

Abstract
T-cell receptor (TCR) recognition of antigens is fundamental to the adaptive immune response. With the expansion of experimental techniques, a substantial database of matched TCR–antigen pairs has emerged, presenting opportunities for computational prediction models. However, accurately forecasting the binding affinities of unseen antigen–TCR pairs remains a major challenge. Here, we present convolutional-self-attention TCR (CATCR), a novel framework tailored to enhance the prediction of epitope and TCR interactions. Our approach utilizes convolutional neural networks to extract peptide features from residue contact matrices, as generated by OpenFold, and a transformer to encode segment-based coded sequences. We introduce CATCR-D, a discriminator that can assess binding by analyzing the structural and sequence features of epitopes and CDR3-β regions. Additionally, the framework comprises CATCR-G, a generative module designed for CDR3-β sequences, which applies the pretrained encoder to deduce epitope characteristics and a transformer decoder for predicting matching CDR3-β sequences. CATCR-D achieved an AUROC of 0.89 on previously unseen epitope–TCR pairs and outperformed four benchmark models by a margin of 17.4%. CATCR-G has demonstrated high precision, recall and F1 scores, surpassing 95% in bidirectional encoder representations from transformers score assessments. Our results indicate that CATCR is an effective tool for predicting unseen epitope–TCR interactions. Incorporating structural insights enhances our understanding of the general rules governing TCR–epitope recognition significantly. The ability to predict TCRs for novel epitopes using structural and sequence information is promising, and broadening the repository of experimental TCR–epitope data could further improve the precision of epitope–TCR binding predictions.
Keywords: TCR prediction, CATCR framework, structural features, convolutional neural networks, transformer
INTRODUCTION
T cells typically recognize antigen peptides presented by MHC molecules through T-cell receptors (TCRs). This process is critical for immune responses against exogenous pathogens and cancers [1]. During recognition, the complementarity-determining regions (CDRs) of the TCR interact with and bind to specific antigen epitopes. Among the different regions of the CDR, CDR3 plays a pivotal role in TCR diversity. CDR3 regions exhibit remarkably high diversity through the V(D)J recombination mechanism, allowing them to adapt to a wide range of existing and potential antigens [2]. The potential number of TCR variants that can be generated is estimated to reach 1018 or more [3]. The importance of TCR diversity in disease monitoring, autoimmune diseases and anticancer immunity has driven research on the rules governing CDR3 epitope binding. Experimental methods, including antigen-directed approaches and TCR-directed approaches [4], have been used to detect the binding between CDR3s and epitopes. However, due to technical barriers and cost limitations, the currently available experimental evidence for specific CDR3–epitope pairs represents only a small fraction of the overall repertoire [1].
Therefore, some studies have focused on computational methods, particularly machine learning approaches [5–7]. However, computational methods still present significant challenges. The ideal model should possess strong generalization capabilities, meaning that it should both effectively predict epitopes detected during training and learn general patterns for application to unseen CDR3–epitope pairs. In certain diseases, the prediction of epitope-specific binding has driven drug and vaccine development [8–12]. However, the performance of existing models diminishes with unfamiliar epitopes [13]. Therefore, developing models that can comprehend the general principles of CDR3 epitope binding is necessary to enhance unseen epitope prediction.
The CDR3 region interacts with the amino acid residues on the peptide through various non-covalent interactions, such as hydrogen bonding, ionic interactions and van der Waals forces, to establish stable binding. The three-dimensional (3D) structure, determined by these amino acid sequences, underlies the specific binding of CDR3 to different epitopes [14, 15]. Thus, individual sequence data alone cannot capture all the information governing CDR–epitope binding. The incorporation of 3D structural information may be crucial for improving prediction accuracy. Innovative protein structure prediction methods, such as AlphaFold2 [16], provide non-experimental approaches for predicting protein structures and have demonstrated promising performance in short peptides [17]. Some studies have utilized methods such as AlphaFold2 for multimeric predictions to predict interactions by constructing 3D structural models; however, these approaches have not consistently shown clear advantages over sequence-based predictions [18]. Consequently, further investigation is required to determine how structural information can effectively predict TCR and epitope binding.
Therefore, in this study, we utilized OpenFold [19] (the PyTorch version of AlphaFold2) to predict the 3D structures of peptides. We used a convolutional neural network (CNN) to extract structural features from these predictions via residue contact matrices (RCMs). We established a segment-based encoding scheme for the sequence information and employed a transformer encoder for feature extraction. We trained a discriminator (convolutional self-attention TCR discriminator, CATCR-D) to predict the binding of epitopes to the CDR3-β region. Building on this, we developed a generative model (convolutional self-attention TCR generator, CATCR-G) that fuses features from the epitope encoder with those of predicted CDR3-β structures and inputs them into a transformer decoder to generate the corresponding sequence. The CDR3-β structural features were derived using an RCM–based transformer (RCMT) informed by epitope features. Additionally, we leveraged the pretrained discriminator to refine the generative model training. We aimed to establish a robust method for predicting the TCR binding to unseen antigen epitopes.
RESULTS
The 3D structure of CDR3β and its epitope sequences were predicted using OpenFold
The model architecture utilized in this research is illustrated in Figure 1. The sequences and pairing information for CDR3-β and the epitopes were obtained from the VDJdb [20], the Immune Epitope Database (IEDB) [21] and the McPAS-TCR [22] databases. The three databases contain 128 259 unique CDR3-β-epitope pairs, including 127 507 CDR3-β sequences and 1176 epitope sequences. Following data cleaning (Supplementary Figure 1), 65 069 records were included in the training and testing sets (Supplementary Table S1).
Figure 1.
