

Abstract
Tuberculosis (TB) drug discovery and development has undergone nothing short of a revolution over the past 20 years. Successful public–private partnerships and sustained funding have delivered a much-improved understanding of mycobacterial disease biology and pharmacology and a healthy pipeline that can tolerate inevitable attrition. Preclinical and clinical development has evolved from decade-old concepts to adaptive designs that permit rapid evaluation of regimens that might greatly shorten treatment duration over the next decade. But the past 20 years also saw the rise of a fatal and difficult-to-cure lung disease caused by nontuberculous mycobacteria (NTM), for which the drug development pipeline is nearly empty. Here, we discuss the similarities and differences between TB and NTM lung diseases, compare the preclinical and clinical advances, and identify major knowledge gaps and areas of cross-fertilization. We argue that applying paradigms and networks that have proved successful for TB, from basic research to clinical trials, will help to populate the pipeline and accelerate curative regimen development for NTM disease.
Introduction
Until the emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), tuberculosis (TB) was the leading cause of infectious disease mortality worldwide, with approximately 1.6 million deaths in 2021 (ref. 1), and is expected to regain the lead owing to the loss of focus and global health-care disruptions caused by the coronavirus disease 2019 (COVID) pandemic2. TB disease largely affects the developing world. As disease burden is strongly associated with socio-economic conditions3, its incidence rates follow different trajectories in different parts of the world4. In 2019, 30 countries with high TB burden accounted for 87% of new TB cases, and eight countries accounted for two-thirds of the total cases5.
Treatments for TB have markedly improved over the past 20 years but still remain intensive. Uncomplicated drug-susceptible TB (DS-TB) is treated daily with four drugs for 4–6 months. Multidrug therapy results in complex patterns of multidrug-resistant (MDR) and extensively drug-resistant (XDR) TB, which unsurprisingly impact treatment options, duration, tolerability and cure rates6,7 (Table 1). Why is such an intensive treatment required to cure TB while other pulmonary infections are successfully treated with a single antibiotic for 1–2 weeks? Despite being caused by a single causative agent, Mycobacterium tuberculosis (Mtb), the disease is increasingly recognized as a heterogeneous infection sometimes referred to as ‘polymicrobial’8. Bacterial subpopulations consist of replicating, dormant (slow-growing or non-growing) and reactivated bacteria, which are present both extracellularly and within various immune cell types. Some of the bacteria are in lesions that antibiotics can reach via the vascular system whereas others are sequestered in niches poorly accessible to drugs and crucial immune factors, particularly the necrotic core of lesions and cavities. The relative proportions of the infected cells can change in response to host immune responses and antibiotic therapy. Consequently, the range of phenotypic antibiotic susceptibility in a patient can be extreme. In addition, antibiotic-induced drug tolerance can form a relapsing reservoir of bacteria. These factors collectively encompass issues of poor drug access and drug action that are the signature of TB disease and treatment and one major reason for the protracted and poorly effective chemotherapy.
Table 1 |.
Epidemiology, treatment and cure rates of TB and NTM pulmonary diseases
| Variable | DS-TB | MDR-TB | XDR-TB | MAC-PDa | MAB-PDa |
|---|---|---|---|---|---|
| Incidence | ~10 million in 2021 (ref. 5) | 127,000 reported in 2021 (estimated 450,000) | 27,000 reported in 2021 | 5 per 100,000 person-years in 2015 in the USA | |
| Prevalence | 14 million in 2022 (ref. 232) | 25–40 per 100,000 in eastern Asia and Hawaii; 1–15 per 100,000 in the European Union and continental USA233 | Insufficient global data; 5–10% prevalence in patients with CF and increasing234 | ||
| Mortality | 1.6 million in 2021 | Global 5-year all-cause mortality >25% (10–48%)235 | South Korea: 5-, 10- and 15-year cumulative mortality 11%, 30% and 50% (ref. 236)b | ||
| ~5% | 15–20% globally | Up to 40% but decreasing with introduction of bedaquiline | |||
| Treatment regimens | Until 2022: 2 months of isoniazid, rifampicin, pyrazinamide, ethambutol then 4 months of isoniazid, rifampicin6; from 2022: 4-month regimen of rifapentine, isoniazid, pyrazinamide and moxifloxacin1 | Until 2022: patient-tailored regimen including a fluoroquinolone and an aminoglycoside or bedaquiline for 18 months; from 2022: 6 months of bedaquiline–pretomanid–linezolid with or without moxifloxacin72 | Until 2022: patient-tailored regimen (4–7 drugs) for 18–24 months; from 2022: 6 months of bedaquiline–pretomanid–linezolid with or without moxifloxacin72 | Macrolide (azithromycin or clarithromycin), rifamycin (rifampicin or rifabutin), ethambutol for non-cavitary disease; adding intravenous amikacin for cavitary disease140 | 3–6 antibiotics with a macrolide (azithromycin or clarithromycin) as backbone, selected on the basis of drug susceptibility profile, including amikacin and/or cefoxitin as injectables237 |
| Cure rates | 85% for uncomplicated TB and 76% for HIV-associated TB, treated with 6-month regimen1 | Average 60% if enrolled on an adequate regimen (2019)1 | Variable (30–50%)c, dependent on availability of new drugs such as bedaquiline238,239 | 39% (ref. 240) to 68% (refs. 241,242) in meta-analyses | 30–60%243, average ~45%; largely influenced by macrolide susceptibility. |
CF, cystic fibrosis; DS-TB, drug-susceptible tuberculosis; MAB-PD, Mycobacterium abscessus pulmonary disease; MAC-PD, Mycobacterium avium complex pulmonary disease; MDR-TB, multidrug-resistant tuberculosis; NTM, nontuberculous mycobacteria; PD, pulmonary disease; TB, tuberculosis; XDR-TB, extensively drug-resistant tuberculosis.
Numbers are incidence and prevalence estimates with the following limitations: reports often use different measurements of incidence and prevalence, which impedes data pooling; NTM-PD is not a reportable disease in most states and countries; and NTM case reports are based on varying diagnostic tests22.
Probability of 5-year mortality ranging from 2% to 82% depending on body mass index, age, cavity, erythrocyte sedimentation rate and sex.
Up to 73% in clinical trial settings with bedaquiline-based regimens and up to 90% in clinical trial settings with bedaquiline–pretomanid–linezolid.
Unfortunately, this bleak picture only gets worse for pulmonary disease caused by nontuberculous mycobacteria (NTM-PD). NTMs are environmental mycobacteria and opportunistic pathogens closely related to Mtb and can cause progressive, fatal pulmonary disease. NTM-PD occurs in patients with immunodeficiencies, structural lung damage or both9. The number of cases is climbing at an alarming rate in higher- and middle-income countries. From 2008 to 2015, the annual incidence and prevalence of NTM lung disease in the USA increased from 6.8 to 11.7 per 100,000 persons, with significant geographical differences10,11. NTM-PD is not a reportable disease in many US states and several higher- and middle-income countries, an additional challenge for epidemiologists and disease modelling experts. In resource-poor regions where TB is endemic, NTM infection is often misdiagnosed as TB and reliable statistics are lacking11,12. NTM-PD is treated for at least a year with multiple antibiotics until sputum cultures remain negative for 12 months. These drugs include injectables, agents with serious side effects13 and antibiotics that can cause pharmacological drug–drug interactions with treatments for the frequent comorbidities, all leading to compliance issues. Despite such stringent guidelines, the cure rates remain generally worse than for MDR-TB and XDR-TB (Table 1). For Mycobacterium abscessus pulmonary disease (MAB-PD), there is no reliable cure14.
TB has been the focus of research and development efforts over the past 20 years, which has resulted in a rather healthy late preclinical and clinical development pipeline (Working Group on New Drugs). In contrast, NTM-PD is on the rise and is largely neglected, as reflected by the remarkably thin clinical development pipeline, populated by only a few repurposed antibiotics15. This is despite NTM microbiology having become an increasingly active research area16 over the past three decades17. The treatment of TB and NTM mycobacterial lung diseases share key shortcomings: high pill burden, long duration, unsatisfactory cure rates, toxicity, the use of injectable agents (although these are being phased out against TB in many parts of the world) and drug–drug interactions. Resistance emerges for multiple reasons associated with diagnostic accuracy, prescription patterns, poor compliance associated with poor drug tolerability, and pathogen and pathology characteristics. Therefore, the target product profiles of more efficacious and shorter regimens to treat active TB and NTM-PD are largely aligned.
Here, we discuss the evolving paradigms of small-molecule antibacterial and regimen development, highlight promising development candidates and preclinical agents, describe the respective benefits and caveats of old and new drug targets, and review conventional and innovative clinical trial designs. Host-directed therapeutic approaches for TB18–20 and NTM-PD21, phage therapy22 and prevention of latent TB reactivation23 have been comprehensively reviewed elsewhere. We focus on the pulmonary system as the primary site of TB and NTM infection (in approximately 80% of all cases), for which pathology, disease progression and response to therapy are best understood and most extensively described. Throughout the Review, we highlight how knowledge acquired by the TB community could inspire and accelerate the development of curative regimens for NTM-PD.
TB and NTM pulmonary diseases
The Mycobacterium genus contains more than 200 species, most of which are saprophytic environmental bacteria. Among these, a small percentage are opportunistic pathogens that can cause pulmonary infections24, with MAB and Mycobacterium avium complex (MAC) being responsible for 80–85% of lung disease worldwide, and Mycobacterium kansasii and Mycobacterium xenopi being less prominent but clinically relevant species11. The notable exceptions are Mtb and Mycobacterium leprae, both obligate pathogens with no significant biological reservoir outside of humans. M. leprae causes a skin disease, and Mtb and NTMs can infect several organs other than the lungs25,26. The most common extrapulmonary Mtb infections are osteomyelitis, meningitis and infection of thoracic lymph nodes27,28.
TB disease and treatment
The pathological hallmarks of pulmonary TB are immune cell aggregates called granulomas, which follow different progression trajectories and are collectively called lung lesions. As they expand, damaging inflammatory processes can lead to several events: necrosis from the granuloma centre outwards and fusion of the necrotic granuloma with airways; bronchogenic spread of necrotic material containing high bacterial burden; tissue destruction; and formation of open cavities or lesions filled with necrotic debris. A spectrum of lesion types is found within a single host, creating microniches and various environmental conditions to which the infectious agent must adapt to survive, which in turn results in differential susceptibility to chemotherapy and immune control29–31. To cure such a heterogeneous disease, current treatment requires four drugs (rifampicin, isoniazid, pyrazinamide, ethambutol) taken daily for 2 months of intensive phase, followed by a continuation phase during which rifampicin and isoniazid are taken daily for 4 months1. Multidrug therapy results in complex patterns of drug resistance: rifampicin or isoniazid mono-resistant TB; MDR-TB, resistant to both rifampicin and isoniazid; XDR-TB; and totally drug-resistant (TDR)-TB, in which increasing numbers of second-line antibiotics are lost to drug resistance7 (Table 1).
The concepts of drug and regimen development have evolved tremendously since 1946, when the first controlled clinical trial of streptomycin was added to bed rest therapy for patients with pulmonary TB (Fig. 1 and Box 1). Although more effective strategies to accelerate the discovery of better and shorter therapies are still needed, two recent approvals are a testament to the progress achieved. First, a 4-month regimen, containing rifapentine and moxifloxacin, was found non-inferior to the standard 6-month regimen that had been used for more than four decades in the treatment of DS-TB32. Second, a 6-month regimen was approved in 2019 for the treatment of MDR and XDR TB comprising only three drugs — bedaquiline, pretomanid and linezolid — which between them have two novel mechanisms of action33. Guidelines for the treatment of TB have therefore started to change recently (Table 1). Many clinical trials aimed at treatment shortening are in progress or just completed, with promising results20.
Fig. 1 |. Timeline and evolution of tuberculosis drug and regimen development concepts.
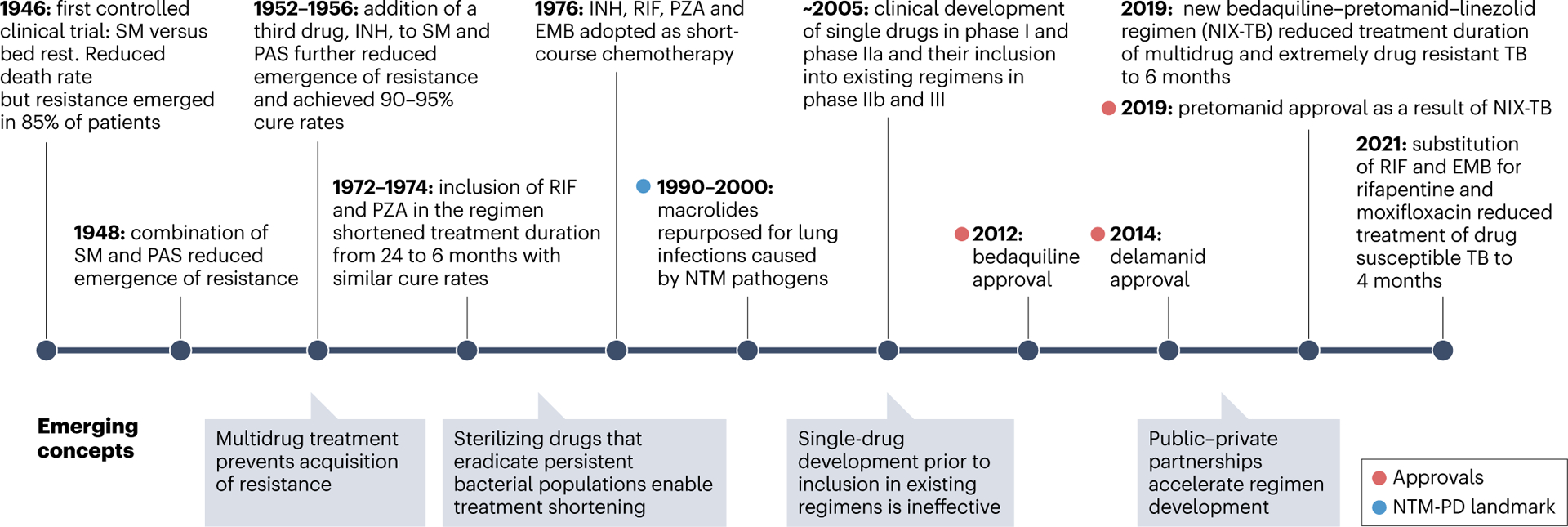
Landmark events are shown above the timeline. A landmark for nontuberculous mycobacteria (NTM) is highlighted in blue — the repurposing of macrolides (azithromycin and clarithromycin) for lung disease caused by Mycobacterium avium complex in the 1990s and by other NTM pathogens in the 2000s. Emerging concepts and new paradigms are summarized below the timeline. EMB, ethambutol; INH, isoniazid; PAS, p-aminosalicylic acid; PD, pulmonary disease; PZA, pyrazinamide; RIF, rifampicin; SM, streptomycin; TB, tuberculosis.
Box 1. Evolution of drug and regimen development for TB.
Using streptomycin to treat tuberculosis (TB) was one of the first randomized controlled chemotherapy trials ever conducted (Fig. 1). Despite significant reduction in deaths and radiological improvement, streptomycin monotherapy led to the emergence of resistance in 80–85% of the patients. This failure rapidly triggered testing of two- and three-drug combinations as new candidates emerged, demonstrating as early as the 1950s that multidrug treatment protects against resistance development. TB is thus one of the first diseases to be treated with combination therapy. Over a couple of decades, massive trials were conducted by the British Medical Research Council with numerous combinations of approximately a dozen drugs. A breakthrough occurred in the mid-1970s when inclusion of rifampicin and pyrazinamide enabled treatment shortening from at least 18 months to 6 months252 and delivered the ‘short-course’ chemotherapy still in use today.
As more stringent regulatory guidelines emerged, the paradigm of combining drugs in early stages of clinical trials shifted towards single-drug development along the entire clinical trial process. After conventional phase I trials, new agents were tested as monotherapy for 14 days in phase IIa. If successful, they were added to the optimized background regimens of patients with multidrug-resistant (MDR) disease, versus placebo, to detect an additive effect as the primary outcome. Although bedaquiline and delamanid followed this path and were approved in 2012 and 2014, this strategy was not sustainable for several reasons: the process was exceptionally lengthy owing to the long treatment duration and follow-up period to detect relapses; the design was not adequate to identify candidates with treatment-shortening potential; new agents were exclusively added to MDR background regimens regardless of potential pharmacokinetic and pharmacodynamic interactions; and finally, the efficacy of these optimized regimens kept improving, raising the bar for the investigational drug. Trials in which a single repurposed antibiotic was substituted in the first-line regimen required very large cohorts to establish non-inferiority given the high cure rates (>95%) of first-line treatment in clinical trial settings253,254, and they failed. Overall, replacing or adding one drug in either drug-sensitive or MDR-TB regimens only delivered incremental improvement of clinical outcome, at best.
With these realizations came another paradigm shift, towards combining drug candidates with approved drugs and other novel agents in early phases of clinical development. The bedaquiline–pretomanid–linezolid combination is the best example of this approach, leading to treatment reduction from 24 to 6 months to cure MDR-TB. In recent and ongoing studies, bedaquiline has been the backbone of numerous combination trials195,255 to identify regimens that further reduce treatment duration and circumvent existing or acquired drug resistance, with a few promising but preliminary results205,224. The major challenge inherent to such a strategy is the sheer number of possible combinations and how to rationally prioritize those with the best potential for treatment shortening4. Public–private partnerships have been established to accelerate regimen development (Box 2). By breaking down barriers between competing institutions and reducing redundant efforts, these partnerships can address bottlenecks and accelerate the identification of promising regimens.
NTM-PD disease and treatment
Although TB shows little predilection, NTM-PD preferentially affects specific patient populations who fall into two major categories: those with impaired immune functions or those with structural abnormalities of the lung or pre-existing lung damage9. Patient numbers are rising globally, although the picture is incomplete because NTM-PD is not a reportable disease in most countries and states; reports often use different measures of incidence and prevalence, which impedes their pooling; and the distinction between asymptomatic colonization and disease is blurred34. Improved diagnostic and culture techniques, combined with an increase in the number of computed tomography (CT) scans obtained for unrelated conditions, contribute to the growing numbers of NTM cases detected. NTM species present in municipal water systems have been enriched through decades of broad chlorine use, to which they are resistant, unlike most bacterial species. Natural disasters and global warming have also been proposed as potential drivers of the rising numbers35.
