

Abstract
Background
FCSK‐congenital disorder of glycosylation (FCSK‐CDG) is a recently discovered rare autosomal recessive genetic disorder with defective fucosylation due to mutations in the fucokinase encoding gene, FCSK. Despite the essential role of fucokinase in the fucose salvage pathway and severe multisystem manifestations of FCSK‐CDG patients, it is not elucidated which cells or which types of fucosylation are affected by its deficiency.
Methods
In this study, CRISPR/Cas9 was employed to construct an FCSK‐CDG cell model and explore the molecular mechanisms of the disease by lectin flow cytometry and real‐time PCR analyses.
Results
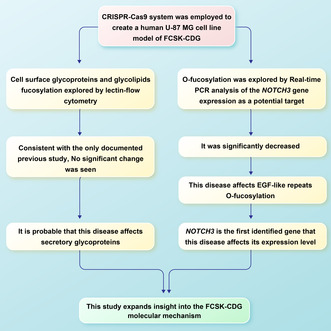
Comparison of cellular fucosylation by lectin flow cytometry in the created CRISPR/Cas9 FCSK knockout and the same unedited cell lines showed no significant change in the amount of cell surface fucosylated glycans, which is consistent with the only documented previous study on different cell types. It suggests a probable effect of this disease on secretory glycoproteins. Investigating O‐fucosylation by analysis of the NOTCH3 gene expression as a potential target revealed a significant decrease in the FCSK knockout cells compared with the same unedited ones, proving the effect of fucokinase deficiency on EGF‐like repeats O‐fucosylation.
Conclusion
This study expands insight into the FCSK‐CDG molecular mechanism; to the best of our knowledge, it is the first research conducted to reveal a gene whose expression level alters due to this disease.
Keywords: congenital disorder of glycosylation, CRISPR/Cas9, FCSK, fucokinase
The current study exploring FCSK‐CDG through a CRISPR‐generated cell model demonstrated the effect of this disease on EGF‐like repeats O‐fucosylation by revealing NOTCH3 as the first identified gene whose expression level alters due to FCSK‐CDG. In addition, it suggested a probable effect of this disease on secretory glycoproteins.
1. INTRODUCTION
Congenital disorders of glycosylation (CDG), initially identified in 1980 (Jaeken et al., 1980) and still hot in 2023, are a large and rapidly growing group of rare inherited metabolic diseases arising from pathogenic variants in the genes associated with glycosylation (Ng & Freeze, 2018).
Glycosylation is a comprehensive enzymatic process that involves the activation of monosaccharides (glucose, galactose, xylose, N‐acetylgalactosamine, N–acetylglucosamin, sialic acid, glucuronic acid, mannose, and fucose) and the subsequent addition of glycans to proteins and lipids through several pathways, including N‐linked, O‐linked, glycosylphosphatidylinositol (GPI), glycosaminoglycan (GAG) and glycolipids (Varki et al., 2022). Due to the intercellular and intracellular critical roles of this process, it is an important health and disease regulator (Reily et al., 2019).
Fucosylation is a common form of glycosylation that is dependent on guanosine diphosphate L‐fucose (GDP‐fucose), its specific transporters, and a group of fucosyltransferase enzymes to incorporate L‐fucose (6‐deoxy‐L‐galactose) into specific proteins and lipids. Two distinct mechanisms generate GDP‐fucose: de novo and salvage pathways. The de novo pathway relies on converting cytoplasmic mannose and glucose to fucose, whereas salvage pathway uses free fucose which is supplied from the environment or recovered from glycoconjugates (Schneider et al., 2017; Figure 1).
FIGURE 1.
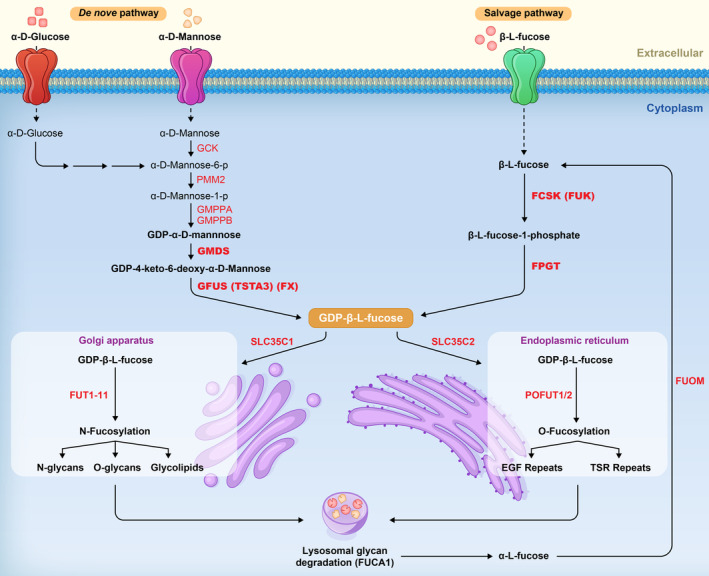
The schematic depiction of fucosylation. GDP‐L‐Fucose synthesis by the de novo and salvage pathways and its utilization in N‐fucosylation and O‐fucosylation.
It has many biologically relevant functions as involvement in ABO and Lewisx blood group systems (de Mattos, 2016; Mondal et al., 2018); leukocyte extravasation (Nimrichter et al., 2008); fertilization (Pang et al., 2011); development, particularly neural development (Fenderson et al., 1990; Ohata et al., 2009); immunological modulation (Marth & Grewal, 2008); cancer metastasis (Blanas et al., 2018; Keeley et al., 2019); inflammation (Li et al., 2014; Thompson et al., 1989); and cognitive processes (Mountford et al., 2015; Tosh et al., 2019).
Impairment in each fucosylation‐related enzyme can lead to CDG with defective fucosylation; although only five types have been described to date, they are named by the official symbol of the impaired gene followed by the suffix “‐CDG”: SLC35C1‐CDG, POFUT1‐CDG, FUT8‐CDG, FCSK‐CDG, and GFUS‐CDG (Hullen et al., 2021).
FCSK‐CDG, congenital disorder of glycosylation with defective fucosylation 2 (CDGF2), MIM# 618324, is an autosomal recessive genetic disease resulting from pathogenic variants in FCSK (MIM# 608675), the gene that encodes fucokinase (EC 2.7.1.52), an essential enzyme in the salvage pathway (Ishihara et al., 1968; Stelzer et al., 2016; see Figure 1). This extremely rare disease was first characterized in 2018 by Ng et al. in two unrelated patients with severe multisystem manifestations such as brain anomalies, intellectual disability, developmental delay, intractable seizures accompanied by epileptic encephalopathy, muscle contractures, hypotonia, walking disability, poor feeding, difficulty in gastric emptying, and ocular disorders (Ng, Rosenfeld, et al., 2018). Subsequently, three other patients with different pathogenic variants in the FCSK gene have been reported, including one by our team (Al Tuwaijri et al., 2023; Manoochehri et al., 2022; Ozgun & Sahin, 2022). Table 1 displays the characteristics of all FCSK‐CDG patients documented thus far (Al Tuwaijri et al., 2023; Manoochehri et al., 2022; Ng, Rosenfeld, et al., 2018; Ozgun & Sahin, 2022).
TABLE 1.
