

Abstract
Unveiling the key pathways underlying postnatal beta-cell proliferation can be instrumental to decipher the mechanisms of beta-cell mass plasticity to increased physiological demand of insulin during weight gain and pregnancy. Using transcriptome and global Serine Threonine Kinase activity (STK) analyses of islets from newborn (10 days old) and adult rats, we found that highly proliferative neonatal rat islet cells display a substantially elevated activity of the mitogen activated protein 3 kinase 12, also called dual leucine zipper-bearing kinase (Dlk). As a key upstream component of the c-Jun amino terminal kinase (Jnk) pathway, Dlk overexpression was associated with increased Jnk3 activity and was mainly localized in the beta-cell cytoplasm. We provide the evidence that Dlk associates with and activates Jnk3, and that this cascade stimulates the expression of Ccnd1 and Ccnd2, two essential cyclins controlling postnatal beta-cell replication. Silencing of Dlk or of Jnk3 in neonatal islet cells dramatically hampered primary beta-cell replication and the expression of the two cyclins. Moreover, the expression of Dlk, Jnk3, Ccnd1 and Ccnd2 was induced in high replicative islet beta cells from ob/ob mice during weight gain, and from pregnant female rats. In human islets from non-diabetic obese individuals, DLK expression was also cytoplasmic and the rise of the mRNA level was associated with an increase of JNK3, CCND1 and CCND2 mRNA levels, when compared to islets from lean and obese patients with diabetes. In conclusion, we find that activation of Jnk3 signalling by Dlk could be a key mechanism for adapting islet beta-cell mass during postnatal development and weight gain.
Electronic supplementary material
The online version of this article (10.1007/s00018-020-03499-7) contains supplementary material, which is available to authorized users.
Keywords: Beta-cell mass, Mapk, Obesity, Pregnancy, Postnatal development
Introduction
Pancreatic beta-cell mass expansion is fundamental for achieving appropriate glucose control in response to physiological and pathophysiological conditions [1]. In rodent models of obesity and pregnancy, the expansion of the functional beta-cell mass relies mainly on increased beta-cell proliferation [2, 3]. Although proliferation of human islet cells is still debated, there is some evidence for an increase in pancreatic beta-cell replication in obese individuals [4, 5]. During the postnatal development, the elevated replication rate of pancreatic beta cells that precedes the acquisition of glucose competence is a key process permitting the necessary expansion of the beta-cell mass [6]. In rats, beta-cell replication is substantial from birth until the 10th postnatal day [7]. Thereafter, the replication rate decreases overtime and remains more or less constant throughout the adult life [7]. In human, a similar pattern of beta-cell growth associated with postnatal beta-cell mass expansion has been postulated [8, 9]. The greatest beta-cell replication is observed before 2 years of age with a progressive decline continuing until 5 years [8, 9]. Therefore, the postnatal proliferative capacity is critical for achieving an appropriate adult beta-cell mass, which is important for preventing type 2 diabetes development.
Several independent studies have reported that stimulation of cell cycle gene expression cyclin D2 (Ccnd2) and D1 (Ccnd1) regulating the cyclin dependent kinases 4 and 6 is a key for postnatal beta-cell growth [7, 10]. The upstream regulation of the two cyclins usually rely on serine threonine kinases (STKs) activation, a group of enzymes comprising hundreds of members including mitogen activated protein kinases (MAPKs) [11]. The c-Jun N terminal kinases (Jnks) group of the MAPKs driven by the Map3k12 also called Dual leucine Zipper Kinase (Dlk) regulates cell proliferation in a mechanism evolving the stimulation of Ccnd1 [12]. In pancreatic beta cells, several STKs have been identified as key players in the regulation of both proliferation and mass in response to physiological and pathophysiological conditions in adults [13]. Some of them are therapeutic targets of the Glucagon-like peptide 1 (GLP-1) receptor agonists, current antidiabetic medicines that potentiate glucose-induced insulin secretion, stimulate beta-cell proliferation and protect beta cell against apoptosis [14]. The identification of STK regulating Ccnd2 and Ccnd1 and thereby beta-cell proliferation during postnatal development would be insightful for identifying new targets for improving beta-cell mass in diabetes. In the present study, using transcriptome analysis and whole STK activity measurement, we found that Map3k12 (Dlk)/Mapk10 (Jnk3) signaling is stimulated in neonate islets and is required for postnatal beta-cell mass proliferation and expansion by inducing Ccnd2 and Ccnd1. In addition, we found that this signaling is likely involved in beta-cell mass adaptation in response to weight gain and pregnancy.
Materials and methods
Animals
Male and pregnant female Sprague–Dawley rats were obtained from Janvier Laboratories (Le Genest-Saint-Isle, France) and housed under a 12 h/12 h light/dark cycle in a climate-controlled and pathogen-free facility. All procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health and were approved by the local committee and Veterinary Office. At birth, litters were adjusted to 10–12 pups per dam to standardise the mothers’ milk availability. Newborn males and females were nursed until they were euthanised at postnatal day (P)10. Control female mice and obese (ob/ob) female mice of the C57BL/6J background were obtained from Elevage Janvier (Le Genest-Saint-Isle, France).
Cell culture
Pancreatic islets from adult and newborn rats were isolated by collagenase digestion. In adults, after ligation at the common bile duct, 10 ml of 2 mg/ml of collagenase solution (Roche, Indianapolis, IN, USA) was injected via the ampulla of Vater. In newborn rats, total pancreas was isolated and was incubated in a 5 ml of 1.5 mg/ml of collagenase for 5 min for lysis. Isolated islets from adult and newborn rats were then purified on a Histopaque density gradient. Islets were dissociated by trypsinization in Trypsin–EDTA 0.5% (Thermofisher) at 37 °C for 8 min. The insulin-secreting cell line (INS-1E) and dispersed rat islet cells were maintained as previously described [7]. Isolated human islets were obtained from the biotherapies for diabetes unit from the “Centre Hospitalier Régional et Universitaire de Lille”. Human pancreases were harvested from adult brain-dead donors in accordance with French regulations and with the local Institutional Ethical Committee from the “Centre Hospitalier Régional et Universitaire de Lille”. Isolation and preparation of islets in Lille were conducted as described previously [15].
