

Abstract
Biofluids contain molecules in circulation and from nearby organs that can be indicative of disease states. Characterizing the proteome of biofluids with DIA-MS is an emerging area of interest for biomarker discovery; yet, there is limited consensus on DIA-MS data analysis approaches for analyzing large numbers of biofluids. To evaluate various DIA-MS workflows, we collected urine from a clinically heterogeneous cohort of prostate cancer patients and acquired data in DDA and DIA scan modes. We then searched the DIA data against urine spectral libraries generated using common library generation approaches or a library-free method. We show that DIA-MS doubles the sample throughput compared to standard DDA-MS with minimal losses to peptide detection. We further demonstrate that using a sample-specific spectral library generated from individual urines maximizes peptide detection compared to a library-free approach, a pan-human library, or libraries generated from pooled, fractionated urines. Adding urine subproteomes, such as the urinary extracellular vesicular proteome, to the urine spectral library further improves the detection of prostate proteins in unfractionated urine. Altogether, we present an optimized DIA-MS workflow and provide several high-quality, comprehensive prostate cancer urine spectral libraries that can streamline future biomarker discovery studies of prostate cancer using DIA-MS.
Keywords: mass spectrometry, data-independent acquisition, data-dependent acquisition, prostate cancer, spectral library, urine
Introduction
Biofluids such as blood and urine contain proteins in circulation of, for example, genitourinary organs, and are ideal for biomarker discovery as they enable noninvasive identification and quantification of proteins that reflect varying disease states in human tissues.1,2 It is of great interest to enable rapid and deep proteomic profiling of biofluids in large-cohort studies consisting of hundreds to thousands of patients for statistical power and generalizability that facilitate biomarker discovery and validation.3 Mass spectrometry (MS)-based bottom-up proteomics is a powerful tool for characterizing the proteome and quantifying protein abundances.4−6 However, profiling large biofluid cohorts is challenging for mainly two reasons. First, the high dynamic range of protein concentrations in fluids can mask the detection of low-abundance proteins. Second, profiling large cohorts is challenging due to the low sample throughput of many currently used methods such as DDA-MS.7
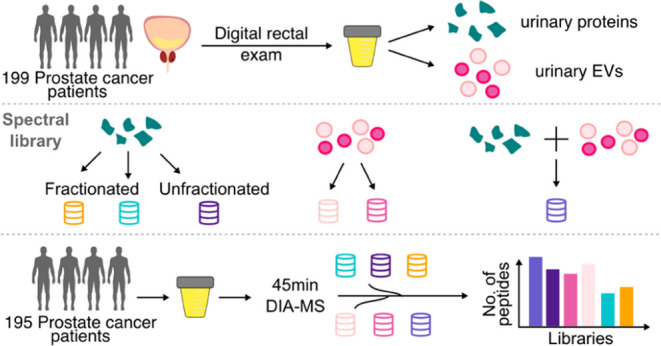
Data-independent acquisition (DIA)-MS is an emerging tool for characterizing proteomes for its ability to increase throughput and reproducibility,8−12 yet there is limited consensus on an ideal downstream data analysis approach for the highly convoluted data generated.13 To investigate the utility of DIA-MS workflows for clinical biofluid cohorts, we evaluated various commonly used DIA-MS data analysis approaches14−17 using a large clinically heterogeneous cohort of urines from 199 patients with prostate cancer. These patients spanned the risk spectrum, from clinical ISUP grade Group 1 disease (associated with better prognosis) to clinical ISUP grade Group 5 disease (associated with worse prognosis).18 In particular, we performed proteomics analysis on patient-derived expressed prostatic secretions (EPS)-urine.19 EPS-urine is a form of prostate proximal fluid collected following a digital rectal exam (DRE), which we and others have previously shown to be enriched in prostate-derived molecules.19,20 Enabling large-scale proteomics studies on EPS-urine with DIA-MS would be appealing for biomarker discovery efforts to answer unmet needs in prostate cancer.21
In this study, several published DIA-MS acquisition schemes10 were compared against a well-established DDA-MS workflow to determine an optimal method that optimizes sample throughput and peptide detection. We show that a sample-specific spectral library resulted in the highest number of peptides detected compared to a library-free approach16 and a pan-human library.15 Our group has previously shown that analysis of extracellular vesicles (EV) isolated from EPS-urine further increases the number of prostate proteins in EPS-urine.22 We leveraged these data sets to generate an enhanced urine spectral library that includes EV proteins. This enhanced spectral library further increased the detection of prostate proteins in unenriched urine without the need for laborious EV isolation. Furthermore, we highlighted the trade-offs between the various data analysis approaches to guide future research. Although generating a cohort-specific library is labor-intensive and often not feasible, it maximizes the detection depth across heterogeneous clinical cohorts to an extent that is not captured by pooled samples. Altogether, we have generated several comprehensive, high-quality EPS-urine spectral libraries as a resource for the community that can enable the in-depth detection of proteins in urine for future studies of prostatic disease.
Experimental Section
Human Subjects
Samples were obtained from patients following informed consent and use of Institutional Review Board-approved protocols at Eastern Virginia Medical School (EVMS, Norfolk, Virginia, IRB# 06-12-FB-0343) and the Research Ethics Review Board at the University Health Network (UHN, Toronto, Ontario, Canada, 10-0159 and 19-5009). Clinical details are listed in Table S1.
Urine Collection
First-catch urine (15 mL) was collected after digital rectal exam (EPS-urine) as previously described.19,20,23 EPS-urine was centrifuged at 2000g for 15 min at 4 °C to pellet cellular debris, and the resulting urine supernatant was stored at −80 °C.
Protein Digestion
EPS-urine was prepared for proteomics using the MStern protocol.24 For each sample, 2 pmol of Saccharomyces cerevisiae invertase (SUC2) (Sigma-Aldrich) was added as a sample processing control. Proteins in each sample were reduced with 5 mM dithiothreitol (DTT) (BioShop Canada) and incubated for 30 min at 60 °C, then alkylated with 25 mM iodoacetamide (IAA) (Sigma-Aldrich) followed by incubation at room temperature for 30 min in the dark. The MStern membrane (Millipore Sigma, MSIP4510) was equilibrated with 50 μL of 70% ethanol (Commercial Alcohols) and then washed twice with 100 mM ammonium bicarbonate (ABC) (BioShop Canada). 250 μL of EPS-urine was added to individual wells and passed through the membrane by vacuum suction. Each well was washed twice with 100 μL of ABC to remove salts, then proteins were digested with 1 μg of mass spectrometry-grade Trypsin/Lys-C enzyme mix (Promega) in 50 μL of digestion buffer (100 mM ABC, 1 mM CaCl2 (Bioshop Canada), 5% acetonitrile [ACN (Fisher Scientific)], pH 8.0). The digestion buffer was passed through the membrane by centrifugation, and the flow-through was reapplied on top of the membrane. Protein digestion was performed at 37 °C for 4 h. Samples were resuspended in each well by gentle pipetting every 2 h. Peptides were then collected by centrifugation, and membrane-bound peptides were eluted with 50 μL of 50% ACN and combined with the flow-through from the previous step. Samples were then dried in a SpeedVac vacuum concentrator (Thermo). Dried peptides were resuspended in 0.1% trifluoroacetic acid in water and desalted using in-house solid phase extraction stage tips containing 3 plugs of 3 M Empore C18 membrane.25 Peptides were reconstituted in 0.1% formic acid (FA) (Millipore Sigma) in water, and peptide concentration was determined by NanoDrop (Thermo).
