

Abstract
The aim of the present study was to determine the role of Akt isoforms in insulin signaling and resistance in neuronal cells. By silencing Akt isoforms individually and in pairs, in Neuro-2a and HT22 cells we observed that, in insulin-sensitive condition, Akt isoforms differentially reduced activation of AS160 and glucose uptake with Akt2 playing the major role. Under insulin-resistant condition, phosphorylation of all isoforms and glucose uptake were severely affected. Over-expression of individual isoforms in insulin-sensitive and resistant cells differentially reversed AS160 phosphorylation with concomitant reversal in glucose uptake indicating a compensatory role of Akt isoforms in controlling neuronal insulin signaling. Post-insulin stimulation Akt2 translocated to the membrane the most followed by Akt3 and Akt1, decreasing glucose uptake in the similar order in insulin-sensitive cells. None of the Akt isoforms translocated in insulin-resistant cells or high-fat-diet mediated diabetic mice brain cells. Based on our data, insulin-dependent differential translocation of Akt isoforms to the plasma membrane turns out to be the key factor in determining Akt isoform specificity. Thus, isoforms play parallel with predominant role by Akt2, and compensatory yet novel role by Akt1 and Akt3 to regulate neuronal insulin signaling, glucose uptake, and insulin-resistance.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-021-03993-6.
Keywords: Akt1, Akt2, Akt3, AS160, Neuronal insulin signaling, Neuronal insulin resistance
Introduction
Neuronal insulin signaling is the primary pathway that regulates glucose homeostasis in the brain, with PI3K-Akt signaling pathway playing a principal role. Akt acts as a central node in neuronal insulin signaling and regulates cell proliferation and survival, cytoskeletal and vesicle trafficking, cellular metabolism, and glucose transport [1–3]. Any hindrance in this pathway paves the way for insulin-resistance like phenotype [3, 4]. We have observed hyperinsulinemia mediated insulin-resistance in neuronal cells in vitro, regulating a number of kinases and phosphatases [5, 6]. Frequently this has been linked to diseases like Alzheimer (Type-III diabetes) as observed by other laboratories and our [7, 8], including other diseases, like Huntington’s disease [9], spinocerebellar ataxia type 1 [10], schizophrenia [11], Parkinson’s disease [12], etc.
Akt is a serine/threonine kinase that belongs to AGC family. Akt comprises of three isoforms, Akt1, Akt2, and Akt3. They have been extensively studied in all insulin-responsive tissues, like skeletal muscles [13], adipocytes [14], and hepatocytes [15]. Akt1 expression varies across tissues, Akt2 is highly expressed in insulin-responsive tissues and Akt3 is expressed in brain and testis [16]. All three isoforms show high sequence similarity and common domain structure, with an N-terminal PH domain, a central kinase domain, and a C-terminal regulatory domain [1]. Akt1 plays a predominant role in cellular growth [17, 18], Akt2 plays a role in glucose homeostasis [19, 20], and Akt3 plays a role in neuronal development [21]. Studies have established that different isoforms have some overlapping yet diverse functions which can be attributed to differential expression [22], differential subcellular localization [23], regulatory partners [24], and different upstream stimulus [25].
In recent years there have been controversy regarding how the three Akt isoforms regulate glucose metabolism [26]. These studies have been conducted in vitro/in vivo, in normal and obese/diabetic condition, with single and/or double knockout or over-expressed samples. Akt2 has emerged as the predominant isoform regulating glucose metabolism in all metabolic tissues [19, 20, 27–29]. Majority of the reports say that Akt1 does not play any role in this regulation [13, 17, 18, 30]. However, there are others that report role of Akt1 in regulating insulin signaling and glucose transport [31] with some even supporting redundancy of Akt1 and Akt2 [14, 32, 33]. Akt3 is the least studied of the isoforms when it comes to glucose metabolism. Majority of the studies on Akt3 negate its role in regulating glucose metabolism [28].
All these studies confined to peripheral insulin-sensitive tissues indicate the role of Akt isoforms in insulin signaling is still ambiguous and could be tissue specific. There is hardly any information on Akt isoforms in regulating neuronal insulin signaling and resistance. All three Akt isoforms are expressed in the brain [16, 34]. Activation of Akt isoforms post-insulin stimulation in brain has also been studied [35]. However, majority of currently available studies focuses on the role of individual isoform in neuronal development, neuronal maturation and outgrowth, polarization and axon branching, synapse formation, and neuronal survival [36, 37]. Thus, no information is yet available to clarify the isoform specific role of Akt in neuron in insulin signaling and resistance and the underlying mechanisms that determine their functional specificity.
In our current study, we utilized neuronal cells like Neuro-2a and HT22, their insulin-resistant versions, generated in our laboratory [6, 8, 38, 39] and insulin-resistant diabetic mice whole brain tissues to study Akt isoform interplay. We elucidated this by isoform specific silencing [single (silencing of one isoform at a time) and double (silencing of two isoforms at a time)] as well as isoform specific over-expression, and elucidated how Akt isoforms regulate AS160 and ultimately glucose uptake in insulin-sensitive and resistant condition.
For clear understanding of the experimental objectives, a flowchart of the experimental design is depicted in Fig. S1.
Results
Effect of inhibition of Akt on neuronal insulin signaling and glucose uptake
To determine whether functional Akt protein exists in N2A cells, we inhibited Akt activity by MK2206, a well-known inhibitor of Akt activation [34], with or without insulin stimulation and determined its activation by assaying Akt phosphorylation and glucose uptake. To execute that, differentiated N2A cells were treated with or without 30 μM MK2206 for 30 min [34] and then stimulated with or without 100 nM insulin for 30 min. Cell lysates were western immunoblotted and probed with pan-anti-Akt antibody. No change was observed in the expression of Akt under all the conditions tested (Fig. 1A). However, the activation of Akt, as determined by phosphorylation at Ser-473, was decreased by 96.13% (Fig. 1A, lane 2 vs lane 4) due to MK2206 treatment. Inhibition of Akt activation led to a decrease in neuronal glucose uptake by 75.05% (Fig. 1B, lane 2 vs lane 4). Data indicate the presence of functional Akt in differentiated N2A cells participating in insulin signaling and glucose uptake.
Fig. 1.

Effect of Akt inhibition on neuronal insulin signaling and glucose uptake; and effect of insulin on Akt and its isoforms. A N2A cells were differentiated in 2% DMSO for 3 days and stimulated with or without 30 µM MK2206 and/or 100 nM insulin for 30 min as indicated. Cells were lysed and subjected to western blotting, followed by probing with relevant primary antibodies. Bar represents relative change in pAkt (Ser-473) probed with anti-Akt antibody. B Differentiated N2A cells were serum starved for 2 h, followed by 100 nM insulin for 30 min. Uptake of 2-NBDG was then measured. Bar represents relative change in uptake of 2-NBDG. C–E N2A cells were differentiated in 2% DMSO for 3 days and stimulated with or without 100 nM insulin for 30 min. Cells were lysed and subjected to western blotting, followed by probing with relevant primary antibodies. C Bar represents relative change in pAkt1 (Ser-473) probed with anti-Akt1 antibody. D Bar represents relative change in pAkt2 (Ser-474) probed with anti-Akt2 antibody. E Post-insulin stimulation, lysates were subjected to immunoprecipitation using anti-Akt3 antibody. Bar represents relative change in pAkt (Ser-473) probed with anti-Akt3 antibody. F–H HT22 cells were differentiated in neurobasal media containing 2 mmol/L glutamine and 1 × N2 supplement for 48 h and stimulated with or without 100 nM insulin for 30 min. Cells were lysed and subjected to western blotting, followed by probing with relevant primary antibodies. F Bar represents relative change in pAkt1 (Ser-473) probed with anti-Akt1 antibody. G Bar represents relative change in pAkt2 (Ser-474) probed with anti-Akt2 antibody. H Post-insulin stimulation, lysates were subjected to immunoprecipitation using anti-Akt3 antibody. Bar represents relative change in pAkt (Ser-473) probed with anti-Akt3 antibody. GAPDH has been used as a loading control (A, C, D, F, G). IgG band was used as loading control (E, H). Experiments were executed three times and a representative result is shown. Data expressed are mean ± SE. ***P < 0.001, **P < 0.01 compared to lane 1, ###P < 0.001, ##P < 0.01 compared to lane 2. IP Immunoprecipitation; IB Immunoblot; A.U Arbitrary Units
Effect of insulin on the expression and activation of Akt1, Akt2, and Akt3 in neuronal cells
Next, we sought out to determine whether all the isoforms of Akt exists in N2A cells and, if so, do they undergo activation by specific phosphorylations due to insulin stimulation. To test that lysates of differentiated N2A cells, with or without insulin stimulation, were subjected to western immunoblotting probed with isoform specific and phospho-isoform specific antibodies. Due to unavailability of a specific phospho-Akt3 antibody to measure phosphorylation levels of Akt3, Akt3 protein was immunoprecipitated using anti-Akt3 antibody, and probed with anti-phospho-Akt antibody. Anti-IgG conformational antibody was used to mask remaining IgG band. No change was observed in the expression of Akt1, Akt2 (GAPDH was used as a loading control) or Akt3 (because of the reasons, as mentioned above, IgG band was used as a loading control) (Fig. 1C–E). However, the activation of Akt1, Akt2 or Akt3 as determined by their phosphorylations at Ser-473, Ser-474, and Ser-472, respectively, was increased by tenfold as a function of insulin stimulation (Fig. 1C–E, lane 1 vs lane 2). To determine whether the above mentioned effects were N2A cell specific we tested the effect of insulin stimulation on individual Akt isoforms in differentiated HT22 cells, a hippocampal neuronal cell line [39, 40]. Ten fold increase in phosphorylation of individual isoforms were obtained as a function of insulin stimulation (Fig. 1 F–H). Data show that all Akt isoforms are expressed in N2A and HT22 cells and activated similarly as a function of insulin stimulation.
Effect of silencing of one isoform, on expression and phosphorylation of the others
To determine the effect of silencing of one isoform on the expression and activation of the others, individual isoform was silenced at the optimized dose, and its effect on expression and activation were tested on the other isoforms, with or without insulin stimulation, probing with isoform specific antibodies. Silencing was optimized and found to be 200 nM for Akt1, 100 nM for Akt2, and 150 nM for Akt3 required to obtain optimal silencing (Fig. S2, S3 and S4). Akt1 silencing (Akt−1) led to a decrease in phosphorylation and expression of Akt1 by 53.11% and 77.19%, respectively, (Fig. 2A, lane 2 vs lane 4; Fig. S2B, lane 1 vs lane 4, respectively); however, it did not affect expression or phosphorylation of Akt2 (Fig. 2B) and Akt3 (Fig. 2C). Similarly, Akt2 silencing (Akt−2) led to a decrease in phosphorylation and expression of Akt2 by 40.71% and 57.99%, respectively, (Fig. 2E, lane 2 vs lane 4; Fig. S3B, lane 1 vs lane 4, respectively) without affecting expression or phosphorylation of Akt1 (Fig. 2D) or Akt3 (Fig. 2F). Akt3 silencing (Akt−3) led to a decrease in phosphorylation and expression of Akt3 by 57.70% and 80.42%, respectively, (Fig. 2I, lane 2 vs lane 4; Fig. S4B, lane 1 vs lane 4, respectively) without affecting expression or phosphorylation of Akt1 (Fig. 2G) or Akt2 (Fig. 2H). Thus, data show that silencing of one isoform does not affect the others. These data also validate specificity of anti-Akt isoform specific antibodies. Similarly, to determine the effect of silencing of two isoforms at a time on the other, two isoforms were silenced in pairs (e.g., Akt−2–3 where Akt2 and Akt3 were silenced, Akt−1–3 where Akt1 and Akt3 were silenced and Akt−1–2 where Akt1 and Akt2 were silenced) and its effect on expression were tested on the other isoform. As expected, silencing of two isoforms in pairs did not affect the expression of the third isoform (Fig. S5).
