

Abstract
Despite the global impact and advances in understanding the pathophysiology of cerebrovascular diseases, the term “stroke” is not consistently defined in clinical practice, in clinical research, or in assessments of the public health. The classic definition is mainly clinical and does not account for advances in science and technology. The Stroke Council of the American Heart Association/American Stroke Association convened a writing group to develop an expert consensus document for an updated definition of stroke for the 21st century. Central nervous system infarction is defined as brain, spinal cord, or retinal cell death attributable to ischemia, based on neuropathological, neuroimaging, and/or clinical evidence of permanent injury. Central nervous system infarction occurs over a clinical spectrum: Ischemic stroke specifically refers to central nervous system infarction accompanied by overt symptoms, while silent infarction by definition causes no known symptoms. Stroke also broadly includes intracerebral hemorrhage and subarachnoid hemorrhage. The updated definition of stroke incorporates clinical and tissue criteria and can be incorporated into practice, research, and assessments of the public health.
Keywords: AHA Scientific Statements, cerebral hemorrhage, cerebral infarction, stroke, subarachnoid hemorrhage, transient ischemic attack
Stroke is classically characterized as a neurological deficit attributed to an acute focal injury of the central nervous system (CNS) by a vascular cause, including cerebral infarction, intracerebral hemorrhage (ICH), and subarachnoid hemorrhage (SAH), and is a major cause of disability and death worldwide. Despite its global impact, the term “stroke” is not consistently defined in clinical practice, in clinical research, or in assessments of the public health. Advances in basic science, neuropathology, and neuroimaging have improved the understanding of ischemia, infarction, and hemorrhage in the CNS. The Stroke Council of the American Heart Association (AHA)/American Stroke Association (ASA) published a scientific statement in 2009 to update and clarify the definition of transient ischemic attack (TIA), which in turn requires a reevaluation of the broader definition of stroke.1 Classic definitions of stroke are decades old and have become outdated, but modern definitions have not been formalized and officially adopted by the AHA, ASA, or any other major organization.
The leadership of the AHA/ASA reached out to colleagues from the American Academy of Neurology, the American Association of Neurological Surgeons and Congress of Neurological Surgeons, the US Food and Drug Administration, the US Centers for Disease Control and Prevention, the National Institute of Neurological Disorders and Stroke, and others to establish a universal definition of stroke based on the current understanding of pathophysiology, as well as implications for clinical practice, research, and public policy.* The writing group was composed of experts in neurology, neurosurgery, neuroradiology, neuropathology, clinical research methods, epidemiology, biomarkers, policy, and global public health.
A similar approach has been used to generate a multisocietal universal definition of myocardial infarction (MI).2 Notably, important differences between MI and stroke warrant definitions of these 2 entities that are somewhat over-lapping yet also distinct, and the universal definition of MI cannot fully apply to the approach to stroke. Unlike heart disease, stroke is more of a heterogeneous disease that includes cerebral hemorrhages and several pathogenic subtypes of ischemic stroke.3 There are also differences in the relative importance of risk factors. Because of these important differences between strokes and heart disease, a common definition may not be appropriate.
This document represents the final expert consensus, summarized in Table 1, which has been peer reviewed as well as reviewed by the endorsing/affirming organizations. This document will be updated in the future as the science of the field advances.
Table 1.
Definition of Stroke
| The term “stroke” should be broadly used to include all of the following: |
Definition of CNS infarction: CNS infarction is brain, spinal cord, or retinal cell death attributable to ischemia, based on
|
| Definition of ischemic stroke: An episode of neurological dysfunction caused by focal cerebral, spinal, or retinal infarction. (Note: Evidence of CNS infarction is defined above.) |
| Definition of silent CNS infarction: Imaging or neuropathological evidence of CNS infarction, without a history of acute neurological dysfunction attributable to the lesion. |
|
Definition of intracerebral hemorrhage: A focal collection of blood within the brain parenchyma or ventricular system that is not caused by trauma. (Note: Intracerebral hemorrhage includes parenchymal hemorrhages after CNS infarction, types I and II—see “Hemorrhagic Infarction.”) |
| Definition of stroke caused by intracerebral hemorrhage: Rapidly developing clinical signs of neurological dysfunction attributable to a focal collection of blood within the brain parenchyma or ventricular system that is not caused by trauma. |
| Definition of silent cerebral hemorrhage: A focal collection of chronic blood products within the brain parenchyma, subarachnoid space, or ventricular system on neuroimaging or neuropathological examination that is not caused by trauma and without a history of acute neurological dysfunction attributable to the lesion. |
| Definition of subarachnoid hemorrhage: Bleeding into the subarachnoid space (the space between the arachnoid membrane and the pia mater of the brain or spinal cord). |
| Definition of stroke caused by subarachnoid hemorrhage: Rapidly developing signs of neurological dysfunction and/or headache because of bleeding into the subarachnoid space (the space between the arachnoid membrane and the pia mater of the brain or spinal cord), which is not caused by trauma. |
| Definition of stroke caused by cerebral venous thrombosis: Infarction or hemorrhage in the brain, spinal cord, or retina because of thrombosis of a cerebral venous structure. Symptoms or signs caused by reversible edema without infarction or hemorrhage do not qualify as stroke. |
| Definition of stroke, not otherwise specified: An episode of acute neurological dysfunction presumed to be caused by ischemia or hemorrhage, persisting ≥24 hours or until death, but without sufficient evidence to be classified as one of the above. |
CNS indicates central nervous system.
Brief History of Definitions of Stroke and TIA
The word “stroke” was likely first introduced into medicine in 1689 by William Cole in A Physico-Medical Essay Concerning the Late Frequencies of Apoplexies.4 Before Cole, the common term used to describe very acute nontraumatic brain injuries was “apoplexy.” Apoplexy was used by Hippocrates circa 400 BC.5 For >2000 years, physicians have struggled to define the term “stroke.” During the 1950s, physicians felt the need to also introduce a term for temporary vascular-related episodes of brain dysfunction that would not qualify as strokes, and “transient ischemic attack” came into use.
Why the struggle to arrive at generally agreed on consensus definitions of stroke and TIA? Information about the brain and its anatomy, functions, and blood supply has advanced substantially during the past 200 years. Neurologists and other specialists in vascular diseases of the brain have proliferated during the past 50 years. The ability to safely and quickly image the brain and its blood-supplying vessels in patients has become a reality during the past 25 years. And, in the past 10 years, modern brain and vascular imaging has become generally available in community medical centers, although many still today do not have this capability. As knowledge, personnel, and technology evolve, we continue to learn about the nature, causes, and clinical and imaging findings in patients with cerebrovascular diseases.
The current World Health Organization definition of stroke (introduced in 1970 and still used) is “rapidly developing clinical signs of focal (or global) disturbance of cerebral function, lasting more than 24 hours or leading to death, with no apparent cause other than that of vascular origin.”6 During the 40 years since this definition was formulated, advances have been made in knowledge about the nature, timing, clinical recognition of stroke and its mimics, and imaging findings that require an updated definition.
During the Second Princeton Cerebrovascular Disease Conference, C.M. Fisher presented an extensive characterization of what he termed “transient ischemic attacks,” which “may last from a few seconds up to several hours, the most common duration being a few seconds up to 5 or 10 minutes.”7 At the Fourth Princeton Cerebrovascular Disease Conference in 1965, the attendees agreed on “transient ischemic attack” as the preferred term for temporary episodes of brain and eye ischemia.8 In 1975, an Ad Hoc Committee on Cerebrovascular Disease published the following definition: “Transient ischemic attacks are episodes of temporary and focal dysfunction of vascular origin, which are variable in duration, commonly lasting from 2 to 15 minutes, but occasionally lasting as long as a day (24 hours). They leave no persistent neurological deficit.”9 The 24-hour duration was arbitrarily chosen without data. When this definition was formulated, diagnostic techniques were unavailable that could determine the presence of brain infarction, and effective treatments of brain ischemia were not established.
The definition of TIA that was used in the 1975 report was universally cited until the beginning of the 21st century, when data accumulated that prompted attempts at redefinition. These data fell into 2 categories: duration of TIAs and imaging findings. The new data ignited controversies, which remain to the present day, about redefining the duration of TIAs and the need for incorporating brain and vascular imaging data into the definition. In 2002, an expert committee proposed a new definition: “A TIA is a brief episode of neurologic dysfunction caused by focal brain or retinal ischemia, with clinical symptoms typically lasting less than one hour, and without evidence of acute infarction.”10
In 2009, an expert committee of the AHA/ASA published a scientific statement defining TIA and recommending evaluation. The definition proposed was “transient ischemic attack (TIA): a transient episode of neurological dysfunction caused by focal brain, spinal cord, or retinal ischemia without acute infarction.”1
The International Classification of Diseases (ICD) system aims to standardize diagnostic classification for most diseases. Recent iterations, including the 10th revision along with its clinical modification (ICD-10-CM) published in 2010,11 classified cerebrovascular disorders chiefly as TIA, cerebral ischemic stroke, ICH, or SAH.
Deficiencies and the Need for Updated Definitions
The World Health Organization’s definition of stroke is obsolete. Based on advances including modern brain imaging, the 24-hour inclusion criterion for cerebral infarction is inaccurate and misleading, because permanent injury can occur much sooner. Furthermore, global cerebral dysfunction is seldom caused by cerebrovascular disease. There are several different definitions of TIA in use with no single agreed-on definition. Advances in evaluation, treatment, and prevention mandate that common definitions be used. This is especially important in epidemiological studies and therapeutic trials. Comparing and contrasting studies in which different definitions are used for inclusion of cases or ascertainment of outcomes is difficult. The advent of thrombolysis and other hyperacute treatments has added to the need to redefine stroke and TIA, because many current guidelines differentiate treatment strategies for these 2 entities. Treatment of patients with CNS ischemia should be directed to the cause and not governed only by whether infarction has developed. However, the location and extent of infarction is one variable to consider when choosing treatment.
Time and Imaging
Early definitions of stroke and TIA focused on the duration of symptoms and signs. More recent studies, using clinical observation and modern brain imaging, have shown that the duration and reversibility of brain ischemia are variable. Brain tissue that is deprived of needed nutrients can, in some patients, survive without permanent injury for a considerable period of time—several hours or even, rarely, days—while in most other individuals, irreversible damage (infarction) occurs quickly. Modern imaging now aims at separating brain tissue that is already infarcted from tissue that is underper-fused but not yet irreversibly injured. Because of the variability of duration, there is now general agreement that a fixed time designation should not be the primary distinguishing factor between stroke and TIA. Time should be a secondary consideration when adequate imaging is unavailable. Time range frequencies could be a part of commentaries on these definitions.
The word “transient” indicates a lack of permanence. Modern brain imaging has shown that many patients in whom symptoms and signs of brain ischemia are clinically transient have evidence of brain infarction. If the ischemia caused death of the tissue, it is misleading to designate the ischemia as transient. Similarly, ischemia may produce symptoms and signs that are prolonged (and so qualify in older definitions as strokes), and yet no permanent brain infarction has occurred. Optimally, all patients with brain ischemia (persistent or transient) would have thorough evaluations that show the presence, nature, and extent of brain damage (infarction and hemorrhage) and the cardiac, cerebrovascular, and/or hematological causes of the brain lesions. However, this is unlikely in the foreseeable future. Definitions are needed that are qualified by how the determinations were established. Stroke would be the term classically used if the means of classification were purely clinical. In contrast, infarction and hemorrhage involving the CNS are terms defined both clinically and by modern imaging.
Tools for the Diagnosis of Stroke
Clinical Diagnosis
Knowledge of neuroanatomy and vascular anatomy is important for the clinical diagnosis of stroke and transient CNS ischemia. Brain injuries attributable to vascular causes are nearly always focal, unless they lead to increases in intracranial pressure that cause global cerebral hypoperfusion, as in SAH, or massive infarcts and ICHs. Consideration of where the process occurs in the brain helps to determine whether the cause is vascular and to identify the potential vessels involved. During clinical diagnosis, 3 questions require an answer: (1) Is the process vascular or a stroke-like mimic? If a vascular process, then (2) where in the CNS is the abnormality, and which blood vessels supply that area? and (3) What is the disease mechanism (eg, ischemia or hemorrhage)?
Before distinguishing among stroke mechanisms, clinicians should first ask whether the findings could be caused by a nonvascular process, such as a brain tumor, metabolic disorder, infection, demyelination, intoxication, or traumatic injury that mimics stroke.
The history and knowledge of general systemic diseases tell the clinician what is wrong (ie, pathophysiology); the neurological examination tells more where the disease process is located. Different data are used to answer the “what and where” questions. Diagnosis of stroke location is most often made by integrating all available information from the neurological symptoms and findings and from neuroimaging.
In determining the stroke mechanism, these clinical bedside data are considered: the past and present personal and family illnesses; the presence and nature of past strokes and/or TIAs; activity at the onset of the stroke; temporal course and progression of the focal symptoms and findings; and accompanying symptoms such as headache, vomiting, and decreased level of consciousness. Information about these items is obtained from a thorough history from the patient, a review of records, and data collected from observers, family members, and friends. These data are primarily historical. The general physical examination, which may uncover findings not known from the history, adds to the data used for diagnosing stroke mechanism. Elevated blood pressure, cardiac enlargement or murmurs, and vascular bruits are examples of physical findings that influence identification of the stroke mechanism.
Retinal infarction is a clinical diagnosis in a patient with acute painless visual loss, typically associated with ischemic whitening of the retina observed on funduscopic examination. A “cherry red spot” may be evident in the macula in patients with central retinal artery occlusion. Retinal infarction rarely requires additional testing to confirm the diagnosis, although occasionally fluorescein angiography is used in atypical cases.
Radiographic Diagnosis
Radiographic imaging studies and other laboratory testing are aimed at answering these questions in the evaluation of acute stroke: (1) Is the lesion(s) in the CNS caused by ischemia or hemorrhage, or is it related to a nonvascular stroke mimic? (2) Where is the lesion(s)? What is its size, shape, and extent? (3) What is the nature and severity of the vascular lesion(s), and how do the vascular lesion(s) and brain perfusion abnormalities relate to the lesion(s)? and (4) Are abnormalities of blood constituents causing or contributing to ischemia or hemorrhage?
Confirmation that the patient has had a stroke and not a stroke mimic depends heavily on brain imaging. Computed tomography (CT) scanning, which is now and in the foreseeable future will be more readily available in most medical centers than magnetic resonance imaging (MRI), is usually able to exclude stroke mimics such as brain tumors and subdural hematomas and to separate brain ischemia from hemorrhage. Brain imaging with CT or MRI can localize the regions of brain infarction and hemorrhage. Imaging of the cervical and intracranial arteries and veins, focusing on those that supply the region of vascular injury, can identify occlusive vascular lesions and show vascular malformations and aneurysms. Vascular imaging can be performed using ultrasound (duplex Doppler imaging of the blood vessels in the neck and transcranial Doppler study of intracranial arteries), or by CT or magnetic resonance angiography or by catheter angiography. Traditional ideas that a strict brain time window exists for acute stroke differ from modern imaging findings obtained by methods such as MRI diffusion-weighted imaging (DWI), which highlights tissue changes after several minutes to days after transient or permanent ischemic events.12,13 A recent Cochrane review of CT and MRI for the diagnosis of acute cerebral infarction within 12 hours of symptom onset showed that the pooled estimates for CT sensitivity and DWI MRI sensitivity were 0.39 and 0.99, respectively, using a clinical diagnosis as the reference standard.14
Today, attention is focused on multisequence use of rapid MRI as a biomarker for acute identification of permanent tissue injury as well as viable tissue at risk, widely known as the penumbra.15 Multimodal magnetic resonance angiography, DWI, fluid-attenuated inversion recovery (FLAIR), and perfusion-weighted MRI are used to detect “mismatch,” which identifies the area of potentially reversible injury. These methods compare favorably with corresponding CT “mismatches” of CT hypodensity, CT angiography, and CT perfusion.16 The use of all of these imaging studies is based on the underlying hypothesis that if the blood supply is not restored, the penumbra will succumb to permanent injury eventually and result in a negative clinical outcome. Advances in assessment of perfusion or flow mapping methods aim to define a threshold to exclude benign oligemia from penumbra, while simultaneously distinguishing the ischemic core from penumbra as an accurate determination of the volume of potentially salvageable tissue.15,17
Mismatch of tissue volumes has been used as a radiographic index of the ischemic penumbra.18–21 Ideally, radiographic assessment will identify patients who have relatively smaller volumes of irreversibly infarcted core and large volumes of salvageable penumbra and will benefit from intensive reperfusion therapy. The optimal tool would characterize the presence, territory, and extent of hemorrhage; the size and location of an ischemic core destined to infarction; the size and volume of a penumbra; and the geographic distribution of vascular occlusion or flow. However, no imaging parameters have yet been proven to achieve this goal sufficiently for use in selecting patients for specific therapies.15,22–25
Interpretations of acute stroke neuroimaging studies are also complicated by abnormalities that may mimic acute stroke by causing brain water protons to experience altered DWI patterns because of changes in the cellular microenvironment.26 Such mimics are most commonly conditions such as infections, cysts, or abscesses that exhibit lower-than-normal apparent diffusion coefficient values (which are “gold standard” signs of acute stroke with sensitivity and specificity >95% when clinical symptoms are considered).27–29 The DWI examinations, including apparent diffusion coefficient maps, should be read together with corresponding T2 or cerebrospinal fluid (CSF)–suppressed FLAIR examinations to exclude nonstroke events. The classic depiction of acute stroke as hyperintense lesions on DWI may be commonly confused with “T2 shine-through” when the apparent diffusion coefficient is not read together with the DWIs.30,31 Certain metabolic abnormalities or diseases may occur in children with atypical MRI findings that may mimic stroke. Trauma may create shear-induced reductions in apparent diffusion coefficient corresponding to some hindrance to normal water proton brain diffusion. In addition, the DWI-observed tissue changes may occur in tissue ischemia-to-necrosis processes that may appear as “pseudonormalization” of the apparent diffusion coefficient.
Because most of the rapid magnetic resonance (MR) DWI, perfusion-weighted MRI, and functional MR images are acquired today with single-shot echo-planar image methods, presence and extent of calcifications, air, and deoxygenated hemoglobin to hemosiderin conversions may mimic or confuse the MR findings by creating regional signal loss attributable to magnetic susceptibility artifacts in affected areas. The sensitivity of echo-planar imaging to iron and air gives MR a valuable ability to detect and depict various forms of hemorrhage from SAH to microbleeds, yet makes these lesions notoriously difficult to quantify, requiring a set of conventional or fast spin-echo images to rule out such signal dropout artifacts.
Imaging of the spinal cord is less well established for the diagnosis of infarction. The sensitivity of MRI is limited, ranging from 45% to 73%, particularly when performed early.32–34 Even with repeated imaging, a substantial fraction of MRIs (14%) will still be normal.32 Moreover, a finding of T2 signal abnormality, even with restricted diffusion, is not specific for infarction and can be seen with demyelination and other disorders. Imaging evidence of vertebral body infarction adjacent to a cord signal abnormality on MRI is a specific indicator of ischemia and a useful confirmatory sign if present, although found in a minority of cases.35
Serum Biomarkers
Although troponin and creatine kinase values are often used to diagnose and quantify MI, biomarkers have not entered the mainstream of diagnosis of brain infarction. Biomarkers have been explored mostly in research on patients who have sustained global brain ischemia, for example, related to cardiac arrest, and in patients with head injuries. Commonly measured markers include S100 calcium binding protein B or S100B, glial fibrillary acidic protein, brain natriuretic peptide, and matrix metalloproteinase-9. None of these substances are routinely measured by hospital laboratories in the time frame needed to make acute care decisions but are a focus of clinical research.
