

Abstract
Replication Protein A (RPA), the replicative single-strand DNA binding protein from eukaryotic cells, is a stable heterotrimeric complex consisting of three polypeptides. Cytological studies have investigated the subcellular distribution and association characteristics of the three RPA subunits during different stages of the cell cycle with varying results. In this study, various HeLa cell fractions were subjected to separation by either immunoprecipitation or velocity sedimentation. These separations were evaluated by immunoblotting for specific RPA subunits to determine whether the RPA in these fractions retains its heterotrimeric association. Immunoprecipitation of either the large (RPA70) or middle-sized (RPA32) subunit of RPA followed by immunoblotting for the other subunits demonstrate that RPA remains complexed throughout the G1, S and G2 phases of the cell cycle. Immunoprecipitation and sedimentation separations of both the nucleosolic and chromatin-bound RPA populations from both cycling and nocodazole-blocked cells showed that the majority of RPA remains complexed under all conditions examined. Consistent with previous reports, hypotonic extracts from 293 cells were shown to contain some RPA32 not complexed with RPA70. These results indicate that in some cell types, extracts may contain small amounts of RPA32 free of RPA70; however, in HeLa cells the majority of RPA clearly remains complexed as a heterotrimer throughout the cell cycle.
INTRODUCTION
Replication Protein A (RPA) is a single-strand DNA (ssDNA) binding protein (SSB) complex first identified as a human protein required to support simian virus 40 DNA replication in vitro. RPA has been shown to be a central protein involved in multiple DNA metabolism processes including DNA replication, DNA repair, and recombination (1,2). This complex consists of three polypeptides designated RPA14, RPA32 and RPA70, based on their molecular weights. These subunits are conserved in all eukaryotes and in archeabacteria (1–3), and the genes encoding all three RPA subunits have been shown to be essential for viability in yeast (4). RPA70 contains the major ssDNA-binding activity of the complex, while RPA32 contains a secondary ssDNA-binding domain (2,5–11). The role of RPA14 remains unknown (1,2).
While the RPA heterotrimer is exceedingly stable (up to 6 M urea) (2), and the majority of RPA in non-synchronized cells is found in the heterotrimeric complex (12), several cytological studies have reported that all the detectable RPA within cells may dissociate into its constituent subunits at specific stages of the cell cycle. One study reported that upon transformation of myotubes with SV40 large T antigen (driving cells to re-enter S phase), that RPA70 was found to co-localize with DNA replication sites while RPA32 did not (13). Conversely, a second study showed that RPA70 and RPA32 co-localize throughout the cell cycle (14). Another report used individual subunit antibodies to show that the three RPA subunits co-localized throughout most of the cell cycle, but that the three subunits of all the detectable RPA apparently dissociate from one another to mutually exclusive subcellular locations when HeLa cells were blocked in mitosis with nocodazole (15).
RPA32 has been shown to be phosphorylated both during the cell cycle and in response to DNA damage (1,2,12,16–22). Recently, it has been shown that apoptosis also leads to hyper-phosphorylation of RPA32 (23). It has been reported that normal cell-cycle phosphorylation may lead to decreased ionic stability of the heterotrimer (24). Whether the DNA damage-dependent or apoptosis-dependent hyper-phosphorylation of RPA32 have this same effect on RPA stability remains to be determined.
Clarification of the issue of RPA heterotrimer association during the cell cycle has been further complicated by recent findings that there are two distinct pools of RPA in cells. The larger pool of RPA can be extracted from cells using either 0.5% non-ionic detergent (25,26), or hypotonic dounce and low salt extraction (24). If cells are treated with cross-linking agents prior to washing and are subsequently immunostained for RPA, the entire nucleus is labeled. Conversely, if cells are washed with non-ionic detergent prior to cross-linking, most of the RPA is removed, and the remaining detergent-stable RPA co-localizes to active S-phase DNA replication foci (25,26). Similarly, while the majority of RPA is removed from cells upon hypotonic conditions, another smaller pool of ‘chromatin-associated’ RPA is extracted from the nuclei with higher levels of salt (0.4–0.5 M) (24). The larger, loosely associated RPA pool is referred to as nucleosolic RPA; while the smaller, more salt-stable and replication foci-associated RPA pool is referred to as chromatin-associated RPA.
This study addresses the issue of whether an appreciable portion of the cellular RPA heterotrimer dissociates at any stage of the cell cycle using combined immunoprecipitation–immunoblotting and glycerol gradient–immunoblotting approaches. Cellular RPA in extracts from either elutriated or nocodazole-blocked HeLa cells were subjected to exhaustive immunoprecipitation with monoclonal antibodies against either RPA70 or RPA32. These precipitates were then immunoblotted for the presence and relative levels of the other RPA subunits. Extracts from both cycling and mitotic HeLa cells were also subjected to glycerol gradient sedimentation, and the subsequent fractions immunoblotted for RPA. We have also examined whether there are substantive differences between the nucleosolic and chromatin-bound RPA fractions with regard to subunit association. Using these biochemical approaches we have found no evidence for subunit dissociation of the majority of any of the RPA pools at any stage of the cell cycle, in either normal cycling cells, or in nocodazole-blocked cells.