Molecular and clinical characterization of CDGF2 patients (Al Tuwaijri et al., 2023; Manoochehri et al., 2022; Ng, Rosenfeld, et al., 2018; Ozgun & Sahin, 2022).
| ID | Patient 1 | Patient 2 | Patient 3 a | Patient 4 | Patient 5 a | |
|---|---|---|---|---|---|---|
| Age | 6 years | 7 years | 18 months | 4.5 years | 2 years | |
| Age at onset of symptoms | 3 years | 4 years | Infancy | Infancy | Infancy | |
| Sex | Male | Female | Female | Male | Male | |
| Descent | Hispanic | Qatari (Middle Eastern) | Somalian | Iranian (Middle Eastern) | Saudi (Middle Eastern) | |
| FCSK pathogenic variant(s) | c.667T>C, p.Ser223Pro; c.2047 C>T, p.Arg683Cys | c.2980 A>C, p.Lys994Gln | c.993_1011del, p.Glu335Hisfs*55 | c.379 C>A, p.Leu127Met; c.394G>C, p.Asp132His | c.3013G>C, p.Val1005Leu | |
| Mutation type | Compound heterozygous | Homozygous | Homozygous | Homozygous | Homozygous | |
| Consanguinity | No | Yes | No | Yes | Yes | |
| Pregnancy complications | No | Born premature at 2 weeks (10 g) | No | No | No | |
| Intellectual disability | 4/5 | Severe | Severe | Mild | Severe | No |
| Developmental delay | 4/5 | Yes | Yes | Yes | Yes | No |
| Gait problems | 4/5 | Nonambulatory | Nonambulatory | Nonambulatory | Nonambulatory | No |
| Hypotonia | 4/5 | Central hypotonia | Central hypotonia with spasticity of extremities | Central hypotonia | Central hypotonia | No |
| Abnormalities in brain MRI | 4/5 | Corpus callosum dysplasia and delay in myelination of deep white matter | Atrophy of cerebellum, corpus callosum agenesis, abnormalities in white matter presented with severe periventricular leukomalacia and paucity of white matter | Periventricular white matter hyperintensities, enlargement of ventricle with encephalomalastic areas | Hypoplasia of cerebellar and vermis with racing car sign, corpus callosum dysplasia | Not available |
| Seizures/Epilepsy | 4/5 | Generalized tonic–clonic seizures due to multifocal epileptic activity in the bilateral hemispheres | Seizures with hypsarrhythmia in EEG and initially consistent with infantile spasms | No | Unprovoked generalized tonic–clonic seizures | Infantile spasms |
| Speech disorder | 1/5 | No | No | No | Yes | No |
| Ocular disorders | 4/5 | Symmetric maculopathy with severe visual dysfunction | Optic nerve atrophy strabismus, cortical blindness, nystagmus | Cortical blindness | Complete loss of vision | No |
| Dysmorphic face | 0/5 | No | No | No | No | No |
| Feeding problems | 3/5 | Yes | Yes | No | Yes | No |
| Hepatopathy | 1/5 | Elevated gamma‐glutamyl transferase (GGT) | No | No | No | No |
| Contractures | 3/5 | Yes | Yes | No | Yes | No |
| Stature | 0/5 | Normal for age | Normal for age | Normal for age | Normal for age | Normal for age |
| Respiratory problems | 3/5 | Recurrent respiratory infections | Respiratory difficulties | No | Respiratory difficulties | No |
| Recurrent infections | 2/5 | Yes | No | No | Yes | No |
| Reference | Ng, Rosenfeld, et al. (2018) | Ng, Rosenfeld, et al. (2018) | Ozgun and Sahin (2022) | Manoochehri et al. (2022) | Al Tuwaijri et al. (2023) | |
It is notable that these patients' mild clinical condition is probably due to their young age, as the onset of symptoms was at 3 and 4 years in patients 1 and 2, respectively.
FCSK‐CDG patient samples and the disease cell model in Ng et al.'s study, despite the significant decrease in fucokinase, have shown no detectable fucosylation change (Ng, Rosenfeld, et al., 2018). It was thought that the de novo pathway generated the majority of GDP‐fucose (Ng, Rosenfeld, et al., 2018; Yurchenco & Atkinson, 1975, 1977); however, given the vast abnormalities in FCSK‐CDG patients, accepting the minimal or negligible impact on cellular fucosylation due to loss of salvage pathway is challenging. However, recent insight into fucosylation support the importance of the salvage pathway (Lau et al., 2015; Smith et al., 2002; Sosicka et al., 2020, 2022).
In this study, we employed CRISPR/Cas9 system to construct a human U‐87 MG cell line model of FCSK‐CDG disease to survey the cellular fucosylation in the absence of the salvage pathway. The utilized cell line, glioblastoma, was selected considering the putative cell type‐dependent contribution of salvage pathway in fucosylation (Ng, Rosenfeld, et al., 2018; Sosicka et al., 2022) and due to prevalent brain anomalies and neurological manifestations of FCSK‐CDG patients.
2. MATERIALS AND METHODS
2.1. Ethical compliance
This study was approved by the ethics committee of Shahrekord University of Medical Sciences (IR.SKUMS.REC.1400.250).
2.2. Guide RNAs design and cloning
Utilizing four online design tools, CHOP CHOP (http://chopchop.cbu.uib.no), CRISPOR (http://crispor.tefor.net), Cas‐Designer (http://www.rgenome.net/cas‐designer/), and CRISPick (http://portals.broadinstitute.org/gppx/crispick//public), and based on the FCSK gene sequence (Ensembl ID ENSG00000157353.17, 16: 70454595–70480274), we designed two guide RNAs (gRNAs) to target both sides of the gene and delete almost its entire length. The following gRNAs were used in this study:
Guide 1 with PAM: 5’‐TCACACGAACCTATCTCCGGAGG −3′ (Intron 1; forward strand).
Guide 2 with PAM: 5’‐ATCATACGCCGCACAGTCAGGGG‐3′ (Exon 23; reverse strand).
Each gRNA was inserted into the pSpCas9 (BB)‐2A‐GFP (PX458) vector using BbsI restriction enzyme (Thermo Fisher Scientific). The plasmid constructs were transformed into separate DH5α competent Escherichia coli (E. coli) cells and extracted using Plasmid Extraction Maxi Plus Kit (Favorgen) after increase through bacterial culture.
2.3. Validation of the cloned guides
To verify the correct insertion of each gRNA into the PX458 vector, we performed PCR, and the PCR product was evaluated by Sanger sequencing.
The used primers were as follows: Forward, 5’‐TTCTTGGGTAGTTTGCAGTTTTAA‐3′ and Reverse, 5′‐CACGCGCTAAAAACGGACTA‐3′ (Dara et al., 2021).
2.4. Cell culture
U‐87 MG (Uppsala 87 malignant glioma) human cell line was purchased from Pasteur Institute of Iran and cultured in a DMEM‐F12 medium including 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin (Thermo Fisher Scientific), at 37°C with 5% CO2.
2.5. Transfection and fluorescence‐activated cell sorting (FACS)
U‐87 MG cells were seeded in a 6‐well cell culture plate, and 80% confluency was achieved after 24 h; they were co‐transfected by guide‐1 and guide‐2 CRISPR/Cas9 constructs utilizing Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer's instruction. In the subsequent 2–8 h, the cells were examined by fluorescence microscopy.
After 48 h, FACS was employed by FACSAria™III (BD Bioscience) to select the transfected cells due to GFP expression in PX458 constructs; GFP‐positive cells were collected and cultured for post‐transfection analyses.
2.6. Single‐cell isolation, DNA extraction, and PCR‐based analyses
To acquire FCSK homozygous knockout (KO) cells (monoclonal cell population), serial dilution was done on a population of transfected cells to reach 10 cells/mL concentration; a 100 μL aliquot of this cell suspension was added to each well of a 96‐well plate, one cell per well; single cells were incubated and the plate was scanned regularly to transfer the single cells that had been proliferated to a larger culture plate, one by one. 96 well plate → 24 well plate → 6 well plate.
Genomic DNA of each monoclonal cell population was extracted (Blood/Cultured Cell Genomic DNA Extraction Mini Kit, Favorgen) and evaluated by performing multiplex PCR with two sets of primers, set #1 and set #2, amplifying wild‐type and CRISPR‐edited DNA, respectively.
