


Abstract
The checkpoint mechanisms that delay cell cycle progression in response to DNA damage or inhibition of DNA replication are necessary for maintenance of genetic stability in eukaryotic cells. Potential targets of checkpoint-mediated regulation include proteins directly involved in DNA metabolism, such as the cellular single-stranded DNA (ssDNA) binding protein, replication protein A (RPA). Studies in Saccharomyces cerevisiae have revealed that the RPA large subunit (Rfa1p) is involved in the G1 and S phase DNA damage checkpoints. We now demonstrate that Rfa1p is phosphorylated in response to various forms of genotoxic stress, including radiation and hydroxyurea exposure, and further show that phosphorylation of Rfa1p is dependent on the central checkpoint regulator Mec1p. Analysis of the requirement for other checkpoint genes indicates that different mechanisms mediate radiation- and hydroxyurea-induced Rfa1p phosphorylation despite the common requirement for functional Mec1p. In addition, experiments with mutants defective in the Cdc13p telomere-binding protein indicate that ssDNA formation is an important signal for Rfa1p phosphorylation. Because Rfa1p contains the major ssDNA binding activity of the RPA heterotrimer and is required for DNA replication, repair and recombination, it is possible that phosphorylation of this subunit is directly involved in modulating RPA activity during the checkpoint response.
INTRODUCTION
The importance of cell cycle regulation in maintaining genetic stability is clearly illustrated by the high incidence of cancer in patients with defects in any one of the several ‘checkpoints’ that operate to delay cell cycle progression upon cellular damage. These regulatory pathways are thought to provide the time necessary for repair processes to occur before genetic alterations are rendered irreversible through cell cycle events such as DNA replication or mitosis (1). Despite the likelihood that proteins involved in DNA metabolism are important targets of checkpoint-mediated control in humans, molecular mechanisms underlying such regulation have not been characterized. One protein that plays an essential role in DNA replication, repair and recombination and that is also involved in checkpoint processes is replication protein A (RPA), the evolutionarily conserved heterotrimeric single-stranded DNA (ssDNA) binding protein (2).
The three subunits of RPA have molecular weights of ∼70, 32 and 14 kDa, and the largest of these contains the major ssDNA binding activity of the protein. In addition to interacting with nucleic acid, the large subunit directly associates with other proteins that are involved in replication, repair and recombination (2). The large subunit also interacts with various transcription factors, including the tumor suppressor p53 (3,4). Direct evidence that RPA is a checkpoint protein has been provided by the generation of a mutant in the Saccharomyces cerevisiae large subunit (Rfa1p) that exhibits defective cell cycle delay following DNA damage sustained during the G1 or S phases of the cell cycle (5). Other studies have shown that Rfa1p is involved in the adaptation to cell cycle arrest that accompanies irreparable DNA damage (6) and that the large subunit of fission yeast RPA is involved in recovery from inhibition of DNA replication (7). Therefore, the RPA large subunit appears to play an important role in various cell-cycle regulatory processes.
The RPA middle subunit is also implicated in cell cycle function, as this polypeptide becomes phosphorylated periodically during the normal cell cycle and in response to genotoxic insult (8–11). An RPA phosphorylation reaction resembling the cell-cycle-regulated reaction has been shown to occur during SV40 DNA replication in vitro (12). Our previous studies demonstrated that this DNA replication-dependent RPA phosphorylation reaction requires the catalytic subunit of DNA-activated protein kinase (DNA-PKcs) (13), a nuclear serine/threonine protein kinase that is necessary for DNA double-strand break repair and V(D)J recombination (14). We also demonstrated that DNA-PKcs-mediated RPA phosphorylation does not affect DNA replication activity in vitro, leading to the conclusion that RPA phosphorylation is not directly involved in the mechanics of DNA replication in the SV40 system (13).
Experiments with cells from ataxia-telangiectasia (A-T) patients have indicated that a homolog of DNA-PKcs is required for ionizing radiation (IR)-induced phosphorylation of the RPA middle subunit. A-T is an autosomal recessive disorder characterized by cerebellar degeneration, ocular telangiectasia, immune dysfunction, premature aging and cancer susceptibility (15). Despite these disparate disease manifestations, A-T is caused by mutation of a single protein, ATM (16), which is similar in primary sequence to DNA-PKcs (17). A-T cells are hypersensitive to killing by ionizing radiation but not ultraviolet (UV) radiation (18) and they exhibit a significant delay in IR-induced but not UV-induced RPA middle subunit phosphorylation (9). Thus, the ATM protein is specifically required for RPA middle subunit phosphorylation upon IR exposure. A-T cells are defective for checkpoint-mediated cell cycle delay during the G1, S or G2 phases of the cell cycle (19,20) and the G1 checkpoint defect has been linked to a deficiency in accumulation of the tumor suppressor p53 (21). Recent reports have shown that purified ATM can directly catalyze both p53 and RPA middle subunit phosphorylation (22,23). Therefore, p53 and RPA, which directly interact with each other, both appear to occupy positions downstream of ATM in the DNA damage response. Although these results would suggest that RPA phosphorylation plays a role in the checkpoint response, a strict correlation between RPA middle subunit phosphorylation and checkpoint function has not been observed in human cells (24).
As yet, the roles of cell-cycle-regulated and radiation-induced RPA phosphorylation have not been clearly defined. However, RPA from human cells treated with different DNA damaging agents is incapable of supporting SV40 DNA replication in vitro, suggesting that DNA damage-induced RPA phosphorylation could be involved in regulating the relative replication and repair functions of RPA (10,25–27). Because RPA phosphorylation reactions have been conserved through evolution (8–11), we have turned to the genetically tractable eukaryote S.cerevisiae to further understand both the mechanisms and the functions of these modification events. Our previous studies demonstrated that Mec1p is required for both cell-cycle-regulated and DNA damage-induced phosphorylation of the S.cerevisiae RPA middle subunit (Rfa2p) (11). Mec1p is a key regulator of the DNA damage and DNA replication checkpoints in yeast and, unlike most known eukaryotic checkpoint proteins, is essential for viability (28–31). The homology of Mec1p to ATM and DNA-PKcs (16,17,29) suggests that the mechanisms and functions of RPA middle subunit phosphorylation are similar in human and yeast cells.