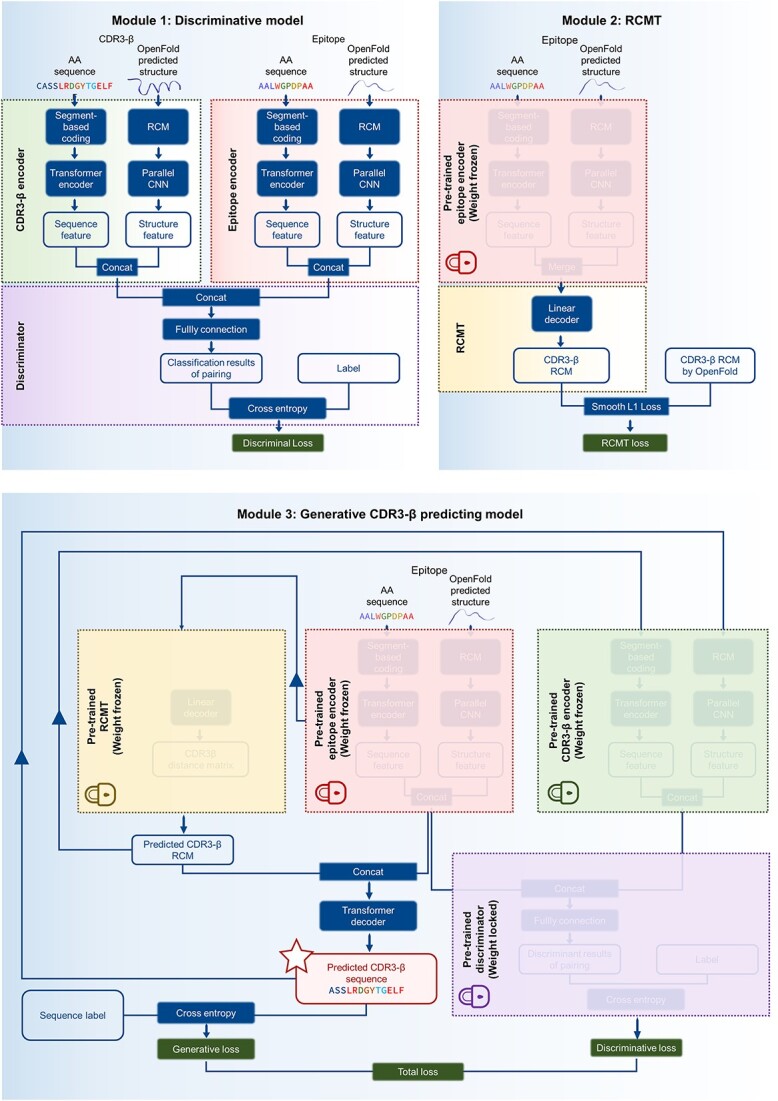
Model architecture. The model comprises three modules. The first module, the discriminator module (CATCR-D), employs a CNN to extract structural features as predicted by OpenFold and a transformer encoder for sequence feature extraction. Structures are represented using RCMs, and sequences are encoded using a segment-based coding scheme before embedding. The features extracted from the epitope and CDR3-β are concatenated and fed into a classifier to determine the binding outcome. The second module, the RCMT, leverages the pretrained epitope encoder from the first module to extract epitope features and utilizes a linear decoder to predict the RCM for CDR3-β. The third module, the generator module, integrates the predicted CDR3-β RCM with epitope features from the epitope encoder and employs the transformer decoder to generate the CDR3-β sequence. This predicted sequence and the predicted CDR3-β RCM are reintroduced into the discriminator to refine the generative model via feedback from the discriminator loss. AA: amino acid; RCM: residue contact matrices; RCMT: residue contact matrices transformer.
We employed OpenFold to predict the 3D structure of the peptide chains to incorporate structural information into the training data. The 3D structure of the peptide chains was characterized using RCMs. We evaluated the accuracy of the structures predicted by OpenFold by comparing them with the experimentally determined protein structures available in the RCSB Protein Data Bank (RCSB PDB) [23] (Figure 2A). The results highlight OpenFold’s robust predictive capability for epitopes, exhibiting a root mean square deviation (RMSD) of 0.4 ± 0.24 Å between the experimentally determined and predicted structures (Figure 2B). However, OpenFold exhibited a slight increase in the RMSD when predicting the CDR3-β structure because of its inability to predict the folding of the middle segment of CDR3-β (Figure 2C). This folding involves interactions of other amino acids in the TCR α and β segments. Therefore, relying solely on sequence information from the CDR3-β region is insufficient for prediction. Nevertheless, OpenFold can accurately predict the structures of individual CDR3-β subsegments, including the N-terminus, C-terminus and complex structure in the middle segment (Figure 2C). Furthermore, OpenFold’s predictions can reflect structural distinctions for similar sequences that differ by only one amino acid (Figure 2D).
Figure 2.
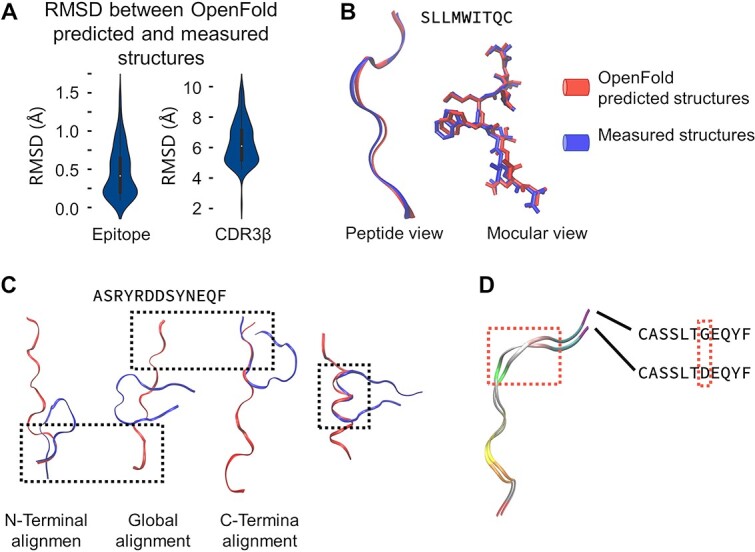
OpenFold predictions of CDR3-β and epitope structures. (A) The RMSD between OpenFold’s predicted structures for epitopes and CDR3-β and the corresponding structures obtained from the RCSB PDB. (B) OpenFold prediction results for epitopes, with the left subfigure depicting the predicted amino acid chain and the right subfigure illustrating the predicted molecular conformation. (C) OpenFold predictions for the CDR3-β structures. OpenFold faces challenges in accurately predicting the conformation of the CDR3-β central segment due to the absence of certain contextual clues that guide protein folding; however, OpenFold has good predictive accuracy for various subsegments. (D) OpenFold’s predictions for sequences with high similarity (differing by a single amino acid) show its ability to discern subtle structural differences. RMSD: root mean square deviation.
Discriminative model for predicting the binding of CDR3 to an epitope
We developed a discriminative model to predict the binding between CDR3-β sequences and epitopes. The discriminative model comprises three principal components: a CDR3-β encoder and an epitope encoder, both dedicated to feature extraction, and a multilayer linear discriminator responsible for generating binding predictions (Module 1 in Figure 1). Within the encoder modules, we extracted both sequence and structural features from the peptide chains, subsequently concatenating these features for comprehensive representation. For the sequence features, certain contiguous short amino acid segments (two to five residues) frequently occur within the CDR3-β sequence and epitope, forming specific secondary structures and providing crucial recognition information for binding. Therefore, highlighting the differences between these similar sequences is meaningful. In addition to traditional single amino acid-based coding methods such as BLOSUM62, we innovated a segment-based coding strategy in which frequently occurring amino acid segments (two to five residues) are treated as one character for coding (Figure 3A). In contrast, infrequent amino acids are encoded separately. Segments appearing more than 1000 times in the dataset were considered frequent segments (Supplementary Table S2).