Western blotting
Cells were incubated for 30 min on ice in the lysis buffer (20 mM Tris acetate, 0.27 mM Sucrose, 1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 50 mM Sodium Fluoride, 10 mM BetaGlycérophosphate, 1 mM DTT) supplemented with proteases and phosphatases inhibitors (Roche). For nuclear extracts preparation, the cells were scraped into 1.5 ml of cold phosphate-buffered saline (PBS), pelleted for 10 s and resuspended in 400 µl cold Buffer A (10 mM HEPES–KOH pH 7.9 at 4 °C, 1.5 mm MgCl2, 10 mM KC1, 0.5 mM dithiothreitol, 0.2 mM PMSF) by flicking the tube. The cells were allowed to swell on ice for 10 min, and then vortexed for 10 s. Samples were centrifuged for 10 s, and the supernatant fraction discarded. The pellet was resuspended in 20–100 µl (according to the starting number of cells) of cold Buffer C (20 mM HEPES–KOH pH 7.9, 25% glycerol, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM dithiothreitol, 0.2 mM PMSF) and incubated on ice for 20 min for high-salt extraction. Cellular debris were removed by centrifugation for 2 min at 4 °C and the supernatant (containing nuclear proteins) was stored at − 70 °C. For Western blotting experiments, total protein extract was separated on 10% SDS–Polyacrylamide gel and electrically blotted to nitrocellulose membrane. The proteins were detected after an overnight incubation of the membrane at 4 °C with the specific primary antibodies (all the antibodies are listed in the Table S1). Proteins were visualized with IRDye800 or IRDye700 (Rockland Immochemicals) as secondary antibodies. Quantification was performed using the Odyssey Infrared Imaging System (Rockland Immochemicals) or ImageJ software.
Immunofluorescence
10-day-old and adult rats were killed, and their pancreases were harvested, fixed in 4% formaldehyde, and processed for paraffin embedding and preparation of 10-μm sections. Beta and alpha cells were visualized by insulin staining using guinea pig anti-insulin antibody (1:100, Dako) and mouse anti-glucagon (1:100, Sigma), respectively. Dlk was detected using the rabbit anti-Dlk primary antibody (1:1000; Genetex) in PBST supplemented with 3% BSA at 4 °C overnight. After being rinsed three times for 10 min each in PBST, slides were incubated with secondary antibody Alexa Fluor (1:500 diluted in PBST plus 1% BSA, Thermofisher) at room temperature for 1 h and were then thoroughly rinsed again. The slides were mounted using a mounting kit containing DAPI (Vectashield. Eurobio-Ingen, H-1200). Cells were observed under a fluorescence microscope. For immunofluorescence of insulin, glucagon and Dlk, whole embryos of littermates control C57BL/6N and KO mice (resulting from the LacZ replacement of the coding exons 1, 2, and a part of 3 of the Map3K12 gene in C57BL/6N mouse) were fixed overnight in 4% paraformaldehyde (PFA) at 4 °C and embedded in paraffin as previously described [16]. Fluorescence images were taken under an Olympus BX51 microscope equipped with DP70 digital camera and the DPManager (DPController) software.
Immunoprecipitation (IP)
INS-1E cells were transfected with the plasmid coding for Dlk cDNA (pcDlk) or small interfering RNA directed against Dlk (siDlk) using Lipofectamime 2000 (Invitrogen) as described [17]. 48 h after transfection, cells were washed with cold PBS and were lysed in 20 mM Tris pH 7.5, 150 mM NaCl 0.1% Triton X-100 buffer (plus protease and phosphatase inhibitors) for 30 min at 4 °C. After centrifugation, the amount of proteins was quantified using bicinchoninic acid protein assay (BCA) reagent (Thermo Fisher Scientific). 500 µg of proteins were then immunoprecipitated with anti-Dlk (1/1000; Genetex) or anti-Jnk3 (1/1000, Cell Signaling) antibodies that have been pre-coupled to protein G Sepharose (GE Healthcare). Proteins were eluted with SDS sample buffer and subjected to SDS/PAGE and subsequent immunoblotting. Approximately, 5% of the protein was run as input, whereas 30% of the pull-down was run in each lane of the Western blots and blotted with anti-Dlk or anti-Jnk3 antibodies.
Apoptosis and proliferation
Dispersed rat islet cells were seeded on poly-l-lysine-coated glass coverslips. After the treatment, the coverslips were washed with PBS and the cells were fixed with methanol. For measuring proliferation with BrdU, the cells were incubated with BrdU (Sigma) 10 µM for 24 h. After permeabilization in PBS-saponin solution, non-specific binding was blocked in a solution of PBS-saponin containing 1% BSA. The cells were then incubated with primary antibodies for 1 h (Ki67, BrdU, Insulin, Glucagon) followed by exposure to secondary antibodies. Coverslips were mounted using a mounting kit containing DAPI (Vectashiled. Eurobio-Ingen, H-1200) and images were analysed with axioscan, an immune slide automatized scanner. Apoptosis was measured by TUNEL assays according to manufacturer's protocol (R&D systems).
Insulin secretion
Dissociated rat islet cells were preincubated with KREBS buffer (127 mM NaCl, 4.7 mM KCl, 1 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 25 mM HEPES, 5 mM NaHCO3) containing 2 mM glucose (basal condition) during 30 min at 37 °C. The medium was then replaced by either the same buffer (basal condition) or a KREBS buffer supplemented with 20 mM of glucose (stimulatory condition). After 45 min, the supernatants were collected and cellular insulin contents were recovered in EtOH acid (75% EtOH, 1.5% HCl). The amount of insulin in the samples was determined by ELISA (Mercodia).
Transfection
INS-1E and dispersed rat islet cells were transfected with small interfering RNA directed against Dlk (siDLK) or JNK3 (siJNK3) using Lipofectamime RNAimax (Thermofisher) as described in [17]. The siRNAs were purchased from Microsynth. The Map3k12 (Dlk) and MAPK10 (Jnk3) mRNA target sequences were 5′-AUA CGA UGU GGU GAA GAU CUC dTdT-3′ and 5′ AGC GGA UGU ACU AGC UTT-3′, respectively.