Samples for DIA Method Evaluation
For the evaluation of DIA methods (Figure 1), 50 mL of EPS-urine from a single clinical ISUP grade Group 1 patient was used. The urine was prepared for proteomics using the MStern protocol described above. Desalted peptides were reconstituted in 0.1% FA in water and quantified by NanoDrop prior to mass spectrometry analysis.
Figure 1.

Schematic of the DIA-MS method evaluation of pooled EPS-urine.
Pooled Samples for Fractionated Spectral Libraries
For generating fractionated spectral libraries, 50 μL of EPS-urine from 159 patients were pooled and processed with the MStern protocol as described above. Peptides were reconstituted in 0.1% FA in water and quantified by NanoDrop prior to fractionation.
High-pH Reversed-Phase Fractionation
The high-pH reversed-phase fractionated urine spectral library was generated from the pooled urine described above (159 patients) using the Pierce high-pH reversed-phase fractionation kit (Thermo). 100 μg of peptides dissolved in 300 μL of 0.1% trifluoroacetic acid were loaded onto the spin column following column equilibration. Ten fractions were collected with 10 elution buffers of increasing concentrations of ACN (5, 7.5, 12.5, 15, 17.5, 20, 30, 50, and 80% ACN) in 0.1% triethylamine. All peptides were dried by SpeedVac and reconstituted in 0.1% FA in water. 2 μg of peptides per fraction was then analyzed by DDA-MS.
Liquid Chromatography
All LC-MS data were acquired using an EASY-nanoLC 1000 system (Thermo), equipped with 1.5 cm trap column (1.5 cm x 75 μm) (Thermo) and an EASY-Spray reversed-phase HPLC analytical column (500 mm x 75 μm inner diameter x 2 μm, C18 beads) (Thermo), coupled to a Q-Exactive HF mass spectrometer with an EASY-Spray ion source (Thermo). iRT peptides (Biognosys) were spiked into each sample prior to LC-MS analysis at 1:10 concentration prior to acquisition. 2 μg of peptides was used. Solvent A (0.1% FA in water), and Solvent B (0.1% FA in ACN) were used for liquid chromatography. For samples acquired by DDA-MS, peptides were eluted on a nonlinear gradient of 130 min, starting with 5% B for 5 min, 5 to 25% B over 100 min, then from 25 to 48% B in 10 min, followed by a wash step of 48 to 95% B for 4 min and 95% B for 10 min. For samples acquired by DIA-MS, chromatographic gradients were first optimized using single-patient pooled EPS-urine (Table S2). The main cohort of EPS-urines was acquired using the following DIA method (45 min with 16 m/z staggered windows).
Mass Spectrometry
For samples acquired with DDA-MS, MS1 and MS2 scans were acquired at a resolution of 60,000 and 17,500, respectively. Data was acquired in Top 15 mode with HCD fragmentation at NCE 27, maximum injection time of 110 ms, and dynamic exclusion 40 s. For samples acquired with DIA-MS, details of evaluated methods are documented in Table S2, with each method acquired in technical triplicates. The selected DIA-MS method was then used to acquire the entire EPS-urine cohort. The selected DIA-MS method acquires a full MS1 scan from 350–1800 m/z (60,000 resolution, 40 ms max IT, AGC 3 × 106), interspersed between every 38 MS2 scans of 30,000 resolution, 55 ms maxIT, and AGC target 1 × 106. Window sizes were optimized by Skyline.26
Gas-Phase Fractionation
The gas-phase fractionated EPS-urine spectral library was generated in DIA-MS mode as previously described.27 Peptides were reconstituted in 0.1% FA in water, and 2 μg of peptides were loaded on a column for each fraction. Six DIA-MS runs were acquired with a mass range of 100 m/z each, sequentially covering the overall mass range from 395 m/z to 1005 m/z (395–495, 495–605, 595–705 m/z, etc.). Each DIA-MS acquisition was acquired with two types of MS1 scans (60,000 resolution, AGC target 1 × 106, max IT 60 ms, 27 NCE), a wide range spectrum of 395–1000 m/z followed by a narrow range spectrum of 100 m/z corresponding to the mass range fraction of each acquisition, interspersed every 25 MS2 scans acquired using 4 m/z DIA overlapping windows (30,000 resolution, AGC target 1 × 106, max IT 60 ms, 27 NCE).
Spectral Library Generation
Spectral libraries were generated using FragPipe (v.18.1)16 with the preloaded workflow “DIA_SpecLib_Quant”. The platform included MSFragger28 (v.3.5), Philosopher29 (v.4.5.1-RC21), IonQuant30 (v.1.8.10), Python (v.3.9.7), EasyPQP (v.0.1.36), and DIA-NN31 (v.1.8.1). MS2 spectral matching was done in MSFragger against the human UniProt-SwissProt database (2020-06-02, 42,042 sequences from canonical and isoform). Database search was performed with the default settings (precursor mass tolerance = 20 ppm, enzyme = trypsin, missed cleavages = 2, minimum peaks = 15). Search results were then statistically validated through Philosopher and MSBooster.32 PSMs with FDR < 1% were processed in EasyPQP for the generation of consensus spectrum of confident peptides (RT loess fraction = 0.05, CiRT for RT alignment for gas-phase fractionation library). Individual spectral libraries were generated using the .raw files from three single-shot label-free DDA data sets, one from this study (uEPS, 199 urine samples) and two from previously published cohorts22 (uEV-P20:153 samples, uEV-P150:151 samples).