In this report, we demonstrate that the large subunit of S.cerevisiae RPA becomes phosphorylated under conditions of genotoxic stress that induce checkpoint-mediated cell cycle delay, such as DNA damage or inhibition of DNA replication. As we previously observed for phosphorylation of the middle subunit, Rfa1p phosphorylation is dependent on Mec1p. Examination of the reaction under different stress conditions and in different checkpoint mutants indicates that multiple mechanisms are involved despite the shared Mec1p requirement. In addition, experiments with cdc13 mutants have provided evidence that Rfa1p phosphorylation is dependent on generation of ssDNA upon DNA damage. These results indicate that the ssDNA binding subunit is a target of checkpoint-mediated modification, providing a possible mechanism for the regulation of RPA activities in response to cellular insult.
MATERIALS AND METHODS
Yeast strains, plasmids and growth conditions
The S.cerevisiae strains used in this study (Table 1) were kindly provided by Drs Ted Weinert (University of Arizona, AZ), David Lydall (University of Manchester, UK) and Heidi Feldmann (University of Munich, Germany). Plasmids used were pRS316 (32), pTEL1 (pDM197) (33) and pMEC1 (pDM207) (11). Yeast cells harboring plasmid were grown in synthetic complete medium lacking uracil, and all other cells were grown in rich medium (YPD) (34). Unless otherwise indicated, cells were grown at 25°C.
Table 1. Strains used in this study.
| Strain | Genotype | Reference |
|---|---|---|
| TWY397 | MATa ura3 his7 leu2 trp1 | 28 |
| TWY308 | MATα mec1-1 ura3 trp1 | 28 |
| TWY312 | MATa mec2-1 ura3 his7 trp1 | 28 |
| TWY316 | MATa mec3-1 ura3 his3 trp1 | 28 |
| TWY398 | MATa rad9Δ::LEU2 ura3 his7 leu2 trp1 | 28 |
| TWY146 | MATα cdc13-1 ura3 his7 | 28 |
| TWY158 | MATα mec1-1 cdc13-1 ura3 his7 | 28 |
| TWY149 | MATα mec1-2 cdc13-1 ura3 his7 | 28 |
| TWY150 | MATα mec1-3 cdc13-1 ura3 his7 | 28 |
| DLY285 | MATa mec1-1::HIS3 ura3 his3 leu2 trp1 | 31 |
| YDM937 | MATa mec1-1 tel1Δ1::HIS3 ura3-52 his3Δ200 leu2Δ1 lys2-801 trp1Δ1 | 33 |
| DLY408a | MATa cdc13-1 cdc15-2 | 40 |
| DLY409 | MATa cdc13-1 cdc15-2 rad9::HIS3 | 40 |
| DLY410 | MATa cdc13-1 cdc15-2 rad24::TRP1 | 40 |
| DLY411 | MATa cdc13-1 cdc15-2 rad9::HIS3 rad24::TRP1 | 40 |
| DLY418 | MATa cdc15-2 | 40 |
| DLY419 | MATa cdc15-2 rad9::HIS3 | 40 |
| W303-1A | MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 | 61 |
aDLY strains 408–411, 418 and 419 are congenic and have the following additional genotype: bar1::hisG ade2-1 can1-100 ura3 leu2-3,112 his3-11 trp1-1.
Antibodies
Antisera directed against the yeast RPA large and middle subunits were generously provided by Dr Steven Brill (Rutgers University). Additional antisera were generated using glutathione-S-transferase fusion versions of the large and middle subunits as antigens. For Rfa1p, two fragments of the RFA1 gene were amplified by PCR and cloned separately into pGEX-4T-1 (Amersham Pharmacia Biotech). The recombinant plasmids were transformed into E.coli DH5α and protein expression was induced with IPTG. Soluble fusion protein was purified by glutathione–agarose chromatography and the two purified protein preparations were combined. The pooled protein was supplied to Covance, Inc. for antiserum production in rabbits. A similar protocol was used for Rfa2p except that a single PCR fragment encompassing all but the initiating methionine of RFA2 was employed. The recombinant plasmids pJM113 and pJM215 containing the full-length RFA1 and RFA2 genes, respectively (35), were kindly provided by Dr Steven Brill.
Analysis of RPA phosphorylation
A western blot assay was used to detect phosphoisomers of RPA that have reduced mobility during SDS–PAGE, as described previously (11). Briefly, denatured or native extracts were electrophoresed through denaturing gels [150:1 (w/w) acrylamide:bis-acrylamide] and transferred to nitrocellulose. Immobilized RPA was detected by autoradiography employing anti-RPA primary antibodies, horseradish peroxidase-linked goat anti-rabbit secondary antibody (Amersham Pharmacia Biotech or Pierce) and chemiluminescence reagents (Amersham Pharmacia Biotech or Pierce).
Hydroxyurea (HU) treatment and UV irradiation
DNA replication inhibition experiments were performed by treating cells with 0.1 M HU for 2 h. The UV irradiation experiment was conducted on asynchronous cell populations. Cultures that had been grown overnight in YPD were diluted to an OD600 of 0.2 in YPD and further incubated for 4 h. Cells were pelleted from 4.5 ml of each culture by centrifugation for 5 min at 1900 g at room temperature and resuspended in ∼0.5 ml of supernatant. Aliquots of these suspensions (200 µl) were spread on YPD plates and the cells were then irradiated at 60 J/m2 using a Stratagene UV Stratalinker 2400. After further incubation at 25°C for 1 h, the yeast were harvested from the plates with cold water and RPA phosphorylation was analyzed by the western blot assay.