Primer set #1: Forward primer (5′‐GGCTCCTTGGTCAACAGATAGA‐3′, located in exon 23 downstream of Guide 2), Reverse primer (5′‐AAGGTGGAGGTAGAAGAGGTCA‐3′, located in exon 21).
Primer set #2: Forward primer (5′‐GGCTCCTTGGTCAACAGATAGA‐3′, located in exon 23 downstream of Guide 2), Reverse primer (5′‐AAATCACACCCATCCACATTTT‐3′, located in intron 1 upstream of Guide 1).
To confirm the desired deletion, Sanger Sequencing was conducted on the obtained FCSK homozygous deleted cells' genomic DNA PCR product.
2.7. Quantitative real‐time RT‐PCR assay
Total RNA was extracted from FCSK KO and unedited U‐87 Cells using Blood/Cultured cell total RNA Purification Mini Kit (Favorgen) and reverse transcribed into cDNA by PrimeScript™ 1st strand cDNA Synthesis Kit (Takara Bio). cDNA amplification to detect expression levels of FCSK (NM_145059.3), NOTCH3 (NM_000435.3), and ACTB (NM_001101.5) was performed by RT‐PCR using SYBR green and specific primers on a Quantstudio™ 3 system (Thermo Fisher Scientific). The results were normalized to the reference housekeeping gene ACTB and calculated using the 2−∆∆CT method. Each experiment was performed in triplicate. The used primers are listed in Table 2.
TABLE 2.
Primers for real‐time PCR.
| Targeted gene | Primers sequence | Product size (bp) | |
|---|---|---|---|
| ACTB (Ramezani, 2021) | Forward | 5′‐GCCTTTGCCGATCCGC‐3′ | 90 bp |
| Reverse | 5′‐GCCGTAGCCGTTGTCG‐3′ | ||
| FCSK | Forward | 5′‐CCTCGTTGGCCGTCTGGA‐3′ | 93 bp |
| Reverse | 5′‐CGAACACCTGTCCTCTTGAAGAAT‐3′ | ||
| NOTCH3 | Forward | 5′‐GGCATCAACCGCTACGACTG‐3′ | 117 bp |
| Reverse | 5′‐CCCATCCACACAGGAACCTC‐3′ | ||
2.8. Western blot assay
To study FCSK expression at the protein level, we extracted the total protein of the FCSK KO and unedited U‐87 cells, separated by SDS PAGE; subsequently, they were transferred to the PVDF (polyvinylidene difluoride) blotting membrane and blocked with 2% non‐fat milk in TBST (tris‐buffered saline with Tween‐20) buffer for 75 min at room temperature. Then, the membranes were incubated with fucokinase antibody (Fucokinase (F‐9): sc‐377371, 1:300) (Santa Cruz Biotechnology), as the primary antibody, at 4°C for 1 h. The membranes were washed and incubated with mouse anti‐rabbit IgG‐HRP: sc‐2357, 1:1000 (Santa Cruz Biotechnology), horseradish peroxidase‐conjugated secondary antibody, for 75 min at room temperature. Following another washing step, protein bands were visualized using the enhanced chemiluminescence reagent. ß‐actin antibody (ß‐Actin (C4): sc‐47778, 1:300) (Santa Cruz Biotechnology) was used as the internal control.
2.9. Lectin flow cytometry assay
Unedited U‐87 and FCSK KO U‐87 cells were centrifuged; each cell pellet was resuspended in PBS, aliquoted, and incubated with 10 μg/mL AAL‐FITC (Vector Laboratories), or 70 μg/mL UEA I‐FITC (Vector Laboratories) for 30 min in 4°C and protected from light. They were further washed twice, resuspended in PBS, and subjected to flow cytometry. As running a negative control in parallel with each assay, lectins were preabsorbed with 300 Mm L‐fucose (Sigma‐Aldrich), substituted into the procedure in place of the unabsorbed lectin, and incubated under the same conditions. All analyses were done using BD FACSCalibur (BD Biosciences) flow cytometer and BD CellQuest Pro software (BD Biosciences).
2.10. Statistical analysis
The results of each assay, which was performed in triplicate, were analyzed in GraphPad Prism software (version 9; GraphPad Prism) using the student's t‐test and displayed as mean ± standard deviation (SD). p‐value <0.05 was considered statistically significant.
3. RESULTS
3.1. Cloning
After cloning and bacterial culture, E.coli DH5α colonies carrying plasmid constructs appeared in Amp+ LB agar plates due to PX458's Amp resistance marker. Engineered colonies containing each gRNA were detected by observation of the 241 bp PCR product in colony PCR, and Sanger sequencing confirmed the correct insertion of gRNAs into PX458 vectors (Figure 2).
FIGURE 2.
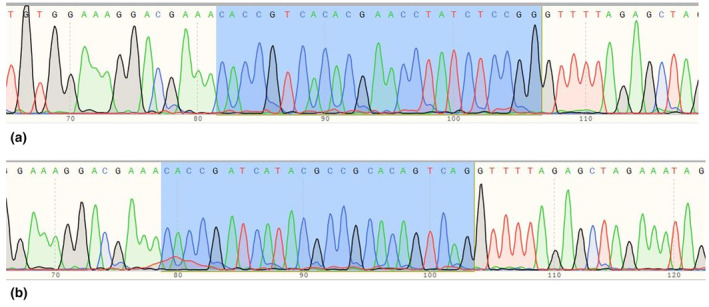
Sanger sequencing results indicating correct guide RNAs cloning into PX458 vectors using BbsI restriction enzyme. Highlighted 25 nucleotides include overhang complementary to the overhang in digested PX458 (5′‐CACC), an extra G nucleotide to promote optimal expression by the hU6 promoter expressing the gRNA in PX458, and the 20 nucleotide gRNA. (a) Electropherogram of inserted Guide 1 in PX458 vector. (b) Electropherogram of inserted Guide 2 in PX458 vector.
3.2. Transfection, single‐cell isolation, and PCR‐based analyses
48 h after transfection, the transfected cells were observed by fluorescence microscope; then, we sorted and collected the cells by FACS. Multiplex PCR on the total genomic DNA extracted from the transfected cells population detected CRISPR‐edited DNA by amplifying the desired 217 bp product in addition to the 1012 bp one, which was the only detected amplified product in the untransfected negative control U‐87 cells. In the case of the FCSK homozygous knockout cells, obtained by means of single‐cell isolation, only the 217 bp product was amplified, because of having no wild‐type DNA (Figure 3). Sanger sequencing confirmed the fine cutting of the designed positions on FCSK gene intron 1 and exon 23 by cas9, 17,341 bp deletion, and joining cut sites (Figure 4).
FIGURE 3.
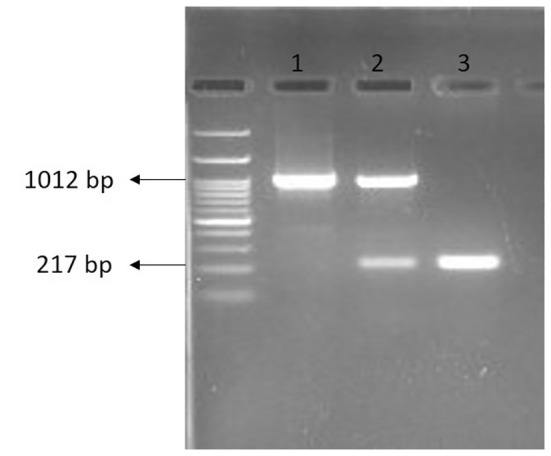
PCR Products of 1012 bp and 217 bp on agarose gel electrophoresis indicating wild‐type and CRISPR‐edited DNA, respectively. 1: Untransfected U‐87 cells, 2: Transfected U‐87 cells population, 3: Homozygous CRISPR‐edited U‐87 cells.