MATERIALS AND METHODS
Cell culture and mitotic block
HeLa cells were maintained in monolayer cultures in Dulbecco’s Modified Eagle Medium (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (Life Technologies) at 37°C and an atmosphere of 5% carbon dioxide. To obtain mitotic cells, cell cultures of 60–70% confluence were switched to media supplemented with 1 µg/ml methyl[5-(2-thienylcarbonyl)-1H-benzimidazol-2-yl]carbamate (nocodazole) (Sigma, St Louis, MO) for 16–17 h. To ensure that cells were in mitosis, cultures were checked microscopically at various time intervals for the rounded morphology characteristic of mitotic HeLa cells. Nocodazole-treated cells were also stained with 4,6-diamidino-2-phenylindole (DAPI) (Sigma) and observed under fluorescence microscopy for the condensed and segregating chromosomes characteristic of mitotic cells. Following nocodazole treatment, ~60% of the cells appeared to be in mitosis, in comparison to the 2% observed in non-treated cells. There was no significant difference in the number of cells undergoing apoptosis between non-treated and nocodazole-treated cells. Human 293 cells were maintained as suspension cultures in Joklik’s suspension modified MEM (SMEM) (Life Technologies) supplemented with 5% (v/v) calf serum (Life Technologies) at 37°C.
Cell culture and preparation of elutriated cell extracts
HeLa S3 cells were grown in suspension in SMEM (Life Technologies) supplemented with 5% (v/v) calf serum (Life Technologies) at 37°C. Two liters of cells (at 5 × 105 cells/ml) were harvested and resuspended in 75 ml of ice cold EB [phosphate buffered saline (PBS) supplemented with 1% (v/v) calf serum, 0.3 mM Na2EDTA and 0.1% (w/v) glucose]. The cells were then loaded into a Beckman elutriator rotor (model JE10X) at 1550 r.p.m. at a pump setting of #270 (∼65 ml/min). After washing with 800 ml of EB, 12 fractions of cells were elutriated by stepwise increases in the pump setting by #30 for each elutriation step. For the twelfth elutriated fraction the rotor speed was decreased to 100 r.p.m. to collect all the largest cells. Immediately following elutriation, ice cold MgCl2 was added to each fraction to 2 mM final concentration. Cells from each fraction were harvested, resuspended in 50 ml of EB with 2 mM MgCl2, and the cells were counted. Cells (1 × 106) were removed for flow cytometric analysis. The remainder of the cells in each fraction were washed once with HB (20 mM HEPES–KOH pH 7.5, 5 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT), harvested, and then resuspended with a volume of HB equivalent to the volume of the cell pellet (to generate extracts with approximately equal protein concentrations). The cells were resuspended and then lysed by 30 passages in a Dounce homogenizer. NaCl was added to 0.1 M final concentration, PMSF to 0.1 mM final concentration, and the lysates were incubated on ice for 15 min. The lysates were spun at 13 000 g for 15 min at 4°C. The supernatants were aliquoted, frozen in dry ice/ethanol, and stored at –70°C.
Preparation of antibody-linked protein A beads
Protein A Sepharose beads (Amersham Pharmacia Biotech, Piscataway, NJ) (1.5 ml) were resuspended in an equal volume of MB (3 M NaCl, 200 mM sodium borate pH 8.0), and poured into a 1.0 cm diameter column. Approximately 8 mg monoclonal antibody [either anti-RPA32 or anti-RPA70 mouse monoclonal antibody (12) in cell culture supernatant] was concentrated by 50% saturation with ammonium sulfate and collected by centrifugation (3000 g for 35 min at 4°C). The pellet was resuspended in a minimal volume of PBS. The volume of this solution was increased to one-tenth of original volume with the addition of 5 M NaCl to 3 M final concentration, and 1 M sodium borate pH 8.0 to 200 mM final concentration. The concentrated antibody solution (~0.5 mg/ml) was applied to the protein A column and cycled twice to allow for efficient binding of the antibodies. The column was washed with 40 ml of MB, and the beads were resuspended in 10 ml of MB. SDS–PAGE was then performed to ensure that similar levels of antibody were bound prior to cross-linking. Dimethylpimelidate (Sigma) was added to 0.2 M and the suspension was incubated at room temperature for 30 min with gentle agitation. The beads were rinsed and then tumbled overnight at 4°C in 400 mM Tris pH 8.0. The slurry was resuspended in PBS and stored at 4°C.
Preparing cell extracts for immunoprecipitations
To obtain cellular extracts, HeLa cell cultures were first treated with 2 ml 0.05% trypsin–EDTA (Life Technologies) per plate, pelleted, and rinsed twice with PBS. Cells (1 × 107) were then resuspended in 1 ml CSK buffer [10 mM HEPES–KOH pH 7.5, 300 mM sucrose, 3 mM MgCl2, 100 mM NaCl, 0.5% (v/v) Triton X-100, 1 mM PMSF, 1 ng/µl leupeptin, 0.01 mM benzamidine and 0.01 mM sodium bisulfite] on ice for 15 min. The suspensions were subjected to centrifugation at 12 000 g at 4°C for 15 min. The supernatant (containing nucleosolic RPA) was removed and saved for immunoprecipitation. The pellet was resuspended and incubated in 1 ml CSK buffer at 4°C for 15 min with agitation, and then centrifuged as before. This was repeated twice, the supernatant collected each time as separate nucleosolic RPA washes. The pellet was then resuspended in 1 ml CSK buffer containing an additional 400 mM NaCl (final concentration 500 mM NaCl), and incubated with agitation at 4°C for 15 min. The suspension was centrifuged as before, and the supernatant collected as the chromatin-associated RPA fraction. The resulting pellet was resuspended and the CSK with 500 mM NaCl wash repeated three times. Each individual wash was retained for later analysis. The remaining pellet was resuspended in SDS–PAGE loading buffer, boiled for 5 min, and saved for immunoblot analysis.