Figure 3.

CATCR-D Prediction of Epitope-CDR3-β Binding. (A) The segment-based coding method, where high-frequency amino acid segments are coded as single characters. (B) Training and validation set losses. (C) Confusion matrix visualizing classification results for the internal and external test sets. (D) Comparison of the AUROC of CATCR-D on the external test set, which includes unseen epitopes and CDR3 sequences, against those of the benchmark methods (TEINet, EPIC-TRACE, epiTCR and TITAN). (E) External test set AUROC comparison between CATCR-D and methods using only a CNN for RCM feature extraction and only a transformer for sequence feature extraction. (F) Performance of segment-based coding versus BLOSUM62 in terms of AUROC on the external test set. (G) AUROC of CATCR-D on the external test set across epitopes of varying lengths and frequencies of occurrence. AUROC: area under the receiver operating characteristic curve.
Subsequently, we employed a transformer encoder to systematically extract features from the encoded sequence data, optimizing the representation of sequence characteristics. We employed RCMs, which represent the distances between amino acids, to extract structural features. We utilized two CNNs with distinct kernel sizes to derive features from the RCMs. These extracted features were then integrated with the sequence features obtained from the transformer encoder, forming a comprehensive input for the discriminator. The discriminator consisted of four progressively shrinking fully connected layers that output the discrimination result after being activated by the sigmoid function, and then, the cross-entropy loss is calculated. We constructed the negative sample set by employing a random selection approach [24], where we chose CDR3-β sequences from the dataset that were confirmed not to bind with the given epitope. The ratio of negative to positive samples was 1:1. During the training process, the cross-entropy loss of both the training and validation sets showed a significant initial decrease. The training set loss stabilized after 160 epochs. In contrast, the validation set loss stabilized after 320 epochs (Figure 3B).
The current challenge is to predict whether an unseen epitope can bind to a given TCR. To rigorously evaluate our model, we established two distinct test sets: the internal test set, comprising epitopes from the training dataset paired with novel CDR3-β sequences, and the external test set, featuring entirely unseen combinations of epitopes and CDR3-β sequences (Supplementary Figure 3). The internal test set achieved a precision of 92.8%, a recall of 98.9% and an F1 score of 0.958. The external test set achieved an accuracy of 84.8%, a recall of 82.8% and an F1 score of 0.837 (Figure 3C).
The area under the receiver operating characteristic curve (AUROC) of the internal and external test sets were 0.965 ± 0.003 and 0.891 ± 0.006, respectively. We selected four classic or newly reported models as controls: TITAN (2021) [25], epiTCR (2023) [26], TEINet (2023) [27] and EPIC-TRACE (2023) [13]. TITAN utilizes convolution and contextual attention to embed epitopes from SMILES and complete TCR from BLOSUM62 [28] embedding. epiTCR employs random forest to predict CDR3 from BLOSUM62 embedding. EPIC-TRACE utilizes ProtBERT embedding to represent the amino acid sequences of both chains and epitopes while employing a combination of convolution and multihead attention structures. TEINet utilizes transfer learning to address the prediction problem. In a comparative analysis, our CATCR-D model demonstrated superior AUROC metrics in internal and external test sets, outperforming the referenced models (Supplementary Table S3). Particularly in the external test set, CATCR-D demonstrated a notable enhancement in generalization performance for unseen antigen epitopes (Figure 3D and Supplementary Table S3).
We further investigated the key factors contributing to the improved performance of CATCR-D. We found that when using only a transformer model, the predicted AUROC for unseen epitope-CDR3-β pairing was only 0.548 ± 0.013, similar to earlier related studies [25, 26]. However, when only the CNN was used to extract features from RCMs, the predicted AUROC improved to 0.756 ± 0.008 (Figure 3E). This result suggests that both sequence and 3D structures provide crucial information for predicting epitope-CDR3-β binding. In terms of the impact of sequence coding methods on prediction performance, we found that segment-based coding significantly improved predictive performance compared to BLOSUM62 (0.779 ± 0.008) (Figure 3F). In the training and testing data we used, the length of the epitope peptides ranged from 7 to 24, and the frequency of epitope occurrence (corresponding to the number of TCRs in the database) ranged from a minimum of 1 to 500. We analyzed the model’s predictive performance for unseen epitope-CDR3-β pairing across epitopes of different lengths and frequencies of occurrence. The AUROC ranged between 0.673 and 0.949. The AUROC demonstrated an increasing trend as the length of the epitope peptide chain increased. Furthermore, epitopes with higher occurrence frequencies exhibited greater predictive accuracy for pairings (Figure 3G).
Training RCM transformer
The results of CATCR-D suggest that our encoder can capture generalized features of epitopes or CDR3-β. We formulated a generative model employing a decoder that utilizes features extracted by the CATCR-D encoder to facilitate the prediction of CDR3 sequences binding to novel epitopes. Previous results suggest that the structural data represented by the RCM contain crucial information about epitope–TCR binding. Given our objective of integrating structural data within the decoder framework of our generative model, a significant challenge arises because the structural data for the target sequence remain undetermined. Therefore, we pretrained the RCMT to estimate the RCM of CDR3-β based on the epitope sequence and its RCM. This approach involves utilizing the feature set outputted by the CATCR-D encoder as input to a linear decoder, which then predicts the RCM for CDR3-β sequences.
The loss of the training and validation sets both decreased with increased epochs during the training process. The training set loss stabilized after 200 epochs. In contrast, the validation set loss stabilized after 300 epochs (Figure 4A). By comparing the RCMT predictions with those labeled by OpenFold, we noted an average discrepancy in the RCMs of 1.695 ± 2.040 Å. Figure 4B shows the average differences between the two prediction methods at each position. The predicted differences in distance at different positions range from 0.010 to 6.195 Å. The distance deviations at positions 12 and 13 are relatively large, while those at other positions are relatively small. Multiple CDR3-β label sequences may exist in the dataset for a given epitope, while the RCMT can output only a single predicted matrix. Subsequently, we analyzed the relationship between the distance distribution of the label matrix at each position and the predicted values of the RCMT. Figure 4C shows the results for three epitopes with many paired CDR3-β label data (external test). The distances predicted by the RCMT are close to the median value of the label distance set at most positions, indicating that the RCMT can reflect the landscapes of the corresponding CDR3-β structure through the epitope sequence and structure.
Figure 4.

