

SUMMARY
Changes in gene regulation have been linked to the expansion of the human cerebral cortex and to neurodevelopmental disorders, potentially by altering neural progenitor proliferation. However, the effects of genetic variation within regulatory elements on neural progenitors remain obscure. We use sgRNA-Cas9 screens in human neural stem cells (hNSCs) to disrupt 10,674 genes and 26,385 conserved regions in 2,227 enhancers active in the developing human cortex and determine effects on proliferation. Genes with proliferation phenotypes are associated with neurodevelopmental disorders and show biased expression in specific fetal human brain neural progenitor populations. Although enhancer disruptions overall have weaker effects than gene disruptions, we identify enhancer disruptions that severely alter hNSC self-renewal. Disruptions in human accelerated regions, implicated in human brain evolution, also alter proliferation. Integrating proliferation phenotypes with chromatin interactions reveals regulatory relationships between enhancers and their target genes contributing to neurogenesis and potentially to human cortical evolution.
In brief
Geller et al. use CRISPR knockout screens in human neural stem cells (hNSCs) to identify genes and transcriptional enhancers required for hNSC proliferation. Their results provide insight into the comparable effects of genetic disruptions in regulatory elements and protein-coding genes on human neurodevelopment.
Graphical Abstract
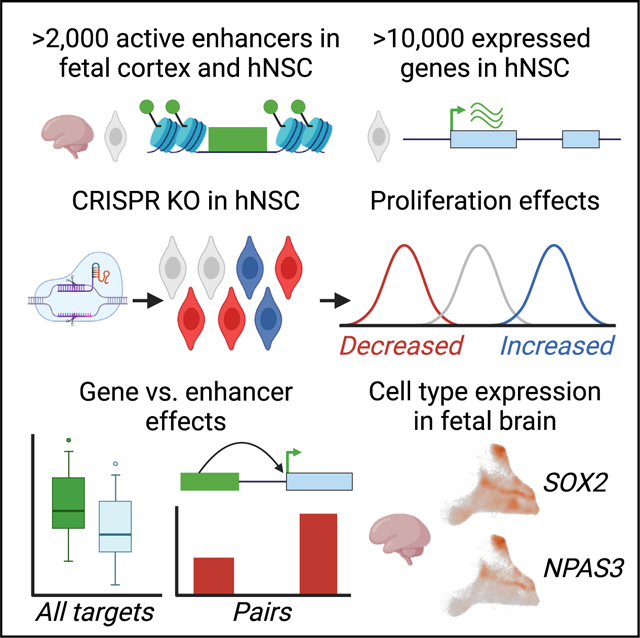
INTRODUCTION
The development of the human cerebral cortex depends on the precise spatial, temporal, and quantitative control of gene expression by transcriptional enhancers.1 Genetic variants with the potential to alter gene expression in the developing brain have been implicated both in neurodevelopmental disorders and in the expansion and elaboration of the cortex during human evolution.2–13 Despite growing evidence showing that regulatory variation influences human brain phenotypes, the biological effects of genetic changes that occur within enhancers active during neurodevelopment have not been systematically studied. Previous screens have focused on characterizing regulatory disruptions at specific loci or have not extensively targeted candidate enhancers that are known to be active in the developing human brain.14–21 Moreover, the effect size distribution of enhancer disruptions in neural cell types is poorly understood, and addressing this question would require disrupting thousands of enhancers across the genome in a single screen.
Here we employed a massively parallel single-guide RNA-Cas9 (sgRNA-Cas9) genetic screen in H9-derived human neural stem cells (hNSCs) to identify enhancers required for normal hNSC proliferation (Figure 1A).14–16,22 We disrupted 26,385 potentially functional conserved regions within 2,227 candidate enhancer sequences that are marked by histone H3 lysine 27 acetylation (H3K27ac), which is associated with enhancer activity both in the developing human cortex and in hNSCs (Figure S1; STAR Methods).23 We also disrupted 10,674 expressed protein-coding genes to compare the effects of gene and enhancer disruptions on hNSC proliferation.24 The candidate enhancers we targeted are specifically active during human corticogenesis compared with other human cell types and tissues (Figure S1). Characterizing the effects of disruptions within these enhancers may thus be of particular relevance for understanding the impact of regulatory variants on human cortical development.
Figure 1. Identifying genes and enhancers that alter human neural stem cell (hNSC) proliferation following genetic disruption.

(A) Top: summary of conserved regions within enhancers, genes, and background control regions targeted in the sgRNA-Cas9 genetic screen. Putative TFBSs within enhancers were identified based on the 46-species Placental Mammal PhastCons element annotation (GRCh37/hg19; STAR Methods). Each PhastCons element was divided into approximately 30-bp conserved regions and targeted for genetic disruption by sgRNAs. Genes were targeted based on evidence of expression in hNSCs. Genomic background controls are non-coding regions with no detectable evidence of regulatory activity based on epigenetic signatures (STAR Methods). Bottom: schematic transduction of sgRNA-Cas9 lentiviral libraries into hNSCs at a low MOI (~0.3) to ensure that cells were infected by a single virion, and, on average, 1,000 cells were infected per sgRNA. Changes in the abundance of each sgRNA over time and classification of proliferation phenotypes was carried out as described in the main text.
(B and C) Scatterplots illustrating beta scores (β) for two biological replicates resulting from genetic disruption of genes (B) and conserved regions within enhancers (C), measured at 12 cell divisions following transduction. Each point shows the effect of individual sgRNAs assigned to a protein-coding gene or conserved region within an enhancer relative to non-targeting controls for each biological replicate, as indicated in the legend shown in (B). Gene and enhancer disruptions affecting proliferation in regions with known biological roles in NSCs are identified by name (orange). Examples of genes were selected based on NSC maintenance or association with neurodevelopmental disorders. Enhancer examples were selected based on evidence of a regulatory interaction with genes with roles in chromatin modification and association with neurodevelopmental disorders or their identification as an HAR or HGE. Spearman correlations between replicates for gene (B) and conserved-region disruptions (C) are shown at the bottom left.
(D) Distribution of proliferation phenotypes resulting from disruption of genomic background control regions (light gray), enhancers (red) and genes (dark gray) at 4, 8, and 12 cell divisions. Boxplots illustrate the lower (25th percentile), middle (50th percentile), and upper (75th percentile) of beta scores for each category. Each point shows the jointly modeled effect across biological replicates for all sgRNAs assigned to a protein-coding gene or conserved region within an enhancer relative to non-targeting controls. Error bars indicate an estimated 95% confidence interval. *p < 2.2 × 10–16, Mann-Whitney U test.
We chose hNSCs as our screening platform because of the fundamental role of the NSC niche in neurogenesis and the specification of cortical size.25,26 Regulatory variants that alter NSC proliferation and self-renewal could result in changes to the number, type, and proportion of cortical neurons generated during cortical development, and these changes may contribute to disorders of brain development and function.27 In addition, the human cortex exhibits an expanded number of progenitor cells during development compared with other primates, suggesting that modification of hNSC proliferation programs contributed to the increase in cortical size during human evolution.26,27
Our screen identified more than 2,000 genes and more than 1,000 disruptions within enhancers that significantly affected hNSC proliferation in vitro. The set of gene targets with significant effects was enriched for genes associated with risk for multiple neurodevelopmental and neuropsychiatric disorders and showed enriched expression in specific neuronal progenitors in the developing human brain, including outer radial glia. Although gene disruptions overall had stronger effects on hNSC proliferation than enhancer disruptions, we identified enhancer disruptions with severe phenotypes. Using chromatin interaction data, we were able to link a subset of enhancers with proliferation phenotypes, including enhancers implicated in human brain evolution, to target genes and compare their effects. Collectively, our findings identify genes and enhancers required for hNSC proliferation as well as insight into the effects of genetic perturbation across thousands of enhancers active in the developing human cortex.
RESULTS
Screen design and approach
We used a commercial H9-derived hNSC line generated by Life Technologies (STAR Methods) These cells are not immortalized and express multiple NSC markers, including NES, SOX2, PAX6, and HES1, and are stated by the supplier to be multipotent, capable of differentiating into neurons, oligodendrocytes, and astrocytes (STAR Methods).24 This line doubles approximately every 40–50 h. These cells do not express anterior NSC markers such as FOXG1, suggesting a generic NSC-like cell state.24 However, because we are targeting putative enhancers specifically active during both human corticogenesis and in these hNSCs, our screen aims to provide insight into the biological role of these cortex-specific regulatory elements in hNSC self-renewal. To determine the competency of these cells to generate neurons, we used two commonly used differentiation protocols: undirected differentiation via removal of recombinant human fibroblast growth factor (bFGF; growth factor reduced [GFR]); and B27-driven differentiation into neurons (STAR Methods).28,29 In both cases, after 20 days, cells exhibited a neuron-like morphology as well as expression of neuronal marker genes (Figure S2). We observed upregulation of neuronal markers, including TUJ1, NEUROD1, and DCX1, as well as the deep and upper cortical layer markers TBR1 and CUX1 (Figure S2B) in both GFR and B-27 driven conditions.27,30 In the GFR condition, we found strong activation of ALDH1, consistent with the supplier’s claim that this protocol also yields astrocytes.31 We observed downregulation of the oligodendrocyte markers PGDFRA and CSPG4, and we did not detect expression of OLIG1, suggesting a lack of oligodendrocyte production (Figure S2).32,33 We did not detect attenuation of expression of PAX6 or SOX2, which may be due to the production of astrocytes expressing these genes in the mixed cultures we generated using the GFR condition.34,35 It may also suggest that the neurons generated by each protocol are relatively immature after a 20-day induction, with some cells remaining in a progenitor-like state. However, in general, our results support the hypothesis that these hNSCs have neurogenic potential.
To validate that the hNSC line was competent for CRISPR perturbation screens, we individually targeted three genes, TFRC, GRN, and UBQLN2, which have been used previously to validate other cell types for CRISPR screening.36 We independently transduced cells with a lentivirus carrying Cas9:GFP and sgRNAs targeting each gene at an MOI of 0.30 and sorted GFP-positive cells to ensure that each cell was infected exactly once. Western blot analysis showed a significantly reduced level of UBQLN2 protein expression in targeted cells compared with non-targeting controls (Figures S3A and S3B). Using RT-qPCR, we found significantly reduced expression of TFRC and GRN compared with non-targeting controls (Figure S3C). These results support the hypothesis that the hNSC line we used is suitable for CRISPR perturbation screening.
To disrupt regions likely to encode transcription factor binding sites within enhancers, we targeted 26,385 conserved regions (47,978 total sgRNAs) across the 2,227 enhancers included in our screen.37 We selected these enhancers based on two criteria. First, the candidate enhancers are marked by H3K27ac, a histone modification associated with enhancer activity, both in hNSCs and in human cortical tissue during developmental periods that include the expansion of proliferative zones and the onset of neurogenesis.2,24 Second, we filtered these enhancers for evidence of strong H3K27ac marking in the developing cortex relative to other human tissues (Figure S1; STAR Methods) to enrich for enhancers with cortex-specific activity.
The enhancers we targeted also included representatives from two classes of enhancers implicated in human brain evolution: 93 human accelerated regions (HARs) and 129 human gain enhancers (HGEs). HARs harbor an excess of human-specific substitutions and exhibit human-specific activities during development.4,6,7,38,39 HGEs have increased levels of enhancer-associated chromatin marks in the developing human cortex relative to other species.2,40 Chromosome conformation studies in the developing cortex suggest that both HARs and HGEs interact with genes involved in neurogenesis, axon guidance, and synaptic transmission.8–10 However, a functional role for HARs and HGEs in regulating human neurogenesis has not been established.
To directly compare the effects of enhancer and gene disruptions, we also targeted 10,674 protein-coding genes (21,663 sgRNAs) that are actively transcribed in hNSCs (Figure 1A).24 Additionally, we included 2,624 genomic background regions (4,497 sgRNAs) and 500 non-targeting sgRNA controls to account for non-specific effects of inducing small genetic lesions that require repair and background effects, respectively, of lentiviral transduction (STAR Methods). We defined genomic background regions as non-coding sequences that exhibited no evidence of function based on epigenetic marking in human tissues and cells (STAR Methods). In total, this yielded a library of 74,138 sgRNAs that we transduced into hNSCs across 8 sub-libraries (Figure S4). Two independent biological replicate transductions were carried out for each sub-library. The abundance of each integrated sgRNA was determined using PCR followed by high-throughput sequencing, initially after lentiviral transduction and then at 4, 8, and 12 cell divisions (Figure 1A; STAR Methods). Modeling the change in abundance of each sgRNA across the time series provided a quantitative basis for measuring effects of enhancer and gene disruption on cellular proliferation.41 We hypothesize that alterations in hNSC proliferation may encompass diverse cellular changes, including disrupted cell cycle regulation, differentiation of hNSCs into derived cell types, and reduced cell survival.
Quantifying gene and enhancer disruption phenotypes
We first quantified the biological effects of targeted disruption on hNSC proliferation, the beta score (β), for each gene, conserved region, or genomic background control relative to a set of non-targeting controls (Figures 1B–1D; STAR Methods). These biological effects were determined using reproducible sgRNA read abundances collected across both replicates and multiple time points (Pearson correlation > 0.9) (Figures S5–S7; Table S1) and demonstrate high levels of on-target specificity (Figure S8). We then performed linear discriminant analysis (LDA) to partition gene, enhancer, and background control disruptions into proliferation-decreasing, proliferation-increasing, or neutral classes (Figure S9; STAR Methods). We performed this analysis on a training set including known proliferation-decreasing genes, background controls, and the top proliferation-increasing effects at each time point (empirical false discovery rate [FDR] < 0.05).42 The trained classifier was then applied to the full dataset and filtered for consistency in classification and the direction of the effect across time points to identify genetic disruptions resulting in proliferation phenotypes (Figure 1A).
