

Abstract
Alzheimer’s disease (AD), as the most common type of dementia, has two pathological hallmarks, extracellular senile plaques composed of β-amyloid peptides and intracellular neurofibrillary tangles containing phosphorylated-tau protein. Amyloid precursor protein (APP) and tau each play central roles in AD, although how APP and tau interact and synergize in the disease process is largely unknown. Here, we showed that soluble tau interacts with the N-terminal of APP in vitro in cell-free and cell culture systems, which can be further confirmed in vivo in the brain of 3XTg-AD mouse. In addition, APP is involved in the cellular uptake of tau through endocytosis. APP knockdown or N-terminal APP-specific antagonist 6KApoEp can prevent tau uptake in vitro, resulting in an extracellular tau accumulation in cultured neuronal cells. Interestingly, in APP/PS1 transgenic mouse brain, the overexpression of APP exacerbated tau propagation. Moreover, in the human tau transgenic mouse brain, overexpression of APP promotes tau phosphorylation, which is significantly remediated by 6KapoEp. All these results demonstrate the important role of APP in the tauopathy of AD. Targeting the pathological interaction of N-terminal APP with tau may provide an important therapeutic strategy for AD.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-023-04774-z.
Keywords: β-Amyloid, Alzheimer’s disease, Amyloid precursor protein, Endocytosis, Human tau transgenic mouse model, Tau protein
Introduction
Alzheimer’s disease (AD) is the most common type of dementia, characterized by synapse dysfunction, neuronal death, and cognitive deficiency [1]. AD has two pathological hallmarks: deposition of neurotoxic β-amyloid (Aβ) peptide which forms extracellular senile plaques and accumulation of phosphorylated-tau protein (ptau) which forms intracellular neurofibrillary tangles [2]. Aβ is produced from the amyloid precursor protein (APP) by sequential cleavages mediated by β- and γ-secretase [3]. Under normal physiological condition, ~ 90% of APP is cleaved by α-secretase as a default pathway, yielding a soluble N-terminal ectodomain of APP (sAPPα), which is secreted into the extracellular space and play neurotrophic and AD-ameliorating roles [4, 5]. Tau as a microtubule-associated protein consists of at least six isoforms generated by alternate splicing and self-assembles into toxic filamentous structures under pathological conditions [6, 7]. The cellular uptake or intercellular spread of tau pathology in the central nervous system follows various predictable patterns. It has been reported that tau protein can be transferred enclosed in exosomes or microvehicles [8–12]. Recent studies have also found that tau protein can be propagated across plasma membranes in vehicle-free forms [13, 14].
Interestingly, exosomes transport not only tau protein but also APP and its metabolites into extracellular space and thus contribute to the cell-to-cell spread of AD pathologies [15, 16]. In addition, since Aβ plaques and neurofibrillary tangles can be observed concomitantly in the AD brain, it has been suggested that these two AD-related proteins are linked. Initially, it has been reported that tau can bind directly to the intracellular C-terminal region of APP (between amino acid residues 713–730) or indirectly to APP cytodomain via Fe65 [17] and that both APP and tau co-exist in Aβ plaques and neurofibrillary tangles [18, 19]. Moreover, it has been shown that APP can accelerate intracellular tau aggregation in tau-expressing human neuroblastoma SH-SY5Y cells in a seed-dependent manner [20]. Cellular models have also shown that overexpression of APP and PSEN1 increases not only Aβ production but also tau aggregation [21]. Using stem cell-derived neurons from familial AD cases, it has been reported that presenilin-1 mutation and APP duplication increased the intracellular levels of total tau and ptau, independent of extracellular Aβ levels [22]. In addition, APP has been reported to be required for intraneuronal uptake of oligomeric tau (oTau) in cultured cells and mediated the memory and synaptic impairment induced by oTau in mice [23]. Altogether, the findings of the above studies suggest that APP may play critical roles in AD pathogenesis and progression as not only the precursor for Aβ formation but also a surface protein responsible for cellular uptake of tau seeds and intercellular propagation of tau pathology. This hypothesis might be further supported by clinical observations showing tau pathogenesis in Down’s syndrome patients who express 1.5-fold higher levels of APP as well as in patients with APP duplication [24, 25].
In the present study, we first found that both exogenous and endogenous soluble tau physically interacts with the N-terminal region of APP in cell-free and cell culture systems. These in vitro data are further confirmed in vivo in the brains of the transgenic AD mouse model. Moreover, APP knockdown by siRNA or blockage of N-terminal APP by specific antagonist 6KApoEp [26] prevents cellular tau uptake and results in the extracellular accumulation of tau in cultured neuronal cells in vitro. In addition, our data further indicated that the APP-mediated tau internalization into host cells is most likely through endocytosis. Much more interestingly, we found that the overexpression of human wild-type (WT) APP in human tau transgenic mice exacerbates ptau pathology in the brain in parallel with an increased intracellular tau accumulation, which is significantly ameliorated by 6KApoEp. All these results demonstrate the important role of APP in the tauopathy of AD. Targeting the pathological interaction of N-terminal APP with tau may provide a promising strategy for AD therapy.
Results
Tau physically interacts with N-terminus of APP in vitro
To quantify the interaction of full-length tau (f-tau) with N-terminal APP, localized surface plasmon resonance (LSPR) assay was performed via injecting various concentrations of f-tau (10–80 nM) over the LSPR chip surface to which recombinant N-terminal APP (18–612) (APP18–612) fragment was coupled. As shown in Fig. 1A, data from the LSPR assay confirmed the interaction of f-tau to APP18-612 fragment in a concentration-dependent manner, with the equilibrium dissociation constant (KD) of 6.38 nM (Fig. 1B).
Fig. 1.

Tau physically interacts with N-terminus of APP in vitro and in vivo. A APP18–612 fragment was loaded on Octet® streptavidin sensor and tested for real-time association and dissociation with various concentrations of f-tau (0-80 nM). Global fit curves of different concentrations of f-tau were shown as color solid lines. B Binding affinity parameters including kinetic association/disassociation constant (Ka and Kd) and affinity constant (KD) were calculated by Trace Drawer software. C Cell lysates prepared from human embryonic kidney 293 cells (HEK293) overexpressing wild-type APP695 (HEK293/APP695) or both wild-type APP695 and full-length tau (HEK293/APP695/tau) were immunoprecipitated using an anti-tau antibody (tau46), and then full-length APP and tau proteins were determined using western blot (WB) with Y188 (anti-C-terminal APP; up panel), or with tau46 (anti-tau; lower panel). D Cell lysates prepared from HEK293 cells overexpressing full-length tau protein (HEK293/tau) or HEK293/APP695/tau cells were immunoprecipitated using Y188, and then tau and APP proteins were determined using WB with tau46 (anti-tau; up panel) or Y188 (anti-APP; lower panel). E N2a cells overexpressing wild-type APP695 (N2a/APPwt) and N2a cells overexpressing full-length tau protein (N2a/tau) were co-cultured with the transwell co-culture system. After 24 h co-culture, cells were fixed in 4% paraformaldehyde solution in PBS, and then the mixture of anti-APP (Y188) and anti-tau (tau46) primary antibodies was used to analyze the co-localization of surface APP and tau under a confocal microcope. Alexa Fluor 594 donkey anti-mouse IgG was used to detect human tau (Top right, red), whereas Alexa Fluor 488 goat anti-rabbit IgG was used to detect cellular APP (Bottom left, green) as secondary antibodies. Co-localization (yellow) of cellular APP and endogenously generated tau are shown in the bottom right image; F N2a cells stably co-overexpressing wild-type APP695 and full-length tau protein (N2a/APPwt/tau) were cultured for 24 h, fixed in 4% paraformaldehyde solution in PBS, and then incubated with the mixture of anti-APP and anti-tau primary antibodies. Alexa Fluor 594 donkey anti-mouse IgG was used to detect human tau (top right, red), whereas Alexa Fluor 488 goat anti-rabbit IgG was used as secondary antibodies to detect cellular APP (bottom left, green). Co-localized signals (yellow) of cellular APP and tau are shown in the bottom right image. DAPI signals show nuclear DNA (blue). G Brain tissue sections from 1-, 6- and 12-month-old 3XTg-AD mice were stained with primary anti-N-terminal APP (22C11) and anti-tau (tau12) antibodies at 4 °C overnight, followed by IF488-Tyramide to detect APP (green) or IF555-Tyramide to detect tau (red) with TSAPLus Fluorescent double staining kit (Servicebio, G1226). Clear co-localizations (yellow) of cellular APP and tau can be observed in the hippocampus of 3XTg-AD mice at 12-month of age (top right image). DAPI staining was used to label nuclear DNA (blue). H Whole brain homogenates were prepared from 3XTg-AD mice at 1-, 6-, and 12-month of age (two female mice per age group). The expression levels of APP and tau proteins were analyzed by WB assay using anti-N-terminal APP (22C11) and anti-tau (tau12) antibodies, respectively. The β-actin is included as an internal loading control. I The interactions between cellular APP and tau were further confirmed in vivo using immunoprecipitation (IP) analysis. Protein samples prepared from brain of 3XTg-AD mice at 12 months of age were immunoprecipitated using anti-tau antibody (tau46) or anti-N-terminal APP antibody (22C11) with mouse sera as a negative control, and then APP and tau proteins were determined using WB with 22C11 (left panel), or with tau46 (right panel), respectively
The interactions between APP and tau were also evaluated using immunoprecipitation (IP) analysis. Cell lysates prepared from human embryonic kidney 293 (HEK293) cells stably overexpressing human WT APP695 (HEK293/APP695) or HEK293 cells overexpressing both APP695 and tau (HEK293/APP695/tau) were immunoprecipitated using an anti-tau antibody (tau46), and then APP and tau proteins were determined using western blot (WB) with Y188 (anti-C-terminal APP; Fig. 1C, up panel) or tau46 (anti-tau; Fig. 1C, lower panel) antibodies; Clear interactions between cellular APP and tau can be observed. Similarly, the IP analysis using cell lysates prepared from HEK293 cells overexpressing human tau (HEK293/tau) or HEK293/APP695/tau cells with anti-APP antibody also clearly revealed the interaction between cellular APP and tau (Fig. 1D).