Phosphatase treatment
HU-treated TWY397 cells and temperature-shifted DLY409 cells were harvested by centrifugation for 5 min at 1900 g at 4°C. The cell pellets were washed once with cold water and collected by centrifugation again. The TWY397 cells were resuspended with 50 mM HEPES pH 7.8, 2 mM EDTA, 2 mM DTT, 20% glycerol, 0.2 mM PMSF and the DLY409 cells were resuspended with 50 mM Tris–Cl pH 7.8, 250 mM NaCl, 0.1% Nonidet P-40, 5 mM EDTA, 50 mM NaF, 10% glycerol, 1 mM DTT, 10 mM PMSF, 10 mM benzamidine, 5 µg/ml aprotinin, 5 µg/ml chymostatin, 5 µg/ml leupeptin, 5 µg/ml pepstatin A. Acid-washed glass beads (425–600 microns; Sigma Chemical Co.) were added to the suspensions and the cells were disrupted by vortexing at 4°C. The glass beads were removed by centrifugal filtration and the resulting extracts were clarified by centrifugation for 5 min (TWY397) or 10 min (DLY409) at 14 000 g at 4°C. Extracts were treated with 400 U λ protein phosphatase (λPPase; New England Biolabs) at 30°C for 30 min in the presence of 50 mM Tris–HCl pH 7.5, 2 mM MnCl2, 0.1 mM Na2EDTA, 5 mM DTT and 0.01% Brij 35. Where indicated, sodium orthovanadate was added to a final concentration of 10 mM.
DNA-PKcs catalyzed RPA phosphorylation
DNA-PKcs and human RPA were purified from HeLa cells as described previously (13,36) and yeast RPA was purified from W303-1A cells by the method of Sung (37). Reaction mixtures (25 µl) contained 40 mM Tris–Cl pH 7.8, 7 mM MgCl2, 5 µM ATP and 5 µCi [γ-32P]ATP (carrier-free). Variable components added to specific reaction mixtures included activated salmon sperm DNA (2 µg/ml final; USB), RPA (160 ng) and DNA-PKcs (15 ng). Reaction mixtures were incubated at 30°C for 15 min and reactions were stopped with SDS–PAGE sample loading buffer. The samples were then subjected to SDS–PAGE and the resulting gel was dried and analyzed by autoradiography.
RESULTS
Inhibition of DNA replication induces phosphorylation of Rfa1p
During the course of our investigation into Rfa2p phosphorylation, we observed that Rfa1p was also modified under certain conditions. Specifically, treatment of wild-type S.cerevisiae with HU, an inhibitor of DNA replication, induces the generation of an Rfa1p isoform with reduced mobility during SDS–PAGE (Fig. 1A). Because phosphorylation of Rfa2p reduces its mobility during SDS–PAGE in a similar manner (8,11), we suspected that the modified form of Rfa1p observed under these conditions was a phosphoprotein. Treatment of extract from HU-exposed cells with λPPase abolishes the HU-induced form of Rfa1p and inclusion of the λPPase inhibitor vanadate in the reaction mixture inhibits this demodification (Fig. 1B). Therefore, the reduction in mobility of Rfa1p during SDS–PAGE is due to phosphorylation. Unlike Rfa2p phosphorylation, Rfa1p phosphorylation is not observed during the normal cell cycle, but only in response to HU and other genotoxic insults (see below).
Figure 1.
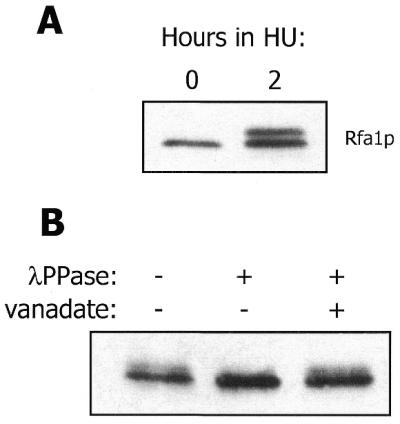
HU induces Rfa1p phosphorylation. (A) Western blot analysis of Rfa1p from TWY397 cells harvested immediately before and after HU exposure. (B) Western blot analysis of Rfa1p from native extract of HU-treated TWY397 cells treated with λPPase and sodium orthovanadate as indicated. For both experiments, TWY397 cells were incubated at 30°C.
MEC1 and TEL1 are required for HU-dependent Rfa1p phosphorylation
Our previous studies indicated that MEC1 is required to achieve the level of HU-induced Rfa2p phosphorylation observed in wild-type cells. However, the MEC1 homolog TEL1, which is not a bona fide checkpoint gene but functionally overlaps with MEC1 (33), can partially rescue HU-induced Rfa2p phosphorylation in mec1 mutants (11). Analysis of HU-induced Rfa1p phosphorylation in a series of checkpoint mutants has revealed a specific MEC1/TEL1 dependence identical to that observed for Rfa2p. While none of the checkpoint mutants tested has a dramatic defect in Rfa1p phosphorylation, the level of phosphorylated Rfa1p is slightly reduced in the mec1 strain (Fig. 2A). As previously observed with HU-induced Rfa2p phosphorylation, mutation of both MEC1 and TEL1 completely abolishes HU-induced Rfa1p phosphorylation (Fig. 2B). Transformation of the double mutant with episomal MEC1 or TEL1 rescues this defect, indicating an overlap in function of these two genes as previously observed for Rfa2p phosphorylation.
Figure 2.