Fig. 2.

Effect of silencing of one isoform, on expression and phosphorylation of other isoform in N2A cells. Three days post-proliferation, N2A cells were transfected with Akt1, Akt2 or Akt3 specific siRNA and then differentiated in 2% DMSO for 3 days. Cells were stimulated with or without 100 nM insulin for 30 min prior to cell lysis. Lysate was subjected to western blotting, followed by probing with relevant primary antibodies. A, D, G Bar represents relative change in pAkt1 (Ser-473) when probed with anti-Akt1 antibody. B, E, H Bar represents relative change in pAkt2 (Ser-474) when probed with anti-Akt2 antibody. C, F, I Bar represents relative change in pAkt (Ser-473) when probed with anti-Akt3 antibody. GAPDH has been used as a loading control (A, B, D, E, G, H). IgG band was used as loading control (C, F, I). Experiments were executed three times and a representative result is shown. Data expressed are mean ± SE. ***P < 0.001, **P < 0.01 as compared to lane 1; ###P < 0.001, ##P < 0.01 as compared to lane 2. IP Immunoprecipitation; IB Immunoblot
Effect of silencing of Akt isoforms, on expression and activation of AS160 (Akt Substrate of 160 kDa) (Ser-588 and Thr-642) and glucose uptake
Ser-588 and Thr-642 are two phosphorylation sites on AS160 those have been reported to be regulated by Akt and are indications of AS160 activation. Postsingle or double silencing, differentiated N2A cells with or without insulin stimulation (100 nM, 30 min), were subjected to western immunoblotting probed with anti-AS160 antibody and anti-phospho AS160 Ser-588 or anti-phospho AS160 Thr-642 antibodies. There was no change in expression of AS160 under all the conditions tested (Fig. 3, A–H). However, post-single silencing, activation of AS160 as determined by phosphorylation at Ser-588, was down-regulated by insulin stimulation post-Akt−1 by 15.75% (Fig. 3A, lane 2 vs lane 4), Akt−2 by 24.45% (Fig. 3B, lane 2 vs lane 4), and Akt−3 by 26.98% (Fig. 3C, lane 2 vs lane 4) silencing as compared to scrambled siRNA transfected cells. Similarly, post-double silencing, activation of AS160 was down-regulated by insulin stimulation post-Akt−2–3 by 32.74% (Fig. 3D, lane 2 vs lane 4), Akt−1–3 by 26.60% (Fig. 3D, lane 2 vs lane 6) and Akt−1–2 by 27.57% (Fig. 3D, lane 2 vs lane 8) silencing as compared to scrambled siRNA transfected cells with insulin stimulation. Correspondingly, the activation of AS160 as determined by phosphorylation at Thr-642 was down-regulated by insulin stimulation post-Akt−1 by 15.20% (Fig. 3E, lane 2 vs lane 4), Akt−2 by 63.41% (Fig. 3F, lane 2 vs lane 4) and Akt−3 by 50.47% (Fig. 3G, lane 2 vs lane 4) silencing as compared with scrambled siRNA transfected cells. Similarly, post-double silencing, activation of AS160 was down-regulated by insulin stimulation post-Akt−2–3 by 55% (Fig. 3H, lane 2 vs lane 4), Akt−1–3 by 43.09% (Fig. 3H, lane 2 vs lane 6), and Akt−1–2 by 50.35% (Fig. 3H, lane 2 vs lane 8) silencing as compared with scrambled siRNA transfected cells by insulin stimulation. The AS160 threonine phosphorylation was more severely affected as compared to corresponding serine. Previous studies have reported Thr-642 phosphorylation as being more important for AS160 function as compared to its Ser-588 [41, 42]. Thus, silencing the isoforms demonstrates that all three isoforms down-regulate AS160 activation at both Ser-588 and Thr-642, without any effect on its expression. Highest down-regulation was caused by Akt2, followed by Akt3 and then Akt1. When one isoform is absent other isoforms differentially regulate AS160 phosphorylation. However, when two isoforms are absent AS160 phosphorylation is regulated in a kind of a compensatory manner. Data strongly suggest that Akt isoforms does participate in insulin signaling in N2A cells via AS160.
Fig. 3.

Effect of Akt isoform silencing on AS160 in N2A cells. Three days post-proliferation, N2A cells were transfected with single (Akt−1, Akt−2, and Akt−3) or double (Akt−2–3, Akt−1–3 or Akt−1–2) specific siRNA and then differentiated in 2% DMSO for 3 days. Cells were stimulated with or without 100 nM insulin for 30 min prior to cell lysis. Lysate was subjected to western blotting, followed by probing with relevant primary antibodies. (A–D) Bar represents relative change in pAS160 (Ser-588) when probed with anti-AS160 antibody. (E–H) Bar represents relative change in pAS160 (Thr-642) when probed with anti-AS160 antibody. Experiments were executed three times and a representative result is shown. Data expressed are mean ± SE. ***P < 0.001, **P < 0.01, *P < 0.05 as compared to lane 1; ###P < 0.001, ##P < 0.01, #P < 0.05 as compared to lane 2. IB Immunoblot
Having observed this we sought out to determine how does those activations reflect on neuronal glucose uptake. Therefore, single or double silenced differentiated N2A cells were stimulated with or without insulin and subjected to glucose uptake assay. As shown in Fig. 4 A and B, all three isoforms, irrespective of single or double silencing, contributed in glucose uptake in N2A cells. Under single silenced condition, Akt−1, Akt−2 or Akt−3 silencing caused a decrease of 35.98% (Fig. 4A, lane 2 vs lane 4), 53.64% (Fig. 4A, lane 2 vs lane 6), and 48.53% (Fig. 4A, lane 2 vs lane 8), respectively, in insulin stimulated 2-NBDG uptake when compared to their respective scrambled siRNA transfected cells. Under double silenced condition, it was observed that Akt−2–3 silenced cells showed a maximum decrease of 76.73% in insulin stimulated 2-NBDG uptake as compared to scrambled siRNA transfected cells (Fig. 4B, lane 2 vs lane 4). Similarly, Akt−1–2 and Akt−1–3 silenced cells also caused 61.13% (Fig. 4B, lane 2 vs lane 8) and 54.19% (Fig. 4B, lane 2 vs lane 6) decrease, respectively, in insulin stimulated 2-NBDG uptake as compared with scrambled siRNA transfected cells. Thus, data demonstrate that Akt2 contributes most to the glucose uptake, followed by Akt3 and then Akt1, suggesting that there is an isoform specific regulation of the degree of glucose uptake in N2A cells. A summary of outcome of silencing is presented in Table S1.
Fig. 4.

Effect of Akt isoform silencing on glucose uptake. Three days post-proliferation, cells were transfected with (A) Single (Akt−1, Akt−2 or Akt−3) (N2A) (B) Double (Akt−2–3, Akt−1–3, Akt−1–2) (N2A) (C) Double (Akt−2–3, Akt−1–3, Akt−1–2) (HT22) specific siRNA and then differentiated for 3 days. Differentiated cells were serum starved for 2 h, followed by 100 nM insulin for 30 min. Uptake of 2-NBDG was then measured. Bar represents relative change in uptake of 2-NBDG. Experiments were executed three times and a representative result is shown. Data expressed are mean ± SE. ***P < 0.001, **P < 0.01 as indicated
In HT22 cells, it was found that Akt−1–3 silenced cells caused a decreased of 55.85% in insulin stimulated 2-NBDG uptake as compared to scrambled siRNA transfected cells (Fig. 4C, lane 2 vs lane 6). Similarly, Akt−2–3 and Akt−1–2 silenced cells also showed 76.73% and 65.91% decrease, respectively, in insulin stimulated 2-NBDG uptake as compared to scrambled siRNA transfected cells (Fig. 4C, lane 2 vs lane 4; and lane 2 vs lane 8, respectively). Therefore, data strongly indicated that in neuronal cells all Akt isoforms contribute in regulating neuronal glucose uptake and Akt2 contributes to the maximum, followed by Akt3 and Akt1.
Effect of insulin on subcellular translocation of Akt1, Akt2, Akt3, and AS160 in neuronal system
Recent reports in adipocytes and skeletal muscle cells demonstrate that, insulin stimulated subcellular translocation and membrane localization of Akt isoforms [23, 43] and AS160 [43] determines Akt isoform specific signaling. One of the first things that the translocated Akt isoform at the membrane does is that it interacts with AS160 to undertake the function of glucose uptake [24, 44, 45]. In neuronal system, there is no study of insulin-dependent translocation of Akt isoforms to plasma membrane. To address this, differentiated N2A cells stimulated with or without insulin (100 nM, 30 min) were lysed and fractionated into cytoplasmic and membrane fractions. Expression of Caveolin-1 is considered as a plasma membrane marker [46]. Thus, expression of Caveolin-1 was tested to establish purity of membrane fractions. Expression of Caveolin-1 was only observed in membrane fraction as compared to cytoplasm (Fig.S6, lane 1 vs lane 2). However, in all membrane translocation experiments GAPDH was used as a loading control, to perform densitometric analysis, as it is present in both “cytoplasm” and “membrane”, as compared to only membrane-specific markers (which cannot be used for densitometry analysis of “cytoplasm” fractions) [47]. Our data show that in cytoplasmic fraction, post-insulin stimulation expression of Akt1 was down-regulated by 12.77% (Fig. 5A, Panel 1 and 4, lane 1 vs lane 2), Akt2 by 13.02% (Fig. 5A, Panel 2 and 4, lane 1 vs lane 2), and Akt3 by 12.53% (Fig. 5A, Panel 3 and 4, lane 1 vs lane 2). In contrast to that, post-insulin stimulation in the membrane fraction, Akt1 translocated and accumulated on the plasma membrane by 59.64% (Fig. 5A, Panel 1 and 4, lane 3 vs lane 4), Akt2 by 230.18% (Fig. 5A, Panel 2 and 4, lane 3 vs lane 4), and Akt3 by 152.51% (Fig. 5A, Panel 3 and 4, lane 3 vs lane 4). Data show that as a function of insulin stimulation all Akt isoforms translocated to the plasma membrane, with Akt2 translocating the most, followed by Akt3 and Akt1 as compared to respective cytoplasmic expression without insulin. In HT22 cells, in cytoplasmic fractions, insulin stimulation down-regulated Akt1 expression by 12% (Fig. 5B, Panel 1 and 4, lane 1 vs lane 2), Akt2 by 12.44% (Fig. 5B, Panel 2 and 4, lane 1 vs lane 2), and Akt3 by 11.05% (Fig. 5B, Panel 3 and 4, lane 1 vs lane 2). Post-insulin stimulation, in the membrane fraction, Akt1 translocated and accumulated on the plasma membrane by 48.16% (Fig. 5B, Panel 1 and 4, lane 3 vs lane 4), Akt2 by 137% (Fig. 5B, Panel 2 and 4, lane 3 vs lane 4), and Akt3 by 80.29% (Fig. 5B, Panel 3 and 4, lane 3 vs lane 4). The trend of this membrane translocation correlates with the trend as observed in glucose uptake (Fig. 4 A, B). Data show that all isoforms translocate to the plasma membrane post-insulin stimulation to different extents. Akt2 comes out as the predominant isoform on the plasma membrane, followed by Akt3 and Akt1 in both the cell lines tested. Therefore, data prove that it is the abundance of a specific Akt isoform in the membrane due to translocation from cytoplasm as a function of insulin stimulation that regulate glucose uptake.