In addition, the interaction between APP and tau was further confirmed in vitro by using immunofluorescence (IF) staining in a transwell co-culture system of murine neuroblastoma Neuro-2a (N2a) cells stably overexpressing human WT APP695 (N2a/APPwt) and N2a cells overexpressing human tau (N2a/tau), or in the culture of N2a cells overexpressing both WT APP695 and tau (N2a/APPwt/tau). After being co-cultured for 24 h co-culture of N2a/APPwt and N2a/tau cells, as expected, clear co-localizations of cellular APP and tau were presented (Fig. 1E, bottom right image), suggesting direct interactions between these two proteins. Similar co-localizations signals of cellular APP and tau protein were also observed in N2a/APPwt/tau cell line that simultaneously co-overexpressing both human WT APP695 and tau proteins (Fig. 1F, bottom right image).
Despite using those transgenic cell lines overexpressing exogenous APP and/or tau proteins, to further confirm the interactions between endogenous tau and APP, cell lysates of SH-SY5Y human neuroblastoma cells, which endogenously express both APP and tau proteins, were harvested and co-immunoprecipitation (Co-IP) assay was performed. As expected, consistent with those findings in transgenic cell lines, interactions between endogenous APP and tau can be observed (suppl Fig. 1A and 1B). Moreover, the colocalization of these two proteins was also observed by IF staining in SH-SY5Y cells. Endogenous APP and tau showed interactions as evidenced by the co-localizations between these two proteins (yellow fluorescence, suppl Fig. 1C).
Colocalization of APP and tau in 3XTg-AD mouse brain in vivo
Interactions between APP and tau were further confirmed in vivo in brain tissue sections from 1-, 6- and 12-month-old 3XTg-AD mice. After being stained with primary anti-N-terminal APP (22C11) and anti-tau (tau12) antibodies followed by fluorescence dye-labeled secondary antibodies, clear co-localizations of APP and tau can be observed in the hippocampus of 3XTg-AD mice, especially at 12-month of age (Fig. 1G, top right image).
Mouse brain homogenates of both WT and 3XTg-AD were prepared and used for WB and IP (Fig. 1H). Consistent with in vitro data, the interactions between APP and tau were further confirmed in vivo using IP analysis (Fig. 1I).
N-terminal APP mediates tau uptake
To investigate whether and how the N-terminal APP interacts with tau and modulates tau uptake, Chinese hamster ovary cells (CHO) and CHO cells overexpressing WT APP695 (CHO/APPwt) were treated with different concentrations of exogenous human tau441, and the cellular tau uptake was determined by quantifying cytosolic tau (c-tau). As shown in suppl Fig. 2A and B, the intracellular c-tau level was increased in a concentration-dependent manner. Importantly, densitometry analysis further disclosed that, after tau441 exposure, c-tau level was significantly higher in CHO/APPwt cells than that in CHO cells at each concentration (suppl Fig. 2B). These results confirm our hypothesis that APP might, in some extent, mediate the cellular uptake of tau.
To further confirm this hypothesis, expression of APP protein in SH-SY5Y cells was down-regulated with human APP siRNA oligo duplexes, and extracellular secreted-tau (s-tau) and intracellular c-tau were analyzed by WB with tau12 antibody in conditioned media and cell lysates, respectively. The densitometry analysis of WB bands clearly showed that APP expression was down-regulated with the treatment of siRNA, whereas the s-tau level as well as the ratio of s-tau to c-tau was significantly increased, indicating the decreased tau uptake (Fig. 2A, B). These results indicate a close relationship between APP level and tau uptake.
Fig. 2.

N-terminal APP specifically mediates cellular tau uptake. A Human neuroblastoma SH-SY5Y cells were cultured for overnight and then transfected with human APP siRNA oligo duplexes (lanes A, B and C) or universal scrambled negative control (Ctrl) siRNA duplex for 48 h. The supernatants were collected and cell lysates were prepared, and then subjected to WB to detect levels of total APP (t-APP, top), secreted tau (s-tau, middle) and cytosolic tau (c-tau, bottom) with 6E10 and tau12 antibodies. The β-actin was included as an internal loading control. B The densitometry analysis was performed to calculate ratios of s-tau to c-tau as well as t-APP to β-actin. The data are presented as the mean (± s.d.). Asterisk indicates p < 0.05 as compared with Ctrl siRNA as determined by t test. These results as presented are representative of three independent experiments with n = 3 for each condition. C SH-SY5Y cells were treated with N-terminal APP blocking peptide, 6KApoEp, at 10 μM for 0–24 h. Immediately following treatment at different time points, extracelluar s-tau in conditioned media as well as intracellular c-tau and β-actin in cell lysates were analyzed by WB with tau12, tau13, and β-actin antibodies. The β-actin is included as an internal loading control. D SH-SY5Y cells and HEK293 cells overexpressing human WT tau (HEK293/htau) were treated with 6KApoEp at 0–10 µM for 24 h. Immediately following treatment, extracelluar s-tau in conditioned media as well as phospho-tau Thr181 and Thr231 (ptau181 and ptau231), total-tau (t-tau) in cell lysates were analyzed by WB using tau12, ptau181, ptau231, and tau12 antibodies, respectively. E SH-SY5Y cells (SH) or SH-SY5Y cells with APP-knockdown (SH/APP-KD) were cultured in 3D-Matrigel system for at least 3 weeks. On day 7, day 14 and day 21, IF staining was performed to locate the intracellular tau and secreted tau (red). DAPI (blue) was used to visualize nuclear DNA and track cells. The merged purple (red + blue) indicated the cell clusters containing intracellular tau (red) and DAPI (blue); the merged yellow (red + green, co-localization of tau and β-actin) indicates intracellular tau; the red puncta in merged images indicated the accumulated extracellular tau in matrices. F, G On day 7, day 14 and day 21, cell lysates and matrices (cultured media) were separated from the 3D cultured SH-SY5Y cells (SH) expressing endogenous WT APP (SH/APPwt) or SH/APP-KD. F The yielded cell lysates on day 21 were subjected to WB to determine the expression of APP and intracellular c-tau using anti-N-terminal APP (22C11) and anti-tau (tau12) antibodies, respectively. G The yielded 3D culture media on day 7, 14 and 21 were collected for quantification of s-tau protein using ELISA. Asterisk indicates p < 0.05 as compared with SH-SY5Y cells as statistically analyzed by t-test. These results as presented are representative of two independent experiments with n = 3 for each condition. H Schematic illustration of pHrodo™ dye-based detection of tau endocytosis. Tau protein labeled with pHrodo™ dye are added to cells and first internalized into mildly acidic early endosomes (pH ~ 6.3), where the pHrodo-tau are either recycled back to the cell surface or delivered to late endosomes. Late endosomes (pH ~ 5.5) mediate the delivery of pHrodo-tau to acidic lysosomes (pH ~ 4.7) for degradation. This acidification of endocytic compartments, a hallmark of endocytosis, is exploited by pHrodo, which is nonfluorescent at a neutral extracellular environment pH ~ 7.4 and exhibit increasing fluorescence as the pH becomes more acidic. I Tau uptake in SH-SY5Y cells overexpressing APP695 (SH-SY5Y/APP695) as assessed by confocal microscopy. Tau protein was labeled by the pHrodo dye. SH-SY5Y cells overexpressing APP695 (SH-SY5Y/APP695 cells were cultured under normal condition and incubated with 1.65 μg/mL pHrodo-labeled tau at 37 °C for 5, 15 and 30 min. Assessment of intracellular tau was performed with a fluorescence confocal microscopy (Olympus SpinSR10, Japan) with the excitation and emission maxima of approximate 488 and 561 nm (scale bar 20 μm)
Considering the interactions between N-terminal APP and tau protein (Fig. 1 and suppl Fig. 1), the N-terminal ectodomain of APP might be the responsible domain for the connection between APP level and tau uptake. To clarify this possibility, CHO cells expressing APP695 with truncations of the N-terminal domain (i.e., E1 domain deletion, APPdelE1; E2 domain deletion, APPdelE2; or E1/E2 domain deletion, APPdelE1/2) were utilized (i.e., CHO/APPdelE1, CHO/APPdelE2, or CHO/APPdelE1/2 cells). CHO or CHO/APPwt cells in addition to CHO/APPdelE1, CHO/APPdelE2, or CHO/APPdelE1/2 cells were treated with human tau441, and the amount of s-tau and c-tau was analyzed by WB with tau12 antibody in conditioned media and cell lysates, respectively. Our results demonstrated that the c-tau amount in cell lysates from CHO/APPwt cells was increased than that in control CHO cells, whereas the c-tau amount in cell lysates from CHO/APPdelE1, CHO/APPdelE2, or CHO/APPdelE1/2 cells was not increased (suppl Fig. 2C, up panel). As expected, the extracellular s-tau content in conditioned media exhibited the opposite profile (suppl Fig. 2C, lower panel). Taken together, these results indicate that deletion of E1, E2, or E1/E2 domain of N-terminal APP markedly attenuates cellular tau uptake and consequently leads to accumulation of extracelluar tau, indicating the involvement of N-terminal APP in tau uptake.