Mec1p and Tel1p are required for HU-induced Rfa1p phosphorylation. (A) Western blot analysis of Rfa1p from exponentially growing (–) or HU-treated (+) cells. Strains employed were: TWY397 (wt), TWY308 (mec1), TWY312 [rad53 (= mec2)], TWY316 (mec3) and TWY398 (rad9). P-Rfa1p, phosphorylated form of Rfa1p. (B) YDM937 (mec1-1 tel1Δ1) cells transformed with pRS316 (vector), pRS316 containing TEL1 (pTEL1) or pRS316 containing MEC1 (pMEC1) were analyzed as in (A). *, immunoreactive band that migrates faster than full-length Rfa1p and is presumed to be an Rfa1p degradation product. The intensity of this band varies between experiments.
DNA damaging agents induce checkpoint-dependent Rfa1p phosphorylation
In addition to HU, agents that cause various forms of DNA damage were tested for their capacity to induce phosphorylation of the RPA large subunit. We found that Rfa1p becomes phosphorylated upon exposure of wild-type cells to UV radiation, IR or the alkylating agent methyl methane sulfonate (Fig. 3 and data not shown). Furthermore, we observed that UV radiation-induced Rfa1p phosphorylation is significantly reduced not only in mec1 mutant cells, but also in the other three checkpoint-deficient strains that we tested (Fig. 3). A similar checkpoint-dependent Rfa1p phosphorylation reaction was observed with populations arrested in G1 by alpha mating factor and exposed to either UV radiation or IR. However, the severity of the defects in the G1-arrested checkpoint mutants was difficult to assess because only weak Rfa1p phosphorylation was observed in the wild-type cells under these conditions (data not shown). It is interesting that the checkpoint dependence of Rfa1p phosphorylation is distinct from that of radiation-induced Rfa2p phosphorylation, which requires MEC1 and MEC3 but is independent of RAD9 and RAD53 (11). Thus, in contrast to the HU-induced phosphorylation reactions described above, RPA phosphorylation in response to DNA damage does not appear to proceed by a single mechanism.
Figure 3.

Radiation induces checkpoint-dependent Rfa1p phosphorylation. Wild-type and checkpoint mutant cells were untreated (–) or challenged with UV radiation (+; 60 J/m2). Rfa1p in denatured extracts of these cells was detected by western blot analysis. The strains employed were the same as those indicated in Figure 2A, except DLY285 (MATa mec1-1) was used instead of TWY308 (MATα mec1-1).
Inactivation of Cdc13p induces Mec1p-dependent Rfa1p phosphorylation
Cdc13p is an ssDNA-binding protein that interacts preferentially with the G-rich strand of telomeric DNA (38). In the absence of Cdc13p function, long tracts of ssDNA are formed at the ends of the chromosomes and these DNA structures induce the DNA damage response (28,39,40). The checkpoint roles of several genes, including MEC1, were originally identified in a synthetic lethality screen employing the temperature-sensitive cdc13-1 mutation (28). At the restrictive temperature, checkpoint-proficient cells arrest reversibly with a G2 DNA content, whereas checkpoint-deficient cells progress through the cell cycle and subsequently die.
RPA phosphorylation was investigated in cdc13-1 cells at both the permissive and restrictive temperatures (Fig. 4). During normal cell cycle progression (25°C), the cdc13-1 mutant exhibits a low level of phosphorylated Rfa1p, suggesting that some DNA damage might be present even at the permissive temperature. However, a marked induction of Rfa1p phosphorylation is observed upon shifting cells to the restrictive temperature (37°C). There is also a detectable induction of Rfa2p phosphorylation, but this increase is more subtle due to the high level of phosphorylated Rfa2p observed during the normal cell cycle. The RPA phosphorylation reactions induced by inactivation of Cdc13p were also analyzed in three mutant alleles of mec1 that were originally identified in the cdc13-1-based synthetic lethality screen (28) (Fig. 4). At the permissive temperature, the mec1-1 mutant is devoid of phosphorylated RPA, while mec1-3 exhibits no detectable phosphorylated Rfa1p but elevated levels of phosphorylated Rfa2p relative to the control cdc13 strain. Upon temperature shift, mec1-1 cells are completely defective for induction of either Rfa1p or Rfa2p phosphorylation, while the mec1-3 mutant exhibits a low level of phosphorylated Rfa1p and a high level of phosphorylated Rfa2p. The mec1-2 mutant has no apparent defect in phosphorylation of either subunit at either temperature. These data indicate that Mec1p is required for RPA phosphorylation induced upon inactivation of Cdc13p. It is interesting that the severity of the telomere damage-induced RPA phosphorylation deficiency in the cdc13 mec1 mutants correlates well with the reported phenotypic penetrance of the mec1 alleles: mec1-1 cells are hypersensitive to killing by HU, IR and MMS, mec1-3 cells are hypersensitive to killing by HU and MMS but not IR and mec1-2 cells are not hypersensitive to killing by any of these agents (28). Nonetheless, we have confirmed that all three cdc13 mec1 mutants die upon transient Cdc13p inactivation (28) (data not shown), indicating that the extent of RPA phosphorylation does not correlate with viability under these conditions.
Figure 4.

RPA phosphorylation induced by telomeric damage is dependent on Mec1p. Western blot analysis of Rfa1p and Rfa2p from cdc13 cells harvested immediately before and after incubation at the restrictive temperature for 2 h. Strains employed were: TWY146 (cdc13), TWY158 (cdc13 mec1-1), TWY149 (cdc13 mec1-2) and TWY150 (cdc13 mec1-3). P-Rfa2p, phosphorylated form of Rfa2p.