Fig. 5.


Mechanism underlying Akt isoform specificity in neuronal cells. A N2A cells were differentiated in 2% DMSO for 3 days. Three days post-differentiation, membrane and cytosol fraction were isolated and subjected to western blotting, followed by probing with relevant primary antibodies. Bar represents relative change in Akt1, Akt2 or Akt3 when probed with anti-Akt isoform specific antibody. B HT22 cells were differentiated in neurobasal media containing 2 mmol/L glutamine and 1 × N2 supplement for 48 h. Three days post-differentiation, membrane and cytosol fraction were isolated and subjected to western blotting, followed by probing with relevant primary antibodies. Bar represents relative change in Akt1, Akt2 or Akt3 when probed with anti-Akt isoform specific antibody. C, D Bar represents relative change in AS160 when probed with anti-AS160 antibody (N2A/HT22 as indicated). Bar represents relative change in pAS160 (Thr-642) when probed with anti-AS160 antibody (N2A/HT22 as indicated). GAPDH has been used as a loading control. Experiments were executed three times and a representative result is shown. Data expressed are mean ± SE. ***P < 0.001, **P < 0.01, *P < 0.05 compared to lane 1 or as indicated. IB Immunoblot
As did earlier, we tested the compartmentalised Akt isoform interaction(s) with AS160 in the cytoplasm and plasma membrane fractions. Phosphorylation of AS160 at Thr-642 showed increase post-insulin stimulation in cytoplasmic fraction by 71.71% (Fig. 5C, Panel 1 and 2, lane 1 vs lane 2) and membrane fraction by 437.31% (Fig. 5C, Panel 1 and 2, lane 3 vs lane 4) as compared to their respective controls without insulin. Expression of AS160 increased in cytoplasm post-insulin stimulation by 17.06% (Fig. 5C, Panel 2 and 3, lane 1 vs lane 2). However, there was a sharp decrease by 67.46% in membrane bound AS160 post-insulin stimulation as compared to samples without stimulation (Fig. 5C, Panel 2 and 3, lane 3 vs lane 4). In HT22 cells, phosphorylation of AS160 at Thr-642 showed similar increase in cytoplasm by 78.17% (Fig. 5D, Panel 1 and 2, lane 1 vs lane 2) and membrane fraction by 479.96% (Fig. 5D, Panel 1 and 2, lane 3 vs lane 4) as compared to their respective controls without insulin. Expression of AS160 increased in cytoplasm, post-insulin stimulation by 17.67% (Fig. 5D, Panel 2 and 3, lane 1 vs lane 2). As in N2A, there was a sharp decrease in membrane bound AS160 post-insulin stimulation as compared to control by 63.57% (Fig. 5D, Panel 2 and 3, lane 3 vs lane 4). Thus, data show the regulation of AS160 by phosphorylation at Thr-642 by activated Akt in neuronal cells.
To further investigate this insulin-dependent redistribution of AS160 in neuronal cells, and how this redistribution correlates with GLUT4 translocation, we examined membrane translocation and possible co-localization of AS160 and GLUT4. Differentiated N2A cells were stimulated with or without insulin, immunostained with anti-GLUT4 and anti-AS160 antibody and were subjected to confocal microscopy (Fig. 6). Immunocytochemical analysis revealed that under basal conditions, GLUT4 was localized inside the cell (Fig. 6I, Panel C, red arrows), and AS160 was distributed just below the membrane (Fig. 6I, Panel D, green arrows), possibly tethering GLUT4 to the GSVs. Co-localization of AS160 and GLUT4 inside the cell was observed (Fig. 6I, Panel E, yellow arrows). The two proteins co-localized with a Pearson’s coefficient of 0.53 (Fig. 6II). Post-insulin stimulation, GLUT4 was found to be present on the membrane (Fig. 6I, Panel H, red arrows), considerably more than unstimulated cells (Fig. 6I, Panel C vs Panel H). Alternatively, AS160 was found dispersed inside the cytoplasm (Fig. 6I, Panel I, green arrows) as compared to unstimulated cells (Fig. 6I, Panel D vs Panel I). Co-localization of AS160 and GLUT4 was observed (Fig. 6I, Panel J, yellow arrows) with a Pearson's coefficient of 0.37 (Fig. 6II), however, GLUT4 was seen at the membrane independently as well, thus decreasing the co-localization (Fig. 6I, Panel J, red arrows) by a significant 30% with or without insulin stimulation (Fig. 6II). Data strongly show insulin-dependent redistribution of AS160 in neuronal cells, and how this redistribution correlates with GLUT4 translocation and glucose uptake.
Fig. 6.

Effect of insulin on subcellular translocation of GLUT4 and AS160. I Three days post-proliferation, N2A cells differentiated in 2% DMSO for 3 days. Cells were stimulated with or without 100 nM insulin for 30 min followed by fixation and permeabilization and probed with anti-GLUT4 and anti-AS160 antibody. Cells were subjected to immunofluorescence microscopy using anti-goat Alexa 555 and anti-rabbit CFL 488 secondary antibodies, respectively. Bar corresponds to 10 μm. Images were captured from different fields and a representative image of 3 images is presented. Red arrow indicates GLUT4, green arrow indicates AS160 and yellow arrow indicates co-localized GLUT4 and AS160. II Graphical representation of co-localization by Pearson’s coefficient (*P < 0.05)
Subcellular translocation of Akt1, Akt2, Akt3, and AS160 in mice whole brain tissue
Having observed in two different neuronal cells, we wished to assess whether the translocation of Akt isoforms is indeed critical, we tested it in High-Fat-Diet (HFD) fed diabetic mice against Normal Diet fed mice (ND) (Kind gift from Dr. Prosenjit Mondal, Indian Institute of Technology—Mandi, Himachal Pradesh, India). Our data show that in cytoplasmic fractions of HFD mice Akt1 expression was down-regulated by 11.07% (Fig. 7A, Panel 1 and 4, lane 1 vs lane 2), Akt2 expression was down-regulated by 13.71% (Fig. 7A, Panel 2 and 4, lane 1 vs lane 2), and Akt3 expression was down-regulated by 13.55% (Fig. 7A, Panel 3 and 4, lane 1 vs lane 2) as compared to respective ND mice controls. In contrast to this, in the membrane fraction of HFD mice, none of the Akt isoforms translocated to the plasma membrane (Fig. 7B, lane 3 vs lane 4). This is an interesting result as this not only corroborates with our results in neuronal cells, but also points to novel role of all three Akt isoforms in neuronal insulin resistance.
Fig. 7.

Mechanism underlying Akt isoforms specificity in mice whole brain tissue. A Subcellular translocation of Akt isoforms in mice whole brain tissue. Mice whole brain was lysed, membrane and cytosol fraction were isolated and subjected to western blotting, followed by probing with relevant primary antibodies. Bar represents relative change in Akt1, Akt2 or Akt3 when probed with anti-Akt isoform specific antibody. Experiments were executed with three independent animals and a representative result is shown. B Subcellular redistribution of AS160 post-insulin stimulation. Bar represents relative change in AS160 when probed with anti-AS160 antibody. Bar represents relative change in pAS160 (Thr-642) when probed with anti-AS160 antibody. GAPDH has been used as a loading control. Experiments were executed with three independent animals and a representative result is shown. Data expressed are mean ± SE. ***P < 0.001, **P < 0.01, *P < 0.05 compared to lane 1 or as indicated. (ND Normal Diet; HFD High-Fat-Diet). IB Immunoblot
Testing expression and phosphorylation of AS160 Thr-642 in the cytoplasm and plasma membrane fractions of ND and HFD mice, as described above (Fig. 7A), phosphorylation of AS160 at Thr-642 decreased in cytoplasmic fraction by 19.96% (Fig. 7B, Panel 1 and 2, lane 1 vs lane 2), and in membrane fraction by 46.70% (Fig. 7B, Panel 1 and 2, lane 3 vs lane 4) in HFD as compared to ND mice. Expression of AS160 decreased in cytoplasm by 29.28% (Fig. 7B, Panel 2 and 3, lane 1 vs lane 2) and plasma membrane by 51.47% (Fig. 7B, Panel 2 and 3, lane 3 vs lane 4) in ND as well as HFD mice.
This is the first study to report differential subcellular translocation of Akt isoforms under ND and HFD condition in mice whole brain tissue. This result prompted us to study the role of Akt isoforms in insulin-resistant neuronal cells.
Expression and activation of Akt1, Akt2, and Akt3 in insulin-resistant neuronal cells
We had previously generated an insulin-resistant diabetic neuronal cell model by differentiating N2A in the chronic presence of insulin (100 nM) in serum-free medium (MFI) [6, 8, 48–50]. This model cellular system was used in the present study to investigate the role of Akt isoforms in insulin-sensitive (MF) and insulin-resistant (MFI) condition. No change was observed in the expression of Akt1, Akt2 or Akt3 post-insulin stimulation in MF and MFI condition (Fig. 8A, B, C). However, phosphorylation of Akt1, Akt2 or Akt3 at Ser-473, Ser-474, and Ser-472, respectively, was decreased differentially post-insulin stimulation in MF and MFI condition. Insulin stimulation led to a decrease in Akt1 phosphorylation by 43.36% (Fig. 8A, lane 2 vs lane 4), in Akt2 phosphorylation by 72.79% (Fig. 8B, lane 2 vs lane 4) and in Akt3 phosphorylation by 52.45% (Fig. 8C, lane 2 vs lane 4). Previous studies in skeletal muscles and adipocytes of diabetic obese patients, Akt2 phosphorylation was reported to be affected [51, 52]. Our data point to differential role of all three Akt isoforms, as phosphorylation of all was affected in neuronal insulin-resistant condition, with Akt2 phosphorylation affected the most, followed by Akt3 and then Akt1.
Fig. 8.