We identified 2,263 genes (21.2% of all targeted genes) that alter hNSC proliferation at 12 cell divisions (Figure 1B and 1D; Table 1; Table S2). Of these, nearly all gene disruption phenotypes showed decreased proliferation, while only 8 gene disruptions resulted in increased proliferation (Figure 1B; Table 1; Table S2). Many gene disruptions that altered proliferation have known roles in NSC biology relating to the balance between self-renewal and neuronal differentiation (e.g., CCND2 and SOX2) or response to growth factor signaling (e.g., FGFR1 and TCF7L1) (Figure 1B).43–46 Disruption of genes associated with microcephaly (e.g., ASPM, CEP135, and MCPH1) decreased hNSC proliferation, consistent with their known roles in human cortical development (Figure 1B).47 Disruptions of genes associated with risk for other neurodevelopmental disorders, notably autism spectrum disorder (e.g., DYRK1A, DIP2A, and CHD8) and X-linked intellectual disability (e.g., UBE2A), also resulted in significant alterations of hNSC proliferation.47–49
Table 1.
Observed proliferation phenotypes following quality control filtering for gene and enhancer disruptions in hNSCs
| 4 cell divisions |
8 cell divisions |
12 cell divisions |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Decrease | Neutral | Increase | Decrease | Neutral | Increase | Decrease | Neutral | Increase | |
| Expressed genes | 1,385 | 8,460 | 0 | 1,916 | 7,928 | 1 | 2,255 | 7,582 | 8 |
| Conserved regions | 331 | 24,711 | 24 | 581 | 24,412 | 73 | 987 | 23,891 | 188 |
| HGE conserved regions | 19 | 949 | 1 | 26 | 940 | 3 | 39 | 923 | 7 |
| HAR conserved regions | 7 | 402 | 0 | 11 | 398 | 0 | 15 | 394 | 0 |
We found 1,175 conserved regions (4.5% of all conserved regions) across 708 cortex-associated enhancers (31.7% of all targeted enhancers) that alter hNSC proliferation by 12 cell divisions. (Figures 1C and 1D; Table 1; Table S3). In contrast to genes, a greater proportion (16%) of disruptions in enhancers increased proliferation (188 proliferation increasing versus 987 proliferation decreasing; Table 1). We also discovered 46 conserved regions within HGEs and 15 within HARs that alter proliferation when disrupted at 12 cell divisions (Figure 1C; Table 1), supporting the hypothesis that HARs and HGEs contribute to human neurodevelopment. Disruption of HGEs affecting proliferation has been identified as proximal to genes with molecular functions in chromosome segregation (e.g., NSL1) or associated with intellectual disability (e.g., PTDSS1).50,51 Disruption of conserved regions within three HARs that contained human-specific substitutions also altered hNSC proliferation; these HARs were located in introns of genes with known functions in brain development (e.g., NPAS3) and cognitive function (e.g., USH2A) (Figure 1C).52,53
Globally, disruptions within enhancers had comparatively weaker effects on proliferation than gene disruptions (Figure 1D) (Wilcoxon rank-sum test, p < 2.2 × 10−16). Although many enhancer disruptions resulted in biological effects of a magnitude comparable with gene disruptions, we observed differences in the onset of their biological effects (Figure 1D). The majority of gene disruption phenotypes (61% of total gene phenotypes) were detected by 4 cell divisions. In contrast, fewer enhancer disruption phenotypes (30% of all conserved-region phenotypes) were detected at this early time point. The total number of phenotypes within enhancers approximately doubled at 8 cell divisions and doubled again at 12 cell divisions (Table 1).
To validate significant hits from the screen, we individually targeted 2 enhancers with proliferation-increasing effects and 2 enhancers with negative proliferation effects (Figure S10; STAR Methods).54,55 We also targeted 2 genes with negative proliferation effects. For each target, we carried out two independent replicate transductions (MOI = 0.3) using a lentivirus delivering lenti-CRISPRv2GFP and a single guide RNA used in the main screen. We maintained the cells in a 24-well culture format and monitored the proportion of GFP-positive cells at multiple time points spanning 10 cell divisions (STAR Methods). Although we observed variability in the effect of guide RNAs targeting the same gene or enhancer, at least one guide RNA for each target resulted in a proliferation phenotype consistent with the phenotypes detected in the massively parallel screen (Figure S10). We also found that disruption of some of the negative-proliferation targets was poorly tolerated by the entire cell population in each well, including GFP-negative cells. This resulted in general cell death in the entire population at later time points (Figure S10). We did not observe such population-wide cell death for non-targeting controls or the positive-proliferation targets. This may reflectnon-cell-autonomous effects due to disruption of each negative-proliferation effect target, possibly due to release of cellular contents into the culture environment or a reduction in overall cell density leading to sparse culture conditions poorly tolerated by untransduced cells. Such effects may be more evident in individual disruptions targeting the relatively small number of cells we used in these experiments as opposed to the massively parallel screen, where cells with negative proliferation phenotypes are a minority in a very large population of unaffected cells.
To further evaluate the performance of our screen, we then compared our significant gene hits with gene hits reported previously by Tian et al.36 as affecting viability in human induced pluripotent stem cells (hIPSCs) and neurons at three post-differentiation time points (Table S4; STAR Methods). This study targeted 2,325 genes representing the “druggable genome.” We found that 96 gene hits with negative effects on hNSC proliferation in our screen were also detected as having negative effects on hIPSCs, which is ~29% of the unique genes reported by Tian et al.36 We also found that 128 of our gene hits with negative effects were detected as having negative effects at one or more of the neuronal time points, or ~22% of the unique genes reported in that study. Even given that our screen and the screens by Tian et al.36 were conducted in different cell types and used different criteria for selecting gene targets, our screen still detected a substantial fraction of genes identified in that study.
Linking gene disruptions to biological processes
Measuring the effect of gene and enhancer disruptions across multiple time points allowed us to distinguish the overall magnitude of the effect on proliferation from temporal changes across cell divisions. Principal-component analysis (PCA) of the observed proliferation changes revealed that the first principal component (PC1 = 94.3% of total variance) is correlated with the severity of the effect on cellular proliferation (Figure 2A; Figure S11). This analysis enabled us to assign a proliferation score to each disruption, which we could then use to rank disruptions based on their cumulative effect on cellular proliferation across multiple time points. The second component (PC2 = 3.9% of total variance) correlated with effect changes across time points. Examples include the continued increase in proliferation resulting from genetic disruption of the X-linked intellectual disability gene UBE2A (Figure 2A) and the decrease in proliferation due to disruption of KIF20B (Figure 2A), a gene implicated in microcephaly.47,49 These findings support the hypothesis that both proliferation-increasing and proliferation-decreasing phenotypes, revealed by massively parallel screening in hNSCs, provide insight into human neurodevelopmental disorders. Together, these two components explain nearly all of the variability in the biological effects of disruption of genes and enhancers (>98% of total variance) and were further utilized to dissect the functional characteristics of hNSC proliferation (Figure 2A; Tables S2 and S3).
Figure 2. Characterization of gene proliferation phenotypes.

(A) Left: principal-component analysis (PCA) of all gene disruptions, with a 2D density overlay illustrating the distribution of proliferation-decreasing (blue), neutral (gray), and proliferation-increasing (green) phenotypes and individual gene disruptions (orange) with biological roles in the maintenance of NSCs or human neurodevelopment; see text for details. Right: temporal dynamics captured by PCA shown at 4, 8, and 12 cell divisions.
(B) Top: histogram of proliferation scores obtained from PCA for gene disruptions (black) and genomic background controls (gray). Bottom: partitioning of all gene disruptions that decrease proliferation into severe (top 25%) and strong (top 50%) categories.
(C) Fold enrichment of GO biological processes and risk gene sets within the partitions shown in (B). n.s., not significant. Hypergeometric BH-corrected p values are as follows: *p < 0.05, **p < 0.005, ***p < 0.0005.
(D) FGF signaling pathway (Reactome: R-HSA-1226099); see text for details. Genes disrupted within this pathway are shown in black, and a significant subset (BH-corrected p = 2.8 × 10−2) results in proliferation phenotypes (red).
We first examined whether gene disruptions converge on specific biological pathways and known human disease phenotypes. We hypothesized that gene disruptions with stronger effects might be functionally distinct from disruptions with weaker effects. We therefore grouped the proliferation phenotypes into categories based on their proliferation scores and performed overrepresentation testing of proliferation phenotypes across neurodevelopmental and neuropsychiatric disorder risk loci, Gene Ontology biological processes, and biological signaling pathways (Figures 2B and 2D; Table S5–S8). We found that genes associated with neurodevelopmental disorders were significantly overrepresented among gene disruptions that decreased hNSC proliferation (Table S5). Genes associated with microcephaly were enriched in all three categories and were most strongly enriched in the most severe set (hypergeometric test, p = 3.4 × 10−4) (Figure 2C). This is consistent with known disease processes that impair cell division in the developing human cortex.47 Genes located within large copy number variants (CNVs) associated with autism spectrum disorder (ASD) also showed strong enrichment in severe phenotypes (hypergeometric test, p = 1.1 × 10−3), providing the means to identify new potential candidate genes in these regions (Table S5).48 CNVs associated with developmental disorders (hypergeometric test, p = 1.6 × 10−3), as well as constrained genes intolerant to loss-of-function mutations (hypergeometric test, p = 5.1 × 10−17) exhibited moderate enrichment across all phenotypes (Figure 2C).48,56,57 Many risk genes associated with developmental disorders and ASD have been identified based on a significant excess of gene-disrupting loss-of-function mutations in affected individuals.56,58,59 These genes are also significantly enriched among proliferation-altering gene disruptions (hypergeometric test p = 3.2 × 10−6 for genes associated with developmental disorders and hypergeometric test p = 9.8 × 10−3 for genes associated with ASD) (Figure 2C), suggesting that loss-of-function mutations in these genes may contribute to developmental disorders in part by altering hNSC proliferation. Finally, we also examined sets of genes recently implicated in risk for schizophrenia (SCZ), bipolar disorder (BD), attention deficit hyperactivity disorder (ADHD), and major depressive disorder (MDD).60 We found that risk genes associated with each of these disorders were significantly overrepresented among gene disruptions that decreased hNSC proliferation in our screen (Figure 2C; Table S6).
We then used Gene Ontology analysis to identify biological processes enriched among proliferation-altering gene disruptions (Table S7). In severe proliferation phenotypes, we observed the strongest fold-enrichment for gene functions related to histone acetyltransferase activity (modified exact test, Benjamini-Hochberg [BH]-corrected p = 2.1 × 10−3) and translational initiation (modified exact test, BH-corrected p = 1.4 × 10−12) (Figure 2C). Additional processes exhibiting elevated fold enrichment in severe phenotypes include sister chromatid cohesion (modified exact test, BH-corrected p = 1.7 × 10−6), mRNA splicing (modified exact test, BH-corrected p = 4.3 × 10−12), transcriptional elongation (modified exact test, BH-corrected p = 2.4 × 10−5), and DNA replication (modified exact test, BH-corrected p = 1.3 × 10−8), demonstrating that genetic disruption of a wide variety of biological processes leads to severe proliferation phenotypes in hNSCs.
To gain insight into the biology associated with changes in hNSC proliferation, we utilized a public database of manually curated and peer-reviewed pathways (Reactome Project; STAR Methods) to test the enrichment of gene proliferation phenotypes within signaling pathways (Table S8).61 We found that gene disruption phenotypes are also enriched for the WNT signaling pathway (Reactome: R-HSA-201681; hypergeometric test, BH-corrected p = 4.5 × 10−2) (Table S8), which contributes to cortical patterning via a signaling center in the cortical hem.62 We also observed enrichment for ROBO signaling (Reactome: R-HSA-9010553; hypergeometric test, BH-corrected p = 8.8 × 10−10), supporting evidence from mammalian genetic models showing that this pathway alters hNSC self-renewal.63 In addition, proliferation-decreasing gene disruption phenotypes are significantly enriched for the fibroblast growth factor (FGF) signaling pathway (Reactome: R-HSA-1226099; hypergeometric test, BH-corrected p = 2.8 × 10−2) (Figure 2D; Table S8), consistent with its role in anterior forebrain cortical patterning.45,64 During human corticogenesis, neural progenitors are influenced by signaling molecules released from nearby patterning centers, and these morphogenetic gradients result in the specification of neuronal cell types and cortical areal identity.62 Genetic disruption impacting these signaling pathways may alter neurogenesis and possibly result in changes to the specification of cortical size and areal identities.45,62
Identifying transcription factor binding site disruptions in enhancers that alter proliferation
To explore genetic disruptions altering enhancer function, we used PCA to isolate the magnitude and temporal effects of disruption for all conserved enhancer regions included in our screen (Figure 3A; Table S3). We combined this analysis with genome-wide predictions of transcription factor binding sites (TFBSs) to identify binding sites enriched in proliferation-altering enhancer disruptions (Table S9). Most conserved regions included in our screen (90.7% of total conserved regions) include a predicted TFBS, with a total of 152,110 motifs predicted across all targeted regions.65 To obtain an initial view of the effect of genetic disruption on predicted TFBS motifs, we individually interrogated 8 conserved regions targeted in our screen, including a subset exhibiting proliferation phenotypes. We performed high-throughput amplicon sequencing on the targeted conserved regions after genetic disruption to determine the molecular effects on enhancer TFBS motif content (Figures S12 and S13; Table S10). In most cases (87.5% of replicated sgRNAs), we observed genetic variation at the sgRNA-Cas9 target site. The proportion of alleles modified following disruption ranged from 33% to 96% (Table S10), and deletions were the most common type of genetic variation observed (average deletion size of 6–7 bp). The 8 individually targeted conserved regions overlap 50 predicted TFBS motifs, and 41 motifs were likely disrupted due to proximity (within 10 bp) to the predicted sgRNA-Cas9 cleavage site. One proliferating-decreasing disruption destroyed a putative TBX2/TBX20 TFBS motif that includes human-specific substitutions within HACNS96 (Figures 3A and 3C; Figure S12). TBX2 mediates regulation of FGF signaling during anterior neural cell specification.66 Transgenic assays demonstrate that HACNS96, which is located within the intron of the transcription factor NPAS3, acts as a transcriptional enhancer during vertebrate neurodevelopment.67 NPAS3 is expressed in the developing brain, and genetic disruption of NPAS3 and HACNS96 results in a proliferation-decreasing phenotype (Figure 2A; Figure S12D), suggesting that disruption of the TBX2/TBX20 motif within HACNS96 may lead to the proliferation-decreasing phenotype we observed by altering NPAS3 regulation.
