


Abstract
In addition to binding DNA in a sequence-specific manner, the p53 tumour suppressor protein can interact with damaged DNA. In order to understand which structural features in DNA the C-teminal domain recognises we have studied the interaction of p53 protein with different types of DNA oligonucleotides imitating damaged DNA. Here we show that one unpaired nucleotide within double-stranded (ds)DNA is sufficient for recognition by the p53 C-terminus, either as a protruding end or as an internal gap in dsDNA. C-terminal interaction with DNA ends facilitated core domain binding to DNA, whereas interaction with gaps prevented core domain–DNA complexing, implying that p53 might adopt distinct conformations upon binding to different DNA lesions. These observations suggest that both single-strand and double-strand breaks can serve as a target for p53 C-terminal recognition in vivo and indicate that p53 might recruit different repair factors to the sites of damaged DNA depending on the type of lesion.
INTRODUCTION
The tumour suppressor protein p53 becomes activated in cells under various stress conditions, including DNA damage induced by ionising radiation, UV or chemical agents, hypoxia, oncogene activation and others (reviewed in 1). Induced p53 triggers growth arrest or cell death by apoptosis via transcriptional activation of a set of genes containing the consensus p53 binding site and transcriptional repression of another set of genes. In this manner, p53 serves as a gatekeeper which prevents propagation of cells that are at risk of aquiring tumourigenic mutations.
Accumulating in vitro and in vivo data suggest that p53 might also function as a caretaker, preserving genomic integrity by regulating DNA repair (for a review see 2). Loss of p53 function leads to the deregulation of DNA repair and causes gross structural changes in the genome. For instance, cells from various organs of 4–6-week-old p53 null mice display aneuploidy and frequent gene amplification, suggesting inefficeint DNA repair in the absence of p53 (3). Mutations in p53 were statistically correlated with the replication error phenotype in mucosa-associated lymphoid tissue lymphomas (4). In addition, it was recently found that p53 is able to reduce the frequency of chemically induced point mutations (5). Disruption of p53 function in some systems resulted in a deficiency in the rate and extent of nucleotide excision repair (NER) (5–7).
It appears that p53 can facilitate DNA repair due to different activities. p53 up-regulates transcription of GADD45, xeroderma pigmentosum gene p48 and a rate limiting factor in DNA repair ribonucleotide reductase (8–11). There is also evidence that p53 modulates DNA repair in a transcription-independent way (12,13). It is possible that p53 can participate in DNA repair processes directly via its ability to bind damaged DNA and to interact with several components of the excisional and recombinational repair mashinery, including XPD, XPB, RPA and RAD51 (14–16).
DNA strand breaks appear to be sufficient to trigger p53 activation in cells and p53 induction is temporally correlated with the appearance of DNA breaks (17,18). Induction of p53 in response to, for instance, DNA damage appears to involve activation of specific DNA binding by the core domain along with protein accumulation (19). Short single-stranded (ss)DNA oligonucleotides added in trans can stimulate sequence-specific binding of the p53 core domain in vitro, indicating that recognition of DNA lesions by the p53 C-terminus might trigger p53 activation in vivo (20,21).
The ability of the C-terminal domain of p53 to bind ssDNA ends (22,23), insertion/deletion mismatches (24), recombination intermediates (25) and γ-irradiated DNA in vitro (26) implies that p53 could recognise DNA damage and/or DNA repair intermediates in vivo. In line with this notion are confocal microscopy studies that demonstrated co-localisation of p53 with sites of damaged DNA in human skin exposed to UV irradiation (27). p53 was also shown to enhance the rejoining of non-homologous DNA ends in living cells in a C-terminal-dependent and transactivation-independent manner (12). Taken together, these observations hint at involvement of the direct recognition of DNA lesions by p53 in exertion of the p53 ‘guardian of the genome’ function. However, the role of DNA damage intermediates in the p53 response is only starting to emerge (28).
In the present study we show that the p53 C-terminal domain binds both staggered ends and gaps of different length within double-stranded oligonucleotides, whereas the core domain did not recognise the single-stranded stretch in protruding ends or gaps. C-terminal domain complexing with DNA ends facilitated the core domain interaction with the same DNA molecule. In contrast, C-terminal interaction with gaps blocked DNA binding by the core. Our findings provide new insights into the possible biological consequences of p53 interaction with different types of DNA lesions.
MATERIALS AND METHODS
Purification of p53 proteins
Sf9 insect cells were infected with human p53 recombinant baculovirus provided by M. Oren (Weizmann Institute, Israel). Cells were harvested 72 h after infection and p53 protein was purified as described (29). The sequence encoding p53 core domain residues 94–292 was subcloned into the BamHI and HindIII sites of vector pQE30 (Qiagen). His-tagged core domain protein was purified using Ni–NTA resin as described by the manufacturer (Qiagen). Purification of the GST–p53 fusion proteins was performed as previously described (21).