Expression and activation of Akt1, Akt2, and Akt3, glucose uptake and subcellular translocation under insulin-resistant condition in neuronal cells. (A–C) N2A cells were differentiated in serum-free medium in the absence of (MF) or chronic presence of 100 nM insulin (MFI) for 3 days. Cells were lysed and subjected to western blotting, followed by probing with relevant primary antibodies. Bar represents relative change when probed with anti-Akt isoform specific antibody. A Bar represents relative change in pAkt1 (Ser-473) probed with anti-Akt1 antibody. B Bar represents relative change in pAkt2 (Ser-474) probed with anti-Akt2 antibody. C Post-insulin stimulation, lysates were subjected to immunoprecipitation using anti-Akt3 antibody. Bar represents relative change in pAkt (Ser-473) probed with anti-Akt3 antibody. D Three days post-proliferation, N2A cells were transfected with double (Akt−2–3, Akt−1–3 or Akt−1–2) specific siRNA and then differentiated under MF MFI condition for 3 days. Differentiated N2A cells were serum starved for 2 h, followed by 100 nM insulin for 30 min. Uptake of 2-NBDG was then measured. E Subcellular translocation of Akt isoforms post-insulin stimulation. N2A cells were differentiated under MF MFI condition for 3 days and stimulated with or without 100 nM insulin for 30 min as indicated. Cells were lysed, and membrane and cytosol fraction were isolated and subjected to western blotting, followed by probing with relevant primary antibodies. Bar represents relative change in Akt1, Akt2 or Akt3 when probed with anti-Akt isoform specific antibody. F Subcellular translocation of AS160 post-insulin stimulation. Bar represents relative change in pAS160 (Thr-642) when probed with anti-AS160 antibody. GAPDH has been used as a loading control. Experiments were executed three times and a representative result is shown. Data expressed are mean ± SE. ***P < 0.001, **P < 0.01, *P < 0.05 compared to lane 1 or as indicated; ###P < 0.001, ##P < 0.01, #P < 0.05 compared to lane 2. IP Immunoprecipitation; IB Immunoblot; A.U Arbitrary Units
Effect of silencing of Akt isoforms in insulin-resistant neuronal cells
To determine whether there is any isoform specific role in regulating neuronal glucose uptake under insulin-resistant condition, Akt isoforms were silenced in pair (Akt−2–3, Akt−1–3 and Akt−1–2). It was found that, in MF condition, the pattern was similar as observed previously in N2A (Fig. 4B) as well as in HT22 cells (Fig. 4C). Under MF condition, Akt−1–3 silenced N2A cells showed 50.70% decrease in insulin stimulated 2-NBDG uptake as compared to scrambled siRNA transfected cells (Fig. 8D, lane 2 vs lane 6). Similarly, Akt−2–3 and Akt−1–2 silenced cells also showed 71.28% and 61.86% decrease, respectively, in insulin stimulated 2-NBDG uptake as compared to scrambled siRNA transfected cells (Fig. 8D, lane 2 vs lane 4; and lane 2 vs lane 8, respectively). Akt2 contributes most to glucose uptake, followed by Akt3 and then Akt1 in MF N2A cells. However, interestingly, in MFI condition, Akt−2–3, Akt−1–3, and Akt−1–2 silenced N2A cells showed similar decrease in insulin stimulated 2-NBDG uptake, indicating that all Akt isoforms are affected in insulin-resistant neuronal cells. Akt−1–3 silenced N2A cells showed 83.64% decrease in insulin stimulated 2-NBDG uptake as compared to scrambled siRNA transfected cells (Fig. 8D, lane 2 vs lane 14). Similarly, Akt−2–3 and Akt−1–2 silenced cells also showed 83.14% and 82.95% decrease, respectively, in insulin stimulated 2-NBDG uptake as compared to scrambled siRNA transfected cells (Fig. 8D, lane 2 vs lane 12; and lane 2 vs lane 16, respectively). Data consolidate that all Akt isoforms contribute in neuronal glucose uptake in insulin-sensitive as well as insulin-resistant cells.
Effect of insulin stimulation on the translocation of Akt isoforms and AS160 in insulin-resistant neuronal cells
Having established the involvement of all Akt isoforms in regulating neuronal insulin-resistance we proceeded to test their subcellular translocation post-insulin stimulation. Our data show that, under MF condition, Akt1, Akt2, and Akt3 maintained a similar trend as in Fig. 5A, that is, Akt2 translocated and accumulated to the plasma membrane the most by 245.47% (Fig. 8E, Panel 2 and 4, lane 3 vs lane 4), with Akt1 and Akt3 by 93.42% (Fig. 8E, Panel 1 and 4, lane 3 vs lane 4) and 155.94% (Fig. 8E, Panel 3 and 4, lane 3 vs lane 4), respectively, as compared to their respective controls without insulin. In contrast to this, none of the Akt isoforms translocated to the plasma membrane post-insulin stimulation under resistant condition (Fig. 8E, lane 7 vs lane 8). This is similar to our data observed in mice whole brain tissue, where translocation of all isoforms were affected in HFD diabetic mice. Data show that, translocation of all isoforms is affected under insulin-resistant condition.
Testing of expression and phosphorylation of AS160 Thr-642 in the cytoplasm and plasma membrane fractions in insulin-sensitive and insulin-resistant cells showed the expression of AS160 increased in cytoplasmic fraction in MF by 21.46% but remained unchanged in MFI condition (Fig. 8F, Panel 2 and 3, lane 1 vs lane 2, and lane 5 vs lane 6, respectively). However, in contrast to MF condition, where expression of AS160 decreased in membrane fraction by 46.39% (Fig. 8F, Panel 2 and 3, lane 3 vs lane 4), there was only a 27.51% decrease under MFI condition (Fig. 8F, Panel 2 and 3, lane 7 vs lane 8). Thus, insulin-dependent AS160 dissociation from plasma membrane reduces in insulin-resistant condition, possibly due to inefficient upstream Akt phosphorylation under MFI condition. Phosphorylation of AS160 at Thr-642, under MF condition, showed increase in cytoplasmic fraction by 157.03% (Fig. 8F, Panel 1 and 2, lane 1 vs lane 2) and membrane fraction by 291.44% (Fig. 8F, Panel 1 and 2, lane 3 vs lane 4). Under MFI condition, phosphorylation of AS160 in cytoplasmic fraction increased by 193.50% (Fig. 8F, Panel 1 and 2, lane 5 vs lane 6), but this increase was nominal as compared to MF condition which was 47.40% (Fig. 8F, Panel 1 and 2, lane 2 vs lane 6). However, there was no significant change in phosphorylation of AS160 in membrane fraction under MFI condition (Fig. 8F, Panel 1 and 2, lane 7 vs lane 8). Data strongly show that in insulin-resistant condition, improper translocation of Akt isoforms to plasma membrane regulates GLUT4 translocation and thereby reduced glucose uptake.
Effect of Akt1, Akt2 and Akt3 over-expression on expression and activation of AS160 under insulin-resistant condition
Having seen for the first time that all isoforms are differentially affected under insulin-resistant condition, we wished to understand whether over-expression of specific isoforms in resistant condition can cause reversal of resistance. To undertake this experiment, we over-expressed each isoform individually and transfected cells were then subjected to MF and MFI conditions, and the effect of over-expression on specific isoform was tested with or without insulin. As compared to controls (respective empty plasmid backbones), Akt+1, Akt+2 or Akt+3 over-expression led to a ~ tenfold increase in expression of Akt1 (Fig. 9A), Akt2 (Fig. 9C) or Akt3 (Fig. 9E), respectively. There was no change in expression of AS160 under all the conditions tested (Fig. 9B, D, F). However, under all control conditions (respective empty plasmid backbone), activation of AS160, as determined by phosphorylation at Thr-642, was down-regulated from MF to MFI conditions (Fig. 9B, D, E lane 2 vs lane 4). Due to over-expression of Akt+1, activation of AS160 was up-regulated by insulin stimulation by 30.70% (Fig. 9B, lane 2 vs lane 6), and by 172.15% (Fig. 9B, lane 4 vs lane 8) under MF and MFI conditions, respectively. Similarly, post-Akt+2 over-expression activation of AS160 was up-regulated by insulin stimulation by 48.66% (Fig. 9D, lane 2 vs lane 6), and by 331.84% (Fig. 9D, lane 4 vs lane 8) under MF and MFI conditions, respectively. Post-Akt+3 over-expression activation of AS160 was up-regulated by insulin stimulation by 31.62% (Fig. 9F, lane 2 vs lane 6), and by 247.55% (Fig. 9F, lane 4 vs lane 8) under MF and MFI conditions, respectively. Activation of AS160 by insulin stimulation under MFI condition was affected most by Akt+2 by 38.26% (Fig. 9D, lane 2 vs lane 8), as compared to Akt+1 by 0.12% (Fig. 9B, lane 2 vs lane 8) or Akt+3 by 11.50% (Fig. 9F, lane 2 vs lane 8). Thus, over-expression of all Akt isoforms was able to up-regulate AS160 phosphorylation in insulin-sensitive (MF) and insulin-resistant (MFI) condition.
Fig. 9.

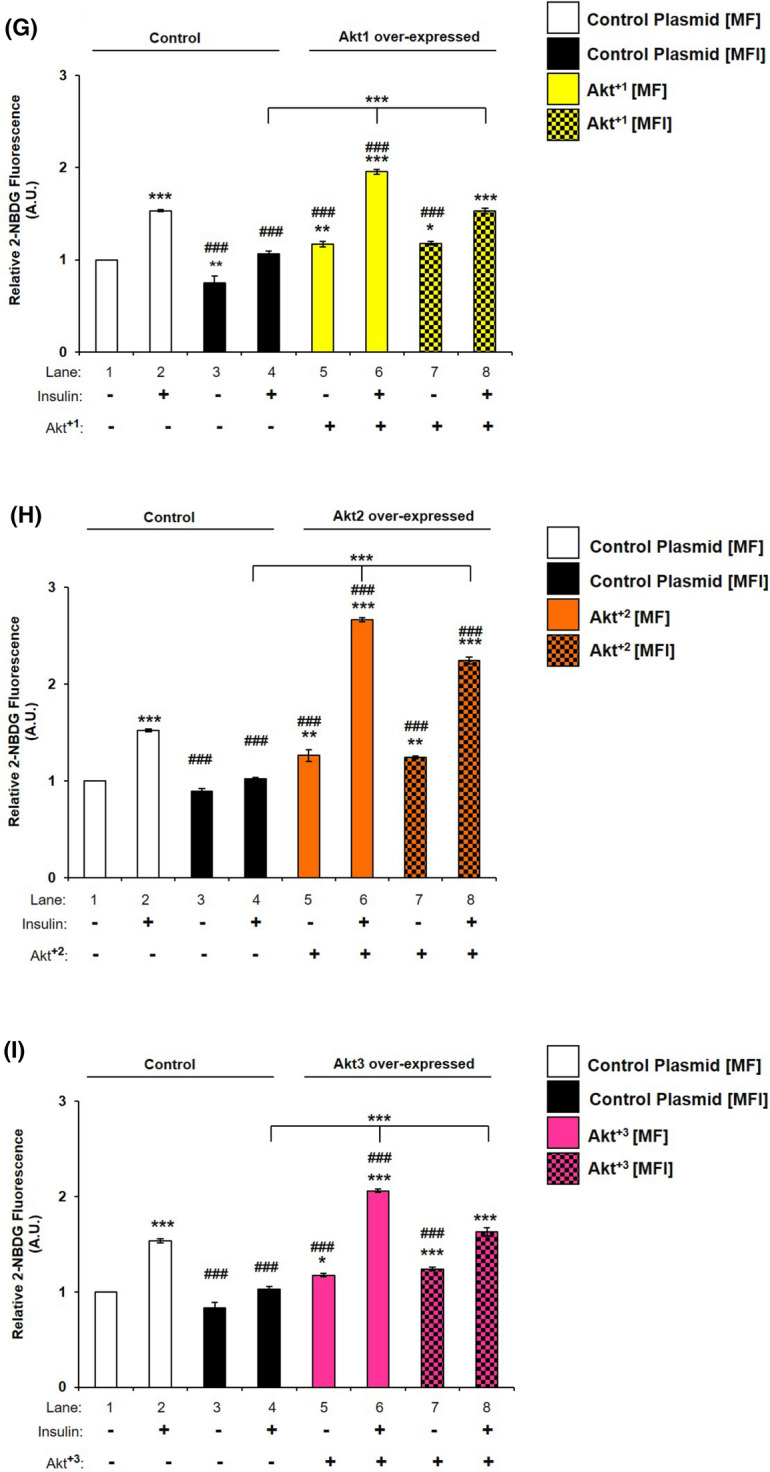
Effect of Akt1, Akt2, and Akt3 over-expression on expression and activation of AS160, and neuronal glucose uptake under insulin-resistant condition in neuronal cells. A–F Three days post-proliferation, Akt1, Akt2 or Akt3 were over-expressed using isoform specific plasmids. N2A cells were differentiated in serum-free medium in the absence of (MF) or chronic presence of 100 nM insulin (MFI) for 3 days. Cells were lysed and subjected to western blotting, followed by probing with relevant primary antibodies. Bar represents relative change when probed with anti-Akt isoform specific antibody. A, C, E Bar represents relative change in Akt1/Akt2/Akt3 probed with anti-Akt isoform specific antibody. GAPDH was used as loading control B, D, F Bar represents relative change in pAS160 (Thr-642) when probed with anti-AS160 antibody. G–I Three days post-proliferation, Akt1, Akt2 or Akt3 were over-expressed using isoform specific plasmids. N2A cells were differentiated in serum-free medium in the absence of (MF) or chronic presence of 100 nM insulin (MFI) for 3 days. N2A cells were serum starved for 2 h, followed by 100 nM insulin for 30 min. Uptake of 2-NBDG was then measured. Experiments were executed three times and a representative result is shown. Data expressed are mean ± SE. ***P < 0.001, **P < 0.01, *P < 0.05 compared to lane 1 or as indicated, ###P < 0.001, ##P < 0.01, #P < 0.05 compared to lane 2. IB Immunoblot; A.U Arbitrary Units
Effect of Akt1, Akt2, and Akt3 over-expression on neuronal glucose uptake under insulin-resistant condition
To determine how isoform specific over-expression regulates neuronal glucose uptake in insulin-sensitive and insulin-resistant condition, Akt1, Akt2 or Akt3 were over-expressed and subjected to glucose uptake assay. Under MF condition, Akt+2 over-expressed N2A cells showed 75.05% increase in insulin stimulated 2-NBDG uptake as compared to control-transfected cells (Fig. 9H, lane 2 vs lane 6). Similarly, Akt+1 or Akt+3 over-expressed N2A cells showed 27.50% and 34.06% increase, respectively, in insulin stimulated 2-NBDG uptake as compared to respective control-transfected cells (Fig. 9G, lane 2 vs lane 6; Fig. 9I and lane 2 vs lane 6, respectively). Similarly, in MFI condition, Akt+2 over-expressed N2A cells showed 118.85% increase in insulin stimulated 2-NBDG uptake as compared to control-transfected cells (Fig. 9H, lane 4 vs lane 8). Akt+1 or Akt+3 over-expressed N2A cells showed 44.12% and 58.82% increase, respectively, in insulin stimulated 2-NBDG uptake as compared to respective control-transfected cells (Fig. 9G, lane 4 vs lane 8; Fig. 9I lane 4 vs lane 8, respectively). However, important point to note that only Akt+2 over-expression up-regulated neuronal glucose uptake under MFI condition by 47.43% as compared to MF condition (Fig. 9H, lane 2 vs lane 8). Overall, Akt+2 over-expression influenced neuronal glucose uptake most, followed by Akt3 or Akt1 in MF as well as MFI condition. Data prove that Akt isoforms over-expression reverses insulin-resistance in neuronal cells. Although Akt isoforms were differentially silenced as presented above, but the functional trend was revalidated through the over-expression data.