Figure 3. Characterization of enhancer proliferation phenotypes.

(A) Left: PCA of all gene disruptions with a 2D density overlay illustrating the distribution of proliferation-decreasing (blue), neutral (gray), and proliferation-increasing (green) phenotypes and individual conserved region disruptions (orange) with biological roles in the maintenance of NSCs or human neurodevelopment; see text for details. Right: temporal dynamics captured by PCA shown at 4, 8, and 12 cell divisions.
(B) Top: histogram of proliferation scores obtained from PCA for enhancer disruptions (black) and genomic background controls (gray). Bottom: partitioning of all enhancer disruptions that decrease proliferation into severe (top 25%) and strong (top 50%) categories.
(C) Top: genomic alignments for human, chimpanzee, and mouse for a conserved region within an HAR enhancer (HACNS96). Human-specific substitutions (red) indicate genetic changes occurring within the conserved regions targeted for genetic disruption in the screen. Bottom: examples of deletion alleles introduced by Cas9 in this locus, determined by long-read amplicon resequencing and alignment to the reference genome (GRCh37/hg19).
(D) Predicted TFBSs were obtained from the JASPAR 2018 TFBS prediction database and are significantly enriched in proliferation-altering enhancer disruptions within the partitions shown in (B). Hypergeometric BH-corrected p values are as follows: *p < 0.05, **p < 0.005, ***p < 0.0005.
To identify predicted TFBS motifs that are overrepresented in proliferation-altering enhancer disruptions, we partitioned the proliferation-decreasing phenotypes within enhancers into severe, strong, and all decreasing categories (Figure 3B). Conserved regions exhibiting severe proliferation-decreasing phenotypes are enriched in E2F4 and E2F6 binding motifs (hypergeometric test, BH-corrected p = 2.6 × 10−2 and p = 5.9 × 10−4, respectively) (Figure 3D), consistent with the role of E2F factors in the transcriptional control of cell cycle dynamics and cell specification.68 We also observed enrichment of ZNF263, SP1, and SP2 (hypergeometric test, BH-corrected p = 6.8 × 10−12, p = 6.3 × 10−4, p = 4.7 × 10−3, respectively), indicating that binding of broadly expressed general transcription factors is important in facilitating normal enhancer function. We did not observe enrichment for TFBSs within proliferation-increasing disruptions, potentially due to the smaller number of conserved regions involved in this cellular phenotype.
Distribution of proliferation-altering phenotypes across enhancers
To describe the proliferation-altering phenotypes at the level of whole enhancers, we summarized the number of conserved-region disruptions impacting proliferation within each targeted enhancer (Table S11). While many enhancers include only one disrupted site that results in a proliferation phenotype (66.9% of proliferation-altering enhancers), we also identified many enhancers that included multiple conserved regions impacting proliferation (Figure 4A). On average, 15% of conserved regions within proliferation-altering enhancers were associated with a proliferation phenotype (Figure 4B). The cumulative burden of proliferation-altering disruptions within whole enhancers scales linearly with the number of targeted sites (Figure 4C), supporting the hypothesis that the total proliferation phenotype burden is proportional to the number of conserved regions disrupted within each enhancer.
Figure 4. Frequency of proliferation-altering disruptions in enhancers, HARs, and HGEs.

(A) The number of disrupted conserved regions leading to a proliferation phenotype within each enhancer (left, shown in gray) or in each HAR or HGE (right, shown in red).
(B) The proportion of proliferation-altering disruptions compared with all targeted sites in all enhancers (gray) and HARs or HGEs (red). The dashed line indicates the mean density of proliferation phenotypes within all enhancers in the screen.
(C) The total number of conserved regions disrupted within each enhancer (x axis) compared with the cumulative proliferation phenotype burden within each enhancer (y axis). Mutation-sensitive enhancers identified by permutation analysis (see D and main text for details) are labeled based on the most proximal gene. HARs and HGEs are labeled in red. The results of the permutation analysis, including the number of disrupted sites with a phenotype compared with the total number of targeted sites and the enrichment p values, are provided in Table S11.
(D) A mutation-sensitive enhancer near SPRY2, a regulator of the FGF signaling pathway. Top: genomic coordinates indicate position on the reference genome (GRCh37/hg19). Three enhancers (i, ii, and iii), shown by the extent of H3K27ac marking (light blue bars) in hNSCs, were disrupted in this locus. Enhancer iii includes a significant excess of proliferation-altering enhancer disruptions. Bottom: H3K27ac signal profile (light blue), mammalian PhastCons elements (orange bars), and corresponding conserved regions that have either neutral effects (gray bars) or proliferation-decreasing phenotypes (dark blue bars) when disrupted in the screen are shown for enhancer iii. The signal track at the bottom displays normalized beta scores (gray) at 12 cell divisions averaged across a sliding window.
We then identified regulatory elements that we termed “mutation-sensitive enhancers,” which are a subset of enhancers found by permutation analysis to contain a significant excess of conserved regions yielding a proliferation phenotype (Table S11; STAR Methods). One mutation-sensitive enhancer (permutation test, BH-corrected p = 7.4 × 10−3) is proximal to SPRY2 (Figure 4D), a known regulator of FGF signaling, illustrating the potential vulnerability of hNSCs to variation influencing this developmental signaling pathway.69 We also identified two mutation-sensitive enhancers that include the HARs HACNS610 (permutation test, BH-corrected p = 4.4 × 10−3) and HAR122 (permutation test, BH-corrected p = 4.4 × 10−3), located within introns of SOX5 and NPAS3, respectively. The identification of mutation-sensitive enhancers containing HARs suggests candidates for human-specific regulatory activity at important positions within regulatory networks impacting hNSC self-renewal.
Comparing enhancer-target gene disruption phenotypes
To link enhancers exhibiting proliferation phenotypes with their putative target genes, we used a high-resolution map of long-range chromatin contacts ascertained from human neural precursor cells (Table S12; STAR Methods).70 Chromatin interaction maps (Figure 5A) identified 180 enhancer-gene pairs between enhancers and genes that exhibit proliferation phenotypes in hNSCs (Table S12). These maps capture a diverse range of regulatory interactions, including interactions between a single enhancer and a single gene target (82, 45.5% of the total), interactions between multiple enhancers converging on a single gene target (59, 32.7% of the total), and a single enhancer that interacts with multiple target genes (39, 21.6% of the total). Likely because of this diversity, we did not observe a correlation (Pearson correlation r = 0.01 for all 180 pairs; r = −0.14 for 82 single gene-single enhancer pairs) between the effect of enhancer and target gene disruptions, and we elaborate this point under Discussion and Limitations of the study. However, individual enhancer-gene interactions provided insight into the relative effects of enhancer versus target gene disruption on proliferation.
Figure 5. Identifying target genes of enhancers contributing to hNSC proliferation using chromatin contact maps.

(A) Chromatin contact maps of human neural precursor cells were used to identify interactions between enhancers and genes that, when disrupted, result in a proliferation phenotype. For each enhancer-gene pair, the absolute value of the gene proliferation score (x axis) and the absolute value of the strongest proliferation score within the enhancer (y axis) is shown. Highlighted genes have known roles in NSC biology or are associated with risk for developmental disorders. Interactions between HARs or HGEs and target genes are labeled in red.
(B) An example of an enhancer (boxed) regulating a single target gene associated with microcephaly. Top: the genomic coordinates (in GRCh37/hg19) of the enhancer are shown relative to the two detected target genes. Chromatin contacts are shown as black arcs. Bottom: the effects of enhancer and target gene disruptions on hNSC proliferation at 4, 8, and 12 cell divisions.
(C) An example of an enhancer (boxed) regulating multiple target genes with roles in hNSC proliferation (labeled as in B).
(D) An example of a human gain enhancer (HGE; boxed) interacting with the nearby gene NSL1 (labeled as in B).
The microcephaly-associated gene CEP135 interacts with a single enhancer active during human corticogenesis (Figure 5B).47 Disruption of the enhancer and of CEP135 results in comparable negative effects on hNSC proliferation, suggesting that cortical phenotypes may arise through changes in the regulation of neurodevelopmental risk genes. We also identified a human cortical enhancer (Figure 5C) interacting with two target genes with known roles in cell proliferation, STARD13 and PDS5B.71,72 Genetic disruption of the enhancer results in a stronger effect on proliferation than disruption of either target gene (Figure 5C, bottom), suggesting that genetic variation within a single enhancer can lead to severe biological effects through dysregulation of multiple genes.
We also utilized this chromatin interaction map to identify gene targets for 9 HARs and 8 HGEs that contribute to hNSC proliferation. One example, shown in Figure 5C, is an HGE that targets NSL1, which functions in neural progenitor cell division and has been associated with cognitive phenotypes in humans.51,73 Disruption of NSL1 negatively affects hNSC proliferation (Figure 5D, bottom). Disruption of the interacting HGE also negatively alters proliferation, resulting in a weaker but significant biological effect compared with the gene disruption (beta score = −0.5485, FDR < 3.6 × 10−4 at 12 cell divisions; Table S3). These findings provide the basis for determining whether gains in activity in HARs and HGEs alter expression of specific gene targets during neurogenesis.
Characterizing cell-type-specific expression of gene hits in the developing human cortex
The developing human cortex contains a diversity of neural progenitor types. To determine whether genes exhibiting proliferation phenotypes are expressed in particular classes of cortical progenitors, we used a previously published fetal human cortex single-cell RNA sequencing (scRNA-seq) atlas that included samples from Carnegie stage 14 (CS14) to gestational week 25 (GW25).74,75 We first identified progenitor types based on expression of known marker genes: intermediate progenitor cells (IPCs) expressing EOMES, outer radial glia (ORG) expressing HOPX and PTPRZ1, and radial glia (RG) that lacked expression of these marker genes (STAR Methods).76,77 We further sub-divided these populations according to their inferred cell cycle phase based on expression of TOP2A and MKI67, which mark cells in G2/M phase.78 We then clustered the average expression of the gene hits we identified across each progenitor type and cell cycle phase to identify groups of gene hits with expression profiles biased toward specific cell types or cell cycle phases (Figure 6). We identified multiple gene clusters that showed biased expression profiles (Figure 6A; Table S13). For example, we identified a cluster including 129 gene hits that showed biased expression in ORG and a cluster including 68 gene hits showing biased expression in IPCs. ORG-biased gene hits included NPAS3, which, as supported by our earlier findings, is regulated in part by the HAR HACNS96 (Figure 5; Figure 6B; Figure S12). IPC-biased gene hits included the ASD risk genes CHD8, CUL3, and DYRK1A.58 We also identified gene clusters with biased expression at particular cell cycle phases, including a G2/M-biased cluster (145 genes) comprised of genes showing high expression across progenitor types specifically at this cell cycle phase (Figure 6A). This cluster included genes associated with microcephaly, such as CEP135 and CEP152 (Figure 6B; Table S13).47 Together, these results identified subsets of genes with proliferation phenotypes in our screen that have expression profiles unique to specific cortical progenitor populations and at specific stages of the cell cycle.
Figure 6. Progenitor-type-specific expression of gene hits in the developing human cortex.

(A) Left: UMAP representation of the developing human cortex single-cell gene expression atlas used for our analysis. Five cortical progenitor types are annotated: outer radial glia (ORG) cells in G1/S phase, intermediate progenitor cells (IPCs) in G1/S or G2/M phase, and radial glia (RG) in G1/S or G2/M phase. Right: heatmaps showing the average scaled expression for genes with proliferation phenotypes that exhibit progenitor-type-specific expression patterns. Three clusters are presented: one with genes showing ORG-biased expression (129 genes, cluster 2 in Table S13), one with genes showing G2/M-biased expression (145 genes, cluster 6 in Table S13), and one with genes showing IPC-biased expression (68 genes, cluster 1 in Table S13). The proliferation phenotype for each gene at 4, 8, and 12 cell divisions is shown above each heatmap. The inferred cell cycle phase for each progenitor cell type is shown on the right, as is the average expression of all genes in each cluster. See the legend at the bottom right for details.
(B) Left: UMAP representation of cortical progenitors in the developing human cortex gene expression atlas with other cell types removed. Progenitor cell types and inferred cell cycle phases are labeled and colored as in (A). Right: UMAP representations displaying the scaled density of expression for individual genes in each cluster shown in (A).
We next sought to determine whether genes within the progenitor-type- or cell-cycle-phase-specific clusters were associated with specific biological functions. To this end, we performed Gene Ontology (GO) biological process and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses (Table S13).79,80 Genes in the G2/M-biased gene cluster were enriched for KEGG cell cycle, DNA replication, and mismatch repair pathways and GO biological processes associated with chromosome segregation during mitosis, mitotic spindle organization, and other cell-cycle- and cell-division-related categories. IPC-biased genes were associated with KEGG pathways and GO biological processes associated with the regulation of RNA splicing. Genes in the ORG-biased cluster were enriched in multiple KEGG pathways associated with metabolic processes, including oxidative phosphorylation, glycolysis, and fatty acid metabolism. Finally, genes in the RG (G1/S) cluster were enriched in KEGG pathways associated with DNA replication and oxidative phosphorylation and biological processes also associated with DNA replication as well as aerobic respiration. Collectively, these findings support the hypothesis that the gene hits we identified act within pathways essential to cellular viability in vitro, but the specific pathways impacted in vivo may vary by cell cycle phase and progenitor subtype.