The risk factors associated with NTM disease also contribute to the rise of NTM-PD24. These risk factors include immunosuppression caused by treatment with TNF blockers, some anticancer treatments, post-organ transplantation treatment and ageing — with an estimated prevalence of 47.5 per 100,000 among individuals aged 65 years or older10. NTM prevalence is significantly higher in elderly female patients with a low body mass index, but the mechanisms behind gender differences remain enigmatic36. Bronchiectasis due to cystic fibrosis (CF) or other causes, and chronic obstructive pulmonary disease (COPD) increase the likelihood of developing NTM disease. For instance, chronic respiratory disease is associated with a 17-fold increased risk of NTM-PD37,38, and NTM infection is the most important emerging threat to patients with CF, with 5–20% prevalence, which is increasing39,40. Owing to modern medicine and increasing life expectancy, these emerging infections thus target a growing patient population in the USA and other higher- and middle-income countries.
Global geographical differences of both disease incidence and the relative distribution of NTM species have been reported, in part driven by ecological factors that influence environmental NTM niches41. The slow-growing MAC encompasses three species, M. avium, Mycobacterium intracellulare and Mycobacterium chimaera, and it is treated with a macrolide, a rifamycin and ethambutol. Differences in pathogenicity and drug susceptibility have been reported between MAC species, but whether these differences affect cure rates globally is unclear. The MAB complex includes three subspecies: M. abscessus subspecies abscessus, M. abscessus subspecies massiliense and M. abscessus subspecies bolletii. Subspecies abscessus and bolletii usually exhibit inducible macrolide resistance conferred by expression of the erm41 gene, whereas massiliense does not, and macrolide susceptibility is the key determinant of treatment outcome for MAB-PD. The optimal approach to treatment of MAB-PD is undefined. The need for new therapies against MAC, as well as MAB, for which there are essentially no active oral therapies beyond clofazimine for subspecies abscessus42,43, cannot be understated44.
There are several reasons why NTM-PD is so difficult to cure. First, NTM species in general and particularly MAB are intrinsically resistant to many drug classes16. NTM species have evolved to resist environmental insults such as antiseptics, biocides and disinfectants, and so can resist human-made antibiotics by interfering with drug uptake45, forming biofilms46,47, enabling intrabacterial biotransformation and inactivation, or decreasing affinity for the drug target. Second, most antibiotics available to clinicians and patients with NTM are underachieving agents that were repurposed from other infectious diseases rather than optimized to eradicate the most common NTM-PD pathogens. Third, immune deficiency disorders predispose to NTM disease, and the immune response is weakened in a sizeable proportion of patients with NTM-PD, therefore normal host defence systems are compromised. These three factors and others negatively impact durable cure of NTM-PD.
How TB and NTM pulmonary diseases intersect and differ
To determine how knowledge of TB can inform NTM drug development, cross-fertilizing opportunities must be identified while being mindful of notable disease differences. In immunocompromised patients with NTM-PD, disease manifestations are reminiscent of the TB that developed in patients with HIV before widespread adoption of combination antiretroviral therapy48–50. In immunocompetent patients with NTM-PD with bronchiectasis conditions — COPD and CF being the most common — cavitary pathology and worsening bronchiectasis are common51–53. These presentations have similarities to TB in immunocompetent individuals54. Cavities in NTM-PD are similar to pulmonary TB cavities, and although cavitary disease is overall less frequent in patients with NTM-PD than in those with TB55, the presence of large cavities is associated with disease progression leading to respiratory failure, poor treatment response and high mortality56. There is a clinical consensus that differentiating NTM-PD from pulmonary TB should not be based solely on radiological findings, owing to considerable overlap in the clinical and radiographic features of pulmonary TB and NTM-PD57–59.
Rethinking the biology of latent TB with modern imaging and ‘omics’ platforms led to the realization that the latent versus active TB dichotomy is oversimplistic and that TB presents as a continuous spectrum from quiescent infection to active cavitary disease60. In the NTM field, infection is distinguished from colonization (also known as ‘indolent disease’) on the basis of well-established clinical, radiological and microbiological criteria61. Although these TB and NTM manifestations are symptomatically different, the concept of a continuum from colonization to fibro-cavitary NTM-PD is likely to apply and is supported by the substantial proportion of patients with MAB-PD62, MAC-PD63 or TB64 who achieve spontaneous sputum culture conversion, around 30%.
Both TB and NTM diseases exhibit clinical persistence when treated with drugs that are reasonably active in vitro. Bacterial subpopulations become dormant in selected niches and phenotypically resistant to drug therapy, a response orchestrated by the DosR dormancy survival regulator and essential to Mtb persistence in vivo65. The DosR regulon is largely conserved across NTM species66, and chemical inhibition of DosR-mediated hypoxic signalling in MAB is bactericidal in chronically infected mice67. Thus, it is likely that the physiology of persistence is similar in TB and NTM-PD, and that therapeutic approaches to eradicate Mtb persister cells would be a good starting point against NTM persisters. A unique feature of NTM that might not be observed in TB (or is a matter of debate) is the formation of biofilm. Both MAC and MAB grow as biofilms in vitro and in environmental reservoirs46. NTM biofilms have been detected in patients with difficult-to-cure NTM infections, such as in the resected lung cavity of a patient with MAB-PD47, as well as embedded in the alveolar walls of several patients with CF and chronic pulmonary MAB who received lung transplantation68. An elegant study on the mechanopathology of biofilm-like Mtb cords69 could help to draw parallels with NTM biofilms and their impact on immune signalling and drug tolerance. Overall, the similarities of TB and NTM sites of infection point to similar challenges faced by drugs to reach and kill the resident bacteria.
The most notable differences between TB and NTM with a clear impact on drug development are: first, the multi-pathogen nature of NTM-PD, whereas TB is caused by one highly conserved aetiological agent, Mtb; second, the genomic heterogeneity of NTM species, which is associated with their environmental lifestyle, whereas the evolution of Mtb has been constrained to the human host as its exclusive reservoir; and third, the large portion of immunocompromised patients with NTM-PD70. The high genome plasticity of NTM pathogens compared with the high degree of conservation of the Mtb pangenome has a profound impact on their minimum inhibitory concentration (MIC) distributions, which need to be established early in drug discovery programmes with representative species.
The NTM-PD global health emergency has captured the attention of higher- and middle-income countries, where the increasing lifespan of immunocompromised individuals and patients with impaired lung function has enabled opportunistic infections. Although these countries have better resources for accurate diagnoses and complex treatment modalities of NTM-PD than poorer countries can apply to TB, this does not translate into better cure rates for NTM-PD compared with TB. DS-TB is generally curable, whereas the lack of reliable cure is characteristic of MAB-PD and the cure rates of most forms of NTM-PD are comparable to those of MDR-TB and XDR-TB (Table 1). In addition to the intrinsic resistance mechanisms of NTM bacteria described above, several important TB drugs including isoniazid, pyrazinamide, ethionamide and the newly approved pretomanid and delamanid require intrabacterial prodrug conversion by a nitroreductase and are largely inactive against most NTM species71 (see later).
In TB, several excellent clinical trial networks coordinate efforts with consortia that aim to accelerate development of shorter drug regimens (Box 2). Unique cooperations between drug developers, funding agencies and clinicians have been instrumental in establishing the rich development pipeline and enabling access to the large numbers of infected patients still required for clinical trials, despite the advances in trial design. The first 4-month regimen for DS-TB32 and the deployment of bedaquiline–pretomanid–linezolid, which reduces treatment duration of MDR-TB and XDR-TB from ~18–24 months to 6 months33,72, are a testament to what can be achieved by public–private partnerships, funding of basic and translational research and clinical trial platforms, which should pave the way for similar initiatives in NTM drug development. Because NTM-PD is more fragmented with fewer centres specialized in diagnosis, evaluation and management, the formation of similar networks will be more challenging.
Box 2. Public–private partnerships for developing TB drug regimens.
The following consortia leverage members’ collective assets, resources and scientific expertise to identify and evaluate drug combinations that have the potential to treat both drug-sensitive and drug-resistant tuberculosis (TB) and are better tolerated, shorter in duration and simpler to use than existing options.
The Critical Path to TB Drug Regimen (CPTR) was initiated in March 2010 by C-Path, the Bill & Melinda Gates Foundation (BMGF) and the Global Alliance for TB Drug Development (TB Alliance), to accelerate the development of novel TB drug regimens. CPTR’s work is led by cross-sector teams that span 26 academic institutions, 20 non-governmental organizations, seven pharmaceutical and 18 diagnostic industry partners, and five global government bodies.
The TB Drug Accelerator (TBDA) is a multi-partner, multidisciplinary network formed in 2012 to facilitate collaboration in TB drug discovery256 and is primarily funded by the BMGF to mobilize resources across three interfaces: academia–industry, competitor–competitor and basic–applied research.
The European Accelerator of Tuberculosis Regime Project (ERA4TB) was launched in 2020 for 6 years. It integrates more than 30 organizations from the European Union and the USA to substantially reduce the time required for development of new TB treatment regimens.
Founded in 2020 and supported by the Foundation of the NIH (FNIH), the Project to Accelerate New Treatments for Tuberculosis (PAN-TB) consortium brings together philanthropic, non-profit and private sector organizations (Bill & Melinda Gates Medical Research Institute, Evotec, GSK, Johnson & Johnson, Otsuka and the TB Alliance) to accelerate the development of novel, shorter drug regimens to treat all forms of TB. The consortium focuses on advancing research through phase II clinical efficacy studies to identify promising regimens for further development.
The TBDA, PAN-TB and ERA4TB consortia work closely together such that new molecular entities identified by the TBDA and ERA4TB and those showing promise in initial human studies can be incorporated into the later-stage, clinical research of the PAN-TB collaboration. Several organizations, including Evotec, GSK and Johnson & Johnson, are members of all three projects, which ensures coordination towards the common goal of advancing TB drug and regimen development. Finally, several excellent clinical trial networks help to support TB drug development: the Aids Clinical Trial Group (ACTG), the TB Trial Consortium (TBTC), UNITE4TB, the International Maternal Paediatric Adolescent AIDS Clinical Trials (IMPAACT) Network and the Pan-African Consortium for the Evaluation of Anti-Tuberculosis Antibiotics (PanACEA). PanACEA is funded by the European and Developing Countries Clinical Trials Partnership (EDCTP) and has established a dynamic network of 11 sub-Saharan clinical trial sites and four European research institutions257.
Preclinical and clinical development pipelines
The healthy status of the TB drug pipeline (Table 2 and Fig. 2) is the result of the judicious combination of multiple complementary drug discovery approaches, each with its strengths and limitations. These strategies rely on ‘old’ but clinically validated targets as well as novel mechanisms of action. This combination mitigates attrition of de novo drug discovery and optimizes chances of success.
Table 2 |.
Selected ongoing clinical trials of new agents for TB and NTM-PD
| Candidate | New MoA? | Trial design | Sponsor | Study identifier |
|---|---|---|---|---|
| TB | ||||
| TBI-223a | No, re-engineered oxazolidinones targeting the ribosome | Phase I (SAD and MAD) to evaluate safety, tolerability and PK in healthy subjects | TB Alliance | NCT03758612, NCT04865536 |
| Sutezolidb | Phase IIb to identify the optimal dose in combination with bedaquiline, delamanid and moxifloxacin for 3 months | Michael Hölscher, University of Munich | NCT03959566 | |
| Delpazolid | Phase IIb dose finding to evaluate exposure–response relationship in combination with bedaquiline, delamanid and moxifloxacin for 4 months | LegoChem Biosciences | NCT04550832 | |
| Sanfetrinem cilexetila | No, first-in-class tricyclic carbapenem targeting peptidoglycan biosynthesis | Phase IIa dose finding to evaluate the EBA, safety and tolerability, including in combination with rifampicin and/or amoxicillin–clavulanate | TASK Applied Science | NCT05388448 |
| TBI-166 (pyrifazimine) | No, rhiminophenazine analogue of clofazimine, inhibitor of redox cycling yielding toxic reactive oxygen species244 | Phase IIa to evaluate EBA, safety and tolerability | Beijing Chest Hospital | NCT04670120 |
| TBAJ-587a | No, diarylquinoline analogues of bedaquiline targeting ATP synthase | Phase I (SAD and MAD) to evaluate safety, tolerability and PK in healthy adults | TB Alliance | NCT04890535 |
| TBAJ-876a | Phase I (SAD and MAD) to evaluate safety, tolerability and PK in healthy adults | TB Alliance | NCT04493671 | |
| WX-081 (sudapyridine) | Phase IIa to evaluate EBA, safety and tolerability in patients with drug-susceptible and drug-resistant TB | Shanghai Jiatan Pharmatech Co. Ltd | NCT04608955 | |
| OPC-167832 (quabodepistat)b | Yes, DprE1 (arabinogalactan biosynthesis) | Phase IIb/c dose finding to evaluate safety and efficacy in combination with delamanid and bedaquiline for 4 months in patients with drug-susceptible disease | Otsuka Pharmaceutical | NCT05221502 |
| TBA-7371a | Phase IIa to evaluate safety, EBA and PK in adults with rifampicin-sensitive TB | Bill & Melinda Gates Medical Research Institute | NCT04176250 | |
| BTZ-043 | Phase Ib/IIa to evaluate safety, tolerability, PK, drug–drug interaction and EBA in patients with drug-susceptible TB | Michael Hölscher (University of Munich) | NCT04044001 | |
| GSK3036656a | Yes, leucyl-tRNA synthetase | Phase IIa dose escalation to evaluate EBA, safety and tolerability in subjects with rifampicin-susceptible TB | GlaxoSmithKline | NCT03557281 |
| SQ109 | Yes, MmpL3 (mycolic acid biosynthesis) and proton motive force | Phase IIb to assess efficacy, safety and tolerability in combination with a standard regimen for MDR-TB treatment | Sequella, Inc. | NA245 |
| Q203 (telacebec) | Yes, cytochrome bcc complex (respiratory chain) | Phase IIa dose ranging to evaluate EBA, safety, tolerability and PK in patients with drug-susceptible TB | Qurient Co.246 | NCT03563599 |
| BVL-GSK098 (alpibectir) | Yes, promotes intrabacterial conversion of ethionamide prodrug | Phase IIa to evaluate EBA, safety and tolerability of ethionamide alone and in combination with BVL-GSK098 in patients with drug-susceptible TB | TASK Applied Science | NCT05473195 |
| GSK2556286a | Yes, adenylyl cyclase (cholesterol catabolism) | Phase I (SAD and MAD) to evaluate safety, tolerability, PK and food effect in healthy adults | GlaxoSmithKline | NCT04472897 |
| GSK839 | Yes, tryptophan synthetase TrpAB | Preclinical | GlaxoSmithKline | NA |
| DDD02049209 | Yes, lysyl-tRNA synthetase | Preclinical | University of Dundee | NA |
| MBX-4888A (spectinamide 1810) | No, semi-synthetic spectinamycin analogue targeting the ribosome | Preclinical | Microbiotix, Richard Lee | NA |
| NTM-PD | ||||
| SPR720 | Yes, new chemical entity inhibiting ATPase domain of GyrB (DNA replication) | Phase II dose ranging to evaluate efficacy, safety, tolerability and PK for 2 months in patients with MAC-PD | Spero Therapeutics | NCT05496374 |
| Omadacycline | No, tetracycline analogue inhibiting the ribosome | Phase II to evaluate the efficacy, safety and tolerability for 3 months in patients with MAB-PD | Paratek Pharmaceuticals | NCT04922554 |
| ALIS (amikacin liposomal inhalation suspension) | No, reformulated amikacin for inhalation, inhibiting the ribosome | Phase III to evaluate efficacy and safety for NTM-PD for up to 15 months in patients with newly diagnosed NTM-PD | Insmed | NCT04677569 |
| Clofazimine | No, repurposed from leprosy | Phase II pharmacokinetic study in NTM-PD | Radboud University Medical Center | NCT05294146 |
| Phase II to evaluate safety, culture conversion, changes in lung function and quality of life for 6 months in MAC-PD | Oregon Health and Science University | NCT02968212 | ||
| Hypertonic saline inhalation | Unknown, nonspecific anti-mycobacterial activity | Phase IV to evaluate effect combined with best supportive care for 3 months in patients with MAC-PD | Radboud University Medical Center | NCT05192057 |
EBA, early (7–14 days) bactericidal activity; MAB-PD, Mycobacterium abscessus pulmonary disease; MAC-PD, Mycobacterium avium complex pulmonary disease; MAD, multiple ascending doses; MDR-TB, multidrug-resistant tuberculosis; MoA, mechanism of action; NA, not applicable; NTM-PD, pulmonary disease caused by nontuberculous mycobacteria; PK, pharmacokinetics; SAD, single ascending dose; TB, tuberculosis.
Identified by the TB drug accelerator.
Part of PAN_TB consortium.
Fig. 2 |. Selected anti-mycobacterial drug candidates and their mechanism of action.

