

Abstract
There is significant contemporary interest in the application of enzymes to replace or augment chemical reagents toward the development of more environmentally sound and sustainable processes. In particular, copper radical oxidases (CRO) from Auxiliary Activity Family 5 Subfamily 2 (AA5_2) are attractive, organic cofactor-free catalysts for the chemoselective oxidation of alcohols to the corresponding aldehydes. These enzymes were first defined by the archetypal galactose-6-oxidase (GalOx, EC 1.1.3.13) from the fungus Fusarium graminearum. The recent discovery of specific alcohol oxidases (EC 1.1.3.7) and aryl alcohol oxidases (EC 1.1.3.47) within AA5_2 has indicated a potentially broad substrate scope among fungal CROs. However, only relatively few AA5_2 members have been characterized to date. Guided by sequence similarity network and phylogenetic analysis, twelve AA5_2 homologs have been recombinantly produced and biochemically characterized in the present study. As defined by their predominant activities, these comprise four galactose 6-oxidases, two raffinose oxidases, four broad-specificity primary alcohol oxidases, and two non-carbohydrate alcohol oxidases. Of particular relevance to applications in biomass valorization, detailed product analysis revealed that two CROs produce the bioplastics monomer furan-2,5-dicarboxylic acid (FDCA) directly from 5-hydroxymethylfurfural (HMF). Furthermore, several CROs could desymmetrize glycerol (a by-product of the biodiesel industry) to d- or l-glyceraldehyde. This study furthers our understanding of CROs by doubling the number of characterized AA5_2 members, which may find future applications as biocatalysts in diverse processes.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-021-03981-w.
Keywords: Copper radical oxidase, Alcohol oxidase, Galactose oxidase, Biocatalysis, Auxiliary Activity Family 5
Introduction
Global climate change due to human activities is a pressing issue confronting our society (https://climate.nasa.gov/scientific-consensus/) [1]. One avenue to decrease reliance on petroleum is to increase the utilization of plant biomass as a renewable carbon feedstock [2, 3]. Plant biomass is an abundant source of complex polysaccharides, polyaromatics (i.e., lignin), and low molecular weight extractives (e.g., lipids, terpenes, etc.), each of which can potentially be valorized via (bio)catalytic routes. Foremost, the development of the so-called “biofuels,” e.g., ethanol fermented from sugar feedstocks, has received the widest attention, but faces certain economic and scalability challenges [4–6]. In parallel, there is a growing appreciation that upgrading plant biomass components to produce high-value biochemicals and biomaterials may provide solutions to lower reliance on fossil petroleum beyond the energy sector [7–11].
The utilization of plant biomass poses many challenges due to inherent chemical and structural complexity, which make fractionation and chemical transformation difficult. Yet, a wide diversity of microorganisms have developed specialized biocatalytic systems of hydrolytic and oxidative enzymes to unlock this natural resource [12–22]. Informed by decades of fundamental biochemical research [23–26], industrial enzyme “cocktails” have been developed for complete saccharification of plant biomass, especially starch and cellulose fractions, for fermentation to biofuels [27–29]. Spurred by a growing track record of successful applications and significant industrial infrastructure for scalable production, enzyme-based biocatalysis is enjoying a period of sustained and growing interest [30–32]. In particular, there is strong demand to replace chemical oxidants, many of which produce hazardous waste, with “greener” alternatives, making the study of oxidative enzymes attractive in this context [14, 32].
One class of oxidases that has shown promise for the valorization of plant biomass is the copper radical oxidases (CROs) that comprise Auxiliary Activity Family 5 (AA5) in the Carbohydrate-Active Enzymes (CAZy) classification [17]. These enzymes contain a mono-nuclear copper radical center coordinated by a distinct crosslinked tyrosine-cysteine active-site residue, which catalyzes two one-electron oxidations with concomitant reduction of oxygen to hydrogen peroxide [33, 34]. Within AA5, two subfamilies have been identified: Characterized subfamily 1 (AA5_1) enzymes have predominant activity on glyoxal and methylglyoxal (GlyOx, EC 1.2.3.15) [35]. Subfamily 2 (AA5_2) contains the archetypal CRO, galactose 6-oxidase (GalOx, EC 1.1.3.9) [33, 36], as well as the more recently discovered general alcohol oxidases (AlcOx, EC 1.1.3.13) [37, 38], and aryl alcohol oxidases (AAO, EC 1.1.3.7) [39, 40]. Both AA5_1 and AA5_2 have been speculated to be involved in the oxidative degradation of lignocellulose [34]; however, recently, AA5_2 alcohol oxidases have been linked to oxidizing plant cuticular long-chain alcohols into aldehyde products during fungal infection [41].
The archetypal galactose oxidase from the phytopathogenic fungus Fusarium graminearum (FgrGalOx) and its engineered variants have found widespread biotechnological applications, including in glycoprotein labeling, biosensor development, polysaccharide functionalization, and other applications involving the oxidation of carbohydrates or other alcohols [42–54]. Of particular note, a highly evolved variant of FgrGalOx was utilized recently in the biocatalytic synthesis of the HIV drug islatravir [55]. Furthermore, CgrAlcOx has been demonstrated to be a promising green catalyst for the oxidation of long-chain aliphatic alcohols for the production of flavor and fragrance compounds [56], whereas CgrAAO has been shown to be effective at oxidizing HMF, which is important in the context of biopolymer manufacturing [39, 57, 58]. Thus, there is sustained interest to expand the utilization of CROs industrially, through the discovery of homologs with inherently diverse catalytic specificities [14, 59, 60].
However, very few (currently thirteen) CROs from AA5_2 have been biochemically characterized [37–40, 61–66], the majority of which originate from two fungal genera: Fusarium [33, 40, 63–65] and Colletotrichum [37–39, 62] (for an updated list see http://www.cazy.org/AA5_characterized.html, [17]). Nonetheless, recent studies have revealed homologs in these fungi with distinct specificities for alkyl and aryl alcohols, indicating that the large sequence space of uncharacterized AA5 members presents an opportunity to uncover new biocatalysts [37, 39, 40, 60]. Inspired by these results, we were keen to continue mapping activities across the AA5_2 subfamily, drawing upon a growing array of new fungal genomes.
Hence, in the present study we have combined molecular phylogeny and sequence similarity network analysis to guide the selection of 40 AA5_2 members from diverse fungi for recombinant expression in the yeast Pichia pastoris. Fourteen of these were successfully produced and twelve were fully biochemically characterized, revealing new galactose 6-oxidases, raffinose oxidases, broad-specificity primary alcohol oxidases, and non-carbohydrate alcohol oxidases.
Results
Phylogeny of the AA5_2 subfamily
A manually curated multiple protein sequence alignment of over 623 AA5_2 catalytic modules, with signal peptides and additional domains removed, was used to generate a Maximum Likelihood (ML) phylogeny and a sequence similarity network (SSN) (Fig. 1). Using AA5_1 glyoxal oxidases [67, 68] as an outgroup, 38 subgroups were identified based on tree topology and generally high supporting bootstrap values (Fig. 1A). As indicated, the previously characterized AA5_2 members are broadly dispersed in the tree, with some enzymes clustered into more well-represented clades. Specifically, GalOxs are located in clade 15, general AlcOxs cluster into clade 26, while AAOs are found in clade 14 and clade 29 (Fig. 1). The corresponding SSN at an alignment score (bit score) cut-off of 550 was consistent with the grouping observed in the phylogenetic tree and subsequently used to aid sequence selection and mapping of biochemical data (Fig. 1B). In Table 1 and subsequently in the text, the targets are named according to their predominant activities as determined by subsequent kinetic assays (vide infra).
Fig. 1.

Sequence relationships of 623 AA5_2 catalytic modules. a Phylogenetic tree with bootstrap values supporting the 38 subgroups are indicated at each node/branch. For branches exhibiting low bootstrap values (< 50), values supporting the nodes are indicated. b Sequence similarity network (SSN). Each node corresponds to one of the 623 catalytic modules used as an input to build the SSN created in Cytoscape with yFiles Organic layout [104]. Edges represent an alignment bit score threshold of 550, which clusters the sequences into groups analogous to those observed in (a) and color coded accordingly. In both panels, AA5 members with previously available biochemical data are colored in black and indicated as (methyl)glyoxal oxidases (UmaGlyOx [67], PchGlyOx1 and PchGlyOx2 [68], PciGlyOx1 and PciGlyOx2 [110]) (comprising the outgroup), galactose oxidases (FgrGalOx [66, 73, 92], FoxGalOx [65], FsaGalOx [64], FsuGalOx [63], FveGalOx, and FsuGalOx [111]), alcohol oxidases (CgrAlcOx and CglAlcOx [37], ChiAlcOx and PorAlcOx [38], raffinose oxidase (CgrRafOx) [62], AA5_2 oxidase (PruAA5_2A (PruAlcOx)) [61], and the aryl alcohol oxidase (CgrAAO) [39]. Enzymes first characterized in the present study are colored in neon green. Two selected sequences for which characterization was published contemporaneously, PruAA5_2A (PruAlcOx) [61] and PorAlcOx [38], are colored in grey
Table 1.
Key amino acid residues in AA5_2 proteins compared to other characterized members

Non-conserved amino acids in relation to FgrGalOx are bolded. Fungal species: Fgr Fusarium graminearum, Cgr Colletotrichum graminicola, Exe Exophiala xenobiotica, Mre Mytilinidion resinicola, Fox Fusarium oxysporum, Afl Aspergillus flavus, Pru Penicillium rubens, Pfe Penicillium fellutanum, Por Pyricularia oryzae, Cgl Colletotrichum gloesporoides , Uma Ustilago maydis, Phu Pseudozyma hubeiensis, Asy Aspergillus sydowii, Sto Stagonospora sp, Cca Corynespora cassiicola. Modularity: Carbohydrate-binding domain (CBM), catalytic domain (CAT), PAN_1 (PF00024), WSC (PF01822), unknown domain (UNK)
Enzymes previously characterized in aref [36, 92], bref [37], cref [39], dref [40]
To further inform target selection, we mapped the variability of the amino acids involved in catalysis and substrate recognition, as determined by mutagenesis and/or structural analysis of AA5_2 members [69–77], onto the SSN (Table 1, Table S1 Figure S1). Thus, 40 targets were selected to represent the maximum diversity of sequence identity and fungal ecology, such as saprotrophic and phytopathogenic members, in this sample size (Table 1 and Figure S1).
Protein production and initial biochemical characterization
cDNAs corresponding to the full-length sequence of 40 candidate enzymes, i.e., including any carbohydrate-binding modules (CBM [78]), PAN_1 domains (PFAM, PF00024), WSC domains (PFAM, PF01822), or unknown domains (UNK) in tandem with the catalytic AA5_2 module, were cloned into the pPICZα-A or pPICZα-C vectors in-frame with the Saccharomyces cerevisiae α-factor secretion signal and a C-terminal hexa-histidine tag. Fourteen proteins were thus produced successfully in Pichia pastoris X33 and purified via Immobilized Metal Affinity Chromatography (IMAC) followed by buffer exchange using size exclusion chromatography and SDS-PAGE analyses (Fig. S2). All proteins were obtained in full-length form except FoxGalOxB (species abbreviations were previously defined in Table 1), for which a truncated construct was purified. Furthermore, enzymatic deglycosylation indicated that AflAlcOx, PruAA5_2A (PruAlcOx), ExeGalOx, and PfeGalOx were N-glycosylated (Fig. S2). Typical protein yields varied between 2.2 and 65 mg L−1 based on the original volume of buffered complex methanol medium (Table S3). Overall, protein production levels were comparable to previous AA5_2 preparations from P. pastoris, e.g., FgrAAO and FoxAAO (2.5 mg L−1) [40], FoxGalOx (10.6 mg L−1) [65], CgrAlcOx (30–40 mg L−1) [37], CgrAAO (43 mg L−1) [39], and the wild-type and M1 variants of FgrGalOx (120 mg L−1 and 110 mg L−1, respectively [79]).
The fourteen successfully produced recombinant proteins were initially screened for activity using three compounds representing efficient substrates of previously characterized AA5_2 members, viz., galactose and raffinose for galactose 6-oxidases and benzyl alcohol for general alcohol oxidases (Table S4). As expected, a range of specificities was observed, with some enzymes clearly displaying higher specific activity on benzyl alcohol (AflAlcOx and CglAlcOx) or galactosyl moieties (FoxGalOxB, MreGalOx, and ExeGalOx), while for others this preference was equivocal (PfeGalOx and PruAA5_2A (PruAlcOx). Notably, several targets had relatively low specific activities on all of the three substrates tested: UmaRafOx, PhuRafOx, CcaAA5, AsyAlcOx, FoxAlcOx, PorAlcOx, and StoAA5. P. pastoris constructs of StoAA5 and CcaAA5 produced soluble protein but the specific activities of these enzyme were not consistent between productions; hence, no further work was conducted on these two homologs.
Based on the initial activity screen results, either galactose or benzyl alcohol was chosen as the substrate for the determination of the pH-rate profile and temperature stability for each target. Exceptionally, glycerol was used as substrate for UmaRafOx and PhuRafOx following further substrate screening (specific activities > 3.0 µmol min−1 mg−1) since these enzymes displayed very low specific activities on the three initial substrates (< 0.25 µmol min−1 mg−1). All recombinant enzymes were active between pH 6.0 and 8.5 (Table 2) and most exhibited bell-shaped pH-rate profiles with optima between 7.0 and 8.5 in sodium phosphate buffer (Fig. S5), akin to other characterized AA5_2 enzymes [37, 39, 64, 65, 80]. Distinctly, MreGalOx and ExeGalOx had optimum values at pH 6.0 and 6.5, respectively, while UmaRafOx and PhuRafOx were most active in glycine–NaOH buffer at pH 8.5.