DISCUSSION
We describe the effects of genetic disruption of more than 20,000 conserved regions in more than 2,200 putative enhancers specifically active in the developing human cortex using a hNSC model. Previous studies of gene-regulatory perturbation have largely focused on particular loci, have not targeted large numbers of enhancers implicated in human corticogenesis, and have employed immortalized cell lines not related to neurodevelopment.14–21 Our results revealed disruptions in 708 enhancers that altered hNSC proliferation, pointing to enhancers that may play important roles in regulating human cortical neurogenesis. These enhancers also constitute a resource for the interpretation of noncoding variation associated with human neurodevelopmental phenotypes. We note that disruptions in HARs and HGEs also altered hNSC proliferation, providing evidence that regulatory elements linked to human brain evolution play an important functional role in neurogenesis. We also targeted all expressed genes in our hNSC model and found that genes associated with neurodevelopmental and neuropsychiatric disorders disproportionately impacted proliferation. This suggests that biological disruption of neural progenitors may contribute both to early-onset neurodevelopmental disorders, such as ASD, and disorders that are diagnosed later in the life-span, such as SCZ.
We note that the changes in proliferation we observed in our screen may be the result of several underlying mechanisms. These include changes in cellular viability or growth as well as aberrant induction of neuronal differentiation or other effects that alter cellular fate. We did not design our screen to distinguish among these potential phenotypes. This will require screening approaches that incorporate additional readouts, such as Perturb-Seq, which would reveal changes in transcriptional signatures associated with gene and enhancer disruptions in each cell.81
Because we included both gene and enhancer disruptions in our screen, we were able to compare their relative effects. We found that enhancer disruptions generally had weaker effects on hNSC proliferation than gene disruptions, although we did identify individual enhancer disruptions with strong effects comparable with gene disruptions. Although we observed a higher frequency of enhancer disruptions with positive effects on hNSC proliferation compared with gene disruptions, enhancer disruptions were strongly biased toward negative effects on hNSC proliferation overall. Integrating enhancer and gene proliferation phenotypes with enhancer-gene interactions provided further insight into how the biological effects of individual enhancer disruptions compared with disruption of their target genes. As described under Results, we observed a diversity of patterns. For some enhancers, disruptions had comparable effects as disruption of their target gene. For others, disruptions had weaker effects. In some cases, we identified enhancers that targeted multiple genes and that, when disrupted, had greater effects than disruption of any of their gene targets.
We did not observe a significant correlation between the magnitude of enhancer and target gene disruptions overall. However, there are multiple technical and biological mechanisms that could account for this finding. First, the genes we targeted are likely regulated by multiple enhancers, and it is well established that enhancers often have redundant regulatory functions.82 We may therefore expect that enhancer disruptions may show weaker effects compared with their target genes, in part because other enhancers we have not disrupted would compensate for the effects of the enhancer disruption we did introduce. The magnitude of these effects may still not be correlated across enhancer-gene pairs because of variation in the robustness and redundancy of the regulatory architecture across genes. Second, as we show under Results, enhancers can target multiple genes, and disrupting those enhancers may have larger effects on proliferation compared with single-gene disruptions. Third, as we discuss further below, our screen design involves introducing small deletions in potential TFBSs within enhancers rather than deleting entire enhancers. The effects of those deletions are likely to vary due to redundancy within enhancers, and the degree of that redundancy may vary across enhancers as well, which would also contribute to a lack of correlation between gene and enhancer effects. Fourth, the chromatin interaction data available for our analysis is sparse and likely only captures a subset of the enhancer-gene interactions that are present, even for enhancers we disrupted. This limits the number of enhancers and genes we can meaningfully compare and our ability to estimate how many enhancers may regulate the genes we targeted. This also applies to cases where we assigned one enhancer to one gene. There are likely other enhancers regulating genes we did not disrupt and that we cannot detect and that may compensate for the disruptions we introduced. Additional insight into these questions will require denser chromatin contact maps and combinatorial disruption of enhancers for multiple genes.
The targeted genetic disruption approach we chose provides distinct insights into enhancer function compared with alternative strategies, such as CRISPRi, which would potentially silence entire enhancers.83 By introducing small deletions in conserved regions, we were able to identify enhancers that were particularly prone to disruption. We were also able to identify TFBS motifs enriched in proliferation-altering regions. These findings highlighted the importance of specific transcription factors and individual binding sites in transcriptional regulation within the NSC niche. Our approach also directly measured the effect size distribution of small mutations in enhancers, which is relevant for understanding the impact of genetic variation in regulatory elements in human disease and evolution. However, in contrast to CRISPRi, disruption of one region in an enhancer is unlikely to completely inactivate most of the enhancers in our target set. Instead, the single genetic disruptions we introduced may alter enhancer activity in more complex ways; they may decrease enhancer activity, increase it, or potentially alter interactions between the enhancer and its target genes. Enhancer disruptions may consequently have more diverse biological effects as well, including altering gene expression to produce an increase in cellular proliferation.
We found that genes exhibiting significant phenotypes showed biased expression in specific progenitor subtypes and at particular cell cycle phases in the developing human cortex. These genes were enriched in essential functional pathways, although the specific pathways and processes varied across progenitor subtypes. This suggests that disruption of these genes may have cell-type specific effects and that the pathways and processes that would be altered may be cell-type-specific. We also note that gene hits showing cell-type-specific biased expression included genes associated with ASD and microcephaly. Our findings may therefore help identify the specific cell types in the developing human brain that are affected in these and other neurodevelopmental disorders and thus yield insight into their etiology.
Collectively, our findings provide an empirical view of the effects of genetic variation on enhancer function and the relative overall impact of enhancer versus target gene disruption. We identified 708 enhancers with at least one disruption that altered hNSC proliferation, demonstrating a functional role of specific regulatory elements in human neurodevelopment. The set of enhancers and genes we report here will enable further studies of gene regulatory variation in human brain development, neurodevelopmental disorders, and human brain evolution.
Limitations of the study
Our study has several limitations that we wish to emphasize. As we discussed above, redundancy in enhancer function and the sparsity of interaction data make it difficult to correlate the effect of enhancer disruption with disruption of target genes, except in individual cases. In addition, because we are using CRISPR knockouts to introduce small deletions in enhancers rather than knocking out entire enhancers, many of the disruptions we introduced may result in partial loss of enhancer function or, potentially, increases in enhancer activation due to loss of binding sites for repressive transcription factors. Our results may therefore not be directly comparable with screens using other approaches, such as CRISPRi. Because we are using hNSC proliferation as our readout, we are not able to determine whether proliferation phenotypes are due to cell death, reduced proliferation, or neuronal differentiation, all of which could result in reduced sgRNA representation. Finally, although we designed our screen to minimize detection of false positive hits by including 500 positive control genes plus 500 negative controls and more than 4,000 sgRNAs targeting more than 2,000 background control regions, we carried out two iterations of the entire screen. We recognize that some of our hits may be false positives, although we think it possible that our screen may also suffer from an increased number of false negatives, particularly in detecting disruptions that increase proliferation. This may account for the bias toward proliferation-decreasing hits we observe in our data.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, James P. Noonan (james.noonan@yale.edu).
Materials availability
Materials used in this study will be provided upon request and available upon publication.
Data and code availability
Sequencing data for massively parallel sgRNA-Cas9 disruption and individual replicate disruption are available through the Gene Expression Omnibus as of the date of publication. Accession numbers are listed in the key resources table. This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
All original code generated for the manuscript is available at Zenodo as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-UBQLN2 1:1000 | Cell Signaling | Cat. #85509S; RRID:AB_2800056 |
| Anti-Actin 1:1000 | Abcam | Cat. #ab216070 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| E. Coli DH10B | Life Technologies | Cat. #18290015 |
| NEB Stable Competent E. Coli. Strain | New England Biolabs | Cat. #C3040H |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Extreme Gene 9 transfection reagent | Millipore-Sigma | Cat. #6365787001 |
| GlutaMax-I | ThermoFisher | Cat. #A1286001 |
| Chloroquine | Millipore-Sigma | Cat. #C6628 |
| Bovine serum albumin | Millipore-Sigma | Cat. #A8022 |
| Sodium butyrate | Millipore-Sigma | Cat. #303410 |
| GlutaMax-supplemented OptiMem | ThermoFisher | Cat. #51–985-034 |
| Recombinant human epidermal growth factor | ConnStem | Cat. #E1000 |
| Recombinant human fibroblast growth factor | ConnStem | Cat. #F1001 |
| StemPro neural supplement | Life Technologies | Cat. #A1050801 |
| Corning Matrigel GFR membrane matrix | Fisher Scientific | Cat. #CB-40230C |
| Polybrene | Millipore-Sigma | Cat. #TR-1003-G |
|
| ||
| Critical commercial assays | ||
|
| ||
| Gibson assembly Master Mix | New England Biolabs | Cat. #E2611L |
| BioRad Calirty Max Western ECL Substrate, | BioRad | Cat. #175063 |
| LabForce HyBlot CL | Thomas Scientific | Cat. # 114J51 |
| Qiagen RNeasy Kit | Qiagen | Cat. #74304 |
| Invitrogen SuperScript III First Strand Synthesis SuperMix |
ThermoFisher | Cat. #18080400 |
| Endo-Free Maxi Prep Isolation Kits | Qiagen | Cat. #12362 |
| Mouse Neural Stem Cell Nucleofection Kit | Lonza | Cat. # VPG-1004 |
| SYBR Green I reagents | Roche Diagnostics | Cat. #04707516001 |
| Gibco CELLstart CTS | Thermo Fisher | Cat. #A10142–01 |
| Gibco neurobasal media | Thermo Fisher | Cat. #21103 |
| Gibco serum-free B27 | Thermo Fisher | Cat. #17504 |
|
| ||
| Deposited data | ||
|
| ||
| Sequencing data for massively parallel sgRNA-Cas9 disruption and individual replicate disruption | This paper | GSE138823 |
| Sequencing data for RNA-seq and H3K27ac enrichment collected from H9-dervived human neural stem cells | Cotney et al.24 | GSE57369 |
| H3K27ac enrichment data in human embryonic limb | Cotney et al.40 | GSE42413 phs001226.v1.p1 |
| H3K27ac enrichment data in human embryonic cortex | Reilly et al.2 | GSE63649 phs001226.v1.p1 |
| H3K27ac data in human H1 ESCs and adult tissues profiled by the Roadmap Epigenomics Project | Kundaje et al.84 | GSE16368 |
| PsychEncode Hi-C Data from human fetal brain | Rajarajan et al.70 | syn22343893 |
| Single-cell human fetal brain RNA-sequencing data | Bhaduri et al.74 Eze et al.75 | NeMO identifier: nemo:dat-0rsydy7 |
| JASPAR 2018 database | Khan et al.65 | https://jaspar2018.genereg.net/ |
| Epilogos | https://epilogos.altius.org | N/A |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| H9-derived human neural stem cells | Life Technologies | Cat. #N7800–1000 |
| HEK293FT | Invitrogen | Cat. #R70007 |
| HEK293T | Yale Cell Preparation and Analysis Core | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Oligonucleotides used to clone sub-libraries and in validation and RT-qPCR assays are listed in Table S14. | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| LentiCRISPRv2GFP | Addgene | Cat. #82416 |
| pCMV-VSV-G | Addgene | Cat. #8454 |
| pCMV-dR8.2 dvpr | Addgene | Cat. #8455 |
|
| ||
| Software and algorithms | ||
|
| ||
| Bowtie v. 1.1.2 | Langmead et al.85 | https://sourceforge.net/projects/bowtie-bio/files/bowtie/1.1.2/ |
| CRISPResso2 | Clement et al.86 | http://crispresso2.pinellolab.org/submissiongreat.stanford.edu |
| GREAT version 3.0.0 | McLean et al.87 | |
| MAGeCK version 0.5.8 | Li et al.88 | https://sourceforge.net/projects/mageck/files/0.5/ |
| MASS (v7.3–54) | Venables and Ripley89 | https://www.stats.ox.ac.uk/pub/MASS4/ |
| DAVID v6.8 | Huang et al.90 Huang et al.91 |
https://david.ncifcrf.gov/ |
| ReactomePA package (v1.14.0) | Fabregat et al.61 | https://bioconductor.org/packages/release/bioc/html/ReactomePA.html |
| Seurat R package (v4.3.0) | Hao et al.92 | https://satijalab.org/seurat/ |
| Batchelor R package (v1.8.1) | Haghverdi et al.93 | https://www.bioconductor.org/packages/release/bioc/html/batchelor.html |
| ComplexHeatmap R package (v2.11.2) | Gu94 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| Cutadapt version 1.16 | Martin95 | https://cutadapt.readthedocs.io/en/v1.16/index.html |
| Juicer | Durand et al.96 | https://github.com/aidenlab/juicer/ |
| ShinyGO | Ge et al.80 | http://bioinformatics.sdstate.edu/go/ |
| Nebulosa v1.2.0 | Alquicira-Hernandez and Powell97 | https://www.bioconductor.org/packages/release/bioc/html/Nebulosa.html |
| ImageJ | Schneider et al.98 | https://imagej.net/ |
| Original code for analyses performed in this study | This paper | https://doi.org/10.5281/zenodo.10258136 |
|
| ||
| Other | ||
|
| ||
| Amicon Ultra-15 100kD filters | Millipore-Sigma | Cat. # UFC901008 |
| CustomArray 90K oligonucleotide synthesis array | CustomArray | N/A |
| Proliferation-decreasing controls, described in the Methods | Wang et al.22 | N/A |
| Accuri C6 Flow Cytometer | BD Biosciences | N/A |
| S3e Cell Sorter | BioRad | N/A |
| Cytoflex LR Flow Cytometer | Beckman Coulter | N/A |
| Roche LightCycler 480 PCR Thermal Cycler | Roche Diagnostics | N/A |
| HiSeq 4000 | Illumina | N/A |
| MiSeq | Illumina | N/A |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines and vectors
Materials were obtained from the following sources: H9-derived human neural stem cells from Life Technologies (N7800–1000), HEK293FT cells from Invitrogen, LentiCRISPRv2GFP from Addgene (Plasmid #82416, provided by David Feldser; this plasmid provides the S. pyogenes Cas9), pCMV-VSV-G (Addgene, Plasmid #8454), and pCMV-dR8.2 dvpr (Addgene, Plasmid #8455). The identity of H9-derived human neural stem cells was confirmed by analysis of hNSC markers via RNA-sequencing and antibody staining for multipotency markers (SOX2, NES).24
Human neural stem cell culture reagents
H9-derived human neural stem cells were cultured in Knockout DMEM/F-12 (Life Technologies, N7800–100) supplemented with EGF and FGF (ConnStem), GlutaMax-I and StemPro neural supplement (Life Technologies) as recommended by the manufacturer. In addition, cells were cultured on Matrigel–coated flasks seeded at ~50,000 cells per square centimeter. The doubling time of H9-derived human neural stem cells is approximately 48 h.28
Lentiviral sgRNA plasmid libraries
Oligonucleotides were synthesized on a CustomArray 90K array (CustomArray, Inc). The first round of PCR amplified sub-library specific sgRNA sequences (S01-S08). The second round of PCR introduced overhangs compatible for Gibson assembly (New England Biolabs) into LentiCRISPRv2GFP linearized with BsmBI. PCR reactions were monitored using SYBR green to ensure each reaction was terminated in the linear amplification phase. Gibson Assembly reaction products were purified and transformed into E. Coli DH10B (Life Technologies). To preserve the diversity of the library, at least 500-fold coverage in library representation was recovered in each transformation, and each transformed library was grown in liquid culture until OD 0.8–1.0 (~8 h). Individual sgRNA representation within each plasmid library (S01-S08) was determined by high-throughput 2×100 bp sequencing on the HiSeq 4000 instrument (Illumina) (Figure S4).