LC-MS/MS Data Analysis
DDA-MS .raw files were searched using the “LFQ-MBR” workflow with FragPipe (v.18.1). MSFragger search parameters were the same as above, with match-between runs (MBR) default settings. PSMs, peptides, and proteins with FDR < 1% were exported in the combined_modified_peptide.tsv, combined_peptide.tsv, combined_ion.tsv, and combined_protein.tsv. DDA-MS peptide intensities from the combined_peptide.tsv file were median normalized and then grouped to proteins using an in-house pipeline that utilizes the Abacus algorithm.33 Proteins mapped with only one peptide were removed from further analysis. Protein intensity was calculated using the iBAQ method.34
DIA-MS data was analyzed with the “DIA_SpecLib-Quant” workflow on FragPipe (v.18.1). Following conversion of the DIA-MS .raw files to mzML format using ProteoWizard MSConvert (v.3.0),17 DIA-MS.mzML files were searched with FragPipe. Data generated for method evaluation was searched with a library-free workflow,31 and other analyses were performed with either library-free or against spectral libraries from this study as specified in the Results and Discussion section. For the library-free workflow, the data was searched with MSFragger with the same parameters as indicated above, then quantified individually by DIA-NN (v.1.8.1) (quantification strategy = AnyLC (high precision), unrelated runs = TRUE). For searches performed with spectral libraries, MSFragger, Philosopher, and EasyPQP were disabled. Spectral libraries were assigned with the parameter “—lib library.tsv” for quantification in DIA-NN with the same quantification parameters as described above. Precursors and proteins with FDR < 1% were reported in the output tables diann-output.tsv files individually. Results from all runs were concatenated from individual diann-output.tsv files. Ion intensity values from the “Precursor.Normalised” column were summed per peptide sequence as peptide intensity. Peptide intensities were normalized by median normalization. Subsequent protein grouping and filtering were the same as above.
Data Quality Control
To assess the quality of our data, we considered the intensity and retention time of spike-in yeast protein SUC2 and synthetic iRT peptides, respectively. Any large shifts in the abundance of SUC2 peptides, large shifts in the retention time of iRT peptides (>10 min), or low peptide counts (<500 peptides per sample) may indicate a poor-quality sample. We did not identify any low-quality samples in our data. All samples were retained for further analysis.
Bootstrapping for Library Generation
Bootstrapped libraries were generated by randomly drawing (with replacement) a defined number of EPS-urine DDA-MS samples for library generation. The number of samples for library generation input was set at 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 40, 60, 80, 100, 120, 140, 160, 180, and 199. File names of the DDA samples were randomly drawn using Python (v.3.9) with Python package numpy (v.1.24.2) per sample increment for 25 iterations. Filenames selected for each step and each iteration were piped into FragPipe (v.18.1) for spectral library generation as described above. Spectral libraries with the median number of peptide entries per sample increment were then used for DIA-MS analysis with DIA-NN (v.1.8.1). Smaller cohorts were subsetted by selecting 10, 20, 50, 100, and 150 samples at random from the full cohort. Spectral libraries were generated by subsampling each cohort with 3 iterations each. The list of number of samples as input for library generation is listed in Table S2.
Bioinformatics and Statistical Analysis
Where appropriate, the quantitative analyses are described in the relevant sections of the Experimental Section. Unless stated otherwise, bioinformatic or statistical analyses and plotting were performed using R (v.4.3.0 and 4.2.1) and RStudio (v.2022.02.0 and v.2022.07.2 + 576). Data were visualized using R packages BoutrosLab.plotting.general (v.7.0.3), ggplot2 (v.3.4.0), ggbeeswarm (v.0.6.0), ggpubr (v.0.4.0), cowplot (v.1.1.1), eulerr (v.7.0.0), ComplexHeatmap (v.2.16.0), ggrepel (v.0.9.3), corrplot (v.0.92), and tidyverse (v.2.0.0). For all pairwise comparisons, unless stated otherwise, correlation coefficients were determined by Spearman’s method (cor.test), with P-values calculated by asymptotic t approximation using an Edgeworth series.
Pathway Analysis
Pathway analysis was performed in g:profiler (v.0.2.1)35,36 against Gene Ontology: Cellular Component gene sets. Over-represented, significant terms (FDR < 0.05) from library-unique gene sets from Figure 4 were visualized in Cytoscape (v.3.9.1) using the EnrichmentMap app (v.3.3.4; Jaccard overlap combined index: 0.375) and annotated with the AutoAnnotate (v.1.3.5) and clusterMaker App (v.2.2; cluster algorithm: MCL Cluster).
Figure 4.

Evaluation of sample-relevant spectral libraries for data analysis of unfractionated EPS-urine DIA-MS data. (A) Schematic of spectral library generation from the aforementioned EPS-urine samples, and the two populations of urinary extracellular vesicles (uEV) isolated by ultracentrifugation at 20,000g (lib-EVP20) and 150,000g (lib-EVP150).22 (B) Library size and (C) intersection of proteins of the generated spectral libraries. (D) Schematic of DIA-MS data analysis using each of the generated libraries. (E) Number of detected proteins and (F) intersection of proteins detected in EPS-urine data using each spectral library. (G) Percent observations of the uniquely detected proteins using lib-EVP20 in the DDA-MS data.22 (H) Pathway analysis of genes uniquely detected by each spectral library. (I) Number of uniquely detected proteins mapped against prostate cancer tissue data sets.43,44
Results and Discussion
Evaluation of DIA-MS Method for Rapid EPS-Urine Analysis
To determine a suitable DIA-MS method that increases sample throughput and peptide detection, we evaluated various published methods10 in technical triplicate against a standard DDA-MS method with a 120 min chromatographic gradient using a pooled EPS-urine sample. Parameters such as window placements (variable, staggered, and static), window sizes, mass range, and chromatographic gradients were considered (Figure 1). Resulting peptide and protein detection rates, number of points across peak elution, and coefficient of variation (CV) were ranked per method tested (Table S2). Between the chromatographic gradients and methods tested, the 45 min method with 16 m/z staggered windows (45 min–1) resulted in increased detection rates with comparable CVs as the standard DDA-MS method (Table 1). We decided that the 45 min method would be suitable for doubling of sample throughput without compromising proteome coverage when compared to a standard DDA-MS.