FIGURE 4.
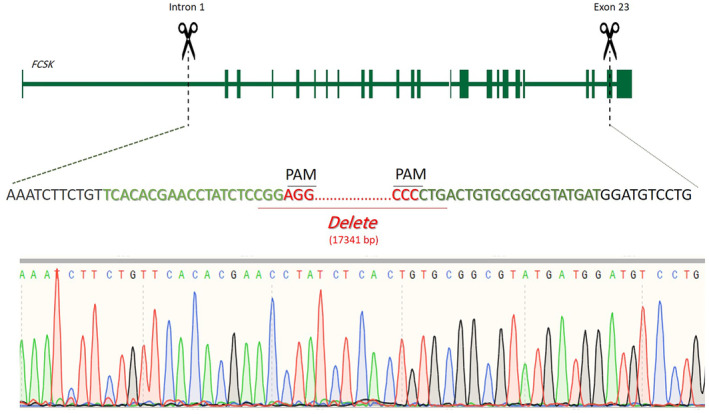
Schematic depiction of the CRISPR/Cas9 targeted sequence and the electropherogram of that region in the CRISPR‐edited U‐87 cells. The double cuts in Intron 1 and Exon 23 resulted in 17,341 bp elimination from the FCSK gene.
3.3. Real‐time RT‐PCR and western blot analyses
RT‐PCR and western blot assays confirmed FCSK KO as no FCSK expression was observed at the mRNA or protein levels in CRISPR‐edited U‐87 cells, whereas it was detected at both levels in unedited U‐87 cells. The result of Western blot is shown in Figure 5a.
FIGURE 5.
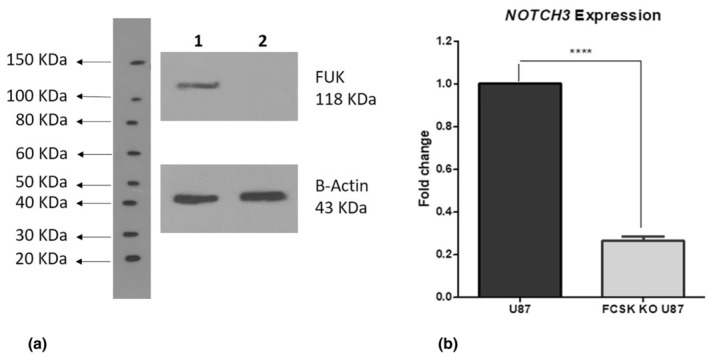
The results of Western blot and Real‐time PCR assays. (a) Western blot analysis confirms FCSK Knockout; lanes 1 and 2 are untransfected U‐87 cells, and homozygous CRISPR‐edited U‐87 cells (FCSK Knockout), respectively. (b) Demonstrates a significant decrease in the NOTCH3 expression in the FCSK KO compared to unedited U‐87 cells based on real‐time PCR analysis. ****: p‐value < 0.0001, Bars and error bars represent mean ± SD.
Real‐time RT‐PCR analysis of NOTCH3 mRNA expression revealed a significant decrease in the FCSK KO U‐87 cells compared with unedited cells (0.26 ± 0.02 fold change; p‐value <0.0001) after normalization to a control (Figure 5b).
3.4. Lectin flow cytometry analyses
The mean of MFIs (mean fluorescence intensity) in the FCSK KO versus unedited U‐87 cells analyzed by lectin flow cytometry were 407.55 ± 55.72 vs.463.58 ± 85.47 and 23.87 ± 3.53 vs. 26.91 ± 1.49 in AAL and UEA I staining, respectively; there was no statistically significant change in the cell surface fucosylation. Figure 6 represents overlay histograms of lectin‐binding in unedited and FCSK KO U‐87 cells.
FIGURE 6.
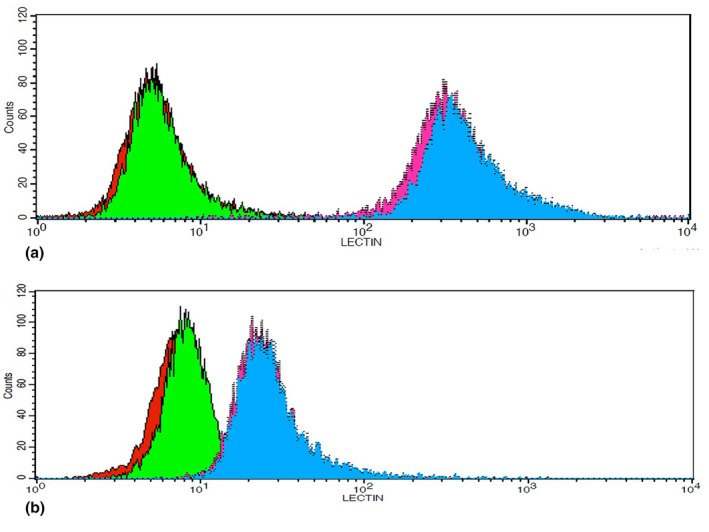
The results of lectin flow cytometry analyses indicate no significant difference between unedited and FCSK KO U‐87 cells in AAL‐binding nor UEA I‐binding. (a) A representative overlay histogram of AAL‐binding in unedited and FCSK KO U‐87 cells. (b) A representative overlay histogram of UEA I‐binding in unedited and FCSK KO U‐87 cells. In both (a and b) Green: Unedited U‐87 cells stained by preabsorbed AAL‐FITC (a) or UEA I‐FITC (b) as a negative control. Red: FCSK KO U‐87 cells stained by preabsorbed AAL‐FITC (a) or UEA I‐FITC (b) as a negative control. Blue: Unedited U‐87 cells stained by AAL‐FITC (a) or UEA I‐FITC (b). Pink: FCSK KO U‐87 cells stained by AAL‐FITC (a) or UEA I‐FITC (B).
4. DISCUSSION
In 2018, Ng et al. described FCSK‐congenital disorder of glycosylation as the first genetic disease identified specifically within the de novo or salvage pathways of fucosylation (Ng, Rosenfeld, et al., 2018); however, FUT8‐CDG, SLC35C1‐CDG, and POFUT1‐CDG, as disorders due to fucosylation related to downstream components of its pathway, had already been identified (Basmanav et al., 2015; Li et al., 2013; Lübke et al., 2001; Luhn et al., 2001; Ng, Xu, et al., 2018).
Despite the identification of FCSK‐CDG (Ng, Rosenfeld, et al., 2018) and GFUS‐CDG (Feichtinger et al., 2021), as well as investigation of the involvement of de novo pathway enzymes GMDS and TSTA3 (GFUS) and salvage pathway enzyme FUK (FCSK) in modifying the invasive and metastatic features of several types of cancers in the last decade (Keeley et al., 2018; Kizuka et al., 2017; Lau et al., 2015; Nakayama et al., 2013; Wei et al., 2018), the specific contribution of each of the two distinct fucosylation pathways has not been well elucidated yet.
According to studies conducted in the mid‐1970s, it was thought that the de novo pathway supplied over 90% of GDP‐fucose and that less than 10% came from salvage (Yurchenco & Atkinson, 1975, 1977). Given that despite their meticulous work, studies were done only in a single cell line and with a single fucose concentration, and rethinking old concepts based on recent studies, a more considerable contribution to fucosylation is suggested for the salvage pathway (Lau et al., 2015; Sosicka et al., 2020). A recent study indicated a comparable contribution of salvage and de novo pathways in the fucosylation of HepG2 and Huh7 cells (Sosicka et al., 2022).