Quantitative PCR
Total RNA was extracted using Aurum Total RNA Mini Kit (Bio-Rad) according to the manufacturer’s protocol. Total RNA was collected from the pools of pancreatic islets of Postnatal day 1 (P1, four pups), P5 (four pups) and P10 (two pups), while in P15, P20, P23, P31 and adult rats, total RNA was extracted from islets of individual animal. The reverse transcription reaction was performed as previously described [18]. Real-time quantitative PCR assays were carried out on the Bio-Rad MyiQ Real-Time PCR Detection System using iQ SyBr Green Supermix (Bio-Rad) as previously described [18]. The primer sequences for rat and mouse used are: Ccnd1 (sense 5′-AGCAGAAGTGCGAAGAGGAG-3′ and antisense 5′-GAGCTTGTTCACCAGAAGCA-3′); Rplp0 (sense 5′-ACCTCCTTCTTCCAGGCTTT-3′ and antisense 5′-CCTCTTTCTTCCAAGCTTT-3′); Tbp (sense 5′-CGTGCCAGAAATGCTGAATA-3′ and antisense 5′-CTGGTGGGTCAACACAAGG-3′); Mapk10 (Jnk3) (sense 5′-CGTGGACATATGGTCTGTGG-3′ and antisense 5′-AGGTGAGGCCTGCATACTTG-3′); Map3k12 (Dlk) (sense 5′-TTAAATCCCAGGCAGAGTGG-3′ and antisense 5′-GAGGGCATTCAGTTCCATGT-3′); Ccnd2 (sense 5′-GACTGCGGAAAAGCTGTGTA-3′ and antisense 5′-TGCTCAATGAAGTCGTGAGG-3′). The primer sequences for Human genes are: MAP3K10 (JNK3) (sense: 5′-GTGGCATTAAGCACCTCCAT-3′ and antisense (5′-ATGACCTCAGGGGCTCTGTA-3′); MAP3K12 (DLK) (sense 5′-GTACTCTCCACACCCCAGGA-3′ and antisense 5′-GGCTCTCTCCAGCTTCCTTT-3′); RPL27 (sense 5′-ATCGCCAAGAGATCAAAGATAA-3′ and antisense 5′-TCTGAAGACATCCTTATTGACG-3′); CCND1 (sense 5′-TGAAGGAGACCATCCCCCTG-3′ and antisense 5′-TGTTCAATGAAATCGTGCGG-3′); CCND2 (sense 5′-TCCAAACTCAAAGAGACCAGC-3′ and antisense 5′-TTCCACTTCAACTTCCCCAG-3′).
PamChip peptide microarrays for kinome analysis
See supplementary methods.
Statistical analyses
The experiments including more than two groups were analyzed by ANOVA or with the non-parametric equivalent Kruskal–Wallis test followed by post-hoc Bonferroni test (Dunnett’s test) when experiments included more than two groups.
Results
Map3k12 (Dlk) expression is involved in beta-cell proliferation during the postnatal development of newborn rats
We previously identified through transcriptome analysis a 70-fold higher expression of Map3k12 in pancreatic islets of 10-day-old rats compared to those of adult animals [7]. Map3k12 encodes the mitogen activated protein 3 kinase 12, a STK also called dual leucine zipper-bearing kinase (Dlk). The expression changes of Map3k12 (Dlk) during the postnatal period was further studied by quantitative PCR and Western Blotting experiments. We found a 2–4-fold increase of the Map3k12 (Dlk) expression between p1 and p10 postnatal days, and declines thereafter, suggesting a transient postnatal activation (Fig. 1a, b). Map3k12 (Dlk) has a multitude of roles that rely on the cellular and environmental context, which may even appear paradoxical. In beta cells, Map3k12 (Dlk) plays a dual role according to its subcellular localization [19]. While the presence of Map3k12 (Dlk) in the nucleus is associated with beta-cell death, the cytosolic localization of the kinase drives opposite cell outcome [19]. In addition, plasmid-based overexpression of Dlk has been associated with increased apoptosis in immortalized insulin-producing cells including HIT cells [19] and INS-1 cells (Fig. S1a and S1b). Therefore, we investigated the subcellular localization of overexpressed Map3k12 (Dlk) in islets of 10-day-old rats. Immunofluorescence (IF) and Western Blotting experiments showed that Map3k12 (Dlk) overexpression is enriched in the cytosol fraction of isolated islet cells of neonate rats (Fig. 1c–i). The cytosolic localization of the kinase observed in islet cells from adult rats (Fig. S2), suggests that overexpression of Map3k12 (Dlk) is not associated with a potential change in the nuclear–cytoplasmic distribution of the protein during postnatal development. As beta cells of neonate rats are highly replicative and have immature glucose responsiveness [7], we hypothesized that the transient overexpression of Map3k12 (Dlk) plays a role for beta-cell replication.
Fig. 1.

Postnatal islet induction and beta-cell nuclear localization of Map3k12 (Dlk) in neonate rats. a The expression of the Map3k12 (Dlk) was measured by a RT-qPCR and b Western blotting in isolated islet of newborn rats between p1, p6, p10 and p20 postnatal days and young adult rats (p32). The mRNA levels of Map3k12 (Dlk) were normalized to those of Tata box binding protein (Tbp) and were expressed as % changes. The expression of Map3k12 (Dlk) at p1 was set to 100%. The data are the mean ± SEM of 3 independent experiments performed in triplicate (**p < 0.01, by ANOVA). b Western blotting was performed using total proteins (20 μg/lane) isolated from islets of 10- and 32-day-old rats using an anti-Dlk (1/1000; Genetex) and anti-αTubulin (1/5000; Sigma) antibodies. The intensities of the bands were quantified using ImageJ, taking care to avoid saturation and subtracting the background. Values were expressed as the integrals (area × mean density) of each band (normalized to αTubulin band). Values from p32 were set to 100%. c–h Representative immunofluorescence images for Map3k12 (Dlk), insulin and glucagon in pancreatic islets of 10-day-old rat (×40 magnification) using anti-Dlk (1/1000; Genetex), anti-insulin (1/100, Dako) and anti-glucagon (1/100, Sigma) antibodies. The scale bars in each picture row correspond to 20 μm. c Map3k12 (Dlk) (green; d, g), and insulin (red; f, h) performed on fixed pancreas of newborn rats of 10 days old. Merged images were achieved to show the potential co-localization of Map3k12 (Dlk) with glucagon (e) or insulin (h). Yellow indicates the co-localization. Blue, DAPI. i Detection of Map3k12 (Dlk) protein in islets nuclear-enriched fraction of 10-day-old rats. Nuclear and cytosol (Cyto) proteins (20 μg/lane) were loaded onto 10% SDS-PAGE and then detected by Western blotting using an anti-Dlk (1/1000; Genetex), anti-Lamin a/c (1/1000; Abcam) and anti-Calreticulin (1/1000; Abcam) antibodies
To assess a possible involvement of the kinase in beta-cell mass expansion, we measured the proliferation rates of dispersed islet cells from 10 day-old rats in which the expression of Map3k12 (Dlk) was reduced using small interfering RNAs (Fig. 2a). Map3k12 (Dlk) silencing resulted in a marked reduction in beta-cell proliferation, as monitored by ki67 or BrdU and insulin co-staining (Fig. 2b, c and Fig. S3). The loss of beta-cell proliferation by Map3k12 (Dlk) silencing was associated with the drop of Ccnd2 and Ccnd1 mRNA levels (Fig. 2d). Decreased beta-cell proliferation by siDlk was not associated with a change in beta-cell apoptosis (Figs. 2e, S4) and insulin secretion (Fig. 2f). Moreover, the expression of key beta-cell genes including Glut2, Pdx1, MafA, Ins2, GCK, Znt8, Kir6.2 and Nkx6.1 were unaltered by Map3k12 (Dlk) silencing (Fig. S5), confirming that the kinase is not involved for specialized beta-cell function. Taken together, our data support the hypothesis that increased Map3k12 (Dlk) expression is required for beta-cell proliferation during postnatal development in a mechanism involving Ccnd2 and Ccnd1.
