltage-gated chloride channel family, are involved in all three major types. CLCN7 mutations affect the differentiation of ameloblasts, odontoblasts, and dental follicle cells and also the interaction between dental follicle cells and osteoclasts via the RANKL/OPG pathway, resulting in delayed tooth eruption owing to lack of or reduction of bone resorption.
Delayed eruption and shedding of primary teeth have also been studied in CMD (Chen et al., 2014). A hallmark of autosomal dominant CMD is the progressive thickening of craniofacial bones. Hyperostosis affects all craniofacial bones and leads to obstruction of cranial foramina, which then results in progressive loss of vision, sensorineural or conductive hearing loss, and facial palsy. Dense and hyperostotic mandibular and maxillary bone causes an approximately three-year delay in tooth eruption, and sometimes the insufficient bone remodeling results in non-erupting primary or secondary teeth. ANKH is a transporter of small molecules including adenosine triphosphate and citrate (Szeri et al., 2020; Szeri et al., 2022) and an important regulator of mineralization in all tissues. CMD mutations in ANKH are clustered toward the C-terminus and include single amino acid changes, in-frame insertions, and in-frame amino acid deletions. These mutations cause reduced levels of pyrophosphate, a potent inhibitor of mineralization, and disrupt osteoclast formation. The few osteoclasts that form have reduced ability to resorb bone. Concerns about obstructed secondary teeth have caused dentists to extract primary teeth prematurely in children with CMD. Considering the approximately three-year delay in tooth development, deferral of extracting the deciduous teeth is recommended in most cases (Chen et al., 2014). Orthodontic treatment is needed in many children with CMD owing to tooth misalignment, and special attention should be given to potential root resorption caused by orthodontic force (Chen et al., 2014).
Treatment of delayed primary tooth eruption is mostly observational. Sometimes the physical obstruction needs to be removed with or without exposure of the affected tooth; orthodontic treatment can be needed, or the involved tooth has to be extracted (Flaitz & Hicks, 2001; Stephen et al., 2001). Some surgical approaches have been recommended for uncovering impacted teeth. These include gingivectomy, apically positioned flap, flap/closed eruption, and preorthodontic uncovering. When a deciduous tooth becomes a physical barrier to the eruption of the permanent tooth (Seehra et al., 2018; Xu et al., 2022), removing it allows the successor to erupt spontaneously. When arch length deficiency creates a physical obstruction, either expansion of the dental arches or extraction can be necessary to create the required space. Either the affected tooth or adjacent teeth can be extracted. Helfrich et al. reported that hematopoietic stem cell transplantation (HSCT) in osteopetrosis patients can improve tooth eruption by normalizing osteoclast function (Helfrich, 2005); however, there are caveats to consider such as the timing of HSCT because this therapy can depend upon the age of the patient (Detailleur et al., 2016).
2.2 |. Congenitally missing teeth
Congenitally missing teeth can cause malocclusion, periodontal injury, insufficient alveolar bone growth, reduced chewing ability, inarticulate pronunciation, and unfavorable appearance. Tooth agenesis can occur as part of a disease spectrum or be non-syndromic and result from molecular disturbances during early stages of development (Aktan et al., 2010). Hypodontia has been described as mild dysplasia of the ectoderm (Fekonja, 2005, 2017; Graber, 1978). The mode of inheritance is probably polygenic, with epistatic genes and environmental factors exerting some influence on the phenotypic expression of genes involved in tooth development by disturbing the tooth germ during the initial stage of formation (Thesleff, 2000, 2006; Varela et al., 2009). Mutations in multiple genes cause non-syndromic tooth agenesis, such as MSH homeobox 1 (MSX1), paired box gene 9 (PAX9), axis inhibition protein 2 (AXIN2), and ectodysplasin A (EDA) (Nieminen, 2009; Ye & Attaie, 2016). Generally, the missing tooth pattern correlates with the causative gene. Second bicuspids and third molars are found in MSX1-associated tooth agenesis (Lidral & Reising, 2002). PAX9 mutations lead to agenesis of second bicuspids, second molars, and some central incisors (Kim et al., 2006). AXIN2 aberrations cause multiple missing teeth, and EDA-associated tooth agenesis is more likely to include multiple anterior teeth (Han et al., 2008). Congenitally missing teeth are syndromic with more than 50 genetic disorders (AlShahrani, 2013) including cherubism, ectodermal dysplasia, Fraser syndrome (FS), oculodentodigital dysplasia (ODDD), oligodontia (Figure 2), osteogenesis imperfecta (OI), osteopetrosis, and trichorhinophalangeal syndrome (Table 1). We will discuss two of these in detail.
FIGURE 2.
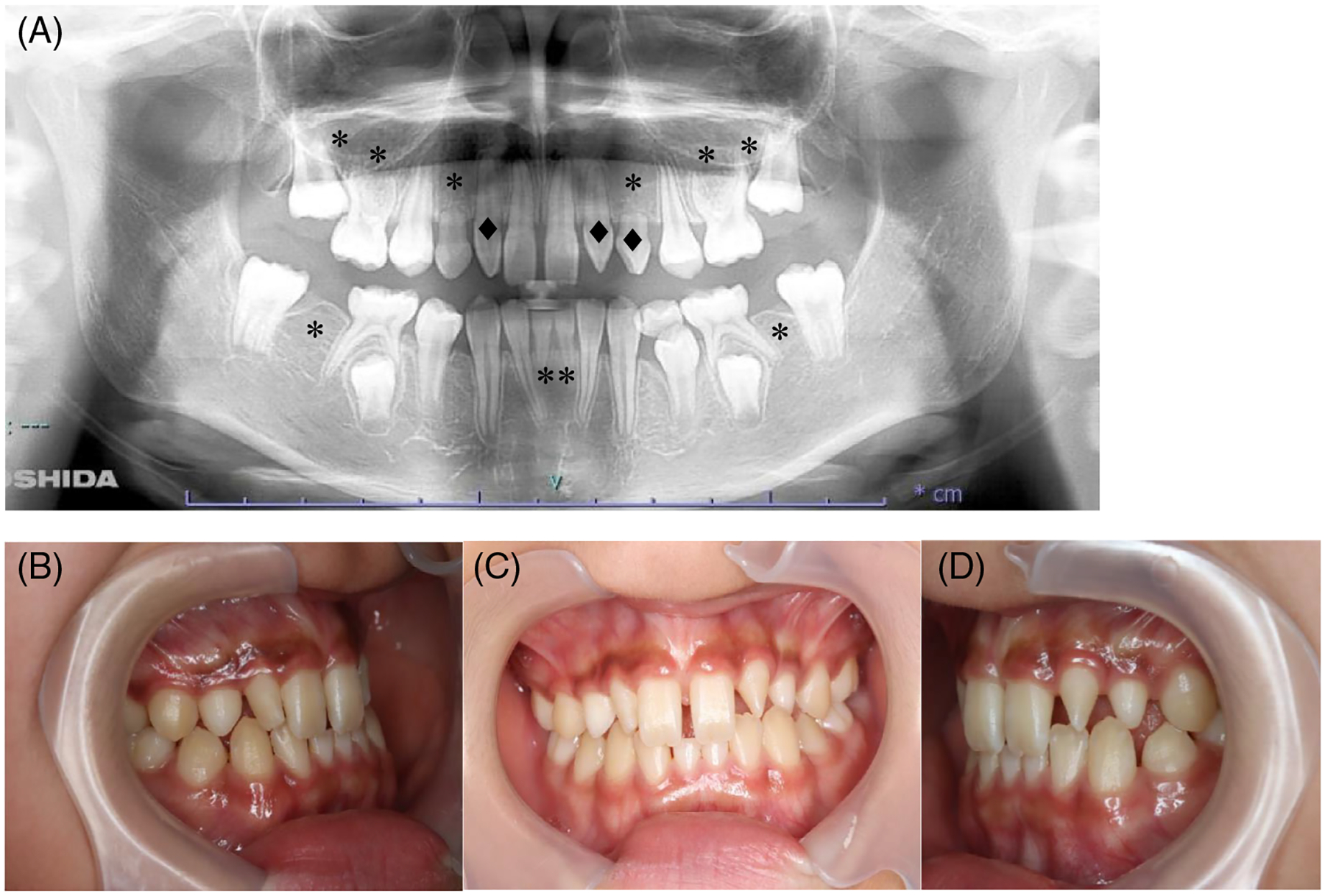
Intraoral images of a patient with oligodontia. (A) Orthopantomogram showing ten congenitally missing teeth (*) and three conical teeth (♦) of a 10-year-old patient. (B) Intraoral photographs.
OI (OMIM 166200, 166210, 259420, 166220) is the general term for a group of hereditary disorders affecting the composition and organization of type I collagen, mostly COL1A1 and COL1A2, which leads to compromised bone and collagen-rich tissues. The incidence is approximately one in 15,000–20,000 births and race is irrelevant (Hari Gopal & Adams, 2023). OI patients are classified as types I, II, III, and IV using the Sillence classification (Sillence & Rimoin, 1978). OI type I is mild, type II is pre- or peri-natally lethal, type III is the more severe and progressively deteriorating type, and type IV is typically of moderate severity (Rauch & Glorieux, 2004). OI patients often have short stature, osteopenia, progressive bone deformities, and fractures. There can also be craniofacial and extra-skeletal manifestations such as blue sclera, hyperlaxity of skin, and ligaments, underdeveloped nasomaxillary complex, progressive hearing loss, dentinogenesis imperfecta, congenitally missing teeth, or malocclusion (Ibrahim et al., 2019; Taqi et al., 2021; Wieczorek & Loster, 2013). Missing or unerupted teeth are common in patients with OI and a previous report showed that the teeth most frequently missing were the premolars, second molars, first molars, and canines (Taqi et al., 2021).