Rfa1p phosphorylation induced by Cdc13p inactivation correlates with ssDNA formation
There is evidence that certain checkpoint proteins regulate the generation of ssDNA upon inactivation of Cdc13p (40). In particular, the rate of telomeric ssDNA formation at the restrictive temperature is greater in a cdc13 rad9 mutant than in a cdc13 mutant. Conversely, very little ssDNA is generated in cells bearing cdc13 and rad24 mutations and a combination of rad9 and rad24 with cdc13 results in a rate of ssDNA formation nearly identical to that observed in the cdc13 rad24 strain. We examined these mutants and found that Rfa1p phosphorylation is induced upon shift to the restrictive temperature in the cdc13 and cdc13 rad9 cells, but is greatly reduced in the cdc13 rad24 strain and undetectable in cdc13 rad9 rad24 (Fig. 5A). Therefore, Rfa1p phosphorylation resulting from temperature-induced Cdc13p inactivation correlates with the extent of telomeric ssDNA that is generated under these conditions (40). In contrast to Rfa1p phosphorylation, induction of Rfa2p phosphorylation upon temperature shift is not diminished in any of the mutants (Fig. 5A). In fact, the cdc13 rad9 mutant gives rise to a species of Rfa2p that migrates even slower during SDS–PAGE than the previously characterized phosphoprotein. Further analysis has revealed that at least three isomers of Rfa2p are generated under these conditions (Fig. 5B) and treatment with λPPase indicates that these reduced mobility forms are phosphoproteins (Fig. 5C). The hyperphosphorylation of Rfa2p under these special conditions is reminiscent of the hyperphosphorylation of the human RPA middle subunit that occurs upon exposure of cells to radiation (9,10,41). As previously demonstrated with RPA from HU-treated cells (Fig. 1B), the modified form of Rfa1p generated in the cdc13 rad9 mutant is also a phosphoprotein (Fig. 5C).
Figure 5.

Rfa1p and Rfa2p phosphorylation induced by telomeric damage exhibit different genetic dependencies. (A) Western blot analysis of RPA from cells harvested immediately before and after incubation at the restrictive temperature for 2 h. Strains employed were DLY408 (cdc13), DLY409 (cdc13 rad9), DLY410 (cdc13 rad24), DLY411 (cdc13 rad9 rad24), DLY418 (- - -) and DLY419 (rad9). These strains contain a mutant allele of CDC15 that allows for mitotic arrest of checkpoint-deficient strains at the restrictive temperature (40). (B) Western blot analysis of Rfa2p from DLY409 cells incubated at the restrictive temperature for 1 h. (C) Phosphatase treatment of native extract from DLY409 cells incubated at the restrictive temperature for 2 h.
DNA-PKcs catalyzes Rfa2p but not Rfa1p phosphorylation in vitro
Our previous studies have demonstrated that the DNA-activated protein kinase (DNA-PK) catalyzes phosphorylation of human RPA during cell-free SV40 DNA replication. Because Mec1p is required for yeast RPA phosphorylation in vivo and is similar in primary sequence to DNA-PKcs and ATM, it is possible that Mec1p is also an RPA kinase. As yet, purified Mec1p is not available to directly test this hypothesis in vitro. However, there is evidence that members of the ATM family of protein kinases have overlapping substrate specificities. For example, purified preparations of DNA-PK, ATM and ATM-related protein (ATR) all catalyze phosphorylation of p53 at Ser15 (22,23,42,43). Therefore, DNA-PKcs-catalyzed phosphorylation of yeast RPA with purified proteins was investigated as a possible model for Mec1p-dependent catalysis. As shown in Figure 6, DNA-PKcs can efficiently catalyze Rfa2p phosphorylation in the presence of ‘activated’ DNA containing both double- and single-strand character. The phosphorylated species of Rfa2p generated through this reaction has reduced mobility during SDS–PAGE relative to the unphosphorylated protein (data not shown). Neither the hyperphosphorylated Rfa2p detected in cdc13 rad9 cells nor phosphorylated Rfa1p appears to be generated to a significant extent under these conditions. A similar pattern of phosphorylation is revealed in the control reaction with human RPA.
Figure 6.

DNA-PKcs catalyzes phosphorylation of yeast RPA. Human and yeast RPA were compared as substrates of DNA-PKcs employing a radioassay with purified proteins (see Materials and Methods). Bands corresponding to phosphorylated RPA are indicated, as well as the approximate positions of the unphosphorylated subunits. h, human; y, yeast; RPA1, human RPA large subunit; RPA2, human RPA middle subunit.
DISCUSSION
As the cellular ssDNA binding protein, RPA plays a central role in the DNA metabolism of eukaryotic cells. Therefore, it is possible that modulation of RPA through post-translational modification could be an important mechanism by which the relative replication, repair and recombination activities within the cell are regulated. In fact, evidence for such control has been provided by studies on the cell-free SV40 DNA replication system and has led to the hypothesis that RPA phosphorylated on the middle subunit in response to UV irradiation is preferentially active in DNA repair (10). We now demonstrate that the large subunit of yeast RPA is also a phosphoprotein, suggesting that more than one polypeptide of the RPA heterotrimer could be involved in DNA metabolic regulation.
Phosphorylation of Rfa1p requires Mec1p and is therefore a reaction in the S.cerevisiae checkpoint response. Unlike the Mec1p-dependent phosphorylation of Rfa2p that we previously reported, Rfa1p phosphorylation does not occur during normal cell cycle progression, but only under genotoxic conditions. Further examination into the genetic requirements of Rfa1p phosphorylation has revealed that the HU-induced reaction is also supported to some extent by Tel1p, another ATM homolog that is not a checkpoint protein but functionally overlaps with Mec1p. This Mec1p/Tel1p dependence is identical to that of HU-induced Rfa2p phosphorylation. In response to radiation, Rfa1p phosphorylation does not involve Tel1p, but does require several checkpoint proteins in addition to Mec1p. While a dependence on Mec3p was also previously observed for radiation-induced Rfa2p phosphorylation, the requirement for functional Rad9p and Rad53p is specific to the large subunit. These results indicate that RPA phosphorylation operates by different mechanisms despite the common requirement for a functional Mec1p checkpoint pathway (Table 2).