Pathology
Neuropathological evaluation of brain (or spinal cord) tissue remains the definitive means to detect ischemic necrosis (an infarct). However, the need for this in the modern era is diminished by the high accuracy of MRI sequences (as described previously) that can accurately define the boundaries of necrotic neural tissue in vivo. Furthermore, the opportunities for directly examining brain tissue are becoming increasingly rare: Relatively small numbers of autopsies are performed, even in academic medical centers, although this decline is offset by the potentially valuable information that can be obtained in highly selected necropsies.36 Biopsy tissue showing ischemic necrosis often comes as a surprise to the neuropathologist, usually when a neurosurgeon samples a space-occupying lesion (often causing severe life-threatening edema) that was thought to be a neoplasm or abscess, but instead finds a subacute infarct with extensive associated swelling. A neuropathologist performing a postmortem examination in a stroke patient is charged with 2 tasks: defining the vascular disease (systemic and/or cerebral) and systemic factors (eg, hypotension) that contributed to the stroke; and establishing (to the extent possible) the distribution of necrosis, as well as its severity and age, that is, how long it was present before the patient’s demise.37–39
The histopathological criteria for recognizing acute irreversible ischemic neuronal injury (necrosis) have been recognized for decades: An affected neuron loses its basophilic cytoplasm (the result of Nissl substance, or rough endoplasmic reticulum) and prominently nucleolated nucleus, which are replaced by a neuronal cell body showing brightly eosinophilic neuronal cytoplasm lacking identifiable substructure, and a pyknotic or collapsed nucleus; the tinctorial change in the cytoplasm may precede nuclear change (Figure 1). The precise time taken between the cessation of oxygenated blood flow (to a given brain/spinal cord region) and this histopathological picture is debated but is widely estimated to be ≈6 to 10 hours. Put differently, if a patient experiences irreversible cerebral necrosis and dies within 1 to 2 hours, there will be no visible neuropathological abnormality (by light microscopy) in affected brain tissue.40 Changes in the neuronal cytoplasm and membrane visible only by electron microscopy doubtless occur in a much shorter time frame, as suggested by animal models, but ultrastructural examination of ischemic brain is almost never carried out, even in the most detailed postmortem examination, because of issues related to rapid tissue autolysis. An intriguing feature in the brain and/or spinal cord of someone who has experienced profound and prolonged hypoxia is the occurrence of intact and essentially normal-appearing neurons immediately adjacent to “red dead” (brightly eosinophilic) cell bodies (Figure 1). Extreme diffuse anoxic-ischemic injury can be difficult to distinguish from a cerebral infarct; usually the latter shows confluent neuronal ischemic change over a defined region of brain/spinal cord, and this change is accompanied by extreme pallor and variably severe vacuolization of the neuropil.38
Figure 1.
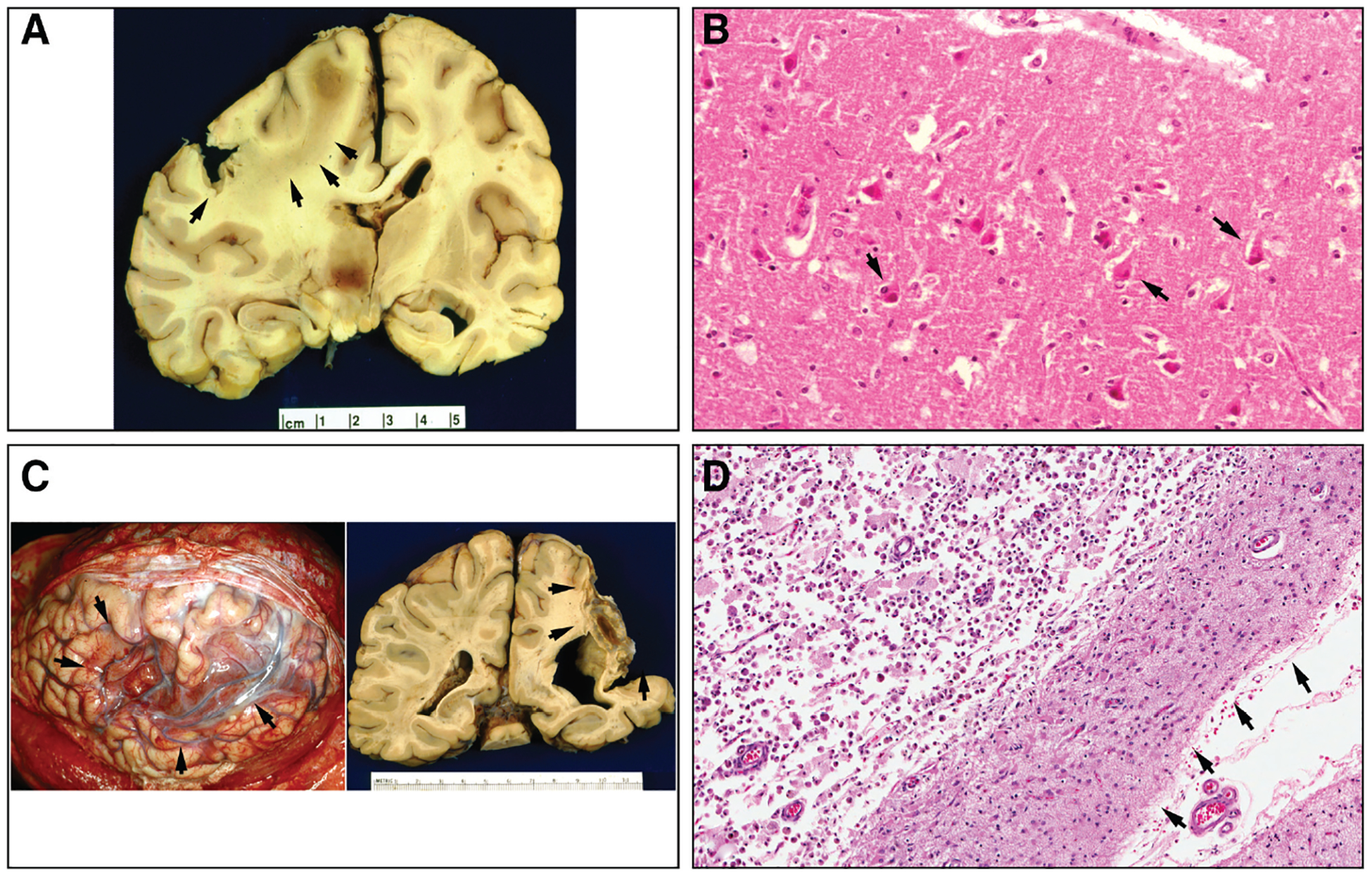
Pathology of cerebral infarction. A, Subacute cerebral infarction involving left cerebral hemisphere (indicated by arrowheads) that had occurred ≈3 to 4 days before death. Note the pronounced cerebral edema with left-to-right shift of midline structures, including subfalcine herniation of the cingulate gyrus, and marked central diencephalic herniation. B, Subacute infarct, microscopic features. Note the pronounced eosinophilia of neurons (indicated by arrows) and subtle vacuolization of the neuropil. C, Old cystic cerebral infarcts (observed at autopsy) in 2 different patients. Brain at left shows appearance of left cerebral hemisphere immediately after calvarium has been removed. Arrowheads indicate a large cavity in the middle cerebral artery territory. Brain (coronal section) at right shows a large right MCA territory infarct (indicated by arrowheads) in a patient who had experienced stroke many years previously. D, Characteristic microscopic appearance of edge of an old cystic infarct. Arrowheads indicate pial surface and subpial regions of preserved, extremely gliotic rim of cortex, comprising largely layer I. Underlying cystic cavity (upper left of micrograph) shows abundant residual macrophages. B and D are from hematoxylin-and-eosin–stained sections. MCA indicates middle cerebral artery. (Courtesy of HV Vinters, Department of Pathology and Laboratory Medicine [Neurology], University of California at Los Angeles, Los Angeles, CA.)
A subacute/acute infarct within white matter, a structure devoid of neurons, can be more difficult to identify. It usually shows well-demarcated pallor of the tissue, within which are abundant neuroaxonal spheroids that may be highlighted using antibodies to neurofilament or amyloid precursor protein.
Other types of neuronal death may occur and may even be the consequence of anoxic-ischemic brain injury. These include apoptotic death, which may result from any one of a large number of insults to the CNS that can accompany ischemia (eg, increased intracellular calcium). The morphological features of apoptosis include the genesis of intranuclear chromatin masses and (eventually) apoptotic bodies.41 A third mechanism, probably less important in ischemic brain injury, is free-radical–induced damage and autophagocytosis, which shows up histologically as condensed cytoplasm, large vacuoles, and a clumped nucleus.41
The sequence of cellular events that follows irreversible ischemic brain injury occurs in a fairly stereotypical progression, although not necessarily in consistently well-defined time frames. There may be extravasation of polymorphonuclear neutrophils from capillaries in or adjacent to an infarct, usually occurring within 1 to 2 days of the onset of necrosis. Macrophages move into the region of necrosis and represent both transformation of monocytes that have originated in the circulation as well as resident microglia within the CNS; this occurs at ≈5 to 6 days after the onset of necrosis, but monocyte migration into infarcted brain can persist for 4 to 5 weeks.37 Not surprisingly, macrophages and microglia have almost identical immunohistochemical markers (eg, CD68 and Iba1+).42 Many macrophages, including lipid-laden cells, will persist in an infarct for the life of the affected patient. New capillary formation (neovascularization) occurs in and adjacent to the infarct (Figure 1), usually in a time frame of 5 to 10 days after the onset of necrosis. Finally, the infarct undergoes cystic cavitation, the cavity being marginated by abundant reactive astrocytes (easily highlighted by immunohistochemistry using primary antibodies to glial fibrillary acidic protein). Such a cystic cavity is traversed by randomly oriented gliovascular bundles.
An interesting phenomenon within infarcted neocortex is the persistence of a very gliotic subpial layer I of the cortex. Neurons and axons that die in and adjacent to an area of necrosis may become encrusted with calcium and iron, that is, they are described as being “mummified,” or “ferruginized.” The cystic cavity marginated by astroglia is characteristic of many microinfarcts and virtually all lacunar and larger cystic infarcts, and typically persists for the life of the patient. Microinfarcts do not always undergo cavitation, but rather appear as collapsed, linear, or triangular scars within brain, lesions that are easily highlighted using immunohistochemistry with primary antibodies directed against glial fibrillary acidic protein and/or a macrophage/microglial marker.43,44 Indeed, these immunohistochemical methods can be used to quantify the number of microinfarcts in a given brain section.
CNS Infarction
CNS Infarction Is Brain, Spinal Cord, or Retinal Cell Death Attributable to Ischemia, Based on Pathological, Imaging, Other Objective Evidence, and/or Clinical Evidence
Cerebral infarction is fundamentally a neuropathological term as described previously. Given that pathological confirmation of CNS infarction is rarely obtained in living patients, a tissue-based definition of CNS infarction must rely on other available information, including clinical and neuroimaging data. Neuroimaging is not perfect, and its use in establishing a tissue-based definition of CNS infarction has many factors that may influence the ability to provide evidence of ischemia, including the time from symptom onset to image acquisition, the sensitivity of the imaging modality for detecting the lesion, and other characteristics of the clinical setting.
The timing of the neuroimaging in relation to the onset of ischemia may impact whether imaging evidence of stroke is seen, since signs of ischemia on noncontrast head CT are seen within the first few hours of CNS infarction in 31% to 60% of cases.45–48 Therefore, within the first 12 hours of an acute stroke, a tissue-based diagnosis of CNS infarction is not possible with the use of routine noncontrast head CT alone but could be if MRI were widely used. Because noncontrast head CT remains the most commonly used imaging modality in the acute setting,49 a patient may have a clear clinical vascular syndrome supporting a diagnosis of CNS infarction but not meet a tissue-based definition of CNS infarction if only CT is used. In addition, the type of imaging modality selected may determine if a tissue-based diagnosis of CNS infarction is made based on the location of the stroke. For example, a patient with focal neurological symptoms localizing to the brainstem may have no imaging evidence of CNS infarction on plain head CT because of image degradation from “streaking artifact” in the brainstem but would be more likely to have imaging evidence of CNS infarction if MRI were used. Ultimately, even MRI lacks sensitivity for some small lesions, particularly in the brainstem.50,51
The selection of the neuroimaging modality may also depend on factors such as availability of the imaging modality, with rural hospitals and developing regions less likely to have access to imaging, especially MRI,49 thereby decreasing the likelihood of making a tissue-based diagnosis of CNS infarction. In addition, patient factors such as contraindication to a particular imaging modality (ie, implanted device or severe claustrophobia) may preclude MRI and reduce the likelihood of a tissue-based diagnosis of CNS infarction. Furthermore, despite the common impression that neuroimaging provides a more objective diagnosis of CNS infarction, physician preferences or bias in the use of neuroimaging may still impact the diagnosis of CNS infarction using a tissue-based definition. For example, a patient in an unblinded clinical trial who has mild symptoms of CNS infarction might not undergo an MRI to diagnose the stroke if the physician has a bias toward one treatment arm, and thus, the patient would not meet a tissue-based definition of CNS infarction, despite clinical stroke symptoms. Finally, given that there is no gold standard for the diagnosis of CNS infarction52 and that studies designed to test the sensitivity and specificity of neuroimaging modalities such as CT and MRI have used a clinical diagnosis of stroke as the reference standard, the requirement of neuroimaging evidence of ischemia in a definition of CNS infarction results in a somewhat circular argument.
A definition of CNS infarction must therefore allow for clinical criteria when neuropathological or neuroimaging data either do not provide evidence of infarction or such data are inadequate or unavailable. The presence of persistent clinical signs or symptoms is not necessary to define cerebral infarction but provides an alternative means to establish that diagnosis.
In the same manner that a TIA is defined by “transient” stroke symptoms and the absence of objective evidence of infarction, a clinical definition of cerebral infarction can be established based on persistent symptoms caused by cerebral ischemia. The duration of time that constitutes “persistent” must be defined for situations in which the neuroimaging is negative or inadequate. To define the threshold of persistent stroke symptoms that most reliably correlates with the presence of pathological cerebral infarction, the most logical approach is to study a large population and examine what event duration best correlates with other objective evidence of infarction. Furthermore, that threshold should be practical and timely to establish the diagnosis of ischemic stroke. In other words, the time threshold that defines “persistent” should be short enough to allow a rapid diagnosis of ischemic stroke but not so short as to include symptoms that are likely to be transient. The likelihood of permanent injury is related to both the severity and duration of ischemia. Without neuroimaging data, measurement of the severity of ischemia is impossible, and therefore, time is by necessity the primary defining factor, although it remains an approximation at best. Several studies suggest that most transient stroke symptoms resolve in <24 hours,53,54 supporting the classic 24-hour threshold as a fallback in the absence of direct and objective evidence of infarction. An earlier time threshold for defining “persistent” stroke symptoms, such as >1 hour, would result in the misclassification of patients with transient symptoms lasting 1 to 24 hours and no imaging evidence of ischemia as “stroke,” rather than “TIA” using the new definition, because up to 50% of patients with transient stroke symptoms lasting 1 to 24 hours have negative DWI MRI.54 Conversely, selecting a later time threshold to define “persistent” stroke symptoms, such as >72 hours, would prolong the diagnosis of ischemic stroke without increasing the likelihood that the symptoms would not be transient, given that in patients with stroke symptoms lasting ≥24 hours, 97% lasted >7 days while only 3% have symptoms that last 1 to 7 days.53 Currently, there are no compelling data showing that an alternative time threshold is superior to 24 hours, although further research may provide a more precise estimate. In the absence of such data, the persistence of symptoms for ≥24 hours remains a reasonable threshold for inferring the presence of permanent injury and therefore infarction.
There may be reasonable exceptions to this definition. For example, a patient who presents with rapidly developing neurological symptoms and is treated with thrombolytic agents or other acute therapies, and whose symptoms completely resolve before the 24-hour threshold, might be considered to have an infarction even if subsequent imaging does not show evidence of injury.
Definition of Ischemic Stroke Should Be Limited to Focal Ischemia and Not Include Global Ischemia
There are several reasons to limit the definition of ischemic stroke to focal ischemia alone. First, there are significant differences in the pathology and mechanisms of ischemia between focal and global ischemia. Focal ischemia occurs within the perfusion territory of an artery that is stenosed or occluded, and cell death is localized to this region. In focal cerebral ischemia, cell death is maximal in the ischemic focus and may extend to the penumbra, with all cellular elements including both neurons and supportive cells affected. In contrast, global ischemia results from decreased cerebral perfusion as a result of decreased blood pressure (eg, in shock or cardiac arrest) or severely increased intracranial pressure (eg, severe head trauma). In global ischemia, selective neuronal loss appears to occur in vulnerable areas of the hippocampus, cerebral neocortex, thalamus, cerebellum, and basal ganglia55 and is not isolated to particular vascular distributions. Furthermore, applying the definition of prolonged cell death attributable to global ischemia in the CNS would include sources of injury such as anoxia caused by airway or lung diseases and some metabolic injuries, which are quite distinctly nonvascular in origin. In addition, survivors of global ischemia (eg, patients resuscitated after cardiac arrest) will always have reperfusion of the ischemic cerebral tissue. This results in a larger role for injury because of the pathological effects of reperfusion in global ischemia than in focal ischemia.
In addition to pathophysiological differences, the treatment of global ischemia differs significantly from the treatment of focal ischemia, and because of these treatment differences, global ischemia should not be included with focal ischemia in the definition of ischemic stroke. Although the duration of ischemia is important in both focal and global ischemia, focal ischemia is acutely treated with reperfusion strategies to improve flow in an artery. In distinct contrast, global ischemia is acutely treated by correcting the systemic disorder that is the underlying cause of hypoperfusion. The evaluation of patients with focal and global ischemia also differs. Focal ischemia typically requires assessment of the cervical and cerebral arteries, investigation of a possible cardiac source of emboli, and evaluation of risk factors for atherosclerosis, whereas the evaluation of global ischemia is focused on identifying the underlying cause of hypoperfusion.
Global ischemia also typically differs from focal ischemia with respect to the initial clinical presentation and prognosis, providing additional rationale for excluding global ischemia from the definition of stroke. Patients with focal ischemia present with neurological deficits that are localizable to a particular vascular distribution and rarely have a depressed level of consciousness. However, patients with global ischemia most commonly present with diffuse nonfocal neurological symptoms, particularly diminished consciousness. The prognosis also differs between focal and global ischemia, because mortality for focal ischemia is ≈12%,56 while for global ischemia >80% of patients do not survive hospitalization,57 with two thirds of the deaths attributable to neurological injury.58 Finally, widely accepted definitions of infarction in other organs, such as liver and lungs, are limited to focal rather than global ischemia.59–61
The writing group recognizes that the universal definition of MI more simply allows for myocardial necrosis without explicit specification of focal versus global ischemia.2 However, in the setting of global ischemia as may occur with cardiac arrest, the universal definition of MI includes “symptoms suggestive of myocardial ischemia, and accompanied by new ST elevation, or new left bundle-branch block, and/or evidence of fresh thrombus by coronary angiography and/or at autopsy,” all of which imply a focal arterial occlusion. Our revised definition of stroke is consistent with this approach and requires symptoms or signs of focal brain dysfunction and/or neuroimaging or pathological evidence of acute infarction. If acute brain imaging or pathological examination is performed that demonstrates focal (or multifocal) infarction in an arterial or watershed territory, then this focal injury would meet the revised definition of CNS infarction and, if accompanied by clinical symptoms, ischemic stroke.
There are marked differences between focal and global ischemia with respect to clinical presentation, treatment, pathophysiology, and prognosis. These differences are sufficient to limit the definition of stroke to focal ischemia.