a, A simplified version of the cell wall and the cytoplasmic membrane of Mycobacterium tuberculosis illustrating drug targets involved in oxidative phosphorylation (NADH:quinone oxidoreductase, cytochrome oxidase and ATP synthase) or cell wall biosynthesis (MmpL3 transporter, DPR epimerase and transpeptidase). b, Late preclinical and clinical tuberculosis candidates that target intracellular pathways, as well as SPR720 and omadacycline in the nontuberculous mycobacteria pulmonary disease (NTM-PD) pipeline. SPR720 inhibits the ATPase activity of the mycobacterial gyrase complex; BVL-GSK098 inhibits the transcriptional repressor EthR to allow expression of the monooxygenase EthA and boost activation of ethionamide (ETH); GSK286 is an agonist of adenylyl cyclase that causes increases cAMP levels, interfering with growth in the presence of cholesterol; GSK839 inhibits tryptophan synthase TrpAB; GSK656 (ganfeborole) and DDD209 are inhibitors of leucyl-RNA and lysyl-tRNA synthetase (LeuRS, LysRS), respectively; the oxazolidinones sutezolid, TBI-223 and delpazolid prevent peptide bond formation; the spectinamide MBX-4888A blocks ribosome translocation; and omadacycline prevents binding of aminoacyl-tRNA. Notably, six compound classes inhibit steps involved in protein synthesis. c, Targeted protein degradation principles in eukaryotic cells and in bacteria. In the eukaryotic proteolysis-targeting chimera (PROTAC) approach (top), a target ligand (TL) and an E3 ligase ligand (EL) are covalently joined by a linker to generate proximity of a protein of interest (POI) and the E3 ligase, resulting in ubiquitylation and tagging of the POI, which leads to proteasomal degradation. In the bacterial BacPROTAC approach (bottom), a TL and a ligand for the caseinolytic protease complex ClpC–ClpP (PL) are covalently linked to generate proximity of the POI and ClpC–ClpP, facilitating degradation. Panel a adapted from ref. 231, Springer Nature Limited.
Repurposing approved antibiotics
Moxifloxacin and linezolid are among the most notable success stories of antibiotic repurposing for TB. Both are broad-spectrum agents developed to treat Gram-positive and/or Gram-negative infections. Their activity against Mtb was discovered in the late 1990s73,74 and they both achieve pharmacokinetic–pharmacodynamic (PK–PD) targets at standard doses, with higher doses anticipated to increase the probability of target attainment75,76. Moxifloxacin, a fourth-generation fluoroquinolone, is part of the new 4-month first-line regimen to treat drug-susceptible TB and one of the pillars of MDR-TB treatment. It is included in many treatment-shortening MDR-TB clinical trials. Although less prominently used, levofloxacin has a similar role in MDR-TB therapy and is often favoured to treat paediatric TB. Linezolid, the first oxazolidinone antibacterial agent77, is part of the new 6-month three-drug regimen recommended by the WHO and the Centers for Disease Control and Prevention (CDC) against MDR-TB and XDR-TB72. Linezolid is occasionally included in the treatment of NTM-PD14, but its clinical utility and PK–PD target attainment have not been established.
Such repurposing approaches have the potential to shorten treatments and decrease attrition rates, but they come with inherent limitations and caveats. Fluoroquinolone use is a correlate of successful MDR-TB treatment, and loss of fluoroquinolone susceptibility results in significantly worse outcome78,79. Not surprisingly, pre-existing resistance is an acute and rapidly growing issue for moxifloxacin and fluoroquinolones in general80,81, particularly given that systematic fluoroquinolone susceptibility testing of Mtb isolates is uncommon and broad-spectrum antibiotic use is insufficiently regulated in many countries with a high disease burden82. Pre-existing resistance to linezolid is not as frequent in Mtb83 for reasons that are yet poorly understood, but this drug comes with another liability. Like most antibiotics, it was developed to cure bacterial infections that typically require short-term therapy84. Prolonged linezolid administration beyond a month leads to peripheral neuropathy and myelosuppression in a large proportion of patients85, resulting in treatment discontinuation86. In the highly successful NIX-TB trial33, combination of bedaquiline and pretomanid with linezolid led to treatment reduction from 24 to 6 months for MDR-TB; however, peripheral neuropathy and myelosuppression developed in 81% and 48% of the patients, respectively. Therefore, these relatively rapid repurposing successes brought new challenges: the need for systematic fluoroquinolone susceptibility testing87 and the development of next generation oxazolidinones with a broader therapeutic window such as sutezolid, delpazolid and TBI-223 (refs. 88,89) (Table 2). In the interim, a lower linezolid dose has been identified to minimize toxic effects while maintaining acceptable efficacy90.
NTM species in general and particularly MAB are intrinsically resistant to many TB drug classes, primarily owing to intrabacterial drug inactivation but also because of their notorious cell wall impermeability and efflux mechanisms16. With the notable exception of macrolides for MAC-PD91 and macrolide-susceptible MAB-PD (Table 1), antibiotic repurposing to treat NTM-PD has not been fruitful, for multiple reasons. Anti-TB agents such as isoniazid, pyrazinamide, pretomanid, delamanid, ethionamide and p-amino-salicylic acid are prodrugs that undergo bioactivation within the bacterial cell and are generally inactive against most NTM species92–95. It has been speculated that activation enzymes for the drugs could be absent, inactive or poorly conserved in NTM, although an association between differential sensitivity and differences in prodrug activation has been shown only for isoniazid96. These anti-TB drugs also exhibit wide MIC distributions, which compromises their clinical utility97,98. For example, the MIC of isoniazid for M. kansasii, one of the closest relatives of Mtb, ranges from 0.5 to 32 μg ml−1 (ref. 99) Except for p-aminosalicylic acid, these drugs have multi-target mechanisms of action often involving the generation of reactive metabolites that trigger complex, lethal cascades that are not fully understood100–103. Comparative mechanistic studies have identified steps in these cascades that are not sufficiently conserved across NTM species to allow efficacious drug action. This knowledge can guide pharmacological approaches to improve bactericidal activity of anti-TB drugs against NTM. Other candidates appear to be potent in vitro at concentrations achieved clinically, yet their clinical utility remains to be established. For example, clofazimine is a repurposed antibiotic approved for the treatment of M. leprae (leprosy) infections and used off-label against MAC-PD in the USA. Meta-analyses of the contribution of clofazimine have delivered inconsistent but sufficiently promising results to justify its inclusion in multiple randomized controlled trials in defined populations of patients with MAB-PD or MAC-PD of varying disease severity (Table 2). In combination trials, clofazimine replaces either ethambutol or rifamycin in the standard of care (Table 3).
Table 3 |.
Selected clinical trials of drug regimens for pulmonary TB and NTM-PD
| Indication | Duration (months) | Trial acronym or sponsor | Agents | Primary end pointa | Study identifier |
|---|---|---|---|---|---|
| Completed, with results indicative of treatment shortening | |||||
| DS-TB | 4 | TBTC Study 31 (non-inferiority phase III) | Isoniazid, rifapentine, moxifloxacin, pyrazinamide for 2 months, followed by isoniazid rifapentine for 2 months | Disease-free survival 12 months after end of treatment; outcome: 4-month treatment non-inferior to standard 6-month regimen (HRZE)32 | NCT02410772 |
| MDR-TB | 6 | NExT (phase III) | Bedaquiline, linezolid, levofloxacin, pyrazinamide, ethionamide/high-dose isoniazid | Favourable treatment outcome 24 months after treatment initiation; outcome: 6-month treatment non-inferior to >9-month injectable-based regimen222 | NCT02454205 |
| MDR-TB | 6 | TB-PRACTECAL (non-inferiority phase II/III) | Bedaquiline, pretomanid, linezolid, moxifloxacin for 6 months | Death, treatment failure or discontinuation; outcome: 6-month regimen safer and non-inferior to the 9- to 20-month standard-care treatment205 | NCT02589782 |
| MDR-TB | 9 | MDR-END (non-inferiority phase II) | Delamanid, linezolid, levofloxacin, pyrazinamide for 9 months | Treatment success; outcome: first 9-month all-oral regimen non-inferior to 20- to 24-month WHO-recommended treatment206 | NCT02619994 |
| DS-TB | 2 | TRUNCATE-TB (non-inferiority phase III) | Isoniazid, pyrazinamide, ethambutol, high-dose rifampicin, bedaquiline, linezolid | Unsatisfactory outcome (death or active disease at 12 months) leads to termination of a treatment arm; outcome: 2-month bedaquiline- and linezolid-containing regimen non-inferior to standard treatment224 | NCT03474198 |
| DS- and MDR-TB | 4 (DS) 6 (MDR) | SimpliciTB (phase IIc) | BPaMZ | Time to culture conversion over 8 weeks, rate of relapse 12 and 24 months after end of therapy with BPaMZ compared with HRZE; outcome: BPaMZ achieves treatment shortening, but side effects precluded completion in ~10% of patients247 | NCT03338621 |
| Dose-finding combination studies | |||||
| Pre-XDR and XDR-TB | 6 | ZeNIX (phase III) | Bedaquiline, pretomanid, linezolid | 600 mg linezolid instead of 1,200 mg in NIX-TB achieves similar cure rates with reduced adverse events (completed90) | NCT03086486 |
| MDR-TB | 6 | Opti-Q or TBTC study 32 (phase II) | Levofloxacin, OBR | Dose-finding study to evaluate efficacy, PK and tolerability of increasing levofloxacin doses in combination with OBR in patients with MDR-TB | NCT01918397 |
| DS-TB | 3 | SUDOCU (phase IIb) | Bedaquiline, delamanid, moxifloxacin, sutezolid | Dose-finding study to evaluate safety, tolerability, PK and exposure–response relationship of sutezolid in combination with bedaquiline, delamanid and moxifloxacin | NCT03959566 |
| DS-TB | 4 | DECODE (phase IIb) | Bedaquiline, delamanid, moxifloxacin, delpazolid | Dose-finding study to evaluate safety, tolerability, PK and exposure–response relationship of delpazolid in combination with bedaquiline, delamanid and moxifloxacin248 | NCT04550832 |
| Conventional non-inferiority trials | |||||
| DS-TB | 4 | RIFASHORT (phase III, completed) | Rifampicin, isoniazid, pyrazinamide, ethambutol | Open-label study: 4 months HRZE with 1,200 mg or 1,800 mg rifampicin was not non-inferior to the standard 6-month regimen (HRZE with 600 mg rifampicin), measuring combined rate of failure up to 12 months after end of therapy249 | NCT02581527 |
| MDR-TB | 9 | endTB (phase III) | 5 study arms comprising 4 or 5 of the following agents: bedaquiline, delamanid, clofazimine, linezolid, moxifloxacin or levofloxacin, pyrazinamide | Randomized, controlled, non-inferiority open-label clinical trial evaluating efficacy and safety of five 9-month treatment regimens containing recently approved drugs for MDR-TB compared with current standard of care for MDR-TB250 | NCT02754765 |
| XDR-TB | 6–9 | endTB-Q (phase III) | BeDeCLi | Randomized, controlled, non-inferiority open-label trial evaluating efficacy of BeDeCLi for 6 or 10 months according to disease phenotype, compared with WHO-recommended treatment | NCT03896685 |
| MDR- and XDR-TB | 6–9 | BEAT-TB (phase III) | Bedaquiline, delamanid, clofazimine, linezolid, levofloxacin for MDR-TB; bedaquiline, delamanid, clofazimine, linezolid for XDR-TB | Design and outcome measures similar to endTB-Q with secondary end points as PK–PD models of drug and metabolite exposure versus efficacy and toxicity | NCT04062201 |
| Ongoing adaptive trials and biomarker studies | |||||
| DS-TB | 4 | Predict-TB (phase II) | Isoniazid, rifampicin, pyrazinamide, ethambutol | Using biomarker combinations to predict treatment duration | NCT02821832 |
| DS-TB, RR/MDR-TB | 2–4 | DBOSPBOS (phase IIb/c) | Bedaquiline, OPC-167832, sutezolid, pretomanid, delamanid | Phase IIb/c, multi-arm, 2-stage, duration randomized trial of efficacy and safety for 2–4 months with bedaquiline, OPC-167832 and sutezolid, plus either pretomanid or delamanid | NCT05971602 |
| NTM-PD | 12 | Shanghai Pulmonary Hospital (phase IV) | Bedaquiline, clofazimine, linezolid + 2 or 3 drugs | Bacteriological and clinical treatment outcome | NCT05494957 |
| MAC-PD | 12 from sputum conversion | RedHill Biopharma | RHB-204 fixed-dose oral capsule containing clarithromycin, rifabutin and clofazimine | Sputum culture conversion after 6 months of treatment (3 consecutive negative sputum cultures at months 4, 5, 6 compared with placebo) | NCT04616924 |
| MAC-PD in patients with AIDS | 18 | National Institute of Allergy and Infectious Diseases | Macrolide, ethambutol, rifabutin or clofazimine | Sputum culture conversion at 1, 2 and 4 months and every 4 months thereafter for minimum of 1.5 years | NCT00001047 |
| NTM-PD | 3 | Medical University of South Carolina (phase II) | Inhaled nitric oxide | Sputum negative culture | NCT03748992 |
BeDeCLi, bedaquiline, delamanid, clofazimine, linezolid; BPaMZ, bedaquiline, pretomanid, moxafloxacin, pyrazinamide; DS-TB, drug-susceptible tuberculosis; HRZE: isoniazid, rifampicin (600 mg or 10 mg kg−1), pyrazinamide and ethambutol for 2 months followed by isoniazid and rifampicin (600 mg or 10 mg kg−1) for 4 months; RR, rifampicin resistant; MDR-TB, multidrug-resistant tuberculosis; NTM-PD, pulmonary disease caused by nontuberculous mycobacteria; OBR, optimized background regimen; PD, pharmacodynamic; PK, pharmacokinetic; TB, tuberculosis; XDR-TB, extensively drug-resistant tuberculosis.
Outcome described for completed trials only.
The intrinsic drug resistance mechanisms of NTM are increasingly being studied and understood, but PK–PD relationships are generally underappreciated. Indeed, the lack of correlation between the MIC of recommended drugs and clinical outcome in MAC-PD is likely associated with the wide MIC distribution largely lying on the high side of the clinical breakpoint (Fig. 3). For example, the MIC distribution of rifampicin for MAC largely overlaps the rifampicin-resistant region for Mtb, but MIC variability in MAC is not associated with mutations in the rpoB target104,105, in stark contrast to Mtb106,107. In other words, clinical breakpoints lie at the lower end of the MIC distributions for RpoB wild-type MAC isolates (Fig. 3a), which agrees with in vitro experiments in which rifampicin did not enhance the effects of currently used MAC drugs108. Similar comparisons for other antibiotics used in the treatment of MAC-PD and MAB-PD reveal an identical trend (Fig. 3b), which likely contributes to the poor cure rates despite long treatment duration109. Bedaquiline emerges as a notable exception, with MIC distributions for MAC-PD and MAB-PD below the clinical breakpoints established for pulmonary TB. The similar potency range of bedaquiline against Mtb and various NTM species has been known since 2005b (ref. 110) but the drug is not bactericidal against MAC and is only bacteriostatic in two mouse models of MAC infection111. Given the positive and often synergistic interactions between bedaquiline and clofazimine112, and the reasonable activity of clofazimine against MAC compared with Mtb, clinical investigations of the combination to treat MAC-PD could be worth considering. However, bedaquiline should be paired judiciously within drug regimens because it antagonizes the bactericidal activity of β-lactams113 and potentially other antibiotic classes that require a burst of ATP to achieve cell death. Overall, the NTM field still lacks reliable clinical breakpoints based on MIC distributions measured with standardized methods, as well as PK–PD studies in adequate models and treatment outcome data114.
Fig. 3 |. Susceptibility ranges of TB drugs repurposed for the treatment of pulmonary disease caused by nontuberculous mycobacteria, relative to clinical breakpoints.

a, Schematic comparison of rifampicin minimum inhibitory concentration (MIC) distribution for Mycobacterium tuberculosis (Mtb) and Mycobacterium avium complex (MAC), showing the overlap between genetically rifampicin-resistant (RIFR) Mtb isolates (rpoB mutated) and rpoB wild-type (WT) but phenotypically resistant MAC isolates. The graphs show the frequency (y axis) of rifampicin MIC (x axis) for Mtb (top) and MAC (bottom). The red vertical line indicates the clinical breakpoint (CB) of rifampicin for tuberculosis (TB). Mtb isolates with an MIC greater than 0.5 mg l−1 are considered rifampicin resistant, generally carry mutations in rpoB and are not treated with rifampicin. Applying this CB to MAC (red dotted line) shows that the MIC range of rpoB WT MAC isolates exceeds the CB, highlighting the intrinsic resistance of MAC to rifampicin, and predicting poor efficacy of rifampicin against MAC-PD. b, Comparison of MIC distributions of drugs included in treatment recommendations for lung disease caused by Mycobacterium abscessus (MAB) and MAC, relative to the CB established for TB at the WHO-recommended doses (CB for linezolid is at a dose of 1,200 mg daily). Blue boxes, MIC distributions for Mtb; pink boxes, MIC distributions for MAC; red boxes, MIC distributions for MAB; red dotted line, CB for pulmonary TB; R, resistant; S, susceptible.
The discouraging MIC values of potential drugs for NTM (Fig. 3b) nevertheless offer an opportunity to search for synergistic drug combinations. Bactericidal antibiotics that target the same cellular process often achieve heightened killing when combined115. One such process is mycobacterial peptidoglycan biosynthesis, which involves partially redundant and complementary transpeptidases and carboxypeptidases116,117, several of which are inhibited by various classes of β-lactam. Indeed, several β-lactam synergies have been reported against MAB118. Motivated by these observations, a systematic screen of the bioactive forms of oral β-lactam prodrugs against MAB revealed strong bactericidal synergies between amoxicillin and either sulopenem, tebipenem or cefuroxime, in the presence of avibactam119. These combinations decreased the bacterial burden by up to 4-log in vitro at concentrations that are clinically achievable. Sulopenem and tebipenem are in phase III clinical trials, and cefuroxime is an FDA-approved oral cephalosporin, thus allowing immediate clinical investigations into potential salvage therapies for patients with MAB-TB with limited to no therapeutic options.
