

Abstract
In demyelinated lesions, astrocytes, activated microglia and infiltrating macrophages secrete several factors regulating oligodendrocyte precursor cells’ behaviour. What appears to be the initiation of an intrinsic mechanism of myelin repair is only leading to partial recovery and inefficient remyelination, a process worsening over the course of the disease. This failure is largely due to the concomitant accumulation of inhibitory cues in and around the lesion sites opposing to growth promoting factors. Here starts a complex game of interactions between the signalling pathways controlling oligodendrocytes migration or differentiation. Receptors of positive or negative cues are modulating Ras, PI3K or RhoGTPases pathways acting on oligodendrocyte cytoskeleton remodelling. From the description of this intricate signalling network, this review addresses the extent to which the modulation of the global response to inhibitory cues may pave the route towards novel therapeutic approaches for myelin repair.
Keywords: Myelin repair, Signalling pathways, Oligodendrocytes, Multiple sclerosis, Therapy, Migration
Repairing myelin in multiple sclerosis
Multiple sclerosis (MS) is the most common auto-immune disease of the central nervous system and affects 2.5 million people worldwide. This pathology is characterized by the destruction of the myelin sheaths by the immune system. This drives to axonal weakness which disturbs severely neuronal communication and leads to motor and sensory disorders. Main feature of the disease starts with relapse/remitting phases (RRMS) followed after several years (15–20 years) by a progressive phase in the majority of patients. The remaining MS patients (10–15%) have a primary progressive form of the disease, without RRMS, characterized by a slow and progressive neurodegeneration in which the symptoms worsen and disability accumulates from the onset. In early stage of the lesion, disturbance of brain–blood barrier is accompanied by moderate T-cell (CD8 + and CD4 +) and B-cell infiltration which will drive to oligodendrocyte destruction only after a profound invasion of inflammatory cells [1, 2]. Like other neuroinflammatory diseases, infiltration at the site of tissue damage in multiple sclerosis is regulated by cytokines [3, 4]. This deleterious inflammation is characterized by increase of T-cell and B-cell, macrophage infiltration and activation of resident microglia and macrophages. In later stages, granzyme B as well as immunoglobulin and activated complement deposition can be observed in lesions [5, 6]. In classical active lesion architecture, normal-appearing white matter surrounding the lesion contains scattered perivascular inflammatory infiltrates and microglia nodules whose density increases at the border of the lesion. Loss of oligodendrocytes occurs in lesion area where activated macrophages phagocyte myelin debris [7]. Tissue composition of the lesion is ultimately altered by reactive astrocytes and microglia which drives to glial scar formation, supposed to prevent tissue destruction spreading by retaining immune cells and toxic metabolites. Moreover, angiogenesis occurs inside and around the demyelinating lesion, [8]. This profound tissue remodelling is the source of a molecular and cellular barrier impeding myelin repair thereby installing over time severe motor and sensitive deficits (Fig. 1).
Fig. 1.

Opposing molecular factors in demyelinated lesions. In the context of multiple sclerosis, the myelin sheaths are attacked by autoimmune reactive cells. The concomitant pro-inflammatory situation is leading to a glial activation of astrocytes and microglial cells contributing to the secretion of various molecular signals. On the one side, growth factors and chemokines are positively acting on the recruitment of oligodendrocyte precursor cells (OPCs) attracted towards the lesion in order to repair myelin. On the other side, repulsive growth inhibitory molecules accumulating in this perturbed microenvironment create a molecular barrier preventing OPCs to reach the centre of the lesion hence precluding myelin repair
Strikingly, remyelination is observed in lesions but is generally incomplete, giving rise to “shadow” plaques where newly generated myelin sheaths are thinner than pre-existing ones [9, 10]. This regenerative process decreases and disappears with chronicity and entrance in progressive phase of the disease. In progressive form of MS, chronic inflammation is maintained in slowly expanding lesions despite a closed brain blood barrier with involvement of activated microglia, T-cells and B-cells. High production of reactive oxygen species contributes to mitochondrial and axonal damages [11].
While the current therapies aim at controlling the inflammatory component of MS, better comprehension of the reasons of remyelination failure in MS would help to develop new therapeutic approaches. In chronic lesions, oligodendrocyte precursor cells (OPCs) derived from neural precursor cells of subventricular zone or coming from perilesional environment are recruited to regenerate the myelin sheet [12, 13] and accumulate at the border rather in the core of the lesion [14, 15]. Whereas many factors promoting OPC recruitment are secreted in demyelinating lesions [16], several repulsive cues contribute to remyelination failure. The massive changes occurring in the lesion regarding matrix and cell composition as well as their activation state produce this particular environment where negative cues outperform remyelinating cues. The counteraction of negative cues or the promotion of positive cues is offering the possibility to develop a therapeutic strategy which could stem on natural capacity myelin repair. To this end, it is important to understand the machinery of this repulsive barrier. In this review, we will highlight signalling pathways involved in remyelination and analyse how repulsive cues alter them.
Intracellular pathways controlling remyelination
Spontaneous remyelination occurring in the early stages of the disease involves proliferation and migration of OPC, followed by their differentiation in mature oligodendrocytes for wrapping up of axons by new myelin sheaths [17, 18]. These steps involve three canonical signalling pathways controlling numerous basic cellular pathways. Ras GTPase, PI3K-Akt pathway and MAPK pathway are central regulators of myelination with already a well-depicted role in developmental myelination (Fig. 2).
Fig. 2.

Involvement of Ras/PI3K/MAPK pathway in remyelination. Migration: the proteoglycan NG2 contributes to front-rear polarity establishment in OPCs in response to growth factors and chemokines. NG2 maintains high RhoA activity at the rear of the cell whereas it stimulates Rac at the migration front via its switch of activity induced by RTK/GPCR dependent PKCα activation. Ras/Raf/MEK/ERK and PI3K/Akt pathways regulate this Rac activation. Conversely, removal of GSK3β mediated inhibition of p190A by Akt probably favours local RhoA inhibition. Proliferation/differentiation: Akt dependent regulation of mTORC1 and β-catenin plays a parallel role to ERK1/2 in proliferation and differentiation of oligodendrocytes. However, β-catenin can have opposite effects on this differentiation depending of its nuclear level. Arrows show activation and truncated red arrows show inhibition. PI3K-Akt pathway is shown in orange boxes, MAPK pathway in blue boxes and GSK3β/β-catenin in purple boxes. Factors shown in green favour establishment of the migration front
The members of the Ras GTPase family (HRas, NRas, KRas) cycle between an inactive form bound to GDP and an active form bound to GTP [19]. Activation of receptor tyrosine kinase RTKs by growth factors and chemokines leads to their tyrosine trans-autophosphorylation, serving as docking sites for Grb2-Sos complex which activates Ras. G-protein coupled receptors (GPCR) also activate Ras via Ras-GEF activation [20]. Among well-characterized downstream pathways, Ras is known to stimulate PI3K-Akt pathway, Raf-MEK-ERK pathway (also known as MAP Kinases) and Ral/Exocyste complex. R-Ras1−/− and R-Ras2−/− null mice exhibit a diminished oligodendrocyte population with higher proportion of immature oligodendrocytes [21]. In these mice PI3K-Akt and Erk1/2-MAPK pathways are less activated.
The PI3K family is composed of regulatory subunits (p85, p87, p110) and p110 catalytic subunit catalysing the phosphorylation of phosphatidylinositol PtdIns(4,5)P2 into PtdIns(3,4,5)P3. It can be directly activated by RTK or via either Ras or Grb2/GAB [22]. PIP3 recruits proteins with PH (pleckstrin homology) domains like PDK1 and Akt allowing Akt activation by PDK1 and mTORC2 phosphorylation. Akt has multiple functions. First it favours survival by inhibiting proapoptotic Bcl-2 proteins, phosphorylating Mdm2 or inhibiting transcription factor NF-kappaB. It regulates also metabolism and differentiation by activating mTORC1 and inhibiting GSK3β [23, 24]. Knock-out of the PI3K-Akt pathway inhibitor PTEN in oligodendrocyte lineage resulted in significant hypermyelination throughout the CNS of transgenic mice associated with increased PIP3 levels and Akt phosphorylation [25, 26]. Similarly, expression of constitutively active Akt in oligodendrocytes resulted in hypermyelination increasing as mice aged up to reach a pathological level [27]. Treatment with the mTOR inhibitor rapamycin inhibited hypermyelination suggesting its involvement downstream Akt [28].
Canonical MAPK pathway activation starts with Raf phosphorylation by Ras. Then Raf phosphorylates MEK which activates ERK [20]. Phosphorylated ERK moves into the nucleus to regulate expression of target genes. MAPK pathway can be activated either by RTK or GPCR via Ras, adenylate cyclase or Rac-dependent mechanism. As well as Akt, ERK activates mTORC1 [29]. Knock-out of b-raf in neural progenitor cells impaired oligodendrocyte differentiation in developing mice [30]. Erk2 deletion from GFAP-expressing radial glial cells impaired their differentiation in oligodendrocyte in vitro [31]. However, this effect was not found in vivo where Erk1 and Erk2 deletion in oligodendrocyte lineage did not affect OPC differentiation but rather reduced myelin sheath thickness [32]. Therefore, MAPK pathway seems to play a more prevalent role in myelin growth compared to cell differentiation.
The interplay between these pathways is adding another complexity in the signalling machinery controlling OPC differentiation. Fine tuning of these pathways also depends on the integrin-dependent activation of Src family kinases like Fyn. Fyn activation by α6β1 integrin can, therefore, amplify PDGF signalling and switch neuregulin signalling from PI3K to MAPK pathway [33]. Among targets of Fyn identified in OPC, Cdk5 has been shown to transmit PDGF-A induced migration [34] and Fyn also interacts with p130cas to activate Rac [35, 36]. Hence, the concomitant activation of RTK, GPCR and integrins in response to environmental cues is creating intracellular competitions of signalling pathways whose biological consequence on OPC relates on the relative strength of each components [37]. Consequently, the dominance of one or the other pathways impacts the different stages towards remyelination.
Pathways involved in migration
To reach the site of lesions, OPCs must migrate directionally and thus acquire antero-posterior polarity regulated by proteins acting specifically at one cell pole. Phosphatidylinositol 3-kinase (PI3K) and its products, phosphatidylinositol triphosphate (PIP3), are preferably located forward in protrusions [38]. PIP3 recruits at the plasma membrane the Arp2/3/WASP complex which regulates the connection of the actin cytoskeleton and the guanine-nucleotide exchange factors (GEF) which then activate the Rho GTPases [39]. Rho GTPases Rac and Cdc42 stimulate the polymerization of actin at the migration front, and at the back RhoA stimulates the contraction of actin to advance the cellular body. The receptor tyrosine kinase (RTK) and GPCR of growth factors and chemokines contribute to raise the PIP3 levels locally and define the migration front by activating the PI3K directly or via the GTPase Ras [40]. Downstream Ras, Ral/exocyste pathway coordinates membrane trafficking and actin polymerization contributing also to the front-rear polarity [41]. All these proteins involved in actin-driven protrusions have been found in oligodendrocytes from OPC stage to myelin sheath preparations [42]. Some specificities exist with predominance of actin nucleator WAVE1 in oligodendrocytes, whereas WAVE2 is more expressed in Schwann cells [42]. Loss of WAVE1 leads to a decreased number of OPC processes and hypomyelination in corpus callosum and optic nerve [43].