Definition of CNS Infarction Should Be Limited to CNS Tissue Including Brain, Spinal Cord, and Retina
The brain, spinal cord, and retina derive from neural tube tissue and therefore constitute the CNS, while the cranial and peripheral nerves derive from neural crest tissue.62 There are differences in the mechanisms of ischemia, treatment, and recovery between CNS and peripheral nervous system (PNS) ischemia that warrant limitation of the definition of infarction to the CNS. CNS ischemia, as previously described, results from stenosis or occlusion of both large vessels and small vessels attributable to local thrombosis or embolization from other vascular regions or from critical hypoperfusion in border-zone regions. PNS ischemia typically results from small-vessel occlusion of the vasa nervorum presenting as mononeuropathies,63 most commonly related to vasculitis or diabetes mellitus. Isolated cranial neuropathies have previously been attributed to a mechanism similar to PNS ischemia but are now believed to result more commonly from MRI-defined microinfarcts in the brainstem64 and are thus more similar to small-vessel infarctions. As a result of the differences in pathogenesis of ischemia between CNS and PNS ischemia, treatments for ischemia of the PNS and CNS differ. For CNS ischemia, the treatment is focused on establishing reperfusion in the acute setting and then secondary prevention of ischemia. For PNS ischemia, treatments are focused on the underlying condition (ie, steroids for vasculitis or glucose control for diabetes mellitus), and acute reperfusion treatments are not available. The CNS and PNS also differ with respect to potential for recovery after ischemic injury. The PNS has a greater regenerative capacity than the CNS because of innate differences between the neurons and supportive cells in these locations, allowing for PNS axonal regeneration after injury.
Because of the differences in the mechanisms of ischemia, treatment, and recovery between CNS and PNS ischemia, as well as structural and embryological differences, the definition of CNS infarction should be limited to the brain, spinal cord, and retina and should include isolated cranial nerve syndromes only when other confirmatory evidence of brainstem ischemia is present.
Definition of CNS Infarction Should Include Neurological Conditions Resulting From Focal Ischemia to the CNS That May Be Atypical in Presentation
Ischemia of the CNS may not always manifest as the sudden onset of focal neurological symptoms. Atypical or “somatic” symptoms (eg, headache, fatigue, malaise) have been reported in 73% of women and 65% of men presenting with acute stroke symptoms,65 suggesting that focal neurological symptoms might not be the only manifestation of CNS infarction in many cases. In some rare conditions, small areas of ischemia are initially asymptomatic but later become symptomatic as multiple ischemic lesions accumulate. For example, in CNS vasculitis or cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), atypical symptoms such as depressed level of consciousness, seizures, headache, or dementia may be the initial presentation of cerebral ischemia.66 Conversely, some conditions may present with stroke-like episodes and neurological symptoms that mimic CNS infarction (eg, mitochondrial encephalopathy, lactic acidosis, and stroke [MELAS],67,68 posterior reversible encephalopathy syndrome,69,70 and transient global amnesia).71–73 However, although these conditions may have a component of ischemia, at this time, they are thought to have other primary mechanisms of cerebral damage and may be reversible without standard acute stroke therapies. The definition of CNS infarction may include atypical neurological symptoms when the symptoms are primarily attributed to focal (or multifocal) ischemia of the CNS.
Definition of CNS infarction:
CNS infarction is brain, spinal cord, or retinal cell death attributable to ischemia, based on
pathological, imaging, or other objective evidence of cerebral, spinal cord, or retinal focal ischemic injury in a defined vascular distribution; or
-
clinical evidence of cerebral, spinal cord, or retinal focal ischemic injury based on symptoms persisting ≥24 hours or until death, and other etiologies excluded.
(Note: CNS infarction includes hemorrhagic infarctions, types I and II; see “Hemorrhagic Infarction.”)
Ischemic Stroke
A comprehensive definition of ischemic stroke requires clinical symptoms and evidence of infarction to provide an accurate description of the process of ischemia occurring in a given patient (Figure 2). Conversely, focal arterial ischemia with transient symptoms (lasting <24 hours) and without evidence of infarction by pathology or imaging should be considered a TIA.
Figure 2.
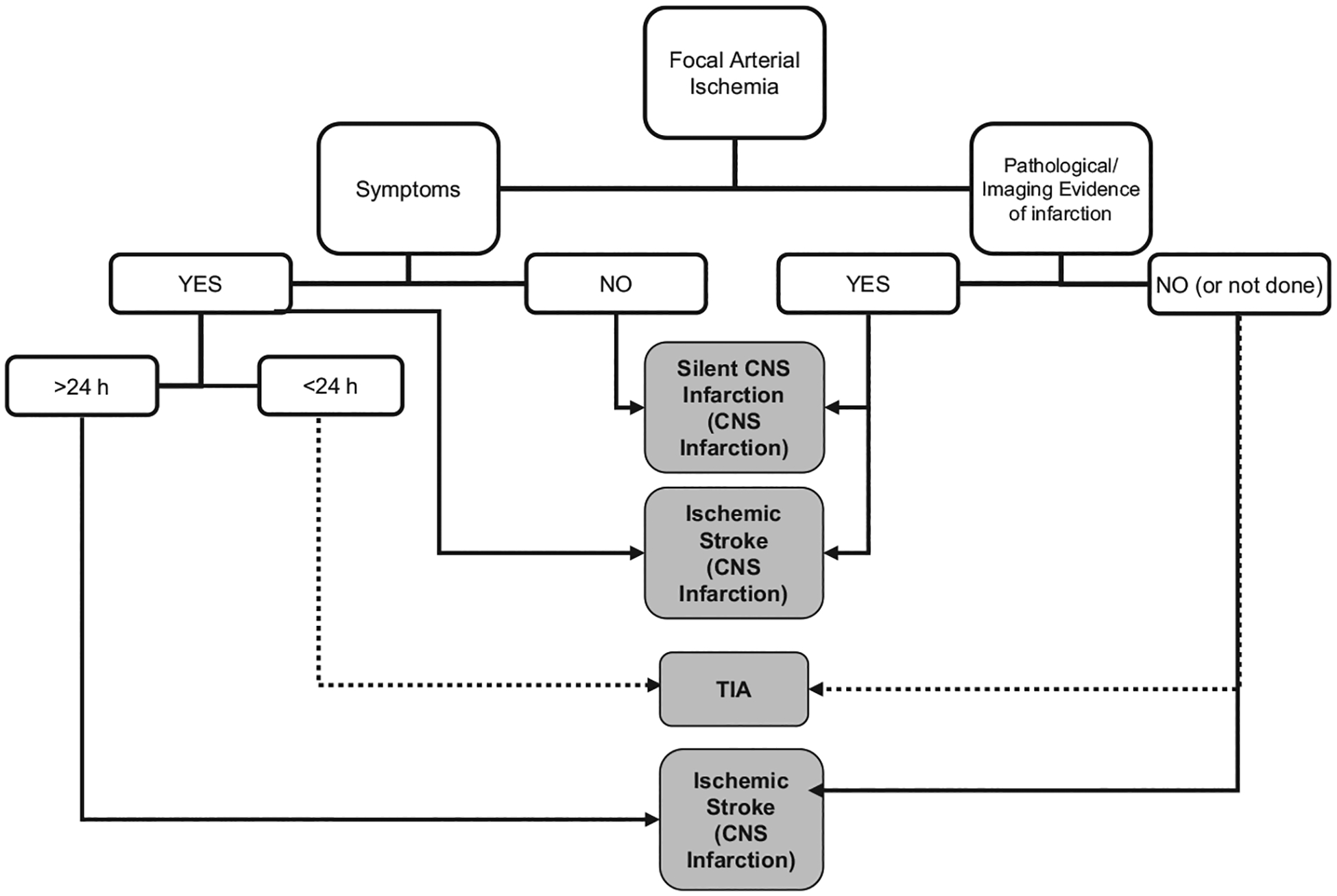
Flow chart depicting a proposed decision tree for determination of a cerebrovascular event definition. Cerebrovascular events are depicted in shaded boxes and are defined by the composite of both features of symptoms on the left and pathological/imaging evidence of infarction on the right. For example, the cerebrovascular event defined as “silent CNS infarction” requires focal arterial ischemia, no symptoms, and pathological or imaging evidence of infarction. CNS indicates central nervous system; and TIA, transient ischemic attack.
Definition of ischemic stroke:
An episode of neurological dysfunction caused by focal cerebral, spinal, or retinal infarction. (Note: Evidence of CNS infarction is as defined previously.)
Silent, Subclinical, or Prior CNS Infarction
The shift to a structural, rather than purely clinical, diagnosis of stroke1 requires a critical reappraisal of the frequently used terms “silent stroke” and “silent infarction.”74 (Silent hemorrhages and microbleeds are dealt with in a subsequent section of this document.) The development of the concept of silent cerebral infarction reflects the recognition that brain abnormalities, consistent with ischemic injury, can be identified pathologically or on neuroimaging in patients without a history of stroke or TIA. Fisher, for example, reported in 1965 that small infarcts occurred in the deep structures of the patients’ brains without known symptoms.75 Silent superficial cortical lesions, more often in the right cerebral hemisphere, were noted among patients presenting with stroke in the National Institute of Neurological and Communicative Disorders and Stroke (NINCDS) Stroke Data Bank.76 These lesions are commonly considered silent because they are unaccompanied by classically defined stroke syndromes. However, they may not be entirely asymptomatic, because patients with these lesions may still have evidence of cognitive, gait, or other functional impairment, as discussed later in this document. Such patients could be considered to have subacute or chronic symptoms and/or signs of stroke in the absence of an initial rapidly developing stroke syndrome.77,78
Definition
No standard or commonly accepted definition for silent infarction exists, partly because of a lack of a clear consensus regarding what is meant by “silence.” “Silence” depends on one’s vantage point and may differ between the patient and physician. Patients may not be aware that some prior constellation of symptoms was related to an imaged abnormality, or they may not have been evaluated for it at the time so that a diagnosis of stroke was never made. Thus, a silent infarction might be a cerebral infarction that was unnoticed, overlooked, or disregarded. The physician, however, may appreciate the relationship between a remote episode of, for example, vertigo and diplopia, previously attributed to some other cause, and a newly imaged abnormality consistent with infarction. Even from the physician’s perspective, the interpretation of silence may differ to reflect the interests of the clinician or investigator. Asymptomatic patients with incidentally discovered infarcts may have subtle examination findings such as mild facial paresis, pronator drift, reflex abnormality, visual field deficit, or other abnormalities.79 Once the clinical abnormality is detected by a physician, it may be inappropriate to continue to refer to the lesion as truly silent. Taken a step further, it is unclear how to classify infarcts unassociated with either symptoms or signs on neurological examination in patients with more subtle cognitive deficits detectable on detailed neuropsychological testing. Because detection of these infarcts depends on the sensitivity of the observer, some have suggested the use of the word “covert,” “subclinical,” or simply “prior” infarction rather than “silent” to represent the fact that they do indeed have clinical consequences.
The definition of silent infarction depends on the detection of structural tissue damage. The likelihood of finding these infarcts will, by necessity, depend on the imaging or other modality used. Even high-resolution MRI may not detect “microinfarcts” visible at postmortem examination and that may be clinically significant in large numbers. Therefore, an autopsy will be more sensitive for the detection of silent infarcts than MRI, which will in turn be more sensitive than CT. Some small studies even provide evidence that measurable functional abnormalities in response to provocative maneuvers may occur in brains of patients with transient ischemia but no imaging evidence of structural damage.80 A review of MRI diagnostic criteria for silent brain infarcts found substantial variability among 45 studies of this issue, but found that the majority used a threshold size of ≥3 mm with excellent reliability.81
The presence of a silent infarction therefore depends on both how hard one looks for evidence of sequelae of the event as well as evidence of brain injury caused by ischemia. Is the absence of a physician’s diagnosis of stroke adequate? Absence of symptoms? Is bedside examination adequate? Or is a normal comprehensive neuropsychological test required? Should CT or MRI be required? If MRI, which sequences are required, as technical developments allow the detection of ever-subtler abnormalities?
The definition of silent infarction is complicated, moreover, by the recognition that many patients and participants in observational studies may have confluent areas of white matter disease in the brain, referred to as “white matter hyper-intensities” or “leukoaraiosis.”82 These areas are readily identified on CT and MRI scans and are often considered to be secondary to ischemia.83 They are also associated with vascular risk factors, particularly age and hypertension, and appear to be associated with stroke risk.84–86 However, they may also reflect nonischemic pathologies,87 including edema, inflammation, demyelination, and gliosis, and therefore will not be considered further here, although further research into their relationship to cerebrovascular disease is warranted.
A reasonable approach to “silent infarction” would be to provide specific information about the nature of the symptoms and findings in any patient with evidence of infarction, as discussed below.
Location
Structurally identified cerebral infarcts may take as many forms as clinical strokes, including small, deep (ie, lacunar) infarcts, superficial cortically located lesions, or microinfarcts. Superficial lesions without symptoms are likely to be smaller than clinically identified lesions, because most large cortical strokes will produce some clinical symptoms or signs. For those in whom silent strokes occurred at a young age (eg, before 6 years of age), early brain plasticity may leave little or no clinical sequelae from even a large infarction. Silent infarcts may be located throughout the CNS, including the brainstem, cerebellum, and spinal cord. Infarcts that are silent are more likely to be located in the right cerebral hemisphere, because symptoms attributable to right hemispheric injury may not be as easily detected by patient or physician.76,88
Prevalence
An autopsy study in Japan found that ≈18% of those without a clinical history of stroke (mean age, 69 years) had evidence of silent infarction.89 The use of modern brain imaging techniques, including CT but particularly MRI, has permitted the routine identification of silent infarcts in populations of living patients, and such studies have permitted estimation of the prevalence and incidence of silent infarction in a more representative population of patients (Table 2). These lesions are quite common, necessitating serious consideration of their place in cerebrovascular nosology. CT studies among patients admitted with acute stroke but no history of prior stroke, for example, have demonstrated that 10% to 38% of such patients have evidence of prior infarction.76,88,90 In 1 study, silent infarcts on CT were seen among 15% of patients with asymptomatic carotid stenosis.91
Table 2.
Estimated Prevalence of Silent Infarcts in Selected Groups of Patients
| Type of Population | Method of Ascertainment | Prevalence of Silent Infarction, % | Reference |
|---|---|---|---|
| General population | Autopsy | 18 | Shinkawa et al89 |
| General population | MRI | 8–28 | Vermeer et al74 |
| Acute stroke | CT | 10–33 | Brott et al91 |
| Acute stroke | MRI | 57 | Adachi et al92 |
| TIA | CT | 0–35 | Brott et al91 |
| Asymptomatic carotid atherosclerosis | CT | 6–28 | Brott et al91 |
| Asymptomatic carotid atherosclerosis | MRI | 23 | Mathiesen et al93 |
| Atrial fibrillation | CT | 11–48 | Brott et al91 |
| Atrial fibrillation | MRI | 32 | Hara et al94 |
| Coronary artery disease | CT | 28 | Tanaka et al95 |
| Coronary artery disease | MRI | 17–60 | Vermeer et al74 |
CT indicates computed tomography; MRI, magnetic resonance imaging; and TIA, transient ischemic attack.
Vermeer et al74 reviewed the literature on MR-detected silent infarcts (n=105 original papers) in 2007. Most studies defined infarcts as T1 hypointense lesions of ≥3 mm in size; some excluded larger cortical infarcts, limiting comparability of studies. Lesions representing small infarcts were generally distinguished from dilated Virchow-Robin, or perivascular, spaces, which tend to be <3 mm in size, have round or linear appearance, and be located in the lower basal ganglia.96,97 Prevalence estimates of silent brain infarcts across the studies utilizing MRI range from 8% to 28%.74 These differences are largely accounted for by age. In a Japanese cohort of mean age 59 years, the prevalence was 10%,98 while in the Cardiovascular Health Study (mean age 75 years, oversampled for blacks), the prevalence was 28%.79
The prevalence of silent infarcts provides a measure of their importance. In fact, MRI-defined silent infarcts are up to 5 times as prevalent as clinically apparent strokes, which are found in ≈3% of the population.99 A wholesale redefinition of stroke that includes these lesions, therefore, would immediately swell the ranks of those with stroke to ≈15% to 20% of the population, suggesting that the burden of cerebrovascular disease is enormous and requires greater attention.
Silent infarcts are 30% to 40% more prevalent in women than men.100–102 This could represent an increase in risk of these often smaller infarcts in women, greater survival among women with silent infarcts, or a difference in the approach to diagnosis and treatment of neurological symptoms among women compared with men.74 Silent infarcts, like clinical strokes, are also more common among non-Hispanic blacks than among whites and Hispanics, although data are limited.103 Silent infarcts may also occur at earlier ages among blacks.103 These demographic differences suggest that a redefinition of stroke to include all silent infarcts might have a differential effect on estimates of the total burden of disease among women compared with men and across race-ethnic groups.
Incidence
Incidence provides another measure of the importance of these lesions. In studies using serial MRI scans, the incidence of silent infarcts was ≈3% annually among elderly participants in 2 observational cohorts.104,105 Incidence was lower in a third, smaller cohort.106 Incidence, unlike prevalence, was similar for men and women, providing evidence to support the hypothesis that women with silent infarcts survive longer than men. Incidence also increased with age, prior brain infarction, and hypertension.74 Incidence, like prevalence, also outnumbered clinical stroke by a factor of 5.
Prognosis
Silent infarcts are well recognized to be associated with several adverse neurological and cognitive consequences, albeit these are difficult to detect in routine circumstances.104 These consequences include impaired mobility, physical decline, depression, cognitive dysfunction, dementia, and clinical stroke. Silent brain infarcts increase the risk of clinical infarction by 2 to 4 times in population-based studies.86,107 This increased risk appears to be independent of other vascular and stroke risk factors, providing further evidence that silent infarcts may serve as a marker of propensity for stroke that is not captured by existing measures.
Silent brain infarcts increase the risk of mild cognitive impairment, and they also may double the risk of frank dementia.108,109 Observational studies have found lower levels of cognition among participants with evidence of silent brain infarction, and they appear to be associated with cognitive decline.110 Of note, silent infarcts are associated with risk of Alzheimer disease as well as of vascular dementia. Cerebral amyloid angiopathy, a microvasculopathy commonly found in the brains of individuals with Alzheimer disease, is now increasingly recognized as a likely contributing cause to cerebral microinfarcts and microbleeds.43,111,112 Although microbleeds can be detected by special MRI sequences, their size may be overestimated; however, there is no reliable way to identify microinfarcts on neuroimaging. Microinfarcts can be detected in autopsy brain specimens, especially with the use of special immunohistochemical methods to detect collections of microglia/macrophages or astrocytes, a fairly reliable “footprint” of microfoci of ischemic change.44 This finding supports the concepts that vascular risk factors and ischemic injury contribute to the pathology of Alzheimer disease, that Alzheimer disease develops earlier in those who have already experienced vascular injury to the brain, and that microinfarcts in the aging brain may result from an Alzheimer disease–related microvasculopathy, cerebral amyloid angiopathy. There is also evidence that silent infarcts are associated with both prevalence and severity of parkinsonism.113 For all these reasons, it is reasonable to conclude that many “silent infarctions” are not truly silent, even though the associated findings may be so subtle as to elude routine neurological evaluation.
Are Silent Infarcts “Strokes”?
There are several arguments in favor of including silent infarcts within the broadest definition of stroke.
First, insofar as silent infarctions are indeed infarctions, pathologically defined, it would appear to be counterintuitive not to include them within the rubric of CNS infarction. The same reasoning used to define “MI” in the “Third Universal Definition of Myocardial Infarction,”2 representing the combined efforts of several cardiology-related professional organizations, would indicate that a lesion is an infarction if it can be pathologically defined as such, independent of the presence or absence of any symptoms, signs, or neuropsychological findings. The expert consensus document uses the term “prior myocardial infarction” rather than “silent myocardial infarction,” avoiding any question of the clinical significance of the finding.2 Thus, according to that expert consensus document, the definition of a “prior myocardial infarction” includes “imaging evidence of a loss of viable myocardium that is thinned and fails to contract, in the absence of a non-ischemic cause.”2 Using the same logic, “prior cerebral infarctions” may be defined as “Imaging evidence of cerebral infarction in the absence of a non-ischemic cause.” Prior cerebral infarctions would be present when imaging is consistent with a cerebral infarction, independent of signs or symptoms. The use of the term “prior” in this context should be understood to refer to a remote or incidentally discovered event. However, “silent infarction” has become more widely used in the stroke lexicon.