Re-engineering antibiotic classes
Following the success of linezolid treatment in patients with XDR-TB who do not respond to chemotherapies120, and the more recent approval of a 6-month course of bedaquiline–pretomanid–linezolid for MDR-TB, re-engineered oxazolidinones with an improved tolerability profile have gained traction. Their increased tolerability is due to increased potency, decreased mitochondrial protein synthesis (MPS) inhibition or both. Although the magnitude of improvement required for NTM-PD is markedly higher than for TB, the strategy is applicable to both disease indications. Three members of the class — TBI-223 (ref. 121), delpazolid122 and sutezolid89 — have emerged among numerous oxazolidinone optimization programmes in recent years and are in clinical development for TB (Table 2). TBI-223 has attractive attributes compared with linezolid, including reduced MPS inhibition123 and reduced bone marrow progenitor cell toxicity in vitro and in animal models124. In a phase IIa trial, delpazolid did not show an advantage over linezolid88. Sutezolid has two favourable properties: reduced MPS inhibition and an abundant active metabolite that complements activity of the parent compound against intracellular and extracellular bacteria. It is being tested in combination with bedaquiline, delamanid and moxifloxacin in a dose-ranging study (Table 2) to assess its 3-month safety profile and adjunctive efficacy. It is one of the PAN-TB consortium (Box 2) drug candidates included in the two novel, rationally designed, combinations that have entered phase II trials. Recently, systematic measurements of MPS inhibition and activity against clinically relevant mycobacterial pathogens uncovered sutezolid as a promising candidate against NTM-PD caused by M. kansasii. The next few years will reveal whether further re-engineered oxazolidinones with markedly improved safety profiles, potency against Mtb and NTM species, and activity against linezolid-resistant isolates can have a determining role in the treatment of DS-TB and drug-resistant TB.
Bedaquiline has been an exemplary drug against MDR-TB and XDR-TB but has two liabilities. First, its extreme hydrophobic and cationic amphiphile properties lead to extensive tissue distribution, phospholipidosis and long terminal half-life. Lingering concentrations have been measured in plasma and at the site of disease for several months after the end of therapy125,126, conditions that are prone to acquisition of resistance127,128. Second, bedaquiline inhibits the cardiac potassium hERG channel, resulting in QTc interval prolongation in humans. Although an association with cardiac arrhythmia has not been seen clinically, bedaquiline analogues that avoid hERG inhibition would be welcome, particularly as moxifloxacin has a similar potential for cardiotoxicity that complicates their combined use. TBAJ-587 and TBAJ-876 are less hydrophobic, 5- to 10-fold more potent second-generation analogues that do not inhibit hERG at the highest concentration tested129 and retain a similar potency against the dominant bedaquiline-resistant isolates in the MmpS5–MmpL5 transporter, an efflux system involved in resistance to several antibiotics130. Substituting TBAJ-587 or TBAJ-876 for bedaquiline has reduced the emergence of resistance to the drug class and to pretomanid in mice treated with combinations similar to those used in the NIX-TB trial131,132. Likewise, WX-081 (sudapyridine) does not inhibit hERG or cause QT prolongation in vivo and delivers efficacy comparable to that of bedaquiline in mice133,134. The three agents are rapidly progressing through clinical development (Table 2) and collectively address the major liabilities of bedaquiline. TBAJ-876 is as efficacious as bedaquiline in a mouse model of MAB infection135 and therefore has promise as a repurposed agent for MAB-PD. Other notable success stories of rebranded or re-engineered agents are the phase II candidate sanfetrinem, developed as an oral tricyclic carbapenem stable to clinically relevant β-lactamases and rapidly lethal to Gram-positive and Gram-negative bacteria136, and the spectinamide MBX-4888A, optimized to overcome efflux by Mtb137,138.
Rifampicin and rifapentine are at the centre stage of TB therapy and treatment shortening; however, this has not translated to NTM-PD treatment. Rifampicin and rifabutin are recommended for MAC-PD but confidence in their clinical utility is not established14, unsurprisingly given their MIC distribution relative to clinical breakpoints (Fig. 3a). The rifamycins are even less active against MAB, despite their highly conserved RNA polymerase (RNAP) target139, and are thus not recommended for treatment140. However, the mechanisms that underlie this pronounced intrinsic resistance have been sufficiently well understood to guide medicinal chemistry efforts139,141,142. Rifamycins are ideal re-engineering candidates as a pragmatic strategy to rapidly develop potent and treatment-shortening antibiotics against NTM-PD, and they have some key attributes for potentially successful NTM-PD treatment. Because rifamycins have not been used to treat MAB infections, the caveat of pre-existing resistance does not apply. Also, rifamycins have bactericidal activity against TB non-replicating persister cells and excellent penetration into all lung lesion compartments143,144. These properties are important for NTM-PD, given the decreased oxygen tension found deep in the airway mucus of patients with CF — conditions that are prone to the emergence of persister cells and in the case of MAB to biofilm-like communities145. Rifamycins are among the few antibiotic classes that retain bactericidal activity under these conditions of hypoxia-induced non-replication. Rifamycins are largely inactive against MAB because their C23-OH group is ADP-ribosylated by the bacterium’s ADP-ribosyltransferase (Arr), preventing the drug from interacting with RNAP. The ribosyltransferase is found in many bacterial species141, and deletion of the Arr-encoding gene overcomes the intrinsic rifamycin resistance of MAB139,142. Two recent drug optimization programmes have delivered rifamycin analogues that block enzymatic inactivation by Arr while maintaining binding to the RNAP target146,147. The vast preclinical and clinical knowledge of rifamycin pharmacology and tolerability should facilitate fast-track development of these novel analogues.
Identifying novel chemical scaffolds and their targets
Following the painful realization that genomics-derived, target-based approaches to screen for new classes of drug with novel modes of action had not delivered on their promise148, pharmaceutical and academic groups embraced large-scale phenotypic screens against whole cells, followed by whole-genome sequencing for target deconvolution. Bedaquiline is the ‘poster child’ of modern phenotypic TB drug discovery and illustrates the power of genomics to identify novel scaffold–target couples110. The successes of phenotypic drug discovery are multifactorial: interrogation of a biological system in a target-agnostic fashion enables expansion of the ‘druggable’ target space, the opportunity to identify molecules that engage multiple targets (or polypharmacology) and ensures that physicochemical properties of the molecules are compatible with intrabacterial uptake and metabolic stability149. Around a dozen candidate molecules with novel mechanisms of action and proof-of-concept efficacy in mouse models have emerged from phenotypic drug discovery in recent years and are in advanced preclinical or clinical development150 (Table 2). A comprehensive review of the medicinal chemistry, in vitro activity and in vivo efficacy of these and earlier programmes was published recently151,152. Candidates that target cell wall biosynthesis pathways are strongly represented in the TB pipeline. Interestingly, whole Mtb cell screens have revealed two highly promiscuous cell wall drug targets: MmpL3, involved in export of trehalose monomycolate, a mycolic acid component; and the DPR epimerase DprE1, which is required for arabinogalactan synthesis (Fig. 2a). These targets are both unique to mycobacteria and vulnerable in NTM species153,154. Three DprE1 inhibitors belonging to three different chemical scaffolds152 are in clinical development for TB but lack in vitro and/or in vivo activity against NTM species154: OPC-167832 (quabodepistat), TBA-7371 and BTZ-043. There is only one MmpL3 inhibitor in clinical development, SQ109, but numerous chemically diverse series155 are in various stages of lead optimization and preclinical development, a subset of which are active against NTM species153.
Several other novel targets involved in the synthesis and remodelling of the mycobacterial cell wall have ligands that show efficacy in in vivo systems. The following targets are required for the synthesis of mycolic acids, unique to mycobacteria: the β-ketoacyl-acyl carrier protein KasA, part of the fatty acid elongation system, which interacts with compound JSF3285 (ref. 156); the fatty acid degradation protein FadD32, which catalyses the activation of long-chain fatty acids as acyl-adenylates and binds to quinoline-2-carboxamides157; and the polyketide synthase Pks13, which, with FadD32, forms the initiation module of the mycolic condensation system and binds to the benzofuran class inhibitor TAM16 (ref. 158). Thus, cell wall synthesis has provided an extremely fertile ground for anti-TB drug discovery over the decades, with the caveat that many (but not all159) of these pathways are not vulnerable in non-replicating bacteria160. These cell wall targets have homologues in MAB, where they are essential, as shown by a genome-wide Tn-seq approach117, but systematic investigations of their vulnerability across clinically relevant NTM pathogens and in vivo proof-of-concept studies are largely lacking. In Mtb, the cas9-based CRISPR interference system has found broad utility for functional genomics, genetic interaction mapping and drug–target profiling161 and could be similarly leveraged in NTM to validate drug–target couples, uncover mechanisms of intrinsic drug resistance and discover potential targets for synergistic drug combinations.
The viability of replicating and non-replicating Mtb depends on energy generated by components of its respiratory chain, which are attractive drug targets. Indeed, compound Q203 targeting the QcrB unit of the cytochrome bc1–aa3 oxidase is in phase II trials. However, elegant genetic studies have revealed that the cytochrome bc1–aa3 and bd oxidases of Mtb are functionally redundant162, protecting Mtb from Q203-induced death. Consistent with this observation, the cytochrome bd inhibitors ND-011992 and CK-2–63 are ineffective on their own, but when combined with Q203 they kill replicating and antibiotic-tolerant, non-replicating mycobacteria by efficiently inhibiting respiration and ATP homeostasis163,164. Combination of Q203 and ND-011992 achieved increased efficacy relative to single-drug treatment in a TB mouse model164, validating the approach of inhibiting multiple components of the branched respiratory chain (Fig. 2a). Nitroimidazoles pretomanid and delamanid illustrate the power of polypharmacology, as they target both cell wall synthesis and respiration pathways. They exhibit a dual mode of action under low and normal oxygen tension, poison multiple essential pathways through the release of nitric oxide and generation of reactive intermediates and thus kill both replicating and non-replicating mycobacteria165. Despite chemical and functional similarities, they are not interchangeable, but clinical trials that compare them side by side in otherwise equivalent regimens are lacking100. Chemical validation and activity of drug–target couples discovered in Mtb have been comprehensively reviewed15.
Several novel chemical entities target mycobacterial cytosolic enzymes (Fig. 2b). Among these, aminoacyl-tRNA synthetases are essential enzymes for protein synthesis and a new source of attractive targets in bacteria166. GSK3036656, in phase II, and DDD02049209, in preclinical development, inhibit the mycobacterial leucyl-tRNA and lysyl-tRNA synthetases, respectively167,168. GSK839 is a new chemical entity and a preclinical development candidate that selectively targets Mtb tryptophan synthase (TrpAB), a heterotetrameric complex that catalyses the conversion of indole-3-glycerol phosphate into l-tryptophan169. GSK2556286 modulates a pathway that appears uniquely vulnerable in mycobacteria when cholesterol is a source of carbon. The compound affects cAMP signalling through interactions with a membrane-bound adenylyl cyclase, leading to repression of the conditionally essential cholesterol catabolic pathway170. Owing to the abundance of cholesterol in foamy macrophages and necrotic lesions where the pathogen resides171,172, GSK2556286 shortens treatment duration relative to standard-of-care drugs and effectively replaces linezolid in the NIX-TB regimen in mouse models173. The ubiquitous caseinolytic protease (Clp) system, a protector from stresses induced by host immunity and other environmental insults, has emerged as a high-priority antimicrobial target174,175. Although targeting the mycobacterial ClpC1–ClpP1P2 protease complex is still in early days, lead compounds exhibit attractive biological profiles as exemplified by depsipeptide natural product analogues that specifically inhibit mycobacterial protein degradation and exhibit rapid bactericidal activity against replicating, hypoxic non-replicating, intracellular and extracellular Mtb176 and in a zebrafish model of infection. Active medicinal chemistry programmes seek to optimize the pharmacological properties of a drug-like series. Finally, BVL-GSK098 (alpibectir) has reached phase II trials and acts via a newly discovered mechanism of transcriptional regulation in mycobacteria177. This elegant mechanism of action was originally discovered with the chemical analogue SMARt751, a N-acylated 4-phenylpiperidine that unleashes an otherwise cryptic regulation system, stimulating ethionamide bioactivation pathways and thus increasing ethionamide susceptibility178. Ethionamide is an essential component of MDR-TB treatment in resource-limited regions but can cause gastrointestinal toxicity. A model extrapolating animal PK–PD parameters to humans predicted that a 25 mg daily dose of SMARt751 would allow a fourfold reduction in the dose of ethionamide while retaining the same efficacy and reducing side effects179, thus markedly improving the clinical utility of an exceptionally affordable TB drug180.
How can the seemingly well-supplied TB drug pipeline be leveraged to populate the very thin NTM-PD pipeline (Table 2)? An early but promising strategy relies on screening focused libraries of compounds that are active against Mtb, to validate both old and new TB targets in NTM181. This approach exploits chemical matter with established structure–activity relationships and desirable pharmacological and tolerability properties, thereby bypassing target deconvolution, reducing attrition and accelerating transition phases from hit to lead to proof-of-concept efficacy in vivo. Anti-TB compounds identified as promising, in vivo efficacious lead candidates for MAB-PD include inhibitors of MmpL3, F-ATP synthase, leucyl-tRNA synthetase, DNA gyrase, the DNA sliding clamp DnaN and the DosRS dormancy response regulator181. For example, SPR720 (fobrepodacin) inhibits the ATPase activity of the mycobacterial gyrase complex (Fig. 2b) and is in phase II trials for the treatment of MAC-PD. Such compounds might be attractive chemical starting points that overcome the notorious intrinsic drug resistance of NTM pathogens182 and have the potential to fast-track drug development and provide a sustainable source of preclinical candidates for the underpopulated NTM-PD pipeline.
Next generation drug discovery: targeted protein degradation
A fundamentally new modality recently entered the field of anti-mycobacterial drug discovery: targeted protein degradation (TPD)183. This approach is gaining interest across various disease areas owing to its potential to modulate proteins that have proved difficult to target with traditional small molecules, because either they lack active sites or their active sites are shallow and poorly druggable. The major class of molecules enabling TPD are known as proteolysis-targeting chimera (PROTAC) protein degraders. PROTACs are heterobifunctional molecules that consist of two chemically linked ligands, with one ligand binding to the protein of interest while the other binds to an E3 ubiquitin ligase. Dual binding induces ubiquitylation of the target protein and its subsequent degradation by the proteasome, after which the PROTAC molecule becomes available for further rounds of covalent modification and degradation of the target protein. This catalytic degradation mechanism of action distinguishes PROTACs from traditional inhibitors. However, bacteria do not harbour a mammalian E3 ligase–proteasome system homologue and thus this specific approach has so far been applied to non-infectious diseases only183. In an elegant structure- and biochemistry-driven approach, Clausen and colleagues184 achieved degradation of model proteins of interest in bacteria by ‘generation of proximity’ using bifunctional bridges (BacPROTACs) that directly link a target to the degradative bacterial caseinolytic protease complex, thus bypassing the E3 ligase requirement (Fig. 2c). Building on these advances, the authors generated BacPROTACs that inhibit growth and kill Mtb185, delivering proof of concept and a strategy for the rational design of degraders that target any bacterial protein and paving the way for the discovery of BacPROTAC antibiotics.
Preclinical models of Mtb and NTM infection
The BALB/c mouse models of acute and chronic TB have been the workhorses to evaluate drug efficacy and have delivered comprehensive databases with single-drug and regimen quantitative assessments186. The relapse mouse model (RMM) is an extension of the chronic model and is used to estimate the treatment duration required to achieve cure187. The C3HeB/FeJ mouse overcomes the lack of large necrotic lesions (one of the hallmarks of human pathology) in mice188. Larger animal models are required to reproduce cavitary TB: the rabbit model of active TB has proved a useful tool to quantify and model drug penetration at the site of disease189. Non-human primate models best recapitulate human disease progression and immunopathology190,191 but come with intrinsic ethical and resource considerations (Mtb is a BioSafety level 3 pathogen). Immunocompetent mice spontaneously and rapidly clear NTM pathogens. Numerous immunocompromised mouse models of MAB infection have been tested with mixed success192,193. Currently missing in the NTM drug discovery field are two reproducible and widely adopted mouse models of TB: one of acute infection that offers robust growth for ~7–10 days and another that presents a chronic phase of infection with stable bacterial burden kinetics for up to 28 days. Each model needs to be adapted for infection with MAB and MAC representative strains. Key features are lab-to-lab reproducibility, a wide dynamic range to differentiate the efficacy of single drugs or drug regimens and establish PK–PD correlates of efficacy, and systematic validation with standard-of-care therapies. The GM–CSF−/− mouse model of acute MAB infection194 and the C57Bl/6 mouse model of intratracheal inoculation with agar bead-embedded MAB appear to be the most promising acute and chronic candidates, respectively. However, much remains to be done to optimize and validate these models across laboratories.
Concepts and challenges of regimen development
It has taken more than 40 years to reduce the duration of TB treatment from 6 to 4 months32, underscoring the formidable barrier to shortening treatment duration and the need for innovation in regimen development programmes. On the positive side, the healthy and growing pipeline of anti-TB drug candidates offers vast opportunities to combine approved and clinical development agents to treat DS-TB or MDR-TB. But this triggers a new challenge: how should novel and existing drugs be rationally combined to bring forward the most promising, treatment-shortening regimens into clinical trials? And how can novel translational and computational approaches be leveraged to accelerate the progression of drug combinations through clinical development?
Prioritizing regimens
So far, the ranking of regimens heading for clinical trials has been driven by limited preclinical data and could be optimized195. In vitro and preclinical data generation is not a particular bottleneck. For example, a recent model-based meta-analysis of data for 17 unique regimens obtained from a total of 1,592 mice across 28 RMM studies generated robust and quantitative metrics of interest, such as the treatment duration required to achieve <10% and <50% relapse. This approach not only provides a framework to analyse emerging preclinical data in the context of historical data and aid in selecting drug combinations for clinical evaluation but has also been instrumental in refining the design of RMM studies to increase precision196. However, the major obstacle for successful regimen development is knowledge integration and quantification across in vitro, animal model and clinical platforms to identify drug combinations that shorten treatment. Algorithms have been developed that integrate efficacy in mouse models, drug penetration into major lesion compartments in larger animal models, drug potency against bacterial subpopulations at the sites of disease and host immunity. These models can simulate lesion sterilization197 and prioritize combinations predicted to accelerate cure187–189,196,198–201. Using an iterative strategy, researchers can identify specific features that predict treatment shortening and further refine computational tools. Although many more in vitro and preclinical models remain to be calibrated against clinical outcome, this translational approach holds promise to spare years of clinical development for new pan-TB drug regimens.