The contribution of Ras, PI3K-Akt and MAPK pathways to the growth of OPC pool and their involvement in the final steps of differentiation and myelination have overshadowed their involvement in migration, explaining sparse studies about their intrinsic contribution during the remyelination sequence. However, the transmission of promigratory signal obviously involves these pathways. Taking the example of CXCL12 dependent migration of OPCs, its binding to its receptor CXCR4 activates both PI3K-Akt pathway and MAPK pathway whose inhibition reduces OPC migration in vitro [44]. Activation of PKC, known to interact with these pathways, stimulated also OPC protruding activity [45]. Work in zebrafish demonstrated that knockdown of NF1 improved OPC migration presumably via the loss of its GAP inhibitory activity on Ras [46]. One frequent outcome of these pathways ends up on regulation of Rho GTPases like Rac. Whereas growth-factor induced Ras signalling activates Rac via PI3K, Ras-Raf-MAPK pathways can also downregulate expression of the Rac GEF activator Tiam1 [47]. Reciprocally, Rac contributes to the recruitment of PI3K at the leading edge of migrating cells [48] and Rac regulates formation of MEK-ERK1 complexes via PAK [49]. A bFGF dose-dependent activation of Rac determines the switch between directed and random migration of OPCs [50, 51]. The specificity of NG2 proteoglycan expression in OPCs has to be considered to fully understand their migration properties. NG2 intrinsically maintains high RhoA activity at the periphery of the cell via the RhoA GEF Syx [51], resulting in contact inhibition of locomotion and explaining the self-repulsion of OPCs observed in vivo in adult brain [52]. However, the phosphorylation of NG2 by PKCα switches downstream signalling from RhoA to Rac stimulation via recruitment of PAR and CRB complex proteins and activation of the Rac GEF Tiam1 [51, 53]. The activation of PKCα, notably by PLC activity downstream RTK and GPCR, favors a polarized shape where NG2 itself contributes to the high Rac activity at the migrating front and maintain RhoA at the rear of the cell (for extensive review see [50]).
Pathways involved in proliferation/survival
In experimentally induced demyelination, a dramatic increase of OPCs is observed [54]. Several mitogens have been involved in lesion-associated remyelination such as PDGF-A, bFGF, NT-3, IGF-1, CNTF, IL-6, LIF [16]. The consecutive Ras, PI3K-Akt and MAPK cascade controls cell proliferation [55]. It was shown that bFGF and IGF1 synergistically promotes cell cycle progression via enhanced cdk2 and cdk1 activity [56, 57]. This effect was impeded by rapamycin, involving mTOR in oligodendrocyte cell progression. Connexin 47 upregulation in OPC induced by coculture with astrocytes, possibly via heterotypic gap junctions, stimulates OPC proliferation via ERK1/2 [25]. Growth factors promote also oligodendrocyte survival like IGF1 which protects OPCs against TNFα-induced damage via PI3K/Akt dependent phosphorylation of BAD [58]. Knockout in oligodendrocyte lineage of PIKE, an upstream activator of PI3K-Akt, impaired remyelination of corpus callosum treated with lysolecithin. In this model, neural precursor from the subventricular zone usually recruited to repair corpus callosum demyelination had lower proliferation because of PIKE knock-out [59].
Pathways involved in differentiation and myelin sheath formation
Transcription factors, like Olig1, Ascl1, Nkx2.2, Sox10, YY1 and Tcf4, are required for the generation of mature oligodendrocytes [60]. Oligodendrocyte differentiation control by the Wnt/β-catenin pathway is ambivalent since it is mainly described as inhibitor for oligodendrocyte differentiation, whereas its transient activation seems crucial for initiating terminal differentiation [61, 62, 63]. This positive effect has been proposed to be dependent of β-catenin level in the nucleus and its association with the transcription factor Tcf7l2 [64]. GSK3β is a key actor of this balance since it regulates β-catenin availability by controlling its phosphorylation-dependent targeting to proteasome. Indeed pharmacological inhibition or knockdown of GSK3β can either promote or supress OPC differentiation [63, 65].
PI3K-Akt and MAPK pathways play an important role in oligodendrocyte differentiation [66–68]. Their parallel role has been demonstrated by the myelination rescue of Erk1/2 deficient mice by PI3K/Akt constitutive activation [67]. One common target of these pathways is TSC2 whose phosphorylation by Akt or Erk1/2 results in mTORC1 complex activation [69]. Even parallel, these pathways could have sequential roles since Erk1/2 regulates differentiation in early stages and mTOR in later stages [70]. The function of mTORC1 is interesting since it coordinates myelin protein expression with lipid synthesis and via transcription factors SREBP [69]. Regarding studies focusing on remyelination of lesions, a focal LPC-induced demyelination with CNP- dependent deletion of Cdk5 impaired OPC differentiation and decreased levels of activated AKT [71]. Erk2 conditional knockout in oligodendrocytes resulted in delayed remyelination of LPC-induced lesion in the corpus callosum accompanied by impaired MBP translation [72]. Similarly, sustained activation of ERK1/2 by expression of constitutively active MEK in oligodendrocyte lineage accelerated remyelination of LPC-induced lesions with improved myelin thickness compared to wild-type littermates [73].
Acting downstream of Ras, Ral GTPase and exocyst impact several aspects of cell polarity through vesicular trafficking, The massive membrane remodelling necessary to myelination is, therefore, impaired when these pathways are ablated [74, 75].
The process of myelin membrane wrapping requires a coordination of the cytoskeleton dynamics. Indeed, in the inner tongue, it has been shown that the polarized growth of myelin is determined by elevated levels of PIP3 at the growing edge [68]. Downstream Rho GTPase effectors use this signal to structure cytoskeleton organisation. Indeed, loss of Cdc42 and Rac1 leads to accumulation of cytoplasm of myelin sheaths and myelin out folding, revealing their role in coordinating myelin sheath wrapping [76, 77]. Ultimately, F-actin presents in the leading edge and outermost myelin layer is disassembled in the rest of the myelin sheath with MBP coming [78, 79].
Remyelinating factors secreted in the lesions, a rheostat controlling oligodendrocyte behaviour
Despite the neurotoxic effect consecutive to the proinflammatory role of astrocytes and macrophages/microglia, they can be beneficial for remyelination. Density of O4-positive OPCs is corelated with increased debris-laden macrophages in human MS lesions [80]. Elevated density of HLA-DR macrophages and microglia at the lesion border in MS also correlated with more extensive remyelination [81]. Indeed, depletion of pro-inflammatory macrophages/microglia at early time points post-demyelination impairs OPC proliferation whereas depletion of M2 polarized cells at later points impairs OPC differentiation [82]. This could be explained by the release of several growth factors important for OPC recruitment, proliferation and differentiation: IGF-1, activin-A, endothelin-2, HGF, PDGF-A, bFGF, galectin 3, TNF, IL-1β, IL4, CXCL1, 8, 10, 12 [83–85].
Analysis of MS lesions revealed selective expression of bFGF within active lesion and in the peri plaque of chronic lesions [86]. The stimulation level of factors such as PDGF-A or bFGF affects the oligodendrocytic cell response [50, 51]. Thus PDGF activates the migration of OPCs at low concentrations via PI3K and proliferation at high concentrations via PLCγ [87]. Therefore, the gradient of growth factors and chemokines around MS lesions [88] suggests a concentration-dependent mechanism of oligodendrocyte response.
Neurotrophic factors also modulate remyelination. In demyelinating lesions of the spinal cord, NRG1 promotes remyelination through generation of oligodendrocytes [89] and presumably through stimulation of migration by activation of Rac1 and Cdc42 via the GEF Dock7 [90]. Among neurotrophins, NT3 enhances Schwann cell migration via Rac1 and Cdc42 activation [91], whereas BDNF binding to p75NTR inhibits Schwann cell migration by activation of the GEF Vav2 and RhoA [92]. Conversely, NT3 inhibits peripheral neural system myelination [93, 94] whereas BDNF promotes it [95, 96].
Hence, a gradient of signalling molecules emanating from the lesion probably controls the transition of oligodendrocyte behaviours between migration, proliferation and differentiation. Thus, depending on OPCs or Schwann cells, these lesion-associated factors can specifically favour one of these behaviours or induce different effects depending on their level of activity. Besides positive factors, inhibitory molecules arise from the lesion and block remyelination.
Inhibitors of remyelination, a reminiscence of developmental guidance mechanisms
As usual with biological processes, positive/activating pathways are counterbalanced by negative/inhibitory signals aiming at providing to the cell the right equilibrium adapted to the function. Indeed, facing the numerous factors favouring myelination, several types of guidance molecules known for their critical roles during brain development are impacting OL and OPC functions by triggering complex signalling pathways controlling cytoskeleton remodelling (Fig. 3).
Fig. 3.
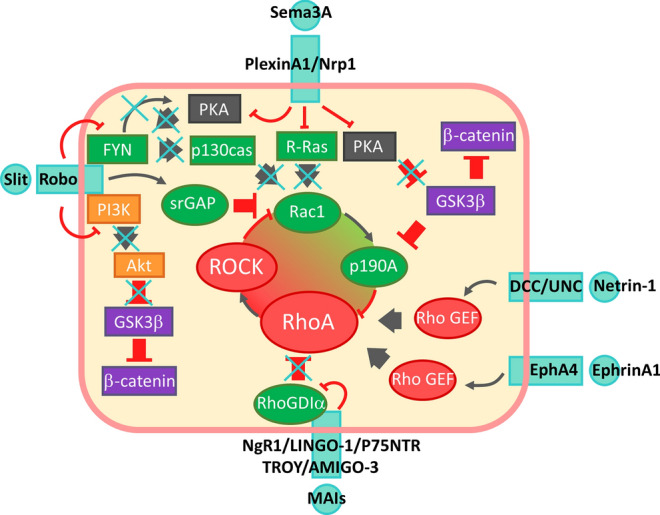
Signalling pathways affected by inhibitors of remyelination. Rac1/RhoA balance is commonly found altered by all inhibitors of remyelination to the benefit of RhoA. MAIs induce sequestration of Rho-GDI by p75 or TROY, releasing activable RhoA-GDP. Ephrin/Eph association as well as DCC/UNC activation by netrin-1 activates RhoA through RhoGEF. Reading of Sema3 effect on RhoGTPases is complexified by the kinetic of its receptor activation relying on an initial stimulation of Rac1 to subsequently activate its RasGAP domain. However downstream signalling unambiguously favours RhoA signalling. Indeed, PlexinA1 inhibits Ras activity and simultaneously removes RhoA inhibiting activity of p190A through removal of PKA dependent inhibition of GSK3β. Arrows show activation and truncated red arrows show inhibition. PI3K-Akt pathway is shown in orange boxes, MAPK pathway in blue boxes and GSK3β/β-catenin in purple boxes. Elements in favour of Rac activity are shown in green and elements in favour of RhoA activity in red. Crosses show inhibitory effect of inhibitors of remyelination on signalling. Signalling pathways presented have been deciphered in neurons and/or oligodendrocytes
Sema3A/PlexinA1/Nrp1
Downstream signalling deciphering of Sema3A was mainly carried out in neurons [97–102]. Sema3A stimulates the Ras GAP activity of PlexinA1 which directly inhibits Ras [97, 98]. Binding of Sema3A to PlexinA1/Nrp1 complex receptor leads to the release of Rac-GEF FARP2 which activates Rac. GTP-bound Rac is then sequestrated by PlexinA1 and allows the recruitment of Rnd1 which activates the GAP domain of PlexinA1 [99, 100]. Collapsus induced by localized addition of Sema3A involves RhoA activation and retrograde waves of active Cdc42 [101] showing disorganized cell polarity which could be potentially linked to Ras/PI3K pathway inhibition. Sema3A is also responsible of decreased activity of PKA and reduced phosphorylation of its targets GSK3β and LKB1 [102].