RCMT Module. (A) Loss curves for the training and validation sets. (B) The discrepancy, as measured by the RMSD, between the RCMs for CDR3 predicted using features extracted by the epitope encoder and those predicted by OpenFold. (C) For epitopes associated with multiple CDR3 sequences, the residue distances at each position predicted by the RCMT closely align with the median distances from OpenFold’s predictions. RMSD: root mean square deviation.
Generator for predicting CDR3-β sequences that bind to a given epitope
We previously trained an encoder and an RCMT leveraging both the sequence and epitope structural information. Subsequently, we designed a generative model, CATCR-G, integrating the pretrained weights to facilitate the prediction of binding CDR3-β sequences for a given epitope. This approach involves utilizing a transformer decoder to generate CDR3-β predictions, formulated on the combined input from the epitope encoder and the RCM, as produced by the RCMT. The predicted CDR3-β sequence, the epitope sequence and structural data are then fed into the pretrained discriminator to refine the generator loss using the discriminator loss. During training, we froze the weights of the encoder and RCMT to preserve their pretrained states.
Initially, the training and validation losses declined quickly. However, while the training loss continued to decrease up to 300 epochs, the validation loss plateaued after 100 epochs and became more variable after 200 epochs (Figure 5A). Consequently, we concluded the training process at 300 epochs. In testing, we applied a beam search to yield the top seven CDR3-β sequence predictions. We evaluated them against reference sequences using the BERTscore metric (Figure 5B), which leverages contextual embeddings from the BERT model. The external test set yielded a BERTscore precision of 0.959 ± 0.013, recall of 0.955 ± 0.018 and F1 score of 0.957 ± 0.014. This indicates that CATCR-G can generate CDR3-β sequences that are highly similar to the reference sequences.
Figure 5.

Evaluation of the generative module (CATCR-G). (A) Loss curves for the training and validation sets. (B) The BERT score method, which assesses the semantic similarity between token embeddings, was used to compare the CDR3 sequences predicted by CATCR-G for unseen epitopes with a reference sequence set. (C) The performance of the complete CATCR-G is compared with that of models that do not use pretrained encoder weights, do not use weights from the RCMT module, or neither. This comparison is shown across different epochs for accuracy, recall and F1 scores, with evaluations based on the BERT score method. (D) The maximum similarity observed at different positions between the predicted CDR3 sequences and reference sequences indicate alignment accuracy. (E) The ROUGE-L metric, which evaluates the longest common subsequence between two sequences, and the skip-thought method, which assesses the semantic coherence of sentence embeddings, were used to score the predicted CDR3 sequences by CATCR-G against reference sequences.
We utilized the BERTscore for evaluation in our examination of the alignment between the predicted and reference CDR3-β sequences at each amino acid position. Despite the typical length of CDR3-β sequences ranging from 8 to 12 amino acids, we extended shorter sequences to 25 amino acids using placeholders where necessary for evaluation. This approach was used to ensure that all sequences had the same length when calculating the BERTscore, enabling a more accurate comparison and assessment of their similarity and alignment. Our analysis revealed that the use of a pretrained encoder significantly improved early training performance, whereas models devoid of this component exhibited diminished BERTscore R, P and F1 values. Furthermore, while models employing only the pretrained encoder matched the combined approach in the initial training, those also incorporating RCMT eventually outperformed in BERTscore evaluations, suggesting a synergistic benefit from using both (Figure 5C). This confirmed high similarity across corresponding positions, consistent with placeholder regions indicating that CATCR-G can appropriately determine CDR3-β length (Figure 5D). Additional evaluations with alternative metrics, ROUGE-L and skip-thought, yielded similarity scores of 0.580 ± 0.145 and 0.959 ± 0.040, respectively, further substantiating the effectiveness of the generative predictions of CATCR-G.
Discussion
TCR recognition and epitope binding, particularly within the CDR3 region, are pivotal for initiating immune responses and are crucial in T-cell therapy development. The high diversity of the CDR3 region is a major determinant of TCR binding specificity [29], and a comprehensive analysis of TCR repertoires can offer insights into the clonal and diverse nature of immune responses [2]. Immunoinformatics methodologies have made significant advances in the specific inference of observed epitopes. For example, in a study of SARS-CoV-2, Rakib et al. [10, 30, 31] screened for potential optimal epitopes that bind to MHC-I to guide vaccine development in response to rapid virus mutation. Despite advancements in our understanding, accurately predicting TCR specificity remains challenging due to the extensive variability resulting from V(D)J recombination, the limited availability of negative samples and other factors that constrain model performance.
A variety of machine learning methods have been applied, ranging from clustering-based approaches such as TCRdist (2022) [32], GLIPH (2017) [33] and TCRMatch (2021) [34] to random forest algorithms such as epiTCR (2023) [26] to address these challenges. The influx of data has inspired deep learning techniques, including gated recurrent unit (GRU) and transformer models, which have been adapted from their success in natural language processing to improve TCR–epitope binding predictions with models such as ERGO (2020) [35], ImRex (2021) [36], TITAN (2021) [25], DeepTCR (2021) [37], TEINet (2023) [27], PanPep (2023) [38] and TEIM-Seq (2023) [39]. These models have improved TCR–epitope binding prediction accuracy. However, despite their generalizability, their predictive performance significantly decreases with TCR–unseen antigen pairs compared to unseen TCR–seen antigen pairs. Therefore, enhancing the predictive efficacy of TCR–antigen binding for unseen TCR–antigen pairs is a major challenge.
From the perspective of binding mechanisms, the structural diversity of the CDR regions, especially the CDR3-β region, forms the basis for TCR diversity [1]. The simulation of 3D structures has been used for molecular docking analysis of epitopes [10]. Bradley [18] utilized an advanced complex structure prediction model, AlphaFold-Multimer, to embed TCR, MHC and epitope sequences for prediction, achieving an AUROC of 82% for eight seen epitope sequences. Compared to sequence-based methods, this approach did not show a clear advantage, possibly because AlphaFold-Multimer is a generative model, which increases model complexity when used for discrimination tasks. While the structure of peptides is determined by their amino acid sequences, the sequence does not contain experiential knowledge of how peptides should fold. OpenFold (or AlphaFold2), predicts the structure of CDR3-β or epitopes based on learning from many known protein structures, essentially providing CATCR with additional knowledge. In this study, we chose to represent structural data through the RCM. Using the RCM to characterize structural data avoids the direct interpretation of predicted complex structures, reduces the requirements for structural prediction accuracy and captures subtle differences in peptide structures for characterization. Our results indicate that embedding peptide structural data can improve model predictive performance, emphasizing the value of integrating structural information to enhance TCR–epitope binding prediction accuracy. Using only structural data, as opposed to only sequence data, achieved better predictive performance in evaluating the contribution of structural and sequence features to model predictive outcomes, further confirming the importance of structural data.