FS (OMIM 219000, 617666, 617667) is a rare autosomal recessive multiple congenital malformation syndrome characterized by cutaneous syndactyly, renal agenesis, ambiguous genitalia, and craniofacial phenotypes such as cleft lip and palate, congenital deafness, and dental abnormalities including congenitally missing teeth, microdontia, and short roots (Gallottini et al., 2018; Keene & Day, 2011; Kunz et al., 2020) (Table 2). The estimated prevalence is around 200,000 live-born infants and 1:10,000 stillbirths (medlineplus.gov). FS is caused by compound heterozygous or biallelic mutations of the Fraser extracellular matrix complex subunit 1 (FRAS1), related extracellular matrix protein 2 (FREM2), and glutamate receptor interacting protein 1 (GRIP1) genes, encoding components of a protein complex that mediates embryonic epithelial-mesenchymal interactions during dental crown and root development. Kunz et al. reported hypodontia in 10 unrelated patients with FS of different genetic etiologies (Kunz et al., 2020).
Orthodontic treatment is needed in many instances of missing teeth to maintain functionality and prevent malocclusion, and for esthetic reasons (Nunn et al., 2003). While treatment is normally initiated during adolescence, interim treatment can be advisable at an earlier age if the psychological well-being of the child is affected. The edentulous space can be either left open for prosthetic restoration or closed by orthodontic means (Kokich & Kokich, 2006). Alternatively, auto-transplantation or protraction of developing third molars should be considered in treatment plans for orthodontic space management (Bauss et al., 2002; Chung et al., 2007). Treatment of more complex cases requires the cooperation of an interdisciplinary team that includes pediatric dentists, orthodontists, prosthodontists, and oral maxillofacial surgeons (Nunn et al., 2003; Tuverson, 1980).
2.3 |. Supernumerary teeth
Supernumerary teeth or tooth-like structures can erupt or remain unerupted in addition to the 20 primary or 32 permanent teeth. The incidence has been reported as 0.1%–3.8% and the condition is approximately twice as common in males as females (Parolia et al., 2011); it was reported to be four times higher in a recent hospital-based study (Cheng et al., 2022). Supernumerary teeth can be morphologically normal but supplemental, conical, or tuberculate in appearance, or display as odontomata. Several molecular signaling pathways known to be involved in normal tooth germ development give rise to supernumerary teeth if inappropriately regulated, such as the fibroblast growth factor (FGF), bone morphogenetic protein (BMP), sonic hedgehog (SSH), and wingless-related (WNT) pathways. Supernumerary teeth are syndromic, with numerous disorders including CCD (Figure 1), orofaciodigital syndrome, Robinow syndrome autosomal dominant, and trichorhinophalangeal syndrome. We will discuss one of those disorders in detail.
The autosomal dominant form of Robinow syndrome is a rare genetic bone disease caused by missense mutations in the secreted protein WNT5A (OMIM 180700) or in five other genes in the WNT/planar cell polarity signaling pathway (Zhang et al., 2022). Robinow syndrome has an incidence of 1:500,000 and a 1:1 male-to-female ratio. Despite its rarity, the prevalence is lower because of infant or early childhood mortality (Soman & Lingappa, 2015). WNT5A regulates growth, patterning, and odontoblast differentiation during odontogenesis by modulating Wnt/β-catenin canonical signaling (Lin et al., 2011). Robinow syndrome is associated with a spectrum of skeletal, neurological, dermatological, genitourinary, and cardiovascular symptoms. Patients also have short stature and a characteristic facial appearance such as macrocephaly, hypertelorism, wide and depressed nasal bridge, and dental phenotypes such as oligodontia and supernumerary teeth (Cammarata-Scalisi et al., 2018; Jain et al., 2017; Nualart Grollmus et al., 2007) (Table 2). Some patients have infranumerary teeth, while approximately 50% with WNT5A mutations have supernumerary teeth (Beiraghi et al., 2011; Zhang et al., 2022).
Treatment of supernumerary teeth depends on the type and position of those teeth and on their effect on adjacent teeth. Removal of them is recommended when: (1) central incisor eruption has been delayed or inhibited; (2) displacement of central incisors is evident; (3) they are accompanied by dentigerous cyst formation; (4) there is active orthodontic alignment of an incisor in close proximity to a supernumerary; (5) their presence would compromise secondary alveolar bone grafting in cleft lip and palate patients; (6) they are present in bone designated for implant placement; (7) they have erupted spontaneously (Garvey et al., 1999; Parolia et al., 2011; Shah et al., 2008). Removal of supernumerary teeth preventing permanent tooth eruption usually results in eruption of the tooth, provided adequate space is available in the arch to accommodate it (Mitchell & Bennett, 1992). Di Biase reported that 75% of incisors erupted spontaneously after a supernumerary tooth was removed (Di Biase, 1971). If there is adequate space in the arch for the unerupted incisor following supernumerary removal, the space can be maintained by fitting a simple removable appliance. If it is inadequate, the adjacent teeth need to be moved distally to create space for incisor eruption.
2.4 |. Enamel hypoplasia
Enamel formation is a complex developmental process. Enamel hypoplasia (Figure 3) is syndromic with a significant number of genetic disorders (Wright et al., 2015). Characteristic of this dental phenotype are irregular-shaped teeth with reduced quantity of enamel, which can be pitted or thinner, or the teeth can be smaller. Organic components of enamel are produced by ameloblasts and consist mostly of amelogenin (AMEL), ameloblastin (AMBN), enamelin (ENAM), amelotin (AMTN), and odontogenic ameloblast-associated protein (ODAM). Mutations in or deletions of genes encoding proteins such as AMEL, AMBN, ENAM, and matrix metalloproteinase 20 (MMP20; enamelysin), which are secreted by ameloblasts, have been reported to cause enamel hypoplasia (Aldred et al., 1992; Gasse et al., 2013; Nakayama et al., 2015; Poulter et al., 2014; Rajpar et al., 2001). Ectodermal dysplasia, Ellis-Van Creveld syndrome, Kenny-Caffey syndrome, ODDD, orofaciodigital syndrome, and PYCD are some of the disorders with enamel hypoplasia in their phenotype spectrum. We will discuss two of those disorders in detail.
FIGURE 3.
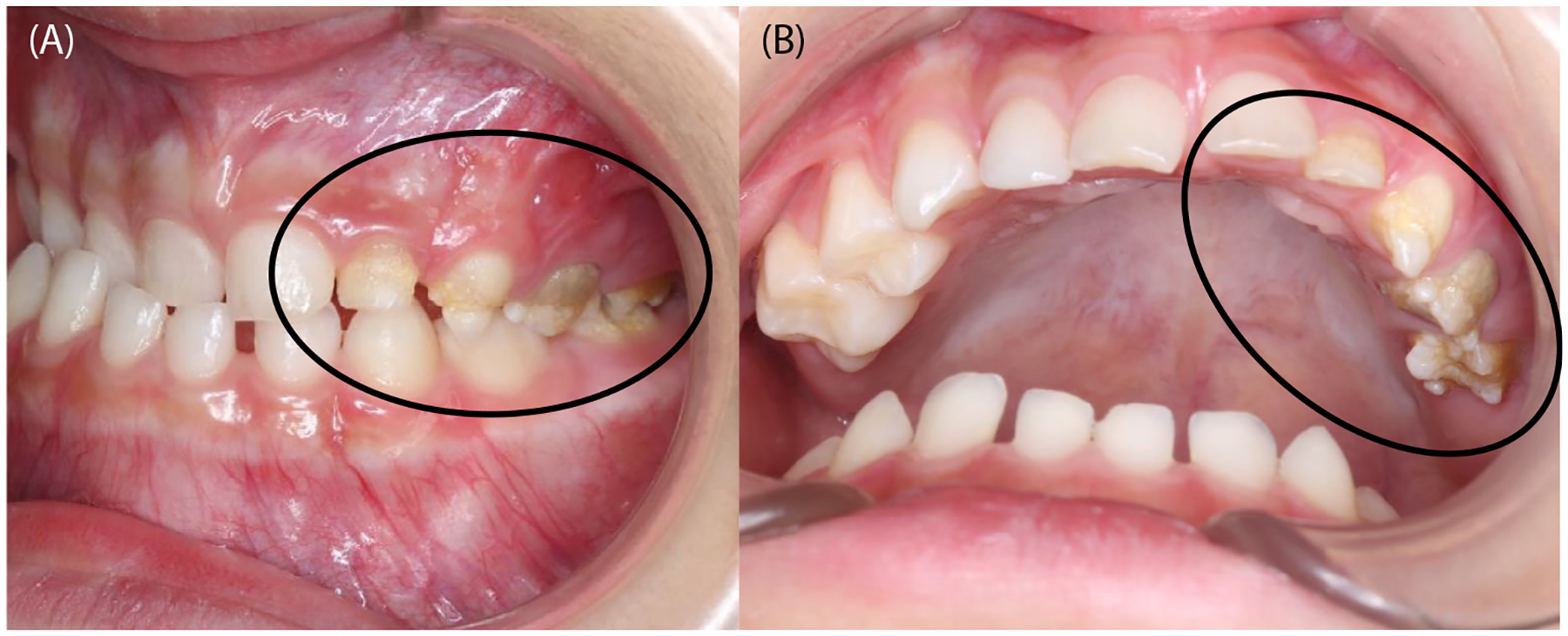
Intraoral images of a patient with enamel hypoplasia (circles).
