


Abstract
Werner syndrome (WS) is an autosomal recessive disease characterized by early onset of many features of aging, by an unusual spectrum of cancers, and by genomic instability. The WS protein (WRN) possesses 3′→5′ DNA helicase and associated ATPase activities, as well as 3′→5′ DNA exonuclease activity. Currently, WRN is the only member of the widely distributed RecQ DNA helicase family with documented exonuclease activity. It is not known whether deficiency of the exonuclease or helicase/ATPase activities of WRN, or all of them, is responsible for various elements of the WS phenotype. WRN exonuclease has limited homology to Escherichia coli RNaseD, a tRNA processing enzyme. We show here that WRN preferentially degrades synthetic DNA substrates containing alternate secondary structures, with an exonucleolytic mode of action suggestive of RNaseD. We present evidence that structure-dependent binding of WRN to DNA requires ATP binding, while DNA degradation requires ATP hydrolysis. Apparently, the exonuclease and ATPase act in concert to catalyze structure-dependent DNA degradation. We propose that WRN protein functions as a DNA processing enzyme in resolving aberrant DNA structures via both exonuclease and helicase activities.
INTRODUCTION
Werner syndrome (WS) is a recessive inherited disease that is manifested by premature aging discernible in early adulthood. Patients with WS frequently succumb to heart diseases and to a limited spectrum of malignancies. Cells from WS patients generally exhibit genomic instability (1,2). The WS gene (WRN) encodes two distinct biochemical activities, a 3′→5′ DNA helicase/ATPase (3–5) and a 3′→5′ DNA exonuclease (6–8). The helicase domain of WRN is homologous to others in the RecQ family of DNA helicase, which includes: RecQ of Escherichia coli (9), Sgs1 of Saccharomyces cerevisiae (10,11), Rqh1 of Schizosaccharomyces pombe (12), FFA-1 of Xenopus (13), as well as BLM [the gene product of Bloom syndrome (BS)] (14), RecQL (15,16), RecQ4 [the gene product of Rothmund-Thomson syndrome (RTS)] (17) and RecQ5 (18) of human. Of these, only WRN has been shown to encode a 3→5′ exonuclease.
The biological role of E.coli RecQ has long been considered to be involved in the recombinational pathway RecF (19) and has recently been suggested to function in processes of the re-initiation of stalled replication forks caused by DNA damage (20). In the later situation, RecQ recognizes the replication fork structure and in association with RecJ, RecA and SSB processes the stalled fork, rendering it susceptible to DNA repair enzymes. In S.cerevisiae, mutations in the RecQ homolog Sgs1 suppress the slow growth phenotype of top III mutants (10). Moreover, Sgs1 physically interacts with DNA topoisomerase II and III (10,11), suggesting a DNA topology- or structure-related function for the Sgs1 helicase.
Recent in vitro studies on the E.coli RecQ helicase indicates that it unwinds 3- and 4-way DNA junctions, the symbolic DNA recombinational intermediates, and functions in concert with RecA and SSB to initiate and/or disrupt DNA recombinations (21). This observation is in accord with an earlier report that RecQ helicase is a suppressor of illegitimate recombinations in E.coli (22). RecQ also stimulates Topo III to catenate covalently closed circular DNA molecules (23). Furthermore, yeast Sgs1 protein binds to and unwinds a variety of non-canonical DNA structures, e.g., 3- or 4-way junctions, nicked and gapped DNA (24). In accord with these studies, WRN has been shown to unwind quadruplex formed by d(CGG)n repeats, a tetrahelical DNA stabilized by guanine–guanine non-Watson–Crick hydrogen bonds (25). BLM and yeast Sgs1 also can efficiently unwind other quadruplex DNA in vitro (26,27). These combined studies suggest that an important function of RecQ helicases is to unwind alternative DNA structures that may impede DNA metabolic events.
Amongst the RecQ helicases identified so far, WRN is the only one that contains an integral exonuclease. The conserved motifs of WRN exonuclease are located in the N-terminus, whereas the RecQ helicase motifs are in the center (2). Activities of WRN exonuclease on a 3′-recessed dsDNA include excision of the 3′ nucleotide with or without a 3′-PO4 and the preferential removal of a 3′-terminal mismatch (8). Sequence alignment of the conserved motifs with known exonucleases indicated that the WRN exonuclease is similar to the proofreading exonuclease of E.coli DNA polymerase I and E.coli RNaseD (28,29). The characterized WRN exonuclease activities with a 3′→5′ direction and the removal of 3′-terminal mismatch are consistent with the prediction, however, WRN is unable to hydrolyze single-stranded DNA (8) suggesting that this exonuclease may have a different function in cells.
We used a variety of DNA templates with defined structures to gain understanding of the function of this exonucleolytic activity. Here we report that WRN protein preferentially binds to DNA containing an open helical structure such as a bubble, a loop or a stem–loop, and these structures stimulate the WRN exonuclease to excise the nascent DNA terminus 3′→5′ in a structure-dependent manner.
MATERIALS AND METHODS
Purification of WRN protein
Recombinant hexahistidine-tagged WRN was purified from baculovirus-infected insect cells by sequential chromatographic steps to >90% homogeneity (7).