Table 2.
Initial rate kinetics of active AA5_2 enzymes*
| Enzyme | Substrate | KM (mM) | kcat (s−1) | kcat/KM (M−1.s−1) | Condition |
|---|---|---|---|---|---|
| ExeGalOx | Galactose | 25 ± 4.0 | 130 ± 3.7 | (5.2 ± 0.8) × 103 | pH 6.0, 49 °C |
| Melibiose | 25 ± 0.6 | 37 ± 0.2 | (1.5 ± 0.03) × 103 | ||
| MreGalOx | Galactose | 64 ± 4.8 | 32 ± 0.7 | (5.0 ± 0.4) × 102 | pH 6.0, 55 °C |
| Melibiose | n.d.§ | n.d.§ | (1.8 ± 0.8) × 102 | ||
| FoxGalOxB | Galactose | 24 ± 1.3 | 81 ± 1.0 | (3.4 ± 0.2) × 103 | pH 8.0, 39 °C |
| Melibiose | n.d.§ | n.d.§ | (5.9 ± 1.7) × 103 | ||
| PfeGalOx | Glycerol | 69 ± 1.6 | 140 ± 1.1 | (2.0 ± 0.1) × 103 | pH 8.5, 30 °C |
| Galactose | 28 ± 6.7 | 140 ± 4.5 | (5.0 ± 1.2) × 103 | ||
| UmaRafOx | Glycerol | 2900 ± 720 | 130 ± 24 | 45 ± 14 | pH 8.5, 30 °C |
| Raffinose | 450 ± 94 | 130 ± 15 | 290 ± 69 | ||
| PhuRafOx | Glycerol | 2900 ± 390 | 56 ± 3.8 | 19 ± 2.9 | pH 8.5, 30 °C |
| Raffinose | 410 ± 33 | 8.8 ± 0.5 | 21 ± 2.1 | ||
| AflAlcOx | Benzyl Alcohol | 19 ± 1.9 | 410 ± 11 | (2.1 ± 0.2) × 104 | pH 8.5, 39 °C |
| HMF | 5.0 ± 1.3 | 190 ± 20 | (3.8 ± 1.1) × 104 | ||
| Galactose | 980 ± 61 | 400 ± 10 | (4.1 ± 0.3) × 102 | ||
| PruAA5_2A (PruAlcOx) | Glycerol | 34 ± 1.7 | 170 ± 3.0 | (4.9 ± 0.3) × 103 | pH 7.5, 35 °C |
| Galactose | 110 ± 18 | 220 ± 8.6 | (2.0 ± 0.3) × 103 | ||
| Benzyl Alcohol | 9.2 ± 1.8 | 110 ± 5.0 | (1.2 ± 0.2) × 104 | ||
| FoxAlcOx | Benzyl Alcohol | 230 ± 70 | 24 ± 4.5 | 100 ± 37 | pH 8.5, 45 °C |
| Galactose | 2000 ± 650 | 8.3 ± 1.7 | 4.1 ± 1.6 | ||
| PorAlcOx | Benzyl Alcohol | 1.3 ± 0.10 | 8.6 ± 0.15 | (6.6 ± 0.5) × 103 | pH 7.5, 30 °C |
| Butanol | 4.6 ± 0.30 | 7.9 ± 0.09 | (1.7 ± 0.1) × 103 | ||
| Glycerol | 57 ± 8.1 | 8.5 ± 0.32 | (1.5 ± 0.2) × 102 | ||
| 1,4-Butanediol | 4.3 ± 0.56 | 9.5 ± 0.51 | (2.2 ± 0.3) × 103 | ||
| CglAlcOx | Benzyl Alcohol | 3.6 ± 0.8 | 350 ± 20 | (9.7 ± 2.2) × 104 | pH 8.0, 30 °C |
| HMF | 0.09 ± 0.01 | 240 ± 16 | (2.7 ± 0.3) × 106 | ||
| 1,4-Butanediol | 3.9 ± 0.4 | 330 ± 13 | (8.5 ± 0.9) × 104 | ||
| AsyAlcOx | Benzyl Alcohol | 79 ± 22 | 17 ± 1.8 | (2.2 ± 0.6) × 102 | pH 8.0, 30 °C |
| Glycerol | 700 ± 80 | 38 ± 2.0 | 54 ± 6.8 |
*Error bars represent the standard deviation from the mean values
§ Individual KM and kcat values not determinable; kcat/KM values obtained from slope of linear v0 versus [S] plots (see panels B in Figs. S7–S18)
Most AA5_2 enzymes were stable at 30 °C, similar to some previously characterized members [37, 39]. Three enzymes (FoxGalOxB, AflAlcOx and PruAA5_2A (PruAlcOx)) were stable between 35 and 40 °C, similar to FoxGalOx (40 °C) [65], and three enzymes (ExeGalOx, MreGalOx and FoxAlcOx) exhibited higher thermostabilities than previously characterized CROs (45 °C–55 °C) (Table 2 and Fig. S6). Cumulatively, these results are consistent with the mesophilic nature of their natural hosts. Due to instrument limitations, the substrate screening was performed at ambient temperature, while subsequent Michaelis–Menten kinetic assays were performed at temperatures that balanced optimal enzyme activity with short-term stability.
Extended substrate screening and Michaelis–Menten kinetics
After establishing preferred assay parameters for the 12 demonstrably active AA5_2 targets, these enzymes were screened in parallel, using a multi-well plate assay, against a broader panel of primary alcohol-containing substrates comprising carbohydrates, alkanols, and aryl alcohols with relevance to biomass utilization, pharmaceutical preparation, or biomaterial synthesis (Fig. 2). Based upon the outcome of this extended screen, key substrates were selected based on specific activity values for detailed Michaelis–Menten kinetic analysis (Table 2 and Fig. S7–S18). Taken together, these assays enabled us to group enzymes by their predominant activities, which are discussed below in the context of their molecular phylogeny and SSN clustering.
Fig. 2.

Extended activity screening of AA5_2 enzymes. Measurements were performed in triplicate at room temperature in 100 mM buffer, using the HRP/ABTS assay. Activities were monitored using 300 mM for carbohydrates, polyols, diols, and primary alcohols, 30 mM for benzyl alcohol and galactitol, 2.5 mM for aldehydes, aryl alcohols, and furans. Reactions were started with the addition of 30 pmol–0.26 nmol of purified enzyme
Galactose 6-oxidases
Enzymes in this group predominantly possess the canonical galactose 6-oxidase activity (EC 1.1.3.9) that first defined the CRO family [81–83]. As such, they have minimal to no activity on other polyols, diols, and other non-carbohydrate primary alcohols. The activity profile of these enzymes is comparable to the archetype, FgrGalOx [59, 84].
ExeGalOx
ExeGalOx is located alone within clade 19 in the phylogenetic tree and it possesses low sequence similarity to its closest homologs FoxGalOxB and the archetypal FgrGalOx (40% and 47%, respectively) (Fig. 1 and Table S2). ExeGalOx displayed moderate specific activity on galactose and showed lower but equivalent specific activities on raffinose and melibiose; hence, we categorized this enzyme as a galactose oxidase (EC 1.1.3.9) (Table S5 and Fig. S7). Compared to FgrGalOx, ExeGalOx had a threefold lower catalytic efficiency on galactose due to a lower kcat even though its KM was fourfold lower (Tables 2, S11, and Fig. S7). In addition, ExeGalOx had a threefold lower catalytic efficiency on melibiose compared to galactose, which reflects a lower turnover rate since the KM for both substrates is the same (Table 2 and Fig. S7).
MreGalOx
MreGalOx also is located alone in clade 3 and shares low sequence identity with the closest neighboring homologs: 35% vs. AsyAlcOx and 39% vs. AflAlcOx (Fig. 1 and Table S2). MreGalOx exhibited low production yields and lower activity compared to other enzymes presented in this study. No activity above the limit of detection of our assay (9 × 10–4 µmol min−1 mg−1 using 0.85 µM of purified enzyme) was observed with many substrates. Nonetheless, MreGalOx demonstrated low specific activity on galactose and comparable specific activities on raffinose and melibiose; thus, we designated this enzyme as a galactose oxidase (EC 1.1.3.9) (Fig. S8 and Table S5). Compared to FgrGalOx, MreGalOx had a 20-fold lower catalytic efficiency on galactose (Tables 2, S11, and Fig. S8). Comparing its activity on melibiose versus galactose, MreGalOx had a threefold higher catalytic efficiency on melibiose; however, KM and kcat parameters could not be reliably determined due to apparent substrate inhibition (Table 2 and Fig. S8).
FoxGalOxB
FoxGalOxB is located alone in clade 16 next to clade 15, which contains the archetypal FgrGalOx and other characterized galactose 6-oxidases (Fig. 1). Only 40% identity is shared between FoxGalOxB and FgrGalOx, despite the close phylogenetic association (Table S2). The full substrate profile of FoxGalOxB led us to designate it as a galactose oxidase (EC 1.1.3.9). Since a galactose oxidase has already been characterized in Fusarium oxysporum [65], we have named this homolog FoxGalOxB [85]. FoxGalOxB showed high specific activity on galactose and melibiose, followed by raffinose and then lactose, whereas low specific activity was observed on polyols, diols, and primary and aryl alcohols (Fig. S9 and Table S6). In addition, FoxGalOxB showed low specific activity on both HMF and HMFCA (3.3 µmol min−1 mg−1), low specific activity on DFF, and no activity on FFCA (Fig. S9 and Table S6). Compared to the archetypal FgrGalOx, FoxGalOxB had a threefold lower catalytic efficiency on galactose (Tables 2, S11, and Fig. S9). Substrate inhibition was also observed on melibiose with FoxGalOxB and an overall twofold decrease in catalytic efficiency compared to galactose was found (Table 2 and Fig. S9).
PfeGalOx
PfeGalOx is located in clade 8, which also contains PruAA5_2A (PruAlcOx) (56% identity) and is located next to the clade containing AflAlcOx (51% identity) (Fig. 1 and Table S2). Analogous to FoxGalOxB, PfeGalOx displayed high specific activity on galactose; however, other galactosylated carbohydrates showed moderate specific activity, with its highest activity observed on raffinose, followed by lactose, and lastly melibiose (Fig. S10 and Table S6). Interestingly, PfeGalOx also showed prominent specific activity on xylose (Fig. S11 and Table S6), which is a novel activity among CROs. Furthermore, PfeGalOx displayed high specific activity on glycerol, and moderate activities on benzyl alcohol, butanol, and 1,2-propanediol/1,3-propanediol, yet low activity on other primary alcohols (Fig. S10 and Table S6). Considering the full substrate profile of PfeGalOx, we have classified this enzyme as a galactose oxidase (EC 1.1.3.9). PfeGalOx had a twofold lower catalytic efficiency on galactose than FgrGalOx [72] attributable to both an eightfold decrease in kcat and a fourfold decrease in KM (Tables 2, S11, and Fig. S10). Furthermore, PfeGalOx showed a twofold higher catalytic efficiency on glycerol than CgrAlcOx [37]; however, it had a 2.5-fold higher catalytic efficiency on galactose compared to glycerol (Tables 2, S12, and Fig. S10).
Raffinose oxidases
Raffinose oxidases (EC 1.1.3.-), from the perspective of specificity, can be considered as a subclass of the galactose 6-oxidases, because these enzymes oxidize the hydroxyl group located on the carbon 6 of the galactosyl residue of this trisaccharide. Nonetheless, these enzymes exhibit a distinct specificity for raffinose over galactose and other galactosides, as has been demonstrated for CgrRafOx [62]. These enzymes are also comparably proficient at oxidizing glycerol, some diols, and glycolaldehyde dimer.
UmaRafOx
UmaRafOx is located in a distinct clade (clade 30; Fig. 1 and Table S2), which also contains PhuRafOx (81% identity) and the previously characterized CgrRafOx (43% identity). UmaRafOx showed low specific activity on glycerol, compared to other enzymes in the present study. Nonetheless, this polyol is the substrate for which the highest specific activity was observed for this enzyme (Fig. S11 and Table S7). UmaRafOx showed an overall low specific activity on carbohydrates; however, the substrate for which UmaRafOx had the second highest specific activity was raffinose (Fig. S12 and Table S7). UmaRafOx also displayed a similar specific activity to raffinose on glycolaldehyde dimer; however, no activity on methyl glyoxal was observed (Fig. S12 and Table S7). UmaRafOx had a 20-fold lower catalytic efficiency on glycerol compared to CgrAlcOx [37], and a 15-fold higher catalytic efficiency on glycerol compared to CgrRafOx (Tables 2 and S12). The catalytic efficiency of UmaRafOx on raffinose is sixfold higher than CgrRafOx [62] (Table 2). Based on these kinetic results, we designated UmaRafOx as a raffinose oxidase (EC 1.1.3.-).
PhuRafOx
PhuRafOx has a substrate profile similar to UmaRafOx, displaying high activity on glycerol followed by raffinose (Fig. S12 and Table S7). PhuRafOx also showed low equivalent specific activities on glycolaldehyde dimer and 1,3-propanediol (Fig. S12 and Table S7). PhuRafOx had a 50-fold lower catalytic efficiency on glycerol compared to CgrAlcOx [37], and a sixfold higher catalytic efficiency on glycerol compared to CgrRafOx (Tables 2 and S12). The catalytic efficiency of PhuRafOx on raffinose is twofold lower than CgrRafOx; however, considering the fully distinct substrate profile we classified PhuRafOx as a raffinose oxidase (EC 1.1.3.-).