The roles of Semaphorins on oligodendrocyte lineage and its relevance in the context of multiple sclerosis has been well-documented [103, 104].
Sema3A expression has been found in MS lesions as well as EAE and cuprizone experimental demyelination models [105–107]. Sema 3A has been originally identified as a repulsive cue for OPCs during optic nerve development [108, 109]. Sema3A exerts its inhibitory action on oligodendrocytes in multiple ways. Sema3A inhibits process extension [110], migration [105] and also oligodendrocyte differentiation in vitro resulting in remyelination impairing when sema3A is infused in demyelinating lesions [111]. Loss of function of Sema3A receptor Nrp1 rescues OPC recruitments into the demyelinating lesion [112]
NOGO and myelin associated inhibitors
Neurite outgrowth inhibitor (Nogo-A), myelin-associated glycoprotein (MAG) and oligodendrocytes myelin glycoprotein (Omgp) are myelin-associated inhibitors (MAIs) largely described for their inhibitory function on neurite extension in neurons. MAIs are expressed in oligodendrocytes and accumulate in demyelinated plaques exhibiting oligodendrocytes debris. MAIs inhibit neurite outgrowth by interaction with Nogo receptor 1 (NgR1), a GPI-anchored receptor activating RhoA pathway. Since NgR1 has no catalytic domain, it associates with LINGO1 [113], AMIGO3 [114] or some of the tumor necrosis factor super family receptors (TNFSFR): p75NTR or TROY to trigger signalling [113, 115, 116]. These receptors assemble into a signalling platform in which LINGO-1 in association with WNK1 [117] facilitates the binding of Rho-GDiα to p75 or TROY, leading to the release of RhoA-GDP [118, 119]. RhoA-GDP will then be converted into RhoA-GTP by Rho-GEFs (Guanine nucleotide Exchange Factors) to activate RhoA [118]. Interestingly, LINGO-1 homodimerization similarly leads to RhoA activation [120, 121]. LINGO-1 also reduces EGFR phosphorylation by a direct association downregulating MAPK and PI3K pathways [121, 122]. Another study reports that the direct binding of Lingo-1 to ErbB2 blocks its translocation in lipid rafts thereby inhibiting OPC differentiation [123]. TROY has been reported to activate cJunk and PKCα phosphorylation in OPC [124] but the precise mechanism remains unclear.
Netrin/DCC/UNC5
Earliest description of unc-6 (ortholog of netrin-1) in C. elegans development suggested its duality, since it performed repulsive effect on axons extending dorsally and attraction on axons extending ventrally [125]. This relies on receptor complexes transmitting netrin-1 signal: homodimers of DCC induce an attraction response, whereas heterodimers DCC/UNC5 induce repulsion [126]. Membrane receptor level, secondary messengers such as PKCα [127, 128] or ligand concentration can affect this response. This later parameter appeared in netrin-1 gradient where lower concentrations are attractive and higher repulsive [129]. This could be explained by the higher affinity of netrin-1 for DCC homodimer compared to DCC/UNC5 heterodimer [126] which would, therefore, bind repulsive heterodimer only at highest concentrations. Intracellular signalling of both receptors relies on similar actors like SFK and Rho GEFs. DCC homodimer drives to Rac1 activation [130] whereas DCC/UNC5 heterodimer drives to RhoA activation [131] (For extensive review on netrin-1 signalling, see [132]). This regulation of RhoA has been shown to be mediated by a Rho GEF in C. elegans [133].
In oligodendrocyte lineage, netrin-1 acts similarly and displays a chemorepulsive effect on OPCs mediated by RhoA activation and its downstream effector ROCK. Interestingly, netrin seems later necessary for differentiated oligodendrocytes branching [134, 135]. This remyelinating effect relies in fact on Fyn recruitment to DCC receptor resulting in RhoA inhibition [136]. Presence of Netrin in MS sample is attested and could regulate OPC recruitment to the lesions [137].
Slit/Robo
Studies on Drosophila embryo revealed that binding of Slit to transmembrane receptor Robo is a chemorepulsive cue stopping midline crossing of the CNS [138, 139]. Slit could display its action by inducing molecular tension in Robo allowed by ECM-immobilization of Slit [140]. This leads to exposure of metalloproteinase cleavage site and ectodomain shedding [141]. This cleavage is required for recruitment of downstream signalling molecules [142]. Slit-Robo signalling involves GSK3β/β-catenin pathways and Rho GTPases. Binding of Slit2 induces the binding of the PI3K subunit p85 and blocks Akt activation. As a consequence, GSK3-β inhibition by Akt is released and β-catenin translocation is blocked [143]. Slit-Robo-GTPase activating proteins (srGAPs) inhibit Rho-GTPases like srGAP2 which inhibits Rac1 [144] (for review: [145]). Interestingly, Slit/Robo pathway can cross react with the other inhibitory pathways. Indeed, SlitC cleaved fragment can interact with PlexinA1 and induces growth cone collapse [146]. In the presence of Slit, Robo1 can silence Netrin/DCC attraction signalling [147]. On the contrary, Robo3 which has lost its ability to bind Slit during evolution, collaborate with DCC for Netrin-1 attraction signalling [148].
Slit2 has been involved in the dispersal of OPCs. Slit2 binding with Robo1 induces Fyn recruitment and inactivation of Fyn as well as RhoA activation [149].
Ephrin/Eph
Ephrin/Eph signalling is involved in direct cell interactions signalling and regulates axon guidance [150]. Ephrin and Eph receptors are both membrane-anchored and from their interaction results a bi-directional signalling with a forward signalling relying on Eph tyrosine phosphorylation and a reverse signalling in ephrin expressing cells. EphrinA binding to EphA4 triggers the RhoA GEF ephexin activation in neurons [151] or Vsm-RhoGEF in vascular smooth cells [152].
Several ephrin and Eph have been found in MS lesion samples [153]. OPC express ephrinB2 and are repealed by EphB2 [154].Ephrin-A1 interaction with OPC EphA4 activates RhoA/ROCK signalling and causes inhibition of oligodendrocyte process extension [155] probably via a RhoGEF. EphrinB3 found in MS lesion is also responsible for RhoA activation in OPCs probably via EphA4 binding, and its antibody-mediated neutralization favours remyelination in an experiment model [156].
Targeting the inhibitory signals to promote remyelination
Few therapeutic approaches are attacking the problem of myelin repair (Fig. 4). The most promising ones are the use of miconazole [157], MD1003 [158] a highly concentrated oral formulation of biotin (also known as Vitamin H or Vitamin B7), the retinoid X receptor Gamma [159], Clemastine fumarate [160] or Sobetirome [161]. The molecules are acting by favouring the differentiation of OPC but do not address the inhibitory molecular barrier. As described above, one common feature of the inhibitory/repulsive molecules signalling overexpressed in MS lesions relies on RhoA activation. This convergence in the signalling pathway mirrors what is seen in CNS nerve lesions where the glial scar being formed upon lesion is also exhibiting a variety of inhibitory factors [162] including ECM components (CSPGs, Tenascin or NG2), myelin-derived growth inhibitors NOGO, MAG, OMgp, and other membrane bound or secreted repulsive cues (Semaphorins, Ephrins). In this situation, many therapeutic options have been developed to antagonize the inhibitory factors and favour neuron-intrinsic regeneration capability to restore axon regrowth [163]. The use of small molecules, antibodies or peptides indeed showed significant improvement of axon regrowth and in some cases retargeting with improved functional recovery [164]. Strikingly, the inhibition of one single inhibitory factor is commonly sufficient to circumvent the deficit of growth in preclinical studies but it so far poorly translated into clear benefits in clinical studies. However, the beneficial effect obtained relies on resetting of the growth capacity by direct or indirect modulation of axon growth-associated signalling pathways. By analogy, it appears interesting in the context of demyelination diseases to modulate the response of OPCs to environmental inhibitory signals in order to promote remyelination. The promotion of pro-migratory/pro-differentiating factors by the selective blockade of inhibitory signals should allow a reinforcement of the intrinsic myelin repair mechanism.
Fig. 4.

Therapeutic strategies to promote remyelination. The left panel shows therapeutic strategies to activate OPC differentiation. MD1003 increases fatty acid synthesis trough acetyl CoA carboxylase activation. Miconazole drives ERK phosphorylation. 9-Cis retinoic acid and Sobetirome act on nuclear receptor Retinoic X receptor gamma and Thyroid hormone receptor, respectively. The right panel illustrates alternative strategies blocking inhibitory signals. Clemastine drives remyelination by antagonising muscarinic receptor activation. Monoclonal antibodies directed against Nogo-A or the receptor LINGO-1 cancel the inhibitory signalling. A polyclonal antibody directed against ephrinB3 prevents its association with EphA4. MTP-PlexA1 promotes both OPC differentiation and migration by blocking Sema3A-induced NRP1/Plexin-A1 dimerization
To test this strategy, studies have been conducted with anti-Nogo-A antibody [165] up to clinical phases. On the same line, the antagonism of the Nogo-Receptor subunit -Lingo1 favouring MAPK and glucocorticoid receptors is showing remyelination capability with a quite good tolerance in human [166]. An anti-SEMA4D antibody (VX15/2503) is currently being investigated [167] to counteract SEMA4D inhibitory effects on remyelination. More recently, an experimental therapeutic approach was designed to block Sema3A inhibitory signalling by inhibiting its Plexin-A1 signalling receptor [105]. This approach is based on the use of a peptide targeting the transmembrane domain of Plexin-A1 (MTP-PlexA1) that disrupts the dimerization necessary to trigger Sema3A signalling pathway [168]. This strategy of inhibiting dimerization of the Plexin-A1 receptor has already been used for the inhibition of glioblastoma progression in PDX models [168] and is a validated approach for other receptors such as NRP1 [169–171] or ErbB2 [172]. The choice to target Plexin-A1 is further strengthened by the demonstration of overexpression in the oligodendrocytes of MS patients. This increase is concomitant with transient re-expression of Sema3A in demyelinated regions. The administration of MTP-PlexA1 peptide showed a positive effect on remyelination which results in a restoration of myelin sheaths and the disappearance of locomotor disorders in the cuprizone but also in the EAE demyelinating in vivo models. The observed effects are due to a disruption of the dimerization of NRP1 and Plexin-A1 which counters the anti-migratory and anti-differentiation effect of Sema3A as shown by in vitro and in vivo approaches using in particular the proximity ligation assay (PLA). The therapeutic benefit observed in mice does not appear to be related to modulation of inflammation but to a direct effect on repair capacity.