Fig. 2.

Effect of Map3k12 (Dlk) silencing in beta-cell proliferation, basal beta-cell death, Ccnd1 and Ccnd2 mRNA levels. The expression of the Map3k12 (Dlk) was measured in dispersed islet cells from 10-day-old rats by a Western Blotting experiments in dispersed islet cells of newborn rats of 10 days old, which were transfected with control siRNA (siCtl; siRNA directed against GFP) or siRNAs against Map3k12 (Dlk) (siDlk) for 48 h. beta cell proliferation was quantified by determining the percentage of insulin + (ins+) and b Ki67+ cells (n = 4) or c BrdU+ (n = 3). d Effect of siDLK on the expression of Ccnd1 and Ccnd2 in the islet cells of 10-day-old rats. The mRNA levels were normalized to those of Tbp and were expressed as % changes. The data are presented as means ± SEM of 5 independent experiments performed in triplicate as determined by ANOVA, (*p < 0.05, ***p < 0.001). e Beta-cell death was assessed by scoring the percentage of insulin positive cells labelled with TUNEL (n = 3). f insulin secretion after 45 min exposure to 2 or 20 mM glucose (n = 4). The data are the mean ± SEM of 3 independent experiments performed in triplicate (***p < 0.001, by ANOVA)
Map3k12 (Dlk) is a strong activator of Mapk signaling including c-Jun amino terminal kinases (Jnk) in different cell types [19, 20], suggesting that Mapk/Jnk activity links Map3k12 (Dlk) to beta-cell proliferation in islets of 10-day-old rats. In support of this hypothesis, the measurement of the phosphorylation of hundreds of validated STK substrates by Pamgene technology [18] confirmed the increased activity of the three Mapks/Jnks, including Mapk8 (Jnk1), Mapk9 (Jnk2) and Mapk10 (Jnk3) among the 25 STKs activity that are stimulated in islets of 10-day-old rats when compared to adult rats (Fig. S6). Increased Mapks/Jnk activity was associated with Mapks/Jnks activation as shown by Western blotting using antibodies recognizing the phosphorylated forms of the three Jnk isoforms (Fig. 3a, b). However, in beta cells, Mapk10 (Jnk3) triggers antiapoptotic and trophic effects, whereas Mapk8 (Jnk1) and Mapk9 (Jnk2) are pro-apoptotic [21]. While they are antagonists, the three Mapks were released from the kinome analysis, because they phosphorylate similar peptide targets. Therefore, we suggested that only Mapk10 (Jnk3) activation couples the effect of Map3k12 (Dlk) to beta-cell proliferation in neonate islets. Several lines of evidences support this hypothesis. First, in agreement with previous data [7], basal beta-cell apoptosis is similar between 10-day-old and adult rats (Fig. S7). Second, Jnk1 and Jnk2 phosphorylation at functional Thr183/ Tyr185 residues was not induced in islet cells from 10-day-old when compared to adult rats (Fig. 3c, d). Therefore, we hypothesized that Map3k12 (Dlk) overexpression directly leads to Mapk10 (Jnk3) activation. In addition, we postulated that the mechanism does not rely on the changes in the expression of Mapk10 (Jnk3) as the Mapk10 (Jnk3) mRNA level was similar in islets of 10-day-old and adult rats (Fig. 3e). To assess the regulation of Mapk10 (Jnk3) by Map3k12 (Dlk), we performed coimmunoprecipitation experiments using the rat INS-1E as unlimited beta-cell source mimicking beta cells of neonate rats. As previously shown in HIT cells [22], Map3k12 (Dlk) regulates Mapk/Jnk activation in INS-1E cells (Fig. S8). Immunoprecipitation experiments confirmed that Map3k12 (Dlk) associates with Mapk10 (Jnk3) in INS-1E cells (Fig. 3f). Association of Dlk and Jnk3 was further validated by immunoprecipitation of Dlk and Jnk3 in HeLa that overexpressed both proteins (Fig. S9). Moreover, as expected, silencing or overexpression of Dlk modulated the phosphorylation of Mapk10 (Jnk3) in INS-1E cells (Fig. 3g, h). All these results indicate that Map3k12 (Dlk) activates Mapk10 (Jnk3) in beta cells.
Fig. 3.