Table 2. Genetic requirements of RPA phosphorylation in yeast.
| Condition | Rfa1p phosphorylation | Rfa2p phosphorylation |
|---|---|---|
| Cell cycle |
– |
MEC1 |
| HU |
MEC1/TEL1 |
MEC1/TEL1 |
| Radiation | MEC1, MEC3, RAD9, RAD53 | MEC1, MEC3 |
The existence of different checkpoint-dependent mechanisms that direct phosphorylation of the two RPA subunits is supported by our studies employing a temperature-sensitive allele of CDC13. When this mutant is grown at the permissive temperature, it exhibits the expected cell-cycle-regulated Rfa2p phosphorylation and a slight degree of Rfa1p phosphorylation that is presumably due to a low level of telomeric damage resulting from the defective Cdc13p. In a cdc13 mec1-1 double mutant, both of these phosphorylation reactions are abolished. However, only phosphorylation of Rfa1p is eliminated in cdc13 mec1-3 at the permissive temperature, demonstrating that the Rfa1p and Rfa2p kinase activities can be separated (Fig. 4). Such differences in the behavior of the two RPA subunits are further apparent when cdc13 is combined with other checkpoint gene mutations. The cdc13 rad9 double mutant exhibits normal Rfa1p phosphorylation at the restrictive temperature, but a hyperphosphorylation of Rfa2p. In contrast, cdc13 rad24 exhibits normal Rfa2p phosphorylation, but greatly reduced phosphorylation of Rfa1p (Fig. 5A). Therefore, distinct Mec1p-dependent sub-pathways operate to direct Rfa1p and Rfa2p phosphorylation. The homology of Mec1p to DNA-PKcs and ATM would suggest that Mec1p is directly involved in catalyzing phosphorylation of the two RPA subunits, but further experimentation will be necessary to define the exact role that Mec1p plays in the two reactions.
Our analysis of mutants defective in CDC13 has provided some insight into the biochemistry of Rfa1p phosphorylation. These experiments suggest that ssDNA must be generated for the large subunit to become modified. We have observed that radiation-induced Rfa1p phosphorylation is more robust in cycling cells than in G1-arrested cells (data not shown) and it is possible that ssDNA generated during DNA replication in the proliferating population contributes to this difference in catalytic efficiency. It is interesting that DNA damage-induced phosphorylation of the RPA middle subunit in human cells, mediated by members of the ATM family, is greatly stimulated when DNA synthesis is allowed to proceed (44). Despite the correlation between Rfa1p phosphorylation and ssDNA formation, the absence of Rfa1p phosphorylation during normal cell cycle progression suggests that ssDNA alone is not sufficient to support the reaction but must be accompanied by genotoxic stress. One possibility is that Mec1p directly catalyzes both Rfa1p and Rfa2p phosphorylation in the presence of ssDNA, but Rfa1p phosphorylation requires a cofactor that is induced by DNA damage. It is also possible that Mec1p specifically catalyzes Rfa2p phosphorylation, but induces a second protein kinase in response to DNA damage that catalyzes Rfa1p phosphorylation in an ssDNA-dependent reaction. Consistent with either scenario, we have found that purified DNA-PKcs, a homolog of Mec1p, can catalyze Rfa2p but not Rfa1p phosphorylation in the presence of DNA containing single-stranded regions (Fig. 6).
Both DNA damage and ssDNA generation are required for induction of the SOS response, an adaptive process in prokaryotes that enhances cell survival upon environmental stress (45). An E.coli cell incapable of repairing a UV-generated DNA lesion cannot mount an SOS response upon UV treatment unless it is undergoing DNA replication and, therefore, generating ssDNA (46,47). In contrast, UV repair-proficient cells are capable of SOS induction in the absence of DNA replication because ssDNA is generated through processing of the lesion. A recent study in yeast has demonstrated that the UV-induced Mec1p-dependent phosphorylation of Rad53p, an important checkpoint regulator in S.cerevisiae (28,31,48), is dependent on DNA replication in mutants that are incapable of excising UV dimers (49). Therefore, induction of certain Mec1p-dependent activities, such as phosphorylation of Rad53p and Rfa1p, is remarkably similar to induction of the SOS response. Some members of the yeast DNA damage checkpoint apparatus are involved in UV-induced mutagenesis (50), which is an important process of the SOS response in bacterial cells. It will be interesting to determine whether checkpoint-dependent RPA phosphorylation is required for any processes that are characteristic of SOS adaptation.
The involvement of ssDNA in Rfa1p phosphorylation could explain the broader checkpoint gene requirements for radiation-induced than HU-induced Rfa1p phosphorylation. While HU-arrested yeast cells contain DNA double-strand breaks (51) and are likely to have extensive regions of unwound DNA due to replication fork stalling, radiation treatment only generates local stretches of ssDNA that are formed once the DNA damage is processed. It is possible that checkpoint proteins other than Mec1p are required in response to radiation to generate sufficient ssDNA template for Rfa1p phosphorylation to occur. Evidence that Mec3p, Rad9p and Rad24p regulate telomeric ssDNA formation upon Cdc13p inactivation by influencing the putative exonuclease activity of Rad17p supports this hypothesis (40). The physical association of the phosphorylated forms of Rad9p and Rad53p, which are both generated in a Mec1p-dependent manner (52–56), suggests that Rad53p could also be involved in ssDNA maturation. It should be noted that immunoprecipitates of Rad53p can catalyze protein phosphorylation (57). Therefore, it is conceivable that Rad53p is an Rfa1p kinase induced by Mec1p upon radiation exposure. However, a low level of radiation-induced Rfa1p phosphorylation is observed in the rad53 mutant (Fig. 3), arguing against this possibility. It should also be noted that the roles of checkpoint proteins will vary with cell cycle phase and the nature of the insult, as observed previously (1,58,59). In fact, the studies presented here provide another striking example of variance in function of Rad9p (50,60), which is involved in radiation-induced Rfa1p phosphorylation but is not required for Rfa1p phosphorylation induced by telomeric damage.