Immunoprecipitations
Prepared extracts were incubated with 10 µl of protein A beads (previously saturated and crosslinked with either anti-RPA32 or anti-RPA70 monoclonal antibody) for 2 h with gentle agitation at 4°C. The suspensions were then centrifuged at 3000 g. The supernatants were saved for further immunoprecipitations. The nucleosolic RPA fractions were immunoprecipitated three separate times with protein A beads crosslinked with either anti-RPA32 or anti-RPA70 monoclonal antibody. A fourth immunoprecipitation was then performed with beads crosslinked with the other primary antibody. The chromatin-associated RPA fractions were immunoprecipitated twice with either anti-RPA32 or anti-RPA70 monoclonal antibody, before a last immunoprecipitation was performed with the other antibody. The pelleted protein A beads were rinsed twice in CSK buffer, resuspended in SDS–PAGE sample buffer, boiled for 5 min, and subjected to immunoblot analysis.
Preparing cell extracts for glycerol gradient sedimentation
HeLa cell extracts were prepared as described above with the following modifications: 2.5 × 107 cells were rinsed twice in PBS and resuspended in 150 µl CSK buffer on ice for 15 min. The suspensions were subjected to centrifugation at 12 000 g for 15 min at 4°C. The supernatant was collected as the nucleosolic extract, and adjusted to 500 mM NaCl concentration. The remaining pellet was rinsed in 500 µl CSK buffer and resuspended in 150 µl CSK buffer plus 400 mM NaCl (to 500 mM final concentration) on ice for 15 min. Centrifugation was performed as above, and the supernatant was designated the chromatin-associated fraction. Extracts from suspension-cultured 293 cells were prepared as described previously (27). The 293 nucleosolic extracts were also adjusted to 500 mM NaCl final concentration before being applied to the glycerol gradients.
Glycerol gradient sedimentation
Extracts prepared from either HeLa or 293 cells (150 or 100 µl, respectively) as described above were sedimented through 5 ml gradients of 5–35% (v/v) glycerol in CSK buffer with 500 mM NaCl final concentration, for 40 h at 237 000 g and 4°C. The gradients were collected drop-wise as ∼200 µl fractions. Samples were taken from each sedimentation fraction of the nucleosolic and chromatin-associated extract separations (10 and 20 µl, respectively), boiled with SDS–PAGE sample buffer, and subjected to immunoblotting and densitometric analysis.
Immunoblotting
Samples were loaded onto 10 or 12.5% polyacrylamide gels, electrophoresis was performed, and the proteins were electrophoretically transferred onto nitrocellulose membranes. These membranes were rinsed in TBS (25 mM Tris–HCl, 137 mM NaCl, 3 mM KCl, pH 7.4) supplemented with 0.1% (v/v) Nonidet P-40 (TBST), and then blocked in TBST containing 5% (w/v) dry milk and 2% (v/v) fetal calf serum (FCS) for 30 min. Membranes were rinsed three times in TBST for 5 min each and incubated in either anti-RPA rabbit polyclonal antibody (12) (final concentration 1/5000), or a combination of anti-RPA32 and anti-RPA70 monoclonal antibodies (hybridoma supernatants diluted 1/200 and 1/100 respectively), in TBST containing 0.5% (w/v) dry milk and 0.2% (v/v) FCS, for 1 h. The nitrocellulose membranes were then rinsed three times in TBST for 15–20 min each, and then incubated for 30 min with horseradish peroxidase-conjugated anti-rabbit or anti-mouse antibody (final concentration 1/6000 in TBST). The membrane was rinsed as before in TBST, and rinsed in TBS for 15 min. The membrane was then incubated in Supersignal chemiluminescent substrate (Pierce, Rockford, IL), as directed by the manufacturer, and exposed to film. The films were subjected to 2D densitometric analysis using a BioRad GS-700 2D densitometer and the BioRad Molecular Analyst 2.1 software.
RESULTS
RPA is heterotrimeric in extracts from HeLa cells in G1, S and G2 phases
The majority of RPA in HeLa cells is known to be extracted from hypotonically lysed cells by low ionic strength buffers (28). Previously, levels of the three RPA subunits have been shown to be relatively constant throughout the cell cycle, and the majority of the RPA present in extracts from asynchronously growing HeLa cells has been shown to be present as heterotrimers (12). However, these experiments did not address whether RPA was predominantly heterotrimeric from cells in all cell-cycle stages. To test this, extracts were prepared from suspension HeLa cells separated into G1, S and G2 phase fractions based on cell size using centrifugal elutriation (as described in Materials and Methods). The various cell-cycle extracts were immunoprecipitated using monoclonal antibodies against either the large (RPA70) or middle (RPA32) subunits of RPA. These precipitates were subjected to denaturing polyacrylamide gel electrophoresis (SDS–PAGE) to separate the constituent polypeptides. The proteins on the gels were then immunoblotted for all three RPA subunits. As can be seen in Figure 1, all three RPA subunits co-precipitated at relatively constant ratios with monoclonal antibodies against either RPA32 (Fig. 1B) or RPA70 (Fig. 1D) in cell extracts from all cell-cycle stages. While the relative intensity of the three bands differ in the immunoblots, this is not due to different levels of the three subunits, but rather to different levels of immune response to the three RPA subunits in the polyclonal antiserum used for immunodetection (Fig. 1, see control lane T). Densitometric analyses of lighter exposures showed that the relative intensity of all three RPA subunits remained consistent in the immunoprecipitates from all cell-cycle fractions, and that the relative signals were consistent with signals seen with bacterially purified heterotrimeric RPA (data not shown).