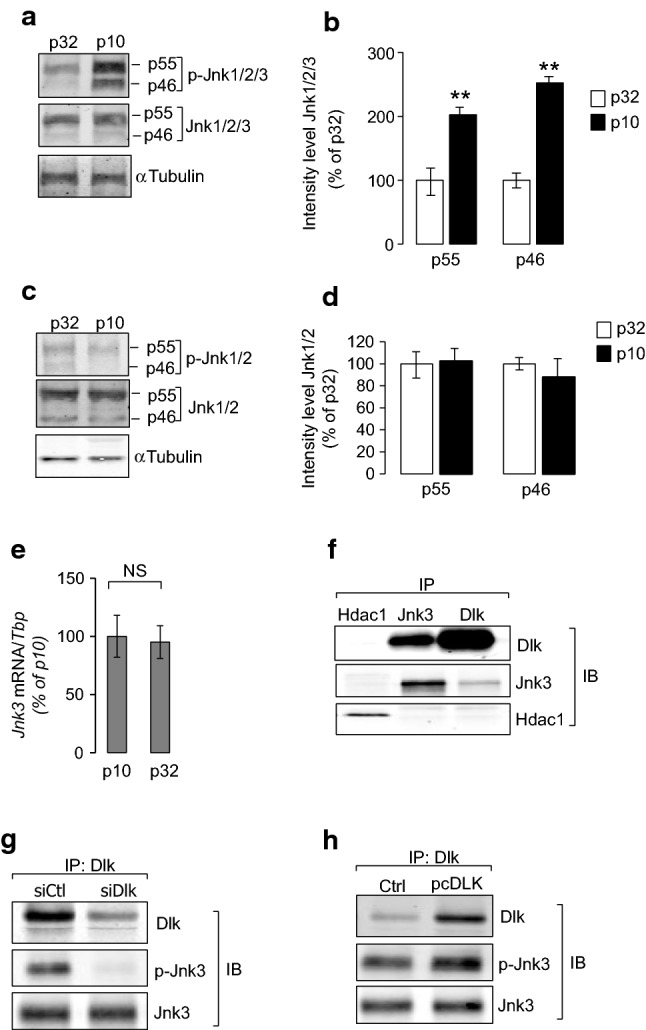
Mapk10/Jnk3 activation by Map3k12 (Dlk) in neonate islet beta cells. Western Blotting analysis of a total and phosphorylated Mapks/Jnks (Jnk1/Jnk2 and Jnk3) and c total and phosphorylated Jnk1 and Jnk2 in islets of 10 (p10) and 32 (p32)-day-old rats. Western blotting was achieved loading total proteins from islets of neonate (p10) and adult rats (p32) using either anti-total Jnk1/Jnk2 and p-Jnk1/Jnk2 (phospho Thr183/Tyr185; 1/1000; Thermofisher), or anti-total Jnk1/Jnk2/Jnk3 and p-Jnk1/Jnk2/Jnk3 (phospho Thr183 + Thr183 + Thr221, Abcam, dilution 1:1000), or anti-αTubulin (1/5000; Sigma). b–d The intensities of bands were measured using ImageJ, taking care to avoid saturation and after subtracting the background. Values are expressed as the integrals (area × mean density) of each band corresponding to p-Jnk1/2/3 or p-Jnk1/2 (p55 and p46), normalized to total Jnk1/2/3 or Jnk1/2. Values from p32 were set to 100% (**p < 0.01, by ANOVA). d The expression of Mapk10/Jnk3 was measured by RT-qPCR in the islets of rats of p10 and p32. The results were normalized against Tbp and the expression levels from p10 were set to 100%. Data are the mean ± SEM of 3 independent experiments performed in triplicate (NS, p > 0.05). e Co-immunoprecipitation of Map3k12 (Dlk) and Mapk10 (Jnk3) in INS-1E cells. Dlk and Jnk3 proteins are pulled down by the anti-Dlk (1/1000; Eurogentec) or anti-Jnk3 antibodies (1/1000, Cell Signaling) but not with control anti-Hdac1 antibody (1/1000; Santa Cruz). Phosphorylation level of Jnk3 (Thr183/Tyr185 residue) after Jnk3 immunoprecipitation in response to f Map3k12 (Dlk) silencing (siDlk) or g Map3k12 (pcDlk) overexpression in INS-1E cells. Immunoblotting were achieved using either anti-Dlk (1/1000; Eurogentec), or anti-Jnk3 (1/1000, Cell Signaling) or anti phospho-Jnk3 (Thr183/Tyr185, 1/1000, Cell Signaling). The figures show the result of a representative experiment out of three
Silencing of Mapk10 (Jnk3) in islets of 10-day-old rats reduced beta-cell proliferation as monitored by Ki67 and BrdU (Fig. 4a–c and Fig. S10), suggesting that Mapk10 (Jnk3) activation by Map3k12 (Dlk) accounts for beta-cell proliferation during postnatal development. Moreover, the diminished beta-cell proliferation caused by Mapk10 (Jnk3) silencing was associated with the reduction of Ccnd2 and Ccnd1 mRNA (Fig. 4d). These results indicate that the Map3k12 (Dlk)/Mapk10 (Jnk3) signaling are required for beta-cell proliferation by controlling the expression of Ccnd2 and Ccnd1.
Fig. 4.

Effect of Mapk10 (Jnk3) silencing in beta-cell proliferation and Ccnd1 and Ccnd2 mRNA levels a Newborn rat islet cells were transfected with control siRNA (siCtl; siRNA directed against GFP) or siRNAs against Jnk3 for 48 h and Jnk3 mRNA levels were measured by RT-qPCR. beta cell proliferation was quantified by determining the percentage of insulin + (ins+) and b Ki67 + cells (n = 3) and c BrdU. d Effect of siJnk3 on the expression of indicated genes in the islet cells of 10-day-old rats. Data are the means ± SEM of 3 independent experiments performed in triplicate (*p < 0.05, ***p < 0.001; ANOVA)
Map3k12 expression is increased in islet cells in response to weight gain and pregnancy conditions
Induction of Map3k12 (Dlk) in highly proliferative beta cells suggests that Map3k12 (Dlk) signaling contributes to beta-cell mass expansion during body growth and development. To verify this hypothesis, we analysed embryos of Dlk-Knockout (KO), in which the Mapk (Jnk) activity including Mapk10 (Jnk3) is totally impaired [16]. Although the homozygous mutant animals (KO) survived during embryonic stages without showing any gross tissue abnormality, they died perinatally, and no KO mutant survived until weaning [16]. Dlk gene disruption results in abnormalities in brain development that are characterized by major defects in axon growth and radial migration of neocortical pyramidal neurons [16]. Besides brain development abnormalities, we observed that beta-cell mass in the KO embryos was reduced (Fig. S11). To further gain insights into the contribution of Map3k12 (Dlk) in the beta-cell expansion, we next measured its expression in insulin-secreting cells from rodent models of pregnancy and obesity. We first measured the expression of Map3k12 (Dlk), Ccnd1 and Ccnd2 in islet cells from pregnant rats at 14 days of gestation that display increased beta-cell replication [23]. In pregnant rats, it was previously shown that the islet beta-cell replication increases after 10 days of gestation (G10), peaks at 14 days (G14), and decreases thereafter until delivery [3, 24]. We found that the expression of Map3k12 (Dlk), Mapk10 (Jnk3), Ccnd1 and Ccnd2 raises compared to age-matched non-pregnant female rats starting from G10 and reaches its maximum at G14 (Fig. 5a). As expected from beta-cell replication studies [3, 24], the expression of the three mRNA level decreased after G14 (Fig. 5a).
Fig. 5.