Construction of oligonucleotide substrates
Oligonucleotides (Table 1), purchased from Operon Technology (Alameda, CA), were purified by HPLC and polyacrylamide gel electrophoresis. Oligomers were 5′-end labeled by incubation with [γ-32P]ATP (NEN, Boston, MA) and polynucleotide kinase (New England Biolabs, Beverly, MA). Annealing to the unlabeled strand (1:1) was carried out in 10 mM Tris–HCl pH 7.4, 1 mM EDTA by incubation at 100°C for 3 min followed by slow cooling to room temperature. For 3′-end labeling, oligonucleotides were annealed to the unlabeled strand; the resulting partial duplex contained a 3′-recessed terminus was used to incorporate four residues of [α-32P]dCTP and two residues of dGTP by the Klenow fragment (exo–) of E.coli DNA polymerase I (New England Biolabs).
Table 1. Sequence of synthetic oligonucleotides.

Exonuclease assay
32P-labeled DNA substrate (0.1 pmol) was incubated with 12.5 or 25 fmol of recombinant WRN at 37°C in a 10 µl reaction mixture containing 40 mM Tris–HCl pH 7.4, 4 mM MgCl2, 5 mM DTT, 100 µg/ml BSA and 1 mM ATP [or 1 mM ATPγS (Roche Molecular Biochemicals, Indianapolis, IN) where specified]. Reactions were terminated by adding 2 µl of 40% glycerol, 50 mM EDTA, 2% SDS, 3% bromophenol blue, 3% xylene cyanol. An aliquot of 6 µl of denaturing loading buffer (76% formamide, 20 mM EDTA, 0.05% bromophenol blue, 0.05% xylene cyanol) was added to 6 µl of the stopped reaction mixture prior to electrophoresis in a 7 M urea/14% polyacrylamide gel in 1× TBE (90 mM Tris base, 90 mM boric acid, 1 mM EDTA). Gels were vacuum dried and reaction products were visualized by autoradiography and quantified by PhosphorImager (Molecular Dynamics, Amersham Pharmacia Biotech, Piscataway, NJ).
Gel mobility shift assay
5′-32P -labeled DNA (100 fmol) was incubated with increasing concentrations of recombinant WRN in 10 µl of 40 mM Tris–HCl pH 7.4, 4 mM MgCl2, 5 mM DTT, 100 µg/ml BSA, 1 mM ATPγS at room temperature for 30 min and then at 4°C for 10 min. Loading buffer (2 µl of 40% glycerol, 0.25% bromophenol blue) was added and reaction products were resolved by 6% polyacrylamide gel electrophoresis in 1× TBE at 10 V/cm for 4 h; gel temperature was maintained at ∼9°C. Gels were vacuum dried and band-shifts were visualized by autoradiography and quantified by PhosphorImager (Molecular Dynamics).
RESULTS
DNA containing a ‘bubble’ stimulates WRN exonuclease activity
WRN exonuclease hydrolyzes double-stranded DNA (dsDNA) with a 3′-recessed end in a 3′→5′ direction, but does not degrade terminal nucleotides at a blunt end (8). To examine the effect of alternative DNA structures on WRN-catalyzed DNA hydrolysis, we measured the extent of WRN exonuclease activity using a blunt-ended dsDNA containing a region of non-complementary base-pairings (for oligonucleotide sequence and DNA construct, see Tables 1 and 2, respectively). As shown in Figure 1A, a blunt-ended dsDNA containing a single-stranded bubble (oligos 3 + 2) is extensively degraded by WRN, while a cognate, fully base-paired duplex (Fig. 1B; oligos 1 + 2) is hydrolyzed little, if at all. Degradation of bubble DNA by WRN is exonucleolytic and proceeds in the 3′→5′ direction. Thus, as illustrated in Figure 1A, cleavage of substrate radiolabeled at the 5′-terminus yields time-dependent accumulation of progressively smaller products differing by a single nucleotide. In accord, cleavage of substrate containing labeled nucleotides at the 3′-terminus (Fig. 1C; oligos 4 + 2) yields time-dependent release of label migrating as mononucleotide, without detectable accumulation of products of any endonuclease activity. The stepwise degradation of bubble DNA from the 3′-terminus seen in Figure 1A slows markedly 5 nt from the bubble (position 32). The observed, structure-dependent degradation of bubble DNA requires ATP hydrolysis; either omission of ATP (data not shown) or substitution of the non-hydrolysable analog ATPγS (Fig. 1D) eliminates activity.
Table 2. WRN binds to and degrades various alternative DNA structures.

1WRN exonuclease activity was determined as amounts of 3′-terminal nucleotides released by WRN (5 nM) from each DNA substrate (10 nM) in a 20 min incubation at 37°C. ‘1’ corresponds to 86.7 fmol of 3′-terminal nucleotides released from the original 100 fmol of DNA substrate under the reaction conditions.
2WRN–DNA binding was determined using gel-shift assay, by which amounts of DNA that forms a stable complex with WRN in an incubation including 10 nM of DNA and 5 nM of WRN were scored. ‘1’ corresponds to 4.9 fmol of DNA that forms WRN–DNA complex from the original 100 fmol of DNA substrate under the reaction conditions.
3*, 5′-32P label.
Figure 1.