Oligonucleotides
Synthetic 29mer to 31mer DNA oligonucleotides were purchased from Pharmacia Biotech (Stockholm, Sweden). The sequences of the DNA oligonucleotides used in this study are shown in Table 1. The dsDNA oligonucleotides were constructed by annealing the corresponding complementary ssDNA, followed by purification of double-strand forms on a 15% non-denaturing polyacrylamide gel. The integrity of dsDNA oligonucleotides containing internal gaps (MNG1, MNG2 and MNG8) was verified as follows. Aliquots of the purified double-strand forms were denatured by heat treatment at 100°C for 3 min, labelled with T4 polynucleotide kinase (Gibco BRL, Grand Island, NY) and subjected to electrophoresis on 10% non-denaturing acrylamide gels together with the non-denatured, labelled, double-strand form of the same DNA oligonucleotides. The gel was then autoradiographed and the intensity of the bands with mobilities corresponding to ssDNA was evaluated by densitometry. DNA oligonucleotides that showed equal distribution of the signal among the three DNA strands of which they were composed were used in electrophoretic mobility shift assays.
Table 1. DNA oligonucleotides.

Band shift assays
Band shift experiments were performed essentially as described (21), except that 5 mM MgCl2 was added to the reaction mixture. Five nanograms of purified p53 protein, 0.5 ng 32P-end-labelled probe and, where indicated, 100 ng antibodies and 1–10 ng unlabelled competitor DNA were mixed in 20 µl of reaction mixture and incubated for 30 min at room temperature. Samples were subjected to electrophoresis on 4% native polyacrylamide gels containing 0.1% Triton X-100 at 200 V for 100 min at 4°C. Gels were fixed for 5 min in water/acetic acid/methanol (8:1:1) mixture, dried and autoradiographed.
RESULTS
p53 binds dsDNA oligonucleotides containing single-strand gaps as short as 1 nt
Several types of DNA lesions can arise in cells exposed to DNA damaging agents. dsDNA breaks with staggered or, less frequently, blunt DNA ends occur in cells exposed to ionising radiation (30). ssDNA gaps can be generated as a consequence of NER or mismatch repair (31,32) or replicational bypass of a DNA lesion (33). We addressed the question of whether p53 is able to recognise DNA lesions such as single-strand gaps.
We analysed the interaction between the double-strand oligonucleotides with different structure and recombinant p53 protein produced in Sf9 insect cells in a band shift assay, as previously described (21). Unlabelled 29mer or 30mer dsDNA oligonucleotides were used as competitors for p53 binding to the labelled ssDNA oligonucleotide NGC: the blunt end double-strand oligonucleotide MNGC and the double-strand MNG1, MNG2 and MNG8 oligonucleotides which have the same sequence as MNGC, but contain 1, 2 and 8 nt internal gaps, respectively (Table 1).
In accordance with our previous results (21), p53 formed a complex with the single-strand probe via its C-terminus (complex I, Fig. 1A, lane 1), as verified by competition with ssDNA, but not dsDNA (NGC versus MNGC, Fig. 1A, lanes 10 and 11 versus 2 and 3, respectively). Gap-containing oligonucleotides MNG1, MNG2 and MNG8 (Table 1) competed efficiently with the single-strand probe for binding to p53 (Fig. 1A, lanes 4–9). MNG8 was an even more efficient competitor than NGC itself in this assay (compare lanes 8 and 10). In Figure 1B binding of the isolated p53 C-terminal domain, GST–p53(320–393) protein, to the labelled MNG8 probe is shown. Interestingly, the interaction with MNG8 is inhibited by PAb421 antibody when used at high concentrations (Fig. 4B, lanes 11 and 12, 200 and 300 ng, respectively). This probably occurs because the DNA binding basic region (residues 363–372) overlaps the PAb421 epitope (residues 372–382). Control antibody PAb1801, which recognises N-terminal residues 46–55 in p53, did not affect complex formation. The C-terminus–MNG8 complex was supershifted by anti-GST antibody, demonstrating involvement of the p53 fusion protein in complex formation. From these experiments we conclude that the p53 C-terminal domain can recognise gaps in dsDNA.
Figure 1.


(A) p53 recognises internal gaps in dsDNA oligonucleotides as short as 1 nt. Aliquots of 1 and 10 ng of unlabelled dsDNA oligonucleotides with internal single-strand gaps efficiently competed for p53 binding to the labelled ssDNA oligonucleotide NGC. Complex I between p53 and NGC is indicated by an arrow. (B) The full-length GST–p53–NGC (lanes 1–8) and GST–p53(320–393)–MNG8 (lanes 9–13) complexes were inhibited by the C-terminal-specific PAb421 antibody in a dose-dependent manner (lanes 2–4 and 10–11, respectively). N-terminal-specific antibody PAb1801 did not affect complex formation (lanes 2–4 and 10). p53 C-terminus–DNA complexes were supershifted by anti-GST antibody (lanes 8 and 13).