PYCD (OMIM 265800) is a rare autosomal recessive skeletal dysplasia characterized by generalized progressive osteosclerosis due to the absence of active CTSK (Hald et al., 2023). The incidence of this disease is 11 in 1.7 million births with a male to female ratio of 1:1. Thirty percent of cases arise in consanguineous marriages (Aziz et al., 2022). Patients often have facial dysmorphia, a long wide-based nose, and a thin mandible, along with enamel hypoplasia, delayed tooth eruption, or malocclusion (Alves & Cantin, 2014; O’Connell et al., 1998). CTSK is a member of the cysteine proteinase family and is predominantly expressed in osteoclasts for degrading bone matrix proteins. It also takes part in mineralization during odontogenesis. Jiang et al. found that CTSK can hydrolyze AMEL, which is one of the most important matrix proteins in developing enamel (Jiang et al., 2017). Soliman and co-authors reported that seven out of eight pediatric patients with PYCD developed enamel hypoplasia (Soliman et al., 2001).
ODDD(OMIM 162400) is a rare congenital autosomal dominant disease characterized by developmental abnormalities of the face, eyes, teeth, and limbs. Jensen (2021) reported that the dental phenotype includes enamel hypoplasia (40%) with subsequent caries, microdontia (21%), congenitally missing teeth (7%), and pulp stones (2%). ODDD has been diagnosed in fewer than 300 people worldwide, with an estimated incidence of one in 10 million (Doshi et al., 2016). It is caused by mutations in the gap junction alpha1 gene, which encodes the gap junction protein connexin 43 (Cx43) (Paznekas et al., 2009). Gap junctions are important for direct cell–cell communication, propagation of electric signals in cardiac tissue, and regulation of cell growth and differentiation in general. In human teeth, Cx43 is expressed in epithelial cells including pre-ameloblasts, stratum intermedium, stellate reticulum, and differentiating odontoblasts at the bell stage (About et al., 2002). In addition, a mouse model of ODDD expressing mutant Cx43 exhibits a disorganized ameloblast layer and abnormal expression of AMEL (Toth et al., 2010).
For treating enamel hypoplasia, vital bleaching, micro-abrasion, and resin infiltration are not usually appropriate because the main issue is not the color but the tooth shape, or susceptibility to dental caries secondary to enamel breakdown. In anterior teeth, if the teeth are asymptomatic and the patient has no esthetic concerns, treatment is not necessarily indicated (Patel et al., 2019). However, hypoplastic teeth can appear smaller and could be an esthetic concern for the patient. In this case, the addition of direct composite restorations could be considered as this is minimally invasive. In molars, sealants or composite restorations can be an option. If the whole tooth surface is significantly affected, preformed metal crowns, composite restorations, or adhesive onlays can be considered. If teeth have severe caries, extraction can be necessary.
3 |. INFLUENCE OF THESE DISEASES ON DENTAL TREATMENT
The dental treatment of many rare genetic bone disorders that affect oral and craniofacial features is not well established because of their low prevalence and the variety of signs and symptoms experienced by patients. Some of these rare disorders entail emotional or psychological stress, delayed or reduced intellectual development, loss of vision or hearing, and other often serious comorbidities. Treatment challenges increase when patient cooperation in pediatric patients or syndromic medical conditions require special attention (Beltrame et al., 2017; Muhney & Campbell, 2007). Unfortunately, some dentists deny treatment owing to lack of knowledge or fear of causing injury. As a result, patients and their families can have difficulties in finding appropriate dental care. There are very few publications on dental treatment of patients with rare diseases; however, most reports describe the requirement for a team approach involving an orthodontist, prosthodontist, and an oral surgeon, with good cooperation and communication from the patient (Paul et al., 2015). Herein, we elaborate dental treatment methods for some representative diseases.
3.1 |. Cleidocranial dysplasia
As the dental phenotype, delayed tooth eruption, and supernumerary teeth are well known (Table 2, Figure 1). Dental treatment involves restoration of the deciduous teeth when they develop cavities, because extraction of the affected teeth does not necessarily induce the eruption of permanent teeth. Orthodontic treatment is usually indicated to direct the eruption of the malpositioned and often impacted teeth. Treatment is likely to involve orthognathic surgery to address maxillary hypoplasia, and it can be necessary to extract some supernumerary teeth. Impacted teeth can be removed surgically in association with orthodontic and/or prosthetic therapy. Dentures can be fabricated over unerupted teeth. Considering the foregoing, Paul et al. proposed a dental treatment protocol (Paul et al., 2015). In short, tooth extraction is proposed for retained deciduous teeth, supernumerary teeth, and abnormal permanent teeth. Surgical exposure or orthodontic eruption is proposed for unerupted teeth, and surgical translocation and/or auto-transplantation is proposed for malocclusion (Paul et al., 2015).
3.2 |. Osteogenesis imperfecta
Patients with OI frequently present with congenitally missing teeth, unerupted teeth, and malocclusions (Table 2). Taqi et al. reported an analysis of 144 patients with OI showing that the prevalence of missing teeth was much higher than in the general population (6.4%) and was associated with disease severity. Indeed, patients with moderate to severe OI had a much higher prevalence of missing teeth (type IV 52%, type III 61%) than those with OI type I (11%) (Taqi et al., 2021). They also reported that 33% of OI patients had at least one unerupted tooth and that the prevalence of tooth eruption was associated with disease severity because type III patients showed the highest prevalence of unerupted teeth (70%), followed by type IV (40%), and type I (30%) (Taqi et al., 2021). Thus, treatment is challenging because the dental phenotypes vary according to the type of OI (Marini & Dang Do, 2000; Okawa et al., 2017; Rapoport et al., 2023). Compared to patients with type I OI, types III, and IV patients require more special intervention in their primary dentition because of more severe dentinogenesis imperfecta. Caries progression is initially slow owing to the smaller amount and irregular nature of dentinal tubules or fast abrasion of the exposed dentine. Early tooth rehabilitation prevents significant tooth abrasion and restores function and esthetics to the teeth (Beltrame et al., 2017). Direct composite restorations or indirect fabricated composite resin restorations have been recommended (Sanches et al., 2005). There are malocclusions in many OI patients and the incidence of class III malocclusion is especially high (70%–80%) in types III and IV OI (Ríos-Rodenas et al., 2015). Because orthognathic surgery can be needed, early intervention by orthodontic specialists is crucial for treating malocclusion in OI patients. In instances of ectopic eruptions, extraction of the corresponding primary teeth is recommended. In recent years, many OI patients have been treated with bisphosphonates (BPs) to reduce pain and improve mobility. BPs slow the rate of bone resorption by promoting an increase in bone mass and thus decrease bone fragility. However, prolonged BP treatment carries the risk of BP-related osteonecrosis of the jaw (BRONJ) or even drug-induced osteopetrosis (Khan et al., 2015; Reyes et al., 2016; Whyte et al., 2023). Fortunately, there are no reports of BRONJ after extractions in children with OI taking BPs, probably because of the lack of bone tissue surrounding the primary teeth (Okawa et al., 2017). It is therefore proposed that BP treatment does not need to be suspended when OI patients undergo primary tooth extraction (Okawa et al., 2017).
3.3 |. Oculodentodigital dysplasia
Enamel hypoplasia is the major dental phenotype in ODDD (Jensen, 2021; Thomsen et al., 1998) (Table 2). Unfortunately, there are few case reports of dental treatment for patients with this condition (Aminabadi et al., 2010; Dean et al., 1986) (Tables 1 and 2). In many cases, the management of odontodysplasia including enamel hypoplasia is controversial: should extraction or conservative therapy be pursued? Although many dentists prefer to extract as soon as a diagnosis of odontodysplasia is made, some prefer to retain the teeth in children until skeletal growth is complete, provided the teeth are free of infection (Dean et al., 1986). Aminabadi et al. reported conservative therapy in a patient whose permanent teeth all encountered enamel hypoplasia, enlarged pulp cavities, thin dentinal walls, and multiple pulp exposures with open apices. In the anterior teeth, to protect the defective enamel and dentine and to enhance esthetics, complete crown coverage was selected using direct composite restorations. In teeth with pulpal involvement, pulp therapy was used with Mineral Trioxide Aggregate as an apical seal and subsequent final obturation. Final treatment of the posterior teeth was more challenging. Stainless steel crowns (SSC) were used to protect against their susceptibility to caries and fracture. Because the dentinal walls were thin, it was not feasible to prepare the teeth for cast restoration. Also, full coverage with composite restoration is prone to wear so it was not a suitable final restoration. Jensen (2021) also reported dental treatment with SSC and showed that a combination of enamel hypoplasia, poor oral hygiene, and parental neglect can lead to extensive destruction of tooth structure, so treatment options become limited. Because variability is expected in view of the nature of genetic mutations and individual disease progression, thorough knowledge of the respective rare genetic disorders and the expected treatment outcomes are essential for developing treatment plans adapted to the needs of individual patients.
4 |. INFLUENCE OF THESE DISEASES ON PATIENTS’ OHR-QOL
Finally, we focus on the oral health-related quality of life (OHR-QoL) of patients to understand their needs better. The effect of QoL on disease outcome is receiving increased attention in medical fields. However, poor health or presence of disease does not inevitably mean poor QoL. This can also apply to dental phenotypes and their consequences for OHR-QoL. The OHR-QoL term is defined as the functional and psychosocial outcomes of oral disorders. Its measurement covers four aspects: (1) functional factors; (2) mastication, utterance, and psychological factors; (3) esthetic, social, and communication-related factors; and (4) pain and discomfort-related factors (Naito et al., 2006).