Figure 1.
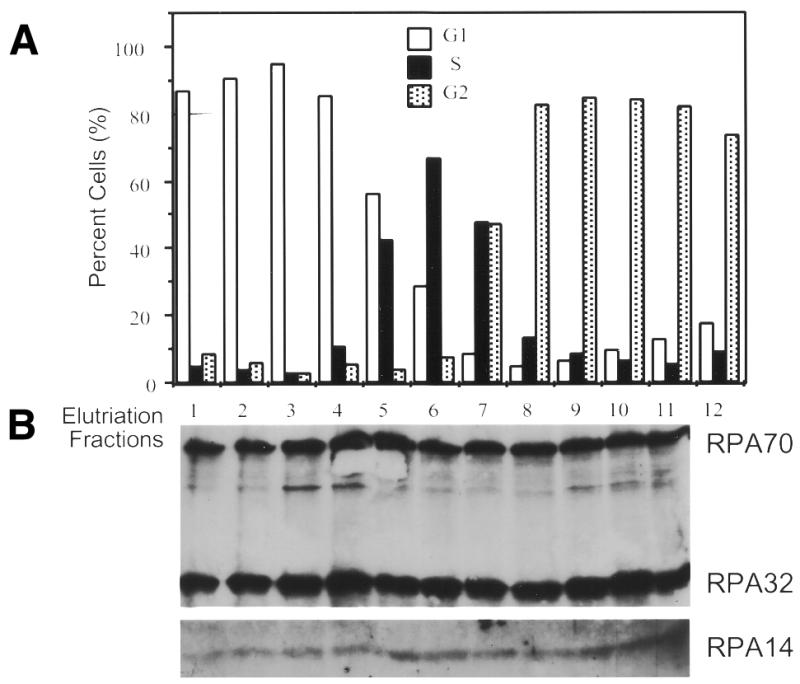
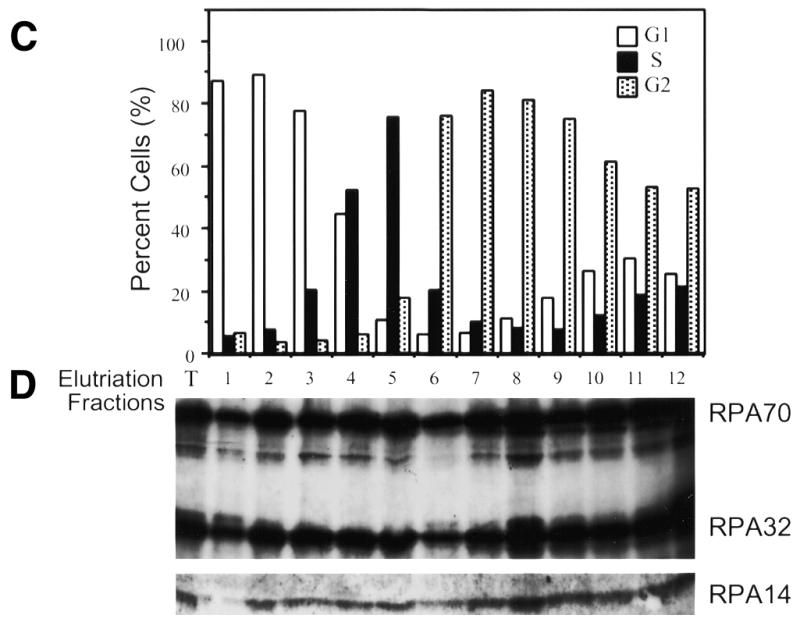
RPA subunit association throughout the cell cycle. HeLa S3 suspension cells were subjected to centrifugal elutriation. Either unfractionated cells (T) or cells from the various fractions (numbered) were analyzed by fluorescence activated cell sorting for cell cycle stage (A and C). Extracts were prepared from elutriated cell fractions and 0.1 ml of each was subjected to immunoprecipitation with either the RPA32 monoclonal antibody (B), or the RPA70 monoclonal antibody (D). The precipitates were immunoblotted using polyclonal antibody against the RPA complex. The weaker RPA14 signal required longer exposures for detection of RPA14. The apparently stronger RPA14 signal in lane 11 of (B) was an artifact caused by difficulties with overexposure required to reproduce the RPA14 signal. Shorter exposures showed no appreciable differences in RPA14 levels between lane 11 and lanes 8–10.