Figure 4.

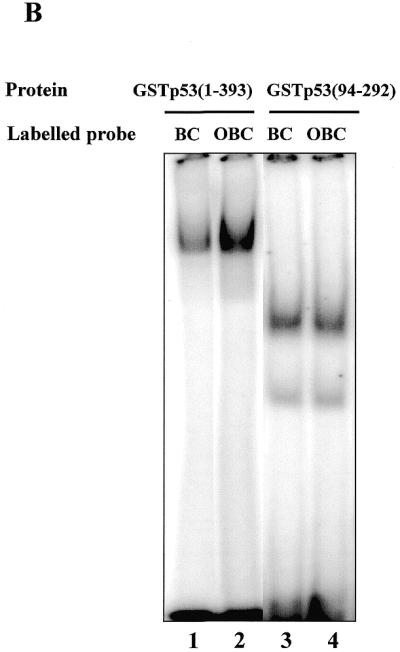

(A) Interaction of the C-terminus with protruding DNA ends induces the core domain–DNA interaction. Labelled dsDNA oligonucleotides MNGC (blunt end) and MN1, MN2 and MN4, with 1, 2 and 4 nt single-stranded ends, respectively, were used. C-terminal-mediated complexes I and II and PAb421-induced core domain-mediated complex IV are indicated by arrows. Efficiency of the p53 core domain–DNA interaction with dsDNA increases with the length of protruding ends on the DNA probe. (B) The presence of protruding single-stranded ends enhances specific DNA binding by the core domain in cis. Interaction of full-length p53 with labelled blunt end probe BC, containing the p53 binding site (lane 1), was less efficient than interaction with the staggered end OBC probe which has the same sequence (lane 2). The isolated p53 core domain protein (residues 94–292) bound both oligonucleotides with the same efficiency (lanes 3–4). In order to visualise the p53–BC complex in the absence of PAb421 antibody, 50 ng of p53 protein was used. (C) Isolated core domain protein p53(98–292) does not recognise protruding ends or gaps on dsDNA. (Upper) Specific oligonucleotide BC was used as a labelled probe and cold double-stranded oligonucleotides with different structures were used as competitors. (Lower) PhosphorImager-assisted quantitation of the series of three competition experiments. No statistically significant difference in efficiency of challenging core–BC complexing was observed between double-stranded oligonucleotides with internal gaps (MNG1, MNG2 or MNG8), staggered ends (MN1, MN2 or MN4) or a blunt end (MNGC).
A comparison of complexing of the full-length p53 protein with labelled dsDNA revealed that while p53 did not bind to double-stranded oligonucleotides MNGC and BC, containing the p53 binding site (34; Fig. 2, lanes 1 and 4), complex formation with the 2 nt gap-containing MNG2 was detected (Fig. 2, lane 10, bracketed). Binding to the MNG8 oligonucleotide with an 8 nt gap was even stronger (data not shown). A p53–MNG1 complex was visible after longer exposure. This result argues that the longer the stretch of unpaired single-strand nucleotides in dsDNA, the better substrate it makes for p53 C-terminal binding.
Figure 2.

Single-strand gaps are the targets for C-terminal binding, but inhibit the core domain interaction with DNA. As labelled probes the following dsDNA oligonucleotides were used: BC, containing the p53 binding site, negative control MNGC and gap-containing MNG1 and MNG2 (1 and 2 nt gaps, respectively). Complex IV formed via the p53 core domain induced by PAb421 is indicated by an arrow. Faster migrating complexes between the p53 C-terminus and MNG2 are indicated with a bracket.
The addition of either PAb421 or PAb1801 p53-specific antibodies did not supershift p53 complexes with MNG1, MNG2 and MNG8 probes formed via the C-terminal domain (Fig. 2, lanes 8, 9, 11 and 12, and data not shown). Likewise, no supershift of the p53–NGC complex by these antibodies was observed (Fig. 1B, lanes 2–7). Instead, PAb421 inhibited p53–NGC binding in a dose-dependent manner (lanes 2–4, 100–300 ng antibody/sample), analogous to inhibition of the p53(320–393)–MNG8 complex. PAb1801 did not affect complexing at any concentration. Presence of the GST–p53 fusion protein in a complex was verified by anti-GST antibody supershift (lane 8).
Notably, PAb421 induced formation of a new slowly migrating complex IV when double-stranded, but not single-stranded, oligonucleotides were used as the labelled probes (compare Fig. 2, lanes 2, 5, 8 and 11, and Fig. 1B, lane 2). The strongest complex was formed with labelled BC oligonucleotide containing the p53 consensus binding site. This complex was formed via p53 core domain interaction with DNA, since it was efficiently competed by BC but not by non-specific MNGC oligonucleotide (data not shown). MNGC–p53 complex IV was competed more efficiently by the specific, but not by the non-specific, oligonucleotides, demonstrating involvement of the core domain in this complex (data not shown). The presence of single-strand gaps in dsDNA significantly reduced core domain complexing with DNA (Fig. 2, compare lanes 8 and 11 with lane 5). Low efficiency of core domain binding to double-stranded oligonucleotides containing single-strand gaps was confirmed in band shift experiments using isolated p53 core domain protein (Fig. 4C).