Although Ng et al. have confirmed the significant decrease of fucokinase, FCSK‐encoded protein, in patients and the created CRISPR/Cas9 FCSK knockout HAP1 cell line, they have not seen any detectable fucosylation change in the fibroblast and serum samples of patients, and FCSK knockout cells (Ng, Rosenfeld, et al., 2018). Their two reported cases and the subsequent reported ones, FCSK‐CDG patients, manifested intellectual disability, developmental delay, seizure, epilepsy, gait problems, hypotonia, contractures, abnormal brain MRI, feeding difficulty, and ophthalmological disorders (Al Tuwaijri et al., 2023; Manoochehri et al., 2022; Ng, Rosenfeld, et al., 2018; Ozgun & Sahin, 2022).
Probable cell type‐dependent contribution of salvage and de novo pathways in fucosylation is a notion that can reveal the reason for this observed unchanged fucosylation despite such severe manifestations due to fucokinase defect; in other words, although some cell types may require the salvage pathway critically, others can function well without it (Ng, Rosenfeld, et al., 2018; Sosicka et al., 2022). We hypothesized that brain cells were presumably dependent on the salvage pathway due to important prevalent neurological manifestations of FCSK‐CDG patients, and selected the U‐87 MG cell line to create a new cell model of this disease.
Another notable notion is the distinct preference between salvage and de novo pathways‐originated GDP‐fucose among various fucosyltransferases (Keeley et al., 2018; Lau et al., 2015; Sosicka et al., 2022). N‐glycosylation performs in Golgi apparatus by the use of 11 known fucosyltransferases (FUTs): FUT1,2 are responsible for adding fucose to the terminal galactose of glycans in α1‐2 orientation; FUT3‐7 and FUT9‐11 mediate α1‐3 and α1‐4 fucosylation of terminal GlcNAc in glycans; and FUT8, the only core fucosylation enzyme, adds fucose to the initial GlcNAc residue on N‐glycans in α1‐6 linkages (Becker & Lowe, 2003). Suggesting an inhibitory role on melanoma invasion and metastasis for fucose salvage pathway by inhibition of invadopodia formation, Keeley et al. indicated that this inhibitory effect was mediated through α1‐2 fucosylation, but not the α1‐3 or α1‐4; they propose that although GDP‐fucose is the fucosylation donor for all fucosyltransferases, salvage pathway‐originated GDP‐fucose might be preferentially provided for certain fucosyltransferases (Keeley et al., 2018). According to a recent study, each fucose linkage added by distinct fucosyltransferase, situated in separate Golgi compartments, depends on different GDP‐fucose pools since they have different access to salvage or de novo pathways‐originated GDP‐fucose (Sosicka et al., 2022).
Given the latter notion, investigating fucose in different linkages was our second assumed key to the mystery. Lectins are carbohydrate‐binding proteins whose affinity to particular carbohydrates can be as specific as that of antibody–antigen or enzyme‐substrate. Regarding this potency, they are being widely used to recognize specific carbohydrate complexes in cellular glycoproteins and glycolipids. (Minko, 2004) We utilized two lectins, not only AAL as the most common fucose‐specific lectin that preferentially recognizes α1–6 and α1–3 linked fucose while recognizing all types of N‐linked fucose with variable efficiencies but also UEA I which prefers (α1‐2) linked fucose residues (Sosicka et al., 2022).
Comparing cellular fucosylation by AAL and UEA I lectin flow cytometry in the created CRISPR/Cas9 FCSK knockout and the unedited U‐87 MG cell lines showed no significant change in cell surface glycoproteins and glycolipids. It can be probably assumed that the salvage pathway preferentially provides GDP‐fucose for secretory glycoproteins since our data showed unchanged cell surface fucosylation in another FCSK KO cell line; on the other hand, HepG2 and Huh7, two cell lines that have previously shown comparable contribution of salvage and de novo pathways by GC–MS, are both professional secretory cells. (Sosicka et al., 2022) However, it is notable that lectin flow cytometry showed low amounts of α1‐2 fucosylation on the cell surface of our created model; according to the results and suggestions of previous studies (Keeley et al., 2018; Lau et al., 2015; Sosicka et al., 2022); well‐documented involvement of α1‐2 fucose in neuronal growth and development molecular mechanisms (Kalovidouris et al., 2005; Mountford et al., 2015; Tosh et al., 2019); and prevalent brain anomalies and neurological manifestations of FCSK‐CDG patients, it seems that this type of fucosidic‐linkage is predominantly incorporated in secretory glycoproteins. After all, fucosylation, as a glycosylation type, is too complex to be elucidated clearly; some have termed glycobiology “glycophobia” due to longevity and hesitation of working on it (Ng & Freeze, 2018). However, the contribution of cytoplasmic GDP‐fucose to fucosylation process is determined by its heritage, and the cells possess the ability to distinguish GDP‐fucose of different origins (Sosicka et al., 2022).
Next, we investigated the NOTCH3 gene expression, as a potential target of O‐fucosylation (Schneider et al., 2017). O‐fucosylation, occurring in the endoplasmic reticulum, is carried out by two known GDP‐fucose protein O‐fucosyltransferase (POFUT) enzymes: POFUT1 attaches fucose to Ser/Thr residues in epidermal growth factor (EGF)‐like repeats, while POFUT2 attaches fucose to Ser/Thr residues in ThromboSpondin type 1 Repeats (TSRs) (Schneider et al., 2017). Notch proteins are among more than 100 proteins that have EGF‐like repeats required for POFUT1 fucosylation. In mammals, the Notch signaling pathway has four Notch receptor proteins (Notch1‐Notch4) and five Notch ligands (JAG1, JAG2, DLL1, DLL3, and DLL4) (Stahl et al., 2008). This important conserved signaling pathway influences an amazing range of cell fate decisions and is involved in so many developmental and biological processes that it is impossible to think of a tissue or growing organ that does not rely on it at some point (Sachan et al., 2023). Notch fucosylation, as an important post‐translational modification, is implicated in its receptor‐ligand binding (Rana & Haltiwanger, 2011). Subsequent to ligand‐mediated activation of Notch receptors, they undergo a series of proteolytic cleavages, and the Notch intracellular domain (NICD) translocates to the nucleus. Within the nucleus, NICD constitutes a complex with DNA‐binding protein RBPJ and transcriptional co‐activator mastermind; this transcription activation complex regulates the expression of Notch pathway target genes, including Notch receptors and ligands genes themselves. In the absence of NICD in the nucleus, RBPJ interacts with multiple transcriptional repressors and represses the transcription of target genes (Bocci et al., 2020; Bray, 2016).
Sosicka et al. in a recent study on the Huh7 cell line have indicated that POFUT1 relies more on salvage pathway‐originated GDP‐fucose, compared with POFUT2, which prefers the GDP‐fucose of the de novo pathway (Sosicka et al., 2022). The RT‐PCR assay revealed a significant decrease in NOTCH3 expression level in our FCSK KO U‐87 cells compared with the same unedited cells. This reduced expression level of the NOTCH gene indicates reduced Notch signaling activity due to defects in Notch protein fucosylation; this result not only proves the contribution and importance of the salvage pathway in O‐fucosylation but also is compatible with Sosicka et al.'s findings (Sosicka et al., 2022).
As to clinical phenotypes, the majority of over 30 reported patients with autosomal recessive inherited CDGs with defective fucosylation (SLC35C1‐CDG, FUT8‐CDG, FCSK‐CDG, and GFUS‐CDG) had similar manifestations as developmental delay, intellectual disability, feeding problems, hypotonia, and abnormal brain MRI (for those whose data were available). Short stature and dysmorphic face were the two other common clinical features in all FUT8‐CDG, SLC35C1‐CDG (except one case), and GFUS‐CDG patients, but it is notable that none of the FCSK‐CDG patients presented with this clinical picture; on the other hand, seizure/epilepsy has been reported in almost all of the patients but not the GFUS‐CDG patient (Hullen et al., 2021). These findings are compatible with “distinct GDP‐fucose sources and different fucosylation reactions” as defective fucosylation of salvage and the de novo pathways, respectively due to FCSK and GFUS mutations, may result in different manifestations. However, the number of diagnosed patients is too low to conclude. POFUT1‐CDG, the only CDG with defective fucosylation that has been reported with autosomal dominant inheritance, manifests a completely different clinical presentation with just skin involvement, known as Dowling‐Degos Disease‐2. It is attributed to the haploinsufficiency effect of POFUT1 on the role of the Notch signaling pathway in controlling proliferation and differentiation of melanocytes and keratinocytes (Li et al., 2013).