In the sequence feature representation, one-hot encoding is the most intuitive method; however, it struggles to reflect the differences between various amino acids. Amino acid substitution matrices, including PAM, BLOSUM and PSSM, as well as feature matrices based on physicochemical properties such as NLF and VHSE8, have been used to encode amino acid sequences. Among these, BLOSUM62 [28] has been widely applied in deep learning tasks due to its effective representation of amino acid sequences. According to our CDR3-β sequence data, we detected a significant presence of frequently occurring short sequences, such as ‘CASS’ and ‘CASR’ at the N-terminus and ‘QYF’ and ‘YTF’ at the C-terminus. These conserved fragments show similarities across different sequences, resulting in closely related features when encoded using traditional methods. Therefore, we employed a fragment-based coding approach to differentiate these similar sequences. Our results indicate that fragment-based coding methods can enhance the performance of the model compared to BLOSUM62. However, the applicability of this coding approach to tasks beyond antigen–TCR binding prediction requires further investigation.
It is well established that an antigen can be recognized by multiple TCRs [39]. According to our dataset, certain epitopes, such as KLGGALQAK, are associated with many TCR pairs. However, most epitopes are linked to relatively few TCR pairs. We limited our dataset to 500 records for any epitope associated with more than 500 CDR3-β sequences to ensure a more balanced representation. The predictive accuracy of CATCR-D diminishes for epitopes with scant paired CDR3-β records or shorter peptide chains, emphasizing the requirement for enhanced prediction strategies for these associations. In previous studies, PanPep [38] improved predictions for novel antigens through a synergistic meta-learning approach and a neural Turing machine to incorporate external memory. Moreover, embedding CDR3-β and epitope sequences into template sequences as part of complex structure prediction is promising for overcoming this challenge [18].
Given the generalization performance of CATCR-D in discrimination tasks, we experimented with generative models. Unlike discriminative models, generative models face more complex challenges: they require a powerful decoder and accurate and comprehensive information during the encoding phase for effective sequence generation. Numerous studies have demonstrated that pretrained encoders can achieve good predictive performance within the encoder–decoder framework, with representative models being BERT [40] and its biomedical variants, such as DNABERT [41]. Hence, we employed pretrained encoder weights in the encoding process and observed that the use of pretrained encoders enhances model performance and allows for earlier convergence. Moreover, we recognized the importance of structural information in the evaluation of CATCR-D, embodied through embedding RCMs. We aimed to integrate similar information into the generative model. Through RCMT, we generated predictions for the RCM of CDR3-β. An epitope might interact with multiple CDR3-β domains. Our results indicate that the RCMT-predicted matrices closely resemble the median conformation of the CDR3-β domain. Therefore, the output of RCMT should be interpreted as an overview of the potential CDR3-β conformations for an epitope rather than a precise structural prediction.
Evaluating generative models is challenging due to the absence of definitive reference standards and the need for task-relevant metrics. We leveraged BERTscore, a tool commonly used in natural language prediction, to gauge the quality of our sequence predictions. The BERTscore measures token similarity between the generated and reference sequences using contextual embeddings and applies standard performance metrics such as precision, recall and F1 scores. We also included additional metrics in our analysis, such as Rouge-L, which evaluates longer sequence similarity and sentence embeddings as per the skip-thought approach. These metrics suggest that CATCR-G can produce CDR3-β sequences with a high resemblance to the reference TCR dataset. Nevertheless, we acknowledge that validating the accuracy of CATCR-G-predicted CDR3-β sequences through wet-lab methods is essential, particularly through confirmation with the recombinant antigen–MHC multimer assay [42]. Moreover, expanding the dataset size through experimental means enhances model performance and makes employing predictive models with more parameters feasible.
This study has limitations. First, we did not incorporate CDR3-α and MHC sequences. In contrast, previous studies [6, 12, 27] have indicated that structural and statistical analyses suggest equal contributions of the α and β chains to specificity, and including CDR3-α and MHC information enhances the predictive accuracy of the model. Models limited to using the β chain CDR3 loop and VDJ gene encodings can only partially reveal the record of antigen recognition [1]. This is because antigens with complete CDR3-α, CDR3-β and MHC records are scarce. Retaining only CDR3-β data is a compromise due to sample size. Moreover, the ideal model is expected to embed fundamental knowledge about antigen presentation, TCR recognition and environment-dependent activation to predict whether the TCR binding to the antigen–MHC complex can effectively induce an immune response.
In conclusion, this study developed the CATCR framework for predicting the TCR binding to antigens. This model can extract sequence and structural information concurrently to generate descriptions of CDR3-β and antigen epitope features. Its discriminator component, CATCR-D, has shown robust performance in predicting interactions between CDR3-β and antigen epitopes, particularly in previously unseen CDR3-β and epitope pairs. Additionally, CATCR encompasses a generative component, CATCR-G, designed for the predictive generation of CDR3-β sequences that are likely to bind to specified epitopes. The predictions from this generative component require further validation through wet-lab experiments. Expanding the TCR–epitope–MHC pairings database is anticipated to significantly enhance the model’s performance and widen its applicability. This model is promising for facilitating advances in personalized immunotherapy.
Methods and materials
Dataset
The CDR3 α and β regions work together to recognize antigens; however, the data recorded in currently available databases are primarily focused on CDR3-β [20–22]. Currently available databases primarily document paired samples of CDR3-β chains. The quantity of paired data for both CDR3-α and -β is limited. To ensure adequate data for training and testing, we included only the CDR3-β sequences and their paired epitope sequences in this study. To construct a dataset with sufficient quantity and diversity, we referred to the databases used in previous studies [6, 13, 25–27, 35] (Supplementary Table S4) and included data from the VDJdb [20], the IEDB [21] and the McPAS-TCR [22] databases in our study.
The McPAS database encompasses 39 986 TCR data records, including 36 620 human TCR data entries. Among these, 12 256 data records included both CDR3-β and antigen epitope sequences, resulting in 10 942 non-redundant entries after duplicate removal. In addition to peptide antigens, the IEDB database records non-amino acid compounds, totaling 219 333 entries. Among them, 113 080 data entries included both CDR3-β and antigen peptide epitopes, resulting in 113 038 non-redundant entries. In VDJdb, we selected CDR3-β–epitope pairs with VDJ scores ≥1, resulting in 5706 entries. After merging the three databases, we obtained 128 259 non-redundant data records, including 127 507 unique CDR3-β sequences and 1176 distinct epitope sequences. We removed all sequences containing ambiguous amino acids (B, J, O, U, X). Among the CDR3-β sequences, 752 sequences corresponding to more than one epitope were eliminated. The number of CDR3-β sequences corresponding to each epitope ranged from 1 to 11 899 (Supplementary Figure 2), with a median of 3 and an average of 109. For epitopes with more than 500 corresponding CDR3 sequences, we randomly selected and retained 500 records. Additionally, we removed records with peptide chain lengths <5 amino acids, resulting in a dataset containing 65 069 CDR3β–epitope pairs.