RPA is complexed in extracts from nocodazole-blocked M phase cells
One study showed that in mitotic cells (specifically, HeLa cells blocked with nocodazole), all detectable RPA was apparently separated into its three constitutive subunits; and that those three subunits localized to three different subcellular regions of the cell (15). To test whether this M phase dissociation could be discerned in extracts from nocodazole-treated cells, extracts were prepared from either exponentially growing (non-treated) or nocodazole-blocked (treated) HeLa cells, and the extracts were subjected to immunoprecipitation-immunoblot analysis (as described in Materials and Methods). These mitotic and control cell extracts were immunoprecipitated with monoclonal antibodies against either RPA32 or RPA70. The immunoprecipitates were subjected to SDS–PAGE and immunoblotted using either RPA32 and/or RPA70 monoclonal antibodies for detection. The results show that RPA70 was co-precipitated with RPA32, and vice versa, from both control and nocodazole-treated cell extracts (Fig. 2).
Figure 2.
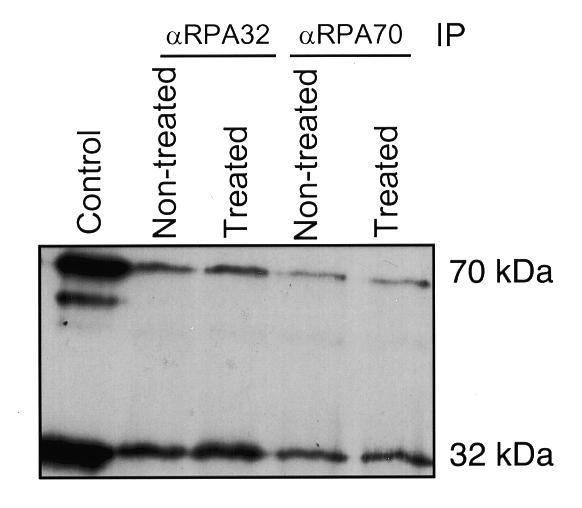
RPA subunit association in mitotic (nocodazole-blocked) cells. Lysates from 4 × 106 cells of either cycling (non-treated) or nocodazole-treated mitotic HeLa cells (treated) were subjected to immunoprecipitation with monoclonal antibody against either RPA32 or RPA70 (as indicated). Each precipitate was subjected to immunoblotting using the monoclonal antibodies against both RPA32 and RPA70. The control lane contained bacterially expressed, purified, human RPA heterotrimer.
RPA is complexed in both the nucleosolic and chromatin fractions
Recent reports that RPA exists in two separable nuclear fractions, a non-ionic detergent-soluble nucleosolic fraction (N) and a non-ionic detergent-stable/ionically eluted chromatin-bound fraction (C) (24–26,29), led us to question whether the reports of wholesale RPA dissociation and differential subcellular localization of the RPA subunits might be due to dissociation of only one of these two RPA subpopulations. [If fixation is performed without washing with non-ionic detergent, the larger nucleosolic pool will be the predominant RPA signal; while if cells are washed with non-ionic detergent, the nucleosolic pool will be removed, leaving the chromatin-bound pool as the only RPA signal (26).] Therefore, both nucleosolic and chromatin RPA fractions were prepared from HeLa cells (as outlined in Fig. 3A). These fractions were subjected to immunoprecipitation for either RPA70 or RPA32 and the precipitates were immunoblotted for the presence of all three subunits. As can be seen in Figure 3B, in both the nucleosolic and chromatin RPA pools, RPA70 co-precipitates with RPA32. Likewise, in Figure 3C, in both the nucleosolic and chromatin RPA pools, RPA32 co-precipitates with RPA70. RPA14 was also detected in these co-precipitations at relative levels consistent with those of purified RPA heterotrimer (data not shown). For both the nucleosolic and chromatin protein fractions, once RPA is depleted from the fraction using three successive immunoprecipitations with anti-RPA32 antibody, further immunoprecipitation with antibody against RPA70 shows no free RPA70 in the remaining supernatant. (The low levels of precipitable RPA70 bring down comparable levels of RPA32.) Likewise, if RPA is depleted from the extract using three successive immunoprecipitations with anti-RPA70 antibody, any further RPA immunoprecipitated from the resultant supernatant with anti-RPA32 is also present as a complex. Interestingly, following successive depletion of RPA using the RPA70 antibody, some RPA complex remains that is precipitable by the RPA32 antibody (data not shown). This is possibly due to ‘masking’ of the recognition epitope for the anti-RPA70 monoclonal antibody under physiological conditions. It has been shown that most of the proteins that interact with RPA interact through the N-terminal region of RPA70, which is where the RPA70 monoclonal antibody binds (2). These results demonstrate that RPA in both the nucleosolic and chromatin associated fractions from HeLa cells can only be detected in the heterotrimeric form.
Figure 3.