Second, inclusion of prior infarctions within estimates of the burden of cerebrovascular disease would be consistent with the growing recognition of their clinical impact. The fact that these lesions are associated with an adverse prognosis for cognitive and functional decline provides the rationale for their inclusion with frank symptomatic stroke as one important measure of health in populations. Relegation of silent strokes to a second-tier category, as if they were simply of incidental or academic importance, or merely markers of susceptibility to stroke, no longer seems appropriate. Moreover, this consideration gains in significance as the population ages and more individuals reach the ages at which strokes tend to occur.
Third, the inclusion of prior infarctions as strokes would emphasize the importance of evaluating and treating these individuals for secondary prevention as aggressively as patients with clearly symptomatic infarcts. As discussed previously, recognition of the increased risk of stroke and other outcomes in this population represents an opportunity to intervene and prevent future asymptomatic and symptomatic strokes. Future trials will be needed, however, to prove that treatment of those with silent strokes will reduce the occurrence of symptomatic strokes or other adverse outcomes. For example, current guidelines for primary prevention of stroke in patients with atrial fibrillation recommend consideration of whether the individual has experienced a stroke in deciding on optimal treatment.114 The CHADS2 (Congestive Heart Failure, Hypertension, Age ≥75, Diabetes, and Stroke/TIA) scoring system gives 2 points to patients with stroke or TIA (using older definitions), and it is generally recommended that patients with a score of ≥2 receive treatment with anticoagulation rather than aspirin.115 However, it is unclear whether an imaging-defined silent infarction should be considered an indication for anticoagulation. Similarly, one may wonder whether patients with carotid stenosis and silent infarctions should be considered to have a higher absolute benefit from anticoagulation than asymptomatic patients with no imaging evidence of infarction. Future studies may address these questions.
There are also reasonable arguments against including silent infarctions within the current definition of stroke. The use of imaging to determine the presence or absence of infarction, it may be argued, necessarily relies on a technology that has limited and ever-changing measurement characteristics (eg, sensitivity, specificity). More sensitive MR techniques may find ever-smaller infarcts, leading to a continual increase in the burden of disease. Such arguments reflect the evolution of medicine and knowledge in general, and they do not detract from the principle that an infarction be defined according to some objective measurement independent of clinical signs or symptoms. Measurement error, or development of more sensitive technologies, should be regarded as distinct from questions about the actual existence of the lesions. Specific consensus recommendations for imaging techniques to determine the presence of prior or silent infarcts lie outside the scope of this discussion, but general guidelines appear earlier in this document.
The inclusion of clinically silent lesions within the rubric of stroke may become problematic in the evaluation of invasive therapies that leave imaging signals of injury without clinical sequelae. Studies among patients undergoing carotid endarterectomy or stenting, for example, have identified MR-detectable but clinically silent infarction in ≈25% of patients, with a range from 0 to 50%.74 Including such imaged events as strokes may unnecessarily inflate the assessment of risk of those procedures without a measurable clinical advantage to doing so. One way to address this problem is to define categories of stroke representing degrees of clinical activity, such as obvious symptoms and signs, subtle signs, and so on. Secondary outcomes could include subtle neuropsychological findings in association with evidence of ischemia or imaged infarction without more overt clinical sequelae. Future research will be needed to determine whether small procedure-related infarctions lead to impaired long-term cognitive or functional performance.
In summary, CNS infarctions occur along an axis of silence or clinical activity, and these in turn depend on the method of observation. Our definitions, therefore, must balance recognition of the spectrum of clinical activity that occurs with cerebral infarction with the awareness that no terminology can account for every individual possibility.
Definition of silent CNS infarction:
Imaging or neuropathological evidence of CNS infarction, without a history of acute neurological dysfunction attributable to the lesion.
This definition is intended to capture those infarcts discovered incidentally, when an autopsy or imaging study is performed outside of a setting consistent with cerebral ischemia (Table 3). For example, silent cerebral infarction may be diagnosed in a patient who undergoes an MRI scan for the evaluation of headache or trauma and is discovered to have imaging evidence of necrosis.
Table 3.
Silent Cerebral Infarctions and Cerebral Microbleeds
| Domain | Silent Cerebral Infarctions | Cerebral Microbleeds |
|---|---|---|
| Clinical features | Any of the following: None Absence of focal symptoms attributable to lesion |
Any of the following: None Absence of focal symptoms attributable to the lesion Cognitive impairment |
| Imaging | CT: Focal areas of hypodensity MRI: acute DWI abnormality, focal T1/FLAIR hypointense, T2 hyperintense lesions (similar to cerebrospinal fluid) |
CT: rarely seen MRI: Focal hypointensity on T2, gradient echo, and/or susceptibility-weighted sequences |
| Size | ≥3 mm | Any size |
| Location | Cortical or subcortical | Cortical or subcortical |
| Number | Single or multiple | Single or multiple |
CT indicates computed tomography; DWI, diffusion-weighted imaging; FLAIR, fluid-attenuated inversion recovery; and MRI, magnetic resonance imaging.
Cerebral Hemorrhage
Hemorrhagic subtypes of stroke, although less common than ischemic stroke and TIA, still have a significant public health impact because of the higher mortality and morbidity associated with them. ICH alone has a nearly 40% case-fatality rate at 30 days.116,117 Although it may seem straightforward to define hemorrhagic subtypes of stroke, a number of issues should be considered, including traumatic injury or secondary causes of bleeding, and the impact of newer technologies on the diagnosis of hemorrhage, among others. Hemorrhages in the CNS should be classified as stroke if they are nontraumatic, caused by a vascular event, and result in injury to the CNS. In contrast, traumatic hemorrhages should not be characterized as stroke. The diagnoses included in this section are ICH, SAH (both aneurysmal and nonaneurysmal), and intraventricular hemorrhage.
Intracerebral Hemorrhage
Nontraumatic ICH is associated with significant morbidity and mortality.118 Symptoms of ICH, however, are not always focal in nature and may be somewhat diffuse and nonspecific. Isolated headache is the presenting symptom in almost 30% of patients with ICH.119 Although ICH has a significantly higher case-fatality rate than other stroke subtypes, it still likely represents ≈5% of mild stroke cases on presentation.120 Therefore, the definition of ICH must be largely based on brain imaging, and the diagnosis cannot be established only on clinical grounds. In the acute setting, CT and MR imaging have extremely high sensitivity and specificity and have been shown to be 96% concordant with each other.121 Some have advocated that although detection of blood on MRI and CT is similar, early MRI can help with diagnosing the underlying pathogenesis of hemorrhage slightly better than CT.122 Catheter angiography can also be useful mainly for diagnosing the specific pathogenesis of hemorrhage rather than initial detection.123
Spontaneous ICH has an entirely different mechanism of injury, affected demographic population, and outcome when compared with traumatic ICH.124 Therefore, an attempt is made to differentiate traumatic ICH when defining ICH. This is not always an easy distinction. For example, if a patient falls and hits his or her head, it is possible that the patient fell because of an ICH before the fall or, alternatively, the fall precipitated a traumatic hemorrhage. In general, traumatic hemorrhages tend to be associated with other types of intracranial bleeding, such as subdural or epidural hematomas. In addition, there is often a coup-contrecoup pattern of injury with traumatic ICH, external signs of trauma, or multiple simultaneous bleeding sites.
Bleeding caused by rupture of a vascular malformation would be considered an ICH,118 and diagnosing an underlying vascular lesion is important for treatment decisions, as well as eventual outcome. The prognosis of an ICH related to an underlying vascular malformation is better than a spontaneous ICH without an underlying lesion.125,126 However, this observation must be interpreted with caution, because patients with arteriovenous malformation–associated ICH are, on average, younger than the patients with non–arteriovenous malformation ICH.
Subdural and epidural hematomas are not included in the definition of stroke. Although subdural hematomas can appear to occur spontaneously and can cause compression of brain structures if large enough, they are typically associated with acute or subacute trauma127 and represent bleeding external to the brain and subarachnoid space. Given the differences in pathology and most likely causes, subdural and epidural hematomas are not considered “strokes.”
Intraventricular hemorrhage is considered a subtype of ICH. Isolated intraventricular hemorrhage is common among premature infants128 and is rare among adults.129 Many times, what may be an isolated intraventricular hemorrhage actually has a small parenchymal ICH adjacent to the ventricle, often in the head of the caudate nucleus or medial thalamus, with intraventricular rupture. Pure intraventricular hemorrhage does occur and can be caused by hypertension, an occult vascular malformation, and, rarely, moyamoya disease and dural arteriovenous fistulas.129
Definition of intracerebral hemorrhage:
A focal collection of blood within the brain parenchyma or ventricular system that is not caused by trauma. (Note: Intracerebral hemorrhage includes parenchymal hemorrhages after CNS infarction, types I and II—see “Hemorrhagic Infarction.”)
Definition of stroke caused by intracerebral hemorrhage:
Rapidly developing clinical signs of neurological dysfunction attributable to a focal collection of blood within the brain parenchyma or ventricular system that is not caused by trauma.
Silent Cerebral Hemorrhage
Chronic small parenchymal hemorrhages, or “microbleeds,” are present in the general population in up to 6% of healthy elderly individuals and in substantially higher rates among those with prior stroke.130 These small hypointense regions, as seen on gradient-echo sequences on MRI, detect breakdown products of blood and most likely represent macrophages containing hemosiderin next to small intraparenchymal blood vessels. Microbleeds appear to share the same underlying pathophysiology as macrohemorrhages and most commonly are observed in patients with cerebral amyloid angiopathy and/or chronic hypertension. Because hemorrhage in the brain is always abnormal, there is no size threshold for microbleeds, unlike small infarct-like lesions. These microbleeds typically are not associated with a clinical event and likely represent “subclinical disease.” There are suggestions that higher volumes of microbleeds are associated with cognitive decline131,132 as well as a higher rate of ICH and ischemic stroke.133 As with silent cerebral infarctions, the clinical impact may depend on the sensitivity of the observer, and classification of these lesions as silent ICH would be consistent and reasonable (Table 3).
Definition of silent cerebral hemorrhage:
A focal collection of chronic blood products within the brain parenchyma, subarachnoid space, or ventricular system on neuroimaging or neuropathological examination that is not caused by trauma and without a history of acute neurological dysfunction attributable to the lesion.
Subarachnoid Hemorrhage
Spontaneous SAH is defined as a stroke because it is a CNS hemorrhage with a vascular cause that commonly results in permanent injury to the CNS. SAH is associated with 20% to 45% mortality134–136 and 10% severe disability,136 although more comprehensive assessments demonstrate much higher rates of cognitive, social, and health disability.137,138
SAH is not a diagnosis based on symptoms alone but rather is defined by the presence of bleeding in the subarachnoid space, confirmed by either imaging or by sampling of the CSF that occupies and circulates within the subarachnoid space. The imaging diagnosis of SAH is by noncontrast head CT imaging or brain MRI. With modern imaging equipment and techniques, the sensitivity of head CT in detecting SAH is >95% in the first 5 days after SAH and 99.7% overall.139 In 1 study with fifth- generation multidetector CT scanners, the sensitivity of head CT was reportedly 100% for detecting SAH,140 although older scanners may be less sensitive. MRI is another imaging modality used to diagnose SAH, with some suggestion that FLAIR sequences may be more sensitive than head CT.141 Artifactual findings may occur with FLAIR and may lead to misinterpretation of normal conditions as SAH or other pathological conditions.142 Sampling of CSF, usually by lumbar puncture, is another method of detecting SAH for the relatively uncommon situation in which neuroimaging is normal or equivocal but clinical suspicion is high. Visual inspection of CSF for xanthochromia, yellowish discoloration that occurs with SAH caused by breakdown of heme from red blood cells in the CSF, was associated with a sensitivity, specificity, positive predictive value, and negative predictive value of 93%, 95%, 72%, and 99%, respectively, for detecting SAH in 1 study,143 while another study found sensitivity to be only 47.3%.144 In a prospective study of patients who presented with acute headache to 2 tertiary care centers, the combination of negative CT and negative lumbar puncture was sufficient to rule out SAH with a sensitivity of 100%.145
Although SAH may be caused by trauma, only spontaneous nontraumatic SAH is considered under the definition of stroke. Causes of nontraumatic SAH are cerebral aneurysm rupture, arteriovenous malformation, intracranial artery dissections, mycotic aneurysms, bleeding disorders, substance abuse,146 reversible cerebral vasoconstriction syndrome, vasculitis, moyamoya, and cerebral amyloid angiopathy.147 Nontraumatic SAH rarely occurs without any of the previously mentioned causes. Perimesencephalic SAH is a type of SAH with a characteristic pattern of blood collected only in the pretruncal cisterns, the absence of an identifiable aneurysm, and associated with a benign prognosis and natural history.148,149 The pathogenesis of this type of SAH remains uncertain, although tearing of the venous structures at the tentorial edge or of the vasa vasorum has been suggested.
Approximately 11% to 60% of patients with SAH report that they had a sudden severe headache during the days to weeks before the SAH, often referred to as sentinel headache.150–152 However, the true existence of sentinel headache as a warning leak has been called into question and challenged by a prospective study,150 because such headaches may be largely a phenomenon of recall bias.150,152 A sudden headache with imaging and CSF studies negative for bleeding in the subarachnoid space is not an SAH and therefore is not a stroke.
Definition of subarachnoid hemorrhage:
Bleeding into the subarachnoid space (the space between the arachnoid membrane and the pia mater of the brain or spinal cord).
Definition of stroke caused by subarachnoid hemorrhage:
Rapidly developing signs of neurological dysfunction and/or headache because of bleeding into the subarachnoid space (the space between the arachnoid membrane and the pia mater of the brain or spinal cord), which is not caused by trauma.
Hemorrhagic Infarction
The term “hemorrhagic stroke” is confusing because it could mean hemorrhage after infarction or primary ICH or SAH. The use of this term should be discontinued.
Hemorrhage may occur after infarction, either spontaneously or caused by antithrombotic or thrombolytic therapy.153,154 Although there are prominent differences between primary CNS infarctions and hemorrhages with respect to the mechanism of damage and prevention of recurrent events, hemorrhage after infarction ranges in severity from minor petechial bleeding to hemorrhage causing mass effect and secondary injury. This has been referred to as “hemorrhagic infarction,” “hemorrhagic transformation of infarction,” “hemorrhagic conversion of infarction,” and “intracerebral hemorrhage,” which leads to confusion among clinicians.
A more standardized approach has been used in clinical trials155 that is well suited for clinical practice as well: hemorrhagic infarction and parenchymal hemorrhage. Hemorrhagic infarction is characterized by its lack of mass effect. Specifically, hemorrhagic infarction type I is defined by petechiae of blood along the margins of the infarction, whereas type II has confluent petechiae within the infarction but without a space-occupying effect. These hemorrhagic infarctions typically present with clinical manifestations similar to nonhemorrhagic infarctions and are often treated according to typical ischemic stroke recommendations and therefore should be considered cerebral infarctions. In contrast, parenchymal hemorrhage is defined by the presence of mass effect, similar to the ICH definition of a focal collection of blood. Parenchymal hemorrhage type I is a confluent hemorrhage limited to ≤30% of the infarcted area with only mild space-occupying effect, and type II is >30% of the infarcted area and/or exerts a significant space-occupying effect. These parenchymal hemorrhages may present with signs and symptoms of mass effect and may require reversal of antithrombotic therapy, aggressive antihypertensive therapy, and/or anti-edema therapy, all of which are distinctly atypical for infarctions but are common recommendations for the treatment of ICH. Therefore, parenchymal hemorrhages should be considered ICHs.
Cerebral Venous Thrombosis
Cerebral venous thrombosis (CVT) can involve the intracranial venous sinuses, the deep venous system, and cortical veins that drain into the major intracranial sinuses.156–161 The occlusion of the venous structures can lead to several mechanisms of brain injury. Tissue ischemia and infarction may result from venous stasis. Secondary petechial or frank hemorrhage may occur within the brain parenchyma (ICH) or in the subarachnoid space, generally localized to cortical sulci of the cerebral convexity.162,163 Lesser degrees of venous congestion cause focal edema, generally vasogenic, in the area of the brain drained by the thrombosed venous structure, without associated infarction. Finally, thrombosis of the venous sinuses may cause no focal brain abnormalities (infarction, hemorrhage, or edema), but impaired venous drainage from the cranial cavity causes increased intracranial pressure without other neurological abnormalities.156–160,162–164
The clinical presentation mirrors these various scenarios, although some symptoms such as headache occur with high frequency (≈80%) across the spectrum of presentations.165 Other symptoms reflect the topography and course of the venous occlusion. Venous infarction, hemorrhage, or edema in the vicinity of an occluded sinus and/or tributary vein results in a presentation that often includes partial seizures and focal neurological deficits accompanied by headache, and potentially by a decreased level of consciousness.160 In contrast, thrombosis of sinuses without parenchymal lesions (infarction, hemorrhage, or edema) can present with a syndrome of “isolated intracranial hypertension,” usually with a subacute diffuse headache and papilledema, although permanent vision loss may occur if this is insufficiently treated. Several other presentations can occur with thrombosis of specific sinuses, including proptosis, chemosis, and ophthalmoplegia in cavernous sinus thrombosis160; involvement of the cranial nerve 5 or 6 in thrombosis of the lateral sinus with extension to the superior or inferior petrosal sinus, respectively; and compromise of cranial nerves 9, 10, and 11 from extension of lateral sinus thrombosis into the jugular bulb.166
Among the many clinical presentations of CVT, there are some that clearly are classifiable as forms of stroke, because they present with persistent focal neurological deficits of acute onset that reflect parenchymal brain damage with a vascular cause; these are the instances in which CVT results in infarction and/or hemorrhage in a localized area of the brain, generally adjacent to an occluded sinus, with or without the accompaniment of tributary cortical vein thrombosis.
Other presentations of CVT do not fit the definition of stroke because they do not reflect persistent focal damage to the CNS. These include (1) instances of transient focal vasogenic edema that, although likely to present with focal neurological deficits and seizures, are not associated with permanent injury in the form of cerebral infarction or hemorrhage, but rather with reversible vasogenic edema; (2) the syndrome of “isolated intracranial hypertension” in which increased intracranial pressure without focal neurological deficits or imaging evidence of infarction or hemorrhage presents with subacute headache and papilledema, at times with an associated transient cranial nerve 6 palsy reflecting increased intracranial pressure; and (3) asymptomatic occlusion of venous sinuses, in the absence of imaging evidence of infarction or hemorrhage. These presentations of CVT pose a risk of permanent nonischemic injury and warrant thorough evaluation to identify potential mechanisms of the CVT to prevent worsening/recurrence and possibly to guide therapy.167–174
Definition of stroke caused by cerebral venous thrombosis:
Infarction or hemorrhage in the brain, spinal cord, or retina because of thrombosis of a cerebral venous structure. Symptoms or signs caused by reversible edema without infarction or hemorrhage do not qualify as stroke.
Implications of the Updated Definition
The revised definition of stroke will impact clinical practice, research, and assessments of the public health. This new definition also builds on the new tissue definition of TIA previously proposed by the AHA/ASA1 and harmonizes with the current definition of MI. Additional AHA statements are needed to improve the clarity and consistency of stroke subtype classification and measurement of stroke severity.