It is quite certain that CDGs with defective fucosylation are not as rare as it is thought to be. They cannot be detected by the transferrin glycosylation status‐based standard screening method for CDG, due to very low amounts of fucose in transferrin; without the use of newer screening technology based on whole plasma proteins‐derived N‐glycans mass spectrometry and more importantly next‐generation sequencing, many patients passed away without diagnosis. (Hullen et al., 2021; Lefeber et al., 2011) It is notable that four of the five known types of fucosylation defects have been discovered quite recently, (Hullen et al., 2021) and it cannot be ignored that more cases will be identified after describing a disorder. Given FCSK‐CDG specifically, it is noteworthy that three of the five reported patients have Middle Eastern ancestry, (Al Tuwaijri et al., 2023; Manoochehri et al., 2022; Ng, Rosenfeld, et al., 2018; Ozgun & Sahin, 2022) which is well explainable by the recessive inheritance nature of this disease and the high rate of consanguineous marriage in some nations in the Middle East and Western Asia (Piedade et al., 2022). Since these are generally less‐developed countries, a high underdiagnosis of FCSK‐CDG and other fucosylation‐related CDGs with autosomal recessive inheritance is expected.
5. CONCLUSION
The present study utilized a CRISPR‐generated cell model expanding insight into the molecular mechanism of this disorder. Our data indicated the significant effect of salvage pathway on EGF‐like repeats O‐fucosylation by a significant decrease in NOTCH3 gene expression; it also suggests a probable preferential contribution of salvage pathway‐originated GDP‐fucose to secretory glycoproteins. Certainly, further studies are required to be conducted to reveal the underlying mechanisms of the disease.
AUTHOR CONTRIBUTIONS
Maryam Fazelzadeh Haghighi: conceptualization, methodology, investigation, writing—original draft. Hossein Jafari Khamirani: conceptualization, investigation. Jafar Fallahi: methodology. Ali Arabi Monfared: methodology. Korosh Ashrafi Dehkordi: conceptualization, validation, funding acquisition, writing—review and editing. Seyed Mohammad Bagher Tabei: conceptualization, supervision, writing‐ review and editing. All contributing authors approved the final manuscript.
FUNDING INFORMATION
This study was financially supported by Shahrekord University of Medical Sciences (grant number: 3834). The funders have no role in the study design, data collection, analyses, decision to publish, or manuscript writing.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ETHICS STATEMENT
This article does not contain any studies with human participants or animals performed by any of the authors.
ACKNOWLEDGMENTS
The authors would like to thank Shahrekord University of Medical Sciences for the financial support of this study (SKUMS‐3834) and Dr. Nasrin Shokrpour at the Research Consultation Center of Shiraz University of Medical Sciences for her valuable editorial assistance.
Fazelzadeh Haghighi, M. , Jafari Khamirani, H. , Fallahi, J. , Monfared, A. A. , Ashrafi Dehkordi, K. , & Tabei, S. M. B. (2024). Novel insight into FCSK‐congenital disorder of glycosylation through a CRISPR‐generated cell model. Molecular Genetics & Genomic Medicine, 12, e2445. 10.1002/mgg3.2445
Seyed Mohammad Bagher Tabei and Korosh Ashrafi Dehkordi should be considered Joint senior author.
Contributor Information
Korosh Ashrafi Dehkordi, Email: ashrafi.k@skums.ac.ir.
Seyed Mohammad Bagher Tabei, Email: tabeismb@sums.ac.ir.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Al Tuwaijri, A. , Alyafee, Y. , Umair, M. , Alsubait, A. , Alharbi, M. , AlEidi, H. , Ballow, M. , Aldrees, M. , Alam, Q. , Al Abdulrahman, A. , Alrifai, M. T. , & Alfadhel, M. (2023). Congenital disorder of glycosylation with defective fucosylation 2 (FCSK gene defect): The third report in the literature with a mild phenotype. Molecular Genetics & Genomic Medicine, 11(4), e2117. 10.1002/mgg3.2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basmanav, F. B. , Fritz, G. , Lestringant, G. G. , Pachat, D. , Hoffjan, S. , Fischer, J. , Wehner, M. , Wolf, S. , Thiele, H. , Altmüller, J. , Pulimood, S. A. , Rütten, A. , Kruse, R. , Hanneken, S. , Frank, J. , Danda, S. , Bygum, A. , & Betz, R. C. (2015). Pathogenicity of POFUT1 in Dowling‐Degos disease: Additional mutations and clinical overlap with reticulate acropigmentation of kitamura. The Journal of Investigative Dermatology, 135(2), 615–618. 10.1038/jid.2014.406 [DOI] [PubMed] [Google Scholar]
- Becker, D. J. , & Lowe, J. B. (2003). Fucose: Biosynthesis and biological function in mammals. Glycobiology, 13(7), 41R–53R. 10.1093/glycob/cwg054 [DOI] [PubMed] [Google Scholar]
- Blanas, A. , Sahasrabudhe, N. M. , Rodríguez, E. , Van Kooyk, Y. , & Van Vliet, S. J. (2018). Fucosylated antigens in cancer: An alliance toward tumor progression, metastasis, and resistance to chemotherapy. Frontiers in Oncology, 8, 39. 10.3389/fonc.2018.00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocci, F. , Onuchic, J. N. , & Jolly, M. K. (2020). Understanding the principles of pattern formation driven by Notch signaling by integrating experiments and theoretical models. Frontiers in Physiology, 11, 929. 10.3389/fphys.2020.00929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray, S. J. (2016). Notch signalling in context. Nature Reviews. Molecular Cell Biology, 17(11), 722–735. 10.1038/nrm.2016.94 [DOI] [PubMed] [Google Scholar]
- Dara, M. , Razban, V. , Talebzadeh, M. , Moradi, S. , & Dianatpour, M. (2021). Using CRISPR/Cas9 system to knock out exon 48 in DMD gene. Avicenna Journal Of Medical Biotechnology, 13(2), 54–57. 10.18502/ajmb.v13i2.5517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mattos, L. C. (2016). Structural diversity and biological importance of ABO, H, Lewis and secretor histo‐blood group carbohydrates. Revista Brasileira de Hematologia e Hemoterapia, 38(4), 331–340. 10.1016/j.bjhh.2016.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feichtinger, R. G. , Hullen, A. , Koller, A. , Kotzot, D. , Grote, V. , Rapp, E. , Hofbauer, P. , Brugger, K. , Thiel, C. , Mayr, J. A. , & Wortmann, S. B. (2021). A spoonful of L‐fucose‐an efficient therapy for GFUS‐CDG, a new glycosylation disorder. EMBO Molecular Medicine, 13(9), e14332. 10.15252/emmm.202114332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenderson, B. A. , Eddy, E. , & Hakomori, S. I. (1990). Glycoconjugate expression during embryogenesis and its biological significance. BioEssays, 12(4), 173–179. 10.1002/bies.950120406 [DOI] [PubMed] [Google Scholar]