WRN exonuclease preferentially hydrolyzes DNA containing a single-stranded bubble. Degradation products resulting from incubation of synthetic DNA oligomers (10 nM) with recombinant WRN (1.25 nM) were resolved in 7 M Urea/14% denaturing polyacrylamide gels. Lanes are, from left to right: No WRN control (S), and 0, 5, 10, 20, 30, 40, 50 and 60 min incubation times. (A) A blunt-ended, partially duplex 46mer oligonucleotide containing 8 purine–pyrimidine mismatches (bubble DNA) is degraded in a time-dependent reaction. Degradation of 5′-32P-labeled substrate proceeds step-wise from the 3′-terminus, halting 5 bases from the bubble at position 32. (B) A blunt-ended, fully duplex 46mer oligonucleotide corresponding to bubble DNA is not an effective substrate. (C) WRN degrades 3′-32P-labeled bubble DNA exonucleolytically from the 3′-terminus. Accumulation of 32P-dCMP from the 3′-end is observed, without appearance of mid-sized products. (D) Degradation of bubble DNA requires hydrolysis of ATP, evidenced by elimination of activity upon substitution of ATPγS. ATP hydrolysis may be essential for maintaining WRN in the appropriate conformational and/or oligomeric state.
The structure-dependent exonuclease activity of WRN is paralleled by the structure-dependent binding of WRN to synthetic DNA substrates, demonstrated in gel band-shift assays. As indicated in Figure 2A, WRN binds to bubble DNA in the presence of 1 mM ATPγS in a concentration-dependent reaction. In contrast, WRN does not retard migration of fully base-paired duplex DNA (Fig. 2C). The band shifting seen in Figure 2A is reduced by omission of ATPγS (Fig. 2B), indicating that structure-dependent binding of WRN to bubble DNA is stimulated upon binding of ATP.
Figure 2.
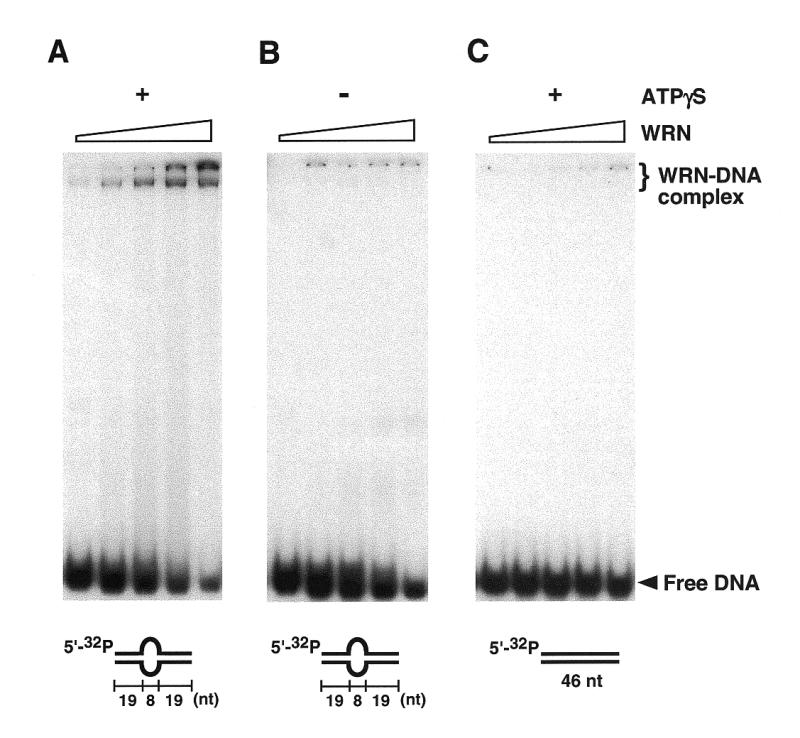
WRN binds tightly to DNA containing a single-stranded bubble, but not to fully duplex DNA. Products resulting from incubation of bubble DNA (10 nM) with WRN (1, 2.5, 5, 10 or 25 nM) in the presence of 1 mM ATPγS were resolved in 6% non-denaturing polyacrylamide gels. (A) WRN forms stable complexes with bubble DNA. Two slowly migrating bands are observed, the faster of which forms at lower WRN concentrations; these bands may represent different oligomeric states of WRN, whose subunit structure is currently unknown. (B) ATPγS is required for formation of stable WRN–DNA complexes. (C) WRN does not form detectable complexes with a corresponding, fully base-paired DNA.
The single-stranded bubble also stimulates WRN exonuclease activity acting on a 3′-recessed terminus (Fig. 3A; oligos 3 + 16), the locus previously reported as degradable by WRN exonuclease (6–8). The degradation patterns are similar to those for the blunt-ended bubble DNA (Fig. 1A), the step-wise hydrolysis of the 5′-labeled loop-bearing strand is reduced 5 nt from the loop (position 32). The partial duplex with a 3′-recessed end (Fig. 3B; oligos 1 + 16), in contrast, is hydrolyzed by WRN but at a lesser extent. Further quantification of the hydrolytic products indicates that WRN hydrolyzes a bubble DNA more progressively than a partially duplex DNA (Fig. 3C). Only the first two to three 3′-terminal nucleotides of the partial duplex are degraded in the 20 min incubation, contrasting to patterns of progressive hydrolysis on the bubble DNA. The extent of stimulation average over the 14 nt is as great as 6-fold in favor of the bubble DNA. The structure-selective DNA binding by WRN to bubble DNA substantiates that WRN exonuclease is a structure-dependent enzyme. As shown in a band-shift assay (Fig. 3D), in the presence of 1 mM ATPγS in a concentration-dependent reaction, WRN selectively binds to the 3′-recessed bubble DNA but not to the partial duplex lacking a bubble.