One protruding nucleotide on dsDNA is sufficient for recognition by the p53 C-terminal domain
We have shown previously that p53 can bind protruding ends of double-stranded oligonucleotides (21). Here we addressed the question of what is the minimal length of the protruding end on dsDNA that p53 can recognise. The double-stranded MN1, MN2 and MN4 oligonucleotides, containing 1, 2 and 4 nt long protruding single-strand ends, respectively (Table 1), were assessed in a competition assay for their ability to interact with p53.
dsDNA oligonucleotides with protruding single-strand ends were potent competitors of p53–NGC complex formation, in contrast to blunt end double-stranded oligonucleotides (Fig. 3, compare lanes 6–13 and 2–5). The ability to compete did not depend on the length of the protruding end: MN1 with only a 1 nt protruding end competed as efficiently as MN4 with a 4 nt protruding end. Thus, the p53 C-terminal domain is able to recognise even one protruding nucleotide on dsDNA. This correlates with the recognition of a gap in dsDNA as short as 1 nt.
Figure 3.

p53 can bind protruding ends on dsDNA as short as 1 nt. dsDNA oligonucleotides MN1, MN2 and MN4 with single-strand overhangs of 1, 2 and 4 nt, respectively, compete with ssDNA for binding to p53. EMSA experiments were carried out with labelled ssNGC as probe. Aliquots of 1 and 10 ng ‘cold’ competitors indicated at the top of the figure were added to the reaction mixture in lanes 2–13.
C-terminal interaction with protruding DNA ends induces core domain–DNA binding
Next, the effect of the C-terminal interaction with different terminal structure oligonucleotides on core domain DNA binding was examined. As shown in Figure 4A, p53 readily formed complexes with labelled staggered end probes MN1, MN2 and MN4, but very weakly with blunt end MNGC (lanes 4, 7, 10 and 1). Similar to the absence of a supershift of complexes between the p53 C-terminus and single-stranded or gap-containing DNA, the addition of either PAb421 or PAb1801 p53-specific antibodies did not supershift p53 complexes with staggered end probes (Fig. 4A, lanes 5, 6, 8, 9, 11 and 12).
Notably, formation of PAb421-induced complex IV via the core domain was facilitated by the presence of protruding ends on a labelled probe. The efficiency of complex formation was significantly increased with increasing length of the protruding ends of a labelled probe (Fig. 4A, compare lanes 2, 5, 8 and 11). Quantitation of the amount of bound DNA using a phosphorimager revealed that p53 formed complex IV with MN4 at least 20 times more efficiently than with MNGC (lane 2) and ∼10 times more strongly than complex IV with MN1 (Fig. 4A, lanes 11, 2 and 5, respectively).
The isolated p53 core domain protein (residues 94–292) showed very weak binding to DNA oligonucleotides lacking a p53 binding site, irrespective of their terminal structure or the presence of gaps, as verified in a competition assay and in a band shift assay (Fig. 4C and data not shown). Thus, the core domain protein cannot differentiate between the blunt end or staggered end DNA probes. Therefore, the increased binding to staggered end DNA probes via the core domain should be attributable to the C-teminal interaction with protruding ends.
We have compared the binding of p53 to double-stranded oligonucleotides containing a consensus p53 binding site with blunt (BC) or staggered (OBC) ends (see Table 1). As can be seen in Figure 4B (lanes 1 and 2), specific binding by p53 was more effecient when the probe had protruding ends: p53 bound 34% of labelled staggered end oligonucleotide OBC, in contrast to 14% of bound BC. The effect was PAb421 independent, since it was observed in its absence. The specificity of binding was verified by competition assay (data not shown). In contrast, the isolated core domain protein p53(94–292) bound both probes with the same efficiency (Fig. 4B, lanes 3 and 4).
No C-terminus–DNA complexes (complexes I and II ) were detected on a gel when staggered end OBC oligonucleotide was used as the labelled probe. This suggests that although the C-terminus makes initial contact with the DNA, the binding equilibrium is shifted towards the core domain interaction with DNA. However, it is not clear whether the C-terminus is involved in core domain–DNA complex formation. This question is of particular interest since the two p53 DNA binding domains might have interrelated functions.
Cooperation between the C-terminal and core domains in binding to DNA
To gain insight into the interrelationship between p53 domains in DNA binding, we addressed the question of whether the C-terminal domain is involved in DNA binding upon core domain–DNA complexing.