Blocking N-terminal APP markedly promotes secreted-tau accumulation
Our previous study has reported that the 6KApoEp peptide strongly binds and blocks N-terminal APP [26]. In our present study, 6KApoEp was used to further confirm the involvement of N-terminal APP in the cellular uptake of tau protein. When CHO/APPwt cells were treated with exogenous tau441 and 6KApoEp, tau441 was accumulated in extracellular space and less intracellular tau441 was detected (suppl Fig. 2D). This is likely the result of competitively inhibiting the binding of N-terminal APP to tau by 6KApoEp treatment. Consistent results were obtained when CHO/APPwt cells were treated with f-tau and 6KApoEp (suppl Fig. 2F). In addition, Aβ1-40 levels in conditioned media were decreased (46% or 48%) after treatment with tau441 or f-tau in the presence of 6KApoEp as compared with those after treatment with tau441 or f-tau alone (suppl Fig. 2E, G).
To further confirm the impact of APP on tau uptake in different cell lines expressing human tau, SH-SY5Y cells were treated with 6KApoEp for ascending time durations up to 24 h, and extracellular s-tau in conditioned media was examined with tau12 and tau13 antibodies. The results showed that extracellular s-tau was further accumulated after 6KApoEp treatment with time in conditioned media (Fig. 2C, suppl Fig. 2H). In addition, immediately after treatment with 6KApoEp, an increased s-tau accumulation in conditioned media was found in both SH-SY5Y cells and HEK293/tau cells, in a concentration-dependent manner (Fig. 2D, suppl Fig. 2I). Furthermore, ptau status was examined with antibodies recognizing ptau at the Thr181 (ptau181) and Thr231 (ptau231). As expected, 6KApoEp treatment decreased ptau levels in both cell lines, especially the SH-SY5Y cells (Fig. 2D, suppl Fig. 2I).
APP mediates tau trafficking as assessed in 3D and 2D cell culture systems
3D cell culture is an invaluable tool in cellular biology research. By mimicking crucial features of in vivo environment, including cell–cell and cell-extracellular matrix interactions, 3D cell culture enables proper structural architecture and differentiated function of normal tissues in vitro. In our study, 3D-cultures of neuroblastoma SH-SY5Y cells, which express endogenous human WT APP, or SH-SY5Y cell with APP-knockdown (SH-SY5Y/APP-KD) were further used to confirm the APP-mediated cellular uptake of tau. SH-SY5Y cells or SH-SY5Y/APP-KD cells were cultured in 3D-Matrigel for at least three weeks. On days 7, 14 and 21, IF staining was performed to locate the c-tau in intracellular space and s-tau accumulated in extracellular matrices. Our data indicated a time-dependent development of SH-SY5Y cells in 3D gels, as shown by the enlarged cell aggregates and increased protein fluorescence (Fig. 2E). WB assay of cell lysates from 3D-culture on day 21 confirmed the knockdown of APP in SH-SY5Y/APP-KD cells (Fig. 2F, upper bands). Interestingly, when seeded in a 3D culture system, SH-SY5Y/APP-KD cells showed a blocked tau protein trafficking compared to SH-SY5Y cells, as evidenced by a relatively reduced intracellular c-tau level on day 21 (Fig. 2F, middle bands) and an increased extracellular accumulation of s-tau in matrices, especially on day 14 and 21 (Fig. 2G). All these findings suggested a disrupted cellular reuptake of tau and an accumulation of extracellular s-tau in SH-SY5Y/APP-KD cells 3D culture, indicating the important role of APP in the cellular trafficking of tau.
Since previous studies have found that LRP1 could mediate the cellular uptake of tau [27, 28], in the present study, we also detected the s-tau level in SH-SY5Y cells with LRP1 knockdown (SH-SY5Y/LRP1-KD). Consistent with previous findings, LRP1 knockdown (suppl Fig. 2 J) yielded a significant increase of s-tau level in the culture medium (suppl Fig. 2L). Moreover, as shown in suppl Fig. 2K and 2L, knockdown of APP in SH-SY5Y/APP-KD cells also resulted in a significant accumulation of s-tau, while no significant alteration of LRP1 expression in this cell line was found. These findings further support the involvement of APP in tau uptake in the LRP1-independent manner.
The acidification of endocytic compartments (early endosomes (pH ~ 6.3), late endosomes (pH ~ 5.5) and lysosomes (pH ~ 4.7), a hallmark of endocytosis, can be exploited by pHrodo, which are nonfluorescent at neutral pH (extracellular environment pH ~ 7.4) and exhibit increasing fluorescence as the pH becomes more acidic (Fig. 2H). In the present study, to confirm the involvement of APP-related endocytosis in tau-uptake, tau protein was labeled by the pHrodo dye and its cellular uptake was assessed by confocal microscopy in SH-SY5Y and SH-SY5Y/APP695 cells. Our data revealed that exogenous pHrodo-labeled tau protein internalized into acidic compartments, as shown by the time-dependently increase of pHrodo signal in both cell lines (Fig. 2I), indicating an endocytosis-related mechanism. Moreover, as expected, the SH-SY5Y/APP695 cells presented a much stronger fluorescence density than SH-SY5Y cells, confirming the involvement of APP-related endocytosis in tau entries into cells (Fig. 2I; suppl Video clip 1–3).
Tau triggers APP entries into host cells through endocytosis
The tau-triggered cell surface APP entries into host cells were further confirmed using SH-SY5Y and HEK293 cell lines expressing S1-GFP-APP, in which the internalization of cell surface APP can be captured through the fusion of fluorescent APP sensor to pairs of self-assembling peptides to generate stable “signaling reporter islands” (SiRIs). As shown in Fig. 3A, obvious intracellular green puncta can be observed in SH-SY5Y cells, indicating the APP entries and the successful transfection of SiRIs. Much more interestingly, as for HEK293 cells in the absence of endogenous tau expression, exogenous tau protein exposure (60 nM recombinant human f-tau) can also activate green fluorescence signal (Fig. 3B), whereas no green fluorescence was observed in control cells without tau exposure (suppl Fig. 3), further supporting the tau-triggered APP internalization.
Fig. 3.
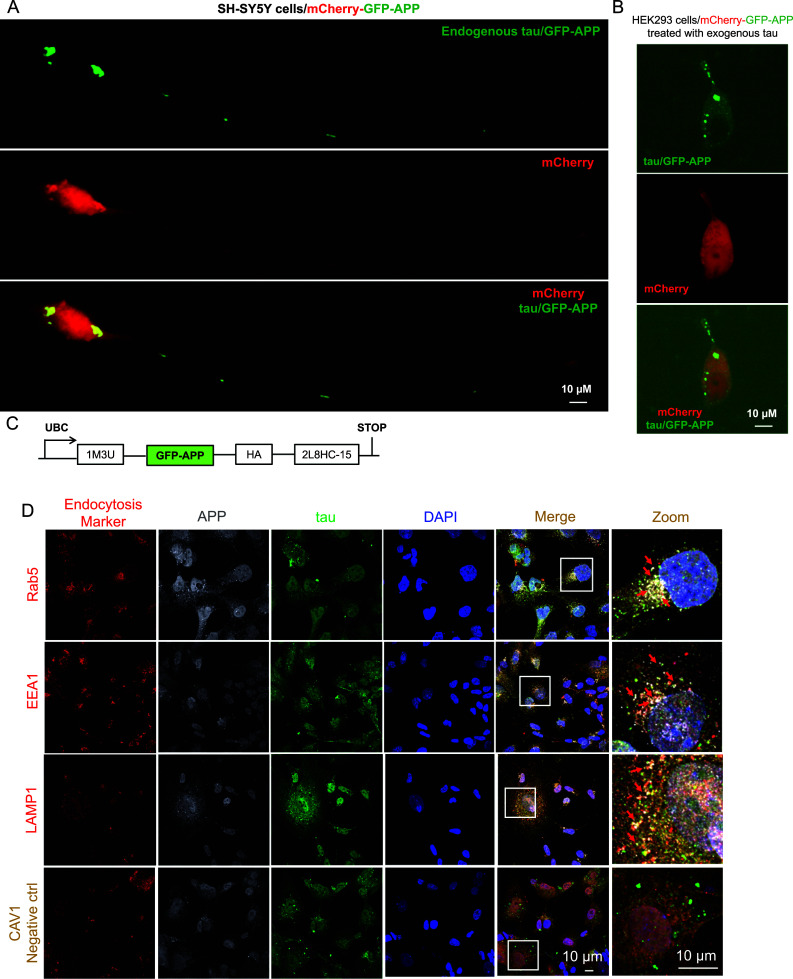
Tau facilitates APP entry into host endocytosis system. A–C Spatial imaging of fluorescent APP reporter clustered by protein scaffolds. Representative confocal images of SH-SY5Y cell line (A) and human embryonic kidney 293 (HEK293) cell line (B) expressing S1-GFP-APP and pAAV[Exp]-CAG > mCherry:WPRE as a cell morphology marker (scale bar = 10 μM). C Construct design of S1-GFP-APP, GFP-APP, GFP-based fluorescent APP Reporter; 1M3U, self-assembling subunit of 1M3U filament assembly; HA, influenza hemagglutinin epitope; 2L8HC4-15, a homo-tetramer. D SH-SY5Y cells were transfected with Flag-tau plasmid and cultured for 48 h. Then the colocalizations of APP (dark gray), tau (green) and endocytosis-related proteins (red) were visualized by confocal microscopy. As expected, clear co-localizations of tau with those APP endocytosis-related proteins (light gray), including LAMP1, EEA1 and Rab5, but not CAV1 were observed.
In addition, after incubation with the indicated antibodies against APP, tau and the indicated endocytosis-related proteins, co-localizations of tau, APP and endocytosis-related proteins (LAMP1, EEA1 and Rab5, but not CAV1) were found by confocal microscope observation (light grey, as indicated by the arrowhead, Fig. 3D). These findings suggest an endocytosis-related mechanism in the tau-triggered APP entries into host cells.