Figure 3.
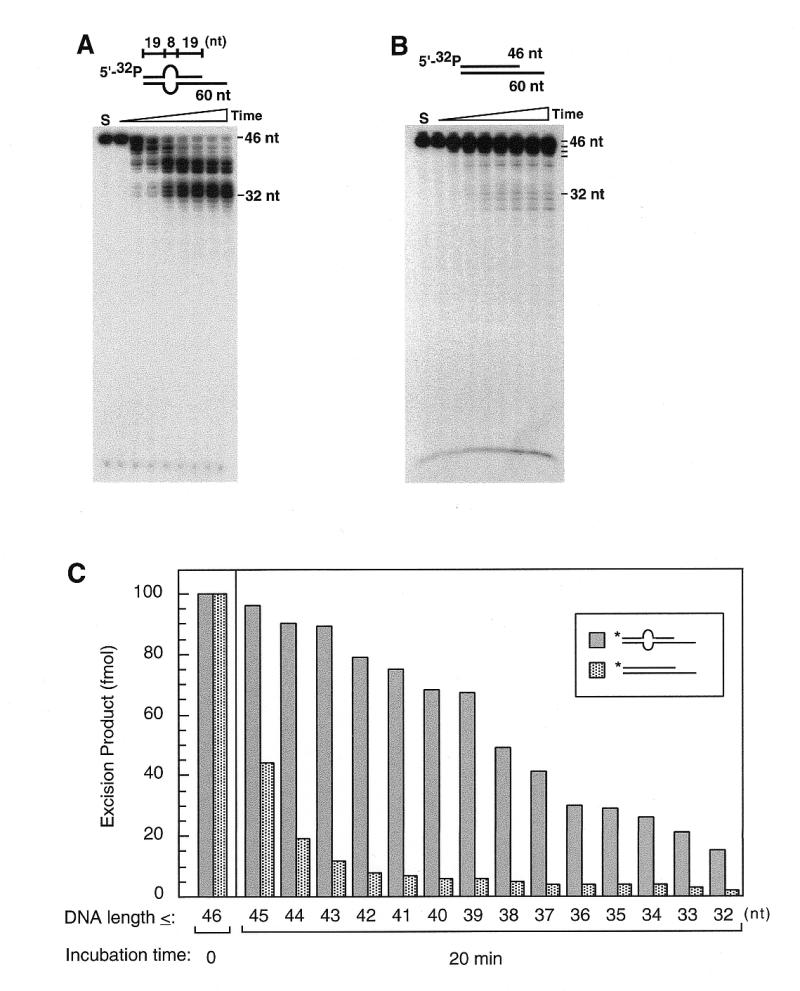

WRN preferentially binds and degrades 3′-recessed DNA containing a bubble structure. Products of degradation from incubation of DNA substrates (10 nM) and WRN (2.5 nM) were resolved in denaturing polyacrylamide gels (A–C). Results of the time-dependent reaction are presented in lanes from left to right: No WRN control (S), and 0, 5, 10, 20, 30, 40, 50 and 60 min of incubation. WRN–DNA binding was observed as band-shifts on a 6% polyacrylamide gel (D). Increasing concentrations of WRN (0, 1, 2.5, 5, 10 and 25 nM) were incubated with DNA substrates (10 nM) in the presence of 1 mM ATPγS and reaction products analyzed by gel-shift assay. (A) WRN readily degrades the 5′-32P-labeled, 3′-recessed strand from the 3′-terminus in a bubble-bearing partially duplex DNA. Progressive hydrolysis is observed in a time-dependent reaction, stalling 5 bases from the bubble at position 32. (B) In contrast, WRN degrades regular 3′-recessed DNA in a less efficient mode. (C) Quantification of reaction products at 20 min incubation in (A) and (B) by integrating intensities of bands which are equal to and smaller than (≤) the numbered position indicates that WRN degrades bubble DNA more rapidly and progressively than does the partially duplex DNA. The bubble DNA is hydrolyzed efficiently to the stalling site (position 32), whereas the partially duplex DNA is slowly degraded and only the first two or three nucleotides from the 3′ terminus can be effectively excised. (D) WRN forms a stable complex with bubble DNA bearing a 5′-tail but not the 5′-overhang partially duplex DNA. WRN–DNA complexes are observed in lanes where bubble DNA was incubated with increasing concentrations of WRN, by contrast, under the same reaction conditions, no significant band-shifts appear in lanes with 3′-recessed DNA.
DNA with single-stranded loop also stimulates WRN exonuclease activity
Blunt-ended duplex DNA containing a single-stranded loop is also hydrolyzed by WRN (Fig. 4; oligos 5 + 2). The cleavage patterns observed for loop DNA are comparable to those for bubble DNA, demonstrating exonucleolytic degradation of both strands from the 3′-terminus. Thus, step-wise degradation of the 5′-labeled, loop-bearing strand (Fig. 4A) slows 5 nt from the loop, analogous to the case of bubble DNA. Degradation of 3′-labeled loop-bearing strand (Fig. 4C; oligos 6 + 2) results in accumulation of label migrating as monomer, without appearance of mid-sized products. Hydrolysis of the opposite, non-loop-bearing strand (Fig. 4B) also slows 5 nt from the loop. The small amount of label migrating as monomer in Figure 4B reflects either a minor 5-exonucleolytic activity (30) of WRN or a residual, contaminating activity.