Broad-spectrum alcohol oxidases
Enzymes in this group exhibit a broad catalytic range, i.e., a lack of selectivity, for carbohydrates, including galactose and galactosides (EC 1.1.3.9), linear alcohols, including diols and polyols (EC 1.1.3.13), as well as diverse aryl alcohols, including benzyl alcohols and furan derivatives (EC 1.1.3.7 and EC 1.1.3.47). At the same time, these enzymes demonstrate distinct substrate preferences within these compound classes. The activity profiles of these enzymes are reminiscent of FgrAAO[40] and CgrAAO[39].
AflAlcOx
AflAlcOx is located alone in clade 7 in the phylogenetic tree and shares moderate sequence identity with its closest homologs PfeGalOx and PruAA5_2A (PruAlcOx) (51% and 48%, respectively) (Fig. 1 and Table S2). AflAlcOx showed high specific activity on benzyl alcohol and moderate specific activities on galactose, glycerol, diols, butanol, HMF, and other aryl alcohols (Fig. S13 and Table S8). Due to this broad range of activities, we have designated AflAlcOx as a general alcohol oxidase (EC 1.1.3.13 and EC 1.1.3.7). AflAlcOx showed a sevenfold lower catalytic efficiency compared to CgrAlcOx on benzyl alcohol (Table S12) and showed a higher catalytic efficiency on HMF than CgrAAO, bacterial HMFO (a Glucose-Methanol-Choline (GMC) superfamily oxidoreductase) and FgrAAO (twofold, fivefold, and 20-fold, respectively) (Table S13). Even though AflAlcOx displayed prominent specific activity on galactose, its catalytic efficiency on benzyl alcohol was 50-fold higher than on galactose (Table 2 and Fig. S13). Furthermore, AflAlcOx had a 25-fold lower catalytic efficiency on galactose than the archetypal FgrGalOx, due to a tenfold increase in KM and a threefold decrease in kcat compared to FgrGalOx (Table S11).
PruAA5_2A (PruAlcOx)
PruAA5_2A (PruAlcOx) is located in the same clade as PfeGalOx (56% identity) and shares 48% identity with AflAlcOx located in the neighboring clade (Fig. 1 and Table S2). PruAA5_2A (PruAlcOx) displayed high specific activity on glycerol, moderate activities on 1,2-propanediol, and 1,3-propanediol and showed decreased specific activities with increasing chain length on other diols (Fig. S14 and Table S8). PruAA5_2A (PruAlcOx) also showed high specific activity on galactose, but low activities were observed for other carbohydrates. Furthermore, PruAA5_2A (PruAlcOx) showed high specific activity on benzyl alcohol and moderate specific activity on HMF with lower activity observed on HMFCA, very low activity on DFF, and no activity on FFCA (Fig. S14 and Table S8). Based on this wide substrate scope, we have classified PruAA5_2A (PruAlcOx) as a general alcohol oxidase (EC 1.1.3.13 and EC 1.1.3.7). PruAA5_2A (PruAlcOx) displayed catalytic efficiencies fivefold higher on glycerol and tenfold lower on benzyl alcohol than CgrAlcOx [37] (Table S12). Despite PruAA5_2A (PruAlcOx) possessing a high specific activity on galactose, similar to AflAlcOx, the catalytic efficiency of PruAA5_2A (PruAlcOx) on benzyl alcohol was 25-fold higher than that on galactose (Table S11). PruAA5_2A (PruAlcOx) has a similar KM value on galactose and shows a fivefold lower turnover rate compared to FgrGalOx (Table S11).
During the course of the present study PruAA5_2A (PruAlcOx) was characterized as exhibiting an activity profile combining those of AA5_1 and AA5_2 members [61]. Here, we observed generally higher activities on all tested substrates and a different specificity profile. In our hands, PruAA5_2A (PruAlcOx) possessed a higher specific activity on galactose rather than raffinose [61] (ninefold) and the highest specific activity was observed on glycerol rather than the glycolaldehyde dimer [61] (twofold) at the equivalent concentrations as the previous study. Furthermore, our enzyme exhibited substantial specific activity on benzyl alcohol (84 µmol min−1 mg−1 at 30 mM), while the previous study did not observe any activity on this substrate at 300 mM [61]. The reasons for these discrepancies are unclear, but we note that P. pastoris SMD1168H and fed-batch production were used previously vs. P. pastoris X33 shake-flask production here.
FoxAlcOx
FoxAlcOx is located in the same clade as the recently characterized FgrAAO and FoxAAO (clade 14) (67% identity) (Fig. 1 and Table S2). This clade is located beside the classical galactose oxidases, and FoxAlcOx shares 58% identity with FgrGalOx. FoxAlcOx has generally low specific activities on all substrates tested and displayed the lowest specific activity out of all the enzymes presented in this study (Fig. S15 and Table S9). FoxAlcOx showed the highest specific activity on benzyl alcohol, while the second highest specific activities were observed on galactose and the aldehyde compounds, i.e., methyl glyoxal and glycolaldehyde dimer (Fig. S15 and Table S9). The catalytic efficiency of FoxAlcOx on benzyl alcohol is 1400-fold lower than CgrAlcOx [37] and fourfold lower than FgrGalOx[86] (Table S12). Due to this broad substrate range, which includes carbohydrates, aryl alcohols, and aldehydes (which likely react with the enzyme in their hydrated forms [34, 35]), we have classified FoxAlcOx as a general alcohol oxidase (EC 1.1.3.13 and EC 1.3.3.7). Since the second highest specific activity for FoxAlcOx was observed on galactose, Michaelis–Menten kinetics were performed for this substrate and showed that FoxAlcOx displayed a 2500-fold lower catalytic efficiency on galactose than FgrGalOx (Table S11).
PorAlcOx
PorAlcOx is located in the same distinct clade as CglAlcOx (57% identity), CgrAlcOx (55% identity), and other previously characterized AlcOxs (clade 26) (Fig. 1 and Table S2). Similar to MreGalOx, PorAlcOx was produced with low yields (Table S3). Nevertheless, comparable moderate specific activities were measured on polyols, diols, primary alcohols, and aryl alcohols with notable activity on benzyl alcohol and HMFCA (Fig. S16 and Table S10). In contrast to AflAlcOx and PruAA5_2A (PruAlcOx), PorAlcOx showed higher activity on HMFCA versus HMF while also displaying low activities on DFF and FFCA (Fig. S16 and Table S10). The broad substrate profile of PorAlcOx is indicative of a general alcohol oxidase (EC 1.1.3.13) (Fig. S16 and Table S10). PorAlcOx had lower catalytic efficiencies compared to CgrAlcOx [37], i.e., kcat/KM values were 20-fold less on benzyl alcohol, 40-fold less on 1,4-butanediol, 80-fold less on butanol, and sixfold less on glycerol (Table 2 and S12). During the course of our study, PorAlcOx was characterized using ethanol, butanol, 1,3-butanediol, and glycerol as substrates. Although we did not obtain kinetic parameters on all of the aforementioned substrates, our catalytic efficiencies on butanol and glycerol are comparable to the previously reported values [38].
Non-carbohydrate alcohol oxidases
Enzymes in this group clearly appear to reject mono-, di-, and trisaccharides, notably including galactose and raffinose, in favor of alkanols and aryl alcohols (EC 1.1.3.7 and EC 1.1.3.47). At the same time, these enzymes are generally proficient on linear diols and polyols. The activity profiles of these enzymes are reminiscent of CgrAlcOx and CglAlcOx [37].
CglAlcOx
CglAlcOx is located in the same clade as PorAlcOx, CgrAlcOx, CglAlcOx, and ChiAlcOx (clade 26) (Fig. 1). Furthermore, it shares 79% identity with CgrAlcOx (Table S2) and 99% identity with CglAlcOx from C. gloesporoides-14/ Colletotrichum fructicola Nara gc5. The CglAlcOx characterized in this study is from C. gloesporoides-23 and contains a single point mutation compared to the previously characterized CglAlcOx [37]. CglAlcOx displays higher activity under acidic conditions, lower thermostability and the same catalytic efficiency on benzyl alcohol (Table 2, and Figs. S5, S6, S17) compared to its ortholog. CglAlcOx showed equally high specific activities on all diols, glycerol, primary alcohols, benzyl alcohols, and substituted aryl alcohols; hence, we characterized this enzyme as a general alcohol oxidase (EC 1.1.3.7 and EC 1.1.3.13) (Fig. S17 and Table S10). CglAlcOx had catalytic efficiencies comparable to CgrAlcOx [37] on benzyl alcohol and 1,4-butanediol and that are 50-fold and 40-fold higher, respectively, compared to CgrAAO [39] (Tables 2 and S12). Moreover, CglAlcOx displayed high specific activity on HMF while showing almost no activity on DFF and FFCA (Fig. S17 and Table S10). CglAlcOx had the highest reported catalytic efficiency to date on HMF with a 140-fold higher kcat/KM value than CgrAAO [39] and a 380-fold higher kcat/KM value than the bacterial HMFO [87] (a GMC superfamily oxidoreductase) (Table S13). As such, CglAlcOx may also be classified as an HMF oxidase (EC 1.1.3.47).
AsyAlcOx
AsyAlcOx is located alone in clade 6 and shares only 45% identity with AflAlcOx in the neighboring clade (Fig. 1 and Table S2). AsyAlcOx possessed moderate specific activities on aryl alcohols and diols, but notable specific activity on benzyl alcohol; thus, we designated this enzyme as a general alcohol oxidase (EC 1.1.3.13 and EC 1.3.3.7). AsyAlcOx showed low activity on diols while displaying enhanced specific activity with increased carbon chain length, culminating with 1,6-hexanediol (Fig. S18 and Table S9). Michaelis–Menten kinetics indicated that AsyAlcOx had over 600-fold and 17-fold lower catalytic efficiencies on benzyl alcohol and glycerol, respectively, than CgrAlcOx [37] (Tables 2, S11 and Fig. S18).
Application to bioproduct conversion
CROs from AA5 are finding increasing applications in the conversion of small molecules to valuable chemical intermediates [46, 47, 50, 55, 56, 59, 88, 89]. Among these, HMF from the thermochemical breakdown of plant biomass [57, 58] and glycerol, a by-product of the biodiesel industry [90], are of particular interest. On one hand, HMF can be converted into the bi-functional polymer precursors DFF and FDCA by controlled oxidation (Fig. 3a) [57, 58]. On the other hand, glycerol is an exemplar prochiral compound and its valorization through desymmetrization has garnered significant interest (Fig. 3b). Relatedly, an evolved variant of FgrGalOx was used for an essential desymmetrization of a key 2-ethynyl-glycerol intermediate in the biocatalytic cascade synthesis of the antiviral drug islatravir [55].
Fig. 3.

Oxidation pathways of HMF and Glycerol. a HMF oxidation pathway. The symbols next to each compound are used to denote the corresponding peaks in the NMR spectra comprising Figs. S19–S23. b Glycerol oxidation pathway showing l- and d-glyceraldehyde. [O] represents a generic oxidant
HMF
Enzymes with specific activities above 3 µmol min−1 mg−1 on any furan substrate were incubated with either 10 mM HMF, HMFCA, DFF, or FFCA, followed by direct quantitation of the product(s) by 1H NMR (Table 3). Previous work has shown that CgrAAO oxidized HMF to DFF, while FgrAAO produced a mixture of DFF and FFCA as the terminal products of HMF oxidation [39, 40].
Table 3.
Percent conversions of HMF, HMFCA, DFF, and FFCA by PruAA5_2A (PruAlcOx), AflAlcOx, CglAlcOx, PorAlcOx, and FoxGalOxB
| AA5 CRO | Substrate | |||
|---|---|---|---|---|
| HMF | HMFCA | DFF | FFCA | |
| FoxGalOxB | DFF (29%), FFCA (4%) | FFCA (17%) | FFCA (12%) | FDCA (7%) |
| AflAlcOx | DFF (52%), FFCA (48%) | FFCA (100%) | FFCA (40%) | 0% |
|
PruAA5_2A (PruAlcOx) |
DFF (52%), FFCA (48%) | FFCA (100%) | FFCA (38%) | FDCA (5%) |
| CglAlcOx | FFCA (91%), FDCA (9%) | FFCA (92%), FDCA (8%) | FFCA (87%), FDCA (13%) | FDCA (24%) |
| PorAlcOx | DFF (49%), FFCA (2%) | FFCA (83%), FDCA (17%) | FFCA (72%), FDCA (28%) | FDCA (49%) |
CglAlcOx showed 100% conversion of HMF to a 91:9 mixture of FFCA and FDCA, respectively (Table 3 and Fig. S19). Based on experiments with the HMF oxidation pathway intermediates HMFCA and DFF, CglAlcOx is able to oxidize both furans efficiently with the major product consisting of FFCA with some FDCA observed (Table 3 and Fig. S19). PorAlcOx, which belongs to the same cluster as CglAlcOx, showed incomplete conversion of 10 mM HMF producing predominantly DFF with 2% FFCA observed (Table 3 and Fig. S20). Interestingly, PorAlcOx showed full conversion of HMFCA and DFF to mostly FFCA, and partial conversion to FDCA was also observed (Table 3 and Fig. S20).
PruAA5_2A (PruAlcOx) oxidized 100% of 10 mM HMF to DFF (including its hydrated form) and FFCA(Table 3 and Fig. S21). AflAlcOx showed similar conversion percentages to PruAA5_2A (PruAlcOx) for HMF, HMFCA, and DFF (Table 3 and Fig. S22). However, no conversion was observed with FFCA for AflAlcOx. FoxGalOxB displayed low percent conversion of HMF and had a similar product profile to PruAA5_2A (PruAlcOx) (Table 3 and Fig. S23).
In previous work, the aryl oxidase FgrAAO, was shown to produce similar ratios of products during overnight incubations with furan-containing substrates compared to PruAA5_2A (PruAlcOx), AflAlcOx, and FoxGalOxB [40]. It was noted for FgrAAO, and other CRO enzymes, that perhaps the low degree of hydration of FFCA compared to DFF limits further conversion [87]. In contrast, the increased production of FDCA observed for both CglAlcOx and PorAlcOx may be caused by their active-site architecture being able to accommodate the aldehyde form (or its hydrate) of FFCA, which results in higher percent conversions to FDCA compared to other CROs.