Training and testing samples
One of the main tasks of this study was to predict unseen epitope–CDR3-β pairs. Therefore, we employed a sample partitioning method based on epitopes using 10-fold cross-validation. The epitopes were divided into ten relatively balanced subsets based on the number of paired CDR3-β sequences. One subset was selected each time as the external testing set, ensuring that it did not contain any epitope or CDR3-β sequence information from the training set. Additionally, 5% of the combined nine subsets were used as an internal testing set, which included epitope sequences from the training set but not CDR3-β sequences. Another 5% of the data were used as a validation set. CATCR-D, RCMT and CATCR-G were trained and tested using the same set of training and testing data during each cross-validation fold to ensure information confidentiality.
The epitope-CDR3-β dataset contains only positive samples. Generating negative samples, where for a positive sample 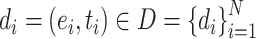 ,
, 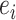 and
and 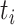 are the epitope and TCR interacting in sample
are the epitope and TCR interacting in sample 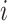 , respectively is necessary to train a robust supervised model. We generated negative samples using a random CDR3-β approach [24, 27]. For this sampling method, the negative CDR3-β for
, respectively is necessary to train a robust supervised model. We generated negative samples using a random CDR3-β approach [24, 27]. For this sampling method, the negative CDR3-β for 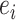 was uniformly sampled from the CDR3-β set
was uniformly sampled from the CDR3-β set 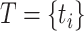 of positive binding pairs, while excluding their known true TCR binding partners. Subsequently, the negative samples for
of positive binding pairs, while excluding their known true TCR binding partners. Subsequently, the negative samples for 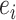 were represented as
were represented as 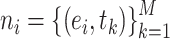 , where
, where 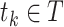 and
and 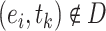 were different subsets. Negative samples were generated within their respective subsets after training and testing sample partitioning to avoid potential data leakage [27].
were different subsets. Negative samples were generated within their respective subsets after training and testing sample partitioning to avoid potential data leakage [27].
Predicting the 3D structure of CDR3-β and epitopes using OpenFold
OpenFold [19] is an open-source PyTorch reimplementation of AlphaFold2 [16] trained from scratch. Like AlphaFold2, it employs sequence alignment and deep learning algorithms. OpenFold offers lower deployment complexity and hardware requirements than AlphaFold2. In this study, we utilized the pretrained model ‘fineturning_ptm_1’ provided by the OpenFold developers to predict the 3D structures of CDR3β and its antigenic epitopes. To evaluate the ability of OpenFold to predict peptide chain structures, 112 matched sequences obtained from the RCSB PDB [23] were selected by aligning the amino acid sequences with those in the dataset. The actual structural coordinates corresponding to the predicted sequences were extracted during the evaluation. VMD software was then used to align the predicted sequences with the actual sequences and compute the RMSD values.
Model architecture
The model architecture is shown in Figure 1. We employed a model consisting of three modules. The first module involved constructing a discriminator based on structural and sequence data (CATCR-D) to determine whether an epitope can recognize a specific CDR3-β sequence. For the structural data, we represented structural features using RCMs, taking the positions of carbon atoms of amino acids from OpenFold’s predicted results as the positions of the amino acids and obtaining their coordinates in Euclidean space. The RCM 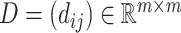 , where
, where 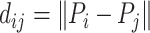 ,
, 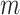 is the length of the amino acid sequence and
is the length of the amino acid sequence and 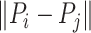 represents the Euclidean distance between the carbon atoms at position
represents the Euclidean distance between the carbon atoms at position 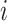 and position
and position 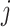 (Supplementary Figure 3). Two CNNs with kernel sizes of 3 and 5 were used to extract features from the RCMs. Each CNN consisted of two convolutional layers, followed by a max pooling layer after the second convolutional layer. The features extracted by the two CNNs were merged and passed through two linear fully connected layers to output a feature vector. For the amino acid sequences, we developed a segment-based coding method. This is because we observed a significant number of highly similar segments among CDR3 (epitope) sequences, such as ‘CASS’ and ‘CASR’ at the N-terminus and ‘QYF’ and ‘YTF’ at the C-terminus. In sequence data processing, we treated these frequently occurring segments as single characters for subsequent processing to enhance the discrimination of these similar segments. Segments that appeared in the dataset more than 1000 times were identified as high-frequency segments and assigned separate coding. During the coding process, longer segment coding is prioritized over shorter segment coding. For example, ‘CASS’ would be assigned a separate coding, while the shorter high-frequency block contained within it, ‘AS’, would not be encoded separately. The encoding for all high-frequency segments is presented in Supplementary Table S2. Subsequently, an embedding method was used to encode the sequences further. We employed a transformer encoder with six layers and eight heads to extract features from the encoded sequence information. Each inner layer contains 1024 nodes. Finally, a feature vector was output through a linear fully connected layer. After extracting and merging features from CDR3-β and epitopes separately using the aforementioned method, a classifier containing four linear fully connected layers was used to output the prediction results.
(Supplementary Figure 3). Two CNNs with kernel sizes of 3 and 5 were used to extract features from the RCMs. Each CNN consisted of two convolutional layers, followed by a max pooling layer after the second convolutional layer. The features extracted by the two CNNs were merged and passed through two linear fully connected layers to output a feature vector. For the amino acid sequences, we developed a segment-based coding method. This is because we observed a significant number of highly similar segments among CDR3 (epitope) sequences, such as ‘CASS’ and ‘CASR’ at the N-terminus and ‘QYF’ and ‘YTF’ at the C-terminus. In sequence data processing, we treated these frequently occurring segments as single characters for subsequent processing to enhance the discrimination of these similar segments. Segments that appeared in the dataset more than 1000 times were identified as high-frequency segments and assigned separate coding. During the coding process, longer segment coding is prioritized over shorter segment coding. For example, ‘CASS’ would be assigned a separate coding, while the shorter high-frequency block contained within it, ‘AS’, would not be encoded separately. The encoding for all high-frequency segments is presented in Supplementary Table S2. Subsequently, an embedding method was used to encode the sequences further. We employed a transformer encoder with six layers and eight heads to extract features from the encoded sequence information. Each inner layer contains 1024 nodes. Finally, a feature vector was output through a linear fully connected layer. After extracting and merging features from CDR3-β and epitopes separately using the aforementioned method, a classifier containing four linear fully connected layers was used to output the prediction results.