Figure 4.

WRN degrades blunt-ended dsDNA bearing an extra-helical loop from 3′→5′. Degradation products resulting from incubation of DNA substrates (10 nM) with WRN (1.25 nM) were resolved in denaturing gels. Lanes from left to right are: No WRN control (S), and 0, 5, 10, 20, 30, 40, 50 and 60 min incubation times. (A) A blunt-ended, partially duplex oligonucleotide containing an 8 nt single-stranded loop (loop DNA) is hydrolyzed in a time-dependent reaction. Degradation of the 5′-32P-labeled loop-bearing strand proceeds step-wise from the 3′-terminus, exhibiting a pause site 5 bases from the loop at position 35. (B) The strand opposite the loop-bearing strand is also degraded in the 3′→5′ direction, and likewise exhibits a major pause site 5 bases from the loop, at position 29. (C) WRN degrades 3′-32P-labeled loop DNA from the 3′-terminus. Accumulation of 32P-dCMP excised from the 3′-end is observed, without appearance of endonucleolytic products.
Not unexpectedly, an extra-helical loop structure also stimulates the hydrolytic activity of WRN acting on a 3′-recessed terminus (Fig. 5A; oligos 5 + 17). The 3′→5′ degradation of the loop-containing strand is progressive and slows 5 nt from the loop (position 35), the same as in the case of bubble DNA (Fig. 3A). The structure-dependent hydrolysis by WRN can be efficiently initiated from a single-strand nick 3′ downstream from the loop (Fig. 5B; oligos 5 + 17 + 18), with degradation patterns similar to those of the 3′-recessed substrate (Fig. 5A). By contrast, WRN exonuclease is very inefficient in hydrolyzing DNA from a single-stranded nick without an adjacent loop structure (Fig. 5C; oligos 1 + 17 + 18). Apparently, the 3′→5′, nick excision activity of WRN is stimulated by a nascent extra-helical loop.
Figure 5.

Extra-helical loop structure stimulates WRN in initiating 3′→5′ exonucleolytic reaction from a single-strand nick. Reaction products from incubation of synthetic oligonucleotide substrates (10 nM) and WRN (2.5 nM) were resolved in denaturing gels. Lanes from left to right are: No WRN control (S), and 0, 5, 10, 20, 30, 40, 50 and 60 min incubation times. (A) DNA loop structure stimulates WRN in degradation of 3′-recessed termini in a time-dependent reaction. Degradation of the 3′-recessed, 5′-32P-labeled loop-bearing strand proceeds step-wise from the 3′-terminus, slowing 5 bases from the loop at position 35. (B) WRN degrades 5′-32P-labeled loop-bearing strand from the 3′-terminus within a single-strand nick. Step-wise degradation proceeds from the 3′-terminus, slowing down 5 bases from the loop, at position 35. (C) In contrast, without the stimulation by an adjacent loop structure, WRN 3′→5′ exonuclease is much less efficient in degradation of DNA from a single-strand nick.
Other aberrant DNA structures that stimulate WRN exonuclease activity
Stem–loop DNA (Fig. 6; oligos 7 + 8) is also hydrolyzed by WRN. In the presence of Mg2+, the 5′-32P -labeled strand opposite the stem–loop in Figure 6A is degraded stepwise from the 3′-terminus with relatively little pausing, up to 3 nt from the stem (position 35), whereas the stepwise degradation is more extensive in the presence of Mn2+. The stem–loop strand, 5′-32P -labeled in Figure 6B, is also progressively degraded; in the presence of Mg2+, the relatively weak hydrolysis may reflect the greater distance of the scissionable bonds from the stem–loop. However, in the presence of Mn2+, the progressive degradation enhances and it slows firstly at position 42, 10 nt from the stem, and secondly at position 35, 3 nt from the stem.
Figure 6.

WRN exonucleolytically degrades blunt-ended dsDNA bearing a stem–loop structure. Reaction products from incubation of DNA substrates (10 nM) and WRN (1.25 nM) were visualized on autoradiograms taken from resolving gels. Lanes exhibit from left to right: No WRN control (S), and 0, 5, 10, 20, 30, 40, 50 and 60 min incubation time. (A) The 5′-32P-labeled strand complementary to a stem–loop bearing strand is degraded step-wise from 3′-terminus in a time-dependent reaction in the presence of 4 mM MgCl2, stalling 3 bases from the stem, at position 35. Similarly but more extensively, degradations also occur in the presence of 4 mM MnCl2. (B) Although less efficiently due to the distance between the stem and the terminus, 5′-32P-labeled stem–loop strand is also degraded 3′→5′ by WRN in the presence of MgCl2. Degradation proceeds slowly, halting at position 42. However, in the presence of MnCl2, degradation is greatly enhanced, proceeding more rapidly, passing the first pause and halting 3 bases from the stem at position 35.