Glycerol
It has been previously reported that the wild-type FgrGalOx and FgrAAO oxidize glycerol stereoselectively to l-glyceraldehyde, while CgrAlcOx produces the natural d-glyceraldehyde isomer [40, 91]. To explore the stereoselectivity of CROs in the present study, those with specific activities toward glycerol greater than 3 µmol min−1 mg−1 were incubated with 0.54 M glycerol for 24 h, followed by chiral HPLC analysis of the resulting 2,4-dinitrophenyl glyceraldehyde-hydrazones (Table 4 and Fig. S24).
Table 4.
Stereochemical outcome of glycerol oxidation by AA5 CROs
| AA5 CRO | Product | |
|---|---|---|
| L-glyceraldehyde hydrazone (%) | D-glyceraldehyde hydrazone (%) | |
| FgrGalOxa | 96 | 4 |
| CgrAlcOxa | 10 | 90 |
| FoxGalOxB | 79 | 21 |
| PfeGalOx | 71 | 29 |
| UmaRafOx | 84 | 16 |
| PhuRafOx | 82 | 18 |
| AflAlcOx | 88 | 12 |
| PruAA5_2A (PruAlcOx) | 96 | 4 |
| CglAlcOx | 3 | 97 |
| PorAlcOx | 11 | 89 |
| AsvAlcOx | 77 | 23 |
aEnzymes previously characterized in ref [40]
CglAlcOx and PorAlcOx showed a preference for producing d-glyceraldehyde with an er of 3:97 and 11:89 (l-:d-), respectively. This may be expected, since both share higher identity with CgrAlcOx (79% and 55%, respectively) than with FgrGalOx (38% and 34%) (Table S2). On the other hand, all of the other enzymes tested for the stereoselective oxidation of glycerol showed a strong preference for the formation of l-glyceraldehyde (Table 4 and Fig. S24). These enzymes all have higher identities to FgrGalOx (52–36%) than CgrAlcOx (33–23%) (Table S2), providing further evidence that the different active-site amino acid scaffolds of FgrGalOx and CgrAlcOx direct the stereoselective oxidation of achiral glycerol.
Discussion
Biocatalysis will play an increasingly important role in the development of a sustainable economy, and the discovery of novel enzymes, particularly oxidases, is essential to support this transition. Only recently it has been appreciated that the range of native catalytic activities found among the copper radical oxidases from Auxiliary Activity Family 5 Subfamily 2 members extends beyond the founding, classical galactose 6-oxidases [37–39]. In the present study, we have biochemically characterized twelve AA5_2 members dispersed across the AA5_2 phylogenetic tree, covering hitherto unexplored clades. These comprise four canonical galactose-6-oxidases (ExeGalOx, MreGalOx, FoxGalOxB, and PfeGalOx), two raffinose-specific galactosyl oxidases (UmaRafOx and PhuRafOx), four broad-spectrum alcohol oxidases (AflAlcOx, PruAA5_2A (PruAlcOx), FoxAlcOx, and PorAlcOx), and two alcohol oxidases that notably do not target carbohydrates (CglAlcOx and AsyAlcOx). Mapping these activities onto the current phylogenetic landscape of AA5_2 indicated that although a number of clades demonstrate conserved substrate specificities, such as clade 15 with GalOxs, clade 30 with RafOxs, and clade 26 with AlcOxs, galactosyl versus general alcohol oxidase activity was not monophyletic. However, detailed analysis of active-site residues, supported by structural homology modeling based on FgrGalOx (PDB ID 1GOF) [92] and computational docking of galactose (Figs. 4 and S25–S27), allows us to rationalize some structure–function relationships among the CROs in the absence of experimental tertiary structures of these homologs.
Fig. 4.
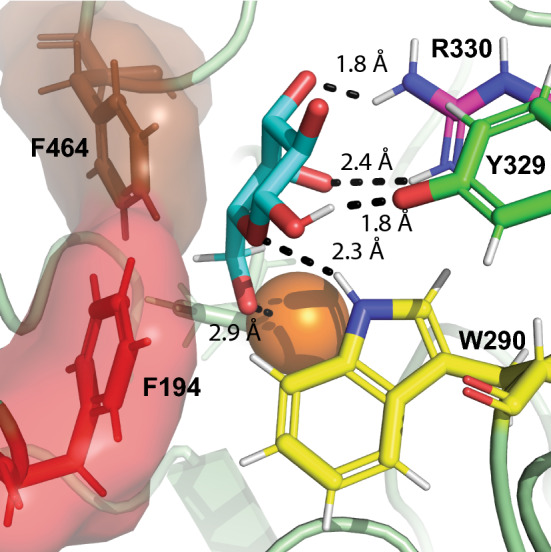
Molecular docking of galactose (cyan) into the experimental tertiary structure of FgrGalOx (PDB ID 1GOF) [92]. The copper atom is depicted as a dark orange sphere, while W290, Y329, R330, F494, and F194 are depicted as yellow, green, magenta, brown, and red sticks, respectively, including surface representation for F194 and F464. Ligand coordination is indicated in black
Four CROs displayed similar substrate profiles to the archetypal galactose oxidase; however, ExeGalOx, MreGalOx, and FoxGalOxB have a distinct difference with a phenylalanine at the position corresponding to Y329 in FgrGalOx (Table 1). Although this residue has been suggested to form a hydrogen bond with the 1-OH of galactose [77] (Fig. 4), ExeGalOx, MreGalOx, and FoxGalOxB all have lower KM values than the archetypal enzyme (Table S11), thus indicating that this tyrosine is dispensable to maintain efficient galactose binding. Computational docking suggests that other predicted hydrogen bonds to active-site arginine and tryptophan residues may be sufficient for galactose binding in these enzymes, such as observed in ExeGalOx (Fig. S25-B).
Consistent with this hypothesis, three broad-spectrum alcohol oxidases (FoxAlcOx, AflAlcOx and PruAA5_2A (PruAlcOx)) also showed prominent activity on galactose. Of these, the enzymes with KM values equal to, or lower than, FgrGalOx on galactose retained an arginine at the corresponding position (Arg330, Tables 1 and S10). This amino acid has been speculated to make two hydrogen bonds with the 3-OH and 4-OH of galactose [69], which is supported by docking analysis of FgrGalOx and ExeGalOx (Figs. 4 and S26-B). However, enzymes with a lysine (FoxAlcOx) or a histidine (AflAlcOx) at this position have higher KM values, possibly caused by the loss of hydrogen bonding interactions. FoxAlcOx possesses a lysine that only forms one predicted hydrogen bond to galactose (2.7 Å) (Fig. S26-C), and the limited activity of this homolog is similar to previously studied AA5_2 members with this substitution [40, 72] (Table S11). Likewise, the longer effective distance between the histidine and galactose modeled in AflAlcOx likely diminishes the potential of hydrogen bonding (Fig. S26-D). With the exception of CgrAAO [39] and AsyAlcOx, other enzymes with no activity on galactose feature a methionine or a tyrosine residue at the position corresponding to Arg330 in FgrGalOx (Table 1).
Another residue that appears to be important in determining galactose binding is F194, which together with F464 is speculated to form a hydrophobic wall in FgrGalOx [69]. Our models indeed support that this wall may interact with the comparatively hydrophobic bottom face (defined by the axial H-1, H-3, and H-5) of β-galactopyranoside, as well as the exocyclic CH2 group (Fig. 4). Enzymes with a substitution at the position corresponding to F194 in FgrGalOx and demonstrable galactose oxidase activity (> 60 µmol min−1 mg−1, viz., PfeGalOx, AflAlcOx, PruAA5_2A (PruAlcOx) with tyrosine and FoxGalOxB with methionine) all have lower kcat values and overall lower catalytic efficiencies, despite some having lower KM values (PfeGalOx and PruAA5_2A (PruAlcOx) (Table S11). Modeling of PfeGalOx, AflAlcOx, and PruAA5_2A (PruAlcOx) with galactose also suggests that the corresponding tyrosine can also form a hydrophobic wall with the bridging phenylalanine. However, this tyrosine is also computed to be within hydrogen bonding distance to the ring oxygen of galactose and, consequently, may orient the 6-OH further away from the copper center compared to FgrGalOx, thereby attenuating activity (Fig. S27-B–D).
Confoundingly, FoxGalOxB, has a methionine in the position corresponding to position 194 in FgrGalOx (Table 1). Despite the obvious disruption of the hydrophobic wall (Fig. S27-E), FoxGalOxB possesses predominant GalOx activity, albeit with a low turnover rate. Although this modeled complex precludes definitive analysis in the absence of site-directed mutagenesis and kinetic analysis of variants, it is notable that the low turnover rate observed for FoxGalOxB is matched by a low KM value. The same hydrogen bonds between galactose, arginine, and tryptophan indicated in other enzymes are maintained in FoxGalOxB, consistent with this observation (Table S11).
In addition to the obvious residues lining the active site pocket, W290 in FgrGalOx is a second shell amino acid which strongly affects catalysis in AA5_2 CROs. FgrGalOx W290 stacks with the tyrosine-cysteine crosslinked radical cofactor that is central to the mechanism of CROs, and potentially makes a hydrogen bond to the ring oxygen of galactose (Fig. 4). Mutagenesis of W290 significantly diminishes catalytic efficiency [73]. The homologs CgrAAO and CgrAlcOx possess, respectively, a tyrosine and a phenylalanine at this position, and site-directed mutagenesis of either to tryptophan modulates the relative activities toward carbohydrates versus other primary alcohols [37, 39]. On the other hand, the recently characterized FgrAAO and FoxAAO both possess a tryptophan at the same position, but yet have much higher catalytic efficiencies on aryl alcohols than galactose [40]. As such, the presence of this tryptophan as a galactose specificity determinant is somewhat ambiguous. However, we note that three general alcohol oxidases (PorAlcOx, CglAlcOx, and AsyAlcOx) and the two raffinose oxidases (UmaRafOx, PhuRafOx), all of which showed low or no activity on galactose (< 3 µmol min−1 mg−1) do not carry a tryptophan at this position. PorAlcOx, CglAlcOx, and AsyAlcOx all have a phenylalanine at this position, while the raffinose oxidases have a tyrosine (Table 1). Other enzymes in our study capable of galactose oxidation all have retained this tryptophan residue, like the archetypal FgrGalOx. In this context, it is notable that a CRO from Streptomyces lividans is poorly active on galactose, yet contains a corresponding secondary shell tryptophan whose presence is critical to the formation of the cysteine-tyrosine crosslink [93, 94]. In this enzyme, the indole ring of the tryptophan is oriented differently compared to FgrGalOx, which may affect specificity through electronic effects and/or altered hydrogen bonding with the substrate [93, 94].
The last amino acid that warrants comment is C383, which lies at the back of the active site pocket but does not directly interact with galactose in FgrGalOx. This residue has been shown previously to affect FgrGalOx catalytic efficiency on galactose [71], as its replacement with a serine increased the activity of the resulting mutant while an asparagine substitution decreased the activity tenfold [40, 71]. X-ray crystallography demonstrated that the C383S mutant of FgrGalOx participates in a H-bonded water network, stabilizing the backbone nitrogen between residues Tyr495 and His496 and possibly increasing its activity [74]. In our study, AflAlcOx, PruAA5_2A (PruAlcOx), and PfeGalOx have some of the highest catalytic efficiencies from all enzymes produced and they all natively have this C383S substitution (Table 1). Furthermore, MreGalOx has the same active site structure as ExeGalOx but displays a tenfold decreased catalytic efficiency on galactose (Table 2). Contrary to ExeGalOx, MreGalOx has an asparagine instead of a cysteine at the position corresponding to C383 in FgrGalOx which, due to a bulkier side chain, could disrupt the hydrogen bonding network, thereby lowering the activity of the enzyme.
Although the contributions of individual active-site amino acids to the specificity of AA5_2 CROs is not completely unambiguous, we can nonetheless discern some general trends based on kinetic observations for the complete set of enzymes characterized to date (based on FgrGalOx residue numbering): the presence of R330 is crucial for low KM values on galactose and galactosides, while its replacement generally favors non-carbohydrate oxidation. The neighboring amino acid, Y329, is not a key determinant of galactose oxidase binding/activity and a variety of amino acid residues are found at this position in CROs (Fig. S1-F). In contrast, F194 is important for efficient turnover rates toward galactose and although this amino acid is mostly conserved among AA5_2 sequences (Fig. S1-D), a majority of other sequences, regardless of carbohydrate or non-carbohydrate activity, contain an aromatic residue at this position likely to maintain the hydrophobic wall with F464. Lastly, the presence of W290 allows for prominent galactose oxidase activity but does not absolutely dictate enzyme specificity, possibly because spatial orientation of the indole ring in the secondary shell may be an addition contributor to substrate specificity [93, 94], which cannot be assessed on the basis of sequence alone.
Conclusion
A growing body of functional information on AA5_2 members, including the detailed biochemical characterization of twelve fungal orthologs in the present study, continues to reveal a surprisingly broad catalytic diversity within this family of CROs. In this context, we note that automated genome annotations often label AA5_2 members as “galactose oxidases” based on sequence similarity to the archetype FgrGalOx, which misrepresents the wider potential of these enzymes. Thus, such annotations should be viewed with caution, and we would generally advocate that these proteins should be more conservatively annotated as “copper radical oxidases” [34] until specificity can be reasonably established. We also note that the true physiological substrates of these enzymes are generally unknown, so specificities determined in vitro—including those here—may only be indicative. Nonetheless, it is revealing that AA5_2 comprises apparent galactose oxidases, raffinose oxidases, broad-spectrum alkyl- and aryl-alcohol oxidases, and non-carbohydrate alcohol oxidases of potential use in the specific biocatalytic transformation of both carbohydrate and non-carbohydrate substrates, including bioproducts, such as HMF and glycerol.