METHOD DETAILS
Lentivirus production
Lentiviral work was performed using BSL-2 Plus safety procedures, including production of lentiviral sgRNA-Cas9 libraries, culturing of transduced cells, and extraction of genomic DNA. Lentivirus was produced by co-transfecting the sgRNA-Cas9-GFP library vector with pCMV-VSV-G and pCMV-dR8.2 dpvr packaging plasmids into HEK293FT cells using Extreme Gene 9 transfection reagent (Millipore-Sigma) in serum-free media supplemented with GlutaMax-I (ThermoFisher) and 25uM chloroquine (Millipore-Sigma). After 8 h, media was replaced with high bovine serum albumin (Millipore-Sigma) (1.1g/100mL) in GlutaMax-supplemented OptiMem (ThermoFisher) with 10uM sodium butyrate (Millipore-Sigma). The virus-containing supernatant was collected 48 h after replacement. Viral supernatant was passed through a 0.45μM low-binding filter and immediately concentrated using Amicon Ultra-15 100kD filters. Concentrated virus was aliquoted, flash-frozen over dry ice and stored at −80C.
sgRNA library design: Defining genomic background controls
A set of background controls were defined by initially shuffling the location of randomly selected subset of targeted enhancers (n = 500). Next, the PhastCons elements underlying the original enhancers were shuffled within the shuffled enhancers; these pseudo-PhastCons elements residing in shuffled enhancers were termed ‘genomic background controls.’ Individual sgRNAs targeting the genomic background controls were scored and filtered in the same manner as the enhancers described above. In addition, these regions were filtered for possible regulatory function based on evidence of epigenetic activity across stem cell and brain tissue-types. Specifically, Dnase-hypersensitive sites (DHSs) identified in H9-derived neural progenitor cells were extended by 1,000 bp; genomic background control regions overlapping the extended DHSs by 1 bp were excluded from downstream analyses.84 In addition, a compendium of epigenetic atlases (Epilogos, https://epilogos.altius.org) was utilized to filter remaining genomic background controls for evidence of gene regulatory function based on chromatin state across a variety of human tissue- and cell-types. Chromatin states across ‘All 127 Roadmap Epigenomes’, ‘Brain’, and ‘ESC derived’ were used to filter genomic background controls based on cumulative evidence across the shuffled enhancers. The following criteria was applied to filter shuffled enhancers: evidence across all chromatin states (not including ‘Quiescent’ states) was set to ‘All 127 Roadmap Epigenomes’ < 5.0, ‘Brain’ < 0.5, and ‘ESC derived’ < 0.5. Genomic background regions within shuffled enhancers passing the filtering criteria for DHSs and chromatin state (described above) were included in downstream analyses.
sgRNA library design: Defining proliferation-decreasing genes
Proliferation-decreasing controls were identified from Wang et al. (2015).22 As sgRNAs exhibiting essential gene scores across a panel of cancer cell lines including KBM7, K562, Jiyoye, and Raji. Genes identified as proliferation-decreasing control targets exhibit a CRISPR-score (‘CS’) < −2.0 and ‘adjusted p value’ < 0.05 across all 4 cell lines performed by Wang et al. (2015). Individual sgRNAs (up to 10 sgRNAs per gene) targeting genes that meet these criteria were used as a control for proliferation-decreasing genes in this study.
sgRNA library design: Scoring and filtering sgRNAs
For enhancer regions, sgRNAs were designed and scored across PhastCons elements (46-species Placental Mammal Conserved Elements obtained from human genome version GRCh37/hg19 at the UCSC Genome Browser).37 For protein-coding regions, sgRNA designs were included from Wang et al. (2015).22 For enhancer, gene, and background control (see below) sgRNAs, the scoring metric was incorporated from Gilbert et al. 2014.83 Bowtie version 1.1.2 was used to score mismatches across the genome (version GRCh37/hg19); a score of e29m1 was used as a cutoff for potential sgRNAs.85 The scoring scheme is summarized as follows: the sgRNA sequence is extended to a 23-mer including the PAM motif NGG. Genome-wide mapping with Bowtie was used to score each sgRNA based on the matrix [9,9,9,9,9,9,9,9,19,19,19,19,19,28,28,28,28,28,28,28,0,19,40] where the PAM motif is represented at the end of the matrix. Then, using the filtering criteria -e29m1 allows sgRNAs with mismatches unlikely to result in cleavage, then excludes sgRNAs if more than 1 genome mapping event is reported. All sgRNAs were then filtered based on GC-content 20–80% and excluding poly-T sequences greater than 4 in length.
sgRNA library design: Defining non-targeting control sgRNAs
Non-targeting controls (n = 500) were generated from random sgRNA sequences that were processed by the same scoring procedure as enhancer regions described above, including filtering based on GC-content 20–80% and poly-T sequences greater than 4 in length. As with enhancer-targeting sgRNAs, Bowtie was used to filter sgRNAs based on the scoring matrix [10,10,10,10,10,10,10,10,10,10,10,10,10,10,10,10,10,10,10,10,0,19,40] where the PAM motif is represented at the end of the matrix. The following mapping criteria were used: -e39m1, –max, and –un; sgRNAs that yield no mapping with up to 3 mismatches permitted across the reference genome (GRCh37/hg19) were reported as unmapped; this subset of sgRNAs was included as non-targeting controls.
sgRNA library design: sgRNAs targeting enhancers and genomic background controls
To select sgRNAs, an R script processed filtered sgRNAs with the following procedure: each targeted PhastCons element was extended by 15 bp and the extended PhastCons element was partitioned into 30bp windows. Then, sgRNAs were randomly drawn with uniform probability from the filtered sgRNA set for each window such that up to 2 unique sgRNAs were selected per window, and at least 2 sgRNAs were selected per PhastCons element. Gencode (v19) was utilized to exclude conserved regions overlapping gene promoters (+/− 1 kb from TSS) and exons for all coding transcripts with evidence of level 1 or 2 support (validated or manual annotation).99
sgRNA library design: sgRNAs targeting protein-coding regions
For protein-coding regions, genes were selected for targeting based on expression levels measured by RNA-sequencing in two biological replicates of H9-derived human neural stem cells (FPKM >1 across two replicates); this yielded 10,674 expressed genes.24 All protein-coding sgRNAs from Wang et al. (2015) were processed through the scoring and filtering procedure described above.22 Next, two filtered protein-coding sgRNAs were randomly selected for each gene.
sgRNA library design: Specificity controls
To assess the proportion of on-target activity resulting from sgRNAs with mismatches in the targeting sequence, we generated a set of specificity controls. Gene targeting sgRNAs for CCND2 SOX2, and SRSF1 were included as a reference for on-target activity. The 20 nt PAM-adjacent targeting sequence was divided into 3 regions based on the tolerance of Cas9 to mismatches (Region 1 is the PAM-adjacent region (1–7 nt), Region 2 (8–12 nt), and Region 3 (13–20 nt) is distal to the PAM-adjacent region.83 To determine the sensitivity of on-target activity to mismatches (MM), single nucleotide mismatches were introduced into the target sequence within each region or spanning multiple regions and the number of mismatches ranged from 1 MM to 4MM.
sgRNA library design: Assigning sub-libraries for genetic screening
All sgRNAs were divided across 8 sub-libraries (‘subpools’) for large-scale transduction into hNSCs (6 enhancer-targeting subpools and 2 gene-targeting subpools). For each enhancer targeted by the screen, the enhancer was randomly assigned to one of the six subpools and all sgRNAs targeting that enhancer were assigned to the same subpool. For each gene targeted by the screen, the gene was assigned to one of the two subpools and all sgRNAs targeting that gene were assigned to the same subpool. In addition, each subpool included identical sets of non-targeting control sgRNAs (described above) and proliferation-decreasing sgRNAs identified in previous sgRNA-Cas9 genetic screens.22
Large-scale human neural stem cell transduction
Target cells in 25 cm tissue culture flasks at 250,000 cells per sq cm were transduced in low volume media containing 8ug/mL polybrene (Millipore-Sigma); 24 h after infection virus was removed and cells were passaged to a density of 50,000 cells per sq cm. To establish lentiviral titer, serial dilutions of concentrated virus were added to 25 cm tissue culture flasks and incubated for 24 h. Cells were then passaged to a density of 50,000 cells per sq cm and infection rate was determined 48 h later using GFP expression measured by flow cytometry (BD Accuri C6). For high-throughput screening, each sub-library was transduced by plating 50 million cells across eight 25 cm flasks and adding the appropriate volume of lentivirus to each flask. Initial multiplicity-of-infection was ~0.3–0.4 to achieve >1000-fold library coverage and infection was monitored after 24 h by flow cytometry for GFP expression over the course of the experiment (at 4, 8, and 12 cell divisions). Cells were harvested at each passage and stored as a cell pellet at −20C for genomic DNA extraction.
sgRNA library readout by high-throughput sequencing
Each sgRNA subpool library readout was performed using two steps of PCR as described in Chen et al. (2015).100 Second round PCR products were purified using column-based cleanup (New England Biolabs). Second round PCR products containing Illumina adapters at each timepoint belonging to a single subpool (e.g., S01) and biological replicate were combined and submitted for sequencing on the same channel(s) of a single sequencing run. Diluted libraries were spiked in with whole-exome libraries and sequenced using 2×100 bp reads on the HiSeq 4000 system (Illumina).
Mutation spectrum of individual conserved region sgRNAs
Individual sgRNA-Cas9-GFP plasmids were cloned, propagated in Stable Competent E. Coli. strains (NEB), and isolated using Endo-Free Maxi Prep Isolation Kits (Thermo-Fisher). Transient transfection of 4 million hNSC per construct was achieved using the Mouse Neural Stem Cell Nucleofection Kit (Amaxa). At 96 h post-transfection, GFP-positive cells were separated on an S3e Cell Sorter (BioRad) followed by DNA extraction. Amplicons from individual sgRNA-mediated were analyzed by high-throughput sequencing on an Illumina MiSeq instrument followed by insertion, deletion, and substitution analysis using CRISPResso2 (Figures S12 and S13).86
Validation assay of CRISPR-mediated proliferation phenotypes and comparison with published screens
sgRNAs for individual validation were cloned by annealing complementary oligonucleotide pairs (Integrated DNA technologies) and ligating into BsmBI-digested pLentiCRISPRv2. The pLentiCRISPRv2-GFP vector is the same one used in the CRISPR library screening, encoding the expression of a sgRNA, Cas9 and GFP. For enhancer and protein-coding targets, two sgRNAs were selected from the pooled CRISPR library. The resulting sgRNA expression vectors were individually packaged into lentivirus by transfection into HEK293T cells (Yale Cell Preparation and Analysis Core). Internally controlled competition assays to evaluate sgRNA proliferation phenotypes were performed as follows. First, human neural stem cells (hNSC) were seeded in 24-well plates on Corning Growth-Factor Reduced Matrigel at a density of 30k cells/cm2 and transduced at a low multiplicity-of-infection (MOI <0.5, 15–30% GFP-positive). Cells were resuspended in hNSC culture media and an initial proportion of GFP-positive cells was measured using a Cytoflex LR flow cytometer. Additional timepoints of GFP-positive cell proportions was collected at 4, 8 and 10 culture passages. All sgRNA sequences used, as well as the backbone sequence, are included in Table S14. Enhancers with negative effects were chosen based on the strength of the observed phenotype and whether they had a putative gene target based on chromatin interactions detected in the PsychEncode Hi-C dataset described above.70 Proliferation increasing enhancer targets were selected based on the strength of the phenotype across timepoints and whether they were located near a gene of potential biological interest (TNIK, which is implicated in WNT signaling and DLGAP1, which has been implicated in risk for obsessive-compulsive disorder).54,55 The smaller number of enhancer disruptions with positive effects on proliferation coupled with the sparsity of the PsychEncode Hi-C dataset required us to use a nearest-gene approach for these candidates.