Implications for Clinicians
The current approach to the evaluation and management of acute stroke, both ischemic and hemorrhagic, stresses the value of rapid clinical and imaging diagnosis and prompt treatment. A tissue-based definition of ischemic stroke and TIA enhances diagnostic criteria and relies on utilization of various imaging techniques in the acute phase of the stroke. In this regard, stroke has approached a status similar to that of acute myocardial ischemia, in which the term “acute coronary syndrome” is used when patients present with symptoms of coronary ischemia but before it can be determined whether there is infarction, as assessed by electrocardiography or biomarkers. Nevertheless, these patients receive urgent evaluations and treatments intended to avert or minimize permanent myocardial tissue damage. The parallel can thus be established with stroke, with “acute cerebrovascular syndromes” corresponding to the potential diagnoses of cerebral infarction, TIA, and hemorrhage in patients presenting within the first 24 hours from onset and before the completion of imaging tests. The concept of acute cerebrovascular syndromes would be analogous to that of acute coronary syndrome as far as both imply a common mechanism of acute vascular injury. Ultimately, diagnostic techniques will help define a specific diagnostic category, infarct or hemorrhage based on positive imaging, or TIA in the absence of positive imaging and resolution of symptoms within 24 hours from onset. This approach has been steadily gaining favor over the past 2 decades in industrialized nations, where the availability of imaging technology and treatment options makes rapid diagnosis achievable and treatments available. Even in areas that are remote from major academic centers with stroke expertise, there is the option of telemedicine, which contributes to enhanced, real-time access to state-of-the-art management of patients with acute cerebrovascular syndromes. The main challenge for the future will be the achievement of access to these technological advances in the developing world, where a substantial portion of the global burden of stroke occurs.
This document has emphasized the need to address the subclinical forms of cerebrovascular disease, in particular “silent” infarcts and microhemorrhages. Because these are often detected as a result of the widespread use of MRI for presentations that may or may not be related to clinically apparent cerebrovascular symptoms, how should the clinician approach the finding of such “silent” lesions? These “silent” lesions are not necessarily innocent imaging findings even in asymptomatic patients because they are associated with potentially serious consequences, including cognitive and functional decline and increased future risk of ischemic or hemorrhagic stroke. As a result, the clinician should consider such patients, even in the absence of previous clinical stroke events, as having evidence of cerebrovascular disease. They should be evaluated in terms of prevalence and severity of stroke risk factors and be treated accordingly with measures that are of proven value for the prevention of stroke. For the category of silent infarction, it seems reasonable for the clinician to apply measures of primary stroke prevention114 for patients harboring such asymptomatic lesions at presentation, because guidelines for secondary stroke prevention175 have been generated from clinical trials that have included only patients with “symptomatic” cerebrovascular disease using older definitions that have not included “silent infarcts.” To the best of our knowledge, no studies have addressed the safety and efficacy of secondary prevention measures in patients who only have silent infarction. For example, it is debatable whether in addition to control of vascular risk factors, the clinician should routinely include the use of anti-platelet agents in this population of asymptomatic patients, because there are no data to indicate that such agents prevent further development of “silent” infarcts.
Another aspect related to silent cerebral infarcts that merits discussion is whether the finding of such lesions in imaging studies changes the previously determined asymptomatic character of an internal carotid artery stenosis not associated with TIA or clinical stroke. How should the clinician use the finding of a silent cerebral infarct in the vascular territory of a stenotic extracranial internal carotid artery without prior symptoms? Does this finding have any bearing on the potential benefit of revascularization procedures, which are known to benefit symptomatic patients substantially more than asymptomatic ones? The current evidence from randomized trials for internal carotid artery stenosis does not apply in this circumstance because such patients were not separately evaluated. Future studies and guidelines are needed to determine whether revascularization is warranted for patients with silent cerebral infarction.
The situation is also challenging for those patients who have microhemorrhages in gradient-echo MRI sequences. How should the clinician address this finding in a patient who has no clinical evidence of cerebrovascular symptoms? Again, the currently available evidence suggests that microhemorrhages are markers of cerebrovascular disease and are most notably associated with hypertension and cerebral amyloid angiopathy. Their presence in an asymptomatic patient should place the patient in the group at risk for cerebrovascular events, both ischemic and hemorrhagic, and the appropriate measures of primary stroke prevention should be implemented on the basis of the prevalence and severity of the vascular risk factors. However, it is still uncertain what the potential benefits of primary stroke prevention measures are in asymptomatic patients with microhemorrhages. Although this is still an area of many uncertainties, it is appropriate for the clinician who detects microhemorrhages in an asymptomatic patient to consider them as evidence of subclinical vascular disease and thus institute appropriate measures of primary stroke prevention, being mindful of the potential, but not yet proven, increased risk of hemorrhagic events in such patients when treated with antithrombotic agents. Furthermore, clinicians and patients should be aware of the relationship between amyloid angiopathy and dementia and consider further evaluation.
Finally, clinicians may be faced with a patient who has atrial fibrillation and is at low risk for cardioembolism (ie, CHADS2 score of 0) in whom imaging studies show a “silent” cortically based infarct. Should this information be considered in the decision of whether to use anticoagulation therapy? The original data used to develop the CHADS2 score did not include “silent” infarcts in the definition of stroke. Although most clinicians would consider this finding as potentially indicative of prior “silent” cerebral embolism, guidelines have not formally recommended an approach to silent infarcts in patients with atrial fibrillation.
In conclusion, prompt and timely diagnostic evaluation and treatment should be implemented in patients with acute cerebrovascular syndromes, including ischemic stroke, TIA, and hemorrhagic stroke. The detection of silent vascular lesions, including infarcts and microhemorrhages, implies the presence of cerebrovascular disease despite the absence of symptoms, and these findings should be followed by the assessment and management of vascular risk factors for the purpose of stroke prevention. Future guidelines will address the available evidence for treatment in patients with silent infarctions and hemorrhages.
Implications for Clinical Research and Administrative Databases
Consistent definitions of clinical stroke, stroke subtypes, TIA, and silent cerebral infarction are critical for interpretation of clinical trials, administrative databases, and those studies that examine temporal trends in stroke incidence, prevalence, and mortality in defined populations. The current document recognizes the dramatic impact of brain imaging on the diagnosis of stroke and stroke subtypes over the past 40 years. CNS infarction and TIAs present the greatest challenge in nomenclature because the definitions are intended to be tissue-based but also depend on the nature and duration of clinical symptoms. CT, MRI diffusion, and pathological examination of the brain are progressively more sensitive to ischemic damage, but even those tools have limitations. The linkage of a clinical presentation to characteristic imaging changes provides the greatest confidence in the diagnosis of ischemic stroke. This approach has been used successfully in several major stroke trials to date,176,177 and the updated definition herein further supports this approach. The challenge for the definition of TIA is that mimics, such as focal migraine, exacerbation of prior stroke deficits, and focal seizures, may be difficult to differentiate from a TIA.
Definitions of stroke and TIA in clinical research should always reflect the goals of a given research study and should be carefully specified before initiating the trial. The updated definition of ischemic stroke, stroke caused by ICH, and stroke caused by SAH can be used in studies of primary prevention, secondary prevention, acute treatment, cardiovascular and cerebrovascular procedural trials, and epidemiological studies as the primary outcome. Ideally, imaging protocols should be standardized for these studies to ensure consistency.
TIA with negative brain imaging results has an inherent weakness as a primary study outcome because of confusion with stroke mimics and because of a lack of clinical and functional impact. TIA is an important warning of a subsequent clinical stroke, and it can be used as a secondary outcome measure, but the duration and nature of the symptoms should be carefully characterized and differentiated from stroke mimics when possible. Adjudication of potential TIA events in clinical trials is extremely challenging because the diagnosis rests predominantly on the history and the exclusion of mimics. Furthermore, the evaluation of TIAs in unblinded studies is particularly difficult since referral for clinical and radiographic assessment may be biased.
Silent infarction implies a lack of clear clinical symptoms or signs as well as an inability to temporally fix the stroke occurrence. However, diffusion MRI has the ability to define the time window of the cerebral infarction within several weeks, and within hours or days, if a prior MRI study is negative. In the setting of cardiovascular procedures such as carotid stenting and valve replacement, diffusion changes can indicate an acute cerebral infarction, with or without clinical symptoms. In the immediate postprocedure time period, transient confusion or other symptoms may be attributed to anesthesia or sedation if the MRI is not done. If a clinical trial is comparing 2 interventional approaches and diffusion-positive stroke by MRI is an important outcome measure, it is critical to obtain standardized imaging in both treatment groups at the same time point.
A cerebral infarct on brain imaging of unclear timing is referred to as a silent infarct. This is less likely to be used as an outcome in a trial but could be used as a baseline patient characteristic that may affect risk of subsequent stroke, cognitive decline, or functional outcome at a later point in the trial. New, clinically silent cerebral infarctions of undetermined onset (diffusion negative on MRI) are not recommended as a primary or secondary outcome in most stroke studies unless all study patients undergo standardized imaging at specific time points according to the study protocol. Under such conditions, new silent infarctions could be considered as secondary outcomes rather than events equivalent to ischemic strokes.
Preventative therapies should be specifically tested in patients with silent infarctions, as at present it is unclear whether recommendations for secondary stroke prevention175 should apply to this population.
Notably, a study looking at temporal trends in stroke incidence over a long time period may face substantial changes in the type and availability of brain imaging in clinical practice over time and therefore might adopt several operational approaches to the diagnosis of stroke. In such studies, the primary definition of stroke may need to remain clinical stroke symptoms or signs lasting ≥24 hours. Those patients with focal symptoms of <24 hours but with positive imaging are not considered to have had an ischemic stroke using this clinical definition because imaging is likely inconsistently performed and with varying methods. A second definition in these studies could be the tissue-based definition proposed in the current statement in which both of these patients would be considered to have had a CNS infarction. Using both definitions provides a measure of the impact of brain imaging on temporal trends in stroke incidence and mortality.
Implications for Public Health
Changing the definition of disease can have significant effects on disease surveillance and prognosis. Case definitions are often adjusted as greater knowledge of the disease is achieved or better testing is available. The Will Rogers phenomenon is often used to describe the effect of changes in diagnostic criteria on classification groups and could be applicable to this updated definition of stroke.178 In general, the Will Rogers phenomenon refers to a paradox that occurs when adding or removing a group from one class causes a rate in that class to increase or decrease while at the same time it also affects the rate in the second related class in an unexpected or paradoxical way. In the case of adding a large number of asymptomatic stroke cases to the existing number of stroke cases, this will increase the total number of stroke cases while possibly decreasing the mortality rate because of the addition of a number of minor cases. Will Rogers, an American humorist, is believed to have said about migration during the 1930s, “When the Okies left Oklahoma and moved to California, they raised the average intelligence level in both states.” The Will Rogers phenomenon similarly has been observed in cancer epidemiology to describe how more sensitive diagnostic tests have led to finding disease at earlier stages; as such, the disease prevalence increases while the case mortality risks fall in parallel.179 When case definitions are updated, tracking a disease over time requires attention to the timing of definition change for accurate interpretation of the data.180 Updating the definition of stroke could result in reclassification of stroke cases for incidence, prevalence, and mortality. In addition, the updated definition will have implications for public health, including surveillance and reporting, national and international statistics, disease classification coding systems, and existing health surveys.
Public health surveillance of stroke can be done with various methods, each yielding a different perspective.180 For example, the prevalence of stroke can be determined in several ways, including surveys using self-report, surveys using clinical and/or imaging evidence, from the review of claims databases, or from reviews of hospital records. All of these data sources have trade-offs in terms of sensitivity and specificity. Several health surveys ask respondents about self-reported history of stroke. Currently, the possibility of underdiagnosing or overdiagnosing stroke exists with self-report measures. Patients may not identify themselves as having a history of stroke or TIA when one has occurred or may attribute symptoms to a stroke when one has not occurred. As the definition of stroke is updated, the potential for misclassification may increase among self-report surveys.
One of the overarching concerns regarding a shift to an imaging-based definition for stroke is the potential to bias stroke surveillance reporting based on the availability of technology to contribute to image-based case ascertainment. MR brain imaging is more sensitive for detecting cerebral infarction and hemorrhage than CT and is not as widely available in developing countries. In many countries with more limited access to state-of-the-art diagnostic equipment, no timely brain imaging may be available. Furthermore, some administrative data systems rely on data from the ICD. Table 4 maps the proposed updated stroke definitions with ICD, Ninth Revision, Clinical Modification (ICD-9-CM) and ICD-10-CM codes. Analysis of existing ICD-10-CM codes and the definition of stroke put forward in this document reveals a gap in coding for silent infarction and silent hemorrhages. Silent infarctions or hemorrhages, if documented by the provider, would be coded as infarcts or hemorrhages. If identified retrospectively, codes representing the late effects of stroke would likely be assigned. Addressing this gap would minimize coding bias across regions that currently lack resources to make image-based diagnoses of silent cerebral events for reporting or surveillance. In these regions, acute clinical symptoms may continue to be used to identify most stroke cases, rather than relying on imaging evidence, and may not capture all cases.
Table 4.
Mapping of Stroke Definitions, Including TIA, From ICD-9-CM to ICD-10-CM
| ICD-9-CM Description | ICD-9-CM | ICD-10-CM | ICD-10-CM Description | AHA Updated Definition |
|---|---|---|---|---|
| CNS infarction | ||||
| Occlusion and stenosis of precerebral and cerebral arteries with infarction | 433.01, 433.11, 433.21, 433.31, 433.81, 433.91 (unspecified), 434.01, 434.11, 434.91 (unspecified) | All I63: I63.0, I63.1, I63.2, I63.4, I63.5, I63.6, I63.8 (other), I63.9 (unspecified) | Cerebral infarction | CNS infarction |
| Silent cerebral infarction* | NA | NA | Silent cerebral infarction | Silent CNS infarction |
| Acute infarction of spinal cord (embolic) (nonembolic) | 336.1 | G95.11 | Acute infarction of spinal cord (embolic) (nonembolic) | CNS infarction |
| Retinal vascular occlusion: central retinal artery occlusion, arterial branch occlusion | 362.31, 362.32 | H34.1, H34.23 | Central retinal artery occlusion, retinal artery branch occlusion | CNS infarction |
| Cerebral thrombosis with infarction | 434.01 | I63.3 | Infarct caused by cerebral venous thrombosis, nonpyogenic | Stroke caused by cerebral venous thrombosis |
| CNS hemorrhage | ||||
| Intracerebral hemorrhage | 431 | All I61: I61.0 – I61.4, I61.8-I61.9 | Nontraumatic intracerebral hemorrhage | Intracerebral hemorrhage |
| Intracerebral hemorrhage | 431 | I61.5 | Nontraumatic intracerebral hemorrhage, intraventricular | Intracerebral hemorrhage |
| Intracerebral hemorrhage | 431 | I61.6 | Nontraumatic intracerebral hemorrhage, multiple, localized | Intracerebral hemorrhage |
| Silent cerebral hemorrhage* | NA | NA | Silent cerebral hemorrhage | Silent cerebral hemorrhage |
| Subarachnoid hemorrhage | 430 | All I60: I60.0 – I60.9 | Nontraumatic subarachnoid hemorrhage | Subarachnoid hemorrhage |
| Nontraumatic intracranial hemorrhage, unspecified | 432.9 | I62.9 | Nontraumatic intracranial hemorrhage, unspecified | … |
| Acute ill-defined cerebrovascular disease | 436 (Included in ischemic stroke for GWTG and TJC PSC) | I67.89 | Other acute cerebrovascular insufficiency NOS and cerebral ischemia (chronic) | … |
| TIA | 435.0, 435.1, 435.2, 435.3, 435.8., 435.9 | All G45 except G45.3 (amaurosis fugax) | Transient cerebral ischemic attacks and related syndromes | TIA |
| Transient retinal arterial occlusion (amaurosis fugax) | 362.34 | G45.3 | Amaurosis fugax | TIA |
AHA indicates American Heart Association; CNS, central nervous system; GWTG, Get With The Guidelines; ICD-9-CM, International Classification of Diseases, Ninth Revision, Clinical Modification; ICD-10-CM, International Classification of Diseases, Tenth Revision, Clinical Modification; NA, not applicable; NOS, not otherwise specified; PSC, primary stroke center; TIA, transient ischemic attack; and TJC, The Joint Commission.
Specific codes for these diagnoses do not currently exist.
Another concern for stroke surveillance is the inclusion of spinal and retinal arterial infarctions within the definition of stroke. Typically, these are not included in epidemiological studies. We propose that, for global reporting of the public health impact of stroke, symptomatic CNS infarction and symptomatic hemorrhage be recorded and, where available, silent subgroups recorded. Having separate recording of these subgroups would allow for more valid analysis of temporal and geographic trends in the surveillance of stroke.