The replacement of chemical oxidants with redox enzymes is a topic of considerable contemporary interest, for which the palette of CROs constitutes an alternative to cofactor-dependent oxidases [60]. For example, CglAlcOx has the highest catalytic efficiency on HMF currently reported, compared to CgrAAO [39] and FgrAAO [40], the bacterial HMF oxidase (a GMC superfamily oxidoreductase) [95], and some glyoxal oxidases from AA5_1 (Table S12) [87, 96, 97]. On the other hand, AflAlcOx may find use in multiple biocatalytic processes because it has high activity on both carbohydrate and non-carbohydrate substrates. The optimal choice of enzyme for a specific biocatalytic application will of course be substrate dependent, for which the current palette of AA5_2 CROs serves as an immediate resource and a starting point for future enzyme engineering efforts.
Materials and Methods
Chemicals and enzymes
Ultrapure water was used for the preparation of all buffers and stock solutions unless stated otherwise. Catalase from bovine liver (2000–5000 units per mg protein, Sigma) and horseradish peroxidase (Rz > 3, 300 units per mg, Bio Basic Canada Inc.), obtained as lyophilized powders, were used as received. Other substrates and reagents were purchased from commercial sources (Sigma-Aldrich, VWR or Fisher) and used without further purification.
Sequence analysis and bioinformatics
A total of 247 bacterial and 103 fungal AA5_2 sequences were collected from the CAZy database in January 2019 [17]. Sequences of characterized AA5_1 members were also retrieved from the CAZy database. In addition, full-length sequences of FgrGalOx (Uniprot P0CS93), CgrAlcOx (GenBank EFQ30446), and CgrAAO (GenBank EFQ27661), as well as their corresponding catalytic domains, were used as templates for BLAST analysis against all Fungi in the Joint Genome Institute MycoCosm portal (https://genome.jgi.doe.gov/programs/fungi/index.jsf) [98, 99]. Where present, signal peptides and additional modules, such as carbohydrate-binding modules, were removed to isolate the catalytic modules for subsequent analysis. Catalytic modules sharing 100% identity were down-sampled to one sequence to eliminate redundancy. A multiple sequence alignment was created with MAFFT v.7.402 using the L-INS-I algorithm [100], on the CIPRES Science Gateway (www.phylo.org) [101]. Using this alignment, catalytic modules having deletions/substitutions at key active-site residues, namely, Cys272 and Tyr316 (which form the crosslinked thioether-tyrosyl cofactor) and other copper coordinating residues of FgrGalOx [36] were removed. Moreover, obviously erroneous sequences generated by incorrect splicing predictions were removed through identification of unusually long deletions/insertions.
With this curated multiple sequence alignment, comprising 623 AA5_2 catalytic modules, a maximum likelihood phylogenetic tree was produced using RAxML v.8, with 100 bootstrap replications [102], on the CIPRES Science Gateway portal. The resulting tree was visualized using FigTree and monophyletic groups were supported by bootstrap values > 60 and tree topology. SSNs were generated by computing BLASTP [103] all-versus-all local alignments of the curated 623 AA5_2 catalytic domains using SSNpipe (https://github.com/ahvdk/SSNpipe), which generated the E-value, bit score, alignment length, sequence identity, and sequence similarity for all sequence pairs. The data were filtered using a bit score threshold between 500 and 600, with increments of 25, to generate the final SSNs. A bit score threshold of 550, clustering the sequences into groups, which resolves the same monophyletic groups as those observed in the phylogenetic tree, was retained. The SSNs were visualized with Cytoscape using yFiles Organic Layout [104], and coloring of each node was based on monophyletic groups inferred from the phylogenetic tree.
In addition, Phyre2 [105] was used to make three-dimensional homology models for AflAlcOx, PruAA5_2A (PruAlcOx), PfeGalOx, ExeGalOx, FoxGalOxB, FoxAlcOx, and MreGalOx. The structural models were aligned to the crystal structures of FgrGalOx (PDB ID 1GOF) [92] and CgrAlcOx (PDB ID 5C86) [37] for spatial amino acid comparison to give further support to the alignment.
DNA cloning
cDNA encoding genes of interest without the predicted native signal peptide, and including a C-terminal His6 tag-encoding sequence, were commercially synthesized in a codon-optimized form (BioBasic, Markham, Canada) and cloned directly into pPICZα-A or pPICZα-C using the EcoRI, SfiI, ClaI, XbaI, and NotI restriction sites flush with the sequence encoding the S. cerevisiae α-factor signal peptide. The resultant constructs were transformed into chemically competent E. coli DH5α by heat shock.
Recombinant strain production
All recombinant proteins were produced using the in-house 3PE Platform (Pichia Pastoris Protein Express www.platform3pe.com) as described in Haon et al., [106]. The transformations of competent P. pastoris X33 cells with 5 µg of PmeI-linearized pPICZα-A plasmids (Invitrogen, Cergy-Pontoise, France) containing the targeted sequences were done by electroporation. Each construction was spread on plates containing either 100 or 500 µg mL−1 of Zeocin, and three P. pastoris transformants were isolated from each plate. Zeocin-resistant P. pastoris transformants were then screened for protein expression in 24-deepwell plates with the following conditions: induction of expression was done for 3 d at 25 °C in BMMY supplemented with CuSO4 (500 µM) and methanol (3%, v/v), with daily addition of 1% (v/v) methanol. The recombinant proteins were then purified from supernatant by affinity chromatography on Ni–NTA resin using automated procedure as described previously [106]. Finally, the analysis of purified recombinant protein expression by SDS-PAGE (8% polyacrylamide pre-cast E-PAGE gels, Invitrogen) using Coomassie blue staining was used to select the best-producing transformants.
Small-scale protein production and purification
The best-producing transformants were streaked on yeast extract peptone dextrose (YPD) agar plates containing Zeocin (100 µg mL−1) and allowed to grow 3 d (30 °C). One colony of the selected transformants served to inoculate 5 mL of YPD broth in 50 mL sterile conical tubes and were incubated for 5 h at 30 °C and 160 rpm in an orbital shaker. The preculture was used to inoculate (0.2%, v/v) 500 mL of BMGY, before incubation for 16 h (30 °C) until the OD600 nm reached 4–6. Then, the biomass was harvested by centrifugation (10 min, 16 °C, 5000g) and resuspended in 100 mL of BMMY medium supplemented with 500 µM of CuSO4 and 3% methanol (v/v) and incubated for 3 d in an orbital shaker (200 rpm, 25 °C), with daily feeding of 1% methanol (v/v). For PorAlcOx, StoAA5, and CcaAA5 clones, proteins expression was further improved by lowering induction temperature to 20 °C and increasing daily methanol feed to 3% (v/v). The cells were then harvested by centrifugation (10 min, 4 °C, 5000g); the supernatants were filtered on 0.45 µm membrane (Millipore, Burlington, Massachusetts, USA) and stored at 4 °C prior to purification.
The filtered supernatants were adjusted to pH 7.8 just before purification, filtered on 0.22 µm filters (Millipore, Molsheim, France), and loaded onto a HisTrap HP-5 mL column (GE Healthcare, USA), equilibrated with buffer A (Tris–HCl 50 mM, pH 7.8, NaCl 150 mM, imidazole 10 mM) and connected to an Äkta Xpress system (GE Healthcare). (His)6-tagged recombinant proteins were eluted with 50% buffer B (Tris–HCl 50 mM pH 7.8, NaCl 150 mM, imidazole 500 mM). After SDS-PAGE (10% polyacrylamide pre-cast gel, Bio-Rad) analysis, fractions containing the recombinant enzymes were pooled, concentrated, and exchanged to 50 mM Tris–HCl pH 7.0. The concentrated proteins were incubated overnight at 4 °C with 1 mM CuSO4 to ensure full copper loading and then buffer exchanged in 50 mM Tris–HCl buffer pH 7.0 to remove CuSO4 excess. The protein concentration was determined by UV absorption at 280 nm using a Nanodrop ND-200 spectrophotometer (Thermo Fisher Scientific, USA) and calculated using theoretical molecular weight and molar extinction coefficients determined with ProtParam web tool (https://web.expasy.org/protparam/) (ExeGalOx ε = 142,522 M−1 cm−1; MreGalOx ε = 153,842 M−1 cm−1; FoxGalOxB ε = 140,907 M−1 cm−1; PfeGalOx ε = 131,967 M−1 cm−1; UmaRafOx ε = 109,062 M−1 cm−1; PhuRafOx ε = 105,052 M−1 cm−1; AflAlcOx ε = 134,885 M−1 cm−1; PruAA5_2A (PruAlcOx) ε = 126,802 M−1 cm−1; FoxAlcOx ε = 131,967 M−1 cm−1; PorAlcOx ε = 89,645 M−1 cm−1; CglAlcOx ε = 95,527 M−1 cm−1; AsyAlcOx ε = 123,947 M−1 cm−1; CcaAA5 ε = 77,475 M−1 cm−1; StoAA5 ε = 69,455 M−1 cm−1).
Large-scale protein production and purification
The large-scale protein production and purification were performed as previously published [40]. Briefly, single colonies of P. pastoris X33 expressing clones were streaked onto agar plates containing Zeocin and grown for 2 d in the dark in a 30 °C incubator. Precultures with 5 mL of YPD and 0.4 mg mL−1 Zeocin were inoculated using a single colony and shaken at 30 °C at 250 rpm for 9 h. Biomass production was initiated by the addition of the preculture into 1 L of BMGY media and shaken in 4 L flasks at 250 rpm overnight at 30 °C. Once the BMGY cultures reached an OD600 of 6–12, the cells were harvested by centrifugation at 3000g for 15 min at room temperature. Subsequently, the cells were resuspended using 400 mL of BMMY media, supplemented with 0.5 mM copper sulfate, containing 3% methanol and transferred to 1 L flasks. The flasks were shaken at 250 rpm at 16 °C for 3 d. The cultures were fed 1% (v/v) methanol every 24 h and on day three, the secreted proteins were separated from the cells by centrifugation at 8000g for 15 min at 4 °C. The supernatant was decanted, filtered through 0.45 µm membrane and the pH of the liquid medium was raised to 7.5–8.0 by dropwise addition of 1 M NaOH to the stirred solution. Subsequently, the liquid media which contained the proteins of interest were filtered again through a 0.45 µm membrane and allowed to equilibrate for at least 12 h at 4 ˚C.
The supernatant was loaded into a 5 mL pre-packed Ni–NTA column, pre-equilibrated with 50 mM sodium phosphate buffer at pH 7.5, containing 300 mM NaCl and 10 mM imidazole, at 5 mL min−1. The column was washed with 10 column volumes of equilibration buffer (50 mM sodium phosphate, 300 mM NaCl, 10 mM imidazole at pH 7.5) at 5 mL min−1. Proteins were eluted with a linear gradient from 2 to 100% of 500 mM imidazole in 50 mM sodium phosphate buffer, 300 mM NaCl, and pH 7.5 at 5 mL min−1. The total elution volume was 125 mL collected in 1 mL fractions. The fractions of interest were pooled and further desalted using a Sephadex G-25 column pre-equilibrated with 50 mM sodium phosphate buffer at pH 7.5 at 5 mL min−1. A total volume of 200 mL of equilibration buffer was passed through the column at 5 mL min−1. The fractions of interest were pooled and concentrated using a 30,000 MWCO Vivaspin centrifugal concentrator. The proteins were aliquoted, flash frozen in liquid nitrogen, and stored at −70 °C. SDS-PAGE was performed using pre-cast 4–20% (w/v) polyacrylamide gel in the presence of 2% (w/v) SDS under reducing conditions. Proteins were visualized using Coomassie blue R-250 and molecular weights were estimated using a standard curve of the log (MW) versus Rf of the protein ladder (BLUelf). Protein concentrations were determined by measuring A280. The extinction coefficients were calculated using the ProtParam tool on the ExPASy server.
Analytical protein deglycosylation
The presence of protein glycosylation on UmaRafOx, PhuRafOx, AflAlcOx, PruAA5_2A (PruAlcOx), ExeGalOx, MreGalOx, PfeGalOx, FoxGalOxB, FoxAlcOx, CglAlcOx, AsyAlcOx, PorAlcOx, StoAA5, and CcaAA5 was assessed by treatment with N-glycosidase F from Flavobacterium meningosepticum (PNGaseF, New England Biolabs) according to the manufacturer’s protocol. Briefly, deglycosylation experiments were performed under denaturing condition by adding 5 µg of protein to 10X Glycoprotein Denaturing Buffer and heated for 10 min at 100 °C. The samples were subsequently diluted to 20 µL with GlycoBuffer 2 and tertigol-type NP-400 detergent. Finally, 1 µL of PNGaseF was added to the sample and incubated for 1 h at 37 °C. Changes in protein mobility were assessed by SDS-PAGE stained with Coomassie blue R-250. Molecular weights of proteins were estimated using a standard curve of the log (MW) versus Rf of the protein ladder (BLUelf).
Initial activity test
The colorimetric HRP–2,2’-azinobis(3-ethylbenzthiazoline-6-sulfonic acid (ABTS)-coupled assay was used to determine the kinetics of the enzymatic oxidation of substrate. The oxidation of the alcohol group on the substrates by AA5 enzymes consumes 1 equivalent of O2 and produces 1 equivalent of H2O2. The oxidation of ABTS (λmax = 514 nm, ε—36,000 M−1 cm−1) [107] is catalyzed by the enzyme HRP using 2 equivalents of H2O2. This is a standard assay used to monitor initial rate kinetics for AA5 enzymes [79]. This assay was optimized for sensitivity and linearity. The enzymes that expressed well from the production screen were tested for activity on 300 mM galactose, 300 mM raffinose, and 30 mM benzyl alcohol. Enzyme activity was measured using the HRP-ABTS-coupled assay in 100 mM sodium phosphate buffer at pH 8.0, 0.46 mM ABTS, and 21 U mL−1 of HRP at 30 °C using a Cary 60 UV–Vis spectrometer.