To identify overlapping genes detected in this study and in Tian et al. 2019, we intersected our gene hits with hits reported at each time point and cell type in Table S1 of that paper, selecting genes with p values < 0.05.36 The values reported in the main text are genes that show consistent negative effects on proliferation and viability in both studies.
Western Blot analysis of CRISPR-mediated knockdown
To quantify the extent of protein knockdown after targeting with the LentiCrisprv2GFP vector, we obtained guide sequences targeting UBQLN2. hNSCs were transduced with lentivirus prepared as described above at MOI 0.30 with either non-targeting vector or UBQLN2-targeting vector and sorted for GFP presence after 12 days. Cells were flash frozen and protein was extracted using RIPA lysis buffer and 5 min of sonication. Protein was run on a 10% gel and wet-transferred to PVDF. Western blots were probed with the following primary antibody dilutions: UBQLN2 primary antibody, 1:1,000 (Cell Signaling #85509S), Actin 1:1,000 (Abcam ab216070). Blots were imaged using ECL (Biorad Calirty Max Western ECL Substrate, #175063) on X-ray film (LabForce HyBlot CL 114J51). Protein knockdown was quantified using ImageJ.98
Real-time quantitative PCR analysis of CRISPR-mediated knockdown
LentiCRISPRv2GFP vectors were designed with guides targeting GRN and TFRC using sequences previously described.36 hNSCs were transduced with lentivirus prepared as described above in parallel, along with non-targeting controls, at MOI 0.30 and sorted for GFP positivity after 12 days. mRNA was extracted using Qiagen RNeasy kit (ID 74304) and converted into cDNA using Invitrogen SuperScript III First Strand Synthesis SuperMix (#18080400). Transcript levels were quantified using Roche LightCycler 480 PCR Thermal Cycler and SYBR Green I reagents (Roche Diagnostics 04707516001) using primers for GRN, TFRC, and GAPDH housekeeping control as previously described.36
Human neural stem cell neuronal differentiation
To assess the competency of the Gibco hNSCs to form neurons, we compared two methods for differentiation. For both, cells were plated at a density of 2.5 × 104 cells/cm2 on Gibco CELLstart CTS (# A10142–01). We utilized the manufacturer’s recommended protocol, in which the bFGF and EGF growth factors are removed from the growth media to induce neural differentiation. In addition, we performed a second differentiation protocol using Gibco neurobasal media (# 21103) supplemented with Gibco serum-free B27 (#17504).28,29 Cell cultures were visually assessed and lysed at 21 days, when RNA was collected and purified using the Qiagen RNeasy Plus mini kit (# 74034). Marker genes were assessed by qPCR using a Roche LightCycler.
QUANTIFICATION AND STATISTICAL ANALYSIS
K-means clustering for identification of cortical-enriched enhancers
H3K27ac data from human cortex was compared with publicly available H3K27ac datasets as follows: A single composite multi-sample enhancer annotation from developing cortex, limb, embryonic stem cells, and select adult tissues profiled by the Roadmap Epigenomics Project was generated by merging replicate peaks across all samples using a 1bp overlap.101 The level of H3K27ac signal in each region for each sample was quantified by averaging read counts per kilobase per million mapped reads (RPKM) in each region from each replicate. Each region was represented by a vector of a length equal to the total number of tissues considered, with each point representing the RPKM value of marking in that region for a single tissue. Each vector was normalized by subtracting the mean of all tissue quantifications from each individual tissue quantification, divided by the standard deviation of values for that vector. The matrix of these normalized tissue quantification values was then subjected to k-means clustering using R to identify sets of sites exhibiting the strongest marking in each tissue compared to all other samples in the comparison. We used GREAT version 3.0.0 (http://great.stanford.edu/) to identify biological functions and processes showing significant enrichment for each set of enhancers.87
Identification of proliferation phenotypes
To quantify the biological effects of disruption on cell proliferation, we utilized a model-based analysis of genome-wide CRISPR-Cas9 knockout (MAGeCK) which models read counts using the negative binomial distribution.41 First, each sgRNA is assigned to a target representing either a gene or conserved region. Each target can contain one or more sgRNAs that can be jointly modeled in the MAGeCK analytical pipeline. Sequencing reads were initially filtered using CutAdapt version 1.16 and options (-j 20, -l 20, -g GACGAAACACCG, –trimmed only).95 Trimmed reads were used as input into MAGeCK version 0.5.8 which was performed for replicates individually and jointly. The following options were used: –norm-method control, –control-sgrna NTC_sgRNA_ID.txt. MAGeCK analysis was conducted for each sub-library independently, and the same panel of non-targeting controls was included in each sub-library and used to normalize read counts. The results of each sub-library were combined using custom R scripts. For the replication plots (Figures 1B and 1C), MAGeCK was performed independently for each replicate. For all subsequent analysis and values reported in tables, the results are from jointly modeling the biological effects across two replicates. MAGeCK provided an estimate of the biological effect following genetic disruption on cellular proliferation termed the β score. For each conserved region, the β score is associated with a permutation-based p value determined by permuting sgRNAs targeting each conserved region and evaluating the probability of observing the biological effect within the set of genomic background control sgRNAs.
Cell proliferation phenotypes were identified using linear-discriminant analysis (LDA) on a training set then applying the learned classifier to the full dataset. Similar approaches have been used to characterize phenotypes following high-throughput editing experiments.42 LDA produces a classification that maximizes the separability of the input groups. The training set was defined as follows: the ‘decreasing’ population includes 500 genes decreasing proliferation across a panel of cancer cell lines (Wang et al. 2015), the ‘neutral’ population is all regions within the genomic background, and the ‘increasing’ population is comprised of the top 1% of proliferation increasing regions identified at each time point (4, 8, and 12 cell divisions) (Figure S9).22 All sgRNAs for the training set were included in the hNSC proliferation screening experiments.
A composite score was then obtained by multiplying the MAGeCK β score by the permutation-based p value for phenotype classification using LDA. LDA was performed jointly across timepoints, and each disrupted region (gene and conserved regions within enhancers) was classified based on a composite of the regression-based effect size and p value. LDA was performed using the R package MASS (v7.3–54) and initialized using a uniform probability distribution for class membership. The resulting proliferation phenotype classifications (labeled as ‘negative, ‘neutral’, or ‘positive’ in Tables S2 and S3) were filtered for reproducible effect sign across biological replicates and consistent phenotype classification across 4, 8, and 12 cell divisions (e.g., proliferation-decreasing at 4, 8 and 12 cell divisions). Disruptions that did not meet this filter (e.g., that showed inconsistent effects across time points), were labeled as “dynamic” in Tables S2 and S3. An empirical false-discovery rate (FDR) was estimated as the proportion of genomic background control regions that were called as proliferation-increasing or proliferation-decreasing relative to regions defined as proliferation-increasing or proliferation-decreasing in the training set.
MAGeCK β scores and proliferation phenotype classifications at 4, 8, and 12 cell divisions are available for visualization in the UCSC Genome Browser (GRCh37/hg19): http://noonan.ycga.yale.edu/noonan_public/Geller_Enhancer_Screen/hub.txt.
Principal component analysis and Pearson correlation analysis
To extract latent factors and perform correlation analyses, we constructed a single composite annotation of all β scores for each ‘subpool’ across multiple timepoints. These Beta scores were assembled into a single data matrix using custom R scripts. Each row in this matrix represented a single conserved region or gene disruption, and each column represented the Beta score at 4, 8, or 12 cell divisions. Principal component analysis was performed on this matrix using R (Figure 2A; Figure 3A). Pearson correlation analysis was also carried out using R (Figure S7).
Gene Ontology and pathway analysis
We performed Gene Ontology enrichment analyses for protein-coding genes displaying proliferation-decreasing phenotypes using the Database for Annotation, Visualization, and Integrated Discovery (DAVID v6.8).90,91 Default settings for functional annotation were utilized. For pathway analysis, we used the ReactomePA package (v1.14.0) for R using default settings.
Transcription factor binding site enrichment analysis
We collected 572 transcription factor binding site (TFBS) predictions from the JASPAR 2018 database (https://jaspar2018.genereg.net/) that overlap with at least one conserved region included in this study.65 We used motifs from the 2018 JASPAR CORE Vertebrate collection that were already mapped to human genome version GRCh37/hg19 at the UCSC Genome Browser (score threshold of 400 or greater and p less than or equal to 10−4). To identify TFBS that are enriched within conserved regions that have biological effects on proliferation, we conducted hypergeometric tests using custom R scripts. The hypergeometric test was conducted for each TFBS independently. Each test was constructed to compare the abundance of TFBS in the category of interest (‘severe’, ‘strong’, ‘all’, or ‘positive’) and compared to ‘neutral’ phenotype conserved regions. The hypergeometric p value for assessing the enrichment of each TFBS for all sites were then corrected for multiple-testing using the Benjamini-Hochberg procedure.102
Enhancer proliferation phenotype permutation analysis
We used permutation analysis to identify enhancers containing a significant excess of proliferation phenotypes. The procedure was implemented using custom R scripts. For each permutation, proliferation phenotypes were randomly shuffled across all conserved regions. We performed 100,000 permutations of the full dataset. The significance of proliferation phenotypes within an enhancer was assessed based on the fraction of permutations where the number of proliferation phenotypes was greater than or equal to the number of proliferation phenotypes observed within the enhancer. The resulting permutation-based p values were then corrected for multiple-testing using the Benjamini-Hochberg procedure.102
Identification of enhancer-gene interactions
Hi-C data from human neural precursor cells were generated by the PsychEncode Consortium.70 High-confidence loop calls and 50-kb topologically associating domains (TADs) were made available by the authors through the Synapse repository. The Juicer tool suite was utilized to identify contact domains using the default settings and the ‘arrowhead’ algorithm.96 Custom R scripts implemented the following procedure to generate enhancer-gene interactions. Enhancers were defined as regions containing at least one proliferation phenotype. Gencode (v19) was utilized to define gene regions including the promoter (+10 kb), transcription start site, and gene body.99 Genes harboring a proliferation phenotype were used to identify enhancer-gene interactions. For loop calls, anchor points were used to identify enhancer-gene interactions. For contact domains, enhancers were associated with each gene harboring a phenotype within the contact domain. High-confidence interactions were reported as enhancer-gene interactions derived from loop calls and contact domains. In addition, topologically associated domains (TADs) were used to identify enhancer-gene interactions. For TADs, enhancers with at least one phenotype within the TAD were associated with all genes harboring proliferation phenotypes within the TAD.
Cell type-specific expression of gene hits in fetal human cortex
Single cell RNA sequencing (scRNA-seq) datasets collected from embryonic and fetal human cortex were obtained from Bhaduri et al. 2021 and Eze et al. 2021.74,75 We collected processed counts matrices associated with samples ranging from Carnegie stage 14 (CS14) to gestational week 25 (GW25) across all represented regions of the cortex. These datasets were loaded into the Seurat R package (v4.3.0) and each dataset was filtered to retain only cells with a) a minimum of 750 represented features and b) features that were expressed in at least 50 cells.92 Datasets with fewer than 100 cells after filtering were considered low quality and discarded. Additionally, cells with read or feature counts greater than two standard deviations from the mean and cells with greater than 10% mitochondrial read percentage were considered doublets, empty, or dying cells and were filtered out.
Resulting high-quality cells in each dataset were then merged into a single Seurat object. This merged counts matrix was normalized (NormalizeData), subset to the top 2000 highly variable features (FindVariableFeatures), and then scaled (ScaleData). Datasets were then integrated with FastMNN (RunFastMNN in SeuratWrappers v0.3.0 (https://github.com/satijalab/seurat-wrappers), params: k = 30, d = 50) to remove batch-related technical variation.93 We then calculated the 30 nearest neighbors (FindNearestNeighbors, params: k = 30, reduction = ‘mnn’) for each cell using the 50 integration features calculated from FastMNN and this was used to cluster similar cells with the Louvain neighborhood aggregation algorithm in Seurat (FindClusters, resolution = 0.85). Clusters were assigned a cell type classification by comparing known markers of cortical cell types with per-cluster marker genes calculated in Seurat (FindAllMarkers, params: min.pct = 0.25, only.pos = TRUE). Marker gene expression and cluster assignments were visualized with a UMAP embedding calculated in Seurat (RunUMAP, params: reduction = ‘mnn’, k = 30).
Cortical progenitor populations were then isolated, re-normalized, and re-scaled as above. These included three progenitor types: intermediate progenitor cells (IPC) expressing EOMES, outer radial glia (ORG) expressing HOPX and PTPRZ1, and radial glia (RG) which lacked expression of these marker genes.76,77 Additionally, the cell cycle phase of each progenitor cluster was assigned using the expression of TOP2A and MKI67 marking G2/M cells.78 The average expression of each gene that showed a proliferation phenotype after enhancer disruption was extracted using Seurat (AverageExpression), resulting in a cell-type-by-gene matrix of average scaled gene expression values. We clustered the average scaled expression values for each gene using the k-means algorithm in the stats R package (kmeans, params: centers = 10). The average expression values for each cluster were visualized separately using ComplexHeatmaps and labeled with their respective cell type-specific expression profiles.94 Clusters with relevant expression patterns were isolated and visualized with ComplexHeatmaps alongside cell type annotations of cell cycle phase and average expression across all genes in the cluster and gene annotations of proliferation phenotypes at T4, T8, and T12. Expression profiles of candidate genes for each cluster were visualized on the UMAP embedding using density plots (plot_density from Nebulosa v1.2.0 R package).97
Gene ontology (GO) analyses were performed using the GUI-based R tool ShinyGO.80 The background gene set was constructed by intersecting all gene targets in our proliferation assay with the set of genes detected in the developing human cortical progenitor scRNA-seq atlas. The foreground query gene sets were composed of genes in all clusters with a shared progenitor-type bias: IPC-biased genes (clusters 1, 9), ORG-biased genes (clusters 2, 7, 8), G2/M-biased genes (clusters 4, 6), and RG (G1/S)-biased genes (cluster 10). Enriched KEGG pathways and enriched biological process GO terms were reported for each gene set respectively.79
Supplementary Material
Highlights.