Children and adult OHR-QoL measures are different (Broder et al., 2007; Slade & Sanders, 2011). There are various measurement methods for determining OHR-QoL in children. One of the first of these used for adolescents is the Oral Health Impact Profile-14 (OHIP-14) (Slade, 1997). In 2002, the Child Perception Questionnaire (CPQ) was developed and subsequently elaborated in the Child Oral Health Impact Profile (COHIP) (Broder et al., 2007). Then in 2004, the Child Oral Impact of Daily Performances (OIDP) was derived from its adult form (Gherunpong et al., 2004). In 2007, a measurement method for very young children was developed, the Early Childhood Oral Health Impact Scale (ECOHIS) (Pahel et al., 2007). So far, at least eight reports have investigated OHR-QoL in patients with rare genetic bone diseases (Aarts et al., 2023; Gjørup et al., 2021; Hanisch, Bohner, et al., 2019; Hanisch, Sielker, et al., 2019; Najirad et al., 2018, 2020; Nguyen et al., 2019; Oelerich et al., 2020) (Table 3). The most frequently used method for measuring OHR-QoL is OHIP-14, a shortened form of the original OHIP-49 that captures seven domains of OHR-QoL with two items per domain: functional limitation, physical pain, psychological discomfort, physical disability, psychological disability, social disability, and handicap. All items are presented with a five-category rating scale of frequency. The higher the OHIP-14 value, the lower the OHR-QoL. The next most frequently used methods are age-specific versions (8–10 years and 11–14 years) of CPQ. The CPQ8–10 comprising 25 questions was used for children between eight and ten years of age (Jokovic et al., 2004) and the CPQ11–14 comprising 37 questions was used for individuals aged 11–14 years (Jokovic et al., 2002). These measurement methods comprised four health domains: oral symptoms, functional limitation, emotional well-being, and social well-being related to oral health conditions. All questions relate to the frequency of events in relation to the condition of the mouth or teeth over the previous four weeks (CPQ8–10) or three months (CPQ11–14). According to Najirad and colleagues, the severity of OI affects OHR-QoL in adolescents aged 11–14 years, but not in children aged 8–10 years (Najirad et al., 2018). In addition, adolescent OI patients aged 11–14 years with posterior crossbite or open bite had statistically significantly higher CPQ11–14 scores than those without (Najirad et al., 2020). X-linked hypophosphatemia (XLH; OMIM 307800) is caused by a mutation in the X-linked phosphate-regulating neutral endopeptidase (PHEX) gene found on chromosome Xp22 and characterized by an insufficient mineralization of bones and dental tissues due to abnormal renal phosphate wasting. XLH is usually characterized by bone deformities, small body size, and dental anomalies (enamel hypoplasia, delayed tooth eruption). This is due to reduced renal phosphate reabsorption, which results in hypophosphatemia and reduced bone and tooth mineralization (Gaucher et al., 2009). According to Hanisch, the OHIP-14 scores of XLH patients were higher than those obtained from the general population (Hanisch, Bohner, et al., 2019). Moreover, Gjørup et al. reported that adults with XLH experience a more negative effect on their OHR-QoL than adults with OI (Gjørup et al., 2021). As in other diseases, the OHIP-14 scores of the patients were lower than in the general population; the difficulty in finding a dentist for treatment, or dissatisfaction with the health system, contributed to the negative effect on OHR-QoL in some of the aforementioned studies (Gjørup et al., 2021; Hanisch, Bohner, et al., 2019; Hanisch, Sielker, et al., 2019; Nguyen et al., 2019; Oelerich et al., 2020).
TABLE 3.
Summary of the studies reporting OHR-QoL of the patients with rare genetic bone diseases.
| Author/year | Disease | OHR-QoL | Patient number | Results |
|---|---|---|---|---|
| (Najirad et al., 2018) | Osteogenesis imperfecta | CPQ8–10 and CPQ11–14 | 138 | Significantly worse in patients with type III, IV than those with type I (only CPQ11–14) |
| (Najirad et al., 2020) | Osteogenesis imperfecta | CPQ8–10 and CPQ11–14 | 138 | Significantly worse in patients with malocclusion than those without (only CPQ11–14) |
| (Hanisch, Bohner, et al., 2019) | X-linked hypophosphatemia | OHIP-14 | 43 |
|
| (Gjørup et al., 2021) | X-linked hypophosphatemia and Osteogenesis imperfecta | OHIP-49 | 35 and 71 | Significantly worse in patients with XLH than those with OI |
| (Oelerich et al., 2020) | Ehlers-Danlos syndrome | OHIP-14 | 46 |
|
| (Hanisch, Sielker, et al., 2019) | Ectodermal dysplasia | OHIP-14 | 110 | Significantly worse than general population |
| (Nguyen et al., 2019) | Loeys-Dietz syndrome | OHIP-14 | 33 | Significantly worse than general population |
| (Aarts et al., 2023) | Oligodontia | FACE-Q Dental | 62 | Significantly worse than general population |
5 |. CONCLUSIONS
The aim of this report is to provide a systematic review of the variability of dental phenotypes associated with rare genetic disorders and their effects on dental treatment. A good understanding of such rare diseases is important for dentists to prevent delayed diagnosis or treatment and to achieve a higher OHR-QoL for the patients affected.
ACKNOWLEDGEMENTS
This work was supported by NIH/NIDCR grant R01DE025664 to IPC.
REFERENCES
- Aarts M, Mettenberger S, Bronkhorst EM, & Ongkosuwito EM (2023). Oral health-related quality of life in patients with oligodontia: A FACE-Q assessment. Journal of Dentistry, 135, 104544. 10.1016/j.jdent.2023.104544 [DOI] [PubMed] [Google Scholar]
- About I, Proust JP, Raffo S, Mitsiadis TA, & Franquin JC (2002). In vivo and in vitro expression of connexin 43 in human teeth. Connective Tissue Research, 43(2–3), 232–237. 10.1080/03008200290000952 [DOI] [PubMed] [Google Scholar]
- Aktan AM, Kara S, Akgünlü F, & Malkoç S (2010). The incidence of canine transmigration and tooth impaction in a Turkish subpopulation. European Journal of Orthodontics, 32(5), 575–581. 10.1093/ejo/cjp151 [DOI] [PubMed] [Google Scholar]
- Aldred MJ, Crawford PJ, Roberts E, & Thomas NS (1992). Identification of a nonsense mutation in the amelogenin gene (AMELX) in a family with X-linked amelogenesis imperfecta (AIH1). Human Genetics, 90(4), 413–416. 10.1007/bf00220469 [DOI] [PubMed] [Google Scholar]
- AlShahrani I (2013). A review of hypodontia: Classification, prevalence, etiology, associated anomalies, clinical implications and treatment options. World Journal of Dentistry, 4, 117–125. [Google Scholar]
- Alves N, & Cantin M (2014). Clinical and radiographic maxillofacial features of pycnodysostosis. International Journal of Clinical and Experimental Medicine, 7(3), 492–496. [PMC free article] [PubMed] [Google Scholar]
- Alves Pereira D, Berini Aytés L, & Gay Escoda C (2008). Pycnodysostosis. A report of 3 clinical cases. Medicina Oral, Patologia Oral y Cirugia Bucal, 13(10), E633–E635. [PubMed] [Google Scholar]
- Aminabadi NA, Pourkazemi M, Oskouei SG, & Jamali Z (2010). Dental management of oculodentodigital dysplasia: A case report. Journal of Oral Science, 52(2), 337–342. 10.2334/josnusd.52.337 [DOI] [PubMed] [Google Scholar]
- Athanasiadou E, Vlachou C, Theocharidou A, Tilaveridis I, Vargiami E, Antoniadis K, & Arapostathis K (2020). When a pedodontic examination leads to the diagnosis of osteopetrosis: A case report. Special Care in Dentistry, 40(1), 113–120. 10.1111/scd.12427 [DOI] [PubMed] [Google Scholar]
- Aziz F, Farida F, & Bashir N (2022). Pycnodysostosis; A rare disease case report. Journal of Ayub Medical College Abbottabad, 34(1), 216–219. 10.55519/JAMC-01-10336 [DOI] [PubMed] [Google Scholar]
- Baart JA, & van Hagen JM (2000). Syndromes 21: Ellis-Van Creveld syndrome. Nederlands Tijdschrift voor Tandheelkunde, 107(6), 242–243. [PubMed] [Google Scholar]
- Baroncelli GI, Angiolini M, Ninni E, Galli V, Saggese R, & Giuca MR (2006). Prevalence and pathogenesis of dental and periodontal lesions in children with X-linked hypophosphatemic rickets. European Journal of Paediatric Dentistry, 7(2), 61–66. [PubMed] [Google Scholar]
- Bauermeister S, & Letts M (1992). The orthopaedic manifestations of the Langer-Giedion syndrome. Orthopedic Reviews, 21(1), 31–35. [PubMed] [Google Scholar]