Thus, it would seem that alteration of the balance between remyelination promoter and inhibitor signals may offer an interesting avenue for the development of what could become, in combination with anti-inflammatory drugs, the beginning of curative therapeutic approaches for MS. However, the exact therapeutic window of anti-inhibitory molecules has to be correctly defined because RhoA activity appears involved in final steps of remyelination. Indeed, RhoA GEF Vav3 knockdown mice display impaired remyelination in lysolecithin and cuprizone demyelination models and produce thinner myelin sheaths [173]. This can be attributed to the necessary fine tuning of actin cytoskeleton during myelination or differentiation program regulation as shown in Schwann cells where RhoA allows differentiation by inhibiting JNK pathway [174]. Generally, it appears that signalling pathways promoted by growth factors and chemokines like Ras, PI3K and Rho GTPases are inversely regulated by repulsive molecules. These inhibitory molecules can, therefore, modulate signalling pathways induced around the lesion and contribute to behavioural transitions. After recruitment of new OPCs thanks to migration and proliferation, cells obviously need a dramatical change of signalling pathways to induce differentiation and produce the myelin sheaths. Repulsive molecules could play a role in this final step. However, we can argue that their pathological secretion in demyelinating pathologies reach a too high level and impairs all steps required for proper remyelination. The next challenge will be to demonstrate the clinical benefit of counteracting the inhibitory molecular barriers to promote remyelination.
Box 1: Negative loop between Rac and RhoA GTPases.
Studies addressing specifically cell signalling in oligodendrocytes are still sparse. However, we can hypothesize that ubiquitous intrinsic regulation of RhoGTPases applies in oligodendrocytes. Cytoskeleton dynamics are under the control of RhoGTPases which cycle between an inactive GDP-bound form and an active GTP-bound form. Their activity is tightly regulated by the coordinated action of regulators: guanine nucleotide exchange factors (GEFs) improve GTP loading, GTPase-activating proteins (GAPs) promote GTPase inactivation by enhancing GTP hydrolysis and GDIs inhibit RhoGTPases by sequestering them in the cytoplasm [175]. Rac promotes actin polymerization at migration front while RhoA ensures contractility of actomyosin predominantly through its effector Rho-associated protein kinase (ROCK). The balance between these antagonistic activities is critical for coordination of cell motility.
Rac negatively regulates RhoA through the RhoGAP p190A. Active Rac induces ROS production which inactivates the tyrosine phosphatase LMW-PTP through direct oxidation of cysteines in the catalytic pocket, relieving the inhibition of p190 [176]. P190A GAP specificity for RhoA is enhanced by PKCα which dissociates p190A from plasma membrane acidic phospholipids and blocks its RacGAP activity [177]. Subcellular targeting of p190A controls its spatial regulation of RhoA [178]. Rnd1 [179], Src, FAK, p120-catenin [180] and cortactin [181] allow targeting of p190A to actin protrusions. The polarity complex proteins also contribute to inhibit RhoA at the migration front via Par6/aPKC dependent activation of p190A [182].
RhoA negatively regulates Rac through ROCK. Tiam1, a main Rac GEF, is recruited by the PAR complex proteins Par3 and Par6, resulting in Rac activation at the migration front [183, 184]. ROCK disrupts Par complex by phosphorylating Par3 and impairs Rac activation in the rear of the cell [185]. Other effects of ROCK have been found such as dissociation of the Rac GEF β-PIX from integrin-based adhesions [186] and Rac GAPs activation [187, 188].
It should be noted that interactions between Rac and Rho are more complex since another effector of RhoA, the formin mDia1, antagonizes ROCK effect by stimulating Rac through Src [189] and could drive RhoA activity found in initial events of membrane ruffling [190].
Box 2: The concept of membrane domain targeting peptides.
Most of the current drugs target extra- or intracellular domains of membrane receptors to modulate downstream signalling. Alternatively, protein–protein interaction (PPI) inhibitors are conceived to alter the dimerization /oligomerization of receptors to inhibit their activity [191]. Membrane domain targeting peptides (MTP) are indeed a new class of small interfering peptides mimicking the transmembrane domains of receptors to act as decoy acting by direct competitive binding with the target domain leading to abnormal oligomerization of the receptor [192]. The consequence is a partial or total shutdown of downstream signals blocking the subsequent biological functions. This approach turns out to be a potent therapeutic strategy in the context of brain tumor or breast cancer and also recently finds an application in the context of Multiple Sclerosis. More than 20 years’ research [193, 194] was needed to solve issues related to the high insolubility of MTPs and to demonstrate the specificity of the approach which is nowadays applicable to GPCR receptors [195–198].
MTPs are 20–30 amino acids long peptides.
MTPs are often containing a GxxxG motif or GAS motif defining the dimerization interface.
MTPs activity is in the range of 0.01–0.1 µM in vitro and µg/kg in vivo.
MTPs exhibit long-lasting biodistribution profiles markedly different from soluble peptides.
MTPs exhibit good tolerance profiles compatible with chronic administrations.
The strength of the strategy is the mechanism of action inducing a prolonged inhibition of receptors which was seen with a single dose up to 48 h in vitro and 72 h in vivo. Initially described as antagonist peptides blocking the dimerization interfaces, studies are now demonstrating a more profound impact by inducing conformational changes of extracellular domains of target receptors. The interference of the dimerization capacity is providing a way to attack both the activation of the receptor but also potential compensatory or redundant pathways when neutralizing the heterodimerization capability of receptors. None of these MTPs reached so far a clinical validation. A global effort is needed to promote the use of MTPs which should not stay at the level of bench tools.
Acknowledgements
The authors wish to thank Dr V. Jolivel for helpful discussion on the manuscript.
Abbreviations
- Akt
Protein kinase B
- AMIGO-3
Amphoterin-induced protein 3
- ARP2
Actin-related protein-2
- Bcl2
B-cell lymphoma 2
- BDNF
Brain derived neurotrophic factor
- bFGF
Basic fibroblast growth factor
- Cdc45
Cell division control protein 45
- Cdk5
Cyclin-dependent kinase 5
- CNP
2′,3′-Cyclic nucleotide 3′ phosphodiesterase
- CNS
Central nervous system
- CNTF
Ciliary neurotrophic factor
- EAE
Experimental autoimmune encephalomyelitis
- ECM
Extracellular matrix
- FARP2
FERM, ARH/RhoGEF and Pleckstrin domain protein 2
- CSPG
Chondroitin sulfate proteoglycan
- CXCL2
C-X-C motif chemokine ligand 2
- GAB
GRB2-Associated binding protein
- GAP
GTPase, activating protein
- GDP
Guanosine diphosphate
- GEF
Guanine–nucleotide exchange factor
- GFAP
Glial fibrillary acid protein
- GPCR
G-protein coupled receptor
- GRB2
Growth receptor factor-bound protein 2
- GSK3 β
Glycogen synthase kinase-3
- GTP
Guanosine triphosphate
- GTPase
Guanosine triphosphatase
- IGF-1
Insulin-like growth factor 1
- IL-6
Interleukin 6
- LIF
Leukemia inhibitory factor
- LINGO-1
Leucine-rich repeat and Ig domain-containing 1
- LKB1
Liver kinase B1
- LPC
Lysophosphatidylcholine
- NgR1
Nogo receptor 1
- Nogo-A
Neurite outgrowth inhibitor
- NT-3
Neurotrophin-3
- MAG
Myelin-associated glycoprotein
- MAIs
Myelin-associated inhibitors
- MAPK
Mitogen-activated protein
- MBP
Myelin basic protein
- MDM2
Murine double-minute 2 homolog
- MS
Multiple sclerosis
- MTP-PlexA1
Membrane domain-targeting peptide of Plexin-A1
- mTOR
Mammalian target of rapamycin
- NF1
Neurofibromin 1
- NG2
Neuron glial antigen 2
- NRP1
Neuropilin 1
- OL
Oligodendrocyte
- Omgp
Oligodendrocyte myelin glycoprotein
- OPC
Oligodendrocyte precursor cell
- P75NTR
Neurotrophin receptor p75
- PDGF
Platelet-derived growth factor
- PDK1
Protein kinase phosphoinositide-dependent kinase1
- PDX
Patient-derived xenograft
- PH
Pleckstrin homology
- PI3K
Phosphatidylinositol 3-kinase
- PIP3
Phosphatidylinositol triphosphate
- PKC α
Protein kinase C α
- PLA
Proximity ligation assay
- PLC
Phospholipase C
- PlexA1
Plexin A1
- PPI
Protein–protein interaction
- PTEN
Phosphatase and tensin homolog
- RRMS
Relapsing/remitting multiple sclerosis
- RTK
Receptor tyrosine kinase
- Sema3A
Semaphorin 3A
- SEMA4D
Semaphorin 4D
- SOS
Son of sevenless homolog
- srGAPs
Slit-Robo-GTPase activating proteins
- SREBP
Sterol regulatory element-binding protein
- TNFSFR
Tumour necrosis factor super family receptors
- TSC2
Tuberous sclerosis complex 2
- WASP
Wiskott-Aldrich syndrome protein
- WNK1
WNK lysine-deficient protein kinase
Funding
This work was conducted in the framework of the Labex Medalis and Idex unistra and by SFRI StratUS (Grant number ANR-10 Idex-002 ANR-20-SFRI-0012) (ANR-10-LABX-0034_Medalis and received a financial support from French government managed by “Agence National de la Recherche” under “Programme d’investissement d’avenir”.