Overall, all isoforms participate in reversal, however, the effect is differential between the isoforms.
Discussion
Compensatory role of Akt isoforms in regulating neuronal glucose uptake
In the present study, utilizing two different neuronal cell lines we are reporting for the first time that in neuronal system, all isoforms of Akt contribute to neuronal glucose uptake. It turns out that Akt2 contributes most, followed by Akt3 and then Akt1 (Fig. 4 A–C). However, from single isoform silenced condition to double, we noticed an additional decrease in glucose uptake, but not additive, although maintaining the trend under both conditions (Table S1). This points to a functional compensatory role of Akt isoforms under double isoform silenced condition. This compensation has previously been reported in neuronal system with requirement of all Akt isoforms in regulating Tau phosphorylation [53]. Interestingly, in insulin-resistant condition, double silencing decreased glucose uptake further (Fig. 8D). However, this decrease was not isoforms specific, pointing to functional compensation yet again. In addition to this, even post-double silencing, the sustained glucose uptake points to additional pathways working in regulating glucose metabolism. These results were further corroborated by isoform specific over-expression studies under insulin-resistant condition. While over-expression of all three isoforms increased neuronal glucose uptake in insulin-sensitive as well as insulin-resistant condition, only Akt2 over-expression increased glucose uptake in insulin-resistant condition to a level comparable to its respective insulin-sensitive condition (Fig. 9H, lane 2 vs lane 8). Our study reports a predominant role of Akt2, novel role of Akt1 and Akt3, and functional compensation by all Akt isoforms in regulating neuronal glucose uptake.
Subcellular translocation determines Akt isoform specificity
In the present study, we examined translocation of Akt isoforms post-insulin stimulation in N2A (Fig. 5A) and HT22 (Fig. 5B) cell lines, and all isoforms translocated to plasma membrane post-insulin stimulation. Interestingly, Akt2 translocated to the plasma membrane the most, followed by Akt3 and Akt1. This has been explained in Fig. 10B. Previous studies by McGraw and Gonzalez (in adipocytes) and Zheng and Cartee (in rat skeletal muscles) have discussed only Akt2 translocates post-insulin stimulation, with very limited translocation of Akt1 [23, 43]. Akt3 has not been studied. We also studied subcellular translocation of Akt isoforms in insulin-resistant diabetic mice whole brain tissue lysate [Normal Diet vs High-Fat-Diet] (Fig. 7A) and in N2A (Fig. 8E). Resistance caused due to high-fat-diet or hyperinsulinemia, caused all Akt isoforms to be affected, such that none of the isoforms translocated to plasma membrane. This has been explained in Fig. 10D. To our knowledge, this is the first report to study isoform specific translocation of all isoforms in neuronal system, more so, in both insulin-sensitive as well as in insulin-resistant condition. Thus, preferential accumulation on plasma membrane is the key determining factor for Akt isoform specific activation, which translates into differential glucose uptake.
Fig. 10.

Schematic diagram depicting role of Akt isoforms in regulating neuronal insulin signaling. A In an insulin-sensitive, under unstimulated condition, Akt (Akt1, Akt2, and Akt3) are present in the cytoplasm. AS160 binds to GSVs (GLUT4 Storage Vesicles) and tethers GLUT4. This does not allow GLUT4 exocytosis under basal conditions. B In an insulin-sensitive, insulin stimulated condition, Akt translocates to plasma membrane in an isoform specific insulin-dependent way (1), getting phosphorylated there (2). In neuronal system, all Akt isoforms translocate in the order Akt2 > Akt3 > Akt1. An activated Akt phosphorylates AS160, hence inactivating it. Phosphorylated and thus inactivated AS160 translocated to cytoplasm (3), promoting GLUT4 dissociation from GSVs and allowing glucose uptake (4). C In an insulin-resistant, unstimulated condition, hyperinsulinemia occurs due to defects in insulin signaling. D In an insulin-resistant, insulin stimulated condition, hyperinsulinemia triggers insulin receptor down-regulation, not allowing Akt isoform specific translocation to the plasma membrane (1). This leads to inadequate phosphorylation (2), leading to Akt’s inability to phosphorylate AS160. Unphosphorylated, thus, active AS160 continues tethering GLUT4 to GSVs, not allowing GLUT4 exocytosis, hence affecting neuronal glucose uptake. (Created with BioRender.com)
We also tested the redistribution of AS160 post-insulin stimulation in N2A (Fig. 5C) and HT22 (Fig. 5D) cells. In the present study, phosphorylation of AS160 was detected in cytoplasm and membrane fraction. It is reported that AS160 gets phosphorylated at the plasma membrane due to a small pool of it being present attached to both the plasma membrane and the GSVs via its second PTB domain [54]. Akt, once phosphorylated and activated at the plasma membrane, in-turn phosphorylates AS160 there. This has been explained in Fig. 10A, B. Thus, phosphorylated AS160 was detected at the membrane. In the cytoplasm, phosphorylated AS160 at Thr-642 acts as binding site for 14-3-3 proteins that mediate AS160’s subcellular localization and GLUT4 translocation [44, 54, 55]. We also detected phosphorylated AS160 in the cytoplasm. We observed a sharp decrease in plasma membrane bound AS160 post-insulin stimulation (Fig. 5C, D 7B, 8F). This was also reestablished using confocal microscopy, where we demonstrated decrease in AS160 and GLUT4 co-localization post-insulin stimulation (Fig. 6). This is in coherence with previous study in adipocytes [45] but first time being reported in neuronal system, where insulin stimulation leads to redistribution of AS160 from the GSVs and plasma membrane to cytoplasm. Our data show that post-insulin stimulation AS160 undergoes phosphorylation on the plasma membrane by activated Akt. This leads to redistribution of AS160 from near the membrane to the cytoplasm, allowing GLUT4 translocation. This has been explained in Fig. 10A, B. All these have never been reported before in neuronal cell. Data prove that Akt isoforms interact with downstream insulin signaling molecule, AS160, thereby creating a pathway eventually leading to part of the glucose uptake in neuronal cells. We have observed phosphorylated AS160 in the cytosolic fraction. As insulin stimulated glucose uptake is regulated by a variety of mechanisms running side-by-side, therefore, it is possible that due to insulin stimulation cytosolic interaction(s) between phosphorylated AS160 and 14-3-3 proteins help in the GLUT4 translocation and Akt isoform specific interaction with AS160 dictates total amount of glucose uptake by a neuronal cell. Overall, the data prove that Akt2 despite being lesser in expression as compared to other two isoforms, translocates to the membrane more than the other isoforms, gets phosphorylated and interact with AS160 thereby phosphorylating it, and regulates glucose uptake in neuronal system (Fig. 10A, B).
Akt isoforms differentially regulate activation of AS160
AS160 is a RabGAP (Rab GTPase-activating protein) that provided an early link between Akt and GLUT4 translocation by vesicular traffic [41, 56–58]. It is a negative regulator of insulin signaling as knockdown of AS160 causes increase in GLUT4 translocation in basal state whereas over-expression of its phosphorylation-defective mutant decreases it [44, 59–62]. There is no information about how individual Akt isoforms regulate activation of AS160, more so, in a neuronal system. Here, we studied two AS160 phosphorylation sites, Ser-588 and Thr-642. Activation determined by both showed down-regulation, with Thr-642 getting more severely affected by Akt isoform specific knockdown than Ser-588 (Fig. 3). Previously, a cluster analysis study has showed that the threonine and serine sites on AS160 clustered separately, with threonine site grouped with other Akt substrates [42]. Further analysis also showed that both of these sites are mutually exclusive with Ser-588 phosphorylation also reported downstream of PKCζ [42]. Other studies have pointed to Thr-642 as better measure of AS160 activation as point mutation of threonine to alanine leads to a significantly major decrease in GLUT4 translocation, followed by Ser mutation leading to further decrease [41]. AS160 Thr-642 phosphorylation also acts as a binding site for 14–3–3 proteins, ultimately affecting GLUT4 translocation [54, 55]. Our data suggested that expression of AS160 did not change post-single/ double silencing (Fig. 3). However, all three Akt isoforms contribute to AS160 regulation, with Akt2 contributing maximum, followed by Akt3 and then Akt1 (Fig. 3). Interestingly, Akt isoforms showed functional compensation in regulating AS160 at both the sites post-silencing. While over-expression of all Akt isoforms up-regulated phosphorylation of AS160 both under insulin-sensitive and insulin-resistant condition, over-expression of Akt2 affected AS160 phosphorylation the most (Fig. 9D). This is explained in Fig. 10. This is the first study reporting regulation of AS160 by all Akt isoforms in neuronal system. McGraw and Gonzalez studied AS160 regulation in adipocytes and reported that Akt2 specifically regulates activation of AS160 [14]. Similar studies have been reported in skeletal muscles [43, 61]. These studies specifically reported no regulation by Akt1. Akt3 was not addressed in either of these studies. This difference may be partly attributed to tissue specific regulation of isoform specific functionality of Akt.