Table 1. Parameters of the Best DIA-MS Method Per Chromatographic Gradient Tested.
| method ID | gradient (min) | window size (m/z) | placement | mass range (m/z) | cycle time (s) | points per peak | peptide count | peptide CV | protein count | protein CV |
|---|---|---|---|---|---|---|---|---|---|---|
| 120 min–1 | 120 | 16 | staggered | 450–950 | 2.91 | 5.14 (±3.42) | 10,590 (±218.09) | 15.42 (±20.78) | 1678 (±10.44) | 14.94 (±25.24) |
| 120 min–11 | 120 | 1.4 | DDA | 350–1800 | 1.2 | 7486 (±187.09) | 16.05 (±22.27) | 1323 (±24.33) | 19.36 (±25.43) | |
| 60 min–1 | 60 | 16 | staggered | 450–950 | 2.91 | 3.86 (±2.16) | 9053 (±114.9) | 17.6 (±20.92) | 1452 (±12.12) | 17.52 (±23.68) |
| 45 min–1 | 45 | 16 | staggered | 400–1000 | 3.37 | 3.56 (±1.76) | 7951 (±54.01) | 22.52 (±23.55) | 1337 (±19.63) | 22.75 (±28.1) |
| 30 min–1 | 30 | 24 | staggered | 450–950 | 1.95 | 3.88 (±2.06) | 6091 (±18.33) | 17.87 (±22.53) | 1125 (±10.6) | 18.13 (±27.6) |
Comparisons of DIA Data Analysis Strategies
To evaluate the performance of spectral libraries generated from commonly used approaches, we generated three spectral libraries from EPS-urine: two libraries from a pooled sample (159 patients) that was fractionated by high-pH reverse-phase fractionation14 (lib-HighpH) or gas-phase fractionation17 (lib-GPF), and a third library from individual EPS-urines (lib-IS; 199 patients) (Figure 2A). All EPS-urines used for generating the spectral libraries were from patients with prostate cancer that spanned the risk spectrum of the localized disease (Table S1). Of the three EPS-urine libraries, the library generated from single-shot DDA of 199 individual EPS-urines (lib-IS) was the largest (68,208 peptides, 5238 proteins), with 39,642 peptides (58.11%) unique to lib-IS (Figure S1A). In line with other studies, spectral libraries generated from individually analyzed samples better characterize the proteome of a diverse cohort than libraries generated from extensive fractionation of pooled samples.37,38
Figure 2.

Evaluation of commonly used DIA-MS data analysis approaches for EPS-urines. (A) Schematic of the sample-specific spectral libraries. (B) Library size of the generated sample-specific spectral libraries. (C) Schematic of DIA-MS data analysis using the generated libraries, DPHLv2,15 and library-free approaches.16 (D) Number of peptides detected in more than 10% of samples in total (top) and per sample (bottom). (E) Number of peptides of the generated libraries from subsampling of the DDA-MS EPS-urine cohort. (F) Percent of peptides detected in DIA-MS data using the subsampled spectral libraries.
Next, to benchmark the performance of these libraries, we acquired DIA-MS data for 195 EPS-urines and searched the data against each of the EPS-urine spectral libraries (Figure 2C). We also compared our EPS-urine libraries to a published pan-human library (DPHLv2) that includes various tissues and biofluids,15 and a commonly used library-free approach that uses MSFragger and DIA-NN.16 Between the search approaches, DPHLv2 detected the most peptides (68,428) (Figure S1B), and the highest number of unique peptides (19,880) (Figure S1C). However, when considering only peptides that are reproducibly detected in more than 10% of samples, only 32.9% of peptides detected by DPHLv2 were reproducibly detected. In contrast, lib-IS resulted in the highest number of urine peptides that were reproducibly detected, closely followed by the library-free approach (Figure 2D). The lib-GPF and lib-HighpH libraries resulted in the least number of uniquely detected peptides.
These observations suggest that while repository-scale libraries (e.g., DPHLv2, pan-human library, etc.) increase detection rates, many peptides are not consistently detected across samples. This was also observed in Muntel et al.,39 where authors showed that for urines, using sample type-specific libraries resulted in more reproducible peptide detection and quantification compared to a pan-human library. This is likely due to matching against a large fraction of peptides in the library that are not present in the sample, introducing false positives and challenging error-rate control.40 Similar to other studies,38,39 among the different approaches for generating sample-specific libraries, the library using individually analyzed samples (lib-IS) can maximize detection rates compared to the libraries using pooled samples, likely by increasing the detection of patient-specific proteins that can be masked by pooling. Thus, we continued to investigate the use of lib-IS for the EPS-urine analysis.
However, generating such a library from large clinical cohorts is time- and resource-intensive, and access to large clinical cohorts may be limited. To investigate the influence of library size on peptide detection, we generated spectral libraries by subsampling the EPS-urine DDA-MS samples with bootstrapping (Experimental Section) and evaluated the performance of these libraries using our DIA-MS cohort. Both the library size and detection rates increased with the number of samples used for library generation (Figure 2E,F). Peptide detection reached 81.3% when the DIA-MS data was matched against a library generated from 60 samples (∼1/3 of the cohort). Matching against smaller libraries resulted in biases toward more frequently detected and slightly more abundant peptides (Figure S1D–E). To investigate the influence of library size on other cohort sizes of urine samples, we subset our cohort into smaller sizes and generated subsampled spectral libraries. We observed that smaller cohorts (n = 10, 20) require a larger fraction (60%) of the samples for library generation to achieve ∼80% peptide coverage (Figure S1F). This suggests for smaller urine cohorts (n ∼ 20), proteome coverage will benefit from the use of a spectral library generated by DDA analysis of the full cohort. On the contrary, generating a spectral library from at least one-third of the cohort in larger cohorts (n > 50) would provide a balance between depth of detection and instrument time. These observations illustrate that it is less laborious generating spectral libraries using a smaller subset of samples, but searching against smaller libraries results in lower proteome coverage. Here, we show that lib-IS can maximize the detection of urinary peptides compared to pan-human and fractionated library approaches.
Matched Cohort Comparisons of DIA and DDA
Many studies have extensively benchmarked DIA-MS against DDA-MS,8,37,38,41,42 but many of them are limited to cell lines or small patient cohorts with reduced biological or interindividual heterogeneity. To investigate how data acquired by DIA- or DDA-MS differ in a large clinical heterogeneous cohort, we compared the number of proteins detected in EPS-urines (195 patients) that were acquired by DIA-MS and DDA-MS. Overall, more peptides and proteins were detected by DIA-MS than by DDA-MS (Figures 3A and S2A), adding an additional 17.3 to 57.3% of proteins per patient (Figure 3B). Of the 3449 proteins observed in both DIA- and DDA-MS, 3109 were detected more consistently by DIA-MS (Figure 3C). This phenomenon was more pronounced for lower-abundance proteins, as quantified by DDA-MS (Figure S2B). Median protein intensities across samples and per-sample protein intensities were highly correlated between both scan modes (Figures 3D and S2C). We show that our 45 min DIA-MS method outperforms our 120 min DDA-MS method by increasing peptide and protein detections per sample and reducing missing values while maintaining high quantitative correlation with DDA-MS.
Figure 3.

Comparing matched EPS-urine samples acquired by DDA- or DIA-MS. (A) Number of proteins detected in matched samples (Wilcoxon’s signed-rank test). (B) Percentage of proteins unique to DDA-MS (orange), DIA-MS (blue), or shared (gray) per patient. (C) Frequency of detection for each protein between DDA- and DIA-MS. Spearman’s correlation and its P-value. (D) Spearman’s correlation of the median log2 protein intensities of shared proteins between the scan modes.