Declarations
Conflict of interests
The authors declare no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Henderson APD, Barnett MH, Parratt JDE, Prineas JW. Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann Neurol. 2009;66:739–753. doi: 10.1002/ana.21800. [DOI] [PubMed] [Google Scholar]
- 2.Marik C, Felts PA, Bauer J, et al. Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain. 2007;130:2800–2815. doi: 10.1093/brain/awm236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conductier G, Blondeau N, Guyon A, et al. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J Neuroimmunol. 2010;224:93–100. doi: 10.1016/j.jneuroim.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 4.Semple BD, Kossmann T, Morganti-Kossmann MC. Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J Cereb Blood Flow Metab. 2010;30:459–473. doi: 10.1038/jcbfm.2009.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merkler D, Ernsting T, Kerschensteiner M, et al. A new focal EAE model of cortical demyelination: multiple sclerosis-like lesions with rapid resolution of inflammation and extensive remyelination. Brain. 2006;129:1972–1983. doi: 10.1093/brain/awl135. [DOI] [PubMed] [Google Scholar]
- 6.Neumann H, Medana IM, Bauer J, Lassmann H. Cytotoxic T lymphocytes in autoimmune and degenerative CNS diseases. Trends Neurosci. 2002;25:313–319. doi: 10.1016/s0166-2236(02)02154-9. [DOI] [PubMed] [Google Scholar]
- 7.Lassmann H. Review: the architecture of inflammatory demyelinating lesions: implications for studies on pathogenesis. Neuropathol Appl Neurobiol. 2011;37:698–710. doi: 10.1111/j.1365-2990.2011.01189.x. [DOI] [PubMed] [Google Scholar]
- 8.Lengfeld J, Cutforth T, Agalliu D. The role of angiogenesis in the pathology of multiple sclerosis. Vasc Cell. 2014;6:23. doi: 10.1186/s13221-014-0023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blakemore WF. Pattern of remyelination in the CNS. Nature. 1974;249:577–578. doi: 10.1038/249577a0. [DOI] [PubMed] [Google Scholar]
- 10.Ludwin SK, Maitland M. Long-term remyelination fails to reconstitute normal thickness of central myelin sheaths. J Neurol Sci. 1984;64:193–198. doi: 10.1016/0022-510X(84)90037-6. [DOI] [PubMed] [Google Scholar]
- 11.Faissner S, Plemel JR, Gold R, Yong VW. Progressive multiple sclerosis: from pathophysiology to therapeutic strategies. Nat Rev Drug Discov. 2019 doi: 10.1038/s41573-019-0035-2. [DOI] [PubMed] [Google Scholar]
- 12.Picard-Riera N, Decker L, Delarasse C, et al. Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc Natl Acad Sci USA. 2002;99:13211–13216. doi: 10.1073/pnas.192314199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zawadzka M, Rivers LE, Fancy SPJ, et al. CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell. 2010;6:578–590. doi: 10.1016/j.stem.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyd A, Zhang H, Williams A. Insufficient OPC migration into demyelinated lesions is a cause of poor remyelination in MS and mouse models. Acta Neuropathol. 2013;125:841–859. doi: 10.1007/s00401-013-1112-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stangel M, Kuhlmann T, Matthews PM, Kilpatrick TJ. Achievements and obstacles of remyelinating therapies in multiple sclerosis. Nat Rev Neurol. 2017;13:742–754. doi: 10.1038/nrneurol.2017.139. [DOI] [PubMed] [Google Scholar]
- 16.Franklin RJM, Ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 2008;9:839–855. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- 17.Baxi EG, DeBruin J, Jin J, et al. Lineage tracing reveals dynamic changes in oligodendrocyte precursor cells following cuprizone-induced demyelination. Glia. 2017;65:2087–2098. doi: 10.1002/glia.23229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crawford AH, Chambers C, Franklin RJM. Remyelination: the true regeneration of the central nervous system. J Comp Pathol. 2013;149:242–254. doi: 10.1016/j.jcpa.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 19.Castellano E, Downward J. RAS interaction with PI3K. Genes Cancer. 2011;2:261–274. doi: 10.1177/1947601911408079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jain R, Watson U, Vasudevan L, Saini DK. Chapter three—ERK activation pathways downstream of GPCRs. In: Shukla AK, editor. International review of cell and molecular biology. Academic Press; 2018. pp. 79–109. [DOI] [PubMed] [Google Scholar]
- 21.Sanz-Rodriguez M, Gruart A, Escudero-Ramirez J, et al. R-Ras1 and R-Ras2 are essential for oligodendrocyte differentiation and survival for correct myelination in the central nervous system. J Neurosci. 2018;38:5096–5110. doi: 10.1523/JNEUROSCI.3364-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pawson T. Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell. 2004;116:191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- 23.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 24.Moore SF, van den Bosch MTJ, Hunter RW, et al. Dual regulation of glycogen synthase kinase 3 (GSK3)α/β by protein kinase C (PKC)α and Akt promotes thrombin-mediated integrin αIIbβ3 activation and granule secretion in platelets. J Biol Chem. 2013;288:3918–3928. doi: 10.1074/jbc.M112.429936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goebbels S, Oltrogge JH, Kemper R, et al. Elevated phosphatidylinositol 3,4,5-trisphosphate in glia triggers cell-autonomous membrane wrapping and myelination. J Neurosci. 2010;30:8953–8964. doi: 10.1523/JNEUROSCI.0219-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harrington EP, Zhao C, Fancy SPJ, et al. Oligodendrocyte PTEN required for myelin and axonal integrity not remyelination. Ann Neurol. 2010;68:703–716. doi: 10.1002/ana.22090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flores AI, Narayanan SP, Morse EN, et al. Constitutively active Akt induces enhanced myelination in the CNS. J Neurosci. 2008;28:7174–7183. doi: 10.1523/JNEUROSCI.0150-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narayanan SP, Flores AI, Wang F, Macklin WB. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J Neurosci. 2009;29:6860–6870. doi: 10.1523/JNEUROSCI.0232-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol. 2013;15:555–564. doi: 10.1038/ncb2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galabova-Kovacs G, Catalanotti F, Matzen D, et al. Essential role of B-Raf in oligodendrocyte maturation and myelination during postnatal central nervous system development. J Cell Biol. 2008;180:947–955. doi: 10.1083/jcb.200709069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fyffe-Maricich SL, Karlo JC, Landreth GE, Miller RH. The ERK2 mitogen-activated protein kinase regulates the timing of oligodendrocyte differentiation. J Neurosci. 2011;31:843–850. doi: 10.1523/JNEUROSCI.3239-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishii A, Fyffe-Maricich SL, Furusho M, et al. ERK1/ERK2 MAPK signaling is required to increase myelin thickness independent of oligodendrocyte differentiation and initiation of myelination. J Neurosci. 2012;32:8855–8864. doi: 10.1523/JNEUROSCI.0137-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Colognato H, Ramachandrappa S, Olsen IM, Ffrench-Constant C. Integrins direct Src family kinases to regulate distinct phases of oligodendrocyte development. J Cell Biol. 2004;167:365–375. doi: 10.1083/jcb.200404076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyamoto Y, Yamauchi J, Tanoue A. Cdk5 phosphorylation of WAVE2 regulates oligodendrocyte precursor cell migration through nonreceptor tyrosine kinase Fyn. J Neurosci. 2008;28:8326–8337. doi: 10.1523/JNEUROSCI.1482-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gonsior C, Binamé F, Frühbeis C, et al. Oligodendroglial p130Cas is a target of Fyn kinase involved in process formation, cell migration and survival. PLoS ONE. 2014;9:e89423. doi: 10.1371/journal.pone.0089423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharma A, Mayer BJ. Phosphorylation of p130Cas initiates Rac activation and membrane ruffling. BMC Cell Biol. 2008;9:50. doi: 10.1186/1471-2121-9-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Meara RW, Michalski J-P, Kothary R. Integrin signaling in oligodendrocytes and its importance in CNS myelination. J Signal Transduct. 2011;2011:354091. doi: 10.1155/2011/354091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maeda YT, Inose J, Matsuo MY, et al. Ordered patterns of cell shape and orientational correlation during spontaneous cell migration. PLoS ONE. 2008 doi: 10.1371/journal.pone.0003734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362–374. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 40.Dawes AT, Edelstein-Keshet L. Phosphoinositides and rho proteins spatially regulate actin polymerization to initiate and maintain directed movement in a one-dimensional model of a motile cell. Biophys J. 2007;92:744–768. doi: 10.1529/biophysj.106.090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zago G, Biondini M, Camonis J, Parrini MC. A family affair: a Ral-exocyst-centered network links Ras, Rac, Rho signaling to control cell migration. Small GTPases. 2017;10:323–330. doi: 10.1080/21541248.2017.1310649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bacon C, Lakics V, Machesky L, Rumsby M. N-WASP regulates extension of filopodia and processes by oligodendrocyte progenitors, oligodendrocytes, and Schwann cells—implications for axon ensheathment at myelination. Glia. 2007;55:844–858. doi: 10.1002/glia.20505. [DOI] [PubMed] [Google Scholar]
- 43.Kim H-J, DiBernardo AB, Sloane JA, et al. WAVE1 is required for oligodendrocyte morphogenesis and normal CNS myelination. J Neurosci. 2006;26:5849–5859. doi: 10.1523/JNEUROSCI.4921-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tian Y, Yin H, Deng X, et al. CXCL12 induces migration of oligodendrocyte precursor cells through the CXCR4-activated MEK/ERK and PI3K/AKT pathways. Mol Med Rep. 2018;18:4374. doi: 10.3892/mmr.2018.9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paez PM, Fulton DJ, Spreur V, et al. Multiple kinase pathways regulate voltage-dependent Ca2+ influx and migration in oligodendrocyte precursor cells. J Neurosci. 2010;30:6422. doi: 10.1523/JNEUROSCI.5086-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee J-S, Padmanabhan A, Shin J, et al. Oligodendrocyte progenitor cell numbers and migration are regulated by the zebrafish orthologs of the NF1 tumor suppressor gene. Hum Mol Genet. 2010;19:4643–4653. doi: 10.1093/hmg/ddq395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zondag GCM, Evers EE, ten Klooster JP, et al. Oncogenic Ras downregulates Rac activity, which leads to increased rho activity and epithelial-mesenchymal transition. J Cell Biol. 2000;149:775–782. doi: 10.1083/jcb.149.4.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Srinivasan S, Wang F, Glavas S, et al. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J Cell Biol. 2003;160:375–385. doi: 10.1083/jcb.200208179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eblen ST, Slack JK, Weber MJ, Catling AD. Rac-PAK signaling stimulates extracellular signal-regulated kinase (ERK) activation by regulating formation of MEK1-ERK complexes. Mol Cell Biol. 2002;22:6023–6033. doi: 10.1128/mcb.22.17.6023-6033.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Binamé F. Transduction of extracellular cues into cell polarity: the role of the transmembrane proteoglycan NG2. Mol Neurobiol. 2014;50:482–493. doi: 10.1007/s12035-013-8610-8. [DOI] [PubMed] [Google Scholar]