As summarized in Table 2, a variety of alternative DNA structures such as 3- and 4-way junctions (substrates 7 and 8, respectively) in addition to the above structures stimulate WRN exonuclease activity acting on the blunt ends. Binding by WRN to those alternative DNA structures can also be observed in different degrees by mobility-shift assays in non-denaturing gels, as exemplified in Figure 7 for substrates 3–8. A comparison of relative exonuclease- and DNA-binding activities of WRN acting on different structures is tabulated in Table 2. It suggests that WRN protein prefers binding to DNA structures bearing disruption of Watson–Crick base-pairing (for example, substrates 2 and 10) or discontinuity of the duplex conformation (for example, substrates 3–8 and 11) and hydrolyzing the nascent 3′ terminus. Thus, a linear duplex DNA with blunt ends (substrate 1) is not a substrate for WRN, even the one bearing a 3′-recessed end (substrate 9) is not as effective as any other bearing an aberrant DNA structure.
Figure 7.
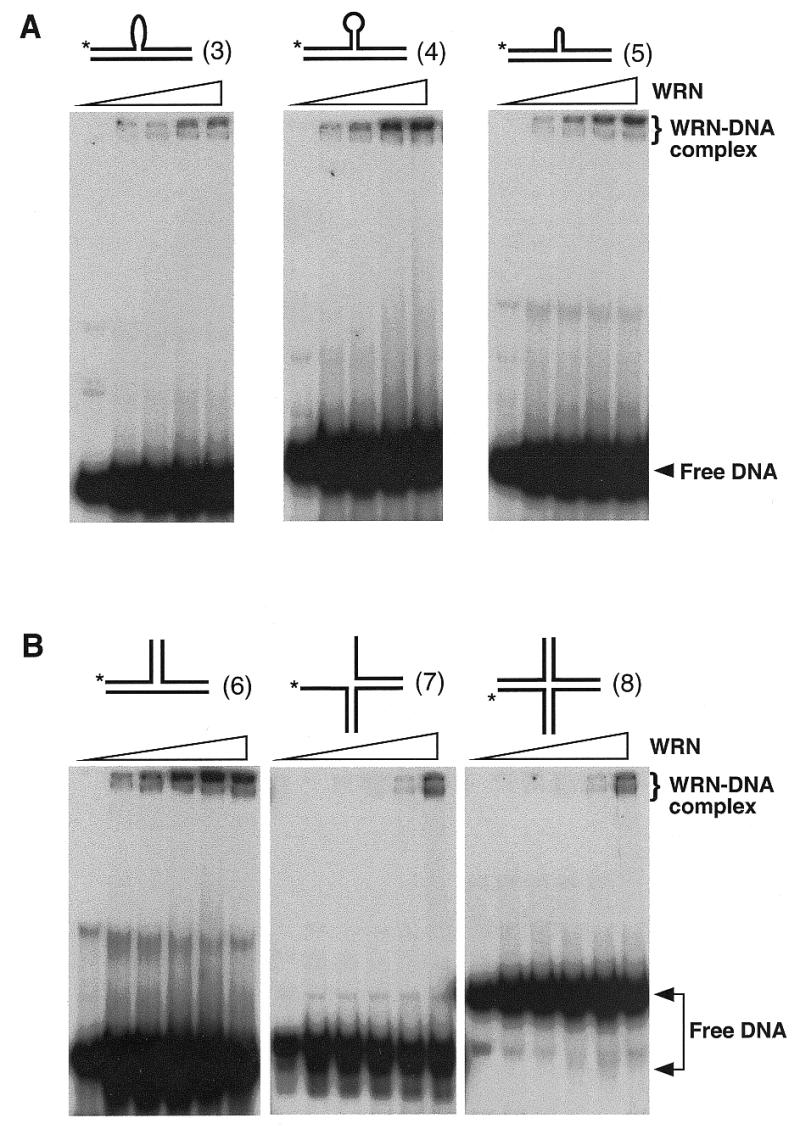
WRN binds to alternative DNA structures. Products resulting from incubation of alternative DNA substrates with increasing concentrations of WRN in the presence of 1 mM ATPγS were resolved in 6% non-denaturing polyacrylamide gels. (A) Stable WRN–DNA complexes appear in reactions of substrate 3, 4 and 5 (10 nM) incubated with increasing concentrations of WRN (0, 1, 2.5, 5 and 10 nM). (B) Stable WRN–DNA complexes appear in reactions of substrate 6, 7 and 8 (10 nM) incubated with increasing concentrations of WRN (0, 1, 2.5, 5, 10 and 25 nM).
DISCUSSION
We have presented data indicating that alternative DNA structures, particularly those containing disruptions of canonical base-pairings in DNA duplex (Table 2), stimulate the hydrolytic activity of WRN on the DNA substrates. This structure-selective activity is ATP-stimulated (Fig. 1), suggesting that the ATPase domain, and probably the entire helicase domain also, are required for a serial mechanistic steps in the hydrolytic reaction, such as structure recognition, DNA binding and the 3′-terminal hydrolysis.
WRN protein is able to form a stable complex with almost all the tested alternative DNA structures, particularly the ones bearing a bubble (Table 2). In contrast, linear duplex DNA with a blunt or 3′-recessed end is not as effective a substrate for WRN-binding. Notably, as observed on native polyacrylamide gels, WRN helicase does not efficiently unwind these DNA structures under the assay conditions (data not shown). With both helicase and exonuclease activities in the same polypeptide, WRN may partially unwind alternative DNA structures to facilitate the action of the exonuclease. The correlation between WRN exonuclease activity and binding to different DNA substrates (Table 2) is not perfect. For example, the exonuclease activity on substrate 2 and 10 (the bubble DNA) correlates with the binding, while the activity on substrate 7 and 8 does not. The data in Table 2 represents the hydrolysis of 3′-terminal nucleotides; thus may be sequence biased and does not take into account the processivity of the exonuclease with different substrates.