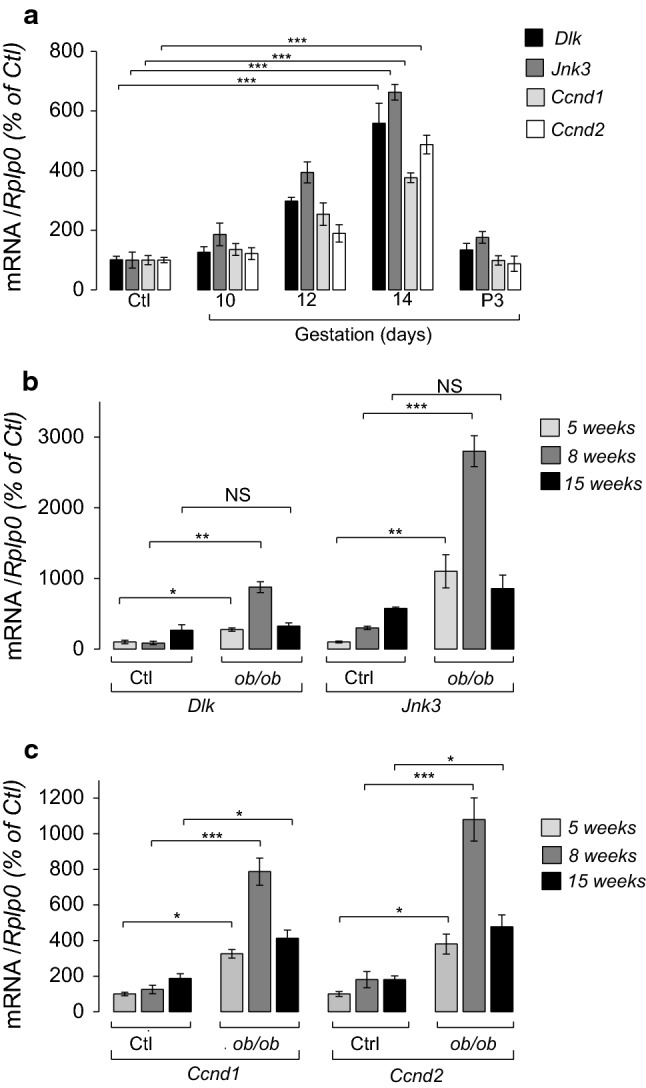
Measurement of islets Map3k12 (Dlk), Ccnd1 and Ccnd2 expression changes in mice model of pregnancy and obesity. a The expression of Map3k12 (Dlk), Mapk10 (Jnk3), Ccnd1 and Ccnd2, was measured by RT-qPCR in the islets of female rats at indicated gestational days 10 (G10), 12 (G12), 14 (G14), and 3 days post-partum (P3). The results are expressed as % of the level in (Ctl). b Measurement of Map3k12 (Dlk) and Mapk10 (Jnk3) and c Ccnd1 and Ccnd2 in islets isolated from ob/ob mice at the indicated ages and of age-matched controls. The data represent the mean ± SEM of 4 animals per group. (Significantly different non-pregnant rats ***p < 0.001; **p < 0.01; *p < 0.05 by Dunnett’s post-hoc test). NS Not significant
In obesity rodent models, such as ob/ob, the beta-cell proliferation largely contributes to the expansion of the beta-cell mass and rises in response to insulin resistance [2, 25]. In ob/ob mice, islet beta-cell growth is prominent in 5–8-week-old animals [26]. We found that the expression of Map3k12 (Dlk) is increased in 5- and 8-week-old ob/ob mice compared to controls mice (Fig. 5b). The subcellular localization of Map3k12 (Dlk) in islets of 8-week-old ob/ob and age-matched control mice were similar, supporting a role for Map3k12 (Dlk) in the beta-cell proliferation during weight gain (Fig. S12). Moreover, induction of Map3k12 (Dlk) in 5- and 8-week-old ob/ob mice was associated with the rise of MAPK10 (Jnk3), Ccnd1 and Ccnd2 (Fig. 5b, c). Beta-cell replication in ob/ob mice was associated with induction of genes involved in the adaptive unfolded protein response (UPR) [27–29]. Quantitative PCR analysis revealed that the expression of UPR genes including Atf4, Atf6, Dnacj3 (p58), Edem1 and Ero1 was unchanged in islet cells from 10-day-old rats in which the expression of Map3K12 (Dlk) was silenced (Fig. S13). This result suggests that Map3K12 (Dlk) overexpression is not involved in the activation of adaptive UPR during weight gain.
We next analyzed human islet cells from non-diabetic donors with severe obesity (Table S2) and found also higher MAP3K12 (DLK), MAPK10 (JNK3), CCND1 and CCND2 mRNA levels, when compared to normal-weight non-diabetic individuals (Fig. 6). Immunofluorescence pictures that were obtained from pancreatic sections of obese individuals without diabetes and patients with diabetes showed no apparent difference in the DLK localization (Fig. S14). This observation supports that induction of MAP3K12 (DLK) mainly account for the role of kinase and its pathway for the control of human beta-cell mass and thereby, glucose homeostasis during severe weight gain.
Fig. 6.

MAP3K12 (DLK) in isolated human islet cells from lean and obese donors. The mRNA levels of MAP3K12 (DLK) MAPK10 (JNK3), CCND1 and CCND2 were measured in isolated human islets from lean donors (N = 7), obese individuals without diabetes donors (N = 7) and obese patients with diabetes (N = 5). The data were normalized against the RPL27 and were expressed as the fold changes over the lean controls. Data are the mean of ± SEM (significantly different from lean individuals without diabetes ***p < 0.001; **p < 0.01; by Dunnett’s post-hoc test). NS Not significant
Discussion
Postnatal pancreatic development is characterized by an increase in beta-cell mass, enabling adequate insulin supply for early life development. In rodents and possibly in humans, the rise in beta-cell mass before weaning results from an elevated proliferation rate [7, 8]. It has been previously shown that the robust proliferative capacity of beta cells is maximal within the first 10 postnatal days [7]. Here we found that beta-cell Map3k12 (Dlk) mRNA levels raise after birth, reach a peak at the 10th postnatal day, and then decline at the 20th postnatal day. Several evidences have led us to postulate that the stimulation of Map3k12 (Dlk) mRNA levels accounts for the beta-cell proliferation in neonate rats. Map3k12 (Dlk) has multiple and paradoxical roles upon cell types and environmental context. A trophic role for Map3k12 (Dlk) has been previously postulated to induce hepatocytes proliferation upon partial hepatectomy [12, 30]. In addition, Map3k12 (Dlk) triggers fibroblast cell proliferation by regulating the expression of cell cycle regulatory proteins including Ccnd1 [12]. In beta cells, Map3k12 (Dlk) may trigger opposite effect according to its subcellular localization [19]. In contrary to the cytosolic presence of Map3k12 (Dlk), the nuclear expression of the kinase is pro-apoptotic [19]. Herein, we show that Map3k12 (Dlk) is mainly localized in the cytosol of neonate and adult rat islet cells. This observation suggests that the subcellular localization of the kinase is not modified during the postnatal development and that its overexpression is required for beta-cell proliferation to regulate the level of the key cyclins involved in postnatal beta-cell proliferation. Several lines of evidence support this hypothesis. Silencing of Map3k12 (Dlk) in isolated islets from newborn rats (10 days old postnatal) dramatically reduced beta-cell proliferation in a mechanism involving the reduction of Ccnd1 and Ccnd2. Importantly, the reduction of Map3k12 (Dlk) level in isolated islets of 10-day-old rats did not change basal beta-cell apoptosis This indicates that the observed reduction in proliferation is not the consequence of increased beta-cell death, which is in line with previous results showing that basal apoptosis of newborn and adult rat islet cells is similar [7]. In addition, Map3k12 (Dlk) silencing did not enhance glucose-induced insulin secretion, nor the expression of key genes of beta-cell identity, pointing to the peculiar role of Dlk in the control of beta-cell replication during postnatal development.