Akt isoform specific role in neuronal insulin-resistance
In the present study, we report differential decrease in phosphorylation of all Akt isoforms in insulin-resistant neuronal cells post-insulin stimulation. Akt2 phosphorylation was affected most, followed by Akt3 and Akt1 (Fig. 8A–C). This is in contrast to peripheral insulin-sensitive tissue systems, where role of Akt isoforms and their interplay has been paradoxical. Gosmanov et al. (2004) reported that in skeletal muscles of obese patients with severe hyperglycemia and atypical diabetes, only Akt2 phosphorylation was affected, with no effect on Akt1 phosphorylation [63]. Akt 3 was not addressed in this study. Brozinick et al. (2003) also reported impaired Akt2 as well as Akt3 phosphorylation in skeletal muscles from lean and obese insulin-resistant humans, without any effect on Akt1 phosphorylation [52]. Similarly, Cozzone et al. (2008) studied primary myotubes from healthy control participants and Type-II diabetic patients and reported decrease in activation of all three isoforms [51]. Kim et al. (2000) reported redundant role of Akt1 and Akt2 in insulin-resistant tissue systems [64]. Addressing in the neuronal system, Gabbouj et al. (2019) reported decrease in Akt2 phosphorylation APP/PS1 Alzheimer mice hippocampus as compared to normal mice post-insulin stimulation, without any effect on Akt1 [35]. Akt3 was not addressed in this study. Previously, involvement of all Akt isoforms has been established with a special emphasis on regulating Tau phosphorylation with reference to Alzheimer’s disease and not on insulin signaling or insulin resistance. Wang et al. (2015) reported that only Akt 3 isoform conditional knockout (but not single or double knockouts) in cortical and cerebellar samples in mice brain affected Tau phosphorylation, pointing to requirement of all Akt isoforms and possible compensation in neuronal systems [53]. To the best of our knowledge, our study is the first to have undertaken all Akt isoform in the neural cells in the study and reported role of all Akt isoforms in regulating neuronal insulin signaling and resistance.
Conclusions
Our study is the first to report isoform specific role of all Akt isoforms in regulating neuronal insulin signaling and resistance. We find that in neuronal cells (a) all Akt isoforms regulate AS160 activation and glucose uptake; Akt2 plays a predominant role, with Akt1 and Akt3 playing significant role as well; (b) Activation of all isoforms decreased differentially under insulin-resistance with Akt2 being affected most, followed by Akt3 and Akt1; (c) Insulin-resistance is reversed by over-expression of any isoform of Akt, but predominantly by Akt2; (d) Insulin-dependent translocation on plasma membrane determines isoform specificity with Akt2 translocating the most, followed by Akt3 and then Akt1; (e) Insulin-resistance hampered this insulin-dependent translocation of all Akt isoforms to plasma membrane, irrespective of isoform; (f) Akt3, despite being neuron specific isoform contributed substantially to AS160 regulation, neuronal glucose uptake, and insulin-resistance. However, Akt2 was still the predominant isoform in regulating all the above functions. This points to a novel, differential yet compensatory interplay of all Akt isoforms in neuronal insulin signaling and resistance. These findings are fundamentally important on their own right for deeper understanding of insulin signaling and resistance in neuronal cells. This may in future help in solving a spectrum of problems associated with a diabetes, diabetes complications and neurodegenerative disorders.
Materials and methods
Materials
Minimum essential media (MEM), Dulbecco’s Minimum essential media (DMEM), foetal bovine serum (FBS), trypsin–EDTA, Opti-MEM, 2-(N-(7-nitrobenz-2-oxa1,3-diazol-4-yl) amino)-2 deoxy glucose (2-NBDG) (Cat. No. N13195), Lipofectamine 2000 (Cat. No. 11668019) and anti-Akt3 antibody (Cat. No. 41700) were procured from Thermo Fisher Scientific Inc. (USA). MCDB 201 medium, nutrient mixture F-12 Ham, albumin from bovine serum (cell culture grade), anti-GAPDH antibody (Cat. No. 9545), and dimethyl sulfoxide (DMSO) were purchased from Sigma–Aldrich Co. (USA). Anti-phospho-Akt (serine-473) (Cat. No. 4058), anti-Akt antibody (Cat. No. 9272), anti-phospho-Akt1 (Serine-473) (Cat. No. 9018), anti-phospho-Akt2 (Serine-474) (Cat. No. 8599), anti-Akt1 antibody (Cat. No. 2938), anti-Akt2 antibody (Cat. No. 3063), anti-phospho-AS160 (serine-588) (Cat. No. 8730), anti-phospho-AS160 (threonine-642) (Cat. No.8881), anti-AS160 antibody (Cat. No. 2670), IgG conformational (Cat. No. 3678) were purchased from Cell Signalling Technology Inc. (USA). Protein A/G agarose beads (Cat no. sc:2003), anti-GLUT-4 (Cat no. sc:1608) were purchased from Santa Cruz Biotechnology (USA). Anti-Caveolin-1 (Cat no.: 894) was kindly gifted by Dr. Chinmoy Mukhopadhyay, Jawaharlal Nehru University, New Delhi. Recombinant insulin (Cat. No. 407709) was purchased from Calbiochem (Germany).
Cell culture
N2A (mice neuroblastoma cell line) and HT22 (mice hippocampal cell line) cells were proliferated in MEM and DMEM, respectively, supplemented with 10% FBS, and antibiotics-penicillin 100 IU/ml and streptomycin 100 mg/ml, at 37 °C in 5% CO2. For N2A cells, 3 days post-proliferation, cells were subjected to differentiation in MEM supplemented with 1% FBS and 2% DMSO at 37 °C in 5% CO2 for 3 days [8]. For HT22, 2 days post-proliferation, cells were subjected to differentiation in neurobasal media containing 2 mmol/L glutamine and 1 × N2 supplement for 2 days [39, 40]. Insulin-resistance in N2A cells was generated as reported earlier from our laboratory [6]. Briefly, cells were kept in an equal mixture of two serum-free media (MCDB 201 medium and nutrient mixture F-12 Ham) in the absence (MF; insulin-sensitive) or in chronic presence of 100 nM insulin for 3 days (MFI; insulin-resistant). Media were replaced after every 12 h. Differentiated neuronal cells were subjected to 30 min insulin stimulation as per standard assay conditions as reported previously from other as well as from our laboratory [8, 48, 49, 65, 66].
Gene silencing by siRNA transfection
Akt isoforms in the cells were silenced as previously reported [39]. To select the specific siRNA among 4 different siRNAs against each isoform provided by the manufacture (Qiagen), specific siRNA for each isoform was chosen based on the one that provided maximum silencing (Fig. S2A, S3A and S4A). Selected isoform specific siRNA sequences were: Akt1:5ʹ ATGCTGTTCAGAGACATTTA3ʹ; Akt2:5ʹAACATTTCTCTGTAGCAGAA3ʹ; Akt3:5ʹGATTGATAATATATAGGAGGA3ʹ. Selection of specific siRNA was followed by optimization of dose of selected siRNA (Fig. S2B, S3B and S4B). For double silencing, optimized doses as selected above for the single silencing were used additively. Scrambled (non-specific) and Akt isoform specific siRNAs were transfected in Opti-MEM using Lipofectamine 2000. Six hours post-transfection, Opti-MEM was changed to respective proliferation media depending on the cell line used. Twelve hours post-media change, cells were subjected to differentiation as explained above.
Plasmid DNA transfection using Akt isoform specific plasmids
Akt isoform specific constructs were purchased from Addgene [Akt1 (#86631) and Akt2 (#86593)] and Origene [Akt3 (NM_RC221051)]. Specific plasmid constructs were transfected in the cells as previously reported [39]. Briefly, N2A cells were transiently transfected with either Akt isoform specific construct or control (respective empty backbone) constructs in Opti-MEM using Lipofectamine 2000 according to manufacturer’s instructions.
Immunoprecipitation
Because a specific phospho-Akt3 antibody was not available to measure phosphorylation levels of Akt3, Akt3 protein was immunoprecipitated using Akt3 specific antibody (Thermo, PA-41700), and probed with Anti-phospho-Akt (serine-473) (CST, Cat. No. 4058). Anti-IgG conformational antibody (CST, Cat No. 3678) was used to mask remaining IgG bands. Cells were proliferated and differentiated as described above. 500 µg of protein lysate with certain amount of primary antibody (as referred in datasheet of the manufacturer) was added and incubated overnight at 4 °C in a microfuge rotator at 10 rpm (Test tube Rotator, Tarsons, Cat. No.: 3070). Next day, protein A/G beads were added for 4 h, along with lysis buffer and protease inhibitors. After 4 h of incubation, cells were centrifuged at 5000 rpm (Centrifuge, Sigma Cat. No.: 2-16KL) for 5 min. Supernatant was discarded and pellet was washed 3 times with lysis buffer along with protease inhibitors. Pellet was suspended in SDS–PAGE sample buffer and heated at 90 °C. The protein samples were resolved on SDS–PAGE [38].
Cell lysis and immunoblotting
After differentiation, cells were stimulated with or without 100 nM insulin for 30 min as indicated above. Cells were lysed in lysis buffer (50 mM HEPES, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10 mM Na4P2O7, 50 mM NaF, 50 mM β-glycerophosphate, 1 mM Na3VO4 and 1% triton X-100) as described previously from our laboratory for 30 min at 4 °C [39]. Supernatants were collected and protein was estimated by Bicinchoninic Acid (BCA) method [67]. Lysates were boiled with Laemmli sample buffer (Tris‐HCl 62.5 mM, pH 6.7; glycerol 10% (v/v); SDS 2% (w/v); bromophenol blue 0.003% (w/v) containing β‐mercaptoethanol 14.3 M) for 5 min, resolved by SDS–PAGE gel electrophoresis and transferred to nitrocellulose membrane. Membranes were blocked with BSA (5%) and incubated with the indicated primary antibodies for 12–16 h, followed by 2 h incubation with HRP-conjugated secondary antibody. The protein bands were visualized with Pierce ECL Plus Western Blotting substrate (Thermo, Cat No. 32132) on Typhoon FLA 9500 (GE Healthcare Bio-Sciences AB). Quantification of all blots was done using Quantity One 1-D Analysis Software (Bio-Rad Laboratories, Inc.). Blots were normalized with the relative expression of the (non-phospho) protein being tested or a housekeeping protein (GAPDH) under the same conditions [39, 48].
Membrane fractionation
N2A/HT22 cells were differentiated in 100 mm tissue culture plates. After respective treatment, cells were lysed in lysis buffer (20 mM Tris, pH 7.5; 250 mM sucrose; 2 mM EDTA; 2 mM EGTA; 1 mM PMSF; 10 µg/ml aprotinin and leupeptin), as reported earlier from our laboratory for 5 min [68]. Lysate was homogenized for 10 min with 30 strokes in Glass homogenizer at 4 °C. Lysates were subjected to ultra-centrifugation, at 200,000×g for 1 h. Supernatant was collected and termed as ‘Cytosolic fraction’ and pellet was re-suspended in lysis buffer with 1% NP-40 (20 mM Tris, pH 7.5; 1% NP-40; 150 mM NaCl; 1 mM EDTA; 1 mM EGTA; 1 mM PMSF; 10 µg/ml aprotinin and leupeptin) at 4 °C for 30 min. Centrifuged supernatant was collected and termed as ‘Membrane fraction’. The presence of membrane-specific marker like Caveolin-1 was tested and GAPDH was used as a loading control in all membrane translocation experiments [47].