Plate assays—proof of concept
The enzyme AflAlcOx was used for the proof-of-concept experiments for the development of a medium throughput plate-based HRP-ABTS-coupled assay. The pH profile, temperature stability profile, and partial substrate screen for AflAlcOx were performed initially using the standard HRP-ABTS-coupled assay using a Cary 60 UV–Vis spectrometer with plastic 1 mL cuvettes (Fig. S4). The same experiments using the same conditions were subsequently performed using a 96-well plate assay with BioTek Epoch microplate spectrophotometer. All experiments with AflAlcOx performed on the Cary 60 UV–Vis spectrometer were equivalent to experiments performed using the BioTek Epoch microplate spectrophotometer (Fig. S4), validating the plate assay. This assay was optimized for sensitivity and linearity. A standard curve for the production of ABTS radical was performed by incubating 7 mM ABTS with 12.25–22.05 nmol potassium persulfate for 16 h in the dark to correctly assess the pathlength of the 96-well plate [107] (Fig. S3).
pH-activity profile
Enzyme activity across a wide range of pH values was determined using phosphate-citrate (pH 4.0–7.0), sodium phosphate (pH 5.5–8.5), glycine–NaOH (pH 8.5–11.0), and CHES (pH 8.5–10.50) buffers. Enzyme activity was measured using the HRP-ABTS-coupled assay in a 96-well plate assay at RT in 100 mM buffer with either 300 mM of galactose, 30 mM benzyl alcohol, or 300 mM of glycerol depending on the initial activity screen test.
Temperature stability
Temperature stability of candidate enzymes was determined by first diluting the stock protein in 50 mM sodium phosphate buffer at the previously optimum pH determined for each enzyme in the presence of 0.1 mg ml−1 of bovine serum albumin (BSA). The diluted protein was then pre-incubated in a thermocycler at 30 °C, 39 °C, 49 °C, 60 °C, 69 °C, and 79 °C. Samples were taken out at different time intervals and the activity of the proteins was measured using the HRP-ABTS-coupled assay in a 96-well plate assay at RT with 300 mM galactose, 30 mM benzyl alcohol, or 300 mM glycerol as the substrate based on previous experiments.
Substrate screen
The activity of candidate enzymes was surveyed on a variety of substrates using the HRP-ABTS-coupled assay in 100 mM sodium phosphate buffer at pH value determined by the pH profile, 0.46 mM ABTS, and 21 U mL−1 of HRP at RT with the 96-well plate assay method. The initial substrate screen included carbohydrates, polyols, diols, and primary alcohol substrates at 300 mM, benzyl alcohol and galactitol at 30 mM, methyl glyoxal and glycolaldehyde dimer at 5 mM, and aryl alcohols and furans at 2.5 mM. One unit of AA5 enzyme activity was defined as the amount of enzyme required to oxidize 2 µmol of ABTS per minute, which is equivalent to the consumption of 1 µmol of oxygen per minute.
Michaelis–Menten Kinetics
To determine Michaelis–Menten parameters of candidate enzymes, different concentrations of substrate solutions were used over the range of 0.1–1,500 mM for galactose, 0.5–750 mM for melibiose, 0.1–285 mM for benzyl alcohol, 0.5–200 mM for butanol, 5–6000 mM glycerol, 0.1–200 mM 1,4-butanediol, and 0.01–25 mM HMF. The reactions were performed using the HRP-ABTS-coupled assay with 0.46 mM ABTS, 21 U mL−1 of HRP at different temperatures in 100 mM buffer at different pH informed by the temperature stability assays and pH profile experiments using the Cary 60 UV–VIS spectrometer with plastic 1 mL cuvettes. More detailed information for the experimental conditions of each Michaelis–Menten experiment is given in Table 4. Data were fit with the Michaelis − Menten equation using OriginPro software (OriginLab 9.55).
Computational docking studies
Molecular docking simulations were performed using the CHIMERA software from UCSF Resource for Biocomputing, Visualization, and Informatics [108]. The FgrGalOx crystal structure (PDB ID 1GOF) and a Phyre model of AflAlcOx, PruAA5_2A (PruAlcOx), PfeGalOx, ExeGalOx, FoxGalOxB, and FoxAlcOx were used to generate the receptors for simulations. Galactose was extracted from a different PDB file (PDB ID 1SO0). Ligands and receptors were first prepared for docking in chimera by adding hydrogens and assigning proper protonation states. The docking simulation itself was performed using Autodock VINA, run within CHIMERA, with the AMBER03 force field [109]. Appropriate simulation cells were defined for the respective docking simulations. For docking of galactose, a 7 Å, 6 × 6 × 7 Å, or a 6 Å cell with the copper atom bordering the z-coordinate edge was chosen. Galactose gave reasonable binding poses with all enzymes used. The modelled receptor–ligand complex structures were analyzed using the PyMOL software (Schrodinger LLC) to determine ligand–receptor interactions, including bond distances.
Enzyme product analysis
Oxidation of HMF, DFF, HMFCA, and FDCA
Oxidation of furan-containing compounds with AA5_2 enzymes was performed as previously published [40]. Ten mM of substrate (HMF, DFF, HMFCA, and FDCA) and 1 mg mL−1 of both catalase and HRP were combined and the reaction was initiated by adding 60 µg of purified AA5_2 enzyme to a vial with 50 mM buffer in a final volume of 1 mL. Reactions were stirred at 400 rpm at room temperature for 17 h followed by enzyme removal through ultrafiltration (5 kDa cut-off Vivaspin Sartorius, Stonehouse, UK). D2O was added to the filtrate to a final composition of 10% (v/v). NMR experiments were performed on a Bruker AVANCE 600 MHz spectrometer. 1H NMR spectra were collected with water suppression (4.7 ppm) using a standard pre-saturation pulse sequence. Chemical shifts were calibrated to the internal HOD peak (4.7 ppm). Standards of all substrates were used to identify distinct chemical shifts for each molecule. Integration values of relevant peak areas were used to determine conversion percentages.
Oxidation of glycerol
Oxidation of glycerol and subsequent product analysis was performed as previously published [40]. 0.54 M glycerol and 1 mg mL−1 of both catalase and HRP were combined and 700 µg of purified AA5_2 enzyme in a final volume of 1 mL (50 mM buffer) was added to start the reaction. Reactions were stirred at 400 rpm at room temperature for 24 h, and subsequently, the enzymes were removed by ultrafiltration (5 kDa cut-off Vivaspin Sartorius, Stonehouse, UK). 20 mg of 2,4-dinitrophenyl hydrazine was added to the reaction mixtures, which were incubated in a heat block at 50 ℃ for 6 h with intermittent shaking. A TLC plate was used to check formation of desired product (Rf = 0.46, 100% EtOAc). The solution was purified via preparatory TLC and the desired hydrazone was mechanically isolated and dissolved in MeOH. Subsequently, the solution was filtered and concentrated and the composition of the purified glyceraldehyde-hydrazones was analyzed by HPLC (3 µL injection, Chiracel® IA-3). Eluents used for HPLC methods, i.e. water with 0.1% formic acid (A) and methanol (B), were LC–MS grade (Optima, Fisher). For separation between l-and d-glyceraldehyde-hydrazones an isocratic method using 60% A, 40% B with a flow rate of 0.65 mL min−1 was used with a 12 min stop time with UV detection at 360 nm. l-glyceraldehyde-hydrazone eluted at 2.30 min and d-glyceraldehyde-hydrazone eluted at 2.76 min. ESI mass spectra were also collected in positive mode scan for m/Z 95–500 running at 0.8 s/cycle, drying gas = 5.0 L min−1, nebulizer pressure = 50 psi, gas temperature = 300 °C, and capillary voltage = 4000 V.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors thank Dr. Maria Ezhova and Dr. Zhicheng (Paul) Xia (UBC Department of Chemistry) for support with NMR data acquisition.
Author contributions
MC performed enzyme cloning, biochemistry, enzyme kinetic and product analysis, all corresponding data analysis, and drafted the manuscript; YM performed sequence analysis, bioinformatics, and enzyme cloning, wrote portions of manuscript, and revised the manuscript; DR and MH performed recombinant protein production and initial kinetic measurements; PM and JH assisted with the glycerol oxidation experiments; ML and JGB assisted with data analysis; and HB coordinated research, analyzed data, and revised the manuscript with input from all the authors.
Funding
Funding is gratefully acknowledged from: The Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grants RGPIN 435223-13 and RGPIN-2018–03892; NSERC Strategic Partnership Grant NETGP 451431-13 for the “NSERC Industrial Biocatalysis Network”; NSERC Strategic Partnership Grant (STPGP 493781–16) and joint Agence Nationale de la Recherche grant (ANR-17-CE07-0047), “FUNTASTIC—Fungal copper radical oxidases as new biocatalysts for the valorization of biomass carbohydrates and alcohols”; and Genome Canada/Genome BC/Ontario Genomics/Genome Quebec for the Large-Scale Applied Research Project (LSARP, project #10405), “SYNBIOMICS—Functional genomics and techno-economic models for advanced biopolymer synthesis”).
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files. All nucleotide sequence, protein sequence, and protein structural information used in this work were extracted from existing accessions in public databases, i.e., GenBank, Joint Genome Institute MycoCosm portal, and the Protein Data Bank.
Declarations
Conflict of interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Cook J, Oreskes N, Doran P, Anderegg W, Verheggen B, Maibach E, Carlton J, Lewandowsky S, Skuce A, Green S, Nuccitelli D, Jacobs P, Richardson M, Winkler B, Painting R, Rice K. Consensus on consensus: a synthesis of consensus estimates on human-caused global warming. Environ Res Lett. 2016;11:048002. doi: 10.1088/1748-9326/11/4/048002. [DOI] [Google Scholar]
- 2.Tollefson J. Can the world kick its fossil-fuel addiction fast enough? Nature. 2018;7702:422–425. doi: 10.1038/d41586-018-04931-6. [DOI] [PubMed] [Google Scholar]
- 3.Fabian N. Economics: Support low-carbon investment. Nature. 2015;519:27–29. doi: 10.1038/519027a. [DOI] [PubMed] [Google Scholar]
- 4.Zhang YHP. What is vital (and not vital) to advance economically-competitive biofuels production. Process Biochem. 2011;46(11):2091–2110. doi: 10.1016/j.procbio.2011.08.005. [DOI] [Google Scholar]
- 5.Naik SN, Goud VV, Rout PK, Dalai AK. Production of first and second generation biofuels: a comprehensive review. Renewable Sustainable Energy Rev. 2010;14(2):578–597. doi: 10.1016/j.rser.2009.10.003. [DOI] [Google Scholar]
- 6.Hayes DJM. Second-generation biofuels: Why they are taking so long. WIREs Energy Environ. 2013;2(3):304–334. doi: 10.1002/wene.59. [DOI] [Google Scholar]
- 7.Bender TA, Dabrowski JA, Gagné MR. Homogeneous catalysis for the production of low-volume, high-value chemicals from biomass. Nat Rev Chem. 2018;2(5):35–46. doi: 10.1038/s41570-018-0005-y. [DOI] [Google Scholar]
- 8.Fatih DM. Biorefineries for biofuel upgrading: a critical review. Appl Energy. 2009;86(Supplement 1):S151–S161. doi: 10.1016/j.apenergy.2009.04.043. [DOI] [Google Scholar]
- 9.Ragauskas AJ, Williams CK, Davison BH, Britovsek G, Cairney J, Eckert CA, Frederick WJ, Hallett JP, Leak DJ, Liotta CL, Mielenz JR, Murphy R, Templer R, Tschaplinski T. The path forward for biofuels and biomaterials. Science. 2006;311(5760):484–489. doi: 10.1126/science.1114736. [DOI] [PubMed] [Google Scholar]
- 10.Dessbesell L, Xu C, Pulkki R, Leitch M, Mahmood N. Forest biomass supply chain optimization for a biorefinery aiming to produce high-value bio-based materials and chemicals from lignin and forestry residues: a review of literature. Can J For Res. 2017;47(3):277–288. doi: 10.1139/cjfr-2016-0336. [DOI] [Google Scholar]
- 11.Allais F, Coqueret X, Farmer T, Raverty W, Rémond C, Paës G (2020) Editorial: From biomass to advanced bio-based chemicals & materials: a multidisciplinary perspective. Front Chem 8(131). 10.3389/fchem.2020.00131 [DOI] [PMC free article] [PubMed]