APP overexpression results in exacerbated tau propagation in APP/PS1 mice
To investigate the possible tau spreading mediated by APP in vivo, APP/PS1 transgenic mouse with overexpression of human APP and their age-matched wild-type littermates (APP expressions were shown in suppl Fig. 4) were intracranially injected into the hippocampus with AAVs encoding mcherry-p2A-hTauP301L (2.0 μL unilaterally). hTauP301L can express and spread from donor cells (mCherry+/tau+) to recipient cells (mCherry−/tau+) (Fig. 4A). Twenty-one days after virus injection, IF staining revealed a clear tau expression and spreading from the location of injection (Fig. 4B). Interestingly, as expected, APP overexpression can accelerate this process, as indicated by the increased green fluorescence (Fig. 4B, left panel). Wirth noting, no significant difference in tau in the cortex compare to WT mice under a low-power lens (top panel). In addition, even under high-power lens condition (bottom panel), the increasing tendency is still not obvious as that in the hippocampus. Different responses of the cortex and hippocampus to exogenous tau stimuli might be due to the different vulnerability of brain regions to tau pathology. Previously, it is reported that cortex has the highest tau expression level and a higher susceptibility to tau pathology than other brain regions including hippocampus. Consistently, our data indicated that 21 days after AAV injection, the fluorescence density of tau was much stronger in the cortex of WT mice than other brain regions, suggesting a higher susceptibility of the cortex to develop tau pathology. This higher exogenous tau baseline may lead to a counteraction of APP-induced tau spreading in the cortex. Moreover, although the WT mice have a relatively lower APP level, other tau-uptake-responsible proteins such as LRP1 could also contribute to this elevated tau level in the cortex [27].
Fig. 4.

APP overexpression exacerbates human-tau propagation in APP/PS1 mouse brain. A Diagram of AAV construction and injection to evaluate tau propagation in mouse brain in vivo. Briefly, AAV vector that leads to the expression of full-length human P301L tau (hTauP301L) and mCherry as a cell transduction marker was designed and constructed (mCherry-P2A-hTauP301L). Viruses were intracerebrally injected into the hippocampus of female APP/PS1 mice at 8 weeks of age or their age-matched wild-type littermates as control. B Twenty-one days after AAV injection, all mice were anesthetized and perfused using 4% paraformaldehyde. Brains were isolated immediately and immersion-fixed for 24 h. Immunofluorescence staining was performed to detect hTauP301L expression and distribution in mouse brain using anti-tau12 against human tau as the primary antibody and secondary antibodies conjugated with Alexa Fluor 488
APP overexpression results in increased phosphorylated-tau pathology in Htau+/− mice
To investigate the possible involvement of APP in promoting tau pathology in vivo, we crossed human WT tau transgenic (Htau+/−) mice [29] with human WT APP transgenic (TgAPPwt+/−) mice [30]. The obtained four genotypes of F1 mice (i.e., Htau−/−/TgAPPwt−/−, Htau−/−/TgAPPwt+/−, Htau+/−/TgAPPwt−/−, and Htau+/−/TgAPPwt+/− littermates) were utilized at 9 months of age in this study. Total tau and ptau status were analyzed by immunohistochemistry (IHC) with antibodies recognizing ptau at Thr231 (ptau231) and Ser202 (CP13). The results showed that Htau+/−/APPwt+/− mice exhibit an augmentation of ptau in the CA3 region of hippocampus and the entorhinal cortex as compared with Htau+/−/APPwt−/− mice (suppl Fig. 5A). Of note, Htau+/−/APPwt−/− mice euthanized at 11 months of age also showed less ptau than Htau+/−/APPwt+/− mice at 9 months of age, although the difference was not as apparent as Htau+/−/APPwt−/−mice of 9 months of age (data not shown).
In addition, extracellular and intracellular total-tau and ptau in brain homogenates were analyzed by WB with tau46 and ptau231 antibody, respectively. The results showed that the expression of both ptau231 and tau46 was increased in extracellular and intracellular proteins from Htau+/−/TgAPPwt+/− and Htau+/−/TgAPPwt−/− mouse brains as compared with those from Htau−/−/TgAPPwt+/− and Htau−/−/TgAPPwt−/− mouse brains (suppl Fig. 5B and 5C). Furthermore, the densitometry analysis disclosed that Htau+/−/TgAPPwt+/− mice showed decreases in extracellular ptau231 and tau46 levels, together with increases in intracellular ptau231 and tau46 as compared with Htau+/−/TgAPPwt−/− mice (suppl Fig. 5D). Together, these results demonstrate that overexpressing WT human APP in Htau+/− mice increases tau uptake and, most importantly, exacerbates ptau pathology (suppl Fig. 5B–D).
Intranasal administration of 6KApoEp remediates phosphorylated-tau pathology
Given that APP overexpression enhances ptau pathology, we then investigated whether blocking the interaction between N-terminal APP and tau could result in the amelioration of ptau pathology in vivo. To do this, 6KApoEp was used as an antagonist to inhibit the binding of tau protein to N-terminal APP. Transgenic female Htau+/− mice at 10 weeks of age were intranasally treated with 6KApoEp (10 µg/mouse/once daily) for 28 weeks. Following 6KApoEp treatment, we examined ptau pathology in coronal brain sections (suppl Fig. 6A) by IHC and in brian homogenates by WB (suppl Fig. 6B and 6C) with CP13 and ptau231 antibodies. The results revealed that 6KApoEp-treated Htau+/− mice showed a dampened ptau in the CA3 region of hippocampus and the EC as compared with 6 K-treated Htau+/− mice (suppl Fig. 6A). The densitometry analysis further confirmed that the ratio of ptau (ptau231, ptau202) to total tau was significantly decreased after 6KApoEp treatment. Taken together, these results provide preclinical evidence that intranasal administration of 6KApoEp remediates ptau pathology in human tau transgenic mouse brains.
Discussion
APP plays a critical initiating role in AD pathogenesis. The APP-derived Aβ together with hyperphosphorylated-tau are the main constituents of senile plaques and neurofibrillary tangles, respectively. These major pathognomonic hallmarks of AD have been the focus of intense research efforts to understand their physiological roles and how they cause clinical symptoms of AD [31]. In the present study, we found that APP facilitates the cellular uptake of tau protein and exacerbates tau pathology by a mechanism involving an interaction of APP with tau protein and the subsequently initiated APP-mediated endocytosis. Down-regulation of APP with human APP siRNA oligo duplexes or blockade of N-terminal APP binding by 6KApoEp resulted in the extracellular accumulation of tau. Interestingly, the N-terminal APP-blocking peptide 6KApoEp exposure also induced a significant reduction of Aβ40 (suppl Fig. 2E and 2G), which was consistent with our previous findings [26]. We found 6KApoEp can block the endocytosis of APP induced by extracellular ligands, such as ApoE, and maintain APP on cell membrane to be enzymatically cleaved through non-amyloidogenic processing mediated by α-secretase, rather than the intracellular amyloidogenic processing by β-/γ-secretase, thereafter leads to a reduced Aβ production [26]. Moreover, in our present study, we found that the enhancement of tau uptake upon APP overexpression was abolished when N-terminal APP was truncated, further showing that the tau uptake is mediated by the N-terminus of APP. These findings confirmed that the enhancement of tau uptake is caused by the ectodomain of N-terminal APP. Our findings do not preclude a possible role of Aβ in tau pathology but do propose an indirect mechanism for the exacerbation of tau pathology by APP. For example, previous findings suggest that aggregating Aβ causes membrane-associated oxidative stress and consequent dysregulation of cellular Ca2+ levels, resulting in the hyperphosphorylation and aggregation of neural tau [32, 33].
Our results may propose dual implications. First, given our evidence that soluble tau physically interacts with N-terminal APP, it has emphasized the importance of APP itself as a cell surface recipient protein, regulating tau uptake through its normal physiological functions. Consistently, other studies have also demonstrated the importance of APP in promoting AD pathogenesis. Puzzo and colleagues (2017) showed that oTau or oligomeric Aβ (oAβ) could cause deficits in memory and long-term potentiation which were both APP-dependent. It has been reported that efficient intra-neuronal uptake of oAβ and oTau requires expression of APP and that both oAβ and oTau bind APP [23]. Additionally, Takahashi and colleagues found that the incorporation of extracellular tau fibrils into cells is dependent on the expression of APP [20]. Our results, together with these studies, raise the possibility that APP might influence the cell-to-cell spreading of tau pathology by serving as a cell surface recipient protein.
Second, tau pathology has always been recognized as the downstream consequence of Aβ. However, this view has been challenged by recent studies that Aβ is not the sole causal factor inducing tau pathology. For example, in genetic forms of AD carrying either mutated APP or overexpressed APP, significant increases in intracellular total-tau and ptau have been demonstrated [34]. Specifically, since PSEN1 and APP V717I mutated neurons displayed different intracellular total-tau and ptau levels but comparable levels of total extracellular Aβ and Aβ40 to Aβ42 ratio, the authors conclude that membrane-bound or intracellular products of APP processing may modulate tau proteostasis. In a comparison of the effects of acute γ- or β-secretase inhibition on tau levels, it has been found that inhibition of either γ- or β-secretase significantly reduces extracellular Aβ levels. Most interestingly, γ-secretase inhibition led to an increase in tau levels as well as APP-C83/C99 accumulations. By contrast, levels of tau were decreased by β-secretase inhibition and pharmacological intervention for reducing APP-C99 generation. Together, one previous study suggests a link between APP processing and tau proteostasis which is independent of extracellular Aβ38, Aβ40, and Aβ42 [34]. Consistently, a 3-D human neural cell culture model has shown that overexpression of APP and PSEN1 increases tau phosphorylation and aggregation [21]. In another study, the supernumerary copy of the APP gene was deleted in trisomic Down syndrome induced pluripotent stem cells (iPS)-derived neurons [35]. Interestingly, while Aβ levels were decreased in the differentiated neurons, neurofibrillary tangle-like structures, and ptau did not [35]. Thus, the importance of not solely focusing on anti-Aβ therapeutics as a means of reducing tau pathology becomes to be evident.