Increasing Proteome Coverage Using Sample-Relevant Libraries
We have shown that DIA-MS can improve protein detections, but lower-abundance and less frequently detected prostate-derived proteins may be missed in spectral libraries derived from unfractionated urine (lib-IS) due to the high dynamic range of the urinary proteome. Our group has previously shown that more prostate proteins can be detected in urinary extracellular vesicles (uEV) isolated from EPS-urine compared to unfractionated EPS-urine.22 uEVs were isolated by differential ultracentrifugation at 20,000g and 150,000g. However, since uEV isolation is time- and resource-intensive, we sought to determine if uEV proteins can be detected in unfractionated EPS-urine via DIA-MS data utilizing spectral libraries of published uEV data sets (Figure 4A). Of the three urine-derived spectral libraries, the lib-EVP20 was the largest (93,800 peptides, 7649 proteins), whereas the size of the lib-EVP150 (64,663 peptides, 5462 proteins) was similar to that of lib-IS (Figure 4B). The EV libraries also covered an additional 3628 proteins compared to lib-IS (Figure 4C), providing a more in-depth prostate cancer urinary proteome.
We then matched the EPS-urine DIA-MS data against the two EV libraries and compared them to the lib-IS results to evaluate whether protein detection in unfractionated EPS-urine can be improved (Figure 4D). We observed that the lib-IS resulted in the highest number of proteins and peptides detected (Figure S3A, Figure 4E), but the majority of proteins (71.9%, 2869) were also detected by the EV libraries (Figure 4F). On the contrary, searching against the EV libraries resulted in the detection of an additional 761 proteins. These results indicate that querying the unfractionated EPS-urine data against libraries generated from EV proteomes can increase protein detection without the need for EV isolation.
To determine if the additional proteins were EV-derived, we compared the frequency of detection of the uniquely detected proteins to their corresponding fractions in the aforementioned DDA-MS data set.22 Proteins unique to lib-EVP20 were more frequently detected in the uEV-P20 fraction of EPS-urine in DDA data (Figure 4G). As expected, proteins uniquely detected by each of the EV libraries were also more frequently detected in the corresponding urinary fraction by DDA-MS (Figure S3C–E), indicating that they are likely EV-derived proteins. Compared to the lib-IS unique proteins detected, the uEV-library-unique proteins were enriched in pathways related to extracellular vesicles and intracellular trafficking, while EPS-urine-unique proteins were enriched in pathways related to the plasma membrane and extracellular space (Figure 4H). To investigate whether these unique proteins are prostate-derived, we compared them to two published prostate cancer tissue data sets.43,44 Of the uniquely detected proteins by the EV libraries, 548 were found in tissues, suggesting that the use of the EV libraries can better represent the prostate proteome in urine (Figure 4I). These observations illustrate that using sample-relevant subproteomes enables the detection of prostate- and EV-derived proteins that would otherwise be missed in unfractionated EPS-urine.
Combined EPS-Urine Library for Comprehensive Peptide Detection
We have shown that the protein coverage of unenriched EPS-urine can be further maximized by searching against EV spectral libraries. To provide an extensive spectral library specific to EPS-urine, we generated an enhanced prostate cancer urine spectral library using all of the described EPS-urine and EV samples (Figure 5A). To demonstrate the performance of the lib-EPS against the other search approaches, we compared the search results against the DPHLv2 library and library-free approach described above. The lib-EPS detected the highest number of urine peptides in total (68,794) (Figure S4A) and per sample (median 13,671) compared to the DPHLv2 library and library-free approach (Figure 5B). Consistent with the observations above, lib-EPS resulted in the highest number of reproducibly detected urine peptides (Figure S4B). Although the number of proteins detected was comparable between the search approaches, lib-EPS improved protein sequence coverage significantly and thus improved confidence of the protein detection (Figure 5C). These observations illustrate that the lib-EPS library can maximize peptide detection in EPS-urines compared to a publicly available resource library and the library-free. Here, we present an extensive resource library specific to the EPS-urine proteome that can allow scalable biomarker discovery of prostate cancer using hundreds of samples with DIA-MS.
Figure 5.

Evaluation of the combined EPS-urine library. (A) Schematic of generation of a combined EPS-urine and EVs spectral library. (B) Number of peptides detected using lib-EPS, library-free, and DPHLv2. (C) Protein sequence coverage of proteins reported by FragPipe with protein group Global Q Value <0.01.
While DIA-MS has advantages for biomarker discovery, there remains no consensus as to how to best select a data analysis approach. When applying DIA-MS to large cohorts, sample processing time, computational runtime, and resulting proteome coverage are important factors to consider, and the ideal search strategy would maximize all of these factors (Table 2). For example, although a library-free approach does not require de novo spectral library generation, it requires almost 10 times the computational runtime compared to that of a spectral library approach (Table S2). On the other hand, DPHLv2 is the largest publicly available spectral library for analysis of human samples, but it resulted in less peptides detected in EPS-urine compared to our prostate urine library that is one-third its size. Furthermore, spectral libraries generated from pooled samples with commonly used fractionation protocols is a viable option when the sample amount is limited but did not result in high proteome coverage in our cohort. Given these trade-offs, our study delivers a comprehensive prostate urine library (lib-EPS) that contains spectral data from unfractionated EPS-urines and urine-derived EVs to increase the detection of prostate proteins in unfractionated EPS-urines acquired by DIA-MS. The generated lib-EPS reduces the overhead time and resources for spectral library generation for future studies applying DIA-MS on EPS-urines and facilitates the expansion of large-scale studies for prostate cancer biomarker discovery.
Table 2. Summary of the Pros and Cons for the DIA-MS Data Analysis Approaches Evaluated (+: Least Advantageous, ++: Moderately Advantageous, +++: the Most Advantageous).
| approach | sample processing time | computational runtime | depth of detection | data consistency |
|---|---|---|---|---|
| cohort library (lib-IS, lib-EPS) | + | +++ | +++ | +++ |
| subset cohort library | ++ | +++ | ++ | ++ |
| pooled sample library (lib-GPF, lib-HighpH) | ++ | +++ | + | +++ |
| library-free | +++ | + | ++ | +++ |
| publicly available resource library (DPHLv2) | +++ | ++ | +++ | + |
Conclusions
DIA-MS has the potential to enable large-scale clinical proteomics studies of hundreds of patient urine samples for the study of prostatic diseases such as prostate cancer. Here, we evaluated commonly used DIA-MS data analysis workflows and generated several sample-specific EPS-urine libraries from a large, diverse cohort of prostate cancer patients. We showed that sample-specific libraries generated from large numbers of individuals (lib-IS) resulted in the highest and most reproducible peptides detected. By adding data acquired from previously isolated urinary EVs22 to our EPS-urine library (lib-IS), we were able to further increase the number and sequence coverage of proteins that can be detected in unfractionated EPS-urine. We also showed that at least 60 patient samples were required for library generation to achieve moderate proteome coverage, in which the diversity and amounts of samples can be difficult to collect clinically. To enable future large-cohort studies that use EPS-urines for prostate cancer research, we present a comprehensive prostate fluid library (lib-EPS) generated from 503 EPS-urines and urine-derived EVs consisting of 115,801 peptide sequences (8151 proteins). The generated library can maximize peptide detection in EPS-urines and enable expansion of more reproducible proteomics analysis for urine prostate cancer data.