We performed competition assays in a more complex setting, allowing simultaneous interaction with a labelled DNA probe via both the C-terminal and core domains. The OBC probe containing both the specific p53 binding site and protruding ends was not used for these experiments since C-terminus–DNA complexes were not detected in this case (Fig. 4B). Instead, p53 was incubated with the labelled MN1 double-stranded oligonucleotide in the presence of activating antibody PAb421.
In order to distinguish between C-terminal and core domain interactions with DNA in trans (i.e. with different DNA molecules) and in cis (i.e with the same DNA molecule) the competitor DNAs were chosen so that the pattern of competition should differ depending on the mode of interaction. The following two scenarios could be envisioned. First, if the C-terminal and core domains bind to DNA exclusively in trans, only the C-terminal-mediated complexes and not the core domain–DNA complexes should be competed by the C-terminal-specific oligonucleotides (illustrated in Fig. 5A, I). In contrast, if the C-terminus induces a core domain interaction with the same DNA molecule, staggered end oligonucleotides will compete for core domain complexing, because they can bind both the C-terminus and core domain (Fig. 5A, II). Gap-containing or single-stranded oligonucleotides, in contrast, will not compete with the core domain–DNA complex.
Figure 5.

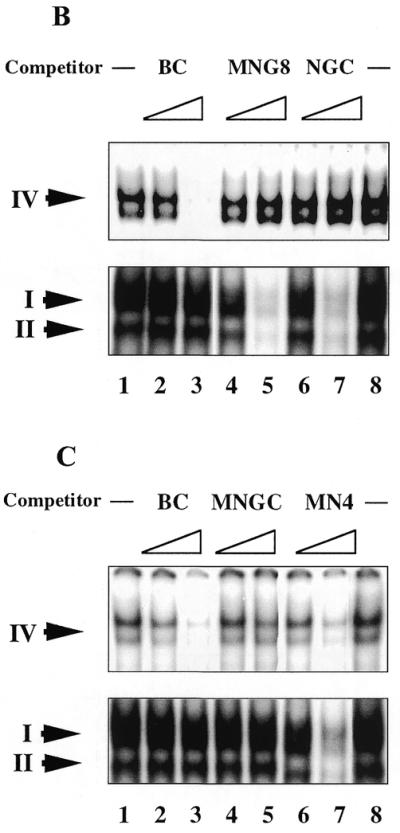
(A) The ability of different oligonucleotides to compete for core domain binding depends on the mode of the C-terminal and core domain interactions with DNA. Only DNA binding domains of dimeric p53 are shown. The core domain is depicted as a horizontally oriented oval, the C-terminal domain as a smaller vertically oriented oval. The labelled staggered end probe is shown in bold. Two possible modes of interrelationship of two p53 domains upon interaction with DNA are presented. (I) If the C-terminal and core domains bind DNA in trans, then C-terminal-specific oligonucleotides will not differ in their ability to compete with complex formation (no competition with the core domain–DNA complex). (II) Both DNA binding domains interact with the same labelled DNA molecule. Only staggered end double-stranded oligonucleotide which is able to bind the core domain will compete with core domain complexing. (B) Differential competition of p53–MN1 complexes I, II and IV by dsDNA oligonucleotides containing internal single-strand gaps. Aliquots of 1 and 10 ng unlabelled competitors (indicated at the top) were added to the reaction mixtures in lanes 2–7. Complex IV (induced by PAb421; upper panel) and complexes I and II (not affected by PAb421; lower panel) are indicated with arrows. Only C-terminal-mediated complexes were competed by MNG8 and NGC, but not the core domain-mediated complexes. (C) Competition of p53–MN1 complexes I, II and IV by dsDNA oligonucleotide with protruding ends. Staggered end oligonucleotide MN4 efficiently competed both C-terminal- and core domain-mediated complexes (lanes 6–7).
The upper and lower panels in Figure 5B and C show the p53–MN1 complexes formed through the core domain (complex IV) and the C-terminus (complexes I and II), respectively. C-terminus–DNA complexes were competed by gap-containing and staggered end dsDNA as well as by single-strand NGC, but not by blunt end dsDNA, in accordance with previous results (Fig. 5B and C, lower panels). Notably, we observed competition of the MN1–p53 core domain complex by oligonucleotide MN4 containing the C-terminal binding site (Fig. 5C, upper panel). Not all of the C-terminal target oligonucleotides compete, only oligonucleotide MN4, which can also bind the core domain. In contrast, single-stranded NGC and gap-containing MNG8, which did not bind the core, did not compete (Fig. 5B, upper panel). Such selective competition argues that the interaction with DNA by both domains can occur in cis.
DISCUSSION
p53 has several biochemical activities which might point to a possible role in DNA repair, involving the recognition of DNA lesions. In the present study we examined the ability of p53 to interact with DNA oligonucleotides mimicking damaged DNA. Our results demonstrate that the p53 C-terminus can recognise protruding ssDNA ends as short as 1 nt. p53 did not discriminate between 3′ and 5′ DNA ends, since no difference in binding to double-stranded MN oligonucleotides with protruding 3′ or 5′ DNA ends was observed in band shift assays (data not shown). In addition, our previous electron microscopy experiments allowed visualisation of p53 bound to both ends of a single-strand oligonucleotide (21).