- 51.Binamé F, Sakry D, Dimou L, et al. NG2 regulates directional migration of oligodendrocyte precursor cells via Rho GTPases and polarity complex proteins. J Neurosci. 2013;33:10858–10874. doi: 10.1523/JNEUROSCI.5010-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hughes EG, Kang SH, Fukaya M, Bergles DE. Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat Neurosci. 2013;16:668. doi: 10.1038/nn.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Makagiansar IT, Williams S, Mustelin T, Stallcup WB. Differential phosphorylation of NG2 proteoglycan by ERK and PKCalpha helps balance cell proliferation and migration. J Cell Biol. 2007;178:155–165. doi: 10.1083/jcb.200612084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Redwine JM, Armstrong RC. In vivo proliferation of oligodendrocyte progenitors expressing PDGFαR during early remyelination. J Neurobiol. 1998;37:413–428. doi: 10.1002/(SICI)1097-4695(19981115)37:3<413::AID-NEU7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 55.Zhong J. RAS and downstream RAF-MEK and PI3K-AKT signaling in neuronal development, function and dysfunction. Biol Chem. 2016;397:215–222. doi: 10.1515/hsz-2015-0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frederick TJ, Wood TL. IGF-I and FGF-2 coordinately enhance cyclin D1 and cyclin E–cdk2 association and activity to promote G1 progression in oligodendrocyte progenitor cells. Mol Cell Neurosci. 2004;25:480–492. doi: 10.1016/j.mcn.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 57.Min J, Singh S, Fitzgerald-Bocarsly P, Wood TL. Insulin-like growth factor I regulates G2/M progression through mammalian target of rapamycin signaling in oligodendrocyte progenitors. Glia. 2012;60:1684–1695. doi: 10.1002/glia.22387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pang Y, Zheng B, Fan L-W, et al. IGF-1 protects oligodendrocyte progenitors against TNFalpha-induced damage by activation of PI3K/Akt and interruption of the mitochondrial apoptotic pathway. Glia. 2007;55:1099–1107. doi: 10.1002/glia.20530. [DOI] [PubMed] [Google Scholar]
- 59.Chan CB, Liu X, Zhao L, et al. PIKE is essential for oligodendroglia development and CNS myelination. Proc Natl Acad Sci USA. 2014;111:1993–1998. doi: 10.1073/pnas.1318185111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wegner M. A matter of identity: transcriptional control in oligodendrocytes. J Mol Neurosci. 2008;35:3–12. doi: 10.1007/s12031-007-9008-8. [DOI] [PubMed] [Google Scholar]
- 61.Emery B. Regulation of oligodendrocyte differentiation and myelination. Science. 2010;330:779–782. doi: 10.1126/science.1190927. [DOI] [PubMed] [Google Scholar]
- 62.Azim K, Butt AM. GSK3β negatively regulates oligodendrocyte differentiation and myelination in vivo. Glia. 2011;59:540–553. doi: 10.1002/glia.21122. [DOI] [PubMed] [Google Scholar]
- 63.Zhou L, Shao C-Y, Xu S, et al. GSK3β promotes the differentiation of oligodendrocyte precursor cells via β-catenin-mediated transcriptional regulation. Mol Neurobiol. 2014;50:507–519. doi: 10.1007/s12035-014-8678-9. [DOI] [PubMed] [Google Scholar]
- 64.Fu H, Kesari S, Cai J. Tcf7l2 is tightly controlled during myelin formation. Cell Mol Neurobiol. 2012;32:345–352. doi: 10.1007/s10571-011-9778-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.González-Fernández E, Jeong H-K, Fukaya M, et al. PTEN negatively regulates the cell lineage progression from NG2+ glial progenitor to oligodendrocyte via mTOR-independent signaling. Elife. 2018 doi: 10.7554/eLife.32021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gaesser JM, Fyffe-Maricich SL. Intracellular signaling pathway regulation of myelination and remyelination in the CNS. Exp Neurol. 2016;283:501–511. doi: 10.1016/j.expneurol.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ishii A, Furusho M, Macklin W, Bansal R. Independent and cooperative roles of the Mek/ERK1/2-MAPK and PI3K/Akt/mTOR pathways during developmental myelination and in adulthood. Glia. 2019;67:1277–1295. doi: 10.1002/glia.23602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Snaidero N, Möbius W, Czopka T, et al. Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell. 2014;156:277–290. doi: 10.1016/j.cell.2013.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Figlia G, Gerber D, Suter U. Myelination and mTOR. Glia. 2018;66:693–707. doi: 10.1002/glia.23273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guardiola-Diaz HM, Ishii A, Bansal R. Erk1/2 MAPK and mTOR signaling sequentially regulates progression through distinct stages of oligodendrocyte differentiation. Glia. 2012;60:476–486. doi: 10.1002/glia.22281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luo F, Burke K, Kantor C, et al. Cyclin-dependent kinase 5 mediates adult OPC maturation and myelin repair through modulation of Akt and GsK-3β signaling. J Neurosci. 2014;34:10415–10429. doi: 10.1523/JNEUROSCI.0710-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Michel K, Zhao T, Karl M, et al. Translational control of myelin basic protein expression by ERK2 MAP kinase regulates timely remyelination in the adult brain. J Neurosci. 2015;35:7850–7865. doi: 10.1523/JNEUROSCI.4380-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fyffe-Maricich SL, Schott A, Karl M, et al. Signaling through ERK1/2 controls myelin thickness during myelin repair in the adult central nervous system. J Neurosci. 2013;33:18402–18408. doi: 10.1523/JNEUROSCI.2381-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bolis A, Coviello S, Visigalli I, et al. Dlg1, Sec8, and Mtmr2 regulate membrane homeostasis in Schwann cell myelination. J Neurosci. 2009;29:8858–8870. doi: 10.1523/JNEUROSCI.1423-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ommer A, Figlia G, Pereira JA, et al. Ral GTPases in Schwann cells promote radial axonal sorting in the peripheral nervous system. J Cell Biol. 2019;218:2350–2369. doi: 10.1083/jcb.201811150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Feltri ML, Suter U, Relvas JB. The function of RhoGTPases in axon ensheathment and myelination. Glia. 2008;56:1508–1517. doi: 10.1002/glia.20752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thurnherr T, Benninger Y, Wu X, et al. Cdc42 and Rac1 signaling are both required for and act synergistically in the correct formation of myelin sheaths in the CNS. J Neurosci. 2006;26:10110–10119. doi: 10.1523/JNEUROSCI.2158-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nawaz S, Sánchez P, Schmitt S, et al. Actin filament turnover drives leading edge growth during myelin sheath formation in the central nervous system. Dev Cell. 2015;34:139–151. doi: 10.1016/j.devcel.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zuchero JB, Fu M, Sloan SA, et al. CNS myelin wrapping is driven by actin disassembly. Dev Cell. 2015;34:152–167. doi: 10.1016/j.devcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wolswijk G. Oligodendrocyte precursor cells in the demyelinated multiple sclerosis spinal cord. Brain. 2002;125:338–349. doi: 10.1093/brain/awf031. [DOI] [PubMed] [Google Scholar]
- 81.Patani R, Balaratnam M, Vora A, Reynolds R. Remyelination can be extensive in multiple sclerosis despite a long disease course. Neuropathol Appl Neurobiol. 2007;33:277–287. doi: 10.1111/j.1365-2990.2007.00805.x. [DOI] [PubMed] [Google Scholar]
- 82.Miron VE, Boyd A, Zhao J-W, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16:1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Domingues HS, Portugal CC, Socodato R, Relvas JB. Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Front Cell Dev Biol. 2016 doi: 10.3389/fcell.2016.00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lloyd AF, Miron VE. The pro-remyelination properties of microglia in the central nervous system. Nat Rev Neurol. 2019;15:447–458. doi: 10.1038/s41582-019-0184-2. [DOI] [PubMed] [Google Scholar]
- 85.Rawji KS, Mishra MK, Yong VW. Regenerative capacity of macrophages for remyelination. Front Cell Dev Biol. 2016 doi: 10.3389/fcell.2016.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Clemente D, Ortega MC, Arenzana FJ, de Castro F. FGF-2 and Anosmin-1 are selectively expressed in different types of multiple sclerosis lesions. J Neurosci. 2011;31:14899–14909. doi: 10.1523/JNEUROSCI.1158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McKinnon RD, Waldron S, Kiel ME. PDGF alpha-receptor signal strength controls an RTK rheostat that integrates phosphoinositol 3’-kinase and phospholipase Cgamma pathways during oligodendrocyte maturation. J Neurosci. 2005;25:3499–3508. doi: 10.1523/JNEUROSCI.5049-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tanuma N, Sakuma H, Sasaki A, Matsumoto Y. Chemokine expression by astrocytes plays a role in microglia/macrophage activation and subsequent neurodegeneration in secondary progressive multiple sclerosis. Acta Neuropathol. 2006;112:195–204. doi: 10.1007/s00401-006-0083-7. [DOI] [PubMed] [Google Scholar]
- 89.Kataria H, Alizadeh A, Shahriary GM, et al. Neuregulin-1 promotes remyelination and fosters a pro-regenerative inflammatory response in focal demyelinating lesions of the spinal cord. Glia. 2018;66:538–561. doi: 10.1002/glia.23264. [DOI] [PubMed] [Google Scholar]
- 90.Yamauchi J, Miyamoto Y, Chan JR, Tanoue A. ErbB2 directly activates the exchange factor Dock7 to promote Schwann cell migration. J Cell Biol. 2008;181:351–365. doi: 10.1083/jcb.200709033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yamauchi J, Chan JR, Shooter EM. Neurotrophin 3 activation of TrkC induces Schwann cell migration through the c-Jun N-terminal kinase pathway. Proc Natl Acad Sci USA. 2003;100:14421–14426. doi: 10.1073/pnas.2336152100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yamauchi J, Chan JR, Shooter EM. Neurotrophins regulate Schwann cell migration by activating divergent signaling pathways dependent on Rho GTPases. Proc Natl Acad Sci USA. 2004;101:8774–8779. doi: 10.1073/pnas.0402795101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gentry JJ, Barker PA, Carter BD. The p75 neurotrophin receptor: multiple interactors and numerous functions. Prog Brain Res. 2004;146:25–39. doi: 10.1016/S0079-6123(03)46002-0. [DOI] [PubMed] [Google Scholar]
- 94.Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chan JR, Cosgaya JM, Wu YJ, Shooter EM. Neurotrophins are key mediators of the myelination program in the peripheral nervous system. Proc Natl Acad Sci USA. 2001;98:14661–14668. doi: 10.1073/pnas.251543398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cosgaya JM, Chan JR, Shooter EM. The neurotrophin receptor p75NTR as a positive modulator of myelination. Science. 2002;298:1245–1248. doi: 10.1126/science.1076595. [DOI] [PubMed] [Google Scholar]
- 97.Rohm B, Rahim B, Kleiber B, et al. The semaphorin 3A receptor may directly regulate the activity of small GTPases. FEBS Lett. 2000;486:68–72. doi: 10.1016/S0014-5793(00)02240-7. [DOI] [PubMed] [Google Scholar]
- 98.Yang T, Terman JR. Regulating small G protein signaling to coordinate axon adhesion and repulsion. Small GTPases. 2013;4:34–41. doi: 10.4161/sgtp.22765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Oinuma I, Katoh H, Negishi M. Molecular dissection of the semaphorin 4D receptor Plexin-B1-stimulated R-Ras GTPase-activating protein activity and neurite remodeling in hippocampal neurons. J Neurosci. 2004;24:11473–11480. doi: 10.1523/JNEUROSCI.3257-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Toyofuku T, Yoshida J, Sugimoto T, et al. FARP2 triggers signals for Sema3A-mediated axonal repulsion. Nat Neurosci. 2005;8:1712–1719. doi: 10.1038/nn1596. [DOI] [PubMed] [Google Scholar]