- Baujat G, & Le Merrer M (2007). Ellis-van Creveld syndrome. Orphanet Journal of Rare Diseases, 2, 27. 10.1186/1750-1172-2-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauss O, Sadat-Khonsari R, Engelke W, & Kahl-Nieke B (2002). Results of transplanting developing third molars as part of orthodontic space management. Part 1: Clinical and radiographic results. Journal of Orofacial Orthopedics, 63(6), 483–492. 10.1007/s00056-002-0131-4 [DOI] [PubMed] [Google Scholar]
- Beiraghi S, Leon-Salazar V, Larson BE, John MT, Cunningham ML, Petryk A, & Lohr JL (2011). Craniofacial and intraoral phenotype of Robinow syndrome forms. Clinical Genetics, 80(1), 15–24. 10.1111/j.1399-0004.2011.01683.x [DOI] [PubMed] [Google Scholar]
- Belengeanu V, Marian D, Hosszu T, Ogodescu AS, Belengeanu AD, Samoila C, Freiman P, & Lile IE (2019). A comprehensive evaluation of an OFDI syndrome from child to teenager. Romanian Journal of Morphology and Embryology, 60(2), 697–706. [PubMed] [Google Scholar]
- Beltrame AP, Rosa MM, Noschang RA, & Almeida IC (2017). Early rehabilitation of incisors with dentinogenesis imperfecta type II—Case report. Journal of Clinical Pediatric Dentistry, 41(2), 112–115. 10.17796/1053-4628-41.2.112 [DOI] [PubMed] [Google Scholar]
- Broder HL, McGrath C, & Cisneros GJ (2007). Questionnaire development: Face validity and item impact testing of the child oral health impact profile. Community Dentistry and Oral Epidemiology, 35(Suppl 1), 8–19. 10.1111/j.1600-0528.2007.00401.x [DOI] [PubMed] [Google Scholar]
- Callea M, Fattori F, Yavuz I, & Bertini E (2012). A new phenotypic variant in cleidocranial dysplasia (CCD) associated with mutation c.391C>T of the RUNX2 gene. BMJ Case Reports, 2012, bcr1220115422. 10.1136/bcr-12-2011-5422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarata-Scalisi F, Avendaño A, & Callea M (2018). Main genetic entities associated with supernumerary teeth. Archivos Argentinos de Pediatria, 116(6), 437–444. 10.5546/aap.2018.eng.437 [DOI] [PubMed] [Google Scholar]
- Chen IP, Tadinada A, Dutra EH, Utreja A, Uribe F, & Reichenberger EJ (2014). Dental anomalies associated with craniometaphyseal dysplasia. Journal of Dental Research, 93(6), 553–558. 10.1177/0022034514529304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng FC, Chen MH, Liu BL, Liu SY, Hu YT, Chang JY, & Chiang CP (2022). Nonsyndromic supernumerary teeth in patients in National Taiwan University Children’s Hospital. Journal of Dental Sciences, 17(4), 1612–1618. 10.1016/j.jds.2022.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KR, Cho JH, Kim SH, Kook YA, & Cozzani M (2007). Unusual extraction treatment in Class II division 1 using C-orthodontic mini-implants. The Angle Orthodontist, 77(1), 155–166. 10.2319/020106-35r.1 [DOI] [PubMed] [Google Scholar]
- Cooper SC, Flaitz CM, Johnston DA, Lee B, & Hecht JT (2001). A natural history of cleidocranial dysplasia. American Journal of Medical Genetics, 104(1), 1–6. 10.1002/ajmg.10024 [DOI] [PubMed] [Google Scholar]
- da Silva Dalben G, Richieri-Costa A, & de Assis Taveira LA (2008). Tooth abnormalities and soft tissue alterations in patients with G/BBB syndrome. Oral Diseases, 14(8), 747–753. 10.1111/j.1601-0825.2008.01457.x [DOI] [PubMed] [Google Scholar]
- Dean JA, Jones JE, & Vash BW (1986). Dental management of oculodentodigital dysplasia: Report of case. ASDC Journal of Dentistry for Children, 53(2), 131–134. [PubMed] [Google Scholar]
- Demir T, Kecik D, & Cehreli ZC (2007). Kenny-Caffey Syndrome: Oral findings and 4-year follow-up of overlay denture therapy. Journal of Dentistry for Children, 74(3), 236–240. [PubMed] [Google Scholar]
- Detailleur V, Vansteenkiste G, Renard M, & Verdonck A (2016). Dental care approach in patients with osteopetrosis. European Archives of Paediatric Dentistry, 17(6), 435–443. 10.1007/s40368-016-0251-y [DOI] [PubMed] [Google Scholar]
- Dhiman NK, Singh AK, Sharma NK, & Jaiswara C (2014). Cleidocranial dysplasia. National Journal of Maxillofacial Surgery, 5(2), 206–208. 10.4103/0975-5950.154838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Biase DD (1971). The effects of variations in tooth morphology and position on eruption. Dental Practice and Dental Records, 22(3), 95–108. [PubMed] [Google Scholar]
- Doshi DC, Limdi PK, Parekh NV, & Gohil NR (2016). Oculodentodigital dysplasia. Indian Journal of Ophthalmology, 64(3), 227–230. 10.4103/0301-4738.180191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driva T, Franklin D, & Crawford PJ (2004). Variations in expression of oral-facial-digital syndrome (type I): Report of two cases. International Journal of Paediatric Dentistry, 14(1), 61–68. 10.1111/j.1365-263x.2004.00503.x [DOI] [PubMed] [Google Scholar]
- Dror Y, Donadieu J, Koglmeier J, Dodge J, Toiviainen-Salo S, Makitie O, Kerr E, Zeidler C, Shimamura A, Shah N, Cipolli M, Kuijpers T, Durie P, Rommens J, Siderius L, & Liu JM (2011). Draft consensus guidelines for diagnosis and treatment of Shwachman-Diamond syndrome. Annals of the New York Academy of Sciences, 1242, 40–55. 10.1111/j.1749-6632.2011.06349.x [DOI] [PubMed] [Google Scholar]
- Dutra EH, Chen IP, & Reichenberger EJ (2013). Dental abnormalities in a mouse model for craniometaphyseal dysplasia. Journal of Dental Research, 92(2), 173–179. 10.1177/0022034512468157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faircloth WJ Jr., Edwards RC, & Farhood VW (1991). Cherubism involving a mother and daughter: Case reports and review of the literature. Journal of Oral and Maxillofacial Surgery, 49(5), 535–542. 10.1016/0278-2391(91)90185-o [DOI] [PubMed] [Google Scholar]
- Fekonja A (2005). Hypodontia in orthodontically treated children. European Journal of Orthodontics, 27(5), 457–460. 10.1093/ejo/cji027 [DOI] [PubMed] [Google Scholar]
- Fekonja A (2017). Prevalence of dental developmental anomalies of permanent teeth in children and their influence on esthetics. Journal of Esthetic and Restorative Dentistry, 29(4), 276–283. 10.1111/jerd.12302 [DOI] [PubMed] [Google Scholar]
- Flaitz CM, & Hicks J (2001). Delayed tooth eruption associated with an ameloblastic fibro-odontoma. Pediatric Dentistry, 23(3), 253–254. [PubMed] [Google Scholar]
- Forys-Dworniczak E, Zajdel-Cwynar O, Kalina-Faska B, Malecka-Tendera E, & Matusik P (2019). Trichorhinophalangeal syndrome as a diagnostic and therapeutic challenge for paediatric endocrinologists. Pediatric Endocrinology Diabetes and Metabolism, 25(1), 41–47. 10.5114/pedm.2019.84708 [DOI] [PubMed] [Google Scholar]
- Gallottini M, Llanos AH, Romito GA, Romano MM, de Oliveira FB, & de Rezende NPM (2018). Oral manifestations and rehabilitation in Fraser syndrome: A case report. Special Care in Dentistry, 38(4), 249–254. 10.1111/scd.12297 [DOI] [PubMed] [Google Scholar]
- Garvey MT, Barry HJ, & Blake M (1999). Supernumerary teeth—An overview of classification, diagnosis and management. Journal of the Canadian Dental Association, 65(11), 612–616. [PubMed] [Google Scholar]
- Gasse B, Karayigit E, Mathieu E, Jung S, Garret A, Huckert M, Morkmued S, Schneider C, Vidal L, Hemmerle J, Sire JY, & Bloch-Zupan A (2013). Homozygous and compound heterozygous MMP20 mutations in amelogenesis imperfecta. Journal of Dental Research, 92(7), 598–603. 10.1177/0022034513488393 [DOI] [PubMed] [Google Scholar]
- Gaucher C, Walrant-Debray O, Nguyen TM, Esterle L, Garabédian M, & Jehan F (2009). PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets. Human Genetics, 125(4), 401–411. 10.1007/s00439-009-0631-z [DOI] [PubMed] [Google Scholar]
- Gherunpong S, Tsakos G, & Sheiham A (2004). Developing and evaluating an oral health-related quality of life index for children; the CHILD-OIDP. Community Dental Health, 21(2), 161–169. [PubMed] [Google Scholar]
- Gjørup H, Beck-Nielsen SS, Hald JD, & Haubek D (2021). Oral health-related quality of life in X-linked hypophosphataemia and osteogenesis imperfecta. Journal of Oral Rehabilitation, 48(2), 160–168. 10.1111/joor.13114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber LW (1978). Congenital absence of teeth: A review with emphasis on inheritance patterns. Journal of the American Dental Association, 96(2), 266–275. 10.14219/jada.archive.1978.0054 [DOI] [PubMed] [Google Scholar]