Moreover, we show that p53 can bind internal single-strand gaps in dsDNA as short as 1 nt. Interaction with single-strand gaps, similar to ssDNA end recognition, was mediated by the C-terminal domain, whereas the core domain bound neither gaps nor ssDNA ends. Although a 1 nt gap was recognised by p53, interaction with longer gaps was more efficient.
What is the common structural component that is present in DNA substrates recognised by the p53 C-terminal domain? Data presented in this study indicate that p53 might recognise a distortion of the normal geometry of DNA, i.e. phosphate backbone deformation (an altered angle and/or spacing between phosphates) within regions of DNA lesions. The observation that the p53 C-terminal domain is able to bind even one protruding/unpaired nucleotide in dsDNA indicates the high potential of DNA lesion recognition by p53.
More than one p53–DNA complex formed via the C-terminal domain was observed on band shift gels. This might reflect the different oligomeric state of p53 (mono-, di- or tetrameric) in these complexes. Alternatively, more than one DNA molecule could be involved in the p53 tetramer–DNA complex. Further studies are required to define the nature of different C-terminus–DNA complexes.
The presence of the protruding ends on double-stranded probes enhanced DNA binding via the core domain (Fig. 4A and B). The efficiency of core domain DNA binding increased with increasing length of the protruding ends. Since the presence of staggered ends on DNA did not affect binding of the isolated core domain, the effect appears to be entirely C-terminus dependent. Furthermore, here we provide evidence that the C-terminal and core domains can bind one DNA molecule simultaneously.
The interrelationship between the two p53 DNA binding domains is a subject of ongoing discussion. According to the reciprocal interference model DNA binding by the C-terminal and core domains is mutually exclusive, whereas the allosteric model does not rule out the possibility of simultaneous binding of both domains to DNA (35–37).
The results presented in this study argue that C-terminal and core domain binding to DNA is not mutually exclusive. First, our experiments show that probes containing binding sites for both the C-terminal and core domains (for instance, OBC versus BC oligonucleotide) were the best substrates for p53 DNA binding via the core domain. Second, we observed competition of the MN1–p53 core domain complex by oligonucleotides containing C-terminal binding sites. Notably, not all of the C-terminal target oligonucleotides competed, only those which could bind the core domain as well (staggered end oligonucleotides); single-stranded NGC and gap-containing MNG8 DNA, which did not bind the core, did not compete (Fig. 5). Such selective competition argues that both domains can interact with the same DNA molecule (model shown in Fig. 5A).
Simultaneous binding of both domains to DNA might have a cooperative effect. This notion is consistent with the model proposed by Nagaich et al. (38). Comparison of the p53–DNA complexes demonstrated that DNA bending and twisting angles are significantly larger in the full-length p53–DNA complex than in the core domain–DNA complex. This implies that the p53 domain(s) flanking the DNA binding region, in particluar the C-terminal domain, is also involved in complexing with specific DNA (38). C-termini located on the concave surface of bent DNA might facilitate bending of the DNA and thus enhance interaction with the core domain. This arrangement may be advantageous for stabilisation of the entire protein–DNA complex. The co-crystal structure of full-length p53 protein in a complex with DNA will be required to give a definite answer as to the contribution of the C-terminal domain to DNA complexing by the core domain.
What could be the biological significance of p53 interaction with DNA ends? It was recently reported that p53 enhances the non-homologous end joining (NHEJ) pathway of double-strand break (DSB) repair in vivo (12). NHEJ appears to be a major DSB repair pathway in higher eukaryotes during the G0, G1 and early S phases of the cell cycle (39). DSB repair through homologous recombination, in contrast, operates mainly in the late S, G2 and M phases, when sister chromatids are positioned optimally for homologous crossover. This is probably because during other stages of the cell cycle a high probability of homologous recombination between non-homologous chromosomes exists, due to the presence of repetitive sequences, representing a substantial fraction of the genome (for a review see 39). p53 was shown to suppress homologous recombination in vivo. In the absence of wild-type p53 function increased rates of homologous recombination were observed, leading to intra- and interchromosomal translocations, gene conversions and deletions (40–43). Taken together, these data suggest that by controlling recombinational repair via suppression of homologous recombination and enhancement of the NHEJ pathway, p53 might prevent chromosomal aberrations, thus maintaining the integrity of the genome.
The molecular mechanism underlying the control of recombinational repair by p53 is presently unknown. It was shown that enhancement of rejoining of DSBs was C-terminus dependent, but transcription activation independent (12). In addition, blunt end rejoining was facilitated by p53 much less efficiently than protruding end rejoining. These results are in line with our in vitro data showing that the C-terminus bound protruding DNA ends more efficiently than blunt ends. The findings presented here imply that p53 might mediate NHEJ through direct binding to protruding DNA ends.