The evidence that WRN unwinds G-quadruplex DNA (25) in combination with the data in Table 2 that WRN degrades termini of 3- and 4-way junction DNA may provide clues to the cellular function of WRN. On the basis of structure-dependency, it is also anticipated that WRN degrades G-quadruplex DNA from its nascent breaks including blunt ends, 3′-recessed ends and single-strand nicks, particularly in light of the finding that a synthetic 4-way junction DNA can be unwound by WRN (J.-C.Shen and L.A.Loeb, unpublished data). Thus, WRN may play an essential role in resolving aberrant DNA structures in living cells by using both DNA unwinding and nucleotide excision activities. However, the question remains: how do the two activities co-ordinate at sites of DNA biosynthetic intermediates?
The exonuclease domain of WRN is homologous to E.coli RNaseD (28,29), a tRNA processing enzyme that exonucleolytically trims the 3′-end of precursor molecules to create a -CCA terminus for addition of the cognate amino acid (31; Scheme 1). RNaseD slows greatly upon reaching the appropriate CCA sequence, thus permitting aminoacylation. The structure-dependent, exonucleolytic mode of action of WRN observed here (Scheme 1), especially the pausing before alternate secondary structure elements, appears similar to that of RNaseD, and is not surprising, given the homology. Although WRN exonuclease activity is separable (6,7), it is unlikely that the exonuclease domain alone reserves similar structure-selective function and conducts the structure-dependent DNA hydrolysis. As we have suggested here, a full-length protein is required for WRN to perform this structure-dependent activity. Mice have been created that harbor a deletion of helicase motifs III and IV in the endogenous WRN homolog (32), presumably leaving the ATPase domain in motifs I and II intact. It would be interesting to determine if these mutants produce a truncated protein and if this protein exhibits structure-dependent or -independent exonucleolytic activity.

Scheme 1. The mode of action of WRN on DNA containing alternate structure appears similar to the action of E.coli RNaseD.
Instead of activation by DNA substrates, WRN exonuclease (3′→5′) can reportedly be trans-activated by an interacting protein, the Ku86/70 complex (33). This finding suggests an important role for WRN in processing DNA breaks. Interestingly, Ku86/70 does not stimulate WRN helicase activity, implicating a delicate co-ordination between helicase and exonuclease activities while at sites of action. Both examples of activity stimulation address the importance of exonuclease in the biological roles of WRN protein.
All known disease-associated WS alleles are nonsense mutations (34), resulting in loss of a C-terminal nuclear localization signal and apparent exclusion of all WRN catalytic activities from their site(s) of action in the nucleus (35). Hence, depending on the levels of functionally redundant catalytic activities, deficiency of WRN exonuclease and/or helicase/ATPase activities may underlie different elements of the WS phenotype (e.g., genomic instability and premature aging). Notably, the helicase and nuclease activities of yeast Dna2, an enzyme involved in DNA replication (36), are both important for the cellular functions. Relevant functional redundancy is exemplified by Sgs1, the S.cerevisiae WRN homolog, and the helicase Srs2; Sgs1 becomes essential for DNA replication when Srs2 is absent (37). This functional redundancy may account for the late onset of the manifestation of Werner syndrome.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Ann Blank for her valuable suggestions and critical reading of this manuscript. We are also grateful to Dr Ashwini Kamath-Loeb for the protein purification and constructive suggestions. This work was supported by grant CA-77852 from the National Institute of Health.
REFERENCES
- 1.Martin G.M., Oshima,J., Gray,M.D. and Poot,M. (1999) J. Am. Geriatr. Soc., 47, 1136–1144. [DOI] [PubMed] [Google Scholar]
- 2.Shen J.-C. and Loeb,L.A. (2000) Trends Genet., 16, 213–220. [DOI] [PubMed] [Google Scholar]
- 3.Yu C.-E., Oshima,J., Fu,Y.-H., Wijsman,E.M., Hisama,F., Alisch,R., Matthews,S., Nakura,J., Miki,T., Ouais,S., Martin,G.M., Mulligan,J. and Schellenberg,G.D. (1996) Science, 272, 258–262. [DOI] [PubMed] [Google Scholar]