Optimizing treatment duration in clinical trials
Recent clinical trials and retrospective analyses increasingly demonstrate that treatment duration can be tailored to individual patients using risk stratification algorithms that integrate patient and pathogen characteristics202,203. For example, an algorithm in the form of a Risk Stratification Tool for Tuberculosis Clinical Trial Design that integrates HIV status, baseline bacterial burden in sputum, sex, baseline cavitary disease, body mass index and month 2 culture status can stratify TB patients in late-stage clinical trials into risk groups and inform the selection of optimal treatment duration for each group. In another example, a retrospective analysis showed that a subset of patients with DS-TB with less severe disease could be cured at 4 months. This analysis was applied to a prospectively designed trial (Predict TB) that is being used to identify this patient subset and validate the corresponding biomarkers. It is exploring quantitative radiological responses measured by positron emission tomography (PET)–CT combined with measurements of Mtb DNA in sputum (GeneXpert). If successful, this study could inform new treatment guidelines for low- versus high-risk patients, help to stratify patients in clinical trials and validate markers associated with durable cure to establish milestones for shorter regimens204. Applying these insights in practice might require further innovations in clinical trial design, such as more extensive use of multiple durations during phases II and III. Recent treatment-shortening successes for DS-TB and MDR-TB such as Study 31 (ref. 32), NIX-TB33, TB-PRACTECAL205, MDR-END206 and TRUNCATE-TB207 (Table 3) provide an invaluable collective clinical trial database to identify correlates of treatment shortening in patients, refine translational tools and optimally integrate exceptional science into clinical trial design.
Identifying regimens to minimize emergence of resistance
Clinical resistance to bedaquiline, delamanid and pretomanid, the latest additions to the TB drug arsenal, emerged more quickly than anticipated127,208 and appears to be on the rise. Developing models and computational tools to identify regimens that can revert this trend and pre-empt resistance acquisition to novel drug candidates is a key priority. The dynamics of emergence of resistance in patient lesions as a function of fluctuating drug concentrations in plasma and at the site of disease cannot be recapitulated in a test tube or even an animal model. Therefore, translational models require multiple biological assumptions, affecting model input certainty. Drug–drug interactions, fitness cost of resistance, suboptimal patient adherence owing to side effects and less predictable factors add to the complexity of the problem. For example, some of the clinical isolates resistant to bedaquiline pre-date its launch and are thought to have emerged in patients receiving clofazimine, which shares genetic determinants of resistance with bedaquiline127. Despite these hurdles, preliminary models have started to emerge209–211, but much remains to be done to select drug regimens that better protect novel clinical candidates from rapid acquisition of resistance212.
Optimizing clinical trial design
After decades of incremental changes in TB clinical trial design, radically innovative concepts are increasingly being adopted to accelerate the progression of drug combinations213,214. In addition, trials that incorporate these concepts tend to provide extensive patient samples taken throughout the course of treatment and follow-up, which are crucial to establish well-characterized biobanks for biomarker investigation and validation.
Biomarkers of long-term outcome and relapse.
Biomarkers are objective characteristics that indicate a pharmacological response to a therapeutic intervention and can be surrogate end points in short clinical trials with few patients, where they can predict clinically meaningful events and accelerate clinical research215. In TB drug development, relapse-free cure is the primary end point and requires follow-up for at least 12 months and ideally 24 months after the end of therapy. Predictors of relapse-free cure that can be quantified during the early phases of treatment are needed to improve the speed and quality of new regimen evaluation, in both TB and NTM infection. The need for rapid and predictive markers is particularly acute to ensure the success of adaptive design trials. Unfortunately, validation in prospectively designed clinical trials has not kept up with scientific and technological advances, despite applications becoming commercially available for some markers. To date, only sputum culture conversion at 2 months is considered a reasonably predictive surrogate marker of relapse-free cure that could inform on treatment duration216,217. Although this was a matter of debate until recently218,219, empirical meta-regression models suggest that early bacteriological results can predict relapse and have been prospectively validated against the outcome of three large randomized controlled trials in DS-TB203. Promising novel biomarkers of treatment response, cure and relapse risk (Box 3) are being investigated in prospective clinical trials20. Biomarkers acquire their true value only in the context of clinical pharmacology, defining crucial relationships between drug exposure and effect (or toxicity), so it is important to engage multidisciplinary teams with expertise in clinical pharmacology, pharmacometrics and mycobacteriology to collect and analyse longitudinal PK–PD data.
Box 3. Candidate biomarkers of treatment response and duration.
Quantitative sputum microbiology
In addition to 2-month culture conversion, the slope of decline in sputum bacterial burden and the time to negative sputum conversion are the most common bacteriological end points. Bacterial burden in expectorated sputum is quantified daily until viable bacteria are no longer detected. These end points are included in most trials, providing a growing reference database to compare their respective predictive power as surrogates of treatment response258.
Molecular surrogates of bacterial burden
The widely deployed GeneXpert assay amplifies DNA from both live and dead bacteria in clinical sputum samples. Although it has been extensively validated as a diagnostic test, prediction of long-term treatment will require adaptation to distinguish viable from non-viable Mycobacterium tuberculosis (Mtb) bacilli. The tuberculosis (TB) molecular bacterial load assay (quantitative TB-MBLA) quantifies 16S RNA by reverse transcription–quantitative PCR (RT–qPCR) in sputum259. It predicts long-term unfavourable outcomes and identifies at-risk patients earlier than the conventional time to positivity (TTP) in liquid culture. Quantification of Mtb surface lipid lipoarabinomannan by enzyme-linked immunosorbent assay (ELISA) appears promising as a strong predictor of Mtb burden in sputum260,261. The remarkably short turnaround time of the assay is compatible with adaptive trial design to prioritize regimens in real time.
Quantifying persisters
Quantifying persisting bacterial populations is an attractive approach that assesses the effect of a drug on the metabolically less-active population. Limited clinical investigations suggest that this approach can help to predict treatment duration and relapse. Although not strictly speaking a measurement of persisting populations, TTP in liquid culture (the number of days required to detect growth after inoculation of liquid medium with sputum) is thought to better capture organisms in an altered metabolic state and predict sterilizing activity. Caution should be exercised in studies that include mixed patient populations with drug-sensitive and multidrug-resistant TB, owing to increased TTP associated with the fitness of drug-resistant isolates262. Culture-based methods with resuscitation-promoting factors improve the detection of otherwise non-culturable mycobacteria that become more frequent in sputum as treatment progresses263,264. This differentially culturable population is valuable to study persistence, but the validation of markers has been particularly challenging. The least advanced but potentially most exciting of the surrogate markers of drug-tolerant bacteria is the rRNA synthesis ratio265 or the abundance of pre-rRNA relative to mature rRNA, as a measurement of ongoing rRNA synthesis and metabolic activity of the bacterial cell. This ratio distinguished between sterilizing and non-sterilizing drugs during early treatment phases in mice and in a limited patient cohort.
Host response
Positron emission tomography–computed tomography combined with early bactericidal activity (EBA) might be a better predictor of the clinical response at 6 months than EBA alone and could improve new TB drug evaluation before phase III clinical trials. It is also a valuable tool to visualize the heterogeneity of drug response at the lesion level, capture sterilizing activity and better understand the respective contribution of various antibiotic classes during early clinical evaluation30. However, cost and repeated radiation doses preclude its use as a biomarker in global public health.
Dose finding and optimization.
Historically, 14-day trials of monotherapy that measured EBA in sputum were used as a dose-finding mechanism but failed to detect the activity of drugs that now play a key role in TB treatment, such as pyrazinamide, bedaquiline and linezolid. Although it is economical, the major limitations of EBA are the short treatment duration, small sample size and that positive results are neither sufficient nor necessary for progression. To improve the predictive value of EBA, PK–PD readouts, novel culture-based techniques and molecular assays have been added to discriminate different patterns of response. Although several EBA trials are in progress to measure the dose–response relationships of novel drug candidates administered alone (Table 2), alternative dose-finding trial designs are gaining momentum. The DprE1 inhibitor OPC-167832 is being tested in combination with bedaquiline and delamanid to identify the tolerated dose that maximizes the proportion of patients achieving sputum culture conversion at predefined time points. Similarly, dose optimization of sutezolid is being investigated in combination with bedaquiline, delamanid and moxifloxacin in the SUDOCU study. In opti-Q Study 32, levofloxacin is being added to an optimized background regimen at increasing doses to identify the minimum exposure and corresponding dose that achieves the shortest time to sputum culture conversion in patients with MDR-TB220. RIFASHORT221 and other high-dose rifampicin trials aim to optimize the rifampicin or rifapentine dose and exposure, within various regimens, to achieve the greatest reduction in Mtb burden or enable treatment shortening. In ZeNIX, investigators identified the lowest efficacious dose of linezolid combined with bedaquiline and pretomanid90 to address the frequent adverse events observed in NIX-TB that led to linezolid discontinuation. This dose-finding strategy integrates drug–drug interactions, tolerability, safety and efficacy in the context of a relevant regimen.
Regimen comparison using conventional non-inferiority designs.
Several landmark non-inferiority trials were recently completed, delivering treatment-shortening regimens for both DS-TB and MDR-TB (Table 3). Under such design, investigational and control regimens, treatment duration and non-inferiority margins are set up-front. Realistic timelines can become crucial in situations in which an investigational drug or regimen is approved and recommended by the WHO while the trial is in progress, as seen in the NExT trial, which was interrupted prematurely when bedaquiline-based therapy became the standard of care in South Africa222. Longitudinal statistical modelling of quantitative bacteriology (time to positivity in liquid culture, or time-to-culture conversion data) are increasingly adopted by investigators, offering economy in trial designs with reduced sample sizes compared with the binary 2-month sputum culture readout. This conventional design will likely remain the phase III gold standard until biomarkers that can reliably predict long-term outcome are validated in large phase III or IIb registration studies. Optimistically, it could be another decade until a biomarker is approved by regulatory authorities and deployed.
Regimen comparison with adaptive trial designs.
Adaptive development pathways offer the opportunity to test a broader range of combinations and durations without prohibitively increasing the number of patients enrolled and are key for contemporary TB drug development. Multi-arm multistage (MAMS) parallel group trials, developed in oncology, evaluate several regimens with the objective of rapidly identifying regimens with insufficient evidence of benefit according to prespecified criteria (Fig. 4a). In Bayesian response adaptive randomization (BAR) phase IIc, the efficacy of each experimental arm is estimated weekly, and randomization probabilities are weighted in favour of better-performing arms. In fully seamless phase II/III or phase IIb/c designs, adaptive evaluation of regimens in the first stage is followed in the second stage by enrichment of the successful arms with additional participants to achieve appropriate power for comparisons on long-term outcomes (Fig. 4b). The first such trial for TB is recruiting participants to assess the efficacy, safety, optimal duration and PK of regimens containing bedaquiline, OPC-167832 and sutezolid, plus either pretomanid (PBOS combination) or delamanid (DBOS) for 4 months, in adults with pulmonary TB (NCT05971602). The most successful of the two regimens will be assessed in the second stage to identify the shortest treatment duration that delivers an acceptable outcome. Trial simulations of these major adaptive strategies for TB indicate that the MAMS design can afford a reduction in patient enrolment of up to 25%, and seamless phase IIb/Iic development pathways can reduce trial duration by 2–3 years223. So far, only the sequential MAMS design has been successfully adapted to the context of TB216,224, although variations of the seamless MAMS designs are in development. Given the turnaround time of validated microbiological readouts, BAR designs remain challenging for TB but a worthy goal considering there have been promising results with non-culture-based microbiological markers225.
Fig. 4 |. Emerging adaptive trial designs for TB.

a, In the classic multi-arm multistage (MAMS) design, several novel regimens are tested in parallel in small cohorts and enrolment is stopped for regimens with insufficient evidence of benefit, according to prespecified criteria251. In a variation of the MAMS design, ultrashort treatment arms (currently 8 weeks) are coupled with a stop-treatment-and-watch strategy to extend treatment of persistent disease or safely re-treat patients whose disease has relapsed224. b, Examples of seamless designs adapted to the needs of tuberculosis (TB) trials. Novel regimens are compared side by side with the standard of care (control), leading to go/no go decisions and if appropriate, the selection of best performing new combination for the next stage. In stage 2, either enrolment is continued for the best performing regimens (top) or different durations of one selected regimen are tested to identify the shortest treatment duration that delivers non-inferiority results (bottom), with a 12-month follow-up period.
The recent success of the TRUNCATE-TB trial illustrates the power of an unprecedented strategy consisting of ultrashort treatment arms coupled with a stop-treatment-and-watch algorithm (Fig. 4a). Four 8-week regimens were tested initially and extended in cases of persistent clinical disease. During a 96-week follow-up period, patients whose disease relapsed were promptly retreated with adequate regimens. The bedaquiline and linezolid-containing regimen was non-inferior to standard treatment and reduced overall treatment duration without safety concerns. The trial set an important ethical precedent for safely stopping treatment with ultrashort regimens and was generally accepted by investigators and participants. Importantly, the design allows for a failure rate compatible with the validation of biomarkers of treatment response and predictors of relapse, without compromising sustained cure of the participants. Inspired by TRUNCATE-TB, A5409 is a planned randomized, adaptive, dose-ranging trial of novel regimens for TB (RAD-TB) that uses sophisticated model-based approaches to select multidrug combinations and allows regimens to be added during the study. It will compare early efficacy (by liquid culture time to positivity) and safety of various new drug combinations and dosages. Sponsored by the CDC, the trial allows inclusion of new chemical entities based on preclinical data and optimizes the link from phase IIc to phase III226.
Challenges for NTM clinical regimens
In contrast to the clinical TB pipeline, only a few drugs — mostly repurposed — are currently being evaluated in clinical trials for NTM-PD: liposomal amikacin, clofazimine, omadacycline, epetraborole and SPR720, with only the latter two being new chemical entities (Tables 2 and 3). The thin drug pipeline is partially the result of poor incentives to invest in antibacterial drug discovery, which is further exacerbated by the fact that NTM disease burden is split across several mycobacterial species, leading to the perception that incidence and prevalence are relatively low. Despite the fragmented nature of NTM-PD across multiple pathogen species, the collective NTM-PD disease burden constitutes a large population of patients who are left without reliable therapeutic options. Although consensus definitions exist for microbiological cure and relapse, standardized end points that correlate with durable cure have not been defined in NTM-PD227. It is likely that a composite of established microbiological and radiographical readouts, combined with novel patient-reported outcomes (PROs)228 are required to comprehensively evaluate treatment response and predict long-term outcome. Standardized PROs are under development as measurements of treatment performance for NTM-PD, inspired by oncology and other disease areas229. Frequent comorbidities complicate the design of trials that rely on homogeneous patient cohorts. Given these limitations and the paucity of prospective clinical trial data for NTM therapeutics, systematic EBA trials combined with rich sputum sampling and modern readouts might be required to assert the clinical utility of drug candidates, define PK–PD relationships and clinical breakpoints of standard-of-care antibiotics, and evaluate correlates of treatment response. Conventional EBA trials for TB are 14-day dose-finding studies performed with small cohorts of treatment-naive patients, to follow the daily reduction in sputum bacterial burden. However, for NTM-PD, ‘extended EBA’ trials (Table 2) or experimental multidrug therapy are subject to less-stringent ethical hurdles because it is treated with underperforming agents and the decision to initiate treatment is more flexible than for TB given that it is rarely transmitted between humans230. This could be an economical and expeditious strategy to generate efficacy benchmarks, optimize the design of fully fledged clinical trials and revisit standard-of-care assumptions.
Outlook
In 2013, Zumla, Nahid and Cole231 ended their review on TB stating, “There is an urgent need for increased coordination and enhanced collaboration among drug developers, funding agencies and clinical trial networks… to develop shorter, more effective TB treatment regimens for the therapy of both DS-TB and drug-resistant TB and can be progressed more quickly using newer clinical trial designs guided by more specific biomarkers.” Encouragingly, much of this has occurred or has been successfully initiated over the past 10 years (Boxes 2 and 3) for TB. Multiple trials are evaluating novel agents, repurposed agents, adjunctive host-directed therapies and novel treatment strategies that will increase the probability of successful future clinical trials. We expect that these amazing advances will deliver dramatically shorter therapy duration for DS-TB and drug-resistant TB within the next 10 years.
Yet, the development of TB treatment regimens has been slow, often hindered by poor preclinical to clinical translation. There is a need for validated in vitro and preclinical models to prioritize treatment-shortening therapies while minimizing resistance; translational tools to identify combinations that minimize adverse events and drug–drug interactions; and biomarkers that are predictive of relapse-free cure including non-sputum-based tests. To extend advances in TB therapeutics to those who need them the most, clinical trials that include under-represented paediatrics, pregnant women and populations with TB–HIV are needed. The road ahead is longer for NTM-PD, but now is a great time to seize cross-fertilizing opportunities and apply lessons learned from TB drug discovery and development, while being mindful of notable disease differences. Exploitation of the fast-growing TB knowledge and successful implementation of similar partnerships and clinical trial networks by the NTM community could spare many years in developing curative regimens for NTM lung diseases.
Progress remains constrained by the overall level of investment in TB, less than half the global target of US$ 2 billion per year that was set for the period 2018–2022 at the first UN high-level meeting on TB1. The total falls even further short of the estimated requirement of the Stop TB Partnership’s Global Plan to End TB, 2023–2030, which is $5 billion per year. Considering the pace at which new drugs and vaccines have been developed and deployed against COVID, we should be more ambitious and raise expectations for TB and NTM-PD drug development.
Acknowledgements
The authors thank A. Diacon for stimulating discussions on present and future biomarkers of TB drug response. T.D. and V.D. receive support from the National Institute of Allergy and Infectious Diseases of the NIH, Award Numbers R01AI132374 and U19 AI142731, and from the Bill & Melinda Gates Foundation, award number INV-004704, respectively. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Glossary
- Clinical breakpoint
The minimum inhibitory concentration above which an antimicrobial agent is considered to have a low probability of treatment success in the clinic.
- Minimum inhibitory concentration (MIC)
The lowest concentration that inhibits bacterial growth in vitro.
- Non-inferiority trials
Designed to show that the effect of a new treatment is not worse than that of an active control by more than a specified margin.
- Pharmacokinetic–pharmacodynamic (PK–PD)
The relationship between concentrations achieved in the body or in the tissue of interest (PK) and the concentrations required to exert antibacterial effect (PD).