Mice whole brain tissue lysis and Immunoblotting
Insulin resistance was generated in 16 weeks old high-fat-diet (HFD) fed Swiss Albino male mice in the laboratory of Dr. Prosenjit Mandal, Indian Institute of Technology—Mandi, over 10 weeks, as previously reported [69, 70], and whole brain tissues of those mice, which were kind gifts from Dr. Mandal, Indian Institute of Technology—Mandi, were used for our experiments. Glucose tolerance test and insulin tolerance tests were performed; however, perfusion was not done before brain collection. Mice were divided into two groups: normal diet (ND) and high-fat-diet (HFD), each group containing three animals. Levels of triglyceride, cholesterol, serum glutamic-oxaloacetic transaminase (SGOT), serum glutamic-pyruvic transaminase (SPGT), and fasting blood glucose were elevated by 48%, 29%, 50%, 53%, and 40%, respectively, in HFD mice as compared to ND mice. Mice whole brain were lysed in a homogenizer in lysis buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10 mM sodium pyrophosphate, 50 mM sodium fluoride, 50 mM β-glycerophosphate, 1 mM Na3VO4, 1% Triton X-100 supplemented with 2 mM PMSF, 10 μg/ml each of leupeptin and aprotinin) at 4 °C for 15 min. Mice whole brain tissues were homogenized as described previously [68]. Supernatants were collected and protein was estimated by Bicinchoninic acid test [67]. For plasma membrane fractionation of whole brain lysates, 150 μl of whole brain lysates were ultra-centrifuged as described above in “Membrane fractionation” step. All experiments were performed following the guidelines prescribed by CPCSEA (Committee for the Purpose of Control and Supervision of Experiments on Animals) with the approval of the Internal Animal Ethics Committee, Visva-Bharati (IAEC/VB/2017/01). The legal requirements/guidelines in the country for the care and use of animals have been followed.
Glucose uptake assay
Glucose uptake assays were performed as described previously from our laboratory [38]. Briefly, differentiated N2A and HT22 were washed and subjected to glucose starvation for 2 h. After respective treatment, cells were treated with 50 μM 2-NBDG for 1 h and lysed in buffer containing 20 mM Tris–HCl pH 7.4, 40 mM KCl, 1% sodium deoxycholate and 1% NP-40 for 15 min at 25 °C with gentle shaking. Cells were then scraped off and centrifuged at 13,000×g for 20 min at 4 °C. The fluorescence was then measured using a fluorescence spectrophotometer LS55 (Perkin Elmer, USA) at the excitation and emission wavelengths of 485 nm and 535 nm, respectively.
Confocal-microscopy
Immunofluorescence studies of N2A cells carried out as described previously [66]. Briefly, N2A cells were treated with 100 nM insulin for 30 min at 37 °C. Cells were fixed with 4% paraformaldehyde for 15 min. Cells were permeabilized by incubating with 0.5% Triton X-100 for 5 min, followed by washing with PBS. Cells were washed in PBS and blocked using blocking buffer (BSA 1% in PBS) for 1 h. Cells were incubated with anti-GLUT4 antibody overnight at 4 °C, washed with PBS solution, followed by incubation with anti-AS160 antibody overnight at 4 °C. Cells were washed using PBS and incubated with Donkey anti-Goat Alexa 555 (for GLUT4) and Mice anti-Goat Alexa 488 (for AS160) secondary antibodies for 45 min at room temperature. Cells were washed further with PBS and mounted using ProLong™ Diamond Antifade Mountant with DAPI on to glass slides. Images were taken using Carl Zeiss LSM 880 microscope using 63 X (with oil) confocal objective. For Pearson’s coefficient, images were run in triplicates and the calculations are performed on the entire population of pixels, without selecting any specific ROI (Region of Interest), using Zen 2.6 (Blue/Black edition) software. Images were taken at Regional Center for Biotechnology, Faridabad, Haryana. Images were processed using Zen 2.6 (Blue/Black edition) software.
Replicates and statistical analysis
Data presented depict three or more biological replicates and are depicted as mean ± SE, with error bars indicating standard error. One-way (Fig. 1C–H, Fig.S6) or two-way (Figs. 1A, B 2, 3, 4, 5, 7, 8, 9, Figs. S2–S5) analysis of variance (ANOVA) followed by Bonferroni post hoc test was used for multiple comparison. p < 0.05 was deemed significant.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We are grateful to Indian Institute of Technology-Delhi for their support. We would like to thank Dr. Prosenjit Mondal, Indian Institute of Technology—Mandi, Himachal Pradesh, India, for helping us in providing with normal diet and high-fat-fed diet mice whole brain samples. We would like to thank Dr. Nishi Raj, Jamia Hamdard-Institute of Molecular Medicine (JHIMM), New Delhi, India for providing pCMV6 Empty vector backbone. We would like to thank Dr. Chinmoy Mukhopadhyay, Jawaharalal Nehru University, New Delhi, for providing us with Caveolin-1 antibody. We would like to thank Dr. Pallavi Varshney for her assistance in training some methodologies. We are grateful to Advanced Technology Platform Centre, Regional Centre for Biotechnology, Faridabad, Haryana, India, for Confocal Microscopy.
Author contributions
MS performed all the experiments, analyzed the data and wrote the original drafts, approved the final version of the manuscript. CSD conceived the idea, provided resources, acquired the funding, written, reviewed and edited the manuscript and approved the final version of the manuscript.
Funding
MS is a recipient of Senior Research Fellowship (09/086 (1256)/2016-EMR-1) from Council of Scientific and Industrial Research (CSIR), Government of India. CSD is supported by a grant from Department of Biotechnology, Government of India, New Delhi, India (PR32795/MED/122/229/2019).
Data availability
All the data related to this manuscript are available with the first author and corresponding author.
Code availability
Not applicable.
Declarations
Conflict of interest
The authors declare no conflicts of interest.
Ethical approval
Mice whole brain tissues were kind gift from Dr. Prosenjit Mondal, Indian Institute of Technology—Mandi, HP, India. All experiments were performed following the guidelines prescribed by CPCSEA (Committee for the Purpose of Control and Supervision of Experiments on Animals) with the approval of the Internal Animal Ethics Committee, Visva-Bharati (IAEC/VB/2017/01). The legal requirements/guidelines in the country for the care and use of animals have been followed.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Gonzalez E, McGraw TE. The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle. 2009;8:2502–2508. doi: 10.4161/cc.8.16.9335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hay N. Akt isoforms and glucose homeostasis—the leptin connection. Trends Endocrinol Metab. 2011;22:66–73. doi: 10.1016/j.tem.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381–405. doi: 10.1016/j.cell.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang X, Liu G, Guo J, Su Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int J Biol Sci. 2018;14:1483–1496. doi: 10.7150/ijbs.27173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta A, Bisht B, Dey CS. Focal adhesion kinase negatively regulates neuronal insulin resistance. Biochim Biophys Acta. 2012;1822:1030–1037. doi: 10.1016/j.bbadis.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 6.Gupta A, Dey CS. PTEN, a widely known negative regulator of insulin/PI3K signaling, positively regulates neuronal insulin resistance. Mol Biol Cell. 2012;23:3882–3898. doi: 10.1091/mbc.E12-05-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de la Monte SM, Wands JR. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol. 2008;2:1101–1113. doi: 10.1177/193229680800200619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gupta A, Bisht B, Dey CS. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology. 2011;60:910–920. doi: 10.1016/j.neuropharm.2011.01.033. [DOI] [PubMed] [Google Scholar]
- 9.Humbert S, Bryson EA, Cordelières FP, Connors NC, Datta SR, Finkbeiner S, et al. The IGF-1/Akt pathway is neuroprotective in huntington’s disease and involves huntingtin phosphorylation by Akt. Dev Cell. 2002;2:831–837. doi: 10.1016/s1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- 10.Chen H-K, Fernandez-Funez P, Acevedo SF, Lam YC, Kaytor MD, Fernandez MH, et al. Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell. 2003;113:457–468. doi: 10.1016/s0092-8674(03)00349-0. [DOI] [PubMed] [Google Scholar]
- 11.Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat Genet. 2004;36:131–137. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- 12.Furlong RM, Lindsay A, Anderson KE, Hawkins PT, Sullivan AM, O’Neill C. The Parkinson’s disease gene PINK1 activates Akt via PINK1 kinase-dependent regulation of the phospholipid PI(3,4,5)P3. J Cell Sci. 2019;132:jcs233221. doi: 10.1242/jcs.233221. [DOI] [PubMed] [Google Scholar]
- 13.Cleasby ME, Reinten TA, Cooney GJ, James DE, Kraegen EW. Functional studies of Akt isoform specificity in skeletal muscle in vivo; maintained insulin sensitivity despite reduced insulin receptor substrate-1 expression. Mol Endocrinol. 2007;21:215–228. doi: 10.1210/me.2006-0154. [DOI] [PubMed] [Google Scholar]
- 14.Kajno E, McGraw TE, Gonzalez E. Development of a new model system to dissect isoform specific Akt signalling in adipocytes. Biochem J. 2015;468:425–434. doi: 10.1042/BJ20150191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu M, Wan M, Leavens KF, Chu Q, Monks BR, Fernandez S, et al. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat Med. 2012;18:388–395. doi: 10.1038/nm.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tschopp O, Yang Z-Z, Brodbeck D, Dummler BA, Hemmings-Mieszczak M, Watanabe T, et al. Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development. 2005;132:2943–2954. doi: 10.1242/dev.01864. [DOI] [PubMed] [Google Scholar]
- 17.Chen WS, Xu P-Z, Gottlob K, Chen M-L, Sokol K, Shiyanova T, et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the akt1 gene. Genes Dev. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBα is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349–38352. doi: 10.1074/jbc.C100462200. [DOI] [PubMed] [Google Scholar]
- 19.Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 20.Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. 2003;112:197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, et al. Role for Akt3/protein kinase bγ in attainment of normal brain size. Mol Cell Biol. 2005;25:1869–1878. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee RS, House CM, Cristiano BE, Hannan RD, Pearson RB, Hannan KM. Relative expression levels rather than specific activity plays the major role in determining in vivo AKT isoform substrate specificity. Enzyme Res. 2011;2011:720985. doi: 10.4061/2011/720985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonzalez E, McGraw TE. Insulin-modulated Akt subcellular localization determines Akt isoform-specific signaling PNAS. Nat Acad Sci. 2009;106:7004–7009. doi: 10.1073/pnas.0901933106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding J, Du K. ClipR-59 interacts with Akt and regulates Akt cellular compartmentalization. Mol Cell Biol. 2009;29:1459–1471. doi: 10.1128/MCB.00754-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma M, Dey CS. AKT ISOFORMS-AS160-GLUT4: the defining axis of insulin resistance. Rev Endocr Metab Disord. 2021 doi: 10.1007/s11154-021-09652-2. [DOI] [PubMed] [Google Scholar]