It was suggested that p53 might facilitate the initial stages of DNA repair in a transcription-independent manner, probably through protein–protein interactions (7). The ability to interact with gapped DNA might provide the basis for a possible role of p53 as a recruitment factor in NER, especially when taking into consideration the ability of p53 to bind to NER helicases XPB, XPD and CSB (14). This notion is bolstered by the observation that p53 co-localised with sites of gapped DNA produced during NER after UV irradiation in living cells (C.Rubbi, A.Okorokov, S.Metcalfe and J.Milner, personal communication).
The ability of p53 to interact with ssDNA ends and gaps suggests that both single-strand breaks (SSBs) and DSBs might serve as targets for the p53 C-terminus interaction in vivo. Notably, C-terminus interaction with DNA ends facilitated core domain binding to DNA, whereas interaction with gaps prevented the core domain–DNA interaction. This implies a different mutual positioning of p53 domains upon binding to SSBs versus DSBs. Thus, it is conceivable that p53 can recruit different repair factors depending on availability of its domains upon binding to different types of lesions. In the future it will be essential to learn how p53 interacts with excision/recombinational repair factors upon binding to DNA ends and gaps.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Moshe Oren (Weizmann Institute, Israel) for the recombinant p53 baculovirus and Thierry Soussi (Institut Curie, France) for the p53 purification protocol. We are also thank Klas G. Wiman and all members of his group for fruitful discussions. S.B.Z. was a recipient of a fellowship from the Karolinska Institutet. This work was supported by grants to G.S. from the Swedish Cancer Society (Cancerfonden), Swedish Medical Research Council (MFR) and Concern Foundation for Cancer Research.
REFERENCES
- 1.Ko L.J. and Prives,C. (1996) Genes Dev., 10, 1054–1072. [DOI] [PubMed] [Google Scholar]
- 2.Albrechtsen N., Dornreiter,I., Grosse,F., Kim,E., Wiesmuller,L. and Deppert,W. (1999) Oncogene, 18, 7706–7717. [DOI] [PubMed] [Google Scholar]
- 3.Fukasawa K., Wiener,F., Van de Woude,G.F. and Mai,S. (1997) Oncogene, 15, 1295–1302. [DOI] [PubMed] [Google Scholar]
- 4.Peng H., Chen,G., Du,M., Singh,N., Isaacson,P.G. and Pan,L. (1996) Am. J. Pathol., 148, 643–648. [PMC free article] [PubMed] [Google Scholar]
- 5.Courtemanche C. and Anderson,A. (1999) Oncogene, 18, 4672–4680. [DOI] [PubMed] [Google Scholar]
- 6.Ford J.M. and Hanawalt,P.C. (1997) J. Biol. Chem., 272, 28073–28080. [DOI] [PubMed] [Google Scholar]
- 7.Zhu Q., Wani,M.A., El-Mahdy,M. and Wani,A.A. (2000) J. Biol. Chem., 275, 11492–11497. [DOI] [PubMed] [Google Scholar]
- 8.Kastan M.B., Zhan,Q., el-Deiry,W.S., Carrier,F., Jacks,T., Walsh,W.V., Plunkett,B.S., Vogelstein,B. and Fornace,A.J.,Jr (1992) Cell, 71, 587–597. [DOI] [PubMed] [Google Scholar]
- 9.Smith M.L., Ford,J.M., Hollander,M.C., Bortnick,R.A., Amundson,S.A., Seo,Y.R., Deng,C.X., Hanawalt,P.C. and Fornace,A.J.,Jr (2000) Mol. Cell. Biol., 20, 3705–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hwang B.J., Ford,J.M., Hanawalt,P.C. and Chu,G. (1999) Proc. Natl Acad. Sci. USA, 96, 424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanaka H., Arakawa,H., Yamaguchi,T., Shiraishi,K., Fukuda,S., Matsui,K., Takei,Y. and Nakamura,Y. (2000) Nature, 404, 42–49. [DOI] [PubMed] [Google Scholar]
- 12.Tang W., Willers,H. and Powell,S.N. (1999) Cancer Res., 59, 2562–2565. [PubMed] [Google Scholar]
- 13.Dudenhoffer C., Kurth,M., Janus,F., Deppert,W. and Wiesmuller,L. (1999) Oncogene, 18, 5773–5784. [DOI] [PubMed] [Google Scholar]
- 14.Wang X., Yeh,H., Schaeffer,L., Roy,R., Moncollin,V., Egly,J.-M., Wang,Z., Frieberg,E., Evans,M., Taffe,B., Bohr,V., Weeda,G., Hoeijmakers,J., Forrester,K. and Harris,C. (1995) Nature Genet., 10, 188–195. [DOI] [PubMed] [Google Scholar]