- Probability of target attainment (PTA)
PTA analysis evaluates the plasma exposure of an antibiotic in a patient population (pharmacokinetics) against a target exposure required for efficacy and calculates the likelihood of achieving a specific pharmacokinetic–pharmacodynamic criterion (‘target′) expressed relative to the minimum inhibitory concentration for a pathogen in that patient population.
- Sputum culture conversion
Conversion of sputum from which Mycobacterium tuberculosis 9MTB0 can be grown under standardized condition to sputum from which no M. tuberculosis can be cultured. This conversion from positive to negative is the best way to determine whether a patient is responding to treatment. Spontaneous conversion was seen in a fraction of patients during the pre-antibiotic era.
Footnotes
Competing interests
The authors declare no competing interests.
Related links
ClinicalTrials.gov: https://clinicaltrials.gov
MARK-TB: https://www.mark-tb.org/
Risk Stratification Tool for Tuberculosis Clinical Trial Design: http://saviclab.org/tb-risk/
STOP-TB partnership: https://www.stoptb.org/
The Critical Path to TB Drug Regimen: https://c-path.org/programs/cptr/
The European Accelerator of Tuberculosis Regime Project: https://era4tb.org/
The Project to Accelerate New Treatments for Tuberculosis: https://fnih.org/our-programs/project-accelerate-new-treatments-tuberculosis-pan-tb
The TB Drug Accelerator: https://www.tbdrugaccelerator.org/
Working Group on New Drugs: https://www.newtbdrugs.org/
References
- 1.WHO. Global Tuberculosis Report https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2022 (Geneva, 2022). [Google Scholar]
- 2.Pai M, Kasaeva T & Swaminathan S Covid-19’s devastating effect on tuberculosis care – a path to recovery. N. Engl. J. Med 386, 1490–1493 (2022). [DOI] [PubMed] [Google Scholar]
- 3.Marais BJ, Hesseling AC & Cotton MF Poverty and tuberculosis: is it truly a simple inverse linear correlation? Eur. Respir. J 33, 943–944 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Dartois VA & Rubin EJ Anti-tuberculosis treatment strategies and drug development: challenges and priorities. Nat. Rev. Microbiol 20, 685–701 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.WHO. Global Tuberculosis Report https://www.who.int/publications/i/item/9789240037021(Geneva, 2021). [Google Scholar]
- 6.WHO. WHO Consolidated Guidelines on Tuberculosis. Module 4: Drug-resistant Tuberculosis Treatment https://www.who.int/publications/i/item/9789240007048 (Geneva, 2020). [Google Scholar]
- 7.Motta I et al. Recent advances in the treatment of tuberculosis. Clin. Microbiol. Infect 10.1016/j.cmi.2023.07.013 (2023). [DOI] [PubMed] [Google Scholar]
- 8.Evangelopoulos D & McHugh TD Improving the tuberculosis drug development pipeline. Chem. Biol. Drug Des 86, 951–960 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Cowman S, van Ingen J, Griffith DE & Loebinger MR Non-tuberculous mycobacterial pulmonary disease. Eur. Respir. J 54, 1900250 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Winthrop KL et al. Incidence and prevalence of nontuberculous mycobacterial lung disease in a large U.S. managed care health plan, 2008–2015. Ann. Am. Thorac. Soc 17, 178–185 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prevots DR & Marras TK Epidemiology of human pulmonary infection with nontuberculous mycobacteria: a review. Clin. Chest Med 36, 13–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raju RM, Raju SM, Zhao Y & Rubin EJ Leveraging advances in tuberculosis diagnosis and treatment to address nontuberculous mycobacterial disease. Emerg. Infect. Dis 22, 365–369 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sawka A & Burke A Medications and monitoring in treatment of nontuberculous mycobacteria lung disease. Clin. Chest Med 44, 815–828 (2023). [DOI] [PubMed] [Google Scholar]
- 14.Daley CL et al. Treatment of nontuberculous mycobacterial pulmonary disease: an official ATS/ERS/ESCMID/IDSA clinical practice guideline. Clin. Infect. Dis 71, 905–913 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; A comprehensive review by US and European physicians of evidence-based recommendations for the treatment of NTM-PD.
- 15.Egorova A, Jackson M, Gavrilyuk V & Makarov V Pipeline of anti-Mycobacterium abscessus small molecules: repurposable drugs and promising novel chemical entities. Med. Res. Rev 41, 2350–2387 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; A comprehensive review of the clinical and preclinical pipeline for MAB-PD, by mechanism of action and target.
- 16.Johansen MD, Herrmann JL & Kremer L Non-tuberculous mycobacteria and the rise of Mycobacterium abscessus. Nat. Rev. Microbiol 18, 392–407 (2020). [DOI] [PubMed] [Google Scholar]; A review covering the biology, virulence factors, host interactions and drug resistance mechanisms of M. abscessus, one of the most antibiotic-resistant mycobacteria.
- 17.Griffith DE & Aksamit TR Understanding nontuberculous mycobacterial lung disease: it’s been a long time coming. F1000Res 5, 2797 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Young C, Walzl G & Du Plessis N Therapeutic host-directed strategies to improve outcome in tuberculosis. Mucosal Immunol 13, 190–204 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tiwari D & Martineau AR Inflammation-mediated tissue damage in pulmonary tuberculosis and host-directed therapeutic strategies. Semin. Immunol 65, 101672 (2022). [DOI] [PubMed] [Google Scholar]
- 20.Lee A, Xie YL, Barry CE & Chen RY Current and future treatments for tuberculosis. BMJ 368, m216 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Anidi IU & Olivier KN Host-directed therapy in nontuberculous mycobacterial pulmonary disease: preclinical and clinical data review. Clin. Chest Med 44, 839–845 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hatfull GF Phage therapy for nontuberculous mycobacteria: challenges and opportunities. Pulm. Ther 9, 91–107 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sterling TR et al. Guidelines for the treatment of latent tuberculosis infection: recommendations from the National Tuberculosis Controllers Association and CDC, 2020. MMWR Recomm. Rep 69, 1–11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar K & Loebinger MR Nontuberculous mycobacterial pulmonary disease: clinical epidemiologic features, risk factors, and diagnosis: the nontuberculous mycobacterial series. Chest 161, 637–646 (2022). [DOI] [PubMed] [Google Scholar]
- 25.Sharma SK, Mohan A & Kohli M Extrapulmonary tuberculosis. Expert. Rev. Respir. Med 15, 931–948 (2021). [DOI] [PubMed] [Google Scholar]
- 26.Shih DC et al. Extrapulmonary nontuberculous mycobacterial disease surveillance – Oregon, 2014–2016. MMWR Morb. Mortal. Wkly Rep 67, 854–857 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilkinson RJ et al. Tuberculous meningitis. Nat. Rev. Neurol 13, 581–598 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Ganchua SKC, White AG, Klein EC & Flynn JL Lymph nodes — the neglected battlefield in tuberculosis. PLoS Pathog. 16, e1008632 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin PL et al. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat. Med 20, 75–79 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie YL et al. Fourteen-day PET/CT imaging to monitor drug combination activity in treated individuals with tuberculosis. Sci. Transl. Med 13, eabd7618 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu J, Liu YJ & Fortune SM Spatiotemporal perspectives on tuberculosis chemotherapy. Curr. Opin. Microbiol 72, 102266 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dorman SE et al. Four-month rifapentine regimens with or without moxifloxacin for tuberculosis. N. Engl. J. Med 384, 1705–1718 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; The first successful trial in more than four decades of treatment shortening in patients with DS-TB, reducing therapy duration from 6 to 4 months.
- 33.Conradie F et al. Treatment of highly drug-resistant pulmonary tuberculosis. N. Engl. J. Med 382, 893–902 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; A landmark clinical trial that successfully reduced treatment duration from 18–24 months to 6 months for patients with drug-resistant TB, showing the power of novel mechanisms of action.
- 34.Dahl VN et al. Global trends of pulmonary infections with nontuberculous mycobacteria: a systematic review. Int. J. Infect. Dis 125, 120–131 (2022). [DOI] [PubMed] [Google Scholar]
- 35.Honda JR, Bernhard JN & Chan ED Natural disasters and nontuberculous mycobacteria: a recipe for increased disease? Chest 147, 304–308 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mirsaeidi M & Sadikot RT Gender susceptibility to mycobacterial infections in patients with non-CF bronchiectasis. Int. J. Mycobacteriol 4, 92–96 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Andrejak C et al. Chronic respiratory disease, inhaled corticosteroids and risk of non-tuberculous mycobacteriosis. Thorax 68, 256–262 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Chan ED & Iseman MD Underlying host risk factors for nontuberculous mycobacterial lung disease. Semin. Respir. Crit. Care Med 34, 110–123 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Abidin NZ et al. Trends in nontuberculous mycobacteria infection in children and young people with cystic fibrosis. J. Cyst. Fibros 20, 737–741 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brugha R & Spencer H Mycobacterium abscessus in cystic fibrosis. Science 372, 465–466 (2021). [DOI] [PubMed] [Google Scholar]
- 41.Honda JR, Virdi R & Chan ED Global environmental nontuberculous mycobacteria and their contemporaneous man-made and natural niches. Front. Microbiol 9, 2029 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim DH et al. In vitro activity and clinical outcomes of clofazimine for nontuberculous mycobacteria pulmonary disease. J. Clin. Med 10, 4581 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pfaeffle HOI et al. Clofazimine for treatment of multidrug-resistant non-tuberculous mycobacteria. Pulm. Pharmacol. Ther 70, 102058 (2021). [DOI] [PubMed] [Google Scholar]
- 44.Holt MR & Baird T Treatment approaches to Mycobacterium abscessus pulmonary disease. Clin. Chest Med 44, 785–798 (2023). [DOI] [PubMed] [Google Scholar]; A concise but complete review of treatment guidelines, side effects, novel and repurposed therapeutic options for MAB-PD.
- 45.van Ingen J, Boeree MJ, van Soolingen D & Mouton JW Resistance mechanisms and drug susceptibility testing of nontuberculous mycobacteria. Drug Resist. Updat 15, 149–161 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Falkinham JO III Ecology of nontuberculous mycobacteria — where do human infections come from? Semin. Respir. Crit. Care Med 34, 95–102 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Fennelly KP et al. Biofilm formation by Mycobacterium abscessus in a lung cavity. Am. J. Respir. Crit. Care Med 193, 692–693 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Henkle E & Winthrop KL Nontuberculous mycobacteria infections in immunosuppressed hosts. Clin. Chest Med 36, 91–99 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Connell ML et al. Lung manifestations in an autopsy-based series of pulmonary or disseminated nontuberculous mycobacterial disease. Chest 141, 1203–1209 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klein JL, Corbett EL, Slade PM, Miller RF & Coker RJ Mycobacterium kansasii and human immunodeficiency virus co-infection in London. J. Infect 37, 252–259 (1998). [DOI] [PubMed] [Google Scholar]
- 51.Tomashefski JF, Stern RC, Demko CA & Doershuk CF Nontuberculous mycobacteria in cystic fibrosis. An autopsy study. Am. J. Respir. Crit. Care Med 154, 523–528 (1996). [DOI] [PubMed] [Google Scholar]
- 52.Swenson C, Zerbe CS & Fennelly K Host variability in NTM disease: implications for research needs. Front. Microbiol 9, 2901 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koh WJ, Hong G, Kim K, Ahn S & Han J Pulmonary sequestration infected with nontuberculous mycobacteria: a report of two cases and literature review. Asian Pac. J. Trop. Med 5, 917–919 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Merckx JJ, Soule EH & Karlson AG The histopathology of lesions caused by infection with unclassified acid-fast bacteria in man. Report of 25 cases. Am. J. Clin. Pathol 41, 244–255 (1964). [DOI] [PubMed] [Google Scholar]
- 55.Yuan MK et al. Comparative chest computed tomography findings of non-tuberculous mycobacterial lung diseases and pulmonary tuberculosis in patients with acid fast bacilli smear-positive sputum. BMC Pulm. Med 14, 65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oshitani Y et al. Characteristic chest CT findings for progressive cavities in Mycobacterium avium complex pulmonary disease: a retrospective cohort study. Respir. Res 21, 10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jeong YJ et al. Nontuberculous mycobacterial pulmonary infection in immunocompetent patients: comparison of thin-section CT and histopathologic findings. Radiology 231, 880–886 (2004). [DOI] [PubMed] [Google Scholar]
- 58.Kwon YS & Koh WJ Diagnosis of pulmonary tuberculosis and nontuberculous mycobacterial lung disease in Korea. Tuberc. Respir. Dis 77, 1–5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jain D, Ghosh S, Teixeira L & Mukhopadhyay S Pathology of pulmonary tuberculosis and non-tuberculous mycobacterial lung disease: facts, misconceptions, and practical tips for pathologists. Semin. Diagn. Pathol 34, 518–529 (2017). [DOI] [PubMed] [Google Scholar]; A review of the radiological and immunopathological similarities and differences between pulmonary tuberculosis and NTM lung disease.
- 60.Barry CE III et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat. Rev. Microbiol 7, 845–855 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Griffith DE et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am. J. Respir. Crit. Care Med 175, 367–416 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Jo KW, Park YE, Chong YP & Shim TS Spontaneous sputum conversion and reversion in Mycobacterium abscessus complex lung disease. PLoS ONE 15, e0232161 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim B, Yu JY & Jhun BW Spontaneous cultural conversion rate of Mycobacterium avium complex pulmonary disease based on baces severity. J. Clin. Med 12, 7125 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.National Tuberculosis Institute. Tuberculosis in a rural population of South India: a five-year epidemiological study. Bull. World Health Organ 51, 473–488 (1974). [PMC free article] [PubMed] [Google Scholar]
- 65.Mehra S et al. The DosR regulon modulates adaptive immunity and is essential for Mycobacterium tuberculosis persistence. Am. J. Respir. Crit. Care Med 191, 1185–1196 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gerasimova A, Kazakov AE, Arkin AP, Dubchak I & Gelfand MS Comparative genomics of the dormancy regulons in mycobacteria. J. Bacteriol 193, 3446–3452 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Belardinelli JM et al. Therapeutic efficacy of antimalarial drugs targeting DosRS signaling in Mycobacterium abscessus. Sci. Transl. Med 14, eabj3860 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Qvist T et al. Chronic pulmonary disease with Mycobacterium abscessus complex is a biofilm infection. Eur. Respir. J 46, 1823–1826 (2015). [DOI] [PubMed] [Google Scholar]
- 69.Mishra R et al. Mechanopathology of biofilm-like Mycobacterium tuberculosis cords. Cell 186, 5135–5150.e28 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ankomah P & Levin BR Exploring the collaboration between antibiotics and the immune response in the treatment of acute, self-limiting infections. Proc. Natl Acad. Sci. USA 111, 8331–8338 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brown-Elliott BA & Woods GL Antimycobacterial susceptibility testing of nontuberculous mycobacteria. J. Clin. Microbiol 57, e00834–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.>WHO. Rapid Communication: Key Changes to the Treatment of Drug-resistant Tuberculosis. https://www.who.int/publications/i/item/WHO-UCN-TB-2022-2 (2022).