CRISPR screens identify enhancers and genes required for hNSC proliferation
Genes implicated in hNSC proliferation are linked to neurodevelopmental disorders
Enhancer disruptions can have effects comparable to gene disruptions
Disruptions in human accelerated regions alter hNSC proliferation
ACKNOWLEDGMENTS
We thank R. Muhle for comments on the manuscript; S. Mane, K. Bilguvar, and C. Castaldi at the Yale Center for Genome Analysis for sequencing data; B. Fontes and R. Airo at Yale Environmental Health and Safety for supporting the lentiviral work; A. Bhaduri for assistance with accessing and interpreting the human fetal brain single-cell transcriptome data; and the members of the PsychENCODE Consortium for providing chromosome conformation capture data to the research community. This work was supported by National Institutes of Health awards R01 GM094780 (to J.P.N.) and R01 HD102030 (to J.P.N.), a Research Award from the Simons Foundation (to J.P.N.), a Deutsche Forschungsgemeinschaft (DFG) Research Fellowship UE 194/1-1 (to S.U.), an NSF Graduate Research Fellowship (to M.A.N.), and an Autism Speaks Predoctoral Fellowship (to E.G.). This research program and related results were also made possible by support from the NOMIS Foundation (to J.P.N.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.113693.
REFERENCES
- 1.Nord AS, Pattabiraman K, Visel A, and Rubenstein JLR (2015). Genomic Perspectives of Transcriptional Regulation in Forebrain Development. Neuron 85, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reilly SK, Yin J, Ayoub AE, Emera D, Leng J, Cotney J, Sarro R, Rakic P, and Noonan JP (2015). Evolutionary changes in promoter and enhancer activity during human corticogenesis. Science 347, 1155–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reilly SK, and Noonan JP (2016). Evolution of Gene Regulation in Humans. Annu. Rev. Genomics Hum. Genet. 17, 45–67. [DOI] [PubMed] [Google Scholar]
- 4.Prabhakar S, Noonan JP, Pääbo S, and Rubin EM (2006). Accelerated Evolution of Conserved Noncoding Sequences in Humans. Science 314, 786. [DOI] [PubMed] [Google Scholar]
- 5.Pollard KS, Salama SR, Lambert N, Lambot M-A, Coppens S, Pedersen JS, Katzman S, King B, Onodera C, Siepel A, et al. (2006). An RNA gene expressed during cortical development evolved rapidly in humans. Nature 443, 167–172. [DOI] [PubMed] [Google Scholar]
- 6.Capra JA, Erwin GD, McKinsey G, Rubenstein JLR, and Pollard KS (2013). Many human accelerated regions are developmental enhancers. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20130025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boyd JL, Skove SL, Rouanet JP, Pilaz L-J, Bepler T, Gordân R, Wray GA, and Silver DL (2015). Human-Chimpanzee Differences in a FZD8 Enhancer Alter Cell-Cycle Dynamics in the Developing Neocortex. Curr. Biol. 25, 772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de la Torre-Ubieta L, Stein JL, Won H, Opland CK, Liang D, Lu D, and Geschwind DH (2018). The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 172, 289–304.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Won H, de la Torre-Ubieta L, Stein JL, Parikshak NN, Huang J, Opland CK, Gandal MJ, Sutton GJ, Hormozdiari F, Lu D, et al. (2016). Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Won H, Huang J, Opland CK, Hartl CL, and Geschwind DH (2019). Human evolved regulatory elements modulate genes involved in cortical expansion and neurodevelopmental disease susceptibility. Nat. Commun. 10, 2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, Sunkin SM, Li Z, Shin Y, Zhu Y, et al. (2018). Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362, eaat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.An J-Y, Lin K, Zhu L, Werling DM, Dong S, Brand H, Wang HZ, Zhao X, Schwartz GB, Collins RL, et al. (2018). Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 362, eaat6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schork AJ, Won H, Appadurai V, Nudel R, Gandal M, Delaneau O, Revsbech Christiansen M, Hougaard DM, Bækved-Hansen M, Bybjerg-Grauholm J, et al. (2019). A genome-wide association study of shared risk across psychiatric disorders implicates gene regulation during fetal neurodevelopment. Nat. Neurosci. 22, 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanjana NE, Wright J, Zheng K, Shalem O, Fontanillas P, Joung J, Cheng C, Regev A, and Zhang F (2016). High-resolution interrogation of functional elements in the noncoding genome. Science 353, 1545–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, Chen DD, Schupp PG, Vinjamur DS, Garcia SP, et al. (2015). BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shalem O, Sanjana NE, and Zhang F (2015). High-throughput functional genomics using CRISPR–Cas9. Nat. Rev. Genet. 16, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korkmaz G, Lopes R, Ugalde AP, Nevedomskaya E, Han R, Myacheva K, Zwart W, Elkon R, and Agami R (2016). Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat. Biotechnol. 34, 192–198. [DOI] [PubMed] [Google Scholar]
- 18.Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, Kane M, Cleary B, Lander ES, and Engreitz JM (2016). Systematic mapping of functional enhancer–promoter connections with CRISPR interference. Science 354, 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR, Anyoha R, Doughty BR, Patwardhan TA, et al. (2019). Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 51, 1664–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reilly SK, Gosai SJ, Gutierrez A, Mackay-Smith A, Ulirsch JC, Kanai M, Mouri K, Berenzy D, Kales S, Butler GM, et al. (2021). Direct characterization of cis-regulatory elements and functional dissection of complex genetic associations using HCR–FlowFISH. Nat. Genet. 53, 1166–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris JA, Caragine C, Daniloski Z, Domingo J, Barry T, Lu L, Davis K, Ziosi M, Glinos DA, Hao S, et al. (2023). Discovery of target genes and pathways at GWAS loci by pooled single-cell CRISPR screens. Science 380, eadh7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, and Sabatini DM (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorkin DU, Barozzi I, Zhao Y, Zhang Y, Huang H, Lee AY, Li B, Chiou J, Wildberg A, Ding B, et al. (2020). An atlas of dynamic chromatin landscapes in mouse fetal development. Nature 583, 744–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cotney J, Muhle RA, Sanders SJ, Liu L, Willsey AJ, Niu W, Liu W, Klei L, Lei J, Yin J, et al. (2015). The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat. Commun. 6, 6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anthony TE, Klein C, Fishell G, and Heintz N (2004). Radial Glia Serve as Neuronal Progenitors in All Regions of the Central Nervous System. Neuron 41, 881–890. [DOI] [PubMed] [Google Scholar]
- 26.Geschwind DH, and Rakic P (2013). Cortical Evolution: Judge the Brain by Its Cover. Neuron 80, 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lui JH, Hansen DV, and Kriegstein AR (2011). Development and Evolution of the Human Neocortex. Cell 146, 18–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shin S, and Vemuri M (2009). Culture and Differentiation of Human Neural Stem Cells. In Protocols for Neural Cell Culture Springer Protocols Handbooks., Doering. and Laurie C, eds. (Humana Press; ), pp. 51–73. [Google Scholar]
- 29.Brewer GJ (1995). Serum-free B27/neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum, and dentate gyrus. J. Neurosci. Res. 42, 674–683. [DOI] [PubMed] [Google Scholar]
- 30.Lodato S, and Arlotta P (2015). Generating Neuronal Diversity in the Mammalian Cerebral Cortex. Annu. Rev. Cell Dev. Biol. 31, 699–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zuo H, Wood WM, Sherafat A, Hill RA, Lu QR, and Nishiyama A (2018). Age-Dependent Decline in Fate Switch from NG2 Cells to Astrocytes After Olig2 Deletion. J. Neurosci. 38, 2359–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elbaz B, and Popko B (2019). Molecular Control of Oligodendrocyte Development. Trends Neurosci. 42, 263–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sock E, and Wegner M (2021). Using the lineage determinants Olig2 and Sox10 to explore transcriptional regulation of oligodendrocyte development. Dev. Neurobiol. 81, 892–901. [DOI] [PubMed] [Google Scholar]
- 34.Sakurai K, and Osumi N (2008). The neurogenesis-controlling factor, Pax6, inhibits proliferation and promotes maturation in murine astrocytes. J. Neurosci. 28, 4604–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bani-Yaghoub M, Tremblay RG, Lei JX, Zhang D, Zurakowski B, Sandhu JK, Smith B, Ribecco-Lutkiewicz M, Kennedy J, Walker PR, and Sikorska M (2006). Role of Sox2 in the development of the mouse neocortex. Dev. Biol. 295, 52–66. [DOI] [PubMed] [Google Scholar]
- 36.Tian R, Gachechiladze MA, Ludwig CH, Laurie MT, Hong JY, Nathaniel D, Prabhu AV, Fernandopulle MS, Patel R, Abshari M, et al. (2019). CRISPR Interference-Based Platform for Multimodal Genetic Screens in Human iPSC-Derived Neurons. Neuron 104, 239–255.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prabhakar S, Visel A, Akiyama JA, Shoukry M, Lewis KD, Holt A, Plajzer-Frick I, Morrison H, FitzPatrick DR, Afzal V, et al. (2008). Human-Specific Gain of Function in a Developmental Enhancer. Science 321, 1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dutrow EV, Emera D, Yim K, Uebbing S, Kocher AA, Krenzer M, Nottoli T, Burkhardt DB, Krishnaswamy S, Louvi A, and Noonan JP (2022). Modeling uniquely human gene regulatory function via targeted humanization of the mouse genome. Nat. Commun. 13, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cotney J, Leng J, Yin J, Reilly SK, DeMare LE, Emera D, Ayoub AE, Rakic P, and Noonan JP (2013). The Evolution of Lineage-Specific Regulatory Activities in the Human Embryonic Limb. Cell 154, 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li W, Köster J, Xu H, Chen C-H, Xiao T, Liu JS, Brown M, and Liu XS (2015). Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol. 16, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, Janizek JD, Huang X, Starita LM, and Shendure J (2018). Accurate classification of BRCA1 variants with saturation genome editing. Nature 562, 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Graham V, Khudyakov J, Ellis P, and Pevny L (2003). SOX2 Functions to Maintain Neural Progenitor Identity. Neuron 39, 749–765. [DOI] [PubMed] [Google Scholar]
- 44.Mirzaa G, Parry DA, Fry AE, Giamanco KA, Schwartzentruber J, Vanstone M, Logan CV, Roberts N, Johnson CA, Singh S, et al. (2014). De novo CCND2 mutations leading to stabilization of cyclin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Nat. Genet. 46, 510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stevens HE, Smith KM, Rash BG, and Vaccarino FM (2010). Neural Stem Cell Regulation, Fibroblast Growth Factors, and the Developmental Origins of Neuropsychiatric Disorders. Front. Neurosci. 4, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuwahara A, Sakai H, Xu Y, Itoh Y, Hirabayashi Y, and Gotoh Y (2014). Tcf3 Represses Wnt–b-Catenin Signaling and Maintains Neural Stem Cell Population during Neocortical Development. PLoS One 9, e94408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jayaraman D, Bae B-I, and Walsh CA (2018). The Genetics of Primary Microcephaly. Annu. Rev. Genom. Hum. Genet. 19, 177–200. [DOI] [PubMed] [Google Scholar]
- 48.Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. (2015). Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nascimento RMP, Otto PA, de Brouwer APM, and Vianna-Morgante AM (2006). UBE2A, Which Encodes a Ubiquitin-Conjugating Enzyme, Is Mutated in a Novel X-Linked Mental Retardation Syndrome. Am. J. Hum. Genet. 79, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sousa SB, Jenkins D, Chanudet E, Tasseva G, Ishida M, Anderson G, Docker J, Ryten M, Sa J, Saraiva JM, et al. (2014). Gain-of-function mutations in the phosphatidylserine synthase 1 (PTDSS1) gene cause Lenz-Majewski syndrome. Nat. Genet. 46, 70–76. [DOI] [PubMed] [Google Scholar]
- 51.Petrovic A, Keller J, Liu Y, Overlack K, John J, Dimitrova YN, Jenni S, van Gerwen S, Stege P, Wohlgemuth S, et al. (2016). Structure of the MIS12 Complex and Molecular Basis of Its Interaction with CENP-C at Human Kinetochores. Cell 167, 1028–1040.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brunskill EW, Ehrman LA, Williams MT, Klanke J, Hammer D, Schaefer TL, Sah R, Il GWD, Potter SS, and Vorhees CV (2005). Abnormal neurodevelopment, neurosignaling and behaviour in Npas3-deficient mice. Eur. J. Neurosci. 22, 1265–1276. [DOI] [PubMed] [Google Scholar]