In addition to defining the mechanisms that control RPA phosphorylation, an important direction of our future experiments will be to identify Mec1p-dependent RPA phosphorylation sites so that mutation analyses ideally suited for studies in yeast can be performed. Generation of phosphorylation site mutants will allow for complete functional characterization of RPA phosphorylation in yeast and information gained from these studies is likely to be applicable to higher eukaryotes as well. Although none of the amino acids that become phosphorylated in yeast RPA have been reported, phosphopeptide mapping of the human RPA middle subunit has demonstrated that two serines near the N-terminus are phosphorylated in normal cycling cells and four additional serines and one threonine in this same region become phosphorylated upon UV irradiation (41). The studies presented here demonstrate that the RPA phosphoproteins generated during the normal cell cycle and in response to DNA damage also differ in yeast and indicate that the large subunit should be considered as a potential regulatory component of RPA.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs Ted Weinert, David Lydall and Heidi Feldmann for their generous gifts of yeast strains, and Dr Steven Brill for kindly providing plasmids and antibodies. We also thank Deborah Tien for her excellent technical assistance and Dr Grant Brown for critically reviewing the manuscript. This work was supported by grants from the National Institutes of Health.
REFERENCES
- 1.Elledge S.J. (1996) Science, 274, 1664–1672. [DOI] [PubMed] [Google Scholar]
- 2.Wold M.S. (1997) Annu. Rev. Biochem., 66, 61–92. [DOI] [PubMed] [Google Scholar]
- 3.He Z., Brinton,B.T., Greenblatt,J., Hassell,J.A. and Ingles,C.J. (1993) Cell, 73, 1223–1232. [DOI] [PubMed] [Google Scholar]
- 4.Dutta A., Ruppert,J.M., Aster,J.C. and Winchester,E. (1993) Nature, 365, 79–82. [DOI] [PubMed] [Google Scholar]
- 5.Longhese M.P., Neecke,H., Paciotti,V., Lucchini,G. and Plevani,P. (1996) Nucleic Acids Res., 24, 3533–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee S.E., Moore,J.K., Holmes,A., Umezu,K., Kolodner,R.D. and Haber,J.E. (1998) Cell, 94, 399–409. [DOI] [PubMed] [Google Scholar]
- 7.Parker A.E., Clyne,R.K., Carr,A.M. and Kelly,T.J. (1997) Mol. Cell. Biol., 17, 2381–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Din S., Brill,S.J., Fairman,M.P. and Stillman,B. (1990) Genes Dev., 4, 968–977. [DOI] [PubMed] [Google Scholar]
- 9.Liu V.F. and Weaver,D.T. (1993) Mol. Cell. Biol., 13, 7222–7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carty M.P., Zernik-Kobak,M., McGrath,S. and Dixon,K. (1994) EMBO J., 13, 2114–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brush G.S., Morrow,D.M., Hieter,P. and Kelly,T.J. (1996) Proc. Natl Acad. Sci. USA, 93, 15075–15080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fotedar R. and Roberts,J.M. (1992) EMBO J., 11, 2177–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brush G.S., Anderson,C.W. and Kelly,T.J. (1994) Proc. Natl Acad. Sci. USA, 91, 12520–12524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith G.C. and Jackson,S.P. (1999) Genes Dev., 13, 916–934. [DOI] [PubMed] [Google Scholar]
- 15.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC, pp. 662–668.
- 16.Savitsky K., Bar-Shira,A., Gilad,S., Rotman,G., Ziv,Y., Vanagaite,L., Tagle,D.A., Smith,S., Uziel,T., Sfez,S. et al. (1995) Science, 268, 1749–1753. [DOI] [PubMed] [Google Scholar]
- 17.Hartley K.O., Gell,D., Smith,G.C., Zhang,H., Divecha,N., Connelly,M.A., Admon,A., Lees-Miller,S.P., Anderson,C.W. and Jackson,S.P. (1995) Cell, 82, 849–856. [DOI] [PubMed] [Google Scholar]
- 18.Taylor A.M., Harnden,D.G., Arlett,C.F., Harcourt,S.A., Lehmann,A.R., Stevens,S. and Bridges,B.A. (1975) Nature, 258, 427–429. [DOI] [PubMed] [Google Scholar]
- 19.Painter R.B. and Young,B.R. (1980) Proc. Natl Acad. Sci. USA, 77, 7315–7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyn M.S. (1995) Cancer Res., 55, 5991–6001. [PubMed] [Google Scholar]
- 21.Kastan M.B., Zhan,Q., el-Deiry,W.S., Carrier,F., Jacks,T., Walsh,W.V., Plunkett,B.S., Vogelstein,B. and Fornace,A.J.,Jr (1992) Cell, 71, 587–597. [DOI] [PubMed] [Google Scholar]
- 22.Smith G.C., Cary,R.B., Lakin,N.D., Hann,B.C., Teo,S.H., Chen,D.J. and Jackson,S.P. (1999) Proc. Natl Acad. Sci. USA, 96, 11134–11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chan D.W., Son,S.C., Block,W., Ye,R., Khanna,K.K., Wold,M.S., Douglas,P., Goodarzi,A.A., Pelley,J., Taya,Y., Lavin,M.F. and Lees-Miller,S.P. (2000) J. Biol. Chem., 275, 7803–7810. [DOI] [PubMed] [Google Scholar]