- 73.Ji B et al. In vitro and in vivo activities of moxifloxacin and clinafloxacin against Mycobacterium tuberculosis. Antimicrob. Agents Chemother 42, 2066–2069 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zurenko GE et al. In vitro activities of U-100592 and U-100766, novel oxazolidinone antibacterial agents. Antimicrob. Agents Chemother 40, 839–845 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yun HY et al. Model-based efficacy and toxicity comparisons of moxifloxacin for multidrug-resistant tuberculosis. Open Forum Infect. Dis 9, ofab660 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alghamdi WA et al. Population pharmacokinetics of linezolid in tuberculosis patients: dosing regimen simulation and target attainment analysis. Antimicrob. Agents Chemother 64, e01174–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Leach KL, Brickner SJ, Noe MC & Miller PF Linezolid, the first oxazolidinone antibacterial agent. Ann. N. Y. Acad. Sci 1222, 49–54 (2011). [DOI] [PubMed] [Google Scholar]
- 78.Ahmad N et al. Treatment correlates of successful outcomes in pulmonary multidrug-resistant tuberculosis: an individual patient data meta-analysis. Lancet 392, 821–834 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Falzon D et al. Resistance to fluoroquinolones and second-line injectable drugs: impact on multidrug-resistant TB outcomes. Eur. Respir. J 42, 156–168 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dheda K et al. The Lancet Respiratory Medicine Commission: 2019 update: epidemiology, pathogenesis, transmission, diagnosis, and management of multidrug-resistant and incurable tuberculosis. Lancet Respir. Med 7, 820–826 (2019). [DOI] [PubMed] [Google Scholar]
- 81.Dreyer V et al. High fluoroquinolone resistance proportions among multidrug-resistant tuberculosis driven by dominant L2 Mycobacterium tuberculosis clones in the Mumbai Metropolitan Region. Genome Med. 14, 95 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Agrawal D, Udwadia ZF, Rodriguez C & Mehta A Increasing incidence of fluoroquinolone-resistant Mycobacterium tuberculosis in Mumbai, India. Int. J. Tuberc. Lung Dis 13, 79–83 (2009). [PubMed] [Google Scholar]
- 83.Mbelele PM et al. Whole genome sequencing-based drug resistance predictions of multidrug-resistant Mycobacterium tuberculosis isolates from Tanzania. JAC Antimicrob. Resist 4, dlac042 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ament PW, Jamshed N & Horne JP Linezolid: its role in the treatment of Gram-positive, drug-resistant bacterial infections. Am. Fam. Physician 65, 663–670 (2002). [PubMed] [Google Scholar]
- 85.Thwaites G & Nguyen NV Linezolid for drug-resistant tuberculosis. N. Engl. J. Med 387, 842–843 (2022). [DOI] [PubMed] [Google Scholar]
- 86.Tse-Chang A, Kunimoto D, Der E & Ahmed R Assessment of linezolid efficacy, safety and tolerability in the treatment of tuberculosis: a retrospective case review. Can. J. Infect. Dis. Med. Microbiol 24, 535616 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Van Rie A et al. Balancing access to BPaLM regimens and risk of resistance. Lancet Infect. Dis 22, 1411–1412 (2022). [DOI] [PubMed] [Google Scholar]
- 88.Kim JS et al. Early bactericidal activity of delpazolid (LCB01–0371) in patients with pulmonary tuberculosis. Antimicrob. Agents Chemother 66, e0168421 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wallis RS et al. Mycobactericidal activity of sutezolid (PNU-100480) in sputum (EBA) and blood (WBA) of patients with pulmonary tuberculosis. PLoS ONE 9, e94462 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Conradie F et al. Bedaquiline–pretomanid–linezolid regimens for drug-resistant tuberculosis. N. Engl. J. Med 387, 810–823 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wallace RJ Jr et al. Initial clarithromycin monotherapy for Mycobacterium avium–intracellulare complex lung disease. Am. J. Respir. Crit. Care Med 149, 1335–1341 (1994). [DOI] [PubMed] [Google Scholar]
- 92.Li G et al. Antimicrobial susceptibility of standard strains of nontuberculous mycobacteria by microplate Alamar Blue assay. PLoS ONE 8, e84065 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zheng H et al. In vitro activity of pretomanid against nontuberculous mycobacteria. Antimicrob. Agents Chemother 66, e0181021 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yu X et al. In vitro activities of bedaquiline and delamanid against nontuberculous mycobacteria isolated in Beijing, China. Antimicrob. Agents Chemother 63, e00031–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Heifets L, Higgins M & Simon B Pyrazinamide is not active against Mycobacterium tuberculosis residing in cultured human monocyte-derived macrophages. Int. J. Tuberc. Lung Dis 4, 491–495 (2000). [PubMed] [Google Scholar]
- 96.Reingewertz TH et al. Differential sensitivity of mycobacteria to isoniazid is related to differences in KatG-mediated enzymatic activation of the drug. Antimicrob. Agents Chemother 64, e01899–19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cowman S, Burns K, Benson S, Wilson R & Loebinger MR The antimicrobial susceptibility of non-tuberculous mycobacteria. J. Infect 72, 324–331 (2016). [DOI] [PubMed] [Google Scholar]
- 98.Kim DH et al. In vitro activity of bedaquiline and delamanid against nontuberculous mycobacteria, including macrolide-resistant clinical isolates. Antimicrob. Agents Chemother 63, e00665–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Boorgula GD et al. Isoniazid pharmacokinetics/pharmacodynamics as monotherapy and in combination regimen in the hollow fiber system model of Mycobacterium kansasii. Tuberculosis 138, 102289 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mudde SE, Upton AM, Lenaerts A, Bax HI & De Steenwinkel JEM Delamanid or pretomanid? A Solomonic judgement! J. Antimicrob. Chemother 77, 880–902 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vilcheze C & Jacobs WR Jr The isoniazid paradigm of killing, resistance, and persistence in Mycobacterium tuberculosis. J. Mol. Biol 431, 3450–3461 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gopal P, Gruber G, Dartois V & Dick T Pharmacological and molecular mechanisms behind the sterilizing activity of pyrazinamide. Trends Pharmacol. Sci 40, 930–940 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ushtanit A et al. Molecular determinants of ethionamide resistance in clinical isolates of Mycobacterium tuberculosis. Antibiotics 11, 133 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Guerrero C et al. Evaluation of the rpoB gene in rifampicin-susceptible and -resistant Mycobacterium avium and Mycobacterium intracellulare. J. Antimicrob. Chemother 33, 661–663 (1994). [DOI] [PubMed] [Google Scholar]
- 105.Moon SM et al. Relationship between resistance to ethambutol and rifampin and clinical outcomes in Mycobacterium avium complex pulmonary disease. Antimicrob. Agents Chemother 66, e0202721 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hombach M, Somoskovi A, Homke R, Ritter C & Bottger EC Drug susceptibility distributions in slowly growing non-tuberculous mycobacteria using MGIT 960 TB eXiST. Int. J. Med. Microbiol 303, 270–276 (2013). [DOI] [PubMed] [Google Scholar]
- 107.Schon T et al. Evaluation of wild-type MIC distributions as a tool for determination of clinical breakpoints for Mycobacterium tuberculosis. J. Antimicrob. Chemother 64, 786–793 (2009). [DOI] [PubMed] [Google Scholar]
- 108.Schildkraut JA et al. The role of rifampicin within the treatment of Mycobacterium avium pulmonary disease. Antimicrob. Agents Chemother 67, e0087423 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schon T & Chryssanthou E Minimum inhibitory concentration distributions for Mycobacterium avium complex — towards evidence-based susceptibility breakpoints. Int. J. Infect. Dis 55, 122–124 (2017). [DOI] [PubMed] [Google Scholar]
- 110.Andries K et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307, 223–227 (2005). [DOI] [PubMed] [Google Scholar]
- 111.Lounis N, Gevers T, Van den Berg J, Vranckx L & Andries K ATP synthase inhibition of Mycobacterium avium is not bactericidal. Antimicrob. Agents Chemother 53, 4927–4929 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ruth MM et al. A bedaquiline/clofazimine combination regimen might add activity to the treatment of clinically relevant non-tuberculous mycobacteria. J. Antimicrob. Chemother 74, 935–943 (2019). [DOI] [PubMed] [Google Scholar]
- 113.Lindman M & Dick T Bedaquiline eliminates bactericidal activity of β-lactams against Mycobacterium abscessus. Antimicrob. Agents Chemother 63, e00827–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Froberg G et al. Towards clinical breakpoints for non-tuberculous mycobacteria – determination of epidemiological cut off values for the Mycobacterium avium complex and Mycobacterium abscessus using broth microdilution. Clin. Microbiol. Infect 29, 758–764 (2023). [DOI] [PubMed] [Google Scholar]
- 115.Roemhild R, Bollenbach T & Andersson DI The physiology and genetics of bacterial responses to antibiotic combinations. Nat. Rev. Microbiol 20, 478–490 (2022). [DOI] [PubMed] [Google Scholar]
- 116.Gupta R et al. The Mycobacterium tuberculosis protein LdtMt2 is a nonclassical transpeptidase required for virulence and resistance to amoxicillin. Nat. Med 16, 466–469 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rifat D, Chen L, Kreiswirth BN & Nuermberger EL Genome-wide essentiality analysis of Mycobacterium abscessus by saturated transposon mutagenesis and deep sequencing. mBio 12, e0104921 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nguyen DC et al. “One-Two Punch”: synergistic ss-lactam combinations for Mycobacterium abscessus and target redundancy in the inhibition of peptidoglycan synthesis enzymes. Clin. Infect. Dis 73, 1532–1536 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Negatu DA, Zimmerman M, Dartois VA & Dick T Strongly bactericidal all-oral β-lactam combinations for the treatment of Mycobacterium abscessus lung disease. Antimicrob. Agents Chemother 66, e0079022 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lee M et al. Linezolid for treatment of chronic extensively drug-resistant tuberculosis. N. Engl. J. Med 367, 1508–1518 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ignatius EH & Dooley KE New drugs for the treatment of tuberculosis. Clin. Chest Med 40, 811–827 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cho YL & Jang J Development of delpazolid for the treatment of tuberculosis. Appl. Sci 10, 2211 (2020). [Google Scholar]
- 123.Negatu DA, Aragaw WW, Cangialosi J, Dartois V & Dick T Side-by-side profiling of oxazolidinones to estimate the therapeutic window against mycobacterial infections. Antimicrob. Agents Chemother 67, e0165522 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mdluli KC et al. TBI-223: a safer oxazolidinone in pre-clinical development for tuberculosis. In ASM Microbe 2017 (ASM, 2017). [Google Scholar]
- 125.McLeay SC, Vis P, van Heeswijk RP & Green B Population pharmacokinetics of bedaquiline (TMC207), a novel antituberculosis drug. Antimicrob. Agents Chemother 58, 5315–5324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Keutzer L, Akhondipour Salehabad Y, Davies Forsman L & Simonsson USH A modeling-based proposal for safe and efficacious reintroduction of bedaquiline after dose interruption: a population pharmacokinetics study. CPT Pharmacomet. Syst. Pharmacol 11, 628–639 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mallick JS, Nair P, Abbew ET, Van Deun A & Decroo T Acquired bedaquiline resistance during the treatment of drug-resistant tuberculosis: a systematic review. JAC Antimicrob. Resist 4, dlac029 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Brown TS et al. Genotype-phenotype characterization of serial Mycobacterium tuberculosis isolates in bedaquiline-resistant tuberculosis. Clin. Infect. Dis 10.1093/cid/ciad596 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sutherland HS et al. Variations in the C-unit of bedaquiline provides analogues with improved biology and pharmacology. Bioorg. Med. Chem 28, 115213 (2020). [DOI] [PubMed] [Google Scholar]
- 130.Briffotaux J, Huang W, Wang X & Gicquel B MmpS5/MmpL5 as an efflux pump in Mycobacterium species. Tuberculosis 107, 13–19 (2017). [DOI] [PubMed] [Google Scholar]
- 131.Xu J et al. Contribution of pretomanid to novel regimens containing bedaquiline with either linezolid or moxifloxacin and pyrazinamide in murine models of tuberculosis. Antimicrob. Agents Chemother 63, e00021–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Almeida D et al. Comparative efficacy of the novel diarylquinoline TBAJ-876 and bedaquiline against a resistant Rv0678 mutant in a mouse model of tuberculosis. Antimicrob. Agents Chemother 65, e0141221 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yao R et al. Sudapyridine (WX-081), a novel compound against Mycobacterium tuberculosis. Microbiol. Spectr 10, e0247721 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Huang Z et al. Discovery and preclinical profile of sudapyridine (WX-081), a novel anti-tuberculosis agent. Bioorg. Med. Chem. Lett 71, 128824 (2022). [DOI] [PubMed] [Google Scholar]
- 135.Sarathy JP et al. TBAJ-876, a 3,5-dialkoxypyridine analogue of bedaquiline, is active against Mycobacterium abscessus. Antimicrob. Agents Chemother 64, e02404–19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Di Modugno E et al. In vitro activity of the tribactam GV104326 against Gram-positive, Gram-negative, and anaerobic bacteria. Antimicrob. Agents Chemother 38, 2362–2368 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lee RE et al. Spectinamides: a new class of semisynthetic antituberculosis agents that overcome native drug efflux. Nat. Med 20, 152–158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Robertson GT et al. Spectinamides are effective partner agents for the treatment of tuberculosis in multiple mouse infection models. J. Antimicrob. Chemother 72, 770–777 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rominski A, Roditscheff A, Selchow P, Bottger EC & Sander P Intrinsic rifamycin resistance of Mycobacterium abscessus is mediated by ADP-ribosyltransferase MAB_0591. J. Antimicrob. Chemother 72, 376–384 (2017). [DOI] [PubMed] [Google Scholar]
- 140.Kumar K, Daley CL, Griffith DE & Loebinger MR Management of Mycobacterium avium complex and Mycobacterium abscessus pulmonary disease: therapeutic advances and emerging treatments. Eur. Respir. Rev 31, 210212 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Baysarowich J et al. Rifamycin antibiotic resistance by ADP-ribosylation: structure and diversity of Arr. Proc. Natl Acad. Sci. USA 105, 4886–4891 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ganapathy US et al. Blocking bacterial naphthohydroquinone oxidation and ADP-ribosylation improves activity of rifamycins against Mycobacterium abscessus. Antimicrob. Agents Chemother 65, e0097821 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Prideaux B et al. The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat. Med 21, 1223–1227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sarathy JP et al. Extreme drug tolerance of Mycobacterium tuberculosis in caseum. Antimicrob. Agents Chemother 62, e02266–e02317 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kolpen M et al. Biofilms of Mycobacterium abscessus complex can be sensitized to antibiotics by disaggregation and oxygenation. Antimicrob. Agents Chemother 64, e01212–19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lan T et al. Redesign of rifamycin antibiotics to overcome ADP-ribosylation-mediated resistance. Angew. Chem. Int. Ed. Engl 61, e202211498 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Paulowski L et al. C25-modified rifamycin derivatives with improved activity against Mycobacterium abscessus. PNAS Nexus 1, pgac130 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Payne DJ, Gwynn MN, Holmes DJ & Pompliano DL Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug. Discov 6, 29–40 (2007). [DOI] [PubMed] [Google Scholar]
- 149.Vincent F et al. Phenotypic drug discovery: recent successes, lessons learned and new directions. Nat. Rev. Drug. Discov 21, 899–914 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; A review discussing the successes and challenges of modern phenotypic — as opposed to target-based — drug discovery, which combines original concepts with modern tools and strategies.
- 150.Working Group on New TB Drugs. Clinical Pipeline https://www.newtbdrugs.org/pipeline/clinical (2023).
- 151.Working Group on New TB Drugs. 2023 Global New TB Drug Discovery Pipeline https://www.newtbdrugs.org/pipeline/discovery (2023).
- 152.Fernandes GFS, Thompson AM, Castagnolo D, Denny WA & Dos Santos JL Tuberculosis drug discovery: challenges and new horizons. J. Med. Chem 65, 7489–7531 (2022). [DOI] [PubMed] [Google Scholar]; A recent update of preclinical anti-TB compounds with in vivo efficacy against TB, and the global pipeline of drug candidates in clinical development, with a focus on mechanism of action.
- 153.Sethiya JP, Sowards MA, Jackson M & North EJ MmpL3 inhibition: a new approach to treat nontuberculous mycobacterial infections. Int. J. Mol. Sci 21, 6202 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sarathy JP, Zimmerman MD, Gengenbacher M, Dartois V & Dick T Mycobacterium tuberculosis DprE1 inhibitor OPC-167832 is active against Mycobacterium abscessus in vitro. Antimicrob. Agents Chemother 66, e0123722 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Li W et al. Direct inhibition of MmpL3 by novel antitubercular compounds. ACS Infect. Dis 5, 1001–1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rudraraju RS et al. Mycobacterium tuberculosis KasA as a drug target: structure-based inhibitor design. Front. Cell Infect. Microbiol 12, 1008213 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Fang C et al. Discovery of heterocyclic replacements for the coumarin core of anti-tubercular FadD32 inhibitors. Bioorg. Med. Chem. Lett 28, 3529–3533 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Aggarwal A et al. Development of a novel lead that targets M. tuberculosis polyketide synthase 13. Cell 170, 249–259 e225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hugonnet JE, Tremblay LW, Boshoff HI, Barry CE III & Blanchard JS Meropenem–clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science 323, 1215–1218 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Vilchèze C Mycobacterial cell wall: a source of successful targets for old and new drugs. Appl. Sci 10, 2278 (2020). [Google Scholar]
- 161.Li S et al. CRISPRi chemical genetics and comparative genomics identify genes mediating drug potency in Mycobacterium tuberculosis. Nat. Microbiol 7, 766–779 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; A research article that describes the application of CRISPR interference to bacterial gene silencing to discover mechanisms of intrinsic and acquired resistance and targets for synergistic drug combinations.
- 162.Beites T et al. Plasticity of the Mycobacterium tuberculosis respiratory chain and its impact on tuberculosis drug development. Nat. Commun 10, 4970 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jeffreys LN et al. Identification of 2-aryl-quinolone inhibitors of cytochrome bd and chemical validation of combination strategies for respiratory inhibitors against Mycobacterium tuberculosis. ACS Infect. Dis 9, 221–238 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lee BS et al. Dual inhibition of the terminal oxidases eradicates antibiotic-tolerant Mycobacterium tuberculosis. EMBO Mol. Med 13, e13207 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Singh R et al. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 322, 1392–1395 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kwon NH, Fox PL & Kim S Aminoacyl-tRNA synthetases as therapeutic targets. Nat. Rev. Drug. Discov 18, 629–650 (2019). [DOI] [PubMed] [Google Scholar]
- 167.Li X et al. Discovery of a potent and specific M. tuberculosis leucyl-tRNA synthetase inhibitor: (S)-3-(aminomethyl)-4-chloro-7-(2-hydroxyethoxy)benzo[c][1,2]oxaborol-1(3H)-ol (GSK656). J. Med. Chem 60, 8011–8026 (2017). [DOI] [PubMed] [Google Scholar]
- 168.Green SR et al. Lysyl-tRNA synthetase, a target for urgently needed M. tuberculosis drugs. Nat. Commun 13, 5992 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Abrahams KA et al. Inhibiting mycobacterial tryptophan synthase by targeting the inter-subunit interface. Sci. Rep 7, 9430 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Brown KL et al. Cyclic AMP-mediated inhibition of cholesterol catabolism in Mycobacterium tuberculosis by the novel drug candidate GSK2556286. Antimicrob. Agents Chemother 67, e0129422 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kim MJ et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol. Med 2, 258–274 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Guerrini V et al. Storage lipid studies in tuberculosis reveal that foam cell biogenesis is disease-specific. PLoS Pathog. 14, e1007223 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Nuermberger EL et al. GSK2556286 is a novel antitubercular drug candidate effective in vivo with the potential to shorten tuberculosis treatment. Antimicrob. Agents Chemother 66, e0013222 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lupoli TJ, Vaubourgeix J, Burns-Huang K & Gold B Targeting the proteostasis network for mycobacterial drug discovery. ACS Infect. Dis 4, 478–498 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bhandari V et al. The role of ClpP protease in bacterial pathogenesis and human diseases. ACS Chem. Biol 13, 1413–1425 (2018). [DOI] [PubMed] [Google Scholar]
- 176.Hawkins PME et al. Potent bactericidal antimycobacterials targeting the chaperone ClpC1 based on the depsipeptide natural products ecumicin and ohmyungsamycin A. J. Med. Chem 65, 4893–4908 (2022). [DOI] [PubMed] [Google Scholar]
- 177.Maitre T, Baulard A, Aubry A & Veziris N Optimizing the use of current antituberculosis drugs to overcome drug resistance in Mycobacterium tuberculosis. Infect. Dis. Now 54, 104807 (2023). [DOI] [PubMed] [Google Scholar]
- 178.Blondiaux N et al. Reversion of antibiotic resistance in Mycobacterium tuberculosis by spiroisoxazoline SMARt-420. Science 355, 1206–1211 (2017). [DOI] [PubMed] [Google Scholar]
- 179.Flipo M et al. The small-molecule SMARt751 reverses Mycobacterium tuberculosis resistance to ethionamide in acute and chronic mouse models of tuberculosis. Sci. Transl. Med 14, eaaz6280 (2022). [DOI] [PubMed] [Google Scholar]
- 180.Rubin EJ Reviving a drug for tuberculosis? N. Engl. J. Med 376, 2292–2294 (2017). [DOI] [PubMed] [Google Scholar]
- 181.Ganapathy US & Dick T Why matter matters: fast-tracking Mycobacterium abscessus drug discovery. Molecules 27, 6948 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; A survey of anti-TB agents that have shown in vivo efficacy in MAB mouse models, showing the value of screening advanced TB chemical matter as a means of fast-tracking MAB drug discovery and highlighting the dire state of the pipeline.