- 101.Iseppon F, Napolitano LMR, Torre V, Cojoc D. Cdc42 and RhoA reveal different spatio-temporal dynamics upon local stimulation with Semaphorin-3A. Front Cell Neurosci. 2015;9:333. doi: 10.3389/fncel.2015.00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shelly M, Cancedda L, Lim BK, et al. Semaphorin3A regulates neuronal polarization by suppressing axon formation and promoting dendrite growth. Neuron. 2011;71:433–446. doi: 10.1016/j.neuron.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dulamea AO. The contribution of oligodendrocytes and oligodendrocyte progenitor cells to central nervous system repair in multiple sclerosis: perspectives for remyelination therapeutic strategies. Neural Regen Res. 2017;12:1939–1944. doi: 10.4103/1673-5374.221146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Syed YA, Abdulla SA, Kotter MRN. Studying the effects of semaphorins on oligodendrocyte lineage cells. Methods Mol Biol. 2017;1493:363–378. doi: 10.1007/978-1-4939-6448-2_26. [DOI] [PubMed] [Google Scholar]
- 105.Binamé F, Pham-Van LD, Spenlé C, et al. Disruption of Sema3A/Plexin-A1 inhibitory signalling in oligodendrocytes as a therapeutic strategy to promote remyelination. EMBO Mol Med. 2019 doi: 10.15252/emmm.201910378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gutiérrez-Franco A, Costa C, Eixarch H, et al. Differential expression of sema3A and sema7A in a murine model of multiple sclerosis: implications for a therapeutic design. Clin Immunol. 2016;163:22–33. doi: 10.1016/j.clim.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 107.Williams A, Piaton G, Aigrot M-S, et al. Semaphorin 3A and 3F: key players in myelin repair in multiple sclerosis? Brain. 2007;130:2554–2565. doi: 10.1093/brain/awm202. [DOI] [PubMed] [Google Scholar]
- 108.Spassky N, de Castro F, Le Bras B, et al. Directional guidance of oligodendroglial migration by class 3 semaphorins and netrin-1. J Neurosci. 2002;22:5992–6004. doi: 10.1523/JNEUROSCI.22-14-05992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sugimoto Y, Taniguchi M, Yagi T, et al. Guidance of glial precursor cell migration by secreted cues in the developing optic nerve. Development. 2001;128:3321–3330. doi: 10.1242/dev.128.17.3321. [DOI] [PubMed] [Google Scholar]
- 110.Ricard D, Rogemond V, Charrier E, et al. Isolation and expression pattern of human Unc-33-like phosphoprotein 6/collapsin response mediator protein 5 (Ulip6/CRMP5): coexistence with Ulip2/CRMP2 in Sema3a- sensitive oligodendrocytes. J Neurosci. 2001;21:7203–7214. doi: 10.1523/JNEUROSCI.21-18-07203.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Syed YA, Hand E, Möbius W, et al. Inhibition of CNS remyelination by the presence of semaphorin 3A. J Neurosci. 2011;31:3719–3728. doi: 10.1523/JNEUROSCI.4930-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Piaton G, Aigrot M-S, Williams A, et al. Class 3 semaphorins influence oligodendrocyte precursor recruitment and remyelination in adult central nervous system. Brain. 2011;134:1156–1167. doi: 10.1093/brain/awr022. [DOI] [PubMed] [Google Scholar]
- 113.Mi S, Lee X, Shao Z, et al. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci. 2004;7:221–228. doi: 10.1038/nn1188. [DOI] [PubMed] [Google Scholar]
- 114.Ahmed Z, Douglas MR, John G, et al. AMIGO3 is an NgR1/p75 co-receptor signalling axon growth inhibition in the acute phase of adult central nervous system injury. PLoS ONE. 2013;8:e61878. doi: 10.1371/journal.pone.0061878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shao Z, Browning JL, Lee X, et al. TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron. 2005;45:353–359. doi: 10.1016/j.neuron.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 116.Park JB, Yiu G, Kaneko S, et al. A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron. 2005;45:345–351. doi: 10.1016/j.neuron.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 117.Zhang Y, Zhang YP, Pepinsky B, et al. Inhibition of LINGO-1 promotes functional recovery after experimental spinal cord demyelination. Exp Neurol. 2015;266:68–73. doi: 10.1016/j.expneurol.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 118.Yamashita T, Tohyama M. The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat Neurosci. 2003;6:461–467. doi: 10.1038/nn1045. [DOI] [PubMed] [Google Scholar]
- 119.Lu Y, Liu X, Zhou J, et al. TROY interacts with rho guanine nucleotide dissociation inhibitor α (RhoGDIα) to mediate Nogo-induced inhibition of neurite outgrowth. J Biol Chem. 2013;288:34276–34286. doi: 10.1074/jbc.M113.519744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jepson S, Vought B, Gross CH, et al. LINGO-1, a transmembrane signaling protein, inhibits oligodendrocyte differentiation and myelination through intercellular self-interactions. J Biol Chem. 2012;287:22184–22195. doi: 10.1074/jbc.M112.366179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cobret L, De Tauzia ML, Ferent J, et al. Targeting the cis-dimerization of LINGO-1 with low MW compounds affects its downstream signalling. Br J Pharmacol. 2015;172:841–856. doi: 10.1111/bph.12945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Inoue H, Lin L, Lee X, et al. Inhibition of the leucine-rich repeat protein LINGO-1 enhances survival, structure, and function of dopaminergic neurons in Parkinson’s disease models. Proc Natl Acad Sci USA. 2007;104:14430–14435. doi: 10.1073/pnas.0700901104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lee X, Shao Z, Sheng G, et al. LINGO-1 regulates oligodendrocyte differentiation by inhibiting ErbB2 translocation and activation in lipid rafts. Mol Cell Neurosci. 2014;60:36–42. doi: 10.1016/j.mcn.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 124.Sun L, Liu S, Sun Q, et al. Inhibition of TROY promotes OPC differentiation and increases therapeutic efficacy of OPC graft for spinal cord injury. Stem Cells Dev. 2014;23:2104–2118. doi: 10.1089/scd.2013.0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hedgecock EM, Culotti JG, Hall DH. The unc-5, unc-6, and unc-40 genes guide circumferential migrations of pioneer axons and mesodermal cells on the epidermis in C. elegans. Neuron. 1990;4:61–85. doi: 10.1016/0896-6273(90)90444-k. [DOI] [PubMed] [Google Scholar]
- 126.Finci LI, Krüger N, Sun X, et al. The crystal structure of netrin-1 in complex with DCC reveals the bifunctionality of netrin-1 as a guidance cue. Neuron. 2014;83:839–849. doi: 10.1016/j.neuron.2014.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bartoe JL, McKenna WL, Quan TK, et al. Protein interacting with C-kinase 1/protein kinase Calpha-mediated endocytosis converts netrin-1-mediated repulsion to attraction. J Neurosci. 2006;26:3192–3205. doi: 10.1523/JNEUROSCI.3469-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Williams ME, Wu SC-Y, McKenna WL, Hinck L. Surface expression of the netrin receptor UNC5H1 is regulated through a protein kinase C-interacting protein/protein kinase-dependent mechanism. J Neurosci. 2003;23:11279–11288. doi: 10.1523/JNEUROSCI.23-36-11279.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Taylor AM, Menon S, Gupton SL. Passive microfluidic chamber for long-term imaging of axon guidance in response to soluble gradients. Lab Chip. 2015;15:2781–2789. doi: 10.1039/c5lc00503e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shekarabi M, Kennedy TE. The netrin-1 receptor DCC promotes filopodia formation and cell spreading by activating Cdc42 and Rac1. Mol Cell Neurosci. 2002;19:1–17. doi: 10.1006/mcne.2001.1075. [DOI] [PubMed] [Google Scholar]
- 131.Picard M, Petrie RJ, Antoine-Bertrand J, et al. Spatial and temporal activation of the small GTPases RhoA and Rac1 by the netrin-1 receptor UNC5a during neurite outgrowth. Cell Signal. 2009;21:1961–1973. doi: 10.1016/j.cellsig.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 132.Boyer NP, Gupton SL. Revisiting Netrin-1: one who guides (Axons) Front Cell Neurosci. 2018;12:221. doi: 10.3389/fncel.2018.00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gujar MR, Stricker AM, Lundquist EA. RHO-1 and the Rho GEF RHGF-1 interact with UNC-6/Netrin signaling to regulate growth cone protrusion and microtubule organization in Caenorhabditis elegans. PLoS Genet. 2019;15:e1007960. doi: 10.1371/journal.pgen.1007960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jarjour AA, Manitt C, Moore SW, et al. Netrin-1 is a chemorepellent for oligodendrocyte precursor cells in the embryonic spinal cord. J Neurosci. 2003;23:3735–3744. doi: 10.1523/JNEUROSCI.23-09-03735.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rajasekharan S, Bin JM, Antel JP, Kennedy TE. A central role for RhoA during oligodendroglial maturation in the switch from Netrin-1-mediated chemorepulsion to process elaboration. J Neurochem. 2010;113:1589–1597. doi: 10.1111/j.1471-4159.2010.06717.x. [DOI] [PubMed] [Google Scholar]
- 136.Rajasekharan S, Baker KA, Horn KE, et al. Netrin 1 and Dcc regulate oligodendrocyte process branching and membrane extension via Fyn and RhoA. Development. 2009;136:415–426. doi: 10.1242/dev.018234. [DOI] [PubMed] [Google Scholar]
- 137.Bin JM, Rajasekharan S, Kuhlmann T, et al. Full-length and fragmented netrin-1 in multiple sclerosis plaques are inhibitors of oligodendrocyte precursor cell migration. Am J Pathol. 2013;183:673–680. doi: 10.1016/j.ajpath.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 138.Kidd T, Brose K, Mitchell KJ, et al. Roundabout controls axon crossing of the CNS midline and defines a novel subfamily of evolutionarily conserved guidance receptors. Cell. 1998;92:205–215. doi: 10.1016/s0092-8674(00)80915-0. [DOI] [PubMed] [Google Scholar]
- 139.Seeger M, Tear G, Ferres-Marco D, Goodman CS. Mutations affecting growth cone guidance in Drosophila: genes necessary for guidance toward or away from the midline. Neuron. 1993;10:409–426. doi: 10.1016/0896-6273(93)90330-t. [DOI] [PubMed] [Google Scholar]
- 140.Barak R, Opatowsky Y. Expression, derivatization, crystallization and experimental phasing of an extracellular segment of the human Robo1 receptor. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2013;69:771–775. doi: 10.1107/S1744309113014863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Seki M, Watanabe A, Enomoto S, et al. Human ROBO1 is cleaved by metalloproteinases and gamma-secretase and migrates to the nucleus in cancer cells. FEBS Lett. 2010;584:2909–2915. doi: 10.1016/j.febslet.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 142.Coleman HA, Labrador J-P, Chance RK, Bashaw GJ. The Adam family metalloprotease Kuzbanian regulates the cleavage of the roundabout receptor to control axon repulsion at the midline. Development. 2010;137:2417–2426. doi: 10.1242/dev.047993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chang P-H, Hwang-Verslues WW, Chang Y-C, et al. Activation of Robo1 signaling of breast cancer cells by Slit2 from stromal fibroblast restrains tumorigenesis via blocking PI3K/Akt/β-catenin pathway. Cancer Res. 2012;72:4652–4661. doi: 10.1158/0008-5472.CAN-12-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Fritz RD, Menshykau D, Martin K, et al. SrGAP2-dependent integration of membrane geometry and slit-robo-repulsive cues regulates fibroblast contact inhibition of locomotion. Dev Cell. 2015;35:78–92. doi: 10.1016/j.devcel.2015.09.002. [DOI] [PubMed] [Google Scholar]
- 145.Blockus H, Chédotal A. Slit-Robo signaling. Development. 2016;143:3037–3044. doi: 10.1242/dev.132829. [DOI] [PubMed] [Google Scholar]
- 146.Delloye-Bourgeois C, Jacquier A, Charoy C, et al. PlexinA1 is a new Slit receptor and mediates axon guidance function of Slit C-terminal fragments. Nat Neurosci. 2015;18:36–45. doi: 10.1038/nn.3893. [DOI] [PubMed] [Google Scholar]
- 147.Stein E, Tessier-Lavigne M. Hierarchical organization of guidance receptors: silencing of netrin attraction by slit through a Robo/DCC receptor complex. Science. 2001;291:1928–1938. doi: 10.1126/science.1058445. [DOI] [PubMed] [Google Scholar]
- 148.Zelina P, Blockus H, Zagar Y, et al. Signaling switch of the axon guidance receptor Robo3 during vertebrate evolution. Neuron. 2014;84:1258–1272. doi: 10.1016/j.neuron.2014.11.004. [DOI] [PubMed] [Google Scholar]