Acknowledgments
This study was supported by the National Institutes of Health through awards U01CA214194 and U2CCA271894 to O.J.S., P.C.B., and T.K. This study was supported by Canadian Institutes of Health Research Project Grants to T.K. (PJT156357) and S.K.L. (PJT162384). T.K. was supported through the Canadian Research Chair program. T.K., S.K.L., and D.V. also received support through a Prostate Cancer Canada Discovery Grant (D2019-2113). A.H. was supported by an MBP Excellence OSTOF award, a Paul Starita Graduate Student Fellowship, and a SCACE Graduate Fellowship in Prostate Cancer Research. A.K. was supported by an Ontario Graduate Scholarship and Ontario Student Opportunity Trust Fund Awards. The authors thank Dr. Lydia Liu and Meinusha Govindarajan for technical assistance.
Glossary
Abbreviations
- EPS:
expressed prostatic secretion
- DRE:
digital rectal exam
- MS:
mass spectrometry
- LC-MS/MS:
liquid chromatography-coupled tandem mass spectrometry
- AGC:
automatic gain control
- DDA:
data-dependent acquisition
- DIA:
data-independent acquisition
- EV:
extracellular vesicles
- CV:
coefficient of variation
- NCE:
normalized collision energy
- maxIT:
maximum injection time
- ISUP:
International Society of Urological Pathology
Data Availability Statement
Raw mass spectrometry data, generated spectral libraries, and processed proteomics data are publicly available on MassIVE database with MassIVE ID: MSV000093759 and FTP link: ftp://msv000093759/.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.4c00009.
Details of generated spectral libraries and search results (Figure S1), additional comparisons of DDA and DIA in matched samples (Figure S2), detection results of uEV spectral library searches (Figure S3), and detection results of the lib-EPS spectral library searches (Figure S4) (PDF)
Clinical information of EPS-urine cohort (Table S1) (XLSX)
Optimization parameters for all DIA methods tested (Table S2) (XLSX)
Author Contributions
● A.H. and A.K. contributed equally to this work. A.H., A.K., S.K., and M.W. contributed to data acquisition. J.O.N., O.J.S., D.V., and S.K.L. collected and provided samples. A.H. and A.K. acquired the mass spectrometry data. A.H., A.K., and V.I. analyzed the data. S.K.L., P.C.B., and T.K. supervised the study. A.H., A.K., and T.K. wrote the manuscript with input from all other authors.
The authors declare the following competing financial interest(s): P.C.B. sits on the Scientific Advisory Boards of Sage Bionetworks, Intersect Diagnostics Inc. and BioSymetrics Inc. All other authors have no conflicts of interest to declare.
Special Issue
Published as part of Journal of Proteome Researchvirtual special issue “Canadian Proteomics”.
Supplementary Material
References
- Crocetto F.; Russo G.; Di Zazzo E.; Pisapia P.; Mirto B. F.; Palmieri A.; Pepe F.; Bellevicine C.; Russo A.; La Civita E.; Terracciano D.; Malapelle U.; Troncone G.; Barone B. Liquid Biopsy in Prostate Cancer Management—Current Challenges and Future Perspectives. Cancers 2022, 14 (13), 3272. 10.3390/cancers14133272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayon L.; Cominetti O.; Affolter M. Proteomics of Human Biological Fluids for Biomarker Discoveries: Technical Advances and Recent Applications. Expert Rev. Proteomics 2022, 19 (2), 131–151. 10.1080/14789450.2022.2070477. [DOI] [PubMed] [Google Scholar]
- Nakayasu E. S.; Gritsenko M.; Piehowski P. D.; Gao Y.; Orton D. J.; Schepmoes A. A.; Fillmore T. L.; Frohnert B. I.; Rewers M.; Krischer J. P.; Ansong C.; Suchy-Dicey A. M.; Evans-Molina C.; Qian W.-J.; Webb-Robertson B.-J. M.; Metz T. O. Tutorial: Best Practices and Considerations for Mass-Spectrometry-Based Protein Biomarker Discovery and Validation. Nat. Protoc. 2021, 16 (8), 3737–3760. 10.1038/s41596-021-00566-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson T.; Mann M.; Aebersold R.; Yates J. R.; Bairoch A.; Bergeron J. J. M. Mass Spectrometry in High-Throughput Proteomics: Ready for the Big Time. Nat. Methods 2010, 7 (9), 681–685. 10.1038/nmeth0910-681. [DOI] [PubMed] [Google Scholar]
- Kowalczyk T.; Ciborowski M.; Kisluk J.; Kretowski A.; Barbas C. Mass Spectrometry Based Proteomics and Metabolomics in Personalized Oncology. Biochim. Biophys. Acta, Mol. Basis Dis. 2020, 1866 (5), 165690 10.1016/j.bbadis.2020.165690. [DOI] [PubMed] [Google Scholar]
- Macklin A.; Khan S.; Kislinger T. Recent Advances in Mass Spectrometry Based Clinical Proteomics: Applications to Cancer Research. Clin. Proteomics 2020, 17, 17 10.1186/s12014-020-09283-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajic T.; Liu Y.; Aebersold R. Using Data-independent, High-resolution Mass Spectrometry in Protein Biomarker Research: Perspectives and Clinical Applications. Proteomics Clin Appl. 2015, 9 (3–4), 307–321. 10.1002/prca.201400117. [DOI] [PubMed] [Google Scholar]
- Bruderer R.; Bernhardt O. M.; Gandhi T.; Xuan Y.; Sondermann J.; Schmidt M.; Gomez-Varela D.; Reiter L. Optimization of Experimental Parameters in Data-Independent Mass Spectrometry Significantly Increases Depth and Reproducibility of Results. Mol. Cell. Proteomics 2017, 16 (12), 2296–2309. 10.1074/mcp.RA117.000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T.; Aebersold R. Recent Advances of Data-Independent Acquisition Mass Spectrometry-Based Proteomics. Proteomics 2023, 23 (7–8), e2200011 10.1002/pmic.202200011. [DOI] [PubMed] [Google Scholar]