- 24.Morgan S.E. and Kastan,M.B. (1997) Cancer Res., 57, 3386–3389. [PubMed] [Google Scholar]
- 25.Wang Y., Zhou,X.Y., Wang,H., Huq,M.S. and Iliakis,G. (1999) J. Biol. Chem., 274, 22060–22064. [DOI] [PubMed] [Google Scholar]
- 26.Liu J.S., Kuo,S.R., McHugh,M.M., Beerman,T.A. and Melendy,T. (2000) J. Biol. Chem., 275, 1391–1397. [DOI] [PubMed] [Google Scholar]
- 27.Park J., Park,S., Peng,X., Wang,M., Yu,M. and Lee,S. (1999) J. Biol. Chem., 274, 32520–32527. [DOI] [PubMed] [Google Scholar]
- 28.Weinert T.A., Kiser,G.L. and Hartwell,L.H. (1994) Genes Dev., 8, 652–665. [DOI] [PubMed] [Google Scholar]
- 29.Kato R. and Ogawa,H. (1994) Nucleic Acids Res., 22, 3104–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siede W., Allen,J.B., Elledge,S.J. and Friedberg,E.C. (1996) J. Bacteriol., 178, 5841–5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paulovich A.G. and Hartwell,L.H. (1995) Cell, 82, 841–847. [DOI] [PubMed] [Google Scholar]
- 32.Sikorski R.S. and Hieter,P. (1989) Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morrow D.M., Tagle,D.A., Shiloh,Y., Collins,F.S. and Hieter,P. (1995) Cell, 82, 831–840. [DOI] [PubMed] [Google Scholar]
- 34.Adams A., Gottschling,D.E., Kaiser,C.A. and Stearns,T. (1998) Methods in Yeast Genetics: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 35.Brill S.J. and Stillman,B. (1991) Genes Dev., 5, 1589–1600. [DOI] [PubMed] [Google Scholar]
- 36.Wold M.S. and Kelly,T. (1988) Proc. Natl Acad. Sci. USA, 85, 2523–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sung P. (1997) Genes Dev., 11, 1111–1121. [DOI] [PubMed] [Google Scholar]
- 38.Nugent C.I., Hughes,T.R., Lue,N.F. and Lundblad,V. (1996) Science, 274, 249–252. [DOI] [PubMed] [Google Scholar]
- 39.Garvik B., Carson,M. and Hartwell,L. (1995) Mol. Cell. Biol., 15, 6128–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lydall D. and Weinert,T. (1995) Science, 270, 1488–1491. [DOI] [PubMed] [Google Scholar]
- 41.Zernik-Kobak M., Vasunia,K., Connelly,M., Anderson,C.W. and Dixon,K. (1997) J. Biol. Chem., 272, 23896–23904. [DOI] [PubMed] [Google Scholar]
- 42.Lees-Miller S.P., Sakaguchi,K., Ullrich,S.J., Appella,E. and Anderson,C.W. (1992) Mol. Cell. Biol., 12, 5041–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hall-Jackson C.A., Cross,D.A., Morrice,N. and Smythe,C. (1999) Oncogene, 18, 6707–6713. [DOI] [PubMed] [Google Scholar]
- 44.Shao R.G., Cao,C.X., Zhang,H., Kohn,K.W., Wold,M.S. and Pommier,Y. (1999) EMBO J., 18, 1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC, pp. 407–464.
- 46.Salles B. and Defais,M. (1984) Mutat. Res., 131, 53–59. [DOI] [PubMed] [Google Scholar]
- 47.Sassanfar M. and Roberts,J.W. (1990) J. Mol. Biol., 212, 79–96. [DOI] [PubMed] [Google Scholar]
- 48.Allen J.B., Zhou,Z., Siede,W., Friedberg,E.C. and Elledge,S.J. (1994) Genes Dev., 8, 2401–2415. [DOI] [PubMed] [Google Scholar]
- 49.Neecke H., Lucchini,G. and Longhese,M.P. (1999) EMBO J., 18, 4485–4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paulovich A.G., Armour,C.D. and Hartwell,L.H. (1998) Genetics, 150, 75–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Merrill B.J. and Holm,C. (1999) Genetics, 153, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun Z., Fay,D.S., Marini,F., Foiani,M. and Stern,D.F. (1996) Genes Dev., 10, 395–406. [DOI] [PubMed] [Google Scholar]
- 53.Sanchez Y., Desany,B.A., Jones,W.J., Liu,Q., Wang,B. and Elledge,S.J. (1996) Science, 271, 357–360. [DOI] [PubMed] [Google Scholar]
- 54.Sun Z., Hsiao,J., Fay,D.S. and Stern,D.F. (1998) Science, 281, 272–274. [DOI] [PubMed] [Google Scholar]
- 55.Emili A. (1998) Mol. Cell, 2, 183–189. [DOI] [PubMed] [Google Scholar]
- 56.Vialard J.E., Gilbert,C.S., Green,C.M. and Lowndes,N.F. (1998) EMBO J., 17, 5679–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng P., Fay,D.S., Burton,J., Xiao,H., Pinkham,J.L. and Stern,D.F. (1993) Mol. Cell. Biol., 13, 5829–5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paulovich A.G., Toczyski,D.P. and Hartwell,L.H. (1997) Cell, 88, 315–321. [DOI] [PubMed] [Google Scholar]
- 59.Longhese M.P., Foiani,M., Muzi-Falconi,M., Lucchini,G. and Plevani,P. (1998) EMBO J., 17, 5525–5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paciotti V., Lucchini,G., Plevani,P. and Longhese,M.P. (1998) EMBO J., 17, 4199–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thomas B.J. and Rothstein,R. (1989) Cell, 56, 619–630. [DOI] [PubMed] [Google Scholar]