A large body of research has been tested with tau reduction strategies as a possible therapeutic intervention for AD. For example, reducing endogenous tau levels prevent behavioral deficits in transgenic mice expressing human APP without altering Aβ levels. Furthermore, tau reduction protected both transgenic and non-transgenic mice against γ-aminobutyric acid type A receptor antagonist pentylenetetrazole-induced excitotoxicity [36]. The genetic deletion of tau in APP/PS1/tau−/− mice bearing human APP and PS1 mutant transgenes rescued neuronal loss, synaptic loss, spatial memory deficits, and motor impairments. Intriguingly, the Aβ burden was reduced by about 50%, soluble and insoluble Aβ40/42 levels, and Aβ42 to Aβ40 ratio were all decreased in the cortex [37]. Lowering tau levels have shown to prevent axonal transport deficits [34]. Additionally, the same lowering tau levels have also been shown to increase inhibitory currents and normalize excitation/inhibition balance as well as N-methyl-D-aspartate receptor-mediated currents in hAPP mice [38]. Once tau levels were selectively decreased with antisense oligonucleotides in mice expressing human tau mutant P301S, there are only fewer tau inclusions, and prevention of hippocampal volume loss, neuronal death, and nesting deficits [39]. Lastly, inducible tau transgene reduction protected mice from tau pathology and neuronal loss even in the presence of Aβ [40]. Our study indicates that a specific inhibitor of N-terminal APP, 6KApoEp, reduced the uptake of tau and resulted in its extracellular accumulation. When we treated Htau+/− mice with 6KApoEp, a reduction of ptau was observed in the hippocampal CA3 and entorhinal cortex regions. These results suggest that interfering with the interaction between tau and N-terminal APP may be a potential therapeutic strategy for AD. Importantly, this method could retain the normal physiological functions of tau, while reducing its intracellular accumulation, the first necessary step for neurofibrillary tangle formation. The physiological functions of APP have also emerged as another important topic in AD pathophysiology [31].
It is worth noting, although phosphorylated and/or aggregated intracellular tau was initially proposed to be causative of neuronal death, recent studies have suggested a toxic role of extracellular non-phosphorylated and non-aggregated tau [41, 42], which is released to brain parenchyma via extracellular vesicles, suchas exosomes and ectosomes. In addition, these extracellular tau proteins are also responsible for the inter-neuronal spreading of tau protein pathology [43, 44]. The extracellular tau can be cleared to periphery by glymphatic and lymphatic systems (45. 46). Extracellular tau can also be taken up by glial cells, either being degraded through AELN or propagated via exosomes [47]. In our present study, after blocking tau uptake by using N-terminal APP blocking peptide 6KApoEp, decreased intracellular p-tau level was observed in both SH-SY5Y and HEK293/htau cells (Fig. 2D), indicating a suppressed intracellular p-tau pathology in vitro. Although extracellular p-tau pathology was not determined in vitro in our present study, APP overexpression in Htau+/− mice truly induced a decrease of extracellular tau and p-tau (suppl Fig. 4D), predicting a potential risk of an exacerbated extracellular tau pathology after N-terminal APP blocking. However, in the brain of Htau+/− mice, after 6KApoEp treatment, the ratio of p-tau (ptau231, ptau202) to total tau was decreased significantly, without significant change of total tau (suppl Fig. 5C), suggesting an overall ameliorated but not exacerbated tau pathology after N-terminal APP blockage. This may also predict the predominant roles of intracellular tau in the pathologic neurodegeneration of AD [48]. Nevertheless, potential toxic consequence of extracellular tau accumulation induced by blocking tau uptake should be taken seriously.
In summary, in this study, we found that both exogenous and endogenous soluble tau physically interact with N-terminal APP in both cell-free and cell culture systems. APP knockdown strategy showed that APP plays an important role in mediating tau uptake in cultured neuronal cells. Blockage of the interaction between the N-terminus of APP and tau also prevented cellular tau uptake, resulting in an increased accumulation of tau in the supernatant of cultured neuronal cells. Overexpression of human WT APP in human tau transgenic mice exacerbates ptau pathology in parallel with the increase of intracellular tau accumulation, which is significantly ameliorated by 6KApoEp, an antagonist for the binding of tau to N-terminal APP. These results demonstrate a pathological interaction of N-terminal APP with tau in promoting tau pathology in AD brains and propose an opportunity that targeting this interaction may be a potential therapeutic intervention for AD.
Materials and methods
Localized surface plasmon resonance analysis
LSPR analysis was conducted with an Open SPRTM instrument (Nicoyalife). Briefly, the COOH sensor chip was installed on the Open SPRTM instrument in accordance with the manufacturer’s standard operating procedures. Run the buffer solution (PBS, pH 7.4) at the maximum flow rate (150 μL/min) and evaporate the bubble after reaching the signal baseline. Adjust the flow rate of buffer solution to 20 µL/min, then load 200 µL solution of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide together with N-hydroxysuccinimide (20 μL/min, 4 min) to activate COOH sensor chips. The APP18–612 fragment (50 µg/mL) and the f-tau (200 nM, Novoprotein) were diluted with activation buffer (total 200 µL). The injection port was rinsed with buffer solution and emptied with air. Fill with 200 µL blocking solution (20 µL/min, 4 min), wash the sample ring with buffer solution and empty it with air. APP18-612 fragment was loaded on a sensor. Observe baseline for 5 min to ensure analysis stability. Next, f-tau of different concentrations (0–80 nM) was injected into the chip from low to high concentration. The kinetic parameters of the binding reactions (Ka, Kd and KD) were calculated and analyzed using Trace Drawer software (Ridgeview Instruments AB, Sweden).
Cell lines and cultures
HEK293/APP695 cells, HEK293/tau cells, HEK293/APP695/tau cells, N2a/APPwt cells, N2a/tau cells, and N2a/APPwt/tau cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) with 10% fetal bovine serum (FBS). Human neuroblastoma SH-SY5Y cells, SH-SY5Y cells overexpressing human WT APP695 (SH-SY5Y/APP695), and SH-SY5Y cells with APP knockdown (SH-SY5Y/APP-KD) were cultured in DMEM nutrient mixture [DMEM/F12 (1:1), Gibco)] with 10% FBS.
Immunoprecipitation assay using cell cultures
Cell lysates prepared from HEK293/APP695 or HEK293/APP695/tau were immunoprecipitated using an anti-tau antibody (tau46), and then full-length APP and tau proteins were determined using western blot (WB) with Y188 (anti-C-terminal APP), or with tau46 (anti-tau). Cell lysates total protein (10 µL, 1 µg/µL) prepared from HEK293/tau or HEK293/APP695/tau cells was loaded in input lines as control. Cell lysates (10 µL, 0.78 µg/µL) were immunoprcipiated using Y188, and then tau and APP proteins were determined using WB with tau46 (anti-tau) or Y188 (anti-APP).
Cells co-culture, immunofluorescence staining
N2a/APP695 and N2a/tau cells were co-cultured with a transwell co-culture system (Corning). N2a/APP695 cells (1 × 105) were seeded in the lower compartment of the transwell that was pre-plated with the microscope cover glass (Thermo Fisher Scientific). After the cells were seeded for 24 h, N2a/tau cells (1 × 105) were seeded in the upper compartment of the transwell. The transwell inserts containing N2a/tau cells were moved into the corresponding 6-well plates containing N2a/APP695 to create the N2a/APP695 and N2a/tau co-culture system. After being co-cultured for 24 h, the microscope cover glass was used for IF staining to detect the co-localization of APP and tau.
N2a/APP695/tau or SH-SY5Y cells were plated in the confocal-imaging chambers (Biosharp) at 2 × 105/well for 24 h. Cells were then fixed in a 4% paraformaldehyde solution. Y188 (Abcam, ab32136, 1:200) and tau46 (Cell Signaling Technology, 4019, 1:200) primary antibodies were used to analyze the co-localization of APP and tau under a confocal microcope (SpinSR10, Olympus). Alexa Fluor 488 goat anti-rabbit IgG was used to detect cellular APP (Invitrogen, A-10680, 1:1000), whereas Alexa Fluor 594 donkey anti-mouse IgG was used to detect human tau (Abcam, ab150076, 1:1000) as secondary antibodies. DAPI-Fluoromount-G was used for nuclear DNA staining (SBA, 0100-20).
siRNA transfection for APP knockdown
Three small interfering RNAs (siRNAs) targeting APP695 (i.e., siRNA-A, siRNA-B, or siRNA-C) oligonucleotides (Table 1) were synthesized by Sangon Biotech (Shanghai) Co., Ltd. The transfections of specific siRNAs were performed under the optimized conditions with lipofectamine 2000 reagent (Invitrogen). Briefly, SH-SY5Y cells were cultured in 6-well plates at 2 × 105/well in DMEM/F12 with 10% FBS for overnight. Before transfection, the culture media was replaced by DMEM/F12 with 5% FBS. Lipofectamine 2000 reagent (5 µL) and siRNAs (60 nM) were diluted using Opti-MEM medium (600 µL, Gibco). Subsequently, the diluted siRNA was mixed with lipofectamine 2000 reagent at a ratio of 1:1 and then incubated at room temperature for 20 min before applying it to the cells. The total proteins of transfected cells were extracted 48-h after transfection with siRNA. To confirm the down-regulation of APP695, the cell lysates were subjected to Western blotting (WB) analysis with anti-APP antibody (Abcam, ab32136, 1:3000). GAPDH was used as an internal reference control. The cytosolic tau (c-tau) in intracellular space and secreted tau (s-tau) in extracellular condition media were quantified by WB analysis with anti-tau antibody (tau12, Biolegend). The ratios of t-APP to β-actin and s-tau to c-tau were calculated.