- 27.Bae SS, Cho H, Mu J, Birnbaum MJ. Isoform-specific regulation of insulin-dependent glucose uptake by Akt/protein kinase B. J Biol Chem. 2003;278:49530–49536. doi: 10.1074/jbc.M306782200. [DOI] [PubMed] [Google Scholar]
- 28.Dummler B, Tschopp O, Hynx D, Yang Z-Z, Dirnhofer S, Hemmings BA. Life with a single isoform of Akt: mice lacking Akt2 and Akt3 are viable but display impaired glucose homeostasis and growth deficiencies. Mol Cell Biol. 2006;26:8042–8051. doi: 10.1128/MCB.00722-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304:1325–1328. doi: 10.1126/science.1096706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noda S, Kishi K, Yuasa T, Hayashi H, Ohnishi T, Miyata I, et al. Overexpression of wild-type Akt1 promoted insulin-stimulated p70S6 kinase (p70S6K) activity and affected GSK3 beta regulation, but did not promote insulin-stimulated GLUT4 translocation or glucose transport in L6 myotubes. J Med Invest. 2000;47:47–55. [PubMed] [Google Scholar]
- 31.Chen WS, Peng X-D, Wang Y, Xu P-Z, Chen M-L, Luo Y, et al. Leptin deficiency and beta-cell dysfunction underlie type 2 diabetes in compound Akt knockout mice. Mol Cell Biol. 2009;29:3151–3162. doi: 10.1128/MCB.01792-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang ZY, Zhou QL, Coleman KA, Chouinard M, Boese Q, Czech MP. Insulin signaling through Akt/protein kinase B analyzed by small interfering RNA-mediated gene silencing. Proc Natl Acad Sci USA. 2003;100:7569–7574. doi: 10.1073/pnas.1332633100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hajduch E, Alessi DR, Hemmings BA, Hundal HS. Constitutive activation of protein kinase B alpha by membrane targeting promotes glucose and system A amino acid transport, protein synthesis, and inactivation of glycogen synthase kinase 3 in L6 muscle cells. Diabetes. 1998;47:1006–1013. doi: 10.2337/diabetes.47.7.1006. [DOI] [PubMed] [Google Scholar]
- 34.Levenga J, Wong H, Milstead RA, Keller BN, LaPlante LE, Hoeffer CA. AKT isoforms have distinct hippocampal expression and roles in synaptic plasticity. Elife. 2017 doi: 10.7554/eLife.30640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gabbouj S, Natunen T, Koivisto H, Jokivarsi K, Takalo M, Marttinen M, et al. Intranasal insulin activates Akt2 signaling pathway in the hippocampus of wild-type but not in APP/PS1 Alzheimer model mice. Neurobiol Aging. 2019;75:98–108. doi: 10.1016/j.neurobiolaging.2018.11.008. [DOI] [PubMed] [Google Scholar]
- 36.Diez H, Garrido JJ, Wandosell F. Specific Roles of Akt iso Forms in Apoptosis and Axon Growth Regulation in Neurons. PLoS ONE. 2012;7:e32715. doi: 10.1371/journal.pone.0032715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schrötter S, Leondaritis G, Eickholt BJ. Capillary isoelectric focusing of Akt isoforms identifies highly dynamic phosphorylation in neuronal cells and brain tissue. J Biol Chem. 2016;291:10239–10251. doi: 10.1074/jbc.M115.700138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arora A, Dey CS. SIRT2 negatively regulates insulin resistance in C2C12 skeletal muscle cells. Biochim Biophys Acta. 2014;1842:1372–1378. doi: 10.1016/j.bbadis.2014.04.027. [DOI] [PubMed] [Google Scholar]
- 39.Varshney P, Dey CS. P21-activated kinase 2 (PAK2) regulates glucose uptake and insulin sensitivity in neuronal cells. Mol Cell Endocrinol. 2016;429:50–61. doi: 10.1016/j.mce.2016.03.035. [DOI] [PubMed] [Google Scholar]
- 40.Liu J, Li L, Suo WZ. HT22 hippocampal neuronal cell line possesses functional cholinergic properties. Life Sci. 2009;84:267–271. doi: 10.1016/j.lfs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 41.Sano H, Kane S, Sano E, Mîinea CP, Asara JM, Lane WS, et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- 42.Ng Y, Ramm G, Burchfield JG, Coster ACF, Stöckli J, James DE. Cluster analysis of insulin action in adipocytes reveals a key role for Akt at the plasma membrane. J Biol Chem. 2010;285:2245–2257. doi: 10.1074/jbc.M109.060236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng X, Cartee GD. Insulin-induced effects on the subcellular localization of AKT1, AKT2 and AS160 in rat skeletal muscle. Sci Rep. 2016;6:39230. doi: 10.1038/srep39230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Larance M, Ramm G, Stöckli J, van Dam EM, Winata S, Wasinger V, et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem. 2005;280:37803–37813. doi: 10.1074/jbc.M503897200. [DOI] [PubMed] [Google Scholar]
- 45.Stöckli J, Davey JR, Hohnen-Behrens C, Xu A, James DE, Ramm G. Regulation of glucose transporter 4 translocation by the Rab guanosine triphosphatase-activating protein AS160/TBC1D4: role of phosphorylation and membrane association. Mol Endocrinol. 2008;22:2703–2715. doi: 10.1210/me.2008-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams TM, Lisanti MP. The caveolin proteins. Genome Biol. 2004;5:214. doi: 10.1186/gb-2004-5-3-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sirover MA. Subcellular dynamics of multifunctional protein regulation: mechanisms of GAPDH intracellular translocation. J Cell Biochem. 2012;113:2193–2200. doi: 10.1002/jcb.24113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arora A, Dey CS. SIRT2 regulates insulin sensitivity in insulin resistant neuronal cells. Biochem Biophys Res Commun. 2016;474:747–752. doi: 10.1016/j.bbrc.2016.05.029. [DOI] [PubMed] [Google Scholar]
- 49.Mishra D, Dey CS. Protein kinase C attenuates insulin signalling cascade in insulin-sensitive and insulin-resistant neuro-2a cells. J Mol Neurosci. 2019;69:470–477. doi: 10.1007/s12031-019-01377-x. [DOI] [PubMed] [Google Scholar]
- 50.Varshney P, Dey CS. Resveratrol regulates neuronal glucose uptake and insulin sensitivity via P21-activated kinase 2 (PAK2) Biochem Biophys Res Commun. 2017;485:372–378. doi: 10.1016/j.bbrc.2017.02.070. [DOI] [PubMed] [Google Scholar]
- 51.Cozzone D, Fröjdö S, Disse E, Debard C, Laville M, Pirola L, et al. Isoform-specific defects of insulin stimulation of Akt/protein kinase B (PKB) in skeletal muscle cells from type 2 diabetic patients. Diabetologia. 2008;51:512–521. doi: 10.1007/s00125-007-0913-8. [DOI] [PubMed] [Google Scholar]
- 52.Brozinick JT, Roberts BR, Dohm GL. Defective signaling through Akt-2 and -3 But Not Akt-1 in insulin-resistant human skeletal muscle: potential role in insulin resistance. Diabetes Am Diabetes Assoc. 2003;52:935–941. doi: 10.2337/diabetes.52.4.935. [DOI] [PubMed] [Google Scholar]
- 53.Wang L, Cheng S, Yin Z, Xu C, Lu S, Hou J, et al. Conditional inactivation of Akt three isoforms causes tau hyperphosphorylation in the brain. Mol Neurodegener. 2015;10:33. doi: 10.1186/s13024-015-0030-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tan S-X, Ng Y, Burchfield JG, Ramm G, Lambright DG, Stöckli J, et al. The Rab GTPase-activating protein TBC1D4/AS160 contains an atypical phosphotyrosine-binding domain that interacts with plasma membrane phospholipids to facilitate GLUT4 trafficking in adipocytes. Mol Cell Biol. 2012;32:4946–4959. doi: 10.1128/MCB.00761-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramm G, Larance M, Guilhaus M, James DE. A role for 14-3-3 in insulin-stimulated GLUT4 translocation through its interaction with the RabGAP AS160. J Biol Chem. 2006;281:29174–29180. doi: 10.1074/jbc.M603274200. [DOI] [PubMed] [Google Scholar]
- 56.Bruss MD, Arias EB, Lienhard GE, Cartee GD. Increased phosphorylation of Akt substrate of 160 kDa (AS160) in rat skeletal muscle in response to insulin or contractile activity. Diabetes. 2005;54:41–50. doi: 10.2337/diabetes.54.1.41. [DOI] [PubMed] [Google Scholar]
- 57.Kane S, Sano H, Liu SCH, Asara JM, Lane WS, Garner CC, et al. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem. 2002;277:22115–22118. doi: 10.1074/jbc.C200198200. [DOI] [PubMed] [Google Scholar]
- 58.Zeigerer A, McBrayer MK, McGraw TE. Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on RabGAP AS160. Mol Biol Cell. 2004;15:4406–4415. doi: 10.1091/mbc.E04-04-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brewer PD, Romenskaia I, Kanow MA, Mastick CC. Loss of AS160 Akt substrate causes Glut4 protein to accumulate in compartments that are primed for fusion in basal adipocytes. J Biol Chem. 2011;286:26287–26297. doi: 10.1074/jbc.M111.253880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard GE, et al. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2005;2:263–272. doi: 10.1016/j.cmet.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 61.Kramer HF, Witczak CA, Fujii N, Jessen N, Taylor EB, Arnolds DE, et al. Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes. 2006;55:2067–2076. doi: 10.2337/db06-0150. [DOI] [PubMed] [Google Scholar]
- 62.Lansey MN, Walker NN, Hargett SR, Stevens JR, Keller SR. Deletion of Rab GAP AS160 modifies glucose uptake and GLUT4 translocation in primary skeletal muscles and adipocytes and impairs glucose homeostasis. Am J Physiol Endocrinol Metab. 2012;303:E1273–E1286. doi: 10.1152/ajpendo.00316.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gosmanov AR, Umpierrez GE, Karabell AH, Cuervo R, Thomason DB. Impaired expression and insulin-stimulated phosphorylation of Akt-2 in muscle of obese patients with atypical diabetes. Am J Physiol Endocrinol Metab. 2004;287:E8–15. doi: 10.1152/ajpendo.00485.2003. [DOI] [PubMed] [Google Scholar]
- 64.Kim YB, Peroni OD, Franke TF, Kahn BB. Divergent regulation of Akt1 and Akt2 isoforms in insulin target tissues of obese Zucker rats. Diabetes. 2000;49:847–856. doi: 10.2337/diabetes.49.5.847. [DOI] [PubMed] [Google Scholar]
- 65.Heide LPVD, Hoekman MF, Biessels GJ, Gispen WH. Insulin inhibits extracellular regulated kinase 1/2 phosphorylation in a phosphatidylinositol 3-kinase (PI3) kinase-dependent manner in Neuro2a cells. J Neurochem. 2003;86:86–91. doi: 10.1046/j.1471-4159.2003.01828.x. [DOI] [PubMed] [Google Scholar]
- 66.Bisht B, Dey CS. Focal Adhesion Kinase contributes to insulin-induced actin reorganization into a mesh harboring Glucose transporter-4 in insulin resistant skeletal muscle cells. BMC Cell Biol. 2008;9:48. doi: 10.1186/1471-2121-9-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 68.Goel HL, Dey CS. Insulin stimulates spreading of skeletal muscle cells involving the activation of focal adhesion kinase, phosphatidylinositol 3-kinase and extracellular signal regulated kinases. J Cell Physiol. 2002;193:187–198. doi: 10.1002/jcp.10161. [DOI] [PubMed] [Google Scholar]
- 69.Silva G, Ferraresi C, de Almeida RT, Motta ML, Paixão T, Ottone VO, et al. Insulin resistance is improved in high-fat fed mice by photobiomodulation therapy at 630 nm. J Biophotonics. 2020;13:e201960140. doi: 10.1002/jbio.201960140. [DOI] [PubMed] [Google Scholar]
- 70.Mujawdiya PK, Sharma P, Sharad S, Kapur S. Reversal of increase in intestinal permeability by mangifera indica seed kernel extract in high-fat diet-induced obese mice. Pharmaceuticals (Basel) 2020 doi: 10.3390/ph13080190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the data related to this manuscript are available with the first author and corresponding author.
Not applicable.