The second module is the RCMT, which integrates the predicted structural information of CDR3-β into the generation model. The structural details of CDR3-β are not directly observable in the generation model. We utilized the epitope encoder in Module 1 to extract epitope features and employed a linear generator with expanding dimensions to generate the predicted RCM of CDR3-β. Throughout this process, the weights of the epitope encoder remained locked.
The third module is a generator (CATCR-G) that combines the epitope sequence and the predicted structural information from RCMT to generate predictions for CDR3-β. The fundamental principle involves merging the features produced by the epitope encoder with the predicted RCM of CDR3-β and employing a transformer decoder to generate the predicted sequence.
Model training
CATCR was implemented using the PyTorch deep learning framework and was written in Python 3.9. The model was trained with a batch size of 128. CATCR-D employs stochastic gradient descent (SGD) to optimize binary cross-entropy loss with a learning rate of 0.05. In RCMT, we trained the model using a smooth L1 loss function augmented with L2 regularization and a learning rate of 0.05 while using the predicted CDR3-β structure from OpenFold as the label. CATCR-G utilizes the Adam optimizer to optimize cross-entropy loss. The predicted CDR3-β sequences and the RCM obtained from the RCMT, along with the epitope sequences and structural information, are reintroduced into CATCR-D to obtain the discriminative loss. The final training loss is given by 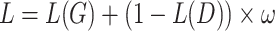 , where
, where 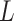 is the total loss,
is the total loss, 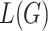 is the loss of the generative model and
is the loss of the generative model and 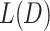 is the discriminative loss. The weights of the discriminator are fixed in the CATCR-G training process. The value of
is the discriminative loss. The weights of the discriminator are fixed in the CATCR-G training process. The value of 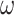 was 0.5. CATCR-G employs a warm-up strategy to adjust the learning rate dynamically, starting at 0.000012 and increasing to 0.00324 after five epochs, then gradually decreasing. The generation model utilizes the BERT score [43], ROUGE-L [44] and skip-thought methods [45] for performance evaluation.
was 0.5. CATCR-G employs a warm-up strategy to adjust the learning rate dynamically, starting at 0.000012 and increasing to 0.00324 after five epochs, then gradually decreasing. The generation model utilizes the BERT score [43], ROUGE-L [44] and skip-thought methods [45] for performance evaluation.
Key Points
We introduced the CATCR framework, which innovatively integrates both the sequence and structural information of peptides for TCR–epitope binding prediction, with the incorporation of structural information significantly enhancing the predictive performance.
A segment-based amino acid encoding method was employed during sequence encoding, which has been shown to outperform traditional encoding methods in its ability to represent amino acid sequences more effectively.
Our newly developed generative model, CATCR-G, predicts TCR sequence binding to an epitope based on the sequence and structural information of the epitope. Robust performance is exhibited in the BERT score metric.
Supplementary Material
ACKNOWLEDGEMENTS
We extend our gratitude to all contributors to VDJdb, IEDB and McPAS-TCR and other TCR-specific datasets for which their data are publicly available.
Author Biographies
Hongchen Ji obtained his MD from the Air Force Medical University in 2015, with a focus on computational biology and personalized immunotherapy.
Xiang-Xu Wang earned his Doctor of Public Health from the Air Force Medical University in 2023, specializing in the etiology and epidemiology of cancer.
Qiong Zhang obtained her MD degree from the Air Force Medical University in 2019, with a primary focus on personalized immunotherapy for cancer and the pathogenesis of pancreatic cancer.
Chengkai Zhang received his Doctor of Clinical Medicine degree in 2023, with a research emphasis on computational biology.
Hong-Mei Zhang is the director of the Oncology Department at Xijing Hospital of the Air Force Medical University. She earned her MD and PhD in Oncology from the Air Force Medical University in 2006, and from December 2007 to May 2010, she served as a postdoctoral fellow at the Health Science Center at the University of Texas, San Antonio. Her primary research focus is on cancer immunotherapy, with a particular emphasis on T-cell immunotherapy.
Contributor Information
Hongchen Ji, Department of Oncology of Xijing Hospital, Air Force Medical University, Xi’an, Shaanxi, China.
Xiang-Xu Wang, Department of Oncology of Xijing Hospital, Air Force Medical University, Xi’an, Shaanxi, China.
Qiong Zhang, Department of Oncology of Xijing Hospital, Air Force Medical University, Xi’an, Shaanxi, China.
Chengkai Zhang, Department of Oncology of Xijing Hospital, Air Force Medical University, Xi’an, Shaanxi, China.
Hong-Mei Zhang, Department of Oncology of Xijing Hospital, Air Force Medical University, Xi’an, Shaanxi, China.
AUTHOR CONTRIBUTIONS
Hongchen Ji (Methodology, Software and Writing—Original Draft), Qiong Zhang (Methodology and Data Curation), Chengkai Zhang (Validation, Investigation and Visualization) and Hong-Mei Zhang (Conceptualization, Supervision, Writing—Review & Editing and Funding acquisition)
FUNDING
This study was supported by the Clinical Key Research Project of Xijing Hospital (XJZT24LZ15).
DATA AVAILABILITY
CATCR was written in Python using the deep learning library PyTorch. All the data and code are available at https://github.com/FreudDolce/CTTCR/.