We also provide some clues supporting a role for Mapk10 (Jnk3) in the mechanism whereby Map3k12 mediates the trophic effects on newborn beta cells (Fig. 7). First, Map3k12 (Dlk) has been primarily identified as an upstream STK component of the signalling cascade leading to the activation Mapk/Jnk pathway including Mapk10 (Jnk3) [21]. In line with this, using a whole STKs activity assessment in islets of neonate rats, we showed that the rise of Map3k12 (Dlk) is associated with increased activity of several Mapk, including Mapk10 (Jnk3) and the two pro-apoptotic Mapk8 (Jnk1) and Mapk9 (Jnk2) isoforms. In beta cells, it has been postulated that in physiological conditions, Mapk10 (Jnk3) activity, may inhibit the pro-apoptotic activities of Mapk8 (Jnk1) and Mapk9 (Jnk2) [21]. Our data showing that neither apoptosis nor Jnk1/Jnk2 activation is modified between neonate and adult islet cells support this hypothesis. In addition, we found that Mapk10 (Jnk3) silencing leads to decreased beta-cell proliferation. Taken together, our observations support the hypothesis that the increased Mapk10 (Jnk3) activity results from the rise in Map3k12 (Dlk) expression. This provides the first proof of concept supporting a physiological role for Mapk10 (Jnk3) in the control of beta-cell proliferation.
Fig. 7.

Schematic representation of beta cell mass regulation by Map3k12 (Dlk)
Beta-cell proliferation also contributes to the adaptation of the functional beta-cell mass under conditions characterized by a diminished insulin sensitivity such as obesity and pregnancy. In this respect, we postulated that Map3k12 (Dlk) signaling may play a pivotal role also in the mechanisms triggered to compensate for insulin resistance in adults. First, we found that the beta-cell number is reduced in Map3k12 KO mice. Second, increased Map3k12 (Dlk) mRNA was accompanied by elevated expressions of Mapk10 (Jnk3), Ccnd1 and Ccnd2 in rodent models of obesity and pregnancy displaying an elevated beta-cell proliferation rate. As the stoichiometry of the MAPKs components is important to activate the signalling cascade [31], it is possible that induction of Mapk10 (Jnk3) in compensatory islets to weight gain is the consequence of the elevated expression of Map3k12 (Dlk). In human, enhanced obesity-driven beta-cell proliferation was monitored in non-diabetic, but not in diabetic donors [5]. This result led to the hypothesis that the lack of proliferation may be causally linked to the reduced beta-cell mass and to diabetes pathogenesis. Our study showing that the MAP3K12 (DLK), MAPK10 (JNK3), CCND1 and CCND2 mRNA levels are upregulated in the islets from non-diabetic obese individuals compared to lean or diabetic obese donors, is consistent with MAP3K12 (DLK) signalling being one of the mechanisms regulating human beta-cell proliferation in severe weight gain conditions.
In conclusion, our study demonstrates that the signalling pathway activated by Map3k12 (Dlk) is a key driver of beta-cell mass expansion during critical life times were the insulin demand is increased such as postnatal development before weaning, pregnancy and obesity in both rodents and human. Abnormalities in this signalling may lead to inadequate functional beta-cell mass and eventually to diabetes development.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We would like to thank Mrs Laure Rolland for the technical assistance and, Antonino Bongiovanni and Meryem Tardivel from the BICeL-Campus HU Facility for access to systems and technical advice.
Abbreviations
- Ccnd
Cyclin D
- Cdk
Cyclin-dependent kinase
- Cdkn
Cyclin-dependent kinase inhibitor
- DLK
Dual leucine Zipper Kinase
- JNK
C-Jun amino terminal kinase
- MAP3K12
Mitogen-activated protein kinase 12
- MAPK10
Mitogen-activated protein kinase 10
- STK
Serine threonine kinase
Author contributions
MT, JKC, FP, RR, AB, PF and AA contributed to the study design and interpretation of the data. MT, SD, AB, PF and AA wrote and edited the manuscript. MT, VPL, VP, CSP, HE, RB, VG, CJ, JB, NB, GP performed the experiments and analysed the data. All authors read and approved the submission of the manuscript.
Funding
This work was supported by grants from “Société Francophone du Diabète (SFD)”, “European Genomic Institute for Diabetes” (E.G.I.D., ANR-10-LABEX-46) and European Commission, European Research Council (GEPIDIAB 294785 to P.F.), and by a grant from the Swiss National Science Foundation (310030-169480 to RR). Human islets were provided by the European Consortium for Islet Transplantation, funded by the Juvenile Diabetes Research Foundation International.
Compliance with ethical standards
Conflict of interest
The authors declare that there is no duality of interest associated with the manuscript.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mathie Tenenbaum, Email: mathie.tenenbaum@univ-lille.fr.
Amar Abderrahmani, Email: amar.abderrahmani@univ-lille.fr.