Disclosures
| Writing Group Disclosures | ||||||||
|---|---|---|---|---|---|---|---|---|
| Writing Group Member | Employment | Research Grant | Other Research Support | Speakers’ Bureau/Honoraria | Expert Witness | Ownership Interest | Consultant/Advisory Board | Other |
| Ralph L. Sacco | University of Miami | NINDS† | None | None | None | None | DSMB (Atrial Fibrillation Trial–institutionally sponsored by PHRI Hamilton, Ontario)* | None |
| Scott E. Kasner | University of Pennsylvania | Gore Associates† | None | None | None | None | AstraZeneca*; BrainsGate (DSMB)*; CardioNet*; Medtronic DSMB*; Novartis*; Parexel*; Pfizer* | None |
| Joseph P. Broderick | University of Cincinnati | NINDS† | None | Oakstone Publishing* | None | None | Genentech*; PhotoThera DSMB* (All consultant fees are placed in educational/research fund for UIC Department of Neurology) | None |
| Louis R. Caplan | Beth Israel Deaconess Medical Center | None | None | AstraZeneca*; Boehringer-Ingelheim*; Bristol-Myers-Squibb*; Otsuka*; Sanofi-Synthelabo* | None | None | ARUBA Trial (Endpoint Adjudication Committee) NINDS*; AstraZeneca*; Avanir Pharmaceuticals*; Bayer Schering Pharma*; Boehringer-Ingelheim*; CoAxia*; Genentech*; Jones & Davis, LLP*; LifeCycle Pharma A/S*; Lytics Lifecycle*; NeuroLogica*; Novo Nordisk*; NovoVision*; Micromedex*; Millennium Pharma*; ReNeuron*; SAMPRISS Trial (Endpoint Adjudication Committee) NINDS*; Takeda Pharma* | Editorial boards: Reviews in Neurological Diseases (Editor)*; Archives of Neurology* |
| J.J. (Buddy) Connors | Vanderbilt University Medical Center | None | None | None | None | None | None | None |
| Antonio Culebras | Upstate Medical University (New York) | None | None | None | J Uriach Foundation* | None | Member WHO Advisory Group for Revision of ICDS-10, sleep disorders* | Editorial boards: Reviews in Neurological Diseases (Associate Editor)*; MedLink (Associate Editor)*; AAN Guideline Update, Atrial Fibrillation panel (Chair)*; AAN Stroke Section (Chair-elect)* |
| Mitchell S.V. Elkind | Columbia University | Bristol-Myers Squibb-Sanofi Partnership*; diaDexus, Inc*; NHLBI†; NIH/NINDS† (NOMAS–PI; LIMITS–PI; NeuSTART–PI; VIPS–Co-I; VERITAS–Co-I; SAMMPRIS–Co-I; SPS3–Co-I; ERICH–Co-I; California Teachers Study–Co-I) | None | Bristol-Myers Squibb-Sanofi Partnership*; Boehringer-Ingelheim*; Genentech* | GlaxoSmithKline* (Avandia defense); Novartis (Zelnorm defense)† | None | GlaxoSmithKline*; Tethys Biosciences* | None |
| Mary G. George | Centers for Disease Control and Prevention | None | None | None | None | None | None | None |
| Allen D. Hamdan | Beth Israel Deaconess Medical Center | None | None | None | None | None | None | None |
| Randall T. Higashida | University of California at San Francisco | None | None | None | None | None | None | None |
| Brian L. Hoh | University of Florida | Brain Aneurysm Foundation†; Micrus Endovascular*; NIH†; Thomas H. Maren Foundation† | None | None | None | None | Actelion Pharmaceutical*; Codman Neurovascular* | None |
| L. Scott Janis | NINDS/NIH/DHHS | None | None | None | None | None | None | None |
| Carlos S. Kase | Boston University Medical Center | NIH† | None | None | None | None | Ferrer International*; Sanofi-Aventis* | None |
| Dawn O. Kleindorfer | University of Cincinnati | AAMC/CDC†; NIH/NINDS†; NIH†; NIH/Yale† | None | Genentech* | None | None | Boehringer-Ingelheim* | None |
| Jin-Moo Lee | Washington University School of Medicine | AGA Medical†; Barnes-Jewish Hospital Foundation†; NIA†; NHLBI†; NINDS† | AstraZeneca† | None | None | None | None | None |
| Michael E. Moseley | Stanford University | NINDS† | None | None | None | None | None | None |
| Eric D. Peterson | Duke University | Bristol-Myers Squibb-Sanofi†; Johnson & Johnson†; Lilly†; NIH†; Merck/Schering† | None | None | None | None | None | None |
| Tanya N. Turan | Medical University of South Carolina | NIH†; NIH/NINDS† | None | None | None | None | Boehringer-Ingelheim Pharma GmbH & Co*; Gore & Associates*; NIH * | None |
| Amy L. Valderrama | Centers for Disease Control and Prevention | None | None | None | None | None | None | None |
| Harry V. Vinters | David Geffen School of Medicine at University of California, Los Angeles, and Ronald Reagan UCLA Medical Center, Los Angeles | National Alzheimer Coordinating Center (NACC) †; NINDS†; State of California, Pediatric Neuropathology Consortium, member†; UCLA Alzheimer Disease Research Center†; UCLA SPOTRIAS grant† | Translational Research Fund, UCLA Department of Pathology* | Occasional CME talks (2–3 annually), Los Angeles–area hospitals* | Beasley & Demos – cerebral cortical malformation case for defense*; Dummit, Buchholz & Trapp – review autopsy slides for wrongful death case for defense*; Kralovec, Jambois & Schwartz – pituitary adenoma case for defense*; Krupnick, Campbell & Malone – incorrect diagnosis of brain tumor case for plaintiff*; LaFollette, Johnson, DeHaas, Fesler & Ames – AVM case for defense*; Lewis, Brisbois, Bisgaard & Smith – age and nature of neuropathologic changes case for defense*; McKeen & Associates, P.C. - review autopsy for fatal cerebral edema with hyponatremia case for defense*; Murphy, Pearson, Bradley & Feeney – subdural hematoma case for defense*; Reback, McAndrews, Kjar, Warford & Stockalper, LLP – autopsy evaluation for Alzheimer disease case for defense*; Steven L. Saldo – spontaneous cerebral hemorrhage case for defense*; Skiver & Associates – wrongful death caused by anesthetic error leading to neurologic complications case for defense*; Snyder & Wenner, P.C. – autopsy review for wrongful death related to seizures case for defense* | Becton Dickinson†; General Electric (medical and imaging equipment)†; GlaxoSmithKline†; Pfizer†; Teva Pharmaceuticals†; 3M (medical equipment and supplies)† | ESAB*; Indiana Alzheimer Center*; Massachusetts ADRC*; HIV Neurobehavioral Research Center (San Diego, UCSD)* | Editorial boards: Neuropathology and Applied Neurobiology*; Neuropathology*; Korean Journal of Pathology*; Journal of Neuroscience Research*; Human Pathology* |
| Reviewer Disclosures | ||||||||
|---|---|---|---|---|---|---|---|---|
| Reviewer | Employment | Research Grant | Other Research Support | Speakers’ Bureau/Honoraria | Expert Witness | Ownership Interest | Consultant/Advisory Board | Other |
| Harold P. Adams | University of Iowa | NINDS*; St. Jude* | None | None | None | None | None | None |
| Gregory W. Albers | Stanford University | None | None | None | None | None | None | None |
| Robert Brown | Mayo Clinic | None | None | None | None | None | None | None |
| Creed Pettigrew | University of Kentucky | None | None | None | None | None | None | None |
| Lee H. Schwamm | Massachusetts General Hospital | None | None | None | None | None | LifeImage*; Lundbeck DIAS-4 International Steering Committee* | Chair, GWTG national steering committee (unpaid)* |
| Babu Welch | UT Southwestern Medical Center | None | None | None | None | None | None | None |
| Philip A. Wolf | Boston University | None | None | None | None | None | None | None |
| Allyson Zazulia | Washington University | None | None | None | None | None | None | None |
This table represents the relationships of writing group members that may be perceived as actual or reasonably perceived conflicts of interest as reported on the Disclosure Questionnaire, which all members of the writing group are required to complete and submit. A relationship is considered to be “significant” if (1) the person receives $10 000 or more during any 12-month period, or 5% or more of the person’s gross income; or (2) the person owns 5% or more of the voting stock or share of the entity, or owns $10 000 or more of the fair market value of the entity. A relationship is considered to be “modest” if it is less than “significant” under the preceding definition.
Modest.
Significant.
This table represents the relationships of reviewers that may be perceived as actual or reasonably perceived conflicts of interest as reported on the Disclosure Questionnaire, which all reviewers are required to complete and submit. A relationship is considered to be “significant” if (1) the person receives $10 000 or more during any 12-month period, or 5% or more of the person’s gross income; or (2) the person owns 5% or more of the voting stock or share of the entity, or owns $10 000 or more of the fair market value of the entity. A relationship is considered to be “modest” if it is less than “significant” under the preceding definition.
Modest.
Footnotes
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the National Institutes of Health or any part of the US federal government.
At the end of deliberations, the final recommendations for the definition of stroke were not acceptable by the leadership of the European Stroke Organization and World Stroke Organization. These organizations declined to participate further in this statement. Their dissent was mainly associated with the inclusion of silent cerebral infarction and silent cerebral hemorrhage within the universal definition of stroke.
The American Heart Association makes every effort to avoid any actual or potential conflicts of interest that may arise as a result of an outside relationship or a personal, professional, or business interest of a member of the writing panel. Specifically, all members of the writing group are required to complete and submit a Disclosure Questionnaire showing all such relationships that might be perceived as real or potential conflicts of interest.
References
- 1.Easton JD, Saver JL, Albers GW, Alberts MJ, Chaturvedi S, Feldmann E, Hatsukami TS, Higashida RT, Johnston SC, Kidwell CS, Lutsep HL, Miller E, Sacco RL. Definition and evaluation of transient ischemic attack: a scientific statement for healthcare professionals from the American Heart Association/American Stroke Association Stroke Council; Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; and the Interdisciplinary Council on Peripheral Vascular Disease. Stroke. 2009;40:2276–2293. [DOI] [PubMed] [Google Scholar]
- 2.Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD; Writing Group on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Universal Definition of Myocardial Infarction. Third universal definition of myocardial infarction. Circulation. 2012;126:2020–2035.22923432 [Google Scholar]
- 3.Sacco RL, Kargman DE, Gu Q, Zamanillo MC. Race-ethnicity and determinants of intracranial atherosclerotic cerebral infarction: the Northern Manhattan Stroke Study. Stroke. 1995;26:14–20. [DOI] [PubMed] [Google Scholar]
- 4.Cole W A Physico-Medical Essay Concerning the Late Frequency of Apoplexies Together With a General Method of Their Prevention and Cure: In a Letter to a Physician. Oxford, United Kingdom; The Theater; 1869. Reprinted by: New York, NY: Classics of Neurology & Neurosurgery Library; 1995. [Google Scholar]
- 5.Hippocrates. The Genuine Works of Hippocrates: Translated From the Greek With a Preliminary Discourse and Annotations by Francis Adams. Adams F, trans-ed. Baltimore, MD: Williams & Wilkins; 1939. [Google Scholar]
- 6.Aho K, Harmsen P, Hatano S, Marquardsen J, Smirnov VE, Strasser T. Cerebrovascular disease in the community: results of a WHO collaborative study. Bull World Health Organ. 1980;58:113–130. [PMC free article] [PubMed] [Google Scholar]
- 7.Fisher CM. Intermittent cerebral ischemia. In: Wright IS, Millikan CH, eds. Cerebral Vascular Diseases. New York, NY: Grune & Stratton; 1958:81–97. [Google Scholar]
- 8.Mohr JP. Historical perspective. Neurology. 2004;62(suppl 6):S3–S6. [DOI] [PubMed] [Google Scholar]
- 9.A classification and outline of cerebrovascular diseases, II. Stroke. 1975;6:564–616. [DOI] [PubMed] [Google Scholar]
- 10.Albers GW, Caplan LR, Easton JD, Fayad PB, Mohr JP, Saver JL, Sherman DG; TIA Working Group. Transient ischemic attack: proposal for a new definition. N Engl J Med. 2002;347:1713–1716. [DOI] [PubMed] [Google Scholar]
- 11.Centers for Disease Control and Prevention Web site. Classification of Diseases, Functioning, and Disability. International Classification of Diseases, Tenth Revision, Clinical Modification (ICD-10-CM). http://www.cdc.gov/nchs/icd/icd10cm.htm#10update. Accessed April 25, 2013. [Google Scholar]
- 12.Furlan M, Marchal G, Viader F, Derlon JM, Baron JC. Spontaneous neurological recovery after stroke and the fate of the ischemic penumbra. Ann Neurol. 1996;40:216–226. [DOI] [PubMed] [Google Scholar]
- 13.Olivot JM, Albers GW. Using advanced MRI techniques for patient selection before acute stroke therapy. Curr Treat Options Cardiovasc Med. 2010;12:230–239. [DOI] [PubMed] [Google Scholar]
- 14.Brazzelli M, Sandercock P, Chappell F, Celani M, Righetti E, Arestis N, Wardlaw J, Deeks J. Magnetic resonance imaging versus computed tomography for detection of acute vascular lesions in patients presenting with stroke symptoms. Cochrane Database Syst Rev. 2009;(4):CD007424. [DOI] [PubMed] [Google Scholar]
- 15.Albers GW, Thijs VN, Wechsler L, Kemp S, Schlaug G, Skalabrin E, Bammer R, Kakuda W, Lansberg MG, Shuaib A, Coplin W, Hamilton S, Moseley M, Marks MP; DEFUSE Investigators. Magnetic resonance imaging profiles predict clinical response to early reperfusion: the diffusion and perfusion imaging evaluation for understanding stroke evolution (DEFUSE) study. Ann Neurol. 2006;60:508–517. [DOI] [PubMed] [Google Scholar]
- 16.Chalela JA, Kidwell CS, Nentwich LM, Luby M, Butman JA, Demchuk AM, Hill MD, Patronas N, Latour L, Warach S. Magnetic resonance imaging and computed tomography in emergency assessment of patients with suspected acute stroke: a prospective comparison. Lancet. 2007;369:293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoo AJ, Verduzco LA, Schaefer PW, Hirsch JA, Rabinov JD, González RG. MRI-based selection for intra-arterial stroke therapy: value of pre-treatment diffusion-weighted imaging lesion volume in selecting patients with acute stroke who will benefit from early recanalization. Stroke. 2009;40:2046–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bang OY. Multimodal MRI for ischemic stroke: from acute therapy to preventive strategies. J Clin Neurol. 2009;5:107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deguchi I, Takeda H, Furuya D, Dembo T, Nagoya H, Kato Y, Ito Y, Fukuoka T, Maruyama H, Tanahashi N. Significance of magnetic resonance angiography-diffusion weighted imaging mismatch in hyperacute cerebral infarction [published correction appears in J Stroke Cerebrovasc Dis. 2012;21:342]. J Stroke Cerebrovasc Dis. 2012;21: 108–113. [DOI] [PubMed] [Google Scholar]
- 20.Kidwell CS, Saver JL, Mattiello J, Starkman S, Vinuela F, Duckwiler G, Gobin YP, Jahan R, Vespa P, Kalafut M, Alger JR. Thrombolytic reversal of acute human cerebral ischemic injury shown by diffusion/perfusion magnetic resonance imaging. Ann Neurol. 2000;47:462–469. [PubMed] [Google Scholar]
- 21.Pedraza S, Puig J, Blasco G, Daunis-i-Estadella J, Boada I, Bardera A, Prats A, Castellanos M, Serena J. Magnetic resonance imaging biomarkers of ischemic stroke: criteria for the validation of primary imaging biomarkers. Drug News Perspect. 2009;22:481–486. [PubMed] [Google Scholar]
- 22.Davis SM, Donnan GA, Parsons MW, Levi C, Butcher KS, Peeters A, Barber PA, Bladin C, De Silva DA, Byrnes G, Chalk JB, Fink JN, Kimber TE, Schultz D, Hand PJ, Frayne J, Hankey G, Muir K, Gerraty R, Tress BM, Desmond PM; EPITHET Investigators. Effects of alteplase beyond 3 h after stroke in the Echoplanar Imaging Thrombolytic Evaluation Trial (EPITHET): a placebo-controlled randomised trial. Lancet Neurol. 2008;7:299–309. [DOI] [PubMed] [Google Scholar]
- 23.Furlan AJ, Eyding D, Albers GW, Al-Rawi Y, Lees KR, Rowley HA, Sachara C, Soehngen M, Warach S, Hacke W; DEDAS Investigators. Dose Escalation of Desmoteplase for Acute Ischemic Stroke (DEDAS): evidence of safety and efficacy 3 to 9 hours after stroke onset. Stroke. 2006;37:1227–1231. [DOI] [PubMed] [Google Scholar]
- 24.Hacke W, Albers G, Al-Rawi Y, Bogousslavsky J, Davalos A, Eliasziw M, Fischer M, Furlan A, Kaste M, Lees KR, Soehngen M, Warach S; DIAS Study Group. The Desmoteplase in Acute Ischemic Stroke Trial (DIAS): a phase II MRI-based 9-hour window acute stroke thrombolysis trial with intravenous desmoteplase. Stroke. 2005;36:66–73. [DOI] [PubMed] [Google Scholar]
- 25.Hacke W, Furlan AJ, Al-Rawi Y, Davalos A, Fiebach JB, Gruber F, Kaste M, Lipka LJ, Pedraza S, Ringleb PA, Rowley HA, Schneider D, Schwamm LH, Leal JS, Söhngen M, Teal PA, Wilhelm-Ogunbiyi K, Wintermark M, Warach S. Intravenous desmoteplase in patients with acute ischaemic stroke selected by MRI perfusion-diffusion weighted imaging or perfusion CT (DIAS-2): a prospective, randomised, double-blind, placebo-controlled study. Lancet Neurol. 2009;8:141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Latchaw RE, Alberts MJ, Lev MH, Connors JJ, Harbaugh RE, Higashida RT, Hobson R, Kidwell CS, Koroshetz WJ, Mathews V, Villablanca P, Warach S, Walters B; on behalf of the American Heart Association Council on Cardiovascular Radiology and Intervention, Stroke Council, and the Interdisciplinary Council on Peripheral Vascular Disease. Recommendations for imaging of acute ischemic stroke: a scientific statement from the American Heart Association. Stroke. 2009;40:3646–3678. [DOI] [PubMed] [Google Scholar]
- 27.Barber PA, Darby DG, Desmond PM, Gerraty RP, Yang Q, Li T, Jolley D, Donnan GA, Tress BM, Davis SM. Identification of major ischemic change: diffusion-weighted imaging versus computed tomography. Stroke. 1999;30:2059–2065. [DOI] [PubMed] [Google Scholar]
- 28.Hakyemez B, Aksoy U, Yildiz H, Ergin N. Intracranial epidermoid cysts: diffusion-weighted, FLAIR and conventional MR findings. Eur J Radiol. 2005;54:214–220. [DOI] [PubMed] [Google Scholar]
- 29.Warach S, Gaa J, Siewert B, Wielopolski P, Edelman RR. Acute human stroke studied by whole brain echo planar diffusion-weighted magnetic resonance imaging. Ann Neurol. 1995;37:231–241. [DOI] [PubMed] [Google Scholar]
- 30.Burdette JH, Elster AD, Ricci PE. Acute cerebral infarction: quantification of spin-density and T2 shine-through phenomena on diffusion-weighted MR images. Radiology. 1999;212:333–339. [DOI] [PubMed] [Google Scholar]