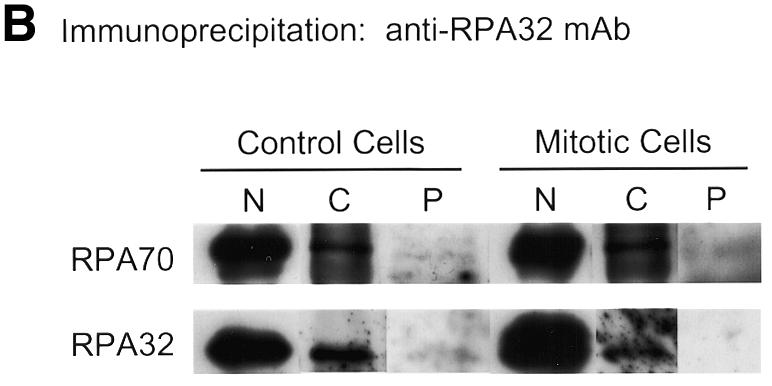
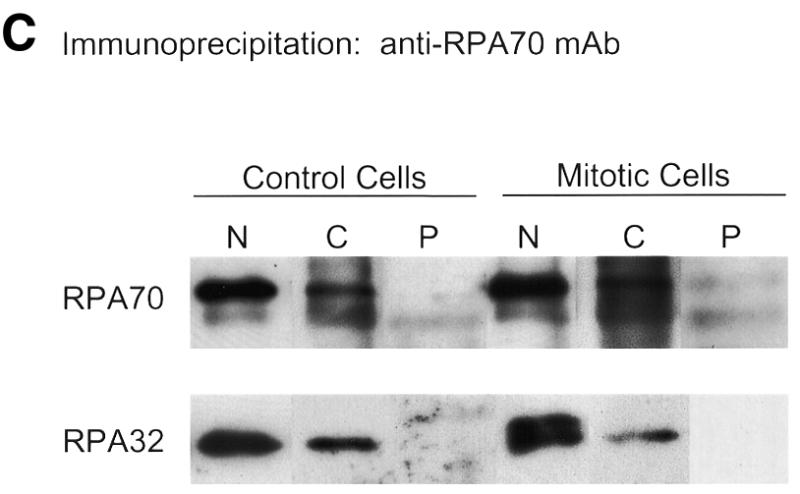
RPA subunit association in nucleosolic and chromatin-associated fractions from both normal and mitotic cells. Cycling and mitotic HeLa cells were separated into three fractions, nucleosolic (N), chromatin-associated (C) and pellet (P) as indicated (A). After preparation of each N and C fraction, the insoluble material was washed three times with the same buffer conditions to minimize any cross-contamination. These washes were all subjected to immunoblotting to confirm clear separation of the N and C RPA fractions (data not shown). The N and C fractions were subjected to multiple immunoprecipitation steps with monoclonal antibody against either RPA32 (B) or RPA70 (C). These immunoprecipitates and the final pellet (P) were subjected to immunoblotting and RPA was detected using RPA polyclonal antibody. Each immunoblotted lane of nucleosolic precipitate represents 8 × 105 cells. Each lane of chromatin-associated precipitate and the pellet preparation represents 4 × 106 cells.
RPA is complexed in the nucleosolic and chromatin fractions from mitotic cells
We also investigated whether both the nucleosolic and chromatin-bound RPA fractions remain complexed after treatment of HeLa cells with nocodazole. HeLa cells were treated with nocodazole as described in Materials and Methods. Visual analysis of DAPI-stained cells showed the nocodazole treated cultures to be 60–65% mitotic (data not shown). The cells were harvested, and both nucleosolic and chromatin-bound RPA pools were prepared as described above. These pools were immunoprecipitated with antibodies against either RPA70 or RPA32 and immunoblotted with the RPA complex antiserum. As was found for the untreated HeLa cell extracts, in the mitotic cell extracts RPA70 was always complexed with RPA32 and vice versa (Fig. 3B and C). Longer exposures showed RPA14 signals consistent with these levels of RPA70 and RPA32 (data not shown). These results indicate that in mitotic cells, the RPA in both nucleosolic and chromatin-bound fractions is only seen in the heterotrimeric form.
Co-sedimentation of RPA subunits
As an alternative method of fractionation, nucleosolic and chromatin-associated RPA pools were also subjected to glycerol gradient sedimentation in the presence of 0.5 M NaCl (to minimize altered sedimentation due to loosely associated proteins). Fractions obtained from glycerol gradient separations were then immunoblotted for the presence of RPA70 and RPA32. It has been noted that hypotonic lysates from human 293 cells contain two populations of RPA32, the RPA heterotrimer and a population not complexed with RPA70 (12). Hence, 293 cell extracts were used for sedimentation markers for both the RPA heterotrimer and for RPA70-free RPA32. As shown in Figure 4, the majority of RPA32 in 293 cell extracts co-sediments with RPA70. We have shown that this peak of co-sedimentation also sediments to the same point as purified recombinant RPA heterotrimer (data not shown). As noted previously, we also detect some RPA32 not complexed with RPA70, and as expected, this RPA70-free RPA32 sediments more slowly than the RPA complex. Densitometric analyses show that the RPA70-free RPA32 represents ~30–40% of the RPA32 present in hypotonically-prepared 293 cell extracts.
Figure 4.

Co-sedimentation of RPA subunits on glycerol gradients. Nucleosolic extracts of cycling (triangles) and mitotically blocked (circles) HeLa cells were sedimented on glycerol gradients of 5–35%, and ∼200 µl fractions were collected. Human 293 cell extract was sedimented similarly as a marker (dashed lines). Samples of these fractions were immunoblotted for the presence of RPA using either monoclonal antibodies against RPA70 (open symbols) or RPA32 (closed symbols). Exposures of the immunoblots were analyzed by densitometry. Values above background were summed as total RPA subunit signal for that gradient. Each fraction was plotted as a percent of total RPA subunit for that gradient.