Table 1.
Sequences of siRNA oligonucleotides
| Name | Sequences of siRNA (5′ to 3′) |
|---|---|
| siRNA-1 | GUUCCUGACAAGUGCAAAUTT; AUUUGCACUUGUCAGGAACTT |
| siRNA-2 | CCUGCAGUAUUGCCAAGAATT; UUCUUGGCAAUACUGCAGGTT |
| siRNA-3 | GCACCAACUUGCAUGACUATT; UAGUCAUGCAAGUUGGUGCTT |
| LRP1-2523 | GCUCACACCGAGACACAUCUUTT; AAGAUGUCUCGGUGUGAGCTT |
| LRP1-9603 | GGUCCAACUACACGUUACUTT; AGUAACGUGUAGUUGGACCTT |
| LRP1-4306 | GCGCAUCGAUCUUCACAAATT; UUUGUGAAGAUCGAUGCGCTT |
| shRNA-APP-Homo-715 | GGCTGAAGAAAGTGACAATGT |
| shRNA-APP-Homo-1551 | CTCGTCACGTGTTCAATATGC |
| shRNA-APP-Homo-533 | GTTCCTGACAAGTGCAAATTC |
| shRNA-APP-Homo-1396 | CCAGGAGAAAGTGGAATCTTT (negative control) |
3D-culture of SH-SY5Y or SH-SY5Y/APP-KD cells and immunofluorescence staining
To obtain SH-SY5Y/APP-KD cells, Lentiviruses containing shRNA [i.e., APP-Homo-715, APP-Homo-1551, APP-Homo-533 or APP-Homo-1396 (negative control)] targeting human APP were transfected into SH-SY5Y to knock down endogenous APP. SH-SY5Y or SH-SY5Y/APP-KD cells pellet was mixed with 5% (W/V) gelatin methacryloyl (GelMA) gels (EFL-GM-60, EFL group) at the density of 3 × 105–5 × 105 cells/mL, embedded into a scaffold (EFL-SCR-3D-24-1, EFL group), and then gelated by 450 nm light source for 25–30 s. The 3D in vitro systems were maintained at 37 °C, 5% CO2 for 3 weeks.
On day 7, 14 and 21, IF staining was performed to locate the cytosolic tau (c-tau) in intracellular space and secreted tau (s-tau) in extracellular matrices. The β-actin is included as an internal control. DAPI was used to visualize nuclear DNA and track cells. Cells in 3D culture were first rinsed with PBS three times and then blocked with 10% goat serum in staining buffer (PBS supplemented with 0.2% TritonX-100, 0.1% BSA, and 0.05% Tween 20). The samples were then incubated with primary antibodies at 4 °C overnight, followed by the incubation with suitable second antibodies at room temperature for 1 h and finally were mounted with DAPI mounting medium (SBA, 0100-20). Images were captured on a confocal microscope (SpinSR10, Olympus).
Determination of secreted-tau, cytosolic-tau, and cytosolic-APP in 3D-culture
GelMA cracking buffer (0.3 mg/mL, EFL-GM-LS-001, EFL group) was added to soak GelMA glue block into a 24-well plate. The glue block was separated from the scaffold via blowing repeatedly using a pipette gun and then sterile lysis was performed in a 37 °C incubator. After full lysis, centrifuge at 1000 rpm for 5 min, s-tau in the supernatant was detected by tau ELISA (Ruixinbio, RX79914). Cells precipitate lysate were mixed with loading buffer, loaded on SDS-PAGE and blotted for the presence of APP (Y188, Abcam, 1:3000), c-tau (tau12, Biolegend, 1:2000) and GAPDH (ab8245, Abcam, 1:7000).
Signaling reporter islands (SiRIs) assay to assess tau entries into host cells
SH-SY5Y and HEK293 cells expressing S1-GFP-APP and red fluorescent protein mCherry were constructed [49] (Fig. 3C) and cultured under standard conditions. Briefly, in each Confocal 35-mm Clear Coverglass-Bottom Petri-Dish (Biosharp), 2–3 μg of the total plasmid of interest (pAAVUBC>IM3U:Linker48:EGFP:hAPP[NM_000484.4](ns)/HA:Linker27:2L8HC4_15 [50–53] and 2 μg of CAG-mCherry-WPRE plasmid as a cell morphology marker were transiently transfected into SH-SY5Y or HEK293 cells using Lipofectamine™ 2000 Transfection Reagent (Invitrogen). SiRIs were generated by self-assembling peptides sensitive to fluorescent APP and transfected into SH-SY5Y or HEK293 cells. SH-SY5Y cells expressing endogenous tau protein were cultured routinely, whereas cultured HEK293 cells were treated on day 2 in vitro with 60 nM recombinant human f-tau (Novoprotein). Cells were washed with DMEM after 40 min incubation to remove human f-tau and then incubated for another 2 h before live cell imaging. Green dots indicated the appearance of puncta formed by clustered fluorescent reporters in live cells.
Tracking internalization in live cells by pHrodo™ Green staining
Tau protein (Novoprotein) was labeled with pHrodo™ Green STP Ester (Invitrogen). Briefly, the pHrodo dye was dissolved in DMSO (500 μg/75 μL DMSO). Tau was resuspended in 0.1 M NaHCO3 (pH 8.4), added into pHrodo-DMSO at a ratio of 1:1 (vol/vol) and incubated for 45 min at room temperature. The labeled tau was spun down (8 min, 12,000 g) and the pellet resuspended in F12/DMEM medium with 5% FBS. The pHrodo-labeled Tau suspension was stored at 4 °C until use. SH-SY5Y or SH-SY5YAPP695 cells were cultured at 2 × 103 density in a 25 mm Confocal dish overnight, then were incubated with 5.05 μg/mL pHrodo™-labeled tau at 37 °C for 30 min. To quantify tau uptake, at various time points (5, 15 and 30 min), cells were washed with PBS to remove extracellular pHrodo in the cell culture medium. Intracellular tau level was observed using fluorescence confocal microscopy (Olympus SpinSR10) with the excitation and emission maxima of approximately 505 and 525 nm.
Determination of the co-localizations of endocytosis-related proteins and APP/tau
SH-SY5Y cells were plated in the confocal-imaging chambers (Biosharp) at 2 × 105/well for 24 h. Before transfection, the culture media was replaced by DMEM/F12 with 5% FBS. Lipofectamine 2000 reagent (5 µL) and Flag-tau 2 µg (1.5 µg/µL) were diluted using Opti-MEM medium (600 µL, Gibco). Subsequently, the diluted Flag-tau was mixed with lipofectamine 2000 reagent at a ratio of 1:1 and then incubated at room temperature for 20 min before applying it to the cells. After 72 h, Cells were then fixed in a 4% paraformaldehyde solution. 6E10 (BioLegend, 803015, 1:200) and tau (Biosensis, C-1691, 1:200), EEA1(Bioworld, BS5707, 1:1000), LAMP1(Bioworld, BS6978, 1:1000) and RAB5A (Bioworld, BS6218, 1:1000) primary antibodies were used to analyze the co-localizations of APP, tau and EEA1 or LAMP1 or RAB5A under a confocal microscope (SpinSR10, Olympus). Alexa Fluor 594 Goat anti-Chicken IgY was used to detect cellular tau (Invitrogen, A-10680, 1:1000), Alexa Fluor 488 Goat anti-mouse IgG was used to detect cellular APP (Abcam, ab150077, 1:1000), whereas Alexa Fluor 647 Goat anti-rabbit IgG was used to detect human EEA1, LAMP1and RAB5A (Abcam, ab150075, 1:1000) as secondary antibodies. DAPI-Fluoromount-G was used for nuclear DNA staining (SBA, 0100-20).
Animals
APP/PS1 transgenic mice, expressing human presenilin 1 (A246E) and Swedish mutant APP (APPSwe) were purchased from Jackson laboratory (B6C3-Tg(APP695)3Dbo Tg(PSEN1)5Dbo/Mmjax, Stock No. 41848-JAX). 3XTg-AD mice (B6;129-Tg(APPSwe, tauP301L)1LfaPsen1tm1Mpm/Mmjax, stock No. 004807) carrying three human transgenes (APPswe, PS1M146V, and tau P301L), were obtained from the Mutant Mouse Resource and Research Center at the Jackson Laboratory. All mice were anesthetized with 2–4% isoflurane (Millipore-Sigma Aldrich), followed by a collection of blood, euthanization by bilateral thoracotomy, transcardial perfusion with physiological saline-containing heparin (10 U/mL; Millipore-Sigma Aldrich), and dissected the brain for biochemical analysis and immunohistochemistry (IHC) staining. Animals were housed under standard specific pathogen-free condition with food and water ad libitum. All animal care and experimental procedures were performed according to the guidelines and were approved by the Animal Care and Use Committee of Guizhou Medical University.