- 53.Lim ET, Raychaudhuri S, Sanders SJ, Stevens C, Sabo A, MacArthur DG, Neale BM, Kirby A, Ruderfer DM, Fromer M, et al. (2013). Rare Complete Knockouts in Humans: Population Distribution and Significant Role in Autism Spectrum Disorders. Neuron 77, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mahmoudi T, Li VSW, Ng SS, Taouatas N, Vries RGJ, Mohammed S, Heck AJ, and Clevers H (2009). The kinase TNIK is an essential activator of Wnt target genes. EMBO J. 28, 3329–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stewart SE, Yu D, Scharf JM, Neale BM, Fagerness JA, Mathews CA, Arnold PD, Evans PD, Gamazon ER, Davis LK, et al. (2013). Genome-wide association study of obsessive-compulsive disorder. Mol. Psychiatry 18, 788–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deciphering Developmental Disorders Study; Gerety SS, Jones WD, Kogelenberg M. van, King DA, McRae J, Morley KI, Parthiban V, Al-Turki S, Ambridge K, et al. (2015). Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, O’Donnell-Luria AH, Hill AJ, Cummings BB, et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An J-Y, Peng M, Collins R, Grove J, Klei L, et al. (2020). Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 180, 568–584.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sey NYA, Hu B, Mah W, Fauni H, McAfee JC, Rajarajan P, Brennand KJ, Akbarian S, and Won H (2020). A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. Nat. Neurosci. 23, 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, et al. (2018). The Reactome Pathway Knowledgebase. Nucleic Acids Res. 46, D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rakic P, Ayoub AE, Breunig JJ, and Dominguez MH (2009). Decision by division: making cortical maps. Trends Neurosci. 32, 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Borrell V, Cárdenas A, Ciceri G, Galcerán J, Flames N, Pla R, Nóbrega-Pereira S, García-Frigola C, Peregrín S, Zhao Z, et al. (2012). Slit/Robo Signaling Modulates the Proliferation of Central Nervous System Progenitors. Neuron 76, 338–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shimogori T, Banuchi V, Ng HY, Strauss JB, and Grove EA (2004). Embryonic signaling centers expressing BMP, WNT and FGF proteins interact to pattern the cerebral cortex. Development 131, 5639–5647. [DOI] [PubMed] [Google Scholar]
- 65.Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Bessy A, Chèneby J, Kulkarni SR, Tan G, et al. (2018). JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46, D260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cho G-S, Park D-S, Choi S-C, and Han J-K (2017). Tbx2 regulates anterior neural specification by repressing FGF signaling pathway. Dev. Biol. 421, 183–193. [DOI] [PubMed] [Google Scholar]
- 67.Kamm GB, Pisciottano F, Kliger R, and Franchini LF (2013). The Developmental Brain Gene NPAS3 Contains the Largest Number of Accelerated Regulatory Sequences in the Human Genome. Mol. Biol. Evol. 30, 1088–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Julian LM, and Blais A (2015). Transcriptional control of stem cell fate by E2Fs and pocket proteins. Front. Genet. 6, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shim K, Minowada G, Coling DE, and Martin GR (2005). Sprouty2, a Mouse Deafness Gene, Regulates Cell Fate Decisions in the Auditory Sensory Epithelium by Antagonizing FGF Signaling. Dev. Cell 8, 553–564. [DOI] [PubMed] [Google Scholar]
- 70.Rajarajan P, Borrman T, Liao W, Schrode N, Flaherty E, Casiño C, Powell S, Yashaswini C, LaMarca EA, Kassim B, et al. (2018). Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science 362, eaat4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang B, Jain S, Song H, Fu M, Heuckeroth RO, Erlich JM, Jay PY, and Milbrandt J (2007). Mice lacking sister chromatid cohesion protein PDS5B exhibit developmental abnormalities reminiscent of Cornelia de Lange syndrome. Development 134, 3191–3201. [DOI] [PubMed] [Google Scholar]
- 72.Petzold KM, Naumann H, and Spagnoli FM (2013). Rho signalling restriction by the RhoGAP Stard13 integrates growth and morphogenesis in the pancreas. Development 140, 126–135. [DOI] [PubMed] [Google Scholar]
- 73.Zollino M, Orteschi D, Murdolo M, Lattante S, Battaglia D, Stefanini C, Mercuri E, Chiurazzi P, Neri G, and Marangi G (2012). Mutations in KANSL1 cause the 17q21.31 microdeletion syndrome phenotype. Nat. Genet. 44, 636–638. [DOI] [PubMed] [Google Scholar]
- 74.Bhaduri A, Sandoval-Espinosa C, Otero-Garcia M, Oh I, Yin R, Eze UC, Nowakowski TJ, and Kriegstein AR (2021). An atlas of cortical arealization identifies dynamic molecular signatures. Nature 598, 200–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eze UC, Bhaduri A, Haeussler M, Nowakowski TJ, and Kriegstein AR (2021). Single-cell atlas of early human brain development highlights heterogeneity of human neuroepithelial cells and early radial glia. Nat. Neurosci. 24, 584–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kowalczyk T, Pontious A, Englund C, Daza RAM, Bedogni F, Hodge R, Attardo A, Bell C, Huttner WB, and Hevner RF (2009). Intermediate Neuronal Progenitors (Basal Progenitors) Produce Pyramidal–Projection Neurons for All Layers of Cerebral Cortex. Cereb. Cortex 19, 2439–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pollen AA, Nowakowski TJ, Chen J, Retallack H, Sandoval-Espinosa C, Nicholas CR, Shuga J, Liu S, Oldham MC, Diaz A, et al. (2015). Molecular identity of human outer radial glia during cortical development 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Klochendler A, Weinberg-Corem N, Moran M, Swisa A, Pochet N, Savova V, Vikeså J, Van de Peer Y, Brandeis M, Regev A, et al. (2012). A Transgenic Mouse Marking Live Replicating Cells Reveals In Vivo Transcriptional Program of Proliferation. Dev. Cell 23, 681–690. [DOI] [PubMed] [Google Scholar]
- 79.Kanehisa M, and Goto S (2000). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ge SX, Jung D, and Yao R (2019). ShinyGO: a graphical enrichment tool for animals and plants. Bioinformatics 36, 2628–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. (2016). Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Osterwalder M, Barozzi I, Tissières V, Fukuda-Yuzawa Y, Mannion BJ, Afzal SY, Lee EA, Zhu Y, Plajzer-Frick I, Pickle CS, et al. (2018). Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Roadmap Epigenomics Consortium; Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Clement K, Rees H, Canver MC, Gehrke JM, Farouni R, Hsu JY, Cole MA, Liu DR, Joung JK, Bauer DE, and Pinello L (2019). CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 37, 224–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, Irizarry RA, Liu JS, Brown M, and Liu XS (2014). MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Venables WN, and Ripley BD (2002). Modern Applied Statistics with S, Fourth edition (Springer; ). [Google Scholar]
- 90.Huang D, Sherman BT, and Lempicki RA (2008). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources 4. [DOI] [PubMed] [Google Scholar]
- 91.Huang DW, Sherman BT, and Lempicki RA (2009). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Haghverdi L, Lun ATL, Morgan MD, and Marioni JC (2018). Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gu Z (2022). Complex heatmap visualization. iMeta 1, e43. [Google Scholar]
- 95.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetJ 17, 10–12. [Google Scholar]
- 96.Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, and Aiden EL (2016). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Alquicira-Hernandez J, and Powell JE (2021). Nebulosa recovers single-cell gene expression signals by kernel density estimation. Bioinformatics 37, 2485–2487. [DOI] [PubMed] [Google Scholar]
- 98.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. (2012). GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, Scott DA, Song J, Pan JQ, Weissleder R, et al. (2015). Genome-wide CRISPR Screen in a Mouse Model of Tumor Growth and Metastasis. Cell 160, 1246–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. (2010). The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol. 28, 1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc.: Ser. B (Methodol.) 57, 289–300. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data for massively parallel sgRNA-Cas9 disruption and individual replicate disruption are available through the Gene Expression Omnibus as of the date of publication. Accession numbers are listed in the key resources table. This paper analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
All original code generated for the manuscript is available at Zenodo as of the date of publication. The DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-UBQLN2 1:1000 | Cell Signaling | Cat. #85509S; RRID:AB_2800056 |
| Anti-Actin 1:1000 | Abcam | Cat. #ab216070 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| E. Coli DH10B | Life Technologies | Cat. #18290015 |
| NEB Stable Competent E. Coli. Strain | New England Biolabs | Cat. #C3040H |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Extreme Gene 9 transfection reagent | Millipore-Sigma | Cat. #6365787001 |
| GlutaMax-I | ThermoFisher | Cat. #A1286001 |
| Chloroquine | Millipore-Sigma | Cat. #C6628 |
| Bovine serum albumin | Millipore-Sigma | Cat. #A8022 |
| Sodium butyrate | Millipore-Sigma | Cat. #303410 |
| GlutaMax-supplemented OptiMem | ThermoFisher | Cat. #51–985-034 |
| Recombinant human epidermal growth factor | ConnStem | Cat. #E1000 |
| Recombinant human fibroblast growth factor | ConnStem | Cat. #F1001 |
| StemPro neural supplement | Life Technologies | Cat. #A1050801 |
| Corning Matrigel GFR membrane matrix | Fisher Scientific | Cat. #CB-40230C |
| Polybrene | Millipore-Sigma | Cat. #TR-1003-G |
|
| ||
| Critical commercial assays | ||
|
| ||
| Gibson assembly Master Mix | New England Biolabs | Cat. #E2611L |
| BioRad Calirty Max Western ECL Substrate, | BioRad | Cat. #175063 |
| LabForce HyBlot CL | Thomas Scientific | Cat. # 114J51 |
| Qiagen RNeasy Kit | Qiagen | Cat. #74304 |
| Invitrogen SuperScript III First Strand Synthesis SuperMix |
ThermoFisher | Cat. #18080400 |
| Endo-Free Maxi Prep Isolation Kits | Qiagen | Cat. #12362 |
| Mouse Neural Stem Cell Nucleofection Kit | Lonza | Cat. # VPG-1004 |
| SYBR Green I reagents | Roche Diagnostics | Cat. #04707516001 |
| Gibco CELLstart CTS | Thermo Fisher | Cat. #A10142–01 |
| Gibco neurobasal media | Thermo Fisher | Cat. #21103 |
| Gibco serum-free B27 | Thermo Fisher | Cat. #17504 |
|
| ||
| Deposited data | ||
|
| ||
| Sequencing data for massively parallel sgRNA-Cas9 disruption and individual replicate disruption | This paper | GSE138823 |
| Sequencing data for RNA-seq and H3K27ac enrichment collected from H9-dervived human neural stem cells | Cotney et al.24 | GSE57369 |
| H3K27ac enrichment data in human embryonic limb | Cotney et al.40 | GSE42413 phs001226.v1.p1 |
| H3K27ac enrichment data in human embryonic cortex | Reilly et al.2 | GSE63649 phs001226.v1.p1 |
| H3K27ac data in human H1 ESCs and adult tissues profiled by the Roadmap Epigenomics Project | Kundaje et al.84 | GSE16368 |
| PsychEncode Hi-C Data from human fetal brain | Rajarajan et al.70 | syn22343893 |
| Single-cell human fetal brain RNA-sequencing data | Bhaduri et al.74 Eze et al.75 | NeMO identifier: nemo:dat-0rsydy7 |
| JASPAR 2018 database | Khan et al.65 | https://jaspar2018.genereg.net/ |
| Epilogos | https://epilogos.altius.org | N/A |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| H9-derived human neural stem cells | Life Technologies | Cat. #N7800–1000 |
| HEK293FT | Invitrogen | Cat. #R70007 |
| HEK293T | Yale Cell Preparation and Analysis Core | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| Oligonucleotides used to clone sub-libraries and in validation and RT-qPCR assays are listed in Table S14. | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| LentiCRISPRv2GFP | Addgene | Cat. #82416 |
| pCMV-VSV-G | Addgene | Cat. #8454 |
| pCMV-dR8.2 dvpr | Addgene | Cat. #8455 |
|
| ||
| Software and algorithms | ||
|
| ||
| Bowtie v. 1.1.2 | Langmead et al.85 | https://sourceforge.net/projects/bowtie-bio/files/bowtie/1.1.2/ |
| CRISPResso2 | Clement et al.86 | http://crispresso2.pinellolab.org/submissiongreat.stanford.edu |
| GREAT version 3.0.0 | McLean et al.87 | |
| MAGeCK version 0.5.8 | Li et al.88 | https://sourceforge.net/projects/mageck/files/0.5/ |
| MASS (v7.3–54) | Venables and Ripley89 | https://www.stats.ox.ac.uk/pub/MASS4/ |
| DAVID v6.8 | Huang et al.90 Huang et al.91 |
https://david.ncifcrf.gov/ |
| ReactomePA package (v1.14.0) | Fabregat et al.61 | https://bioconductor.org/packages/release/bioc/html/ReactomePA.html |
| Seurat R package (v4.3.0) | Hao et al.92 | https://satijalab.org/seurat/ |
| Batchelor R package (v1.8.1) | Haghverdi et al.93 | https://www.bioconductor.org/packages/release/bioc/html/batchelor.html |
| ComplexHeatmap R package (v2.11.2) | Gu94 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| Cutadapt version 1.16 | Martin95 | https://cutadapt.readthedocs.io/en/v1.16/index.html |
| Juicer | Durand et al.96 | https://github.com/aidenlab/juicer/ |
| ShinyGO | Ge et al.80 | http://bioinformatics.sdstate.edu/go/ |
| Nebulosa v1.2.0 | Alquicira-Hernandez and Powell97 | https://www.bioconductor.org/packages/release/bioc/html/Nebulosa.html |
| ImageJ | Schneider et al.98 | https://imagej.net/ |
| Original code for analyses performed in this study | This paper | https://doi.org/10.5281/zenodo.10258136 |
|
| ||
| Other | ||
|
| ||
| Amicon Ultra-15 100kD filters | Millipore-Sigma | Cat. # UFC901008 |
| CustomArray 90K oligonucleotide synthesis array | CustomArray | N/A |
| Proliferation-decreasing controls, described in the Methods | Wang et al.22 | N/A |
| Accuri C6 Flow Cytometer | BD Biosciences | N/A |
| S3e Cell Sorter | BioRad | N/A |
| Cytoflex LR Flow Cytometer | Beckman Coulter | N/A |
| Roche LightCycler 480 PCR Thermal Cycler | Roche Diagnostics | N/A |
| HiSeq 4000 | Illumina | N/A |
| MiSeq | Illumina | N/A |