- 182.Chong SL, Tan JL & Ngeow YF The resistomes of Mycobacteroides abscessus complex and their possible acquisition from horizontal gene transfer. BMC Genomics 23, 715 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bekes M, Langley DR & Crews CM PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug. Discov 21, 181–200 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Morreale FE et al. BacPROTACs mediate targeted protein degradation in bacteria. Cell 185, 2338–2353 e2318 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hoi DM et al. Clp-targeting BacPROTACs impair mycobacterial proteostasis and survival. Cell 186, 2176–2192.e22 (2023). [DOI] [PubMed] [Google Scholar]
- 186.Nuermberger EL Preclinical efficacy testing of new drug candidates. Microbiol. Spectr 5, 10.1128/microbiolspec.TBTB2-0034-2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tasneen R et al. Novel regimens of bedaquiline-pyrazinamide combined with moxifloxacin, rifabutin, delamanid and/or OPC-167832 in murine tuberculosis models. Antimicrob. Agents Chemother 66, e0239821 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Irwin SM et al. Presence of multiple lesion types with vastly different microenvironments in C3HeB/FeJ mice following aerosol infection with Mycobacterium tuberculosis. Dis. Model. Mech 8, 591–602 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Ernest JP et al. Development of new tuberculosis drugs: translation to regimen composition for drug-sensitive and multidrug-resistant tuberculosis. Annu. Rev. Pharmacol. Toxicol 61, 495–516 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Via LE et al. A sterilizing tuberculosis treatment regimen is associated with faster clearance of bacteria in cavitary lesions in marmosets. Antimicrob. Agents Chemother 59, 4181–4189 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lin PL et al. Radiologic responses in cynomolgous macaques for assessing tuberculosis chemotherapy regimens. Antimicrob. Agents Chemother 57, 4237–4244 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Obregon-Henao A et al. Susceptibility of Mycobacterium abscessus to antimycobacterial drugs in preclinical models. Antimicrob. Agents Chemother 59, 6904–6912 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Nicola F, Cirillo DM & Lore NI Preclinical murine models to study lung infection with Mycobacterium abscessus complex. Tuberculosis 138, 102301 (2023). [DOI] [PubMed] [Google Scholar]
- 194.De Groote MA et al. GM-CSF knockout mice for preclinical testing of agents with antimicrobial activity against Mycobacterium abscessus. J. Antimicrob. Chemother 69, 1057–1064 (2014). [DOI] [PubMed] [Google Scholar]
- 195.Libardo J, Boshoff HI & Barry CE III The present state of the tuberculosis drug development pipeline. Curr. Opin. Pharmacol 42, 81–94 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Berg A et al. Model-based meta-analysis of relapsing mouse model studies from the critical path to tuberculosis drug regimens initiative database. Antimicrob. Agents Chemother 66, e0179321 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Bartelink IH et al. New paradigm for translational modeling to predict long-term tuberculosis treatment response. Clin. Transl. Sci 10, 366–379 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Larkins-Ford J, Degefu YN, Van N, Sokolov A & Aldridge BB Design principles to assemble drug combinations for effective tuberculosis therapy using interpretable pairwise drug response measurements. Cell Rep. Med 3, 100737 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Mudde SE et al. Predictive modeling to study the treatment-shortening potential of novel tuberculosis drug regimens, towards bundling of preclinical data. J. Infect. Dis 225, 1876–1885 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Pienaar E et al. A computational tool integrating host immunity with antibiotic dynamics to study tuberculosis treatment. J. Theor. Biol 367, 166–179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Dooley KE, Hanna D, Mave V, Eisenach K & Savic RM Advancing the development of new tuberculosis treatment regimens: the essential role of translational and clinical pharmacology and microbiology. PLoS Med. 16, e1002842 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Davies GR & Wallis RS Methods for selecting regimen duration to prevent relapse in drug-susceptible and drug-resistant TB. Int. J. Tuberc. Lung Dis 20, 13–17 (2016). [DOI] [PubMed] [Google Scholar]
- 203.Imperial MZ, Phillips PPJ, Nahid P & Savic RM Precision-enhancing risk stratification tools for selecting optimal treatment durations in tuberculosis clinical trials. Am. J. Respir. Crit. Care Med 204, 1086–1096 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; A retrospective analyses of large TB clinical trials to develop a risk stratification tool for the selection of patient-specific optimal treatment durations.
- 204.Chen RY et al. Using biomarkers to predict TB treatment duration (Predict TB): a prospective, randomized, noninferiority, treatment shortening clinical trial. Gates Open Res. 1, 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Nyang’wa BT et al. A 24-week, all-oral regimen for rifampin-resistant tuberculosis. N. Engl. J. Med 387, 2331–2343 (2022). [DOI] [PubMed] [Google Scholar]
- 206.Mok J et al. 9 months of delamanid, linezolid, levofloxacin, and pyrazinamide versus conventional therapy for treatment of fluoroquinolone-sensitive multidrug-resistant tuberculosis (MDR-END): a multicentre, randomised, open-label phase 2/3 non-inferiority trial in South Korea. Lancet 400, 1522–1530 (2022). [DOI] [PubMed] [Google Scholar]
- 207.Paton NI et al. Treatment strategy for rifampin-susceptible tuberculosis. N. Engl. J. Med 388, 873–887 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.The CRyPTIC Consortium.Genome-wide association studies of global Mycobacterium tuberculosis resistance to 13 antimicrobials in 10,228 genomes identify new resistance mechanisms. PLoS Biol. 20, e3001755 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Fors J, Strydom N, Fox WS, Keizer RJ & Savic RM Mathematical model and tool to explore shorter multi-drug therapy options for active pulmonary tuberculosis. PLoS Comput. Biol 16, e1008107 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Dheda K et al. Drug-penetration gradients associated with acquired drug resistance in patients with tuberculosis. Am. J. Respir. Crit. Care Med 198, 1208–1219 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Strydom N et al. Tuberculosis drugs’ distribution and emergence of resistance in patient’s lung lesions: a mechanistic model and tool for regimen and dose optimization. PLoS Med. 16, e1002773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Diacon AH Two steps forward, one step back. N. Engl. J. Med 387, 2380–2381 (2022). [DOI] [PubMed] [Google Scholar]
- 213.Lienhardt C et al. Advances in clinical trial design: weaving tomorrow’s TB treatments. PLoS Med. 17, e1003059 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; A review and opinion of present and future advances in clinical trial design to harmonize and accelerate the development of novel TB regimens.
- 214.Davies G, Boeree M, Hermann D & Hoelscher M Accelerating the transition of new tuberculosis drug combinations from phase II to phase III trials: new technologies and innovative designs. PLoS Med. 16, e1002851 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Biomarkers Development Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin. Pharmacol. Ther 69, 89–95 (2001). [DOI] [PubMed] [Google Scholar]
- 216.Boeree MJ et al. A dose-ranging trial to optimize the dose of rifampin in the treatment of tuberculosis. Am. J. Respir. Crit. Care Med 191, 1058–1065 (2015). [DOI] [PubMed] [Google Scholar]
- 217.Wallis RS, Wang C, Meyer D & Thomas N Month 2 culture status and treatment duration as predictors of tuberculosis relapse risk in a meta-regression model. PLoS ONE 8, e71116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Phillips PP et al. Limited role of culture conversion for decision-making in individual patient care and for advancing novel regimens to confirmatory clinical trials. BMC Med. 14, 19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Davies GR Early clinical development of anti-tuberculosis drugs: science, statistics and sterilizing activity. Tuberculosis 90, 171–176 (2010). [DOI] [PubMed] [Google Scholar]
- 220.Bouton TC et al. An optimized background regimen design to evaluate the contribution of levofloxacin to multidrug-resistant tuberculosis treatment regimens: study protocol for a randomized controlled trial. Trials 18, 563 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Aguilar Diaz JM et al. New and repurposed drugs for the treatment of active tuberculosis: an update for clinicians. Respiration 102, 83–100 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Esmail A et al. An all-oral 6-month regimen for multidrug-resistant tuberculosis: a multicenter, randomized controlled clinical trial (the NExT Study). Am. J. Respir. Crit. Care Med 205, 1214–1227 (2022). [DOI] [PubMed] [Google Scholar]
- 223.Chang V, Phillips PPJ, Imperial MZ, Nahid P & Savic RM A comparison of clinical development pathways to advance tuberculosis regimen development. BMC Infect. Dis 22, 920 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Paton NI et al. A treatment strategy for rifampicin-susceptible tuberculosis. N. Engl. J. Med 388, 873–887 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]; A landmark clinical trial based on the pioneering paradigm of ‘stop-treatment-and-watch’ to discover treatment-shortening TB drug regimens.
- 225.Nogueira BMF et al. Diagnostic biomarkers for active tuberculosis: progress and challenges. EMBO Mol. Med 14, e14088 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.CDC. Virtual Meeting of the Advisory Council for the Elimination of Tuberculosis https://www.cdc.gov/faca/committees/pdfs/acet/acet-minutes-20211214-15-508.pdf (2022).
- 227.Vinnard C et al. Assessing response to therapy for nontuberculous mycobacterial lung disease: quo vadis? Front. Microbiol 9, 2813 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Henkle E et al. Patient-reported symptom and health-related quality-of-life validation and responsiveness during the first 6 months of treatment for Mycobacterium avium complex pulmonary disease. Chest 164, 53–64 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Cella D et al. Patient-Reported Outcomes in Performance Measurement (RTI Press, 2015). [PubMed] [Google Scholar]
- 230.Waglechner N et al. Genomic epidemiology of Mycobacterium abscessus in a Canadian cystic fibrosis centre. Sci. Rep 12, 16116 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Zumla A, Nahid P & Cole ST Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug. Discov 12, 388–404 (2013). [DOI] [PubMed] [Google Scholar]; The predecessor of the present Review, showing the progress achieved and road travelled over the past 10 years.
- 232.Houben R, Esmail H, Cobelens F, Williams CML & Coussens AK Tuberculosis prevalence: beyond the tip of the iceberg. Lancet Respir. Med 10, 537–539 (2022). [DOI] [PubMed] [Google Scholar]
- 233.van Ingen J et al. Nontuberculous mycobacterial lung disease caused by Mycobacterium avium complex – disease burden, unmet needs, and advances in treatment developments. Expert Rev. Respir. Med 15, 1387–1401 (2021). [DOI] [PubMed] [Google Scholar]; An overview of treatment guidelines and numerous unmet needs for MAC-PD.
- 234.Adjemian J, Olivier KN & Prevots DR Nontuberculous mycobacteria among patients with cystic fibrosis in the United States: screening practices and environmental risk. Am. J. Respir. Crit. Care Med 190, 581–586 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Diel R, Lipman M & Hoefsloot W High mortality in patients with Mycobacterium avium complex lung disease: a systematic review. BMC Infect. Dis 18, 206 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Jhun BW et al. Prognostic factors associated with long-term mortality in 1445 patients with nontuberculous mycobacterial pulmonary disease: a 15-year follow-up study. Eur. Respir. J 55, 1900798 (2020). [DOI] [PubMed] [Google Scholar]
- 237.Griffith DE & Daley CL Treatment of Mycobacterium abscessus pulmonary disease. Chest 161, 64–75 (2022). [DOI] [PubMed] [Google Scholar]
- 238.Borisov SE et al. Effectiveness and safety of bedaquiline-containing regimens in the treatment of MDR- and XDR-TB: a multicentre study. Eur. Respir. J 49, 1700387 (2017). [DOI] [PubMed] [Google Scholar]
- 239.Ndjeka N et al. High treatment success rate for multidrug-resistant and extensively drug-resistant tuberculosis using a bedaquiline-containing treatment regimen. Eur. Respir. J 52, 1801528 (2018). [DOI] [PubMed] [Google Scholar]
- 240.Xu HB, Jiang RH & Li L Treatment outcomes for Mycobacterium avium complex: a systematic review and meta-analysis. Eur. J. Clin. Microbiol. Infect. Dis 33, 347–358 (2014). [DOI] [PubMed] [Google Scholar]
- 241.Nasiri MJ et al. Antibiotic therapy success rate in pulmonary Mycobacterium avium complex: a systematic review and meta-analysis. Expert Rev. Anti Infect. Ther 18, 263–273 (2020). [DOI] [PubMed] [Google Scholar]
- 242.Kwak N et al. Treatment outcomes of Mycobacterium avium complex lung disease: a systematic review and meta-analysis. Clin. Infect. Dis 65, 1077–1084 (2017). [DOI] [PubMed] [Google Scholar]
- 243.Kwak N et al. Mycobacterium abscessus pulmonary disease: individual patient data meta-analysis. Eur. Respir. J 54, 1801991 (2019). [DOI] [PubMed] [Google Scholar]
- 244.Yano T et al. Reduction of clofazimine by mycobacterial type 2 NADH:quinone oxidoreductase: a pathway for the generation of bactericidal levels of reactive oxygen species. J. Biol. Chem 286, 10276–10287 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Borisov SE et al. Efficiency and safety of chemotherapy regimen with SQ109 in those suffering from multiple drug resistant tuberculosis [Russian]. Tuberculosis Lung Dis. 96, 6–18 (2018). [Google Scholar]
- 246.Kim J et al. Safety, tolerability, pharmacokinetics, and metabolism of telacebec (Q203) for the treatment of tuberculosis: a randomized, placebo-controlled, multiple ascending dose phase 1B trial. Antimicrob. Agents Chemother. 67, e0112322 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Cevik M Abstr. 109. SimpliciTB Results and hepatic safety of pretomanid regimens +/− pyrazinamide. CROI 2023 https://www.croiconference.org/abstract/simplicitb-results-and-hepatic-safety-of-pretomanid-regimens-pyrazinamide/ (2023).
- 248.Dierig A et al. A phase IIb, open-label, randomized controlled dose ranging multi-centre trial to evaluate the safety, tolerability, pharmacokinetics and exposure-response relationship of different doses of delpazolid in combination with bedaquiline delamanid moxifloxacin in adult subjects with newly diagnosed, uncomplicated, smear-positive, drug-sensitive pulmonary tuberculosis. Trials 24, 382 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Jindani A et al. Four-month high-dose rifampicin regimens for pulmonary tuberculosis. N. Engl. J. Med 384, 1705–1718 (2023). [DOI] [PubMed] [Google Scholar]
- 250.Guglielmetti L et al. Evaluating newly approved drugs for multidrug-resistant tuberculosis (endTB): study protocol for an adaptive, multi-country randomized controlled trial. Trials 22, 651 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Boeree MJ et al. High-dose rifampicin, moxifloxacin, and SQ109 for treating tuberculosis: a multi-arm, multi-stage randomised controlled trial. Lancet Infect. Dis 17, 39–49 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Fox W, Ellard GA & Mitchison DA Studies on the treatment of tuberculosis undertaken by the British Medical Research Council tuberculosis units, 1946–1986, with relevant subsequent publications. Int. J. Tuberc. Lung Dis 3, S231–279 (1999). [PubMed] [Google Scholar]
- 253.Dorman SE et al. Substitution of rifapentine for rifampin during intensive phase treatment of pulmonary tuberculosis: study 29 of the tuberculosis trials consortium. J. Infect. Dis 206, 1030–1040 (2012). [DOI] [PubMed] [Google Scholar]
- 254.Gillespie SH et al. Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. N. Engl. J. Med 371, 1577–1587 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.WHO. Global Tuberculosis Report 2020. Report No. ISBN 978-92-4-001313-1 (WHO, 2020). [Google Scholar]
- 256.Aldridge BB et al. The tuberculosis drug accelerator at year 10: what have we learned? Nat. Med 27, 1333–1337 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]; A review of achievements of the Tuberculosis Drug Accelerator, an experiment designed to facilitate collaboration in TB drug discovery by breaking down barriers between competing labs and institutions.
- 257.Mekota AM et al. Building sustainable clinical trial sites in sub-Saharan Africa through networking, infrastructure improvement, training and conducting clinical studies: the PanACEA approach. Acta Trop. 238, 106776 (2022). [DOI] [PubMed] [Google Scholar]
- 258.Koele SE et al. Early bactericidal activity studies for pulmonary tuberculosis: a systematic review of methodological aspects. Int. J. Antimicrob. Agents 61, 106775 (2023). [DOI] [PubMed] [Google Scholar]
- 259.Ntinginya NE et al. Tuberculosis molecular bacterial load assay reveals early delayed bacterial killing in relapse patients. Clin. Infect. Dis 76, e990–e994 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Jones A et al. Sputum lipoarabinomannan (LAM) as a biomarker to determine sputum mycobacterial load: exploratory and model-based analyses of integrated data from four cohorts. BMC Infect. Dis 22, 327 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Kawasaki M et al. Lipoarabinomannan in sputum to detect bacterial load and treatment response in patients with pulmonary tuberculosis: analytic validation and evaluation in two cohorts. PLoS Med. 16, e1002780 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.le Roux SP et al. Resistance-conferring mycobacterial mutations and quantification of early bactericidal activity. Am. J. Respir. Crit. Care Med 203, 635–637 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Mukamolova GV, Turapov O, Malkin J, Woltmann G & Barer MR Resuscitation-promoting factors reveal an occult population of tubercle bacilli in sputum. Am. J. Respir. Crit. Care Med 181, 174–180 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Chengalroyen MD et al. Detection and quantification of differentially culturable tubercle bacteria in sputum from patients with tuberculosis. Am. J. Respir. Crit. Care Med 194, 1532–1540 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Walter ND et al. Mycobacterium tuberculosis precursor rRNA as a measure of treatment-shortening activity of drugs and regimens. Nat. Commun 12, 2899 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]