- 12.Goswami P, Chinnadayyala SSR, Chakraborty M, Kumar AK, Kakoti A. An overview on alcohol oxidases and their potential applications. Appl Microbiol Biotechnol. 2013;97(10):4259–4275. doi: 10.1007/s00253-013-4842-9. [DOI] [PubMed] [Google Scholar]
- 13.Hernández-Ortega A, Ferreira P, Martínez AT. Fungal aryl-alcohol oxidase: a peroxide-producing flavoenzyme involved in lignin degradation. Appl Microbiol Biotechnol. 2012;93(4):1395–1410. doi: 10.1007/s00253-011-3836-8. [DOI] [PubMed] [Google Scholar]
- 14.Hollmann F, Arends IWCE, Buehler K, Schallmey A, Bühler B. Enzyme-mediated oxidations for the chemist. Green Chem. 2011;13(2):226–265. doi: 10.1039/C0GC00595A. [DOI] [Google Scholar]
- 15.Horn SJ, Vaaje-Kolstad G, Westereng B, Eijsink VG. Novel enzymes for the degradation of cellulose. Biotechnol Biofuels. 2012;5(1):45. doi: 10.1186/1754-6834-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johansen KS. Discovery and industrial applications of lytic polysaccharide mono-oxygenases. Biochem Soc Trans. 2016;44(1):143–149. doi: 10.1042/BST20150204. [DOI] [PubMed] [Google Scholar]
- 17.Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels. 2013;6(1):41. doi: 10.1186/1754-6834-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2013;42(D1):D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Artzi L, Bayer EA, Moraïs S. Cellulosomes: bacterial nanomachines for dismantling plant polysaccharides. Nat Rev Microbiol. 2017;15(2):83–95. doi: 10.1038/nrmicro.2016.164. [DOI] [PubMed] [Google Scholar]
- 20.Payne CM, Knott BC, Mayes HB, Hansson H, Himmel ME, Sandgren M, Ståhlberg J, Beckham GT. Fungal cellulases. Chem Rev. 2015;115(3):1308–1448. doi: 10.1021/cr500351c. [DOI] [PubMed] [Google Scholar]
- 21.Murphy C, Powlowski J, Wu M, Butler G, Tsang A. Curation of characterized glycoside hydrolases of fungal origin. Database. 2011;2011:bar020. doi: 10.1093/database/bar020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Basit A, Liu J, Rahim K, Jiang W, Lou H. Thermophilic xylanases: from bench to bottle. Crit Rev Biotechnol. 2018;38(7):989–1002. doi: 10.1080/07388551.2018.1425662. [DOI] [PubMed] [Google Scholar]
- 23.Ryu DDY, Mandels M. Cellulases: biosynthesis and applications. Enzyme Microb Technol. 1980;2(2):91–102. doi: 10.1016/0141-0229(80)90063-0. [DOI] [Google Scholar]
- 24.Tomme P, Warren RAJ, Gilkes NR (1995) Cellulose Hydrolysis by Bacteria and Fungi. In: Poole RK (eds) Advances in microbial physiology. Academic Press, pp 1–81. 10.1016/S0065-2911(08)60143-5 [DOI] [PubMed]
- 25.Davies G, Henrissat B. Structures and mechanisms of glycosyl hydrolases. Structure. 1995;3(9):853–859. doi: 10.1016/S0969-2126(01)00220-9. [DOI] [PubMed] [Google Scholar]
- 26.Møller MS, Svensson B. Structural biology of starch-degrading enzymes and their regulation. Curr Opin Struct Biol. 2016;40:33–42. doi: 10.1016/j.sbi.2016.07.006. [DOI] [PubMed] [Google Scholar]
- 27.Knob A, Terrasan CRF, Carmona EC. β-Xylosidases from filamentous fungi: an overview. World J Microbiol Biotechnol. 2010;26(3):389–407. doi: 10.1007/s11274-009-0190-4. [DOI] [Google Scholar]
- 28.Juturu V, Wu JC. Microbial xylanases: engineering, production and industrial applications. Biotechnol Adv. 2012;30(6):1219–1227. doi: 10.1016/j.biotechadv.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 29.Friedl A. Bioethanol from starch. In: Meyers RA, editor. Encyclopedia of sustainability science and technology. New York, NY: Springer; 2012. pp. 987–1001. [Google Scholar]
- 30.Schoemaker HE, Mink D, Wubbolts MG. Dispelling the myths–biocatalysis in industrial synthesis. Science. 2003;299(5613):1694–1697. doi: 10.1126/science.1079237. [DOI] [PubMed] [Google Scholar]
- 31.Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K. Engineering the third wave of biocatalysis. Nature. 2012;485(7397):185–194. doi: 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]
- 32.Wu S, Snajdrova R, Moore JC, Baldenius K, Bornscheuer UT. Biocatalysis: enzymatic synthesis for industrial applications. Angew Chem Int Ed. 2021;60(1):88–119. doi: 10.1002/anie.202006648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whittaker JW. Free radical catalysis by galactose oxidase. Chem Rev. 2003;103(6):2347–2364. doi: 10.1021/cr020425z. [DOI] [PubMed] [Google Scholar]
- 34.Kersten P, Cullen D. Copper radical oxidases and related extracellular oxidoreductases of wood-decay Agaricomycetes. Fungal Genet Biol. 2014;72:124–130. doi: 10.1016/j.fgb.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 35.Whittaker MM, Kersten PJ, Nakamura N, Sanders-Loehr J, Schweizer ES, Whittaker JW. Glyoxal oxidase from Phanerochaete chrysosporium is a new radical-copper oxidase. J Biol Chem. 1996;271(2):681–687. doi: 10.1074/jbc.271.2.681. [DOI] [PubMed] [Google Scholar]
- 36.Ito N, Phillips SEV, Stevens C, Ogel ZB, McPherson MJ, Keen JN, Yadav KDS, Knowles PF. Novel thioether bond revealed by a 1.7 Å crystal structure of galactose oxidase. Nature. 1991;350(6313):87–90. doi: 10.1038/350087a0. [DOI] [PubMed] [Google Scholar]
- 37.Yin D, Urresti S, Lafond M, Johnston EM, Derikvand F, Ciano L, Berrin J-G, Henrissat B, Walton PH, Davies GJ, Brumer H. Structure–function characterization reveals new catalytic diversity in the galactose oxidase and glyoxal oxidase family. Nat Commun. 2015;6(1):10197. doi: 10.1038/ncomms10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oide S, Tanaka Y, Watanabe A, Inui M. Carbohydrate-binding property of a cell wall integrity and stress response component (WSC) domain of an alcohol oxidase from the rice blast pathogen Pyricularia oryzae. Enzyme Microb Technol. 2019;125:13–20. doi: 10.1016/j.enzmictec.2019.02.009. [DOI] [PubMed] [Google Scholar]
- 39.Mathieu Y, Offen WA, Forget SM, Ciano L, Viborg AH, Blagova E, Henrissat B, Walton PH, Davies GJ, Brumer H. Discovery of a fungal copper radical oxidase with high catalytic efficiency toward 5-hydroxymethylfurfural and benzyl alcohols for bioprocessing. ACS Catal. 2020;10(5):3042–3058. doi: 10.1021/acscatal.9b04727. [DOI] [Google Scholar]
- 40.Cleveland M, Lafond M, Xia FR, Chung R, Mulyk P, Hein JE, Brumer H. Two Fusarium copper radical oxidases with high activity on aryl alcohols. Biotechnol Biofuels. 2021;14(1):138. doi: 10.1186/s13068-021-01984-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bissaro B, Kodama S, Hage H, Ribeaucourt D, Haon M, Grisel S, Simaan A, Beisson F, Forget S, Brumer H, Rosso M-N, O’Connell R, Lafond M, Kubo Y, Berrin J-G (2021) Unravelling the role of alcohol copper radical oxidases in fungal plant pathogens. 10.21203/rs.3.rs-493001/v1
- 42.Roberts GP, Gupta SK. Use of galactose oxidase in the histochemical examination of mucus-secreting cells. Nature. 1965;207(4995):425–426. doi: 10.1038/207425a0. [DOI] [PubMed] [Google Scholar]
- 43.Heitzmann H, Richards FM. Use of the avidin-biotin complex for specific staining of biological membranes in electron microscopy. Proc Natl Acad Sci USA. 1974;71(9):3537–3541. doi: 10.1073/pnas.71.9.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilchek M, Spiegel S, Spiegel Y. Fluorescent reagents for the labeling of glycoconjugates in solution and on cell surfaces. Biochem Biophys Res Commun. 1980;92(4):1215–1222. doi: 10.1016/0006-291X(80)90416-7. [DOI] [PubMed] [Google Scholar]
- 45.Ramya TNC, Weerapana E, Cravatt BF, Paulson JC. Glycoproteomics enabled by tagging sialic acid- or galactose-terminated glycans. Glycobiology. 2012;23(2):211–221. doi: 10.1093/glycob/cws144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Monosik R, Stredansky M, Tkac J, Sturdik E. Application of enzyme biosensors in analysis of food and beverages. Food Anal Methods. 2012;5(1):40–53. doi: 10.1007/s12161-011-9222-4. [DOI] [Google Scholar]
- 47.Schoevaart R, Kieboom T. Application of galactose oxidase in chemoenzymatic one-pot cascade reactions without intermediate recovery steps. Top Catal. 2004;27(1):3–9. doi: 10.1023/B:TOCA.0000013536.27551.13. [DOI] [Google Scholar]
- 48.Mikkonen KS, Parikka K, Suuronen J-P, Ghafar A, Serimaa R, Tenkanen M. Enzymatic oxidation as a potential new route to produce polysaccharide aerogels. RSC Adv. 2014;4(23):11884–11892. doi: 10.1039/C3RA47440B. [DOI] [Google Scholar]
- 49.Leppänen A-S, Xu C, Parikka K, Eklund P, Sjöholm R, Brumer H, Tenkanen M, Willför S. Targeted allylation and propargylation of galactose-containing polysaccharides in water. Carbohydr Polym. 2014;100:46–54. doi: 10.1016/j.carbpol.2012.11.053. [DOI] [PubMed] [Google Scholar]
- 50.Leppänen A-S, Niittymäki O, Parikka K, Tenkanen M, Eklund P, Sjöholm R, Willför S. Metal-mediated allylation of enzymatically oxidized methyl α-d-galactopyranoside. Carbohydr Res. 2010;345(18):2610–2615. doi: 10.1016/j.carres.2010.09.026. [DOI] [PubMed] [Google Scholar]
- 51.Yalpani M, Hall LD. Some chemical and analytical aspects of polysaccharide modifications. II. A high-yielding, specific method for the chemical derivatization of galactose-containing polysaccharides: Oxidation with galactose oxidase followed by reductive amination. J Polym Sci, Polym Chem Ed. 1982;20(12):3399–3420. doi: 10.1002/pol.1982.170201213. [DOI] [Google Scholar]
- 52.Kelleher FM, Bhavanandan VP. Preparation and characterization of beta-d-fructofuranosyl O-(alpha-d-galactopyranosyl uronic acid)-(1–6)-O-alpha-d-glucopyranoside and O-(alpha- d -galactopyranosyl uronic acid)-(1–6)- d-glucose. Carbohydr Res. 1986;155:89–97. doi: 10.1016/S0008-6215(00)90135-6. [DOI] [PubMed] [Google Scholar]
- 53.Xu C, Spadiut O, Araújo AC, Nakhai A, Brumer H. Chemo-enzymatic assembly of clickable cellulose surfaces via multivalent polysaccharides. Chemsuschem. 2012;5(4):661–665. doi: 10.1002/cssc.201100522. [DOI] [PubMed] [Google Scholar]
- 54.Parikka K, Leppänen A-S, Xu C, Pitkänen L, Eronen P, Österberg M, Brumer H, Willför S, Tenkanen M. Functional and anionic cellulose-interacting polymers by selective chemo-enzymatic carboxylation of galactose-containing polysaccharides. Biomacromol. 2012;13(8):2418–2428. doi: 10.1021/bm300679a. [DOI] [PubMed] [Google Scholar]
- 55.Huffman MA, Fryszkowska A, Alvizo O, Borra-Garske M, Campos KR, Canada KA, Devine PN, Duan D, Forstater JH, Grosser ST, Halsey HM, Hughes GJ, Jo J, Joyce LA, Kolev JN, Liang J, Maloney KM, Mann BF, Marshall NM, McLaughlin M, Moore JC, Murphy GS, Nawrat CC, Nazor J, Novick S, Patel NR, Rodriguez-Granillo A, Robaire SA, Sherer EC, Truppo MD, Whittaker AM, Verma D, Xiao L, Xu Y, Yang H. Design of an in vitro biocatalytic cascade for the manufacture of islatravir. Science. 2019;366(6470):1255–1259. doi: 10.1126/science.aay8484. [DOI] [PubMed] [Google Scholar]
- 56.Ribeaucourt D, Bissaro B, Guallar V, Yemloul M, Haon M, Grisel S, Alphand V, Brumer H, Lambert F, Berrin J-G, Lafond M. Comprehensive insights into the production of long chain aliphatic aldehydes using a copper-radical alcohol oxidase as biocatalyst. ACS Sustainable Chem Eng. 2021;9(12):4411–4421. doi: 10.1021/acssuschemeng.0c07406. [DOI] [Google Scholar]
- 57.Rosatella AA, Simeonov SP, Frade RFM, Afonso CAM. 5-Hydroxymethylfurfural (HMF) as a building block platform: biological properties, synthesis and synthetic applications. Green Chem. 2011;13(4):754–793. doi: 10.1039/C0GC00401D. [DOI] [Google Scholar]
- 58.Sousa AF, Vilela C, Fonseca AC, Matos M, Freire CSR, Gruter G-JM, Coelho JFJ, Silvestre AJD. Biobased polyesters and other polymers from 2,5-furandicarboxylic acid: a tribute to furan excellency. Polym Chem. 2015;6(33):5961–5983. doi: 10.1039/C5PY00686D. [DOI] [Google Scholar]
- 59.Siebum A, van Wijk A, Schoevaart R, Kieboom T. Galactose oxidase and alcohol oxidase: scope and limitations for the enzymatic synthesis of aldehydes. J Mol Catal B: Enzym. 2006;41(3):141–145. doi: 10.1016/j.molcatb.2006.04.003. [DOI] [Google Scholar]
- 60.Ribeaucourt D, Bissaro B, Lambert F, Lafond M, Berrin J-G (2021) Biocatalytic oxidation of fatty alcohols into aldehydes for the flavors and fragrances industry. Biotechnol Adv 107787. 10.1016/j.biotechadv.2021.107787 [DOI] [PubMed]