Extracts from HeLa cells were also subjected to glycerol gradient sedimentation and immunoblotted for the presence of the two larger subunits of RPA. For nucleosolic RPA preparations from both cycling and mitotic HeLa cells, all of the detectable RPA32 co-sedimented with all the RPA70 (Fig. 4). Under the conditions used for sedimentation (500 mM NaCl), this peak sedimented to the same point as purified recombinant RPA heterotrimer. No peaks of non-complexed RPA70 or RPA32 were detected. Densitometric analyses of the immunoblot exposures indicated that if the nucleosolic RPA preparations from HeLa cells contained any uncomplexed RPA32 or RPA70, the amount of uncomplexed RPA subunit would have to represent <1% of the total RPA in these fractions. Chromatin-associated RPA preparations (which contain 5–10% of the total cellular RPA) from cycling and mitotic HeLa cells were also subjected to sedimentation and immunoblotting. These results showed that in most cases all detectable RPA32 co-sedimented with RPA70 (data not shown). However, occasionally the chromatin-associated preparations contained some RPA32 that sedimented more slowly than the RPA70 peak, indicating the presence of a small amount of RPA70-free RPA32. Densitometric analyses indicate that when such RPA70-free populations of RPA32 are detected, that they represent only 0–3% of the total cellular RPA32. These findings support the immunoprecipitation results, indicating that the majority of RPA from both cycling and mitotic HeLa cells exists as a heterotrimeric complex. This is true for both the nucleosolic and chromatin-bound subpopulations of RPA.
DISCUSSION
The results presented herein appear to be inconsistent with several previously published cytological studies (13,15); however, since these other studies appear to be inconsistent both with each other, and with another cytological study (14), inconsistency could not be avoided. As to why these two studies detected a supposed dissociation of all detectable RPA at particular stages of the cell cycle remains in question. One possible explanation is that results of immunofluorescent studies can vary widely depending on the cell types, the methods of fixation, and the specific characteristics of the antibodies used. Since these studies used different cells, different fixation techniques, and often different antibodies, it is not altogether surprising that different findings were reported. A possible explanation for the RPA complex dissociation reported by Cardoso et al. (13), first proposed by Brenot-Bosc et al. (14), is that the observed dissociation of RPA might be due to apoptosis of the myotube cells, rather than to transition through a normal cell cycle. This possibility was proposed based on results by the same group showing that transformation of myotubes with simian virus 40 large T-antigen [which was part of the protocol in (13)] results in appreciable levels of apoptosis (30). Although it was not known at that time how RPA responds to apoptosis, it has been recently shown that the RPA complex apparently dissociates when cells are triggered to apoptose (23). Since apoptosis involves the breakdown of many cellular macromolecules through nuclease and protease action, it would not be surprising if apoptosis also triggered the dissociation of otherwise stable protein complexes. If other experimental variables, such as non-optimal cell culture technique or over-treatment with nocodazole, were to trigger cellular apoptotic responses, this could lead to the breakdown of the RPA complex and artifactual conclusions about RPA complex dissociation during normal cellular progression.
It is not surprising that there should exist two different pools of RPA. It has been shown previously that another DNA replication protein (PCNA) also exists in the nucleus in two populations, one population which can be easily washed from the nucleus and a second that remains tightly associated with DNA replication foci (31). Since both of these two DNA replication factors are also required for various DNA repair systems, one would expect that both would be present in the nucleus in places other than just DNA replication foci. Based on both biochemical assays and protein–protein interaction studies (1,2), the heterotrimeric form of RPA is required for DNA repair. This is supportive of our findings that the nucleosolic RPA, which is likely the population from which RPA is recruited for DNA repair, is predominantly present as a heterotrimeric complex.
It has been reported that a small proportion of the RPA32 in hypotonic (nucleosolic) cell extracts is not complexed with RPA70 (12), but presumably is complexed with RPA14 (2). While we have also detected such a population of RPA32 in 293 cells, and infrequently and to a lesser degree in chromatin-associated RPA from HeLa cells (data not shown; J.-S.Liu, S.-R.Kuo, X.Yin, T.Beerman, and T.Melendy, manuscript submitted), it should be noted that the levels of RPA32 not associated with RPA70 in HeLa cells are quite low (only 0–3%) as compared to RPA levels present as a heterotrimer. As previously reported (13,15), Dimitrova and Gilbert (26) recently indicated that there may also be low levels of uncomplexed RPA14 present at the periphery of the nucleolus. The levels of nucleolar RPA14 appear to be very low and we are currently unable to address this issue using these biochemical approaches.
Overall, our findings support the conclusion that the majority of all cellular RPA, both nucleosolic and chromatin-associated, remain complexed as a heterotrimer throughout the cell cycle. This is consistent with the well-established stability of the RPA complex, although inconsistent with some cell staining studies (13,15). In addition, previous cell-staining studies (14), and more recent work by Dimitrova and Gilbert (26) (indicating that all three RPA subunits in the chromatin-associated RPA pool predominantly co-localize with DNA replication foci), lend support to our findings. Although there exist some data indicating that there may be low levels of non-heterotrimeric RPA32 within HeLa cells, such low levels do not compromise our overall conclusion, that the large majority of RPA in actively cycling cells is predominantly present in the heterotrimeric form.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Ron Berezney for scientific discussions and advice, and D. Dimitrova and D. Gilbert for communication of results prior to publication. This work was supported by an American Cancer Society research grant #RPG GMC-87550 and a pilot project grant from the University at Buffalo, Office of the Provost. T.M. is also supported by NIH grant #GM56406 and a Career Development Award, #AI01686, from the NIH National Institutes of Allergy and Infectious Diseases.