- 4.Gray M.D., Shen,J.-C., Kamath-Loeb,A.S., Blank,A., Sopher,B.L., Martin,G.M., Oshima,J. and Loeb,L.A. (1997) Nature Genet., 17, 100–103. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki N., Shimamoto,A., Imamura,O., Kuromitsu,J., Kitao,S., Goto,M. and Furuichi,Y. (1997) Nucleic Acids Res., 25, 2973–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang S., Li,B., Gray,M.D., Oshima,J., Mian,I.S. and Campisi,J. (1998) Nature Genet., 20, 114–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen J.-C., Gray,M.D., Oshima,J., Kamath-Loeb,A.S., Fry,M. and Loeb,L.A. (1998) J. Biol. Chem., 273, 34139–34144. [DOI] [PubMed] [Google Scholar]
- 8.Kamath-Loeb A.S., Shen,J.-C., Loeb,L.A. and Fry,M. (1998) J. Biol. Chem., 273, 34145–34150. [DOI] [PubMed] [Google Scholar]
- 9.Nakayama K., Irino,N. and Nakayama,H. (1985) Mol. Gen. Genet., 200, 266–271. [DOI] [PubMed] [Google Scholar]
- 10.Gangloff S., McDonald,J.P., Bendixen,C., Arthur,L. and Rothstein,R. (1994) Mol. Cell. Biol., 14, 8391–8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watt P.M., Louis,E.J., Borts,R.H. and Hickson,I.D. (1995) Cell, 81, 253–260. [DOI] [PubMed] [Google Scholar]
- 12.Stewart E., Chapman,C.R., Al-Khodairy,F., Carr,A.M. and Enoch,T. (1997) EMBO J., 16, 2682–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan H., Chen,C.Y., Kobayashi,R. and Newport,J. (1998) Nature Genet., 19, 375–378. [DOI] [PubMed] [Google Scholar]
- 14.Ellis N.A., Groden,J., Ye,T.-Z., Straughen,J., Lennon,D.J., Ciocci,S., Proytcheva,M. and German,J. (1995) Cell, 83, 655–666. [DOI] [PubMed] [Google Scholar]
- 15.Puranam K.L. and Blackshear,P.J. (1994) J. Biol. Chem., 269, 29838–29845. [PubMed] [Google Scholar]
- 16.Seki M., Miyazawa,H., Tada,S., Yanagisawa,J., Yamaoka,T., Hoshino,S., Ozawa,K., Eki,T., Nogami,M. and Okumura,K. (1994) Nucleic Acids Res., 22, 4566–4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitao S., Shimamoto,A., Goto,M., Miller,R.W., Smithson,W.A., Lindor,N.M. and Furuichi,Y. (1999) Nature Genet., 22, 82–84. [DOI] [PubMed] [Google Scholar]
- 18.Kitao S., Ohsugi,I., Ichikawa,K., Goto,M., Furuichi,Y. and Shimamoto,A. (1998) Genomics, 54, 443–452. [DOI] [PubMed] [Google Scholar]
- 19.Kowalczykowski S.C., Dixon,D.A., Eggleston,A.K., Lauder,S.D. and Rehrauer,W.M. (1994) Microbiol. Rev., 58, 401–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Courcelle J. and Hanawalt,P.C. (1999) Mol. Gen. Genet., 262, 543–551. [DOI] [PubMed] [Google Scholar]
- 21.Harmon F.G. and Kowalczykowski,S.C. (1998) Genes Dev., 12, 1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanada K., Ukita,T., Kohno,Y., Saito,K., Kato,J.-I. and Ikeda,H. (1997) Proc. Natl Acad. Sci. USA, 94, 3860–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harmon F.G., DiGate,R.J. and Kowalczykowski,S.C. (1999) Mol. Cell, 3, 611–620. [DOI] [PubMed] [Google Scholar]
- 24.Bennett R.J., Keck,J.L. and Wang,J.C. (1999) J. Mol. Biol., 289, 235–248. [DOI] [PubMed] [Google Scholar]
- 25.Fry M. and Loeb,L.A. (1999) J. Biol. Chem., 274, 12797–12802. [DOI] [PubMed] [Google Scholar]
- 26.Sun H., Karow,J.K., Hickson,I.D. and Maizels,N. (1998) J. Biol. Chem., 273, 27587–27592. [DOI] [PubMed] [Google Scholar]
- 27.Sun H., Bennett,R.J. and Maizels,N. (1999) Nucleic Acids Res., 27, 1978–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mushegian A.R., Bassett,D.E.,Jr, Boguski,M.S., Bork,P. and Koonin,E.V. (1997) Proc. Natl Acad. Sci. USA, 94, 5831–5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moser M.J., Holley,W.R., Chatterjee,A. and Mian,I.S. (1997) Nucleic Acids Res., 25, 5110–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki N., Shiratori,M., Goto,M. and Furuichi,Y. (1999) Nucleic Acids Res., 27, 2361–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cudny H., Zaniewski,R. and Deutscher,M.P. (1981) J. Biol. Chem., 256, 5633–5637. [PubMed] [Google Scholar]
- 32.Lebel M. and Leder,P. (1998) Proc. Natl Acad. Sci. USA, 95, 13097–13102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper M.P., Machwe,A., Orren,D.K., Brosh,R.M., Ramsden,D. and Bohr,V.A. (2000) Genes Dev., 14, 907–912. [PMC free article] [PubMed] [Google Scholar]
- 34.Matsumoto T., Imamura,O., Yamabe,Y., Kuromitsu,J., Tokutake,Y., Shimamoto,A., Suzuki,N., Satoh,M., Kitao,S., Ichikawa,K., Kataoka,H., Sugawara,K., Thomas,W., Mason,B., Tsuchihashi,Z., Drayna,D., Sugawara,M., Sugimoto,M., Furuichi,Y. and Goto,M. (1997) Hum. Genet., 100, 123–130. [DOI] [PubMed] [Google Scholar]
- 35.Matsumoto T., Shimamoto,A., Goto,M. and Furuichi,Y. (1997) Nature Genet., 16, 335–336. [DOI] [PubMed] [Google Scholar]
- 36.Budd M.E., Choe,W. and Campbell,J.L. (2000) J. Biol. Chem., 275, 16518–16529. [DOI] [PubMed] [Google Scholar]
- 37.Lee S.K., Johnson,R.E., Yu,S.L., Prakash,L. and Prakash,S. (1999) Science, 286, 2339–2342. [DOI] [PubMed] [Google Scholar]