Tau/APP expressions and interactions in vivo in 3XTg-AD mouse brain as assessed by immunofluorescent staining, Western blotting and immunoprecipitation
Female 3XTg-AD mice at 1-, 6-, and 12 months of age were anesthetized with 2–4% isoflurane (Millipore-Sigma Aldrich), followed by transcardial perfusion with physiological saline-containing heparin (10 U/mL; Millipore-Sigma Aldrich). Brains were immediately and carefully dissected, separated on ice, and cut into two hemispheres for further use. For immunoflurescence staining, the left hemisphere was fixed in 4% paraformaldehyde for 24 h, then dehydrated with 15%, 20%, and 30% sucrose solutions successively (each for 24 h), and finally embedded in an optical cutting temperature compound (OCT) and stored at − 80 °C until sectioning. The frozen brain in OCT was cut into 6-μm sections. TSAPLus fluorescent dual staining kit (Servicebio, G1226) was used for double immunofluorescence staining. Tissue self-fluorescence quench solution A (100 μL) was added to the tissue, incubated for 30 min at room temperature, and wash with PBS for 5 min. The sections were covered with 3% H2O2 drops and incubated at room temperature for 25 min under dark conditions to block endogenous peroxidation enzyme to reduce non-specific background staining. Anti-APP antibody (Biolegend, 4G8, 1:200) was incubated at 4 °C overnight. TSAR-488 staining solution (100 μL) was added to the tissue at room temperature and incubated in the dark for 10 min. For tau, anti-tau antibody (HUABIO, ET1603-2, 1:200) was added and incubated at 4 °C overnight. Corresponding HRP secondary antibody was incubated at room temperature for 50 min. TSA-555 staining solution (100 μL) was added to the tissue at room temperature and incubated in the dark for 10 min. The nuclei were stained with DAPI at room temperature in the dark for 10 min. Tissue self-fluorescence quench solution B was dropped into the tissue and incubated for 5 min at room temperature. Images were observed and captured by a confocal microcope (SpinSR10, Olympus).
For WB, the right hemisphere was homogenated with sample buffer (pH 7.6, 50 mM Tris–HCl, 10 mM dithiothreitol, 2% sodium dodecyl sulfate, 10% glycerol, and 0.2% bromophenol blue) and protease inhibitors cocktail on ice and boiled for 10 min. Proteins (10–50 µg) were separated by 10% SDS-PAGE and transferred to nitrocellulose membranes. The membranes were incubated with primary antibodies (6E10 and tau12 for APP and tau, respectively) overnight at 4 °C followed by incubation with secondary antibodies for 1 h at room temperature. Blots were visualized with the Chemiluminescent HRP Substrates (Millipore) and Chemiluminescence Imaging System (GeneGnome XRQ). The densitometry analysis was performed with Image J software.
The interactions between cellular APP and tau were further confirmed in vivo using immunoprecipitation (IP) analysis. Protein samples (10 µL, 2 µg/µL) prepared from the brain of 3XTg-AD mice at 12 months of age were loaded in input lines as control. Protein samples (10 µL, 1.56 µg/µL) were immunoprecipitated using an anti-tau antibody (tau46) or anti-N-terminal APP antibody (22C11), and then APP and tau proteins were determined using WB with 22C11, or with tau12 (anti-tau).
AAV production and injection
mCherry-P2A-tau(GFP)AAVs (1 × 1013/mL) were generated and collected as previously described [54]. This engineered capsid enables efficient transduction of the central nervous system via the vasculature. Purified viruses were concentrated, washed in PBS, sterile-filtered and titred using quantitative PCR. mCherry-P2A-hTauP301L constructs were packaged into AAV2/8. All procedures were performed with approval from the Institutional Animal Care and Use Committee and in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, the Animal Welfare Act, and guidelines from the Guizhou Medical University. The experiments were performed on APP/PS1 female mice and wild-type (WT) littermates. APP/PS1 double transgenic mice were purchased from The Jackson Laboratory (USA, strain name, B6C3-Tg (APPswe, PS1dE9) 85Dbo/J; stock number, 004462). Mice were maintained in ventilated cages with food and water ad libitum and housed under a 12-h light/dark cycle. Experiments were performed starting at 8 weeks of age on females (APP/PS1) and age-matched wild-type littermates as control. Mice were intracranially injected with AAVs encoding mcherry-p2A-hTauP301L into the hippocampus (2.0 μl unilaterally). Injections were performed under standard aseptic surgery conditions. Mice were anaesthetized and maintained with isoflurane (2%) and injections were made at the coordinates (from bregma A/P − 2.5 mm, M/L − 1.5 mm, D/V − 2 mm). A Hamilton needle was used to inject the AAV solution at speed of 0.5 μL/min. Afterwards, the skin over the injection site was sutured and mice were allowed to recover on a warming pad. Mice received meloxicam for two days after surgery.
Immunofluorescent staining
APP/PS1 mice and their WT littermates were perfused using 4% paraformaldehyde in 0.1 M sodium cacodylate pH 7.4 for 15 min at room temperature. Brains were then immersion-fixed for 24 h at room temperature. Immunofluorescent staining was performed as described elsewhere; in brief, samples were rinsed 3 × 5 min, 1 × 1 h in cold phosphate-buffered saline (PBS; pH: 7.4) and then sectioned at 100 µm using a vibratome (Leica). Sections were immersed in normal goat serum (Jackson ImmunoResearch Laboratories) 1:10 in PBS containing 0.5% BSA, 0.1% Triton-X 100, and 0.1% sodium azide (PBTA) at 4 °C on a rotator with overnight agitation. Next, sections were immersed in primary antibodies, anti-tau12 (Biolegend 39416; 1:200) diluted in PBTA. Subsequently, sections were rinsed in PBTA 5 × 5 min, 1 × 1 h and then placed in corresponding secondary antibodies (Jackson Immuno Research Laboratories; 1:200). Finally, sections were rinsed and mounted using DAPI Fluoromount-G (SounthernBiotech) on a glass slide.
Quantification and statistical analysis
All data were normally distributed. Therefore, in instances of single mean comparisons, Levene’s test for equality of variances followed by t test for independent samples was used to assess significance. In instances of multiple mean comparisons, analysis of variance was used, followed by post hoc comparison using Bonferroni’s method. Alpha levels were set at 0.05 for all analyses. All statistical analysis was performed using SPSS release 23.0 (IBM).
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary methods and figure legends (DOCX 35 KB)
Supplementary key resources table (DOCX 23 KB)
Supplementary video clip 3 (MP4 2053 KB)
Acknowledgements
We would like to thank Drs. Mark P. Mattson, Takashi Mori, Darrell Sawmiller, Ahsan Habib for their helpful discussion. We would like to thank Dr. Lucy Hou and Mr. Jun Tian for their technical supporting.
Author contributions
JC, AF, SL, YF JSC and YX performed the experiments, assisted in the design of the study, analyzed the data and drafted the manuscript. JC YF and JSC performed IHC, WB, IP and ELISA, and contributed to data analysis. AF, LZ, DZ, YX and XX assisted in the design of the study, manuscript composition and editing. JT and SL designed and supervised the study, analyzed the data and assisted in the composition and editing of the manuscript. All the authors discussed the results and commented on the final version of the manuscript.
Funding
This study was supported by the High-level Talent Foundation of Guizhou Medical University (YJ19017, HY2020, J. T.), National Natural Science Foundation of China (NSFC) (82171423, 82060211, J. T.), Anyu Biopharmaceutics, Inc., Hangzhou (06202010204, J. T.), the National Key Technologies R & D Program of China during the 9th Five-Year Plan Period [4008 (2019), J. T.], National Natural Science Foundation of China Project for Innovative Talents (HS22102, J. T.), Guizhou Provincial Health Commission (gzwjkj2019-1-044, J. C.), Scientific Research Project of Higher Education Institutions in Guizhou Province [192(2022), J.C.], Guizhou Province Basic Research Program (Natural Science) Project [Qiankehe Foundation-ZK (2023) General 301, J.C.], and the Guizhou Provincial Health Commission (WT19006, J. C.).
Availability of data and materials
The datasets used and/or analyzed in the current study are available from the corresponding author upon reasonable request.
Declarations
Conflict of interest
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Jun Tan (J.T.) is a cofounder for Anyu Biopharmaceutics, Inc. J.C., S.L., D.Z. and J.T. are inventors on a patent application submitted by Anyu Biopharmaceutics, Inc. All other authors report no biomedical financial interests or potential conflicts of interest.
Ethics approval and consent to participate
All mice were housed and maintained in the Animal Facility at Guizhou Medical University, and all experiments were conducted in compliance with protocols approved by Guizhou Medical University Institutional Animal Care and Use Committee.
Consent for publication
All authors agree for publication.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jiang Chen, Anran Fan and Song Li contributed equally to this work.