- Ludwig C.; Gillet L.; Rosenberger G.; Amon S.; Collins B. C.; Aebersold R. Data-Independent Acquisition-Based SWATH-MS for Quantitative Proteomics: A Tutorial. Mol. Syst. Biol. 2018, 14 (8), e8126 10.15252/msb.20178126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Ge W.; Ruan G.; Cai X.; Guo T. Data-Independent Acquisition Mass Spectrometry-Based Proteomics and Software Tools: A Glimpse in 2020. Proteomics 2020, 20 (17–18), e1900276 10.1002/pmic.201900276. [DOI] [PubMed] [Google Scholar]
- Venable J. D.; Dong M.-Q.; Wohlschlegel J.; Dillin A.; Yates J. R. Automated Approach for Quantitative Analysis of Complex Peptide Mixtures from Tandem Mass Spectra. Nat. Methods 2004, 1 (1), 39–45. 10.1038/nmeth705. [DOI] [PubMed] [Google Scholar]
- Kitata R. B.; Yang J.; Chen Y. Advances in Data-independent Acquisition Mass Spectrometry towards Comprehensive Digital Proteome Landscape. Mass Spectrom. Rev. 2023, 42 (6), 2324–2348. 10.1002/mas.21781. [DOI] [PubMed] [Google Scholar]
- Batth T. S.; Francavilla C.; Olsen J. V. Off-Line High-pH Reversed-Phase Fractionation for In-Depth Phosphoproteomics. J. Proteome Res. 2014, 13 (12), 6176–6186. 10.1021/pr500893m. [DOI] [PubMed] [Google Scholar]
- Xue Z.; Zhu T.; Zhang F.; Zhang C.; Xiang N.; Qian L.; Yi X.; Sun Y.; Liu W.; Cai X.; Wang L.; Dai X.; Yue L.; Li L.; Pham T. V.; Piersma S. R.; Xiao Q.; Luo M.; Lu C.; Zhu J.; Zhao Y.; Wang G.; Xiao J.; Liu T.; Liu Z.; He Y.; Wu Q.; Gong T.; Zhu J.; Zheng Z.; Ye J.; Li Y.; Jimenez C. R.; A J.; Guo T. DPHL v.2: An Updated and Comprehensive DIA Pan-Human Assay Library for Quantifying More than 14,000 Proteins. Patterns 2023, 4 (7), 100792 10.1016/j.patter.2023.100792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F.; Teo G. C.; Kong A. T.; Fröhlich K.; Li G. X.; Demichev V.; Nesvizhskii A. I. Analysis of DIA Proteomics Data Using MSFragger-DIA and FragPipe Computational Platform. Nat. Commun. 2023, 14 (1), 4154 10.1038/s41467-023-39869-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle B. C.; Pino L. K.; Egertson J. D.; Ting Y. S.; Lawrence R. T.; MacLean B. X.; Villén J.; MacCoss M. J. Chromatogram Libraries Improve Peptide Detection and Quantification by Data Independent Acquisition Mass Spectrometry. Nat. Commun. 2018, 9 (1), 5128 10.1038/s41467-018-07454-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein J. I.; Egevad L.; Amin M. B.; Delahunt B.; Srigley J. R.; Humphrey P. A.; The 2014 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Carcinoma: Definition of Grading Patterns and Proposal for a New Grading System. Am. J. Surg. Pathol. 2016, 40 (2), 244–252. 10.1097/PAS.0000000000000530. [DOI] [PubMed] [Google Scholar]
- Drake R. R.; White K. Y.; Fuller T. W.; Igwe E.; Clements M. A.; Nyalwidhe J. O.; Given R. W.; Lance R. S.; Semmes O. J. Clinical Collection and Protein Properties of Expressed Prostatic Secretions as a Source for Biomarkers of Prostatic Disease. J. Proteomics 2009, 72 (6), 907–917. 10.1016/j.jprot.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Principe S.; Kim Y.; Fontana S.; Ignatchenko V.; Nyalwidhe J. O.; Lance R. S.; Troyer D. A.; Alessandro R.; Semmes O. J.; Kislinger T.; Drake R. R.; Medin J. A. Identification of Prostate-Enriched Proteins by in-Depth Proteomic Analyses of Expressed Prostatic Secretions in Urine. J. Proteome Res. 2012, 11 (4), 2386–2396. 10.1021/pr2011236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo A.; Liu L. Y.; Nyalwidhe J. O.; Semmes O. J.; Vesprini D.; Downes M. R.; Boutros P. C.; Liu S. K.; Kislinger T. Proteomic Discovery of Non-Invasive Biomarkers of Localized Prostate Cancer Using Mass Spectrometry. Nat. Rev. Urol. 2021, 18 (12), 707–724. 10.1038/s41585-021-00500-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo A.; Govindarajan M.; Qiu Z.; Liu L. Y.; Ignatchenko V.; Waas M.; Macklin A.; Keszei A.; Main B. P.; Yang L.; Lance R. S.; Downes M. R.; Semmes O. J.; Vesprini D.; Liu S. K.; Nyalwidhe J. O.; Boutros P. C.; Kislinger T. Prostate Cancer Reshapes the Secreted and Extracellular Vesicle Urinary Proteomes. BioRxiv 2023, 2023.07.23.550214 10.1101/2023.07.23.550214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Principe S.; Jones E. E.; Kim Y.; Sinha A.; Nyalwidhe J. O.; Brooks J.; Semmes O. J.; Troyer D. A.; Lance R. S.; Kislinger T.; Drake R. R. In-Depth Proteomic Analyses of Exosomes Isolated from Expressed Prostatic Secretions in Urine. Proteomics 2013, 13 (10–11), 1667–1671. 10.1002/pmic.201200561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger S. T.; Ahmed S.; Muntel J.; Cuevas Polo N.; Bachur R.; Kentsis A.; Steen J.; Steen H. MStern Blotting-High Throughput Polyvinylidene Fluoride (PVDF) Membrane-Based Proteomic Sample Preparation for 96-Well Plates. Mol. Cell. Proteomics 2015, 14 (10), 2814–2823. 10.1074/mcp.O115.049650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak N. A.; Pichler G.; Paron I.; Nagaraj N.; Mann M. Minimal, Encapsulated Proteomic-Sample Processing Applied to Copy-Number Estimation in Eukaryotic Cells. Nat. Methods 2014, 11 (3), 319–324. 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- MacLean B.; Tomazela D. M.; Shulman N.; Chambers M.; Finney G. L.; Frewen B.; Kern R.; Tabb D. L.; Liebler D. C.; MacCoss M. J. Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinformatics 2010, 26 (7), 966–968. 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pino L. K.; Just S. C.; MacCoss M. J.; Searle B. C. Acquiring and Analyzing Data Independent Acquisition Proteomics Experiments without Spectrum Libraries. Mol. Cell. Proteomics 2020, 19 (7), 1088–1103. 10.1074/mcp.P119.001913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A. T.; Leprevost F. V.; Avtonomov D. M.; Mellacheruvu D.; Nesvizhskii A. I. MSFragger: Ultrafast and Comprehensive Peptide Identification in Mass Spectrometry-Based Proteomics. Nat. Methods 2017, 14 (5), 513–520. 