- Gunbay S, Zeytinoglu B, Ozkinay F, Ozkinay C, & Oncag A (1996). Orofaciodigital syndrome I: A case report. Journal of Clinical Pediatric Dentistry, 20(4), 329–332. [PubMed] [Google Scholar]
- Hald JD, Beck-Nielsen S, Gregersen PA, Gjorup H, & Langdahl B (2023). Pycnodysostosis in children and adults. Bone, 169, 116674. 10.1016/j.bone.2023.116674 [DOI] [PubMed] [Google Scholar]
- Han D, Gong Y, Wu H, Zhang X, Yan M, Wang X, Qu H, Feng H, & Song S (2008). Novel EDA mutation resulting in X-linked nonsyndromic hypodontia and the pattern of EDA-associated isolated tooth agenesis. European Journal of Medical Genetics, 51(6), 536–546. 10.1016/j.ejmg.2008.06.002 [DOI] [PubMed] [Google Scholar]
- Hanisch M, Bohner L, Sabandal MMI, Kleinheinz J, & Jung S (2019). Oral symptoms and oral health-related quality of life of individuals with x-linked hypophosphatemia. Head & Face Medicine, 15(1), 8. 10.1186/s13005-019-0192-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch M, Sielker S, Jung S, Kleinheinz J, & Bohner L (2019). Self-assessment of oral health-related quality of life in people with ectodermal dysplasia in Germany. International Journal of Environmental Research and Public Health, 16(11), 1933. 10.3390/ijerph16111933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hari Gopal S, & Adams ME (2023). Osteogenesis Imperfecta. In Orthopaedics for the Newborn and Young Child (pp. 395–403). Springer. [Google Scholar]
- Harting I, Karch S, Moog U, Seitz A, Pouwels PJW, & Wolf NI (2019). Oculodentodigital dysplasia: A hypomyelinating leukodystrophy with a characteristic MRI pattern of brain stem involvement. American Journal of Neuroradiology, 40(5), 903–907. 10.3174/ajnr.A6051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashibara T, Komura T, Sobue S, & Ooshima T (2000). Tooth eruption in a patient with craniometaphyseal dysplasia: Case report. Journal of Oral Pathology & Medicine, 29(9), 460–462. 10.1034/j.1600-0714.2000.290907.x [DOI] [PubMed] [Google Scholar]
- Hejlesen J, Underbjerg L, Gjorup H, Sikjaer T, Rejnmark L, & Haubek D (2019). Dental anomalies and orthodontic characteristics in patients with pseudohypoparathyroidism. BMC Oral Health, 20(1), 2. 10.1186/s12903-019-0978-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfrich MH (2003). Osteoclast diseases. Microscopy Research and Technique, 61(6), 514–532. 10.1002/jemt.10375 [DOI] [PubMed] [Google Scholar]
- Helfrich MH (2005). Osteoclast diseases and dental abnormalities. Archives of Oral Biology, 50(2), 115–122. 10.1016/j.archoralbio.2004.11.016 [DOI] [PubMed] [Google Scholar]
- Ho W, Cheretakis C, Durie P, Kulkarni G, & Glogauer M (2007). Prevalence of oral diseases in Shwachman-Diamond syndrome. Special Care in Dentistry, 27(2), 52–58. 10.1111/j.1754-4505.2007.tb00328.x [DOI] [PubMed] [Google Scholar]
- Ibrahim S, Strange AP, Aguayo S, Shinawi A, Harith N, Mohamed-Ibrahim N, Siddiqui S, Parekh S, & Bozec L (2019). Phenotypic properties of collagen in dentinogenesis imperfecta associated with osteogenesis imperfecta. International Journal of Nanomedicine, 14, 9423–9435. 10.2147/ijn.S217420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itin PH, & Fistarol SK (2004). Ectodermal dysplasias. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 131, 45–51. 10.1002/ajmg.c.30033 [DOI] [PubMed] [Google Scholar]
- Jain PS, Gupte TS, Jetpurwala AM, & Dedhia SP (2017). Robinow syndrome and fusion of primary teeth. Contemporary Clinical Dentistry, 8(3), 479–481. 10.4103/ccd.ccd_622_17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen ED (2021). Generalised hypomineralisation of enamel in oculodentodigital dysplasia: Comprehensive dental management of a case. BMJ Case Reports, 14(1), e238079. 10.1136/bcr-2020-238079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Liu F, Wang WG, Jiang X, Wen X, Hu KJ, & Xue Y (2017). Distribution of cathepsin K in late stage of tooth germ development and its function in degrading enamel matrix proteins in mouse. PLoS One, 12(1), e0169857. 10.1371/journal.pone.0169857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokovic A, Locker D, Stephens M, Kenny D, Tompson B, & Guyatt G (2002). Validity and reliability of a questionnaire for measuring child oral-health-related quality of life. Journal of Dental Research, 81(7), 459–463. 10.1177/154405910208100705 [DOI] [PubMed] [Google Scholar]
- Jokovic A, Locker D, Tompson B, & Guyatt G (2004). Questionnaire for measuring oral health-related quality of life in eight- to ten-year-old children. Pediatric Dentistry, 26(6), 512–518. [PubMed] [Google Scholar]
- Keene EJ, & Day PF (2011). Case report: Hypodontia and short roots in a child with Fraser syndrome. European Archives of Paediatric Dentistry, 12(4), 216–218. 10.1007/bf03262810 [DOI] [PubMed] [Google Scholar]
- Khan AA, Morrison A, Hanley DA, Felsenberg D, McCauley LK, O’Ryan F, Reid IR, Ruggiero SL, Taguchi A, Tetradis S, Watts NB, Brandi ML, Peters E, Guise T, Eastell R, Cheung AM, Morin SN, Masri B, Cooper C, … Compston J (2015). Diagnosis and management of osteonecrosis of the jaw: A systematic review and international consensus. Journal of Bone and Mineral Research, 30(1), 3–23. 10.1002/jbmr.2405 [DOI] [PubMed] [Google Scholar]
- Kim JW, Simmer JP, Lin BP, & Hu JC (2006). Novel MSX1 frame-shift causes autosomal-dominant oligodontia. Journal of Dental Research, 85(3), 267–271. 10.1177/154405910608500312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokich VG, & Kokich VO (2006). Congenitally missing mandibular second premolars: Clinical options. American Journal of Orthodontics and Dentofacial Orthopedics, 130(4), 437–444. 10.1016/j.ajodo.2006.05.025 [DOI] [PubMed] [Google Scholar]
- Kunz F, Kayserili H, Midro A, de Silva D, Basnayake S, Guven Y, Borys J, Schanze D, Stellzig-Eisenhauer A, Bloch-Zupan A, & Zenker M (2020). Characteristic dental pattern with hypodontia and short roots in Fraser syndrome. American Journal of Medical Genetics A, 182(7), 1681–1689. 10.1002/ajmg.a.61610 [DOI] [PubMed] [Google Scholar]
- Lam DK, Sándor GK, Holmes HI, Carmichael RP, & Clokie CM (2007). Marble bone disease: A review of osteopetrosis and its oral health implications for dentists. Journal of the Canadian Dental Association, 73(9), 839–843. [PubMed] [Google Scholar]
- Landa S, Esteban S, Montes E, Santamaria J, Vitoria A, & Santolaya JM (2000). Maxillofacial alterations in a family with pycnodysostosis. Medicine Oral, 5(3), 169–176. [PubMed] [Google Scholar]
- Lee BN, Jung HY, Chang HS, Hwang YC, & Oh WM (2017). Dental management of patients with X-linked hypophosphatemia. Restorative Dentistry & Endodontics, 42(2), 146–151. 10.5395/rde.2017.42.2.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidral AC, & Reising BC (2002). The role of MSX1 in human tooth agenesis. Journal of Dental Research, 81(4), 274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Li L, Liu C, Liu H, He F, Yan F, Zhang Y, & Chen Y (2011). Wnt5a regulates growth, patterning, and odontoblast differentiation of developing mouse tooth. Developmental Dynamics, 240(2), 432–440. 10.1002/dvdy.22550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sun X, Zhang X, Wang X, Zhang C, & Zheng S (2019). RUNX2 mutation impairs osteogenic differentiation of dental follicle cells. Archives of Oral Biology, 97, 156–164. 10.1016/j.archoralbio.2018.10.029 [DOI] [PubMed] [Google Scholar]
- Loddenkemper T, Grote K, Evers S, Oelerich M, & Stögbauer F (2002). Neurological manifestations of the oculodentodigital dysplasia syndrome. Journal of Neurology, 249(5), 584–595. 10.1007/s004150200068 [DOI] [PubMed] [Google Scholar]
- Machuca G, Martinez F, Machuca C, & Bullon P (1997). Craniofacial and oral manifestations of trichorhinophalangeal syndrome type I (Giedion’s syndrome): A case report. Oral Surgery, Oral Medicine, Oral Pathology, and Oral Radiology, 84(1), 35–39. 10.1016/s1079-2104(97)90291-2 [DOI] [PubMed] [Google Scholar]
- Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, Castori M, Cohen H, Colombi M, Demirdas S, De Backer J, De Paepe A, Fournel-Gigleux S, Frank M, Ghali N, … Tinkle B (2017). The 2017 international classification of the Ehlers-Danlos syndromes. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 175(1), 8–26. 10.1002/ajmg.c.31552 [DOI] [PubMed] [Google Scholar]