REFERENCES
- 1.Iftode C., Daniely,Y. and Borowiec,J.A. (1999) Crit. Rev. Biochem. Mol. Biol., 34, 141–180. [DOI] [PubMed] [Google Scholar]
- 2.Wold M.S. (1997) Annu. Rev. Biochem., 66, 61–92. [DOI] [PubMed] [Google Scholar]
- 3.Chedin F., Seitz,E.M. and Kowalczykowski,S.C. (1998) Trends Biochem. Sci., 23, 273–277. [DOI] [PubMed] [Google Scholar]
- 4.Brill S.J. and Stillman,B. (1991) Genes Dev., 5, 1589–1600. [DOI] [PubMed] [Google Scholar]
- 5.Bochkarev A., Pfuetzner,R.A., Edwards,A.M. and Frappier,L. (1997) Nature, 385, 176–181. [DOI] [PubMed] [Google Scholar]
- 6.Bochkareva E., Frappier,L., Edwards,A.M. and Bochkarev,A. (1998) J. Biol. Chem., 273, 3932–3936. [DOI] [PubMed] [Google Scholar]
- 7.Gomes X.V., Henricksen,L.A. and Wold,M.S. (1996) Biochemistry, 35, 5586–5595. [DOI] [PubMed] [Google Scholar]
- 8.Gomes X.V. and Wold,M.S. (1996) Biochemistry, 35, 10558–10568. [DOI] [PubMed] [Google Scholar]
- 9.Kim D.K., Stigger,E. and Lee,S.H. (1996) J. Biol. Chem., 271, 15124–15129. [DOI] [PubMed] [Google Scholar]
- 10.Pfuetzner R.A., Bochkarev,A., Frappier,L. and Edwards,A.M. (1997) J. Biol. Chem., 272, 430–434. [DOI] [PubMed] [Google Scholar]
- 11.Philipova D., Mullen,J.R., Maniar,H.S., Lu,J., Gu,C. and Brill,S.J. (1996) Genes Dev., 10, 2222–2233. [DOI] [PubMed] [Google Scholar]
- 12.Din S., Brill,S.J., Fairman,M.P. and Stillman,B. (1990) Genes Dev., 4, 968–977. [DOI] [PubMed] [Google Scholar]
- 13.Cardoso M.C., Leonhardt,H. and Nadal-Ginard,B. (1993) Cell, 74, 979–992. [DOI] [PubMed] [Google Scholar]
- 14.Brenot-Bosc F., Gupta,S., Margolis,R.L. and Fotedar,R. (1995) Chromosoma, 103, 517–527. [DOI] [PubMed] [Google Scholar]
- 15.Murti K.G., He,D.C., Brinkley,B.R., Scott,R. and Lee,S.H. (1996) Exp. Cell Res., 223, 279–289. [DOI] [PubMed] [Google Scholar]
- 16.Brush G.S., Morrow,D.M., Hieter,P. and Kelly,T.J. (1996) Proc. Natl Acad. Sci. USA, 93, 15075–15080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carty M.P., Zernik-Kobak,M., McGrath,S. and Dixon,K. (1994) EMBO J., 13, 2114–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng X., Cheong,N., Wang,Y. and Iliakis,G. (1996) Radiother. Oncol., 39, 43–52. [DOI] [PubMed] [Google Scholar]
- 19.Liu V.F. and Weaver,D.T. (1993) Mol. Cell. Biol., 13, 7222–7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu J.S., Kuo,S.R., McHugh,M.M., Beerman,T.A. and Melendy,T. (2000) J. Biol. Chem., 275, 1391–1397. [DOI] [PubMed] [Google Scholar]
- 21.Rodrigo G., Roumagnac,S., Wold,M.S., Salles,B. and Calsou,P. (2000) Mol. Cell. Biol., 20, 2696–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shao R.G., Cao,C.X., Zhang,H., Kohn,K.W., Wold,M.S. and Pommier,Y. (1999) EMBO J., 18, 1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Treuner K., Okuyama,A., Knippers,R. and Fackelmayer,F.O. (1999) Nucleic Acids Res., 27, 1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Treuner K., Findeisen,M., Strausfeld,U. and Knippers,R. (1999) J. Biol. Chem., 274, 15556–15561. [DOI] [PubMed] [Google Scholar]
- 25.Dimitrova D.S., Todorov,I.T., Melendy,T. and Gilbert,D.M. (1999) J. Cell Biol., 146, 709–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dimitrova D.S. and Gilbert,D.M. (2000) Exp. Cell Res., 254, 321–327. [DOI] [PubMed] [Google Scholar]
- 27.Melendy T., Sedman,J. and Stenlund,A. (1995) J. Virol., 69, 7857–7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fairman M.P. and Stillman,B. (1988) EMBO J., 7, 1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Treuner K., Eckerich,C. and Knippers,R. (1998) J. Biol. Chem., 273, 31744–31750. [DOI] [PubMed] [Google Scholar]
- 30.Endo T. and Nadal-Ginard,B. (1998) J. Cell Sci., 111, 1081–1093. [DOI] [PubMed] [Google Scholar]
- 31.Bravo R. and Macdonald-Bravo,H. (1987) J. Cell Biol., 105, 1549–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]