10.1038/nmeth.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Veiga Leprevost F.; Haynes S. E.; Avtonomov D. M.; Chang H.-Y.; Shanmugam A. K.; Mellacheruvu D.; Kong A. T.; Nesvizhskii A. I. Philosopher: A Versatile Toolkit for Shotgun Proteomics Data Analysis. Nat. Methods 2020, 17 (9), 869–870. 10.1038/s41592-020-0912-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F.; Haynes S. E.; Nesvizhskii A. I. IonQuant Enables Accurate and Sensitive Label-Free Quantification With FDR-Controlled Match-Between-Runs. Mol. Cell. Proteomics 2021, 20, 100077 10.1016/j.mcpro.2021.100077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demichev V.; Messner C. B.; Vernardis S. I.; Lilley K. S.; Ralser M. DIA-NN: Neural Networks and Interference Correction Enable Deep Proteome Coverage in High Throughput. Nat. Methods 2020, 17 (1), 41–44. 10.1038/s41592-019-0638-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K. L.; Yu F.; Teo G. C.; Li K.; Demichev V.; Ralser M.; Nesvizhskii A. I. MSBooster: Improving Peptide Identification Rates Using Deep Learning-Based Features. Nat. Commun. 2023, 14 (1), 4539 10.1038/s41467-023-40129-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fermin D.; Basrur V.; Yocum A. K.; Nesvizhskii A. I. Abacus: A Computational Tool for Extracting and Pre-Processing Spectral Count Data for Label-Free Quantitative Proteomic Analysis. Proteomics 2011, 11 (7), 1340–1345. 10.1002/pmic.201000650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B.; Busse D.; Li N.; Dittmar G.; Schuchhardt J.; Wolf J.; Chen W.; Selbach M. Global Quantification of Mammalian Gene Expression Control. Nature 2011, 473 (7347), 337–342. 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Kolberg L.; Raudvere U.; Kuzmin I.; Adler P.; Vilo J.; Peterson H. G:Profiler-Interoperable Web Service for Functional Enrichment Analysis and Gene Identifier Mapping (2023 Update). Nucleic Acids Res. 2023, 51 (W1), W207–W212. 10.1093/nar/gkad347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J.; Kull M.; Peterson H.; Hansen J.; Vilo J. G. Profiler--a Web-Based Toolset for Functional Profiling of Gene Lists from Large-Scale Experiments. Nucleic Acids Res. 2007, 35 (Web Server issue), W193–200. 10.1093/nar/gkm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkovits K.; Linden A.; Galozzi S.; Schilde L.; Pacharra S.; Mollenhauer B.; Stoepel N.; Steinbach S.; May C.; Uszkoreit J.; Eisenacher M.; Marcus K. Characterization of Cerebrospinal Fluid via Data-Independent Acquisition Mass Spectrometry. J. Proteome Res. 2018, 17 (10), 3418–3430. 10.1021/acs.jproteome.8b00308. [DOI] [PubMed] [Google Scholar]
- Barkovits K.; Pacharra S.; Pfeiffer K.; Steinbach S.; Eisenacher M.; Marcus K.; Uszkoreit J. Reproducibility, Specificity and Accuracy of Relative Quantification Using Spectral Library-Based Data-Independent Acquisition. Mol. Cell. Proteomics 2020, 19 (1), 181–197. 10.1074/mcp.RA119.001714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntel J.; Xuan Y.; Berger S. T.; Reiter L.; Bachur R.; Kentsis A.; Steen H. Advancing Urinary Protein Biomarker Discovery by Data-Independent Acquisition on a Quadrupole-Orbitrap Mass Spectrometer. J. Proteome Res. 2015, 14 (11), 4752–4762. 10.1021/acs.jproteome.5b00826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger G.; Bludau I.; Schmitt U.; Heusel M.; Hunter C. L.; Liu Y.; MacCoss M. J.; MacLean B. X.; Nesvizhskii A. I.; Pedrioli P. G. A.; Reiter L.; Röst H. L.; Tate S.; Ting Y. S.; Collins B. C.; Aebersold R. Statistical Control of Peptide and Protein Error Rates in Large-Scale Targeted Data-Independent Acquisition Analyses. Nat. Methods 2017, 14 (9), 921–927. 10.1038/nmeth.4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruderer R.; Bernhardt O. M.; Gandhi T.; Miladinović S. M.; Cheng L.-Y.; Messner S.; Ehrenberger T.; Zanotelli V.; Butscheid Y.; Escher C.; Vitek O.; Rinner O.; Reiter L. Extending the Limits of Quantitative Proteome Profiling with Data-Independent Acquisition and Application to Acetaminophen-Treated Three-Dimensional Liver Microtissues. Mol. Cell. Proteomics 2015, 14 (5), 1400–1410. 10.1074/mcp.M114.044305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Costa C.; Martínez-Bartolomé S.; McClatchy D. B.; Saviola A. J.; Yu N.-K.; Yates J. R. Impact of the Identification Strategy on the Reproducibility of the DDA and DIA Results. J. Proteome Res. 2020, 19 (8), 3153–3161. 10.1021/acs.jproteome.0c00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo A.; Liu L. Y.; Sadun T. Y.; Salmasi A.; Pooli A.; Felker E.; Houlahan K. E.; Ignatchenko V.; Raman S. S.; Sisk A. E.; Reiter R. E.; Boutros P. C.; Kislinger T. Prostate Cancer Multiparametric Magnetic Resonance Imaging Visibility Is a Tumor-Intrinsic Phenomena. J. Hematol. Oncol. 2022, 15 (1), 48 10.1186/s13045-022-01268-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha A.; Huang V.; Livingstone J.; Wang J.; Fox N. S.; Kurganovs N.; Ignatchenko V.; Fritsch K.; Donmez N.; Heisler L. E.; Shiah Y.-J.; Yao C. Q.; Alfaro J. A.; Volik S.; Lapuk A.; Fraser M.; Kron K.; Murison A.; Lupien M.; Sahinalp C.; Collins C. C.; Tetu B.; Masoomian M.; Berman D. M.; van der Kwast T.; Bristow R. G.; Kislinger T.; Boutros P. C. The Proteogenomic Landscape of Curable Prostate Cancer. Cancer Cell 2019, 35 (3), 414–427.e6. 10.1016/j.ccell.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw mass spectrometry data, generated spectral libraries, and processed proteomics data are publicly available on MassIVE database with MassIVE ID: MSV000093759 and FTP link: ftp://msv000093759/.