References
- 1.Chen C, Cohrs CM, Stertmann J, et al. Human beta cell mass and function in diabetes: recent advances in knowledge and technologies to understand disease pathogenesis. Mol Metab. 2017;6:943–957. doi: 10.1016/j.molmet.2017.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Golson ML, Misfeldt AA, Kopsombut UG, et al. High fat diet regulation of β-cell proliferation and β-cell mass. Open Endocrinol J. 2010 doi: 10.2174/1874216501004010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baeyens L, Hindi S, Sorenson RL, German MS. β-Cell adaptation in pregnancy. Diabetes Obes Metab. 2016;18:63–70. doi: 10.1111/dom.12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis DB, Lavine JA, Suhonen JI, et al. FoxM1 is up-regulated by obesity and stimulates β-cell proliferation. Mol Endocrinol. 2010;24:1822–1834. doi: 10.1210/me.2010-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanley SC, Austin E, Assouline-Thomas B, et al. β-Cell mass dynamics and islet cell plasticity in human type 2 diabetes. Endocrinology. 2010;151:1462–1472. doi: 10.1210/en.2009-1277. [DOI] [PubMed] [Google Scholar]
- 6.Mezza T, Kulkarni RN. The regulation of pre- and post-maturational plasticity of mammalian islet cell mass. Diabetologia. 2014;57:1291–1303. doi: 10.1007/s00125-014-3251-7. [DOI] [PubMed] [Google Scholar]
- 7.Jacovetti C, Matkovich SJ, Rodriguez-Trejo A, et al. Postnatal β-cell maturation is associated with islet-specific microRNA changes induced by nutrient shifts at weaning. Nat Commun. 2015 doi: 10.1038/ncomms9084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meier JJ, Butler AE, Saisho Y, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57:1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregg BE, Moore PC, Demozay D, et al. Formation of a human β-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab. 2012;97:3197–3206. doi: 10.1210/jc.2012-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kushner JA, Ciemerych MA, Sicinska E, et al. Cyclins D2 and D1 are essential for postnatal pancreatic β-cell growth. Mol Cell Biol. 2005;25:3752–3762. doi: 10.1128/MCB.25.9.3752-3762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bononi A, Agnoletto C, De Marchi E, et al. Protein kinases and phosphatases in the control of cell fate. Enzyme Res. 2011 doi: 10.4061/2011/329098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daviau A, Couture J-P, Blouin R. Loss of DLK expression in WI-38 human diploid fibroblasts induces a senescent-like proliferation arrest. Biochem Biophys Res Commun. 2011;413:282–287. doi: 10.1016/j.bbrc.2011.08.086. [DOI] [PubMed] [Google Scholar]
- 13.Kulkarni RN, Mizrachi E-B, Ocana AG, Stewart AF. Human β-cell proliferation and intracellular signaling: driving in the dark without a road map. Diabetes. 2012;61:2205–2213. doi: 10.2337/db12-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- 15.Vantyghem MC, Kerr-Conte J, Arnalsteen L, et al. Primary graft function, metabolic control, and graft survival after islet transplantation. Diabetes Care. 2009;32:1473–1478. doi: 10.2337/dc08-1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirai S, Cui DF, Miyata T, et al. The c-Jun N-terminal kinase activator dual leucine zipper kinase regulates axon growth and neuronal migration in the developing cerebral cortex. J Neurosci. 2006;26:11992–12002. doi: 10.1523/JNEUROSCI.2272-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brajkovic S, Ferdaoussi M, Pawlowski V, et al. Islet brain 1 protects insulin producing cells against lipotoxicity. J Diabetes Res. 2016 doi: 10.1155/2016/9158562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abderrahmani A, Yengo L, Caiazzo R, et al. Increased hepatic PDGF-AA signaling mediates liver insulin resistance in obesity-associated type 2 diabetes. Diabetes. 2018;67:1310–1321. doi: 10.2337/db17-1539. [DOI] [PubMed] [Google Scholar]
- 19.Wallbach M, Duque Escobar J, Babaeikelishomi R, et al. Distinct functions of the dual leucine zipper kinase depending on its subcellular localization. Cell Signal. 2016;28:272–283. doi: 10.1016/j.cellsig.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 20.Holzman LB, Merritt SE, Fan G. Identification, molecular cloning, and characterization of dual leucine zipper bearing kinase. A novel serine/threonine protein kinase that defines a second subfamily of mixed lineage kinases. J Biol Chem. 1994;269:30808–30817. doi: 10.1016/S0021-9258(18)47353-X. [DOI] [PubMed] [Google Scholar]
- 21.Beeler N, Riederer BM, Waeber G, Abderrahmani A. Role of the JNK-interacting protein 1/islet brain 1 in cell degeneration in Alzheimer disease and diabetes. Brain Res Bull. 2009;80:274–281. doi: 10.1016/j.brainresbull.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 22.Stahnke M-J, Dickel C, Schröder S, et al. Inhibition of human insulin gene transcription and MafA transcriptional activity by the dual leucine zipper kinase. Cell Signal. 2014;26:1792–1799. doi: 10.1016/j.cellsig.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jacovetti C, Abderrahmani A, Parnaud G, et al. MicroRNAs contribute to compensatory β cell expansion during pregnancy and obesity. J Clin Investig. 2012;122:3541–3551. doi: 10.1172/JCI64151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rieck S, Kaestner KH. Expansion of beta-cell mass in response to pregnancy. Trends Endocrinol Metab. 2010;21:151–158. doi: 10.1016/j.tem.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mosser RE, Maulis MF, Moullé VS, et al. High-fat diet-induced β-cell proliferation occurs prior to insulin resistance in C57Bl/6J male mice. Am J Physiol Endocrinol Metab. 2015;308:E573–E582. doi: 10.1152/ajpendo.00460.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindström P. Beta-cell function in obese-hyperglycemic mice [ob/ob Mice] Adv Exp Med Biol. 2010;654:463–477. doi: 10.1007/978-90-481-3271-3_20. [DOI] [PubMed] [Google Scholar]
- 27.Plaisance V, Brajkovic S, Tenenbaum M, et al. Endoplasmic reticulum stress links oxidative stress to impaired pancreatic beta-cell function caused by human oxidized LDL. PLoS ONE. 2016 doi: 10.1371/journal.pone.0163046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan JY, Luzuriaga J, Bensellam M, et al. Failure of the adaptive unfolded protein response in islets of obese mice is linked with abnormalities in β-cell gene expression and progression to diabetes. Diabetes. 2013;62:1557–1568. doi: 10.2337/db12-0701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cnop M, Toivonen S, Igoillo-Esteve M, Salpea P. Endoplasmic reticulum stress and eIF2α phosphorylation: the Achilles heel of pancreatic β cells. Mol Metab. 2017 doi: 10.1016/j.molmet.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Douziech M, Grondin G, Loranger A, et al. Zonal induction of mixed lineage kinase ZPK/DLK/MUK gene expression in regenerating mouse liver. Biochem Biophys Res Commun. 1998;249:927–932. doi: 10.1006/bbrc.1998.9249. [DOI] [PubMed] [Google Scholar]
- 31.Kragelj J, Palencia A, Nanao MH, et al. Structure and dynamics of the MKK7-JNK signaling complex. Proc Natl Acad Sci USA. 2015;112(11):3409–3414. doi: 10.1073/pnas.1419528112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.