- 31.Eastwood JD, Engelter ST, MacFall JF, Delong DM, Provenzale JM. Quantitative assessment of the time course of infarct signal intensity on diffusion-weighted images. AJNR Am J Neuroradiol. 2003;24:680–687. [PMC free article] [PubMed] [Google Scholar]
- 32.Masson C, Pruvo JP, Meder JF, Cordonnier C, Touzé E, De La Sayette V, Giroud M, Mas JL, Leys D; Study Group on Spinal Cord Infarction of the French Neurovascular Society. Spinal cord infarction: clinical and magnetic resonance imaging findings and short term outcome. J Neurol Neurosurg Psychiatry. 2004;75:1431–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Novy J, Carruzzo A, Maeder P, Bogousslavsky J. Spinal cord ischemia: clinical and imaging patterns, pathogenesis, and outcomes in 27 patients. Arch Neurol. 2006;63:1113–1120. [DOI] [PubMed] [Google Scholar]
- 34.Robertson CE, Brown RD Jr, Wijdicks EF, Rabinstein AA. Recovery after spinal cord infarcts: long-term outcome in 115 patients. Neurology. 2012;78:114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Faig J, Busse O, Salbeck R. Vertebral body infarction as a confirmatory sign of spinal cord ischemic stroke: report of three cases and review of the literature. Stroke. 1998;29:239–243. [DOI] [PubMed] [Google Scholar]
- 36.Yin NS, Benavides S, Starkman S, Liebeskind DS, Saver JA, Salamon N, Jahan R, Duckwiler GR, Tateshima S, Vinuela F, Vespa PM, Chute DJ, Vinters HV. Autopsy findings after intracranial thrombectomy for acute ischemic stroke: a clinicopathologic study of 5 patients. Stroke. 2010;41:938–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petito CK. The neuropathology of focal brain ischemia. In: Kalimo H. ed. Pathology & Genetics: Cerebrovascular Diseases. Basel, Switzerland: ISN Neuropath Press; 2005:215–221. [Google Scholar]
- 38.Vinters HV, Farrell MA, Mischel PS, Anders KH. Anoxic-ischemic change and cerebrovascular disease. In: Diagnostic Neuropathology. New York, NY: Marcel Dekker; 1998:51–146. [Google Scholar]
- 39.Hazrati LN, Bergeron C, Butany J. Neuropathology of cerebrovascular diseases. Semin Diagn Pathol. 2009;26:103–115. [DOI] [PubMed] [Google Scholar]
- 40.Vinters HV. Cerebrovascular disease: practical issues in surgical and autopsy pathology. In: Love S, ed. Neuropathology. A Guide for Practising Pathologists. Berlin-Heidelberg, Germany: Springer; 2001:51–99. Current Topics in Pathology; vol 95. [DOI] [PubMed] [Google Scholar]
- 41.Busl KM, Greer DM. Hypoxic-ischemic brain injury: pathophysiology, neuropathology and mechanisms. NeuroRehabilitation. 2010;26:5–13. [DOI] [PubMed] [Google Scholar]
- 42.Wirenfeldt M, Babcock AA, Vinters HV. Microglia: insights into immune system structure, function, and reactivity in the central nervous system. Histol Histopathol. 2011;26:519–530. [DOI] [PubMed] [Google Scholar]
- 43.Soontornniyomkij V, Lynch MD, Mermash S, Pomakian J, Badkoobehi H, Clare R. Cerebral microinfarcts associated with severe cerebral beta-amyloid angiopathy. Brain Pathol. 2010;20:459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vinters HV, Ellis WG, Zarow C, Zaias BW, Jagust WJ, Mack WJ, Chui HC. Neuropathologic substrates of ischemic vascular dementia. J Neuropathol Exp Neurol. 2000;59:931–945. [DOI] [PubMed] [Google Scholar]
- 45.Demchuk AM, Hill MD, Barber PA, Silver B, Patel SC, Levine SR; NINDS rtPA Stroke Study Group, NIH. Importance of early ischemic computed tomography changes using ASPECTS in NINDS rtPA Stroke Study. Stroke. 2005;36:2110–2115. [DOI] [PubMed] [Google Scholar]
- 46.Patel SC, Levine SR, Tilley BC, Grotta JC, Lu M, Frankel M, Haley EC Jr, Brott TG, Broderick JP, Horowitz S, Lyden PD, Lewandowski CA, Marler JR, Welch KM; National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Lack of clinical significance of early ischemic changes on computed tomography in acute stroke. JAMA. 2001;286:2830–2838. [DOI] [PubMed] [Google Scholar]
- 47.Hacke W, Kaste M, Fieschi C, von Kummer R, Davalos A, Meier D, Larrue V, Bluhmki E, Davis S, Donnan G, Schneider D, Diez-Tejedor E, Trouillas P. Randomised double-blind placebo-controlled trial of thrombolytic therapy with intravenous alteplase in acute ischaemic stroke (ECASS II): Second European-Australasian Acute Stroke Study Investigators. Lancet. 1998;352:1245–1251. [DOI] [PubMed] [Google Scholar]
- 48.Wardlaw JM, Dorman PJ, Lewis SC, Sandercock PA. Can stroke physicians and neuroradiologists identify signs of early cerebral infarction on CT? J Neurol Neurosurg Psychiatry. 1999;67:651–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ginde AA, Foianini A, Renner DM, Valley M, Camargo CA Jr. Availability and quality of computed tomography and magnetic resonance imaging equipment in U.S. emergency departments. Acad Emerg Med. 2008;15:780–783. [DOI] [PubMed] [Google Scholar]
- 50.Lövblad KO, Laubach HJ, Baird AE, Curtin F, Schlaug G, Edelman RR, Warach S. Clinical experience with diffusion-weighted MR in patients with acute stroke. AJNR Am J Neuroradiol. 1998;19:1061–1066. [PMC free article] [PubMed] [Google Scholar]
- 51.Oppenheim C, Stanescu R, Dormont D, Crozier S, Marro B, Samson Y, Rancurel G, Marsault C. False-negative diffusion-weighted MR findings in acute ischemic stroke. AJNR Am J Neuroradiol. 2000;21:1434–1440. [PMC free article] [PubMed] [Google Scholar]
- 52.Von Kummer R Imaging of stroke pathology without predefined gold standard. Cerebrovasc Dis. 2002;14:270. [DOI] [PubMed] [Google Scholar]
- 53.Levy DE. How transient are transient ischemic attacks? Neurology. 1988;38:674–677. [DOI] [PubMed] [Google Scholar]
- 54.Shah SH, Saver JL, Kidwell CS, Albers GW, Rothwell PM, Ay H, Koroshetz WJ, Inatomi Y, Uchino M, Demchuk AM, Coutts SB, Purroy F, Alvarez-Sabin JS, Sander D, Sander K, Restrepo L, Wityk RJ, Marx JJ, Easton JD. A multicenter pooled, patient-level data analysis of diffusion-weighted MRI in TIA patients. Stroke. 2007;38:463. Abstract. [Google Scholar]
- 55.Ellison D, Love S, Chimelli L, Harding BN, Lowe JS, Vinters HV, Brandner S, Yong WH. Neuropathology. 3rd ed. London, UK: Mosby Elsevier; 2013. [Google Scholar]
- 56.Rosamond WD, Folsom AR, Chambless LE, Wang CH, McGovern PG, Howard G, Copper LS, Shahar E. Stroke incidence and survival among middle-aged adults: 9-year follow-up of the Atherosclerosis Risk in Communities (ARIC) cohort. Stroke. 1999;30:736–743. [DOI] [PubMed] [Google Scholar]
- 57.Schneider A, Böttiger BW, Popp E. Cerebral resuscitation after cardiocirculatory arrest. Anesth Analg. 2009;108:971–979. [DOI] [PubMed] [Google Scholar]
- 58.Laver S, Farrow C, Turner D, Nolan J. Mode of death after admission to an intensive care unit following cardiac arrest. Intensive Care Med. 2004;30:2126–2128. [DOI] [PubMed] [Google Scholar]
- 59.Lund H, Stewart HL, Lieber MM. Hepatic infarction. Am J Pathol. 1935;11:157–178.5. [PMC free article] [PubMed] [Google Scholar]
- 60.Dalen JE. Pulmonary embolism: what have we learned since Virchow? Natural history, pathophysiology, and diagnosis. Chest. 2002;122:1440–1456. [DOI] [PubMed] [Google Scholar]
- 61.Friedman LS. Ischemic hepatitis, hepatic infarction, and ischemic cholangiopathy. In: UpToDate. Chopra S, ed. Waltham, MA: UpToDate; 2011. http://www.uptodate.com/contents/ischemic-hepatitis-hepatic-infarction-and-ischemic-cholangiopathy. Accessed April 25, 2013. [Google Scholar]
- 62.Moore KL, Persaud TVN. The Developing Human: Clinically Oriented Embryology. 8th ed. Philadelphia, PA: Saunders; 2008:389. [Google Scholar]
- 63.Smith BE, Dyck PJ. Subclinical histopathological changes in the oculo-motor nerve in diabetes mellitus. Ann Neurol. 1992;32:376–385. [DOI] [PubMed] [Google Scholar]
- 64.Thömke F, Gutmann L, Stoeter P, Hopf HC. Cerebrovascular brainstem diseases with isolated cranial nerve palsies. Cerebrovasc Dis. 2002;13:147–155. [DOI] [PubMed] [Google Scholar]
- 65.Stuart-Shor EM, Wellenius GA, DelloIacono DM, Mittleman MA. Gender differences in presenting and prodromal stroke symptoms. Stroke. 2009;40:1121–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Choi JC. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: a genetic cause of cerebral small vessel disease. J Clin Neurol. 2010;6:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oppenheim C, Galanaud D, Samson Y, Sahel M, Dormont D, Wechsler B, Marsault C. Can diffusion weighted magnetic resonance imaging help differentiate stroke from stroke-like events in MELAS? J Neurol Neurosurg Psychiatry. 2000;69:248–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kolb SJ, Costello F, Lee AG, White M, Wong S, Schwartz ED, Messé SR, Ellenbogen J, Kasner SE, Galetta SL. Distinguishing ischemic stroke from the stroke-like lesions of MELAS using apparent diffusion coefficient mapping. J Neurol Sci. 2003;216:11–15. [DOI] [PubMed] [Google Scholar]
- 69.Benziada-Boudour A, Schmitt E, Kremer S, Foscolo S, Rivière AS, Tisserand M, Boudour A, Bracard S. Posterior reversible encephalopathy syndrome: a case of unusual diffusion-weighted MR images. J Neuroradiol. 2009;36:102–105. [DOI] [PubMed] [Google Scholar]
- 70.Bartynski WS. Posterior reversible encephalopathy syndrome, part 2: controversies surrounding pathophysiology of vasogenic edema. AJNR Am J Neuroradiol. 2008;29:1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nakada T, Kwee IL, Fujii Y, Knight RT. High-field, T2 reversed MRI of the hippocampus in transient global amnesia. Neurology. 2005;64:1170–1174. [DOI] [PubMed] [Google Scholar]
- 72.Pantoni L, Lamassa M, Inzitari D. Transient global amnesia: a review emphasizing pathogenic aspects. Acta Neurol Scand. 2000;102:275–283. [DOI] [PubMed] [Google Scholar]
- 73.Winbeck K, Etgen T, von Einsiedel HG, Röttinger M, Sander D. DWI in transient global amnesia and TIA: proposal for an ischaemic origin of TGA. J Neurol Neurosurg Psychiatry. 2005;76:438–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vermeer SE, Longstreth WT Jr, Koudstaal PJ. Silent brain infarcts: a systematic review. Lancet Neurol. 2007;6:611–619. [DOI] [PubMed] [Google Scholar]
- 75.Fisher CM. Lacunes: small, deep cerebral infarcts. Neurology. 1965;15:774–784. [DOI] [PubMed] [Google Scholar]
- 76.Chodosh EH, Foulkes MA, Kase CS, Wolf PA, Mohr JP, Hier DB, Price TR, Furtado JG Jr. Silent stroke in the NINCDS Stroke Data Bank. Neurology. 1988;38:1674–1679. [DOI] [PubMed] [Google Scholar]
- 77.Berguer R, Sieggreen MY, Lazo A, Hodakowski GT. The silent brain infarct in carotid surgery. J Vasc Surg. 1986;3:442–447. [DOI] [PubMed] [Google Scholar]
- 78.Meagher E, Grace PA, Bouchier-Hayes D. Are CT infarcts a separate risk factor in patients with transient cerebral ischaemic episodes? Eur J Vasc Surg. 1991;5:165–167. [DOI] [PubMed] [Google Scholar]
- 79.Price TR, Manolio TA, Kronmal RA, Kittner SJ, Yue NC, Robbins J, Anton-Culver H, O’Leary DH. Silent brain infarction on magnetic resonance imaging and neurological abnormalities in community-dwelling older adults: the Cardiovascular Health Study: CHS Collaborative Research Group. Stroke. 1997;28:1158–1164. [DOI] [PubMed] [Google Scholar]
- 80.Lazar RM, Fitzsimmons BF, Marshall RS, Mohr JP, Berman MF. Midazolam challenge reinduces neurological deficits after transient ischemic attack. Stroke. 2003;34:794–796. [DOI] [PubMed] [Google Scholar]
- 81.Zhu YC, Dufouil C, Tzourio C, Chabriat H. Silent brain infarcts: a review of MRI diagnostic criteria. Stroke. 2011;42:1140–1145. [DOI] [PubMed] [Google Scholar]
- 82.Vernooij MW, Ikram MA, Tanghe HL, Vincent AJ, Hofman A, Krestin GP, Niessen WJ, Breteler MM, van der Lugt A. Incidental findings on brain MRI in the general population. N Engl J Med. 2007;357:1821–1828. [DOI] [PubMed] [Google Scholar]
- 83.Jeerakathil T, Wolf PA, Beiser A, Massaro J, Seshadri S, D’Agostino RB, DeCarli C. Stroke risk profile predicts white matter hyperintensity volume: the Framingham Study. Stroke. 2004;35:1857–1861. [DOI] [PubMed] [Google Scholar]
- 84.Kuller LH, Longstreth WT Jr, Arnold AM, Bernick C, Bryan RN, Beauchamp NJ Jr; Cardiovascular Health Study Collaborative Research Group. White matter hyperintensity on cranial magnetic resonance imaging: a predictor of stroke. Stroke. 2004;35:1821–1825. [DOI] [PubMed] [Google Scholar]
- 85.Wong TY, Klein R, Sharrett AR, Couper DJ, Klein BE, Liao DP, Hubbard LD, Mosley TH; ARIC Investigators: Atherosclerosis Risk in Communities Study. Cerebral white matter lesions, retinopathy, and incident clinical stroke. JAMA. 2002;288:67–74. [DOI] [PubMed] [Google Scholar]
- 86.Vermeer SE, Hollander M, van Dijk EJ, Hofman A, Koudstaal PJ, Breteler MM; Rotterdam Scan Study. Silent brain infarcts and white matter lesions increase stroke risk in the general population: the Rotterdam Scan Study. Stroke. 2003;34:1126–1129. [DOI] [PubMed] [Google Scholar]
- 87.Fazekas F, Kleinert R, Offenbacher H, Schmidt R, Kleinert G, Payer F, Radner H, Lechner H. Pathologic correlates of incidental MRI white matter signal hyperintensities. Neurology. 1993;43:1683–1689. [DOI] [PubMed] [Google Scholar]
- 88.Ricci S, Celani MG, La Rosa F, Righetti E, Duca E, Caputo N. Silent brain infarctions in patients with first-ever stroke: a community-based study in Umbria, Italy. Stroke. 1993;24:647–651. [DOI] [PubMed] [Google Scholar]
- 89.Shinkawa A, Ueda K, Kiyohara Y, Kato I, Sueishi K, Tsuneyoshi M, Fujishima M. Silent cerebral infarction in a community-based autopsy series in Japan: the Hisayama Study. Stroke. 1995;26:380–385. [DOI] [PubMed] [Google Scholar]
- 90.Kase CS, Wolf PA, Chodosh EH, Zacker HB, Kelly-Hayes M, Kannel WB, D’Agostino RB, Scampini L. Prevalence of silent stroke in patients presenting with initial stroke: the Framingham Study. Stroke. 1989;20:850–852. [DOI] [PubMed] [Google Scholar]
- 91.Brott T, Tomsick T, Feinberg W, Johnson C, Biller J, Broderick J, Kelly M, Frey J, Schwartz S, Blum C. Baseline silent cerebral infarction in the Asymptomatic Carotid Atherosclerosis Study. Stroke. 1994;25:1122–1129. [DOI] [PubMed] [Google Scholar]
- 92.Adachi T, Kobayashi S, Yamaguchi S. Frequency and pathogenesis of silent subcortical brain infarction in acute first-ever ischemic stroke. Intern Med. 2002;41:103–108. [DOI] [PubMed] [Google Scholar]
- 93.Mathiesen EB, Waterloo K, Joakimsen O, Bakke SJ, Jacobsen EA, Bønaa KH. Reduced neuropsychological test performance in asymptomatic carotid stenosis: the Tromsø Study. Neurology. 2004;62:695–701. [DOI] [PubMed] [Google Scholar]
- 94.Hara M, Ooie T, Yufu K, Tsunematsu Y, Kusakabe T, Ooga M, Saikawa T, Sakata T. Silent cortical strokes associated with atrial fibrillation. Clin Cardiol. 1995;18:573–574. [DOI] [PubMed] [Google Scholar]
- 95.Tanaka H, Sueyoshi K, Nishino M, Ishida M, Fukunaga R, Abe H. Silent brain infarction and coronary artery disease in Japanese patients. Arch Neurol. 1993;50:706–709. [DOI] [PubMed] [Google Scholar]
- 96.Bokura H, Kobayashi S, Yamaguchi S. Distinguishing silent lacunar infarction from enlarged Virchow-Robin spaces: a magnetic resonance imaging and pathological study. J Neurol. 1998;245:116–122. [DOI] [PubMed] [Google Scholar]
- 97.Jungreis CA, Kanal E, Hirsch WL, Martinez AJ, Moossy J. Normal perivascular spaces mimicking lacunar infarction: MR imaging. Radiology. 1988;169:101–104. [DOI] [PubMed] [Google Scholar]
- 98.Kohara K, Fujisawa M, Ando F, Tabara Y, Niino N, Miki T, Shimokata H; NILS-LSA Study. MTHFR gene polymorphism as a risk factor for silent brain infarcts and white matter lesions in the Japanese general population: the NILS-LSA Study. Stroke. 2003;34:1130–1135. [DOI] [PubMed] [Google Scholar]
- 99.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB; on behalf of the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Howard G, Wagenknecht LE, Cai J, Cooper L, Kraut MA, Toole JF. Cigarette smoking and other risk factors for silent cerebral infarction in the general population. Stroke. 1998;29:913–917. [DOI] [PubMed] [Google Scholar]
- 101.Vermeer SE, Koudstaal PJ, Oudkerk M, Hofman A, Breteler MM. Prevalence and risk factors of silent brain infarcts in the population-based Rotterdam Scan Study. Stroke. 2002;33:21–25. [DOI] [PubMed] [Google Scholar]
- 102.Longstreth WT Jr, Bernick C, Manolio TA, Bryan N, Jungreis CA, Price TR. Lacunar infarcts defined by magnetic resonance imaging of 3660 elderly people: the Cardiovascular Health Study. Arch Neurol. 1998;55:1217–1225. [DOI] [PubMed] [Google Scholar]
- 103.Prabhakaran S, Wright CB, Yoshita M, Delapaz R, Brown T, DeCarli C, Sacco RL. Prevalence and determinants of subclinical brain infarction: the Northern Manhattan Study. Neurology. 2008;70:425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Longstreth WT Jr, Dulberg C, Manolio TA, Lewis MR, Beauchamp NJ Jr, O’Leary D, Carr J, Furberg CD. Incidence, manifestations, and predictors of brain infarcts defined by serial cranial magnetic resonance imaging in the elderly: the Cardiovascular Health Study. Stroke. 2002;33:2376–2382. [DOI] [PubMed] [Google Scholar]
- 105.Vermeer SE, Den Heijer T, Koudstaal PJ, Oudkerk M, Hofman A, Breteler MM; Rotterdam Scan Study. Incidence and risk factors of silent brain infarcts in the population-based Rotterdam Scan Study. Stroke. 2003;34:392–396. [DOI] [PubMed] [Google Scholar]
- 106.Schmidt R, Ropele S, Enzinger C, Petrovic K, Smith S, Schmidt H, Matthews PM, Fazekas F. White matter lesion progression, brain atrophy, and cognitive decline: the Austrian stroke prevention study. Ann Neurol. 2005;58:610–616. [DOI] [PubMed] [Google Scholar]
- 107.Bernick C, Kuller L, Dulberg C, Longstreth WT Jr, Manolio T, Beauchamp N, Price T; Cardiovascular Health Study Collaborative Research Group. Silent MRI infarcts and the risk of future stroke: the Cardiovascular Health Study. Neurology. 2001;57:1222–1229. [DOI] [PubMed] [Google Scholar]
- 108.Breteler MM. Vascular risk factors for Alzheimer’s disease: an epidemiologic perspective. Neurobiol Aging. 2000;21:153–160. [DOI] [PubMed] [Google Scholar]
- 109.Lopez OL, Jagust WJ, Dulberg C, Becker JT, DeKosky ST, Fitzpatrick A, Breitner J, Lyketsos C, Jones B, Kawas C, Carlson M, Kuller LH. Risk factors for mild cognitive impairment in the Cardiovascular Health Study Cognition Study: part 2. Arch Neurol. 2003;60:1394–1399. [DOI] [PubMed] [Google Scholar]
- 110.Vermeer SE, Prins ND, den Heijer T, Hofman A, Koudstaal PJ, Breteler MM. Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med. 2003;348:1215–1222. [DOI] [PubMed] [Google Scholar]