- 61.Mollerup F, Aumala V, Parikka K, Mathieu Y, Brumer H, Tenkanen M, Master E (2019) A family AA5_2 carbohydrate oxidase from Penicillium rubens displays functional overlap across the AA5 family. PLoS One 14(5). 10.1371/journal.pone.0216546 [DOI] [PMC free article] [PubMed]
- 62.Andberg M, Mollerup F, Parikka K, Koutaniemi S, Boer H, Juvonen M, Master E, Tenkanen M, Kruus K. A novel Colletotrichum graminicola raffinose oxidase in the AA5 Family. Appl Environ Microbiol. 2017;83(20):e01383–e1417. doi: 10.1128/AEM.01383-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Faria CB, de Castro FF, Martim DB, Abe CAL, Prates KV, de Oliveira MAS, Barbosa-Tessmann IP. Production of galactose oxidase inside the Fusarium fujikuroi species complex and recombinant expression and characterization of the galactose oxidase GaoA protein from Fusarium subglutinans. Mol Biotechnol. 2019;61(9):633–649. doi: 10.1007/s12033-019-00190-6. [DOI] [PubMed] [Google Scholar]
- 64.Paukner R, Staudigl P, Choosri W, Haltrich D, Leitner C. Expression, purification, and characterization of galactose oxidase of Fusarium sambucinum in E. coli. Protein Expression Purif. 2015;108:73–79. doi: 10.1016/j.pep.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paukner R, Staudigl P, Choosri W, Haltrich D, Leitner C. Galactose oxidase from Fusarium oxysporum - expression in E. coli and P. pastoris and biochemical characterization. PLoS One. 2014;9(6):e100116. doi: 10.1371/journal.pone.0100116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Whittaker JW. The radical chemistry of galactose oxidase. Arch Biochem Biophys. 2005;433(1):227–239. doi: 10.1016/j.abb.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 67.Leuthner B, Aichinger C, Oehmen E, Koopmann E, Müller O, Müller P, Kahmann R, Bölker M, Schreier PH. A H2O2-producing glyoxal oxidase is required for filamentous growth and pathogenicity in Ustilago maydis. Mol Genet Genomics. 2005;272(6):639–650. doi: 10.1007/s00438-004-1085-6. [DOI] [PubMed] [Google Scholar]
- 68.Kersten PJ, Cullen D. Cloning and characterization of cDNA encoding glyoxal oxidase, a H2O2-producing enzyme from the lignin-degrading basidiomycete Phanerochaete chrysosporium. Proc Natl Acad Sci U S A. 1993;90(15):7411–7413. doi: 10.1073/pnas.90.15.7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun L, Bulter T, Alcalde M, Petrounia IP, Arnold FH. Modification of galactose oxidase to introduce glucose 6-oxidase activity. ChemBioChem. 2002;3(8):781–783. doi: 10.1002/1439-7633(20020802)3:8<781::AID-CBIC781>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 70.Sun L, Petrounia IP, Yagasaki M, Bandara G, Arnold FH. Expression and stabilization of galactose oxidase in Escherichia coli by directed evolution. Protein Eng, Des Sel. 2001;14(9):699–704. doi: 10.1093/protein/14.9.699. [DOI] [PubMed] [Google Scholar]
- 71.Deacon SE, McPherson MJ. Enhanced expression and purification of fungal galactose oxidase in Escherichia coli and use for analysis of a saturation mutagenesis library. ChemBioChem. 2011;12(4):593–601. doi: 10.1002/cbic.201000634. [DOI] [PubMed] [Google Scholar]
- 72.Deacon SE, Mahmoud K, Spooner RK, Firbank SJ, Knowles PF, Phillips SEV, McPherson MJ. Enhanced fructose oxidase activity in a galactose oxidase variant. ChemBioChem. 2004;5(7):972–979. doi: 10.1002/cbic.200300810. [DOI] [PubMed] [Google Scholar]
- 73.Rogers MS, Tyler EM, Akyumani N, Kurtis CR, Spooner RK, Deacon SE, Tamber S, Firbank SJ, Mahmoud K, Knowles PF, Phillips SEV, McPherson MJ, Dooley DM. The stacking tryptophan of galactose oxidase: a second-coordination sphere residue that has profound effects on tyrosyl radical behavior and enzyme catalysis. Biochemistry. 2007;46(15):4606–4618. doi: 10.1021/bi062139d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wilkinson D, Akumanyi N, Hurtado-Guerrero R, Dawkes H, Knowles PF, Phillips SEV, McPherson MJ. Structural and kinetic studies of a series of mutants of galactose oxidase identified by directed evolution. Protein Eng, Des Sel. 2004;17(2):141–148. doi: 10.1093/protein/gzh018. [DOI] [PubMed] [Google Scholar]
- 75.Escalettes F, Turner NJ. Directed evolution of galactose oxidase: generation of enantioselective secondary alcohol oxidases. ChemBioChem. 2008;9(6):857–860. doi: 10.1002/cbic.200700689. [DOI] [PubMed] [Google Scholar]
- 76.Rannes JB, Ioannou A, Willies SC, Grogan G, Behrens C, Flitsch SL, Turner NJ. Glycoprotein labeling using engineered variants of galactose oxidase obtained by directed evolution. J Am Chem Soc. 2011;133(22):8436–8439. doi: 10.1021/ja2018477. [DOI] [PubMed] [Google Scholar]
- 77.Lippow SM, Moon TS, Basu S, Yoon S-H, Li X, Chapman BA, Robison K, Lipovšek D, Prather KLJ. Engineering enzyme specificity using computational design of a defined-sequence library. Chem Biol. 2010;17(12):1306–1315. doi: 10.1016/j.chembiol.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 78.Gilbert HJ, Knox JP, Boraston AB. Advances in understanding the molecular basis of plant cell wall polysaccharide recognition by carbohydrate-binding modules. Curr Opin Struct Biol. 2013;23(5):669–677. doi: 10.1016/j.sbi.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 79.Spadiut O, Olsson L, Brumer H. A comparative summary of expression systems for the recombinant production of galactose oxidase. Microb Cell Fact. 2010;9(1):68. doi: 10.1186/1475-2859-9-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Whittaker MM, Ballou DP, Whittaker JW. Kinetic isotope effects as probes of the mechanism of galactose oxidase. Biochemistry. 1998;37(23):8426–8436. doi: 10.1021/bi980328t. [DOI] [PubMed] [Google Scholar]
- 81.Avigad G, Amaral D, Asensio C, Horecker BL. The D-galactose oxidase of Polyporus circinatus. J Biol Chem. 1962;237(9):2736–2743. doi: 10.1016/S0021-9258(18)60220-0. [DOI] [PubMed] [Google Scholar]
- 82.Cooper JAD, Smith W, Bacila M, Medina H. Galactose oxidase from Polyporus circinatus, Fr. J Biol Chem. 1959;234(3):445–448. doi: 10.1016/S0021-9258(18)70223-8. [DOI] [PubMed] [Google Scholar]
- 83.Whittaker MM, Whittaker JW. The active site of galactose oxidase. J Biol Chem. 1988;263(13):6074–6080. doi: 10.1016/S0021-9258(18)68751-4. [DOI] [PubMed] [Google Scholar]
- 84.Xu F, Golightly EJ, Schneider P, Berka RM, Brown KM, Johnstone JA, Baker DH, Fuglsang CC, Brown SH, Svendsen A, Klotz AV. Expression and characterization of a recombinant Fusarium spp. galactose oxidase. Appl Biochem Biotechnol. 2000;88(1):23. doi: 10.1385/ABAB:88:1-3:023. [DOI] [Google Scholar]
- 85.Henrissat B, Teeri TT, Warren RA. A scheme for designating enzymes that hydrolyse the polysaccharides in the cell walls of plants. FEBS Lett. 1998;425(2):352–354. doi: 10.1016/S0014-5793(98)00265-8. [DOI] [PubMed] [Google Scholar]
- 86.Whittaker MM, Whittaker JW. Catalytic reaction profile for alcohol oxidation by galactose oxidase. Biochemistry. 2001;40(24):7140–7148. doi: 10.1021/bi010303l. [DOI] [PubMed] [Google Scholar]
- 87.Carro J, Ferreira P, Rodríguez L, Prieto A, Serrano A, Balcells B, Ardá A, Jiménez-Barbero J, Gutiérrez A, Ullrich R, Hofrichter M, Martínez AT. 5-hydroxymethylfurfural conversion by fungal aryl-alcohol oxidase and unspecific peroxygenase. FEBS J. 2015;282(16):3218–3229. doi: 10.1111/febs.13177. [DOI] [PubMed] [Google Scholar]
- 88.Forget SM, Xia F, Hein JE, Brumer H. Determination of biocatalytic parameters of a copper radical oxidase using real-time reaction progress monitoring. Org Biomol Chem. 2020;18(11):2076–2084. doi: 10.1039/C9OB02757B. [DOI] [PubMed] [Google Scholar]
- 89.Toftgaard Pedersen A, Birmingham WR, Rehn G, Charnock SJ, Turner NJ, Woodley JM. Process requirements of galactose oxidase catalyzed oxidation of alcohols. Org Process Res Dev. 2015;19(11):1580–1589. doi: 10.1021/acs.oprd.5b00278. [DOI] [Google Scholar]
- 90.Pagliaro M, Rossi M (2010) Glycerol: Properties and production. The Future of Glycerol. Royal Society of Chemistry’s, Cambridge, UK. 10.1039/9781849731089
- 91.Klibanov AM, Alberti BN, Marletta MA. Stereospecific oxidation of aliphatic alcohols catalyzed by galactose oxidase. Biochem Biophys Res Commun. 1982;108(2):804–808. doi: 10.1016/0006-291X(82)90900-7. [DOI] [PubMed] [Google Scholar]
- 92.Ito N, Phillips SEV, Yadav KDS, Knowles PF. Crystal structure of a free radical enzyme, Galactose Oxidase. J Mol Biol. 1994;238(5):794–814. doi: 10.1006/jmbi.1994.1335. [DOI] [PubMed] [Google Scholar]
- 93.Chaplin AK, Petrus MLC, Mangiameli G, Hough MA, Svistunenko DA, Nicholls P, Claessen D, Vijgenboom E, Worrall JAR. GlxA is a new structural member of the radical copper oxidase family and is required for glycan deposition at hyphal tips and morphogenesis of Streptomyces lividans. Biochem J. 2015;469(3):433–444. doi: 10.1042/bj20150190. [DOI] [PubMed] [Google Scholar]
- 94.Chaplin AK, Svistunenko DA, Hough MA, Wilson MT, Vijgenboom E, Worrall JA. Active-site maturation and activity of the copper-radical oxidase GlxA are governed by a tryptophan residue. Biochem J. 2017;474(5):809–825. doi: 10.1042/bcj20160968. [DOI] [PubMed] [Google Scholar]
- 95.Dijkman WP, Fraaije MW. Discovery and Characterization of a 5-Hydroxymethylfurfural Oxidase from Methylovorus sp. Strain MP688. Appl Environ Microbiol. 2014;80(3):1082–1090. doi: 10.1128/AEM.03740-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kadowaki MAS, Godoy MOd, Kumagai PS, Costa-Filho AJd, Mort A, Prade RA, Polikarpov I. Characterization of a new glyoxal oxidase from the thermophilic fungus Myceliophthora thermophila M77: Hydrogen peroxide production retained in 5-Hydroxymethylfurfural oxidation. Catalysts. 2018;8(10):476. doi: 10.3390/catal8100476. [DOI] [Google Scholar]
- 97.Daou M, Yassine B, Wikee S, Record E, Duprat F, Bertrand E, Faulds CB. Pycnoporus cinnabarinus glyoxal oxidases display differential catalytic efficiencies on 5-hydroxymethylfurfural and its oxidized derivatives. Fungal Biol Biotechnol. 2019;6(1):4. doi: 10.1186/s40694-019-0067-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Grigoriev IV, Cullen D, Goodwin SB, Hibbett D, Jeffries TW, Kubicek CP, Kuske C, Magnuson JK, Martin F, Spatafora JW, Tsang A, Baker SE. Fueling the future with fungal genomics. Mycology. 2011;2(3):192–209. doi: 10.1080/21501203.2011.584577. [DOI] [Google Scholar]
- 99.Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F, Smirnova T, Nordberg H, Dubchak I, Shabalov I. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 2013;42(D1):D699–D704. doi: 10.1093/nar/gkt1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Miller MA, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. 2010 Gateway Computing Environments Workshop (GCE). 10.1109/GCE.2010.5676129.
- 102.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinf. 2009;10(1):421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Haon M, Grisel S, Navarro D, Gruet A, Berrin J-G, Bignon C (2015) Recombinant protein production facility for fungal biomass-degrading enzymes using the yeast Pichia pastoris. Front Microbiol 6(1002). 10.3389/fmicb.2015.01002 [DOI] [PMC free article] [PubMed]
- 107.Re R, Pellegrini N, Proteggente A, Pannala A, Yang M, Rice-Evans C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radical Biol Med. 1999;26(9):1231–1237. doi: 10.1016/S0891-5849(98)00315-3. [DOI] [PubMed] [Google Scholar]
- 108.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 109.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Daou M, Piumi F, Cullen D, Record E, Faulds CB. Heterologous production and characterization of two glyoxal oxidases from Pycnoporus cinnabarinus. Appl Environ Microbiol. 2016;82(16):4867–4875. doi: 10.1128/AEM.00304-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cordeiro FA, Faria CB, Barbosa-Tessmann IP. Identification of new galactose oxidase genes in Fusarium spp. J Basic Microbiol. 2010;50(6):527–537. doi: 10.1002/jobm.201000078. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files. All nucleotide sequence, protein sequence, and protein structural information used in this work were extracted from existing accessions in public databases, i.e., GenBank, Joint Genome Institute MycoCosm portal, and the Protein Data Bank.