- 15.Dutta A., Ruppert,J.M., Aster,J.C. and Winchester,E. (1993) Nature, 365, 79–82. [DOI] [PubMed] [Google Scholar]
- 16.Sturzbecher H.W., Donzelmann,B., Henning,W., Knippschild,U. and Buchhop,S. (1996) EMBO J., 15, 1992–2002. [PMC free article] [PubMed] [Google Scholar]
- 17.Nelson W.G. and Kastan,M.B. (1994) Mol. Cell. Biol., 14, 1815–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang L.C., Clarkin,K.C. and Wahl,G.M. (1996) Proc. Natl Acad. Sci. USA, 93, 4827–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hupp T.R., Sparks,A. and Lane,D.P. (1995) Cell, 83, 237–245. [DOI] [PubMed] [Google Scholar]
- 20.Jayaraman L. and Prives,C. (1995) Cell, 81, 1021–1029. [DOI] [PubMed] [Google Scholar]
- 21.Selivanova G., Iotsova,V., Kiseleva,E., Strom,M., Bakalkin,G., Grafstrom,R.C. and Wiman,K.G. (1996) Nucleic Acids Res., 24, 3560–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bakalkin G., Yakovleva,T., Selivanova,G., Magnusson,K.P., Szekely,L., Kiseleva,E., Klein,G., Terenius,L. and Wiman,K.G. (1994) Proc. Natl Acad. Sci. USA, 91, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bakalkin G., Selivanova,G., Yakovleva,T., Kiseleva,E., Kashuba,E., Magnusson,K.P., Szekely,L., Klein,G., Terenius,L. and Wiman,K.G. (1995) Nucleic Acids Res., 23, 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee S., Elenbaas,B., Levine,A. and Griffith,J. (1995) Cell, 81, 1013–1020. [DOI] [PubMed] [Google Scholar]
- 25.Dudenhoffer C., Rohaly,G., Will,K., Deppert,W. and Wiesmuller,L. (1998) Mol. Cell. Biol., 18, 5332–5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reed M., Woelker,B., Wang,P., Wang,Y., Anderson,M.E. and Tegtmeyer,P. (1995) Proc. Natl Acad. Sci. USA, 92, 9455–9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coates P.J., Save,V., Ansari,B. and Hall,P.A. (1995) J. Pathol., 176, 19–26. [DOI] [PubMed] [Google Scholar]
- 28.Giaccia A.J. and Kastan,M.B. (1998) Genes Dev., 12, 2973–2983. [DOI] [PubMed] [Google Scholar]
- 29.Selivanova G., Iotsova,V., Okan,I., Fritsche,M., Strom,M., Groner,B., Grafstrom,R.C. and Wiman,K.G. (1997) Nature Med., 3, 632–638. [DOI] [PubMed] [Google Scholar]
- 30.Weaver D. (1995) Trends Genet., 11, 388–392. [DOI] [PubMed] [Google Scholar]
- 31.Sancar A. (1994) Science, 266, 1954–1956. [DOI] [PubMed] [Google Scholar]
- 32.Modrich P. (1994) Science, 266, 1959–1960. [DOI] [PubMed] [Google Scholar]
- 33.Svoboda D.L. and Vos,J.M. (1995) Proc. Natl Acad. Sci. USA, 92, 11975–11979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Halazonetis T.D., Davis,L.J. and Kandil,A.N. (1993) EMBO J., 12, 1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hupp T.R. and Lane,D.P. (1994) Curr. Biol., 4, 865–875. [DOI] [PubMed] [Google Scholar]
- 36.Bayle J.H., Elenbaas,B. and Levine,A.J. (1995) Proc. Natl Acad. Sci. USA, 92, 5729–5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson M.E., Woelker,B., Reed,M., Wang,P. and Tegtmeyer,P. (1997) Mol. Cell. Biol., 17, 6255–6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagaich A.K., Zhurkin,V.B., Durell,S.R., Jernigan,R.L., Appella,E. and Harrington,R.E. (1999) Proc. Natl Acad. Sci. USA, 96, 1875–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lieber M.R. (1999) Genes Cells, 4, 77–85. [DOI] [PubMed] [Google Scholar]
- 40.Wiesmuller L., Cammenga,J. and Deppert,W.W. (1996) J. Virol., 70, 737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mekeel K.L., Tang,W., Kachnic,L.A., Luo,C.M., De Frank,J.S. and Powell,S.N. (1997) Oncogene, 14, 1847–1857. [DOI] [PubMed] [Google Scholar]
- 42.Bertrand P., Rouillard,D., Boulet,A., Levalois,C., Soussi,T. and Lopez,B.S. (1997) Oncogene, 14, 1117–1122. [DOI] [PubMed] [Google Scholar]
- 43.Honma M., Zhang,L.S., Hayashi,M., Takeshita,K., Nakagawa,Y., Tanaka,N. and Sofuni,T. (1997) Mol. Cell. Biol., 17, 4774–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]