- Marini JC, & Dang Do AN (2000). Osteogenesis Imperfecta. In Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, … Wilson DP (Eds.), Endotext. South Dartmouth, MA. [Google Scholar]
- Mitchell L, & Bennett TG (1992). Supernumerary teeth causing delayed eruption—A retrospective study. British Journal of Orthodontics, 19(1), 41–46. 10.1179/bjo.19.1.41 [DOI] [PubMed] [Google Scholar]
- Moussaid Y, Griffiths D, Richard B, Dieux A, Lemerrer M, Leger J, Lacombe D, & Bailleul-Forestier I (2012). Oral manifestations of patients with Kenny-Caffey Syndrome. European Journal of Medical Genetics, 55(8–9), 441–445. 10.1016/j.ejmg.2012.03.005 [DOI] [PubMed] [Google Scholar]
- Muhney K, & Campbell PR (2007). Pediatric dental management of a patient with osteogenesis imperfecta and dentinogenesis imperfecta. Special Care in Dentistry, 27(6), 240–245. 10.1111/j.1754-4505.2007.tb01757.x [DOI] [PubMed] [Google Scholar]
- Naito M, Yuasa H, Nomura Y, Nakayama T, Hamajima N, & Hanada N (2006). Oral health status and health-related quality of life: A systematic review. Journal of Oral Science, 48(1), 1–7. 10.2334/josnusd.48.1 [DOI] [PubMed] [Google Scholar]
- Najirad M, Ma MS, Rauch F, Sutton VR, Lee B, Retrouvey JM, & Esfandiari S (2018). Oral health-related quality of life in children and adolescents with osteogenesis imperfecta: Cross-sectional study. Orphanet Journal of Rare Diseases, 13(1), 187. 10.1186/s13023-018-0935-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najirad M, Madathil SA, Rauch F, Sutton VR, Lee B, Retrouvey JM, & Esfandiari S (2020). Malocclusion traits and oral health-related quality of life in children with osteogenesis imperfecta: A cross-sectional study. Journal of the American Dental Association, 151(7), 480–490.e482. 10.1016/j.adaj.2020.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama Y, Holcroft J, & Ganss B (2015). Enamel hypomineralization and structural defects in amelotin-deficient mice. Journal of Dental Research, 94(5), 697–705. 10.1177/0022034514566214 [DOI] [PubMed] [Google Scholar]
- Nguyen QC, Duverger O, Mishra R, Mitnik GL, Jani P, Frischmeyer-Guerrerio PA, & Lee JS (2019). Oral health-related quality of life in Loeys-Dietz syndrome, a rare connective tissue disorder: An observational cohort study. Orphanet Journal of Rare Diseases, 14(1), 291. 10.1186/s13023-019-1250-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieminen P (2009). Genetic basis of tooth agenesis. Journal of Experimental Zoology Part B: Molecular and Developmental Evolution, 312B(4), 320–342. 10.1002/jez.b.21277 [DOI] [PubMed] [Google Scholar]
- Nualart Grollmus ZC, Morales Chavez MC, & Silvestre Donat FJ (2007). Periodontal disease associated to systemic genetic disorders. Medicina Oral, Patología Oral y Cirugía Bucal, 12(3), E211–E215. [PubMed] [Google Scholar]
- Nunn JH, Carter NE, Gillgrass TJ, Hobson RS, Jepson NJ, Meechan JG, & Nohl FS (2003). The interdisciplinary management of hypodontia: Background and role of paediatric dentistry. British Dental Journal, 194(5), 245–251. 10.1038/sj.bdj.4809925 [DOI] [PubMed] [Google Scholar]
- O’Connell AC, Brennan MT, & Francomano CA (1998). Pycnodysostosis: Orofacial manifestations in two pediatric patients. Pediatric Dentistry, 20(3), 204–207. [PubMed] [Google Scholar]
- Oelerich O, Kleinheinz J, Reissmann DR, Köppe J, & Hanisch M (2020). Correlation between oral health-related quality of life and objectively measured oral health in people with Ehlers-Danlos syndromes. International Journal of Environmental Research and Public Health, 17(21), 8243. 10.3390/ijerph17218243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okawa R, Kubota T, Kitaoka T, Kokomoto K, Ozono K, & Nakano K (2017). Oral manifestations of Japanese patients with osteogenesis imperfecta. Pediatric Dental Journal, 27(2), 73–78. [Google Scholar]
- Pahel BT, Rozier RG, & Slade GD (2007). Parental perceptions of children’s oral health: the Early Childhood Oral Health Impact Scale (ECOHIS). Health and Quality of Life Outcomes, 5, 6. 10.1186/1477-7525-5-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palagano E, Menale C, Sobacchi C, & Villa A (2018). Genetics of osteopetrosis. Current Osteoporosis Reports, 16(1), 13–25. 10.1007/s11914-018-0415-2 [DOI] [PubMed] [Google Scholar]
- Papadaki ME, Lietman SA, Levine MA, Olsen BR, Kaban LB, & Reichenberger EJ (2012). Cherubism: Best clinical practice. Orphanet Journal of Rare Diseases, 7(Suppl 1), S6. 10.1186/1750-1172-7-s1-s6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parolia A, Kundabala M, Dahal M, Mohan M, & Thomas MS (2011). Management of supernumerary teeth. Journal of Conservative Dentistry, 14(3), 221–224. 10.4103/0972-0707.85791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Aghababaie S, & Parekh S (2019). Hypomineralisation or hypoplasia? British Dental Journal, 227(8), 683–686. 10.1038/s41415-019-0782-9 [DOI] [PubMed] [Google Scholar]
- Paul SA, Simon SS, Karthik AK, Chacko RK, & Savitha S (2015). A review of clinical and radiological features of cleidocranial dysplasia with a report of two cases and a dental treatment protocol. Journal of Pharmacy and Bioallied Sciences, 7(Suppl 2), S428–S432. 10.4103/0975-7406.163490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paznekas WA, Karczeski B, Vermeer S, Lowry RB, Delatycki M, Laurence F, Koivisto PA, Van Maldergem L, Boyadjiev SA, Bodurtha JN, & Jabs EW (2009). GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Human Mutation, 30(5), 724–733. 10.1002/humu.20958 [DOI] [PubMed] [Google Scholar]
- Pontes FS, Ferreira AC, Kato AM, Pontes HA, Almeida DS, Rodini CO, & Pinto DS Jr. (2007). Aggressive case of cherubism: 17-year follow-up. International Journal of Pediatric Otorhinolaryngology, 71(5), 831–835. 10.1016/j.ijporl.2007.01.017 [DOI] [PubMed] [Google Scholar]
- Poulter JA, Murillo G, Brookes SJ, Smith CE, Parry DA, Silva S, Kirkham J, Inglehearn CF, & Mighell AJ (2014). Deletion of ameloblastin exon 6 is associated with amelogenesis imperfecta. Human Molecular Genetics, 23(20), 5317–5324. 10.1093/hmg/ddu247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajpar MH, Harley K, Laing C, Davies RM, & Dixon MJ (2001). Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta. Human Molecular Genetics, 10(16), 1673–1677. 10.1093/hmg/10.16.1673 [DOI] [PubMed] [Google Scholar]
- Rapoport M, Bober MB, Raggio C, Wekre LL, Rauch F, Westerheim I, Hart T, van Welzenis T, Mistry A, Clancy J, Booth L, Prince S, & Semler O (2023). The patient clinical journey and socioeconomic impact of osteogenesis imperfecta: A systematic scoping review. Orphanet Journal of Rare Diseases, 18(1), 34. 10.1186/s13023-023-02627-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch F, & Glorieux FH (2004). Osteogenesis imperfecta. Lancet, 363(9418), 1377–1385. 10.1016/s0140-6736(04)16051-0 [DOI] [PubMed] [Google Scholar]
- Regan JP, Szymanski K, Podda S, Gargano F, & Kopiecki A (2017). A surgical approach to the craniofacial defects of Opitz G/BBB syndrome. Journal of Surgical Case Reports, 2017(2), rjx032. 10.1093/jscr/rjx032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis MT, Matias DT, Faria ME, & Martin RM (2016). Failure of tooth eruption and brachydactyly in pseudohypoparathyroidism are not related to plasma parathyroid hormone-related protein levels. Bone, 85, 138–141. 10.1016/j.bone.2016.02.002 [DOI] [PubMed] [Google Scholar]
- Reyes C, Hitz M, Prieto-Alhambra D, & Abrahamsen B (2016). Risks and benefits of bisphosphonate therapies. Journal of Cellular Biochemistry, 117(1), 20–28. 10.1002/jcb.25266 [DOI] [PubMed] [Google Scholar]
- Ríos-Rodenas M, de Nova J, Gutiérrez-Díez MP, Feijoo G, Mourelle MR, Garcilazo M, & Ortega-Aranegui R (2015). A cephalometric method to diagnosis the craniovertebral junction abnormalities in osteogenesis imperfecta patients. Journal of Clinical and Experimental Dentistry, 7(1), e153–e158. 10.4317/jced.52126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanches K, de Queiroz AM, de Freitas AC, & Serrano KV (2005). Clinical features, dental findings and dental care management in osteogenesis imperfecta. Journal of Clinical Pediatric Dentistry, 30(1), 77–82. 10.17796/jcpd.30.1.t53201466660612r [DOI] [PubMed] [Google Scholar]