- 111.Haglund M, Passant U, Sjöbeck M, Ghebremedhin E, Englund E. Cerebral amyloid angiopathy and cortical microinfarcts as putative substrates of vascular dementia. Int J Geriatr Psychiatry. 2006;21:681–687. [DOI] [PubMed] [Google Scholar]
- 112.Haglund M, Sjöbeck M, Englund E. Severe cerebral amyloid angiopathy characterizes an underestimated variant of vascular dementia. Dement Geriatr Cogn Disord. 2004;18:132–137. [DOI] [PubMed] [Google Scholar]
- 113.Reitz C, Trenkwalder C, Kretzschmar K, Roesler A, V Eckardstein A, Berger K. Relation of cerebral small-vessel disease and brain atrophy to mild Parkinsonism in the elderly. Mov Disord. 2006;21:1914–1919. [DOI] [PubMed] [Google Scholar]
- 114.Goldstein LB, Bushnell CD, Adams RJ, Appel LJ, Braun LT, Chaturvedi S, Creager MA, Culebras A, Eckel RH, Hart RG, Hinchey JA, Howard VJ, Jauch EC, Levine SR, Meschia JF, Moore WS, Nixon JV, Pearson TA; on behalf of the American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Epidemiology and Prevention; Council for High Blood Pressure Research, Council on Peripheral Vascular Disease, and Interdisciplinary Council on Quality of Care and Outcomes Research. Guidelines for the primary prevention of stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association [published correction appears in Stroke. 2011;42:e26]. Stroke. 2011;42:517–584. [DOI] [PubMed] [Google Scholar]
- 115.Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA. 2001;285:2864–2870. [DOI] [PubMed] [Google Scholar]
- 116.Palm F, Urbanek C, Rose S, Buggle F, Bode B, Hennerici MG, Schmieder K, Inselmann G, Reiter R, Fleischer R, Piplack KO, Safer A, Becher H, Grau AJ. Stroke incidence and survival in Ludwigshafen am Rhein, Germany: the Ludwigshafen Stroke Study (LuSSt). Stroke. 2010;41:1865–1870. [DOI] [PubMed] [Google Scholar]
- 117.Kleindorfer DO, Khoury J, Moomaw CJ, Alwell K, Woo D, Flaherty ML, Khatri P, Adeoye O, Ferioli S, Broderick JP, Kissela BM. Stroke incidence is decreasing in whites but not in blacks: a population-based estimate of temporal trends in stroke incidence from the Greater Cincinnati/Northern Kentucky Stroke Study. Stroke. 2010;41:1326–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.van Beijnum J, Lovelock CE, Cordonnier C, Rothwell PM, Klijn CJ, Al-Shahi Salman R; SIVMS Steering Committee and the Oxford Vascular Study. Outcome after spontaneous and arteriovenous malformation-related intracerebral haemorrhage: population-based studies. Brain. 2009;132(pt 2):537–543. [DOI] [PubMed] [Google Scholar]
- 119.Kleindorfer DO, Miller R, Moomaw CJ, Alwell K, Broderick JP, Khoury J, Woo D, Flaherty ML, Zakaria T, Kissela BM. Designing a message for public education regarding stroke: does FAST capture enough stroke? Stroke. 2007;38:2864–2868. [DOI] [PubMed] [Google Scholar]
- 120.Lovelock CE, Redgrave JN, Briley D, Rothwell PM. Reliable estimation of the proportion of minor stroke due to intracerebral haemorrhage. Int J Stroke. 2009;4:6–10. [DOI] [PubMed] [Google Scholar]
- 121.Kidwell CS, Chalela JA, Saver JL, Starkman S, Hill MD, Demchuk AM, Butman JA, Patronas N, Alger JR, Latour LL, Luby ML, Baird AE, Leary MC, Tremwel M, Ovbiagele B, Fredieu A, Suzuki S, Villablanca JP, Davis S, Dunn B, Todd JW, Ezzeddine MA, Haymore J, Lynch JK, Davis L, Warach S. Comparison of MRI and CT for detection of acute intracerebral hemorrhage. JAMA. 2004;292:1823–1830. [DOI] [PubMed] [Google Scholar]
- 122.Wijman CA, Venkatasubramanian C, Bruins S, Fischbein N, Schwartz N. Utility of early MRI in the diagnosis and management of acute spontaneous intracerebral hemorrhage. Cerebrovasc Dis. 2010;30:456–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhu XL, Chan MS, Poon WS. Spontaneous intracranial hemorrhage: which patients need diagnostic cerebral angiography? A prospective study of 206 cases and review of the literature. Stroke. 1997;28:1406–1409. [DOI] [PubMed] [Google Scholar]
- 124.Siddique MS, Gregson BA, Fernandes HM, Barnes J, Treadwell L, Wooldridge TD, Mendelow AD. Comparative study of traumatic and spontaneous intracerebral hemorrhage. J Neurosurg. 2002;96:86–89. [DOI] [PubMed] [Google Scholar]
- 125.Stapf C, Mast H, Sciacca RR, Choi JH, Khaw AV, Connolly ES, Pile-Spellman J, Mohr JP. Predictors of hemorrhage in patients with untreated brain arteriovenous malformation. Neurology. 2006;66:1350–1355. [DOI] [PubMed] [Google Scholar]
- 126.Choi JH, Mast H, Sciacca RR, Hartmann A, Khaw AV, Mohr JP, Sacco RL, Stapf C. Clinical outcome after first and recurrent hemorrhage in patients with untreated brain arteriovenous malformation. Stroke. 2006;37:1243–1247. [DOI] [PubMed] [Google Scholar]
- 127.Talalla A, Morin MA. Acute traumatic subdural hematoma: a review of one hundred consecutive cases. J Trauma. 1971;11:771–777. [PubMed] [Google Scholar]
- 128.Stoll BJ, Hansen NI, Bell EF, Shankaran S, Laptook AR, Walsh MC, Hale EC, Newman NS, Schibler K, Carlo WA, Kennedy KA, Poindexter BB, Finer NN, Ehrenkranz RA, Duara S, Sánchez PJ, O’Shea TM, Goldberg RN, Van Meurs KP, Faix RG, Phelps DL, Frantz ID 3rd, Watterberg KL, Saha S, Das A, Higgins RD; Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics. 2010;126:443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Flint AC, Roebken A, Singh V. Primary intraventricular hemorrhage: yield of diagnostic angiography and clinical outcome. Neurocrit Care. 2008;8:330–336. [DOI] [PubMed] [Google Scholar]
- 130.Roob G, Schmidt R, Kapeller P, Lechner A, Hartung HP, Fazekas F. MRI evidence of past cerebral microbleeds in a healthy elderly population. Neurology. 1999;52:991–994. [DOI] [PubMed] [Google Scholar]
- 131.Qiu C, Cotch MF, Sigurdsson S, Jonsson PV, Jonsdottir MK, Sveinbjrnsdottir S, Eiriksdottir G, Klein R, Harris TB, van Buchem MA, Gudnason V, Launer LJ. Cerebral microbleeds, retinopathy, and dementia: the AGES-Reykjavik Study. Neurology. 2010;75:2221–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Werring DJ, Frazer DW, Coward LJ, Losseff NA, Watt H, Cipolotti L, Brown MM, Jäger HR. Cognitive dysfunction in patients with cerebral microbleeds on T2*-weighted gradient-echo MRI. Brain. 2004;127(pt 10):2265–2275. [DOI] [PubMed] [Google Scholar]
- 133.Fan YH, Zhang L, Lam WW, Mok VC, Wong KS. Cerebral microbleeds as a risk factor for subsequent intracerebral hemorrhages among patients with acute ischemic stroke. Stroke. 2003;34:2459–2462. [DOI] [PubMed] [Google Scholar]
- 134.Shea AM, Reed SD, Curtis LH, Alexander MJ, Villani JJ, Schulman KA. Characteristics of nontraumatic subarachnoid hemorrhage in the United States in 2003. Neurosurgery. 2007;61:1131–1137. [DOI] [PubMed] [Google Scholar]
- 135.Biotti D, Jacquin A, Boutarbouch M, Bousquet O, Durier J, Ben Salem D, Ricolfi F, Beaurain J, Osseby GV, Moreau T, Giroud M, Béjot Y. Trends in case-fatality rates in hospitalized nontraumatic subarachnoid hemorrhage: results of a population-based study in Dijon, France, from 1985 to 2006. Neurosurgery. 2010;66:1039–1043. [DOI] [PubMed] [Google Scholar]
- 136.Sacco S, Totaro R, Toni D, Marini C, Cerone D, Carolei A. Incidence, case-fatalities and 10-year survival of subarachnoid hemorrhage in a population-based registry. Eur Neurol. 2009;62:155–160. [DOI] [PubMed] [Google Scholar]
- 137.Scharbrodt W, Stein M, Schreiber V, Böker DK, Oertel MF. The prediction of long-term outcome after subarachnoid hemorrhage as measured by the Short Form-36 Health Survey. J Clin Neurosci. 2009;16:1409–1413. [DOI] [PubMed] [Google Scholar]
- 138.Al-Khindi T, Macdonald RL, Schweizer TA. Cognitive and functional outcome after aneurysmal subarachnoid hemorrhage. Stroke. 2010;41:e519–e536. [DOI] [PubMed] [Google Scholar]
- 139.Cortnum S, Sørensen P, Jørgensen J. Determining the sensitivity of computed tomography scanning in early detection of subarachnoid hemorrhage. Neurosurgery. 2010;66:900–902. [PubMed] [Google Scholar]
- 140.Boesiger BM, Shiber JR. Subarachnoid hemorrhage diagnosis by computed tomography and lumbar puncture: are fifth generation CT scanners better at identifying subarachnoid hemorrhage? J Emerg Med. 2005;29:23–27. [DOI] [PubMed] [Google Scholar]
- 141.da Rocha AJ, da Silva CJ, Gama HP, Baccin CE, Braga FT, Cesare FdA, Veiga JC. Comparison of magnetic resonance imaging sequences with computed tomography to detect low-grade subarachnoid hemorrhage: role of fluid-attenuated inversion recovery sequence. J Comput Assist Tomogr. 2006;30:295–303. [DOI] [PubMed] [Google Scholar]
- 142.Maeda M, Yagishita A, Yamamoto T, Sakuma H, Takeda K. Abnormal hyperintensity within the subarachnoid space evaluated by fluid-attenuated inversion-recovery MR imaging: a spectrum of central nervous system diseases. Eur Radiol. 2003;13(suppl 4):L192–L201. [DOI] [PubMed] [Google Scholar]
- 143.Dupont SA, Wijdicks EF, Manno EM, Rabinstein AA. Thunderclap headache and normal computed tomographic results: value of cerebrospinal fluid analysis. Mayo Clin Proc. 2008;83:1326–1331. [DOI] [PubMed] [Google Scholar]
- 144.Arora S, Swadron SP, Dissanayake V. Evaluating the sensitivity of visual xanthochromia in patients with subarachnoid hemorrhage. J Emerg Med. 2010;39:13–16. [DOI] [PubMed] [Google Scholar]
- 145.Perry JJ, Spacek A, Forbes M, Wells GA, Mortensen M, Symington C, Fortin N, Stiell IG. Is the combination of negative computed tomography result and negative lumbar puncture result sufficient to rule out subarachnoid hemorrhage? Ann Emerg Med. 2008;51:707–713. [DOI] [PubMed] [Google Scholar]
- 146.Rinkel GJ, van Gijn J, Wijdicks EF. Subarachnoid hemorrhage without detectable aneurysm. A review of the causes. Stroke. 1993;24:1403–1409. [DOI] [PubMed] [Google Scholar]
- 147.Cuvinciuc V, Viguier A, Calviere L, Raposo N, Larrue V, Cognard C, Bonneville F. Isolated acute nontraumatic cortical subarachnoid hemorrhage. AJNR Am J Neuroradiol. 2010;31:1355–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.van Gijn J, van Dongen KJ, Vermeulen M, Hijdra A. Perimesencephalic hemorrhage: a nonaneurysmal and benign form of subarachnoid hemorrhage. Neurology. 1985;35:493–497. [DOI] [PubMed] [Google Scholar]
- 149.Beseoglu K, Pannes S, Steiger HJ, Hänggi D. Long-term outcome and quality of life after nonaneurysmal subarachnoid hemorrhage. Acta Neurochir (Wien). 2010;152:409–416. [DOI] [PubMed] [Google Scholar]
- 150.Linn FH, Wijdicks EF, van der Graaf Y, Weerdesteyn-van Vliet FA, Bartelds AI, van Gijn J. Prospective study of sentinel headache in aneurysmal subarachnoid haemorrhage. Lancet. 1994;344:590–593. [DOI] [PubMed] [Google Scholar]
- 151.Jakobsson KE, Säveland H, Hillman J, Edner G, Zygmunt S, Brandt L, Pellettieri L. Warning leak and management outcome in aneurysmal subarachnoid hemorrhage. J Neurosurg. 1996;85:995–999. [DOI] [PubMed] [Google Scholar]
- 152.Linn FH, Rinkel GJ, Algra A, van Gijn J. The notion of “warning leaks” in subarachnoid haemorrhage: are such patients in fact admitted with a rebleed? J Neurol Neurosurg Psychiatry. 2000;68:332–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med. 1995;333:1581–1587. [DOI] [PubMed] [Google Scholar]
- 154.International Stroke Trial Collaborative Group. The International Stroke Trial (IST): a randomised trial of aspirin, subcutaneous heparin, both, or neither among 19435 patients with acute ischaemic stroke. Lancet. 1997;349:1569–1581. [PubMed] [Google Scholar]
- 155.Hacke W, Kaste M, Fieschi C, Toni D, Lesaffre E, von Kummer R, Boysen G, Bluhmki E, Höxter G, Mahagne MH, Hennerici M. Intravenous thrombolysis with recombinant tissue plasminogen activator for acute hemispheric stroke: the European Cooperative Acute Stroke Study (ECASS). JAMA. 1995;274:1017–1025. [PubMed] [Google Scholar]
- 156.Cumurciuc R, Crassard I, Sarov M, Valade D, Bousser MG. Headache as the only neurological sign of cerebral venous thrombosis: a series of 17 cases. J Neurol Neurosurg Psychiatry. 2005;76:1084–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jacobs K, Moulin T, Bogousslavsky J, Woimant F, Dehaene I, Tatu L, Besson G, Assouline E, Casselman J. The stroke syndrome of cortical vein thrombosis. Neurology. 1996;47:376–382. [DOI] [PubMed] [Google Scholar]
- 158.Pfefferkorn T, Crassard I, Linn J, Dichgans M, Boukobza M, Bousser MG. Clinical features, course and outcome in deep cerebral venous system thrombosis: an analysis of 32 cases. J Neurol. 2009;256:1839–1845. [DOI] [PubMed] [Google Scholar]
- 159.Sagduyu A, Sirin H, Mulayim S, Bademkiran F, Yunten N, Kitis O, Calli C, Dalbasti T, Kumral E. Cerebral cortical and deep venous thrombosis without sinus thrombosis: clinical MRI correlates. Acta Neurol Scand. 2006;114:254–260. [DOI] [PubMed] [Google Scholar]
- 160.Stam J Thrombosis of the cerebral veins and sinuses. N Engl J Med. 2005;352:1791–1798. [DOI] [PubMed] [Google Scholar]
- 161.Saposnik G, Barinagarrementeria F, Brown RD Jr, Bushnell CD, Cucchiara B, Cushman M, deVeber G, Ferro JM, Tsai FY; on behalf of the American Heart Association Stroke Council and the Council on Epidemiology and Prevention. Diagnosis and management of cerebral venous thrombosis: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:1158–1192. [DOI] [PubMed] [Google Scholar]
- 162.Benabu Y, Mark L, Daniel S, Glikstein R. Cerebral venous thrombosis presenting with subarachnoid hemorrhage. Case report and review. Am J Emerg Med. 2009;27:96–106. [DOI] [PubMed] [Google Scholar]
- 163.Oppenheim C, Domigo V, Gauvrit JY, Lamy C, Mackowiak-Cordoliani MA, Pruvo JP, Méder JF. Subarachnoid hemorrhage as the initial presentation of dural sinus thrombosis. AJNR Am J Neuroradiol. 2005;26:614–617. [PMC free article] [PubMed] [Google Scholar]
- 164.Quattrone A, Bono F, Oliveri RL, Gambardella A, Pirritano D, Labate A, Lucisano A, Valentino P, Zappia M, Aguglia U, Lavano A, Fera F, Pardatscher K. Cerebral venous thrombosis and isolated intracranial hypertension without papilledema in CDH. Neurology. 2001;57:31–36. [DOI] [PubMed] [Google Scholar]
- 165.Agostoni E Headache in cerebral venous thrombosis. Neurol Sci. 2004;25(suppl 3):S206–S210. [DOI] [PubMed] [Google Scholar]
- 166.Damak M, Crassard I, Wolff V, Bousser MG. Isolated lateral sinus thrombosis: a series of 62 patients. Stroke. 2009;40:476–481. [DOI] [PubMed] [Google Scholar]
- 167.Breteau G, Mounier-Vehier F, Godefroy O, Gauvrit JY, Mackowiak-Cordoliani MA, Girot M, Bertheloot D, Hénon H, Lucas C, Leclerc X, Fourrier F, Pruvo JP, Leys D. Cerebral venous thrombosis 3-year clinical outcome in 55 consecutive patients. J Neurol. 2003;250:29–35. [DOI] [PubMed] [Google Scholar]
- 168.Canhão P, Ferro JM, Lindgren AG, Bousser MG, Stam J, Barinagarrementeria F; ISCVT Investigators. Causes and predictors of death in cerebral venous thrombosis. Stroke. 2005;36:1720–1725. [DOI] [PubMed] [Google Scholar]
- 169.Einhäupl K, Stam J, Bousser MG, De Bruijn SF, Ferro JM, Martinelli I, Masuhr F.; European Federation of Neurological Societies. EFNS guideline on the treatment of cerebral venous and sinus thrombosis in adult patients. Eur J Neurol. 2010;17:1229–1235. [DOI] [PubMed] [Google Scholar]
- 170.Ferro JM, Canhão P, Bousser MG, Stam J, Barinagarrementeria F; ISCVT Investigators. Cerebral vein and dural sinus thrombosis in elderly patients. Stroke. 2005;36:1927–1932. [DOI] [PubMed] [Google Scholar]
- 171.Ferro JM, Canhão P, Stam J, Bousser MG, Barinagarrementeria F; ISCVT Investigators. Prognosis of cerebral vein and dural sinus thrombosis: results of the International Study on Cerebral Vein and Dural Sinus Thrombosis (ISCVT). Stroke. 2004;35:664–670. [DOI] [PubMed] [Google Scholar]
- 172.Ferro JM, Canhão P, Stam J, Bousser MG, Barinagarrementeria F, Massaro A, Ducrocq X, Kasner SE; ISCVT Investigators. Delay in the diagnosis of cerebral vein and dural sinus thrombosis: influence on outcome. Stroke. 2009;40:3133–3138. [DOI] [PubMed] [Google Scholar]
- 173.Gosk-Bierska I, Wysokinski W, Brown RD Jr, Karnicki K, Grill D, Wiste H, Wysokinska E, McBane RD 2nd. Cerebral venous sinus thrombosis: incidence of venous thrombosis recurrence and survival. Neurology. 2006;67:814–819. [DOI] [PubMed] [Google Scholar]
- 174.Martinelli I, Bucciarelli P, Passamonti SM, Battaglioli T, Previtali E, Mannucci PM. Long-term evaluation of the risk of recurrence after cerebral sinus-venous thrombosis. Circulation. 2010;121:2740–2746. [DOI] [PubMed] [Google Scholar]
- 175.Furie KL, Kasner SE, Adams RJ, Albers GW, Bush RL, Fagan SC, Halperin JL, Johnston SC, Katzan I, Kernan WN, Mitchell PH, Ovbiagele B, Palesch YY, Sacco RL, Schwamm LH, Wassertheil-Smoller S, Turan TN, Wentworth D; on behalf of the American Heart Association Stroke Council, Council on Cardiovascular Nursing, Council on Clinical Cardiology, and Interdisciplinary Council on Quality of Care and Outcomes Research. Guidelines for the prevention of stroke in patients with stroke or transient ischemic attack: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:227–276. [DOI] [PubMed] [Google Scholar]
- 176.Mohr JP, Thompson JL, Lazar RM, Levin B, Sacco RL, Furie KL, Kistler JP, Albers GW, Pettigrew LC, Adams HP Jr, Jackson CM, Pullicino P; Warfarin-Aspirin Recurrent Stroke Study Group. A comparison of warfarin and aspirin for the prevention of recurrent ischemic stroke. N Engl J Med. 2001;345:1444–1451. [DOI] [PubMed] [Google Scholar]
- 177.Sacco RL, Diener HC, Yusuf S, Cotton D, Ounpuu S, Lawton WA, Palesch Y, Martin RH, Albers GW, Bath P, Bornstein N, Chan BP, Chen ST, Cunha L, Dahlöf B, De Keyser J, Donnan GA, Estol C, Gorelick P, Gu V, Hermansson K, Hilbrich L, Kaste M, Lu C, Machnig T, Pais P, Roberts R, Skvortsova V, Teal P, Toni D, Vandermaelen C, Voigt T, Weber M, Yoon BW; PRoFESS Study Group. Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N Engl J Med. 2008;359:1238–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Pia Sormani M The Will Rogers phenomenon: the effect of different diagnostic criteria. J Neurol Sci. 2009;287(suppl 1):S46–S49. [DOI] [PubMed] [Google Scholar]
- 179.Gofrit ON, Zorn KC, Steinberg GD, Zagaja GP, Shalhav AL. The Will Rogers phenomenon in urological oncology. J Urol. 2008;179:28–33. [DOI] [PubMed] [Google Scholar]
- 180.Teutsch SM. Considerations in planning a surveillance system. In: Lee L, Teutsch S, Thacker S, St. Louis M, eds. Principles & Practice of Public Health Surveillance. 3rd ed. New York, NY: Oxford University Press; 2010:18–43. [Google Scholar]