- Hullen, A. , Falkenstein, K. , Weigel, C. , Huidekoper, H. , Naumann‐Bartsch, N. , Spenger, J. , Feichtinger, R. G. , Schaefers, J. , Frenz, S. , Kotlarz, D. , Momen, T. , Khoshnevisan, R. , Riedhammer, K. M. , Santer, R. , Herget, T. , Rennings, A. , Lefeber, D. J. , Mayr, J. A. , Thiel, C. , & Wortmann, S. B. (2021). Congenital disorders of glycosylation with defective fucosylation. Journal of Inherited Metabolic Disease, 44(6), 1441–1452. 10.1002/jimd.12426 [DOI] [PubMed] [Google Scholar]
- Ishihara, H. , Massaro, D. J. , & Heath, E. (1968). The metabolism of l‐fucose: III. The enzymatic synthesis of β‐l‐fucose 1‐phosphate. The Journal of Biological Chemistry, 243(6), 1103–1109. 10.1016/S0021-9258(19)56958-7 [DOI] [PubMed] [Google Scholar]
- Jaeken, J. , Vanderschueren‐Lodeweyckx, M. , Casaer, P. , Snoeck, L. , Corbeel, L. , Eggermont, E. , & Eeckels, R. (1980). Familial psychomotor retardation with markedly fluctuating serum prolactin, FSH and GH levels, partial TBG‐deficiency, increased serum arylsulphatase A and increased CSF protein: A new syndrome?: 90. Pediatric Research, 14(2), 179. 10.1203/00006450-198002000-00117 [DOI] [Google Scholar]
- Kalovidouris, S. A. , Gama, C. I. , Lee, L. W. , & Hsieh‐Wilson, L. C. (2005). A role for fucose alpha(1‐2) galactose carbohydrates in neuronal growth. Journal of the American Chemical Society, 127(5), 1340–1341. 10.1021/ja044631v [DOI] [PubMed] [Google Scholar]
- Keeley, T. , Lin, S. , Lester, D. K. , Lau, E. K. , & Yang, S. (2018). The fucose salvage pathway inhibits invadopodia formation and extracellular matrix degradation in melanoma cells. PLoS One, 13(6), e0199128. 10.1371/journal.pone.0199128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeley, T. S. , Yang, S. , & Lau, E. (2019). The diverse contributions of fucose linkages in cancer. Cancers (Basel), 11(9), 1241. 10.3390/cancers11091241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizuka, Y. , Nakano, M. , Yamaguchi, Y. , Nakajima, K. , Oka, R. , Sato, K. , Ren, C. T. , Hsu, T. L. , Wong, C. H. , & Taniguchi, N. (2017). An alkynyl‐fucose halts hepatoma cell migration and invasion by inhibiting GDP‐Fucose‐synthesizing enzyme FX, TSTA3. Cell Chemistry & Biology, 24(12), 1467–1478.e5. 10.1016/j.chembiol.2017.08.023 [DOI] [PubMed] [Google Scholar]
- Lau, E. , Feng, Y. , Claps, G. , Fukuda, M. N. , Perlina, A. , Donn, D. , Jilaveanu, L. , Kluger, H. , Freeze, H. H. , & Ronai, Z.’. A. (2015). The transcription factor ATF2 promotes melanoma metastasis by suppressing protein fucosylation. Science Signaling, 8(406), ra124. 10.1126/scisignal.aac6479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefeber, D. J. , Morava, E. , & Jaeken, J. (2011). How to find and diagnose a CDG due to defective N‐glycosylation. Journal of Inherited Metabolic Disease, 34(4), 849–852. 10.1007/s10545-011-9370-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Hsu, H. C. , Ding, Y. , Li, H. , Wu, Q. , Yang, P. , Luo, B. , Rowse, A. L. , Spalding, D. M. , Bridges, S. L., Jr., & Mountz, J. D. (2014). Inhibition of fucosylation reshapes inflammatory macrophages and suppresses type II collagen‐induced arthritis. Arthritis & Rhematology, 66(9), 2368–2379. 10.1002/art.38711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M. , Cheng, R. , Liang, J. , Yan, H. , Zhang, H. , Yang, L. , Li, C. , Jiao, Q. , Lu, Z. , He, J. , Ji, J. , Shen, Z. , Li, C. , Hao, F. , Yu, H. , & Yao, Z. (2013). Mutations in POFUT1, encoding protein O‐fucosyltransferase 1, cause generalized Dowling‐Degos disease. American Journal of Human Genetics, 92(6), 895–903. 10.1016/j.ajhg.2013.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lübke, T. , Marquardt, T. , Etzioni, A. , Hartmann, E. , von Figura, K. , & Körner, C. (2001). Complementation cloning identifies CDG‐IIc, a new type of congenital disorders of glycosylation, as a GDP‐fucose transporter deficiency. Nature Genetics, 28(1), 73–76. 10.1038/ng0501-73 [DOI] [PubMed] [Google Scholar]
- Luhn, K. , Wild, M. K. , Eckhardt, M. , Gerardy‐Schahn, R. , & Vestweber, D. (2001). The gene defective in leukocyte adhesion deficiency II encodes a putative GDP‐fucose transporter. Nature Genetics, 28(1), 69–72. 10.1038/ng0501-69 [DOI] [PubMed] [Google Scholar]
- Manoochehri, J. , Kamal, N. , Khamirani, H. J. , Zoghi, S. , Haghighi, M. F. , Goodarzi, H. R. , & Bagher Tabei, S. M. (2022). A combination of two novels homozygous FCSK variants cause disorder of glycosylation with defective fucosylation: New patient and literature review. European Journal of Medical Genetics, 65(8), 104535. 10.1016/j.ejmg.2022.104535 [DOI] [PubMed] [Google Scholar]
- Marth, J. D. , & Grewal, P. K. (2008). Mammalian glycosylation in immunity. Nature Reviews. Immunology, 8(11), 874–887. 10.1038/nri2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minko, T. (2004). Drug targeting to the colon with lectins and neoglycoconjugates. Advanced Drug Delivery Reviews, 56(4), 491–509. 10.1016/j.addr.2003.10.017 [DOI] [PubMed] [Google Scholar]
- Mondal, N. , Dykstra, B. , Lee, J. , Ashline, D. J. , Reinhold, V. N. , Rossi, D. J. , & Sackstein, R. (2018). Distinct human alpha(1,3)‐fucosyltransferases drive Lewis‐X/sialyl Lewis‐X assembly in human cells. The Journal of Biological Chemistry, 293(19), 7300–7314. 10.1074/jbc.RA117.000775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountford, C. , Quadrelli, S. , Lin, A. , & Ramadan, S. (2015). Six fucose‐alpha(1‐2) sugars and alpha‐fucose assigned in the human brain using in vivo two‐dimensional MRS. NMR in Biomedicine, 28(3), 291–296. 10.1002/nbm.3239 [DOI] [PubMed] [Google Scholar]