- 149.Liu X, Lu Y, Zhang Y, et al. Slit2 regulates the dispersal of oligodendrocyte precursor cells via Fyn/RhoA signaling. J Biol Chem. 2012;287:17503–17516. doi: 10.1074/jbc.M111.317610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Niethamer TK, Bush JO. Getting direction(s): the Eph/ephrin signaling system in cell positioning. Dev Biol. 2019;447:42–57. doi: 10.1016/j.ydbio.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sahin M, Greer PL, Lin MZ, et al. Eph-dependent tyrosine phosphorylation of ephexin1 modulates growth cone collapse. Neuron. 2005;46:191–204. doi: 10.1016/j.neuron.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 152.Ogita H, Kunimoto S, Kamioka Y, et al. EphA4-mediated Rho activation via Vsm-RhoGEF expressed specifically in vascular smooth muscle cells. Circ Res. 2003;93:23–31. doi: 10.1161/01.RES.0000079310.81429.C8. [DOI] [PubMed] [Google Scholar]
- 153.Sobel RA. Ephrin A receptors and ligands in lesions and normal-appearing white matter in multiple sclerosis. Brain Pathol. 2005;15:35–45. doi: 10.1111/j.1750-3639.2005.tb00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Prestoz L, Chatzopoulou E, Lemkine G, et al. Control of axonophilic migration of oligodendrocyte precursor cells by Eph-ephrin interaction. Neuron Glia Biol. 2004;1:73–83. doi: 10.1017/S1740925X04000109. [DOI] [PubMed] [Google Scholar]
- 155.Harboe M, Torvund-Jensen J, Kjaer-Sorensen K, Laursen LS. Ephrin-A1-EphA4 signaling negatively regulates myelination in the central nervous system. Glia. 2018;66:934–950. doi: 10.1002/glia.23293. [DOI] [PubMed] [Google Scholar]
- 156.Syed YA, Zhao C, Mahad D, et al. Antibody-mediated neutralization of myelin-associated EphrinB3 accelerates CNS remyelination. Acta Neuropathol. 2016;131:281–298. doi: 10.1007/s00401-015-1521-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Najm FJ, Madhavan M, Zaremba A, et al. Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature. 2015;522:216–220. doi: 10.1038/nature14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tourbah A, Lebrun-Frenay C, Edan G, et al. MD1003 (high-dose biotin) for the treatment of progressive multiple sclerosis: a randomised, double-blind, placebo-controlled study. Mult Scler. 2016;22:1719–1731. doi: 10.1177/1352458516667568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chandraratna RA, Noelle RJ, Nowak EC. Treatment with retinoid X receptor agonist IRX4204 ameliorates experimental autoimmune encephalomyelitis. Am J Transl Res. 2016;8:1016–1026. [PMC free article] [PubMed] [Google Scholar]
- 160.Green AJ, Gelfand JM, Cree BA, et al. Clemastine fumarate as a remyelinating therapy for multiple sclerosis (ReBUILD): a randomised, controlled, double-blind, crossover trial. Lancet. 2017;390:2481–2489. doi: 10.1016/S0140-6736(17)32346-2. [DOI] [PubMed] [Google Scholar]
- 161.Hartley MD, Banerji T, Tagge IJ, et al. Myelin repair stimulated by CNS-selective thyroid hormone action. JCI Insight. 2019 doi: 10.1172/jci.insight.126329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sandvig A, Berry M, Barrett LB, et al. Myelin-, reactive glia-, and scar-derived CNS axon growth inhibitors: expression, receptor signaling, and correlation with axon regeneration. Glia. 2004;46:225–251. doi: 10.1002/glia.10315. [DOI] [PubMed] [Google Scholar]
- 163.Curcio M, Bradke F. Axon regeneration in the central nervous system: facing the challenges from the inside. Annu Rev Cell Dev Biol. 2018;34:495–521. doi: 10.1146/annurev-cellbio-100617-062508. [DOI] [PubMed] [Google Scholar]
- 164.Hug A, Weidner N. From bench to beside to cure spinal cord injury: lost in translation? Int Rev Neurobiol. 2012;106:173–196. doi: 10.1016/B978-0-12-407178-0.00008-9. [DOI] [PubMed] [Google Scholar]
- 165.Ineichen BV, Kapitza S, Bleul C, et al. Nogo-A antibodies enhance axonal repair and remyelination in neuro-inflammatory and demyelinating pathology. Acta Neuropathol. 2017;134:423–440. doi: 10.1007/s00401-017-1745-3. [DOI] [PubMed] [Google Scholar]
- 166.Rudick RA, Mi S, Sandrock AW. LINGO-1 antagonists as therapy for multiple sclerosis: in vitro and in vivo evidence. Expert Opin Biol Ther. 2008;8:1561–1570. doi: 10.1517/14712598.8.10.1561. [DOI] [PubMed] [Google Scholar]
- 167.Fisher TL, Reilly CA, Winter LA, et al. Generation and preclinical characterization of an antibody specific for SEMA4D. MAbs. 2016;8:150–162. doi: 10.1080/19420862.2015.1102813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jacob L, Sawma P, Garnier N, et al. Inhibition of PlexA1-mediated brain tumor growth and tumor-associated angiogenesis using a transmembrane domain targeting peptide. Oncotarget. 2016;7:57851–57865. doi: 10.18632/oncotarget.11072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Arpel A, Gamper C, Spenlé C, et al. Inhibition of primary breast tumor growth and metastasis using a neuropilin-1 transmembrane domain interfering peptide. Oncotarget. 2016;7:54723–54732. doi: 10.18632/oncotarget.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nasarre C, Roth M, Jacob L, et al. Peptide-based interference of the transmembrane domain of neuropilin-1 inhibits glioma growth in vivo. Oncogene. 2010;29:2381–2392. doi: 10.1038/onc.2010.9. [DOI] [PubMed] [Google Scholar]
- 171.Roth L, Nasarre C, Dirrig-Grosch S, et al. Transmembrane domain interactions control biological functions of neuropilin-1. Mol Biol Cell. 2008;19:646–654. doi: 10.1091/mbc.E07-06-0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Arpel A, Sawma P, Spenlé C, et al. Transmembrane domain targeting peptide antagonizing ErbB2/Neu inhibits breast tumor growth and metastasis. Cell Rep. 2014;8:1714–1721. doi: 10.1016/j.celrep.2014.07.044. [DOI] [PubMed] [Google Scholar]
- 173.Ulc A, Zeug A, Bauch J, et al. The guanine nucleotide exchange factor Vav3 modulates oligodendrocyte precursor differentiation and supports remyelination in white matter lesions. Glia. 2019;67:376–392. doi: 10.1002/glia.23548. [DOI] [PubMed] [Google Scholar]
- 174.Wen J, Tan D, Li L, et al. RhoA regulates Schwann cell differentiation through JNK pathway. Exp Neurol. 2018;308:26–34. doi: 10.1016/j.expneurol.2018.06.013. [DOI] [PubMed] [Google Scholar]
- 175.Bidaud-Meynard A, Binamé F, Lagrée V, Moreau V. Regulation of Rho GTPase activity at the leading edge of migrating cells by p190RhoGAP. Small GTPases. 2017 doi: 10.1080/21541248.2017.1280584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Nimnual AS, Taylor LJ, Bar-Sagi D. Redox-dependent downregulation of Rho by Rac. Nat Cell Biol. 2003;5:236–241. doi: 10.1038/ncb938. [DOI] [PubMed] [Google Scholar]
- 177.Lévay M, Settleman J, Ligeti E. Regulation of the substrate preference of p190RhoGAP by PKC-mediated phosphorylation of a phospholipid binding site. Biochemistry. 2009;48:8615–8623. doi: 10.1021/bi900667y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Jiang W, Betson M, Mulloy R, et al. p190A RhoGAP is a glycogen synthase kinase-3-beta substrate required for polarized cell migration. J Biol Chem. 2008;283:20978–20988. doi: 10.1074/jbc.M802588200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Oinuma I, Kawada K, Tsukagoshi K, Negishi M. Rnd1 and Rnd3 targeting to lipid raft is required for p190 RhoGAP activation. Mol Biol Cell. 2012;23:1593–1604. doi: 10.1091/mbc.E11-11-0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wildenberg GA, Dohn MR, Carnahan RH, et al. p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell. 2006;127:1027–1039. doi: 10.1016/j.cell.2006.09.046. [DOI] [PubMed] [Google Scholar]
- 181.Binamé F, Bidaud-Meynard A, Magnan L, et al. Cancer-associated mutations in the protrusion-targeting region of p190RhoGAP impact tumor cell migration. J Cell Biol. 2016;214:859–873. doi: 10.1083/jcb.201601063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhang H, Macara IG. The PAR-6 polarity protein regulates dendritic spine morphogenesis through p190 RhoGAP and the Rho GTPase. Dev Cell. 2008;14:216–226. doi: 10.1016/j.devcel.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nishimura T, Yamaguchi T, Kato K, et al. PAR-6-PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat Cell Biol. 2005;7:270–277. doi: 10.1038/ncb1227. [DOI] [PubMed] [Google Scholar]
- 184.Pegtel DM, Ellenbroek SIJ, Mertens AEE, et al. The Par-Tiam1 complex controls persistent migration by stabilizing microtubule-dependent front-rear polarity. Curr Biol. 2007;17:1623–1634. doi: 10.1016/j.cub.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 185.Nakayama M, Goto TM, Sugimoto M, et al. Rho-kinase phosphorylates PAR-3 and disrupts PAR complex formation. Dev Cell. 2008;14:205–215. doi: 10.1016/j.devcel.2007.11.021. [DOI] [PubMed] [Google Scholar]
- 186.Kuo J-C, Han X, Hsiao C-T, et al. Analysis of the myosin-II-responsive focal adhesion proteome reveals a role for β-Pix in negative regulation of focal adhesion maturation. Nat Cell Biol. 2011;13:383–393. doi: 10.1038/ncb2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Sanz-Moreno V, Gadea G, Ahn J, et al. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135:510–523. doi: 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 188.Ohta Y, Hartwig JH, Stossel TP. FilGAP, a Rho- and ROCK-regulated GAP for Rac binds filamin A to control actin remodelling. Nat Cell Biol. 2006;8:803–814. doi: 10.1038/ncb1437. [DOI] [PubMed] [Google Scholar]
- 189.Tsuji T, Ishizaki T, Okamoto M, et al. ROCK and mDia1 antagonize in Rho-dependent Rac activation in Swiss 3T3 fibroblasts. J Cell Biol. 2002;157:819–830. doi: 10.1083/jcb.200112107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kurokawa K, Matsuda M. Localized RhoA activation as a requirement for the induction of membrane ruffling. Mol Biol Cell. 2005;16:4294–4303. doi: 10.1091/mbc.e04-12-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bosch J. PPI inhibitor and stabilizer development in human diseases. Drug Discov Today Technol. 2017;24:3–9. doi: 10.1016/j.ddtec.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 192.Hubert P, Sawma P, Duneau J-P, et al. Single-spanning transmembrane domains in cell growth and cell-cell interactions: more than meets the eye? Cell Adh Migr. 2010;4:313–324. doi: 10.4161/cam.4.2.12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Senes A, Gerstein M, Engelman DM. Statistical analysis of amino acid patterns in transmembrane helices: the GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J Mol Biol. 2000;296:921–936. doi: 10.1006/jmbi.1999.3488. [DOI] [PubMed] [Google Scholar]
- 194.Sharpe HJ, Stevens TJ, Munro S. A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell. 2010;142:158–169. doi: 10.1016/j.cell.2010.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Cai X, Bai B, Zhang R, et al. Apelin receptor homodimer-oligomers revealed by single-molecule imaging and novel G protein-dependent signaling. Sci Rep. 2017;7:40335. doi: 10.1038/srep40335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Evans AE, Tripathi A, LaPorte HM, et al. New insights into mechanisms and functions of chemokine (C-X-C Motif) receptor 4 heteromerization in vascular smooth muscle. Int J Mol Sci. 2016 doi: 10.3390/ijms17060971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Harikumar KG, Wootten D, Pinon DI, et al. Glucagon-like peptide-1 receptor dimerization differentially regulates agonist signaling but does not affect small molecule allostery. Proc Natl Acad Sci USA. 2012;109:18607–18612. doi: 10.1073/pnas.1205227109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Tarasova NI, Rice WG, Michejda CJ. Inhibition of G-protein-coupled receptor function by disruption of transmembrane domain interactions. J Biol Chem. 1999;274:34911–34915. doi: 10.1074/jbc.274.49.34911. [DOI] [PubMed] [Google Scholar]