References
- 1.Anand R, Gill KD, Mahdi AA. Therapeutics of Alzheimer’s disease: past, present and future. Neuropharmacology. 2014;76 Pt A:27–50. doi: 10.1016/j.neuropharm.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 2.Kumar A, Singh A, Ekavali (2015) A review on Alzheimer's disease pathophysiology and its management: an update. Pharmacol Rep 67:195–203 [DOI] [PubMed]
- 3.Deyts C, Thinakaran G, Parent AT. APP receptor? To be or not to be. Trends Pharmacol Sci. 2016;37:390–411. doi: 10.1016/j.tips.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Obregon D, Hou H, Deng J, Giunta B, Tian J, et al. Soluble amyloid precursor protein-α modulates β-secretase activity and amyloid-β generation. Nat Commun. 2012;3:777. doi: 10.1038/ncomms1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Kant R, Goldstein LS. Cellular functions of the amyloid precursor protein from development to dementia. Dev Cell. 2015;32:502–515. doi: 10.1016/j.devcel.2015.01.022. [DOI] [PubMed] [Google Scholar]
- 6.Ayers JI, Giasson BI, Borchelt DR. Prion-like spreading in tauopathies. Biol Psychiatry. 2018;83:337–346. doi: 10.1016/j.biopsych.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pérez M, Cuadros R, Medina M. Tau assembly into filaments. Methods Mol Biol. 2018;1779:447–461. doi: 10.1007/978-1-4939-7816-8_27. [DOI] [PubMed] [Google Scholar]
- 8.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 9.Simón D, García-García E, Royo F, Falcón-Pérez JM, Avila J. Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett. 2012;586(1):47–54. doi: 10.1016/j.febslet.2011.11.022. [DOI] [PubMed] [Google Scholar]
- 10.Saman S, Lee NC, Inoyo I, Jin J, Li Z, et al. Proteins recruited to exosomes by tau overexpression implicate novel cellular mechanisms linking tau secretion with Alzheimer’s disease. J Alzheimers Dis. 2014;40(Suppl 1):S47–70. doi: 10.3233/JAD-132135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruan Z, Pathak D, Venkatesan Kalavai S, Yoshii-Kitahara A, Muraoka S, et al. Alzheimer’s disease brain-derived extracellular vesicles spread tau pathology in interneurons. Brain. 2020;144:288–309. doi: 10.1093/brain/awaa376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Annadurai N, De Sanctis JB, Hajdúch M, Das V. Tau secretion and propagation: perspectives for potential preventive interventions in Alzheimer’s disease and other tauopathies. Exp Neurol. 2021;343:113756. doi: 10.1016/j.expneurol.2021.113756. [DOI] [PubMed] [Google Scholar]
- 13.Merezhko M, Brunello CA, Yan X, Vihinen H, Jokitalo E, et al. Secretion of tau via an unconventional non-vesicular mechanism. Cell Rep. 2018;25(8):2027–2035.e4. doi: 10.1016/j.celrep.2018.10.078. [DOI] [PubMed] [Google Scholar]
- 14.Hellén M, Bhattacharjee A, Uronen RL, Huttunen HJ. Membrane interaction and disulphide-bridge formation in the unconventional secretion of tau. Biosci Rep. 2021;41(8):BSR20210148. doi: 10.1042/BSR20210148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng T, Wu X, Wei X, Wang M, Zhang B. The release and transmission of amyloid precursor protein via exosomes. Neurochem Int. 2018;114:18–25. doi: 10.1016/j.neuint.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 16.Laulagnier K, Javalet C, Hemming FJ, Chivet M, Lachenal G, et al. Amyloid precursor protein products concentrate in a subset of exosomes specifically endocytosed by neurons. Cell Mol Life Sci. 2018;75:757–773. doi: 10.1007/s00018-017-2664-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barbato C, Canu N, Zambrano N, Serafino A, Minopoli G, et al. Interaction of tau with Fe65 links tau to APP. Neurobiol Dis. 2005;18:399–408. doi: 10.1016/j.nbd.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 18.Giaccone G, Pedrotti B, Migheli A, Verga L, Perez J, et al. beta PP and tau interaction. A possible link between amyloid and neurofibrillary tangles in Alzheimer’s disease. Am J Pathol. 1996;148:79–87. [PMC free article] [PubMed] [Google Scholar]
- 19.Smith MA, Siedlak SL, Richey PL, Mulvihill P, Ghiso J, et al. Tau protein directly interacts with the amyloid beta-protein precursor: implications for Alzheimer’s disease. Nat Med. 1995;1:365–369. doi: 10.1038/nm0495-365. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi M, Miyata H, Kametani F, Nonaka T, Akiyama H, et al. Extracellular association of APP and tau fibrils induces intracellular aggregate formation of tau. Acta Neuropathol. 2015;129:895–907. doi: 10.1007/s00401-015-1415-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature. 2014;515:274–278. doi: 10.1038/nature13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore S, Evans LD, Andersson T, Portelius E, Smith J, et al. APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep. 2015;11:689–696. doi: 10.1016/j.celrep.2015.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Puzzo D, Piacentini R, Fa M, Gulisano W, Li Puma DD, et al. LTP and memory impairment caused by extracellular Abeta and tau oligomers is APP-dependent. Elife. 2017;6:e26991. doi: 10.7554/eLife.26991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rumble B, Retallack R, Hilbich C, Simms G, Multhaup G, et al. Amyloid A4 protein and its precursor in Down’s syndrome and Alzheimer’s disease. N Engl J Med. 1989;320:1446–1452. doi: 10.1056/NEJM198906013202203. [DOI] [PubMed] [Google Scholar]
- 25.Wilcock DM, Griffin WS. Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J Neuroinflamm. 2013;10:84. doi: 10.1186/1742-2094-10-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sawmiller D, Habib A, Hou H, Mori T, Fan A, et al. A novel apolipoprotein E antagonist functionally blocks apolipoprotein E interaction with N-terminal amyloid precursor protein, reduces beta-amyloid-associated pathology, and improves cognition. Biol Psychiatry. 2019;86:208–220. doi: 10.1016/j.biopsych.2019.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rauch JN, Luna G, Guzman E, Audouard M, Challis C, et al. LRP1 is a master regulator of tau uptake and spread. Nature. 2020;580(7803):381–385. doi: 10.1038/s41586-020-2156-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cooper JM, Lathuiliere A, Migliorini M, Arai AL, Wani MM, et al. Regulation of tau internalization, degradation, and seeding by LRP1 reveals multiple pathways for tau catabolism. J Biol Chem. 2021;296:100715. doi: 10.1016/j.jbc.2021.100715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- 30.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, et al. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Multhaup G, Huber O, Buee L, Galas MC. Amyloid precursor protein (APP) metabolites APP intracellular fragment (AICD), Abeta42, and tau in nuclear roles. J Biol Chem. 2015;290:23515–23522. doi: 10.1074/jbc.R115.677211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mattson MP, Arumugam TV. Hallmarks of brain aging: adaptive and pathological modification by metabolic states. Cell Metab. 2018;27:1176–1199. doi: 10.1016/j.cmet.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, et al. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ovchinnikov DA, Korn O, Virshup I, Wells CA, Wolvetang EJ. The impact of APP on Alzheimer-like pathogenesis and gene expression in down syndrome iPSC-derived neurons. Stem Cell Rep. 2018;11:32–42. doi: 10.1016/j.stemcr.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 37.Leroy K, Ando K, Laporte V, Dedecker R, Suain V, et al. Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am J Pathol. 2012;181:1928–1940. doi: 10.1016/j.ajpath.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 38.Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9:eaag0481. doi: 10.1126/scitranslmed.aag0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeVos SL, Corjuc BT, Commins C, Dujardin S, Bannon RN, et al. Tau reduction in the presence of amyloid-beta prevents tau pathology and neuronal death in vivo. Brain. 2018;141:2194–2212. doi: 10.1093/brain/awy117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sebastián-Serrano Á, de Diego-García L, Díaz-Hernández M. The neurotoxic role of extracellular tau protein. Int J Mol Sci. 2018;19:998. doi: 10.3390/ijms19040998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gómez-Ramos A, Díaz-Hernández M, Cuadros R, Hernández F, Avila J. Extracellular tau is toxic to neuronal cells. FEBS Lett. 2006;580:4842–4850. doi: 10.1016/j.febslet.2006.07.078. [DOI] [PubMed] [Google Scholar]
- 43.Michel CH, Kumar S, Pinotsi D, Tunnacliffe A, St George-Hyslop P, et al. Extracellular monomeric tau protein is sufficient to initiate the spread of tau protein pathology. J Biol Chem. 2014;289:956–967. doi: 10.1074/jbc.M113.515445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wei Y, Liu M, Wang D. The propagation mechanisms of extracellular tau in Alzheimer’s disease. J Neurol. 2022;269:1164–1181. doi: 10.1007/s00415-021-10573-y. [DOI] [PubMed] [Google Scholar]
- 45.Patel TK, Habimana-Griffin L, Gao X, Xu B, Achilefu S, et al. Dural lymphatics regulate clearance of extracellular tau from the CNS. Mol Neurodegener. 2019;14:11. doi: 10.1186/s13024-019-0312-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ishida K, Yamada K, Nishiyama R, Hashimoto T, Nishida I, et al. Glymphatic system clears extracellular tau and protects from tau aggregation and neurodegeneration. J Exp Med. 2022;219:e20211275. doi: 10.1084/jem.20211275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang S, Bhaskar K. Degradation and transmission of tau by autophagic-endolysosomal networks and potential therapeutic targets for tauopathy. Front Mol Neurosci. 2020;13:586731. doi: 10.3389/fnmol.2020.586731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Congdon EE, Jiang Y, Sigurdsson EM. Targeting tau only extracellularly is likely to be less efficacious than targeting it both intra- and extracellularly. Semin Cell Dev Biol. 2022;126:125–137. doi: 10.1016/j.semcdb.2021.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Linghu C, Johnson SL, Valdes PA, Shemesh OA, Park WM, et al. Spatial multiplexing of fluorescent reporters for imaging signaling network dynamics. Cell. 2020;183:1682–1698.e24. doi: 10.1016/j.cell.2020.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fletcher JM, Boyle AL, Bruning M, Bartlett GJ, Vincent TL, et al. A basis set of de novo coiled-coil peptide oligomers for rational protein design and synthetic biology. ACS Synth Biol. 2012;1:240–250. doi: 10.1021/sb300028q. [DOI] [PubMed] [Google Scholar]
- 52.Grigoryan G, Kim YH, Acharya R, Axelrod K, Jain RM, et al. Computational design of virus-like protein assemblies on carbon nanotube surfaces. Science. 2011;332:1071–1076. doi: 10.1126/science.1198841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gradišar H, Božič S, Doles T, Vengust D, Hafner-Bratkovič I, et al. Design of a single-chain polypeptide tetrahedron assembled from coiled-coil segments. Nat Chem Biol. 2013;9:362–366. doi: 10.1038/nchembio.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu W, Zhang X, Tung YC, Xie S, Liu F, et al. Hyperphosphorylation determines both the spread and the morphology of tau pathology. Alzheimers Dement. 2016;12(10):1066–1077. doi: 10.1016/j.jalz.2016.01.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary methods and figure legends (DOCX 35 KB)
Supplementary key resources table (DOCX 23 KB)
Supplementary video clip 3 (MP4 2053 KB)
Data Availability Statement
The datasets used and/or analyzed in the current study are available from the corresponding author upon reasonable request.