- Schieppati A, Henter JI, Daina E, & Aperia A (2008). Why rare diseases are an important medical and social issue. Lancet, 371(9629), 2039–2041. 10.1016/s0140-6736(08)60872-7 [DOI] [PubMed] [Google Scholar]
- Seehra J, Yaqoob O, Patel S, O’Neill J, Bryant C, Noar J, Morris D, & Cobourne MT (2018). National clinical guidelines for the management of unerupted maxillary incisors in children. British Dental Journal, 224(10), 779–785. 10.1038/sj.bdj.2018.361 [DOI] [PubMed] [Google Scholar]
- Shah A, Gill DS, Tredwin C, & Naini FB (2008). Diagnosis and management of supernumerary teeth. Dental Update, 35(8), 510–512, 514,–516, 519–520. 10.12968/denu.2008.35.8.510 [DOI] [PubMed] [Google Scholar]
- Sillence DO, & Rimoin DL (1978). Classification of osteogenesis imperfect. Lancet, 1(8072), 1041–1042. 10.1016/s0140-6736(78)90763-8 [DOI] [PubMed] [Google Scholar]
- Slade GD (1997). Derivation and validation of a short-form oral health impact profile. Community Dentistry and Oral Epidemiology, 25(4), 284–290. 10.1111/j.1600-0528.1997.tb00941.x [DOI] [PubMed] [Google Scholar]
- Slade GD, & Sanders AE (2011). The paradox of better subjective oral health in older age. Journal of Dental Research, 90(11), 1279–1285. 10.1177/0022034511421931 [DOI] [PubMed] [Google Scholar]
- Sobacchi C, Schulz A, Coxon FP, Villa A, & Helfrich MH (2013). Osteopetrosis: Genetics, treatment and new insights into osteoclast function. Nature Reviews Endocrinology, 9(9), 522–536. 10.1038/nrendo.2013.137 [DOI] [PubMed] [Google Scholar]
- Soliman AT, Ramadan MA, Sherif A, Aziz Bedair ES, & Rizk MM (2001). Pycnodysostosis: Clinical, radiologic, and endocrine evaluation and linear growth after growth hormone therapy. Metabolism, 50(8), 905–911. 10.1053/meta.2001.24924 [DOI] [PubMed] [Google Scholar]
- Soman C, & Lingappa A (2015). Robinow syndrome: A rare case report and review of literature. International Journal of Clinical Pediatric Dentistry, 8(2), 149–152. 10.5005/jp-journals-10005-1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark Z, & Savarirayan R (2009). Osteopetrosis. Orphanet Journal of Rare Diseases, 4, 5. 10.1186/1750-1172-4-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen LX, Hamersma H, Gardner J, & Beighton P (2001). Dental and oral manifestations of sclerosteosis. International Dental Journal, 51(4), 287–290. 10.1002/j.1875-595x.2001.tb00840.x [DOI] [PubMed] [Google Scholar]
- Stock F, Hanisch M, Lechner S, Biskup S, Bohring A, Zschocke J, & Kapferer-Seebacher I (2021). Prepubertal periodontitis in a patient with combined classical and periodontal Ehlers-Danlos syndrome. Bio-molecules, 11(2), 149. 10.3390/biom11020149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri L, Gagari E, & Vastardis H (2004). Delayed tooth eruption: Pathogenesis, diagnosis, and treatment. A literature review. American Journal of Orthodontics and Dentofacial Orthopedics, 126(4), 432–445. 10.1016/j.ajodo.2003.10.031 [DOI] [PubMed] [Google Scholar]
- Szeri F, Lundkvist S, Donnelly S, Engelke UFH, Rhee K, Williams CJ, Sundberg JP, Wevers RA, Tomlinson RE, Jansen RS, & van de Wetering K (2020). The membrane protein ANKH is crucial for bone mechanical performance by mediating cellular export of citrate and ATP. PLoS Genetics, 16(7), e1008884. 10.1371/journal.pgen.1008884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeri F, Niaziorimi F, Donnelly S, Fariha N, Tertyshnaia M, Patel D, Lundkvist S, & van de Wetering K (2022). The mineralization regulator ANKH mediates cellular Efflux of ATP, Not pyrophosphate. Journal of Bone and Mineral Research, 37(5), 1024–1031. 10.1002/jbmr.4528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahseen K, Khan S, Uma R, Usha R, Al Ghanem MM, Al Awadi SA, & Farag TI (1997). Kenny-Caffey syndrome in six bedouin sibships: Autosomal recessive inheritance is confirmed. American Journal of Medical Genetics, 69(2), 126–132. [PubMed] [Google Scholar]
- Taqi D, Moussa H, Schwinghamer T, Vieira AR, Dagdeviren D, Retrouvey JM, Rauch F, & Tamimi F (2021). Missing and unerupted teeth in osteogenesis imperfecta. Bone, 150, 116011. 10.1016/j.bone.2021.116011 [DOI] [PubMed] [Google Scholar]
- Thaweesapphithak S, Saengsin J, Kamolvisit W, Theerapanon T, Porntaveetus T, & Shotelersuk V (2022). Cleidocranial dysplasia and novel RUNX2 variants: Dental, craniofacial, and osseous manifestations. Journal of Applied Oral Science, 30, e20220028. 10.1590/1678-7757-2022-0028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thesleff I (2000). Genetic basis of tooth development and dental defects. Acta Odontologica Scandinavica, 58(5), 191–194. 10.1080/000163500750051728 [DOI] [PubMed] [Google Scholar]
- Thesleff I (2006). The genetic basis of tooth development and dental defects. American Journal of Medical Genetics, 140(23), 2530–2535. 10.1002/ajmg.a.31360 [DOI] [PubMed] [Google Scholar]
- Thomsen M, Schneider U, Weber M, & Niethard FU (1998). The different appearance of the oculodentodigital dysplasia syndrome. Journal of Pediatric Orthopaedics B, 7(1), 23–26. 10.1097/01202412-199801000-00003 [DOI] [PubMed] [Google Scholar]
- Toth K, Shao Q, Lorentz R, & Laird DW (2010). Decreased levels of Cx43 gap junctions result in ameloblast dysregulation and enamel hypoplasia in Gja1Jrt/+ mice. Journal of Cellular Physiology, 223(3), 601–609. 10.1002/jcp.22046 [DOI] [PubMed] [Google Scholar]
- Tuverson DL (1980). Anterior interocclusal relations. Part I. American Journal of Orthodontics and Dentofacial Orthopedics, 78(4), 361–370. 10.1016/0002-9416(80)90018-4 [DOI] [PubMed] [Google Scholar]
- Varela M, Arrieta P, & Ventureira C (2009). Non-syndromic concomitant hypodontia and supernumerary teeth in an orthodontic population. European Journal of Orthodontics, 31(6), 632–637. 10.1093/ejo/cjp046 [DOI] [PubMed] [Google Scholar]
- Vinay C, Uloopi KS, Rao RC, Kumar RS, & Madhuri V (2011). Oligodontia associated with osteopetrosis: A rare case report. Journal of Dentistry for Children, 78(1), 53–56. [PubMed] [Google Scholar]
- Whyte MP, McAlister WH, Dhiman V, Gopinathan NR, & Bhadada SK (2023). Drug-induced osteopetrosis. Bone, 173, 116788. 10.1016/j.bone.2023.116788 [DOI] [PubMed] [Google Scholar]
- Wieczorek A, & Loster J (2013). Dentinogenesis imperfecta type II: Ultrastructure of teeth in sagittal sections. Folia Histochemica et Cytobiologica, 51(3), 244–247. 10.5603/fhc.2013.0035 [DOI] [PubMed] [Google Scholar]
- Wise GE, Frazier-Bowers S, & D’Souza RN (2002). Cellular, molecular, and genetic determinants of tooth eruption. Critical Reviews in Oral Biology & Medicine, 13(4), 323–334. 10.1177/154411130201300403 [DOI] [PubMed] [Google Scholar]
- Wise GE, Huang H, & Que BG (1999). Gene expression of potential tooth eruption molecules in the dental follicle of the mouse. European Journal of Oral Sciences, 107(6), 482–486. 10.1046/j.0909-8836.1999.eos107610.x [DOI] [PubMed] [Google Scholar]
- Wright JT, Carrion IA, & Morris C (2015). The molecular basis of hereditary enamel defects in humans. Journal of Dental Research, 94(1), 52–61. 10.1177/0022034514556708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Wang P, Liu H, & Gu M (2022). Efficacy of three surgical methods for gingivectomy of permanent anterior teeth with delayed tooth eruption in children. Head & Face Medicine, 18(1), 23. 10.1186/s13005-022-00328-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, & Attaie AB (2016). Genetic basis of nonsyndromic and syndromic tooth agenesis. Journal of Pediatric Genetics, 5(4), 198–208. 10.1055/s-0036-1592421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano M, Nikitakis NG, Sanchez-Quevedo MC, Sauk JJ, Sedano H, & Rivera H (2003). Oral and dental manifestations of vitamin D-dependent rickets type I: Report of a pediatric case. Oral Surgery, Oral Medicine, Oral Pathology, and Oral Radiology, 95(6), 705–709. 10.1067/moe.2003.116 [DOI] [PubMed] [Google Scholar]
- Zhang C, Jolly A, Shayota BJ, Mazzeu JF, Du H, Dawood M, Soper PC, Ramalho de Lima A, Ferreira BM, Coban-Akdemir Z, White J, Shears D, Thomson FR, Douglas SL, Wainwright A, Bailey K, Wordsworth P, Oldridge M, Lester T, … Carvalho CMB (2022). Novel pathogenic variants and quantitative phenotypic analyses of Robinow syndrome: WNT signaling perturbation and phenotypic variability. HGG Advances, 3(1), 100074. 10.1016/j.xhgg.2021.10007 [DOI] [PMC free article] [PubMed] [Google Scholar]