- Nakayama, K. , Moriwaki, K. , Imai, T. , Shinzaki, S. , Kamada, Y. , Murata, K. , & Miyoshi, E. (2013). Mutation of GDP‐mannose‐4,6‐dehydratase in colorectal cancer metastasis. PLoS One, 8(7), e70298. 10.1371/journal.pone.0070298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, B. G. , & Freeze, H. H. (2018). Perspectives on glycosylation and its congenital disorders. Trends in Genetics, 34(6), 466–476. 10.1016/j.tig.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, B. G. , Rosenfeld, J. A. , Emrick, L. , Jain, M. , Burrage, L. C. , Lee, B. , Undiagnosed Diseases Network , Craigen, W. J. , Bearden, D. R. , Graham, B. H. , & Freeze, H. H. (2018). Pathogenic variants in fucokinase cause a congenital disorder of glycosylation. American Journal of Human Genetics, 103(6), 1030–1037. 10.1016/j.ajhg.2018.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, B. G. , Xu, G. , Chandy, N. , Steyermark, J. , Shinde, D. N. , Radtke, K. , Raymond, K. , Lebrilla, C. B. , AlAsmari, A. , Suchy, S. F. , Powis, Z. , Faqeih, E. A. , Berry, S. A. , Kronn, D. F. , & Freeze, H. H. (2018). Biallelic mutations in FUT8 cause a congenital disorder of glycosylation with defective fucosylation. American Journal of Human Genetics, 102(1), 188–195. 10.1016/j.ajhg.2017.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimrichter, L. , Burdick, M. M. , Aoki, K. , Laroy, W. , Fierro, M. A. , Hudson, S. A. , von Seggern, C. E. , Cotter, R. J. , Bochner, B. S. , Tiemeyer, M. , Konstantopoulos, K. , & Schnaar, R. L. (2008). E‐selectin receptors on human leukocytes. Blood, 112(9), 3744–3752. 10.1182/blood-2008-04-149641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohata, S. , Kinoshita, S. , Aoki, R. , Tanaka, H. , Wada, H. , Tsuruoka‐Kinoshita, S. , Tsuboi, T. , Watabe, S. , & Okamoto, H. (2009). Neuroepithelial cells require fucosylated glycans to guide the migration of vagus motor neuron progenitors in the developing zebrafish hindbrain. Development, 136(10), 1653–1663. 10.1242/dev.033290 [DOI] [PubMed] [Google Scholar]
- Ozgun, N. , & Sahin, Y. (2022). A case with congenital disorder of glycosylation with defective fucosylation 2 and new mutation in FUK gene. Brain & Development, 44(3), 239–243. 10.1016/j.braindev.2021.11.001 [DOI] [PubMed] [Google Scholar]
- Pang, P.‐C. , Chiu, P. C. , Lee, C.‐L. , Chang, L.‐Y. , Panico, M. , Morris, H. R. , Haslam, S. M. , Khoo, K. H. , Clark, G. F. , Yeung, W. S. , & Dell, A. (2011). Human sperm binding is mediated by the sialyl‐Lewisx oligosaccharide on the zona pellucida. Science, 333(6050), 1761–1764. 10.1126/science.1207438 [DOI] [PubMed] [Google Scholar]
- Piedade, A. , Francisco, R. , Jaeken, J. , Sarkhail, P. , Brasil, S. , Ferreira, C. R. , Rijoff, T. , Pascoal, C. , Gil, A. , Lourenço, A. B. , Abreu, M. , Gomes, M. , Videira, P. A. , & dos Reis Ferreira, V. (2022). Epidemiology of congenital disorders of glycosylation (CDG)—Overview and perspectives. Journal of Rare Diseases, 1(1), 3. 10.1007/s44162-022-00003-6 [DOI] [Google Scholar]
- Ramezani, A. (2021). CtNorm: Real time PCR cycle of threshold (Ct) normalization algorithm. Journal of Microbiological Methods, 187, 106267. 10.1016/j.mimet.2021.106267 [DOI] [PubMed] [Google Scholar]
- Rana, N. A. , & Haltiwanger, R. S. (2011). Fringe benefits: Functional and structural impacts of O‐glycosylation on the extracellular domain of Notch receptors. Current Opinion in Structural Biology, 21(5), 583–589. 10.1016/j.sbi.2011.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reily, C. , Stewart, T. J. , Renfrow, M. B. , & Novak, J. (2019). Glycosylation in health and disease. Nature Reviews. Nephrology, 15(6), 346–366. 10.1038/s41581-019-0129-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachan, N. , Sharma, V. , Mutsuddi, M. , & Mukherjee, A. (2023). Notch signalling: Multifaceted role in development and disease. The FEBS Journal. 10.1111/febs.16815 [DOI] [PubMed] [Google Scholar]
- Schneider, M. , Al‐Shareffi, E. , & Haltiwanger, R. S. (2017). Biological functions of fucose in mammals. Glycobiology, 27(7), 601–618. 10.1093/glycob/cwx034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, P. L. , Myers, J. T. , Rogers, C. E. , Zhou, L. , Petryniak, B. , Becker, D. J. , Homeister, J. W. , & Lowe, J. B. (2002). Conditional control of selectin ligand expression and global fucosylation events in mice with a targeted mutation at the FX locus. The Journal of Cell Biology, 158(4), 801–815. 10.1083/jcb.200203125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosicka, P. , Ng, B. G. , & Freeze, H. H. (2020). Therapeutic monosaccharides: Looking back, moving forward. Biochemistry, 59(34), 3064–3077. 10.1021/acs.biochem.9b00565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosicka, P. , Ng, B. G. , Pepi, L. E. , Shajahan, A. , Wong, M. , Scott, D. A. , Matsumoto, K. , Xia, Z. J. , Lebrilla, C. B. , Haltiwanger, R. S. , Azadi, P. , & Freeze, H. H. (2022). Origin of cytoplasmic GDP‐fucose determines its contribution to glycosylation reactions. The Journal of Cell Biology, 221(10), e202205038. 10.1083/jcb.202205038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl, M. , Uemura, K. , Ge, C. , Shi, S. , Tashima, Y. , & Stanley, P. (2008). Roles of Pofut1 and O‐fucose in mammalian Notch signaling. The Journal of Biological Chemistry, 283(20), 13638–13651. 10.1074/jbc.M802027200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer, G. , Rosen, N. , Plaschkes, I. , Zimmerman, S. , Twik, M. , Fishilevich, S. , Stein, T. I. , Nudel, R. , Lieder, I. , Mazor, Y. , Kaplan, S. , Dahary, D. , Warshawsky, D. , Guan‐Golan, Y. , Kohn, A. , Rappaport, N. , Safran, M. , & Lancet, D. (2016). The GeneCards suite: From gene data mining to disease genome sequence analyses. Current Protocols in Bioinformatics, 54(1), 1.30‐1–1.30‐33. 10.1002/cpbi.5 [DOI] [PubMed] [Google Scholar]
- Thompson, S. , Kelly, C. A. , Griffiths, I. D. , & Turner, G. A. (1989). Abnormally‐fucosylated serum haptoglobins in patients with inflammatory joint disease. Clinica Chimica Acta, 184(3), 251–258. 10.1016/0009-8981(89)90058-2 [DOI] [PubMed] [Google Scholar]
- Tosh, N. , Quadrelli, S. , Galloway, G. , & Mountford, C. (2019). Two new fucose‐alpha (1‐2)‐glycans assigned in the healthy human brain taking the number to seven. Scientific Reports, 9(1), 18806. 10.1038/s41598-019-54933-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki, A. , Cummings, R. D. , Esko, J. D. , Stanley, P. , Hart, G. W. , Aebi, M. , Darvill, A. G. , Kinoshita, T. , Packer, N. H. , Prestegard, J. H. , Schnaar, R. L. , & Seeberger, P. H. (2022). Essentials of Glycobiology (4th ed.). Cold Spring Harbor Laboratory Press. 10.1101/9781621824213 [DOI] [PubMed] [Google Scholar]
- Wei, X. , Zhang, K. , Qin, H. , Zhu, J. , Qin, Q. , Yu, Y. , & Wang, H. (2018). GMDS knockdown impairs cell proliferation and survival in human lung adenocarcinoma. BMC Cancer, 18(1), 600. 10.1186/s12885-018-4524-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yurchenco, P. D. , & Atkinson, P. H. (1975). Fucosyl‐glycoprotein and precursor pools in HeLa cells. Biochemistry, 14(14), 3107–3114. 10.1021/bi00685a011 [DOI] [PubMed] [Google Scholar]
- Yurchenco, P. D. , & Atkinson, P. H. (1977). Equilibration of fucosyl glycoprotein pools in HeLa cells. Biochemistry, 16(5), 944–953. 10.1021/bi00624a021 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.