References
- 1. Hudson D, Fernandes RA, Basham M, et al. Can we predict T cell specificity with digital biology and machine learning? Nat Rev Immunol 2023;23(8):511–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chi X, Li Y, Qiu X. V(D)J recombination, somatic hypermutation and class switch recombination of immunoglobulins: mechanism and regulation. Immunology 2020;160(3):233–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shen Y, Voigt A, Leng X, et al. A current and future perspective on T cell receptor repertoire profiling. Front Genet 2023;14:1159109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Joglekar AV, Li G. T cell antigen discovery. Nat Methods 2021;18(8):873–80. [DOI] [PubMed] [Google Scholar]
- 5. Grazioli F, Mösch A, Machart P, et al. On TCR binding predictors failing to generalize to unseen peptides. Front Immunol 2022;13:1014256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ehrlich R, Kamga L, Gil A, et al. SwarmTCR: a computational approach to predict the specificity of T cell receptors. BMC Bioinformatics 2021;22(1):422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cai M, Bang S, Zhang P, Lee H. ATM-TCR: TCR-epitope binding affinity prediction using a multi-head self-attention model. Front Immunol 2022;13:893247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sami SA, Marma KKS, Mahmud S, et al. Designing of a multi-epitope vaccine against the structural proteins of Marburg virus exploiting the immunoinformatics approach. ACS Omega 2021;6(47):32043–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mahmud S, Rafi MO, Paul GK, et al. Designing a multi-epitope vaccine candidate to combat MERS-CoV by employing an immunoinformatics approach. Sci Rep 2021;11(1):15431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rakib A, Sami SA, Mimi NJ, et al. Immunoinformatics-guided design of an epitope-based vaccine against severe acute respiratory syndrome coronavirus 2 spike glycoprotein. Comput Biol Med 2020;124:103967, 103967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang H, Wang C, Rubelt F, et al. Analyzing the mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nat Biotechnol 2020;38(10):1194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mayer-Blackwell K, Schattgen S, Cohen-Lavi L, et al. TCR meta-clonotypes for biomarker discovery with tcrdist3 enabled identification of public, HLA-restricted clusters of SARS-CoV-2 TCRs. elife 2021;10:e68605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Korpela D, Jokinen E, Dumitrescu A, et al. EPIC-TRACE: predicting TCR binding to unseen epitopes using attention and contextualized embeddings. Bioinformatics 2023;39(12):btad743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koyama K, Hashimoto K, Nagao C, Mizuguchi K. Attention network for predicting T-cell receptor–peptide binding can associate attention with interpretable protein structural properties. Front Bioinform 2023;3:1274599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Henry KA, MacKenzie CR. Antigen recognition by single-domain antibodies: structural latitudes and constraints. MAbs 2018;10(6):815–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021;596(7873):583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang Z, Zeng X, Zhao Y, Chen R. AlphaFold2 and its applications in the fields of biology and medicine. Signal Transduct Target Ther 2023;8(1):115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bradley P. Structure-based prediction of T cell receptor:peptide-MHC interactions. elife 2023;12:e82813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahdritz G, Bouatta N, Kadyan S, et al. OpenFold: Retraining AlphaFold2 yields new insights into its learning mechanisms and capacity for generalization. bioRxiv 2022; bioRxiv: 2022.11.20.517210. [DOI] [PMC free article] [PubMed]
- 20. Goncharov M, Bagaev D, Shcherbinin D, et al. VDJdb in the pandemic era: a compendium of T cell receptors specific for SARS-CoV-2. Nat Methods 2022;19(9):1017–9. [DOI] [PubMed] [Google Scholar]
- 21. Vita R, Mahajan S, Overton JA, et al. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res 2019;47(D1):D339–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tickotsky N, Sagiv T, Prilusky J, et al. McPAS-TCR: a manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics 2017;33(18):2924–9. [DOI] [PubMed] [Google Scholar]
- 23. Berman HM, Westbrook J, Feng Z, et al. The protein data Bank. Nucleic Acids Res 2000;28(1):235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lu T, Zhang Z, Zhu J, et al. Deep learning-based prediction of the T cell receptor–antigen binding specificity. Nat Mach Intell 2021;3(10):864–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weber A, Born J, Rodriguez Martínez M. TITAN: T-cell receptor specificity prediction with bimodal attention networks. Bioinformatics 2021;37(Supplement_1):I237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pham MDN, Nguyen TN, Tran LS, et al. epiTCR: a highly sensitive predictor for TCR–peptide binding. Bioinformatics 2023;39(5):btad284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang Y, Huo M, Li SC. TEINet: a deep learning framework for prediction of TCR–epitope binding specificity. Brief Bioinform 2023;24(2):bbad086. [DOI] [PubMed] [Google Scholar]
- 28. Styczynski MP, Jensen KL, Rigoutsos I, Stephanopoulos G. BLOSUM62 miscalculations improve search performance. Nat Biotechnol 2008;26(3):274–5. [DOI] [PubMed] [Google Scholar]
- 29. Szeto C, Lobos CA, Nguyen AT, et al. TCR recognition of peptide–MHC-I: rule makers and breakers. Int J Mol Sci 2021;22(3):1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Obaidullah AJ, Alanazi MM, Alsaif NA, et al. Immunoinformatics-guided design of a multi-epitope vaccine based on the structural proteins of severe acute respiratory syndrome coronavirus 2. RSC Adv 2021;11(29):18103–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rakib A, Sami SA, Islam MA, et al. Epitope-based Immunoinformatics approach on Nucleocapsid protein of severe acute respiratory syndrome-Coronavirus-2. Molecules 2020;25(21):5088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Olson BJ, Schattgen SA, Thomas PG, et al. Comparing T cell receptor repertoires using optimal transport. PLoS Comput Biol 2022;18(12):e1010681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Glanville J, Huang H, Nau A, et al. Identifying specificity groups in the T cell receptor repertoire. Nature 2017;547(7661):94–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chronister WD, Crinklaw A, Mahajan S, et al. TCRMatch: predicting T-cell receptor specificity based on sequence similarity to previously characterized receptors. Front Immunol 2021;12:640725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Springer I, Besser H, Tickotsky-Moskovitz N, et al. Prediction of specific TCR-peptide binding from large dictionaries of TCR-peptide pairs. Front Immunol 2020;11:1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moris P, De Pauw J, Postovskaya A, et al. Current challenges for unseen-epitope TCR interaction prediction and a new perspective derived from image classification. Brief Bioinform 2021;22(4):bbaa318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sidhom JW, Larman HB, Pardoll DM, Baras AS. DeepTCR is a deep learning framework for revealing sequence concepts within T-cell repertoires. Nat Commun 2021;12(1):1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gao Y, Gao Y, Fan Y, et al. Pan-peptide meta learning for T-cell receptor–antigen binding recognition. Nat Mach Intell 2023;5(3):236–49. [Google Scholar]
- 39. Peng X, Lei Y, Feng P, et al. Characterizing the interaction conformation between T-cell receptors and epitopes with deep learning. Nat Mach Intell 2023;5(4):395–407. [Google Scholar]
- 40. Devlin J, Chang M-W, Lee K, et al. BERT: pre-training of deep bidirectional transformers for language understanding. arXiv e-prints 2018; arXiv:1810.04805.
- 41. Ji Y, Zhou Z, Liu H, Davuluri RV. DNABERT: pre-trained bidirectional encoder representations from transformers model for DNA-language in genome. Bioinformatics 2021;37(15):2112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kurtulus S, Hildeman D. Assessment of CD4+ and CD8+ T cell responses using MHC class I and II tetramers. Methods Mol Biol 2013;979:71–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang T, Kishore V, Wu F, et al. BERTScore: evaluating text generation with BERT. arXiv e-prints 2019; arXiv:1904.09675.
- 44. Lin C-Y. ROUGE: a package for automatic evaluation of summaries. Association for Computational Linguistics, Barcelona, Spain, 2004; 74–81.
- 45. Kiros R, Zhu Y, Salakhutdinov R, et al. Skip-thought vectors. arXiv e-prints 2015; arXiv:1506.06726.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
CATCR was written in Python using the deep learning library PyTorch. All the data and code are available at https://github.com/FreudDolce/CTTCR/.